Energy Transfer and Triplet Exciton Confinement
advertisement

Energy Transfer and Triplet Exciton Confinement in
Polymeric Electrophosphorescent Devices
FANG-CHUNG CHEN,1 SHUN-CHI CHANG,1 GUFENG HE,1 SEUNGMOON PYO,1 YANG YANG,1
MASAYUKI KUROTAKI,2 JUNJI KIDO2
1
Department of Materials Science and Engineering, University of California at Los Angeles, Los Angeles, California 90095
2
Graduate School of Engineering, Yamagata University, Yamagata 992, Japan
Received 21 February 2003; revised 28 May 2003; accepted 28 May 2003
ABSTRACT: Energy transfer and triplet exciton confinement in polymer/phosphorescent
dopant systems have been investigated. Various combinations of host– guest systems
have been studied, consisting of two host polymers, poly(vinylcarbazole) (PVK) and
poly[9,9-bis(octyl)-fluorene-2,7-diyl] (PF), blended with five different phosphorescent
iridium complexes with different triplet energy levels. These combinations of hosts and
dopants provide an ideal situation for studying the movement of triplet excitons
between the host polymers and dopants. The excitons either can be confined at the
dopant sites or can flow to the host polymers, subject to the relative position of the
triplet energy levels of the material. For PF, because of its low triplet energy level, the
exciton can flow back from the dopants to PF when the dopant has a higher triplet
energy and subsequently quench the device efficiency. In contrast, efficient electrophosphorescence has been observed in doped PVK films because of the high triplet energy
level of PVK. Better energy transfer from PVK to the dopants, as well as triplet exciton
confinement on the dopants, leads to higher device performance than found in PF
devices. Efficiencies as high as 16, 8.0, and 2.6 cd/A for green, yellow, and red emissions,
respectively, can be achieved when PVK is selected as the host polymer. The results in
this study show that the energy transfer and triplet exciton confinement have a
pronounced influence on the device performance. In addition, this study also provides
material design and selection rules for the efficient phosphorescent polymer lightemitting diodes. © 2003 Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 41: 2681–2690,
2003
Keywords: triplet; light-emitting; phosphorescence
INTRODUCTION
Triplet excitons of organic materials are generally
nonemissive because the transition to ground
states is forbidden by spin selection rules.1 However, triplets are believed to be the abundant
species when charges recombine within organic
electroluminescence (EL) devices,2 although the
spin statistics predicting a 75% yield of triplet
Correspondence to: Y. Yang (E-mail: yangy@ucla.edu)
Journal of Polymer Science: Part B: Polymer Physics, Vol. 41, 2681–2690 (2003)
© 2003 Wiley Periodicals, Inc.
excitons in EL devices has been challenged for
conjugated polymers.3–5 By the harvesting of triplet excitons, highly efficient organic light-emitting diodes (OLEDs) have been demonstrated via
doping with phosphorescent dyes.6 – 8 Iridium(III)
complexes have been shown to be the most efficient triplet dopants employed in highly efficient
OLEDs.7,8 A green OLED with an internal quantum efficiency of nearly 100% has been demonstrated.8 Moreover, by the accurate control of the
fabrication conditions, the endothermic energy
transfer of triplets has also been applied to fabricating blue phosphorescent OLEDs,9 and this
2681
2682
CHEN ET AL.
Figure 1. Chemical structures of the materials used in this study.
suggests insignificant nonradiative decay of host
triplets. On the other hand, for polymer lightemitting diodes (PLEDs), high efficiencies have
also been achieved by the doping of different
hosts, such as poly[9,9-bis(octyl)-fluorene-2,7diyl] (BOc-PF or PF) and poly(vinylcarbazole)
(PVK), with Pt or Ir complexes.10 –18 However,
there is still a lack of systematic studies on the
relationship between hosts and dopants in electrophosphorescent PLEDs.
Iridium(III) complexes with blue to red emissions
have been reported.19,20 By the alteration of the
chemical structures of the ligands, the triplet properties can be fine-tuned. They provide a suitable
system for investigating the dopant energetic influences on the performance of phosphorescent
PLEDs. In this study, two types of devices, consisting of different hosts (PF and PVK) doped with
different iridium complexes {iridium(III) fac-tris(2phenylpyridine) [Ir(ppy)3 ], iridium(III) bis(2phenylpyridinato-N,C2⬘)(acetylacetonate) (PPIr),
iridium(III)
bis(2-phenyl
benzothiozolatoN,C2⬘)(acetylacetonate) (BtIr), bis(2-[2⬘-benzothienyl)-pyridinato-N,C3⬘]iridium(acetylacetonate) (BtpIr), and iridium(III) fac-tris[2-(4-octyl-phenyl)pyridine] [Ir(Ocppy)3]}, were fabricated. With these
combinations of two polymers and five dopants,
the interactions between the host polymers and
dopants were investigated. This study is particularly important in view of the fact that the harvest of electrophosphorescent PLED is strongly
influenced by the relative energy levels between
the host and the dopant. In this article, we report
this systematic study on the selection of the host
and dopant materials and their device performance.
EXPERIMENTAL
Two polymers, PVK and PF, were chosen as host
materials for the PLEDs. The chemical structures
of these compounds are illustrated in Figure 1.
The highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals
(LUMOs) of the polymers and dopants were deduced from cyclic voltammograms (CVs) or from
the literature,20 –23 and they are listed in Table 1.
All Ir complexes exhibited reversible oxidation
waves, which were assigned as IrIII/IrIV redox
couples.20,24 The CV measurements were conducted with a BAS 100B with a typical threeelectrode configuration. A glass carbon electrode,
a platinum wire, and a silver wire served as the
working, counter, and quasireference electrodes,
respectively. Ferrocene was used as the internal
standard. For optical measurements, such as photoluminescence (PL) spectra, thin films of the
polymers were spin-coated from solutions onto
quartz substrates. The ultraviolet–visible absorption and PL spectra were measured on an HP
8453 spectrophotometer and a Spex Fluorolog-3
double-grating spectrofluorometer, respectively.
The triplet-state energies of dopants were calculated from their highest peak of phosphorescence.25 Cross-polarized optical microscopy was
performed with a Nikon polarizing optical microscope equipped with a charge coupled device
(CCD) camera. Atomic force microscopy (AFM)
images were obtained with a Digital Instrument
multimode scanning probe microscope. The cantilevers were 125-m-long etched Si probes. The
images were automatically plane-fitted to account
for the sample tilt.
For charge balance, two types of PLED structures
were designed for different host materials:15,16 type I,
indium tin oxide (ITO)/3,4-poly(ethylene dioxythiophene)–poly(styrene sulfonate) (PEDOT)/
PVK–2-(4-biphenylyl)-5-(4-tert-butyl-phenyl)-1,3,4oxadiazole (PBD)– dopant/Ca/Al, and type II, ITO/
PEDOT/PVK/doped PF/Ca/Al. Bilayer electrodes,
consisting of ITO emission/glass substrate coated
POLYMERIC ELECTROPHOSPHORESCENT DEVICES
2683
Table 1. Energy Levels of the Materials
PVK
PF
Ir(ppy)3
PPIr
Ir(Ocppy)3
BtIr
BtpIr
Oxidation
Potential
(V vs FOC)
Reduction
Potential
(V vs FOC)
HOMO
(eV vs Vac.)a
LUMO
(eV vs Vac.)a
Triplet
Energy
(eV)
0.74
0.97
0.32
0.40
0.24
0.56
0.36
—
⫺2.72
⫺2.69
⫺2.61
⫺2.60
⫺2.29
⫺2.42
⫺5.54
⫺5.77
⫺5.12
⫺5.20
⫺5.04
⫺5.36
⫺5.16
⫺2.04
⫺2.08
⫺2.11
⫺2.19
⫺2.20
⫺2.65
⫺2.38
2.50
2.15
2.41
2.41
2.41
2.23
2.02
a
Deduced from electrochemical potentials under the assumption that the energy level of ferrocene (FOC) was ⫺4.8 eV versus
vacuum (Vac.).
with a thin layer of the conducting polymer PEDOT,
were used as the anodes in all the devices. For type
I devices, a single layer of the polymer (PVK), consisting of PBD, an electron-transport medium, with
a 1:1 weight ratio and different amounts of the
dopants, was used as the active material. The PVK/
PBD layer was formed via spin coating from a 1,2diclorobenzene solution at a concentration of 2.0 wt
%. For type II devices, bilayer polymer films consisting of a 20-nm-thick hole-transporting layer
(PVK) and a second layer of PF doped with iridium
complexes were used as the emissive active media.15 PF was dissolved in toluene at a concentration of 1.8 wt %. The thickness of these two types of
devices was about 100 nm. Calcium (500 Å) and
aluminum (1000 Å) bilayer cathodes were thermally evaporated in a 1 ⫻ 10⫺6 Torr vacuum. The
devices were characterized with a Keithley 236
source-measure unit. The brightness measurements were carried out with a silicon photodiode
calibrated with a PR-650 spectra colorimeter. All
devices were fabricated and tested under a nitrogen
environment.
energy traps of the dopant and subsequently recombine directly to form either singlets or triplets. By intersystem crossing, singlets in dopants
can convert into triplets with very high efficiency
and decay radiatively by phosphorescence. However, singlets formed on the host can transfer to
the dopant by long-range dipole– dipole coupling
(Forester energy transfer),27 whereas triplets are
capable of transferring energy by electron exchange (Dexter energy transfer).28 Because Forester energy transfer is usually more efficient
than Dexter energy transfer, the most likely exciton loss comes from the nonradiative decay of
3
the host triplets (kNR
in Scheme 1), which is a
process competing with Dexter energy transfer to
the dopant. This indicates that the reduction of
the exciton loss from the nonradiative decay of
hosts or, in other words, the confinement of triplets on dopants, is crucial to harvesting 100% of
the excitons.
The phosphorescent efficiency of Ir complexes
in a PF matrix has been shown to be a function of
RESULTS AND DISCUSSION
Energy Transfer between the Host Polymer and the
Dopant
Two important factors, energy transfer and
charge transfer, are of concern in dye-doped lightemitting diodes.26 In a fluorescent host doped
with phosphorescent dopant OLEDs, several
routes can lead to the phosphorescence of the
dopants, such as direct charge trapping and energy transfer.26 Upon charge injection into an
organic thin film from electrodes, either electrons
or holes (or both) may be caught by the deep
Scheme 1
2684
CHEN ET AL.
Figure 2. PL spectra of (a) undoped PF film and
(b– d) PF films doped with 11 wt % BtpIr, 10 wt % BtIr,
or 10 wt % PPIr, respectively. All spectra were obtained
with pumping at 382 nm. The PL intensity of curve d is
identical to that of PF, but the intensity is one order
smaller.
the dopant triplet energy levels.29 Figure 2(a)
shows the PL spectra of a neat PF thin film. When
the PF film was doped with BtpIr, the PF emission decreased, and a new red emission with a
peak at 616 nm was obtained [Fig. 2(b)]. This
clearly indicates an excitation energy transfer
from PF to BtpIr. For the PF/BtIr blend system,
although the emission of PF was quenched, only a
weak dopant PL emission was observed for highly
BtIr-doped films [Fig. 2(c)]. One possible reason
for different PL efficiencies of BtpIr and BtIr is
the varying aggregation-induced concentration
Figure 3. PL intensities of BtpIr and BtIr in PF films
at different dopant concentrations. The PL contributions from the dopants increase with the concentrations.
Figure 4. PL intensities of PPIr-doped PF films at
different dopant concentrations. No energy transfer
from PF to PPIr can be observed.
quenching. However, as in Figure 3, which demonstrates the intensities of the dopant PL as a
function of concentration, the PL contribution
from dopants increases with the dopant concentrations. For all concentrations, BtpIr has a
higher PL efficiency. In serious concentrationquenching conditions, we usually expect a decrease of PL from the dopant materials.15,22,26,30
In addition, BtpIr and BtIr have very similar
chemical structures and photophysical properties.19,20 Although some extent of concentration
quenching is expected in PF/dopant films, the
large difference in the PL efficiencies shown in
Figure 2 still cannot be explained by this effect
alone. Moreover, in the PPIr-doped PF films, despite the quenching of PL of PF by doping, the PL
efficiency of PPIr in PF films was extremely low;
no apparent emission from PPIr could be observed [Fig. 2(d)]. In Figure 4, which presents the
PL spectra of PF doped with different concentrations of PPIr, PF shows a very low PL efficiency in
heavily PPIr-doped films, and this suggests a very
efficient exciton loss pathway.
Figure 5 shows the absorption spectra of dopants and the PL spectra of PF thin films. To
understand the singlet energy efficiencies from
the host to the dopants, we have calculated the
Forster radii for dopants in a PF matrix on the
basis of the spectral overlap between the dopant
absorption and host emission spectra.26 The deduced Forster radii of PPIr, BtIr, and BtpIr are
27, 32, and 31 Å, respectively. Consequently, all
three dopants have reasonably high Forster energy-transfer efficiencies. The difference in the PL
efficiencies of the dopants in PF films, as observed
POLYMERIC ELECTROPHOSPHORESCENT DEVICES
in Figure 2, cannot be well explained by only the
different Forster energy transfer efficiencies from
PF to the dopants. If the Forster energy transfer
is the dominant factor that determines the PL
efficiencies of dopants in PF films, BtIr should
have the highest PL efficiency. However, as observed from Figure 2, BtpIr emits most efficiently
in the PF matrix.
However, the different photophysical behaviors of Ir complexes in the PF films can be qualitatively explained by the relative positions of the
triplet energy levels of the dopants to that of the
host polymer. For convenience of discussion, we
divide the polymer blends into three different categories based on the positions of their dopant
triplet energy levels.
In case 1, the triplet energy of the dopant is
higher than that of the host polymer. This is the
case of the PF/PPIr blend system. Upon photoexcitation, the singlet excitation energy transfers
from PF to PPIr. Because the triplet energy of
metal-to-ligand charge-transfer (3MLCT) of PPIr
(2.41 eV) is higher than that of 3PF (2.15 eV),31
thermodynamically, the excitation energy tends
to flow to PF triplet energy states. Subsequently,
3
PF decays via nonradiative transition to the
ground state (Scheme 1).
In case 2, the triplet energy of the dopant is
lower than that of the host polymer. This is the
case of the PF/BtpIr system, in which the lowest
triplet energy of BtpIr (2.0 eV) is lower than that
of 3PF. The excitation energy transfers from the
singlet of PF to the triplet of BtpIr, and excitons
tend to stay in BtpIr. The backflow of energy is
unlikely to happen because it is thermodynamically unfavorable. In other words, the triplet ex-
Figure 5. Absorption spectra of Ir complex solutions
(5.0 ⫻ 10⫺5M) and PL spectra of PF and PVK thin
films.
2685
Figure 6. PL spectra of PPIr-doped PVK films at
different dopant concentrations.
citons are confined in BtpIr. Thus, a higher phosphorescent efficiency of BtpIr in PF films was
observed [Fig. 2(b)].
In case 3, the triplet energy level of the dopant
is similar to that of the host polymer. In this case,
there is a competition between the energy transfer from the dopant to the host polymer and the
internal triplet exciton relaxation within the dopant, which is supported by the weak BtIr emission shown in Figure 2(c).
The backward energy transfer has been reconfirmed by the triplet lifetime measurement in solutions.32 The preliminary result shows that the
dopants with different triplet energies exhibit different lifetimes in PF solutions, and this is consistent with the mechanism that we have previously proposed. The lifetime data and Stern–
Volmer analysis will be published elsewhere.32
On the contrary, the energy transfer from PVK
to the dopants is much more efficient. Figure 6
shows the PL spectra of PPIr-doped PVK thin
films. The PL was mainly from PPIr when the
dopant concentration was higher than 1 wt %.
The emission of PVK was quenched efficiently by
doping with PPIr. Other Ir complexes also exhibited efficient phosphorescence in PVK films. The
fluorescence of PVK comes from the excimer and,
therefore, has a longer lifetime (⬃35 ns).33 It was
reported earlier that a longer lifetime of the host
material could facilitate the energy transfer.7 In
addition, the triplet energy of PVK is 2.5 eV,
which has been deduced from PVK phosphorescence at 77 K.34 Even in the PVK/PPIr system, in
which the dopant has a high energy triplet state,
the host triplet energy level is still higher than
the lowest triplet excitation level (3MLCT) of
2686
CHEN ET AL.
Figure 7. EL spectra of PVK devices doped with Ir(ppy)3 (3 wt %), PPIr (3 wt %), BtIr (3 wt %), or BtpIr
(4%).
CIE chromaticity coordinates of the emission of
the PPIr-doped PF device operating at 10, 50, and
100 mA/cm2 are (0.39, 0.56), (0.39, 0.54), and
(0.39, 0.53), respectively. For PF/BtpIr devices
[Fig. 8(b)], very little host emission was observed,
and this implied better exciton confinement,
which is consistent with previous predictions
from PL spectra.
Another interesting observation from doped PF
light-emitting diodes (shown in Fig. 8) is that the
pattern of the EL spectrum contributed from the
dopant complex was not exactly the same as the
one from the doped PVK devices shown in Figure
7 or the PL in dilute solutions shown in Figure
9(a,b). Broader (full width at half-maximum
PPIr. Triplet exciton confinement on the dopants
is considered better than on PF systems. More
efficient energy and better triplet exciton confinement result in a higher efficient phosphorescence
of Ir complexes in the blended PVK films.
Phosphorescent Polymer Light-Emitting Diodes
EL Spectra
The EL spectra of doped PVK devices are shown
in Figure 7. When PVK was used as the matrix,
no host EL was obtained in doped PVK PLEDs,
even at high current densities. Because of the
highly efficient charge/energy transfer, no host
EL was observed at a dopant concentration as low
as 1%. Additionally, the shape of EL of the doped
PVK devices was identical at different dopant
concentrations and under various driving current
densities. In contrast, the EL (Fig. 8) contributed
from both the dopant and host was observed for
PF devices, indicative of the incomplete energy/
charge transfer between the host and the dopant
even at low current densities. Recalling that PL
mainly came from PF in the PPIr-doped PF film
(Fig. 2), we found that the EL was mainly contributed from PPIr [Fig. 8(a)]. This dramatic difference of PL and EL is believed to be due to
carrier trapping at the dopant sites.
In addition, the EL spectrum of the PF devices
also depends on the applied current density. For
example, the emission from the host PF (420 – 480
nm) increases with increasing current density
[Fig. 8(a)]. Such a host emission results in a
change in the Commission Internationale de
L’Eclairage Chromaticity (CIE) coordinate. The
Figure 8. (a) EL spectra of PPIr (3%)-doped PF lightemitting diodes under different current densities. The
residual PF emission can be seen at a higher current
density and suggests incomplete energy transfer. (b).
EL spectra of BtpIr (5%)-doped PF under different current densities. The residual PF emission is less than in
part a.
POLYMERIC ELECTROPHOSPHORESCENT DEVICES
Figure 9. PL spectra of (a) Ir(ppy)3 and (b) BtpIr in
toluene (dashed lines) and the solid state.
⫽ 100 nm) and redshifted EL spectra were observed in the doped PF devices. To understand
the cause of the EL spectral change in the doped
PF device, we examined the PL spectra of different states of Ir complexes (Fig. 9). Consider Ir(ppy)3, for example, in dilute toluene solution (5
⫻ 10⫺5M). The maximum wavelength was at 508
nm, and the full width at half-maximum was 50
nm. The powdered solid sample exhibited an orange emission, which had a maximum wavelength at 560 nm, and the full width at halfmaximum increased to 95 nm. Similar observations for other substitution ligand Ir complexes
have been reported elsewhere.20 Close intermolecular contacts of ligands lead to a redshifted
emission spectrum for the solid sample with respect to its corresponding solution spectrum. For
PPIr and Ir(ppy)3, the broad and featureless solid-
2687
state emission originates from the excimer. For
BtpIr, the structured emission is probably due to
the ground-state dimers.20 From the previous discussion, we infer that the redshift of the EL spectrum is due to some extension of dopant aggregation and hence the emission from dimeric units of
dopants with strong – interactions.
In contrast, the EL spectra of PVK devices
were consistent with the corresponding PL spectra in dilute solutions (Figs. 7 and 9), and this
suggested that the same exciting states were generated during photo and electrical excitation. This
implies few intermolecular interactions between
the dopants in the PVK matrix. The aggregation
of dopants in PF films might result from the polarity difference between the host material and
dopant molecules. Ir complexes have high polarities. PF has a nonpolar repeating unit, and PVK
unit is a carbazole, which has a high polarity.
According to the rule that like solvates like, dopant molecules have higher solubility in PVK
than in PF. Therefore, a phase separation between PF and the dopants could occur.
Cross-polarized optical microscopy was used to
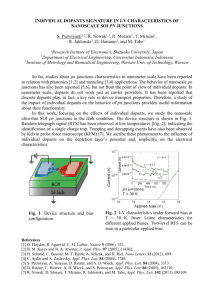
investigate the doped polymer thin films. Figure
10 shows an image of a 5% Ir(ppy)3-doped PF film
under cross-polarized optical microscopy. Some
bright spots shaped like long needles and about 1
m long can be observed. It is known that only
crystalline domains can be observed under crosspolarized light. The bright dots suggest that PPIr
has a high tendency to crystallize in PF films
during the evaporation of the solvent in the spincoating process. For other PF films doped with Ir
complexes, similar phenomena were observed.
This common observation implied low solubility,
Figure 10. Cross-polarized microscopy of a 5% Ir(ppy)3-doped PF film. The needle-shaped crystallinity
of Ir(ppy)3 can be clearly observed.
2688
CHEN ET AL.
main was observed. In addition, the root-meansquare surface roughness of this film, which was
deduced from AFM, was 0.83nm. No phased separation was observed. Thus, it is expected that a
higher efficiency of a device doped with Ir(Ocppy)3
can be achieved by an improved film morphology.
Device Performance
Figure 11. The AFM phase image of a 5 wt % Ir(ppy)3-doped PF film. The image clearly shows the aggregates of the dopant molecules.
which was due to the difference in the polarities
between the host and dopant materials. However,
for doped PVK films, no crystalline domain was
observed, and this suggested a more uniform distribution of the dopants in the PVK films.
The thin-film phase separation was further
confirmed with AFM. Figure 11 shows the AFM
phase image of an Ir(ppy)3-doped PF film. It can
be seen clearly from the image that phase separation existed in the doped PF film. However, a
smoother surface was observed in the neat PF
film. The surface root-mean-square roughness of
the pure PF film was smaller (⬃0.76 nm) than
that of the doped PF film (⬃16.1 nm), and this
indicated that aggregates were caused by the immiscibility of PF and Ir(ppy)3. However, a similar
surface root-mean-square roughness (⬃0.85 nm)
was obtained for the undoped and doped PVK
films that was indicative of undetectable phase
separation within doped PVK films. It was inferred that the Ir complexes could be well dispersed in PVK films because of their similar polarities. The AFM data support the results obtained with cross-polarized optical microscopy.
To improve the compatibility between the host
and dopant materials, we synthesized a highly
soluble Ir complex, Ir(Ocppy)3, which had a lower
polarity, by attaching an octyl side chain to the
ligands. A cross-polarized image of a 5 wt %
Ir(Ocppy)3-doped PF film exhibited an entirely
amorphous phase. No detectable crystalline do-
Figure 12 shows current–light–voltage curves of a
device with PPIr-doped PVK as the emitting medium. The maximum quantum efficiency was 13
cd/A. The device turn-on voltage, defined for a
brightness of 0.1 cd/m2, and the maximum brightness were 5.3 V and 15,000 cd/m2, respectively.
Better energy transfer between the dopant and
PVK, as well as triplet exciton confinement, led to
high performance. Bilayer devices consisting of a
hole-transporting layer of PVK (ca. 200 A) and an
electron-transporting layer of PF, doped with different concentrations of Ir complexes (ca. 1000 A),
were fabricated as well. Figure 13 shows the current–light–voltage curves of the device, and it
turned on at 4.5 V. The quantum efficiency was
4.1 cd/A. Table 2 summarizes the device performance with various combinations of the hosts and
dopants. All PVK-based devices had higher efficiencies than the PF-based ones.
For the confinement of the triplet excitons, host
polymers with higher triplet energy should be
used, and this implies that polymers with much
wider band gaps are favorable. However, the electronic properties of this kind of polymer may be
degraded, for example, and this leads to higher
charge injection barriers. The higher operating
voltage is likely to cause a low power efficiency
even though a high quantum efficiency could be
Figure 12. Current–light–voltage curves of (a,b) a 3
wt % PPIr-doped PVK device and (c,d) a 3 wt %
Ir(Ocppy)3-doped PVK device.
POLYMERIC ELECTROPHOSPHORESCENT DEVICES
Figure 13. Current–light–voltage curves of (a,b) a 3
wt % PPIr-doped PF device and (c,d) a 2 wt %
Ir(Ocppy)3-doped PF device.
2689
a PVK/PPIr device (Fig. 12), we found that the
operating voltage increased in the case of doping
with Ir(Ocppy)3. Because Ir(Ocppy)3 has a higher
HOMO than PPIr (Table 1), it is suspected that
hole trapping was more efficient in the
Ir(Ocppy)3-doped device. Similarly, an increase in
the operating voltages was observed in PF devices
as well (Fig. 13). The highest efficiency of the
Ir(Ocppy)3-doped PF device was 6.2 cd/A. The octyl side chains on the ligands enhanced the dopant solubility in PF and resulted in more uniform dopant distribution in the polymer host. The
improvement of the performance was due to both
better thin-film morphology and more efficient
hole trapping.
CONCLUSIONS
3
obtained. As for kinetics, reducing kNR
(Scheme 1)
of hosts provides an alternative; host materials
with longer triplet lifetimes meet this requirement.35
Figure 14 shows the current–voltage curves of
PPIr-doped PF devices with different concentrations. The driving voltages of the devices increased with the dopant concentration. This suggests that the dopants behaved as carrier traps in
the devices. This observation also supports the
carrier-trapping mechanism previously proposed
from the difference between the PL and EL spectra (Fig. 8).
A PVK device doped with Ir(Ocppy)3 exhibited
a higher efficiency (16 cd/A) than that of other
green devices in which Ir(ppy)3 and PPIr were
used as the dopants. Comparing the current–voltage curves of a PVK/Ir(Ocppy)3 device with that of
Iridium(III) complexes with different triplet exciton energies exhibit various photophysical behaviors in a PF matrix. The performance of phosphorescent PLEDs based on PF has also been shown
to be rather sensitive to the triplet energies of the
dopants. Because of the low triplet energy level,
choosing PF as the host results in inefficient energy transfer and poorer triplet confinement in
comparison with devices in which PVK serves as
the host material. For devices using doped PVK
films as the emitting layers, clear and stable EL
emissions from dopants and higher device efficiency have been demonstrated. Because of more
efficient energy and better exciton confinement,
values as high as 16, 8, and 2.6 cd/A for green,
Table 2. Comparison of Dopant Devices Using
Different Host Materials
Host
PVK
PF
Dopant
(wt % in host)
Turn-On
Voltage
(V)
Efficiency
(cd/A)
max
(nm)
Ir(PPy)3 (3%)
PPIr (3%)
Ir(Ocppy)3 (3%)
BtIr (3%)
BtpIt (4%)
Ir(ppy)3 (3%)
PPIr (3%)
Ir(Ocppy)3 (2%)
BtIr (5%)
BtpIr (5%)
5.5
5.2
9.6
5.5
6.5
4.5
4.5
11.0
5.2
5.0
12.7
13.0
16.0
8.0
2.6
3.9
4.1
6.2
3.0
1.9
516
516
518
560
614
520
526
518
560
614
Figure 14. Current–voltage curves of PPIr-doped PF
devices doped with different concentrations. The increasing driving voltages with the dopant concentration suggest that the dopants behave as carrier traps in
the devices.
2690
CHEN ET AL.
yellow, and red emissions, respectively, were
achieved when PVK was selected as the host,
blended with the electron-transporting material
PBD. The improvement of the triplet exciton confinement, which has been revealed in this study,
provides another direction for improving the efficiency of phosphorescent PLEDs. In light of our
results, a new blue (or even ultraviolet) emission
polymer is needed for future efficient phosphorescent PLEDs. This new blue polymer should have
a broad energy gap, a high triplet energy level,
polar side groups for better solubility of triplet
dopants, and a bipolar charge-transport capability.
This research was sponsored by the National Science
Foundation (ECS-0100611) and the Office of Naval Research (N00014-01-1-0136). The authors thank Mark
E. Thompson (University of Southern California) for
providing the dopants.
14.
15.
16.
17.
18.
19.
20.
21.
REFERENCES AND NOTES
1. Turro, N. J. Modern Molecular Photochemistry;
University Science Books: Mill Valley, CA, 1991.
2. Baldo, M. A.; Forrest S. R. Physical Review B 1999,
60, 14422.
3. Wohlgenannt, M.; Tandon, K.; Mazumdar, S.; Ramasesha, S.; Vardeny, Z. V. Nature 2001, 409, 494.
4. Shuai, Z.; Beljonne, D.; Silbey, R. J.; Bredas, J. L.
Phys Rev Lett 2000, 84, 131.
5. Wilson, J. S.; Dhoot, A. S.; Seeley, A. J. A. B.; Khan,
M. S.; Kohler, A.; Friend, R. H. Nature 2001, 413,
828.
6. Baldo, M. A.; O’Brien, D. F.; You, Y.; Shoustikov,
A.; Sibley, S.; Thompson, M. E.; Forrest, S. R. Nature 1998, 395, 151.
7. Baldo, M. A.; Lamansky, S.; Burrows, P. E.;
Thompson, M. E.; Forrest, S. R. Appl Phys Lett
1999, 75, 4.
8. Adachi, C.; Baldo, M. A.; Thompson, M. E.; Forrest,
S. R. J Appl Phys 2001, 90, 5048.
9. Adachi, C.; Kwong, R. C.; Djurovich, P.; Adamovich, V.; Baldo, M. A.; Thompson, M. E.; Forrest,
S. R. Appl Phys Lett 2001, 79, 2082.
10. Yang, M. J.; Tsutsui, T. Jpn J Appl Phys 2000, 39,
L828.
11. Lee, C. L.; Lee, K. B.; Kim J. J. Appl Phys Lett
2000, 77, 2280.
12. Lamansky, S.; Kwong, R. C.; Nugent, M.; Djurovich, P.; Thompson M. E. Org Electron 2001, 2, 53.
13. O’Brien, D. F.; Giebeler, C.; Fletcher, R. B.; Cadby,
A. J.; Palilis, L. C.; Lidzey, D. G.; Lane, P. A.;
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
Bradley, D. D. C.; Blau, W. Synth Met 2001, 116,
379.
Lane, P. A.; Palilis, L. C.; O’Brien, D. F.; Giebeler,
C.; Cadby, A. J.; Lidzey, D. G.; Campbell, A. J.;
Blau, W.; Bradley, D. D. C. Phys Rev B 2001, 63,
235206.
Guo, T. F.; Chang, S. C.; Yang, Y.; Kwong, R. C.;
Thompson, M. E. Org Electron 2001, 1, 15.
Chang, S. C.; He, G. F.; Chen, F. C.; Guo, T. F.;
Yang, Y. Appl Phys Lett 2001, 79, 2088.
Kawamura, Y.; Yanagida S.; Forrest, S. R. J Appl
Phys 2002, 92, 87.
Vaeth, K. M.; Teng, C. W. J Appl Phys 2002, 92,
3447.
Lamansky, S.; Djurovich, P.; Murphy, D.; AbdelRazzzaq, F.; Lee, H. E.; Adachi, C.; Burrows, P. E.;
Forrest S. R.; Thompson, M. E. J Am Chem Soc
2001, 123, 4304.
Lamansky, S.; Djurovich, P.; Murphy, D.; AbdelRazzzaq, F.; Kwong, R. C.; Tsyba, I.; Bortz, M.;
Mui, B.; Thompson, M. E. Inorg Chem 2001, 40,
1704.
Kolosov, D.; Adamovich, V.; Djurovich, P.; Thompson, M. E.; Adachi, C. J Am Chem Soc 2002, 124,
9945.
Wu, C. C.; Sturm, J. C.; Register, R. A.; Tian, J.;
Dana, E. P.; Thompson, M. E. IEEE Trans Electron
Devices 1997, 44, 1269.
Janietz, S.; Bradley, D. D. C.; Grell, M.; Giebeler,
C.; Inbasekaran, M.; Woo. E. P. Appl Phys Lett
1998, 73, 2453.
King, K. A.; Spellane, P. J.; Watts, R. J. J Am Chem
Soc 1985, 107, 1432.
Baldo, M. A.; Forrest, S. R. Physical Review B
2000, 62, 10958.
Shoustikov, A. A.; You, Y.; Thompson, M. E. IEEE
J Sel Top Quantum Electron 1998, 4, 3.
Forster, T. Discuss Faraday Soc 1959, 27, 7.
Dexter, D. L. J Chem Phys 1953, 21, 836.
Chen, F. C.; He, G. F.; Yang, Y. Appl Phys Lett
2003, 82, 1006.
Shaheen, S. E.; Kippelen, B.; Peyghambarian, N.;
Wang, J.-F.; Anderson, J. D.; Mash, E. A.; Lee,
P. A.; Armstrong, N. R.; Kawabe, Y. J Appl Phys
1999, 85, 7939.
Rothe, C.; Monkman, A. P. Physical Review B 2002,
65, 073201.
Chen, F. C.; Yang, Y.; Djurovich, P. I.; Thompsom,
M. E. J Phys Chem B, to be submitted.
Itaya, A.; Sakai, H.; Masuhara, H. Chem Phys Lett
1998, 146, 570.
Rippen, G.; Kaufmann, W.; Klopffer, W. Chem
Phys 1980, 52, 165.
Kwong, R. C.; Lamansky, S.; Thompson, M. E. Adv
Matter 2000, 12, 1134.