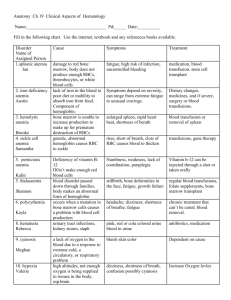
Week 3 Hematopoietic Function Hematopoiesis Process of forming blood Plasma: liquid protein Leukocytes: white blood cells Erythrocytes: red blood cells Hemoglobin: oxygen-carrying component Hematocrit: amount of blood volume occupied by erythrocytes Thrombocytes: platelets Review of Structure and Function blood, bone marrow, lymphoid tissues, mononuclear phagocytic system, and the immune system Components are located in several organs Bone marrow, blood vessels, Spleen, lymph nodes, and thymus Hematopoiesis means blood formation (hemato= blood) https://www.youtube.com/watch?v=RsKZWqsUpw&feature=player_embedded Review of Structure and Function Red blood cells (RBC) aka erythrocytes Responsible for oxygen transport the amount moved is dependent on the amount of hemoglobin in the red blood cell live about 120 days, and are removed by the spleen https://www.youtube.com/watch?v=_ZV5140OykE&feature=player_em bedded Iron Metabolism Transferrin: Transport protein for iron (each can bind to two iron) Ferrokinetics-movement of iron in the metabolic cycle Plasma iron clearance Plasma iron turnover/transport rate Utilization of newly formed RBC’s (RCV) Uptake turnover of iron RBC iron turnover (EIT) Review of Structure and Function Platelets (thrombocytes) Short-lived fragments of a bone marrow cell, responsible for clotting when needed Must be replaced continuously http://youtu.be/_yQD0U3ZtCs Review of Structure and Function White blood cells (WBC) Many different types, they are responsible for inflammatory reactions and fighting infections Commonly called Leukocytes to refer to all types Review: WBC’s Granulocytes: short lived, stay in the blood Neutrophils, Eosinophils, Basophils Lymphocytes: originate in bone marrow, some go to Thymus to become T lymphocytes for cell-mediated immunity, others go to lymphoid tissue to become B Lymphocytes or eventually become plasma Monocytes: most widespread Circulate in blood for anti-inflammatory response Basically become specialized in every tissue/organ in the body Macrophages, histiocytes, RE cells, Kupffer cells. Symptoms, Signs, and Tests Most hematopoietic diseases will cause nonspecific symptoms Physical exam: lymphadenopathy, splenomegaly or hepatomegaly Laboratory exams: Biopsy: used to diagnose hematopoietic cancers and diseases Hemostasis Stoppage of blood flow Normal when it seals a blood vessel to prevent blood loss and hemorrhage Abnormal when it causes inappropriate clotting or when clotting is insufficient to stop blood flow Stages of Hemostasis 1. Vessel spasm 2. Formation of platelet plug 3. Blood coagulation 4. Clot retraction 5. Clot dissolution Organ Failure The result of bone-marrow failure is anemia, leukopenia, and thrombocytopenia These have the potential to increase the likelihood of infection and bleeding Specific Diseases Anemia's with decreased RBC production Anemia of chronic disease is the most common, and most difficult to treat Iron-deficiency anemia Vitamin B12-deficiency anemia (pernicious anemia) Myelophthisic anemia Specific Diseases Disorders of WBC Usually secondary to another illness Leukopenia: too few WBC’s Lymphopenia: too few Lymphocytes Disorders of Platelets Idiopathic Thrombocytopenic Purpura (ITP) Believed to be the result of antibodies to platelets, associated with bleeding from small vessels Specific Diseases Hyperplastic/Neoplastic Disease Leukemia—WBC cancers Lymphomas—WBC cancers Multiple myeloma—cancer of the plasma cells Polycythemia—increased RBC production Disorders of the WBCs Leukocytes: key players in the inflammatory response and in fighting infections Normal range = 5,000 to 10,000 cells/mL3 blood Leukopenia: decreased levels Leukocytosis: increased levels Neutrophils One type of leukocytes Usually the first to arrive at the site of infection Normal range is 2,000–7,500 cells/mL Neutropenia (1 of 2) Neutrophils < 1500 cells/mL Causes Increased usage Drug suppression Radiation therapy Congenital conditions Bone marrow cancers Spleen destruction Vitamin deficiency Neutropenia (2 of 2) Manifestations Depends on severity and cause Infections and ulcerations, especially of the respiratory tract, skin, vagina, and gastrointestinal tract Signs and symptoms of infection (e.g., fever, malaise, and chills) Diagnosis: neutrophil levels and bone marrow biopsy Treatment: antibiotic therapy and hematopoietic growth factors Infectious Mononucleosis (1 of 2) “Kissing disease”—oral transmission. Self-limiting. Most prevalent in adolescents and young adults. Caused by Epstein-Barr virus in the herpes family. EBV infects the B cells by killing the cell or being incorporated into its genome. Those B cells incorporated with EBV produce heterophile antibodies. Once the disease is eliminated, a few B cells remain altered, giving the individual an asymptomatic infection for life and the potential to occasionally spread the EBV to others. Infectious Mononucleosis (2 of 2) Manifestations Insidious onset. Incubation = 4–8 weeks. Initially see anorexia, malaise, and chills. Manifestations intensify to include leukocytosis, fever, chills, sore throat, and lymphopathy. Acute illness usually lasts 2–3 weeks; may not fully recover for 2–3 months. Treatment: symptomatic and supportive Lymphomas Cancers affect lymphatic system Most common hematologic cancer in the US Two main types Hodgkin’s Non-Hodgkin’s Lymphoma Applied to an entire spectrum of malignant diseases involving the lymphocytes Malignant lymphocytes infiltrate the lymph nodes, spleen, thymus, bone marrow Extranodal lymphomas: lymphomas that originate in areas outside the lymph nodes (solid organs, brain, skin, eyes, etc) May go into the blood and present as Leukemia Symptoms: node enlargement, fatigue, malaise, weight loss, fever, sweating, anemia, leukopenia, Dx requires a lymph node biopsy Non-Hodgkin’s Lymphoma More common Poor prognosis Many different types Similar to Hodgkin’s manifestations, staging, and treatment Different in the spread and diagnosis Can originate in the T or B cells No Reed-Sternberg cells Non-Hodgkin’s Lymphoma Follicular: slow growing and most common Tumor cells resemble normal mature lymphocytes Chronic and slow growing, non responsive to chemo Diffuse large B cell: Group of lymphomas that occurs in several forms, responds to chemo Do not resemble normal lymph nodes Infiltrate normal tissues and spread to organs Burkitts: Small B cells divide rapidly and often have apoptosis Extra-nodal masses more prominent than enlarged nodes Most often presents with an abdominal mass Responds well to chemo and can be cured. Hodgkin’s Lymphoma (1 of 4) Least common of the two Solid tumors with the presence of Reed-Sternberg cells Typically originate in the lymph nodes of the upper body Several subtypes Very curable with treatment Manifestations: painless enlarged nodes, weight loss, fever, night sweats, pruritus, coughing, difficulty breathing, chest pain, recurrent infections, and splenomegaly Hodgkin’s Lymphoma (2 of 4) Staging Stage I: The lymphoma cells are in one lymph node group or one part of a tissue or an organ. Stage II: The lymphoma cells are in at least two lymph node groups on the same side of the diaphragm, or the lymphoma cells are in one part of a tissue or an organ and the lymph nodes near that organ. Hodgkin’s Lymphoma (3 of 4) Staging Stage III: The lymphoma cells are in lymph nodes above and below the diaphragm. Lymphoma cells may be found in one part of a tissue or an organ near these lymph node groups. Cells may also be found in the spleen. Stage IV: Lymphoma cells are found in several parts of one or more organs or tissues, or the lymphoma cells are in an organ and in distant lymph nodes. Recurrent: The disease returns after treatment. Hodgkin’s Lymphoma (4 of 4) Diagnosis: physical examination, presence of ReedSternberg cells in a lymph node biopsy, complete blood count, chest X-rays, computed tomography scan, magnetic resonance imaging, positron emission tomography scan, and bone marrow biopsy Treatment: chemotherapy, radiation, and surgery Hodgkin's Lymphoma Proper Staging is the most important factor for tx and prognosis Lymph biopsies of nodes in the liver, spleen, and bone marrow Symptoms: lymph node enlargement, fever, fatigue, etc. Stage 1: single region involved Stage 2: 2 or more regions on the same side of the body Stage 3: both sides of the body with or without the spleen being involved Stage 4: wide spread, 2 + extranodal tissues or nonlymphoid organs Hodgkin’s Lymphoma 4 classical types: Reed-Sternberg cells are binucleated or multinucleated Nodular sclerosis Mixed cellularity Lymphocyte predominance Lymphocyte depletion Non-typical: lymph nodes contain cells with popcorn nuclei. Lymphocyte rich Multiple Myeloma Malignant plasma cells overgrowth in bone marrow Plasma cells are descendants of B lymphocytes Plasma cells secrete immunoglobulin and can be detected in serum Monoclonal gammopathy: spike in immunoglobulin due to increased cells Symptoms: infections, fracture, lytic bone lesions, renal failure, purpura, Raynaud's syndrome, anemia, leukopenia, thrombocytopenia, Bence Jones Protein (proteinuria), renal failure DX: X-rays show bone lesions, blood tests show hypercalcemia, bone marrow biopsy Grim Prognosis: Most patients die within 3-4 years from renal failure or infections, chemotherapy does not work. Polycythemia Also called erythocytosis: too many red blood cells Primary is called polycythemia Vera Uncontrolled production of red blood cells and increased red blood cells mass Secondary can be caused by chronic hypoxia, living in high altitudes, congenital heart disease, renal carcinoma, and chronic lung disease Symptoms: HTN, flushing in the face, headaches, visual and cognitive problems, splenomegaly, thrombi Can lead to leukemia, considered neoplastic disease What does it mean for us: RBC mass P-32 treatment Leukemia ALL leukemia's have the following: Bone marrow is infiltrated with malignant cells (bone marrow biopsy) Peripheral blood contains an increased number of immature blood cells Detected in blood tests, may be an early indicator Neoplastic stem cells with chromosomal changes specific to each type This is how we can tell which type Important for dx, prognosis, and treatment Complications include anemia, recurrent infections, uncontrollable bleeding Malignant cells replace normal cells and disrupt hematopoiesis Leukemia Acute lymphoblastic leukemia (ALL): most common form in children Acute Myelogenous Leukemia (AML): most common type (40%) Chronic Lymphoblastic Leukemia (CLL): almost unknown under 40 yrs old Chronic Myelogenous Leukemia (CMl): rare in children, only 15% are this type Leukemia: All Immature lymphoid cells (precursor to T and B lymphocytes) Peripheral blood contains malignant lymphoid cells 30% of all leukemia's, and most common childhood cancer Symptoms: splenomegaly, enlarged lymph nodes, weakness, recurrent infections, bleeding into the skin and major organs Chemo is preferred, 2/3 can be cured, otherwise, fatal within 3-6 months Leukemia: AML Proliferation of myeloblasts Most common form in adults Patients will die within 6 months without treatment, chemo may induce remission Patients may receive radiation therapy and chemotherapy Four main types AML with recurrent genetic abnormalities AML from multi-lineage dysplasia Therapy related AML AML not otherwise specified This is divided further into 8 subtypes M0-M7 Leukemia: CLL Mostly occurs in adults over 50 Involves lymphoid cells, cells are indistinguishable from normal cells Indication is elevated lymphocytes in blood Confirmed with bone marrow biopsy Does not respond to chemo, most patients die in 9 years Very similar to small cell lymphoma Can transform into more aggressive forms of leukemia or lymphoma Leukemia: CML Malignant stem cells, may differentiate into neutrophilic leukocytes Patients have a high WBC counts Slow onset with mild anemia and hyper metabolism, fatigue, recurrent infections Pts may have splenomegaly and increased clotting 3 phases: Chronic Accelerated Blast crisis Chemotherapy does not work, most die within 3-5 years Bone marrow transplant, chemotherapy and radiation therapy Tyrosine kinase inhibitors Leukemia (1 of 4) Second most common blood cancer Cancer of the leukocytes Leukemia cells abnormally proliferate, crowding normal blood cells Risk factors: exposure to chemical, viral, and radiation mutagens; smoking; use of chemotherapies; certain disease conditions (e.g., Down syndrome); and immunodeficiency disorder Leukemia (2 of 4) Types Acute lymphoblastic leukemia Affects primarily children Responds well to therapy Good prognosis Acute myeloid leukemia Affects primarily adults Responds fairly well to treatment Prognosis somewhat worse than that of acute lymphoblastic leukemia Leukemia (3 of 4) Types Chronic lymphoid leukemia Affects primarily adults Responds poorly to therapy, yet most patients live many years after diagnosis Chronic myeloid leukemia Affects primarily adults Responds poorly to chemotherapy, but the prognosis is improved with allogeneic bone marrow transplant Leukemia (4 of 4) Manifestations: leukopenia, anemia, thrombocytopenia, lymphadenopathy, joint swelling, bone pain, weight loss, anorexia, hepatomegaly, splenomegaly, and central nervous system dysfunction Diagnosis: a history, physical examination, peripheral blood smears, complete blood count, and bone marrow biopsy Treatment: chemotherapy and bone marrow transplant Multiple Myeloma (1 of 2) Plasma cell cancer (third most common) Excessive numbers of abnormal plasma cells in the bone marrow, crowding the blood-forming cells and causing Bence Jones proteins to be excreted in the urine Bone destruction leads to hypercalcemia and pathologic fractures Often well advanced upon diagnosis Multiple Myeloma (2 of 2) Manifestations Insidious onset Include: anemia, thrombocytopenia, leukopenia, decreased bone density, bone pain, hypercalcemia, and renal impairment Diagnosis: serum and urine protein, calcium, renal function tests, complete blood count, biopsy, X-rays, computed tomography, and magnetic resonance imaging Treatment: chemotherapy and complication management Disorders of the RBCs Erythropoiesis Production of erythrocytes Regulated by erythropoietin Occurs in bone marrow Disorders typically result from a deficit or defect in the erythrocytes. Anemia Results from a decreased number of erythrocytes, reduction of hemoglobin, or presence of abnormal hemoglobin Decreases O2-carrying capacity, leading to tissue hypoxia Several types with varying etiology General manifestations: weakness, fatigue, pallor, syncope, dyspnea, and tachycardia Blood-loss anemia Caused by acute or chronic blood loss Where it applies? GI bleed studies Iron-Deficiency Anemia (1 of 2) Very common Iron is necessary for hemoglobin production Causes: decreased iron consumption, decreased iron absorption, or increased bleeding Additional manifestations: cyanosis to sclera, brittle nails, decreased appetite, headache, irritability, stomatitis, pica, and delayed healing Iron-Deficiency Anemia (2 of 2) Diagnosis: complete blood count (low hemoglobin, hematocrit, MCV, and MCHC), serum ferritin, serum iron, and transferrin saturation Treatment: identify and treat cause, increase dietary intake, and administer iron supplements Pernicious Anemia (1 of 2) Vitamin B12 deficiency usually caused by a lack of intrinsic factor. Cause: autoimmune. Vitamin B12 is required for DNA synthesis. Leads to decreased maturation and cell division. May see myelin breakdown and neurological complications. Pernicious Anemia (2 of 2) Manifestations: bleeding gums, diarrhea, impaired sense of smell, loss of deep tendon reflexes, anorexia, personality or memory changes, positive Babinski’s sign, stomatitis, paresthesia, and unsteady gait Diagnosis: serum B12 levels, Schilling’s test, complete blood count, gastric analysis, and bone marrow biopsy Treatment: injectable B12 Aplastic Anemia (1 of 2) Bone marrow depression of all blood cells (pancytopenia). Causes: idiopathic, autoimmune, medications, medical treatments, viruses, and genetic abnormalities. Onset may be insidious, sudden, and severe. Aplastic Anemia (2 of 2) Manifestations: Anemia (e.g., weakness, pallor, dyspnea) Leukocytopenia (e.g., recurrent infections) Thrombocytopenia (e.g., bleeding) Diagnosis: complete blood count and bone marrow biopsy Treatment: identify and manage underlying cause, oxygen therapy, infection control, infection treatment, bleeding precautions, blood transfusions, and bone marrow transplants Hemolytic Anemia Excessive erythrocyte destruction Causes: idiopathic, autoimmune, genetics, infections, blood transfusion reactions, and blood incompatibility in the neonate Several types including sickle cell anemia, thalassemia, and erythroblastosis fetalis Sickle Cell Anemia (1 of 6) Neither recessive nor dominant—codominant. Hemoglobin S causes erythrocytes to be abnormally shaped. Abnormal erythrocytes carry less oxygen and clog vessels, causing hypoxia and tissue ischemia. More common in people of African and Mediterranean descent. Also seen in people from South and Central America, the Caribbean, and the Middle East Sickle Cell Anemia (2 of 6) Forms of sickle cell anemia Sickle cell trait. Heterozygous. Less than half of erythrocytes are sickled. Sickle cell disease Homozygous. Most severe. Almost all erythrocytes are sickled. Sickle Cell Anemia (3 of 6) Manifestations Typically appear around 4 months of age Sickle cell crisis Painful episodes that can last for hours to days Pain caused by tissue ischemia and necrosis Triggered by dehydration, stress, high altitudes, and fever Sickle Cell Anemia (4 of 6) Manifestations include abdominal pain, bone pain, dyspnea, delayed growth and development, fatigue, fever, jaundice, pallor, tachycardia, skin ulcers, angina, excessive thirst, frequent urination, priapism, and vision impairment Sickle Cell Anemia (5 of 6) Diagnosis: hemoglobin electrophoresis, complete blood count, and bilirubin test Life expectancy improving with better management Sickle Cell Anemia (6 of 6) Treatment No cure, palliative Stem cell research showing promise Medications (e.g., Hydrea [hydroxyurea]) Avoid sickling triggers Other strategies: oxygen therapy, hydration, pain management, infection control, vaccinations, blood transfusions, bone marrow transplants, genetic counseling Thalassemia (1 of 2) Autosomal dominant inheritance Abnormal hemoglobin from a lack of one of two proteins that make up hemoglobin (alpha and beta globin) Most common in people of Mediterranean descent Also seen in those of Asian, Indian, and African descent Manifestations: abortion, delayed growth and development, fatigue, dyspnea, heart failure, hepatomegaly, splenomegaly, bone deformities, jaundice Thalassemia (2 of 2) Severe cases can lead to death in childhood. Life expectancy can improve with effective management. Diagnosis: complete blood count (low MCV, MCHC) and iron levels. Treatment: blood transfusion, chelation therapy, and splenectomy. Polycythemia (1 of 2) Abnormally high erythrocytes Rare Considered a neoplastic disease Increased blood volume and viscosity, leading to tissue ischemia and necrosis Complications: thrombosis, hypertension, heart failure, hemorrhage, splenomegaly, hepatomegaly, and acute myeloblastic leukemia Polycythemia (2 of 2) Manifestations: cyanotic or plethoric skin, high blood pressure, tachycardia, dyspnea, headaches, visual abnormalities Diagnosis: complete blood counts, bone marrow biopsy, and uric acid levels Treatment: chemotherapy, radiation, phlebotomy, and managing clotting disorders Disorders of Platelets Normal platelet levels range from 150,000 to 350,000 cells/mL3 Include issues in quantity and quality of platelets Thrombocytosis: increased levels Thrombocytopenia: decreased levels Hemophilia A X-linked recessive bleeding disorder Deficiency or abnormality of clotting factor VIII Varies in severity Manifestations: bleeding or indications of bleeding (e.g., bruising, petechia, etc.) Diagnosis: clotting studies and serum factor VIII levels Treatment: clotting factor transfusions, recombinant clotting factors, desmopressin (DDAVP), and bleed precautions von Willebrand’s Disease (1 of 5) Most common hereditary bleeding disorder Decreased platelet adhesion and aggregation Manifestations: bleeding or indications of bleeding (e.g., bruising, petechia, etc.) von Willebrand’s Disease (2 of 5) Forms of von Willebrand’s disease Type 1 Most common and mildest form Follows autosomal dominant Reduced von Willebrand’s factor levels Can cause significant bleeding with trauma or surgery von Willebrand’s Disease (3 of 5) Forms of von Willebrand’s disease Type 2 Either autosomal dominant or recessive. Five subtypes. von Willebrand’s factor building blocks are smaller than usual or break down easily. von Willebrand’s Disease (4 of 5) Forms of von Willebrand’s disease Type 3 Follows autosomal recessive No measurable von Willebrand’s factor or factor VIII Causes severe bleeding problems Acquired type Occurs with Wilms’ tumor, congenital heart disease, systemic lupus erythematosus, and hypothyroidism von Willebrand’s Disease (5 of 5) Diagnosis: bleeding studies and factor VIII levels Treatment Mild cases usually do not require treatment Cryoprecipitate infusions Administration of desmopressin (DDAVP) Bleeding precautions Measures to control bleeding Disseminated Intravascular Coagulation (1 of 2)complication of many conditions Life-threatening Results from an inappropriate immune response Widespread coagulation followed by massive bleeding because of the depletion of clotting factors Manifestations: tissue ischemia and abnormal bleeding Disseminated Intravascular Coagulation (2 of 2) Complications: shock and multisystem organ failure Diagnosis: complete blood count and bleeding studies Treatment: identify and treat underlying cause, replace clotting components, and prevent activation of clotting mechanisms Idiopathic Thrombocytopenia Purpura (1 of 5) Hypocoagulation resulting from an autoimmune destruction of platelets Acute form More common in children Sudden onset Self-limiting Chronic form More common in adults age 20–50 More common in women Idiopathic Thrombocytopenia Purpura (2 of 5) Causes: idiopathic, autoimmune diseases, immunizations with a live vaccine, immunodeficiency disorders, and viral infections Manifestations: bleeding or indications of bleeding (e.g., bruising, petechia, etc.) Idiopathic Thrombocytopenia Purpura (3 of 5) Diagnosis: complete blood count (platelet levels < 20,000/mL) and bleeding studies Treatment Acute ITP: glucocorticoid steroids, immunoglobulins, plasmapheresis, and platelet pheresis Chronic ITP: glucocorticoid steroids, immunoglobulins, splenectomy, blood transfusions, and immunosuppressant therapy Thrombotic Thrombocytopenic Purpura (4 of 5) Deficiency of enzyme necessary for cleaving von Willebrand’s factor, leading to hypercoagulation. Hypercoagulation depletes platelet levels. Characterized by thromboses, thrombocytopenia, and bleeding. Causes: idiopathic causes, heredity, bone marrow transplants, cancer, medications, pregnancy, and HIV. Thrombotic Thrombocytopenic Purpura (5 of 5) Manifestations: purpura, changes in consciousness, confusion, fatigue, fever, headache, tachycardia, pallor, dyspnea on exertion, speech changes, weakness, and jaundice Diagnosis: complete blood counts, blood smears, and lactate dehydrogenase levels Treatment: plasmapheresis, splenectomy, and glucocorticoid steroids