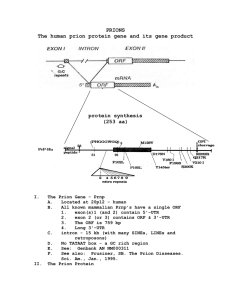
GROUP – 2 PRESENTATION TOPIC: PRESENTER: MEMBERS OF GROUP 2 NO NAME NO NAME 1 Mustapha Alpha – 5697 11 Umaru Barrie - 2 John Joe Sandy – 5478 12 Oredola Williams – 5613 3 Musa L. Kamara – 5557 13 Adetunji Wilson-Taylor - 4 Ahmad T. Garber – 5611 14 Yusuf Marah – 5432 5 John M. Koroma – 5722 15 Amara Conteh – 5839 6 Sukainatu Deen - 7 David F. Godwin – 5426 8 Prince Benjamin – 5721 9 Emmanuel Ucheya – 5470 10 Sylnata Johnson - ORDER OF PRESENTATION 1. Brief overview of Prion Diseases 2. Creutzfeldt-Jakob Disease (CJD) PRION DISEASES Are also called TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES (TSE). They are rare progressive, fatal, and untreatable neurodegenerative disorders that affect both humans & animals. They result from protein changes into an abnormal form called prion. Prions are much smaller than viruses and differ from viruses, bacteria, and all living cells because they do not contain any genetic material. PRION DISEASES Cont. In prion diseases, a normal protein called cellular prion protein (PrPC) changes shape (misfolds) and becomes abnormal. This abnormal substance is now called scrapie prion protein (PrPSc), or prion. PrPC misfolding Irreversible PrPSc (Prion) Scrapie refers to the prion disease first observed in sheep. Mutations in the gene for the prion protein can cause a misfolding of the dominantly alpha helical regions into beta pleated sheets. PRION DISEASES Cont. The newly formed prions resist breakdown by enzymes in the brain. Thus, they slowly accumulate. The body defense system remove these PrPSc aggregates leaving behind holes When samples of brain tissue are viewed through a microscope, they somewhat resemble Swiss cheese or a sponge (hence, the term spongiform). This causes degeneration of the brain cells leading to mental changes and ultimately, death. Types of Prion Diseases Creutzfeldt-Jakob disease (CJD), Gerstmann-SträusslerScheinker syndrome (GSS), Fatal Familial Insomia (FFI), Kuru in humans as a result ritualistic cannibalism in Papua New Guinea – relatives prepared and consumed the tissues (including brain) of deceased family members. Types of Prion Diseases Bovine Spongiform Scrapie in sheep. encephalopathy (BSE mad cow disease) Chronic Wasting Disease (CWD) in elk and deer, Alpers’ Syndrome in infants is also thought to be a transmissible spongiform encephalopathy caused by a prion. (Nwanebu F. C. et al. 2008) Prion Characteristics No antibiotics can cure disease caused by prions. They are not typical of a prokaryotic organism or a eukaryotic organism. All that is present in this pathogen is the protein PrPSc, the mutation of PrPC PrPSc is resistant to any form of digestion. Prions are non-immunogens and do not induce an immune response. Prions are not easy to decompose biologically They are resistant to high temperatures & disinfectants Creutzfeldt-Jakob disease (CJD) Creutzfeldt-Jakob disease (CJD) Bovine Spongiform Encephalopathy (BSE) was first described in the UK in 1985 as a prion-based disease that affected cattle. In 1996, similar condition was detected in a human being. It was then referred to as “Human form of mad-cowdisease” The BSE was later named Creutzfeldt-Jakob disease (CJD) The disease was named after a German neurologist Hans Gerhard Creutzfeldt and Alfons Maria Jakob. CJD is characterized by progressive deterioration of mental function, leading to dementia, involuntary jerking of muscles (myoclonus), and staggering when walking (ataxia). The disease usually occurs spontaneously but may result from eating contaminated beef or from inheriting an abnormal gene. CLASSES OF CJD Sporadic CJD Variant CJD (vCJD) Acquired CJD Kuru Familial CJD Iatrogenic CJD A. SPORADIC CJD – – – – Is the most common form accounting for 85% of cases of CJD. affects about 1 in a million people each year globally. It usually affects people over 40 years old. No cause is known. B. FAMILIAL CJD – Accounts for about 15% of the cases of CJD. – Familial CJD is often inherited and usually starts at an earlier age. – Results from a mutation in the gene for prion protein, which causes normal PrPC to change shape (misfold) and become disease-causing prion . – Lasts longer than sporadic CJD. C. ACQUIRED CJD – CJD can be acquired by eating beef or beef products from cattle who have bovine spongiform encephalopathy (mad cow disease). – The most common types are: kuru, variant & iatrogenic CJD. – Variant CJD usually begins around age 30 or younger, (in contrast to sporadic CJD, which usually begins after 40 yrs of age). i. Variant CJD (vCJD) - caused by the consumption of food contaminated with prions especially from infected cow/sheep. ii. Iatrogenic CJD - from contamination with tissue from an infected person, usually from: Infected blood transfusion, use of human-derived pituitary growth hormones, gonadotropin hormone therapy, corneal and/or meningeal transplants, or Surgical instruments. iii. Kuru - from cannibalism of deceased family members (Papua New Guinea). EPIDEMIOLOGY Countries that have confirmed human cases of vCJD Countries that have BSE cases EPIDEMIOLOGY Although CJD is the most common human prion disease, it is still rare. It occurs worldwide at a rate of about 1 case per million population per year. In the United States, CJD deaths among persons younger than 30 years of age are extremely rare (fewer than five deaths per billion per year). In more than 85% of cases, the duration of CJD is less than 1 year (median: four months) after onset of symptoms. (CDC, 2008) By the end of 2000, nearly 90 people had died from the disease in the UK. Deaths from vCJD have also been reported in France & Italy. PATHOPHYSIOLOGY Normal protein called cellular prion protein (PrPC) changes into scrapie prion protein (PrPSc)/prions. PrPSc has the ability to convert other PrPC to itself. A chain reaction follows, resulting in a cluster of tangled, nonfunctional proteins called plaques, which are aggregates of PrPSc in the brain. The body defences remove these PrPSc aggregates leaving behind holes This causes degeneration of the brain cells leading to mental changes and ultimately, death. CLINICAL MANIFESTATION The symptoms of CJD are caused by the progressive death of the brain cells. The first symptom is rapidly progressive dementia leading to memory loss, This is followed by personality changes and hallucinations. Other frequently occurring features include: Changes in behavior, lack of coordination and the ability to walk straight (ataxia), memory loss and visual impairment. In later stages: Mental deterioration becomes very obvious, with unintentional movements (myoclonus), Blindness, weakness of limbs, and infected persons may even go into a coma. DIAGNOSIS CJD is suspected when there are rapidly progressing dementia with myoclonus. Further investigation can then be performed to support the diagnosis including. 1. 2. 3. 4. 5. Electroencephalography - usually done to check for characteristic abnormalities in the electrical activity of the brain. (However, these changes occur late in the disease ) CSF analysis for 14-3-3 protein - these proteins are often (but not always) present in people with CJD & they may be present in many other disorders. Magnetic Resonance Imaging (MRI) - It can detect characteristic changes in the brain, including some that occur only in people with variant CJD. Blood test for PrPSc - Research have showed that PrPSc could be detected in the blood of animals long before the symptoms appeared. Biopsy of brain tissue is the definitive diagnostic test for all other forms of prion disease. TREATMENT Currently, CJD cannot be cured, and its progress cannot be slowed. However, certain drugs may be given to relieve symptoms: Valproate - an anticonvulsant agent, Clonazepam and benzodiazepine - reduce muscle jerks & anxiety. General support and care for the person and family members are important PREVENTION Don’t feed cattle with animal bi-products. Watch to make sure you are feeding your animals safe feeds. All imported meat must be inspected. Infected countries should not export cattle or meat to uninfected areas. Animals suspected of the disease should be quarantined Active National Disease Surveillance system. 1. REFERENCES Chakraborty C. et al. (April 2005). "Prion disease: a deadly disease for protein misfolding". Current Pharmaceutical Biotechnology 6 (2): 167–77. 2. Obi RK, Nwanebu FC (2008). "Prions And Prion Diseases". African Journal of Clinical and Experimental Microbiology 9 (1): 38–52. 3. Ironside, J. W. et al. (1996). "A new variant of Creutzfeldt–Jakob disease: neuropathological and clinical features.". Cold Spring Harbor symposia on quantitative biology 61: 523–30 4. Niimi Y, et al. (December 2008). "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course". Neuropathology 28 (6): 645–51. 5. WHO. (Feb 2012) "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" 6. Lugaresi E. et al. Oct 16, 1986). "Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei". The New England Journal of Medicine 315 (16): 997–1003. 7. Peden A. H. et al (2004) "Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient". Lancet 364 (9433): 527–9. 8. Emerging Infectious Diseases: "Is Creutzfeldt-Jakob Disease Transmitted in Blood?" (Ricketts et al) vol 3, Jun 1997 9. "CJD (Creutzfeldt–Jakob Disease, Classic)". Centers for Disease Control and Prevention. 2008-02-26. Retrieved 2009-06-20. 10. Corinne Ida Lasmézas et al. (2013). "Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents". Proceedings of the National Academy of Sciences 110 (17): 7044. THANK YOU!!!