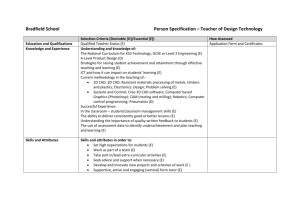
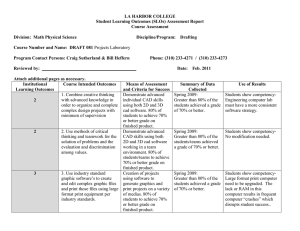
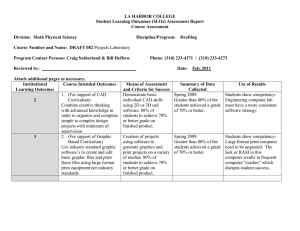
Lack of association between the Trp719Arg
advertisement

Lack of association between the Trp719Arg polymorphism in kinesinlike protein 6 and coronary artery disease in 19 case-control studies Themistocles L Assimes MD PhD1, Hilma Hólm MD2, Sekar Kathiresan MD 3-6, Muredach P Reilly MD7,8, Gudmar Thorleifsson PhD2, Benjamin F Voight PhD4,5,10, Jeanette Erdmann PhD11, Christina Willenborg11,12, Dhananjay Vaidya MBBS PhD MPH13, Changchun Xie PhD14 , Chris C Patterson PhD15, Thomas M Morgan MD16, Mary-Susan Burnett PhD17, Mingyao Li PhD18, Mark A Hlatky MD1, Joshua Knowles MD PhD1, John R Thompson PhD19, Devin Absher PhD20, Carlos Iribarren MD MPH PhD21, Alan Go MD21, Stephen Fortmann MD 1, Steven Sidney MD21, Neil Risch PhD22, Hua Tang PhD23, Richard M Myers PhD20, Klaus Berger MD24, Monika Stoll PhD25, Svati H. Shah MD MHS26, Gudmundur Thorgeirsson MD PhD27, Karl Andersen MD PhD27, Aki S Havulinna MSc28, J. Enrique Herrera MS13, Nauder Faraday MD29, Yoonhee Kim PhD30, Brian J. Kral MD MPH13, Rasika Mathias ScD13, Ingo Ruczinski PhD31, Bhoom Suktitipat MD32, Alexander F Wilson PhD30, Lisa R. Yanek MPH13, Lewis C Becker MD13, Patrick LinselNitschke MD11, Wolfgang Lieb MD11, Inke R König PhD12, Christian Hengstenberg MD33, Marcus Fischer MD33, Klaus Stark MD33, Wibke Reinhard MD33, Janina Winogradow33, Martina Grassl33, Anika Grosshennig11,12, Michael Preuss11,12, Stefan Schreiber MD34 , H-Erich Wichmann MD35-37, Christa Meisinger MD MPH35,38 , Jean Yee BS39,40, Yechiel Friedlander PhD41, Ron Do MSc42, Goran Berglund MD43, James B Meigs MD MPH6,44 , Gordon Williams MD6,45 , David M Nathan MD6,46, Calum A MacRae MD PhD3,6, Liming Qu18, Robert Wilensky MD7,8, William H. Matthai Jr. MD7, Atif Qasim MD8, Hakon H Hakonarson MD PhD47, Augusto D Pichard MD17, Kenneth M Kent MD PhD17, Lowell Satler MD17, Joseph M Lindsay MD17, Ron Waksman MD17, Christopher W Knouff48, Dawn M Waterworth PhD48, Max C Walker48, Vincent Mooser MD48, Jaume Marrugat MD PhD49, Gavin Lucas49, Isaac Subirana49, Joan Sala50, Rafael Ramos51, Nicola Martinelli MD52, Oliviero Olivieri MD52, Elisabetta Trabetti53, Giovanni Malerba53 , Pier Franco Pignatti53 , Aarti Surti5, Candace Guiducci5, Daniel Mirel5, Melissa Parkin5, Noel Burtt5, Stacey B Gabriel PhD5, Joel N Hirschhorn MD PhD5,54, Rosanna Asselta PhD55, Stefano Duga PhD55, Kiran Musunuru MD PhD MPH3-6, Mark J Daly PhD4-6, Shaun Purcell PhD4,5,56, Myocardial Infarction Genetics consortium68, Peter S Braund58, 1 Benjamin J Wright PhD19, Anthony J Balmforth PhD59, Stephen G Ball FRCP59, Wellcome Trust Case Control Consortium68, Willem Ouwehand MD PhD60, Panos Deloukas PhD61, Michael Scholz62, Francois Cambien MD63, Cardiogenics68, Andreas Huge25, Thomas Scheffold PhD64, Veikko Salomaa MD PhD28, Domenico Girelli MD PhD52, Christopher B. Granger MD65, Leena Peltonen MD PhD5,61,66, Pascal P McKeown15, David Altshuler MD PhD4-6,10,54 , Olle Melander MD PhD67, Joseph M Devaney PhD17, Stephen E Epstein MD17, Daniel J Rader MD8,9, Roberto Elosua MD PhD49, James C Engert PhD42,68, Sonia Anand MD PhD14, Alistair S Hall FRCP59, Andreas Ziegler PhD12, Christopher J O’Donnell MD MPH3,6,69, John A Spertus MD MPh70, David Siscovick MD MPH39, Stephen M Schwartz MD PhD39,40, Diane Becker MPH ScD13, Unnur Thorsteinsdottir PhD2,27, Kari Stefansson MD PhD2, 27, Heribert Schunkert MD11, Nilesh J Samani F.Med.Sci. 58, Thomas Quertermous MD1 Members of the writing group for this manuscript are in bold (manuscript preparation, analysts, and Principle Investigators from each of the 19 studies). Running Title: KIF6 Trp719Arg and CAD in 19 case control studies WORD COUNT: Text and References ~5200 List of Affiliations 1 Department of Medicine, Stanford University School of Medicine, Stanford, California 94305, USA. deCODE Genetics, 101 Reykjavik, Iceland. 3 Cardiovascular Research Center and Cardiology Division, Massachusetts General Hospital, Boston, Massachusetts 02114, USA. 4 Center for Human Genetic Research, Massachusetts General Hospital, Boston, Massachusetts 02114, USA. 5 Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA. 6 Department of Medicine, Harvard Medical School, Boston, Massachusetts 02115, USA. 7 The Cardiovascular Institute, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA 8 The Institute for Translational Medicine and Therapeutics, School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. 9 The Cardiovascular Institute, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. 10 Department of Molecular Biology, Massachusetts General Hospital, Boston, Massachusetts, 02114, USA. 11 Medizinische Klinik II, Universität zu Lübeck, 23538 Lübeck, Germany. 12 Institut für Medizinische Biometrie und Statistik, Universität zu Lübeck, 23538 Lübeck, Germany. 13 Department of Medicine, The Johns Hopkins School of Medicine, Baltimore, MD, USA 2 2 14 Population Health Research Institute, Hamilton Health Sciences and Departments of Medicine and Clinical Epidemiology and Biostatistics, McMaster University, Hamilton, L8L 2X2 Ontario, Canada. 15 Centre for Public Health, Queen’s University Belfast, Institute of Clinical Science, Belfast, BT126BJ, Northern Ireland, UK. 16 Department of Pediatrics, Vanderbilt University School of Medicine, Nashville, Tennessee 37232, USA. 17 Cardiovascular Research Institute, MedStar Research Institute, Washington Hospital Center, Washington, DC 20010, USA. 18 Biostatistics and Epidemiology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. 19 Department of Health Sciences, University of Leicester, Leicester, LE1 7RH, UK. 20 HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA 21 Division of Research, Kaiser Permanente, Oakland, CA, USA 22 Institute for Human Genetics, University of California, San Francisco, San Francisco, CA, USA 23 Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA 94305 24 Institute of Epidemiology and Social Medicine, University Münster 48129 Münster, Germany. 25 Leibniz-Institute for Arteriosclerosis Research, University Münster, 48149 Münster, Germany. 26 Department of Medicine and Center for Human Genetics, Duke University Medical Center, Durham, NC, USA 27 Faculty of Medicine, University of Iceland, and Department of Medicine, Landspitali University Hospital,101 Reykjavık, Iceland. 28 Chronic Disease Epidemiology Unit, Department of Health Promotion and Chronic Disease Prevention, National Public Health Institute, Helsinki 00300, Finland. 29 Department of Anesthesia and Critical Care Medicine, The Johns Hopkins School of Medicine 30 Genometrics Section, Inherited Disease Research Branch, National Human Genome Research Institute, National Institutes of Health, 31 Department of Biostatistics, The Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA. 32 Department of Epidemiology, The Johns Hopkins Bloomberg School of Public Health 33 Klinik und Poliklinik für Innere Medizin II,Universität Regensburg, 93042 Regensburg, Germany. 34 Institut für Klinische Molekularbiologie, Christian-Albrechts Universität, 24105 Kiel, Germany 35 Institute of Epidemiology, Helmholtz Zentrum München, German Research Center for Environmental Health, 85764 Neuherberg, Germany. 36 Institute of Medical Informatics, Biometry and Epidemiology, Ludwig-Maximilians-Universität Munich, 81377 Munich,Germany 37 Klinikum Grosshadern, Munich, Germany 38 Klinikum Augsburg, Germany 39 Cardiovascular Health Research Unit, Departments of Medicine and Epidemiology, University of Washington, Seattle, Washington 98101, USA. 40 Department of Epidemiology, University of Washington, Seattle, Washington 98195, USA. 41 Unit of Epidemiology, Hebrew University-Hadassah School of Public Health, Jerusalem 91120, Israel. 42 Department of Human Genetics, McGill University, Montréal, Québec H3A 1A1, Canada. 43 Department of Clinical Sciences, Internal Medicine, University Hospital Malmö, Lund University, Malmö 20502, Sweden. 44 General Medicine Division, Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, 02114, USA. 45 Cardiovascular Endocrinology Section, Division of Endocrinology, Diabetes, and Hypertension, Brigham and Women’s Hospital, Boston, Massachusetts 02115, USA. 46 Diabetes Center, Massachusetts General Hospital, Boston, Massachusetts 02114, USA. 47 The Center for Applied Genomics,Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA. 48 Genetics Division and Drug Discovery, GlaxoSmithKline, King of Prussia, Pennsylvania 19406, USA 49 Cardiovascular Epidemiology and Genetics, Institut Municipal D’investigacio Medica, and CIBER Epidemiologı´a y Salud Pu´ blica, 08003 Barcelona, Spain. 3 50 Servei de Cardiologia i Unitat Corona`ria, Hospital de Girona JosepTrueta and Institut de Investigacio´ Biomedica de Girona, 17007 Girona, Spain. 51 Unitat de Recerca i Unitat Docent de Medicina de Familia de Girona, IDIAP Jordi Gol, Institut Catala` de la Salut, 08007 Barcelona, Spain. 52 Department of Medicine, University of Verona, 37134 Verona, Italy. 53 Department of Mother and Child, Biology and Genetics, Section of Biology and Genetics, University of Verona, 37134 Verona, Italy. 54 Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA. 55 Department of Biology and Genetics for Medical Sciences, University of Milan, 20133 Milan, Italy. 56 Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA. 57 A full list of members is provided in the Supplementary Note online. 58 Department of Cardiovascular Sciences, University of Leicester, Glenfield Hospital, LE39QP, UK. 59 LIGHT Research Institute, Faculty of Medicine and Health, University of Leeds, Leeds, LS1 3EX, UK. 60 Department of Haematology, University of Cambridge,Long Road, Cambridge, CB2 2PT, UK. 61 Wellcome Trust Sanger Institute, CambridgeCB10 1SA, UK. 62 Trium Analysis Online GmbH, 81677 Munich, Germany. 63 INSERM UMR_S 525, Universite Pierre et Marie Curie –Paris 6, Paris 75634, France. 64 Institute for Heart and Circulation Research of the University of Witten/Herdecke, 44227 Dortmund, Germany. 65 Department of Medicine and Duke Clinical Research Institute, Duke University Medical Center, Durham, NC, USA 66 Institute for Molecular Medicine, University of Helsinki, Helsinki 00029, Finland. 67 Department of Clinical Sciences, Hypertension and Cardiovascular Diseases, University Hospital Malmö, Lund University, Malmö 20502, Sweden. 68 Department of Medicine, McGill University, Montreal, H3A 1A1 Quebec, Canada. 69 National, Heart, Lung, and Blood Institute and its Framingham Heart Study, Framingham, Massachusetts 01702, USA. 70 Mid-America Heart Institute and University of Missouri-Kansas City, Kansas City, Missouri 64111, USA. DISCLOSURES The collection of clinical and sociodemographic data in the Dortmund Health Study was supported by the German Migraine & Headache Society (DMKG) and by unrestricted grants of equal share from Astra Zeneca, Berlin Chemie, Boots Healthcare, Glaxo-Smith-Kline, McNeil Pharma (former Woelm Pharma), MSD Sharp & Dohme and Pfizer to the University of Muenster. Recruitment of the Medstar sample was supported by a research grant from GlaxoSmithKline and genotyping of the PennCATH and Medstar samples was supported by GlaxoSmithKline. Four co-authors of this study are employees of deCODE genetics, a for-profit company that develops SNP based diagnostic tests for various diseases including coronary artery disease. Address for Correspondence: 4 Themistocles L. Assimes, MD PhD Falk Cardiovascular Research Building Stanford University School of Medicine Stanford, CA 94305-5406 Tel: 650-498-4154 Fax: 650-475-5650 Email: tassimes@stanford.edu 5 Abstract Objectives We sought to replicate the association between the kinesin-like protein 6 (KIF6) Trp719Arg polymorphism (rs20455) and clinical coronary artery disease (CAD). Background Recent prospective studies suggest that carriers of the 719Arg allele in KIF6 are at increased risk of clinical CAD compared with non-carriers. Methods The KIF6 Trp719Arg polymorphism (rs20455) was genotyped in nineteen case-control studies of non-fatal CAD either as part of a genome-wide association study or in a formal attempt to replicate the initial positive reports. Results Over 17 000 cases and 39 000 controls of European descent as well as a modest number of South Asians, African Americans, Hispanics, East Asians, and admixed cases and controls were successfully genotyped. None of the nineteen studies demonstrated an increased risk of CAD in carriers of the 719Arg allele compared with non-carriers. Regression analyses and fixed effect meta-analyses ruled out with high degree of confidence an increase of ≥2% in the risk of CAD among European 719Arg carriers. We also observed no increase in the risk of CAD among 719Arg carriers in the subset of Europeans with early onset disease (<50 years of age for males and <60 years for females) compared with similarly aged controls as well as all non-European subgroups. Conclusions The KIF6 Trp719Arg polymorphism was not associated with the risk of clinical CAD in this large replication study. ABSTRACT # OF WORDS: 219 MANUSCRIPT WORD COUNT (Text and References): 4951 6 ABBREVIATIONS USED SNP: Single Nucleotide Polymorphism CAD: Coronary Artery Disease MI: Myocardial Infarction CKMB: Creatine Kinase, myocardial band GWAS: Genome Wide Association Study ECG: Electrocardiography OR: Odds Ratio 95% CI: 95% Confidence Interval 7 Introduction Recent prospective observational studies suggest an association between the Trp719Arg SNP in kinesin-like protein 6 (rs20455) and the development of clinical coronary artery disease (CAD)(1-5). Carriers of the 719Arg alleles were found to have a modest increase in the risk in the Atherosclerosis Risk in Communities study (ARIC) (Hazard Ratio and 95% CI for log additive model: 1.11, 1.02-1.21, p = 0.02)(1), the Cardiovascular Health Study (CHS) (HR and 95% CI for dominant model: 1.29, 1.1-1.52, p = 0.005)(5), and the Women’s Health Study (WHS)(HR and 95% CI for dominant model: 1.24, 1.04-1.46, p = 0.01)(4). An increase in the risk of CAD was also observed in the placebo arm of two statin trials: the West of Scotland Coronary Prevention Study (WOSCOPS) (Odds Ratio for incident CAD events and 95% CI for dominant model: 1.55, 1.14-2.09, p = 0.005) and the Cholesterol and Recurrent Events (CARE) trial (HR for recurrent myocardial infarction and 95% CI for dominant model: 1.50 (1.05-2.15), p = 0.03) (3). Curiously, carriers of the 719Arg allele were not at increased risk of CAD or recurrent MI in the pravastatin arms of these two trials. Furthermore, in the Pravastatin or Atorvastatin Evaluation and Infection Therapy Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) trial, carriers in the pravastatin arm were also not at increased risk of CAD while carriers in the atorvastatin arm had a decreased risk compared with non-carriers (adjusted HR 0.65, 95% CI 0.48 to 0.88; p = 0.005)(2). The discrepant results between the two arms of these three statin trials were deemed a consequence of a differential effect of genotype on the benefit derived from the use of statins with carriers benefiting to a larger degree from statin therapy than non-carriers (2,3). Because carriers randomized to the more potent statin were actually at decreased risk compared with non-carriers in the PROVE IT-TIMI 22 trial, it was hypothesized that the degree of incremental benefit from statin use among carriers was a function of the intensity of lipid lowering therapy(2). The results of these initial studies have been used to justify the development of a KIF6 Trp719Arg variant pharmacogenomic test (“Statincheck”)(6). This test is currently being marketed to health care professionals as an aid to identifying subjects at high risk of incident or recurrent CAD events who stand to gain 8 the most from the use of statins(6,7). However, in recent GWAS for CAD and/or MI, SNPs at the KIF6 locus were not among the associations reaching genome wide significance(8-12). Furthermore, in the ongoing Ottawa Heart Genomics Study, no association was found between the KIF6 Trp719Arg SNP and angiographically defined CAD in a subset of 1540 cases and 1455 controls(13). These discrepant results demand examination of this association in additional populations to substantiate the use of the KIF6 test in the management of subjects at risk of clinical CAD. In this study, we report association analyses for the KIF6 Trp719Arg SNP in 19 case-control studies of CAD that have recently genotyped this SNP using various genotyping platforms either as part of a GWA study or in a formal attempt to replicate the initial positive reports (1-5). Methods Study Populations We included subjects participating in 19 different case-control studies of CAD conducted around the world: the Atherosclerotic Disease, VAscular functioN, and genetiC Epidemiology study (ADVANCE)(14) of northern California, USA, the AMI Gene Study/Dortmund Health (AMI Gene)(15) in Germany, the CATHGEN Research Project (CATHGEN)(16) in North Carolina, USA, the deCODE CAD study (deCODE)(10) in Iceland, the National Finrisk study (FINRISK)(17) in Finland, the sibling Genetic Study of Atherosclerosis Risk (GeneSTAR)(18) in Maryland, USA, the German Myocardial Infarction Family studies (GerMIFS I and GerMIFS II)(8) in Germany, the Heart Attack Risk in Puget Sound (HARPS)(19) study in Washington state, USA, the international INTERHEART study (INTERHEART)(20,21) coordinated by Population Health Research Institute of McMaster University in Hamilton, Ontario, the Irish Family Study (IFS)(22) in Ireland, the Malmo Diet and Cancer study (MDC)(23) in Malmo, Sweden, the Medstar/Washington Hospital Center (MEDSTAR)(10) study in Washington, D.C., the Massachusetts General Hospital Premature CAD study (MGH PCAD)(24) in Boston, USA, the Mid-America Heart Institute study (MAHI)(25,26) in Kansas City, Missouri, the University of Pennsylvania Medical Center Cardiac catheterization cohort study (PennCATH)(10) in Philadelphia, the Registre Gironı´ del Cor (REGICOR)(27) study in Gerona, Spain, the Verona Heart Study 9 (VHS)(28) in Verona, Italy, and the Wellcome Trust Case Control Consortium study of CAD (WTCCC CAD) (11) in the United Kingdom. All participants gave written informed consent and local ethics committees approved all studies. Investigators from several of these 19 studies are members of consortia with a primary interest of identifying novel genetic determinants of CAD through the use of GWAS technology. The consortia include the PennCATH/Medstar consortium comprised of the PennCATH and Medstar studies, the Myocardial Infarction Genetics consortium (MIGen)(12) comprised of the HARPS, REGICOR, MGH PCAD, FINRISK, and MDC studies, the Cardiogenics consortium(8) comprised of the WTCCC, GerMIFS I, GerMIFS II studies, and the Coronary ARtery Disease Genome-wide Replication And Meta-analysis consortium (CARDIoGRAM) comprised of the ADVANCE study, the deCODE study, and the Penn/Medstar, MIGen, and CARDIOGENICS consortia. Each study used standard criteria to identify cases with myocardial infarction (MI) established by international organizations during the 1990s or more recently. While some of these studies restricted their enrollment of cases to subjects with at least one MI, others included cases diagnosed with clinically significant coronary atherosclerosis without MI. Elevation of cardiac markers (CKMB or troponin) accompanied with symptoms and/or ECG suggestive of cardiac ischemia were typical criteria used to identify MI cases. Non-MI cases included subjects with angina and confirmatory tests for ischemia, unstable angina based on symptoms and ECG changes without elevation of cardiac enzymes, or revascularization procedures that may have occurred in the presence or absence of symptoms. For the current analyses, both cases with and without a diagnosis of MI were considered in order to emulate the outcome used in the majority of the prospective studies published to date (1-5). Most of the studies either only enrolled subjects of European descent or restricted their genotyping efforts to Europeans. However, in addition to Europeans, the ADVANCE study genotyped a modest number of African Americans, Hispanics, East Asians, and admixed individuals, the CATHGEN and GENESTAR studies genotyped a modest number of African Americans, and the INTERHEART study genotyped a large number of South Asians. 10 More details on the design of each study including the precise criteria used by each of the 19 studies to ascertain cases and controls can be found in the Supplementary Appendix and related references. Genotyping The Trp719Arg polymorphism in kinesin-like protein 6 (rs20455) was directly genotyped in all studies and no imputation of the genotype was necessary. For the GerMIFS I and WTCCC CAD studies, the genotyping data were extracted from the Affymetrix 500k array(29). For the deCODE, ADVANCE, and GeneSTAR studies, the genotyping data were extracted from the Illumina Infinium HumanHap317/370, 550, and 1M chip arrays, respectively(29). For the GerMIFS II, FINRISK, HARPS, MDC, MGH PCAD, PennCATH, Medstar, and REGICOR studies, the genotyping data were extracted from the Affymetrix 6.0 array(29). For the AMI Gene, IFS, INTERHEART, and MAHI studies, the SNP was genotyped using the iPLEX MassARRAY platform (Sequenom) platform(29). Finally, for CATHGEN and VHS, the SNP was genotyped using the Centaurus platform(29). All genotype data generated from the various platforms passed extensive quality control measures including tests of Hardy Weinberg Equilibrium (p > 0.001 in controls). Clinical measures The age of onset of clinical CAD (for cases) and sex for cases and controls were documented in all studies. The presence of other traditional risk factors was documented in most but not all studies. In the deCODE study, risk factor data other than age, sex, and BMI were not collected. Furthermore, the presence of other traditional risk factors was defined in various ways and the timing of enrollment of cases hampered the accurate measurement of these risk factors in some studies. For example, several studies enrolled cases weeks to months (and sometimes years) after their initial CAD event, making it difficult to reliably discern whether certain medications being taken by a participant at the time of enrollment were prescribed to treat risk factors present prior to their first ever clinical manifestation of CAD vs. for secondary prevention reasons or to treat CAD symptoms (e.g. beta blockers, calcium channel blockers, ACE inhibitors, and statins). No attempt was made to standardize risk factor definitions across all studies. 11 Statistical Analysis We first calculated the crude Odds Ratios (OR) and 95% confidence interval (95% CI) based on the 2x2 table of KIF6 Trp719Arg allele-by-trait counts for each case control study. In case control studies with more than one race/ethnic group, we calculated an OR for each race/ethnic group. We then compared the distribution of the raw genotype counts in each case-control stratum with Armitage’s trend test(30). Next, we calculated OR and 95% CI adjusted for age of onset of CAD and sex using standard unconditional logistic regression. Because of the difficulties in ascertaining risk factors other than age and sex, we did not adjust OR for any additional CAD risk factors. Log additive and dominant OR were calculated as both modes of inheritance were reported to date and the 719Trp allele served as the reference allele given the 719Arg allele was identified as the high risk allele in prior publications. Analyses were repeated after excluding cases with an age of onset of CAD ≥50 years for males and ≥60 years for females. Controls for this subgroup analysis were also restricted to those in the same sex specific age range as cases at the time of enrollment. The GeneSTAR family study analyses were performed using generalized linear latent and mixed models (GLLAMM) with the logistic link and family membership as the random effect in order to account for family structure while the Irish Family Study used likelihood based association statistics incorporated in the UNPHASED software package(31) to account for family structure. In the latter family study, only the crude OR for the additive model using all subjects could be calculated. Lastly, we combined crude and adjusted OR, 95% CI, and p values using a Mantel-Haenszel model in which the groups were allowed to have different population frequencies for genotypes but were assumed to have common relative risks(32). We weighed the results from each study by the standard error of the effect derived from the p value of the OR (see Supplementary Appendix for details). Using this meta-analytic approach, we calculated the overall combined OR, 95% CI, and p value separately for all case control studies of Europeans and all case-control studies of African Americans. The meta-analyses were repeated for the subgroup of cases in each study with early onset disease and their respective controls. For each meta-analysis, we also performed two standard heterogeneity tests (Cohran’s Q and Higgin’s I2) to assess the appropriateness of a fixed effects model over a random effects model(33). 12 From previous work, we know that one of the two admixed race/ethnic strata in the ADVANCE study, the admixed non-Hispanics, has significant differences in the degree of admixture between cases and controls(14). Specifically, cases in this stratum are, on average, significantly more European (and less African American) than controls. Therefore, we included a covariate in the multivariate regression model indicating the proportion of white ancestry for each individual estimated by the program STRUCTURE (34,35) in this stratum. The other admixed stratum in the ADVANCE study, the admixed Hispanics, did not show differences in degree of admixture between cases and controls. Therefore, no additional covariates were added to the regression model for this stratum. Some Icelandic affected individuals and controls are related, both within and between groups, causing the chi square test statistic to have a mean >1 and a median larger than 0.6752. The inflation factor, lambda (λ), was estimated at 1.21 using a method of genomic control(36) by calculating the average of the observed chi statistics for the genome-wide SNP set, which accounts for relatedness and for potential population stratification. The 95% CI and p values presented for deCODE are based on adjusting the chi square statistic by dividing it by this inflation factor. The inflation factor was also found to be high in the GerMIFS I study (λ ~1.27), therefore p values and 95% CI for this study were also adjusted in the same manner. For all other studies with genome wide data, a genomic control correction was not applied to the data as the inflation factor was found to be very low (λ ≤1.05). We performed sensitivity analyses in Europeans only by repeating all analyses described above after first excluding non-MI cases in the subset of case-control studies which included both MI and non-MI cases of CAD. Results Table 1 summarizes the ascertainment scheme and the inclusion criteria for phenotype, age, sex, and race stratified by study and case-control status. The distribution and frequency of all traditional risk factors of CAD stratified by study and case-control status can be found in the Supplementary appendix (Table 1S). Out of the 19 case control studies, 12 enrolled only cases with MI and 14 also restricted enrollment or genotyping to early 13 onset disease (when considering the traditional age cutoffs of ≤ 65 years for females and ≤ 55 years for males at the time of first onset of disease). However, the largest study (deCODE) enrolled both MI and non MI cases of CAD and included CAD cases with any age of onset. The proportion of cases that were female varied from study to study and was influenced by the ascertainment scheme and matching (either one to one or frequency matching). The overall weighted average age of onset of CAD was 55.7 years. Table 2 summarizes the genotype counts stratified by study, case control status, and race/ethnic group as well as the adjusted OR, 95% CI, and p values for the association between KIF6 rs20455 SNP and CAD. Crude ORs were not materially different from their respective ORs adjusted for age and sex. Therefore, only ORs adjusted for age and sex are presented. Among Europeans, only one out of the 19 studies (deCODE) demonstrated a nominally significant association between the KIF6 polymorphism and CAD but in the opposite direction of the published literature (the 719Arg allele inversely associated with risk: log additive OR of 0.93, 95% CI 0.88-0.99, log dominant OR of 0.91, 0.85-0.99). The meta-analysis produced a point estimate of the log additive OR near unity with very tight confidence intervals (log additive OR 0.98, 95% CI 0.95-1.02, log dominant OR 0.97, 0.93-1.01). We also found no significant association between the KIF6 rs20455 SNP and CAD among South Asian participants in the INTERHEART study (log additive OR 1.02, 95% CI 0.91-1.14, log dominant OR 1.04, 0.87-1.24), African American participants in the ADVANCE, CATHGEN, and GENESTAR studies (fixed effects meta analysis log additive OR 0.91, 95% CI 0.73-1.13, log dominant OR 0.81, 0.42-1.55), and a smaller number of Hispanic, East Asian, and admixed individuals participating in the ADVANCE study (see Table for details). Table 3 shows genotype counts and association analyses restricted to the subgroup of cases with very early onset disease (<50 years for males and <60 years for females) and controls within the same age range at the time of enrollment. Among this subgroup, we also observed no increase in the risk of CAD in either the European carriers of the 719Arg allele compared with non-carriers (fixed effects meta-analysis log additive OR 0.99, 95% CI 0.95-1.04, log dominant OR 1.01, 0.95-1.08) or the non-European subjects (see Table for details). Lastly, our sensitivity analyses in Europeans restricting cases to those defined on the basis of an MI in all 19 case control studies also revealed no increased risk of CAD in European carriers of the 719Arg allele 14 compared with non-carriers both for the overall analysis (fixed effects meta-analysis log additive OR 0.99, 95% CI 0.96-1.03, log dominant OR 0.98(0.94-1.02) and the subgroup analysis of early onset MI cases (fixed effects meta-analysis log additive OR 1.03, 95% CI 0.98-1.09, log dominant OR 1.03, 0.97-1.11). Additional details for these analyses are provided in Tables 2S and 3S of the Supplementary Appendix. None of the Q tests revealed heterogeneity in our meta-analyses (lowest p value = 0.252) and all metaanalyses in Europeans had an I2 value of zero percent suggesting that the variability in effect sizes in Europeans was due entirely to sampling error within the studies (see Table 4S in Supplementary Appendix). Thus, performing random effects model meta-analyses was not necessary in Europeans as the results would be identical to the fixed-effects model. Only the log dominant model for CAD overall among African Americans demonstrated an I2 value > 0%. For this stratum, the random effect model (DerSimonian and Laird method) revealed an OR that was similar to the fixed effects model but with wider confidence intervals (Table 4S). Discussion The principal finding of this study was the uniform lack of elevated risk of clinical CAD among carriers of the KIF6 719Arg allele compared with non-carriers in 19 case control studies performed around the world. Our meta-analysis involving a very large number of subjects with European ancestry (over 17000 cases and 39000 controls) suggests that the risk of CAD among European carriers of the 719Arg allele is unlikely to be increased by more than 2% compared with non-carriers. Our study has two strengths in addition to the very high power conferred by studying a large number of European subjects. First, we studied a large number of early onset CAD cases (<50 years of age for males and <60 years of age for females) but observed no association with KIF6 Trp719Arg and CAD despite the expectation that susceptibility alleles would be more prevalent in this subgroup(37). In this subgroup with early onset disease, we were able to rule out an increase in risk of ≥8% in Europeans. Second, this is the only study to date that examines several non-European racial/ethnic groups. In each of non-European case-control strata, we also found no significant associations between the KIF6 Trp719Arg polymorphism and CAD. However. the point estimates of the OR for these non-European subgroups have substantially wider confidence intervals 15 than those derived in Europeans due to relatively small sample sizes (particularly in our Hispanic, East Asians, admixed Hispanic, and admixed non Hispanics groups). Thus, further study of all of these non-European race/ethnic groups is needed to rule out more modest effects on risk. Our study has three important limitations related to the case-control design. The first limitation is the potential selection bias by studying only non-fatal cases of CAD. If the 719Arg allele increases the risk of incident fatal CAD more than the risk of incident non-fatal CAD, the exclusion of incident fatal CAD cases could conceivably bias the OR towards the null (i.e. OR = 1). However, the difference in relative risk between these two subgroups of cases would have to be quite large to result in a substantially biased OR in our study given the majority of incident CAD events are not fatal especially among subjects with early onset disease (e.g. in the ARIC surveillance study 20 to 35% of subjects aged 60 years or greater and 10 to 20% of subjects under the age of 60 years died as a consequence of a complication of their initial presentation of CAD(38)). The second potential limitation is our inability to measure traditional risk factors as robustly as they are measured in prospective studies. We therefore made no attempt to fully adjust ORs for all traditional risk factors of CAD. However, prospective studies published to date suggest that traditional risk factors are not correlated with the KIF6 Trp719Arg polymorphism making adjustment unnecessary (1-5). Lastly, our study does not allow us to explore whether statin use modifies the effect of the 719Arg allele on risk as was done in the WOSCOPS, CARE, and PROVE IT TIMI 22 trials as reliable information on the use of statins in relation to the incident event was not available for most studies. However, we believe it unlikely that our null results are a consequence of a high prevalence of the use of statins at the time of the event in cases. In fact, we suspect that the overall prevalence of the use of statins in our set of 19 case control studies is actually lower than the prevalence of use observed at the last follow up for participants in the ARIC, CHS, and WHS studies(39-41) because a majority of our case-control studies restricted their recruitment and/or genotyping efforts to very early onset cases and young controls. For example, in the ADVANCE study, which focused on very early onset CAD (<45 years for males, <55 years for females), access to the electronic pharmacy records confirmed that only a small proportion of cases (~14.4%) and controls (~4.7%) were on statins during the appropriate time window of exposure. 16 Is it possible that other less obvious sources of bias are responsible for the differences in associations observed between case control and cohort studies of KIF6? While we cannot rule out this possibility, we also deem this scenario unlikely given such cryptic biases, if they exist, have not influenced associations between SNPs at the 9p21.3 locus and CAD in the same manner(9,10). In fact, many of the case-control studies included in this report have produced OR of disease for the 9p21.3 locus equivalent to or larger than the hazard ratios derived from cohort studies(9,10,12,14,42-44). These cohort studies include the two largest cohort studies to date (ARIC and WHS) reporting on the association between KIF6 and CAD. Thus, we are left with the real possibility that the initial reports falsely suggested an association between variation in KIF6 and CAD. The reasons for this are unclear but include the possibility of chance findings or inadequate correction for multiple testing(45-47). The lack of biologic studies implicating KIF6 in the pathogenesis of coronary atherosclerosis combined with the lack of consistent evidence of expression of this gene in relevant tissues such as the vasculature (48,49) could also argue for a false positive association. However, many recent population genetic studies of complex traits have clearly demonstrated that such mechanistic data are not necessary to validate highly significant genetic associations uncovered through GWAS (50). Our findings question not only the usefulness of the KIF6 test in identifying subjects at increased risk of incident or recurrent CAD but also its usefulness in identifying subjects most likely to benefit from statins. Although we could not test the latter hypothesis directly, the previously reported interaction between genotype and benefit from statins is largely dependent on the validity of the association among subjects not on statins which could not be replicated in this study. We also call attention to the fact that the interaction term p value between genotype and statin use was only marginally significant in the WOSCOPS (p = 0.021) and PROVEITTIMI22 (p = 0.018) trials and not significant in the CARE trial (adjusted p = 0.39)(2,3). Despite these observations, additional high quality prospective cohort studies of the effect of the KIF6 variant on CAD risk among users and non users of statins are needed before any firm conclusions can be made regarding the validity of this interaction. In conclusion, we were unable to confirm an increased risk of CAD in carriers of the KIF6 719Arg allele compared with non-carriers in a very large number of European subjects. We also observed no compelling 17 evidence of an association between the KIF6 Trp719Arg SNP and CAD in multiple other race/ethnic groups and among subjects with early onset CAD. Our null results are unlikely to be a consequence of selection bias, a high prevalence of use of statins among cases, or other cryptic biases. These findings do not support the clinical utility of testing for the KIF6 Trp719Arg polymorphism in the primary prevention of CAD and indirectly question whether genotype information at this locus is able to identify subjects most likely to benefit from the use of statins. 18 ACKNOWLEDGMENTS ADVANCE study. The ADVANCE study was funded by the US National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program through a grant to T.Q (R01HL087647-03). HARPS. The HARPS study was supported by the grants (R01HL056931, P30ES007033) and a contract (N01HD013107) from US National Institutes of Health. REGICOR. The REGICOR study was partially funded by the Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III (Red HERACLES RD06/0009), the CIBER Epidemiologı´a y Salud Pu´ blica, the FIS and AGAUR Generalitat de Catalunya. Massachusetts General Hospital. The deCODE CAD/MI Study was sponsored by NIH grant, National Heart, Lung and Blood Institute (R01HL089650-02). MIGen: The MIGen consortium study was funded by the US National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program through a grant to D.A (R01HL087676-02). S.K. is supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award, a charitable gift from the Fannie E. Rippel Foundation, the Donovan Family Foundation, a career development award from the NIH and the Department of Medicine and Cardiovascular Research Center at Massachusetts General Hospital. J.B.M. is supported by grant K24 DK080140 from the NIH. Broad Institute. Genotyping was partially funded by The Broad Institute Center for Genotyping and Analysis, which is supported by grant U54 RR020278 from the National Center for Research Resources. FINRISK. V.S. was supported by the Sigrid Juselius Foundation. L.P. was supported by the Center of Excellence in Complex Disease Genetics of the Academy of Finland, the Nordic Center of Excellence in Disease Genetics and the Finnish Foundation for Cardiovascular Research. WTCCC Study. The study was funded by the Wellcome Trust. Recruitment of cases for the WTCCC Study was carried out by the British Heart Foundation (BHF) Family Heart Study Research Group and 19 supported by the BHF and the UK Medical Research Council. N.J.S. and S.G.B. hold chairs funded by the BHF. N.J.S. is supported by the Leicester NIHR Biomedical Research Unit in Cardiovascular Disease. PennCATH/Medstar. Recruitment of the PennCATH cohort was supported by the Cardiovascular Institute of the University of Pennsylvania. Recruitment of the Medstart study sample was supported by the MedStar Research Institute and the Washington Hospital Center as well as a research grant from GlaxoSmithKline. Genotyping was performed at the Center for Applied Genomics at the Children’s Hospital of Philadelphia and supported by GlaxoSmithKline through an Alternate Drug Discovery Initiative research alliance award (to M.P.R. and D.J.R.) with the University of Pennsylvania School of Medicine. D.J.R. was supported by a Doris Duke Charitable Foundation Distinguished Clinical Scientist Award. GeneSTAR. The GeneSTAR study was funded by the US National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program through a grant to L.C. B (R01HL087698-03). Verona Heart Study. The study was supported by a grant from the Italian Ministry of University and Research and grants from the Veneto Region and the Cariverona Foundation, Verona, Italy. Mid-America Heart Institute. T.M. is supported by a career development grant from the NIH. Irish Family Study. We thank the clinical staff members for their valuable contribution to the collection of families for this study. The research was supported by the Northern Ireland Research and Development Office, a Royal Victoria Hospital Research Fellowship, the Northern Ireland Chest, Heart and Stroke Association, and the Heart Trust Fund (Royal Victoria Hospital). GerMIFS I and II. The German Study was supported by the Deutsche Forschungsgemeinschaft and the German Federal Ministry of Education and Research in the context of the German National Genome Research Network. Cardiogenics. Cardiogenics is an EU-funded integrated project (LSHM-CT- 2006-037593). INTERHEART. 20 .We acknowledge the contribution of S. Yusuf who initiated and, together with the Steering Committee, supervised the conduct of the INTERHEART study. We thank members of the Project Office, S. Rangarajan (study coordinator) and K. Hall (laboratory manager), for their assistance in coordinating the genetics component of the INTERHEART project. S.A. holds the Michael G. DeGroote and Heart and Stroke Foundation of Ontario Chair in Population Health and the May Cohen Eli Lilly Endowed Chair in Women’s Health Research, McMaster University Contributions are as follows: Manuscript preparation: Themistocles L Assimes, Hilma Holm, Sekar Kathiresan, Chris C Patterson, Thomas Morgan, Mark Hlatky, Joshua Knowles, Thomas Quertermous, Diane Becker, Heribert Shunkert , Jeanette Erdmann, Stephen M Schwartz, David S Siscovick, Christopher J O’Donnell, Muredach P Reilly , John A Spertus, James C Engert, Pascal P McKeown, Nilesh J Samani Statistical Analysis: Themistocles L Assimes, Gudmar Thorleifsson, Benjamin F Voight, Christina Willenborg, Dhananjay Vaidya, Changchun Xie, Chris C Patterson, Thomas Morgan, Mingyao Li, John R Thompson Atherosclerotic Disease, VAscular functioN, and genetiC Epidemiology. Themistocles L. Assimes, Devin Absher, Joshua Knowles, Carlos Iribarren, Alan Go, Stephen Fortmann, Steven Sidney, Neil Risch, Hua Tang, Richard M. Myers, Mark Hlatky, Thomas Quertermous Acute Myocardial Infarction Gene Study/Dortmund Health Study. Thomas Scheffold, Klaus Berger, Monika Stoll, Andreas Huge CATHGEN. Svati H. Shah, Christopher B. Granger deCODE Study. Hilma Holm, Gudmar Thorleifsson, Gudmundur Thorgeirsson, Karl Andersen, Unnur Thorsteinsdottir, Kari Stefansson FINRISK. Veikko Salomaa, Aki S Havulinna, Leena Peltonen GeneSTAR. Dhananjay Vaidya, Nauder Faraday, J. Enrique Herrera, Yoonhee Kim, Brian J. Kral, Rasika Mathias, Ingo Ruczinski, Bhoom Suktitipat, Alexander Wilson, Lisa R. Yanek, Lewis C Becker, Diane Becker 21 German MI Family Studies I and II. Heribert Schunkert, Jeanette Erdmann, Patrick Linsel-Nitschke, Wolfgang Lieb, Andreas Ziegler, Inke R König, Christian Hengstenberg, Marcus Fischer, Klaus Stark, W. Reinhard, J. Winogradow, M. Grassl, Anika Grosshennig, Michael Preuss, Stefan Schreiber , KORA: H-Erich Wichmann, Christa Meisinger Heart Attack Risk in Puget Sound. Stephen M Schwartz, David S Siscovick, Jean Yee, Yechiel Friedlander INTERHEART. James C Engert, Ron Do, Changchun Xie, Sonia Anand Irish Family Study. Pascal P McKeown, Chris C Patterson Malmo Diet and Cancer Study. Olle Melander, Goran Berglund Massachusetts General Hospital Premature Coronary Artery Disease Study. Sekar Kathiresan, James B Meigs, Gordon Williams, David M Nathan, Calum A MacRae, Christopher J O’Donnell Mid-America Heart Institute. Thomas Morgan, John A Spertus PennCath/Medstar. Muredach P Reilly, Mingyao Li, Liming Qu, Robert Wilensky, William Matthai, Atif Qasim, Hakon H Hakonarson, Joseph M Devaney, Mary-Susan Burnett, Augusto D Pichard, Kenneth M Kent, Lowell Satler, Joseph M Lindsay, Ron Waksman, Christopher W Knouff, Dawn M Waterworth, Max C Walker, Vincent Mooser, Stephen E Epstein, Daniel J Rader Registre Gironi del COR. Roberto Elosua, Jaume Marrugat, Gavin Lucas, Isaac Subirana, Joan Sala, Rafael Ramos Verona Heart Study. Domenico Girelli, Nicola Martinelli, Oliviero Olivieri, Elisabetta Trabetti, Giovanni Malerba, Pier Franco Pignatti Myocardial Infarction Genetics consortium cohorts: Aarti Surti, Candace Guiducci, Lauren Gianniny, Daniel Mirel, Melissa Parkin, Noel Burtt, Stacey B Gabriel, Benjamin F Voight, Sekar Kathiresan, Joel N Hirschhorn, Rosanna Asselta, Stefano Duga, Marta Spreafico, Kiran Musunuru, Mark J Daly, Shaun Purcell, Stephen M Schwartz, Jean Yee, Gavin Lucas, Isaac Subirana, David Siscovick, Christopher J O’Donnell, Nilesh J Samani, Olle Melander, Roberto Elosua, Leena Peltonen, Veikko Salomaa, Stephen M Schwartz, David Altshuler. 22 Wellcome Trust Case Control Consortium study of CAD. Nilesh J Samani, John R Thompson, Peter S Braund, Benjamin J Wright, Anthony J Balmforth, Stephen G Ball, Alistair S Hall CARDIOGENICS consortium: Heribert Schunkert, Nilesh J Samani, Jeanette Erdmann, Willem Ouwehand, Christian Hengstenberg, Panos Deloukas, Michael Scholz, Francois Cambien 23 References 1. Bare LA, Morrison AC, Rowland CM, et al. Five common gene variants identify elevated genetic risk for coronary heart disease. Genet Med 2007;9:682-9. 2. Iakoubova OA, Sabatine MS, Rowland CM, et al. Polymorphism in KIF6 gene and benefit from statins after acute coronary syndromes: results from the PROVE IT-TIMI 22 study. J Am Coll Cardiol 2008;51:449-55. 3. Iakoubova OA, Tong CH, Rowland CM, et al. Association of the Trp719Arg polymorphism in kinesinlike protein 6 with myocardial infarction and coronary heart disease in 2 prospective trials: the CARE and WOSCOPS trials. J Am Coll Cardiol 2008;51:435-43. 4. Shiffman D, Chasman DI, Zee RY, et al. A kinesin family member 6 variant is associated with coronary heart disease in the Women's Health Study. J Am Coll Cardiol 2008;51:444-8. 5. Shiffman D, O'Meara ES, Bare LA, et al. Association of gene variants with incident myocardial infarction in the Cardiovascular Health Study. Arterioscler Thromb Vasc Biol 2008;28:173-9. 6. StatinCheck. Berkeley HeartLab Inc., 2009. 7. Berkeley HeartLab Inc., 2009. 8. Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357:443-53. 9. McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007;316:1488-91. 10. Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007;316:1491-3. 11. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661-78. 12. Kathiresan S, Voight BF, Purcell S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 2009;41:334-41. 24 13. Stewart AF, Dandona S, Chen L, et al. Kinesin family member 6 variant Trp719Arg does not associate with angiographically defined coronary artery disease in the Ottawa Heart Genomics Study. J Am Coll Cardiol 2009;53:1471-2. 14. Assimes TL, Knowles JW, Basu A, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE Study. Hum Mol Genet 2008. 15. Vennemann MM, Hummel T, Berger K. The association between smoking and smell and taste impairment in the general population. J Neurol 2008;255:1121-6. 16. Sutton BS, Crosslin DR, Shah SH, et al. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case-control and family datasets. Hum Mol Genet 2008;17:1318-28. 17. Vartiainen E, Jousilahti P, Alfthan G, Sundvall J, Pietinen P, Puska P. Cardiovascular risk factor changes in Finland, 1972-1997. Int J Epidemiol 2000;29:49-56. 18. Herrera-Galeano JE, Becker DM, Wilson AF, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol 2008;28:1484-90. 19. Meiner V, Friedlander Y, Milo H, et al. Cholesteryl ester transfer protein (CETP) genetic variation and early onset of non-fatal myocardial infarction. Ann Hum Genet 2008;72:732-41. 20. Serre D, Montpetit A, Pare G, et al. Correction of population stratification in large multi-ethnic association studies. PLoS One 2008;3:e1382. 21. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937-52. 22. Meng W, Hughes A, Patterson CC, et al. Genetic variants of complement factor H gene are not associated with premature coronary heart disease: a family-based study in the Irish population. BMC Med Genet 2007;8:62. 25 23. Berglund G, Elmstahl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med 1993;233:45-51. 24. Low AF, O'Donnell CJ, Kathiresan S, et al. Aging syndrome genes and premature coronary artery disease. BMC Med Genet 2005;6:38. 25. Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA 2007;297:1551-61. 26. Morgan TM, Xiao L, Lyons P, Kassebaum B, Krumholz HM, Spertus JA. Investigation of 89 candidate gene variants for effects on all-cause mortality following acute coronary syndrome. BMC Med Genet 2008;9:66. 27. Senti M, Tomas M, Marrugat J, Elosua R. Paraoxonase1-192 polymorphism modulates the nonfatal myocardial infarction risk associated with decreased HDLs. Arterioscler Thromb Vasc Biol 2001;21:415-20. 28. Martinelli N, Girelli D, Malerba G, et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am J Clin Nutr 2008;88:941-9. 29. Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet 2009;10:117-33. 30. Armitage P. Tests for Linear Trends in Proportions and Frequencies. Biometrics 1955;11:375 - 386. 31. Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered 2008;66:87-98. 32. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719-48. 33. Huedo-Medina TB, Sanchez-Meca J, Marin-Martinez F, Botella J. Assessing heterogeneity in metaanalysis: Q statistic or I2 index? Psychol Methods 2006;11:193-206. 34. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics 2000;155:945-59. 26 35. Assimes TL, Knowles JW, Priest JR, et al. Common polymorphisms of ALOX5 and ALOX5AP and risk of coronary artery disease. Hum Genet 2008. 36. Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999;55:997-1004. 37. Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 1994;330:1041-6. 38. Average Annual Incidence Rate Tables (Atherosclerosis Risc in Communities Community Surveillance 1987-2004). Collaborative Studies Coordinating Center, University of North Carolina at Chapel Hill, 2007. 39. Bernick C, Katz R, Smith NL, et al. Statins and cognitive function in the elderly: the Cardiovascular Health Study. Neurology 2005;65:1388-94. 40. Kurth T, Everett BM, Buring JE, Kase CS, Ridker PM, Gaziano JM. Lipid levels and the risk of ischemic stroke in women. Neurology 2007;68:556-62. 41. Truesdale KP, Stevens J, Cai J. Nine-year changes in cardiovascular disease risk factors with weight maintenance in the atherosclerosis risk in communities cohort. Am J Epidemiol 2007;165:890-900. 42. Samani NJ, Raitakari OT, Sipila K, et al. Coronary Artery Disease-Associated Locus on Chromosome 9p21 and Early Markers of Atherosclerosis. Arterioscler Thromb Vasc Biol 2008. 43. Schunkert H, Gotz A, Braund P, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation 2008;117:1675-84. 44. Paynter NP, Chasman DI, Buring JE, Shiffman D, Cook NR, Ridker PM. Cardiovascular disease risk prediction with and without knowledge of genetic variation at chromosome 9p21.3. Ann Intern Med 2009;150:65-72. 45. Freimer N, Sabatti C. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet 2004;36:1045-51. 46. Freimer NB, Sabatti C. Guidelines for association studies in Human Molecular Genetics. Hum Mol Genet 2005;14:2481-3. 27 47. Pe'er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008;32:381-5. 48. Iakoubova O, Shepherd J, Sacks F. Association of the 719Arg variant of KIF6 with both increased risk of coronary events and with greater response to statin therapy. J Am Coll Cardiol 2008;51:2195; author reply 2195-6. 49. Marian AJ. Surprises of the genome and "personalized" medicine. J Am Coll Cardiol 2008;51:456-8. 50. Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science 2008;322:881-8. 28 Supplementary Appendix Lack of association between the Trp719Arg polymorphism in kinesinlike protein 6 and coronary artery disease in 19 case-control studies Themistocles L. Assimes et al. (see main text for full list of authors and affiliations) Address for Correspondence: Themistocles L. Assimes, MD PhD Falk Cardiovascular Research Building Stanford University School of Medicine Stanford, CA 94305-5406 29 SUPPLEMENTARY METHODS Study Populations Detailed description of the eligibility criteria and the source population for all groups in the multi ethnic ADVANCE study can be found elsewhere (1-4). Briefly, between October 28, 2001 and December 31, 2003, 3179 member of the Kaiser Permanente of Northern California health maintenance organization were enrolled into three CAD groups and two control groups. The case groups included subjects with “early onset” CAD (as defined by a CAD qualifying event captured by the electronic databases at any time after Jan 1, 1999 including AMI, angina with at least one angiographic stenosis of >50%, or revascularization procedure in men 18 to 45 or women 18 to 55 years of age at the time of the event), incident stable exertional angina at an older age, and incident non fatal AMI at an older age. The young control group included de novo recruited subjects between the ages of 30 and 45 for men or 30 and 55 for women as well as a subset of 479 participants in the same age range from the Coronary Artery Risk Development in Young Adults (CARDIA) study originally recruited at the Oakland field center and attending the study’s Year 15 examination in 2000–2001(5). Older controls included men and women aged 60–69 years at the time of identification in June, 2001. All controls were free of clinical CAD, cerebrovascular disease (CVD) and peripheral arterial disease (PAD) at the time of recruitment. Thus, the total study sample included 3658 subjects. Only a subset of all subjects was used for the genome wide association study. Cases included all “early onset” CAD cases (n = 470) and a small number of cases (n = 43) in the older incident exertional angina group that met the age criterion for “early onset” CAD but not all other criteria to be included in the “early onset” case group. Controls included a stratified subset of all young controls (n = 527) that best matched the age at first onset of clinically significant CAD, race/ethnic group mix, and gender of cases. The AMI Gene Study/Dortmund Health Study(6) is a prospective multi-centre registry involving 4 heart centres and 6 cardiologic departments in North-Rhine-Westphalia, Lower Saxony, and Mecklenburg-Western Pomerania. Between 2004 and 2006, we enrolled a cohort of 809 consecutive men younger than 65 and suffering from non-ST-segment elevation MI or ST segment elevation MI with an onset of symptoms less than 30 24 hours before admission. All cases underwent cardiac catheterization and interventional or surgical revascularization. After informed consent personal data collection and blood sampling where carried out at admission. Information on history, traditional risk factors and co-morbidities were documented after interventional procedures and clinical stabilization of the patients. Controls came from the Dortmund Health Study(6,7), a population-based survey conducted in the city of Dortmund with the aim to determine the prevalence of headache types, cardiovascular and other chronic diseases and their risk factors in the general population. Sampling for the study was done randomly from the city’s population register stratified by fiveyear age group and gender. History of MI and other cardiovascular conditions was assessed in face-to-face interviews. The study was conducted in 2003-2004. The recruitment protocols and study procedures were approved by the ethics committees of the University of Witten-Herdecke and the University of Muenster, Germany, respectively. CATHGEN study participants were enrolled at Duke University Medical Center (Durham, NC) through the CATHGEN biorepository, consisting of subjects greater than 18 years of age, recruited sequentially through the cardiac catheterization laboratories from 2001-2005(8). Biological samples and extensive clinical, angiographic, and longitudinal follow-up data were collected on all subjects consenting to participation. Blood samples were obtained from the femoral artery at initiation of the procedure. For purposes of this study, cases were defined as those having a history of MI (by self-report and corroborated by review of medical records), or having suffered an MI during the study follow-up period. Controls from this cohort, defined as those with no history of MI prior or subsequent to the index cardiac catheterization, as well as no history of percutaneous or surgical coronary revascularization procedure; no subsequent percutaneous or surgical coronary revascularization procedures; ejection fraction on left ventriculogram greater than 40%; and no or minimal CAD on coronary angiography (defined as a CAD index less than or equal to 23 and no coronary vessel with clinically significant CAD (stenosis greater than 50%)). CAD index is a numerical summary of angiographic data directly related to outcome(9). The GWA study of CAD in Icelanders by deCODE has been previously described (10). However, this report includes an additional 2706 cases of CAD and 18224 controls. Briefly, over the last 10 years, MI 31 patients have been recruited through the cardiovascular disease (CVD) genetics program at deCODE. Individuals who suffered an MI were identified from a registry of over 10,000 individuals who: a) had an MI before the age of 75 in Iceland in the years 1981 to 2002 and satisfy the MONICA criteria(11)or had MI discharge diagnosis from the major hospitals in Reykjavik in the years 2003 to 2005. MI diagnoses of all individuals in the registry follow strict diagnostic criteria based on signs, symptoms, electrocardiograms, cardiac enzymes and necropsy findings(11,12). Additional subjects with coronary artery disease, but that are without known history of MI, were identified from a list of those who have undergone percutaneous coronary intervention (PCI) in the major hospital in Reykjavik in the years 1993 to 2003 or from those who have participated in the CVD genetics program at deCODE and have a self reported history of coronary artery bypass graft (CABG) or PCI. The controls used for the study were recruited as a part of various genetic programs at deCODE. The medical history of the controls were unknown unless they had also participated in any of the CVD genetic programs (i.e. MI, stroke, peripheral vascular disease, type II diabetes, obesity, familial combined hyperlipidemia, coronary restenosis, and hypertension). Individuals with known MI, stroke, peripheral vascular or coronary artery disease were excluded as controls. The 24951 controls used break down approximately as follows: neurologic disorders ~3000, psychiatric disorders ~4900, cancers ~3400, gynecologic disoders ~1500, pulmonary disorders ~2300, psoriasis ~800, type II diabetes ~800, osteoporosis ~2100, preeclampsia ~700, rheumatoid arthritis ~500, longevity ~1000, benign prostatic hyperplasia ~400, enuresis ~751, infectious diseases ~1700, glaucoma 300, population controls ~800. FINRISK(13) contributed cases of early-onset MI and controls. Cases were defined as men aged ≤50 and women aged ≤60 and hospitalized with MI or died of MI, and were obtained from a national populationbased survey of risk factors for cardiovascular disease. FINRISK surveys are conducted every 5 years, and from the 23,188 individuals enrolled in FINRISK 1992, 1997, and 2002, we assembled cases and controls. Hospitalization data and mortality data for MI were obtained from the Finnish National Hospital Discharge Register and the Finnish National Causes-of-Death Register as of December 31, 2004 based on ICD-9 code of 410 and ICD-10 codes of I20 and I21. These registers have excellent validity. Cases included MI prevalent at baseline examination and incident events on follow-up. Controls were randomly selected from participants of 32 the three FINRISK cohorts, who survived until the event date of the case without hospitalization for CHD or for stroke, matched to cases based on age, sex, study area, and FINRISK cohort year. Data collected in both cases and controls at a baseline study examination include risk factors (based on questionnaire and blood samples), anthropometry, and blood pressure measurement. The GeneSTAR (Genetic Study of Atherosclerosis Risk)(14) famly study cases were identified from probands hospitalized in 10 Baltimore area hospitals with documented CHD (myocardial infarction, angina with revascularization, angina with documented obstructive coronary stenoses without revascularization, or documented obstructive coronary stenoses with or without revascularization) prior to 60 years of age. The classification of all cases followed strict diagnostic criteria based on electrocardiograms, cardiac enzymes and necropsy findings. All CHD events were adjudicated by 2 cardiologists using standard criteria applied to data obtained from medical records. The controls used for the study were unaffected siblings, spouses and offspring of CHD cases followed over a 22 year time period (from 1983 to 2006). Medical history of the controls was gathered directly during participation in the GeneSTAR Study. Controls that developed incident CHD (defined as all of the above as well as sudden cardiac death) during the 22 year follow up time period were excluded from the analysis. German MI Family Study I (GerMIFS I) has been recently described(15). The recruitment for the German MI Family Study II (GerMIFS II) was similar to that for German MI Family Study I. All 1,222 patients had a validated MI with a strong genetic component as documented by an early age of onset (prior to the age of 60 years). Moreover, a positive family history for CAD was documented in 726 (59.4 %) of patients. Patients were identified following their admission for acute treatment of MI or in cardiac rehabilitation clinics. Population-based controls were derived from the MONICA/KORA Augsburg survey S4 (n=820)(16) and the PopGen blood donor sample (PopGen-BSP) (n=478)(17). The Heart Attack Risk in Puget Sound (HARPS) (18) study is a population-based case-control study of cases of early-onset MI and controls matched on age and sex. Eligible case patients were men aged <50 and women <60 diagnosed with a first MI between 1991 to 2002. Cases were ascertained through medical record review at all acute care facilities in King, Pierce, and Snohomish counties, Washington, US. Controls were 33 identified using random digit dialing from these counties (1991-2002) and had no history of cardiovascular disease. Data collected through in-person interviews with each case and control include information on medical and lifestyle risk factors, and a self-administered food frequency questionnaire. The INTERHEART(19,20) is a global case/control study of risk factors for acute MI involving 27,098 individuals recruited from 262 centers in 52 countries. Informed written consent to obtain the baseline information and to collect and store the genetic and other biologic specimens was obtained from 21,508 individuals (including all individuals analyzed in this study). To identify incident cases of acute MI, all patients, irrespective of age, admitted to the coronary care unit (or an equivalent cardiology ward) within 24 hours of symptom onset were screened. Cases were eligible if they had characteristic symptoms plus electrocardiogram changes indicative of a new MI (new pathologic Q waves, at least 1 mm ST elevation in any two or more contiguous limb leads or a new left bundle branch block, or new persistent ST-T wave changes diagnostic of a non-Q wave MI) or a plasma level of cardiac troponin level above that considered normal in the hospital/institution where the patient was registered. For each case, at least one control of the same age (±5 years) and sex was recruited from the same centre. Controls were defined as individuals who had no previous diagnosis of heart disease or history of exertional chest pain. Eligible controls were classified as i) hospitalbased, defined as patients attending the hospital or outpatient clinics for the following reasons: refraction and cataracts, physical check-up, routine pap smear, routine breast exam, elective minor surgery for conditions that were not obviously related to CHD or its risk factors, elective orthopedic surgery (eligibility dependent on ability to complete physical measures) or patients attending the hospital or outpatient clinics for: outpatient fractures, arthritic complaints, plastic surgery, hemorrhoids, hernias, hydroceles, routine colon cancer screening, endoscopy, minor dermatologic disorders; or ii) community-based, defined as visitors or relatives of a patient from a non-cardiac ward, or an unrelated (not first-degree relative) visitor of a cardiac patient. Of the controls in INTERHEART, 58% were hospital-based and 36% of controls were community-based, and results were similar with both types of controls. In the remainder of the controls, 3% were from an undocumented source, and 3% were recruited through the WHO MONICA study in Göteborg, Sweden. Exclusion criteria for controls were identical to those described for cases. Structured questionnaires were administered to all cases and controls to 34 obtain information on demographic factors (including self-reported ethnicity) as well as socioeconomic and health status. For this project, we analyzed individuals with self-reported ethnicity defined as “South Asian” or “European” regardless of their geographic locations. Recruitment for the Irish Family Study (IFS)(21) took place between August 1999 and October 2004. All subjects were Caucasian whose four grandparents were born in Ireland. Each family had at least one member affected with early-onset MI (disease onset ≤55 years for males and ≤60 years for females) and at least one unaffected sibling and / or both parents surviving. Unaffected siblings were required to: (i) be older than the affected sibling was at the onset of coronary heart disease; (ii) have no symptoms of angina or possible MI by World Health Organization questionnaire assessments; (iii) have no history of coronary heart disease diagnosed by a doctor; and (iv) have a resting 12 lead ECG record showing no evidence of ischemia or previous MI. The Malmo Diet and Cancer study (MDC)(22) contributed cases of early-onset MI and controls matched for age and sex. Cases were defined as men aged ≤50 and women aged ≤60 and hospitalized with MI, and were obtained from a population-based cohort study of 28,098 men and women living in Malmo, Sweden, between 1991 and 1996. Data on MI were obtained by record linkage to the Swedish National Hospital Discharge Register as of December 31, 2000 based on ICD-9 code 410 and ICD-10 code I21. Cases included MI prevalent at baseline examination and incident events on follow-up. Controls were randomly selected among the 28,098 participants and were free of MI (criteria above) at baseline examination and during follow-up. Data collected in cases and controls at a baseline examination include risk factors (based on questionnaire and blood samples), anthropometry, dietary assessment, and a physical examination. The Washington Hospital Center catheterization (MEDSTAR)(10) study is a Washington Hospital Center based angiographic study of 1,500 subjects specifically designed for biomarker and genetic association studies of acute and chronic coronary atherosclerosis. MedStar is a cross sectional study of coronary atherosclerosis in a consecutive cohort of selected patients undergoing cardiac catheterization at Washington Hospital between August 2004 and March 2007. All subjects have been enrolled in a Washington Hospital Institutional Review Board approved protocol and all subjects gave written informed consent. Enrollment criteria include any clinical indication for cardiac catheterization and ability to give informed consent. The 35 following data were extracted from the medical record; age, gender, race/ethnicity, past medical, social, family and medication history, cardiovascular risk factors (diabetes, smoking, and hypertension), physical exam including vital signs, weight and height (for BMI), and cardiovascular findings. Ethnicity information was self reported. Data from cardiac catheterization including coronary angiography were recorded. Coronary angiograms were scored on the day by the interventional cardiologist who performed the procedure and reviewed by a second cardiologist at a later date. Blood was drawn in a 12-hour fasting state (except in those with acute MI), at the time of the initial catheter insertion prior to the administration of any contrast dye for plasma, serum and buffy coat DNA isolation. All demographic, anthropometric, clinical, cardiac catheterization, laboratory, and genetic data are integrated into study database that exclude personal identifiers. A case-control GWAS similar to PennCATH (see description below) was performed in MedStar (N=1,322 Caucasians) composed of controls (N=447) who on coronary angiography showed no evidence of CAD and CAD cases (N=874) with one or more coronary vessels demonstrating ≥50% stenosis. These cases were divided into stable CAD cases without history of MI and CAD cases with a history of MI. Controls were aged over 45 in men and women. The Massachusetts General Hospital of Premature CAD study (MGH PCAD) (23) is a hospital-based case-control study of cases of early-onset MI and controls matched on age and sex. Eligible case patients were men aged ≤50 and women aged ≤60 who were hospitalized at MGH with MI between 1999 and 2004. An attempt was made to recruit all patients admitted to MGH meeting the case definition. Controls were recruited from Boston and its vicinity through a general newspaper advertisement and eligible if they reported no history of cardiovascular disease or cardiac medications. Data collected through in-person interviews and examination for each case and control include risk factors, anthropometry, and blood pressure. The Mid-America Heart Institute (MAHI)(24,25) patients (N = 811) were recruited in successive prospective cohort studies designed to investigate clinical outcomes among survivors of acute coronary syndrome. Age, sex, and BMI-matched control subjects without known CAD (N = 650) consisted of ambulatory outpatients from the same geographic area presenting for routine laboratory testing. All 1,461 36 subjects submitted for genotyping were of self-reported white/mixed European ancestry. There were no restrictions on age. The Penn-CATH(10) cohort is a University of Pennsylvania Medical Center based angiographic study of over 3,800 subjects that has been used previously for replication of novel genes and risk factors for atherosclerotic cardiovascular disease and type 2 diabetes(26-29). Between July 1998 and March 2003, PennCATH recruited a consecutive cohort of patients undergoing cardiac catheterization at Penn. A total of 3,850 subjects were recruited. Enrollment criteria included any clinical indication for cardiac catheterization and ability to give informed consent. The following data were extracted from the medical record; age, gender, race/ethnicity, past medical (including diabetes, hypertension, dyslipidemia, prior MI and cardiac events), social, family and medication history, cardiovascular risk factors, physical exam including vital signs, weight and height (for BMI). Ethnicity information was self-reported. Data from cardiac catheterization including coronary angiography were recorded. Blood was drawn in a fasting state, DNA (buffy coat) and plasma was isolated, and lipoproteins and glucose were assayed on all samples. A GWAS was recently performed in a subset of PennCATH participants including 468 European subjects who on coronary angiography showed no evidence of CAD (“controls”) and 933 subjects with one or more coronary vessels demonstrating ≥50% stenosis (“cases”). These cases were equally selected for stable CAD cases without history of MI and CAD cases with a history of MI. Controls were aged over 40 in men and 45 in women. The Registre Gironı´ del Cor (REGICOR) study(30) is a population-based case-control study of cases of early-onset MI and controls matched on age and sex. Specifically, eligible case patients were consecutive patients hospitalized with a first MI to the only coronary care unit in the catchment area of Gerona, Spain. Controls were subjects randomly selected from a cross-sectional study of cardiovascular risk factors in the catchment area and were deemed free of MI by history, physical examination, and ECG. Data collected through in-person interviews and examination include risk factors (based on questionnaire and blood samples), anthropometry, and blood pressure. The Verona Heart Study (VHS)(31) is an ongoing study aimed at identifying new risk factors for CAD and MI in a population of subjects with angiographic documentation of their coronary vessel. The CAD group 37 had angiographically documented severe coronary atherosclerosis, the majority of them being candidates for coronary artery bypass grafting or percutaneous coronary intervention. Control subjects were selected such that they had normal coronary arteries, being submitted to coronary angiography for reasons other than CAD. Controls with history or clinical evidence of atherosclerosis in vascular territories beyond the coronary bed were excluded. Information on MI diagnoses was gathered through medical records showing diagnostic electrocardiogram and enzyme changes, and/or the typical sequelae of MI on ventricular angiography and on echocardiography. The local Ethical Committee approved the study. Informed consent was obtained from all the patients after a full explanation of the study. Wellcome Trust Case Control CAD study (WTCCC CAD) (15) (32) has been recently described. Briefly, cases comprised subjects with a validated history of MI or CAD before the age of 66 years and also a family history of premature CAD in a first degree relative. Controls were derived in equal numbers from two sources: (i) the 1958 UK Birth Cohort and (ii) the National Blood Service Collection (NBS) recruited specifically for the WTCCC Study. Statistical Analysis Fixed meta analysis for the additive model were performed using the following R script: pv<-c(P1, P2, P3, etc…) rr<-c(OR1, OR2, OR3, etc…) zscore<-abs(qnorm(pv)) lrr<-log(rr) wt<-zscore/lrr lr<-sum(wt**2*lrr)/(sum(wt**2)) r<-exp(lr) z<-sum(wt*zscore)/sqrt(sum(wt**2)) z1<-abs(z) p<-2*pnorm(-z1) se<-lr/z 38 ci<-exp(c(lr-1.96*se,lr+1.96*se)) r<-exp(lr) p r ci where P1, P2, P3, etc represent the P-values from the respective studies that have been changed to one-sided pvalues, i.e. p/2 if OR > 1, else 1 - p/2. The different cohorts are weighed by the standard error of the OR represented by OR1, OR2, OR3 etc…. Informed consent. All participants in the 19 studies gave written informed consent in accordance with the guidelines of local ethical committees. 39 Membership of the Wellcome Trust Case Control Consortium Management Committee: Paul R Burton1, David G Clayton2, Lon R Cardon3, Nick Craddock4, Panos Deloukas5, Audrey Duncanson6, Dominic P Kwiatkowski3,5, Mark I McCarthy3,7, Willem H Ouwehand8,9, Nilesh J Samani10, John A Todd2, Peter Donnelly (Chair)11 Data and Analysis Committee: Jeffrey C Barrett3, Paul R Burton1, Dan Davison11, Peter Donnelly11, Doug Easton12, David M. Evans3, Hin-Tak Leung2, Jonathan L Marchini11, Andrew P Morris3, Chris CA Spencer11, Martin D Tobin1, Lon R Cardon (Co-chair)3, David G Clayton (Co-chair)2 UK Blood Services & University of Cambridge Controls: Antony P Attwood5,8, James P Boorman8,9, Barbara Cant8, Ursula Everson13, Judith M Hussey14, Jennifer D Jolley8, Alexandra S Knight8, Kerstin Koch8, Elizabeth Meech15, Sarah Nutland2, Christopher V Prowse16, Helen E Stevens2, Niall C Taylor8, Graham R Walters17, Neil M Walker2, Nicholas A Watkins8,9, Thilo Winzer8, John A Todd2, Willem H Ouwehand8,9 1958 Birth Cohort Controls: Richard W Jones18, Wendy L McArdle18, Susan M Ring18, David P Strachan19, Marcus Pembrey18,20 Bipolar Disorder (Aberdeen): Gerome Breen21, David St Clair21; (Birmingham): Sian Caesar22, Katherine Gordon-Smith22,23, Lisa Jones22; (Cardiff): Christine Fraser23, Elaine K Green23, Detelina Grozeva23, Marian L Hamshere23, Peter A Holmans23, Ian R Jones23, George Kirov23, Valentina Moskvina23, Ivan Nikolov23, Michael C O’Donovan23, Michael J Owen23, Nick Craddock23; (London): David A Collier24, Amanda Elkin24, Anne Farmer24, Richard Williamson24, Peter McGuffin24; (Newcastle): Allan H Young25, I Nicol Ferrier25 Coronary Artery Disease (Leeds): Stephen G Ball26, Anthony J Balmforth26, Jennifer H Barrett26, D Timothy Bishop26, Mark M Iles26, Azhar Maqbool26, Nadira Yuldasheva26, Alistair S Hall26; (Leicester): Peter S Braund10, Paul R Burton1, Richard J Dixon10, Massimo Mangino10, Suzanne Stevens10, Martin D Tobin1, John R Thompson1, Nilesh J Samani10 Crohn’s Disease (Cambridge): Francesca Bredin27, Mark Tremelling27, Miles Parkes27; (Edinburgh): Hazel Drummond28, Charles W Lees28, Elaine R Nimmo28, Jack Satsangi28; (London): Sheila A Fisher29, Alastair Forbes30, Cathryn M Lewis29, Clive M Onnie29, Natalie J Prescott29, Jeremy Sanderson31, Christopher G Mathew29; (Newcastle): Jamie Barbour32, M Khalid Mohiuddin32, Catherine E Todhunter32, John C Mansfield32; (Oxford): Tariq Ahmad33, Fraser R Cummings33, Derek P Jewell33 Hypertension (Aberdeen): John Webster34; (Cambridge): Morris J Brown35, David G Clayton2; (Evry, France): G Mark Lathrop36; (Glasgow): John Connell37, Anna Dominiczak37; (Leicester): Nilesh J Samani10; (London): Carolina A Braga Marcano38, Beverley Burke38, Richard Dobson38, Johannie Gungadoo38, Kate L Lee38, Patricia B Munroe38, Stephen J Newhouse38, Abiodun Onipinla38, Chris Wallace38, Mingzhan Xue38, Mark Caulfield38; (Oxford): Martin Farrall39 Rheumatoid Arthritis: Anne Barton40, Ian N Bruce40, Hannah Donovan40, Steve Eyre40, Paul D Gilbert40, Samantha L Hider40, Anne M Hinks40, Sally L John40, Catherine Potter40, Alan J Silman40, Deborah PM Symmons40, Wendy Thomson40, Jane Worthington40 Type 1 Diabetes: David G Clayton2, David B Dunger2,41, Sarah Nutland2, Helen E Stevens2, Neil M Walker2, Barry Widmer2,41, John A Todd2 Type 2 Diabetes (Exeter): Timothy M Frayling42,43, Rachel M Freathy42,43, Hana Lango42,43, John R B Perry42,43, Beverley M Shields43, Michael N Weedon42,43, Andrew T Hattersley42,43; (London): Graham A Hitman44; (Newcastle): Mark Walker45; (Oxford): Kate S Elliott3,7, Christopher J Groves7, Cecilia M Lindgren3,7, Nigel W Rayner3,7, Nicholas J Timpson3,46, Eleftheria Zeggini3,7, Mark I McCarthy3,7 Tuberculosis (Gambia): Melanie Newport47, Giorgio Sirugo47; (Oxford): Emily Lyons3, Fredrik Vannberg3, Adrian VS Hill3 Ankylosing Spondylitis: Linda A Bradbury48, Claire Farrar49, Jennifer J Pointon48, Paul Wordsworth49, Matthew A Brown48,49 AutoImmune Thyroid Disease: Jayne A Franklyn50, Joanne M Heward50, Matthew J Simmonds50, Stephen CL Gough50 Breast Cancer: Sheila Seal51, Michael R Stratton51,52, Nazneen Rahman51 Multiple Sclerosis: Maria Ban53, An Goris53, Stephen J Sawcer53, Alastair Compston53 40 Gambian Controls (Gambia): David Conway47, Muminatou Jallow47, Melanie Newport47, Giorgio Sirugo47; (Oxford): Kirk A Rockett3, Dominic P Kwiatkowski3,5 DNA, Genotyping, Data QC and Informatics (Wellcome Trust Sanger Institute, Hinxton): Suzannah J Bumpstead5, Amy Chaney5, Kate Downes2,5, Mohammed JR Ghori5, Rhian Gwilliam5, Sarah E Hunt5, Michael Inouye5, Andrew Keniry5, Emma King5, Ralph McGinnis5, Simon Potter5, Rathi Ravindrarajah5, Pamela Whittaker5, Claire Widden5, David Withers5, Panos Deloukas5; (Cambridge): Hin-Tak Leung2, Sarah Nutland2, Helen E Stevens2, Neil M Walker2, John A Todd2 Statistics (Cambridge): Doug Easton12, David G Clayton2; (Leicester): Paul R Burton1, Martin D Tobin1; (Oxford): Jeffrey C Barrett3, David M Evans3, Andrew P Morris3, Lon R Cardon3; (Oxford): Niall J Cardin11, Dan Davison11, Teresa Ferreira11, Joanne Pereira-Gale11, Ingeleif B Hallgrimsdóttir11, Bryan N Howie11, Jonathan L Marchini11, Chris CA Spencer11, Zhan Su11, Yik Ying Teo3,11, Damjan Vukcevic11, Peter Donnelly11 PIs: David Bentley5,54, Matthew A Brown48,49, Lon R Cardon3, Mark Caulfield38, David G Clayton2, Alistair Compston53, Nick Craddock23, Panos Deloukas5, Peter Donnelly11, Martin Farrall39, Stephen CL Gough50, Alistair S Hall26, Andrew T Hattersley42,43, Adrian VS Hill3, Dominic P Kwiatkowski3,5, Christopher G Mathew29, Mark I McCarthy3,7, Willem H Ouwehand8,9, Miles Parkes27, Marcus Pembrey18,20, Nazneen Rahman51, Nilesh J Samani10, Michael R Stratton51,52, John A Todd2, Jane Worthington40 1 Genetic Epidemiology Group, Department of Health Sciences, University of Leicester, Adrian Building, University Road, Leicester, LE1 7RH, UK; 2 Juvenile Diabetes Research Foundation/Wellcome Trust Diabetes and Inflammation Laboratory, Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Wellcome Trust/MRC Building, Cambridge, CB2 0XY, UK; 3 Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford OX3 7BN, UK; 4 Department of Psychological Medicine, Henry Wellcome Building, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK; 5 The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK; 6 The Wellcome Trust, Gibbs Building, 215 Euston Road, London NW1 2BE, UK; 7 Oxford Centre for Diabetes, Endocrinology and Medicine, University of Oxford, Churchill Hospital, Oxford, OX3 7LJ, UK; 8 Department of Haematology, University of Cambridge, Long Road, Cambridge, CB2 2PT, UK; 9 National Health Service Blood and Transplant, Cambridge Centre, Long Road, Cambridge, CB2 2PT, UK; 10 Department of Cardiovascular Sciences, University of Leicester, Glenfield Hospital, Groby Road, Leicester, LE3 9QP, UK; 11 Department of Statistics, University of Oxford, 1 South Parks Road, Oxford OX1 3TG, UK; 12 Cancer Research UK Genetic Epidemiology Unit, Strangeways Research Laboratory, Worts Causeway, Cambridge CB1 8RN, UK; 13 National Health Service Blood and Transplant, Sheffield Centre, Longley Lane, Sheffield S5 7JN, UK; 14 National Health Service Blood and Transplant, Brentwood Centre, Crescent Drive, Brentwood, CM15 8DP, UK; 15 The Welsh Blood Service, Ely Valley Road, Talbot Green, Pontyclun, CF72 9WB, UK; 16 The Scottish National Blood Transfusion Service, Ellen’s Glen Road, Edinburgh, EH17 7QT, UK; 17 National Health Service Blood and Transplant, Southampton Centre, Coxford Road, Southampton, SO16 5AF, UK; 18 Avon Longitudinal Study of Parents and Children, University of Bristol, 24 Tyndall Avenue, Bristol, BS8 1TQ, UK; 19 Division of Community Health Services, St George’s University of London, Cranmer Terrace, London SW17 0RE, UK; 20 Institute of Child Health, University College London, 30 Guilford St, London WC1N 1EH, UK; 21 University of Aberdeen, Institute of Medical Sciences, Foresterhill, Aberdeen, AB25 2ZD, UK; 22 Department of Psychiatry, Division of Neuroscience, Birmingham University, Birmingham, B15 2QZ, UK; 23 Department of Psychological Medicine, Henry Wellcome Building, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK; 24 SGDP, The Institute of Psychiatry, King's College London, De Crespigny Park Denmark Hill London SE5 8AF, UK; 25 School of Neurology, Neurobiology and Psychiatry, Royal Victoria Infirmary, Queen Victoria Road, Newcastle upon Tyne, NE1 4LP, UK; 26 LIGHT and LIMM Research Institutes, Faculty of Medicine and Health, University of Leeds, Leeds, LS1 3EX, UK; 27 IBD Research Group, Addenbrooke's Hospital, University of Cambridge, Cambridge, CB2 2QQ, UK; 28 Gastrointestinal Unit, School of Molecular and Clinical Medicine, University of Edinburgh, Western General Hospital, Edinburgh EH4 2XU UK; 29 Department of Medical & Molecular Genetics, King's College London School of Medicine, 8th Floor Guy's Tower, Guy's Hospital, London, SE1 9RT, UK; 30 Institute for Digestive Diseases, University College London Hospitals Trust, London, NW1 2BU, UK; 31 Department of 41 Gastroenterology, Guy's and St Thomas' NHS Foundation Trust, London, SE1 7EH, UK; 32 Department of Gastroenterology & Hepatology, University of Newcastle upon Tyne, Royal Victoria Infirmary, Newcastle upon Tyne, NE1 4LP, UK; 33 Gastroenterology Unit, Radcliffe Infirmary, University of Oxford, Oxford, OX2 6HE, UK; 34 Medicine and Therapeutics, Aberdeen Royal Infirmary, Foresterhill, Aberdeen, Grampian AB9 2ZB, UK; 35 Clinical Pharmacology Unit and the Diabetes and Inflammation Laboratory, University of Cambridge, Addenbrookes Hospital, Hills Road, Cambridge CB2 2QQ, UK; 36 Centre National de Genotypage, 2, Rue Gaston Cremieux, Evry, Paris 91057.; 37 BHF Glasgow Cardiovascular Research Centre, University of Glasgow, 126 University Place, Glasgow, G12 8TA, UK; 38 Clinical Pharmacology and Barts and The London Genome Centre, William Harvey Research Institute, Barts and The London, Queen Mary’s School of Medicine, Charterhouse Square, London EC1M 6BQ, UK; 39 Cardiovascular Medicine, University of Oxford, Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, UK; 40arc Epidemiology Research Unit, University of Manchester, Stopford Building, Oxford Rd, Manchester, M13 9PT, UK; 41 Department of Paediatrics, University of Cambridge, Addenbrooke’s Hospital, Cambridge, CB2 2QQ, UK; 42 Genetics of Complex Traits, Institute of Biomedical and Clinical Science, Peninsula Medical School, Magdalen Road, Exeter EX1 2LU UK; 43 Diabetes Genetics, Institute of Biomedical and Clinical Science, Peninsula Medical School, Barrack Road, Exeter EX2 5DU UK; 44 Centre for Diabetes and Metabolic Medicine, Barts and The London, Royal London Hospital, Whitechapel, London, E1 1BB UK; 45 Diabetes Research Group, School of Clinical Medical Sciences, Newcastle University, Framlington Place, Newcastle upon Tyne NE2 4HH, UK; 46 The MRC Centre for Causal Analyses in Translational Epidemiology, Bristol University, Canynge Hall, Whiteladies Rd, Bristol BS2 8PR, UK; 47 MRC Laboratories, Fajara, The Gambia; 48 Diamantina Institute for Cancer, Immunology and Metabolic Medicine, Princess Alexandra Hospital, University of Queensland, Woolloongabba, Qld 4102, Australia; 49 Botnar Research Centre, University of Oxford, Headington, Oxford OX3 7BN, UK; 50 Department of Medicine, Division of Medical Sciences, Institute of Biomedical Research, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK; 51 Section of Cancer Genetics, Institute of Cancer Research, 15 Cotswold Road, Sutton, SM2 5NG, UK; 52 Cancer Genome Project, The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK; 53 Department of Clinical Neurosciences, University of Cambridge, Addenbrooke’s Hospital, Hills Road, Cambridge CB2 2QQ, UK; 54 PRESENT ADDRESS: Illumina Cambridge, Chesterford Research Park, Little Chesterford, Nr Saffron Walden, Essex, CB10 1XL, UK. 42 Membership of the Cardiogenics consortium Associazione per lo Studio della Trombosi in Cardiologia, 27100 Pavia, Italy: Diego Ardissino EUROIMMUN AG, 23560 Lübeck, Germany: Christian Krüger, Lars Komorowski, Christian Probst, Ulf Steller Helmholtz Institut Zentrum München, 85764 Neuherberg, Germany: Thomas Meitinger, Erich Wichmann, Peter Lichtner Institut National de la Santé et de la Recherche Médicale (INSERM), 75654 Paris, France: Francois Cambien, Laurence Tiret Johannes Gutenberg-University of Mainz, 55099 Mainz, Germany: Stefan Blankenberg, Karl Lackner, Thomas Münzel Queen’s University of Belfast, Belfast BT71NN, UK: Alun Evans, Frank Kee Sanquin Blood Supply Foundation, 1066 CX Amsterdam, The Netherlands: Ellen van der Schoot, Jaap Jan Zwaginga Technische Universität München, 80333 München, Germany: Dietlind Zohlnhoefer TRIUM Analysis Online GmbH, 81677 München, Germany: Martin Daumer, Michael Scholz The Chancellor, Master and Scholar of the University of Cambridge, Cambridge CB2 1TS, UK: Willem Ouwehand, Richard Farndale, Nicholas Watkins University of Leeds, Leeds LS2 9JT Leeds, UK: Alistair Hall, Stephen Ball, Tim Bishop University of Leicester, Leicester LE1 7RH, UK: Nilesh Samani, Alison Goodall, John R Thompson University of Lübeck, 23538 Lübeck, Germany: Heribert Schunkert, Jeanette Erdmann, Andreas Ziegler University of Regensburg, 93053 Regensburg, Germany: Christian Hengstenberg, Gerd Schmitz University of Uppsala, 75105 Uppsala, Sweden: Ann-Christin Syvänen, Tomas Axelsson, Ulrika Liljedahl Wellcome Trust Sanger Institute, Hinxton Cambridge CB10 1SA, UK: Panos Deloukas, Kate Rice 43 References 1. Go AS, Iribarren C, Chandra M, et al. Statin and beta-blocker therapy and the initial presentation of coronary heart disease. Ann Intern Med 2006;144:229-38. 2. Iribarren C, Go AS, Husson G, et al. Metabolic syndrome and early-onset coronary artery disease: is the whole greater than its parts? J Am Coll Cardiol 2006;48:1800-7. 3. Taylor-Piliae RE, Norton LC, Haskell WL, et al. Validation of a New Brief Physical Activity Survey among Men and Women Aged 60-69 Years. Am J Epidemiol 2006. 4. Assimes TL, Knowles JW, Basu A, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE Study. Hum Mol Genet 2008. 5. Hughes GH, Cutter G, Donahue R, et al. Recruitment in the Coronary Artery Disease Risk Development in Young Adults (Cardia) Study. Control Clin Trials 1987;8:68S-73S. 6. Vennemann MM, Hummel T, Berger K. The association between smoking and smell and taste impairment in the general population. J Neurol 2008;255:1121-6. 7. Happe S, Vennemann M, Evers S, Berger K. Treatment wish of individuals with known and unknown restless legs syndrome in the community. J Neurol 2008;255:1365-71. 8. Sutton BS, Crosslin DR, Shah SH, et al. Comprehensive genetic analysis of the platelet activating factor acetylhydrolase (PLA2G7) gene and cardiovascular disease in case-control and family datasets. Hum Mol Genet 2008;17:1318-28. 9. Smith LR, Harrell FE, Jr., Rankin JS, et al. Determinants of early versus late cardiac death in patients undergoing coronary artery bypass graft surgery. Circulation 1991;84:III245-53. 10. Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007;316:1491-3. 11. Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology/World Health Organization task force on standardization of clinical nomenclature. Circulation 1979;59:607-9. 44 12. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined--a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol 2000;36:959-69. 13. Vartiainen E, Jousilahti P, Alfthan G, Sundvall J, Pietinen P, Puska P. Cardiovascular risk factor changes in Finland, 1972-1997. Int J Epidemiol 2000;29:49-56. 14. Herrera-Galeano JE, Becker DM, Wilson AF, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol 2008;28:1484-90. 15. Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357:443-53. 16. Holle R, Happich M, Lowel H, Wichmann HE. KORA--a research platform for population based health research. Gesundheitswesen 2005;67 Suppl 1:S19-25. 17. Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet 2006;9:55-61. 18. Meiner V, Friedlander Y, Milo H, et al. Cholesteryl ester transfer protein (CETP) genetic variation and early onset of non-fatal myocardial infarction. Ann Hum Genet 2008;72:732-41. 19. Serre D, Montpetit A, Pare G, et al. Correction of population stratification in large multi-ethnic association studies. PLoS One 2008;3:e1382. 20. Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937-52. 21. Meng W, Hughes A, Patterson CC, et al. Genetic variants of complement factor H gene are not associated with premature coronary heart disease: a family-based study in the Irish population. BMC Med Genet 2007;8:62. 45 22. Berglund G, Elmstahl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med 1993;233:45-51. 23. Low AF, O'Donnell CJ, Kathiresan S, et al. Aging syndrome genes and premature coronary artery disease. BMC Med Genet 2005;6:38. 24. Morgan TM, Krumholz HM, Lifton RP, Spertus JA. Nonvalidation of reported genetic risk factors for acute coronary syndrome in a large-scale replication study. JAMA 2007;297:1551-61. 25. Morgan TM, Xiao L, Lyons P, Kassebaum B, Krumholz HM, Spertus JA. Investigation of 89 candidate gene variants for effects on all-cause mortality following acute coronary syndrome. BMC Med Genet 2008;9:66. 26. Helgadottir A, Manolescu A, Helgason A, et al. A variant of the gene encoding leukotriene A4 hydrolase confers ethnicity-specific risk of myocardial infarction. Nat Genet 2006;38:68-74. 27. Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006;38:320-3. 28. Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 2007;39:770-5. 29. Helgadottir A, Thorleifsson G, Magnusson KP, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet 2008;40:21724. 30. Senti M, Tomas M, Marrugat J, Elosua R. Paraoxonase1-192 polymorphism modulates the nonfatal myocardial infarction risk associated with decreased HDLs. Arterioscler Thromb Vasc Biol 2001;21:415-20. 31. Martinelli N, Girelli D, Malerba G, et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am J Clin Nutr 2008;88:941-9. 32. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661-78. 46 47 Table 1. Study design of case control studies included in the meta-analysis of the Trp719Arg polymorphism (rs20455) in kinesin-like protein 6 and basic demographic characteristics of participants stratified by case control status ADVANCE AMI Gene CATHGEN deCODE FinRisk GENESTAR GerMIFS I GerMIFS II HARPS INTERHEART (Europeans) N Country of study Ascertainment scheme Qualifyin g event 505 U.S. population-based CAD * Study Controls 514 U.S. population-based -- 45.6±5.7 61.3 39.5 Cases 793 Germany hospital-based MI men <65 52.2±8.2 0 0.0 Controls 1121 Germany hospital-based -- -- 52.6±13.7 53.1 0.0 Cases 1575 U.S. hospital-based MI ³18 men or women 61.9±11.9 28.9 17.6 Controls 970 U.S. hospital-based -- ³18 men or women 56.6±11.8 52.4 24.7 Cases 4313 Iceland population-based CAD no age criterion 68.9±12.1 30.8 0.0 Controls 24952 Iceland population-based -- Cases 167 Finland MI Controls 172 Finland drawn from population-based cohort nested case-cohort -- men £50 or women £ 60 -- Cases 378 U. S. hospital-based CAD Controls 2652 U. S. unaffected siblings of cases -- Cases 722 Germany hospital-based MI Controls 1,643 Germany population-based -- Cases 1182 Germany hospital-based MI Controls 1280 Germany hospital-based -- 45.4±6.5 61.5 45.7 49.2±21.7 63.2 0.0 47.1 ± 6.2 33.5 0.0 47.1 ± 6.0 31.4 0.0 men and women < 60 46.9±7.0 26.2 24.6 -- 47.2±13.1 58.2 40.5 men £60 or women £ 65 -- 50.2±7.9 32.5 0.0 62.5±10.1 50.5 0.0 51.3±7.6 20.3 0.0 51.2±11.9 47.9 0.0 46.0 ± 6.9 51.1 0.0 -- men £60 or women £ 65 -- U.S. community-based 559 U.S. community-based -- men £50 or women £ 60 -- 45.2 ± 7.3 55.5 0.0 789 U.S. and Europe hospital-based MI no age criterion 61.6±12.3 29 0.0 859 U.S. and Europe hospital and community-based -- 61.2±12.2 30.7 0.0 hospital-based MI no age criterion 51.4±10.8 10.1 100.0 hospital and community-based -- -- 49.8 ± 11.0 9.1 100.0 46.0±6.3 20.1 0.0 55.2±8.0 55.2 0.0 48.5 ± 4.4 41.9 0.0 48.7 ± 4.6 42.4 0.0 Cases 505 Controls Cases Controls MI Cases 482 Indian subcontinent** Indian subcontinent** Northern Ireland hospital-based MI Controls 622 Northern Ireland older siblings of cases -- Cases 86 Sweden MI Controls 99 Sweden drawn from population-based cohort nested case-cohort Cases 1092 (South Asians) Controls 1187 MDC non Mean age in Female European years ± sd sex (%) (%) men £45 or women £ 55 -- Cases INTERHEART IFS Age and sex criterion -- men £55 or women £ 60 -men £50 or women £ 60 -- MedStar MGH PCAD MAHI PennCATH REGICOR VHS WTCCC CAD Cases 875 U.S. hospital-based CAD men and women <65 48.9 ± 6.4 18.2 0.0 Controls 447 U.S. hospital-based -- men and women ≥45 59.8 ± 8.9 48.8 0.0 Cases 204 U.S. hospital-based Controls 260 U.S. hospital-based -- men £50 or women £ 60 -- 53.8 ± 11.1 33.5 0.0 Cases 807 U.S. hospital-based MI no age criterion 61.5±12.7 32.1 0.0 Controls 637 U.S. outpatients -- -- 60.7±12.4 39 0.0 Cases 933 U.S. hospital-based CAD ≤66 men and women 52.7 ± 7.6 24.3 0.0 Controls 468 U.S. hospital-based -- ≥45 men and women 61.7 ± 9.6 51.7 0.0 45.9 ± 5.8 20.2 0.0 Cases 312 MI 47.0 ± 6.1 29.9 0.0 Spain hospital-based MI -- men £50 or women £ 60 -- 46.0 ± 5.6 21.5 0.0 CAD no age criterion 61.4±10.0 20.4 0.0 -- 58.0±12.3 35 0.0 <66 years 49.3±7.9 20.2 0.0 -- 44.7±9.3 49.2 0.0 Controls 317 Spain Cases 1106 Italy drawn from community-based cohort hospital-based Controls 383 Italy hospital-based Cases 1,926 U.K. community-based Controls 2,954 U.K. community-based CAD *total number of subjects with successful genotyping of rs20455 CAD = coronary artery disesae, MI = myocardial infarction, ADVANCE = Atherosclerotic Disease, VAscular functioN, and genetiC Epidemiology, AMI Gene = AMI Gene Study/Dortmund Health, CATHGEN = CATHGEN Research Project, deCODE = deCODE genetics CAD study, FINRISK = National Finrisk study (FINRISK), GeneSTAR = Genetic Study of Atherosclerosis Risk, GerMIFS I and GerMIFS II = German Myocardial Infarction Family studies I and II, HARPS = Heart Attack Risk in Puget Sound, INTERHEART = international INTERHEART study coordinated by Population Health Research Institute of McMaster University, IFS = Irish Family Study, MDC = Malmo Diet and Cancer, MEDSTAR = Washington Hospital Center/Medstar angiographic CAD study, MGH PCAD = Massachusetts General Hospital of Premature CAD study, MAHI = Mid-America Heart Institute, PennCATH = University of Pennsylvania Medical center angiographic CAD study, REGICOR = Registre Gironı´ del Cor study, VHS = Verona Heart Study, **India, Pakistan, Bangladesh, Sri Lanka, Nepal Table 2. Kinesin-like protein 6 Trp719Arg polymorphism (rs20455) allele Frequencies, genotypes counts, and Odds Ratios adjusted for age and sex in 19 case-control studies of CAD, stratified by race/ethnic group CASES (CAD) No. subjects Europeans ADVANCE 275 AMI Gene 793 CATHGEN 1298 4313 deCODE† FinRisk 167 GeneSTAR 285 GerMIFS I* 722 GerMIFS II 1182 HARPS 505 INTERHEART 789 482 IFS†‡ MDC 86 MedStar 875 MGH PCAD 204 MAHI 807 PennCath 933 REGICOR 312 VHS 1106 WTCCC CAD 1922 Total - Meta Analysis 17056 Non Europeans ADVANCE admixed Hisp. ADVANCE admixed non Hisp. ADVANCE Afr.Am. ADVANCE East Asians ADVANCE Hisp. CathGEN Afr.Am. 34 74 49 45 28 277 CONTROLS Odds Ratios, 95% confidence intervals and P v Allele Allele 719Trp/ 719Arg/ 719Arg/ No. 719Trp/ 719Arg/ 719Arg/ Freq Freq 719Trp 719Trp 719Arg subjects 719Trp 719Trp 719Arg 719Arg 719Arg Log Additive Mode of Inheritance P 0.345 0.369 0.361 0.300 0.374 0.368 0.367 0.368 0.404 0.362 0.344 0.372 0.349 0.353 0.367 0.379 0.348 0.378 0.356 119 311 545 2131 64 106 293 472 187 335 203 35 370 89 322 359 134 437 792 122 379 570 1779 81 148 328 551 228 337 226 38 399 86 377 441 139 501 890 34 103 183 403 22 31 101 159 90 117 53 13 106 29 108 133 39 168 240 311 1121 730 24952 172 1579 1643 1280 559 859 622 99 447 260 637 468 317 383 2933 39372 0.378 0.381 0.355 0.312 0.355 0.365 0.368 0.360 0.381 0.354 0.346 0.394 0.364 0.348 0.359 0.358 0.339 0.372 0.355 122 430 297 11813 73 626 662 522 216 354 261 33 174 114 256 194 141 145 1242 143 528 347 10689 76 752 753 595 260 402 292 54 221 111 304 213 137 191 1299 46 163 86 2450 23 201 228 163 83 103 69 12 52 35 77 61 39 47 392 0.87(0.69-1.1) 0.89(0.75-1.04) 1.02(0.89-1.18) 0.93(0.88-0.99) 1.09(0.8-1.49) 0.99(0.82-1.21) 0.97(0.84-1.13) 1.01(0.89-1.14) 1.1(0.92-1.31) 1.03(0.9-1.19) 1.03(0.81-1.30) 0.91(0.6-1.39) 0.99(0.8-1.22) 1.04(0.82-1.31) 1.04(0.89-1.21) 1.04(0.86-1.26) 1.02(0.78-1.34) 1.03(0.87-1.22) 1.02(0.93-1.12) 0.98(0.95-1.02) 0.249 0.142 0.749 0.018 0.596 0.939 0.742 0.893 0.279 0.671 0.835 0.667 0.919 0.878 0.647 0.666 0.747 0.757 0.624 0.349 0.412 0.419 0.745 0.356 0.375 0.762 12 27 4 19 11 16 16 32 17 20 13 100 6 15 28 6 4 161 22 37 87 35 22 240 0.455 0.622 0.816 0.486 0.364 0.783 7 10 5 9 8 9 10 8 22 18 12 86 5 19 60 8 2 145 0.77(0.34-1.67) 0.83(0.44-1.57) 0.7(0.39-1.23) 0.6(0.3-1.13) 1.07(0.45-2.58) 0.89(0.66-1.21) 0.507 0.555 0.207 0.114 0.876 0.465 GENESTAR Afr.Am. 93 0.796 2 34 57 1073 0.786 53 353 INTERHEART (South Asians) 1092 0.451 351 498 243 1187 0.449 389 531 Afr.Am. Total-Meta Analysis 419 1400 Please see footnote of table 1 for resolution of study acronym. Afr.Am. = African Americans Hisp. = Hispanics † Adjusted for relatedness *P values adjusted by genomic control method as λ=1.27 ‡Approach to analysis unable to produce dominant model odds ratios 667 267 1.05(0.72-1.51) 0.813 1.02(0.91-1.14) 0.791 0.91(0.73-1.13) 0.380 or age and sex in 19 case-control 95% confidence intervals and P values Log Dominant Mode of Inheritance P 0.84(0.6-1.16) 0.9(0.71-1.13) 0.96(0.79-1.17) 0.91(0.85-0.99) 1.18(0.76-1.82) 1.09(0.83-1.43) 0.97(0.88-1.08) 0.99(0.91-1.08) 1.07(0.83-1.37) 0.95(0.78-1.15) 0.73(0.4-1.33) 0.91(0.68-1.22) 1.03(0.69-1.52) 1.01(0.82-1.25) 1.03(0.79-1.33) 1.06(0.77-1.45) 0.93(0.73-1.19) 1.09(0.96-1.24) 0.97(0.93-1.01) 0.289 0.349 0.674 0.020 0.458 0.543 0.661 0.770 0.602 0.573 0.305 0.527 0.904 0.919 0.850 0.716 0.568 0.189 0.137 0.73(0.21-2.36) 1.44(0.49-4.41) 0.7(0.18-3) 0.48(0.18-1.25) 0.88(0.27-2.82) 0.58(0.25-1.37) 0.607 0.506 0.619 0.135 0.831 0.213 2.35(0.56-9.83) 0.241 1.04(0.87-1.24) 0.669 0.81(0.42-1.55) 0.520 Table 3. Kinesin-like protein 6 Trp719Arg polymorphism (rs20455) allele Frequencies, genotypes counts, and Odds Ratios adjusted for age and sex in 19 case-control of CAD, stratified by race/ethnic group and restricted to early onset disease (age of onset of CAD <50 years of age for males and <60 years of age for females) and sim aged controls CASES (CAD) CONTROLS Odds Ratios, 95% confidence intervals and No. subjects Allele Allele 719Arg 719Trp/ 719Arg/ 719Arg/ No. 719Trp/ 719Arg/ Freq Freq /719Ar 719Trp 719Trp 719Arg subjects 719Trp 719Trp 719Arg 719Arg g Log Additive Mode of Inheritance P Europeans ADVANCE AMI Gene CATHGEN 275 296 585 0.345 0.380 0.366 119 117 233 122 133 276 34 46 76 311 193 295 0.378 0.415 0.359 122 68 121 143 90 136 46 35 38 0.87(0.69-1.1) 0.025 0.98(0.74-1.3) 0.875 0.99(0.78-1.28) 0.990 deCODE† 750 0.292 375 312 63 12548 0.312 5955 5366 1227 0.91(0.72-1.16) 0.450 FinRisk GeneSTAR GerMIFS I* GerMIFS II HARPS INTERHEART IFS†‡ MDC MedStar MGH PCAD MAHI PennCath REGICOR VHS WTCCC CAD Total - Meta Analysis 204 201 468 621 875 195 371 167 601 312 227 354 505 197 1085 8289 0.353 0.358 0.372 0.364 0.349 0.364 0.342 0.374 0.351 0.348 0.392 0.383 0.404 0.409 0.359 89 79 190 253 370 85 154 64 255 134 81 134 187 71 444 86 100 208 284 399 78 180 81 270 139 114 169 228 91 503 29 22 70 84 106 32 37 22 76 39 32 51 90 35 138 260 1579 923 869 447 216 309 172 151 317 190 128 559 113 2612 22192 0.348 0.365 0.362 0.361 0.364 0.343 0.353 0.355 0.358 0.339 0.363 0.359 0.381 0.420 0.355 114 626 373 359 174 92 129 73 61 141 77 53 216 33 1101 111 752 432 393 221 100 142 76 72 137 88 58 260 65 1167 35 201 118 117 52 24 38 23 18 39 25 17 83 15 344 1.09(0.8-1.49) 0.95(0.76-1.18) 0.99(0.8-1.23) 0.97(0.81-1.16) 1.1(0.92-1.31) 1.09(0.82-1.44) 0.86(0.63-1.16) 0.91(0.6-1.39) 0.98(0.74-1.29) 1.04(0.82-1.31) 1.14(0.85-1.51) 1.11(0.82-1.49) 1.02(0.78-1.34) 0.88(0.56-1.38) 1.04(0.94-1.16) 0.99(0.94-1.04) 0.596 0.627 0.949 0.760 0.279 0.559 0.315 0.667 0.878 0.878 0.386 0.508 0.747 0.570 0.444 0.729 34 74 49 45 0.412 0.419 0.745 0.356 12 27 4 19 16 32 17 20 6 15 28 6 22 37 87 35 0.455 0.622 0.816 0.486 7 10 5 9 10 8 22 18 5 19 60 8 0.77(0.34-1.67) 0.83(0.44-1.57) 0.7(0.39-1.23) 0.6(0.3-1.13) 0.507 0.555 0.207 0.114 Non Europeans ADVANCE admixed Hisp. ADVANCE admixed non Hisp. ADVANCE Afr.Am. ADVANCE East Asians ADVANCE Hisp. 28 0.375 11 13 4 22 0.364 8 12 2 1.07(0.45-2.58) 0.876 CathGEN Afr.Am. 157 0.755 7 63 87 128 0.781 5 46 77 1.02(0.64-1.61) 0.950 GENESTAR Afr.Am. 70 0.807 2 23 45 1073 0.786 53 353 667 1.15(0.75-1.76) 0.532 INTERHEART (South Asians) 515 0.458 163 232 120 633 0.453 203 286 144 1.03(0.88-1.21) 0.701 Afr.Am. Total-Meta Analysis 276 1288 1.06(0.76-1.49) 0.721 Please see footnote of table 1 for resolution of study acronym. Afr.Am. = African Americans Hisp. = Hispanics Controls are restricted to males < 50 years of age and females < 60 years of age at the time of blood draw to match the sex specific age range of cases. † Adjusted for relatedness *P values adjusted by genomic control method as λ=1.27 ‡Approach to analysis unable to produce dominant model odds ratios age and sex in 19 case-control studies ars of age for females) and similarly 95% confidence intervals and P values Log Dominant Mode of Inheritance P 0.84(0.6-1.16) 0.289 1.02(0.67-1.54) 0.929 0.98(0.7-1.37) 0.900 0.95(0.69-1.29) 0.730 1.18(0.76-1.82) 0.99(0.73-1.35) 0.99(0.87-1.16) 0.97(0.86-1.1) 1.07(0.83-1.37) 0.96(0.65-1.42) 0.458 0.951 0.999 0.611 0.602 0.837 0.73(0.4-1.33) 0.305 0.91(0.62-1.33) 0.612 1.03(0.69-1.52) 0.904 1.12(0.8-1.79) 0.374 1.16(0.73-1.83) 0.539 1.06(0.77-1.45) 0.716 0.72(0.38-1.37) 0.320 1.10(0.95-1.27 0.216 1.01(0.95-1.08) 0.767 0.73(0.21-2.36) 1.44(0.49-4.41) 0.7(0.18-3) 0.48(0.18-1.25) 0.607 0.506 0.619 0.135 0.88(0.27-2.82) 0.831 0.89(0.23-3.46) 0.860 1.79(0.43-7.5) 0.427 1.04(0.81-1.34) 0.735 1.05(0.87-1.26) 0.609 range of cases. Table 1S. Summary statistics of traditional risk factors of CAD in each of the 19 studies included in the Trp719Arg polymorphism in kinesin-like protein 6 meta analysis, stratified by case control status * Study ADVANCE AMI Gene CATHGEN deCODE FinRisk GENESTAR GerMIFS I N Cases 506 Ever Diabetes Hyperchol Body mass Hyperten index cigarette mellitus esterolemi sion (%) smoking (%) (%) a (%) (kg/m2) 58.3 34.2 21.7 28.7 31.7±7.1 Controls 514 34.8 19.3 6.6 19.8 28.0±6.8 Cases 809 65.9 71.8 14.6 60.6 28.0±4.7 Controls 1132 55.9 62.6 8 68.1 27.5±4.9 Cases 1575 65.7 72.8 30.7 71.4 NA Controls 970 43.8 61.9 21.3 45.4 NA Cases 4313 34.8 N/A N/A N/A 27.8±4.7 Controls 24952 34.8 N/A N/A N/A 26.9±5.4 Cases 167 74.4 72.5 17.7 75.2 29.6 ± 5.0 Controls 172 58.2 68 5.9 48.2 27.7 ± 4.0 Cases 378 82 83.1 43.3 80.4 30.8±6.3 Controls 2652 52.6 37 10.2 33.4 29.9±7.0 Cases 875 70.3 86.5 12.3 76.1 27.4±3.6 Controls 1,644 49.3 62.7 11 78 28.1±4.5 Cases 1222 64.7 88.1 15.5 80.8 27.9±4.0 Controls 1298 55.6 41.7 3.5 67.3 27.4±4.6 Cases 505 73.9 50.5 14.9 43.7 29.2 ± 6.8 Controls 559 41.7 30.8 3 26 26.9 ± 5.7 Cases 789 67.1 44.4 15 21.6 27.3±4.7 Controls 859 52.2 31.8 7.1 15.8 26.9±4.1 Cases 1092 61.4 28.3 20.4 21.7 24.9±4.3 (South Asians) Controls 1187 44.3 13.4 8.9 13.9 24.8±4.1 IFS Cases 482 82.5 27.9 9.4 85.8 28.5±4.3 Controls 622 57.6 28.9 5.6 8.6 28.2±5.0 GerMIFS II HARPS INTERHEART (Europeans) INTERHEART MDC MedStar MGH PCAD MAHI PennCATH REGICOR VHS WTCCC CAD Cases 86 87.2 81.4 4.7 37.2 26.9 ± 4.2 Controls 99 61.6 62.6 1 1 25.7 ± 4.3 Cases 875 52.6 76.3 30.1 88.4 31.7 ± 6.8 Controls 447 49.8 60.6 19 65.1 31.7± 7.9 Cases 204 74.9 33.5 19.2 79 30.0 ± 7.0 Controls 260 57.3 25.3 0.4 31.3 27.9 ± 6.5 Cases 811 71 60.1 23.9 58.8 29.4 Controls 650 50.2 51.1 11.9 50.4 27.9 Cases 933 45.8 61.2 24.4 80.1 29.7 ± 5.6 Controls 468 34.8 49.2 11.2 61.3 28.9 ± 6.4 Cases 312 82.8 38 14.8 48.9 27.5 ± 4.2 Controls 317 61.9 31.5 6.1 33.1 27.0 ± 3.9 Cases 1106 68.7 66.1 19.4 74.8 26.8±3.5 Controls 383 43.5 40.1 7 66 25.4±3.4 Cases 1,414 78.3 41.1 10.9 79.3 27.6±4.3 Controls 2,938 NA NA NA NA NA *total number of subjects with successful genotyping of rs20455 CAD = coronary artery disesae, MI = myocardial infarction, ADVANCE = Atherosclerotic Disease, VAscular functioN, and genetiC Epidemiology, AMI Gene = AMI Gene Study/Dortmund Health, CATHGEN = CATHGEN Research Project, deCODE = deCODE genetics CAD study, FINRISK = National Finrisk study (FINRISK), GeneSTAR = Genetic Study of Atherosclerosis Risk, GerMIFS I and GerMIFS II = German Myocardial Infarction Family studies I and II, HARPS = Heart Attack Risk in Puget Sound, INTERHEART = international INTERHEART study coordinated by Population Health Research Institute of McMaster University, IFS = Irish Family Study, MDC = Malmo Diet and Cancer, MEDSTAR = Washington Hospital Center/Medstar angiographic CAD study, MGH PCAD = Massachusetts General Hospital of Premature CAD study, MAHI = Mid-America Heart Institute, PennCATH = University of Pennsylvania Medical center angiographic CAD study, REGICOR = Registre Gironı´ del Cor study, VHS = Verona Heart Study, WTCCC Table 2S. Kinesin-like protein 6 Trp719Arg polymorphism (rs20455) allele Frequencies, genotypes counts, and Odds Ratios adjusted for age and sex in 19 casecontrol studies of CAD, restricted to European subgroup of cases with documented myocardial infarction CASES (MI) CONTROLS Odds Ratios, 95% confidence intervals and P values Allele Allele Log Additive 719Trp/ 719Arg/ 719Arg/ No. 719Trp/ 719Arg/ 719Arg/ Freq Freq Mode of P 719Trp 719Trp 719Arg subjects 719Trp 719Trp 719Arg 719Arg 719Arg Inheritance ADVANCE 151 0.341 67 65 19 311 0.378 122 143 46 0.98(0.69-1.4) 0.927 AMI Gene 793 0.369 311 379 103 1121 0.381 430 528 163 0.89(0.75-1.04) 0.142 CATHGEN 1298 0.361 545 570 183 730 0.355 297 347 86 1.02(0.89-1.18) 0.749 0.93 (0.87deCODE† 3577 0.299 1771 1475 331 24952 0.312 11813 10689 2450 0.023 0.99) FinRisk 167 0.374 64 81 22 172 0.355 73 76 23 1.09(0.8-1.49) 0.596 GeneSTAR 164 0.399 52 93 19 1579 0.365 626 752 201 1.14 (0.89-1.45) 0.301 GerMIFS I* 722 0.367 293 328 101 1643 0.368 662 753 228 0.97(0.84-1.13) 0.742 GerMIFS II 1182 0.368 472 551 159 1280 0.360 522 595 163 1.01(0.89-1.14) 0.893 HARPS 505 0.404 187 228 90 559 0.381 216 260 83 1.1(0.92-1.31) 0.279 INTERHEART 789 0.362 335 337 117 859 0.354 354 402 103 1.03(0.9-1.19) 0.671 †‡ 482 0.344 203 226 53 622 0.346 261 292 69 1.03(0.81-1.30) 0.835 IFS MDC 86 0.372 35 38 13 99 0.394 33 54 12 0.91(0.6-1.39) 0.667 MedStar 421 0.356 171 200 50 447 0.364 174 221 52 1.16(0.88-1.51) 0.299 MGH PCAD 204 0.353 89 86 29 260 0.348 114 111 35 1.04(0.82-1.31) 0.878 MAHI 807 0.367 322 377 108 637 0.359 256 304 77 1.04(0.89-1.21) 0.647 PennCath 468 0.386 175 225 68 468 0.358 194 213 61 1.01(0.81-1.23) 0.900 REGICOR 312 0.348 134 139 39 317 0.339 141 137 39 1.02(0.78-1.34) 0.747 VHS 644 0.392 244 295 105 383 0.372 145 191 47 1.10(0.91-1.33) 0.316 WTCCC CAD 1373 0.359 562 636 175 2933 0.355 1242 1299 392 1.04(0.94-1.15) 0.447 Total - Meta Analysis 14145 39372 0.99(0.96-1.03) 0.625 Please see footnote of table 1 for resolution of study acronym. MI = myocardial infarction For this analysis, the Odds Ratios of 12 out of 19 studies are unchanged compared to the primary analysis shown in the main text for the CAD outcome (Table 2) because cases in these studies has already been defined on the basis of a documented MI. No. subjects † Adjusted for relatedness *P values adjusted by genomic control method as λ=1.27 ‡Approach to analysis unable to produce dominant model odds ratios usted for age and sex in 19 case- 95% confidence intervals and P values Log Dominant Mode of P Inheritance 0.95(0.58-1.53) 0.818 0.9(0.71-1.13) 0.349 0.96(0.79-1.17) 0.674 0.91 (0.84-0.99) 0.028 1.18(0.76-1.82) 1.37 (0.98-1.99) 0.97(0.88-1.08) 0.99(0.91-1.08) 1.07(0.83-1.37) 0.95(0.78-1.15) 0.73(0.4-1.33) 1.18(0.81-1.70) 1.03(0.69-1.52) 1.01(0.82-1.25) 1.02(0.75-1.40) 1.06(0.77-1.45) 1.02 (0.78-1.34) 1.11(0.97-1.28) 0.98(0.94-1.02) 0.458 0.064 0.661 0.770 0.602 0.573 0.305 0.386 0.904 0.919 0.850 0.716 0.875 0.126 0.422 t for the CAD outcome (Table 2) Table 3S. Kinesin-like protein 6 Trp719Arg polymorphism (rs20455) allele Frequencies, genotypes counts, and Odds Ratios adjusted for age and sex in 19 case-contro studies of CAD, restricted to European subgroup of cases with early onset myocardial infarction (age of onset of MI <50 years of age for males and <60 years of age fo females) ans similarly aged controls CASES (MI) CONTROLS Odds Ratios, 95% confidence intervals and P value Allele Allele 719Trp Log Additive 719Trp/ 719Arg/ 719Arg/ No. 719Arg/ 719Arg/ Freq Freq /719Tr Mode of P 719Trp 719Trp 719Arg subjects 719Trp 719Arg 719Arg 719Arg p Inheritance ADVANCE 151 0.341 67 65 19 311 0.378 122 143 46 0.98(0.69-1.4) 0.927 AMI Gene 296 0.380 117 133 46 193 0.415 68 90 35 0.98(0.74-1.3) 0.875 CATHGEN 585 0.366 233 276 76 295 0.359 121 136 38 0.99(0.78-1.28) 0.990 750 0.292 375 312 63 12548 0.312 5955 5366 1227 0.91(0.72-1.16) 0.450 deCODE† FinRisk 204 0.353 89 86 29 260 0.348 114 111 35 1.09(0.8-1.49) 0.596 GeneSTAR 125 0.392 42 68 15 1579 0.365 626 752 201 1.09 (0.83-1.44) 0.521 GerMIFS I* 468 0.372 190 208 70 923 0.362 373 432 118 0.99(0.8-1.23) 0.949 GerMIFS II 621 0.364 253 284 84 869 0.361 359 393 117 0.97(0.81-1.16) 0.760 HARPS 875 0.349 370 399 106 447 0.364 174 221 52 1.1(0.92-1.31) 0.279 INTERHEART 195 0.364 85 78 32 216 0.343 92 100 24 1.09(0.82-1.44) 0.559 371 0.342 154 180 37 309 0.353 129 142 38 0.86(0.63-1.16) 0.315 IFS†‡ MDC 167 0.374 64 81 22 172 0.355 73 76 23 0.91(0.6-1.39) 0.667 MedStar 311 0.360 125 148 38 151 0.358 61 72 18 1.23(0.87-1.72) 0.240 MGH PCAD 312 0.348 134 139 39 317 0.339 141 137 39 1.04(0.82-1.31) 0.878 MAHI 227 0.392 81 114 32 190 0.363 77 88 25 1.14(0.85-1.51) 0.386 PennCath 201 0.410 68 101 32 128 0.359 53 58 17 1.27(0.85-1.87) 0.241 REGICOR 505 0.404 187 228 90 559 0.381 216 260 83 1.02(0.78-1.34) 0.747 VHS 197 0.409 71 91 35 113 0.420 33 65 15 0.88(0.56-1.38) 0.570 WTCCC CAD 826 0.360 333 391 102 2612 0.355 1101 1167 344 1.06(0.94-1.19) 0.325 Total - Meta Analysis 7387 22192 1.03(0.98-1.09) 0.243 Please see footnote of table 1 for resolution of study acronym For this analysis, the Odds Ratios of 13 out of 19 studies are unchanged compared to the primary analysis of the CAD outcome shown in the main text (Table 3) because cases in these studies has already been defined on the basis of a documented MI. Controls are restricted to males < 50 years of age and females < 60 years of age at the time of blood draw to match the sex specific age range of cases. † Adjusted for relatedness *P values adjusted by genomic control method as λ=1.27 No. subjects ‡Approach to analysis unable to produce dominant model odds ratios ed for age and sex in 19 case-control ge for males and <60 years of age for 95% confidence intervals and P values Log Dominant Mode of Inheritance 0.95(0.58-1.53) 1.02(0.67-1.54) 0.98(0.7-1.37) 0.95(0.69-1.29) 1.18(0.76-1.82) 1.27 (0.86-1.88) 0.99(0.87-1.16) 0.97(0.86-1.1) 1.07(0.83-1.37) 0.96(0.65-1.42) 0.818 0.929 0.900 0.730 0.458 0.233 0.999 0.611 0.602 0.837 0.73(0.4-1.33) 1.25(0.79-1.98) 1.03(0.69-1.52) 1.12(0.8-1.79) 1.40(0.81-2.44) 1.06(0.77-1.45) 0.72(0.38-1.37) 1.14(0.97-1.34) 1.03(0.97-1.11) 0.305 0.340 0.904 0.374 0.230 0.716 0.320 0.106 0.263 own in the main text (Table 3) c age range of cases. P Table 4S. Summary of Results of Heterogeneity Tests for Meta Analyses (Q value and I2) Heterogeneity Tests Q-value df (Q) P-value I-squared All CAD Log Additive Model Europeans All CAD Log Dominant Model Europeans All CAD Log Additive Model African Americans All CAD Log Dominant Model African Americans* 11.24 10.98 1.36 2.76 18 17 2 2 0.884 0.858 0.507 0.252 0.0 0.0 0.0 27.4 Early Onset CAD Log Additive Model Europeans Early Onset CAD Log Dominant Model Europeans Early Onset CAD Log Additive Model African Americans Early Onset CAD Log Dominant Model African Americans 7.13 7.23 1.86 0.90 18 17 2 2 0.989 0.980 0.394 0.637 0.0 0.0 0.0 0.0 All AMI Log Additive Model Europeans 13.07 18 0.788 All AMI Log Dominant Model Europeans 14.05 17 0.663 Early Onset AMI Log Additive Model Europeans 7.85 18 0.981 Early Onset AMI Log Dominant Model Europeans 9.47 17 0.924 *Random effects model for this stratum (DerSimonian and Laird method): OR, 95%CI, 0.86(0.39,1.91), p value 0.71 0.0 0.0 0.0 0.0