powerpoint - University of Illinois at Urbana
advertisement

Lecture 35
Vibrational spectroscopy
(c) So Hirata, Department of Chemistry, University of Illinois at Urbana-Champaign. This material has
been developed and made available online by work supported jointly by University of Illinois, the
National Science Foundation under Grant CHE-1118616 (CAREER), and the Camille & Henry Dreyfus
Foundation, Inc. through the Camille Dreyfus Teacher-Scholar program. Any opinions, findings, and
conclusions or recommendations expressed in this material are those of the author(s) and do not
necessarily reflect the views of the sponsoring agencies.
Vibrational spectroscopy
Transition energies between vibrational states
fall in the range of IR photons. IR absorption
spectroscopy can determine vibrational energy
levels and thus molecular structures and
dynamics.
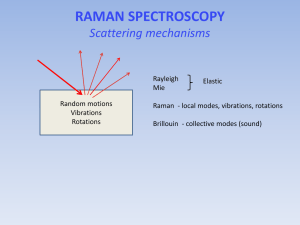
Raman spectroscopy can also be used to study
molecular vibrations.
We will learn the theories of diatomic and
polyatomic molecular vibrations in the harmonic
approximation.
We will discuss the effect of anharmonicity.
Diatomic molecules
in harmonic approximation
Anharmonicity
æ ¶V ö
1 æ ¶2V ö 2 1 æ ¶3V ö 3
V ( x ) = V ( 0) + ç ÷ x + ç 2 ÷ x + ç 3 ÷ x +…
2! è ¶x ø 0
3! è ¶x ø 0
è ¶x ø 0
=0
=k
ì 2 ¶2 1 2 ü
1
+
kx
Y
=
v
+
wY v
(
íý
2)
v
2
ïî 2m ¶x 2
ïþ
w=
k
m
=
k
( )
m1m2
m1 +m2
Selection rules: IR absorption
Energy separations between vibrational
states are in infrared range.
Transition dipole
òe
f
v f xˆ e i vi dt e dt v = ò v f
dipole moment
( ò e xˆe dt )v dt
i
i
e
i
v
mx
= m x ò v f vi dt v = 0 (orthogonality)
Zero
IR absorption does not occur in nature!?
(Global warming solved by orthogonality!?)
Selection rules: IR absorption
Fallacy is the constancy of the dipole moment
during vibration.
Transition dipole
òe
f
dipole moment
v f xˆ e i vi dt e dt v = ò v f
( ò e xˆe dt )v dt
i
i
e
i
v
mx
ìï
üï
æ ¶mx ö
ˆ +… ý v dt
= ò v f í mx
+ç
R
i
v
÷ø
R=0
¶R
è
ïî
ïþ
R=0
Zero
æ ¶mx ö
ˆ dt +…
= m x ò v f vi dt v + ç
v
Rv
è ¶R ÷ø ò f i v
( )
( )
0
Gross selection rule: dipole varies with vibrations
Selection rules: IR absorption
Which molecules have infrared absorption?
N2
NO (zero dipole; zero dipole derivatives)
O2
NO (zero dipole; zero dipole derivatives)
CO2
YES (zero dipole; nonzero dipole derivatives)
H 2O
YES (nonzero dipole; nonzero derivatives)
Selection rules: IR absorption
yH vi vH vi 1
¥
- y2
ˆ
ò v f Rvi dt v µ ò Hv yHv e dy
-¥
f
¥
(
i
)
µ ò H v vi H v -1 + H v +1 e
-¥
f
i
1
2
v f vi 1
Specific selection rule
i
- y2
dy
1
2
H vi 1
Selection rules: Raman scattering
Transition polarizability
e
ò
å
f
v f xˆ e m d t e d t v ò e m yˆ e i vi d t e d t v
E0 - Em ± hn
m
polarizability
æ
e i xˆ e m d t e ò e m yˆ e i dt e ö
ò
= ò vf çå
÷ vi dt v
÷ø
çè m
E0 - Em ± hn
= ò vf
= a xy
a xy
( ) Rˆ +… } v dt
ˆ dt +…
ò v v dt + ( ) ò v Rv
{(a )
xy R=0
+
¶a xy
¶R
i
R=0
¶a xy
i
f
v
¶x
f
i
v
Gross selection rule:
polarizability varies with vibration
0
v
v f vi 1
Specific selection rule
Anharmonicity
Fundamental: v = 1 0
Hot band: v = 2 1; v = 3 2, etc.
Overtone:
v = 2 0; v = 3 0, etc.
Polyatomic molecules
in harmonic approximation
Linear molecules: 3N – 5 modes.
Nonlinear molecules: 3N – 6 modes.
The Schrödinger equation for polyatomic
vibrations (i.e., once assumed to be
separable from rotations) can be solved
exactly in the harmonic approximation.
The wave function becomes the product of
harmonic oscillator wave functions along
normal modes. The energy is the sum of
harmonic oscillators’ energies.
Normal modes
A normal mode is classical motion of nuclei
with well-defined frequency, a set of nuclear
coordinates representable by arrows in the
case of CO2:
The 3N – 6 dimensional classical vibration of
masses connected by harmonic springs can
be decomposed into 3N – 6 separate onedimensional classical harmonic oscillators,
each of which in a normal coordinate.
Classical versus quantum
harmonic oscillators
Classical – Newton
Quantum – Schrödinger
d 2x
-kx = m 2
dt
ì 2 ¶2 1 2 ü
+ kx ý Y v = Ev Y v
í2
ïî 2m ¶x 2
ïþ
Classical – Hamilton
p2 1 2
+ kx = E
2m 2
Y v (x) = N v H v ( y)e
- y 2 /2
æ
1ö
k
Ev = ç v + ÷ w , w =
, v = 0,1,2,…
2ø
m
è
Normal mode analysis
Consider just the in-line motion of CO2:
1
kx
1
2
2
kx
2
2
O1
C
O2
x
We have
All three
coordinates
are coupled
(
(
)
-k xO - xC = mO
) (
1
d 2 xO
1
)
dt
2
-k xC - xO - k xC - xO = mC
(
1
)
2
-k xO - xC = mO
2
d 2 xO
dt
2
2
d 2 xC
dt 2
Normal mode analysis
(
(
)
-k xO - xC = mO
) (
1
d 2 xO
1
)
dt
2
-k xC - xO - k xC - xO = mC
(
1
)
2
-k xO - xC = mO
2
In matrix form:
æ x
æ -k
k
0 ö ç O1
ç
֍ x
k
-2k
k
ç
֍ C
k
-k ø ç xO
è 0
2
è
ö æ m
÷ ç O
÷ =ç 0
÷ ç
÷ø çè 0
d 2 xO
0
mC
0
dt
d 2 xC
dt 2
2
2
æ x
0 ö
O1
2
ç
÷ d
0 ÷ 2 ç xC
÷ dt ç
mO ÷ø
çè xO2
ö
÷
÷
÷
÷ø
Normal mode analysis
The object of the normal mode analysis is to
find linear combinations of the original
coordinates that decouple the equations:
æ -k
k
0
ç
ç k -2k k
k
-k
è 0
so that
æ
ö ç xO1
֍ x
֍ C
ø ç xO
2
è
ö æ m
÷ ç O
÷ =ç 0
÷ ç
÷ø çè 0
0
mC
0
æ x
0 ö
O1
÷ d2 ç
0 ÷ 2 ç xC
÷ dt ç
mO ÷ø
çè xO2
k x1 n ew m 1 n ew
d
2
k x 2 n ew m 2 n ew
k x 3 n ew m 3 n ew
d
d2
Fx = M 2 x
dt
x1 n ew
dt
d
ö
÷
÷
÷
÷ø
2
2
x 2 n ew
dt
2
2
x 3 n ew
dt
2
These are the normal
coordinates
Normal mode analysis
d2
Fx = M 2 x
dt
1
d2
2
M FM
M x = M MM
M
x
2
dt Mass-weighted
Mass-weighted Mass-weighted
1
- 12
- 12
- 12
coordinates
Force constant
Matrix
Mass-weighted
force constant matrix
æ
ç
1
M2 = ç
ç
ç
è
- 12
1
2
coordinates
d2
Fx = 2 x
dt
mO
0
0
0
mC
0
0
0
mO
ö
÷
÷
÷
÷
ø
M
- 12
æ
ç
=ç
ç
ç
çè
1
mO
0
0
1
mC
0
0
0 ö
÷
0 ÷
÷
÷
1
mO ÷
ø
Normal mode analysis
æ
ç
1
1
F = M 2 FM 2 = ç
ç
ç
çè
æ
ç
ç
ç
ç
çè
(
)
-k
mO
k
mO mC
0
k
mO mC
-2k
mC
k
mO mC
0
k
mO mC
-k
mO
(
-k
d2
-xO + xO = 2 -xO + xO
1
2
1
2
mO
dt
Q1 (normal mode)
)
Q1 (normal mode)
-k
mO
k
mO mC
0
k
mO mC
-2k
mC
k
mO mC
0
k
mO mC
-k
mO
ö
÷
÷
÷
÷
÷ø
ö
æ -1 ö
÷ æ -1 ö
÷ ç 0 ÷ = -k ç
÷
0
÷ m ç
֍
÷
O
÷è 1 ø
è 1 ø
÷ø
Normal modes
(
)
(
-k
d2
-xO + xO = 2 -xO + xO
1
2
1
2
mO
dt
Q1 (normal mode)
)
Q1 (normal mode)
Symmetric stretch
ö d2 æ
ö
-k ( 2mO + mC ) æ
mO
mO
xC + xO ÷ = 2 ç xO - 2
xC + xO ÷
ç xO1 - 2
2
1
2
mO mC
mC
mC
è
ø dt è
ø
Q2 (normal mode)
Q2 (normal mode)
Anti-symmetric stretch
æ
ö d2 æ
ö
mC
mC
0 ç xO +
xC + xO ÷ = 2 ç xO +
xC + xO ÷
1
2
1
2
mO
mO
è
ø dt è
ø
Q3 (normal mode)
Q3 (normal mode)
Translation
Classical to quantum transition
-k
d2
Q1 = 2 Q1
mO
dt
k
mO
E1 = ( v1 + 12 )
Symmetric stretch
1285 cm−1
-k ( 2mO + mC )
mO mC
d2
Q2 = 2 Q2
dt
E2 = ( v2 +
1
2
)
k ( 2mO + mC )
mO mC
Anti-symmetric stretch
2349 cm−1
Normal modes
A normal mode transforms as an irreducible
representation of the symmetry group of the
molecule:
A1g
A1u
IR-Raman exclusion rule
Infrared active – nonzero dipole derivatives –
x, y, z irreps.
Raman active – nonzero polarizability
derivatives – xx, yy, zz, xy, yz, zx irreps.
Exclusion rule: if the molecule has the
inversion symmetry, no modes can be both
infrared and Raman active, because x, y, and
z always have character of −1 (ungerade) for
inversion while xx, yy, zz, xy, yz, and zx have
+1 (gerade).
IR and Raman activity: CO2
A1g
A1u
Raman active
IR active
D∞h,
E
…
i
…
A1g
1
…
1
…
x2+y2, z2
…
…
…
…
…
…
A1u
1
…
−1
−1
z
…
…
…
…
…
…
IR and Raman activity: H2O
A1
A2
B1
B2
IR- & Raman-active
C2v, 2mm
E
C2
σv
σv’
h=4
A1
1
1
1
1
z, x2, y2, z2
A2
1
1
−1
−1
xy
B1
1
−1
1
−1
x, zx
B2
1
−1
−1
1
y, yz
Irreducible representation of
vibrational wave functions
v=2
A1
v=3
v=2
v=1
v=0
v=1
B1
v=0
A1
Raman depolarization ratio
ρ = I┴ / III = 0.75 ~ 1.0 (depolarized – non
totally symmetric modes – xy, yz, zx)
++++++++++
x
y
– – – – – – – –
æ a
a xy a xz
xx
ç
ç a yx a yy a yz
ç
çè a zx a zy a zz
ö
÷
÷
÷
÷ø
Summary
We have learned the gross and specific selection rules
of IR and Raman spectroscopy for vibrations.
We have considered the harmonic approximation for
diatomic and polyatomic molecules. In the latter, we
have performed normal mode analysis.
We have studied the effect of anharmonicity on
vibrational spectra.
We have analyzed the symmetry of normal modes and
vibrational wave functions.
On this basis, we have rationalized IR-Raman exclusion
rule and Raman depolarization ratio.