log(K/K o )
advertisement
")
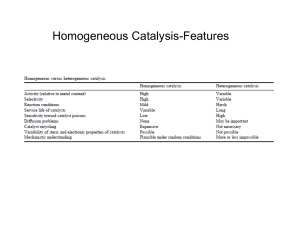
Hammett plots – the most common linear free energy relationship. It is important to know effects of substituents on chemical properties. In particular, this contributes to understanding reaction mechanisms and to predicting rate constants and equilibrium constants. Hammett equation relates rates and equilibria for many reactions of compounds containing substituted phenyl groups. There is a relationship between the acid strengths of substituted benzoic acids and the rates of many other chemical reactions, for instance, the rates of hydrolysis of substituted ethyl benzoates. CO2Et CO2H + EtOH X X ko: rate constant for hydrolysis of ethyl benzoate k: rate constants for hydrolysis of substituted esters Ko: acid dissociation constant of benzoic acid (= Keq for X = H) K: acid dissociation constant of substituted acids (= Keq for X = substituents) log(k/ko) = mlog(K/Ko) DDG‡ = mDDGo Linear free-energy relationship DGo = -RTlogKeq k= kkBT h e + + -DG /RT Hammett linear free-energy relationship log(k/ko) = sr log(K/Ko) = sr s : substituent constant r : reaction constant (should be measured for given reactions) -> sensitivity of a particular reaction to substituent effects measurement of s values r = 1 : reference reaction, the ionization of benzoic acids CO2H CO2+ X H+ log(K/Ko) = sx X s can be determined for specific substituents (measured, known) s values: negative, the substituted benzoic acid is less acidic than benzoic acid itself -> electron donating groups have negative s values positive, the substituted benzoic acid is more acidic than benzoic acid itself -> electron withdrawing groups have positive s values inductive effect inductive and resonance effects s+ : used for reactions in which there is direct resonance interaction between an electron donor substituent and a cationic reaction center s- : used for reactions in which there is direct resonance interaction between an electron donor substituent and the electron-rich reaction site s+ O OMe OMe O- O N sO -O O- Summary using Hammett equation for a particular reaction 1. Measure kx or Kx for substituents 2. Plot log(kx/ko) vs s 3. Determine r and interpret the results log(k/ko) = sxr log(K/Ko) = sxr log(k/ko) Slope = r s 1. When r > 1, the reaction under study is more sensitive to substituents than benzoic acid, and negative charge is building during the reaction. -> an electron withdrawing group stabilizes the developed negative charge. 2. When 0 < r < 1, the reaction is less sensitive to substituents than benzoic acid, but negative charge is still building. 3. When r is equal to or close to 0, the reaction shows no substituent effects. 4. When r is negative, the reaction is creating positive charge. -> electron donating groups increases in rates. 5. lrl large: much positive or negative charge separation Predicting rate constants using Hammett equation CO2- CO2Me k = 2 x 10-4/M s log(km/kH) = sr = 0.70 x 2.38 = 1.69 km/kH = 101.69 = 49 CO2- CO2Me k=? km = 49 x kH = 98 x 10-4/M s NO2 NO2 Cl OH Which reaction is faster? log(kp-Br/kH) = sr = -1.31 x 0.26 Br Br Cl OH log(kp-NO2/kH) = sr = -1.31 x 0.81 NO2 NO2 Cl OH r = -1.31 X sm (NO2) = 0.70 X kp-Br/kp-NO2 = 5.25 SN2; negative r The power of Hammett plots for deciphering mechanisms r = 2.23 not distinguished by r values -> other technique is required to distinguish r = -5.09 -> SN1-like mechanism small r =values due to little or no change in charge during reaction (SN2) SN1: large negative r+ carbocation is developed SN2 치환기에 따라 SN1에서 SN2로 바뀜 Miscellaneous experiments for studying mechanisms Kinetic analysis, isotope effects and structure-function analysis <- reaction mechanism Product identification It is important to identify all the products of a reaction. Sometimes, identification of a minor product will provide a valuable clue as to the kind(s) of mechanistic pathways. OH SN2 O2N expected product Changing the reactant structure to divert or trap a proposed intermediate Mandelate racemase : a carbanion alpha to the carboxylate was proposed as the intermediate formed. To test for the presence of this intermediate, the following substrate was designed. CO2H OH mandelic acid Trapping and competition experiments A common method for intermediate identification is trapping of the intermediate with an added reagent. never seen at room temperature Phosphoranes: are proposed intermediates in the hydrolysis of RNA and DNA trapping this intermediate Checking for a common intermediate possible intermediate Similar ratio of products However, quite different ratio Stereochemical anlaysis SN2 SN1 Double inversion -> retention of stereochemistry Isotope scrambling O OH O H Claisen rearrangement Allyl fragment may be formed during reaction 50 : 50 Techniques to study radicals: clocks and traps Radical clocks are one experimental technique that has received considerable use in the analysis of radical reactions. Most radical clocks involve an intramolecular free radical rearrangement that proceeds with a well-defined rate constant. photolosis ring opening rate constant ~ 108/s -> life time: 1 to 4 ns Three products are formed O CH3 H O. O hv O . O O CH3 OH . O . O CH3 H The use of a spin strap: the addition of a free radical to a nitroso or nitrone group creates a relatively stable spin adduct that can be detected by EPR (electron paramagnetic resonance) spectroscopy. a relatively stable spin adduct that can be detected by EPR CH3CH2. N (I = 1) -> 2H (I = 1/2) -> 3H JCH3 -> JCH2 + N O. N CH2CH3 O 1 : 1 1:2:1 : 1:2:1 1:3:3:1 : 1:3:3:1 : 1 : 1:2:1 : 1:3:3:1 JN Catalysis General principles of catalysis Turnover number: the average number of reactants that a catalyst acts on before the catalyst loses its activity. The catalyst increases rate of reaction (decreases activation energy). The thermodynamics of a catalyzed reaction are unaffected by the catalyst. A heterogeneous catalyst: one that does not dissolve in the solution. eg) Pd/C A homogeneous catalyst: one that dissolves in the solution. All calaysts operate by the same general principle – that is, the activation energy of the rds must be lowered in order for a rate enhancement to occur. Binding the transition state better than the ground state Substrate: any material (reactant) used in a catalytic reaction Activated complex: simply referred to as the transition state Rate enhancement: the ratio of the rate constant for the catalyzed reaction to that for the uncatalyzed one (kcat/kuncat). To achieve catalysis, the catalyst must stabilize the transition state more than it stabilizes the ground state. That is, the transition state must be bound better than the ground state. A spatial temporal approach To achieve catalysis, the catalyst must stabilize the transition state more than it stabilizes the ground state. That is, the transition state must be bound better than the ground state. Another explanation for catalysis: spatial temporal postulate, many intramolecular reactions are often much faster than corresponding intermolecular reactions. -> the rate of reaction between functionalities A and B is proportional to the time that A and B reside within a critical distance. The longer that A and B spend together in the correct geometry for reaction, the greater that probability. When bound to the catalyst, the distance between A and B is closer than when they are free in solution, and inherently the transition state brings A and B close because bonds are beginning to form. In summary, binding is the key element in the most widely accepted theory of how catalysis is achieved. Greater binding of the transition state relative to the ground state is all that needs to be invoked to give a rate enhancement. Forms of catalysis Proximity as binding phenomenon Intermolecular aminolysis (k1; 1/M·s) 1.3 x 10-4 /M·s Intramolecular cyclization (k2; 1/s) 0.17/s -> a rate enhancement of 1200 Effective molarity (E.M.) or intramolecularity; ratio of the first order to second order rate constants for the analogous reactions. -> tell us the effective concentration of one of the components in the intramolecular reaction Entropies of translation and rotation Gem-dimethyl effect: sterically compress two groups together and preorganize two reactants in proximity close decrease of angle R R An angle for R = H is greater than an angle for R = alkyl. E.M. = 1011-1012 Tetrahedral intermediate -> infinitely stable not isolated Twisted amide -> very reactive Electrophilic catalysis Electrophilic catalysis includes simple electrostatics, hydrogen bonding, acid catalysis, and electrophilic metal coordination. Electrostatic interactions Oxyanion hole 150-fold rate enhancement Cation-p interactions Metal ion catalysis 2 x 1016-fold rate enhancement pKa of metal-bound water = 7.2 (108 more acidic than water itself) Acid-base catalysis; see previous section Nucleophilic catalysis Nucleophilic catalysis arise when a nucleophile binds to a reactant and enhances its rate of reaction. -> less common than electrostatic catalysis NMe2 NMe2 N H DMAP; N,N'-dimethylaminopyridine N better electrophile and more reactive than the starting acid halide or anhydride Covalent catalysis Iminium ion enamine N H H N HH O Iminium ion 아래로 공격 Strain and distortion When a substrate binds to a catalyst that is more complementary in structure or electronic characeristics to the transition state, the substrate may distort in order to optimize binding interactions. That is, because the catalyst is designed to optimally bind the transition state, it necessarily is not optimal for the most stable structure of the ground state. -> distortion as a strain on the substrate -> the strain raises the energy of the substrate -> diminish activation energy The chair is distorted into a conformation resembling a half-chair Brønsted acid-base catalysis specific catalysis The specific acid is defined as the protonated form of the solvent in which the reaction is being performed. eg) H3O+, CH3CNH+, CH3SO(H+)CH3 The specific base is defined as the conjugate base of the solvent. eg) HO-, -CH2CN, CH3SOCH2The specific acid catalysis refers to a process in which the reaction rate depends upon the specific acid, not upon other acids in the solution. The specific base catalysis refers to a process in which the reaction rate depends upon the specific base, not upon other bases in the solution. kobs = k[H3O+]/KaRH+ Specific acid catalyzed reactions added acid (AcOH in H2O) A-는 반응에 관여하지 않음. 따라서 rate law에 포함되지 않음 KaHA = [A-][H3O+]/[HA] KaRH+ = [R][H3O+]/[RH+] 앞과 동일 [HA] 항 없음 If the acid catalysis is involved in an equilibrium prior to the rds, and it is not involved in rds, then the kinetics of the reaction will depend solely upon the concentration of the specific acid. This is true even if an added acid (AcOH) is involved in protonating the reactant. Why? When a prior equilibrium is established, [RH+] determines the rate of the reaction. The concentration of RH+ depends solely upon the pH and the pKa of RH+, and does not depend upon the concentration of the acid HA that was added to solution. Specific base catalyzed reactions BH+는 반응에 관여하지 않음. 따라서 rate law에 포함되지 않음 KaRH =[R-][H3O+]/[RH] kobs = kKaRH /[H3O+] [B] 항 없음 23 page에서 다시 설명 Kinetic plots The hallmark of specific acid or specific base catalysis is that the rate depends on the pH and not on the concentration of various acids or bases. This always means that an equilibrium involving the acid or base occur prior to rds, and the acid or base is not involved in rds itself. specific acid catalysis specific base catalysis kobs = k[H3O+]/KaRH+ kobs = kKaRH /[H3O+] logkobs = logk –pH-logKaRH+ logkobs = logk +pH+logKaRH+ added acid added base General catalysis In case that the proton transfer is involved in rds, not in a prior equilibrium -> general catalysis When an acid is involved in rds, -> general acid catalysis When a base is involved in rds, -> general base catalysis The term ‘general’ refers to the fact that any acid or base we added to the solution will affect the rate of the reaction. The term ‘specific’ refers to the fact that just one acid or base, from the solvent, affects the rate of the reaction. General acid catalyzed reactions HA는 반응에 관여함. 따라서 rate law에 포함 Ka = [A-][H3O+]/[HA] kobs = k[HA] or k[H3O+][A-]/Ka - Since the acid is always regenerated after the reaction, its concentration never changes over the course of the reaction -> pseudo-first order - the concentration of either HA or A- is in the rate expression. General base catalyzed reactions B는 반응에 관여함. 따라서 rate law에 포함 kobs = k[B] or k[HO-][HB+]/Kb - Since the base is always regenerated after the reaction, its concentration never changes over the course of the reaction -> pseudo-first order - the concentration of either B or BH+ is in the rate expression. 19 page fast slow 실제로는 첫번째 단계가 rds 따라서 HO-는 general base로 작용 Note: ‘specific’ is used to designate the protonated or deprotonated form of the solvent (H3O+ or HO- for water), but it is also used to designate a mechanism involving an acid or base in an equilibrium prior to rds. However, sometimes hydronium or hydroxide can be involved in rds. In this case the specific acid and base are participating in general-catalysis. Kinetic plots Since the rate for general-acid or general-base catalysis always depends on [HA] or [B] added to the solution, and is not soley dictated by the pH, the experimental observations are quite dufferent from specific-acid-specific-base catalysis. Since [HA] or [B] has been incorporated into kobs, this value is lineraly related to [HA] or [B]. However, the pH depence is more difficult to understand. kobs = k[HA] or k[H3O+][A-]/Ka kobs = k[B] or k[HO-][HB+]/Kb 페이지 20과 비교 general acid catalysis general base catalysis pH < pKa, HA로 주로 존재, 따라서 k의 변화 없음 pH ~ pKa, HA와 A- (이것은 반응에 관여하지 않음)이 공존 pH > pKa, HA가 점차적으로 A-로 바뀌어 k가 지속적으로 감소 C와 mirror image general acid catalysis general base catalysis Concerted or sequential general-acid-general-base catalysis Both a general acid and a general base catalyst are required for a reaction. This is often the case with enzymes. acid base kobs = k[HA][B] The rate dependence on pH is a combination of that observed for general-acid The largest kobs is found at pH where and general-base catalyst. the product of the concentrations of HA and B is at a maximum Enzymatic catalysis physical process chemical process Michaelis-Menten kinetics k1 kcat E + S ← → E·S → P + E k-1 E + S → ← E·S: Michaelis complex → P E·S rate = d[P]/dt = kcat[ES] d[ES]/dt = k1[E][S] – k-1[ES] - kcat[ES] = 0 [E]0 = [E] + [ES] [E] = [E]0 - [ES] k1([E]0-[ES])[S] – k-1[ES] - kcat[ES] = 0 [ES] = [E]0[S] Km + [S] Km = (k-1 + kcat)/k1 rate = d[P]/dt = kcat[ES] = kcat[E]0[S] Michaelis-Menten equation Km + [S] Kinetic parameters to be determined for enzymatic reactions: kcat and Km The meaning of Km, kcat and kcat/Km 1. kcat: catalytic constant turnover number: the maximum number of substrate molecules converted to products per active site per unit time or the rate constant for the conversion of the substrate to the product within the active site of the catalyst (unit; 1/s) -> proximity, acid-base catalysis, electrostatic consideration, covalent catalysis, and the relief of strain will influence kcat. 2. Km : apparent dissociation constant that may be treated as the overall dissociation constants of all enzyme-bound species. k-1 >> kcat -> Km = k-1/k1 the dissociation constant for ES complex. Under these circumstances, Km can provide insights into how good a receptor the catalyst is. The smaller Km means a better receptor. -> Km reflects a physical process (binding) rather than a chemical transformation. k-1 << kcat -> Km = kcat/k1 (a very good catalyst) does not resemble the dissociation constant for ES complex 3. kcat/Km (specific constant) Km >> [S], kcat/Km ; apparent second order rate constant -> this is related to how well the catalyst binds the substrate and how well the catalyst turns over the substrate to product. -> Information on both binding and catalysis Enzyme active sites Ricin A is a potent cytotoxin from 아주까리 Proposed mechanism general acid attacks ribosomes, hydrolyzing the N-glycoside linkage of specific adenosine nucleotides in oligonucleotides general base -> enhances nucleophilicity of water 6-OH 3-OH Rate enhancement: 104-105 Vmax: 2.24 x 10-5 M/s Km: 4.69 x 10-5 M