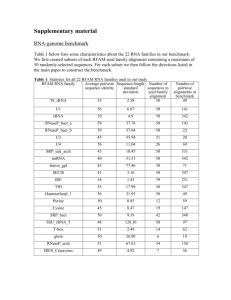
Expected accuracy sequence alignment
advertisement

Expected accuracy sequence
alignment
Usman Roshan
Expected accuracy alignment
• The dynamic programming formulation
allows us to find the optimal alignment
defined by a scoring matrix and gap
penalties. This may not necessarily be the
most “accurate” or biologically informative.
• We now look at a different formulation of
alignment that allows us to compute the
most accurate one instead of the optimal
one.
Posterior probability of
xi aligned to yj
• Let A be the set of all alignments of
sequences x and y, and define P(a|x,y) to be
the probability that alignment a (of x and y) is
the true alignment a*.
• We define the posterior probability of the ith
residue of x (xi) aligning to the jth residue of y
(yj) in the true alignment (a*) of x and y as
P(xi ~ y j a* | x, y) P(a | x, y)1{x i ~ y j a}
Do et. al., Genome Research, 2005
a A
Expected accuracy of
alignment
•
We can define the expected accuracy of an alignment a as
Do et. al., Genome Research, 2005
•
The maximum expected accuracy alignment can be obtained by the
same dynamic programming algorithm
V (i 1, j 1) P( xi ~ y j )
V (i , j ) max
V (i 1, j )
V
(
i
,
j
1
)
Example for expected
accuracy
•
•
•
•
True alignment
AC_CG
ACCCA
Expected accuracy=(1+1+0+1+1)/4=1
•
•
•
•
Estimated alignment
ACC_G
ACCCA
Expected accuracy=(1+1+0.1+0+1) ~ 0.75
Estimating posterior probabilities
• If correct posterior probabilities can be
computed then we can compute the correct
alignment. Now it remains to estimate these
probabilities from the data
• PROBCONS (Do et. al., Genome Research
2006): estimate probabilities from pairwise
HMMs using forward and backward
recursions (as defined in Durbin et. al. 1998)
• Probalign (Roshan and Livesay,
Bioinformatics 2006): estimate probabilities
using partition function dynamic programming
matrices
Posterior probabilities from
HMM
• We need to sum the probabilities of all
alignments where xi is aligned to yj. In
other words we want:
Pr(all alignments of x and y such that x i aligned to y j ) =
Pr(x i aligned to y j | alignments of x and y) =
Pr(alignments of x and y and x i aligned to y j )
Pr(alignments of x and y)
Forward and backward
probabilities
• Define fk(i) as the probability of emitting
x1x2…xi given that the ith hidden state is k.
• Similarly the backward probability bk(i) as the
probability of emitting xi+1xi+2…xn given that
the ith hidden state is k.
• Both fk(i) and bk(i) can be computed quickly
by dynamic programming (see HMM lecture
notes pages 9 to 11)
• Once forward and backward are
computed we can calculate
Pr(all alignments of x and y such that x i aligned to y j ) =
Pr(x i aligned to y j | alignments of x and y) =
Pr(x i
yj )
Pr(alignments of x and y and x i aligned to y j )
fM (i, j)bM (i, j)
f (| x |,| y |)
Pr(alignments of x and y)
Partition function posterior
probabilities
• Standard alignment score:
S (a ) T
ln( M
(i , j ) a
ij
/ f i f j ) ( gap_ penalties)
• Probability of alignment (Miyazawa, Prot. Eng. 1995)
P(a ) e S (a )/ T
• If we knew the alignment partition function then
P(a, T ) e S (a)/ T / Z (T )
Partition function posterior
probabilities
• Alignment partition function (Miyazawa,
Prot. Eng. 1995)
Z(T) e S(a )/T
a A
• Subsequently
Z
M
i, j
e
a Aij
S ij ( a ) / T
s( xi , y j ) / T
S i 1, i 1 ( a ) / T
e
e
a Ai 1 j 1
Partition function posterior
probabilities
• More generally the forward partition
function matrices are calculated as
M
i, j
E
i, j
F
i, j
Z
Z
Z
Zi , j
(Z
Z
Z
Z e Z e
Z e Z e
Z Z Z
M
i 1, j 1
M
g/T
i , j 1
M
g/T
i 1, j
M
E
i, j
i, j
E
F
i 1. j 1
i 1, j 1
E
ext / T
i . j 1
F
ext / T
i 1. j
F
i, j
)e
s( xi , y j ) / T
Partition function matrices vs.
standard affine recursions
ZiM, j
ZiE, j
ZiF, j
Zi , j
( ZiM 1, j 1 ZiE 1. j 1 ZiF 1, j 1 )e
ZiM, j 1e g / T ZiE. j 1eext / T
ZiM 1, j e g / T ZiF 1. j eext / T
ZiM, j ZiE, j ZiF, j
s( xi , y j ) / T
V (i, j) max{E(i, j),F(i, j),M(i, j)
M(i, j) V (i 1, j 1) s(x i , y j )
E(i, j) max{E(i, j 1) ext,V (i, j 1) g}
F(i, j) max{F(i 1, j) ext,V (i 1, j) g}
Posterior probability
calculation
• If we defined Z’ as the “backward”
partition function matrices then
P ( xi ~ y j )
Z
M
i 1, j 1
Z'
Z
M
i 1, j 1
e
s ( xi , y j ) / T
Posterior probabilities using
alignment ensembles
• By generating an ensemble A(n,x,y) of n
alignments of x and y we can estimate
P(xi~yj) by counting the number of times
xi is aligned to yj.. Note that this means
we are assigning equal weights to all
alignments in the ensemble.
P(xi ~ y j a* | x, y) P(a | x, y)1{x i ~ y j a}
a A
Generating ensemble of
alignments
• We can use stochastic backtracking (Muckstein et.
al., Bioinformatics, 2002) to generate a given
number of optimal and suboptimal alignments.
• At every step in the traceback we assign a
probability to each of the three possible positions.
• This allows us to “sample” alignments from their
partition function probability distribution.
• Posteror probabilities turn out to be the same when
calculated using forward and backward partition
function matrices.
Probalign
1.
For each pair of sequences (x,y) in the input set
– a. Compute partition function matrices Z(T)
– b. Estimate posterior probability matrix P(xi ~ yj) for (x,y)
by
M
M
P ( xi ~ y j )
2.
Zi 1, j 1 Z 'i 1, j 1
Z
e
s ( xi , y j ) / T
Perform the probabilistic consistency transformation and
compute a maximal expected accuracy multiple alignment:
align sequence profiles along a guide-tree and follow by
iterative refinement (Do et. al.).
V (i 1, j 1) P( xi ~ y j )
V (i , j ) max
V (i 1, j )
V (i , j 1)
Multiple protein alignment
• Protein sequence alignment: hard problem for
multiple distantly related proteins
• Several standard protein alignment
benchmarks available: BAliBASE,
HOMSTRAD, OXBENCH, PREFAB, and
SABMARK
• Benchmark alignments are based on manual
and computational structural alignment of
proteins with known structure.
Measure of accuracy
• Sum-of-pairs score: number of correctly
aligned pairs divided by number of pairs in
true alignment.
AACAGT
AA_ _GT
AACAGT
AAGT_ _
Blue: correct
Red: incorrect
Acc: 2/4=50%
• Column score: number of correctly aligned
columns
• Statistical significance using Friedman rank
test
Experimental design
• Methods compared:
–
–
–
–
Probalign
PROBCONS
MUSCLE
MAFFT
• Probalign temperature parameter trained on
RV11 subset of BAliBASE 3.0.
• Default (optimized) parameters for remaining
programs
BAliBASE 3.0
Sum-of-pairs and column score accuracies
Data
Probalign
MAFFT
RV11
69.3 / 45.3
67.1 / 44.6
RV12
94.6 / 86.2
93.6 / 83.8
RV20
92.6 / 43.9
92.7 / 45.3
RV30
85.2 / 56.4
85.6 / 56.9
RV40
92.2 / 60.3
92.0 / 59.7
RV50
89.3 / 55.2
90.0 / 56.2
All
87.6 / 58.9
87.1 / 58.6
Friedman rank test P-values
Method
RV11
RV12
MAFFT
NS
< 0.005
Probcons
0.049
0.0233
MUSCLE < 0.005
< 0.005
RV20
NS
NS
0.008
Probcons
67.0 / 41.7
94.1 / 85.5
91.7 / 40.6
84.5 / 54.4
90.3 / 53.2
89.4 / 57.3
86.4 / 55.8
RV30
NS
NS
< 0.005
MUSCLE
59.3 / 35.9
91.7 / 80.4
89.2 / 35.1
80.3 / 38.3
86.7 / 47.1
85.7 / 48.7
82.5 / 48.5
RV40
< 0.005
< 0.005
< 0.005
RV50
NS
NS
NS
All
< 0.005
< 0.005
< 0.005
Heterogeneous length data I
BAliBASE datasets with maximum length and minimum devation
Max length /
Probalign
MAFFT
Probcons
Standard dev.
500 / 100
88.4 / 56.6
88.0 / 58.0
86.7 / 51.6
500 / 200
88.5 / 54.6
87.0 / 51.9
87.2 / 48.9
1000 / 100
91.4 / 58.1
90.4 / 55.7
89.7 / 51.6
1000 / 200
90.7 / 55.0
89.3 / 51.4
89.2 / 48.7
BAliBASE datasets with long extensions
Max length /
Probalign
Standard dev.
RV40 1000 / 100 (25)
1000 / 200 (20)
92.7 / 59.3
93.0 / 57.3
MUSCLE
81.5 / 42.5
81.9 / 42.4
84.3 / 44.1
83.2 / 42.5
MAFFT
Probcons
91.0 / 54.8
90.8 / 52.1
89.9 / 48.2
90.6 / 47.6
Heterogeneous length data II
BAliBASE 2.0 reference 6 datasets with max length and minimum deviation
Max length /
Probalign
MAFFT
Probcons
Standard dev.
500 / 100 (40)
89.1 / 44.9
87.3 / 49.0
87.4 / 38.6
500 / 200 (21)
88.3 / 43.8
85.0 / 46.4
86.7 / 40.0
500 / 300 (9)
95.3 / 61.0
82.6 / 51.3
87.3 / 46.6
500 / 400 (5)
94.6 / 55.0
72.0 / 38.2
79.8 / 38.0
1000 / 100 (15)
90.2 / 43.3
82.4 / 36.9
85.4 / 27.6
1000 / 200 (12)
89.2 / 38.2
79.7 / 32.4
83.6 / 27.7
1000 / 300 (7)
94.5 / 52.8
78.3 / 42.4
83.9 / 34.6
1000 / 400 (5)
94.6 / 55.0
72.0 / 38.2
79.8 / 38.0