Epilepsy and Autism: Dr. Stefanie Berry

Epilepsy and Autism
Stefanie Jean-Baptiste Berry, MD
Pediatric Epileptologist
Northeast Regional Epilepsy Group
What is Autism?
Onset before 3 years of age
Impairment in social interaction
Impairment in communication
Restricted, repetitive and stereotyped behavior, interests and activities
Prevalence 60 in 10,000 children (0.6%)
Higher prevalence in males
High genetic and metabolic contribution
Some genetic syndromes have a high association with autism (Angelman and
Fragile X)
Autism Spectrum Disorder
Subtypes
Autistic disorder
Asperger’s syndrome
Pervasive developmental disorder
Disintegrative disorder
Rett’s syndrome
Autistic Disorder (AD)
Classic autism with incapacitating deficits in all 3 domains: 1. sociability 2. language and imagination 3. cognitive and behavior flexibility
Asperger’s syndrome
Autism without mental retardation or delayed language development
Pervasive Development Disorder
Milder autism
Disintegrative Disorder
Severe autism
Acquired between ages 2 and 10 after entirely normal early development
Regression is more profound
Rett’s Syndrome
X-linked
MeCP2 gene
Postnatal brain growth affected
Severe mental retardation and motor deficits
Clinical diagnosis
Variable causes of autism
Genetics plays a big role (high concordance in monozygotic twins, 4.5% increased risk in siblings)
Epilepsy and autism may share common neurobiological basis
? Environmental factor
Natural history and outcome of autism is variable
IQ less than 70% in most children with autism
1/3 with no communicative speech
75% who fail to show language skills by age 5 years fail to adjust personally or socially
Others make fairly adequate social adjust but retain peculiarities of personality, lack of humor and unawareness of social nuances
Even with treatment programs, only some improve significantly (2 nd half of 1 st decade)
Best outcome in patients with normal intelligence and speak before 5 years old
Applied behavior analyses (ABA) improve chances of functioning independently
What is Epilepsy?
2 or more unprovoked seizures
Incidence <10 years old 5.2 to 8.1 per
1,000 (highest <1 year)
Seizure types:
Generalized
Focal (Partial)
Focal with secondary generalization
Generalized Seizures:
1.) Generalized tonic-clonic (grand mal)-
Unconscious, whole body shaking; variable duration
2.) Absence (petit mal) – Staring, unawareness, brief (seconds)
3.) Myoclonic – Brief jerk of arm or leg
4.) Atonic – Sudden drop
Focal (Partial) Seizures:
1.) Simple – Consciousness preserved; twitching of one side of face or body, numbness, visual
2.) Complex – Impaired consciousness; twitching, head/eye deviation etc.
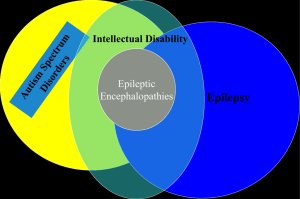
Epilepsy and Autism
Prevalence of epilepsy among all children is 2-3%
5-38% autism with epilepsy
Risk for epilepsy increased with greater intellectual disability, symptomatic vs. idiopathic, age and history of regression
35-65% with EEG abnormalities
Epilepsy in autism increased mortality
Bimodal age distribution
Peak infancy to age 5 years and adolescence
Highest risk for epilepsy in those with severe mental retardation and cerebral palsy
Epilepsy persists in the majority of patients into adult life (remission 16%)
Autistic Disorder - Clinical epilepsy by adolescence in more than 1/3 of patients
Asperger’s syndrome - Estimated 5-10% likelihood of developing epilepsy in early childhood
Pervasive Developmental Disorder - Risk for epilepsy linked to underlying brain dysfunction
Disintegrative Disorder - Risk for epilepsy as high as 70%
Rett’s Syndrome - Risk for epilepsy is more than 90%
Diagnosis complicated because seizures may be mistaken for behaviors (not responding to name)
Unusual repetitive behaviors, common in autism, hard to distinguish from seizures
All seizure types may be seen
Prevalence of epilepsy and types of seizures vary
Swedish study complex partial, atypical absence, myoclonic and tonic-clonic most common
American study tonic clonic and atypical absence most common
Other studies state complex partial with centrotemporal spikes most common
Some studies suggest that epileptiform discharges on EEG without seizures can cause behavioral and cognitive impairment
Epilepsy more common in children who regressed in language after age 3.
Usually treat based on clinical seizures not just EEG findings.
Should anti-epileptic medication be prescribed to children with autism, language regression and subclinical EEG abnormalities?
Long-duration EEGs that include slow wave sleep more likely to show epileptiform abnormalities
Long-duration EEG of children with autism spectrum disorder and regression without clinical seizures – 46% with epileptiform activity
Focal spikes - Centrotemporal spikes and temporoparietal spikes
Landau-Kleffner Syndrome
Overlap with autistic regression
Loss of language is prominent
Language regression after age 3
Acquired aphasia associated with clinical seizures or an epileptiform EEG
Clinical seizures not required for diagnosis
No decline in sociability or repetitive behaviors
EEG abnormalities (spikes, sharp waves and spike wave discharges); mainly over temporoparietal regions
25% do not have clinical seizures
Loss of language attributed to clinical or subclinical epilepsy in cortical areas responsible for language
Children with severe and persistent language disorders at increased risk for seizures and epilepsy.
Medical treatment of seizures in autism similar to treating other children with epilepsy
Limited data on response of children with epileptiform EEG without clinical seizures
Same strategy as LKS – reduce epileptiform activity
Reports that language of LKS and those with autism improved in response to anticonvulsants
Improvements have also been reported in patients treated with corticotropin, steroids, or immunoglobulins
Clinical reports of the use of Depakote in children with autism with and without clinical seizures
Most showed improvement in core symptoms of epilepsy
Absence of clinical trials, therefore no definite recommendations about treatment
Surgical resection in children with autism and intractable epilepsy – may improve seizures +/- autistic symptoms
1 study subpial transections – language and behavior improved