Experiment
1
Purification of an Impure Acetanilide Sample by
Recrystallization and Determination of Purity by
Melting Point.
Objectives
To illustrate the concept of recrystallization and its application
To demonstrate the proper techniques in recrystallizing an impure organic compound
To illustrate the principles and proper techniques in the determination of melting point, one of the
physical properties of organic compounds
Reagents
Acetanilide (N-Phenylacetamide, C8H9NO), Impure Acetanilide (Acetanilide-sodium chloride-congo
red mixture), activated carbon, acetone (2-propanone, C3H6O), Ethyl alcohol (ethanol, C2H6O), and
toluene (Methylbenzene, C7H8).
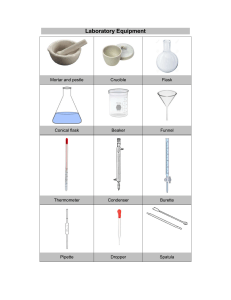
Materials
For Recrystallization
test tubes, beaker, graduated cylinders, medicine dropper, hot plate, Bunsen burner wire gauze,
iron ring, iron stand, test tube holder, stirring rod, short stem funnel, steam bath, Büchner funnel
with stopper, vacuum flask, crucible tongs and an ice water bath,
For Melting Point Determination
Mel-Temp apparatus, Glass plates, Spatula
Procedure
A. Determination of an Appropriate Solvent for Recrystallizing Acetanilide
Perform the succeeding procedure in testing the solubility of acetanilide in water [H 2O],
ethanol [CH3CH2OH], acetone {(CH3)2CO] and toluene [C6H5CH3]. Determine which solvent is most
suitable for recrystallizing acetanilide [C6H5NHCOCH3].
Place approximately 25 mg of acetanilide into a test tube containing 1 mL of solvent being
tested. Shake the tube and observe. If the substance fails to dissolve, heat the test tube gently under
a hot water bath. Do not heat longer than necessary to avoid undue evaporation of the solvent. If
undissolved solid still remains, add 1 mL more solvent and warm. Shake the test tube and observe
for the dissolution of acetanilide. Test for the solubility of acetanilide in water, CH3CH2OH, (CH3)2CO
and C6H5CH3.
Cool the contents of four test tubes (i.e. H2O, CH3CH2OH, (CH3)2CO and C6H5CH3) and
observe for any reappearance of the crystals. If crystals do not appear in any of the test tubes, add
a pin head-size acetanilide crystal into the solution or scratch the inside wall of the test tube beneath
the solvent surface. Which solvent do you observe will be most suitable for recrystallizing
acetanilide? Why? Report your findings to the instructor.
B. Recrystallization of Impure Acetanilide
Accurately weigh 1 g of impure acetanilide. Place it in an Erlenmeyer flask and add 50 mL
of the solvent that was observed to be suitable for recrystallizing the compound. Heat the mixture,
stirring it uniformly until it almost boils. If the solution is colored, add a pinch of activated charcoal
and continue heating to almost boiling. Check if the suspension is still colored by spotting a drop of
the mixture on filter paper. Add more activated charcoal if necessary. Meanwhile, prepare to filter the
hot suspension.
Prepare a filtration set-up (Figure 1) by folding a fluted filter paper and placing it on a funnel.
Use a 125-mL Erlenmeyer flask as the receiver. Pre-heat the set-up just prior to use by pouring hot
solvent into the filter paper. Why? Empty the flask and filter the hot suspension. It is essential that
this operation be completed as quickly as possible in order that cooling is minimized. Why? It is also
advisable to place only a few mL of the hot solution into the funnel, keeping the remainder of the
solution in the flask hot by heating over the Bunsen flame until it is transferred to the funnel.
Figure 1: Filtration Set-up using a Fluted filter paper.
If the filtrate remains to be colored, add a pinch of activated charcoal and heat. Refilter by
gravity filtration.
Allow it to cool slowly to room temperature undisturbed. Observe the appearance of
crystals. To induce crystallization of more acetanilide, chill the flask in an ice-water bath and scratch
the walls of the flask in contact with the solution.
While waiting for most of the acetanilide to recrystallize, assemble the suction filtration
apparatus (Figure 2) as follows: cut a circular piece of filter paper of such diameter that it exactly
2
covers the flat surface of the perforated portion of the Büchner funnel filter plate. Determine the
weight of this paper and place it in the Büchner funnel. Fit the Büchner funnel to a suction flask.
Figure 2: Suction Filtration Apparatus
When the suction filtration apparatus has been set-up, moisten the paper in the funnel
with a few drops of the solvent, (why?) and draw the solvent down through the suction. Now, filter
the crystals out of the solution by pouring it in the Büchner funnel and the solvent will eventually be
drawn inside the vacuum flask through suction. When the crystals are almost dry, release the
vacuum by disconnecting the tube from the suction flask (Do not turn the pump off!) and wash the
crystals with 5-10 mL cold solvent with stirring (use a stirring rod) for half a minute. Reconnect the
tube to dry the crystals.
To dry the crystals, transfer the crystals together with the filter paper into a watch glass,
cover it with another filter paper and keep it in the locker until the next laboratory period and
experiment (melting point determination). Weigh the dried crystals and filter paper to the nearest
0.01 gram. Determine the percentage of the pure crystals recovered from the impure sample.
C. Melting point determination
Determine the melting point of the recrystallized and impure acetanilide from the previous
experiment. Using the melting point apparatus, adjust the temperature setting from 10-20°C. Do not
set the temperature greater than this range. (Why?) Place very few amounts of the acetanilide
sample on the glass plate and place this on the heating plate of the apparatus. Observe the crystals
and record the temperature range at which the crystals start to melt until all of it has melted. Remove
the glass plate from the apparatus and clean it for the next person to use.
NOTE: Do not discard the glass plate. Properly dispose of its contents and return it to the technician.
Questions:
3
1.
Among the following solvents-water, acetone, ethanol, and toluene, which one was observed
to b e most suitable for recrystallization of acetanilide? Why?
2.
Why was a water bath used instead of direct heating in the choice of solvent?
3.
Why was activated charcoal added into the mixture of acetanilide, sodium chloride and
congo red? What is adsorption?
4.
Would it be wrong to dissolve the original impure material in a large excess of solvent?
Why?
5.
In gravity filtration, what is the advantage of using a short-stemmed funnel over a long-stem
funnel?
6.
Why were the funnel and the flask preheated before filtering the impure acetanilide?
7.
Why was the filter paper in the Büchner funnel moistened before pouring in the mixture
containing the acetanilide to be recrystallized?
8.
What is the purpose of washing the acetanilide crystals with a few mL of cold solvent? What
do you think would happen if the washing solvent was not cold?
9.
Why must the rubber tubing of the suction flask be disconnected before turning off the water
aspirator?
10.
Why is it necessary to use thick-walled tubing in connecting the suction flask to the
aspirator?
11.
At what steps in the recrystallization procedure did you lose some of the product? What can
you do in the future to avoid this losses?
12.
What is the effect of impurities on the observed melting point of acetanilide?
13.
Given the solubilities of the following compounds:
Water
Ethanol
Benzene
cold
hot
cold
hot
cold
Acetamide
s
s
s
s
ss
Aspirin
i
s
s
s
ss
Sample Y
i
s
s
s
i
where s means soluble; i means insoluble; and ss means slightly soluble
(A)
(B)
(C)
(D)
hot
s
ss
ss
Which of the solvents would be suitable for recrystallization of acetamide? Of aspirin?
Could acetamide be separated from acetanilide by recrystallization from water? Why or
why not?
Could aspirin be separated from acetanilide by recrystallization from water? Why or why
not?
Devise a recrystallization scheme for the purification of aspirin contaminated with small
amount of acetamide and sample Y.
4
Experiment
2
Separation of a Binary Mixture by Simple and
Fractional Distillation
Objectives
To present the concept of distillation and its applications
To illustrate proper techniques in distillation
Reagents
n-hexane (C6H14), toluene (C7H8)
Materials
50-mL pear-shaped flask, simple and fractional distillation set-ups, thermometer, graduated
cylinder, small test tubes (10x75mm), 2-3 utility clamps, hot plate or burner, oil bath, wire gauze,
beaker, capillary tubes, rubber band, iron ring and iron stand.
Procedure
Separation of Hexane and Toluene by Simple Distillation
Acquaint yourself on the parts of an apparatus for simple distillation (Figure 1). Start with
the burner, followed by an iron ring and wire gauze placed 4-5 cm above the burner. Place a 100mL beaker on top of the wire gauze. Clamp a 100-mL pear-shaped flask (or a round-bottom flask)
to an iron stand protecting the neck of the flask with a few layers of tissue paper. A semimicroscale set-up for distillation may have a one-piece attachment that already includes the air
column, the distilling head, the thermometer holder, the condenser, and the receiver. In this case,
place a thin film of grease crosswise at the male ground-glass joint (upper portion) of the column
and insert it to the female ground-glass joint of the pear-shaped flask. Note: Do not use excess
grease, as it will contaminate the sample. Prior to the attachment of the distilling section, make
sure that the delivery tubes of the condenser are attached properly (water inlet is near the receiving
end and the outlet near the Claisen head, Why?) and securely. Support the condenser with another
utility clamp making sure that the condenser is padded with tissue paper. Place a few drops of oil
at the thermometer holder before placing the thermometer in. Note: these instructions are based on
the available set-up. If you are provided with individual parts, consult any literatures on
experimental organic chemistry for the set-up of a simple distillation apparatus. Make sure that all
connections are secured. Have a slow stream of water circulate through the condenser and use a
clean and dry 10-mL graduated cylinder as a receiving flask to collect the first few mL to be
discarded (forerun). Have the instructor check your apparatus at this point.
5
Figure 1: Actual Apparatus for Simple Distillation
Place 30 mL of n-hexane and 30 mL of toluene into the 100 mL round bottom flask. Make
sure to do this while the round bottom flask is not placed in the set-up, avoiding any spill onto an oil
bath/water bath.
Bring the mixture to a boil. When liquid begins to drop into the receiver, adjust the heat so
that the drops come steadily at a rate of about one second per drop. Discard the first 1mL and
record the temperature every 1 mL fractions as the distillation proceeds until 20 mL of the distillate
are collected. Transfer all distillates having the same boiling point into a test tube.
If distillation is thru, turn off the burner and lower the oil bath. Never heat the distilling flask
to dryness and do not stop the circulating water until the mixture is no longer hot.
6
Separation of Hexane and Toluene by Fractional Distillation
Acquaint yourself on the parts of an apparatus for fractional distillation (Figure 2): Actual
Apparatus for Simple Distillation. The only difference in set-up is the type of column used. Place a
100-mL beaker on top of the wire gauze. Clamp a 100-mL pear-shaped flask (or a round-bottom
flask) to an iron stand protecting the neck of the flask with a few layers of tissue paper. A semimicroscale set-up for distillation may have a one-piece attachment that already includes the
fractional column, the Claisen head, the thermometer holder, the condenser, and the receiver. In
this case, place a thin film of grease crosswise at the male ground-glass joint (upper portion) of the
column and insert it to the female ground-glass joint of the pear-shaped flask. Note: Do not use
excess grease as it will contaminate the sample. Prior to the attachment of the distilling section,
make sure that the delivery tubes of the condenser are attached properly (water inlet is near the
receiving end and the outlet near the Claisen head, Why?) and securely. Support the condenser
with another utility clamp making sure that the condenser is padded with tissue paper. Place a few
drops of oil at the thermometer holder before placing the thermometer in. Note: these instructions
are based on the available set-up. If you are provided with individual parts, consult any literatures
on experimental organic chemistry for the set-up of a simple distillation apparatus. Make sure that
all connections are secured. Have a slow stream of water circulate through the condenser and use
a clean and dry 10-mL graduated cylinder as a receiving flask to collect the first few mL to be
discarded (forerun). Have the instructor check your apparatus at this point.
Figure 2: Actual Apparatus for Fractional Distillation
Place 30 mL of n-hexane and 30 mL of toluene into the 100 mL round bottom flask. Make
sure to do this while the round bottom flask is not placed in the set-up, avoiding any spill onto an oil
bath/water bath.
Bring the mixture to a boil. When liquid begins to drop into the receiver, adjust the heat so
that the drops come steadily at a rate of about one second per drop. Discard the first 1 mL and
7
record the temperature every 1 mL fractions as the distillation proceeds until 20 mL of the distillate
are collected. Transfer all distillates having the same boiling point into a test tube.
If distillation is thru, turn off the burner and lower the oil bath. Never heat the distilling flask
to dryness and do not stop the circulating water until the mixture is no longer hot. Compare data
gathered from simple and fractional distillation.
Questions
1. Plot the boiling point vs volume distilled for both the distillation of hexane-ethyl acetate mixture
with and without column on the same graph. Label the curve On the basis of these curves,
which procedure was more efficient at separating the mixture into its components?
2. Would a longer fractionating column be more efficient in separating mixtures of liquids than a
shorter column? Why? Give advantages and disadvantages of using a longer column over a
shorter one.
3. Why is better separation of two liquids achieved by slow rather than fast distillation?
4. In a fractional distillation, is the composition of the vapor just above the surface of the liquid
the same as the vapor near the thermometer bulb? Explain.
5. Why is it important to have the cooling water enter the condenser jacket at the lower end and
exit at the upper end rather than have it flow in the opposite direction?
6. What is the purpose of adding boiling chips to the distilling mixture?
7. Why should it be dangerous to heat an organic compound in a distilling apparatus that was
closed tightly at every joint and had no vent or opening to the atmosphere or to vacuum
pump?
8. Why should a distilling flask at the beginning of distillation be filled to not more than two-thirds
of its capacity?
8
Experiment
3
Extraction: Determination of its Efficiency & Calculation
of the Distribution Coefficient
Objectives
To illustrate the concept of extraction and its application
Reagents
Acetic Acid (CH3COOH), Toluene, sodium hydroxide, and phenolphthalein
Materials
Burette, Pipette, Aspirator, 125-mL Erlenmeyer flasks, Separatory Funnel, beaker, Iron ring and
Iron stand, volumetric flask.
Procedure
Measure 2mL Acetic Acid and put it in a 50-mL beaker, add 20-mL distilled water and stir.
Place the contents of the beaker in a 250mL volumetric flask then dilute up to the mark with distilled
water. Stopper the flask then mix the solution thoroughly. Into three (3) separate 125mL Erlenmeyer
flasks, pipet 20mL of the Acetic Acid solution into each flask. Label the flasks as 1, 2 and 3.
Determination of the Number of Grams of Acetic Acid in a 20-mL Aliquot
Fill a burette with 0.1 M standard NaOH solution. Withdraw enough solution to remove the
air from the jet tip and bring the liquid into a graduated region of the burette. Record the initial volume
of the NaOH solution in the burette. Add 2 drops of phenolphthalein into flask 1 and then titrate with
the 0.1 M standard NaOH solution. Swirl the flask while titrating. Add the base solution drop by drop
near the end of the operation, until the last drop of base turns the solution in the flask to pink. Record
the final burette reading. Calculate the number of grams of Acetic Acid that are dissolved in the 20mL aqueous solution.
Determination of the Number of Grams of Acetic Acid extracted by one 20-mL portion of Toluene
Place the contents of flask 2 in a separatory funnel (Figure 1), and then add 20-mL
Toluene. Shake the funnel with intermitted release of pressure for several minutes. Place the
funnel upright, with the stopcock closed and the stopper removed. Allow the two layers to separate
completely.
9
s
Figure 1: Separatory Funnel used for Extraction”
Drain the lower aqueous layer into a clean 125-mL Erlenmeyer flask, add 2 drops of
phenolphthalein and again titrate the solution with the 0.1 M standard NaOH solution. Dispose the
other layer from the top of the funnel (Why?) and into the organic waste disposal bottle.
Calculate the number of grams of Acetic Acid that remained in the aqueous layer. By
subtracting the calculated grams of Acetic Acid in the aqueous layer from the results obtained in the
first part, determine the weight of the acid that was extracted into the toluene layer then determine
the distribution coefficient.
Determination of the Number of Grams of Acetic Acid extracted by two 10-mL portions of Toluene
Repeat the procedures from the second part, but this time extract the remaining 20-mL
aliquot of Acetic Acid solution in flask 3, first with only 10-mL Toluene. Collect the lower aqueous
layer in a clean beaker and dispose the other layer from the top of the funnel and into the organic
waste disposal bottle. Put the aqueous layer that is in the beaker back into the separatory funnel and
extract with another 10-mL portion of fresh toluene. Now drain the lower aqueous layer into a clean
125-mL Erlenmeyer flask then add 2 drops of phenolphthalein. Titrate the contents of the flask till the
endpoint (pink). Perform the same calculations that were done in the second part.
10
11
Experiment
4
Identification of Common Analgesic Drugs by Thin
Layer Chromatography
Objective
To perform thin-layer chromatography and calculate Rf values
To utilize thin-layer chromatography to identify the analgesic compound(s) present in an unknown
sample of an over-the-counter painkiller preparation
To learn concepts of chromatography, polarity of molecules and intermolecular forces of attraction.
Reagents
Solutions of analgesics in methanol (aspirin, paracetamol, ibuprofen, caffeine), unknown OTC
analgesic tablet, methanol, solvent mixture (25 parts ethyl acetate: 1 part ethanol: 1 part acetic
acid)
Materials
Small beaker (50-mL), aluminum foil or plastic wrap to cover beaker, test tubes, stirring rod, glass
capillary tubes for spotting, plastic TLC sheet, about 5 cm ⋅ 10 cm, ultraviolet lamp, ruler and pencil
Procedure
Get the TLC plate from the stock room. This plate consists of silica gel on a flexible plastic
or aluminum sheet. Handle it with care (do not touch the silica face, handle it on the sides) and lay it
over a notebook. Mark lightly with a pencil a straight line 0.5 cm from one end. Make a light pencil
mark or a dot on the straight line you drew.
Dip the spotter to the corresponding sample solution so that it partly fills with liquid. Spot
the solution on the dot previously marked on the TLC plate. The spot should not exceed 1mm in
diameter. The amount of liquid coming out of the capillary can be controlled by holding the uppermost
end of the capillary. (General rule: the smaller the spot the better). You can make the spot
concentrated by repeatedly touching the plate. Allow the spot to dry thoroughly. The capillary is
cleaned by spotting the excess liquid on a sheet of tissue paper and rinsed with technical grade
acetone.
Add enough of the solvent mixture (25 Ethyl acetate: 1 Ethanol: 1 Acetic acid) to give a thin
layer of solvent in the bottom of the chromatographic chamber (50-mL beaker). To provide an
atmosphere saturated with solvent inside the container, place a piece of filter paper around the
inside surface of the container, extending into the solvent. Then cover the container with the plastic
wrap, foil or screw cap and set it aside while preparing the chromatographic sheet.
Place the TLC plate in the chromatographic chamber (Figure 1) in such a way that the
spot(s) is/are not immersed in the solvent and the sheet is in an upright position (The pencil mark is
located at the bottom). Return the cover on the mouth of the beaker (Why?). When the solvent front
in the plate is about 0.5 cm from the top, remove the plate from the chamber and mark the solvent
front with a pencil. View the spots on the TLC plate by using the appropriate visualizing agent. (UV
lamp/Iodine chamber). Calculate the Rf value of the spot(s) in your TLC profile.
12
Rf = distance traveled by the substance = distance to center of spot
distance traveled by the solvent
distance to solvent front
Figure 1: Chromatographic chamber for Thin Layer Chromatography
Caution: UV radiation is harmful to your eyes. Do not stare directly at the UV lamp.
QUESTIONS:
1. Suggest possible advantages and disadvantages of using a longer (taller) TLC sheet?
2. Why do you think it was important to use a very small amount of sample when spotting the plate?
3. The relative movement of components is controlled partially by the polarity of the molecules. The
TLC sheet is coated with a highly polar substance, whereas the solvent mixture has a much lower
polarity. From your chromatographic results, predict the relative polarities of aspirin, paracetamol,
ibuprofen and caffeine by arranging them in order of increasing polarity. Explain your reasoning.
13
Experiment
5
Column Chromatography of Food Dye.
Objectives
To illustrate the concepts of chromatography and its applications
To illustrate the proper techniques involved in thin layer chromatography (TLC).
Reagents
Water saturated 2-butanol with acetic acid, ammonia in butanol, 1 part 1-butanol 1 part acetic acid,
2 parts methanol 1 part water, dye mixture (Indigo dye)
Materials
Thin Layer Chromatography plates (Silica plates), capillary tubes, small test tubes (10x75mm), watch
glass, beaker, 10-mL graduated cylinder, TLC plates, medicine droppers, filter paper.
Procedure
To choose an appropriate solvent for Column Chromatography, choose a solvent system
that gives a TLC profile where majority of the spots (or the target spot to be separated) is within a Rf
value of 0.3-0.5 (range). The choice of solvent must then have a lower polarity than this solvent that
gave an Rf value of 0.3-0.5 for its TLC profile. Why?
Separation of Pigments by Column Chromatography with TLC monitoring
Clamp the column upright (Figure 2), and insert a very small wad of cotton and tap it gently
into a constriction. You may add a small amount of sand to level bottom. Place 2 g of alumina/silica
on a piece of filter paper and pour it into the column in a slow stream. (Make sure that no
alumina/silica adheres to the side of the column where it is dry. You may tap the column gently with
an aspirator to make it compact. Make sure that the column bed is flat. Add enough sand to cover
the surface area of the column bed (~3mm).
In separate test tube, place about 10-15 mL of the chosen solvent. In another test tube,
place 10-15 mL of the chose solvent but with higher polarity. To be able to do this, mix 75% of the
chosen solvent with 25% of another substance with higher polarity. Prepare the next set of solvents
by adjusting the percentages of each substance in the mixture. (50:50, 75:25 and 100:0)
Place approximately one-half milliliter (~0.5 mL) of the dye solution into the column when
ready. Drop the sample mixture with a medicine dropper against the side of the column in a circular
motion. Do not directly drop the sample onto the column bed (Why?). Add the chosen solvent in such
a way that as the liquid extract touches the column bed, it is replenished immediately with the solvent,
thereby avoiding drying up of the column. The solvent used for elution should be changed only when
there is little or no separation of bands on the column. Moreover, the solvent must be used in order
of increasing polarity, taking note of the total volume used for each.
14
Figure 2: Actual Apparatus for Column Chromatography
As bands begin to separate and move down the column, place the empty test tube under
the column to collect the separated pigment solution as they are eluted. Change test tube as pigment
start to come out or when a pigment has completely been collected. After collection, take the TLC
profile of the pigments collected in a single TLC plate. Make sure that they are sufficiently apart
(when you spot) so that they don't mix when developing your TLC.
15
Experiment
6A
Alcohols and Phenols
Objectives
To illustrate some general chemical properties of alcohols and phenols.
To present the different tests involved in distinguishing alcohols and phenols from other
organic compounds.
Reagents
95% ethyl alcohol, isopropyl alcohol, tert-butyl alcohol, acetyl chloride, Lucas' reagent,
anhydrous magnesium sulfate, 1% potassium permanganate, litmus paper, sodium metal,
bromine water, 1% potassium dichromate, 1% ferric chloride solution, ceric nitrate, phenol,
resorcinol, hydroquinone, salicylic acid, glacial acetic acid, 6M sodium hydroxide solution,
6M sulfuric acid, 10% sodium bicarbonate solution, chromic acid, acetone
Materials
medicine droppers, test tubes, test tube rack, test tube holder, beakers, graduated cylinder,
crucible tongs, cork stopper, and sand bath.
Procedure
Alcohols and Phenols
The activity of alcohols is mainly due to the relative reactive hydroxyl group rather
than those of the comparatively inert alkyl substituents.
Ceric Nitrate Test for Alcohols: The ceric nitrate test uses cerium ammonium
nitrate in nitric acid to test for alcohols. Alcohols cause the reagent to change from yellow to
red. However, it can only be used for alcohols with ten or fewer carbons.
To each of three small test tubes, add approximately 1 mL of ceric nitrate solution.
Add 10 drops of ethyl alcohol into the first tube and into test tubes two and three, add
isopropyl and tert-butyl alcohols, respectively. Mix thoroughly and note if the yellow color
changes to red. Compare the results with a fourth test tube containing 0.5 mL of water with
1 mL ceric nitrate.
Chromic Acid Test for Distinguishing 1° and 2° Alcohols from Tertiary
Alcohols: The Chromic acid test is a rapid method for distinguishing primary and secondary
alcohols from tertiary alcohols. The reagent is very corrosive and is often used as a cleaning
solvent. It is prepared by combining solutions of sodium dichromate and sulfuric acid. Also
giving positive results with aldehydes and some enols, the reagent gives dark-colored
solutions with phenols.
Transfer 5 drops or 50 mg of test sample into 1 mL of reagent grade acetone.
Cautiously add 4 drops of chromic acid reagent to the resulting solution and observe for the
16
discoloration of the reagent. The appearance of a green to blue coloration within 5 seconds
verifies a positive result indicating the alcohol has been oxidized. Try using phenol as a test
sample instead of the three alcohols. Give chemical equations for the chromic acid oxidation
of primary and secondary alcohols.
Lucas Test for Distinguishing 1° and 2° and 3° Alcohols: The Lucas test makes
use of ZnCl2 in concentrated hydrochloric acid to differentiate primary, secondary and tertiary
alcohols. It only works for samples that are soluble in the reagent. This generally means that
it may not be applied for alcohols exceeding 6 carbons.
To each of three small test tubes add approximately 1 mL of Lucas reagent. Add
into the first tube, 10 drops of ethyl alcohol. Into test tubes two and three, add isopropyl and
tert-butyl alcohols, respectively. Stopper the test tubes and then shake them vigorously.
Note the length of time it takes for the mixture to become cloudy or separate into two layers.
A tertiary alcohol reacts rapidly and thus gives observable results quickly. Secondary
alcohols usually show signs of a reaction within five minutes: while primary alcohols remain
clear for several hours. Present chemical equations for the reactions and explain the rate by
which each alcohol formed an immiscible layer.
Phenols, unlike alcohols, have hydroxyl groups bound to a carbon atom that forms
part of the aromatic ring. This feature greatly alters the properties of the phenolic hydroxyl
and serves to distinguish it from hydroxyl groups of alcohols.
Acidity of Phenols: Dissolve 50 mg or 5 drops of phenol into 0.5 mL of distilled
water and test the solution with litmus. Pour the solution into 1 mL of 10% sodium
bicarbonate solution and observe for the evolution of a gas. Repeat the test using 10 drops
of glacial acetic acid and ethyl alcohol instead of phenol and determine which of the sample
is most acidic.
Ferric Chloride Test for Water Soluble Phenols: Dissolve a drop of phenol in 1
mL of ethyl alcohol. Transfer 4 drops of freshly prepared 1% ferric chloride into this solution
and observe. Repeat the test using resorcinol, salicylic acid, hydroquinone, acetic acid and
ethyl alcohol instead of phenol.
17
Experiment
6B
Aldehydes and Ketones
Objectives
To illustrate the chemical properties of aldehydes and ketones.
To distinguish aldehydes and ketones from other organic compounds by qualitative analysis.
Reagents
95% ethyl alcohol, isopropyl alcohol, tert-butyl alcohol, acetyl chloride, Lucas' reagent, anhydrous
magnesium sulfate, 1% potassium permanganate, litmus paper, sodium metal, 1% ferric chloride
solution, phenol, resorcinol, hydroquinone, acetic acid, concentrated hydrochloric acid, 6M sodium
hydroxide solution, 3M sulfuric acid, 10% sodium bicarbonate solution, 1% sodium dichromate,
chromic acid, acetone, methanol, 2,4-Dinitrophenylhydrazine, Schiff's reagent, Tollen's reagent,
Benedict's solution, Fehling's solution A and B, Iodoform reagent, sodium bisulfite solution, 5%
sodium nitroprusside solution, 3% sodium hydroxide solution, 0.1M potassium permanganate, ferric
chloride, 5% ammonium hydroxide solution, benzaldehyde, cyclohexanone and formaldehyde.
Materials
medicine droppers, test tubes, test tube rack, test tube holder, filter paper, beakers, graduated
cylinder, crucible tongs, cork stopper, hot plate, sand bath, and Bunsen burner.
Procedure
Aldehydes and Ketones
The activity of aldehydes and ketones depend mostly on the reactive carbonyl group and αhydrogen, but the oxidative properties of aldehydes is what differentiates it from ketones. While many
ketones are easily oxidized, various tests are available to distinguish the two carbonyl compounds
from one another. In the succeeding activities, the general properties of aldehydes and ketones will
be illustrated. The test samples will include: acetone, formaldehyde, cyclohexanone and
benzaldehyde.
Oxidation of Aldehydes with Potassium Permanganate: Place 5 drops of 1% potassium
permanganate into a small test tube containing 1 mL of distilled water. To this solution, add 4 drops
of test sample. Check for the discoloration of the permanganate solution after intermittent shaking
for 5 minutes. If the solution remains unreacted, introduce 3 drops of 6M sodium hydroxide. Explain
your observations and write chemical equations for the reactions.
2,4-Dinitrophenylhydrazine Test for Aldehydes and Ketones: Aldehydes and ketones
readily react with 2,4-dinitrophenylhydrazine to give the yellow phenylhydrazone derivative. The
melting points of some of these derivatives are available in handbooks or manuals. These may be
compared with the derivatives of an unknown carbonyl compound to serve as confirmatory test.
Measure 1 mL of 2,4-dinitrophenylhydrazine and transfer it to a small test tube. Add 2 drops
of test sample to the test tube and shake. Observe for the appearance of a yellow precipitate. Repeat
18
the procedure using acetic acid and ethanol as test samples. Write a general equation for the
reaction.
Schiff's Test for Aldehydes: The Schiff's reagent is an aqueous solution of an organic
dye, p-rosaniline hydrochloride, which is then acidified with HCl to form a colorless bis-N-sulfinic
acid. It reacts with the aldehyde to give an unstable complex that liberates a pink or purple dye.
Measure 1 mL of Schiff's reagent into 4 separate small test tubes. Add 4 drops of test
sample to the reagent and swirl. Observe for the appearance of a pink to purple coloration. If the
compound is insoluble or immiscible in the reagent, stopper the tube with cork and shake it
vigorously. Record and interpret the results. Write a general chemical equation for the reaction.
Tollen's Test for Aldehydes: Tollen's reagent is a solution of silver nitrate in concentrated
ammonium hydroxide. Aliphatic and aromatic aldehydes reduce the reagent to metallic silver. A
Silver mirror or a black precipitate of silver constitutes a positive result. Because it decomposes on
standing and deposits as a highly explosive residue, the reagent should be prepared just before use
and it should not be stored.
Examine the reaction of Tollen's reagent with 4 drops of the test sample. If no reaction
occurs at room temperature for 2 minutes, heat the test tube at 60°C. Observe the appearance of a
black precipitate after 3 minutes. Write a chemical equation for the reaction.
Iodoform Test for Methyl Ketones. The reagent for the test contains iodine in potassium
iodide. It gives the yellow iodoform derivative with methyl ketones. However, due to its reactivity
towards acetaldehyde and compounds that are oxidizable to methyl carbinols the test is only valid if
the compound in question is undoubtedly a ketone.
Prepare the iodoform derivative of acetone by introducing 4 drops of the sample into a test
tube containing 2 mL of 6M sodium hydroxide. Add enough iodine-potassium iodide solution to cause
a persistent pink coloration. Heat the tube at 60°C for a minute and check if the solution is
decolorized. Add more of the reagent to keep the solution colored. Continue heating and carefully
observe for any signs of a precipitate. Repeat the test using isopropyl alcohol as the test sample.
Write chemical equations for the reactions.
19