Human Research History and Regulations
advertisement

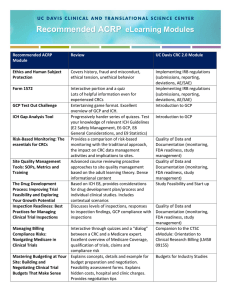
HUMAN RESEARCH HISTORICAL PERSPECTIVE Objectives • Identify the history events that lead to the development of principles, regulations, and guidance to conduct human research • Briefly describe these regulations and guidance • Define the term “Good Clinical Practice” and how is applies to clinical research Human Research Protection Program Historical Perspective AAHRPP 2001 ICH Guidelines 1996 Jesse Gelsinger 1999 Common Rule 1991 Consolidated HHS/FDA Regulations 1981 Belmont Report 1979 1972 The Syphilis Study (Expose’) Declaration of Helsinki 1964 Amendments to the Food Drug and Cosmetic Act 1962 Nuremberg Code 1947 Human Radiation Experiments 1942 The Nazi Experiments The Syphilis Study begins Nuremberg Code The trial verdict adopted ten points constituted the "Nuremberg Code". Legal force of the document was not established and it was not incorporated directly into either the American or German law, the Nuremberg Code and the related Declaration of Helsinki are the basis for the Code of Federal Regulations governing federally-funded research in the United States Nuremberg Code – 10 points 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. The voluntary consent of the human subject is absolutely essential. This means that the person involved should have legal capacity to give consent; should be so situated as to be able to exercise free power of choice, without the intervention of any element of force, fraud, deceit, duress The experiment should be such as to yield fruitful results for the good of society, unprocurable by other methods or means of study The experiment should be so designed and based on the results of animal experimentation and a knowledge of the natural history of the disease or other problem under study that the anticipated results will justify the performance of the experiment. The experiment should be so conducted as to avoid all unnecessary physical and mental suffering and injury. No experiment should be conducted where there is a prior reason to believe that death or disabling injury will occur; except, perhaps, in those experiments where the experimental physicians also serve as subjects. The degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment. Proper preparations should be made and adequate facilities provided to protect the experimental subject against even remote possibilities of injury, disability, or death. The experiment should be conducted only by scientifically qualified persons. The highest degree of skill and care should be required through all stages of the experiment of those who conduct or engage in the experiment. During the course of the experiment the human subject should be at liberty to bring the experiment to an end if he has reached the physical or mental state where continuation of the experiment seems to him to be impossible. During the course of the experiment the scientist in charge must be prepared to terminate the experiment at any stage, if he has probable cause to believe, in the exercise of the good faith, superior skill and careful judgment required of him that a continuation of the experiment is likely to result in injury, disability, or death to the experimental subject. Declaration of Helsinki The fundamental principle is respect for the individual , their right to self determination and the right to make informed decisions. Belmont Report Basic Ethical Principles: • Respect for persons – (informed consent process) • Beneficence – do no harm – (maximum benefit/minimized risk) • Justice- well considered procedures administered fairly – (fairness of the selection of subjects) Common Rule • In 1981, DHHS and FDA issued regulations based on the Belmont Report. – DHHS issued Code of Federal Regulations (CFR) Title 45 (public welfare), Part 46 (protection of human subjects). – The FDA issued CFR Title 21 (food and drugs), Parts 50 (protection of human subjects) and 56 (Institutional Review Boards) • In 1991, the core DHHS regulations were formally adopted = "Common Rule The main elements of the Common Rule include : – requirements for assuring compliance by research institutions – requirements for researchers obtaining and documenting informed consent – requirements for IRB membership, function, operations, review of research, and record keeping. – additional protections for certain vulnerable research subjects-- pregnant women, prisoners, and children Good Clinical Practice (GCP) • Good Clinical Practice offers protection for human subjects in clinical trials. There is no one source of guidance for GCP. They are embodied within laws, regulations and guidance such as: – – – – – – – The Belmont Principles IRB and Consent Regulations Guidelines on the obligations of investigators, sponsors and monitors Code of Federal Regulations retaining to drugs and devices Form FDA 1572 ICH Guidelines Official guidance documents International Conference on Harmonization (ICH) • ICH guidance was published as a worldwide guideline for GCP in an effort to standardize for clinical trials Importance of GCP and ICH • Following GCPs ensures the accuracy and reliability of data generated in the course of a clinical trial. • Compliance with GCPs during clinical trials will ensure that: – The rights and safety of human subjects are not compromised. – Appropriately and adequately trained staff manage the study. – The study is carefully documented. – Protocol is strictly adhered to. Standard Operating Procedures for any Clinical Trial • GCPs are recognized as the standard operating procedures for a clinical trial • GCPs, therefore, encompass all aspects of a clinical trial including (but not limited to: – Obtaining informed consent – Documenting accurate case histories – Maintaining completed “paper trails” for all study documents – Reporting adverse events – Proper record storage and retention • MMC GCP-SOP’s