Prion biology relevant to bovine spongiform encephalopathy1
J. Novakofski*2, M. S. Brewer†, N. Mateus-Pinilla‡, J. Killefer*, and R. H. McCusker*
Departments of *Animal Sciences and †Food Science and Human Nutrition,
University of Illinois at Urbana–Champaign 61801-4737; and ‡Illinois Natural History Survey,
Center for Wildlife and Plant Ecology, Champaign, IL 61820
ABSTRACT: Bovine spongiform encephalopathy
(BSE) and chronic wasting disease (CWD) of deer and
elk are a threat to agriculture and natural resources,
as well as a human health concern. Both diseases are
transmissible spongiform encephalopathies (TSE), or
prion diseases, caused by autocatalytic conversion of
endogenously encoded prion protein (PrP) to an abnormal, neurotoxic conformation designated PrPsc. Most
mammalian species are susceptible to TSE, which, despite a range of species-linked names, is caused by a
single highly conserved protein, with no apparent normal function. In the simplest sense, TSE transmission
can occur because PrPsc is resistant to both endogenous
and environmental proteinases, although many details
remain unclear. Questions about the transmission of
TSE are central to practical issues such as livestock
testing, access to international livestock markets, and
wildlife management strategies, as well as intangible
issues such as consumer confidence in the safety of the
meat supply. The majority of BSE cases seem to have
been transmitted by feed containing meat and bone
meal from infected animals. In the United Kingdom,
there was a dramatic decrease in BSE cases after neural
tissue and, later, all ruminant tissues were banned
from ruminant feed. However, probably because of
heightened awareness and widespread testing, there is
growing evidence that new variants of BSE are arising
“spontaneously,” suggesting ongoing surveillance will
continue to find infected animals. Interspecies transmission is inefficient and depends on exposure, sequence homology, TSE donor strain, genetic polymorphism of the host, and architecture of the visceral
nerves if exposure is by an oral route. Considering the
low probability of interspecies transmission, the low
efficiency of oral transmission, and the low prion levels
in nonnervous tissues, consumption of conventional animal products represents minimal risk. However, detection of rare events is challenging, and TSE literature
is characterized by subsequently unsupported claims
of species barriers or absolute tissue safety. This review
presents an overview of TSE and summarizes recent
research on pathogenesis and transmission.
Key Words: Bovine Spongiform Encephalopathy, Chronic Wasting Disease, Prion
2005 American Society of Animal Science. All rights reserved.
Introduction
Bovine spongiform encephalopathy (BSE) is one of
several transmissible spongiform encephalopathies
(TSE), or prion diseases, which are progressive neurological disorders thought to be caused by conversion of
endogenous, host-encoded prion protein (PrP) to an
abnormal conformation designated PrPsc (Prusiner,
1982; Legname et al., 2004). Prion diseases may be
genetic, sporadic, or transmitted, although in all cases,
progression of the disease is associated with accumula-
1
The authors acknowledge the generous support of Cargill, Incorporated, Minneapolis, MN, in publication of this review.
2
Correspondence: 1503 South Maryland Dr. (phone: 217-333-6181;
e-mail: Jnova@uiuc.edu).
Received November 22, 2004.
Accepted March 7, 2005.
J. Anim. Sci. 2005. 83:1455–1476
tion of abnormal PrPsc conformer in the brain and subsequent neurodegeneration (Prusiner, 2004). Both the
disease process and disease transmission are related
to stability of the PrPsc conformer, which is markedly
resistant to biological or chemical degradation. The abbreviation PrPsc refers to scrapie, the prototypical prion
disease, whereas an alternative, PrPres, refers to proteinase resistance. The mechanisms of prion disease
transmission are of great interest because spontaneous
occurrences of prion disease are rare and most cases in
domestic and wild animals result from transmission.
Moreover, based on DNA analyses, the AA sequence of
PrP is well conserved in mammalian species; therefore,
most, if not all, mammalian species are susceptible to
prion diseases (Table 1) and the disease may be transmitted between species. Most prion diseases may be
transmitted to laboratory animals, and substantial evidence indicates that a variant form of human Creutzfeldt-Jakob Disease (vCJD; sporadic CJD is the proto-
1455
1456
Novakofski et al.
Table 1. Examples of transmissible spongiform encephalopathies (TSE)
TSE disease
Primary occurrence
Known secondary occurrence
Bovine spongiform
encephalopathy (BSE)
Cattle
Bison, eland, gemsbok, kudu, nyala, oryx, (all Bovidae);
cats, goats, lab rodents, mink (repassage), pigs, sheep,
humans, other primatesa,b,c
Scrapie
Sheep, goats
Other primates, cattle, lab rodents, Rocky Mountain elk
Creutzfeldt-Jakob (CJD)
Humans
Other primates, lab rodents
Fatal familial insomnia (FFI)
Humans
Lab rodents
Kuru
Humans
Other primates, lab rodents
Gerstmann-Straussler-Scheinker
syndrome (GSS)
Humans
Other primates, lab rodents
Variant CJD (vCJD)
Humans
Lab rodents
Chronic wasting disease
(CWD)
Mule deer, Rocky Mountain elk,
white tail deer (wild and domestic)
Cattle, ferrets, lab rodents (repassage), mink,
squirrel monkeys, goats
Feline spongiform encephalopathy
(FSE)
House cats, cheetah, ocelot, puma,
tiger
Lab rodents
Transmissible mink encephalopathy
(TME)
Domestic mink
Cattle, ferrets, raccoons, lab rodents
a
Lab rodents include gerbils, guinea pigs, hamsters, mice, and rats.
Repassage means secondary transmission after adaptation in an intermediate species.
Other primates may include chimpanzees, macaque monkeys, marmosets, prosimians, squirrel monkeys.
b
c
typical TSE of humans) was caused by interspecies
transmission following the BSE outbreak in the United
Kingdom. The geographic distributions of BSE and
chronic wasting disease (CWD; TSE of cervids) have
continued to increase in recent years, despite widespread control efforts, indicating the importance of better understanding these diseases. The purpose of this
article is to review characteristics of prion diseases and
transmission as well as to examine recent findings that
suggest it may be necessary to revise common perceptions about the disease.
Basis of Prion Disease
Transmission of TSE seem to result from three unusual characteristics of PrPsc, although many specifics
are unclear. First, PrPsc is autocatalytic in that PrPsc
promotes conversion of additional PrP molecules into
the PrPsc conformation (Raymond et al., 2000; Prusiner,
2004). Second, PrPsc is resistant to proteases (intracellar, intestinal, and environmental) that would normally
destroy the protein (Telling et al. 1996; Riesner 2003).
This results in accumulation of undegraded PrPsc in
vivo and persistence ex vivo. Third, accumulated PrPsc
results in neuronal damage and spongiform changes in
the brain (Unterberger et al., 2005).
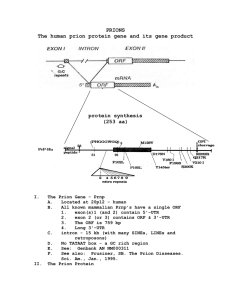
The prnp gene is present in most, if not all, wild-type
mammals and is highly conserved across species (van
Rheede, et al., 2003). Endogenous PrP is encoded by a
single exon of the prnp gene, which codes for a 256to 264-AA precursor of approximately 28 kDa that is
processed by cleavage of a 22- to 24-AA signal peptide
yielding a mature protein of 231 to 253 AA (Riesner,
2003; Van Rheede, et al., 2003). Species differences in
PrP size are primarily a function of differences in the
number of octapeptide repeat regions.
Normal PrP is a glycoprotein primarily found on
nerve and immune cell membranes, although the protein is found at lower concentrations on many cell types.
The PrP protein has O- and N-linked glycosylation sites
in its precursor form (Riesner, 2003) and is found as a
mixture of nonglycosylated, monoglycosylated and diglycosylated forms. Cell surface attachment occurs via
a glycosyl phosphatidyl inositol anchor (Riesner, 2003).
Membrane bound PrP also associates with heparan sulfates (Caughey et al., 1994) and laminin (Graner et al.,
2000). In neurons, PrP is associated with both the cell
body and synaptic vesicles (Collinge et al. 1994; Sales
et al., 1998).
The PrP is a metal-binding protein having at least
one site that binds Cu2+, with a Kd of 10−14 M (Jackson
et al., 2001). Other transition metal ions (Zn, Mn, and
Ni) bind with lower affinity. Metal binding alters PrP
biochemical characteristics, including protease resistance (Brown, et al. 1997; Lehmann, 2002). The PrP
mutations that have additional copies of the metalbinding octapeptide repeat have properties similar to
PrPsc (Lehmann and Harris, 1996). Copper binding
stimulates PrP endocytosis (Pauly and Harris, 1998).
In addition to extracellular function, intracellular PrP
may modify properties of intracellular proteins, including antiapoptotic Bcl-2 (Kurschner and Morgan, 1995),
several heat shock proteins (Edenhofer et al., 1996;
Zanata et al., 2002), neuronal synapsin, and the growth
factor signal adapter Grb2 (Spielhaupter and Schatzl,
2001). Knockout mice without PrP often seem to be
normal, suggesting that normal PrP function may be
redundant with an unidentified protein. Neurological
abnormalities in some strains of PrP knockout mice
result from an incomplete construct, which upregulates
the adjacent dpl gene (Aguzzi, 2003). The dpl gene prod-
BSE Biology
1457
Figure 1. Cellular processing of PrP. 1) Cellular PrP is processed like many other membrane-associated or extracellular
proteins (1). The PrP can be internalized before degradation by proteosome or lysosomal proteases. In contrast, PrPsc
processing results in limited proteolysis (2). Limited degradation produces PrPsc fragments, which accumulate and
presumably play a role in cell death. These fragments lead to propagation of the PrPsc infection in adjacent cells.
Conversion of PrP to PrPsc occurs sequentially. A) Normal PrP can refold into PrPsc in the extracellular space. B)
Fragments of PrPsc may remain within the cell or may be externalized by transport vesicles or by cellular rupture
upon death. C) Intracellular PrPsc could interact with PrP during intracellular processing resulting in conversion of
PrP to PrPsc in the infected cell. D) Intracellular PrP may spontaneously change conformation to PrPsc. Endogenous,
host-encoded prion protein = PrP; abnormal PrP conformation = PrPsc.
uct, doppel, and PrP are approximately 25% homologous.
Pathogenesis
Disease-associated PrPsc has undergone a conformational change from predominantly helical to predominantly β-pleated sheet (Nguyen et al., 1995; Legname
et al., 2004). This modification is posttranslational, so
the sequence of AA in PrP and PrPsc within an individual is identical. Spontaneous conformational change of
PrP to PrPsc is thermodynamically unfavorable (Baskakov et al., 2004). Variation arising from mutations in
the PrP AA sequence may make the conversion more
favorable. The altered conformation renders PrPsc resistant to proteinase K activity, such that it is incompletely
cleaved, resulting in several products with decreased
solubility and increased tendency to aggregate (Telling
et al., 1996; Riesner, 2003). Similarly, the infective
PrPsc conformer is resistant to heat and chemical sterilization processes (see Decontamination section below).
Protease resistance is the basis for most current methods of PrPsc detection.
Conversion of PrP to PrPsc may occur in several steps
(Figure 1). The PrPsc in the extracellular spaces can
interact directly with PrP, causing refolding of the latter to produce additional PrPsc. Strong evidence for this
was provided by studies showing conversion of PrP by
PrPsc even in the absence of cells (Raymond et al., 2000).
In addition, intracellular PrPsc could interact with PrP
during intracellular processing, resulting in conversion
within the infected cell. Either of these processes propagates PrPsc. Experimental models indicate that elevated intracellular PrP results in incomplete degradation of PrP and conversion to PrPsc-like proteins (Chiesa
and Harris, 2001). Improper or elevated retrograde
transport of PrP within the cells or altered proteasomal
degradation can result in accumulation of intracellular
PrP, independent of the presence of PrPsc (Ma et al.,
2003). However, ubiquitination of protease-resistant
PrPsc is probably a late event in transmitted TSE pathogenesis rather than an important early step (Kang et
1458
Novakofski et al.
al., 2004). Extrapolating from these findings, it is possible to speculate that sporadic TSE may arise by a combination of these intracellular events.
The neurotoxic mechanism of PrPsc remains unclear,
with different lines of evidence supporting some combination of PrPsc toxicity and alteration in normal PrP
function. The presence of both PrPsc and synthesis of
PrP are required for pathogenesis. Inoculation of a dose
of PrPsc that is infectious in normal mice does not cause
disease in PrP knockout mice (Prusiner et al., 1993),
indicating that endogenous PrP conversion to PrPsc is
necessary to cause pathology. Loss of normal PrP function also is not the cause of pathology because PrP
knockout mice do not develop disease. Crosslinking of
PrP with antibodies in the absence of any PrPsc results
in rapid onset of neuronal disease (Solforosi et al.,
2004), suggesting that disruption of normal PrP function or accumulation alone may result in a neurotoxic
process.
Clinical signs of TSE are primarily neurological: behavior changes, impaired coordination, muscle spasms,
and repetitive movements. Body wasting in spite of
normal food intake is common, although obesity and
abnormal glucose metabolism are observed in some
strain/host combinations (Ye and Carp, 1995; Wadsworth et al., 2003). Neurological deterioration is progressive after clinical symptoms appear. Specific combinations of symptoms, referred to as TSE phenotype,
reflect different pathogenic TSE strains. The TSE typically have long incubation periods of months (rodents,
cats), years (cattle, sheep, deer), or decades (man), so
clinical signs usually are evident in older animals. Because the sequence of PrP protein is highly conserved
and most of the PrPsc is eventually derived from converted host-encoded PrP, infection does not evoke a
substantial immune response or inflammatory reaction
in host animals (Aguzzi, 2003; Prusiner, 2004); however, chronic inflammatory conditions may modify natural PrPsc transmission (Heikenwalder et al., 2005). All
TSE cause spongiform changes in the brain associated
with some degree of amyloid plaque development. Amyloid plaques consist of PrPsc aggregates, which may be
detected histologically as diffuse, condensed, or florid
plaques in infected brain tissue. Specific combinations
of clinical symptoms reflect the specific regions in the
brain where PrPsc accumulates, the characteristic pattern of damage and the nature of plaque deposition
(Brown et al., 1994; DeArmond et al., 1997).
Animals may develop PrPsc-based disease 1) by ingestion of PrPsc; 2) by peripheral exposure to PrPsc, most
commonly by an iatrogenic route (surgery, cadaveric
growth hormone injection, corneal transplantation); 3)
by hereditary transmission as an autosomal, dominant
trait; or 4) sporadically by unknown origin (Wadsworth
et al., 2003; Prusiner, 2004).
Oral infection by ingestion involves transfer of PrPsc
from the digestive tract to the spleen or lymphoreticular
system, and then to the peripheral nervous system and
eventually to the brain. Infectivity is established in
peripheral lymphoid organs before infective PrPsc is
found in the central nervous system (CNS), indicating
that peripheral conversion of PrP to PrPsc is necessary
step for infection. There seem to be two potential routes
of neuro-invasion by PrPsc via lymphoid tissue: one path
involving the spleen, and the other path involving visceral lymph nodes (Kimberlin and Walker, 1989). Following ingestion, PrPsc may be degraded by digestive
enzymes, leaving a pathogenic fragment similar to that
of a proteinase K-resistant PrPsc fragment. The PrPsc
fragment may be cotransported across the intestinal
epithelial with ferritin (Mishra et al., 2004). The PrPsc
or PrPsc fragments are transported from the intestine
to secondary lymphoid organs by intestinal dendritic
cells, which are specialized to acquire antigen from peripheral tissues (Huang and MacPherson, 2004). Dendritic cells from the intestine present PrPsc to T and B
lymphocytes within lymphoid tissues such as Peyer’s
patches of the intestine or follicular dendritic cells
(FDC) of the spleen, thymus, and tonsils (Mabbott and
Bruce, 2003; Prinz et al., 2003). In the lymphoid tissues,
PrPsc molecules accumulate following conversion from
the PrP conformation. Accumulation of PrPsc and progression to the CNS nervous system requires B-cell
cytokines, although expression of PrP on the B-cells is
not necessary for transmission (Klein et al., 1997; 1998).
The spleen is a rich source of B-cells, and B-cell cytokines enable splenic FDC cells to mature (Prinz et al.,
2003; Aguzzi and Miele, 2004). Splenic FDC may serve
as the source of PrP for peripheral PrPsc formation as
they express a high level of PrP (Aguzzi, 2003). Subsequent transfer of PrPsc from FDC to sympathetic nerves
depends on the distance between the two structures
(Prinz et al., 2003). Chronic lymphocytic inflammation,
which upregulates cyotkines, enables PrPsc accumulation in otherwise PrPsc free tissues (Heikenwalder et
al., 2005).
After oral inoculation, PrPsc can be detected in a portion of the follicles of Peyer’s patches in the distal ileum
of cattle for much of the disease progression (Terry
et al., 2003). Detection of PrPsc in lymphoid tissue of
experimentally infected animals is possible before clinical signs are observed. In the CNS, detectable PrPsc is
first seen in the spinal cord, then the brain stem, and
then in higher brain areas. Levels of PrPsc in the brain
increase exponentially following infection before the development of clinical symptoms. By the onset of clinical
symptoms, PrPsc titers in spleen and lymph tissues have
either plateaued or decreased, although in the later
stages of disease, PrPsc levels are increased in Peyer’s
patches. (Kimberlin and Walker, 1988; Anderson et al.,
1996). Oral infectivity demonstrates that PrPsc is resistant to stomach acids, mammalian digestive proteases,
and in the case of ruminants, bacterial proteases.
Neural invasion by PrPsc does not require exposure
to lymphoid tissue. Peripheral exposure, for example
through damaged skin (Taylor et al., 1996), the tongue
(Bartz et al., 2003), or vascular system (Kimberlin and
Walker, 1989), results in direct PrPsc exposure of the
BSE Biology
nervous system with subsequent retrograde transport
to the CNS and rapid infection. Infection seems to be
particularly rapid and effective by peripheral exposure
of heavily innervated tissue such as the tongue (Bartz
et al., 2003). There is also retrograde transmission by
nerves as PrPsc is found in sensory nerves and neuromuscular junction in the tongue of intracerebrally (i.c.)
inoculated animals (Bartz et al., 2002; Mulcahy et al.,
2004). Iatrogenic infection by medical procedures results from central or peripheral neural inoculation. It
is worth noting that there have been more than twice
as many iatrogenic CJD cases as vCJD cases. Iatrogenic
prion disease transmission has been documented from
exposure to neuro-surgical instruments, from injection
of cadaver-derived growth hormone or gonadotrophin,
and from corneal or dura tissue transplants (Brown et
al., 2000; Weissmann et al., 2002).
Another route of peripheral infection is maternal
transmission to the fetus or newborn. The traditional
view is that scrapie is maternally transmitted to the
fetus; however, it is clear that separating fetal (vertical)
transmission from postnatal lateral transmission is difficult (Ridley and Baker, 1995). Transmission of PrPsc
in utero across the maternal-fetal barrier requires a
placental architecture permitting relocation of infective
PrPsc and genetic susceptibility of the fetus.
Genetics of Prion Disease
Specific TSE phenotypes result from the combined
effects of pathogenic mutations, genetic polymorphisms
of prnp, and the specific strain of infectious PrPsc. Sporadic (spontaneous) and hereditary forms of prion disease share a common pathogenesis except that the initiating events are unknown in the first case and predetermined by a pathogenic mutation in the second.
“Sporadic” refers to development of prion disease with
no familial history or apparent exogenous infection. The
spontaneous conversion of PrP to PrPsc is thermodynamically unfavorable, as indicated by the low incidence of sporadic CJD (Baskakov et al., 2004). Variation
in the PrP AA sequence may make the conversion more
or less favorable, thereby altering susceptibility to prion
disease. There are 86 reported mutations or polymorphisms of the prnp sequence, with at least 25 that increase risk of spontaneous PrPsc formation, predispose
the individual to exogenous infection, or both (Heaton
et al., 2003; Wadsworth et al., 2003). Susceptibility may
be reflected by minimal infectious dose, incubation time
following exposure, or relative statistical risk in a population.
A common protein polymorphism of human PrP, methionine or valine at residue 129 (129M/V), seems to
be predictive of CJD susceptibility (Figure 2). Heterozygosity at 129 protects individuals from CJD, whereas
129V homozygotes are strongly overrepresented in individuals with iatogenic or sporadic CJD (Collinge et al.,
1991; Palmer et al., 1991). All vCJD patients have the
129M homozygous genotype (Collinge et al., 1996). Be-
1459
sides susceptibility, PrP polymorphisms may result in
different symptoms. In sporadic CJD, homozygous
129M individuals developed no amyloid plaque,
whereas homozygous 129V individuals developed extensive plaques (Pickering-Brown et al., 1995).
Approximately 10 to 15% of human prion diseases
are familial, meaning the individual has inherited a
prnp mutation that confers a high susceptibility. All
families identified with inherited forms of prion diseases have either point mutations or insertions in the
prnp gene (Collinge and Palmer, 1994; Prusiner, 2004).
Mutation D178N is found in fatal familial insomnia
and familial CJD (Gambetti et al., 2003). The clinical
and pathological differences between familial CJD and
fatal familial insomnia, which have the same D178N
mutation, are the result of the129M/V polymorphism
(Goldfarb et al., 1992). Mutations responsible for Gerstmann-Straussler-Scheinker syndrome include P102L,
A117V, F198S D202N, Q212P, and Q217R (Piccardo et
al., 2001).
PrPsc Strains: Conformation and Glycoslyation
Besides the AA sequence variation that contributes
to phenotypic variation of TSE, a single PrP sequence
can change into multiple PrPsc conformers, each producing a distinct pathologic phenotype (Telling et al., 1996;
Safar et al., 2000). Strain differences result from variation of the α helix to β sheet conversion within PrPsc,
as well as variation in degree of PrP glycoslyation. In
general, transmitted TSE strains retain their phenotypic fingerprint, so the existence of different strains
of PrPsc is an important key to understanding the risk
for interspecies transmission.
At least 20 strains of scrapie have been differentiated
by their incubation period, clinical symptoms, and brain
lesion pattern (Bruce et al., 1994, 2003). Different
strains of TSE can be serially passaged in inbred mice
with no change in the TSE strain characteristics, demonstrating that strain differences may be independent
of PrP amino acid sequence in the host (Bruce, 2003).
Characteristics of both the hyper and drowsy hamsteradapted scrapie PrPsc strains also can be reproduced
in a cell-free conversion system, indicating the importance of PrPsc conformation (Bessen et al., 1995). Different strains of PrPsc generated from the same PrP precursor are cleaved at distinct sites by proteinase K,
demonstrating differences in tertiary structure of PrPsc.
Variation in glycosylation patterns of PrPsc may play
a role in TSE phenotype (DeArmond et al., 1997; Hill
et al., 1998), and because of the different cleavage sites,
the size distribution of glycosylated proteinase K digestion fragments provides another signature of strain
type (Wadsworth et al., 2003). The BSE PrPsc contains
high levels of diglycosylated and low levels of monoglycosylated protein. Some scrapie strains contain primarily monoglycoslyated protein, whereas the glycosylation
profile in other strains is more similar, but not identical,
to BSE (Baron et al., 2004). Human prion disease also
1460
Novakofski et al.
Figure 2. Sequence alignment of mature prion proteins. AA sequences were obtained from the NCBI (2004) protein
database and the Swiss-Prot/TrEMBL (2004) Protein Knowledge database and consensus sequences within a species
and comparisons between species were generated in ClustalW. Sequence is indicated using single letter AA coding
and a period (.) indicates an AA is not present in that species. Amino acids that are identical to those in ovine PrP
are not shown. Shaded bars indicate notable domains of PrP. Proteinase K digestion of PrPsc removes much of the
octapeptide repeat and a portion of the metal binding domain to leave the resistant PrP 27-30 fragment. The α1 helix
is important as a probable binding site for PrPsc during catalytic conversion. The region at 165-177 is also important
for PrPsc conversion. It may play a role in interspecies transmission and this region binds the putative X protein
chaperone (Kiyotoshi et al., 1997; Gossert, et al., 2005). The H/R/Q polymorphism in this region at AA 171 in
sheep is a major determinant of scrapie resistance. Endogenous, host-encoded prion protein = PrP; abnormal PrP
conformation = PrPsc.
can be phenotyped by glycosylation patterns. Sporadic
and iatrogenic forms of CJD exhibit a high proportion
of monoglycosylated to diglycosylated PrPsc, whereas
vCJD, exhibits a higher proportion of diglycosylated
PrPsc similar to BSE (Collinge et al., 1996; Wadsworth
et al., 2003). Different PrP glycoforms, as well as different amounts of PrP, are expressed in different areas
of the brain and may contribute to TSE variation in
neuropathology (Beringue et al., 2003). In addition, host
PrP genotype has a major influence on the degree of
PrPsc glycosylation contributing to variation in neuropathology on passage (Somerville et al., 2005).
Diagnosis and Potential Treatment
There currently is no definitive antemortem test for
TSE. Initial diagnosis of TSE is achieved through clinical examination. However, observation of neurological
symptoms, such as abnormal motor movement, behavioral, and cognitive changes are indicative but not definitive because they may reflect other neurodegenerative processes (Cockcroft, 2004). In humans, ancillary
techniques that can be employed to support a clinical
diagnosis include electroencephalography, brain imaging, cerebrospinal fluid screening, tonsil biopsy (Kü-
bler et al., 2003; Soto, 2004). Electroencephalography
is used to identify changes in brain wave patterns that
are common to, but not unique to TSE. Imaging brain
structure is primarily useful to exclude other encephalopathies. Elevated 14-3-3 protein in cerebrospinal fluid
is indicative of TSE but also may be increased by other
causes of neurodegeneration.
Postmortem examination of brain samples is required to confirm the presence of TSE (Table 2). Histological techniques can identify the characteristic spongiform lesions, astrocytic gliosis, and (usually) plaques
found in TSE. Although the antibodies used are not
conformer specific, immunohistochemical staining can
identify pathological accumulation of PrP, which is assumed to be PrPsc because it has accumulated. Both
approaches are labor intensive. Biochemical tests using
brain samples are based on differential sensitivity of
PrP vs. PrPsc to proteinase K digestion. After proteinase
K digestion, the remaining immunoreactive PrP is considered to be PrPsc and may be assayed by conventional
techniques such as Western blotting or ELISA (Kübler
et al., 2003; Soto, 2004). Commercial tests that have
been approved for diagnosis of BSE or CWD may be
sold only to federal or state diagnostic laboratories and
are not available for research purposes.
1461
BSE Biology
Table 2. Prion detection methodsa
Method
Immunohistochemistry
Assay
Microscopic examination of brain sections
stained with antibodies for PrP protein.
Antibodies are not conformation-specific
but PrP does not accumulate except
under pathological conditions.
Species
Country
approvalb
All
Comments
Gold standard for TSE detection.
Completion time: 2 to 3 d. Low
sensitivity to postmortem autolysis.
Detects PrPsc deposits in brain before
histopathological identification of
prion-induced vacuoles.
Bioassay
Suspect brain sample injected into the
brain or periphery of test animal
(usually mouse). Infectivity
diagnosed by clinical signs of
infection and incubation period.
Test results confirmed by other
methods.
All
Completion time: several months.
Histopathology
Microscopic examination of brain sections
for changes indicative of spongiform
encephalopathy, vacuoles, or presence
of prion-associated fibrils.
All
Completion time: 2 to 3 d. Proper
fixation required to prevent
postmortem autolysis.
Western blotting
Sample is homogenized, treated with
proteinase K, which degrades normal
PrP, electrophoretically separated
with SDS-PAGE, blotted onto a
membrane, and probed with an
antibody to detect the proteaseresistant fragments (PrPsc).
All
Visualization techniques vary from
chromogenic precipitate to radio
isotopes.
Western blot of protease-treated samples
with PrP antibody
Cattle
Lowest detection limits of all tests.
Sensitivity depends mouse strain
used.
Rapid methods (company)
Prionics-Check Western Test
(Prionics AG, Schlieren,
Switzerland)
European
Union,
United States
Detection via alkaline phosphatasecoupled secondary antibody with a
chemiluminescent substrate, a
reduced molecular weight compared
with native PrP, and glycosylationdependent band pattern of resistant PrP
Prionics-Check LIA
(Prionics AG)
Enzyme-linked immunosorbant assay
(ELISA)
Validated for sheep and goats
Cattle
European
Union,
United States
Uses monoclonal antibodies and
chemiluminescent detection.
Bio-Rad test
(Bio-Rad, Hercules, CA)
ELISA sandwich with colorimetric
substrate.
0.5-g obex sample required.
Dilution accuracy: 1:10 to 1:30.
Detection limit: 1.0 to 5.0 pmol.
Completion time: 4 h.
Cattle
Deer
Sample degradation of normal protein
with proteinase K.
0.5-g obex sample required.
Dilution accuracy: 1:10.c
Detection limit: 5.0 to 20 pmol.
Completion time: 6 to 8 h.
European
Union,
United States,
Japan
0.35-g obex sample.
Dilution accuracy: 1:300.
Detection limit: 0.5 to 2.0 pmol.
Completion time: 6 h.
European
Union,
United States,
Japan
Dilution accuracy: 1:10 to 1:30.
Detection limit: 1.0 to 10 pmol.
Completion time: 4 h
Elk
Detection with two specific monoclonal
antibodies for protease resistant PrP
(colorimetric).
Enfer test
(Abbott Labs, Abbott Park,
IL)
ELISA
One-step sample preparation and
proteinase K treatment, polyclonal
antibodies, chemiluminescence
detection.
Cattle
Continued
1462
Novakofski et al.
Table 2 (Continued). Prion detection methodsa
Method
Country
approvalb
Comments
Cattle
European
Union,
United States,
Japan
Dilution accuracy: 1:30.
Detection limit: 0.5 to 5.0 pmol.
Completion time: 8 h.
Does not involve proteinase K.
Cattle,
deer
United States
Completion time: 4 to 5 h
Does not involve proteinase K.
Assay
Species
CDI test
(Impro Biotechnology, South)
San Francisco, CA)
Conformation-dependent immunoassay.
Herd-check
(IDEXX Laboratories,
Westbrook, ME)
Immunoassay after affinity based
separation of PrPsc in brain
homogenate.
Denatures sample to expose buried
epitope in PrPsc. Uses
two radio-labeled antibody fragments
to capture and detect PrP.
a
See Table 1 for abbreviations. Endogenous, host-encoded prion protein = PrP; abnormal PrP conformation = PrPsc.
Tests approved by the European Commission for BSE detection are based on immunodetection of the pathological PrPsc conformer.
b
Dilution accuracy is a reflection of test accuracy with known positive samples.
b
Animal bioassay remains the most sensitive method
for detection of PrPsc (Soto, 2004). Although time consuming and labor intensive, it is necessary to detect
very low levels of infectivity. Use of animals overexpressing PrP has shortened bioassay time, whereas use
of animals transgenic for another species of prion is
central to interspecies transmission research. Cell culture bioassays with various cell lines, including common fibroblasts, may be used to detect PrPsc that is not
measurable with biochemical techniques (Weissman et
al., 2002; Vorberg et al., 2004).
A variety of substances have been investigated to
prevent transmission or arrest progress of TSE. Inhibitory compounds could interfere with PrP conformational change, PrPsc aggregate formation, or PrPsc protease resistance. In addition, primary therapeutic considerations are toxicity and ability to cross the blood
brain barrier. Early interest focused on compounds similar to Congo red, which is used as a histological stain
for PrPsc. Compounds showing activity in vitro include
Congo red (Caughey and Race, 1992; Rudyk et al.,
2000), porphyrins (Caughey et al., 1998), lysosomotropics or protease inhibitors (Doh-ura et al., 2000), pyridine-based compounds (Perrier et al., 2000), branched
polyamines (Supattapone et al., 2001), and phenothiazines or bis-acridines (May et al., 2003). Several compounds have shown promise in vivo including tetracycline (Forloni et al., 2002) and quinolines (MurakamiKubo et al., 2004). Quinacrine (an acridine) and chlopromazine depress PrPsc aggregation, but they are ineffective in treating animal models (Barret et al., 2003).
Transmissible Spongiform Encephalopathies
Scrapie
Scrapie was first reported in 1732, and it is the prototypical spongiform encephalopathy found in ovines and
caprines. Scrapie refers to the pruritus-induced rubbing
and scratching (scraping) that results in wool loss. It
is an acquired infection with an incubation period of 2
to 5 yr and an average age of onset of clinical symptoms
of 2.5 yr (Schreuder, 1994). Typically, no more than
5% of the animals in a herd show clinical symptoms
of infection.
Scrapie epidemics are self-sustaining because of horizontal transmission (animal-to-animal) and apparent
environmental persistence. Contaminated pastures
have caused numerous outbreaks of scrapie (Chatelain
et al., 1983; Redman et al., 2002). In Iceland, eradication attempts failed when scrapie-free sheep were restocked onto pastures that had been kept free of sheep
for 3 yr (Sigurdarson, 1991) indicating the stability of
PrPsc even in the soil. Infectious material buried in the
soil loses only approximately 50% of its infectivity in 3
yr (Brown and Gajdusek, 1991). There is limited evidence for specific mechanisms of environmental transmission, although it clearly occurs. Likely possibilities
include saliva or feces (Miller et al., 2004), urine
(Shaked et al., 2001), or decay residue of infectious
tissues such as placenta (Table 3). Because PrPsc is
stable in the soil, animals could become infected by
consumption of soil or inhalation of contaminated dust.
Scrapie PrPsc is recoverable from lymphatic tissue,
tonsils, spleen, and thymus, as well as nervous tissue.
Accumulation of PrPsc in placental tissue depends on
susceptible fetal genotype and stage of gestation (Tuo
et al., 2001, 2002; Andreoletti et al., 2002); however,
the presence or absence of PrPsc in placentomes of fetuses does not correlate with time to postnatal infection
(Andreoletti et al., 2002). This suggests that infection
does not occur in utero but is postnatal. Infection may
occur from direct postnatal contact with placental tissue or from pasture contaminated with placental tissue.
Scrapie has been experimentally transmitted (by i.c.
inoculation) to a number of animals, including hamsters, mice, mink, ferrets, goats, monkeys, and cattle.
Oral infection has been successfully demonstrated in
mice (Chandler, 1963), sheep, goats (Pattison et al.,
1972), and squirrel monkeys (Gibbs et al., 1980).
Variation in the coding region of the prnp gene, previously known as the scrapie gene Sip, influences susceptibility to scrapie. The major AA polymorphisms are
A136V, H154R, and H171Q,R (Tranulis, 2002). Ani-
BSE Biology
1463
Table 3. Potential infectivity of food-animal tissuesa,b
High infectivity
Brain, eyes, spinal cord, skull, vertebral column, tonsil,
trigeminal ganglia, dorsal root ganglia, distal ileumb
Medium infectivity
Lymph nodes, proximal colon, spleen, dura mater, pineal gland,
placenta, cerebrospinal fluid, pituitary, adrenal
Low infectivity
Distal colon, nasal mucosa, peripheral nerves, bone marrow,
liver, lung, pancreas, thymus, blood, feces, uterusc,d,e
Minimal infectivity
Heart, kidney, mammary gland, milk, ovary, saliva, salivary
gland, seminal vesicle, serum, skeletal muscle, testis,
thyroid, bile, bone, connective tissue, hair, skin, urine
a
DEFRA (2004b). Based on scrapie-infected sheep and goats.
Tonsil and distal ileum classified by USDA (2004a).
Blood moved from DEFRA infectivity category; Ironside and Head (2003).
d
Feces moved from DEFRA infectivity category; Miller et al. (2004).
e
Uterus moved from DEFRA infectivity category; Tuo et al. (2002).
b
c
mals with prnp genotypes, A136R154Q171/ARQ, ARQ/
VRQ, and VRQ/VRQ seem to be the most susceptible
to scrapie. Heterozygous AHQ or ARR animals are partially susceptible, whereas homozygous ARR sheep are
clinically resistant (Hunter, 2003; Baylis and Goldmann, 2004). Sheep with a R171Q polymorphism are
resistant, but not immune, to scrapie (Goldmann et
al., 1994).
Scrapie is a reportable disease in the United States,
and the Animal and Plant Health Inspection Service
has had a voluntary scrapie-free flock certification program since October 1992. The national prevalence of
scrapie is 0.2% in the United States, with approximately 84% of cases occurring in the Suffolk or Hampshire breeds (USDA, 2004d).
Bovine Spongiform Encephalopathy
First observed as a progressive neural degenerative
disease in the United Kingdom in 1984, BSE was specifically diagnosed in 1987 (DEFRA, 2000). By June
1990, there were some 14,000 confirmed cases out of an
estimated population of 10 million cattle in the United
Kingdom. Since 1986, nearly 200,000 cases of BSE have
been identified in the United Kingdom (DEFRA, 2004a).
The geographic distribution of BSE has increased to 24
countries, including the United States (Office International des Epizooties, 2004).
Symptoms of BSE include abnormal gait and stumbling, hyperresponsiveness to stimuli, tremors, loss of
BW (>75% of cases), aggression, licking or rubbing, and
decreased milk yield (roughly 50% of cases; Cockcroft,
2004). Unlike scrapie, however, pruritus (itching or
scratching) is not a common symptom (Wilesmith et
al., 1988). The median age for clinical signs of natural
BSE infection is 42 mo, with an incubation period of 2
to 8 yr. Animals as young as 20 mo have tested positive
for BSE (DEFRA, 2004a). The anatomical locations and
severity of brain lesions are similar in most cases of
BSE. Vacuolar changes are prominent in the nucleus of
the solitary tract (hindbrain, glossopharyngeal nerves),
nucleus of the spinal tract of V (hindbrain, trigeminal
nerves), vestibular nuclear complex (medulla; the obex
is the portion of medulla or brainstem just distal to
the cerebellum), central gray matter, rostral colliculus
(mesencephalon; orientation reflex), and hypothalamus
(Simmons et al., 1996).
Bovine spongiform encephalopathy is considered a
“common source” epidemic, meaning that animals contract the disease from a common element, most likely
eating feed containing rendered meat and bone meal
(MBM) contaminated with bovine brain and spinal cord
from infected cattle. Development of infection is related
to dose and tissue PrPsc content. Semen, chemicals,
autosomal inheritance, biologics, and pharmaceuticals
have been ruled out as the common source (Wilesmith
et al., 1988, 1991, 1992).
There is little evidence for vertical transmission of
BSE from dam to fetus (Foster et al., 1999; Wrathall
et al., 2002); however, calves born in close contact with
BSE-infected cows may be infected within days after
calving (Hoinville et al., 1995; Donnelly et al., 1997).
Calves and mice fed placenta from confirmed BSE cases
remained healthy (Middleton and Barlow, 1993; Hoinville et al., 1995), suggesting that maternal transfer is
postnatal in cattle, although the route remains unclear.
No PrPsc has been demonstrated in bovine colostrum,
although PrPsc infectivity has been demonstrated in
human colostrum (Tamai et al., 1992).
In the United Kingdom epidemic, BSE occurred predominantly in Friesian and Friesian crossbred cattle,
although this is likely to be a function of management
practices rather than genetics. There are two sequences
of the bovine prnp gene, one with five repeats of an
octapeptide metal binding sequence and one with six
(Goldmann et al., 1991), but this genotype difference
does not influence the occurrence of BSE (Hunter et al.,
1994). An analysis of 370 cattle in Scotland indicated
similar frequencies of the two genotypes between
healthy cattle and animals with BSE; however, the
number of octapeptide repeats may influence rate of
progression or age at onset (Castilla et al., 2004; Croes
et al., 2004). Besides the octapeptide repeat, there is a
23-bp insertion/deletion polymorphism in the noncoding prnp promoter region that is associated with BSE
susceptibility (Sander et al., 2004). Other members of
1464
Novakofski et al.
the Bovidae family, including captive nyala, eland,
kudu, gemsbok, and oryx, also are susceptible to BSE
or very similar diseases (Kirkwood and Cunningham,
1994).
Specified bovine offals, which included brain, spinal
cord, spleen, thymus, tonsils, and intestines, were
banned from use in ruminant feed in the United Kingdom in 1989. The BSE epidemic peaked in 1992 at
almost 1,000 cases per week. Subsequently, the number
of positive animals decreased by 1993, which is consistent with a 2- to 8-yr incubation in cattle. A large part
of the decrease is attributed to the ban on feeding ruminant-derived MBM to other ruminants (Anderson et
al., 1996); however, BSE cases continued to occur at a
lower rate, which was attributed to cross-contamination of ruminant feed. This led to complete exclusion of
MBM from all farm animal feeds in 1996. The United
Kingdom stopped exporting MBM in 1996. Currently,
there are approximately 5 to 10 BSE-positive cases/
wk being confirmed in the United Kingdom (DEFRA,
2004a). Of these cases, more than 95% were born after
the initiation of the ban on feeding ruminant-derived
MBM. The small number of BSE cases continuing to
occur in animals born more than 15 yr after the feed
ban suggests additional routes of infection, such as horizontal transmission or environmental reservoirs.
There are several hypotheses concerning the origins
of BSE. One theory is that BSE existed at very low,
and therefore undetected, levels in the cattle population
before 1988, as a rare sporadic disease, and that its
increase in incidence in the United Kingdom resulted
from coincidental changes in the industry or in the
environment. Given the delay between exposure and
development of clinical symptoms, British cattle were
likely exposed to a TSE that resulted in disease in 1981
or 1982. This exposure corresponded to cessation of
solvent extraction of fat from MBM in most rendering
plants (Wilesmith et al., 1991, 1992; Figure 3). Downer
cattle or cattle with “staggers” may have been infected
with BSE before its recognition as a transmissible disease. A second theory suggests that BSE initially appeared as a new TSE in the 1980s, which was spread
by the contemporary industry practices of low-temperature rendering and inclusion of bovine-derived MBM
in bovine feed. A third theory suggests that scrapie
reached infective levels through recycling in MBM ultimately adapting to the strain currently recognized as
BSE. A fourth theory suggests that a new strain of
scrapie that was more readily transmitted to cattle
arose in sheep (DEFRA, 2004b). In fact, sheep-passaged
BSE has strain characteristics similar to the original
BSE, and sheep infected with BSE are not clinically
distinct from sheep infected with scrapie (Hill et al.,
1998).
Atypical Strains of BSE. Unlike scrapie, most cases
of BSE seem to be a single strain based on three characteristics: 1) a typical BSE case has a high concentration
of lesions in the brain stem with unremarkable olfactory
bulb involvement; 2) a typical BSE case has an overre-
Figure 3. Meat and bone meal production from rendered animal by-products.
presentation of high-molecular-mass/diglycosylated
PrPsc and a lower proportion of monoglycoslyated glycoforms; and 3) a typical BSE case develops very little
amyloid plaque in damaged tissues.
Recently, three cases of BSE in France (Biacabe et
al., 2004) and two cases in Italy (Casalone et al., 2004)
have been described that were atypical based on standard strain typing using Western blotting of proteinase
K-digested brain stem samples. The atypical cases from
Italy also had a distribution of brain lesions different
than expected from BSE. The French atypical BSE
cases occurred in cattle that were older than 24 mo and
that did not have clinical symptoms when tested. In
these cattle, PrPsc was characterized by an unglycosylated proteinase fragment with a greater molecular
weight than typical BSE, a characteristic similar to the
PrPsc from cattle experimentally infected with scrapie.
In contrast, the two Italian atypical cases were characterized by lower-molecular-weight unglycosylated
PrPsc, similar to sporadic CJD. Moreover, mouse passages of BSE PrPsc suggest that two distinct BSE
strains may exist in British cattle as well (Lloyd et al.,
2004). These observations strongly suggest that BSE
of cattle exists in multiple sporadic strains, as do TSE
in other species. Therefore, increased surveillance is
likely to detect additional BSE strains and additional
cases in spite of rigorous preventative measure.
1465
BSE Biology
Chronic Wasting Disease
The first recognition of CWD occurred in 1967 in
captive mule deer and hybrid mule deer × white-tailed
deer at a Colorado research facility (Williams and
Young, 1980). By 1985, it had been identified as a TSE
in free-ranging cervids, including mule deer, white tail
deer, and elk in Colorado and Wyoming. Currently,
CWD is found in 12 states and two Canadian provinces
in both farmed-raised and wild cervid populations.
When clinical signs of CWD infection appear, cervids
present behavioral changes (loss of fear to humans),
progressive weight loss, excessive salivation, polydipsia, polyuria, ataxia, weakness, inability to stand, dehydration, dull hair coat, drooping head and ears, and
emaciation (Williams and Young, 1980; Spraker et al.,
1997). The age at onset of clinical symptoms in elk
ranges from 2 to 8 yr, and the duration of symptoms
ranges from 5 to 12 mo before death (Miller et al., 1998).
The youngest naturally infected mule deer reported was
approximately 17 mo old, whereas the youngest elk
was approximately 24 mo old (Miller et al., 2000; Ball,
2002). Based on dentition, white tail deer may be infected as young as 5 to 7 mo of age (Illinois Natural
History Survey, unpublished data). Amino acid polymorphisms of cervid prnp occur at Q95H, G96S, A116G,
M132L and S138N (O’Rourke et al., 1999, 2004; Johnson et al., 2003). No genotype seems to be resistant,
although Q95G96A116S138 deer are less likely to have
CWD, whereas elk homozygous for M132 are overrepresented among CWD-positive animals.
In CWD, microscopic lesions and pathological
changes are limited to the CNS, characterized by neuronal degeneration and spongiform encephalopathy
with no inflammatory response, intracytoplasmic vacuoles in neurons, and astrocytic hypertrophy and hyperplasia with occurrence of amyloidal plaques (Williams
and Young, 1993). Abnormal PrPsc has been demonstrated by immunohistochemistry in the brain, palatine
tonsils, lymph nodes, along the small and large intestine, Peyer’s patches and spleen of affected deer (Sigurdson et al., 1999). Accumulation in the alimentary
tract and lymphoid tissue precedes accumulation in
brainstem (Williams and Miller, 2002).
The origin of CWD is undetermined. Like BSE, it has
been suggested that CWD originated with the transmission of scrapie from domestic sheep to cervids. Scrapie
inoculation results in a TSE of elk indistinguishable
from CWD (Hamir et al., 2004b); however, countries
such as the United Kingdom, with more prevalent rates
of scrapie, have not reported CWD. Another theory suggests that it is the result of a spontaneous naturally
occurring TSE of cervids (Williams and Young, 1980;
Miller and Williams, 2004).
Like scrapie, CWD epidemics are self-sustaining in
both captive and free-ranging cervid populations. It is
unlikely that the primary route of CWD transmission
occurs by consumption of infected tissue, although artificial feeding stations for cervids could exacerbate the
problem because of environmental contamination. Epidemics in captive deer and elk provide strong evidence
of horizontal transmission similar to scrapie epidemics
(Miller and Williams, 2003; 2004). It is likely that transmission occurs by both direct and indirect contact. Contaminated pastures have resulted in CWD in captive
herds similar to outbreaks of scrapie. Early involvement of lymphoid tissue of the alimentary tract is consistent with reported horizontal transmission from saliva or feces in contaminated pens (Miller et al., 2004).
Placental transmission remains a possible route for vertical transmission of CWD in deer (Miller and Williams,
2003); however, the specific mechanisms for transmission, including source of PrPsc, reservoirs, route of infection, and infective dose, remain unknown.
Sources of Transmissible PrPsc
The degree to which PrPsc in different tissues can
transmit infection is critical to understanding risk to
the livestock industry and human population because
PrPsc is not destroyed by cooking or proteinases and
may be infectious if consumed (Taylor, 1999). Many
nonneuronal tissues, including abomasum, adrenal
gland, heart, intestine, kidney, lung, lymph nodes,
mammary gland, parotid gland, skeletal muscle, tonsils, spleen, and uterus, express PrP; however, PrPsc
accumulation is not a direct function of PrP expression,
nor is it tissue-specific (Table 3). In animals with clinical TSE symptoms, levels of PrPsc are greatest in the
CNS but rarely are found in peripheral tissues other
than lymph nodes, spleen, and tonsil (Horiuchi et al.,
1995; Wadsworth et al., 2001).
Infectious dose usually is expressed in terms of LD50,
the amount of agent required to infect or to kill 50% of
the population exposed, or in terms of infectious
units, the amount required to kill one animal from a
group. One picogram of PrPsc contains approximately
100 infectious units, or 10 LD50 (Brown et al., 2001).
An oral LD50 of brain tissue is in the milligram range
(Bolton et al., 1991). The infectivity of PrPsc-containing
tissue varies depending on the route of exposure and
the PrPsc content, which increases with disease progression, peaking when clinical symptoms appear (Kimberlin and Walker, 1988; Anderson et al., 1996). Intraperitoneal injection is approximately 104 times more efficient, and i.c. inoculation is approximately 109 times
more efficient than oral ingestion (Prusiner, 2004). Intravenous infection requires a five- to sevenfold greater
dose compared with an i.c. inoculation (Brown et al.,
2001).
Meat and Bone Meal
Although there are different hypotheses concerning
the origins of BSE, clearly the BSE infective agent
spread through the cattle population via ruminant feeding of rendered ruminant MBM. Meat and bone meal is
the rendered product from mammal tissues, including
1466
Novakofski et al.
bone, exclusive of any added blood, hair, hoof, horn,
hide, manure, stomach, and ruminal contents. It must
contain a minimum of 4.0% P, and the Ca level must
not exceed 2.2 times the P level. (AAFCO, 2004; CFIA,
2004). Rendering methods are not capable of fully inactivating PrPsc (Taylor et al., 1997), and it is plausible
that changes in the rendering industry concurrent with
a rare event of scrapie adaptation to a bovine host resulted in the BSE epidemic. Specifically, a shift from
batch rendering to continuous processing, elimination
of solvent extraction with steam stripping, or a reduction in rendering temperature may have resulted in
infectious MBM (Taylor et al., 1995b; Anderson et al.,
1996). Before 1970, high-temperature batch rendering
(of sheep carcasses) was the predominant practice in
the United Kingdom. In the mid-1970s, rendering temperature was lowered, and then, during the late 1970s
and early 1980s, solvent extraction, typically applied
to “greaves” after the primary cooking process, was
abandoned on a large scale by the United Kingdom’s
rendering industry (Wilesmith et al., 1992; Taylor et
al., 1997; Figure 3). This may have resulted in lower
inactivation of the infectious PrPsc, allowing it to concentrate through recycling of infected animals in MBM
(Phillips et al., 2000; Horn et al., 2001).
The U.S. Food Safety and Inspection Service (FSIS)
has amended meat inspection regulations to designate
the brain, skull, eyes, trigeminal ganglia, spinal cord,
vertebral column (excluding the vertebrae of the tail,
the transverse processes of the thoracic and lumbar
vertebrae, and the wings of the sacrum), and dorsal
root ganglia of cattle 30 mo of age and older, and the
tonsils and distal ileum of the small intestine of all
cattle, as specified risk materials, which are inedible
and prohibited for human food (USDA, 2004a). Specific
methods for disposition are not prescribed by FSIS,
although processors must outline methods in their
HACCP plans. In addition, protein derived from mammalian tissues, with some exceptions, such as blood,
milk, or any product composed entirely of porcine or
equine protein, must be excluded from ruminant feed
(Code of Federal Regulations, 2000).
Stunning
It has been shown that muscle foods may become
contaminated with CNS tissue or other high-risk tissue
during the slaughter or processing of cattle. Contamination can result from stunning with pneumatic or cartridge-fired penetrating bolt devices, advanced meatrecovery systems (mechanically deboned meat), inadvertent inclusion of nervous tissue during fabrication,
or from contaminated equipment during fabrication
(Kelley et al., 2000; Daly et al., 2002; USDA, 2004c).
Air injection stunners fragment CNS tissues, which can
enter the circulatory system as emboli and lodge in the
capillaries of peripheral tissues, such as lung and heart,
which are heavily vascularized (Schmidt et al., 1999;
Horlacher et al., 2002). Experiments suggest captive-
bolt stunning could distribute CNS material into lymph
nodes, spleen, liver, kidney, and muscle (Daly et al.,
2002). Stunning also has been shown to increase CNS
content of jugular blood samples (Love et al., 2000).
Central nervous system tissue has been detected at low
levels (g/g) on subprimal cuts, in ground meat, and in
tissue derived from advanced meat-recovery systems in
U.S. commercial plants (Schmidt et al., 2001). Nervous
tissue also has been detected in whole muscle cuts from
cattle harvested in European Union commercial facilities (Prendergast et al., 2004). Emboli containing CNS
tissue were detected in just under 1% of slaughtered
cattle (Lucker et al., 2002). Therefore, FSIS prohibited
the use of penetrating captive-bolt stunning devices
that inject air into the cranial cavity (USDA, 2004b).
The sawdust from carcass splitting clearly has the
potential to distribute CNS tissue onto both equipment
and carcasses (Schmidt et al., 2001; Lucker, et al.,
2002). Distribution of CNS tissue onto operators and
equipment also may occur via other routes (Daly et al.,
2002). Removal of cervical vertebrae and spinal canal
from chuck bones before advanced meat-recovery-system processing decreases contamination with CNS tissue (Schmidt et al., 2001). Commercial kits that detect
CNS tissue at levels of 1% or more in meat products
(Hughson et al., 2003) will not be useful for detection
because research suggests levels present in meat from
commercial processing plants are in the range of 0.001%
(Schmidt et al., 2001).
Muscle Tissue
It is generally thought that transmission of prion
disease by muscle foods is unlikely (Table 3). Attempts
at experimental transmission by feeding muscle from
BSE-infected animals have failed (European Commission 2002a,b); however, ground meat products carry
some risk from inadvertent nervous tissue contamination during slaughter or processing. The use of mechanical deboning or advanced meat recovery systems to recover tissue from bones containing nervous tissue represents the greatest risk of transmission.
Like most tissues, muscle contains PrP at roughly 2
to 10% of the concentration in brain (Moudjou et al.,
2001). The PrP is likely produced by the muscle cells
rather than by other cellular components because cultured myoblasts and myotubes express PrP (Brown et
al., 1998). The presence of PrPsc in muscle is unusual.
Muscle containing PrPsc has been reported from i.c.infected transgenic mice that overexpress prnp, from
infected outbred wild-type mice (Bosque et al., 2002),
in orally infected Syrian hamsters (Thomzig et al., 2003;
2004), and in a subset of CJD patients (Glatzel et al.,
2003; Kovacs et al., 2004). Accumulation of PrPsc also
has been reported in the skeletal muscle of tongue from
hamsters following either intralingual or i.c. inoculation with transmissible mink encephalopathy (Mulcahy
et al., 2004). Several muscles (psoas major, supraspinalis, semimembranous) from genetically susceptible
BSE Biology
scrapie-infected sheep have tested positive for PrPsc
(Andreoletti et al., 2004). The PrPsc was located in muscle spindles, part of the neural reflex mechanism in
muscle. Immunodetectable PrPsc has not been found in
muscle from cattle, sheep, elk, or raccoons experimentally infected with scrapie, CWD, or mink encephalopathy by other groups (Hamir et al., 2004a). Furthermore,
in small trials, PrPsc has not been demonstrated in
muscle from BSE-infected cattle (European Commission, 2002b). Other investigators have failed to observe
PrPsc in muscle from either CJD or vCJD patients (Head
et al., 2004).
The issue of possible transmission from muscle foods
is a critical one and needs additional investigation to
define actual risk. Pathogenesis of PrPsc must be established specifically for a host species, PrPsc strain, and
infection route because extrapolation of results from
animal models and across interspecies combinations is
not necessarily predictive of transmission rates (Thomzig et al., 2003). To date, no infectivity has been detected
in skeletal muscle of food animals using bioassays, the
current gold standard for infectivity detection, although
this does not rule out detection of rare events in the
future.
Tallow and Gelatin
Edible tallow is rendered only from the tissue of
healthy bovine animals (Codex Alimentarius, 1998);
however, because tallow might be rendered from infected animals without clinical symptoms, there is reason for concern. The risk seems low because epidemiological studies have failed to connect tallow consumption with BSE occurrence (Wilesmith et al., 1988).
Moreover, rendering of BSE-spiked material produced
tallow that was not infectious, although the MBM produced from the same material was highly infectious
(Taylor et al., 1995b). Continued vigilance is warranted
in light of a report that inactivation of PrPsc by autoclaving is less effective in the presence of lipid (Appel et
al., 2001).
Two-thirds of the gelatin produced worldwide comes
from cattle by-products (Schrieber and Seybold, 1993).
The European Commission evaluated whether changes
in temperature, pH, and fat removal during the manufacturing process produce “safe” gelatin. Both acid and
alkaline processing methods to produce gelatin reduce
BSE infectivity by 102- to 103-fold (Grobben et al., 2003);
however, the European Commission (2002a) concluded
that the process could not be modified and still produce
a functional product. Because questions remain, the
FDA (1997) issued Guidance for Industry. The Sourcing
and Processing of Gelatin to Reduce the Potential Risk
Posed by Bovine Spongiform Encephalopathy (BSE) in
FDA-Regulated Products for Human Use to ensure the
safety of gelatin. The FDA stated that there is no scientific basis for banning the use of gelatin in consumer
products intended for oral consumption or cosmetic use,
even if the gelatin is derived from countries reporting
1467
cases of BSE. However, products regulated by the FDA
that are used for human injection, for use in or on the
eye, or implantation should not be manufactured from
gelatin produced using cattle from BSE-positive
countries.
Milk
Because infective PrPsc is found in the lymphoreticular system and lymphocytes are found in milk, it is
possible that milk may be infective. High concentrations of PrP have been found in the secretory vesicles
of epithelial cells in the digestive tract, suggesting this
also may occur in mammary gland epithelial cells (Fournier et al., 1998). Normal PrP is found in the mammary
gland (Horiuchi et al., 1995). Taylor et al. (1995a) demonstrated that mice receiving milk from BSE-infected
cows (orally, i.c., or intraperitoneally) showed no signs
of neurological disease, spongiform encephalopathy, or
other specific pathology after 2 yr. Colostrum, which
contains different proteins from milk, also might be
considered an increased risk. Human colostrum of a
CJD patient was able to transmit PrPsc to mice (Tamai
et al., 1992), although the presence of PrPsc in bovine
colostrum has not been reported. Milk, colostrum, and
mammary tissue are not currently considered to be infective (DEFRA, 2004b; Table 3). Nonetheless, this conclusion is based on relatively low sensitivity assays, so
investigations began in United Kingdom in 2000 to test
milk and milk components for the presence of PrPsc
using more sensitive methods (SEAC, 2002).
Blood
Experiments to demonstrate the infectivity in blood
from animals with prion disease have produced mixed
results. Not all infected animals exhibit detectable levels of PrPsc in the blood nor does blood from infected
animals consistently transmit disease (Ironside and
Head, 2003). Transmissible PrPsc has been demonstrated in blood and blood components from infected
mice, hamsters, guinea pigs, chimpanzees, and monkeys (Brown et al., 1998; 2001). Experiments examining
BSE infectivity from blood indicate a strong likelihood
that blood or blood components from some, but not all,
donor animals are infectious. Hunter et al. (2002) found
that sheep-adapted BSE was transmitted to host sheep
by transfusion of whole blood in 17% of exposed animals, whereas scrapie transmission by the same route
was approximately 19%. Llewelyn et al. (2004) found
that one of 48 people became infected after receiving a
transfusion from asymptomatic individuals who subsequently died with vCJD. The recipient developed vCJD
6.5 yr after transfusion and 3 yr after the donor developed symptoms. Infectivity of blood components increases with disease progression (Cervenakova et al.,
2003). Transmission in animal models shows that the
greatest infectivity is associated with blood cells and,
in lesser amounts, with plasma fractions (Brown et al.,
1468
Novakofski et al.
1998; Cervenakova et al., 2003). Both PrP and PrPsc
bind plasminogen (Fischer et al., 2000; Maissen et al.,
2001). The potential transmission of prion disease by
blood is clear, although the actual risk to humans under
routine medical circumstances is low (Table 3). The
FDA (2002) has issued a Guidance for Industry. Revised
Preventive Measures to Reduce the Possible Risk of
Transmission of Creutzfeldt-Jakob Disease (CJD) and
Variant Creutzfeldt-Jakob Disease (vCJD) by Blood and
Blood Products, which was intended to decrease the
risk of prion disease transmission by blood transfusion.
Urine
A protease-resistant form of PrP has been found in
urine of animals and humans infected with PrPsc
(Shaked et al., 2001). The protein found in urine, which
seems to differ from brain-derived PrPsc, was entitled
UPrPsc by the authors. The UPrPsc was found in the
urine of hamsters inoculated with scrapie, cattle with
BSE, and humans with CJD but was not found in the
urine of uninfected hamsters, cattle, or humans. The
UPrPsc was found in the urine as early as 17 d after
i.c. inoculation of hamsters with scrapie PrPsc and long
before clinical or pathological changes occurred. Clinical signs appeared at d 105; however, in contrast to the
original brain-derived PrPsc, the UPrPsc did not cause
overt clinical infection, although inoculated hamsters
did have UPrPsc in their urine, implying that UPrPsc
was transmitted and replicated within the new host.
Interspecies Transmission
There is overwhelming evidence that TSE can be
transmitted between species. This occurs primarily because there is a very high homology across all mammalian PrP protein sequences (Figure 4). Glycosylation
sites, disulfide bonds, and trans-membrane regions are
perfectly conserved in 26 mammalian species, spanning
all placental orders, whereas sequences responsible for
secondary structure, processing, and membrane attachment are well conserved (Krakauer et al., 1998; Van
Rheede et al., 2003). The conservation of PrP among
mammals implies that the protein has a useful role,
although its physiological function is unclear (Oesch et
al., 1991).
Interspecies infection is generally less efficient than
intraspecies transmission, which is reflected by a
higher LD50 or delay in onset of symptoms in the first
passage from a donor species to a new host species.
Transmission of BSE to mice requires 1,000 times more
infectivity than transmission to cattle (Bradley, 1999).
These differences are referred to as a “species barrier.”
However, subsequent passages within the new host species are usually more efficient, indicating PrPsc adaptation. Species barriers are dependent on several factors,
including PrP protein sequence, and the physiology of
the digestive tract, immune, and nervous systems. The
species barrier is most protective if the delay in onset
of symptoms approaches the normal life expectancy of
the host species. The likelihood of interspecies transmission may be very low or result in subclinical disease,
but it is unlikely there are absolute barriers between
mammalian species. There may be a substantial species
barrier, but it is important to recognize that the number
of documented species barriers is small, whereas the
demonstrated examples of interspecies transmission
are clearly increasing.
Bovine to Livestock Transmission
Experimental transmission of BSE to a variety of
animals, including pigs, sheep, and goats, has been
demonstrated (Table 1). Exposing pigs to BSE brain
homogenate by intracranial, i.v., and i.p. routes resulted in TSE in approximately 1 to 3 yr, whereas shortterm feeding of infected MBM did not result in disease
(Wells et al., 2003). Not all exposed pigs developed clinical disease, although preclinical histopathology suggested this was a function of a long incubation period
rather than complete resistance. Sheep succumb to experimental BSE challenge in approximately 500 d (Foster et al., 1993). This is similar to the time required for
intraspecies scrapie transmission between sheep where
no PrP sequence difference exists. Ovine PrP differs
from bovine PrP at several AA besides known polymorphic sites, which suggests that susceptibility is not entirely controlled by overall sequence similarity between
donor and recipient PrP. Sheep with the R171Q polymorphism are resistant but not immune to BSE (Jeffrey
et al., 2001; Houston et al., 2003). Moreover, human PrP
contains R171, indicating that this particular residue is
not an important locus for resistance.
Cervid to Livestock Transmission
Circumstantial evidence as well as experimental exposure data (Hamir et al., 2001) indicate the likelihood
of transmitting CWD to cattle or humans is low. Strain
typing indicates that PrPsc from BSE-infected cattle
and CWD-infected deer or elk are different (Stack et
al., 2004); however, interspecies transmission has been
experimentally demonstrated by CWD mule deer brain
tissue inoculation into cattle (Hamir et al, 2001). Similarly, CWD can be transmitted to racoons and ferrets.
Moreover, ferret-passaged CWD demonstrated an increased host range and pathogenicity. Ferret-adapted
CWD PrPsc was subsequently transmissible to hamsters and became more pathogenic with a shorter incubation period as the number of passages increased in
ferrets (Bartz et al., 1998). A comparison of in vitro PrP
conversion to PrPsc suggests the likelihood of cervid-tobovine transmission may be similar to that of bovineto-human transmission (Raymond et al., 2000). Conversion of PrP to PrPsc by cervid PrPsc is most efficient for
cervid PrP, moderate for ovine, and least efficient for
humans or bovine PrP (Raymond et al., 2000). In this
case, the number of cattle tested with CWD may be
BSE Biology
1469
Figure 4. Phylogeny of mature prion protein AA sequence. Amino acid sequences were obtained from the NCBI
(2004) protein database and the Swiss-Prot/TrEMBL (2004) Protein Knowledge database; comparisons were generated
in ClustalW and phylogenetic relationships visualized by PHYLIP (2004) using a bootstrap of 500. Branch points and
horizontal distances reflect amino acid dissimilarity. The break in link to Gallus gallus was added to size the tree to
printable space, as the genetic distance is more than 10 times any of the mammalian differences; fewer than half the
AA are conserved between sheep and chickens. The sheep, cattle, and human sequences included as 1 and 2 reflect
polymorphisms within each species.
too small to be conclusive. Interspecies transmission
of BSE occurs between bovine and human with PrP
sequences that are 88% identical, whereas bovine, porcine, and cervid PrP sequences are 94% identical (Figure 2). The greater sequence similarity of cervid to livestock species, which may share pasture space and ecologies, compared with the similarity between humans
and bovines, suggests the potential for CWD transmission livestock species. The threat that CWD poses was
formalized in a declaration by the U.S. Secretary of
Agriculture that “the possibility [that] CWD from deer
and elk could cause disease in humans or in domestic
livestock … is an emergency that threatens the livestock industry of this country” (Federal Register, 2001).
Livestock to Human Transmission
Transmission of BSE to humans as vCJD may never
be proven unequivocally. It is clear from TSE strain
analyses that BSE can be transmitted to humans, causing vCJD, which is distinct from sporadic CJD (Collinge
et al., 1996; Bruce et al., 1997; Hill et al., 1997). Of the
144 cases of vCJD reported, all the individuals that
have been genotyped were homozygous 129M, which
also is known to increase the risk for CJD. Evidence
from transgenic mice expressing human PrP (129V and/
or 129M) suggests that BSE may also be transmitted
to humans but manifest as sporadic CJD (Asante et al.,
2002; Wadsworth et al., 2004). Transmission of BSE to
1470
Novakofski et al.
humans must be very inefficient considering the number of vCJD cases relative to the number of exposures.
It also is likely that scrapie transmission to man is
extremely inefficient because scrapie is common in
sheep, whereas CJD, the most similar human TSE,
occurs in roughly one in one million individuals.
Decontamination of PrPsc
One of the most striking characteristics of PrPsc is
its resistance to inactivation by common cleaning and
sterilization techniques (Brown et al., 1982; Taylor,
1999). Contamination with PrPsc has two components;
PrPsc in organic material, and PrPsc that is tightly
bound to surfaces such as stainless steel and plastic.
Cleaning with conventional detergents removes organic
material, and any associated PrPsc, which would otherwise inactivate or dilute sterilizing agents. Therefore,
cleaning before disinfection can result in greater decontamination, even though cleaning agents do not inactivate remaining PrPsc. Inactivation of PrPsc has not been
accomplished by treatment with detergents, formaldehyde, hydrogen peroxide, ethanol, ether, or acetone (Rutala and Weber, 2001).
Decontamination of PrPsc requires a one log reduction
greater than the LD50. For example, if the concentration
of PrPsc in infectious brain is 106 i.c. LD50 per gram,
a 107, or seven-log reduction of infectivity would be
required for safe disinfection. Treatment with chlorine
at 1,000 to 5,000 ppm (full strength commercial bleach)
for 30 min produces less than a four-log reduction in
infectivity (Brown et al., 1982). Longer exposure produces minimal improvement in decontamination,
whereas higher levels of chlorine (10,000 ppm) result
in greater than a four-log reduction (Kimberlin et al.,
1983; Rutala and Weber, 2001). Standard steam sterilization (121°C for 15 min) results in less than a threelog reduction. To effect a greater than three-log reduction requires autoclaving at 121°C or higher for more
than 30 min (Kimberlin et al., 1983; Taguchi et al.,
1991). Treatment with sodium hydroxide (1 N) for 15
to 30 min is effective at decontamination (Rutala and
Weber, 2001). High concentrations of phenol or guanidine thiocyanate also inactivate PrPsc (Ernst and Race,
1993; Manuelidis, 1997). If conventional cleaning before
disinfection can produce a four-log reduction, subsequent sterilization must produce an additional threelog reduction to produce minimally adequate decontamination of high-risk material.
Metal Surfaces
Binding of PrPsc to surgical instruments is believed
to be a major source of iatrogenic transmission of CJD
(WHO, 1999). The PrPsc binds to surfaces (metal and
plastic) without losing infectivity (Weissmann et al.,
2002). Instruments that have undergone multiple cycles of cleaning and sterilization have demonstrably
resulted in cases of iatrogenic CJD (Weissmann et al.,
2002). Stainless-steel-bound PrPsc may be more infective than soluble PrPsc. Stainless-steel wire (0.5 × 5
mm) exposed to PrPsc retained 105 LD50 infective units
and required only 5 min of i.c. exposure to cause infection (Flechsig et al., 2001). Metal-bound PrPsc seems
particularly resistant to decontamination. Using a bioassay, wire contaminated with PrPsc remained infectious after cleaning with sodium hydroxide and autoclaving, conditions that would have inactivated PrPsc
in tissue (Yan et al., 2004).
Environmental Persistence
It is apparent that PrPsc is extremely resistant to
degradation and that environmental reservoirs capable
of transmission exist; however, because laboratory detection of infectious PrPsc at low levels is very difficult,
there is little additional experimental information
about environmental persistence of infectious PrPsc.
The first published report of PrPsc persistence is that
of Brown and Gajdusek (1991), showing that scrapieinfected fluid, mixed with soil, packed into perforated
Petri dishes, and embedded within soil-containing pots,
retained infectivity after being buried for 3 yr. Anecdotal evidence suggests that environmental decontamination is exceedingly difficult. Facilities that have been
exposed to scrapie remain infectious for long periods,
and there are no published reports describing successful decontamination of a facility infected with CWD.
Miller et al. (2004) recently demonstrated CWD infection from pens contaminated by carcasses or fecal material. Published risk assessments for environmental
transmission of scrapie and BSE, either within or between species, are based almost exclusively on assumptions because little experimental data are available
(Ridley and Baker, 1999; Cummins et al., 2002).
Implications
Most mammalian species are susceptible to transmissible spongiform encephalopathies caused by aberrant
prions. Transmission between animals and species occurs via exposure to infected tissue or residue. In all
cases, infectivity is concentrated in nervous tissue,
which has been banned from the food and feed supply.
Interspecies transmission is rare, but like intraspecies
transmission, it is more likely to occur between genetically susceptible individuals. Although bovine spongiform encephalopathy may have arisen from interspecies transmission of scrapie, increasing evidence suggests that it continues to arise spontaneously, and
continued surveillance will eventually detect these
cases as well. Similarly, strain phenotype is largely
maintained with passage through different species,
suggesting that many of the multiple strains of transmissible spongiform encephalopathies in domestic animals (>20 scrapie, >3 bovine spongiform encephalopathy) probably arose independently. Together, these observations indicate that transmissible spongiform
BSE Biology
encephalopathies cannot be eradicated. Considering
the low probability of interspecies transmission and the
low prion levels in nonnervous tissues, consumption
of conventional animals products represents minimum
risk to humans.
Literature Cited
AAFFCO. 2004. Official Publication. Assoc. Am. Feed Control Officials, Oxford, IN.
Aguzzi, A. 2003. Prions and the immune system: A journey through
gut, spleen, and nerves. Adv. Immunol. 81:123–171.
Aguzzi, A., and G. Miele. 2004. Recent advances in prion biology.
Curr. Opin. Neurol. 17:337–342.
Anderson, R. M., C. A. Donnelly, N. M. Ferguson, M. E. J. Woolhouse,
C. J. Whatt, H. J. Udy, S. MaWhinney, S. P. Dunstan, T. R. E.
Southwood, J. W. Wilesmith, J. B. M. Ryan, L. J. Hoinville, J.
E. Hillerton, A. R. Austin, and G. A. H. Wells. 1996. Transmission
dynamics and epidemiology of BSE in British cattle. Nature
382:779–788.
Andreoletti, O., C. Lacroux, A. Chabert, L. Monnereau, G. Tabouret,
F. Lantier, P. Berthon, F. Eychenne, S. Lafond-Benestad, J. M.
Elsen, and F. Schelcher. 2002. PrP(Sc) accumulation in placentas
of ewes exposed to natural scrapie: Influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J. Gen. Virol. (Pt.
10) 83:2607–2616.
Andreoletti, O., S. Simon, C. Lacroux, N. Morel, G. Tabouret, A.
Chabert, S. Lugan, F. Corbiere P. Ferre, G. Foucras, H. Laude,
F. Eychenne, J. Grassi, and F. Schelcher. 2004. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat.
Med. 10:591–593.
Appel, T., M. Wolff, F. von Rheinbaben, M. Heinzel, and D. Riesner.
2001. Heat stability of prion rods and recombinant prion protein
in water, lipid and lipid-water mixtures. J. Gen. Virol. (Pt 2)
82:465–473.
Asante, E. A., J. M. Linehan, M. Desbruslais, S. Joiner, I. Gowland,
A. L. Wood, J. Welch, A. F. Hill, S. E. Lloyd, J. D. Wadsworth,
and J. Collinge. 2002. BSE prions propagate as either variant
CJD-like or sporadic CJD-like prion strains in transgenic mice
expressing human prion protein. EMBO J. 21:6358–6366.
Ball, K. 2002. Chronic wasting disease in a Rocky Mountain elk. Can.
Vet. J. 43:880–882.
Baron, T. G., and A. G. Biacabe, 2001. Molecular analysis of the
abnormal prion protein during co-infection of mice with a bovine
encephalopathy and a scrapie agent. J. Virol. 75:107–114.
Barret, A., F. Tagliavini, G. Forloni, C. Bate, M. Salmona, L. Colombo,
A. De Luigi, L. Limido, S. Suardi, G. Rossi, F. Auvre, K. T. Adjou,
N. Sales, A. Williams, C. Lasmezas, and J. P. Deslys. 2003.
Evaluation of quinacrine treatment for prion diseases. J. Virol.
77:8462–8469.
Bartz, J. C., A. E. Kincaid, and R. A. Bessen. 2002. Retrograde transport of transmissible mink encephalopathy within descending
motor tracts. J. Virol. 76:5759–5768.
Bartz, J. C., A. E. Kincaid, and R. A. Bessen. 2003. Rapid prion
neuroinvasion following tongue infection. J. Virol. 77:583–591.
Bartz, J. C., R. F. March, D. I. McKenzie, and J. M. Aiken. 1998. The
host range of chronic wasting disease is altered on passage in
ferrets. J. Virol. 251:297–301.
Baskakov, I. V., G. Legname, Z. Gryczynski, and S. B. Prusiner. 2004.
The peculiar nature of unfolding of the human prion protein.
Protein Sci. 13:586–595.
Beringue, V., G. Mallinson, M. Kaisar, M. Tayebi, Z. Sattar, G. Jackson, D. Anstee, J. Collinge and S. Hawke. 2003. Regional heterogeneity of cellular prion protein isoforms in the mouse brain.
Brain 126:2065–2073.
Bessen, R. A., D. A. Kocisko, G. J. Raymond, S. Nandan, P. T. Lansbury, and B. Caughey. 1995. Non-genetic propagation of strainspecific properties of scrapie prion protein. Nature 375:698–700.
1471
Biacabe, A. G., J. L. Laplanche, S. Ryder, and T. G. Baron. 2004.
Distinct molecular phenotypes in bovine prion diseases. EMBO
Rep. 5(1):110–115.
Bolton, D. C., S. J. Seligman, G. Bablanian, D. Windsor, L. J. Scala,
K. S. Kim, C. M. Chen, R. J. Kascsak, and P. E. Bendheim. 1991.
Molecular location of a species-specific epitope on the hamster
scrapie agent protein. J. Virol. 65:3667–3675.
Bosque, P. J., C. Ryou, G. Telling, D. Peretz, G. Legname, S. J.
DeArmond, and S. B. Prusiner. 2002. Prions in skeletal muscle.
Proc. Natl. Acad. Sci. USA 99:3812–3817.
Bradley, R. 1999. BSE transmission studies with particular reference
to blood. Dev. Biol. Stand. 99:35–40.
Brown, P., and D. C. Gajdusek. 1991. Survival of scrapie virus after
3 years’ interment. Lancet 337:269–270.
Brown, P., R. G. Rohwer, E. M. Green, and D. C. Gajdusek. 1982.
Effect of chemicals, heat, and histopathologic processing on highinfectivity hamster-adapted scrapie virus. J. Infect. Dis.
145:683–687.
Brown, P., C. J. Gibbs, Jr., P. Rodgers-Johnson, D. M. Asher, M. P.
Sulima, A. Bacote, L. G. Goldfarb, and D. C. Gajdusek. 1994.
Human spongiform encephalopathy: The National Institutes of
Health series of 300 cases of experimentally transmitted disease.
Ann. Neurol. 35:513–529.
Brown, D. R., K. Qin, J. W. Herms, A. Madlung, J. Manson, R. Strome,
P. E. Fraser, T. Kruck, A. von Bohlen, W. Schulz-Schaeffer, A.
Giese, D. Westaway, and H. Kretzschmar. 1997. The cellular
prion protein binds copper in vivo. Nature 390:684–687.
Brown, P., R. G. Rohwer, B. C. Dunstan, C. MacAuley, D. C. Gajdusek,
and W. N. Drohan. 1998. The distribution of infectivity in blood
components and plasma derivatives in experimental models of
transmissible
spongiform
encephalopathy.
Transfusion
38:810–816.
Brown, P., M. Preece, J. P. Brandel, T. Sato, L. McShane, I. Zerr, A.
Fletcher, R. G. Will, M. Pocchiari, N. R. Cashman, J. H. d’Aignaux, L. Cervenakova, J. Fradkin, L. B. Schonberger, and S. J.
Collins. 2000. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 55:1075–1081.
Brown P., L. Cervenakova, and H. Diringer. 2001. Blood infectivity
and the prospects for a diagnostic screening test in CreutzfeldtJakob disease. J. Lab. Clin. Med. 137:5–13.
Bruce, M. E. 2003. TSE strain variation. Br. Med. Bull. 66:99–108.
Bruce, M., A. Chree, I. McConnell, J. Foster, G. Pearson, and H.
Fraser. 1994. Transmission of bovine spongiform encephalopathy and scrapie to mice: Strain variation and the species barrier.
Philos. Trans. R. Soc. Lond. (B. Biol. Sci.) 343:405–411.
Bruce, M. E., R. G. Will, J. W. Ironside, I. McConnell, D. Drummond,
A. Suttie, L. McCardle, A. Chree, J. Hope, C. Birkett, S. Cousens,
H. Fraser, and C. J. Bostock. 1997. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature
389:498–501.
Casalone, C., G. Zanusso, P. Acutis, S. Ferrari, L. Capucci, F. Tagliavini, S. Monaco, and M. Caramelli. 2004. Identification of a
second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc.
Natl. Acad. Sci. USA 101:3065–3070.
Castilla, J., A. Gutierrez-Adan, A. Brun, B. Pintado, B. Parra, M. A.
Ramirez, F. J. Salguero, F. Diaz San Segundo, A. Rabano, M.
J. Cano, and J. M. Torres. 2004. Different behavior toward bovine
spongiform encephalopathy infection of bovine prion protein
transgenic mice with one extra repeat octapeptide insert mutation. J. Neurosci. 24:2156–2164.
Caughey, B., K. Brown, G. J. Raymond, G. E. Katzenstein, and W.
Thresher. 1994. Binding of the protease-sensitive form of PrP
(prion protein) to sulfated glycosaminoglycan and congo red. J.
Virol. 68:2135–2141.
Caughey, B., and R. E. Race. 1992. Potent inhibition of scrapie-associated PrP accumulation by congo red. J. Neurochem. 59:768–771.
Caughey, W. S., L. D. Raymond, M. Horiuchi, and B. Caughey. 1998.
Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. USA
95:12117–12122.
1472
Novakofski et al.
Cervenakova., L., O. Yakovleva, C. McKenzie, S. Kolchinsky, L.
McShane, W. N. Drohan, and P. Brown. 2003. Similar levels of
infectivity in the blood of mice infected with human-derived
vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43:1687–1694.
CFIA. 2004. Approved feed ingredients Schedule IV, Part 1, 5.1.7.
Canadian Food Inspection Agency. Available: http://www.
inspection.gc.ca/english/anima/feebet/feebete.shtml. Accessed
Nov. 4, 2004.
Chandler, R. L. 1963. Experimental scrapie in the mouse. Res. Vet.
Sci. 4:269–275.
Chatelain, J., N. Delasnerie-Laupretre, F. Cathala, and P. Brown.
1983. Scrapie in France: Some possible predisposing factors in
the naturally-acquired disease of sheep. Vet. Microbiol. 8:511–
515.
Chiesa, R., and D. A. Harris. 2001. Prion diseases: What is the neurotoxic molecule? Neurobiol. Dis. 8:743–763.
Cockcroft, P. D. 2004. The similarity of the physical sign frequencies
of bovine spongiform encephalopathy and selected differential
diagnoses. Vet. J. 167:175–180.
Code of Federal Regulations. 2000. Substances Prohibited from Use
in Animal Food or Feed. Title 21, Vol. 6, Parts 589, 523–526.
U.S. Government Printing Office, Washington, DC.
Codex Alimentarius. 1998. Food Labelling Complete Texts. Current
Official Standards. Codex Standard 211, Part 2.4.1. FAO, WHO,
United Nations, Geneva, Switzerland.
Collinge, J., and M. S. Palmer. 1994. Molecular genetics of human
prion diseases. Phil. Trans. R. Soc. Lond. Biol. Sci. 343:371–378.
Collinge, J., M. S. Palmer, and A. J. Dryden. 1991. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 337:1441–
1442.
Collinge, J., M. A. Whittington, K. S. Sidle, C. J. Smith, M. S. Palmer,
A. R. Clarke, and J. G. Jefferys. 1994. Prion protein is necessary
for normal synaptic function. Nature 370:295–297.
Collinge, J., K. C. Sidle, J. Meads, J. Ironside, A. F. Hill. 1996. Molecular analysis of prion strain variation and the aetiology of ‘new
variant’ CJD. Nature 383:685–690.
Croes, E. A., J. Theuns, J. J. Houwing-Duistermaat, B. Dermaut, K.
Sleegers, G. Roks, M. Van Den Broeck, B. Van Harten, J. C.
Van Swieten, M. Cruts, C. Van Broeckhoven, and C. M. Van
Duijn. 2004. Octapeptide repeat insertions in the prion protein
gene and early onset dementia. J. Neurol. Neurosurg. Psychiatr.
75:1166–1170.
Cummins, E. J., P. M. Grace, D. J. Fry, K. P. McDonnell, S. F. Colgan,
and S. M. Ward. 2002. Quantitative exposure assessment for
the combustion of meat and bone meal derived from specified
risk material in the context of BSE in Ireland. J. Agric. Saf.
Health 8:365–383.
Daly, D. J., D. M. Prendergast, J. J. Sheridan, I. S. Blair, and D. A.
McDowell. 2002. Use of a marker organism to model the spread
of central nervous system tissue in cattle and the abattoir environment during commercial stunning and carcass dressing.
Appl. Environ. Microbiol. 68:791–798.
DeArmond, S. J., H. Sanchez, F. Yehiely, Y. Qiu, A. Ninchak-Casey,
V. Daggett, A. P. Camerino, J. Cayetano, M. Rogers, D. Groth,
M. Torchia, P. Tremblay, M. R. Scott, F. E. Cohen, and S. B.
Prusiner. 1997. Selective neuronal targeting in prion disease.
Neuron 19:1337–1348.
DEFRA. 2000. The BSE inquiry: The Report. House of Commons
papers 1999–2000. Her Majesty’s Stationary Office, London.
Available: http://www.bseinquiry.gov.uk/report/index.html. Accessed Nov. 4, 2004.
DEFRA. 2004a. Animal Health and Welfare, Bovine Spongiform Encephalopathy of BSE. U.K. Dept. Environ. Food and Rural Affairs. Available: http://www.defra.gov.uk/animalh/bse/index.
html. Accessed Nov. 4, 2004.
DEFRA. 2004b. Animal Health and Welfare, Bovine Spongiform Encephalopathy, Science and Research, Epidemiology of BSE. U.K.
Dept. Environ. Food and Rural Affairs. Available: http://www.
defra.gov.uk/animalh/bse/science-research/epidem.html.
Accessed Nov. 4, 2004.
DEFRA. 2004c. Animal Health and Welfare, Bovine Spongiform Encephalopathy, Science and Research, Transmission of BSE. U.K.
Dept. Environ. Food and Rural Affairs. Available: http://www.
defra.gov.uk/animalh/bse/science-research/transmis.html. Accessed Nov. 4, 2004.
Doh-ura, K., T. Iwaki, and B. Caughey. 2000. Lysosomotropic agents
and cysteine protease inhibitors inhibit scrapie-associated prion
protein accumulation. J. Virol. 74:4894–4897.
Donnelly, C. A., N. M. Ferguson, A. C. Ghani, J. W. Wilesmith, R.
M. Anderson. 1997. Analysis of dam-calf pairs of BSE cases:
Confirmation of a maternal risk enhancement. Proc. R. Soc.
Lond. (B. Biol. Sci.) 264:1647–1656.
Edenhofer, F., R. Rieger, M. Famulok, W. Wendler, S. Weiss, E. L.
Winnacker. 1996. Prion protein PrPc interacts with molecular
chaperones of the Hsp60 family. J. Virol. 70:4724–4728.
Ernst, D. R., and R. E. Race. 1993. Comparative analysis of scrapie
agent inactivation methods. J. Virol. Methods 41:193–201.
European Commission. 2002a. Opinion on TSE infectivity distribution in ruminant tissues. EC Scientific Steering Committee.
Available:
http://www.europa.eu.int/comm/food/fs/sc/ssc/
outcome_en.html. Accessed Nov. 4, 2004.
European Commission. 2002b. Prions in Muscle. Statement Adopted
by the Scientific Steering Committee. 4–5 April. EC Health and
Consumer Protection Directorate. Available: www.europa.eu.
int/comm/food/fs/sc/ssc/out254_en.pdf. Accessed Nov. 4, 2004.
FDA. 1997. Guidance for Industry. The sourcing and processing of
gelatin to reduce the potential risk posed by bovine spongiform
encephalopathy (BSE) in FDA-regulated products for human
use.
Available:
http://www.fda.gov/opacom/morechoices/
industry/guidance/gelguide.htm. Accessed Nov. 4, 2004.
FDA. 2002. Guidance for Industry. Revised Preventive Measures to
Reduce the Possible Risk of Transmission of Creutzfeldt-Jakob
Disease (CJD) and Variant Creutzfeldt-Jakob Disease (vCJD) by
Blood and Blood Products. Available: http://www.fda.gov/cber/
gdlns/cjdvcjd.pdf. Accessed Nov. 4, 2004.
Federal Register. 2001. Declaration of Emergency Because of Chronic
Wasting Disease. Docket No. 01-019-1. Fed. Regis. 66:49342–
49343.
Fischer, M. B., C. Roeckl, P. Parizek, H. P. Schwarz, and A. Aguzzi.
2000. Binding of disease-associated prion protein to plasminogen. Nature 408:479–483.
Flechsig, E., I. Hegyi, M. Enari, P. Schwarz, J. Collinge, and C.
Weissmann. 2001. Transmission of scrapie by steel-surfacebound prions. Mol. Med. 7:679–684.
Forloni, G., S. Iussich, T. Awan, L. Colombo, N. Angeretti, L. Girola,
I. Bertani, G. Poli, M. Caramelli, M. Grazia Bruzzone, L. Farina,
L. Limido, G. Rossi, G. Giaccone, J. W. Ironside, O. Bugiani, M.
Salmona, F. Tagliavini. 2002. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. USA 99:10849–10854.
Foster J. D., J. Hope, and H. Fraser. 1993. Transmission of bovine
spongiform encephalopathy to sheep and goats. Vet. Rec.
133:339–441.
Foster, J., W. McKelvey, H. Fraser, A. Chong, A. Ross, D. Parnham,
W. Goldmann, and N. Hunter. 1999. Experimentally induced
bovine spongiform encephalopathy did not transmit via goat
embryos. J. Gen. Virol. (Pt. 2) 80:517–524.
Fournier, J. G., F. Escaig-Haye, T. Billette de Villemeur, O. Robain,
C. I. Lasmezas, J. P. Deslys, D. Dormont, and P. Brown. 1998.
Distribution and submicroscopic immunogold localization of cellular prion protein (PrPc) in extracerebral tissues. Cell Tissue
Res. 292:77–84.
Gambetti, P., P. Parchi, and S. G. Chen. 2003. Hereditary CreutzfeldtJakob disease and fatal familial insomnia. Clin. Lab. Med.
23:43–64.
Gibbs, Jr., C. J., H. L. Amyx, A. Bacoate, C. I. Masters, and D. C.
Gajdusek. 1980. Oral transmission of Kuru, Creutzfeldt-Jakob
Disease, and Scrapie to non-human primates. J. Infect. Dis.
142:205–208.
Goldfarb, L. G., R. B. Petersen, M. Tabaton, P. Brown, A. C. LeBlanc,
P. Montagna, P. Cortelli, J. Julien, C. Vital C, and W. W. Pendelbury. 1992. Fatal familial insomnia and familial Creutzfeldt-
BSE Biology
Jakob disease: Disease phenotype determined by a DNA polymorphism. Science 258:806–808.
Goldmann, W., N. Hunter, G. Benson, J. D. Foster, and J. Hope.
1991. Different forms of the bovine PrP gene have five or six
copies of a short, G-C-rich element within the protein-coding
exon. J. Gen. Virol. (Pt. 1) 72:201–204.
Goldmann, W., N. Hunter, G. Smith, J. Foster, and I. Hope. 1994.
PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host
of scrapie. J. Gen. Virol. 75(Pt 5):989–995.
Gossert, A. D., S. Bonjour, D. A. Lysek, F. Fiorito and K. Wuthrich.
2005. Prion protein NMR structures of elk and of mouse/elk
hybrids. Proc. Natl. Acad. Sci. USA 18:102:646–650.
Graner, E., A. F. Mercadante, S. M. Zanata, O. V. Forlenza, A. L.
Cabral, S. S. Veiga, M. A. Juliano, R. Roesler, R. Walz, A. Minetti,
I. Izquierdo, V. R. Martins, R. R. Brentani. 2000. Cellular prion
protein binds laminin and mediates neuritogenesis. Brain Res.
Mol. Brain Res. 76:85–92.
Grobben, A. H., P. J. Steele, R. A. Somerville, D. M. Taylor. 2003.
Inactivation of the bovine spongiform encephalopathy (BSE)
agent by the acid and alkaline processes for manufacturing of
bone gelatine. Biotechnol. Appl. Biochem. 39:329–338.
Hamir, A. N., R. C. Cutlip, J. M. Miller, E. S. Williams, M. J. Stack, M.
W. Miller, K. I. O’Rourke, and M. J. Chaplin. 2001. Preliminary
findings on the experimental transmission of chronic wasting
disease agent of mule deer to cattle. J. Vet. Diagn. Invest.
13:91–96.
Hamir, A. N., J. M. Miller and R. C. Cutlip. 2004a. Failure to detect
prion protein (PrPres) by immunohistochemistry in striated
muscle tissues of animals experimentally inoculated with agents
of transmissible spongiform encephalopathy. Vet. Pathol.
41:78–81.
Hamir, A. N., J. M. Miller, R. C. Cutlip, R. A. Kunkle, A. L. Jenny,
M. J. Stack, M. J. Chaplin, and J. A. Richt. 2004b. Transmission
of sheep scrapie to elk (Cervus elaphus nelsoni) by intracerebral
inoculation: Final outcome of the experiment. J. Vet. Diagn.
Invest. 16:316–321.
Head, M. W., D. Ritchie, N. Smith, V. McLoughlin, W. Nailon, S.
Samad, S. Masson, M. Bishop, L. McCardle, J. W. Ironside.
2004. Peripheral tissue involvement in sporadic, iatrogenic, and
variant Creutzfeldt-Jakob disease: An immunohistochemical,
quantitative, and biochemical study. Am. J. Pathol. 164:143–
153.
Heaton, M. P., K. A. Leymaster, B. A. Freking, D. A. Hawk, T. P.
Smith, J. W. Keele, W. M. Snelling, J. M. Fox, C. G. ChitkoMcKown, and W. W. Laegreid. 2003. Prion gene sequence variation within diverse groups of U.S. sheep, beef cattle, and deer.
Mamm. Genome 14:765–777.
Heikenwalder, M., N. Zeller, H. Seeger, M. Prinz, P. C. Klohn, P.
Schwarz, N. H. Ruddle, C. Weissmann and A. Aguzzi. 2005.
Chronic lymphocytic inflammation specifies the organ tropism
of prions. Science 307:1107–1110.
Huang, F. P., and G. G. MacPherson. 2004. Dendritic cells and oral
transmission of prion diseases. Adv. Drug. Deliv. Rev. 56:901–
913.
Hill, A. F., M. Desbruslais, S. Joiner, K. S. Sidle, I. Gowland, J.
Collinge, L. J. Doey, and P. Lantos. 1997. The same prion strain
causes vCJD and BSE. Nature 389:448–450, 526.
Hill, A. F., K. C. Sidle, S. Joiner, P. Keyes, T. C. Martin, M. Dawson,
and J. Collinge. 1998. Molecular screening of sheep for bovine
spongiform encephalopathy. Neurosci. Lett. 255:159–162.
Hoinville, L. J., J. W. Wilesmith and M. S. Richards. 1995. An investigation of risk factors for cases of bovine spongiform encephalopathy born after the introduction of the “feed ban.” Vet. Rec.
136:312–318.
Horiuchi, M., N. Yamazaki, T. Ikeda, N. Ishiguro, and M. Shinagawa.
1995. A cellular form of prion protein (PrPC) exists in many nonneuronal tissues of sheep. J. Gen. Virol. (Pt. 10) 76:2583–2587.
Horlacher. S., E. Lucker, E. Eigenbrodt, and S. Wenisch. 2002. Brain
emboli in the lungs of cattle. Berl. Munch. Tierarztl. Wochenschr.
115:1–5.
1473
Horn, G., M. Bobrow, M. Bruce, M. Goedert, A. McLean, and J. Webster. 2001. Review of the origin of BSE. Review prepared for the
UK Dept. Environ., Food and Rural Affairs. Available: http://
www.defra.gov.uk/animalh/bse/publications/bseorigin.pdf. Accessed Nov. 4, 2004.
Houston, F., W. Goldmann, A. Chong, M. Jeffrey, L. Gonzalez, J.
Foster, D. Parnham, and N. Hunter. 2003. Prion diseases: BSE
in sheep bred for resistance to infection. Nature 423:498.
Hughson, E., P. Reece, M. J. Dennis, and S. Oehlschlager. 2003.
Comparative evaluation of the performance of two commercial
kits for the detection of central nervous system tissue in meat.
Food Addit. Contam. 20:1034–1043.
Hunter, N. 2003. Scrapie and experimental BSE in sheep. Br. Med.
Bull. 66:171–183.
Hunter, N., W. Goldmann, E. Marshall, and G. O’Neill. 2000. Sheep
and goats: natural and experimental TSEs and factors influencing incidence of disease. Arch. Virol. Suppl. 16:181–188.
Hunter, N., J. Foster, A. Chong, S. McCutcheon, D. Parnham, S.
Eaton, C. MacKenzie and F. Houston. 2002. Transmission of
prion diseases by blood transfusion. J. Gen. Virol. 8311:2897–
2905.
Ironside. J. W., and M. W. Head. 2003. Variant Creutzfeldt-Jakob
disease and its transmission by blood. J. Thromb. Haemost.
1:1479–1486.
Jackson, G. S., I. Murray, L. L. Hosszu, N. Gibbs, J. P. Waltho, A.
R. Clarke, and J. Collinge. 2001. Location and properties of
metal-binding sites on the human prion protein. Proc. Natl.
Acad. Sci. USA 98:8531–8535.
Jeffrey, M., S. Ryder, S. Martin, S. A. Hawkins, L. Terry, C. BerthelinBaker, and S. J. Bellworthy. 2001. Oral inoculation of sheep
with the agent of bovine spongiform encephalopathy (BSE). 1.
Onset and distribution of disease-specific PrP accumulation in
brain and viscera. J. Comp. Pathol. 124:280–289.
Johnson, C., J. Johnson, M. Clayton, D. McKenzie and J. Aiken. 2003.
Prion protein gene heterogeneity in free-ranging white-tailed
deer within the chronic wasting disease affected region of Wisconsin. J. Wildlife Dis. 39:576–581.
Kang, S. C., D. R. Brown, M. Whiteman, R. Li, T. Pan, G. Perry,
T. Wisniewski, M. S. Sy, B. S. Wong. 2004. Prion protein is
ubiquitinated after developing protease resistance in the brains
of scrapie-infected mice. J. Pathol. 203:603–608.
Kelley, L. C., S. Hafner, P. C. McCaskey, M. T. Sutton, and K. A.
Langheinrich. 2000. An evaluation of methods for the detection
of spinal cord in product derived from advanced meat recovery
systems. J. Food Prot. 63:1107–1112.
Kimberlin, R. H., and C. A. Walker. 1988. Scrapie. Ciba Foundation
Symposium 135, 37–88.
Kimberlin, R. H., and C. A. Walker. 1989. The role of the spleen in
the neuroinvasion of scrapie in mice. Virus Res. 12:201–211.
Kimberlin, R. H., C. A. Walker, G. C. Millson, D. M. Taylor, P. A.
Robertson, A. H. Tomlinson, A. G. Dickinson. 1983. Disinfection
studies with two strains of mouse-passaged scrapie agent. Guidelines for Creutzfeldt-Jakob and related agents. J. Neurol. Sci.
59:355–369.
Kirkwood, J. K., and A. A. Cunningham. 1994. Epidemiological observations on spongiform encephalopathies in captive wild animals
in the British Isles. Vet. Rec. 135:296–303.
Kiyotoshi, K., L. Zulianello, M. Scott, C. M. Cooper, A. C. Wallace,
T. L. James, F. E. Cohen and S. B. Prusiner. 1997. Evidence for
protein X binding to a discontinuous epitope on the cellular prion
protein during scrapie prion propagation. Proc. Natl. Acad. Sci.
USA 94:10069–10074.
Klein, M. A., R. Frigg, E. Flechsig, A. J. Raeber, U. Kalinke, H.
Bluethmann, F. Bootz, M. Suter, R. M. Zinkernagel, and A.
Aguzzi. 1997. A crucial role for B cells in neuroinvasive scrapie.
Nature 390:687–690.
Klein, M. A., R. Frigg, A. J. Raeber, E. Flechsig, I. Hegyi, R. M.
Zinkernagel, C. Weissmann, and A. Aguzzi. 1998. PrP expression
in B lymphocytes is not required for prion neuroinvasion. Nat.
Med. 4:1429–1433.
1474
Novakofski et al.
Kovacs, G. G., E. Lindeck-Pozza, L. Chimelli, A. Q. Araujo, A. A.
Gabbai, T. Strobel, M. Glatzel, A. Aguzzi, and H. Budka. 2004.
Creutzfeldt-Jakob disease and inclusion body myositis: Abundant disease-associated prion protein in muscle. Ann. Neurol.
55:121–125.
Krakauer, D. C., P. M. Zanotto, and M. Pagel. 1998. Prion’s progress:
Patterns and rates of molecular evolution in relation to spongiform disease. J. Mol. Evol. 47:133–145.
Kübler, E., B. Oesch, and A. J. Raeber. 2003. Diagnosis of prion
diseases. Br. Med. Bull. 66:267–279.
Kurschner, C., and J. L. Morgan. 1995. The cellular prion protein
(PrP) selectively binds to Bcl-2 in the yeast two-hybrid system.
Brain Res. Mol. Brain Res. 30:165–168.
Legname, G., I. V. Baskakov, H. O. Nguyen, D. Riesner, F. E. Cohen,
S. J. DeArmond and S. B. Prusiner. 2004. Synthetic mammalian
prions. Science 305:673–676.
Lehmann, S. 2002. Metal ions and prion diseases. Curr. Opin. Chem.
Biol. 6:187–192.
Lehmann, S., and D. A. Harris. 1996. Two mutant prion proteins
expressed in cultured cells acquire biochemical properties reminiscent of the scrapie isoform. Proc. Natl. Acad. Sci. USA
93:5610–5614.
Llewelyn, C. A., P. E. Hewitt, R. S. G. Knight, K. Amar, S. Cousens,
J. Mackenzie, and R. G. Will. 2004. Possible transmission of
variant Creutzfeldt-Jakob disease by blood transfusion. Lancet
363:417–421.
Lloyd, S. E., J. M. Linehan, M. Desbruslais, S. Joiner, J. Buckell, S.
Brandner, J. D. Wadsworth and J. Collinge. 2004 J. Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J. Gen. Virol.
85:2471–2478.
Love, S., C. R. Helps, S. Williams, A. Shand, J. L. McKinstry, S. N.
Brown, D. A. Harbour, and M. M. Anil. 2000. Methods for detection of haematogenous dissemination of brain tissue after stunning of cattle with captive bolt guns. J. Neurosci. Methods
99:53–58.
Lucker, E., B. Schlottermuller, and A. Martin. 2002. Studies on contamination of beef with tissues of the central nervous system
(CNS) as pertaining to slaughtering technology and human BSEexposure risk. Berl. Munch. Tierarztl. Wochenschr. 115:118–
121.
Ma, J., R. Wollmann, and S. Lindquist. 2002. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science
298:1781–1785.
Mabbott, N. A., and M. E. Bruce. 2003. Prion disease: Bridging the
spleen-nerve gap. Nat. Med. 9:1463–1464.
Maissen, M., C. Roeckl, M. Glatzel, W. Goldmann, and A. Aguzzi.
2001. Plasminogen binds to disease-associated prion protein of
multiple species. Lancet 357:2026–2028.
Manuelidis, L. 1997. Decontamination of Creutzfeldt-Jakob disease
and other transmissible agents. J. Neurovirol. 3:62–65.
May, B. C., A. T. Fafarman, S. B. Hong, M. Rogers, L. W. Deady, S.
B. Prusiner, and F. E. Cohen. 2003. Potent inhibition of scrapie
prion replication in cultured cells by bis-acridines. Proc. Natl.
Acad. Sci. USA 100:3416–3421.
Middleton, D. J., and R. M. Barlow. 1993. Failure to transmit bovine
spongiform encephalopathy to mice by feeding them with extraneural tissues of affected cattle. Vet. Rec. 132:545–547.
Miller, M. W., M. A. Wild, and E. S. Williams. 1998. Epidemiology
of chronic wasting disease in captive Rocky Mountain elk. J.
Wildlife Dis. 34:532–538.
Miller, M. W., and E. S. Williams. 2003. Prion disease: Horizontal
prion transmission in muledeer. Nature 425:35–36.
Miller, M. W., and E. S. Williams. 2004. Chronic wasting disease of
cervids. Curr. Top. Microbiol. Immunol. 284:193–214.
Miller, M. W., E. S. Williams, C. W. McCarty, T. R. Spraker, T. J.
Kreeger, C. T. Larsen, and E. T. Thorne. 2000. Epizootiology of
chronic wasting disease in free-ranging cervids in Colorado and
Wyoming. J. Wildlife Dis. 36:676–690.
Miller, M., E. S. Williams, N. T. Hobbs, and L. L. Wolfe. 2004. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis. 10:1003–1006.
Mishra, R. S., S. Basu, Y. Gu, X. Luo, W. Q. Zou, R. Mishra, R. Li, S.
G. Chen, P. Gambetti, H. Fujioka and N. Singh. 2004. Proteaseresistant human prion protein and ferritin are cotransported
across Caco-2 epithelial cells: Implications for species barrier in
prion uptake from the intestine. J. Neurosci. 24:11280–11290.
Moudjou, M., Y. Frobert, J. Grassi, and C. La Bonnardiere. 2001.
Cellular prion protein status in sheep: Tissue-specific biochemical signatures. J. Gen. Virol. (Pt. 8) 82:2017–2024.
Mulcahy, E. R., J. C. Bartz, A. E. Kincaid, and R. A. Bessen. 2004.
Prion infection of skeletal muscle cells and papillae in the tongue.
J. Virol. 78:6792–6798.
Murakami-Kubo, I., K. Doh-Ura, K. Ishikawa, S. Kawatake, K. Sasaki, J. Kira, S. Ohta, and T. Iwaki. 2004. Quinoline derivatives
are therapeutic candidates for transmissible spongiform encephalopathies. J. Virol. 78:1281–1288.
NCBI. 2004. Protein sequence database. National Center for Biotechnology Information. Available: http://www.ncbi.nlm.nih.gov/.
Accessed Nov. 4, 2004.
Nguyen, J., M. A. Baldwin, F. E. Cohen, and S. B. Prusiner. 1995.
Prion protein peptides induce alpha-helix to beta-sheet conformational transitions. Biochemistry 34:4186–4192.
Office International des Epizooties. 2004. Number of reported cases
of bovine spongiform encephalopathy (BSE) in farmed cattle
worldwide. World Organisation for Animal Health (OIE). Available: http://www.oie.int/eng/info/en_esbmonde.htm. Accessed
Nov. 4, 2004.
O’Rourke, K. I., T. E. Besser, M. W. Miller, T. F. Cline, T. R. Spraker,
A. L. Jenny, M. A. Wild, G. L. Zebarth and E. S. Williams. 1999.
PrP genotypes of captive and free-ranging Rocky Mountain elk
(Cervus elaphus nelsoni) with chronic wasting disease. J. Gen.
Virol. 80:2765–2769.
O’Rourke, K. I., T. R. Spraker, L. K. Hamburg, T. E. Besser, K. A.
Brayton, and D. P. Knowles. 2004. Polymorphisms in the prion
precursor functional gene but not the pseudogene are associated
with susceptibility to chronic wasting disease in white-tailed
deer. J. Gen. Virol. 85(Pt 5):1339–1346.
Oesch, B., D. Westaway, and S. B. Prusiner. 1991. Prion protein
genes: evolutionary and functional aspects. Curr. Top. Microbiol.
Immunol. 172:109–124.
Palmer, M. S., A. J . Dryden, J. T. Hughes, and J. Collinge. 1991.
Homozygous prion protein genotype predisposes to sporadic
Creutzfeldt-Jakob disease. Nature 352:340–342.
Pattison, I. H., M. N. Hoare, J. N. Jebbett, and W. A. Watson. 1972.
Further observations on the production of scrapie in sheep by
oral dosing with foetal membranes from scrapie-infected sheep.
Br. Vet. J. 130:1xv–1xvii.
Pauly, P. C., and D. A. Harris. 1998. Copper stimulates endocytosis
of the prion protein. J. Biol. Chem. 273:33107–33110.
Perrier, V., A. C. Wallace, K. Kaneko, J. Safar, S. B. Prusiner, and
F. E. Cohen. 2000. Mimicking dominant negative inhibition of
prion replication through structure-based drug design. Proc.
Natl. Acad. Sci. USA. 97:6073–6078.
Phillips, N. A., J. Bridgeman, and M. Ferguson-Smith. 2000. Report
of the inquiry into BSE and variant CJD. Vol. 1: Findings and
conclusions. The Stationary Office, London. Available: http://
www.bseinquiry.gov.uk/. Accessed Nov. 4, 2004.
PHYLIP. 2004. The Phylogeny Inference Package. Univ. of Washington and Joseph Felsenstein. Available: http://evolution.gs.washington.edu/phylip.html. Accessed Nov. 4, 2004.
Piccardo, P., J. J. Liepnieks, A. William, S. R. Dlouhy, M. R. Farlow,
K. Young, D. Nochlin, T. D. Bird, R. R. Nixon, M. J. Ball, C.
DeCarli, O. Bugiani, F. Tagliavini, M. D. Benson, and B. Ghetti.
2001. Prion proteins with different conformations accumulate
in Gerstmann-Straussler-Scheinker disease caused by A117V
and F198S mutations. Am. J. Pathol. 158:2201–2207.
Pickering-Brown, S. M., D. M. A. Mann, F. Owen, J. W. Ironside, R.
de Silva, D. A. Roberts, D. J. Balderson, and P. N. Cooper. 1995.
Allelic variations in apolipoprotein E and prion protein genotype
related to plaque formation and age of onset in sporadic Creutzfeldt-Jakob disease. Neurosci. Lett. 187:127–129.
BSE Biology
Prendergast, D. M., J. J. Sheridan, D. J. Daly, D. A. McDowell, and
I. S. Blair. 2004. Dissemination of central nervous system tissue
during the slaughter of cattle in three Irish abattoirs. Vet. Rec.
154:21–24.
Prinz, M., M. Heikenwalder, T. Junt, P. Schwarz, M. Glatzel, F. L.
Heppner, Y. X. Fu, M. Lipp, A. Aguzzi. 2003. Positioning of
follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 425:957–962.
Prusiner, S. B. 1982. Novel proteinaceous infectious particles cause
scrapie. Science 216:136–144.
Prusiner, S. B. 2004. Prion Biology and Diseases. 2nd ed. Cold Spring
Harbor Monograph Series, Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, NY.
Prusiner, S. B., D. Groth, A. Serban, R. Koehler, D. Foster, M. Torchia,
D. Burton, S. L. Yang, and S. J. DeArmond. 1993. Ablation
of the prion protein (PrP) gene in mice prevents scrapie and
facilitates production of anti-PrP antibodies. Proc. Natl. Acad.
Sci. USA 90:10608–10612.
Raymond. G. J., A. Bossers, L. D. Raymond, K. I. O’-Rourke, L. E.
McHolland, P. K. Bryant, III, M. W. Miller, E. S. Williams, M.
Smits, and B. Caughey. 2000. Evidence of a molecular barrier
limiting susceptibility of humans, cattle and sheep to chronic
wasting disease. EMBO J. 19:4425–4430.
Redman, C. A., P. G. Coen, L. Matthews, R. M. Lewis, W. S. Dingwall,
J. D. Foster, M. E. Chase-Topping, N. Hunter, and M. E. Woolhouse. 2002. Comparative epidemiology of scrapie outbreaks in
individual sheep flocks. Epidemiol. Infect. 128:513–521.
Ridley, R. M., and H. F. Baker. 1999. The myth of maternal transmission
of
spongiform
encephalopathy.
Br.
Med.
J.
311(7012):1071–1075.
Riesner, D. 2003. Biochemistry and structure of PrP(C) and PrP(Sc).
Br. Med. Bull. 66:21–33.
Rudyk, H., S. Vasiljevic, R. M. Hennion, C. R. Birkett, J. Hope, and
I. H. Gilbert. 2000. Screening Congo Red and its analogues for
their ability to prevent the formation of PrP-res in scrapie-infected cells. J. Gen. Virol. 81:1155–1164.
Rutala, W. A., and D. J. Weber. 2001. Creutzfeldt-Jakob disease:
Recommendations for disinfection and sterilization. Clin. Infect.
Dis. 32:1348–1356.
Safar, J., F. E. Cohen, and S. B. Prusiner. 2000. Quantitative traits
of prion strains are enciphered in the conformation of the prion
protein. Arch. Virol. Suppl. 2000:227–235.
Sales, N., K. Rodolfo, R. Hassig, B. Faucheux, L. Di Giamberardino,
and M. L. Moya. 1998. Cellular prion protein localization in
rodent and primate brain. Eur. J. Neurosci. 10:2464–2471.
Sander, P., H. Hamann, I. Pfeiffer, W. Wemheuer, B. Brenig, M. H.
Groschup, U. Ziegler, O. Distl, and T. Leeb. 2004. Analysis of
sequence variability of the bovine prion protein gene (PRNP) in
German cattle breeds. Neurogenetics 5:19–25.
Schmidt, G. R., K. L. Hossner, R. S. Yemm, and D. H. Gould. 1999.
Potential for disruption of central nervous system tissue in beef
cattle by different types of captive bolt stunners. J. Food Prot.
62:390–393.
Schmidt, G. R., R. S. Yemm, K. D. Childs, J. P. O’Callaghan, and K.
L. Hossner. 2001. The detection of central nervous system tissue
on beef carcasses and in comminuted beef. J. Food Prot.
64:2047–2052.
Schreuder, B. E. C. 1994. General aspects of spongiform encephalopathies and hypotheses on the agents. Vet. Q. 15:167–174.
Schrieber, R., and U. Seybold. 1993. Gelatine production, the six
steps to maximum safety. Dev. Biol. Stand. 80:195–198.
SEAC. 2002. Determination of abnormal prion protein in the milk
of cattle experimentally infected with the BSE agent. Summary:
Paper No: SEAC/INF/75/22. Spongiform Encephalopathy Advisory Committee. Available: http://www.seac.gov.uk/papers/
milkSEAC75.pdf. Accessed Nov. 4, 2004.
Shaked, G. M., Y. Shaked, Z. Kariv-Inbal, M. Halimi, I. Avraham,
and R. Gabizon. 2001. A protease-resistant prion protein isoform
is present in urine of animals and humans affected with prion
diseases. J. Biol. Chem. 276:31479–31482.
1475
Sigurdson, C. J., E. S. Williams, M. S. Miller, T. R. Spraker, K. I.
O’-Rourke, and E. A. Hoover. 1999. Oral transmission and early
lymphoid tropism of chronic wasting disease PrPres in mule
deer fawns (Odocoileus hemionus). J. Gen. Virol. 80:2757–2764.
Simmons, M. M., P. Harris, M. Jeffrey, S. C. Meek, I. W. Blamire,
G. A. Wells. 1996. BSE in Great Britain: Consistency of the
neurohistopathological findings in two random annual samples
of clinically suspect cases. Vet. Rec. 138:175–177.
Solforosi, L., J. R. Criado, D. B. McGavern, S. Wirz, M. SanchezAlavez, S. Sugama, L. A. DeGiorgio, B. T. Volpe, E. Wiseman,
G. Abalos, E. Masliah, D. Gilden, M. B. Oldstone, B. Conti,
and R. A. Williamson. 2004. Cross-linking cellular prion protein
triggers neuronal apoptosis in vivo. Science 303:1514–1516.
Somerville, R. A., S. Hamilton and K. Fernie. 2005. Transmissible
spongiform encephalopathy strain, PrP genotype and brain region all affect the degree of glycosylation of PrPSc. J. Gen. Virol.
(Pt. 1) 86:241–246.
Soto, C. 2004. Diagnosing prion diseases: needs, challenges and hopes.
Nat. Rev. Microbiol. 2:809–819.
Spielhaupter, C., and H. M. Schatzl. 2001. PrPC directly interacts
with proteins involved in signaling pathways. J. Biol. Chem.
276:44604–44612.
Spraker, T. R., M. W. Miller, E. S. Williams, D. M. Getzy, W. J.
Adrian, G. G. Schoonveld, R. A. Spowart, K. I. O’Rourke, J. M.
Miller, and P. A. Merz. 1997. Spongiform encephalopathy in
free-ranging mule deer (Odocoileus hemionus), white-tailed deer
(Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in north central Colorado. J. Wildlife Dis. 33:1–6.
Stack, M. J., A. Balachandran, M. Chaplin, L. Davis, S. Czub and
B. Miller. 2004. The first Canadian indigenous case of bovine
spongiform encephalopathy (BSE) has molecular characteristics
for prion protein that are similar to those of BSE in the United
Kingdom but differ from those of chronic wasting disease in
captive elk and deer. Can. Vet. J. 45:825–830.
Swiss-Prot/TrEMBL. 2004. Protein Knowledge Base. The Swiss Institute of Bioinformatics and the European Bioinformatics Institute. Available: http://us.expasy.org/. Accessed Nov. 4, 2004.
Supattapone, S., H. Wille, L. Uyechi, J. Safar, P. Tremblay, F. C.
Szoka, F. E. Cohen, S. B. Prusiner, and M. R. Scott. 2001.
Branched polyamines cure prion-infected neuroblastoma cells.
J. Virol. 75:3453–3461.
Taguchi, F., Y. Tamai, K. Uchida, R. Kitajima, H. Kojima, T. Kawaguchi, Y. Ohtani, and S. Miura. 1991. Proposal for a procedure
for complete inactivation of the Creutzfeldt-Jakob disease agent.
Arch. Virol. 119:297–301.
Tamai Y., H. Kojima, R. Kitajima, F. Taguchi, Y. Ohtani, T. Kawaguchi, S. Miura, M. Sato, and Y. Ishihara. 1992. Demonstration
of the transmissible agent in tissue from a pregnant woman
with Creutzfeldt-Jakob disease. N. Engl. J. Med. 327:649.
Taylor, D. M., C. E. Ferguson, C. J. Bostock, and M. Dawson. 1995a.
Absence of disease in mice receiving milk from cows with bovine
spongiform encephalopathy. Vet. Rec. 136:592–596.
Taylor, D. M., and K. Fernie. 1996. Exposure to autoclaving or sodiumhydroxide extends the dose-response curve of the 263K strain
of scrapie agent in hamsters. J. Gen. Virol. 77:811–813.
Taylor, D. M., S. L. Woodgate, and M. J. Atkinson. 1995b. Inactivation
of the bovine spongiform encephalopathy agent by rendering
procedures. Vet. Rec. 136:605–610.
Taylor, D. M., I. McConnell, and H. Fraser. 1996. Scrapie infection
can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J. Gen. Virol.
77:1595–1599.
Taylor, D. M., S. L. Woodgate, A. J. Fleetwood, and R. J. Cawthorne.
1997. Effect of rendering procedures on the scrapie agent. Vet.
Rec. 141:643–649.
Telling, G. C., P. Parchi, S. J. DeArmond, P. Cortelli, P. Montagna,
R. Gabizon, J. Mastrianni, E. Lugaresi, P. Gambetti, and S. B.
Prusiner. 1996. Evidence for the conformation of the pathologic
isoform of the prion protein enciphering and propagating prion
diversity. Science 274:2079–2082.
1476
Novakofski et al.
Terry, L. A., S. Marsh, S. J. Ryder, S. A. Hawkins, G. A. Wells, and
Y. I. Spencer. 2003. Detection of disease-specific PrP in the distal
ileum of cattle exposed orally to the agent of bovine spongiform
encephalopathy. Vet. Rec. 152:387–392.
Thomzig, A., C. Kratzel, G. Lenz, D. Kruger, and M. Beekes. 2003.
Widespread PrPSc accumulation in muscles of hamsters orally
infected with scrapie. EMBO Rep. 4:530–533.
Thomzig, A., W. Schulz-Schaeffer, C. Kratzel, J. Mai, and M. Beekes.
2004. Preclinical deposition of pathological prion protein PrP(Sc)
in muscles of hamsters orally exposed to scrapie. J. Clin. Invest.
113:1465–1472.
Tranulis, M. A. 2002. Influence of the prion protein gene, Prnp, on
scrapie susceptibility in sheep. APMIS 110:33–43.
Tuo, W., K. I. O’Rourke, D. Zhuang, W. P. Cheevers, T. R. Spraker, and
D. P. Knowles. 2002. Pregnancy status and fetal prion genetics
determine PrPSc accumulation in placentomes of scrapie-infected sheep. Proc. Natl. Acad. Sci. USA 99:6310–6315.
Tuo, W., D. Zhuang, D. P. Knowles, W. P. Cheevers, M. S. Sy, and
K. I. O’Rourke. 2001. PrP-C and PrP-Sc at the fetal-maternal
interface. J. Biol. 276:18229–18234.
Unterberger, U., T. Voigtlander, and H. Budka. 2005. Pathogenesis
of prion diseases. Acta Neuropathol. 109:32–48.
USDA. 2004a. Food Safety and Inspection Service. Prohibition of the
use of specified risk materials for human food and requirements
for disposition of non-ambulatory disabled cattle. Fed. Regis.
69:1861–1874.
USDA. 2004b. Food Safety and Inspection Service. Prohibition of the
Use of Certain Stunning Devices Used to Immobilize Cattle
During Slaughter. Fed. Regis. 69:1885–1891.
USDA. 2004c. Food Safety and Inspection Service. Meat Produced by
Advanced Meat/Bone Separation Machinery and Meat Recovery
(AMR) Systems. Fed. Regis. 69:1874–1885.
USDA. 2004d. Phase II: Scrapie: Ovine Slaughter Surveillance Study.
2002–2003. USDA:APHIS:VS, CEAH, National Animal Health
Monitoring System, Fort Collins, CO. Available: http://www.
aphis.usda.gov/vs/ceah/ncahs/nahms/sheep/SOSSphase2.pdf.
Accessed Nov. 4, 2004.
van Rheede, T., N. M. Smolenaars, O. Madsen, W. W. de Jong. 2003.
Molecular evolution of the mammalian prion protein. Mol. Biol.
Evol. 20:111–121.
Vorberg, I., A. Raines, B. Story and S. A. Priola. 2004. Susceptibility
of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J. Infect. Dis. 189:431–439.
Wadsworth, J. D., E. A. Asante, M. Desbruslais, J. M. Linehan, S.
Joiner, I. Gowland, J. Welch, L. Stone, S. E. Lloyd, A. F. Hill,
S. Brandner, and J. Collinge. 2004. Human prion protein with
valine 129 prevents expression of variant CJD phenotype. Science 306:1793–1796.
Wadsworth, J. D., A. F. Hill, J. A. Beck, and J. Collinge. 2003. Molecular and clinical classification of human prion disease. Br. Med.
Bull. 66:241–254.
Wadsworth, J. D., S. Joiner, A. F. Hill, T. A. Campbell, M. Desbruslais,
P. J. Luthert, and J. Collinge. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease
using a highly sensitive immunoblotting assay. Lancet
358:171–180.
Weissmann, C., M. Enari, P. C. Klohn, D. Rossi, and E. Flechsig. 2002.
Transmission of prions. J. Infect. Dis. 186(Suppl. 2):S157–S165.
Wells G. A., S. A. Hawkins, A. R. Austin, S. J. Ryder, S. H. Done, R.
B. Green, I. Dexter, M. Dawson and R. H. Kimberlin. 2003.
Studies of the transmissibility of the agent of bovine spongiform
encephalopathy to pigs. J. Gen. Virol. (Pt. 4) 84:1021–1031.
Wilesmith, J. W., J. B. M. Ryan, and M. J. Atkinson. 1991. Bovine
spongiform encephalopathy: Epidemiological studies on the origin. Vet. Rec. 128:199–203.
Wilesmith, J. W., J. B. M. Ryan, and W. D. Hueston. 1992. Bovine
spongiform encephalopathy: Epidemiological features 1985–
1990. Vet. Rec. 130:90–94.
Wilesmith, J. W., G. A. Wells, M. P. Cranwell, and J. B. Ryan. 1988.
Bovine spongiform encephalopathy: Epidemiological studies.
Vet. Rec. 123:638–644.
Williams, E. S., and M. W. Miller. 2002. Chronic wasting disease in
deer and elk in North America. Rev. Sci. Tech. 21:305–316.
Williams, E. S., and S. Young. 1992. Spongiform encephalopathies
in Cervidae. Rev. Sci. Tech. 11:551–567.
Williams, E. S., and S. Young. 1993. Neuropathology of chronic wasting disease of mule deer (Odocoileus hemionus) and elk (Cervus
elaphus nelsoni). Vet. Pathol. 30:36–45.
WHO. 1999. WHO Infection Control Guidelines for Transmissible
Spongiform Encephalopathies. Report of a WHO Consultation,
Geneva, Switzerland. Available: http://www.who.int/csr/
resources/publications/bse/en/whocdscsraph2003.pdf. Accessed
Nov. 4, 2004.
Wrathall, A. E., K. F. Brown, A. R. Sayers, G. A. Wells, M. M. Simmons, S. S. Farrelly, P. Bellerby, J. Squirrell, Y. I. Spencer, M.
Wells, M. J. Stack, B. Bastiman, D. Pullar, J. Scatcherd, L.
Heasman, J. Parker, D. A. Hannam, D. W. Helliwell, A. Chree,
and H. Fraser. 2002. Studies of embryo transfer from cattle
clinically affected by bovine spongiform encephalopathy (BSE).
Vet. Rec. 150:365–378.
Yan, Z. X., L. Stitz, P. Heeg, E. Pfaff, and K. Roth. 2004. Infectivity
of prion protein bound to stainless steel wires: a model for testing
decontamination procedures for transmissible spongiform encephalopathies. Infect. Control Hosp. Epidemiol. 25:280–283.
Ye, X., and R. I. Carp. 1995. The pathological changes in peripheral
organs of scrapie-infected animals. Histol. Histopathol.
10:995–1021.
Zanata, S. M., M. H. Lopes, A. F. Mercadante, G. N. Hajj, L. B.
Chiarini, R. Nomizo, A. R. Freitas, A. L. Cabral, K. S. Lee, M.
A. Juliano, E. de Oliveira, S. G. Jachieri, A. Burlingame, L.
Huang, R. Linden, R. R. Brentani, and V. R. Martins. 2002.
Stress-inducible protein 1 is a cell surface ligand for cellular
prion that triggers neuroprotection. EMBO J. 21:3307–3316.