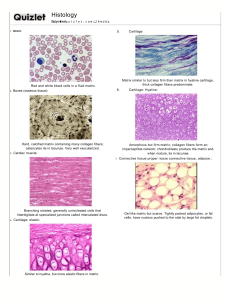
LECTURE NOTES ON CONNECTIVE TISSUE PROTEINS AND DISORDERS BY DR UMAR AMINU ABDULLAHI DEPARTMENT OF CHEMICAL PATHOLOGY AND IMMUNOLOGY, COLLEGE OF HEALTH SCIENSES, USUMANU DANFODIYO UNIVERSITY SOKOTO. INTRODUCTION Types of tissues: Nervous tissue: Communication Epithelial: Boundaries/barriers Connective tissue: Binding and hold things together Muscular tissue: Movements Connective tissue: Tissue that supports, protects, binds/connects, transports, anchored, and provide immunity to cells, tissues and organs (cell & ECM). Underlined features of connective tissue (CT) structural units Cells: Mesenchymal, macrophages, adipocytes, fibroblast etc. INTRODUCTION CONT’D Gels (ground substances): viscous, transparent, varies from fluid to gel that fills the space between cells and fibres in which CT are embedded. Consist of: o Glycoconjugates (polysaccharides) • Glycosaminoglycans (GAGs) • Proteoglycans • Structural (adhesive) glycoproteins o Tissue fluid Fibers: Long, thin extracellular protein polymers secreted by cells in CT Collagen Elastic Reticular Classification of CT: CT proper: dense or loose Dense: regular, irregular and elastic Loose: areolar, reticular & adipose Supportive: bone & cartilage Cartilage: hyaline, elastic & fibrocartilage Specialized: adipose tissue, elastic, hematopoietic & mucous tissue. EXTRACELLULAR MATRIX (ECM) Extracellular matrix: a dynamic three-dimensional macromolecular network that provides stability, signaling and structural support to the organs. Cells are embedded in a jelly of proteins and polysaccharides called the extracellular matrix or the ground substance In epithelial tissues, cells are tightly attached to form sheets; ECM is scanty and forms the basal lamina The basal lamina forms a supporting layer underlying the epithelia and helps prevent the cells from ripping apart In connective tissues ECM forms a larger space and carries the mechanical stress to which the tissue is subjected The ECM serves as a reservoir for many extracellular signalling molecules that control cell growth and differentiation In addition, it provides a network through or on which cells can move during tissue assembly ECM CONT’D Structure: water, proteins & carbohydrates. Functions: Cell support within a tissue Regulation of intercellular adhesion and communication Control of cell migration ECM components: Proteoglycan Fibrous structural proteins: collagen & elastin Fibrous adhesive proteins: fibronectin & Laminin ECM CONT’D The cells in animal tissues are “glued” together by cell adhesion molecules (CAMs), proteins embedded in their surface membranes CAMs mediate cell-cell and cell-ECM interactions CAMs fall into four major families: the cadherins, immunoglobulin (Ig) superfamily, integrins and selectins CAMs mediate, through their extracellular domains, adhesive interactions between cells of the same type (homotypic adhesion) or between cells of different types (heterotypic adhesion) A CAM on one cell can directly bind to the same kind of CAM on an adjacent cell (homophilic binding) or to a different class of CAM (heterophilic binding) Clustered CAMs form cell junctions The cytosolic domains of CAMs form connections with cytoskeletal elements GLYCOSAMINOGLYCAN (GAGs) Glycosaminoglycan (GAGs): a long polysaccharide, highly acidic, negatively charged & unbranched linear polysaccharide. GAGs are located primarily on the surface of cells or ECM but are also found in secretory vesicles in some types of cells. Usually attached to protein to form proteoglycan except in hyaluronan. Structurally is made of repeating disaccharide units, the first sugar is Nacetyl glucosamine or N-acetyl galactosamine, and the second sugar is uronic acid. GAGs are degraded by lysosomal enzymes: glycosidases and sulfatases Various degrees of deficiency of these enzymes leads to different types of mucopolysaccharidoses Rare diseases inherited in an autosomal recessive manner •e.g. Hurler Syndrome - deficiency of α-L- iduronidase • Hunter Syndrome - iduronate sulfatase Classification oHyaluronic acid (HA): synovial fluid, vitreous humour, ECM of loose CT, allows migration of cells during morphogenesis and wound healing. oChondroitin sulfate & dermatan sulfate: cartilage, bone, and heart valves, along with HA give compressibility to the bone and cartilage. oHeparan sulfate and heparin: basement membranes, A hypersulfated form of heparan sulfate called heparin, produced mostly by mast cells, plays a key role in allergic reactions. oKeratan sulfate: cornea, bone, cartilage aggregated with chondroitin sulfates. PROTEOGLYCAN Proteoglycan: are the products of the covalent linkage of core proteins with glycosaminoglycans (GAG). a group of glycoproteins that cushion cells and bind many extracellular molecules Highly hydrated They contain more carbohydrates than glycoproteins the sugars are usually sulfated A proteoglycan = 100 proteoglycan monomers PROTEOGLYCAN CONT’D The glycosylation of core proteins begins in the lumen of the endoplasmic reticulum and continues in the Golgi A link trisaccharide containing xylose and two galactose residues can be the first to attach through O-glycosidic bond with a Serine of the core proteins and then the other sugars are added Or the modified sugars could be directly be attached through Nglycosidic bond with Asparagine residue of the core proteins Once chain formation is over, sulfotransferases add sulfate which is obtained from (PAPS) • The abundance of negative charges increases polarity • The proteoglycans bind large amounts of water and fill the gaps between the other components of the ECM in the form of a hydrated gel Epimerization of glucoronate to iduronate also takes place after the chain is synthesized COLLAGEN Collagen: Are the main insoluble fibrous protein of the connective tissue accounts for around 25-30 % of the total protein content of the body. Major component in ECM Found in connective tissue cells of all multicellular animals Classification oType I Fibrillar collagen: Found in skin, tendons, bones, most tissues, etc. oType II Fibrillar collagen: Found in cartilage, vitreous humor, cornea, intervertebral disc oType III Fibrillar collagen: skin, muscle, soft tissues, etc., frequently occurs together with type I oType IV Non Fibrillar: a sheet-like meshwork, a major part of basal lamina that assembles into a multi-layered network (All basement membrane) oType V Fibrillar: Most interstitial tissues associated with type I oType VI: Most interstitial tissues associated with type I oType VII: Network forming collagen beneath stratified squamous epithelia. oType IX: Fibril-associated, cartilage, oType X: Hypertrophic cartilage oType XII: Fibril-associated, tendon, ligaments, some other tissues. oType XVII: Hemi desmosome oType XIX: Rhabdomyosarcoma All collagens are fibrous proteins made from three polypeptides called collagen α chains Unlike the α helix secondary structure of many proteins, the structure of collagen is a left-handed helix The three chains wrap around each other to form a right-handed triple helix The collagen triple helix can form because of an unusual abundance of three amino acids: glycine, proline and hydroxy proline They make up the characteristic repeating pattern Gly-X-Y, where X and Y can be any amino acid but are often proline and hydroxy proline and less often lysine and hydroxy lysine Glycine is essential because its small side chain, a hydrogen atom, is the only one that can fit into the crowded center of the three stranded helix Although the rigid peptidylproline and peptidyl -hydroxyproline linkages are not compatible with formation of a classic single-stranded helix, they stabilize the distinctive three-stranded collagen helix The hydroxyl group of hydroxyproline (by forming hydrogen bonding with members of other chains) helps hold its ring in a conformation that stabilizes the triple helix The triple helix of collagen IV is interrupted at several points by nonhelical segments that give flexibility to the structure Type IV collagen is a sheet/network forming type • Triple helices associate with each other through the globular domains at the C and N terminals COLLAGEN BIOSYNTHESIS Collagen is first synthesized as preprocollagen which contains a signal sequence In the lumen of the endoplasmic reticulum, preprocollagen is modified: removal of the signal sequence, hydroxylation of proline and lysine residues and glycosylation of some hydroxylysine residues preprocollagen is changed to procollagen In the Golgi apparatus procollagen is associated into a triple helix (initiated through disulfide bonds between C-terminal residues) and released to the extracellular space Extracellular peptidases remove the N- and C-terminal propeptides to give tropocollagen Tropocollagens associate laterally to generate fibrils In fibrils, adjacent collagen molecules are displaced from one another by about one-quarter of their length Striation is observed in electron micrographs The fibrils are strengthened through covalent cross-links Lysine and hydroxylysine side chains are modified by extracellular lysyl oxidases to form aldehydes (allysine and hydoxyallysine) in place of the amine group at the end of the side chain The aldehydes form covalent crosslinks with lysine, hydroxylysine and histidine residues in adjacent molecules The cross-links stabilize the side-by-side packing of collagen molecules and generate a strong fibril Removal of the propeptides and covalent cross-linking take place in the extracellular space to prevent the potentially catastrophic assembly of fibrils within the cell Type I, II and III collagen fibers are characteristic of the ECM of the (skin, bone and tendons); cartilage and arteries, respectively DISORDERS OF COLLAGEN SYNTHESIS Type I collagen deficient (Bone) : Osteogenesis imperfecta – “brittle bone syndrome” Heterogeneous group of inherited disorders distinguished by bones that easily bend and fracture Abnormally Short chains make up the collagen I triple helix ; or other amino acids may take the place of glycine, the structurally abnormal pro – α- chains → prevent folding of the protein into a triple-helical conformation C/F Blue sclera o Types • Type I OI is called Osteogenesis imperfecta tarda: present in early infancy, fractures secondary to minor trauma. • Type II OI is called Osteogenesis imperfecta congenita: more severe, patients die in utero or in the neonatal period of pulmonary hypoplasia, most patients with severe OI have mutations in the gene for either the pro 1- or pro 2- αchain of Type I collagen. Type II (cartilage): Chondrodysplasia Type III (major blood vessels): Ehlers-Danlos syndrome (Rubber man syndrome): is a heterogeneous group of CT disorders lack of peptidases; resistance to peptidases; insufficient synthesis of collagen; rapid degradation;… recurrent dislocation of joints (hypermobility), hyper extensible skin. NB: All types of collagen are defective (type III most commonly). o Types of EDS • Classica EDS: defect in pro α1 or pro α2 chains of collagen Type V in many, but not all, families; autosomal dominant • Hypermobility EDS: defects unknown; autosomal dominant • Vascular EDS: structural defects in pro α1 chain of collagen Type III encoded by the COL3A1 gene; autosomal dominant. • Kyphoscoliosis: deficiency of lysyl hydroxylase, a collagen modifying enzyme; autosomal recessive • Arthrochalasia: deficient processing of N-terminal end of pro α1 or pro α2 chains of collagen type I ; autosomal dominant • Dermatosporaxis: deficiency of procollagen I N-terminal peptidase; autosomal recessive Type IV collagen (basement membrane): Alport’s syndrome C/F haematuria, sensory neural hearing loss (SNHL), anterior lenticornus (excessive curvature of cornea) Autoimmune collagen disorders Good pasture syndrome: Auto-antibodies against basement membrane. o C/F: haematuria, haemoptysis Bullo-vesical disorders Bullous Pemphigus: auto-ab against hemi desmosome Pemphigus Vulgaris: auto-ab against desmosome + acantolysis Menke’s disease (deficiency of lysyl oxidase) Other abnormalities in collagen Homocystinuria • Accumulated homocysteine reacts with lysyl aldehydes to block cross linking • Skeletal deformities, vascular and ocular defects Deficiency of Ascorbic acid • Defective hydroxylation of collagen • Poor wound healing and increase fragility of blood vessels Lathyrism • Ingestion of lathyrus sativa • Contain toxic agent beta oxalyl amino alanine that inhibit lysyl oxidase Certain arterial aneurysms (collagen III) Ulrich muscular dystrophy (collagen VI) Certain chondrodysplasia (collagen IX and XI) Kniest dysplasia (collagen II) FIBRONECTIN Is a large molecular weight (about 440,000 Da) adhesive dimeric glycoprotein found in body fluids and connective tissues, one of the major component of ECM. Synthesized by a variety of cells including liver hepatocytes, lung fibroblasts, endothelial cells, macrophages and type 2 alveolar epithelial cells. The protein is composed of similar but not identical polypeptides Distributed widely in human body, found on cell surfaces, extracellular matrix, and basal laminae. Fibronectins are dimers of two similar polypeptides linked at their C-termini by two disulfide bonds Each chain comprises six functional regions with different ligand-binding specificities Fibrin binding domains, Heparin binding domains, RGD sequences, Cell surface receptor binding domains, Collagen binding domains, Heparin and Fibrin binding domains. Integrins are usually bound by repeats of Arg-Gly-Asp (RGD) sequences of fibronectins Fibronectin has a wide variety of biological functions including cell migration, cell differentiation and apoptosis, these biological activities are mediated through interaction with various members of integrin family and cell surface proteoglycans. Types of Fibronectin Circulating plasma fibronectin (soluble protomeric form): synthesized by hepatocytes. o Most of fibronectin in Adults is plasma fibronectin. o A soluble dimeric form, found in the blood and other body fluids, enhancing blood clotting, wound healing and phagocytosis. o Good indicator of pathological conditions associated with injury of the RES, radiation-induced lung injury, and in patients with Broncho-pulmonary carcinoma o Tumour metastasis, thereby making circulating fibronectin or its isoforms potential Tumour markers, similarly, increased plasma concentrations of fibronectin is associated with various carcinomas e.g. breast, colonic, lung and ovarian carcinomas. Locally synthesized cellular fibronectin (insoluble multimeric form) o Synthesized by several cells including epithelial and connective tissue cells and is deposited into the extra cellular matrix. oLocated on the cell surfaces it helps cell-cell attachment oDerived from endothelium which play an important role in assembling of sub endothelial matrix oPlays key roles in various cellular events that include cell migration and proliferation of fibroblasts and endothelial cells o Physiologically, plays an important role in cell adhesion, motility and tissue repair.