Blood and Body Fluid
advertisement

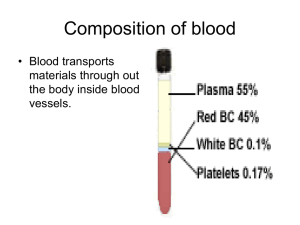
Chapter 3 Blood Physiology • Blood composition and properties • Blood cells – – – – Hematopoiesis RBC: function, anemia. WBC Platelet: function, coagulation and fibrinolysis. • Blood grouping and transfusion Introduction • Blood volume: 7~8% (70-80ml/kg B.W) – Plasma (60%) and cells (40%). • Types of blood cells: – RBC (Erythrocytes), WBC (Leukocytes) and Platelets (Thrombocytes) • Main function: – Maintain homeostasis • Buffering pH • Humoral regulation • Body temperature regulation – Transportation: • Gases, nutrients, hormones, and so on. – Host defense: • Immune reaction, coagulation. Section 1 Components and Characteristic Water: 93-95% Plasma: 50-60% Whole blood Solutes: 5-7% Proteins: Nutrients Products Electrolytes: Others: urea, gases. WBC, Platelet: 1% RBC: 40-50% (male) 37-48% (female) Blood Components • Water: – 93~95% (plasma); 65~68% (RBC); 81~86% (whole blood). – Solvent, humoral balance, osmotic pressure. • Electrolytes: – Na+, K+, Mg2+, Cl-, HCO3-, etc. Cell shape, pH. • Proteins: – Albumin: 40-48g/L. Colloidal osmotic pressure; carrier; buffer pH. – Globulin: 15-30g/L. Immune reaction: antibody; carrier. – Fibrinogen: 2-4g/L. Blood coagulation. – Hemoglobin (Hb): • 120-160g/L (male), 110-150g/L (female) • Function: carry gases. • Others: – carbohydrates, lipids, amino acid, pigments, hormones, gas (O2, CO2), and others like urea, uric acid. Physical and chemical properties • Blood pH: – Normal interval: 7.35~7.45. • Regulated by lung and kidney. • Viscosity: – Friction of molecules and cells in blood. – Relative viscosity: • Whole blood: 4~5 times to water (RBC). • Plasma: 1.6~2.4 times to water (Proteins). • Anemia or body fluid loss. • Osmotic pressure – Definition: • An ability of a liquid to attract and retain water. It drives osmosis. 300mmol/L – Composition and roles: • Crystal osmotic pressure: 298.7 mmol/L. – Maintain shape and size of cells. • Colloid osmotic pressure: 1.3 mmol/L. – Retain blood volume – Decide distribution of water between blood and interstitial fluid. Section 2 Blood Cells • Red blood cell • White blood cell • Platelet Hemopoiesis • The process of blood generation. Cell Lineage Lifespan Daily Production Rate RBC 120 days 2.5 109/L Neutrophil 7 hours 0.85 109/L Platelet 10 days 2. 5 109/L • Ontogeny of Hematopoiesis – Prenatal stages: • First month: yolk sac. • Third month: liver • Fourth month: bone marrow – Postnatal stages: • Bone marrow of almost any bone, predominatantly by flat bones and long bones. 100 100 Hemopoitic activity (%) Yolk Sac Bone morrow Liver Lymph nodes Spleen 0 1 2 3 4 5 6 7 8 9 Prenatal age (months) Proportion of Red Morrow (%) 100 birth 100 Vertebrate Tibia Sternum Femur 10 20 30 Ribs 40 50 Postnatal age (years) 60 70 80 90 – Stage 1, hemopoietic stem cells: pluripotent uncommitted stem cells. – Stage 2, committed progenitor cell: unipotent committed stem cells. Includes: • • • • Erythrocytic progenitor cell Megakaryocytic progenitor cell Granulocytic progenitor cell Lymphocytic progenitor cell – Stage 3, precursors (cell): immature cells, differentiate functional cells. Including: • Ery. progenitor erythrocytes. • Mega. progenitor platelets. • Gran. progenitor granulocytes and monocytes. • Lym. progenitor T and B lymphocytes. • Hematopoietic growth factor and related molecules – Necessary for proliferation and differentation of hematopoietic cells in the marrow. – Colony-stimulating factors (CSF): see a table in next slide. – Cytokines: • IL-1, stem cell factor (SCF), etc. – Extracellular matrix proteins: • Sulfated glycosamimoglycans and heparin sulfate, may concentrate hematopoitic growth factors in local micro environment; • Fibronectin and hemonectin, mediate adhension of cells, and may serve a growth promoting function. Hematopoietic growth factors Growth Factors Function: stimulate progenitor of the followings: GM-CSF (granulocytemacrophage CSF) Granulocyte-monocyte G-CSF (granulocyte CSF) Granulocyte M-CSF (macrophage CSF) Monocyte EPO (Erythropoietin) Erythrocyte IL-1,3,6 (Interleukin-3, 1, 6) Myeloid lineage TPO (Thrombopoietin) Platelet Red blood cells (erythrocytes) • Circular, biconcave discs without nuclei. 7~8m, thickness 1~2.5 m. • Cell count and volume: – Hematocrit: Percentage of blood volume occupied by packed cell volume. – Volume: • 4.5~5.51012/L, average 5.01012/L (male). • 3.8~4.61012/L, average 4.21012/L (female). • Physical properties – Permeability: – Deformation: – Fragility and hemolysis: • Isosmotic solution and lower osmotic solution – Suspension stability: • The erythrocytes are very stable in suspension. • Cause: repelling force of same charge and bigger surface area. • Erythrocytes Sedimentation Rate (ESR): Sedimentated distance of RBC after one hour. – 0~15 mm/h (male), 0~20 mm/h (female). – Ratio of Surface area/Volume of RBC. – Albumin, globulin, fibrinogen, and cholesterol. – Rouleaux: RBC aggregate. • Function of RBC: – The main constituent of RBC is hemoglobin. – To deliver O2 to tissues by hemoglobin. Hemoglobin (HB) • HB is made up of two polypeptide chains and chains. • Each polypeptide has alpha helical segments folded and bent into a globular configuration, with a heme ring within a pocket where the iron molecule can interact with oxygen. • Hb formation materials: – Protein: enough intake from food. – Iron: 3-4g/person. Mainly in Hb (70%). • Degrading Hb: 95%. • Absorbed from small intestine: 1mg/d, 5%. • Microcytic hypochromic anemia: Lack of iron. • RBC Maturation factors: – Vitamin B12: • Cobalamine, 2~5g/d. • Produced by gut bacteria (esp. in ruminants). Good sources include meat, liver, fish, eggs and milk. • Absorbed in terminal ileum with intrinsic factor’ help. • Function: Improve utilization of FA. – Folic acid: • • • • FA is essential for the synthesis DNA. Synthesized by microorganisms and higher plants. Good sources are green leafy vegetables, yeast and organ meats. Absorbed in the proximal jejunum. – Lack of folic acid and vitb12: give rise to immature cells due to DNA synthesis derangement. – Megaloblast anemia. •Regulation of erythropoiesis: Hypoxia: EPO RBC Hemopoitic stem cell (uncommitted progenitor) Erythrocytic progenitor (committed progenitor) EPO Pronormblast (precursor) Normoblast, Reticulocyte Mature RBC (without nucleus) • Erythropoietin (EPO): – A glycoprotein, 34kd. Produced in interstitial cells in cortical kidney such as fibroblast, endothelial cells. – Roles: • Erythrocytic progenitor proliferate and differentiate to precursor. • Accelerate precursor proliferation and differentiation. • Promote bone marrow release reticulocytes. • Renal type anemia: EPO production decrease • Other hormones: – Androgen, thyroid hormone, parathyroid hormone,etc. • RBC destruction: – Life span of RBC is about 120 days. Older cells White blood cells (leucocyte) • WBC: – 4~10109/L, average is 7109/L. – Include: • neutrophil, eosinophil, basophil • monocyte, lymphocyte. – Protection, execute specific and non-specific immune reaction. • Physical and chemical properties – Chemotaxis: attracted by chemical substances released by bacteria and foreign substances. – Movement: Move to chemotaxic source – Phagocytosis: engulf and digest Composition and functions • Neutrophil: – 10~12m, 2.0~7.0109/L, 60-70%. – Function: • Phagocytosis: older cells, becteria, dead tissues, and other foreign substances. • To execute non-specific immune activity in first front. • Monocytes: – 15~30m, 0.12 ~ 0.8109/L, 3 ~ 8%. – Monocytes-macrophages system: • Monocytes (in blood) wander into tissues and become macrophages (50 ~ 80 m). Stronger phagocytosis. • Contain many kinds of cytokines such as CSF, ILs, TNF, INF-a,b. – Roles: • Engulf and clear: bacteria, vermins, older, necrotic tissues, dead neutrophils, dead cells and fragments. • Activate lymphocytes to execute specific immune response. • Recognize and kill cancer cells. • Produce CSF, Ils, TNF, INF-, , regulate growth of granulocytes. • Lymphocytes: – 0.8~4.0109/L, 20 ~ 40%. – Development of lymphocyte: • T lymphocyte: – lymphocytic stem cells T lymphocytes (thymus gland). • B lymphocyte: – lymphocytic stem cells B lymphocytes (lymphoid tissue). – Functions: • T lymphocytes: cellular type of immunity • B lymphocytes: humoral immunity • Eosinophils – 0.02~0.5 109/L, 0.5~5%. – Functions: • Inhibit allergic reaction induced by basophils: – Produce PGE to inhibit secretion of basophils; – Engulf substances secreted by basophils; – Secrete matters to hydrolyze histamine and 5-HT. • Phagocytic action to some worms. • Basophils – 0.0~1.0 109/L, 0~1%. – Large cytoplastmic granules contain heparin, 5hydroxytryptamine and histamine. – Function: • Secrete heparin blood to prevent coagulation. • Wander into tissue and become mast cell. • Induce allergy. Platelet • Hemostasis: – The process of blood clotting and then the subsequent dissolution of the clot. Platelet activation adhension aggregation clot thrombus FDP thrombin vWF ADP and TXA2 fibrin plasmin fibrinogen Blood Coagulation Fibrinolysis • Anatomic physiology of platelet: – 2~4 m, thickness 1m. Fibrinogen GP IIb/III a Phospholipid Va Ca2+ Receptor X GPI vWF – Membrane: • Receptor: For adhension, aggregation and coagulation. • Phospholipid: provides the lipid cofactors needed for coagulation reactions. – Granules in platelet: • -granules: coagulation factors, growth factors (e.g. PDGF). • -granules (dense bodies): Ca2+, ADP and serotonin. – Volume: 100~300 109/L in adult. • Thrombocytopenia: <50 109/L, hemorrhage • Thrombocytosis: >1000109/L, Thrombosis • Physical properties – Adhesion: • Mediated by von Willebrand factor (vWF). • vWF is producted and stored in a-granules of platelets. Also synthesized by megakaryocytes. • Function of vWF: – To act as a bridge between glycoprotein on the surface of platelets (GPIb/IX) and collagen fibrils. – Serves as a carrier protein for factor VIII. • von Willebrand Disease (vWD): deficiency in vWF a patient with long bleeding time, a low level of factor vWF/VIII complex. • Bernard-Soulier Syndrome:deficiency of glycoprotein Ib/IX. – Aggregation: • Activated platelets aggregate together. • Activation of platelets: induced by thrombin. – – – – Thrombin + receptor initiate signal cascade. G-protein, and phospholipase C(PLC-g). PLC-g IP3 and DAG formation. IP3 Ca2+ , and DAG PKC. • Mechanisms: – Ca2+ phospholipase A2 (PLA2) arachidonic acid thromboxane A2 (TXA2) – PKC ADP fibrinogen to adhere to two platelet surface glycoproteins (GPIIb and GPIIIa) fibrinogen-induced platelet aggregation. – Glanzmann-Thrombasthenia, deficiency of glycoprotein IIb/IIIa. • Contractile function: – PLC-g Ca2+ myosin light chain kinase (MLCK) – MLCK phosphorylation of light chain of myosin – Myosin interacts with actin – Platelet morphology, motility, and clot retraction. • Roles of platelet: – Platelet clot formation at the site of vessel injury (primary hemostasis); – Enhance activation of coagulation factors to solidify platelet clot by interlacing with fibrin (secondary hemostasis). • Platelet function disorders: – Disorders of platelet adhesion: • Bernard-Soulier Syndrome: deficiency of glycoprotein Ib/IX. – Disorders of platelet aggregation: • Glanzmann-Thrombasthenia, deficiency of glycoprotein IIb/IIIa. – Disorders of platelet secretion: • Alpha or Dense Granules Deficiency. – Disorders of platelet procoagulant activity: • Platelets fail to promote activation of the blood clotting proteins. – Acquired platelet function disorders: • Drugs like aspirin, non-steroidal anti-inflammatory drugs like indomethacin, ibuprofen. Blood coagulation • A process of blood from liquid to colloid. A serious of enzymes reactions. • Coagulation factors: – Factors involved in the blood coagulation – Attentions: • FIII come from tissue, others from plasma. • FIV is Ca2+, and others are proteins. • FII, VII, IX, XII exist as proenzymes. Factor Trivial Name(s) Pathwa y Prekallikrein Fletcher factor Intrinsi c High molecular weight kininogen (HMWK) contact activation cofactor; Fitzgerald, Flaujeac Williams factor Intrinsi c Characteristic I Fibrinogen Both II Prothrombin Both III Tissue Factor Extrins ic Contains N-term. gla segment - IV Calcium Both V Proaccelerin, labile factor, accelerator (Ac-) globulin Both VI (Va) Accelerin VII Proconvertin, serum prothrombin conversion accelerator (SPCA), cothromboplastin Extrins ic Endopeptidase with gla residues VIII Antihemophiliac factor A, antihemophilic globulin (AHG) Intrinsi c Protein cofactor IX Christmas Factor, antihemophilic factor B,plasma thromboplastin component (PTC) Intrinsi c Endopeptidase with gla residues X Stuart-Prower Factor Both Endopeptidase with gla residues XI Plasma thromboplastin antecedent (PTA) Intrinsi c Endopeptidase XII Hageman Factor Intrinsi c Endopeptidase XIII Protransglutaminase, fibrin stabilizing factor (FSF), fibrinoligase Both Transpeptidase Protein cofactor This is Va, redundant to Factor V clotting cascade Stage 1: Formation of prothrombin activator. Stage 2: Conversion of prothrombin to thrombin. Stage 3: conversion of fibrinogen to fibrin • Difference of stage 1: – Prothrombin-converting enzyme: Xa, Ca2+, V, PL. – Difference of factor Xa: • Intrinsic stage: – Start from XII. The intrinsic pathway requires factors VIII, IX, X, XI, and XII. Also required are the proteins prekallikrein and high-molecular-weight kininogen, as well as Ca2+ and phospholipids secreted from platelets. • Extrinsic stage: – Start from FIII (TF), is initiated at the site of injury in response to the release of TF. – TF is a cofactor in the factor VIIa – Factor VIIa, cleaves factor X to factor Xa Prevention of coagulation • Plasma inhibitors • Fibrinolysis • Role of the endothelial cells Plasma inhibitors Inhibitor Mol. Weight (kD) Action Plasma Conc. (mg/ml) Antithrombin III 50 Antiserine protease 240 2antiplasma 70 Antiplasmin 70 2macroglobulin 725 Antiprotease 2500 Protein c 56 Anti-factor V and Viii 5 • Antithrombin III: – Nonspecific protease inhibitors – Produced in liver and endothelial cells – Inhibit active sites of FIXa,FXa,FXIa,FXIIa, thrombin. • Protein C: – Vitamin K-dependent protein – Is activated to activited protein C (aPC) by thrombin in presence of endothelial cell-derived cofactor thrombomodulin. – aPC inactivates FV and FVIII in presence of another vitamin K-dependent cofactor: protein S. – See next slide. Anticoagulation pathway VIII VIIIa VIIIi PS X X aPC PS V + Va FII PC Vi Thrombin FI Fibrin • Heparin: – – – – – A polysaccharide produced in basophilic mast cells Distributed in the pericapillary tissue. Abundant in lung, heart, liver, muscle tissues. Inhibit thrombin conversion. Promote antithrombin III activity. • Calcium ions precipitants: – Sodium citrate Fibrinolysis • Fibrinolysis: – Process of liquefaction of fibrin Activator plasminogen plasmin Inhibitor fibrin fibrin degradation products Activator: Tissue plasminogen activator (tPA), urokinase. Plasmin, a serine protease, is inhibited by 2-antiplasma. • tPA: – Released from vascular endothelial cells following injury; – Binds to fibrin and is consequently activated. • Urokinase: – Produced as the precursor, prourokinase by epithelial cells lining excretory ducts. – Role: to activate the dissolution of fibrin clots. • plasminogen activator-inhibitors: – PAI-1 and PAI-2 Endothelial cells • Endothelium produces several inhibitors of hemostasis: – Prostaglandin I2: • secreted by endothelial cells and is a potent inhibitor of platelet aggregation. – Thrombomodulin: • Enhances the activiation of protein C by thrombin and results in the inactivation of factor V and VIII. – Heparans: • a heparin-like molecule, produced by endothelial cells. Increase the anticoagulant effect of antithrombin III. – Plasminogen activator: • necessary for dissolution of fibrin clots, such as tPA. Coagulation disorders • Hemophilia A: – Deficiency of FVIII. The disease severity usually parallels the factor VIII levels. – Serve (< 1% VIII): with spontaneous bleeding; – Moderate (1-5% VIII): with occasional bleeding, usually with trauma; – Mild (6-30% VIII): with bleeding only after surgery or trauma. – Therapy: administration of FVIII. • Hemophilia B (Christmas Disease): – FIX deficiency. – Treatment requires IX-rich material: fresh frozen plasma (FFP) or lyophilized concentrates proagulatant proteins. • Decreased production of coagulation factors: – E.g. Liver disease, vitamin K malabsorption, dietary deficiency of vitamin K. • Inactivation of coagulation factors: – e.g. specific inhibitors, excessive activation of coagulation (DIC) and/or enzymatic destruction of coagulation factors. Blood grouping and transfusion • The discovery of blood groups: – 1901, Austrian Karl Landsteiner discovered human blood groups. – Blood agglutination was an immunological reaction. – Awarded the Nobel Prize in Physiology or Medicine in 1930. • Agglutination: – Agglutinogen: antigen on membrane of RBC. – Agglutinin: antibody in the plasma. • RBC grouping: – ABO, Rh, MnSs, lewis – The differences in human blood are due to the presence or absence of certain protein molecules called antigens and antibodies. ABO grouping Blood group A:A antigens on the surface of RBC, B antibodies in blood plasma. Blood group B:B antigens on the surface of RBC, A antibodies in blood plasma. Blood group AB:both A and B antigens on the surface of RBC, no A or B antibodies at all in blood plasma. Blood group O:neither A or B antigens on the surface of RBC, but you have both A and B antibodies in blood plasma • Antigens and antibodies: – Antigens: • A, B. • Carbohydrate – Antibodies: Antibody A and B. • Ig M: congenital, • Bigger Mr. Rh grouping • Original discovery: – Rhesus monkey: Red cells injected into rabbits got serum injected back to Rhesus monkey, or human agglutination happens. • Rh antigen and antibody – Antigen: D, E, C, c, e. • 99% Chinese people are Rh+ • Minority in China 2-5% is Rh• 15% western people are Rh– Antibody: IgG Which blood group do you belong to? A Rh+ B Rh+ AB Rh+ O Rh+ A Rh- B Rh- AB Rh- O Rh- Blood Transfusion • Clinical significance: – ABO and Rh blood groups must be compatible between the donor blood and the patient blood. – Agglutinated RBC clog blood vessels or crack to becomes toxic when HB outside the cell. • Cross-match test: – Main lateral: donor’s RBC and recipient’s serum. – Co-lateral: donor’s serum and recipient’s RBC. • Principle of blood transfusion: – Agglutination of main lateral: absolutely no. – Both of main and co-lateral do not agglutinate: – Co-lateral agglutinates but Main lateral: • slow and less amount of blood transfusion could be recommended. People with blood group O are called “universal donors”. People with blood group AB are called “universal receivers”. • Clinical importance for Rh group: – Blood transfusion between Rh+ and Rh- persons. – A mother who is Rh- woman give birth a baby who is Rh+. – Preventive measure: given an injection of anti-Rh antibodies. Donor RBC Serum Recipient RBC (co-lateral) Serum (main lateral) Cross match test