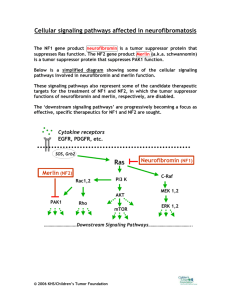
Overview Of Neurofibromatosis Flat patches all over the skin that are darker than the surrounding area, eyes dotted with freckle-like bumps. These are just some visible symptoms of the devastating genetic disorder Neurofibromatosis Type 1. No human being has asked to be born with the altered NF1 tumor suppressor gene. Unfortunately, NF1 does not skip a generation, so the NF1 gene continues to be passed to our future. Identifying NF1 requires awareness of the various symptoms patients experience and why they are experiencing them. Relevant topics that are covered in this review are how the symptoms of an NF1 patient correlate to an altered protein the mutated NF1 gene encodes, what's happening in the cells of a neurofibromatosis patient, and what kind of mutation occurred to create the altered NF1 gene. Nevertheless, understanding the inner workings of the NF1 disease requires knowledge of the genetic cause. The NF1 gene is a tumor suppressor located on chromosome 17 ( Figure 1). The NF1 gene is supposed to regulate cell growth; hence when mutations cause the NF1 gene on chromosome 17 to be inactive, cell growth becomes uncontrolled, resulting in Neurofibromatosis type 1 developing. Neurofibromatosis type 1 does not skip a generation; NF1 has an autosomal dominant inheritance pattern caused by a deletion in the NF1 gene (Figure 2). A parent with NF1 has a fifty percent chance of passing the abnormal NF1 gene to their child (Jett, et al., 2010). Patients with this type of NF1 are born with a mutated copy of the NF1 gene in each cell. In other autosomal dominant conditions, one altered copy is enough to cause that specific disorder. Still, with neurofibromatoses, two copies of the NF1 gene have to be mutated to trigger the formation of tumors in neurofibromatosis type 1 (Jett, et al., 2010). Neurofibromatoses Type 1 is one of the most common Mendelian disorders, which is caused by a heterozygous mutation in the NF1 gene, a mutated allele. The mendelian disorder’s in NF1 patients mean that germline mutations in a single gene are inherited from one of the biological parents. Moreover, the NF1 gene mutations have caused this genetic disorder that is passed down. Analyzing the mutation of NF1 is difficult because of how complex the NF1 gene is (Dong, et al., 2017). The cause of neurofibromatosis in patients is a spontaneous mutation that doesn't happen due to inheritance but from conception. A spontaneous mutation is “the net result of all that can go wrong with DNA during the life cycle of an organism" (Smith, 1992). More than eighty percent of disease-causing NF1 results from truncating mutations (Dong, et al., 2017). That means one gene copy has been inactivated or deleted, and the remaining gene copy cannot produce the needed gene product to maintain normal function. The cause of the NF1 gene being mutated could also be caused by a nonsense mutation, resulting in premature translation by a stop codon (Dong, et al., 2017). Moreover, these truncating mutations then cause the neurofibromin protein to be haploinsufficient. NF1 encodes for neurofibromin, a large protein with a chain of 2818 amino acids. The NF1 gene provides instructions to make the healthy protein neurofibromin. Neurofibromin is a crucial negative regulator in the ras signal transduction pathway (Zwarthoff, 1996). This negative regulator is essential because gene regulation is critical to normal development. The human body requires positive and negative regulators to turn genes on and off when needed (Zwarthoff, 1996). Neurofibromin is a tumor suppressor protein. It prevents cells from growing and dividing too rapidly or uncontrollably. This protein is produced in numerous types of cells. Neurofibromin exhibits its highest concentration in the nervous system in neurons, myelinating cells, non-myelinating Schwann cells, and the adrenal medulla (Bergoug, 2020). NF1 is a helical protein similar in shape to the Ras-GAP protein (Figure 3). Neurofibromin is a multidomain protein. A domain is a functional and structural unit in a protein. Since neurofibromin is a multidomain protein, each part fills its functions independently (Bergoug, 2020). (Zwarthoff, 1996) described that the absence of the healthy neurofibromin protein in cells of NF1 patients leads to an increase in the number of cells growing. This results in benign and, in a few cases- malignant tumors (Zwarthoff,1996). All because the NF1 gene has been mutated from premature termination during translation. Thus, the production of the shortened and, in some cases, absentee neurofibromin protein results in the symptoms of Neurofibromatoses type 1 patients. There are various symptoms NF1 patients experience due to the nonfunctional neurofibromin protein. Such as Lisch nodules, which are pigmented cells that develop on the irises, appear brown and yellow (Figure 4). Another symptom due to the altered neurofibromin protein is cafe au lait spots. These are spots on the body that are pigmented (Figure 4). NF1 patients have these spots due to the increase in melanin content in their cells from the mutated NF1 gene. The increase in melanin also freckles in the armpits and groin area. As NF1 patients age, their cafe au lait spot becomes increasingly more pigmented. The main symptom the alteration of the neurofibromin protein causes is the growth of tumors in the brain, along the nerves, and on the skin, which is called neurofibromas (Figure 4). The neurofibromas on the skin and nerves appear as bumps and can cause bone deformities in NF1 patients (Farshetchi, 2020) (Figure 5). These tumors occur because the altered neurofibromin protein is not functioning and thus can not regulate cell growth. NF1 patients have also been known to have learning disabilities and social and behavioral issues. The deformities in the central nervous system caused by the mutated NF1 gene could be the reason for these social and psychological problems in NF1 patients (Huijbregts, 2015). Incidentally, various treatments help manage these symptoms in neurofibromatosis Type 1 patients. Surgery on tumors that are damaging nearby organs and tissue is being done on neurofibromatosis patients. There are also treatments like stereotactic radiosurgery that don’t require an incision. Drugs such as gabapentin for nerve pain are also prescribed to NF1 patients. New research has led to the FDA approving the first-ever treatment for neurofibromatosis. The drug Koselugo that the FDA has approved is to be used on patients who exhibit neurofibromas. The generational malediction of Neurofibromatosis Type 1 is passed along from parents to children. Neurofibromatosis is a very complex disease that researchers still don’t fully understand. Neurofibromatoses Type 1 patient has multiple symptoms caused by the neurofibromin protein the NF1 gene has encoded for, as well as mutations in the NF1 gene. The more breakthrough research doctors do worldwide, the closer NF1 patients are to relief. References Bergoug, M., Doudeau, M., Godin, F., Mosrin, C., Vallée, B., & Bénédetti, H. (2020). Neurofibromin Structure, Functions and Regulation. Cells, 9(11), 2365. https://doi.org/10.3390/cells9112365 Dong, Y. Y., Zhang, Y. H., Li, H. W., Chen, L. Z., Wang, T. M., Hu, W., Hu, M., She, Q. Y., Liu, D. X., & Deng, Y. H. (2017). A Novel Frameshift Mutation in Neurofibromin 1 Gene in a Chinese Family with Neurofibromatosis Type 1. Chinese medical journal, 130(5), 629–630. https://doi.org/10.4103/0366-6999.200538 Friedman, J. (2019, June 6). Figure 1. [Café au lait macules]. Nih.gov; University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1109/figure/nf1.F1/ Huijbregts, S. C., Loitfelder, M., Rombouts, S. A., Swaab, H., Verbist, B. M., Arkink, E. B., Van Buchem, M. A., & Veer, I. M. (2015). Cerebral volumetric abnormalities in Neurofibromatosis type 1: associations with parent ratings of social and attention problems, executive dysfunction, and autistic mannerisms. Journal of Neurodevelopmental Disorders, 7(1). https://doi.org/10.1186/s11689-015-9128-3 Jett, K., & Friedman, J. M. (2010). Clinical and genetic aspects of neurofibromatosis 1. Genetics in medicine: official journal of the American College of Medical Genetics, 12(1), 1–11. https://doi.org/10.1097/GIM.0b013e3181bf15e3 Karmakar, S., & Reilly, K. M. (2017). The role of the immune system in neurofibromatosis type 1-associated nervous system tumors. CNS oncology, 6(1), 45–60. https://doi.org/10.2217/cns-2016-0024 AboutKidsHealth. (n.d.). Www.aboutkidshealth.ca. https://www.aboutkidshealth.ca/article?contentid=864&language=english Farschtschi, S., Mautner, V. F., McLean, A., Schulz, A., Friedrich, R. E., & Rosahl, S. K. (2020). The Neurofibromatoses. Deutsches Arzteblatt international, 117(20), 354–360. https://doi.org/10.3238/arztebl.2020.0354