Experiment 2: Fractional Distillation of a Mixture of Two Unknowns
advertisement

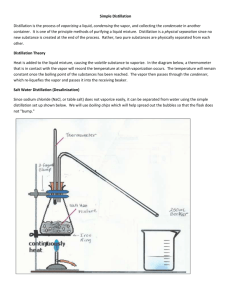
Experiment 2: Fractional Distillation of a Mixture of Two Unknowns Background: Boiling is a process familiar to anyone who has cooked pasta or brewed tea. As heat is applied to a pan of water, the temperature of the water increases until it reaches 100°C (212°F). At this temperature, additional heat causes the water to bubble vigorously as the liquid water is converted into gaseous water, or steam. Most organic liquids will behave in a similar fashion. On heating, the temperature of the liquid increases until the boiling point is reached. Additional heating causes the liquid to vaporize accompanied by vigorous bubbling of the liquid. The boiling point of a substance is a physical property of a substance and can be useful for characterizing that substance. The fact that different substances have different boiling points allows us to separate them. The process of heating a substance until it is vaporized, cooling the vapors, and collecting the condensed liquid is the basis of a commonly used purification technique called distillation. Vapor Pressure and Boiling Point If a liquid is placed in a closed container, some molecules at the surface of the liquid evaporate into the space above the liquid. If this didn’t happen, you wouldn’t be able to smell a liquid. Once vaporized, some of the molecules in the vapor condense back into the liquid in a competing process. As the space above the liquid becomes occupied with molecules of vaporized liquid, the pressure of the vapor above the liquid (the liquid’s vapor pressure) rises until it reaches a certain value. When the pressure stabilizes, the rates of evaporation and condensation are equal and the system is at equilibrium. The equilibrium vapor pressure of a liquid increases with increasing temperature. As the temperature of the liquid is raised, more molecules vaporize and the equilibrium vapor pressure increases. Again, think about boiling water, it steams more and more as it gets hot, and then eventually it boils. When the vapor pressure of a liquid is equal to the pressure of the atmosphere above the liquid, it boils. The normal boiling point of a substance is defined as the temperature at which the vapor pressure of that substance equals standard atmospheric pressure, 760 mmHg. If the barometric pressure is less than 760 mmHg, the temperature at which a substance boils will be less than the normal boiling point. That’s why it takes longer to cook pasta at high altitude. When measuring boiling points, it is important to record the barometric pressure at the time of the measurement. Make sure that you record the pressure in the lab when you do your experiment. In the chemical literature, the prrressure at which a boiling point is measured is often noted as a superscript or subscript. If a boiling point is reported without a pressure it is assumed to be the normal boiling point and the pressure is 760 mmHg. 22 To compare an observed boiling point to one reported in the chemical literature, it is often necessary to compensate for differences in pressure. The following formula provides a good estimate to correct an observed boiling point to a normal boiling point: Dt = 0.00012 (760 - p) (t + 273) where Dt is the correction in degrees Centigrade to be added to the observed boiling point t at barometric pressure p (in mmHg). Another rule of thumb for estimating boiling points near 760 mmHg states that the observed boiling point will differ from the normal boiling point by 0.5° for every 10 mmHg difference in pressure. In most cases, this correction is not necessary if you are carrying out your distillation under atmospheric pressure, whatever that happens to be today. Other sources of error in your temperature measurment should greatly outweigh this effect. This calculation can be helpful for reduced pressure distillations which are commonly used for higher boiling organic compounds. Simple Distillation - Single Volatile Liquid The vaporization of a liquid, relocation, and condensation of the resulting vapor is the basis for a method of purification called distillation. Organic liquids containing very small amounts of high boiling impurities are easily purified by “simple” distillation, that is, an easy distillation with relatively poor separation. The behavior of the measured boiling point during the course of a simple distillation is graphed in the figure below. As the liquid vaporizes and the vapor comes into contact with the thermometer bulb, the temperature rises. The temperature stabilizes at the boiling point and most of the liquid distills. The temperature drops when there is no liquid left in the distillation flask. You should never distill a liquid to dryness. Boiling point of a pure substance as a function of amount of liquid distilled Typical Apparatus for Simple Distillation 23 Distillation of Mixtures - Miscible Liquids Miscible liquids are soluble in each other in all ratios, that is, they mix well together. We will be distilling a mixture of miscible liquids in lab. These mixtures obey Dalton’s law of partial pressures which states that vapor pressure above a mixture is equal to the sum of the vapor pressures of the individual components. For example, for a two component mixture: Ptotal = PA + PB where PA and PB are the partial pressures of components A and B respectively. Remember that partial pressures increase as the temperature of the liquid reaches its boiling point. You can see water begin to steam shortly before it boils. In a mixture of miscible substances, the partial pressure of a component depends on the vapor pressure of the pure component and the relative amount of the component in the mixture. This relationship is stated as Raoult’s law—the partial pressure of a component in an ideal solution is equal to the vapor pressure of the pure component multiplied by its mole fraction: PA = PAo X A The mole fraction of component A, XA is defined as: XA = number of moles of A total number of moles in mixture For a mixture containing only two components, A and B, the mole fraction is: XA = number of moles of A number of moles of A + number of moles of B Combining Dalton’s law of partial pressures and Raoult’s law to a mixture of A and B, the vapor pressure of the mixture is: Ptotal = PAo X A + PBo X B Since the vapor pressures of the pure components increase with temperature, the vapor pressure of each above the mixture will also increase. When the vapor pressure of the mixture reaches 760 mmHg, the mixture will boil. For ideal solutions, i.e., solutions that obey Raoult’s law, the boiling point of the mixture will be between the boiling points of the pure components. The important consequence of Raoult’s law is that the vapor above a boiling mixture is enriched in the lower boiling component. By carrying out a distillation carefully it is possible to collect portions of the distillate which are considerably enhanced in the amount of lower 24 boiling component and higher boiling component. If the difference in the boiling points of the two components is large, a careful distillation can separate the mixture into its two components. In the figure below two curves representing the temperature of the distilling vapor as a function of the volume of distillate. The black curve represents an ideal distillation in which the lower boiling component distills completely and then the higher boiling component distills. The gray curve is a more empirical plot in which the distillate at the beginning of the distillation is enriched in the amount of the lower boiling component and the distillate at the later stages of the distillation is enriched in the higher boiling component. You will be making a curve like this one for your lab report. Distillation of a Two Component mixture 90 Temperature °C 80 70 60 50 40 30 20 10 0 0 10 20 30 40 Volume of Distillate (mL) 50 By careful control of temperature and by using columns designed to increase the surface area that the distilling vapors come in contact with, it is possible to make the empirical curve closely resemble the ideal curve, and thus better separate the components of the mixture. This technique is referred to as fractional distillation. The apparatus for a fractional distillation is shown in the Technique section below. is similar to the apparatus for a simple distillation with an extension added to increase the path the vapor has to travel. This extension is called a fractionating column. There are many types of fractionating columns that are used in fractional distillation. They are all similar in that the surface area, which contacts the distilling vapor, is increased. The larger the surface area contacted by the vapor, the more efficient the column is in separating the components. There are columns that are open, columns with glass indentations called Vigruex columns, and columns which are loosely packed with glass, metal or ceramic material. The fractionating column is often insulated to keep the temperature of the column nearly constant. If the temperature of the column fluctuates widely, it is difficult to maintain a slow, constant 25 distillation rate. Sophisticated fractionating columns have regulated heating coils built into them. Increasing the surface area over which the vapor must travel increases the purity of the distillate because the number of times that the liquid is distilled is effectively increased. As the vapor travels up the column it condenses on the surface and then re-evaporates. Every time this happens, the vapor becomes more pure. The units for measuring how many times the vapor goes through this cycle of condensation and re-evaporation are called “Theoretical Plates”. This term comes from the very old distillation columns that had actual “plates” in the column for the vapor to condense and evaporate from. Techniques: Distillation Apparatus - Fractional Distillation Thermometer Adapter (check for O-Ring!) Distilling Adapter Water Out Water in Open to the Air Air Condenser Acts as Fractionating Column Water Condenser Distillation Flask Recieving Flask Collect Fractions Here Boiling Chips Distillation flask: The distillation flask is a round-bottom flask and is sometimes referred to as “the pot”. The liquid to be distilled should fill the distillation flask to 1/2 to 2/3 of its capacity. If the flask is too small, the liquid is likely to bump or foam over into the receiving flask without vaporizing. If the flask is too large, a substantial amount of the liquid may be lost as vapor filling the flask. This residual volume is called the take-up volume. To promote even boiling of the liquid, boiling chips are added to the liquid before starting to heat the liquid. The irregular surface of the chips provides sites for bubbles of vapor 26 to form. An alternative method for promoting even boiling is to agitate the liquid with a magnetic stirrer as it is being heated. CAUTION: Never add boiling chips or a stir bar to a hot liquid. This can cause a seemingly calm liquid to boil suddenly and violently. Distilling adapter: The adapter connects the distillation flask, the condenser, and the thermometer. This type of adapter is often referred to as a “distillation head”. The ground glass joints must be lined up and connected tightly to avoid leakage of the vaporized liquid. Seal the joints with a little bit of grease. More grease does not make a better seal, use just enought to coat the joint. Leakage will result in loss of some (or all!) of the vapor and will pollute the laboratory environment. The position of the thermometer is adjusted so the bulb is below the adapter sidearm connected to the condenser. (See the diagram) In order to get reliable readings on the thermometer during the distillation, the vapors of the heated liquid must totally surround and contact the thermometer bulb. Condenser. The condenser cools the vapor to a liquid and directs this condensate to the receiving flask. The most common type of condenser is the water-jacketed type shown in the diagram above. The water supply is connected to the condenser with rubber hoses. The water flows in the lower hose connection (most remote from the distillation flask) and out the upper hose connection. Remember: With condensers, the water should flow up-hill. Before turning the water flow on, check the hose connections carefully to ensure that they are secure and will not pop off. An extra margin of security can be gained by twisting wire around the hose connections. The water flow is adjusted so there is a slow, constant flow of cold water to the condenser. You really only need a trickle, more than that will cause the hoses to pop off, spray your neighbors, and cause a flood. The receiving flask. The container to collect the condensed vapor is called the receiver. It may be a round-bottom flask, an Erlenmeyer flask, a bottle, or a graduated cylinder. Use your graduated “reaction vial” as the reciever to help you monitor the volume of your fractions. In the diagram, an adapter leads from the end of the condenser into a small graduated container. In this case the sole purpose of the adapter in this case is to direct the condensate from the condenser to the receiver. One important feature of the setup should be noted at this point—the system is open to the atmosphere. This apparatus is being heated, and you should never heat a closed system. If the liquid being collected has a low boiling point, it is good practice to cool the receiving flask with a cold water bath. Heat is slowly applied to the distillation flask. The amount of heat to apply is determined by the rate of distillation. The liquid should gently bubble and vaporize. As vapor rises from the liquid, it moves up the apparatus raising the temperature of the apparatus. The vapor will fill the distillation flask and most of the distillation head. The thermometer bulb should be completely surrounded by the vapor. If vapor creeps past the thermometer bulb without contacting it, the measured boiling point will be low. The vapor condenses in the condenser and drips into the receiving flask. Typically, the liquid should drip into the receiving flask at a rate of about 10 drops per minute. If the rate of distillation is too rapid, the heat applied to the distillation flask must be decreased. With too rapid a rate, the measured boiling point is likely to be inaccurate and the purity of the distilled liquid will be compromised. 27 The first material that distills before the temperature stabilizes is called the forerun. The forerun will contain any low boiling impurities and is usually discarded after checking its purity. The material collected after the temperature stabilizes is the purified substance. Usually the temperature stabilizes slightly below the literature value for the boiling point and then slowly creeps up. When the temperature begins to drop, the distillation is halted by removing the heat from the distillation flask. As a safety precaution, distillations are never carried out to dryness. The residual liquid plus the liquid from the take-up volume is the price one pays for purity. The boiling point of the collected material is actually a range rather than a point. Note: A technique called Steam Distillation is often used to purify high boiling organic compounds by distilling an immiscible mixture of the impure compound and water. For example, the perfume industry uses this technique to purify many essential oils. Heating Many of the experiments in this course require heating. Although there are numerous methods available, reaction conditions and chemical and physical properties of the materials make certain heating methods preferable to others. Since most organic compounds, especially the commonly used solvents such as hexane and ether, are flammable, a flameless method is generally preferred. In these laboratories, you will usually use either a steam bath or a sand bath. Steam Bath: A steam bath, illustrated below, is often used to heat solutions that boil below about 90°C, or to heat a mixture to approximately 100°C. The laboratories are supplied with steam from a central boiler. Before connecting the steam line to the bath, drain the water out of the steam pipes. Turn the steam on with caution, the knobs are usually quite hot and some hot water will come out before the steam does. Turn it on slowly, it’s quite normal for the pipes to make some odd noises before the steam starts to come out. When you’ve got mostly steam, rather than mostly hot water, connect the steam line to the top inlet on the bath. The bottom inlet is attached to a hose, which drains into the sink. Any water that condenses in the bath while you’re using it will drain out. Think of the top of the steam bath like a stove burner, the whole metal surface will be hot. You usually won’t need to turn the steam up very far to keep the steam bath hot. Having the steam turned up higher than necessarly just makes noise and an odd smelling, jungle-style atmosphere. Moisture is the enemy of many reactions and the lab should be kept as dry as possible. Usually, steam baths have concentric rings as covers which should be in your lab drawer. You can control the “size” of the bath by adding or removing rings. 28 Sand baths or Heating Mantles: A useful source of heat for reactions, especially where controlled temperatures over 100°C are required, is the heating mantle( illustrated in the figure below). The heating mantle used in these laboratories consists of a ceramic shell with embedded electric heating coils. The ceramic bowl will accommodate flasks with volume capacities up to 250 mL. With flasks smaller than 250 mL, the mantle can be used as an air bath or a sand bath, usually the latter. A small layer of sand in the bowl will serve as a medium for conducting heat to the reaction flask. Add just enough sand to nestle your flask into. A depth of about 1 cm should be plenty. Large amounts of sand act more as an insulator than a conductor. If you use more sand than just a thin layer, you will find that the reaction will take a longer time to heat up. The heating mantle is saferr, because it does not produce flames. However, there are still precautions to be taken. Heating mantles must be used with variable transformers. ALWAYS PLUG THE HEATING MANTLE INTO THE VARIAC. NEVER PLUG IT DIRECTLY INTO THE WALL. If a heating mantle is plugged directly into the wall outlet, it will become extremely hot. This is extremely hazardous as it can cause fires and burns and can also cause the heating mantle to burn out. Avoid spilling chemicals into the heating mantle. 29 electric cord which is attached to variable transformer small amount of sand to improve heat conductio Making an NMR Sample: To make an NMR sample of a compound, pipette two drops of your liquid into a vial, or if you have a solid, use a spatula-tip full of compound. With a different, clean pipette add a pipette full (1.5-2 mL) of deuterated chloroform (CDCl3) to the vial. Pipette up and down a few times to mix and then pipette the sample into a clean NMR tube. Cap the tube. NMR tubes are available at the stockroom. The sample should fill the tube at least up to where the bottom of the spinner will be, at least 3 inches. Push the NMR tube into the spinner gently. CAUTION: Always push the tube from close to the spinner! Use the depth gage to measure how far down the tube needs to go. Place the sample in the next available numbered slot in the rack and write your name on the sheet next to the right number. 30 Procedure: Miscible liquids such as a mixture of organic solvents with similar polarities (miscible mixtures) with boiling point differences of at least 10oC can be separated by fractional distillation. In this experiment, you will be given a mixture of two hydrocarbons included in the list given below. You will be distilling this mixture and collecting fractions appropriately. Using the boiling point data, the bromine test for unsaturation, and a test for aromaticity (which will be done in Experiment 3) and spectroscopic data (NMR) you will be able to identify the components of the mixture. Boiling Points of Hydrocarbons (oC) Alkanes hexane bp = 69 cyclohexane bp = 81 2,2,4-trimethylpentane bp = 99 n-octane bp = 126 Alkenes 1-hexene cyclohexene 1-octene cyclooctene bp = 63 bp = 84 bp = 123 bp = 146 Aromatics Toluene bp = 111 Ethylbenzene bp = 136 1,3,5-trimethylbenzene bp = 165 Procedure Set up your fractional distillation apparatus as described in the Techniques section of this experiment, using your air condenser as a fractionating column. Make sure that all of the joints of your distillation apparatus are tightly sealed with a very small amount of grease. Secure all joints with yellow Keck clips and securely clamp the apparatus from the center of the condenser to the laboratory framework. Safety Precaution:Make sure that all your glassware is in good shape (check for cracks or broken tips especially the distilling flask which you will be heating). Place 15 mL your unknown mixture in a 25 mL round-bottom flask equipped with a magnetic bar. Save a small sample (approximately 0.5 mL) of your mixture for Experiment 3 (analysis by gas chromatography and spectroscopic methods). Place this sample in a small vial, cap it tightly and label it. Seal the vial with parafilm. 31 Let your TA check your setup before you start distilling. Distill the mixture slowly to obtain the best result, increase the temperature very gradually. The key to a good separation is to establish liquid-vapor equilibria throughout the column. Too vigorous heating will flood the column. Think about it: Even though your heating mantle is heating, the temperature that the thermometer displays doesn’t change much until after the liquid boils. Why? Collect the distillate in a receiver with volumetric graduations, your “reaction vial” with the conical bottom is good for this. Save fractions about every 2 mL in labeled vials. Cap the vials immediately. You may need an extra pair of hands to help you transfer liquid into the vials, now is the time to make friends with the person down the bench from you! If the boiling point changes or there is any change in the appearance of the distillate start a new fraction immediately. Think about it: Which fractions are the most pure samples of your low boiling substance? Which fractions are the most pure samples of your high boiling substance? Is more fractions better than less? Why? While you’re collecting fractions, you should also be recording temperature vs. volume data so that you can create a graph like the one shown below. As the distillation proceeds, record the temperature and the total volume of the distillate collected in your laboratory notebook. Plot the data on graph paper or using a computer, the volume of distillate on the horizontal(X) axis and the temperature on the vertical(Y) axis . Note on the graph the positions that correspond to the collected fractions. In an ideal fractional distillation with complete separation of the components, the first substance would distill at its boiling point until there was none of it left in the mixture. Then, the temperature of the distilling vapor would rise to the boiling point of the second substance and all of it would distill at its boiling point. In practice, the behavior will depend on the fractionating column, the difference in boiling points, and the rate of distillation. Think about it: You may have observed a drop in temperature in the m iddle of the distillation, in between the low and high boiling substances. Why might this have happened? What does this say about the quality of your separation? After the distillation is complete, use your temperature vs volume data to determine which fractions are likely to contain the most pure samples of the two different hydrocarbons in your unknown mixture. Make NMR samples of the two “most pure” fractions, noting which sample is high boiling and which sample is low boiling. Save these fractions (at least two) to check for the purity by gas chromatography and for chemical test for unsaturation and aromaticity which will be done as part of Experiment 3. Wrap the vials containing the collected fractions and your pre-distilled mixture with parafilm and store them in the refrigerator (this will prevent evaporation of the samples). Make sure to label the vials with your name and section number. 32 In your lab report: Discuss the Think About It questions as part of your discussion of distillation. Make a graph of your Temperature vs. Volume data and evaluate the quality of your distillation. How could the separation have been improved? Identify the possible components in your unknown mixture using the boiling point data given above. If there is more than one possibility corresponding to a certain boiling point, list all of them. If you propose reasonable answers for your data you will not be penalized if your answer is incorrect. You will further test and determine the identity of this component using methods in Experiment 3. Don’t forget to tell your TA which unknown number (or letter) you had. Some sample data for the distillation of methanol (b.p. 64.7oC) and 2-propanol (b.p. 82.5oC) is plotted on the following graph as an example of how to summarize your distillation data. Fractional distillation of methanol and 2-propanol 33