UNIVERSITE PARIS DESCARTES
Ecole Doctorale « Génétique, cellules, Immunologie, Infectiologie, Dévelopment »
Gc2iD
Specialité : Biologie Cellulaire
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE PARIS DESCARTES
Présentée par
Rosamaria Calicchio
Le 27 novembre 2013
High-throughput transcriptional analysis of the endothelial
alterations in preeclampsia identifies
JDP2 (Jun dimerization protein 2)
as a novel actor in hypoxia sensing
Directeur de thèse: Dr. Daniel Vaiman
JURY :
Pr. Vassilis Tsatsaris
Président
Pr. Loïc Sentilhes
Rapporteur
Dr. Zahra Tanfin
Rapportrice
Dr. Daniel Vaiman
Directeur de thèse
Dr. Francisco Miralles
Examinateur
Dr. Claire Francastel
Examinatrice
Pr. Patricia Fauque
Examinatrice
"Considerate la vostra semenza:
fatti non foste a viver come bruti
ma per seguir virtute e canoscenza"
Ulisse,
Dante Alighieri, Divina Commedia, Inferno canto XXVI, 116-120
Remerciements
Je remercie le Dr. Zahra Tanfin et le Pr. Loïc Sentilhes de m’avoir fait l’honneur d’être
rapporteurs de mes travaux de thèse. Je suis profondément reconnaissante du temps et de
l’attention qu’ils ont consacrés à la relecture de mon manuscrit. Je tiens également à remercier
le Pr. Patricia Fauque et le Pr. Vassilis Tsatsaris pour avoir accepté de lire et d’évaluer ce
travail, ainsi que pour leur implication. Je remercie le Dr. Claire Francastel : c’était vraiment
un grand plaisir pour moi d’avoir dans mon jury celle qui m’a accueillie dans son labo au
début de mon M2 et m’a toujours accompagnée et soutenue pendant mon parcours
scientifique, dès le début.
Je tiens à témoigner toute ma reconnaissance et mon affection au Dr. Daniel Vaiman. Je crois
fortement que si c’est important d’avoir des objectifs, il est surtout nécessaire d’avoir un bon
guide, et c’était le cas ! Merci de m’avoir toujours soutenue et motivée pendant ces trois
dernières années, même et surtout dans des moments un peu « difficiles ». Merci pour ton
enthousiasme débordant et ta curiosité, qui vont bien au-delà du domaine scientifique dans
lequel tu excelles déjà. Merci d’avoir toujours été à l’écoute et enclin à la discussion, et
d’avoir participé à mon projet de thèse par tes questions, tes remises en questions et tes points
de réflexion. La tournure de ce projet scientifique s’est par ailleurs révélée complètement
inattendue, et son évolution permanente a rendu ce travail encore plus passionnant. Il faut
avoir un peu d’« Ulysse » en nous pour faire de la recherche, avoir (ou trouver) le courage de
poursuivre des voies pas encore tracées, soulever des questions inédites, ou bien apporter des
nouveaux éléments de réponse à des questions connues, sans arrêt ! C’est l’enseignement le
plus enrichissant que m’ont apporté ces trois années, et qui fait d’une thèse, de ma thèse, une
expérience exceptionnellement formatrice ! Merci infiniment ! Je remercie egalement le Dr.
Francisco Miralles pour son implication dans la première partie de ce travail.
Je tiens à remercier toute mon équipe, pour le soutien et l’encouragement qu’elle m’a donnée
pendant ma thèse. J’ai eu l’impression d’avoir été encadrée par l’équipe entière et je suis sûre
que si je suis parvenue jusque là, c’est parce qu’auprès de vous j’ai toujours trouvé des
chercheurs de très haut niveau, des compétences dont j’ai souvent profité, mais aussi une
vraie famille, au point de se sentir aussi bien au labo que chez soi. Un grand merci à ma
voisine, tout près de moi, Sandrine, qui a été toujours là pour mes moments d’enthousiasme et
aussi de « demotivation », à Céline, RH du labo et une des chercheuse les plus passionnées
que j’ai eu la chance de connaitre et que j’estime profondément, Capu et Michelle, qui ont été
toujours comme des petites mamans pour moi, à Ahmed, pour les « réunion de labo » autour
d’une cigarette ou d’un the à la menthe, et encore Virginie, Jana, Julie, Brigitte, Aminata,
Patrick, Côme, Jean, Marc, Laurence et Florence. Un merci aux étudiants du labo, présents et
passés : Ludivine, bien sûr, avec qui j’ai partagé des projets scientifique et une grande amitié,
que j’espère, prospérera, et encore Aurélien, Lucile, Sandrine, Aurélie, Amélie, Jonathan,
Michael, Marie, Aude, Leila, Louis et Pietro. Merci à Charlotte, qui m’a accordée un peu de
temps pour relire ces lignes, merci pour les soirées passées ensemble, et c’est avec plaisir que
je te confie la tâche de « event organizer » !!! Merci à Elma pour l’aide technique qu’elle m’a
gentillement fournie ces derniers mois, mais aussi et surtout pour toutes les pauses déjeuner,
café, apéro et même si l’on s’est connu seulement vers la fin de mon séjour au labo, j’espère
vivement que l’on aura bien d’autres occasions de se retrouver et de se rappeler des mois
passés ensemble au labo avec un sourire. Un grazie à Elisabetta, con la quale ho condiviso
questi ultimi mesi in lab, pause caffé e aperitivi; peccato che tu non sia arrivata prima, ma
sono sicura che sia solo l’inizio, e che avremo l’occasione di vederci e rivederci, in lab o
altrove !!!!!
Je remercie profondément le Dr. Jacques Mathieu, le Dr. Carole Peyssonnaux et son équipe.
C’était pour moi un grand plaisir de collaborer avec vous et voir des idées et des projets
prendre forme. Merci Jacques pour toutes nos discussions formelles et informelles, pour
m’avoir accordée beaucoup de ton temps pour mes manips, pour mes idées et aussi pour mes
moments d’ « incertitude » et de panique.
Un grazie alla mia famiglia, che mi ha sempre sostenuto nelle mie scelte per tutti questi anni e
soprattutto che é stata presente molto piu’ di quanto io non lo sia stata per loro. Condividere
questo momento con voi mi riempie di gioia, e mostrarvi dove sono arrivata a piccoli passi ha
un gusto tutto particolare ed una soddisfazione che non dimentichero’ mai. Un grazie a
Nicola, compagno e amico di sempre : so che non é stato sempre facile starmi accanto per
tutti questi anni, e senza il tuo supporto e sostegno non sarei andata lontano, cosi’ lontano!
Grazie grazie grazie !!!
Un grand merci à toutes les personnes qui sont passées dans ma vie pendant ces quatre années
parisiennes, qui ont juste fait un coucou ou bien qui sont restées de façon un peu plus
présente: chacune d’entre elles m’a apportée quelque chose que je garderai jalousement dans
mes souvenirs. La vie nous réserve toujours des surprises et des choses inattendues… Et vous
tous, vous étiez une agréable surprise !!!
Voilà voilà, tout ça tout ça
Summary
Preeclamspia is a unique human disorder which affects 3-8% of pregnancies worldwide,
clinically defined as the new onset of hypertension and proteinuria. The root cause of the
disease seems to be linked to a defect of placental vascularization, which enhances cycles of
hypoxia –reoxygenantion, placental ischemia and the release of placental debris into maternal
circulation. The latter ones are responsible for a widespread endothelial activation,
exacerbated pro-coagulable and pro-inflammatory state.
To best characterize the response of endothelial cells to the plasma factors present in maternal
circulation of preeclamptic women, we chose a genome –wide approach in order to evaluate
the gene expression profile of Human Umbilical Vein Endothelial Cells (HUVEC) line
cultivated with preeclamptic plasma, compared to cells cultivated with human plasma coming
from normal pregnancies. This study allows us to identify the gene Jun Dimerization Protein2
(JDP2) which could be responsible for part of transcriptomic modifications. Interestingly
inhibiting JDP2 by the use of siRNA significantly down- regulates VEGF expression, thus
mimicking the effects of preeclamptic plasma on HUVEC.
In the last part of my project we focus specifically on the impact of JDP2 knock down on
hypoxia- induced genes. Low oxygen tension modifies gene expression via the stabilization of
the transcription factor HIF-1α. In fact under hypoxic condition, HIF-1α escapes proteasomal
degradation, it forms heterodimers with ARNT (HIF- 1β) and induces the expression of genes
having a Hypoxia Responsive Element (HRE) in their promoter. One of the first and best
characterized models of the effect of hypoxia on gene expression is the induction of VEGF
expression under hypoxic condition.
In order to evaluate the contribution of JDP2 to VEGF expression, and more generally to
hypoxia target genes, we cultivate HUVEC in normoxic and hypoxic conditions. The same
conditions were used in association with transfection of siRNA against JDP2. In conclusion,
under hypoxic condition, JDP2 down- regulation has a negative impact on VEGF expression.
Moreover, JDP2 seems to be an essential mediator of hypoxia –induced gene expression,
since it is necessary for a full HRE promoter activity.
In conclusion we identified JDP2 as a new gene which may play an important role in
endothelial dysfunction during preeclampsia. Moreover its expression is crucial for hypoxia induced VEGF expression, thus suggesting its crucial function in order to guarantee the full
cellular response against hypoxic stress conditions.
Table of contents
Summary.............................................................................................................................................. 7
Table of contents .............................................................................................................................. 9
Figures .....................................................................................................Erreur ! Signet non défini.
Tables.......................................................................................................Erreur ! Signet non défini.
Abbreviations ................................................................................................................................. 13
ACE angiotensin I converting enzyme .................................................................................... 13
Introduction ....................................................................................................................................... 1
Chapter I.
1.
Normal pregnancy versus Preeclampsia...................................................... 2
Normal pregnancy ...................................................................................................... 2
A.
Implantation ......................................................................................................... 2
B.
Decidualization..................................................................................................... 3
i. Decidual cells differentiation ............................................................................... 4
ii.
Immune cell invasion........................................................................................ 5
iii.
Decidual vascular remodeling: fetal contribution ............................................ 6
iv.
Maternal vascular change ................................................................................. 7
C.
Full term placenta: structure and function.......................................................... 10
Figure 1: Principal feto- maternal exchanges ...................................................................... 11
2.
Preeclampsia............................................................................................................. 11
A.
Introduction ........................................................................................................ 11
B.
Epidemiology ..................................................................................................... 13
C.
Risk factors ......................................................................................................... 14
i. Medical conditions ............................................................................................. 14
ii.
Genetic component, known and unknown ..................................................... 15
Table 1: Putative genes and polymorphisms involved in preeclampsia .................... 16
iii.
Other risk factors ............................................................................................ 18
D.
Management and treatment of preeclampsia ...................................................... 21
E.
Prevention of preeclampsia ................................................................................ 23
F.
Pathophysiology of preeclampsia....................................................................... 24
Chapter II. Maternal syndrome during preeclampsia ................................................ 30
1.
Endothelial health and vascular maintenance .......................................................... 31
A.
Angiogenic imbalance ........................................................................................ 31
i. sFlt-1 and its biological relevance in preeclamptic placenta .............................. 32
ii.
sFlt-1 and kidney damages during preeclampsia ............................................ 33
iii.
VEGF signaling pathway alteration and cerebral edema ............................... 34
iv.
Other factors involved in endothelial permeability perturbation.................... 35
Figure 2: Factors involved in increased permeability in preeclampsia ...................... 39
v.
sEng and its biological relevance in preeclampsia ......................................... 39
vi.
sEng and sFlt-1: impact on vaso-regulation and coagulation state ................ 40
Figure 3: NO synthesis in endothelial cells and effects on smooth muscle cells .... 40
B.
Vasculature contribution to vasodilation and coagulation state ......................... 42
C.
Inflammation ...................................................................................................... 44
D.
Immune system reaction..................................................................................... 45
E.
Early biomarkers of preeclampsia ...................................................................... 46
Table 2: Early circulating biomarkers of preeclampsia .................................................... 48
F.
Animal models for preeclampsia........................................................................ 48
Table 3: Mouse models for preeclampsia .............................................................................. 49
Chapter III. Hypoxia and cellular response ..................................................................... 51
1.
Hypoxia inducible factor 1 (HIF1) ........................................................................... 52
A.
HIF-1α ................................................................................................................ 53
B.
HIF- 1β ............................................................................................................... 54
2.
HIF-2 and HIF-3....................................................................................................... 54
3.
Regulation of HIF-1 protein stability ....................................................................... 56
A.
Canonical pathway: HIF-1α oxygen-dependent regulation ............................... 56
B.
Regulation of HIF transcriptional activity ......................................................... 57
C.
Oxygen–independent mechanisms of HIF-1α stabilization and regulation of
transcriptional activity .................................................................................................. 58
i. Regulation of PHDs and VHL ........................................................................... 58
ii.
Other pathways involved in HIF regulation ................................................... 60
Table4: Oxygen- independent regulation of HIF-α ........................................................... 62
4.
HIFs in placental development ................................................................................. 63
A.
VEGF-A transcription regulation: beyond oxygen ............................................ 66
Figure 5: VEGF promoter: binding sites and principal transcriptional regulators . 67
5.
HIFs and preeclampsia ............................................................................................. 69
A.
HIFs expression in pregnancies complicated by preeclampsia .......................... 69
B.
HIF contribution to preeclampsia ....................................................................... 70
C.
Causes of HIF deregulation ................................................................................ 71
Chapter IV. JDP2: from chromatin organization to regulation of gene expression
73
1.
JDP2 expression ....................................................................................................... 74
2.
JDP2: involvement in chromatin organization and gene regulation ........................ 75
Results ............................................................................................................................................... 93
1.
Paper 1 ...................................................................................................................... 93
Results .............................................................................................................................................102
Discussion ......................................................................................................................................106
2.
Paper 2 (in preparation) .......................................................................................... 127
Results .............................................................................................................................................136
Discussion ......................................................................................................................................144
Discussion and perspectives ...................................................................................................154
1.
Preeclampsia: a vascular perspective ..................................................................... 154
2.
Preeclampsia and beyond: the future maternal health ............................................ 158
3.
JDP2: role in the endothelial response to preeclampsia and in hypoxia sensing ... 160
A.
JDP2: a new supervisor of endothelial hypoxic response ................................ 162
B.
JDP2 involvement in the transcriptional modification of AP-1 members under
hypoxic condition ....................................................................................................... 164
C.
A Possible role of JDP2 on VEGF expression mediated by histone
modifications .............................................................................................................. 165
Conclusion ......................................................................................................................................167
Bibliography ..................................................................................................................................168
Supplemental papers .................................................................................................................220
Figures
Figure 1: Principal feto- maternal exchanges ..........................................................................11
Figure 2: Factors involved in increased permeability in preeclampsia ...................................39
Figure 3: NO synthesis in endothelial cells and effects on smooth muscle cells ....................40
Figure 4: altered NO pathway in preeclampsia. Red arrows show modified molecules in
preeclamptic syndrome .......................................................................................................….42
Figure 5: VEGF promoter: binding sites and principal transcriptional regulators .................67
Figure 6: Factors affecting HIF-1α deregulation in preeclampsia and consequences on
placentation and late maternal syndrome .................................................................................71
Tables
Table 1: Putative genes and polymorphisms involved in preeclampsia ..................................16
Table 2: Early circulating biomarkers of preeclampsia ...........................................................48
Table 3: Mouse models for preeclampsia ................................................................................49
Table 4: Oxygen- independent regulation of HIF-α ............................................................... 62
Abbreviations
ACE angiotensin I converting enzyme
Ang-1 Angiopoietin 1
Ang-2 Angiopoietin 2
AP-1 Activating protein-1
AP-2 Activating protein-2
ARNT aryl hydrocarbon receptor nuclear translocator
AT1 angiotensin II receptor, type 1
AT1-AA angiotensin II receptor, type 1 autoantibodies
bHLH–PAS basic helix-loop-helix–Per-Arnt-Sim
bZIP basic leucine zipper domain
CTAD C-terminal transactivation domain
CTLA4 cytotoxic T-lymphocyte-associated protein 4
E-cadherin epithelial cadherin
EGF Epidermal Growth Factor
EGF epidermal growth factor
Egr-1 Early gene response protein-1
EPAS1 Endothelial PAS domain protein1
ERK extra-cellular signals regulated kinases
F2 coagulation factor 2, or prothrombin
FasL Fas ligand
FGF Fibroblast Growth Factor
FIH-1 Factor inhibiting HIF
FV coagulation factor V
HAT histone-acetyl-transferase
hCG human chorionic gonadotropin
HELLP Hemolysis, Elevated Liver enzymes and Low Platelets
HGF Hepatocyte Growth Factor
HIF Hypoxia inducible factors
HSP90 heat shock protein 90
ICM inner cell mass
IGF Insulin-like Growth Factors
IGF Insulin-like growth factors
IGFBP insulin-like growth factor binding proteins
IGFBP-1 insuline-like growth factor binding protein 1
IGF-I insulin-like growth factor
IL-17 interleukin 17
IL-8 interleukin 8
JNK Jun NH2-terminal kinase
LPL lipoprotein lipase
MAPK mitogen activated protein kinase
MMPs Matrix metalloproteinases
MT1-MMP Membrane type 1 metalloprotease
NLS Nuclear Localization Sequences
NO nitric oxide
NOS3 endothelial NO synthase
NOX NADPH-oxidase
NTAD N-terminal transactivation domain
ODDD O2- dependent degradation domain, ODDD
PAI plasminogen activator inhibitors
PDZ primary decidual zone
PECAM-1 platelet-endothelial cell adhesion molecule-1
PHD prolyl hydrohylase domain proteins
PlGF Placental growth factor
RACK1 receptor of activated kinase 1
ROS reactive oxygen species
SDZ secondary decidual zone
SERPIN1 serin protease inhibitor 1
sFlt-1 soluble fms-like tyrosine kinase-1, Soluble VEGF Receptor 1
SOD superoxide dismutase
Sp1 Specificity protein-1
STAT Signal Transducer and Activator of Transcription
sVEGF-1 Soluble VEGF Receptor 1
TGF-β transforming growth factor β
TIE1 Tyrosine kinase with immunoglobulin-like and EGF-like domains 1
TIMP tissue inhibitor of metalloprotease
TNF- α tumor necrosis factor α
TPO platelet growth factors
TRAIL TNF-related apoptosis inducing ligand
TRE TPA-response elements
uNK uterine Natural killer
uPA urokinase plasminogen activator
uPAR urokinase plasminogen activator receptor
VCAM vascular cell adhesion molecule
VE-cadherin vascular endothelial cadherin
VEGF vascular endothelial growth factor
VHL von Hippel- Lindau protein
Introduction
Introduction
Introduction
From conception, the first stages of human development come in succession within the
maternal uterus and end up with the birth of the offspring through parturition. This dynamic
process, known as pregnancy, starts with the contact of the conceptus with a receptive uterus
and this interaction imposes the adaptation of the maternal body in order to ensure a correct
fetal growth and development.
During pregnancy systemic and local changes alter the maternal vascular, immune and
hormonal system. A series of synchronized events leads to the formation of a transient organ,
the placenta through the process of placentation. Placenta is sensu stricto of fetal origin, it is a
selective barrier with secretory, immunological, endocrine, and exchange functions.
In
humans, the success of a correct fetal development is conditioned to a vascular adaptation in
order to ensure a better exchange between the mother and the fetus. This implies the
fulfillment of an angiogenic program, leading to the creation of passive (non contractile)
vessels and therefore accompanied by a strong vascular remodeling of maternal vessels,
where fetal cells (trophoblasts) play a crucial role.
Perturbation of this program strongly impacts fetal and sometimes maternal health, leading to
miscarriage, preterm birth, intrauterine growth restriction (IUGR), and preeclampsia.
In the case of preeclampsia, defective placentation is associated to a widespread maternal
syndrome, characterized by a stress condition that affects mainly the vascular system, triggers
hypertension and a widespread chronic inflammation.
This prolonged cellular stress that endothelial cells undergo all along pregnancy could
explain, or at least participate, to the increased risk of cardiovascular diseases of women who
suffered preeclampsia, even years after their pregnancies.
1
Introduction
Chapter I. Normal pregnancy versus Preeclampsia
1. Normal pregnancy
A. Implantation
Normal pregnancy is the harmonic succession of 3 physiological processes: implantation,
decidualization and placentation, which install the feto-maternal cross-talk in order to
ensure a correct fetal development (Carson et al., 2000; Dey et al., 2004). Due to accessibility
of the biological materials, knowledge on implantation and decidualization in Humans is
hampered, and mouse models have been thoroughly used to get deeper knowledge into these
physiological events.
Successful implantation imposes the direct interaction of the blastocyst and the maternal
uterus in a specific time span known as window of receptivity, a transient period which is
centered around the mid luteal phase (7 days after ovulation) in Humans. Blastocyst
implantation occurs 7-9 days after fertilization and consists of 4 main steps: apposition,
adhesion, attachment, and penetration (Daikoku et al., 2011; Giudice, 1999). At this moment
the blastocyst consists in an inner cell mass (ICM), which will give rise to the embryo tissues
and organs, a fluid-filled cavity called the blastocoele, all surrounded by a cell layer called
trophectoderm, the source of placental membranes of embryonic origin. Trophectoderm side
in contact with the ICM, called also polar trophectoderm, is the mediator of the first
interaction with the uterine luminal epithelium (Carson et al., 2000; Enders, 1976).
In Humans implantation is an intrusive process: just after the first contact, the blastocyst
creeps into epithelial cells and basal lamina, highly proliferates and embeds between the
uterine stromal layer (Schlafke and Enders, 1975). Furthermore its proteolytic activity on
endometrial tissue triggers a local inflammatory reaction and an increase in vascular
permeability which are both at the basis of the embryo tolerance and further decidualization
and trophoblast invasion (Dey et al., 2004). Indeed at this step two different trophoblast
populations differentiate on the polar trophectoderm: primitive syncytiotrophoblasts, which
are multinucleated cells, and primitive mononuclear cytotrophoblasts (Schlafke and Enders,
2
Introduction
1975). Polar trophectoderm actively proliferates during the implantation process, while the
distal one, called mural trophectoderm, preserves its structure.
B. Decidualization
Decidualization implies the building of decidual tissue from endometrial cells and is driven
mainly by ovarian secretion of 17β-estradiol, progesterone, and syncytiotrophoblast secretion
of a hormone, human chorionic gonadotropin (hCG). Correct decidualization aims at:
Differentiation of stromal cells into decidual cells
Vascular and extracellular matrix remodeling
Migration of immune cell types (macrophages and uNK cells) in the uterine wall
All these steps are crucial to avoid fetal rejection, to bypass the maternal immune system and
later in gestation, to guarantee a correct blood flow allowing an adequate fetal growth.
In Humans, decidualization does not need an implantation event to start, but it needs it to
reach completion (Jackson et al., 1980). Spontaneous decidualization occurs in the secretory
phase of the menstrual cycle and consists in the differentiation of stromal cells into decidual
cells, proliferation and increase in vascularization. Once implantation takes place, this
physiological process gives rise to the decidua basalis (proximal part of the decidua
placentalis), a layer of cells of maternal origin (the reason why the placenta is said to have a
feto-maternal origin) which is dismissed with placenta during parturition. Otherwise, without
an implantation event, the decidualized endometrium is shed during the ending phase of
menstrual cycle.
Vascular landscape must to be adapted to support the growing fetus needs. For this purpose
the fetal placental vascular system and maternal (decidual) system sustain a consistent
vascular adaptation during the first trimester of pregnancy, driven mainly by oxygen gradient
and several growth factors and angiogenic factors. These changes consist in the formation of
new blood vessels (through vasculogenesis and angiogenesis) and in strong modifications of
the pre- existing endometrial vascular bed (due to arterial remodeling).
3
Introduction
i.
Decidual cells differentiation
During the first 2 weeks after implantation, feto-maternal exchanges are mediated by
diffusion across the first place of interaction between the mother ant the fetus, a layer
composed of syncytiotrophoblasts (differentiated from embryonic trophectoderm) and of the
decidua basalis (the decidual tissue underlying the implantation site). The elucidation of the
morphological and physiological role of this restricted decidual area, called primary decidual
zone (PDZ), comes mostly from mouse models. PDZ is less vascularized and rich in tight
junctions (Halder et al., 2000; Wang et al., 2004). Its differentiation in secondary decidual
zone (SDZ) implies a decrease in vessels density in favor of enhanced vascular and luminal
surfaces. The increase in luminal diameter is functionally related to increased blood supply
necessary for the growing fetus (Wang et al., 2004). In a few weeks, decidual formation
covers all the endometrial tissue. This spatial and chronological decidual differentiation
may protect the growing embryo from possible maternal immune system activation and
simultaneously it can create an oxygen gradient at the trophoblastic- decidual interphase
which controls trophoblast invasion, by promoting trophoblast mitogenesis and by limiting
trophoblast invasiveness to the layer of decidua as well (Genbacev, 2001; Genbacev et al.,
1997). Ovarian secretion of 17β-estradiol and progesterone, associated to the production of
hCG from the syncytiotrophoblast stimulates the local production of growth factors which act
in an autocrine and paracrine way on decidualized cells thereby stimulating mitogenesis and
angiogenesis. Fibroblast-like stromal cells undergo a process of differentiation similar to
mesenchymal–epithelial transition and become highly secretory (Healy, 1991; Salamonsen
et al., 2009). They transform into larger and rounded decidual cells, through the storage of
cytoplasmic glycogen, and start to express decidual markers (Dunn et al., 2003; Popovici et
al., 2000). Early literature provides evidences of a process of endoreduplication (polypoidy)
in many decidual cells, according to which cells replicate their genome without cellular
division. This process may increase protein synthesis thus supporting fetal needs and a correct
placental development. Indeed at this step decidual cells have a consistent secretory function,
by producing several growth factors: Epidermal growth factor (EGF), fibroblast growth factor
(FGF), transforming growth factor β (TGF-β), insuline- like growth factor binding protein 1
(IGFBP-1) and several hormones (prolactin, renin) (Healy, 1991). Decidual endoreduplication
is well described in rodents, but a satisfying characterization of this event is still missing in
Humans (Lejeune et al., 1982; Sroga et al., 2012).
Decidualization also encompasses
4
Introduction
important uterine extracellular matrix modifications that make endometrial tissue more
sensible to trophoblastic invasion (hydratation, change in collagen type, proteoglycans).
Decidual cells synthesize several integrins and surface glycoproteins, like Mucin 1, which
control altogether adhesion, invasion and migration of trophoblasts cells (Iwahashi et al.,
1996; Loke et al., 1995; Meseguer et al., 2001; Simón et al., 2000; Staun-Ram and Shalev,
2005). They also participate to the building of a basal membrane of collagen IV, laminin,
proteoglycans and heparan- sulfate.
ii.
Immune cell invasion
Immune cells colonization of decidualized endometrium is a crucial step for a correct
placentation.
Indeed the first trimester of pregnancy is a proinflammatory state, fostered by the
proteolytic activity of blastocyst on endometrial tissue during implantation and invasion, and
by damage on endothelial and smooth muscle cells for correct spiral arteries remodeling
(Dekel et al., 2010). Hence the importance of an active immune cell system, that can manage
the damage and the repair, becomes more and more consistent.
After fertilization, immune cells infiltration increases and at the end of first trimester
decidual immune system can count 65-70% of uterine Natural killer (uNK) (Manaster and
Mandelboim, 2010), 10-20% macrophages and 2-4 % of dendritic cells (Abrahams et al.,
2004; Le Bouteiller and Piccinni, 2008; Nagamatsu and Schust, 2010). Hormones and
trophoblast-derived cytokines and chemokines play a major role in immune cells migration.
In particular prolactin secreted by decidualized cells drives uNK and Hofbauer cells towards
endometrial colonization (Carlino et al., 2008; Jabbour et al., 2002).
Trophoblasts secrete chemokines to attract immune cells at the implantation site, and
cytokines to stimulate their differentiation (Fest et al., 2007; Mor et al., 2005). uNK cells
modulate trophoblast invasion by producing interleukin 8 (IL-8) and interferon-inducible
proteins, chemokines, and angiogenesis and vascular remodeling via the secretion of several
pro-angiogenic factors, such as Angiopoietin 1 and 2 (Ang-1, Ang-2), tyrosine kinase with
immunoglobulin-like and EGF-like domains 1 (TIE1), vascular endothelial growth factor c
5
Introduction
(VEGF-c), placental growth factor (PlGF), urokinase plasminogen activator (uPA) and its
receptor (uPAR), membrane type 1 metalloproteinase (MT1-MMP) (Albertsson et al., 2000;
Lash et al., 2006; Naruse et al., 2009). Macrophages are normal cellular components of the
endometrium, but they increase after insemination and they remain present throughout
pregnancy (Kabawat et al., 1985). A specific macrophage population, known as Hofbauer
cells, is responsible for the production of pro-angiogenic factors like VEGF and interleukin 17
(IL-17) (Cooper et al., 1995; Pongcharoen et al., 2007). In general, the angiogenic role of
macrophages is well established in several disorders, especially in tumors, endometriosis and
vascular diseases (Lewis and Pollard, 2006; Lipinski et al., 2006; Siristatidis et al., 2006). In
pregnancy they have a key function in the regulation of placental vascular architecture, by
balancing the secretion of pro-angiogenic factors like EGF, FGF, TGF-β, platelet growth
factors (TPO), Insulin-like Growth Factors (IGFs), and anti-angiogenic modulators, such as
the soluble form of VEGF receptor-1 (sVEGF-1 or sFlt-1) (Guilbert et al., 1993).
iii.
Decidual vascular remodeling: fetal contribution
Regulated enzymatic digestion of the decidua by the syncytium facilitates its expansion in
fluid filled spaces, called lacunae. Primitive syncytiotrophoblasts organize themselves in
processes (trabeculae) which advance in lacunae and may breach maternal sinusoids
(Benirschke, 1973; Enders, 1989; Herzog, 1909). During the second week of human
gestation, migration of mononuclear cytotrophoblasts into invaginations of the trabeculae
gives rise to the primary villi. Conversion in secondary villi (around day 15-20 post
conception) is encouraged by the invasion of mesenchymal cells, which fill primary villi. At
around day 21 post-implantation, mesenchymal cells proliferate and differentiate in
hemangiogenic precursor cells, thus forming tertiary villi.
The very first placental vascularization consists in the differentiation of pluripotent
mesenchymal cells inside villous core, during a process of de novo local vessels formation
called vasculogenesis (Castellucci et al., 1990). Indeed, it seems that the process of placental
vascularization is mainly driven by de novo formation of new capillaries, rather than
infiltration of embryonic vessels into decidualized tissue.
6
Introduction
Two different types of villi form a complex crowded intervillous space: floating villi and
anchoring villi, the latters being in contact with maternal uterine tissue.
At this step primitive vasculogenesis is strongly regulated by the vascular endothelial growth
factor (VEGF), expressed by cytotrophoblasts and angiogenic precursors (Demir et al., 2004).
By the third week of gestation villi appear composed of two trophoblast layers
(syncytiotrophoblast and cytotrophoblasts), all surrounded by mesenchymal tissue. Hofbauer
cells have been found in tertiary villi surrounding vasculogenic precursor cells and they
probably participate to precursor cell differentiation and proliferation through VEGF secretion
(Demir and Erbengi, 1984; Kaufmann et al., 2004). Villous network and intervillous space are
connected peripherally with maternal sinusoids and maternal spiral arteries, but until 10-12
weeks of gestation extravillous cytotrophoblast plugs obstruct maternal spiral arteries
preventing maternal blood to flow into intervillous space.
Step by step angiogenesis
develops, giving rise to new blood vessels from already existing vessels.
iv.
Maternal vascular change
The first trimester of pregnancy includes also a strong spiral artery remodeling, which is
tightly linked to invasive capacities of some cytotrophoblast cells. Spiral arteries are highresistance vessels with a coiled form that branch out from the uterus and decrease in lumen
diameter as they go towards myometrium, endometrium and decidua. Fetal and placental
development imposes their disorganization and transformation into low-resistance and high
capacitance vessels thanks to the loss of smooth muscle cells and elastic lamina. For this
purpose several mechanisms come into play: migration, changes in cell adhesion, cell dedifferentiation, pro-apoptosis factors release, reorganization in extracellular matrix. All these
processes are interconnected in a tightly regulated manner and orchestrated by a specific
cytotrophoblast sub-population.
Cytotrophoblasts at the top of anchoring villi proliferate and, according to their destination,
they differentiate into interstitial cytotrophoblasts, which invade endometrium until the
inner third of the myometrium, and endovascular cytotrophoblasts, that penetrate into
maternal spiral arteries. Around 10 weeks of gestation endovascular cytotrophoblasts proceed
across the interstitium, reach the wall of spiral arteries and form a plug within the arterial
7
Introduction
lumen, thus preventing maternal blood flow to reach the developing placenta. Starting from
12 weeks of gestation endovascular cytotrophoblasts slide on the vascular walls, invade the
lumen of spiral arteries and stimulate the maternal vascular remodeling.
Vascular remodeling starts with the disorganization of uterine arterial wall even before
trophoblast invasion: the smooth muscle cells layer starts to disorganize, followed by
endothelia basophilia, vacuolization and lumen dilation (Craven et al., 1998). At around 12
weeks of gestation the plug at the distal part of spiral arteries is loosen and endovascular
cytotrophoblasts migrate from the interstitium into the myometrial segments of the spiral
arteries and replace the inner layer of endothelial cells. Trophoblast invasion stimulates also a
fibrinoid deposition on the vascular bed (Brosens et al., 1967)(Whitley and Cartwright, 2010),
mainly composed by fibronectin, collagen IV and laminin, thus forming a new basement
membrane which ensure integrity of remodeled vessels. Trophoblast invasion enhances
apoptosis of endothelial cells and smooth muscle cells by releasing pro-apoptotic factors or
by destabilizing cellular adhesion molecules architecture (Harris, 2010; Whitley and
Cartwright, 2010). Several factors secreted by endovascular cytotrophoblasts may trigger
cellular apoptosis, like tumor necrosis factor alpha (TNF- α), TNF-related apoptosis inducing
ligand (TRAIL) and Fas ligand (FasL) (Hammer and Dohr, 2000; Keogh et al., 2007;
Pijnenborg et al., 1998) .
Even the extracellular matrix is strongly impacted by trophoblast migration. Spiral arteries
walls, like those of other arteries, are organized in three different layers: the intima, the inner
one, composed by a single layer of endothelial cells lying on a basement membrane of
collagen type IV and laminins and in direct contact with the blood flow; the inner elastic
lamina, a layer of collagen type IV, elastin and fibronectin which separates endothelial and
smooth muscle cells and external elastic lamina, which surrounds the smooth muscle cells;
the adventitial layer, the outer one, is composed predominantly of collagen fibers and
fibroblasts. The matrix of elastic lamina is sprinkled by small pores or fenestrae, through
which endothelial cells and smooth muscle cells are connected (Arribas et al., 2006; McGrath
et al., 2005).
Vascular matrix has to be conceived as an architecture that guarantees the stability of
vascular wall and the physiological function of each cell type; alteration of this structure may
8
Introduction
strongly impact cellular interactions, cell survival and cell behavior. And it is exactly what
happens physiologically during the first trimester of pregnancy in order to ensure vascular
remodeling. Disorganization of vascular matrix architecture is initially driven by maternal
leukocytes even before trophoblast invasion, and fully accomplished by interstitial and
endovascular trophoblasts (Craven et al., 1998; Kam et al., 1999; Smith et al., 2009).
Proteolytic enzymes, secreted by trophoblasts, endothelial cells, smooth muscle cells,
macrophages and decidual natural killer cells play a major role in intima and media
disorganization. Serine proteases, with trypsin-like activity, have an active function on
degradation of collagen IV and fibronectin. Matrix metalloproteinases (MMPs), a family of
zinc fingers endopeptidases, participate to whole matrix degradation, and to the release of
molecules affecting vascular cells proliferation and survival. Elastin degradation, for example,
can release peptides responsible for vascular smooth muscle cells de-differentiation (Harris
and Aplin, 2007). TGF-β, released upon the activity of MMP-2, strongly impacts
endovascular trophoblasts migration and their power of invasion (Tse et al., 2002). Survival
and apoptosis of endothelial cells seem to be regulated by the balance between pro-survival
and pro-apoptotic factors, whose induction can be regulated by specific MMPs. Among them,
MMP-9 can stimulate VEGF release, known to be a pro-survival factor; MMP-9 and MMP-2
can induce the cleavage of Collagen-VIII and thus the release of endostatin, a strong inducer
of endothelial cells de-differentiation and apoptosis (Dhanabal et al., 1999; Staun-Ram et al.,
2004). These cellular and extracellular changes contribute altogether to transform spiral
arteries into thick and flaccid vessels, unresponsive to maternal contractility control, with a
diameter that increased three-fold relative to the original, enabling an adequate blood supply
for the developing fetus (Hirano et al., 2002).
This deeper invasion is accentuated around the implantation site (corresponding to the core of
placental bed), and is reduced peripherally, in term of invading cell number and invasiveness
power (Hirano et al., 2002; Lyall, 2005). It has been estimated that virtually around 100-150
arteries are transformed, resulting in a 10-fold increase in blood supply in the uterine wall
during the third trimester of fetal development (Lyall, 2005).
During the first 10 weeks of gestation arteries obstruction induces the creation of a placental
microenvironment with an oxygen tension below 20 mm Hg and, when the plug is bursted,
the oxygen tension rises up to thrice (Jauniaux et al., 2000; Rodesch et al., 1992). Low
9
Introduction
oxygen concentration can protect differentiating organs from free oxygen radicals and oxygen
mediated teratogenesis (Burton et al., 2003; Jauniaux et al., 2003; Nicol et al., 2000),
suggesting a finalist idea for this low first-trimester oxygen pressure. The role of oxygen in
fetal and placental development will be discussed in details in chapter 3.
C. Full term placenta: structure and function
The full-term placenta appears as a discoid structure, mainly of fetal origin, which receives
blood from the fetus and from the mother and regulates blood supply by two different
vascular systems: the utero-placental circulation and the feto-placental circulation.
The utero-placental circulation starts developing at the end of the first trimester when plugs
occluding the decidual segments of spiral arteries are lost and when maternal blood flows into
the intervillous space. Maternal blood accesses the feto-placental unit through the basal plate
endometrial arteries, and flows around tertiary villi, charged in oxygen and nutrients, then the
blood deoxygenated and nutrient depleted returns to the maternal systemic circulation via the
venous system of the basal plate. Blood circulation is facilitated by the spatial organization
of blood vessels and blood pressure across different types of vessels. Indeed perpendicular
orientation of spiral arteries and parallel orientation of veins within the uterine wall avoid
blood squeezing from the intervillous space. At this time a blood pressure gradient has been
established between uterine arteries and intervillous space (80 mm Hg in uterine arteries
versus 10 mmHg in the intervillous space), so that the pressure gradient, together with arterial
low resistance, increase the performance of the utero-placental perfusion.
During the third month tertiary villi concentrate in the chorionic plate (chorion frondosum),
the site of feto-maternal exchanges. In the rest of the chorion (chorion leave), tertiary villi
degenerate and no exchange occurs.
Fetus addresses deoxygenated blood to the placenta through umbilical cord, containing two
umbilical arteries and one umbilical vein, the latter being in this case the conduct of
oxygenated blood towards the fetal heart. Umbilical cord arteries invade chorionic plate in a
pattern of disperse type branching, giving rise to a network of chorionic arteries, which
branch into cotyledon arteries. Cotiledonary vessels begin branching into the tertiary villi
10
Introduction
branches, thus forming an arterio-capillary venous network in which fetal and maternal
bloods are close, but without intermingling.
In Humans, and Primates in general, the placenta has a hemochorial structure: despite this,
like in other mammals whose placentas may be epithelio- or endothelio-chorial, maternal and
fetal blood never mix, since they remain separated by a thin layer of syncytiotrophoblast cells.
As mentioned before, in addition to nutrition and excretion functions, placenta has also
endocrine, and immunological functions, in order to prepare maternal body to guarantee fetal
growth and development and protection of the fetus in utero (Figure 1).
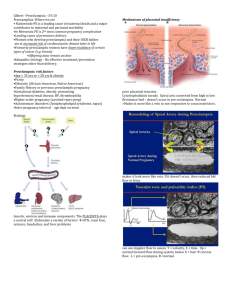
Figure 1: Principal feto- maternal exchanges
2. Preeclampsia
A. Introduction
Preeclampsia is a unique complication of human pregnancy with a great impact on
maternal mortality and perinatal morbidity worldwide.
Clinically preeclampsia is defined by the onset of hypertension (systolic and diastolic blood
exceeding 140 and 90 mm Hg, respectively, on at least 2 occasion 6 hours apart) and
proteinuria (protein excretion above 300 mg in a 24h urine collection) at or after the 20th
week of gestation in normotensive women (ACOG Committee on Practice Bulletins-Obstetrics, 2002).
11
Introduction
According to the onset of the clinical features preeclampsia can be split (quite arbitrarily) into
two entities: early-onset preeclampsia, when preeclampsia develops before 34 weeks of
gestation, and late-onset preeclampsia, if symptoms occur after 34 weeks of gestation. Early
symptoms manifestation is usually associated with the increased severity of the disease.
However this classification is still debated among the scientific community and a global
consensus on the definition of mild and severe preeclampsia is still lacking (Brown et al.,
2001). The main points called into question are: 1) consideration of gestational hypertension
without proteinuria as preeclampsia, 2) definition of early onset preeclampsia before 34
(Canada) or 35 (USA) weeks of gestation, 3) definition of severe hypertension. Diagnostic
discordance reflects the complexity and variability of this disease, which sometime makes it
quite hard to find concordances among different scientific works.
Despite these two well-established and generally accepted cardinal features, several other
symptoms often complicate the clinical picture and, moreover, they could be very variable in
term of onset, presentation and severity. The organs mostly affected during the preeclamptic
syndrome are the kidney, the liver and the brain. Patients suffering preeclampsia may develop
severe headache or visual alteration, pulmonary edema, inferior limbs edema, hemolysis,
hepatic infarction or abruptions, intra-abdominal bleeding, thrombocytopenia, Hemolysis,
Elevated Liver enzymes and Low Platelets syndrome (HELLP) or eclampsia, which in the
worst case lead to maternal death.
Historically edema was a criterion defining preeclampsia, together with hypertension and
proteinuria. Later on, since gain of weight and edema of the feet, hands and face, are a
common trait of women during the last trimester of normal pregnancy, this symptom is not
anymore considered as a diagnostic tool.
HELLP syndrome refers not only to a liver dysfunction, but, also to a more diffused
coagulopathy and thrombotic microangiopathy. The coagulation state is more active in normal
pregnancies, but hyper activated in preeclampsia. Several serum biomarkers of a procoagulant state, can be detected even after the onset of the symptoms (Estellés et al., 1989;
Hsu et al., 1993; Taylor et al., 1991). The same markers of platelet activation are abnormal
before the manifestation of the disease (Ballegeer et al., 1992).
12
Introduction
Eclampsia refers to cerebral seizures that usually occur after the onset of proteinuria, and
rarely 48h up to 1 month post partum. In the latter case, one third of cases refers to
preeclamptic pregnancies without manifested symptoms before parturition (Sibai, 2005).
Eclampsia could be the end outcome of cerebral edema and vasoconstriction and usually
begins with premonitory signs like headache and visual disturbance. Eclampsia, renal failure
cerebrovascular complications, including stroke and cerebral hemorrhage, are the main causes
of maternal death in preeclamptic women.
In some cases even the developing fetus and neonate suffer complications that can strongly
influence their growth. Intrauterine Growth Restriction (IUGR) occurs when the fetus fails to
reach his potential growth (observed in up to a third of preeclampsias), or his survival, in the
case of prematurity, bronchopulmonary dysplasia, and placental abruption.
Usually once symptoms appear, the maternal dysfunction gets worse and worse. The only
way to reverse disease is the delivery of the feto-placental unit, which is the main cause of
prematurity and perinatal death.
B. Epidemiology
Preeclampsia is one of the most common pregnancy disorders, affecting 2-5% of women
worldwide (Goldenberg et al., 2008). It is one of the main causes of maternal death per year,
estimated around 30% of hypertensive pregnancies. In developing countries, where
management and diagnosis of pregnancies complications are more difficult or still lacking,
preeclampsia is responsible for 20-30% of the total number of maternal deaths per year,
estimated around 60 000 (WHO 2005 World Healt Report). Thus, one of the main goals of the
World Health organization (WHO) Nations has been considered to reduce maternal mortality
by 75% between 1990 and 2015 (Osungbade and Ige, 2011).
Considering gravity and severity, 25% of preeclampsia cases are severe, and 5-10% may
evolve in the severe symptoms depicted before.
HELLP syndrome complicates 20% of severe preeclampsia cases (Sibai et al., 1993;
Weinstein, 1982). HELLP syndrome usually arises in late gestation, but in 30% of cases
symptoms can appear post-partum. It could be also associated to other complications, as
13
Introduction
abruptio placenta (9-20%), exaggerated coagulation (5-56%), renal failure (7-36%) (Haram et
al.,2009).
Eclampsia complicates 1-2 % of preeclamptic pregnancies and in 79% of cases neurological
abnormalities develop the week before the first seizure like headache, blurred vision and
temporary loss of vision (Knight and UKOSS, 2007). In rare cases preeclampsia can be
asymptomatic during pregnancy and complications can appear post partum, sometimes in the
form of eclampsia (Sibai and Stella, 2009).
In some cases preeclampsia represents a considerable risk for the fetal health and even
survival. Indeed, according to estimations, 5-18 % to 1/3 of preeclampsia cases are associated
to intra-uterine growth restriction (IUGR), 1-6% to placental abruption and 1-9 % perinatal
mortality (Sibai, 2003). In developed countries fetal mortality has been strongly reduced in
the last 20 years thanks to medical induction of parturition and improvement of medical
follow-up for pre-term birth; still in France for instance, it is estimated that 1/3 of the fetal
deaths is directly or indirectly caused by preeclampsia, which represents around 600 fetal
deaths amongst 1900 in 2010. On the other hand, preeclampsia remains responsible for 15%
of pre-term birth in developing countries.
C. Risk factors
The high variability in symptoms, severity and disease onset reflects the large spectrum of
risk factors that predispose to preeclampsia. Whether different risk factors have to be
considered as a classification tool for different forms of the same disease is still debated.
i.
Medical conditions
Several pre-existing pathologies can increase the risk to develop preeclampsia, including
chronic hypertension, diabetes mellitus, renal diseases, obesity and hypercoagulable state
and chronic infections (Barton and Sibai, 2008; Duckitt and Harrington, 2005; LópezJaramillo et al., 2008).
14
Introduction
All these pathologies share in common a chronic inflammation status; so that stressed and
activated endothelium could be more sensible to the pregnancy charges.
Autoimmune
diseases are a well described risk factor for preeclampsia such as lupus erythematosus and
even more documented in the case of the antiphospholipid syndrome (Clowse et al., 2008;
Heilmann et al., 2011; Salmon et al., 2011).
ii.
Genetic component, known and unknown
A familial history of preeclampsia is one of the risk factors, thus reinforcing the idea of an
important genetic contribution to this disease. The risk of disease is boosted to up to four
folds in women whose first-degree relatives suffered of a preeclamptic syndrome (mother,
sisters, or both) (Carr et al., 2005). Women with antecedents of preeclampsia in their prior
pregnancy have a considerably (~7 fold) increased risk in the next ones, despite the fact that
generally preeclampsia is a disease of the first pregnancy (Duckitt and Harrington, 2005;
Klungsøyr et al., 2012). An increased risk is manifest also in a history of preeclampsia of
father’s relatives (Esplin et al., 2001), owing to the paternal contribution to the building of
placenta and developing fetus.
Ethnical origin, too, may represent a point to keep in consideration. In fact the incidence of
preeclampsia is increased in African women, while it is intermediate in Caucasians and
reduced in Asian women (Cruickshank and Beevers, 1982). The increased incidence in
African countries may be linked to an augmented risk of hypertension, which is a strong
predisposition factor for adverse pregnancies outcomes (Tsikouras et al., 2012).
Women with a fetus affected by trisomy 21 have a higher risk of the disease compared to a
normal pregnancy (Banerjee et al., 2002). This is also the case for the rarer trisomy 13,
leading to speculate that genes on chromosome 13 could be implied in the pathophysiology of
preeclampsia (Tuohy and James, 1992).
The inherited nature of preeclampsia has been investigated in several studies on targeted
genes or genome-wide approach in order to find polymorphisms, genes, or genomic
regions associated to the disease.
15
Introduction
Candidate gene approaches identified several genes that could be part of the causes of the
disease. These genes can be grouped in five main functional categories (vasoactive proteins,
thrombophilia and hypofibrinolysis, oxidative stress and lipid metabolism, endothelial injury,
immunogenetics), according to their physiological role and implication in the disease (Table
1) (Mütze et al., 2008).
Table 1: Putative genes and polymorphisms involved in preeclampsia
Among 22 polymorphisms in 15 genes, found at least in two separate studies, only seven
variants have been confirmed by a recent meta-analysis approach (Buurma et al., 2013): it is
the case of polymorphisms near the genes ACE (angiotensin I converting enzyme), CTLA4
(cytotoxic T-lymphocyte-associated protein 4), LPL (lipoprotein lipase), and SERPIN1 (serin
protease inhibitor 1), F2 (coagulation factor 2, or prothrombin), FV (coagulation factor V).
16
Introduction
Interestingly most of these genes are also associated to an increased risk in cardiovascular
disease.
Unfortunately separate studies often show discordance in term of results, and for the most part
of genes further confirmation and functional validation are still lacking.
It is for example the case for methylenetetrahydrofolate reductase, factor V Leiden variant,
and protrombin, whose roles in the disease were largely investigated in numerous target genes
studies and meta –analyses, with controversial results (Gerhardt et al., 2005; Lin and August,
2005; Mütze et al., 2008; Rodger et al., 2010). Meta-analysis investigation failed to reproduce
relevance of genes involved in the renin-angiotensin system (AGT or SERPINA8, coding for
angiotensinogen, and AT1R, coding for angiotensin II receptor type 1) and NOS3, coding for
endothelial NO synthase. In the specific case of NOS3 a recent meta-analysis showed that one
out of two polymorphisms identified, does have a significant association with preeclampsia
(Dai et al., 2013).
Meta-analysis approaches also identified the T allele of angiotensinogen M235T as implicated
in the disease, but gene candidate approaches, mainly concentrated on ACE (angiotensin
converting enzyme), angiotensin II type 1 and type 2 receptor, failed to confirm this result,
again (Medica et al., 2007; Zafarmand et al., 2008).
The major reason of this disharmony lies presumably in discordance in disease definition and
complexity due to population heterogeneity, which makes it more difficult to reproduce
results in populations of different ethnic origins.
Another interesting approach in order to find putative genetic regions involved in
preeclampsia is the genome-wide linkage screening. Accordingly, remarkable linkage peaks
have been found in the specific genomic regions 2p13 (Arngrímsson et al., 1999), 2p25, 9p13
(Laivuori et al., 2003), and loci on chromosomes 2q, 9p, 10q, 11q, 22q (Lachmeijer et al.,
2001; Moses et al., 2000), but data replication has not been reported at this moment in other
linkage association studies.
Specifically it is the case for four single nucleotides polymorphisms (SNPs) of Activin A
receptor type IIA, which have been found associated with preeclampsia (Roten et al., 2009),
17
Introduction
but contradicted by another study on 74 families from Australia/ New Zealand, which failed
to find the same result (Fitzpatrick et al., 2009). The same approach has been used to find the
gene ROCK2 (rho associated coiled-coil protein kinase 2) in the region 2p25 as a gene
possibly involved in preeclampsia (Ark et al., 2005). Later on a study on ten polymorphisms
within ROCK2 did not highlight any linkage peaks on this region (Peterson et al., 2009).
Further validation and confirmation are waiting for polymorphisms identified in COMT
(cathecol-O-methyltransferase), SERPINA3, HLA-G, CCR5 (chemokine receptor 5), and
genes coding for complement regulatory molecules like MCP (membrane cofactor protein)
and CF1 (complement factor 1) (Chelbi et al., 2012; Gurdol et al., 2012; Lim et al., 2010;
Qing et al., 2011; Salmon et al., 2011; Zhang et al., 2012).
Statistical weakness is the Achilles’s heel of most part of these studies, which increases
results variability and negatively influences reproducibility.
Globally genetic polymorphisms can give their contribution to the onset of preeclampsia, but
the disease remains a complex interconnection among the genetic, immunologic and
environmental components.
iii.
Other risk factors
Other risk factors could be directly linked to the mother, to ther pregnancy, to the couple, and
to the lifestyle.
The mother. Some other factors, linked to the mother status, come into play in developing
preeclampsia. Nulliparity is one of the major risk factors of preeclampsia, which in women at
the first pregnancy rises up to 7.5 % (Duckitt and Harrington, 2005). Moreover 75% of
preeclampsia occurs in nulliparous women. Maternal age, too, has its importance. In fact
over 40 years old aged women have an increased risk of developing the disease (Duckitt and
Harrington, 2005; Seoud et al., 2002). On the opposite side, too young women develop more
frequently gestational hypertension and preeclampsia as well (Tsikouras et al., 2012).
18
Introduction
Pregnancy. Increased placental mass, which occurs in multiple gestations, hydatiform
mole, extrauterine pregnancies and triple gestations, increases the risk to develop
preeclampsia (Coonrod et al., 1995; Roberts and Gammill, 2005; Worley et al., 2008).
Similarily a whole interpregnancy interval can increase the risk to develop preeclampsia
(Skjaerven et al., 2002). Indeed a ten years interval between two consecutive pregnancies
corresponds to a risk very close to nulliparity (Skjaerven et al., 2002).
Association between pregnancies disorders and assisted reproductive technologies is also a
developing field (Thomopoulos et al., 2013). It has been suggested that links with
preeclampsia or growth restriction could reside in some modifications of the epigenetic
landscape inherited from the in vitro culture of gametes which could perturb the subsequent
feto-placental development (Fauque et al., 2007).
The couple. Different ethnical origins within the couple may represent an increased risk to
develop preeclampsia (Caughey et al., 2005), thus emphasizing that immungenetics
background is important for a successful feto-placental development.
Preeclampsia risk is increased also in multiparous women who change partner (Dekker and
Robillard, 2007; Dekker et al., 1998). In this case they reach the same risk level as nulliparous
women. A recent hypothesis suggests that a stable familial structure and paternal care may
represent an evolutionary advantage, supported in some way by preeclampsia, which allows
preeclampsia to escape to Darwinian effect and concur to the global maintenance of
preeclampsia- predisposing alleles throughout populations (Chelbi et al., sous presse).
Sperm exposition could induce a uterine tolerance towards father antigens which could be
“healthy” for later pregnancies with the same partner (Williams, 2012). Indeed it has been
shown that even oral sex can prevent risk of preeclampsia (Koelman et al., 2000), while
barrier contraception (Klonoff-Cohen et al., 1989) and conception by intracytoplasmic sperm
injection (Wang et al., 2004) increases the risk of the disease. This may imply that a limited
exposure to seminal liquid or paternal antigens could be a predisposing factor (Kajino et al.,
1988) and suggests that exposure to paternal antigens favor the implantation and development
of an embryo with a different immunologic and genetic anlagen potentially detected in the
uterus as a hemi- allograft.
19
Introduction
Allelic combination of maternal Killer Immunoglobulin-like receptor AA (KIR-AA) and
fetal genotype HLA-C, inherited from the father, can contribute to the onset of preeclampsia
(Hiby et al., 2004, 2010) In the same study the authors showed that KIR-AA and HLA-C2 are
inversely correlated in term of frequency in different human populations. The adverse
pressure of selection on their combination within the same population may suggest that
reproductive achievement could participate to the human HLA-C and KIR polymorphisms
selection (Hiby et al., 2004, 2010).
All these factors reposition immunologic tolerance as an important element to take into
account among predisposing factors to preeclampsia and a field of research which could
supply a complementary perspective to the feto-maternal cross-talk during preeclamptic
pregnancies.
The lifestyle. According to lifestyle, socioeconomic conditions could also influence mother
and fetus wellbeing. It has been shown that incidence of preeclampsia is increased in
developing countries and in situations that are unfavorable for women education,
alimentation, and globally welfare state (Cerón-Mireles et al., 2001; Funai et al., 2005; LópezJaramillo et al., 2001). Nevertheless these data were not substantiated by a correlation
between preeclampsia and economic status of women in developed countries (Lawlor et al.,
2005). So it is possible that pregnancy, which is a state of stress and important demand to the
maternal body, needs a healthy background to correctly progress (and sometimes maternal
health is underestimated in developing countries). On the other hand, ethnicity could explain
part of the increased risk of preeclampsia in developing countries.
Life in high altitude may represent another important risk factor (Keyes et al., 2003; Palmer
et al., 1999) that could be linked to higher hematocrit (the red volume percentage of blood red
cells) and lower blood oxygen pressure.
Viscous blood is a condition associated to
pregnancy, and blood viscosity is increased in preeclampsia (Kametas et al., 2004). Lower
oxygen pressure could induce a prolonged hypoxic status in the developing placenta which
affects its normal development and it is already described as associated to pathophysiology of
preeclampsia (Palmer et al., 1999).
20
Introduction
Smoking too, can influence the risk of preeclampsia, but, unexpectedly, it seems to reduce the
risk of the disease (Conde-Agudelo et al., 1999). A recent study shows that smokers have an
increased serum level of Placental Growth factor (PlGF), which could favor a correct
placental development and avoid preeclampsia outcome (Llurba et al., 2013).
So complexity of risk factors, genetics and environmental, well reflects the variability of a
disease whose pathophysiology remains partly a mystery and for which a specific treatment
have still to be found.
D. Management and treatment of preeclampsia
Regarding management of preeclampsia there is no universally accepted standards of care,
and moreover it depends on local guidelines. Nevertheless a series of practices, according to
severity and gestational age, seems associated to reduced adverse maternal and perinatal
outcomes. These practices include surveillance of systolic and diastolic blood pressure,
prevention and treatment of eclampsia, assessment of all vulnerable organ systems affected
during preeclampsia, and control of fetal status as well.
Expectant management care could be conceivable in women with a gestational age less than
34 weeks. Indeed treatment of symptoms more than a stabilization and delivery could
improve fetal development without increasing too much the risk for the mother (Magee et al.,
2009). However insufficient data are available to choose between expectant or interventionist
management outcomes in every case (Churchill and Duley, 2002), so it is hard or even
impossible to establish a common good practice. Interventionist management is suitable in
women affected by preeclampsia before 24 weeks of gestation, preeclamptic women at term
and preeclampsia complicated by HELLP syndrome. In the last two cases induction of labor
and expedited delivery are the most common policies. Expectant management does not result
in improvement of fetal conditions for women affected by preeclampsia before 24 weeks of
gestation, and in this case, maternal risk often imposes a sudden intervention (Gaugler-Senden
et al., 2006).
21
Introduction
Expectant management of preeclampsia implies the systemic monitoring of all symptoms
typical of preeclampsia, and, if possible, their control trough pharmacological treatment in
order to keep pregnancy as long as possible without increasing the risks for maternal health.
In fact, once preeclampsia arises, symptoms get worse all along pregnancy and, despite the
great efforts to go deeper in the comprehension of the pathophysiology, delivery of the fetoplacental unit remains the only efficient action to rescue all symptoms of the disease.
Expectant management aims mainly at controlling hypertension and preventing seizures of
eclampsia.
Antihypertensive drugs are prescribed when diastolic and systolic blood pressure exceeds
respectively 160 mmHg and 110 mmHg. Most common drugs used in case of preeclampsia
are alpha and beta blockers (labetalol, oxprenolol), central antihypertensive (methyldopa)
and calcium channels blockers (nifedipin or nicardipin, verapamil) (Duley et al., 2006;
Magee et al., 2011). There is no universal rule to choose an antihypertensive drug rather than
another and in most cases it depends on clinician experiences. The aim of the antihypertensive
treatment is to keep stable blood pressure between 140 and 160 mmHg for the systolic
pressure and 90-110 for the diastolic pressure, and to prevent cerebrovascular complications
(Petit et al., 2009). But sometimes antihypertensive treatments may cause the opposite effect
and induce hypotension which can be dramatic for feto-placental development. It could be the
case for diazoxide treatment (Hennessy et al., 2007). The choice of conversion enzyme
inhibitor (IEC) and angiotensin II receptors antagonists is forbidden in pregnancy because of
their toxicity.
Eclampsia seizures are usually treated and prevented by MgSO4, which is normally used
also for prophylaxis in women with severe preeclampsia, and sometimes associated with
nifedipine. MgSO4 treatment reduces cerebral ischemia and neuronal damage by improving
cerebral vasodilatation (Belfort, 1992; Duley et al., 2003). But this treatment induces
sometimes side effects for the mother and for the fetus, since it can cross the placental barrier.
Magnesium sulfate treatment can be associated in fact to post partum hemorrhage and fetal
hyporeflexia,
respiratory
depression,
flaccidity,
all
symptoms
linked
to
fetal
hypermagnesemia (Lipsitz, 1971; Witlin et al., 1997).
22
Introduction
Today, no pharmacogenomic studies are available on the effects of the cited drugs in
preeclampsia, and most data originate from studies assessing the effects of treatments on
hypertensive patients.
It is the case for example for the allele G of A2996G and allele A of G498A polymorphisms
of eNOS genes: both alleles are associated to an increased efficiency of beta- blocker atenolol
in hypertensive patients (Liljedahl et al., 2003). A recent study also showed the improvement
of hypertension treatment according to the allelic variant of the gene CACNA1A: calcium
channel blockers treatment is more efficient in patients with the rs1051375 A/A allele
composition, while beta blockers act better on a genetic background rs1051375 G/G; finally
in heterozygous individuals both treatment have the same efficiency (Beitelshees et al., 2009).
Nevertheless these studies could offer important guidelines in the treatment of preeclamptic
women. In fact a better “classification” of preeclampsia, in term of causes and clinical
aspects, together with the segregation of patients in drug responders and non-responders,
could be a promising frontier for the so-called P4 medicine (prediction, personalization,
prevention, participation), in order to better predict risks and individualize treatment in a
direct and rational manner according to individual genetic background.
E. Prevention of preeclampsia
Preventing treatments have been the main topic of several studies, in order to find drugs and
treatment that could reduce preeclampsia incidence.
Low-dose acetylsalicylic acid (aspirin) is one of the most promising effective agents in the
prevention of preeclampsia. Low–dose aspirin reduces the synthesis of thromboxane A2 via
acetylation and inhibition of the enzyme Cyclo-oxygenase-1 (COX-1) (Shimokawa and
Smith, 1992). In this way, its effect could be crucial in the reduction of vasoconstriction,
platelet aggregation and thrombosis. Even though its role in prevention and therapy has been
debated for long time (Rossi and Mullin, 2011), a recent meta- analysis confirmed its
beneficial effect if the treatment starts early in pregnancy, before 16 weeks of gestation
(Bakhti and Vaiman, 2011; Roberge et al., 2012a, 2012b). Interestingly efficiency of the
treatment is influenced by the period of daily intake: the treatment seems more efficient if
23
Introduction
aspirin is administered in the evening rather than in the morning (Ayala et al., 2012).
Nowadays, according to the recent guidelines of the National Institute for Health and clinical
Excellence, it is recommended for women with high risk to develop preeclampsia to start a
treatment based on 75 mg daily intake of aspirin from the 12th week of gestation (National
Collaborating Centre for Women’s and Children’s Health (UK), 2010).
Another promising treatment is low-molecular-weight heparin (1mg/kg). In particular, in
association with aspirin treatment, it could prevent the risk of thrombosis and have a
beneficial effect on haemostasis and generalized inflammation (Gris et al., 2011; Kupferminc
et al., 2011; de Vries et al., 2012). Moreover it has been shown that heparin can increase
VEGF expression in vitro (Mello et al., 2005): this leads to speculate on its positive role on
placental development.
In order to reduce oxidative stress, a current hallmark of preeclampsia, different antioxidant
treatments have been tested (such as vitamin C, D, E, NO donors) but insufficient evidence
supports their efficacy in reducing the risk of preeclampsia (Meher and Duley, 2007; Parrish
et al., 2013; Rossi and Mullin, 2011; Thorne-Lyman and Fawzi, 2012; Roberts, 2010).
It should be noted that intervention to prevent preeclampsia is provided only for women at
high risk of preeclampsia, who have already a history of preeclampsia (Pottecher et al.,
2009). And if we consider that 75% of cases of preeclampsia touch nulliparous women, the
necessity to decrypt with high accuracy the risks factors and find the earliest possible
biomarkers of the disease, become more and more important.
F. Pathophysiology of preeclampsia
Symptoms of preeclampsia appear approximately after 18-20 weeks of gestation and affect
mainly the mother. Nevertheless more and more evidences suggest that the real causes of the
disease came earlier, during the first 12 weeks of gestation and are not ascribed to the
maternal organism but rather to the placenta which is a feto-maternal organ..
The first reason is that preeclampsia needs a placenta to develop. As mentioned before, all
maternal disorders disappear generally very fast after the delivery of the placenta. Moreover
preeclampsia may occur even without a fetus in the presence of placenta-like tissue (as in the
24
Introduction
case of hydatiform moles) (Koga et al., 2010). In the case of extrauterine pregnancies,
removal of the placenta (and not only of the fetus) is required to block symptoms progression
(Shembrey and Noble, 1995). In the case of seizures of eclampsia post partum, they are
usually a side effect of placental fragments that are retained within the maternal body after
parturition (Matsuo et al., 2007).
The second finding that prompted research to investigate on a very early cause of disease is
the state of the preeclamptic placenta. Placentas from severe preeclamptic pregnancies
suffer endothelial damage, fibrin deposition, atherosclerosis, necrosis. All these pathological
evidences are putative consequences of placental hypoperfusion and ischemia which may
occur during early placental development (Salafia et al., 1998).
This leads to the elaboration of a “two steps” model of the disease, with different target
organs, different timings, but strictly interconnected (Roberts and Gammill, 2005).
The first step is asymptomatic, occurs during the first weeks of gestation and undermines
normal placental development. It is ascribed to a defect in trophoblast invasion, responsible
for a failure of spiral arteries remodeling which causes placental oxidative stress due to
ischemia and hypoperfusion (Granger et al., 2002; Gupta et al., 2005).
The second one is the symptomatic step, known as the maternal syndrome. Stressed
placentas are thought to release plasma factors and placental debris into the maternal
circulation which induce a generalized inflammatory response, endothelial damage and hyper
activation of the state of coagulation (Redman and Sargent, 2004; Roberts et al., 1989). This
second step will be described in details in the next chapter.
1st step: Abnormal placentation
The hypothesis of a reduced placental perfusion in preeclampsia originates more than 70
years ago (EW The relation between hydatid moles, relative ischemia of the gravid uterus,
and the placental origin of eclampsia. Am J Obstet Gynecol. 1939). Later on, reduced
placental perfusion was assessed by radioactive washout tests to measure intervillous blood
flow (Käär et al., 1980) and more recently by abnormal uterine artery Doppler ultrasound
(North et al., 1994; Papageorghiou et al., 2002). Further confirmation comes from animal
25
Introduction
models in which a preeclamspia –like syndrome can be induced by obstruction of spiral
arteries thus reducing blood flow in uterine cavity (the RUPP model for Restricted Uterine
Perfusion Pressure) (Crews et al., 2000; Khalil and Granger, 2002).
These observations were supported also by morphological alterations of placental tissue.
Preeclamptic placentas biopsies highlight that arteries remodeling touches only 44% of
decidual spiral arteries and 18 % of myometrial spiral arteries against 100% remodeled
decidual arteries and 76% remodeled myometrial arteries in normal placentas (Meekins et al.,
1994). So that in preeclampsia part of arteries is not modified, and even in the case where
remodeling occurs, it is reduced to the uterine decidual region, while in normal pregnancies it
reaches the inner third of maternal myometrium.
On the basis of these findings, the root cause of preeclampsia appears to reside in an early
defect of extravillous cytotrophoblasts differentiation and invasion of uterine vessels.
Reduced utero-placental blood flow is the conclusion of the impaired vascular remodeling of
spiral arteries during the first weeks of gestation. Because of insufficient or absent vasculature
re-organization, blood vessels retain their smooth muscle and elastic lamina component and
instead of high capacitance and unresponsive vessels, they maintain high vascular resistance,
elasticity and response to contractility. This alteration may explain anomalies in Doppler
ultrasounds of uterine vessels in first trimester placentas that later evolve in preeclamptic
syndrome (Carbillon, 2012).
In normal pregnancy invasive trophoblasts undergo a process named “pseudovasculogenesis”
in which they change their phenotype in a process similar to epithelial- endothelial transition.
Trophoblast invasiveness depends on a switch in the expression of adhesion molecules, so
that cytotrophoblasts acquire a typical endothelial-like phenotype.
Cytotrophoblast progenitors leave their epithelial-like phenotype by down-regulation of
adhesion molecules such as epithelial cadherin (E-cadherin) and α6β4 integrin; then they
invade uterine vasculature, acquiring a more similar endothelial-like phenotype, with the
expression of vascular endothelial cadherin (VE-cadherin), vascular cell adhesion molecule-1
(VCAM), platelet-endothelial cell adhesion molecule-1 (PECAM-1), α5β3 integrin (Zhou et
al., 1997), urokinase plasminogen activator (Queenan et al., 1987) and thrombin receptor
26
Introduction
(Even-Ram et al., 1998). Down-regulation of adhesion molecules is of primary importance for
cytotrophoblast migration through extracellular matrix and acquisition of endothelial marks
allows migration toward the decidua and myometrium and replacement of the muscoloendothelial lining of spiral arteries.
In preeclampsia, placental bed shallow invasion is mainly due to a defective switch towards
an invasive cytotrophoblast phenotype: cytotrophoblasts keep an epithelial-like phenotype, as
it is demonstrated by the E-cadherin expression in cytotrophoblasts of the villi, decidua, and
even in uterine arterioles, by a weak expression α5β3 integrin and no expression of VEcadherin in cytotrophoblasts of uterine walls (Zhou et al., 1997). Reduced invasiveness is also
associated with a reduced cytotrophoblast expression of Vascular Endothelial growth Factor
(VEGF) and its receptor Flt-1, a pro-angiogenic factor that regulates vasodilatation and
invasiveness via the up-regulation of nitric oxide (NO) (Papapetropoulos et al., 1997; Zhou et
al., 2002). NO is a potent vasodilator which can, in turn, enhances cytotrophoblast migration
through the up-regulation of MMP2 and MMP9 (Novaro et al., 2001). Its reduced availability
can participate to hypoperfusion and/or intermittent perfusion of the placental bed. Impaired
change in cytotrophoblast phenotype is also associated to a decrease of the number and
density of endovascular cytotrophoblasts in the decidual region observed in preeclamptic
placentas (Noris et al., 2005).
A direct consequence of the altered vasculature remodeling is a perturbed blood flow in
placental bed, leading to cycles of hypoxia–reoxygenation. Variation of oxygen tension has
two main consequences: alteration of feto-placental development and increased oxidative
stress. Oxygen tension has a main role in placental development and fetal programming,
since a subset of genes and pathways are direct targets of the hypoxia-specific transcription
factor, HIF-1, regulated and stabilized under hypoxic condition (Patel et al., 2010). The role
of oxygen tension will be described in details in chapter 3. In the context of cytotrophoblast
behavior, it has been shown that HIF-1alpha is up- regulated in preeclamptic placenta, and
one of its main targets is transforming growth factor beta 3 (TGF-β3), which regulates
negatively trophoblast invasion (Caniggia et al., 1999, 2000; Rajakumar et al., 2001).
Oxidative stress is the result of the unbalance between reactive oxygen species (ROS)
production and insufficient cellular redox capacities. The feto-placental unit is constantly
27
Introduction
exposed to oxidative stress in normal pregnancy, and this is due to the O2 rich blood coming
from the mother, the hyper active placental metabolism and the extensive embryonic cell
divisions. But in preeclampsia, exacerbated oxidative stress puts the mother and the foetus too
in critical conditions. In preeclamptic placentas, increased ROS production and reduced
detoxifying enzymatic activities lead to lipid peroxidation, free radical formation and
production of isoprostanes (Madazli et al., 2002; Serdar et al., 2002; Wang et al., 1992). This
state is triggered by increased levels of NADPH-oxidase (NOX), increased activity of
xanthine oxidase, that contribute to superoxide formation, and decreased antioxidant enzymes
like superoxide dismutase (SOD), glutathione peroxidase, glutathione-S-stransferase (Cui et
al., 2006; Many et al., 2000; Raijmakers et al., 2004; Wang and Walsh, 1996).
The main consequences of oxidative stress affect the cytotrophoblast fate and more generally
the cellular survival during placental development. It has been shown that trophoblast cell
lines exposed to oxidative stress in vitro reproduce the same structural damages and perturbed
gene expression as preeclamptic placentas (Many et al., 2000; Sikkema et al., 2001;
Vanderlelie et al., 2005; Wang and Walsh, 2001). A physiological oxygen supply, which
occurs when maternal vessels perfuse correctly the intervillous space, triggers the
cytotrophoblasts switch towards an invasive phenotype and pseudovasculogenesis. Oxidative
stress is also the leading cause of an increased apoptoticapoptotic events (Ishihara et al., 2002;
Leung et al., 2001), as it was proved by in vitro placental tissue cultivated in hypoxic
conditions or exposed to cycles of hypoxia-reoxygenation (Hung and Burton, 2006; Huppertz
et al., 2003; Levy et al., 2000; Mondon et al., 2005).
Apart from oxidative stress, another type of stress recently described in preeclamptic
placentas is the induction of protein tyrosine nitration. Protein nitration is a physiological
protein modification, which has already been found increased in pathologic conditions like
inflammation and cardiovascular diseases, thus suggesting a putative role in vascular biology
(Peluffo and Radi, 2007; Turko and Murad, 2002). In the preeclamptic placental bed,
nitrotyrosines have been found increased (Bosco et al., 2012; Myatt et al., 1996), but whether
nitration is the cause or the collateral effect of oxidative stress is still debated. In fact
oxidative stress may increase the combination of superoxides and nitric oxide, thus favouring
the formation of peroxynitrite (ONOO-). Peroxynitrite can damage vascular reactivity through
nitration and alteration of NOS, prostacyclin synthase, cyclooxygenase, critical modulation of
28
Introduction
VEGF signaling pathway, and finally reduction of NO bioavailability and of its vasodilators
effects.
Higher pressure blood flow associated to impaired arteries remodeling and stress conditions
may participate to syncytial shedding, and increased apoptosis, which has been observed in
preeclamptic placentas (Lala and Chakraborty, 2003). In the last decades more and more
evidences suggest that placental debris, necrotic cytotrophoblasts and syncytiotrophoblast are
released into the intervillous space and then in the maternal circulation, thus causing the
maternal syndrome of the 3rd trimester (Redman and Sargent, 2000).
Indeed the first step, referring to abnormal placentation, occurs during the first trimester of
pregnancy and, despite the pathological changes that affect placental bed, is asymptomatic.
But at the same time it prepares and participates to the second phase of the disease that makes
of the maternal body the real battlefield.
29
Introduction
Chapter II. Maternal syndrome during preeclampsia
Early causes of preeclampsia evolve into the symptomatic phase of the disease, which
involves the whole maternal organism. Maternal complications include vasoconstriction,
chronic inflammation, hemodynamic changes, edema, glomerular endotheliosis and a
hypercoagulable state. Symptoms are more evident and severe in the most vascularized
organs, like the kidney, the liver and the brain. Globally maternal pathology can be seen as
the ending outcome of a generalized endothelial dysfunction.
In order to decrypt the actors and the consequences of the maternal syndrome, the research
axes in preeclampsia are split in two different and complementary entities.
On one side several studies concentrate on the link between placental abnormalities and the
maternal syndrome. The hypothesis is that some factors are released from the placenta into
the maternal circulation and impact endothelial physiology. So this axis aims to identify,
through a targeted approach, circulating factors (already known to be associated to
hypertension, inflammation, platelet aggregation and alteration in permeability) which could
be modified in maternal circulation and associated to widespread endothelial activation. In
parallel the use proteomic approaches help to identify new candidate molecules that can
complete the maternal pathological landscape. Identifications of such factors have also
encouraged researchers to determine whether some of them are modified even before the
manifestation of the clinical symptoms, in order to be used as putative early biomarkers of the
disease.
On the other side, researchers try to establish the impact of “toxic” placental factors on
endothelial physiology to see how changes in factors concentrations or activity could impact
endothelial functions and provoke the maternal syndrome during preeclampsia. For this
purpose in vitro endothelial cell culture and in some cases animal models help to go deeper in
our understandings of the pathophysiology of preeclampsia.
The aim of this chapter is to analyze endothelial functions and how circulating factors can
influence vascular deregulation in the pathophysiology of preeclampsia.
30
Introduction
1. Endothelial health and vascular maintenance
A lot of efforts have been done to better understand the process of vasculogenesis and
angiogenesis in embryonic tissues and adult organs, in physiological processes and in
diseases. But it is since the last decade that vascular maintenance attracts the interest of
researchers. In fact, experiences on blood stream arrest and consequent vessels regression
(See et al., 1975) , supported for a longtime the idea that blood flow is the main force
involved in vessels stability through a passive mechanism of adaptation to hemodynamic
changes.
Nowadays vessels stability has more the connotation of a dynamic process, finely regulated
by pathways that sometimes involve several cell types, indispensable for tissue homeostasis,
sensible to adaptation to tissue needs in terms of nutrients and oxygen delivery, and,
responsive to tissue injury, when it is necessary.
In this regard, endothelial cells are the first sensors of hemodynamic changes in the blood
stream, the main actors in regulation of organ perfusion via changes in permeability, the
stabilizers of blood pressure through the balance of vasodilators and vasoconstrictors, and the
mediators of response to damages by triggering a pro-coagulation and pro-inflammatory
state.
A. Angiogenic imbalance
If we would consider the most important findings in understanding the pathophysiology of
preeclampsia, the couple of years 2004-2006 represents a breakthrough in research in
preeclampsia, with the discovery of deregulation of VEGF and TGF-β signaling pathways.
Indeed, the preeclamptic syndrome is linked to the so-called “angiogenic imbalance”, which
refers to alteration in circulating levels of active VEGF and TGF-β. This is due to the
increasing levels of their respective soluble receptors Flt-1 and Endoglin.
31
Introduction
i.
sFlt-1 and its biological relevance in preeclamptic placenta
The VEGF pathway is one of the main actors in angiogenesis, cell survival, and vessels
maintenance in adult tissues. The VEGF family includes four VEGF isoforms (VEGF-A, -B,
-C, -D), the placental growth factor (PlGF), and three membrane receptors (VEGFR-1,
VEGFR-2, VEGFR-3).
Human first trimester placentas highly express VEGF family members and their receptors.
The complex expression profile of VEGF ligands and receptors during cytotrophoblasts
differentiation highlights the possibility that VEGF ligands can interact with different
receptors in order to activate separate cell programs. In fact, the coupled expression of VEGFC and VEGFR-2 in cytotrophoblasts stem cells participates to activation of a proliferation
program, while in invasive cytotrophoblasts the transduction of VEGF-C signal is mediated
by VEGFR-3, which is more linked to uterine invasion properties (Zhou et al., 2002).
sFlt-1 is a truncated form of VEGF receptor Flt-1 (VEGFR-1), resulting from a splice variant.
It contains only the extracellular domain and, once in the circulation, it can bind both VEGF
and PlGF thus blocking their interaction with the cell surface receptors (Maynard et al.,
2003). It has been shown that in normal pregnancy sFlt-1 levels increase during the last two
months, with the decrease of VEGF and PlGF: this observation suggests a sort of biological
balance between pro and anti-angiogenic factors in order to regulate placental growth (Levine
et al., 2004a).
In the course of a preeclamptic pregnancy, the physiological increase in sFlt-1 is
exacerbated and anticipated up to the first trimester, well before the manifestation of the
maternal syndrome (Hertig et al., 2004; Levine et al., 2004a; McKeeman et al., 2004); in this
way, it leads to a consequent reduction in free circulating VEGF and PlGF, and is correlated
with the severity of the pathology (Chaiworapongsa et al., 2004; Hertig et al., 2004; Levine et
al., 2004a). The relevance of sFlt-1 in preeclampsia was also confirmed by a rat model which
reproduces a preeclampsia-like phenotype (with hypertension, proteinuria and glomerular
endotheliosis) when treated with exogenous sFlt-1 during gestation (Maynard et al., 2003).
In a normal pregnancy invasive cytotrophoblasts express VEGF-A, PlGF and VEGFR-1,
which act in a autocrine and paracrine way to regulate placental angiogenesis and
32
Introduction
cytotrophoblasts pseudovasculogenesis. In preeclamptic placentas decrease in invasiveness
is associated to a decrease in VEGF and VEGFR-1 expression and an increase in sFlt-1
secretion, associated to an increase in apoptosis in vitro (Levine et al., 2004a; Zhou et al.,
2002). This suggests a fundamental role of the VEGF pathway in cytotrophoblast
survival and invasiveness, which is defective in preeclampsia .
Altered sFlt-1 expression, has already been found in chorionic villi at 11 weeks of gestation
(Farina et al., 2008), and its effect is not limited to the placental bed: increased sFlt1, fostered
by placental ischemia, is released in the maternal circulation and actively participates to
endothelial dysfunction during the maternal syndrome (Rana et al., 2012).
Less is known about the role of sFlt-1 in the regulation of circulating free PlGF. PlGF, is
highly expressed in placenta, mostly in the second and third trimesters, with a peak between
29 and 32 weeks. Tissue damage linked to ischemia, inflammation and wound healing
activates PlGF-induced angiogenesis (Carmeliet et al., 2001). In preeclampsia PlGF inhibition
seems to have a crucial role in endothelial damage, as proven by induction of a preeclampsia–
like syndrome in pregnant rats submitted to PlGF and VEGF blocking (Maynard et al., 2003).
However, PlGF downstream pathway has not been clearly elucidated.
ii.
sFlt-1 and kidney damages during preeclampsia
The term glomerular endotheliosis was coined by Spargo and colleagues (SPARGO et al.,
1959) and refers to a glomerular alteration distinctive of preeclampsia, characterized by large
glomeruli in which the capillary lumen is almost occluded because of the swelling of
endothelial and mesengial cells and fibrin deposition. Endothelial cells lose the fenestration
structure typical of the kidney thus compromising physiological filtration rate (Lafayette et
al., 1998; Nochy et al., 1980). Moreover, preeclamptic plasma can induce a specific increase
in the permeability of human glomerular endothelial cells in vitro (Du et al., 2011).
Glomerular endothelium is a fenestrated lining characterized by trans-cellular holes
concentrated in peripheral cytoplasm, often organized in clusters and surrounded by a
network of actin microfilaments. Alteration of this structure in preeclampsia is partly due to
the reduced availability of VEGF as a consequence of its sequestration by sFlt-1 which is
33
Introduction
increased in the circulation of preeclamptic women. (Maynard et al., 2003). VEGF signaling
pathway is of primary importance in building glomerular endothelial fenestration (Eriksson et
al., 2003), it is expressed in glomerular podocytes and its receptors on glomerular endothelial
cells (Maharaj et al., 2006). Even if the mechanisms are not completely elucidated, it seems
that they can regulate actin rearrangements and (Andrews, 1981; Ioannidou et al., 2006) the
plasmalemma vesicle -associated protein-1 (PV-1 or PL-VAP), a type 2 transmembrane
glycoprotein important for fenestrae organization (Roberts and Palade, 1997; Stan et al.,
2004; Strickland et al., 2005). The role of VEGF in kidney physiology was confirmed by
reproduction of proteinuria and glomerular endotheliosis in animal models after anti-VEGF
treatment (Kitamoto et al., 2001; Sugimoto et al., 2003), in a mouse model in which VEGF is
specifically absent in podocytes (Eremina et al., 2003) and even in humans in case of cancer
therapies based on anti-VEGF antibodies, which induce hypertension, proteinuria and
glomerular endotheliosis as treatment side effects (Zhu et al., 2007).
iii.
VEGF signaling pathway alteration and cerebral edema
Unlike most maternal organs that adapt vasculature during pregnancy in order to increase
blood flow, as for the uteroplacental bed, brain is not able to do so, but must adopt
hemodynamic changes in order to maintain a constant blood flow and oxygen delivery. In
preeclampsia, increased blood pressure induces changes in cerebral vascular resistance, the
disruption of blood brain barrier permeability, and consequently the accumulation of plasma
in cerebral parenchyma, edema and, in the worse cases, eclampic seizure (Cipolla, 2007;
Friedman et al., 2009; Koch et al., 2001).
It is known that circulating levels of sFlt-1 sequester VEGF and PlGF, thus reducing the
amount of active factors in the circulation; however it is still difficult to quantify and decipher
the remaining active VEGF/PlGF effects on endothelial cells.
Exposure to preeclamptic plasma has a strong effect on blood brain barrier permeability, by
increasing permeability by 18.0-fold compared to no plasma exposure (Amburgey et al.,
2010; Neal et al., 2004). The factors responsible for brain barrier modifications are still
unknown. Once again, VEGF seems a good candidate, since a recent study shows that
physiological brain barrier is restored by blocking VEGFR tyrosine kinase activity
34
Introduction
(Amburgey et al., 2010). But since no differences in VEGF concentration between normal and
preeclamptic plasma were detected in this study, the hypothesis is that there should be, in
preeclamptic plasma, some factors that block or limit VEGF activity in normal pregnancy; in
the case of preeclampsia VEGF inactivation is missing, and blood brain barrier permeability
perturbed (Amburgey et al., 2010).
Recently it has been shown that increased permeability in endothelial cells seems to be linked
specifically to the increase of the active isoform VEGF165b and the reduction of soluble
PlGF. The idea is that in preeclamptic conditions, decrease in circulating PlGF (sequestered
by sFlt-1) results in a loss of repression of the active isoform of VEGF165b which triggers an
increase in cellular permeability: physiological endothelial barrier in fact can be restored by
blocking VEGF165 b or increasing the level of PlGF (Bills et al., 2011).
It seems that a fine balance between VEGF, PlGF and sFlt-1 is necessary to regulate
endothelial permeability: any perturbation of this equilibrium can have a deleterious effect on
cellular homeostasis.
iv.
Other factors involved in endothelial permeability perturbation
Organ perfusion is allowed by endothelium permeability. Ranges of permeability change
according to organ needs, but generally, under normal conditions 30% of endothelial
junctions in post capillary venules allow the transfer of 60Å molecules (Simionescu et al.,
1978).
Endothelial integrity is ensured by three types of junctions, with a specific role and a subset of
proteins: adherent junctions, tight junctions, and gap junctions.
In endothelial cells VE-cadherin is the main organizer of adherent junction: it is a
transmembrane protein with an intracellular domain through which the protein interacts with
the cytoskeleton, with p-120 catenin and β-catenin via two specific tyrosine residues (Y658 and
Y731) (Potter et al., 2005; Yamada et al., 2005). This interaction is indispensable for
endothelial permeability and maintenance.
35
Introduction
Occludin is another protein involved in tight junction formation and stability in epithelial
and endothelial cells. It belongs to the tetraspanin protein membrane family with four
transmembrane domains and cytoplasmic C-terminal and N-terminal tails (Furuse et al.,
1993). Tight junctions assembly needs the interaction of occludin C-terminal tail with other
proteins of tight junction like ZO-1, ZO-2, ZO-3 (Furuse et al., 1994; Li et al., 2005).
Integrity of tight junctions depends on the high phosphorylation level of occludin Ser and Thr
residues (Sakakibara et al., 1997; Wong, 1997), while dephosphorylation of the same residues
by protein phosphatases PP2A or PP1 can negatively impact tight junction assembly and
increase cellular permeability (Seth et al., 2007).
Incubation of endothelial cells with preeclamptic plasma induces an increase in permeability
associated to a dowregulation of VE-cadherin and occludin (the major components of tight
junctions), a perturbed localization of both proteins at the cell membranes with the
breaking of the interaction VE-cadherin/β-catenin/p120 complexes, and enlarged gaps at
the cells borders (Wang et al., 2002).
Recently it has been show that VE-cadherin interacts with aPKCλ at the level of cellular
junctions and co-localizes with the protein PARD-3 (Zhao et al., 2011). PARD-3 belongs to
partitioning defective proteins, and their implication has been documented in the alteration of
epithelial cell junctions. PARD-3 can interact with atypical protein kinase C (aPKC), in order
to control asymmetrical cell division, protein distribution, and cell polarity in epithelial cells
(Joberty et al., 2000; Lin et al., 2000; Schmoranzer et al., 2009). PKC proteins are already
known to be important in increased endothelium permeability after preeclamptic plasma
treatment in endothelial cells (Haller et al., 1998). Treatment with preeclamptic plasma blocks
the interactions between VE-cadherin and aPKCλ, favors the formation of complexes VEcadherin- PARD-3 and the internalization of both proteins in the cytosol, thus disturbing
barrier integrity and cell permeability (Zhao et al., 2011).
This observation is consistent with the hypothesis that the mislocalization of VE-cadherin at
the cell membrane could be due to the retention of the protein in subcellular
compartments of the Golgi complexes after preeclamptic plasma treatment, thus preventing
its addressing to the cell membrane (Groten et al., 2000). In this regard, proteolytic activity of
enzymes of the coagulation system, which are known to be up-regulated in preeclamptic
36
Introduction
plasma (Perry and Martin, 1992; Weiner, 1991), can favor the rapid degradation of proteins at
the level of cellular junctions, and the turnover at the cell membrane is impaired by the
retentions of VE-cadherin in intracellular compartments (Groten et al., 2000).
Different studies tried to identify specific plasma factors involved in damages to vascular
integrity, but, despite a lot of efforts, knowledge about deregulated pathways is still very poor.
Increased oxidative stress, documented by increased pro-inflammatory cytokines in the
maternal circulation, like IL-8 or TNF-α, and lipid peroxides, can perturb endothelial barrier,
and in this regard, in vitro treatment with antioxidant seems to restore the integrity of the
endothelial monolayer (Anim-Nyame et al., 2003; Zhang et al., 2003). It is possible that
TNFα mediates an increase in permeability by promoting inflammation and oxidative stress.
In fact increase in circulating TNF-α is associated with the up-regulation of ICAM-1 and
VCAM-1 (Beckmann et al., 1997; Mattila et al., 1992), two markers of endothelial cell
activation which can promote leukocyte migration, interaction with endothelial cells and thus
an increase in microvascular permeability. Moreover increased concentration of TNF-α can
trigger oxidative stress by interfering with the electron transport system of the endothelial
mitochondria, thus increasing free radicals, lipid peroxidation (Stark, 1993) and finally
perturbed endothelial cell permeability.
Circulating oxidized low density lipoprotein (oxLDL), which are increased in preeclamptic
plasma (Belo et al., 2005; Hubel et al., 1998; Qiu et al., 2006) have a destructive effect on
blood brain barrier integrity. oxLDL interact and activate the lectin-like oxLDL receptor-1
(LOX-1), expressed mainly on endothelial cells. oxLDL-LOX1 binding reaches up to 260%
increase in pregnant rats perfused with plasma coming from pregnant women with early-onset
preeclampsia, resulting in blood brain barrier disruption (Schreurs et al., 2013). The activation
of LOX-1 already known to be up-regulated in hypoxic placenta (Lee et al., 2005), induces
the overexpression of chemokines and adhesion molecules, and triggers CD40/CD40L
pathway (which contributes to pro-inflammatory cellular state) and over production of
Reactive Oxygen Species (ROS) (Mitra et al., 2011). ROS interact with nitric oxide (NO),
reducing the availability of a potent vasodilator and increasing the concentration of
peroxynitrite (Schreurs et al., 2013).
37
Introduction
Nitrative stress, in turn, participates to increase the vascular permeability: in fact endothelial
cell treatment with peroxynitrite generators (3-morpholinosydnonimine) induces the same
alterations seen after treatment with preeclamptic plasma: increased vascular permeability,
disorganization of VE-Cadherin and occludin with formation of gaps at the cell membranes
(Zhang et al., 2005).
Peroxynitrite augmentation is described in the vasculature of preeclamptic women
(Roggensack et al., 1999) and its role as a toxic radical for endothelial cell function is well
documented (Beckman and Koppenol, 1996; Buhimschi et al., 1998; Sankaralingam et al.,
2009). Peroxynitrite impacts protein function by oxidation of the thiol group of both
cysteine and glutathione and nitration of tyrosine residues. These modifications can touch
different pathways and impair endothelial cell permeability. Peroxynitrite may perturb
cytoskeletal structure by β-actin nitration, and participate to increased TNF-α induced
permeability (Neumann et al., 2006). Another mechanism that saw peroxynitrite as a motor
of endothelium destabilization involves the nitration of Protein phosphatase type 2A (PP2A)
at the level of its catalytic activity PP2AC. Nitration can activate phosphatase which in turn
dephosphorylates endothelial junction proteins ZO-1 and occludin (Nunbhakdi-Craig et al.,
2002; Seth et al., 2007; Sontag and Sontag, 2006), thus perturbing their membrane
localization with the increase of endothelial cell permeability (Wu and Wilson, 2009).
Among factors released by preeclamptic placenta, the placental releases serine protease such
as chymotrypsin-like protease/chymase (CLP) seems to be a good candidate in
deregulation of endothelial cell barriers: in vitro studies of cell permeability show that culture
of endothelial cells with preeclampsia trophoblasts or preeclamptic serum can alter VEcadherin localization at the level of tight junction. Specifically CLP acts as an inducer of
PAR-2, a G-protein coupled protease receptor, which is activated in stress condition and
mediates the VE-cadherin disorganization at the cell membrane (Gu et al., 2012). Moreover
CLP induces an increase in endothelial permeability in a dose-dependent manner, associated
with the reduction of expression of VE-cadherin and occludin and that normal phenotype can
be restored by chemotrypsin depletion (Gu et al., 2009) (Figure 2).
38
Introduction
Figure 1: Factors involved in increased permeability in preeclampsia
v.
sEng and its biological relevance in preeclampsia
Similarly to VEGF, the TGF-β pathway is compromised by the increase in preeclamptic
maternal circulation of a soluble form of Endoglin (sEng), which competes with membrane
receptors, traps circulating TGFβ-1 and TGFβ-3 and avoids their availability.
Endoglin is expressed in endothelial cells and syncytiotrophoblasts and mediates TGF-β
regulation of proliferation, vessels homeostasis and vasodilation. In preeclamptic pregnancies,
sEng follows sFlt-1 profile and is increased in the maternal circulation several weeks before
the onset of maternal symptoms (Levine et al., 2006). In vitro studies show that sEng perturbs
endothelial tube formation, in vivo sEng exaggerates sFlt-1 effects on endothelial cell
homeostasis, impacts endothelial vaso-regulation (Venkatesha et al., 2006) and increases
microvascular permeability in murine lung liver and kidney. The combined effect of increased
sFlt-1 and sEng has a strong impact on vascular cellular integrity, as it has been demonstrated
by the observation that in pregnant rats it reproduces the characteristics of severe
39
Introduction
preeclampsia, with hypertension, proteinuria, HELLP syndrome, and impaired fetal growth
(Levine et al., 2006).
vi.
sEng and sFlt-1: impact on vaso-regulation and coagulation state
Increased sEng in preeclampsia contributes to the onset of hypertension by perturbing
endothelial cells production of two of the main important vessels vasodilators, Nitric Oxide
(NO) and prostacyclin (PGI2).
NO is a lipophilic gas derived from the enzymatic conversion of L-Arginine in L-Citrulline
(Palmer et al., 1988) or by nitrite degradation. NO synthesis is assigned to a specific subset of
cell specific enzymes, nitric oxide synthase (NOS): NOS-I is expressed in the nervous
system, NOS-II, or inducible NOS (iNOS), in macrophages and neutrophils and NOS-III, or
endothelial NOS (eNOS), in endothelial cells (Figure 3), although these distinctions are not
exclusive.
Figure 3: NO synthesis in endothelial cells and effects on smooth muscle cells
Nitric oxide is produced by endothelial cells in shear stress conditions, in response to
acetylcholine, and arachidonic acid (Pohl et al., 1986; Vanhoutte, 2003), and during
pregnancy in a progesterone-dependent manner (Hayashi et al., 1995; Sladek et al., 1997). It
acts as a vasodilator and anticoagulant in an autocrine and paracrine way in order to
increase vasodilation, smooth muscle relaxation (Ignarro et al., 1987; Tang et al., 2003),
40
Introduction
inhibition of platelet aggregation and adhesion (Mendelsohn et al., 1990), inhibition of
leukocyte adhesion (Kubes et al., 1991) and induction of smooth muscle cells proliferation
and migration (Marks et al., 1995). In large vessels, like aorta, coronary and brain arteries,
endothelial NO is the major actor in promoting vasodilatation (Vanhoutte, 2003), and its
inactivation enhances vasoconstriction, inflammation and thrombosis in humans and animal
models (Moncada et al., 1991).
In preeclampsia contradictory results have been published about NO concentration, with
results showing no changes (Davidge et al., 1996; Silver et al., 1996), reduction (Ranta et al.,
1999; Shaamash et al., 2000) or augmentation (Garmendia et al., 1997; Seligman et al., 1994).
NO alteration is the result of complex mechanisms which interplay to manage NO substrates
availability, NO synthase enzymatic activity, natural NO inhibitors activity, and NO as a
substrate in other pathways.
In a physiological condition VEGF and TGF-β1 mediate eNOS activation respectively
through Thr495 dephosphorylation or Ser1177 phosphorylation (Michell et al., 2001; Mount
et al., 2007).
Studies on NO metabolites show that they are altered in preeclampsia and inversely correlated
to the concentration of sFlt-1 and sEng (Sandrim et al., 2008). Increased levels of sEng and
sFlt-1 can reduce active VEGF and TGF-β1, and impact negatively eNOS enzymatic activity
(Venkatesha et al., 2006). NO production is also perturbed by the decrease of substrate
(arginine) (Noris et al., 2004) and increase of asymmetric dimethyl arginine (ADMA), a
natural NOS inhibitor, both documented in preeclampsia (Fickling et al., 1993; Holden et al.,
1998; Savvidou et al., 2003). Its limited synthesis is also impacted by oxidative stress:
increased ROS production sequesters free NO to produce peroxynitrite and increases nitrative
stress (Roggensack et al., 1999) (Figure 4).
41
Introduction
Figure 4: altered NO pathway in preeclampsia. Red arrows show modified
molecules in preeclamptic syndrome
The blockade of active VEGF and TGF-β signaling pathway is also implied in the reduction
of PGI2 (He et al., 1999; Ristimäki et al., 1990), another important vasodilator and antiplatelet aggregator which is increased, like NO, in normal pregnancy (Ylikorkala et al., 1986).
Reductions in PGI2 concentration have been described in preeclamptic women (Fitzgerald et
al., 1987; Klockenbusch et al., 2000; Wang et al., 1992), and endothelial PGI2 release is
perturbed even before clinical manifestation of preeclampsia (Mills JL et al., 1999).
All these findings suggest that sEng and sFlt-1 are an important node in the onset of the
maternal symptoms, especially those involving hypertension. Indeed the angiogenic
imbalance reverses the physiological vasodilation and anti-thrombotic endothelial
contribution to pregnancy towards vasoconstriction and pro-thrombotic action which
participate and foster endothelial injury and vessels increased permeability.
B. Vasculature contribution to vasodilation and coagulation state
Hypertension is usually associated to endothelial dysfunction. It has been proposed that
endothelial injury is the outcome of the stress imposed by elevated blood pressure on vessels
walls (Moncada and Vane, 1978). Nowadays, the role of damaged endothelium as an active
cause implied in hypertension maintenance is sustained by the findings that treatments for
hypertension do not ameliorate the endothelium status (Panza et al., 1993) and moreover by
42
Introduction
the persistency of endothelial dysfunction without hypertension in the offspring of essential
hypertensive patients (Taddei et al., 1996).
Vasculature secretes several vasodilators and vasoconstrictors that regulate systemic blood
pressure. Preeclamptic hypertension is in part due to the deregulation of this balance. In
fact, a lot of factors, secreted by smooth muscle cells and endothelial cells, have been found
deregulated in preeclamptic women circulation.
The Important actors which complete the clinical picture of hypertension are endothelin 1
(ET-1) and thromboxane A2 (TXA2).
Endothelial cells produce ET-1 in the form of preproendothelin-1, which turns into an active
form after endopeptidase cleavage (Brown et al., 2000; Inoue et al., 1989). In its mature form
it can binds ETA receptors, on smooth muscle cells, or ETB, on endothelial cells, with separate
downstream signaling pathways (Boulanger and Lüscher, 1990; Herrmann and Lerman,
2001). Interaction with ETA receptor is the mediator of ET-1 vasoconstriction effect (Kiely et
al., 1997; Pernow et al., 1996), while vasodilatory contribution refers to endothelindependent induction of NO and PGI2 after interaction of ETB receptor (Giardina et al., 2001;
Molnár and Hertelendy, 1995).
For most investigators, ET-1 is increased in preeclampsia in the maternal circulation but its
levels comes back to the normal after delivery (Clark et al., 1992; Dekker et al., 1991; Nova
et al., 1991; Taylor et al., 1990), even if some investigators did not find an increased level of
ET-1 in the plasma of preeclamptic women (Paarlberg et al., 1998). Increased level of ET-1
participates to generalized spasms and vasoconstriction. Its implication in the disease has
been confirmed by the effects of long term ET-1 treatment in pregnant sheep, which
reproduces a preeclampsia-like syndrome with reduced utero-placenta perfusion, elevated
pressure and proteinuria (Greenberg et al., 1997).
Because of ET-1 double effect as vasoconstrictor and vasodilator, it could be possible that in
preeclampsia, it can counterbalance the increase in vascular resistance by activating ET B
receptor in favor of muscle relaxation (Conrad et al., 1999; Gandley et al., 2001).
43
Introduction
Endothelium and platelets participate to high blood pressure together with the increased
synthesis of TXA2. TXA2 is increased in the circulation of women with preeclampsia,
correlates with the severity of the disease and with platelet activation (Paarlberg et al., 1998),
and returns to physiological concentrations after delivery (Fitzgerald et al., 1990; Friedman,
1988; Wang et al., 1992). Animal model studies show that reduced placental perfusion and
consequent endothelial dysfunction induce increased TXA2 production and that TXA2
antagonists can prevent short term hypertension in a model of PE obtained by surgically
reduced uterine perfusion in pregnant dogs (Woods, 1989).
A pro-coagulant state is physiologically increased during pregnancy, but extremely
unbalanced in preeclampsia, as highlighted by plasma concentration of fibronectin, von
Willebrand factor, thrombomodulin, cellular fibronectin, and Plasminogen Activator Inhibitor
1 (PAI-1) (Chavarría et al., 2002; Estellés et al., 1989; Friedman et al., 1995; Hsu et al., 1993;
Nadar et al., 2004; Taylor et al., 1991). The hypothesis is that factors released in the maternal
circulation enhance hyperactivation of endothelial cells, platelet and leukocytes, with
overexpression of markers of cellular activation like thromboglobulin (Ballegeer et al., 1992),
altered prostacyclin/thromboxane A2
ratio (Wang et al., 1992), P-Selectin, soluble E-
Selectin, and vascular cell adhesion molecule-1 (VCAM-1) expression (Chaiworapongsa et
al., 2002).
All these factors concur to define another important clinical sign of preeclampsia
characterized by thrombocytopenia and coagulopathy derived by exaggerated endothelial
and leukocytes activation and platelet aggregation.
C. Inflammation
The pro-inflammatory state which accompanies physiological pregnancy, is exaggerated in
preeclampsia, as shown by the increased levels of pro-inflammatory cytokines, like tumor
necrosis α (TNF-α) and interleukin 6 (IL-6), in the maternal circulation (Bachour et al., 2008).
Some of these factors are increased even before the onset of clinical symptoms of the disease.
(López-Jaramillo et al., 2008)
44
Introduction
Stressed preeclamptic placentas release pro-inflammatory cytokines in the maternal
circulation like IL-1, IL-6, and TNF α (Lockwood et al., 2008). Pro-inflammatory cytokines,
in turn, activate endothelial cells to produce inflammatory mediators. Moreover they perturb
global cellular homeostasis by interfering with permeability and vaso-regulation, as discussed
before.
Increased apoptosis and necrosis in the placental bed allow the liberation of placental debris
and syncytiotrophoblasts microfragments that have an anti–angiogenic effect and activate
endothelial cells and the immune system as well (Tannetta et al., 2013). During preeclampsia
several markers of syncytial shedding are increased in the maternal circulation, like
cytokeratins (H Schröcksnadel, 1993), soluble fetal DNA (Lo et al., 1999; Zhong et al., 2001),
and cellular syncytial fragments (Johansen et al., 1999) .
Syncytial debris are transcriptionally active and participate to endothelial dysfunction directly
through the synthesis of sFlt-1 (Rajakumar et al., 2012), or indirectly through interaction and
activation of neutrophils and monocytes, which, in turn, release TNFα, IL-1 and IL-2 (Sargent
et al., 2006).
Whether inflammation is a cause or a consequence of endothelial dysfunction is still
debated, but surely endothelial injury has an active role to maintain a pro-inflammatory status
during the maternal syndrome.
D. Immune system reaction
Generalized inflammation and endothelial activation are strictly connected to innate and
non-specific immune response.
Activated immune cells coming from placenta diffuse into the maternal circulation, actively
diffuse inflammatory stimuli and in turn activate maternal leukocytes, neutrophils and
endothelial cells. Th-1 type cytokines, IL-2, IL-12 and interferon-γ (IFN-γ) released by
placental leukocytes and peripheral blood cells (Darmochwal-Kolarz et al., 1999; Rein et al.,
2002; Saito et al., 1999; Sakai et al., 2002) and elastases coming from neutrophils act on
endothelium, which starts to express adhesion molecules like ICAM, VCAM and E-Selectin
45
Introduction
(Lyall et al., 1995), the latter one concurring to activate peripheral leukocytes in a positive
feedback loop, thus maintaining a pro-inflammatory system.
The complement system is also affected in the placental bed and maternal circulation.
Increased levels of C4d, and regulatory element CD55 and CD59 have been found in the
preeclamptic placenta (Buurma et al., 2012); proteomic studies allowed to identify
downregulation in C4a, CB, C7 and C1r in plasma of preeclamptic patients (Auer et al., 2010;
Zhang et al., 2011). In autoimmune diseases (that are known risks factors for preeclampsia
such as lupus erythematosus or antiphospholipid syndrome), genetic polymorphisms of genes
of the complement cascade have been associated with preeclampsia (Salmon et al., 2011).
Alteration of the immune system goes beyond the innate response, as proven by the presence
of circulating agonistic autoantibodies against angiotensin II receptor, type 1 (AT1-AAs)
(Zhou et al., 2008a, 2008b).
The renin-angiotensin-aldosterone system is responsible of an increased blood pressure in
normal pregnancy. Preeclamptic patients suffer of a decreased plasma renin activity, increased
aldosterone release and elevated vascular sensitivity to angiotensin II, a potent vasoconstrictor
(August et al., 1990).
AT1-AA can participates to increased hypertension through the activation of AT1 receptor
(thus partly explaining the hypersensitivity to angiotensin II observed in preeclamptic
women) but also by increased level of sFlt-1 and sEng, as shown by administration of AT1AA to pregnant rats (Parrish et al., 2010) . Apart the regulation of blood pressure, they play an
activating effect on NADPH oxidase and indirectly on ROS production, thereby participating
to oxidative stress, inflammation and pro-coagulation state as well (Dechend et al., 2003).
Moreover AT1-AAs persist in women circulation even after delivery, representing a latent
risk for future cardiovascular disease (Hubel et al., 2007).
E. Early biomarkers of preeclampsia
Late symptoms prompted researchers to orient the field of investigation on the possibility to
predict the disease on the basis of circulation factors, easily detectable and modified before
the clinical complications.
46
Introduction
These putative markers could be the products of a defective placentation and can be free
molecules, microparticles, or free DNA/RNA released into the maternal circulation.
Today, several first trimester markers have been identified and include angiogenic factors
(sFlt-1, PlGF), sEng, P-selectin, inhibitin A, Activin A placental protein 13 (PP13), cell free
nucleic acids.
Circulating angiogenic and anti-angiogenic factors are modified several weeks before the
onset of the maternal syndrome. Longitudinal studies highlight an increase of sFlt1 in the
second trimester, and a diminution of circulating active PlGF since the first trimester of
gestation in women who will later develop preeclampsia (Chaiworapongsa et al., 2005;
Levine et al., 2004a; Romero et al., 2008).
Both are promising early biomarkers for
preeclampsia, but since in some cases preeclampsia do not show alterations in these factors,
their predictive values could be more promising if associated to other biomarkers (Akolekar et
al., 2010; Jacobs et al., 2011).
sEng is also detectable as increased at the end of the first trimester of pregnancy and usually,
like sFlt1, is correlated to the severity of the disease (Baumann et al., 2008; Foidart et al.,
2010), and is more evident in preeclampsia complicated by HELLP syndrome (Venkatesha et
al., 2006).
P-selectin has also been found increased in the first trimester (Bosio et al., 2001; Chavarría et
al., 2008) and even between 11 and 15 weeks of gestation reaching a detection rate of 59%
(Banzola et al., 2007). This molecule is synthesized by activated platelets and endothelial
cells and an increased level in circulation allows predicting a perturbed vascular state before
the widespread endothelial dysfunction.
Inhibitin A, Activin A and Placenta protein 13 (PP13) are of placental origin and they have
been found increased in women who later develop a preeclamptic syndrome (Poon et al.,
2010; Sebire et al., 2000; Yu et al., 2011). PP13 is particularly promising since it has been
found increased in the maternal circulation during the first trimester (Chafetz et al., 2007;
Huppertz et al., 2008; Khalil et al., 2009) and protein alteration, in combination of uterine
artery Doppler, reaches a detection rate for preeclampsia of 90 % with a false positive rate of
6% (Nicolaides et al., 2006).
47
Introduction
Cell free nucleic acids are also promising biomarkers of disease. A recent study confers a
detection rate for preeclampsia around 84% with a false positive rate of 5% to blood
circulating RNA between 15 and 20 weeks of gestation (Purwosunu et al., 2009). Several
studies confirmed also an increased cell free fetal DNA, probably associated to increased
placental necrosis and apoptosis. Cell free fetal DNA is increased from two to up to five fold
starting from 17 weeks of gestation (Levine et al., 2004b).
Even though there is no single marker able to ensure an early and precise detection of the
disease (Table 2), the combination of different markers, associated with uterine artery
Doppler (Carbillon, 2012; Scazzocchio et al., 2013) could offer a more promising and
sensible system to predict preeclampsia (Poon et al., 2013). The main goal for the future could
be to adapt screening of several molecules to a routine use in clinical milieu.
Table 2: Early circulating biomarkers of preeclampsia
F. Animal models for preeclampsia
Early biomarkers are a useful tool to predict preeclamptic syndrome. But to progress in
understanding the early causes of the disease, the use of animal models becomes
indispensable in order to bypass technical and ethical issues concerning experimentations
48
Introduction
during early human pregnancy, and, simultaneously, go deeper in the knowledge of
deregulated molecular mechanisms in preeclampsia.
Since preeclampsia is a specifically human disease: no spontaneous model exists and all
animal models are obtained by the alteration of the main physiological pathways involved in
preeclampsia: vasoregulation, immune system, hypoxic conditions (Table 3).
Table 3: Mouse models for preeclampsia
Recently our laboratory has developed a unique model of severe preeclampsia by
overexpressing the human STOX1 gene in a mouse model (Doridot et al., 2013).
49
Introduction
STOX1 belongs to the extended Forkhead Box transcription factors gene family. In 2005 a
linkage study in Dutch families highlighted its association with preeclampsia (Van Dijk et al.,
2005). It is expressed in extravillous trophoblasts (van Dijk et al., 2010) and overexpressed
during the first trimester in women who later develop preeclampsia, thus suggesting its
implication in the very early placental development (Founds et al., 2009).
Moreover its overexpression in a choriocarcinoma cell line (JEG-3), a model for trophoblasts,
reproduces a gene expression profile similar to that found in the preeclamptic placenta
(Rigourd et al., 2008). STOX1 is also an important regulator of mitochondrial homeostasis
and a regulator of oxidative and nitrative stress in vitro as well as in vivo in placenta
overexpressing human STOX1 (Doridot et al. paper submitted).
All these findings prompted us to generate two lines of transgenic mice that overexpress the
human STOX1 gene (TgSTOX13 and TgSTOX42). Phenotype analysis of pregnant WT
females crossed with transgenic males showed a preeclamptic–like syndrome characterized
by: the pregnancy-dependent onset of hypertension, proteinuria, increased circulating sFlt-1
and sEng, and glomerular endotheliosis. Transgenic mice phenotype is partly reversed by low
doses of aspirin administered all along gestation.
Even if all these models do not reproduce completely human placentation, they represent a
useful tool in order to study the causes of preeclampsia linked to perturbed placentation, to
test new treatments for maternal syndrome, and to evaluate the impact of the preeclamptic
syndrome on future maternal health.
50
Introduction
Chapter III. Hypoxia and cellular response
Life in aerobic condition has evolved in parallel with cellular capacity to use oxygen as a key
component of energetic metabolism. Increase in oxygen needs is proportional to organism
mass and metabolic activity.
Most intracellular oxygen is addressed to mitochondria, the organelle in charge of oxidative
phosphorylation: electrons are transferred through a series of complexes localized on the inner
mitochondrial membrane until oxygen, the last acceptor. The energy derived from redox
reactions is converted in high-energy phosphate bond in ATP.
Beyond its function for cellular metabolic needs, oxygen concentration is an important
promoter and regulator of physiological processes (embryonic development, placentation,
life at high altitudes) and pathological conditions (cancer or ischemic damages).
In mammals, tissues have usually to face an oxygen concentration comprised between 40 and
60 mmHg adjusted in each tissue according to the local network of blood vessels and organ
needs (Semenza, 2004). Any change in oxygen tension results in the expression of a battery of
genes in charge of maintaining cellular homeostasis and to limit cellular damages induced by
reactive oxygen species.
Hypoxia refers to a condition of oxygen penury involved in important cellular processes like
proliferation, differentiation and migration and cellular metabolism.
The cellular response to hypoxia is mediated by the Hypoxia inducible factors family,
composed by three genes coding for HIF-α subunits (HIF-1α, HIF-2α, HIF-3α) and three
genes coding for the HIF-β subunits (HIF-1β/ARNT, ARNT2, ARNT3). In order to be
transcriptionally active, α subunits form heterodimers with HIF-1β/ARNT or ARNT2.
Regulation of Hypoxia inducible factors family members is of primary importance in
response to a stress condition rapidly and efficiently.
51
Introduction
1. Hypoxia inducible factor 1 (HIF1)
Environmental changes often translate into a change of gene expression at the cellular
level. It is what happens during hypoxia, low oxygen pressure associated to some
physiological processes or pathological events.
One of the first studies on cellular response to hypoxia was focused on the erythropoietin
gene expression. Under hypoxic condition erythropoietin is synthesized by the kidney and is
involved in red blood cells survival and red blood cells progenitor differentiation. Analysis of
cis-acting regulatory sequences allows for the identification of a transcription factor
responsible for the mediation of cell response to hypoxia, HIF-1 (Semenza and Wang, 1992;
Wang et al., 1995). The importance of this pathway is strengthened by the fact that it is
conserved in almost all mammalian cell types and higher Eukaryotes (Lee et al., 2004).
Current literature identifies more than 100 genes regulated by oxygen tension via HIF-1
activation. Targeted gene approaches combined with whole-genome techniques, including
microarray and ChIP-seq define a picture of the cellular behavior under hypoxic conditions.
Modified genes belong to different functional categories: cellular proliferation, apoptosis,
cellular differentiation, energy metabolism, cellular adhesion, extracellular matrix
remodeling. Since its implication, better understanding of its regulation, its regulators, and its
targets has become a challenging aim in order to progress in important research fields, like
early development, tumor progression and cellular differentiation.
HIF-1 binds DNA in the form of a heterodimer composed of two different subunits, HIF-1α
and HIF-1β (or ARNT, aryl hydrocarbon receptor nuclear translocator). Both subunits
belongs to the bHLH–PAS (basic helix-loop-helix–Per-Arnt-Sim) transcription factor family
, and recognize a minimal DNA sequence known as HRE (hypoxia responsive element, 5’TACGTGC-3’) located in the minimal or distal promoter of target genes.
52
Introduction
A. HIF-1α
In humans HIF-1α is located on chromosome 14 (14q23). The gene, composed of 15 exons, is
ubiquitously expressed in all cell types, and codes for the subunit HIF-1α. Although the
gene is transcribed constitutively in all tissues (Wenger et al., 1996; Wiener et al., 1996),
protein stabilization is ensured exclusively under hypoxic conditions.
Apart from the bHLH domain and two PAS domains (PAS-A and PAS-B), responsible for
DNA binding and dimerization, the HIF-1α subunit contains a specific domain which
regulates protein stability according to oxygen sensing (O2- dependent degradation domain,
ODDD) and a N-terminal and C-terminal transactivation domain TAD (NTAD and CTAD).
NTAD and CTAD domains act synergistically, and loss of one of these domains, as described
in an HIF-1 α splice variant, strongly impacts HIF-1 activity in hypoxic conditions (Gothié et
al., 2000). HIF-1α contains also an inhibition of transcription domain, located in the Cterminal region between NTAD and CTAD, whose progressive deletion allows an increase of
HIF-1 transcriptional activity in normoxia (Jiang et al., 1997).
HIF-1α nuclear translocation is regulated by two NLS sequences (Nuclear Localization
Sequences). The first one, is a bipartite sequence, close to nucleoplasmin NLS, located near
the bHLH domain; its inhibition by PAS-B is responsible for HIF-1α cytoplasmic
accumulation. The second one, conserved in all three α subunits, near the C-terminal region,
is a monopartite –type sequence similar to the SV40 large T antigen NLS and it is responsible
for nuclear HIF-α internalization in hypoxic conditions (Kallio et al., 1998; Luo and Shibuya,
2001).
HIF-1α plays a crucial role in the first stages of embryogenesis and vascularization.
Transgenic mice KO for HIF-1α are affected by impaired neuronal development, absence of
neuronal tube and cerebral vascularization, defective somite patterning, cardiovascular
malformations, increase of organ hypoxic damages probably due to the absence of vascular
network, and embryonic lethality at E10.5 (Iyer et al., 1998; Ryan et al., 1998).
53
Introduction
B. HIF- 1β
Hif-1β/arnt1 is composed by 22 exons and localized on human chromosome 1 (1q21). The
gene codes for the protein HIF-1β (ARNT1, arylhydrocarbon-receptor nuclear translocator 1)
and it is expressed in all cell types and in an oxygen-independent manner. Three splice
variants have been identified for HIF-1β but their function remains unclear.
Like HIF-1α, HIF-1β contains a bHLH domain, which confers the property to bind DNA
sequence, and two PAS domains (PAS-A and PAS-B) to help dimerization with α subunits.
HIF-1β contains also a TAD domain but it doesn’t seem to be involved in activation in
hypoxic condition .
Transgenic mice lacking hif-1β are affected by impaired neuronal and cardiovascular
development, neocortex hyperplasia, alteration in angiogenesis and embryonic lethality
between E9 and E10, due mainly to alteration in placental vascularization (Kozak et al.,
1997).
2. HIF-2 and HIF-3
Two other α subunits belong to the HIF transcription factors family, HIF-2α and HIF-3α.
They dimerize with HIF-1β in order to form HIF-2 and HIF-3, which regulate cellular
response to hypoxia in a more specific tissue context, according to their expression pattern.
Hif-2α (or EPAS1, Endothelial PAS domain protein1) is located on human chromosome 2
(2p21-p16). The gene is composed of 16 exons and codes for the protein HIF-2α. Protein
organization is similar to that of HIF-1α, with a bHLH domain, two PAS domains, an ODDD,
two transactivation domains, NTAD and CTAD, and a mono–partite type sequence NLS for
nuclear translocation. This homology confers to HIF-2α the same properties as HIF-1α: the
protein is stabilized under hypoxic conditions, it interacts with HIF-1β in order to form HIF-2
and regulates gene expression. HIF-2α protein localization depends on oxygen tension, cell
type and developmental stage: this coordinated regulation confers to HIF-2α specificity for a
subset of genes in specific cell types, in addition to target genes shared in common with HIF1. During embryonic development it is expressed in endothelium, lung, and cellular
54
Introduction
population belonging to neuronal crest (Ema et al., 1997; Flamme et al., 1997; Tian et al.,
1998); after birth it is expressed also in immune system cells (bone marrow macrophages and
uterine decidual cells), parenchyma, cardiomyocytes, and kidney epithelial cells (Wiesener et
al., 2003)
Mice deficient for hif-2α die at E.10: the most important malformations affect vascular,
pulmonary, skeletal and muscle development (Compernolle et al., 2002; Peng et al., 2000;
Scortegagna et al., 2003; Tian et al., 1998).
Hif-3α/Ipas is a 17 exon-gene located on human chromosome 19 (19q13.32). The protein
shares 55% homology in amino acids sequence with other α subunits, and also contains an
ODDD and NTAD domain, but it lacks CTAD domain.
It is expressed in the adult thymus, lung, brain, heart, and kidney (Gu et al., 1998). Less is
known about its function and its regulation. It is also expressed in normoxic conditions and
regulates negatively HIF-1 and HIF-2 in case of oxygen deprivation thanks to the overexpression of HIF-3α splice variants. In hypoxia, an HIF-3α isoform (IPAS) is overexpressed,
sequesters HIF-1α subunit and blocks its transcriptional activity (La Ferla et al., 2002;
Makino et al., 2001, 2007). The same mechanism involves another HIF-3α splice variant,
HIF-3α-4, which is up-regulated in hypoxia and competes with HIF-1β to form heterodimers
with the HIF-2α subunit (Maynard et al., 2007).
HIF-2β HIF-3β and HIF-4β
Three other β subunits complete the HIF family.
Hif-2β/Arnt2 on human chromosome 15 (15q24) codes for the protein HIF-2β or ARNT2,
expressed in kidney, liver and brain. It can dimerize with HIF-α subunits and regulates genes
expression in an organ specific and oxygen specific manner (Maltepe et al., 2000). During the
first stage of development it seems to have a crucial role in neuronal development.
Homozygous Arnt2 gene knockout mouse embryos show embryonic lethality and defects in
hypothalamus and neuronal system development (Keith et al., 2001).
55
Introduction
HIF-3β or ARNT3 and Hif-4β known also as ARNTL2 are the other factors belonging to the
HIF family. No data suggest their implication in the cellular response to oxygen from what is
known today.
3. Regulation of HIF-1 protein stability
Oxygen implication in energetic metabolism and the necessity of a rapid defense strategy
against either hypoxia or hyperoxia, induce the necessity to build a system that is normally
inactive but easily and rapidly available in case of abnormal oxygen pressure.
In normal oxygen pressure HIF-1α is ubiquitously expressed, but the protein half-life is
around 5 minutes (Wang et al., 1995). Reduced oxygenation stabilizes the protein and ensures
the presence of a pool of protein transcriptionally active for a rapid response to environmental
changes.
A. Canonical pathway: HIF-1α oxygen-dependent regulation
HIF-1α degradation in normoxia is a multistep process, which includes hydroxylation,
ubiquitination and degradation.
The first step involves post-translational modifications at the level of the ODDD domain
and it is oxygen-dependent. ODDD is a 200 amino acids region that is hydroxylated by prolyl
hydrohylase domain proteins (PHD) on two prolines, P402 and P564 (P405 and P531 for
HIF-2 α) in normoxic conditions. Protein degradation requires hydroxylation of both prolines:
mutation in one of these sites partly stabilizes protein in normoxic conditions, while mutations
on both residues ensure protein accumulation in normal oxygen concentration (Masson et al.,
2001). PHDs (PHD1, PHD2, PHD3) are part of the 2-oxoglucorate (2OG)-dependent
oxygenases superfamily: they use oxygen and 2OG as co- substrate, plus Fe(II) and ascorbate
as cofactors (Bruick and McKnight, 2001; Epstein et al., 2001).
Hydroxylated prolines are recognized by β- subunit of von Hippel- Lindau protein (VHL),
which in turn interacts with elongin C. Elongin C, together with elongin B, form a protein
platform which interacts with cullin-2 and RBX1 in order to form a multi-proteic complex
56
Introduction
with E3 ubiquitin ligase activity (Ivan et al., 2001; Jaakkola et al., 2001). Because of its
function as mediator of polyubiquitination, VHL is considered as one of the master regulators
of HIF-1 stability.
The last step includes the protein polyubiquitination, translocation and degradation in the 26S
proteasome (Maxwell et al., 1999).
Recently a new regulation pathway has been described for HIF-1, which involves protein
sumoylation. Sumoylation is a post-translational modification on prolines residues, mediated
by SUMO-1, SUMO-2 and SUMO-3.
Hypoxic condition induces the up-regulation of the protein RSUME, small RWD domaincontaining protein. RSUME is up-regulated in hypoxia in vitro, directly interacts with
SUMO-conjugating enzyme UBC9 and HIF-1α and mediates its sumoylation and stabilization
(Carbia-Nagashima et al., 2007).
B. Regulation of HIF transcriptional activity
Expression of HIF target genes requires the translocation of HIF-1α into the nucleus, the
formation of heterodimers with HIF-1β and the binding to HRE sequence on the promoter
or enhancer of target genes.
HIF transcriptional activity also needs the interaction with the cofactor CBP/p300 at the
CTAD domain, near the C-terminal tail (Arany et al., 1996; Lando et al., 2002a). Once again,
this interaction is oxygen dependent.
In normoxia this interaction is prevented by hydroxylation of asparagine 803 in HIF-1α
(asparagine 847 in HIF-2α) in the C-terminal region by Factor inhibiting HIF (FIH-1)
(Freedman et al., 2002; Lando et al., 2002a). FIH-1 is one of the major inhibitors of HIF
activity: mutation in the site of hydroxylation is a sufficient condition to stabilize HIF-1 and
HIF-2 in a normoxic environment. Moreover modulation of FIH-1 expression is proportional
to the expression of HIF target genes. FIH-1 is an asparagynil-hydroxylase ubiquitously
expressed independently from oxygen tension (Stolze et al., 2004). Since its action depends
on the availability of the same cofactors as PHD (oxygen, Fe (II) and 2-OG), its inhibiting
57
Introduction
activity is restricted to normoxic conditions (Lando et al., 2002b).
Furthermore PHD
inhibitors that reduce PHD cofactors and accessibility to their substrates, are able to block
FIH-1 activity in the same way.
In hypoxic conditions, the cysteine/hystidine rich domain 1 (CH1) of CBP/P300 recognizes
and binds the non hydroxylated CAD domain and enhances HIF transcriptional activity
(Freedman et al., 2002).
p300 is a histone-acetyl-transferase (HAT) with the property to acetylate lysines on the
specific histone tails, thus modifying protein compaction and DNA accessibility. Recently it
has been shown that p300 acetylates HIF-1α on lysine 709, resulting in protein stabilization
and accumulation. Acetylation implies the direct interaction between HIF-1α and p300 and
because of lysine 709 has been identified as a putative site of polyubiquitination (Kim et al.,
2011), the balance between acetylation/ polyubiquitination may represent a new regulatory
system of HIF-1α stability which is independent of VHL action (Geng et al., 2012). p300
activity is counterbalanced by Histone deacetylase1 (HDAC1), which can specifically
deacetylate lysine 709 and affect negatively HIF-1α stability and nuclear accumulation (Geng
et al., 2012).
C. Oxygen–independent mechanisms of HIF-1α stabilization and
regulation of transcriptional activity
Apart from oxygen, other factors come into play in order to finely regulate HIF-1α
transcription, stability and accumulation, directly or indirectly, in an oxygen-dependent or
independent fashion.
i.
Regulation of PHDs and VHL
One possibility to stabilize HIF-1α in normoxic conditions is a negative regulation of factors
involved in its degradation. Several factors have been identified as regulators of PHDs and
VHL activity, and therefore, indirectly, of HIF-1α stabilization.
58
Introduction
Sirtuins are NAD+-dependent deacetylases and ADP-ribosyl-transferases involved in
metabolism regulation, stress response, neuronal degeneration, and life span. Different
members have been identified as putative modulators of HIF-1α stabilization. Sirtuin1 is a
target gene of HIF-1 and HIF-2 and is upregulated in hypoxia (Chen et al., 2011). During
hypoxic conditions it deacetylates HIF-1α on lysine 674 and blocks the interaction with p300
(Lim et al., 2010); reciprocally, deacetylation of
HIF-2α by SIRT1 enhances its
transcriptional activity (Dioum et al., 2009). SIRT6 binds directly HIF-1α and acts as a corepressor, as shown by down-regulation of HIF-1 target genes (Zhong et al., 2010).
Recently a new system of regulation managed by SIRT3 has been described, which involved
ROS production. ROS can specifically inactivate PHDs by reducing availability of catalytic
Fe(II), which is converted in Fe(III) (Pan et al., 2007). SIRT3, located in the mitochondria,
enhances antioxidant cellular defense and reduces ROS production (Bell and Guarente, 2011;
Finley et al., 2011). Its negative regulation of ROS guarantees HIF-1 inactivation mediated by
PHD hydroxylation. Angiotensin II, thrombin and platelet derived growth factor (PDGF)
can also increase HIF-1 stabilization in normoxic conditions via ROS–dependent mechanisms
(Chen et al., 2005; Görlach et al., 2001; Pagé et al., 2002; Richard et al., 2000).
Even though increased ROS are associated to reduced HIF-1α hydroxylation and degradation,
the mechanism of regulation is still questionable. In facts FIH seems more sensitive to
peroxides than PHD and could equally participate to HIF-1α inactivation (Masson et al.,
2012). Moreover HIF-1α stabilization after antioxidant treatment (Vassilopoulos and
Papazafiri, 2005) leaves the relation between ROS and HIF-1α still unsolved and a
stimulating field of research.
PHD activity can be also inhibited by iron chelation and divalent metal cation, like Co (II),
Ni (II), Mn (II) (Goldberg et al., 1988), or metabolites products, like the accumulation of
succinate, fumarate, pyruvate and oxaloacetate (Dalgard et al., 2004; Isaacs et al., 2005;
Selak et al., 2005). PHD2 is negatively regulated by TGF-β1 signaling pathway, thereby
fostering HIF-1α stabilization and nuclear accumulation upon environmental oxygen
(McMahon et al., 2006).
59
Introduction
Nitric oxide over-production can affect negatively PHD activity: it competes with oxygen,
interacts with PHD thereby blocking its catalytic activity (Metzen et al., 2003).
Inactivation of VHL, documented in VHL-associated hereditary cancer syndrome and the
Chuvash polycythemia, is responsible for impaired HIF-1 proteasomal degradation, abnormal
normoxic protein accumulation and altered expression of HIF-1 target genes (Kondo and
Kaelin, 2001; Perrotta et al., 2006).
ii.
Other pathways involved in HIF regulation
Other pathway can act directly on HIF-1α by modulating stabilization or degradation without
VHL-mediated polyubiquitination.
Exclusive interaction of heat shock protein 90 (HSP90) and the protein receptor of activated
kinase 1 (RACK1) can regulate HIF-1α stabilization/degradation in normoxic conditions
independently of canonical VHL-mediated degradation (Isaacs et al., 2002; Liu et al., 2007).
HSP90 is a cellular chaperone that binds and stabilizes HIF-1α at the level of the PAS-A
domain in normoxia and hypoxia. HSP90 positive regulation is counterbalanced by RACK1,
which competes with HSP90 for the PAS-A domain, interacts with elongin C and activates
HIF-1α proteasomal degradation.
HIF-α stabilization under normoxic conditions is also ensured by the activation of the
phosphatidylinositol-3-kinase (PI3K) and MAPK signaling pathway (Jiang et al., 2001).
Phosphatidylinositol 3-kinase pathway acts on the 5'-untranslated region of HIF-1α mRNA
increasing protein accumulation (Chen et al., 2005; Pagé et al., 2002). In the context of
cancer, activation of the PI3K pathway after inactivation of tumor suppressor PTEN,
increases HIF-1α nuclear localization in a normoxic environment (Zundel et al., 2000). This
pathway is also activated by insulin (Treins et al., 2002; Zelzer et al., 1998), insulin-like
growth factor (IGF-I) (Chavez and LaManna, 2002; Feldser et al., 1999), and epidermal
growth factor (EGF) stimulation (Jiang et al., 2001). Insulin stimulates also HIF-3α mRNA
and protein synthesis and accumulation (Heidbreder et al., 2007), while IGF-I modulates the
increase and stabilization of HIF-2α, as well. Interestingly IGF-II has an opposite effect
according to the α-type subunit and cell types. IGF-II increases HIF-1α protein stabilization
60
Introduction
in keratinocytes under hypoxic conditions. IGF-II treatment activates the MAP kinase
pathway, which in turns activates MDM2: the results is the down-regulation of p53 and an
increased HIF-1 transcriptional activity (Kwon et al., 2004). IGF-II is also responsible for
decreased HIF-2α mRNA in murine trophoblast in vitro under prolonged hypoxic conditions
(Feldser et al., 1999; Kwon et al., 2004; Pringle et al., 2007).
Pro-inflammatory cytokines can also stabilize HIF-1α. IL1-β can increase protein
transcriptional activity thanks to activation of the ERK1/2 pathway, as shown by the upregulation of VEGF, which is one of the main targets of HIF-1α (Qian et al., 2004). TNF-α
enhances HIF-1α transcriptional activity thanks to the activation of HIF-1α co-factors in a
process mediated by the NFκB pathway (Görlach and Bonello, 2008; Jung et al., 2003).
Endothelin and progesterone can increase HIF-1α protein, while estrogen increases
transcription of Hif-2α (Daikoku et al., 2003; Song et al., 2008). It has been proposed that
endothelin increases HIF-1α accumulation by PHD inactivation, but further studies need to
clarify the molecular mechanism (Spinella et al., 2010). The AKT pathway, activated by
17ß-estradiol, mediates of hormone-dependent HIF-1α accumulation (Feng, 2009).
Methoxyprogesterone acetate (MPA) acts using the same mechanism but with the opposite
effect: it decreases HIF-1α protein level by down-regulating AKT expression (Feng, 2009)
(Table 4).
61
Introduction
Table4: Oxygen- independent regulation of HIF-α
Acetylation and interaction with HDACs have been described as a mechanism of regulation
for HIF-α. The HDAC family includes 18 enzymes which remove the acetyl group from the
lysyl group of histones and more generally proteins, interfering with chromatin compaction,
gene expression and protein function (Ahringer, 2000; Choudhary et al., 2009; Yang and
Seto, 2003). According to their homology with yeast enzymes, HDACs are grouped in four
classes.
HIF-1α directly interacts with HDAC6, which increases its stabilization (Qian et al., 2006);
deacetylation at lysine 532 in the ODDD domain by class I/II HDACs (Jeong et al., 2002) or
in N-terminal region specifically by HDAC4 (Geng et al., 2011) infers on HIF- 1α stability by
protecting from protein degradation. HDAC7 has a cytoplasmic localization in a welloxygenated environment. Under hypoxic conditions it interacts specifically with HIF-1α (any
physical interactions with other HIF-α members have been detected in this study) and
62
Introduction
reinforces the link with the co-factor p300, thereby strengthening the HIF-1 protranscriptional activity (Kato et al., 2004).
Finally, free CBP/p300 could be another limiting factor to ensure HIF-1 transcriptional
activity. Some proteins can interact and sequester p300 thereby impeding the interaction with
HIF-1: it is the case for CITED2 (p35srj) (Bhattacharya et al., 1999) and p53 (Blagosklonny
et al., 1998). Other mechanisms can act on CBP/p300 or HIF-1α in order to stabilize the
complex and support HIF-1 transcriptional activity. Sentrin/SUMO-specific protease SENP3
prevents the binding of SUMO2/3 with p300 and allows the interaction with HIF-1α in order
to form a transcriptional active machinery (Huang et al., 2009). The hepatitis E virus open
reading frame ORF3 promotes phosphorylation of p300, which is necessary for HIF-1 α
binding (Moin et al., 2009). Another necessary condition for transcription of hypoxia target
gene is the binding of mammalian target of rapamycin (mTOR) with the TOS domain on the
N-terminal tail of HIF-1α: blocking this interaction results in a decrease of HIF-1 target genes
expression (Land and Tee, 2007)
4. HIFs in placental development
Oxygen plays a major role in placental development and guarantees a successful pregnancy
by controlling the two major steps of human placental development (the first being hypoxic,
until the end of the first trimester, and then normoxic), through regulating cellular behavior,
stem cells programming, and embryonic development.
In humans, and to a lesser extent, in mice, placental development needs the transition from
a hypoxic condition to a tissue-typical oxygen state (around 10% of partial pressure). In
humans, the switch, which occurs at 11-12 weeks of gestation, coincides with the disruption
of cytotrophoblasts plugs at the tip of spiral arteries located in the intervillous space, leading
to an efficient perfusion of the uterine bed with maternal blood (Burton et al., 1999; Hustin
and Schaaps, 1987). In this context, placental bed passes from an oxygen pressure of less than
18 mmHg (2,5%) to up to 60 mmHg (8,5%) (Burton and Caniggia, 2001; Jauniaux et al.,
2000; Rodesch et al., 1992). The short-term response to this change is a state of placental
oxidative stress (Jauniaux et al., 2003), due to the high metabolic rate of the growing placenta
63
Introduction
and limited expression of fetal genes involved in antioxidant defense at 8-10 weeks of
gestation (Watson et al., 1997, 1998).
However the increase in oxygen tension is essential for the later step of placentation, and in
particular for cytotrophoblasts migration towards decidua and pseudovasculogenesis. Oxygen
gradient all along trophoblast processes that invade decidua modulates cytotrophoblast
behavior. In vitro cultures of early placental villous explants (isolated from placentas at 5-8
weeks of gestation) down-regulate invasive properties in favor of a more proliferative
phenotype in low oxygen milieu (Caniggia et al., 2000a; Genbacev et al., 1996). Hypoxic
condition induces an anti-invasive and a pro-apoptotic effect on later extravillous explants
between 8 and 18 weeks of gestation without any change in proliferation rate (Lash et al.,
2006).
Oxygen organizes the cellular fate through selected pathways activated via HIF proteins.
HIF-1α and HIF-2α mRNA and proteins are expressed in different placental cell types
(Genbacev et al., 2001; Rajakumar and Conrad, 2000). HIF-1α localizes in trophoblast
populations and vascular network, with a peak at 8-10 weeks of gestation and decreases after
perfusion of the maternal bed takes place (Caniggia et al., 2000a, 2000b; Rajakumar and
Conrad, 2000). Subsequently, around 10-12 of weeks of gestation, increase of PHDs and
VHL expression is accompanied by a depression in HIF-1α protein level (Ietta et al., 2006).
HIF-1 and HIF-2 regulate several genes that are implied in key processes of placentation
such as proliferation, invasion and vascularization. Hypoxia and specifically HIF
implication have been highlighted by mice deficient for Hif-1α and Hif-2α: impaired decidual
invasion and altered angiogenesis participate to embryonic lethality in both transgenic lines
(Cowden Dahl et al., 2005).
Trophoblast colonization of decidual regions is the result of a fine balance between pro- and
anti- invasive stimuli that drive trophoblast fate and extracellular matrix reorganization. IGFs
family and TGF-β pathway are directly involved in placentation: both are regulators and
regulated by HIF factors.
The Insulin-like growth factors family includes two genes (IGF-I and IGF-II) that are
highly expressed in the placenta (Han et al., 1996) and involved in fetal growth, and cellular
64
Introduction
cytotrophoblast proliferation and migration (Forbes et al., 2008; Hills et al., 2004; Irving and
Lala, 1995; McKinnon et al., 2001). Circulating IGFs are bound to insulin-like growth factor
binding proteins (IGFBP1-6), which modulate IGFs function according to cell type. IGFBPs
affinity for IGFs depends on cell type and post-translational modifications, like glycosylation
or phosphorylation. Both IGF proteins can interact with several IGFBPs and the interaction
can activate or inhibits them, by sequestering them and/or preventing their interaction with
their cognate membrane receptor. In this context, hypoxia increases the levels of IGF-I, IGFII (Steinbrech et al., 2000) and IGFBP1, 2, 3 in several tissues (Feldser et al., 1999; Slomiany
and Rosenzweig, 2004; Tazuke et al., 1998). Increased maternal concentration of IGFs plays a
fundamental role during the first step of pregnancy by providing proliferation and invasion
stimuli and preventing apoptosis (Forbes et al., 2008).
HIF-1α also plays a role in regulating TGF-β signaling pathway. Of three TGF-β isoforms
identified, all are synthesized by decidual tissue (Caniggia et al., 2000b; Lysiak et al., 1995):
TGF-β1 and TGF-β2 are expressed all along pregnancy, TGF-β3 is more expressed until the
10th week of gestation, following HIF-1α expression profile (Caniggia et al., 1999). TGF-β1
increases the expression of adhesion molecules like cadherin-11 (Getsios et al., 1998) and
integrins (Irving and Lala, 1995), ezrin, E-cadherin, beta-catenin (Karmakar and Das, 2004);
it is also implied in the secretion of fibronectin (Feinberg et al., 1994) and upregulation of
tissue inhibitor of metalloproteinases (TIMP-1 and 2) and SERPINs such as plasminogen
activator inhibitors (PAI-1 and 2, aka SERPINE1 and SERPINB2) (Karmakar and Das,
2002). All these data support an important implication of TGF-β1 in the inhibition of
trophoblast fusion and migration.
TGF-β3, too, impacts negatively on the decidual invasion, probably by reducing proteolytic
activity of matrix metalloproteinases and urokinase plasminogen activator (Lash et al., 2005).
Its contribution in the early stage of placental development is sustained by HIF-1α, which
enhances TGF-β3 expression in hypoxic conditions in vivo and in vitro (Caniggia et al.,
2000a, 2000b; Nishi et al., 2004; Schäffer et al., 2003).
Matrix destabilization also contributes to trophoblast invasive properties. Hypoxia regulates
extracellular matrix organization via regulation of the plasminogen activator system.
urokinase Plasminogen Activator (uPA) ant its receptor (uPAr) are over-expressed under
65
Introduction
hypoxic conditions and promote trophoblast invasive properties through a calcium dependent
signaling
pathway
which
involves
mitogen
activated
protein
kinase
(MAPK),
phosphatidylinositol-3 kinase and phospholipase C (Liu et al., 2003). Plasmin activation,
dependent on uP-uPAr interaction, triggers extracellular matrix degradation via matrix
metalloproteinases proteolytic activity (Kjøller et al., 1997). Recent studies have also shown
that hypoxia can activate Plasminogen Activator Inhibitor-1 and Plasminogen Activator
Inhibitor-2 (PAI-1 and PAI-2) in vitro, which inhibits uPA enzymatic activity and prevents
early gestation invasion (Fitzpatrick and Graham, 1998; Lash et al., 2006; Meade et al., 2007).
All these findings suggest a key role of hypoxia in regulating the fine balance between
stabilizers and destroyers of extracellular matrix, but further studies need to decrypt the
molecular mechanisms involved.
Hypoxia is also a stimulating agent for placental vascularization. Placental vasculogenesis
and angiogenesis ensue from a concert of growing factors, angiopoietins, and VEGF family
members. VEGF-A is a direct HIF-1α target gene. Low oxygen state is associated with an
increase in VEGF expression (Wang and Semenza, 1995) and angiopoietin (Zhang et al.,
2001) which can coordinate the formation of new vessels in placental bed.
A. VEGF-A transcription regulation: beyond oxygen
VEGF-A is one of the main promoters of vascularization in normal and pathological
conditions. Its regulation is committed to several signals that coordinate protein expression in
feto- placental development, in adult angiogenesis and in pathological background like cancer
progression. Human Vegf-a is located on human chromosome 6 (6p21.3) and it codes for nine
VEGF-A isoforms. The most common isoforms are VEGF 121, 165 and 189, and generally,
VEGF 165 is the most abundant. Since its crucial role in developmental stage and adult
tissues, VEGF expression results from the coordination of regulation of transcription, of
mRNA maturation and stability, and of mRNA translation. Here we focused on regulation of
Vegf-a transcription.
The VEGF promoter is a 2400 bp region without a TATA box, but with consensus binding
sites for several transcription factors (Sp1/Sp3, AP-1, AP-2, Egr-1, STAT-3, and HIF-1),
66
Introduction
which are activated by different stimuli: growing factors, cytokines, hormones, oncogenes,
and tumor suppressors genes (Figure 5).
Figure 5: VEGF promoter: binding sites and principal transcriptional regulators
The proximal promoter (-88 base pairs upstream the transcription starting site), a GC reach
sequence, integrates signals from Specificity protein-1 and 3 (Sp1, Sp3), which bind to the
consensus sequence GGCGGG, Activating protein-2 (AP-2), which recognizes the sequence
GGCCGGGG and Early gene response protein-1 (Egr-1), which interacts with the sequence
GCGGGGGCG.
Sp1 and Sp3 belong to Sp/KLF (Kruppel-like factor) family, together with Sp2 and Sp4. They
interact with two binding sites in the region -88/-66. The binding is enhanced by
phosphorylation of serine and threonine residues. In particular phosphorylation on Threonine
453 and 739 by extra-cellular signals regulated kinases (ERK) pathway is necessary for
VEGF-A expression: mutation in these residues strongly affects VEGF-A expression
(Milanini-Mongiat et al., 2002). Sp1 phosphorylation can be induced by TGF-β1 (Benckert et
al., 2003), Neu differentiation factors (NDFs) (Alroy et al., 1999), serum (Jensen et al., 1997),
and growing factors like Hepatocyte Growth Factor (HGF) (Reisinger et al., 2003).
Sp3 is homologous to Sp1, it competes with Sp1 for the same binding site and the binding
requires protein phosphorylation on serine 73 by ERK pathway (Pagès, 2007). It is described
in the literature as a putative inducer or repressor of Vegfa activity, according to cell type, to
oxygen tension and to Sp1/Sp3 ratio (Discher et al., 1998; Hata et al., 1998).
67
Introduction
Ap-2 binds specifically the consensus sequence in the proximal promoter. Other regions have
been described as putative Ap-2 binding site in the promoter region between -88bp and -65 bp
(Gille et al., 1997), but no functional studies have been developed until now. Ap-2 can
stimulate VEGF-A expression as a consequence of damages induced by UVA (Gille et al.,
2000), or by growth factors, like TGF-α (Gille et al., 1997). However, in specific cell types it
can act also as a tumor suppressor. It has been shown that Ap-2 expression decreases in
prostate cancer cells, and also in melanoma, breast, and colorectal cancer: the model proposed
is that Ap-2 can prevent Sp3 binding to Vegf-a promoter and block its transcription (Ruiz et
al., 2004).
Activating protein-1 (Ap-1) belongs to the leucin zipper transcription factors family that
consist in a multigenic family including Fos (c-Fos, FosB, Fra-1 and Fra-2), Jun (c-Jun, JunB,
JunD and), ATF (ATFa, ATF-2 and ATF-3) and JDP (JDP1 and JDP2). It binds, in form of
heterodimers, the consensus sequence 5′-TGAG/CTCA-3′, also known as TPA-response
elements (TREs) and it is involved in cell cycle regulation, proliferation, differentiation and
apoptosis (Angel and Karin, 1991; Hess et al., 2004). In response to hypoxia, Ap-1 can
potentiate HIF-1α-dependent VEGF expression (Alfranca et al., 2002; Michiels et al., 2001;
Salnikow et al., 2002). Other stresses, like oxidative stress, UV rays or hypoglycemia
condition, can induces Fos and Jun over-expression, which in turn activates VEGF-A
expression via HIF-1α (Dong et al., 2012; Gerald et al., 2004; Minet et al., 2001; Textor et al.,
2006).
STAT3 belongs to the Signal Transducer and Activator of Transcription (STAT) family,
which includes seven members: STAT1, STAT2, STAT3, STAT4, STAT5 (STAT5A and
STAT5B), and STAT6, implied in differentiation, growth and cell survival. STAT3
recognizes a consensus sequence located between -848 and 840 bp (TTCCCAAA) on VEGF
promoter (Niu et al., 2002; Wei et al., 2003a). Like AP-1, it can enhance oxygen-dependent
VEGF expression, forming a molecular complex with HIF-1α and CBP/p300 (Gray et al.,
2005). Interestingly, in microvascular endothelial cells, STAT3 is a VEGF target gene, it is
phosphorylated on tyrosine residues and activated (Bartoli et al., 2003). STAT3 also induces
VEGF expression in an oxygen-independent fashion: it mediates VEGF induction due to
oncostatin, which is increased in glioblastomas (Repovic et al., 2003), to IL-6 which exercises
an angiogenic activity on cervical tumor (Wei et al., 2003b) and peroxynitrite (Platt et al.,
68
Introduction
2005). STAT3 blockers can have an inhibiting effect on tumor growth by reducing VEGF
expression: it is the case for caffeic acid and its synthetic derivative CADPE [3-(3,4dihydroxy-phenyl)-acrylic acid 2-(3,4-dihydroxy-phenyl)-ethyl ester] (Jung et al., 2007).
MicroRNAs (miR), as well, play a crucial role in the regulation of VEGF-A mRNA
transcription and RNA stability. Some miRs regulate Vegfa expression by modulating HIF
machinery, while others in an oxygen independent manner. Computational analysis helps to
identify 96 miR that can regulate VEGF expression, and some of these have been validated by
functional studies (Hua et al., 2006) miR20b can block VEGF-A expression by interfering
with HIF-1 binding on the HRE element (Cascio et al., 2010); miR-15b, miR-16, miR-20a,
and miR-20b interact with the 3’ untranslated region of VEGF-A mRNA and block its
expression in vitro, even though the mechanism is still not elucidated (Hua et al., 2006; Liu et
al., 2009). Some miR can act indirectly on HIF-1 regulators: it is the case for miR31, which
binds the 3’ untranslated region of FIH, and indirectly enhances HIF-1 target genes
expression (Liu et al., 2010). miR-210 is one of the most up-regualted miR under hypoxic
conditions, and is also associated with VEGF-A increased expression (Huang et al., 2010;
Quero et al., 2011).
5. HIFs and preeclampsia
Pregnancies complicated by preeclampsia are the result of abnormal placentation as
described in chapter 1. Failed trophoblast invasion is the cause of intermittent placental
oxygenation and cycles of placental hypoxia- reoxygenation that result into oxidative stress
and later maternal dysfunction. This status perturbs HIF expression during pregnancy and
since HIFs target genes are directly involved in the building of the placenta, its deregulation
may participate to the preeclamptic syndrome.
A. HIFs expression in pregnancies complicated by preeclampsia
Preeclamptic placentas overexpress HIF-1α and HIF-2α (Rajakumar et al., 2001, 2004).
Placental HIF-1α and VHL, but not HIF-2α, are also increased in women living at high
altitudes, who have an elevated risk to develop preeclampsia (Zamudio et al., 2007). Recently,
increased circulating HIF-1α mRNA levels have been found in the plasma of preeclamptic
69
Introduction
women (Ashur-Fabian et al., 2012). All these findings suggest a perturbed system of HIF-1
regulation that could have repercussions on downstream HIF pathways.
B. HIF contribution to preeclampsia
The impact of HIF deregulation affects trophoblast invasion and maternal health. As
shown before, one of the main drivers of trophoblast invasive properties is the downregulation
of the TGF-β pathway. In preeclampsia TGF-β3 follows HIF-1α expression profile and is upregulated in preeclamptic placentas, thus regulating negatively decidua colonization by
invading trophoblasts (Nishi et al., 2004). Inhibition of HIF-1α blocks TGF-β3 and restores
trophoblasts invasive capacities of in vitro (Caniggia et al., 2000a). In parallel, impaired
invasion fosters altered blood flow in placental bed and consequent HIF over-expression.
HIF-1α deregulation affects also placental angiogenesis and the secretion of factors
responsible for the maternal syndrome. Preeclampsia is usually associated to an increase of
placental VEGF and sFlt-1, an increase in circulating sFlt-1 and total VEGF, but a decrease of
free VEGF and PlGF (because of sFlt-1 augmentation) (Maynard et al., 2003; Tsatsaris et al.,
2003). Low oxygen environment induces over-expressed
sFlt-1 in cultures of villous
explants, and inhibition of Hif-1α is accompanied by reduced sFlt-1, thus demonstrating that
HIF-1α participates actively to sFlt-1 increase in preeclampsia (Nevo et al., 2006). HIF-1α
participates in the up-regulation of sEng via the TGF-β3 signaling pathway (Yinon et al.,
2008). HIF-1α effects contribute also to vasoconstriction via up-regulation of endothelin-1,
which is a well-known HIF-1α target gene (Minchenko and Caro, 2000; Yamashita et al.,
2001). Interestingly administration of the stabilized form of HIF-1 α in pregnant mice
reproduces a preeclampsia-like syndrome, with hypertension, increased sFlt-1 and s-Eng,
kidney damages, proteinuria and HELLP syndrome (Tal et al., 2010). Recently it has been
shown that hypoxic condition participates to the over-expression of the urotensin-II
receptor, which has been found upregulated in preeeclamptic placentas. Urotensin–II is a
potent vasoconstrictor: syncytiotrophoblasts respond to in vitro hypoxia by up-regulating
urotensin–II receptor and increased levels of sFlt-1 (Gould et al., 2010). Urocortin-2 and
urocortin-3 derive from the corticotrophin-releasing factor (CRF) family and are involved in
the modulation of contractility of the myometrium and vascular resistance: both are found
increased in preeclamptic placentas and regulated via HIF-1α (Imperatore et al., 2010).
70
Introduction
C. Causes of HIF deregulation
During preeclampsia HIF-1α deregulation is the result of different factors which can
cooperate to increase its expression and decrease its degradation in an oxygen–dependent and
-independent fashion (Figure 6).
Figure 6: Factors affecting HIF-1α deregulation in preeclampsia and consequences
on placentation and late maternal syndrome
Hypoxia is due to impaired trophoblast invasion that leads to placental ischemia and cycles of
hypoxia-reoxygenation
that
may
contribute
to
HIF-1α
accumulation.
Persistent
inflammation, usually associated to preeclampsia, also favors HIF-1α increase all along
pregnancy.
Moreover recently it has be shown that HIF-1α degradation machinery is impaired in
preeclamptic placentas, with decreased activity of proteasome (Rajakumar et al., 2008) and
reduced expression of PHD-2 and FIH (Rolfo et al., 2010): all factors that contributes to
HIF-1α persistency. Placental explants from preeclamptic pregnancies show an increased
level of HIF-1α independently of oxygen environmental concentration, thus suggesting that
deregulation of oxygen independent pathways could have an impact on HIF-1α expression
71
Introduction
(Rolfo et al., 2010). 2-methoxyoestradiol (2-ME) is an antiangiogenic and antitumor factor
which down-regulates HIF-1α and blocks HIF-1α-dependent VEGF expression (Mabjeesh et
al., 2003). It is reduced in placenta from pregnancies complicated by preeclampsia and its
implication in the preeclamptic syndrome was shown in a mouse model lacking catechol-Omethyltransferase
(Comt),
the
enzyme
that
converts
hydroxyoestradiol
into
2-
methoxyoestradiol. Pregnant Comt-/- mice suffer from hypertension, proteinuria and
glomerular endotheliosis, angiogenic imbalance, hypoxic placentas and increased HIF-1α: all
the symptoms are attenuated by 2-ME administration (Kanasaki et al., 2008). 2-ME affects
directly HIF-1α expression: trophoblasts cultivated in hypoxic conditions reduce sFlt-1
secretion and recover invasive properties after 2-ME treatment, which enhances HIF-1α
down-regulation and consequently TGF-β3 suppression (Lee et al., 2010).
Another hypothesis for HIF-1α deregulation in preeclampsia is the association with specific
HIF-1α polymorphisms, but recent studies failed to find significant association in Korean
(Kim et al., 2012) and Mexican populations (Nava-Salazar et al., 2011).
72
Introduction
Chapter IV. JDP2: from chromatin organization to regulation of
gene expression
Any change in extracellular and intracellular environment can drive cellular adaptation
through modulation of gene expression and/or modifications of proteins activity, localization
and degradation. Modulation of gene expression requires the physical interaction of
transcription factors with the DNA sequence of target genes, as well as possibly chromatin
reorganization through epigenetic modifications. Indeed, activated transcription factors in the
right place at the right time could be not enough to organize cell response to external stimuli.
In the last 30 years, researches on DNA organization identify another indispensable
requirement for gene expression: the chromatin status. DNA is packaged in a histone core:
post-translational modifications apposed on histones tails (acetylation, methylation,
sumoylation, phosphorylation, ubiquitination) result in changes of chromatin packaging
which allows or prevents transcription factors interaction with DNA.
Association between histone modifications and gene expression is summarized in the so
called “histone code hypothesis”, according to which some histones modifications are more
permissive to transcription and associated to an open chromatin status, at least partially
equated to euchromatin, while others are repressive marks that prevent DNA binding and they
are associated to a more compact chromatin folding, partially equated to heterochromatin .
In this context, Jun Dimerization Protein 2 (JDP2) acts as a transcription factor, able to bind
directly DNA, and as a chromatin modulator, in charge of modifying chromatin structure
and finally allowing or preventing gene expression.
JDP2 encodes for a 18kDa protein. It is part of JDP subfamily with JDP1, and more largely
the AP-1 family, along with Jun proteins (cJun, JunB, JunD), Fos proteins (c-Fos, FosB,
Fra-1 and Fra-2) and ATF (ATF-A, ATF-2 and ATF-3) (Hess et al., 2004; Karin et al., 1997).
All these proteins share in common a basic leucin zipper domain (bZIP) domain, responsible
for dimerization, a necessary condition for DNA binding on TPA-response element (also
known as TRE 5'-TGAG/CTCA-3). Jun proteins can form homodimers or heterodimers,
while Fos proteins are usually associated to Jun proteins. They are implicated in several
73
Introduction
cellular processes like differentiation, cell cycle regulation, apoptosis and cancer progression
(Bossy-Wetzel et al., 1997; Schreiber et al., 1999; Young et al., 1999). According to the
nature of AP-1 dimers and cell types, stimuli coming from cytokines, growth factors and
stress conditions are translated into activation or inhibition of gene expression.
In the same way, JDP2 can form homodimers or heterodimers with proteins of the same
family, like c-Jun (Aronheim et al., 1997), Activating transcription factor 2 (ATF-2) (Jin et
al., 2001), or other families like C/EBP homologous protein- 10 (CHOP-10) (WeidenfeldBaranboim et al., 2008), CCAAT/enhancer binding protein γ (C/EBPγ) (Nakade et al.,
2007), Interferon regulatory factor-2-binding protein-1 (IRF2-BP1) (Kimura, 2008), and
progesterone receptor (Wardell et al., 2002). The composition of the dimer defines JDP2
function as activator or more usually repressor of gene expression. Dimerization is ensured
by a conserved leucine zipper domain, common to all the members of the same family. JDP2
recognizes and binds two kinds of consensus sequence: TRE and Cyclic AMP-Responsive
Element consensus sequence (CRE) thanks to a basic domain adjacent to the leucine zipper
dimerization motif.
JDP2 was found for the first time in association with c-Jun in a screen to detect proteinprotein interaction (Aronheim et al., 1997), and is considered as one of the principal
inhibitors of c-Jun activity: it sequesters c-Jun and blocks its interaction with activating cofactors. To date, 32 articles describe JDP2 function in regulating important cellular processes
like differentiation, cell cycle progression, cellular senescence and tumor progression.
1. JDP2 expression
The JDP2 protein is ubiquitously present in all tissues and cell types and insensible to most
part of stress stimuli. Protein down-regulation has been detected in cells stimulated with 20%
serum or after translation inhibitor anisomycin treatment (Weidenfeld-Baranboim et al.,
2011); controversial studies found either an increase (Piu et al., 2001) or decrease
(Weidenfeld-Baranboim et al., 2011) of JDP2 after cell exposure to UV irradiation: this
divergence maybe linked to the different experimental procedures and the different kinetics
chosen in these studies. In the context of cancer, in a panel of 53 patients, only 5.7% tumors
revealed an increased level of JDP2 (Heinrich et al., 2004). Finally, in the case of pregnancy
74
Introduction
disorders, analysis of transcriptomic data from preeclamptic placentas reveals that JDP2 is
down-regulated in third trimester preeclamptic placentas (Nishizawa et al., 2007).
Nothing is known about molecular mechanism regulating jdp2 mRNA expression, but
recently it has been shown that its degradation depends on post-translational
modifications (Weidenfeld-Baranboim et al., 2011). In fact, Mouse Embryonic Fibroblasts
(MEFs) stimulated with serum, UV rays or translation inhibitor anisomycin, show a decrease
in JDP2 protein level linked to phosphorylation of threonine148 by Jun N-terminal kinase
(JNK) and p38, which addresses protein to proteasome degradation (Weidenfeld-Baranboim
et al., 2011).
2. JDP2: involvement in chromatin organization and gene
regulation
JDP2 participates to chromatin organization since its primary organization until regulation
of the higher-ordered structure.
In vitro studies show that linear DNA incubated with core histones and JDP2 organizes itself
into a coiled structure: the DNA packaging into mononucleosome is proportional to JDP2
protein level. These findings were confirmed by in vivo studies on cells overexpressing JDP2
and treated with micrococcal nucleases: mononucleosomes assembly is more evident on DNA
regions containing JDP2 binding sites (Jin et al., 2006). All these findings are sufficient to
define JDP2 as a histone- chaperone.
JDP2 is also able to bind directly core histones and interacts with enzymes responsible for
histones modifications, in particular histones acetyltransferases (HATs) and deacetylases
(HDACs). Indeed, JDP2 has been found as part of the inhibitor of histone
acetyltransferases complex (INHAT), whose role is to mask histones residues thereby
blocking acetyltransferase (HAT) activity of p300, PCAF, CBP and GCN5 and finally
regulate gene expression (Jin et al., 2006; Seo et al., 2001). Transgenic mice deficient for
JDP2 show impaired adipogenesis (Nakade et al., 2007), osteoclastogenesis, and myeloid
cells differentiation, in particular neutrophils differentiation (Maruyama et al., 2012). In
normal conditions, neutrophil differentiation is favored by down-regulation of ATF3, one of
75
Introduction
the main inhibitors of the TLR signaling pathway, and in vitro studies have shown that JDP2
inhibits ATF3 expression in fibroblasts (Weidenfeld-Baranboim et al., 2009). In JDP2 KO
mice, ATF3 level is strongly increased and associated to hyper-acetylation of the promoter
region, thus prompting to speculate of a putative epigenetic regulation of ATF3 promoter by
JDP2. Moreover these mice are more sensitive to bacterial infection and neutrophils suffer of
stress condition due to increased ROS production. All these findings suggest that JDP2 plays
an active role not only during differentiation but also as a cellular guardian against stress
conditions, in order to balance cellular homeostasis in response to damaging factors. Impaired
adipogenesis, too, is ascribed to impaired acetylation in the promoter of genes involved in the
process of differentiation. In fact JDP2 reduces p300 HAT activity on CEBPγ promoter, one
of the first genes involved into the adipogenesis cascade: promoter hypo-acetylation results in
the repression of gene expression and the altered expression of downstream genes of the same
cascade (Nakade et al., 2007).
Apart from blocking HAT activity, JDP2 is also able to recruit Histones Deacetylase3
(HDAC3) on the genes promoters, thus blocking gene expression: this mechanism is
specifically involved in repression of c-jun expression (Jin et al., 2002).
The mechanism was described for the first time as involved in the differentiation of murine
embryonic carcinoma cells F9 into endoderm-like cells. The endoderm-like phenotype is
associated to increased expression of c-Jun after retinoic acid treatment. The c-Jun promoter
contains a Differentiation Response Element (DRE), which is recognized by the complex
ATF2 and p300 and activates c-Jun expression, which in turn activates the genes cascade
responsible of the endoderm-like phenotype. JDP2 recruits indirectly HDAC3 and interacts
with ATF2. Replacement of the p300/ATF-2 complex by the JDP2/ATF2/HDAC3 complex
results in an inhibition of c-Jun expression and cell retention into an undifferentiated state (Jin
et al., 2002).
JDP2 could also modulate ATF2 regulation in an epithelial-mesenchymal transition, a
cellular model for metastatic processes. In pancreatic cell lines ATF2 enhances cellular
sensibility to TGF-β signaling and promotes a mesenchymal phenotype and invasiveness
properties; on the opposite, JDP2 overexpression seems to have more protective and antiinvasive effects: cells maintain their phenotype, associated to collagen I expression and
76
Introduction
persistency of tight junctions; in vivo studies also show a reduced JDP2 level in pancreatic
cancer tissues (Yuanhong et al., 2010). These data suggest that JDP2 can act as a tumor
suppressor by counterbalancing ATF2 pro-invasive effects (Xu et al., 2012).
The function of JDP2 in cancer biology has not been completely elucidated, and is still
fluctuating between the role of oncogene and tumor suppressor. On one hand the function of
JDP2 as tumor suppressor is reinforced by the finding that it inhibits tumor progression in
severe combined immune-deficient mice (SCID) injected with cells overexpressing JDP2
(Heinrich et al., 2004).
On the other hand JDP2 has been identified as a candidate
oncoprotein in p27-/- lymphoma through a high-throughput strategy based on viral insertional
mutagenesis (Hwang et al., 2002), and its overexpression induces a partial oncogenic
transformation of chicken embryo fibroblasts (Blazek et al., 2003)
Another interesting function of JDP2 is its protective effect against p53-induced apoptosis
after UV irradiation. In fact, the p53 promoter contains a variant of AP-1 site, called PF-1,
which is recognized by AP-1 family members. JDP2 overexpressing cells are more resistant
to UV-induced cell death and this could be due to the down-regulation of p53 expression:
even though the mechanism is still not clarified, one possibility is that JDP2 and c-Jun, both
increased upon UV irradiation, form repressive heterodimers responsible for p53 transcription
modulation (Piu et al., 2001). The same JDP2 anti-apoptotic function has been described in
cardiomyocytes stimulated with TGF-β1: the overexpression of JDP2 reduces hypertrophy
after β- adrenoreceptor agonist isoprenaline and apoptotic rate (Hill et al., 2013).
JDP2 has also been described as an activator of transcription in association with the
progesterone receptor (Wardell et al., 2002) or the protein CHOP-10, a related member of
CEBP family. In this latter case both proteins interacts through the bZIP domain in vivo and
in vitro and activates genes axpression by binding TRE element but not CRE elements on
gene promoters (Weidenfeld-Baranboim et al., 2008).
The best characterized model which involves activating properties of JDP2 is the epigenetic
regulation of replicative senescence (Nakade et al., 2009).
Replicative senescence is associated to cell cycle arrest and expression of specific markers
like p16Ink4a and p19ARF (Arf and p14ARF in humans). Primary cells enter naturally in a
77
Introduction
senescent state after several weeks in culture; but sometimes cells can undergo senescence
after interaction with damaging factors thereby escaping oncogenic transformation. In this
context cellular senescence can be seen as a protective mechanism against uncontrolled
cellular growth. Huang and coworkers studied the effects of oxygen-induced cellular
senescence in wild type MEFs (wt MEFs) and MEFs derived from mice lacking JDP2
expression (JDP2 -/- MEFs). Wt MEFs become senescent after 6 weeks of exposure to
environmental oxygen (20%); on the opposite JDP2 -/- MEFs maintain their proliferative
state; in low oxygen condition (3% oxygen) both cell cultures continue to proliferate.
Expression of p16Ink4a and p19 is detectable only in senescent wt MEFs cultivated in 20 %
oxygen condition: this means that oxidative stress induces cellular senescence through a
molecular mechanism mediated by JDP2.
Studies on the epigenetic background of the p16Ink4a promoter highlight the fundamental role
of JDP2 in chromatin remodeling and gene expression. Cellular proliferation is associated to
H3K27 methylation and the binding of the Polycomb repressive complexes 1 and 2 (PRC1
and PRC2) on the p16Ink4a locus, which results in the inhibition of p16Ink4a expression. JDP2
avoids PRC1 and 2 binding, and reduced H3K27 methylation is a condition compatible with
p16Ink4a expression (Nakade et al., 2009).
In conclusion JDP2 is involved in important cellular changes that require changes of gene
expression pattern: cell cycle, differentiation, tumor progression and cellular senescence. Its
action as activator or repressor depends on its partners, on the cell type, and on the type of
stress. Globally it acts as a cell protector against damaging factors and as a guardian of
cellular homeostasis. The last 15 years of researches helped to understand its implication in
important cellular processes, but further studies need to clarify its regulation and its
participation in normal and pathological conditions.
78
Results
Results
Results
1. Paper 1
PREECLAMPTIC
INVOLVING
PLASMA
THE
AP-1
INDUCES
TRANSCRIPTION
TRANSCRIPTIONAL
MODIFICATIONS
REGULATOR
JDP2
IN
ENDOTHELIAL CELLS
Rosamaria Calicchio1, Christophe Buffat2, Jacques R.R. Mathieu1, Nour Ben Salem1,3, Celine
Mehats1, Sébastien Jacques1, Alexandre Hertig4, Nadia Berkane4, Julie Grevoul-Fresquet5,
Umberto Simeoni2, Carole Peyssonnaux1, Julie Gavard1, Daniel Vaiman1, Francisco Miralles1
1
INSERM U1016-CNRS UMR8104, Université Paris Descartes, Institut Cochin, 24, rue du
Faubourg Saint-Jacques, 75014 Paris, France.
2
Laboratoire de biologie moléculaire, Génétique Oncologique et Endocrinienne, Hôpital de la
Conception- AP-HM, 147 Boulevard Baille, 13385 Marseille, France
3
Laboratoire de biochimie, CHU Farhat Hached, Sousse, Tunisie
4
Service de Gynécologie obstétrique Médecine de la Reproduction, Hôpital Tenon, 4, rue de
la Chine, 75020 Paris, France
5
Service Gynécologie et Obstétrique, AP-HP Hôpital Bicêtre, 78, rue du Général Leclerc,
94275 Le Kremlin-Bicêtre, France
This work has been accepted in The American Journal of Pathology on August 2013
93
Results
Preeclamptic plasma induces transcription modifications involving the AP1 transcriptional regulator JDP2 in endothelial cells
Summary
It is well know from literature that preeclamptic plasma releases soluble factors in the
maternal circulation, which altogether, induce a stress condition for endothelial cells, like
anti- angiogenic factors, pro-inflammatory cytokines, and activated immune cells. All these
factors impact important endothelial functions like permeability, inflammatory response,
coagulation state. A lot of efforts have been undertaken in order to characterize the
endothelial response to factors present in the maternal circulation of preeclamptic women. Up
to now, all these studies were targeted on the alteration of specific endothelial functions, or on
the identification of specific molecules whose concentration is altered in the maternal
circulation and which could impact maternal health.
This project was based on the idea that endothelial cells can be considered as biosensors able
to “perceive” stimuli coming from preeclamptic plasma and to translate them into a modified
expression profile.
The originality of this study lies in the analysis of the response of the whole endothelial
genome to the preeclamptic plasma treatment. For this purpose we exposed an endothelial cell
line, HUVEC, to preeclamptic plasma for a short period and the total RNA was hybridized on
a microarray in order to characterize the whole expression profile in response to the treatment.
This approach helped us to find modified genes, some of them already known as playing a
role in the physiopathology of preeclampsia, like endothelin-1 and apelin, and also new actors
involved in endothelial dysfunction. We decided to focus on one of the most down-regulated
specific genes, JDP2, and try to better characterize its function and its implication in
endothelial cell expression profile. Our last results indicate that JDP2 knock- down is
responsible for part of transcriptional modifications observed at least for three genes,
IGFBP3, BCL2A1 and VEGFA. Future analysis of its implication in various tissues will be an
important perspective of the present study.
94
Results
Abstract
Preeclampsia is a pregnancy disorder characterized by hypertension and proteinuria. In
preeclampsia the placenta releases factors into the maternal circulation which cause a
systemic endothelial dysfunction. Here, we investigated the effects of plasma from women
with preeclamptic and normal pregnancies on the transcriptome of an immortalized human
umbilical vein endothelial cell line (HUVEC). The cells were exposed for 24 hours to
preeclamptic or normal pregnancy plasma and their transcriptome analyzed using Agilent
microarrays. A total of 116 genes were found differentially expressed. 71 were up-regulated
and 45 down-regulated. In silico analysis revealed significant consistency and identified four
functional categories of genes: mitosis and cell cycle progression, anti-apoptotic, fatty acid
biosynthesis and endoplasmic reticulum stress (ERs) effectors. Moreover, several genes
involved in vasoregulation and endothelial homeostasis showed modified expression
including: EDN1, APLN, NOX4 and CBS.
Promoter analysis detected, among the up-
regulated genes, a significant over-representation of genes containing AP-1 regulatory sites.
This correlated with down-regulation of JDP2, a gene encoding a repressor of AP-1. The role
of JDP2 in the regulation of a subset of genes in the HUVECs was confirmed by siRNA
inhibition. We characterized transcriptional changes induced by preeclamptic plasma on
HUVECs, and identified for the first time JDP2 as a regulator of a subset of genes modified
by preeclamptic plasma.
Key Words
Preeclampsia, Plasma, Endothelial Cells, Microarrays, NOX4, CBS, AP-1,
JDP2
95
Results
Introduction
Preeclampsia is a pregnancy complication affecting approximately 5–8% of pregnancies and
capable of causing both maternal and fetal morbidity and mortality 1. The maternal syndrome develops
after 20 weeks of gestational age and is characterized by elevated blood pressure (>140mm Hg/90mm
Hg), proteinuria (>300mg/24h), systemic endothelial cells (EC) dysfunction and inflammation.
Defective placentation is thought to be at the root of the disease. In the developing preeclamptic
placenta, the normal process of trophoblast invasion and remodeling of the uterine maternal spiral
arteries is impaired. This default leads to reduced placental perfusion, oxidative stress and
inflammation, with subsequent release of placental factors and debris into the maternal circulation.
These factors are supposed to cause a widespread EC activation leading to the multisystem
dysfunction characteristic of preeclampsia 2.
Maternal endothelial dysfunction in preeclampsia is revealed by structural modifications of
the ECs of the kidney glomeruli as well as by functional modifications including: changes in the
balance of coagulant and anticoagulant factors, increased circulating concentrations of von Willebrand
Factor (vWF), endothelin-1 (ET-1), soluble adhesion molecules and cytokines. Moreover, increased
levels of the antiangiogenic factors soluble fms-like tyrosine kinase-1 (sFLt1) and soluble endoglin
(sEng) presumably released by the placenta, are found in the plasma of preeclamptic women.
Therefore, endothelial dysfunction can by itself explain many maternal symptoms of preeclampsia.
ECs dysfunction will lead to hypertension due to vasoconstriction, proteinuria due to glomerular
damage, and would be the cause of systemic inflammation and also of edema due to increased
vascular permeability 3, 4.
Targeted studies have shown that the plasma from preeclamptic women is able to elicit
specific responses in human EC in vitro 4. Thus, incubation of EC with preeclamptic plasma increases
the expression and the production of ET-1, platelet derived growth factor (PDGF), vascular cell
adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), cellular fibronectin,
chemokines, cytokines, inducible nitric oxide synthase (iNOS), NADPH oxidase 2 (NOX2),
prostacyclines, nitric oxide (NO), and reactive oxygen species (ROS)
4-14
. These studies support the
idea that preeclamptic plasma contains factors able to trigger EC dysfunction.
In addition to these targeted studies only one study, published in 2005, has investigated the
effects of preeclamptic plasma on the global gene expression profiles of EC
15
. In this work, the
authors used microarrays to analyze the in vitro gene expression profiles of human umbilical vein
endothelial cells (HUVEC) and human glomerular microvascular endothelial cells (hGMEC) after a 24
hours exposition to preeclamptic or matched controls plasma.
However, they could not detect
96
Results
substantially altered gene expression by preeclamptic factors, except for a modest induction of IL-8
(around 1.5 fold). Therefore, they concluded that there are few endothelium-activating factors in the
plasma of preeclamptic patients that can directly activate EC. According to that, the endothelial
activation in PE would result more likely from other mechanisms which would act alone or in
association with the plasma factors. However, as reported above, many studies indicate that the
preeclamptic plasma can by itself induce significant modifications in the EC. The question of the
presence in the preeclamptic plasma of factors able to directly modify the gene expression profile of
EC is an issue of some importance. Such factors could be used as biomarkers or targets for therapeutic
approaches. We made the hypothesis that the technical limitations of the 2005 microarrays were part
of the reason why only very mild modifications were detected, prompting us to carry out a new study.
Therefore, we carried out a transcriptomic study on an immortalized HUVEC line exposed to plasma
from normotensive or preeclamptic pregnancies. We found modifications of gene pathways, that could
be involved in long-term endothelial dysfunction. Most interestingly, several genes in these pathways
present a possible regulation through the Jun family of transcription factors (i.e. an enrichment in AP1 binding sites). Since the AP-1 negative regulator JDP2 (Jun Dimerization Protein 2), was among the
most down-regulated genes by exposure to preeclamptic plasma, we analyzed its direct implication
through siRNA knock-down experiments. Our results suggest that JDP2 is one of the factors
responsible for endothelial transcriptional modifications in the preeclamptic patients.
97
Results
Materials and Methods
Sample collection, processing and validation
Frozen plasma samples from women with normal or preeclamptic pregnancies were obtained from a
previous epidemiological unpublished study. In all cases informed consent was obtained. The local
ethics committee of Federative Research Institute 48 approved the use of the samples for the present
project (agreement number 08-012). Preeclampsia was defined using the criteria of gestational
hypertension and proteinuria. Gestational hypertension was defined as new onset systolic blood
pressure >140 or diastolic pressure > 90 mmHg after 20 weeks gestation. Proteinuria was defined as >
300 mg/24 hour urine collection. Proteinuria was measured using dipstick test. All preeclamptic
plasmas showed traces above 300 mg/dl which corresponds to an approximate of 1-2g/24 h. The blood
samples were collected in 10 ml EDTA-tripotassium Vacutainer tubes (Beckton Dickinson,). Within 1
hour the tubes were centrifuged for 15 minutes at 700g and 4°C to remove blood cells. Plasma samples
were aliquoted and stored at -80°C. The clinical characteristics of the women who provided the blood
used in this study are listed in Table 1. Plasma was obtained from two groups: a group of
normotensive women with uncomplicated pregnancies and a group of preterm pregnant preeclamptic
women. At the moment of blood collection the control group (n = 10) had an average gestational age
of 36 weeks (range 28-40) and had no history of hypertension, diabetes or infection. The preeclamptic
group (n=10) was composed of women with preeclampsia with an average gestational age at the
moment of blood sampling of 32 weeks (range 27-36). On the basis of blood pressure and proteinuria
values most of the women of this group could be categorized as having severe preeclampsia. All the
procedures followed for the plasma and clinical data collection from the patients were in accordance
with institutional guidelines. In addition to the clinical parameters the plasma were further
characterized by measuring the levels of tumor necrosis factor alpha (TNF- using (Quantikine
human TNF- immunoassay, R&D systems), and sVCAM-1 using (Quantikine human sVCAM-1
immunoassay, R&D systems). The levels of both TNF- and sVCAM-1 were found significantly
elevated in the plasmas of preeclamptic women when compared to matched controls.
(Supplementary Figure S1). These analyses corroborate previous studies and indicate increased EC
activation and inflammatory status in the preeclamptic women included in this study.
Endothelial cells culture
The endothelial cell used in our study was the Human Umbilical Vein Endothelial Cell (HUVEC) line
immortalized with the large T-antigen of SV40 and ectopic expression of the hTERT (human
Telomerase Reverse Transcriptase) 16. Since the establishment of this cell line, its phenotype has been
maintained over passages. Cells were routinely cultured in uncoated 75 cm2 tissue culture flasks at
37°C and 5% CO2. Culture medium consisted of DMEN supplemented with 10% (vol/vol) heatinactivated fetal calf serum (FCS), 2mM glutamax, 5 IU/mL heparin, 100 IE/ml penicillin, and 100
mg/mL streptomycin. In addition, we used an immortalized human microvascular cell line, the
HMEC-1, to confirm our results on most modified genes. The culture medium for the HMEC-1
consisted of MCDB-131 supplemented with 10% (vol/vol) heat-inactivated fetal calf serum (FCS), 0.2
μg/ml hydrocortisone, 10 ng/ml EGF, 2mM glutamax, 5 IU/mL heparin, 100 IE/ml penicillin, and 100
mg/mL streptomycin.
98
Results
Endothelial monolayer permeability
Endothelial monolayer permeability was assessed by the passage of FITC-conjugated dextran (0.1
mg/ml, 40kDa, Invitrogen) as previously described 17. Briefly, 100.000 HUVEC were plated onto 6.5
mm Transwell Collagen-coated 3 μm pore PTFE membrane inserts (Costar), left for 3 days to form
mature monolayers and further starved for 16 hours. After 1 hour of stimulation with 10%
preeclamptic or normal plasma, each sample from the bottom chamber was read on a fluorescent plate
reader (FUSION, Packard BioScience Company).
Experimental Design
In all experiments HUVECs were grown until forming 90% confluent monolayers. Then, cells were
washed twice in PBS and serum starved for 12 hours. To study the effects of preeclamptic plasma
factors on endothelial gene expression, serum-starved HUVECs were grown for 24 hours in culture
medium devoid of FCS but supplemented with 10% (vol/vol) plasma from preeclamptic patients or
matched controls. To determine the responsiveness of HUVECs to pro-inflammatory stimulus, cells
were incubated for 5 hours in complete culture medium with tumor necrosis factor-alpha (TNF-α). The
concentrations of TNF-α were (0.1, 0.5, 1 and 5 ng/mL).
RNA isolation
Total RNA was isolated from HUVECs with Trizol (Invitrogen Life Technology) according to the
manufacturer’s instructions and treated with DNase I to eliminate genomic DNA contamination. The
quality of the RNA was analyzed using the Bioanalyser 2100 and the RNA 6000 nano LabChip kit
(Agilent Technologies). Only total RNA samples with a RIN number ≥ 0.8 were used.
Agilent Oligonucleotide Microarrays
For gene expression measurement we used the Agilent whole genome microarray kit: Agilent human
8x60 K (ref G4851A) interrogating a total of 27958 Entrez gene RNAs and 7419 lincRNAs. The
hybridizations were carried out at the genomic and transcriptomic platform of the Cochin Institute,
according to standardized procedures. Normalization and statistics were performed using Partek
Genomic Suite. Raw data were preprocessed using quantile normalization, and log transformed.
Unsupervised analysis on data allowed checking non desired experimental factors and biological
outliers using hierarchical clustering (Pearson's dissimilarity and average linkage) and principal
component analysis (PCA) including all samples. In order to avoid false positive signals we excluded
from the analysis those genes with an average expression level under 100. In our experience,
expression values from probes giving low absolute intensity signals are not reliable. To find
differentially expressed genes, we applied a classical unpaired Student’s t test between compared
groups and computed the fold-change for each gene. Then, we used these two statistics to filter and
select differentially expressed genes. We selected genes with p value < 0.05 and fold-changes < 1.25.
The data sets were prepared according to the guidelines of minimum information for a microarray
experiment (MIAME) and were deposited in the Gene Expression Omnibus (GEO) data base:
http://www.ncbi.nlm.nih.gov/geo/. The GEO accession number for the platform is GSE41681.
99
Results
Quantitative RT-PCR
Quantitative RT-PCR was used to validate a subset of genes that showed differential expression in the
HUVECs treated with preeclamptic plasma. 12 genes were selected based upon their fold differences
and biological relevance. The RNA samples were reverse transcripted according to a standardized
protocol. Briefly, 2 μg of total DNase-treated RNA was reverse transcribed in a volume of 25 μL at
39°C using the Superscript reverse transcriptase (Invitrogen) during 1 hour. Quantitative PCR was
carried out in duplicates on 8 controls, 9 preeclamptic samples individually using the amplification kit
LC480 SYBR Green Master Kit (Roche) and the reaction was performed in a Light Cycler
Thermocycler (Roche). Primers (supplementary Table 2) were designed for the coding sequences
(GENBANK) of the different genes to be analyzed using the PRIMER3 software
(http://frodo.wi.mit.edu/cgi-bin/primer3). The different couples were chosen to cover all of the
previously described isoforms and aligned with basic local alignment search tool software to avoid
nonspecific annealing. 35 cycles were performed with the following 3 temperature steps (95°C for 10
s, 55°C for 15 s, and 72°C for 15s). Finally, samples were submitted to a progressive temperature
elevation (from 65 to 99°C at 0.1°C/s), resulting in a fusion curve, enabling us to check the PCR
products homogeneity. The threshold cycle number (Ct) values were collected with the LightCycler
software (Roche) and analyzed through a second derivative maximum method. These Cts were
normalized by the Ct values obtained for 3 reporter genes, succinate dehydrogenase subunit A
(SDHA), and glyceraldehydes-3-phsphate dehydroenase (GAPDH).
Functional clustering by DAVID
Two lists of genes induced or repressed in the HUVECs cultured with preeclamptic plasma were
submitted to the DAVID database 18, 19. Briefly, DAVID clusterizes genes from a list according to a
series of keywords common to several genes from the list. The proportion of each keyword from the
gene list submitted is compared with the proportion in the whole genome, making it possible to
compute a P value. Enrichment values are then calculated as the geometric mean of the inverse log of
each P value.
Functional clustering and biological pathway Analysis with GENOMATIX
A list of statistically significant differentially expressed genes between HUVECs treated with normal
or preeclamptic plasma was generated. This list was analyzed with several GENOMATIX tools,
including GeneRanker and the Pathway analysis tool GePS (http://www.genomatix.de/en/index.html).
The genes modified more than 1.25 fold were submitted as text files with the level of
induction/repression. This allowed to generate pathways with a threshold for significance in the gene
clustering established at p <0.05.
Pathology association testing
To assess the potential role of the differentially expressed genes in pathology we used the
GENOMATIX diseases MESH (Medical Subject Headings) tool. This analysis allowed to test for an
eventual association between the list of modified genes and specific key words associated with
100
Results
preeclampsia including:
inflammation, etc...
preeclampsia,
hypertension,
inflammation,
cardiovascular
disease,
Promoter analysis
The set of modified genes identified by microarray analysis was submitted to promoter analysis to
identify regulatory mechanisms. The GENOMATIX Gene to Promoter tool was used to analyze the
promoter regions of this genes searching for mammalian transcription factor binding sites. A
background set of promoter sequences was extracted in a similar manner using a set of unmodified
genes from our microarrays study. A sequence-specific over representation was calculated using a ttest for comparing the content of a given promoter in a given putative Transcription Factor Binding
Site (TFBS). The statistical values were corrected by a strict Bonferronni corrections, requesting a pvalue of 2x10-4, given the independent test of 180 putative binding sites (0,05/180).
JDP2 gene silencing
HUVECs were plated in DMEM/Glutamax and 10% fetal bovine serum on day 0, and were
transfected with 10 pmol of siRNA oligonucleotides targeting human JDP2 (Qiagen) or non-targeting
controls using Lipofectamine RNAiMAX Qiagen on day 1, in serum-free medium. The control siRNA
sequence of the sense strand is UAGCAAUGACGAAUGCGUAdTdT. The sense and antisense
sequences of the duplex specific for human JDP2 is 5’-GCCAUGAGUUGCAACCAAATT-3’ and 5’UUUGGUUGCAACUCAUGGCTT -3’. After 6 hours, the media was changed to complete medium.
Total RNA was extracted after 48 hours and processed for quantitative RT-PCR. JDP2 siRNA
silencing was performed also in association with human plasma treatment. In this case after 6 hours of
transfection, the media was changed to complete medium and after 24 hours cells were incubated with
10% (vol/vol) plasma from normal pregnancies for 24 hours and total RNA was extracted.
Statistics
Microarray analysis was performed using DAVID and GENOMATIX 18. GENOMATIX was used
also for promoter analysis. Clinical parameters of the patients were analyzed using the Mann-Whitney
test. Validation performed by quantitative RT-PCR was analyzed by Student’s t test. p values < 0.05
were considered significant.
101
Results
Results
Validation of the endothelial cell line used in the study
An immortalized HUVEC line was used in order to ensure the reproducibility of the results. In PE,
expression of markers of proinflammatory endothelial activation and increased vascular permeability
have been consistently reported. Therefore, to verify that the chosen HUVEC line was appropriate for
our study we determined its response to proinflammatory stimuli and we evaluated its response to
preeclamptic plasma in terms of permeability. We verified that the HUVEC line was responsive to
proinflammatory stimuli by treating it with TNF-α (either 0.1, 0.5, 1 and 5 ng/ml ) and measured the
expression of several endothelial markers after a 5 hours exposition. This resulted in a dose-response
up-regulation of a number of endothelial activation markers including IL-1, IL-8, VCAM-I, ICAM-I
and E-selectin (Supplementary Figure 2). Also, we conducted an assay to compare the permeability
of the HUVECs exposed to preeclamptic or normal pregnant plasma. This analysis showed that the
preeclamptic plasmas significantly increased the permeability of the HUVEC line, confirming that
these cells display another of the characteristic response of EC to PE (Supplementary Figure 3).
Therefore, this HUVEC line meets basic criteria necessary to analyze the transcriptional effects of
preeclamptic plasma factors on the EC.
Microarray analysis of the HUVECs treated with preeclamptic plasma
The HUVECs were incubated for 24 hours with plasma from either preeclamptic or matched normal
pregnant women plasma, and gene-expression profiles were analyzed with DNA microarrays
interrogating 27958 genes. In the preeclamptic plasma-treated HUVECs 71 genes were significantly
up-regulated and 45 down-regulated as compared to HUVECs incubated with normal pregnant
plasma. Table 3 lists a selection of the differentially expressed genes with p < 0.05 (full list of
modified genes is given in supplementary Table S1). We then used the DAVID and GENOMATIX
softwares to perform functional and networks analysis. This allowed us to identify gene classifiers
and pathways that are significantly enriched in HUVECs treated with preeclamptic plasma (Table 4).
Expression levels of genes involved in cell cycle control and progression were increased in HUVECs
treated with preeclamptic plasma, such as PTTG1, CCNB1, CCNA2, TXNIP, PLK1, PSRC1, CDC20,
KPNA2, CKAP2, CDCA8, SPHK1, ID3. Interestingly, we found that JDP2 a member of the activation
protein-1 (AP-1) family known to suppress cell cycle progression, was down- regulated 20. Another
category of up-regulated genes was composed of inhibitors of the apoptotic process (BCL2A1, BIRC5,
102
Results
SPHK1, HSPA5, RTEL1) while the pro-apoptotic genes PMAIP1, PHLPP1, DDIT4, IGFBP3 and
CHAC1 were found down-regulated. CHAC1, as well as five other down-regulated genes (ATF4,
CEBPB, TRIB3, XBP-1 and DDIT4) are components of the unfolded protein response (UPR) pathway.
This pathway constitutes a response to endoplasmic reticulum stress (ER stress), and its activation is a
feature of many chronic inflammatory and autoimmune diseases 21. HSPA5 (also known as BiP) which
is up-regulated in our study has been described as a master regulator of the anti-apoptotic UPR
signaling network
22
.
Several modified genes are involved in fatty acid biosynthesis including
(INSIG1, FASN, AGMO) which are all targets of SREBP (Sterol Regulatory Element Binding Protein).
Moreover, genes involved in cardiovascular development (ADAMTS1, CCNB1, ITGB3, SLIT2,
VEGFA, SPHK1, CITED2, ERF11, EDN1, ID3 and INSR) and endothelial vasoregulation (EDN1,
APLN, CBS, and NOX4) were identified.
Pathology association testing
To further pursue the in silico analysis, the lists of up-regulated and down-regulated genes was
submitted to the GENOMATIX Gene Ranker tool and the resulting Diseases (MESH) table screened
for terms related to preeclampsia (Table 5). This allows the association of modified genes and
disease-associated key words. The test indicates that several MesH annotations are significantly overrepresented in our list of modified genes including: Pregnancy complications, Cardiovascular diseases,
Vascular diseases, Hypertension pregnancy-induced, Inflammation, Preeclampsia, etc…
Validation of differential gene expression by quantitative RT-PCR
Microarray data were validated by quantitative RT-PCR using specific primers for a selected subset of
genes found differentially expressed (Figure 1A and B). The genes were selected according to their
fold change and/or their putative functional relevance. Thus, EDN1 and JDP2 are two of the most
modified genes in our study, with fold changes of 1.88 and -2.67 respectively. Other selected genes are
representative of the biological functions which appear over-represented when performing functional
analysis, using the DAVID or GENOMATIX software (CDC20 is involved in cell cycle, BCL2A1,
ID3 and IGFBP3 in apoptosis regulation, etc..). This analysis was conducted on HUVECs incubated
for 24 hours with plasmas of 10 preeclamptic women and compared to the results obtained with 10
plasmas from normal pregnant women. This way we validated the modified expression of EDN1,
APLN, NOX4, TGM2, CDC20, BCL2A1, BIRC5, ID3, INSIG1, JDP2, IGFBP3 and VEGFA. In
addition we tested the gene modifications in two groups of patients (severe cases, n=5, average time
for plasma collection = 32 weeks, and mild cases, n=7, average time for plasma collection = 36
weeks). We did not find significant differences in gene expression alterations between the groups,
103
Results
except for EDN1 that was increased 1.8 fold in mild PE and 2.7 fold in severe PE (p = 0.002),
suggesting that except for this factor, the modifications observed are general for preeclampsia
(Supplementary Figure S4).
Promoter analysis
To identify master genes of the preeclamptic plasma-induced transcriptomic modifications, we
proceeded to an analysis of the predicted binding sites for transcription factors present in the
promoters of the differentially expressed genes. The occurrence of binding sites for transcription
factors was obtained from the MatInspector Genomatix software for the promoters of 44 most upregulated, 9 most down-regulated genes and a sample of 57 non modified genes. This allowed
identifying over-representation in the modified gene set of putative transcription factor binding sites
for AP-1 (p< 0.0024; p < 0.0005 when only the induced genes were compared with the unmodified
set). Interestingly, JDP2 one of the most down-regulated (-2.67) genes in the HUVECs exposed to
preeclamptic plasma, encodes the Jun dimerization protein 2, a member of the AP-1 family which
functions as a repressor of the AP-1 dependent transcription.
Role of JDP2 in the transcriptomic response of HUVECs to preeclamptic plasma
To focus on the role of JDP2 in the transcriptomic activation of AP-1 regulated genes induced by the
preeclamptic plasma, HUVECs were treated with a siRNA targeting JDP2 (siJDP2). The cells were
transfected with either the siJDP2 or a control (scrambled) siRNA and cultured for 24 hours in a
medium supplemented with 10% normal plasma from pregnant women. Subsequently, quantitative
RT-PCR was used to analyze the expression of 18 genes, containing (8) or not AP-1(10) regulatory
sites in their proximal promoters. As shown in Figure 2, siJDP2 reduced by 75% the levels of JDP2
mRNA in the HUVECs. JDP2 knockdown resulted in significant increased expression levels of 3
genes (CAMK2N1, BCL2A1, CEBPB). Of these only two (BCL2A1 and CEBPB) are known to contain
AP-1 regulatory sites in their promoter. The siRNA JDP2 silencing resulted also in significant downregulation of 11 genes, 6 of them contain AP-1 sites (EDN1, TGM2, CDC20, ID3, E2F1 and CCNA2).
Finally the siRNA JDP2 had no effect on the expression of 9 genes. Of these 3 contain AP-1 sites in
their promoters (NOX4, INSIG1 and ATF4). The same experiment was repeated using another
endothelial cell line the HMEC-1 (Human Dermal Microvascular Endothelial Cells) to determine
whether the effects of JDP2 down-regulation were restricted to the HUVEC line. This analysis
showed that in general the genes modified by the JDP2 siRNA in the HUVECS are overall modified
104
Results
with a very similar expression profile in HMEC-1, albeit with a decreased intensity, some of them
being not modified such as EDN1 (supplementary Figure 5).
TNF-α mediates some effects of preeclamptic plasma on endothelial cells but does not
regulate JDP2 expression
TNF-α is known to induce proinflammatory activation in EC, and increased circulating levels have
been reported in preeclampsia. Indeed, the measurements of cytokine levels in the plasmas of the
women used in our study indicates that the TNF-α levels are significantly higher in the plasmas from
preeclamptic women (1.5 pg/ml) than in the controls (0.5 pg/ml); (supplementary Figure S1). Thus,
we investigated whether some of the gene expression modifications induced by the preeclamptic
plasma were caused by the increased levels of TNF-α, and if they were related to JDP2 downregulation. To this end HUVECs were treated for 24 hours with preeclamptic plasma in the presence
or absence of Etanercept (an inhibitor of TNF-α). Subsequently the expression of some genes (JDP2,
EDN1, APLN, NOX4, BIRC5, ID3, IGFBP3, TGM2, BCL2A1, INSIG1, VEGFA) which were found
significantly modified in the microarray analysis where analyzed by quantitative PCR. We found that
TNF-α inhibition did not block the down-regulation of JDP2 induced by the preeclamptic plasma,
suggesting that this effect is mediated by other factors. Amongst the 12 genes tested we observed that
Etanercept was able to bring the expression levels of one of them, NOX4 (not modulated by JDP2)
back to the level of expression in HUVEC treated with control plasma, while it was induced over twofold by preeclamptic plasma alone (Figure 3). This suggests that NOX4 regulation passes through
TNF-α activation in a JDP2-independent pathway. Reciprocally, it suggests that increased TNF-α
levels account only for a subset of genes modified in preeclamptic plasma-treated ECs.
105
Results
Discussion
In this study we have analyzed the effects of plasma from women with either preeclamptic or normal
pregnancies on the global gene expression profile of immortalized human vein endothelial cell line
(HUVEC) that were exposed for 24 hours to preeclamptic or normal pregnancy plasma. We detected
116 genes with significantly modified expression in cells exposed to plasma from preeclamptic versus
normal pregnancies. Some of these genes are most relevant to EC pathophysiology, and involved in
Vasoregulatory functions, Mitosis and Cell cycle progression, Apoptosis regulation, Fatty acids
biosynthesis, Endoplasmic reticulum stress-response.
The role of these genes in the context of
preeclampsia and EC physiology is discussed below:
Genes involved in vasoregulatory functions, free radical production and angiogenesis are
altered by the preeclamptic plasma: Several genes identified as modified by the preeclamptic
plasma are known to directly modify the properties of the endothelial vessels and thus to be able to
promote hypertension. Amongst those, EDN1, APLN and CBS were significantly modified. EDN1,
encoding endothelin-1 (ET-1), was the most up-regulated gene in the HUVECs treated with
preeclamptic plasma. ET-1, mainly produced by EC, possesses potent vasoactive activity. It has been
implicated in the pathophysiology of many cardiovascular diseases 23, 24. Circulating levels of ET-1 are
increased in the blood of preeclamptic women, and several factors released by the preeclamptic
placenta (including sFlt1, inflammatory cytokines, and agonistic angiotensin II type-1 receptor
autoantibodies) are known to induce ET-1 expression. These factors induce hypertension in animal
models through the production of ET-1, strengthening the possibility that ET-1 may be a mediator in
the genesis of PE syndrome secondary to anti-angiogenic factors released by the placenta.
Another up-regulated gene in our experiment is APLN, encoding Apelin, a biologically active
peptide present in several isoforms that are agonists for the orphan G coupled receptor APJ. In the
cardiovascular system Apelin is present both in the endothelium and vascular smooth muscle cells
(VSMCs). Acting on the endothelium it releases nitric oxide, which mediates vasodilation, while
acting directly on VSMCs, it causes vasoconstriction 25. A possible role of Apelin in hypertension,
initial stages of heart failure and ischaemic heart disease has been suggested
26
. Apelin levels have
previously been found increased in the placenta and plasma of preeclamptic women 27, 28.
Among genes involved in vasoregulatory functions and down-regulated in our experiment we
found CBS. This gene encodes the cystathionine- -synthase, an enzyme catalyzing the first and ratelimiting step of the transsulfuration pathway resulting in the conversion of homocysteine to the
cysteine precursor cystathionine. CBS can also catalyze the condensation of cysteine with
106
Results
homocysteine to form cystathionine and Hydrogen sulfide (H2S). H2S is now known to induce
vasorelaxation by opening ATP sensitive K-channels in smooth muscle cells and up regulating VEGF.
Furthermore, H2S has also an antioxidant activity by directly scavenging NO and ROS 29. Endothelial
dysfunction has been observed in Cbs-deficient mice, both homozygotes or heterozygotes
30, 31
.
Isolated vessels from these animals display reduced dilatory response to endothelium dependent
vasodilators (bradykinin, acetylcholine and methacholine). CBS down regulation has also been
reported in the placental endothelium of early-onset preeclampsias 32. Altogether these data show that
CBS could play a significant role in vasoregulation and its involvement in the endothelial dysfunction
of preeclampsia seems plausible. In sum, the up-regulation of EDN1 and APLN, and the downregulation of CBS in our experiment are consistent with their role in vasoregulatory functions and their
known implication in the preeclamptic syndrome.
In relation with free radical production, and their deleterious effects in the context of
preeclampsia, we found significant up-regulation of NOX4. This gene encodes a nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase isoform. In EC, NOX4, has been identified as a major
source of ROS
33, 34
. Excessive ROS production has been convincingly implicated in vascular
pathologies. Some studies indicate that NOX4 up-regulation activates EC, arrests proliferation and
causes apoptotic and necrotic death
35
. However, NOX4 over-expression promoted EC proliferation
and inhibit apoptosis while NOX4 silencing or expression of a dominant-negative form of NOX4
impaired EC proliferation
36
. Further studies are needed to resolve these conflicting observations.
Notwithstanding, it is likely that increased expression of NOX4 in EC would lead to increased ROS
production. ROS will activate various redox sensitive kinases such Akt, Src, and MAPK, as well as
transcription factors including NF-kβ, AP-1, p53, Ets and HIF-1, thereby increasing redox-sensitive
gene expression, regulating growth, apoptosis regulation, migration, angiogenesis, permeability, and
inflammation 34. A recent study has demonstrated that through ROS production, NOX4 mediates the
expression of plasminogen activator inhibitor-1 (PAI-1) via p38 MAPK pathway in cultured human
EC 37. In this way NOX4 could contribute to the pro-coagulant status observed in preeclampsia. Using
Entanercept we show here that NOX4 transcriptional induction in EC exposed to preeclamptic plasma
is TNF-α dependent, while TNF-α levels are increased in preeclamptic plasma. Our results suggest
therefore that inhibition of NOX4 transcription or activity could be a target for therapeutic approaches.
Another
way
of
modifying
the
access
of
oxygen
to
the
tissue
besides
vasoconstriction/vasodilation is the building of the vascular network. To this respect we found here
that the gene encoding the vascular endothelial growth factor A (VEGFA) -an important actor of
endothelial proliferation, migration, permeability, survival and vasodilation- is reduced in the
HUVECs treated with preeclamptic plasma. In PE, the amount of bioavailable VEGF is decreased
because the ischemic placenta releases soluble fms-like tyrosine kinase-1 (sFLt1), into the maternal
107
Results
circulation. sFLt1 binds free VEGF and makes it unavailable for signaling via membrane-bound
receptors. Endothelial-derived VEGFA could play an essential role in maintaining endothelial
homeostasis by regulating key vascular proteins such as vascular endothelial growth factor receptor-2
(VEGFR2), tyrosine kinase with immunoglobulin and EGF homology domains-2 (Tie2) and vascular
endothelial cadherin (VE-cadherin)
38, 39
. Our results suggest that in preeclampsia the EC might be
confronted to a double lack of bioavailable VEGFA trough the loss of paracrine and autocrine VEGFA
signaling.
Cell proliferation versus apoptosis in EC exposed to preeclamptic plasma: Several of the
up-regulated genes encode proteins that drive cell proliferation, suggesting that the preeclamptic
plasma could be enriched in growth factors compared to normal pregnant plasma. This is consistent
with other studies that have reported that preeclamptic plasma stimulates ECs proliferation 40. Several
factors present in the preeclamptic plasma at increased concentrations could be responsible of this
effect: VEGF, ET-1, oxidized low density lipoprotein (OX-LDL). In addition, increased ROS
production subsequent to NOX4 up-regulation could also increase the expression of genes involved in
EC growth and cell cycle progression, as mentioned before. Consistently, a recent study comparing
the transcriptome of peripheral blood mononuclear cells (PBMCs) from preeclamptic and normal
pregnant women found that many of the up-regulated genes in the preeclamptic PBMCs are involved
in mitosis and cell cycle progression
41
. Strikingly, a number of genes up-regulated in this study are
also found up-regulated in our experiment, including CCNB1, CCNB2, CDC20, CDCA8, NEK2 and
CASC5. While proliferation is favored, anti-apoptotic genes were consistently up-regulated in the
HUVECs treated with preeclamptic plasma such as BCL2A1, BIRC5 and ID3. BCL2A1 encodes a
member of the BCL-2 protein family acting as anti-apoptotic regulators
42, 43
capable of blocking
caspase activation and whose expression has already been found up-regulated in the placenta of severe
preeclampsias 44. BIRC5 encodes the Baculoviral IAP Repeat-Containing Protein (also designated as
survivin), functions as a pivotal regulator of programmed cell death and mitosis. It interacts with the
products of several genes found up-regulated in our study, such as CDCA8, CDC20, CCNB1 and
CCNB2. ID3 is a member of the basic helix-loop-helix (bHLH) family implicated in the pathobiology
of vascular diseases of rodents, pigs and humans
45, 46, 47
. The increase in the expression of anti-
apoptotic genes (BCL2A1, BIRC5, ID3) and concomitant down-regulation of pro-apoptotic genes
(such as IGFBP3)the expression of which is modified in the placenta and in the plasma of
preeclamptic women
48
-, DDIT3 or CHAC1) could be an adaptive response to plasma factors
(inflammatory cytokines, anti-angiogenic factors) which might trigger a stress challenging cell
survival. Regulation of the apoptosis pathway by the preeclamptic plasma is also substantiated by the
down-regulation of the pro-apoptotic branch of the unfolded protein response (UPR) pathway. Indeed,
our transcriptome analysis shows down-regulation of the genes ATF4, CEBPB, CHAC1, TRIB3, XBP-
108
Results
1 and DDIT4 upon exposure of the HUVEC to preeclamptic plasma. The UPR is activated in response
to an overload of misfolded proteins in the endoplasmic reticulum (ER). Stress sensors in the ER such
as ATF6 initiate signals on the cytosolic face of the ER to reduce protein synthesis, promote protein
folding and increase the degradation of misfolded proteins. Failure of this response to alleviate protein
misfolding stress leads to late expression of proteins such as DDIT3 (also designated as CHOP-10) or
CHAC1, culminating in cell death
49
. ER stress and UPR pathways have strong links to major
inflammatory and stress signaling networks, including the activation of the JNK-AP1 and NF-κB-IKK
pathways, as well as production of ROS and NO 50-52. In addition, the chaperone HSPA5(BiP) which is
up-regulated in the present study has been proposed as a master regulator of the of the anti-apoptotic
UPR signaling network
22
.
Increased expression of HSPA5(BiP) is induced by several factors,
including elevated levels of ROS and intracellular homocysteine
53, 54
. Homocysteine-induced ER
stress results in the induction of SREBP-dependent genes involved in the biosynthesis and uptake of
cholesterol and triglycerides and leads to the accumulation of cholesterol in cultured HUVECs 55. We
show here that HUVECs exposed to the preeclamptic plasma up-regulate insulin induced gene
1(INSIG1), a key component of the sterol regulatory element binding protein (SREBP-mediated
regulation of cholesterol biosynthesis)
56
.
INSIG1 binds to the sterol-sensing domain of SCAP
(SREBP cleavage activating protein) and retains the SCAP/SREBP complex in the ER, preventing the
translocation of the N-terminal domain of SREBP into the nucleus. This blocks SREBP from acting as
a transcription factor for the genes involved in lipids and cholesterol biosynthesis (including INSIG1).
A series of studies, have demonstrated that EC inflammatory activation by oxidized phospholipids
depletes cholesterol and activates SREBP nuclear translocation, with a concomitant increase in
INSIG1 mRNA levels57. Increased circulating levels of triglycerides, low-density lipoproteins (LDLs),
and lipid peroxides have been found in the plasma of preeclamptic women 58, 59. In addition to INSIG1,
several other genes involved in fatty acids biosynthesis were found to be up-regulated in our study
including HMGCS1, and FASN that are both targets of SREBP.
In sum, the modifications detected in HUVECs treated with preeclamptic plasma are coherent
with what is currently known on endothelial dysfunction in preeclampsia. Deregulation of genes
encoding vasoregulatory functions (EDN1, APLN and CBS) can be linked to the hypertension resulting
from vasoconstriction. The concomitant up-regulation of genes encoding anti-apoptotic effectors and
down-regulation of pro-apoptotic factors (including those involved in ER stress control) could be an
adaptive response to plasma factors (i.e. inflammatory cytokines, anti-angiogenic factors) which might
trigger a stress challenging cell survival.
109
Results
JDP2 a band master of endothelial stress in preeclampsia?
One of the most striking findings of our analysis was the discovery that many of the modified
genes contain activator protein-1 (AP-1) binding sites in their promoters. This correlates with the fact
that JDP2 was one of the most down regulated genes in the HUVECs treated with preeclamptic
plasma, thus suggesting for the first time an involvement of this regulation cascade in endothelial
defects in preeclampsia. JDP2 encodes JDP2 (Jun dimerization protein-2), a member of the AP-1
family implicated in many cellular processes including carcinogenesis, cell differentiation and cell
proliferation. JDP2 is a 18.7 kDa protein able to homodimerize, or heterodimerize with other AP-1
members, such as c-Jun, JunB, JunD, DDIT-3(CHOP-10), ATF2, and a member of the C/EBP family,
C/EBPγ. JDP2 acts as a repressor of AP-1, cAMP-response element, and TPA responsive elementdependent transcription60, 61. Althougth JDP2 acts generally as a repressor, it has been reported that
depending on the context and cell type, it can alternatively act as a transcriptional activator 62, 63.
In the present experiment it was tempting to speculate that the up-regulation of several genes
could result at least in part from the down-regulation of JDP2. We tested this hypothesis by inhibiting
JDP2 expression in the HUVECs using a JDP2 siRNA, followed by the analysis of the expression of a
subset of genes containing AP-1 sites in their promoters and up-regulated by the exposure to
preeclamptic plasma. JDP2 silencing resulted in the up-regulation of 1 gene (BCL2A1) out of 16 genes
found up-regulated by preeclamptic plasma (and containing AP-1 sites in their promoter regions).
JDP2 siRNA inhibition leads also to the down regulation of IGFBP3, and VEGFA, two genes which
are down-regulated in the HUVECs exposed to preeclamptic plasma. Thus down-regulation of JDP2
by the preeclamptic plasma could directly explain at least the transcriptional modification of BCL2A1,
IGFBP3 and VEGFA. Concerning the other genes it is probable that the up-regulation of these genes
requires not only the down-regulation of JDP2 but also the activation or down-regulation of other
cofactors. Likely, the activity of such cofactors could be dependent on signals which can be provided
by the preeclamptic plasma but not by the normal pregnancy plasma that could adapt the cells by
countering the effect of JDP2 repression. Nevertheless the fact that the single inhibition of JDP2
modifies their expression argues in favor of a role of JDP2 in their regulation at least in the context of
preeclampsia.
In summary, the significant response of HUVECs cells after 24 hours of exposition to
preeclamptic plasma may help to understand the effects of preeclampsia on EC, and allow identifying
the starting point of putative cascades of gene deregulations, that might be the source of long-term
endothelial disorders in the patients. The data generated by our study set a novel base for future
studies aiming to elucidate the effects of preeclampsia circulating factors on the physiology of EC.
110
Results
Acknowledgments
We thank Dr. Catherine Farnarier (Laboratoire d'IMMUNOLOGIE - Hôpital de la Conception,
Marseille, France) for performing the cytokines measurements in the plasma of the pregnant women
used in this study.
Tables
Table 1 : Clinical data of the preeclamptic and matched pregnant control women included in
the study
Data
Normal pregnancy (n=10)
Preeclampsia (n=10)
P value
Maternal age (Years)
Gestational age (Weeks)
Race
Caucasian
African
Others
Nulliparity
31.5 [29 - 34]
38 [32 - 39]
31 [29.5 - 35]
30 [30 - 33.5]
NS
0.03
5
4
1
3
6
3
1
5
Systolic blood pressure (mmHg)
120 [119 - 126]
150 [143 - 167.5]
Diastolic blood pressure (mmHg)
Proteinuria (mg/dL)
70 [70 - 80]
0
100 [94 - 100]
>300
1.2x10-5
4.8x10
-5
Differences between preeclamptic and control group were analyzed using a Mann-Whitney
non parametric test. Data are given as medians and 75% confidence intervals are shown
between brackets, based upon the interquartile range. NS = not significant. Proteinuria was
measured with dipstick test and was above 300 mg/dl (~1-2g/24h).
111
Results
Table 2: Primers used in the quantitative RT-PCR analysis
Gene
Forward primer
Reverse primer
Amplicon size (bp)
NOX4
5'-TGCCATGAAGCAGGACTCTA-3'
5'-GCCACATTCTCACATTTCCA-3'
144
BIRC5
CEBPB
ATF4
VEGFA
BCL2A1
5'-CATAGAGCTGCAGGGTGGAT-3'
5'-CGTGTGTACACGGGACTGAC-3'
5'-CCTGAAAGATTTGATAGAAGAGGTC-3'
5'-GGATCAAACCTCACCAAGG-3'
5'-GGCATCATTAACTGGGGAAG-3'
5'-AAAACCCAGTAGGGTCCACA-3'
5'-AAAACAAAACAAAACATCAACAGC-3'
5'-TGGAACACACAGCTACAGCA-3'
5'-CCTTTCCTCGAACTGATTTTT-3'
5'-TGAAATCTCCTTATAGGTATCCACA-3'
120
110
118
120
114
E2F1
CCNB2
5'-ACGCTATGAGACCTCACTGAA-3'
5'-CCGACGGTGTCCAGTGATTT-3'
5'-TCCTGGGTCAACCCCTCAAG-3'
5'-TGTTGTTTTGGTGGGTTGAACT-3'
249
180
CCNA2
CDC20
ADAMTSL1
ID3
5'-CGCTGGCGGTACTGAAGTC-3'
5'-GCACAGTTCGCGTTCGAGA-3'
5'-GGGGCCTCCTACTCTCTGAG-3'
5'-CATCGACTACATTCTCGACCTG-3'
5'-GAGGAACGGTGACATGCTCAT-3'
5'-CTGGATTTGCCAGGAGTTCGG-3'
5'-AGTCCACATTACTGCATGTTCTG-3'
5'-TCCTTTTGTCGTTGGAGATGAC-3'
120
188
88
128
APLN
TGM2
INSIG1
CAMK2N1
EDN1
5'-GTCTCCTCCATAGATTGGTCTGC-3'
5'-GAGGAGCTGGTCTTAGAGAGG-3'
5'-CCTGGCATCATCGCCTGTT-3'
5'-GACACCAACAACTTCTTCGGC-3'
5'-AACCAGGTCGGAGACCATGA-3'
5'-GGAATCATCCAAACTACAGCCAG-3'
5'-CGGTCACGACACTGAAGGTG-3'
5'-AGAGTGACATTCCTCTGGATCTG
5'-TCATCTTCAATAACAACCCGCTT-3'
5'-CCGAAGGTCTGTCACCAATGT-3'
149
184
103
92
123
GAPDH
IGFBP3
JDP2
SDHA
5'-AACAGCGACACCCATCCT C-3'
5'-CTGTGGCCATGACTGAGGAAAG-3'
5'-GATGCCGGAACAAGAAGAAG-3'
5'-TACAAGGTGCGGATTGATGA-3'
5'-CATACCAGGAAATGAGCTTGACAA-3'
5'-TCCCTGAGCCTGACTTTGCC-3'
5'-GCTTCAGCTCCTCAATCTGG-3'
5'-CAAAGGGCTTCTTCTGTTGC-3'
81
97
105
66
112
Results
Table 3: Differentially expressed genes in the HUVECs treated with preeclamptic plasma.
Partial list of genes displaying significantly modified expression (p < 0.05) between the
HUVECs exposed to preeclamptic plasma and those exposed to plasma from normal pregnant
women.
Gene Symbol
Gene Name
Fold Change
Up-regulated genes
EDN1
ADAMTSL1
ID3
NOX4
INSIG1
BCL2A1
HIST1H1D
CDC20
APLN
FASN
SPARC
CCNB1
PSRC1
CBR3
KIF20A
HMMR
ICAM2
NEK2
HSPA5
BIRC5
CDCA8
CCNA2
LOXL2
KPNA2
PTTG1
TGFBI
Endothelin-1
ADAMTS-like 1
Inhibitor of DNA binding 3
NADPH oxidase 4
Insulin induced gene 1
BCL2-related protein A1
Histone cluster 1, H1d
Cell division cycle 20 homolog
Apelin
Fatty acid synthase
Secreted protein, acidic, cysteine-rich
Cyclin B1
Proline/serine-rich coiled-coil 1
Carbonyl reductase 3
Kinesin family member 20A
Hyaluronan-mediated motility receptor
Intercellular adhesion molecule 2
NIMA (never in mitosis gene a)-related kinase 2
Heat shock 70kDa protein 5
Baculoviral IAP repeat-containing 5
Cell division cycle associated 8
Cyclin A2
Lysyl oxidase-like 2
Karyopherin alpha 2
Pituitary tumor-transforming 1
Transforming growth factor, beta-induced
1.88
1.79
1.74
1.69
1.59
1.53
1.44
1.43
1.43
1.38
1.37
1.37
1.36
1.34
1.34
1.33
1.32
1.32
1.28
1.28
1.27
1.27
1.27
1.26
1.25
1.25
Down-regulated genes
NFIL3
CITED2
STC2
CAMK2N1
ATF4
VEGFA
TXNIP
KLF9
CBS
TSC22D3
IGFBP3
TRIB3
CEBPB
DDIT4
JDP2
CHAC1
Nuclear factor, interleukin 3, regulated
Cbp/p300-interacting transactivator
Stanniocalcin 2
Calcium/calmodulin-dependent protein kinase II inhibitor 1
Activating transcription factor 4
Vascular endothelial growth factor A
Thioredoxin interacting protein
Kruppel-like factor 9
Cystathionine- -synthase
TSC22 domain family, member 3
Insulin-like growth factor binding protein 3
Tribbles homolog 3
CCAAT/enhancer binding protein (C/EBP), beta
DNA-damage-inducible transcript 4
Jun dimerization protein 2
Cation transport regulator homolog 1
-1.34
-1.35
-1.44
-1.47
-1.50
-1.50
-1.53
-1.68
-1.69
-1.86
-1.93
-1.96
-2.02
-2.15
-2.67
-2.88
113
Results
Table 4: Most representative over-represented biological functions identified by the DAVID
and GENOMATIX softwares in HUVECs treated with preeclamptic plasma versus normal
pregnancy plasma
DAVID Functional Annotation Chart
Functional Cathegory
N° of Genes
Genes Symbol
P value
Cell cycle
13
CKAP2 , TXNIP, TRNP1, CASC5, CDC20 , BIRC5 , PTTG1 ,
SMC4 , CCNB1 , CDCA8 , SPAG5 , PLK1 , CCNA2
1.30x10-5
Apoptosis
11
PHLPP1, CKAP2 , TSC22D3, CHAC1, BCL2A1 , TRIB3,
BIRC5 , PMAIP1, RTEL1 , PHLDA1, DDIT4
6.67x10
Anti-apoptosis
8
CEBPB, VEGFA,
RTEL1 , CITED2
4.70x10-4
Regulation of cell migration
7
VEGFA, EDN1 , SPHK1 , ITGB3 , IGFBP3, INSR, CITED2
9.65x10-4
Negative regulation of
apoptosis
9
CEBPB, VEGFA, SPHK1 , BCL2A1 , RAG1, BIRC5 , HSPA5 ,
RTEL1 , CITED2
2.61x10-3
SPHK1 ,
BCL2A1 ,
BIRC5 ,
HSPA5 ,
-5
GENOMATIX Gene Ranker Analyzer
PTTG1 , CCNB1 , CCNA2 , PLK1 , PSRC1 , CDC20 ,
-9
CDCA8 , SPAG5 , SMC4 , KIF20A , EDN1 , INSR, BIRC5 , 8.45x10
CASC5 , CCNB2
M phase of mitotic cell cycle
15
Cell cycle
23
PTTG1 , CCNB1 , CCNA2 , TXNIP, PLK1 , PSRC1 , CDC20 ,
KPNA2 , CKAP2 , CDCA8 , SPHK1 , SPAG5 , GAS2L3 ,
CITED2, SMC4 , TRNP1, KIF20A , EDN1 , ID3 , INSR,
BIRC5 , CASC5 , CCNB2
2.99x10-6
Fatty acid biosynthetic
process
6
AGMO , GGT5, INSIG1 , FASN , EDN1 , TRIB3
1.48x10-4
Regulation of Apoptosis
22
2.38x10-4
SLIT3 , CHAC1, PHLDA1, PMAIP1, TXNIP, SLIT2 , IGFBP3,
GARS, HSPA5 , VEGFA, CKAP2 , SPHK1 , PHLPP1, DDIT4,
RAG1, CITED2, CLN5, CEBPB, ID3 , TRIB3, BCL2A1 , BIRC5
-4
Response to hypoxia
6
CCNB1 , VEGFA, DDIT4, STC2, CITED2, EDN1
6.50x10
Positive regulation of
locomotion
6
ITGB3 , SLIT2 , VEGFA, SPHK1 , EDN1 , INSR
6.50x10-4
Cardiovascular system
development
11
ADAMTS1 , CCNB1 , ITGB3 , SLIT2 , VEGFA,
CITED2, ERRFI1, EDN1 , ID3 , INSR
Anti-apoptosis
6
HSPA5 , SPHK1 , CITED2, CEBPB, BCL2A1 , BIRC5
6.41x10-3
Cellular response to stress
14
PTTG1 , CCNB1 , CCNA2 , ZSWIM7, PMAIP1, PLK1 , ATF4,
HSPA5 , VEGFA, INSIG1 , CBS, STC2, RTEL1 , EDN1
2.00x10-3
SPHK1
, 8.98x10-4
Up-regulated genes in HUVECs treated with preeclamptic plasma are shown in bold
characters and down-regulated genes in plain characters.
114
Results
Table 5: GENOMATIX disease MeSH terms associated with the list of modified genes in
HUVECS exposed to preeclamptic plasma
Genes
MeSH-Term
P value
Observed Expected
Total
List of observed genes
Neovascularization,
Pathologic
2.47x10-9
38
14.09
2238
PTTG1 , ADAMTS1 , CCNB1 , CCNA2 , PMAIP1, ITGB3 , TXNIP, IGFBP3, FAP ,
PLK1 , RNASE1 , ATF4, HSPA5 , AKAP12, VEGFA, FST , NRGN , SPHK1 , DDIT4,
SPARC , STC2, RAG1, CITED2, NOX4 , HMMR , APLN , FASN , EDN1 , TGFBI ,
ICAM2 , AKR1C3 , CEBPB, ID3 , TRIB3, MTHFD2, INSR, BCL2A1 , BIRC5
Pregnancy Complications
1.24x10-7
48
23.9
3795
PTTG1 , CCNB1 , CCNA2 , PMAIP1, ITGB3 , TXNIP, HBZ, SLIT2 , IGFBP3, PLK1 ,
RNASE1 , ATF4, CDC20 , HSPA5 , AKAP12, VEGFA, DCBLD2, INSIG1, MAOA,
KPNA2 , FST , NRGN , CBS, SPHK1 , CLIC3 , HMGCS1 , SPARC , STC2, KLF9,
RAG1, CITED2, RPGR, NOX4 , KIF20A , RTEL1 , APLN, FASN , EDN1 , ICAM2 ,
AKR1C3 , CEBPB, TROAP , ID3 , TRIB3, MTHFD2, INSR, BCL2A1 , BIRC5
Cardiovascular Diseases
3.72x10-7
70
45.05
7153
TRPM2 , TSC22D3, PTTG1 , TMEM195 , SLIT3 , ADAMTS6 , ADAMTS1 , CCNB1 ,
GGT5, PHLDA1, CCNA2 , PMAIP1, ITGB3 , TXNIP, SLIT2 , IGFBP3, FAP , PLK1 ,
RNASE1 , GARS, ATF4, PSRC1 , CDC20 , HSPA5 , AKAP12, LOXL2 , VEGFA,
DCBLD2, HIST1H1D , INSIG1 , PPYR1 , TUBA8 , MAOA, FST , NRGN , CKAP2 ,
JDP2, CBS, SPHK1 , PHLPP1, DDIT4, HMGCS1 , SPARC , STC2, MXD3 , RAG1,
CITED2, ERRFI1, RPGR, NOX4 , HMMR , PDE4DIP , KIF20A , APLN, FASN ,
EDN1 , MARS, TGFBI , ICAM2 , AKR1C3 , CEBPB, ID3 , TRIB3, MTHFD2, INSR,
BCL2A1 , BIRC5 , NFIL3, CBR3 , CASC5
Brain Ischemia
1.72x10-6
32
59
2159
Vascular Diseases
2.06x10-6
62
38.76
6155
ADAMTS1 , CCNB1 , PHLDA1, CCNA2 , PMAIP1, ITGB3 , TXNIP, IGFBP3,
RNASE1 , ATF4, HSPA5 , VEGFA, INSIG1 , MAOA, FST , NRGN , CBS, SPHK1 ,
DDIT4, SPARC , STC2, RAG1, CITED2, NOX4 , HMMR, APLN, EDN1 , CEBPB,
TRIB3, INSR, BCL2A1 , BIRC5
TRPM2 , TSC22D3, PTTG1 , TMEM195 , ADAMTS6 , ADAMTS1 , CCNB 1, PHLDA1,
CCNA2 , PMAIP1, ITGB3 , TXNIP, IGFBP3, FAP , PLK1 , RNASE1 , GARS, ATF4,
PSRC1 , CDC20 , HSPA 5, AKAP12, LOXL2 , VEGFA, DCBLD2, HIST1H1D ,
INSIG1 , MAOA, FST , NRGN , CKAP2 , JDP2, CBS, SPHK1 , PHLPP1, DDIT4,
HMGCS1 , SPARC , STC2, MXD3 , RAG1, CITED2, ERRFI1, RPGR, NOX4 , HMMR ,
KIF20A , APLN , FASN , EDN1 , MARS, TGFBI , ICAM2 , AKR1C3 , CEBPB, ID3 ,
TRIB3, INSR, BCL2A1 , BIRC5 , NFIL3, CASC5
Placental Insufficiency
5.32x10-6
9
1.27
203
Ischemia
4.21x10-5
23
9.4
1494
PTTG1 , ADAMTS1 , CCNA2 , PMAIP1, ITGB3 , IGFBP3, ATF4, HSPA5 , VEGFA,
MAOA, FST , CBS, SPHK1 , RAG1, NOX4 , APLN , FASN , EDN1 , ICAM2 , TRIB3,
INSR, BCL2A1 , BIRC5
Inflammation
4.08x10-4
43
27.05
1405
TRPM2 , TSC22D3, ADAMTS1 , CCNB1 , PMAIP1, ITGB3, TXNIP, SLIT2 , IGFBP3,
RNASE1 , GARS, ATF4, CDC20 , HSPA5 , LOXL2 , VEGFA, LXN , INSIG1 , MAOA,
FST , NRGN , CBS, SPHK1 , DDIT4, SPARC , RAG1, CITED2, ERRFI1, NOX4
,
HMMR , APLN , FASN , EDN1 , MARS, TGFBI , ICAM2 , CEBPB, ID3 , TRIB3, INSR,
BCL2A1 , BIRC5 , CASC5
Hypertension, PregnancyInduced
2.21x10-3
15
6.58
1045
ITGB3, IGFBP3, HSPA5 , VEGFA, MAOA, FST , CBS, CLIC3 , STC2, APLN , EDN1 ,
ICAM2 , AKR1C3 , INSR, BIRC5
Pre-Eclampsia
4.05x10-3
14
6.32
1004
ITGB3, IGFBP3, HSPA5 , VEGFA, MAOA, FST , CBS, CLIC3 , STC2, EDN1 , ICAM2 ,
AKR1C3 , INSR, BIRC5
Kidney Diseases, Cystic
5.20x10-3
12
5.14
817
CCNB1 , CCNA2 , HBZ, IGFBP3, PLK1 , VEGFA, NRGN , SPARC , EDN1 , INSR,
PGP , BCL2A1
Embolism and Thrombosis
2.76x10-3
18
8.84
1405
ADAMTS1 , ITGB3 , IGFBP3, HSPA5 , VEGFA, DCBLD2, CKAP2 , CBS, SPARC ,
RAG1, NOX4 , APLN , EDN1 , MARS, TGFBI , ICAM2 , CEBPB, INSR
CCNB1 , PMAIP1, ITGB3 , IGFBP3, VEGFA, CBS, NOX4 , EDN1 , INSR
115
Results
Figures
Figure 1
3
Co
Relative gene expression
***
PE
***
2,5
2
**
1,5
**
*
1
0,5
0
EDN1
Relative gene expression
2
***
APLN
NOX4
TGM2
CDC20
BCL2A1
***
1,6
**
***
1,2
0,8
**
0,4
0
BIRC5
ID3
INSIG1
JDP2
IGFBP3
VEGFA
Figure 1. Validation of differentially expressed genes in HUVECs exposed to preeclamptic plasma.
Selected differentially expressed genes in HUVECs exposed to preeclamptic plasma were analyzed by
quantitative real time RT-PCR to validate the results obtained with the DNA-microarray experiment (
116
Results
A and B). Relative gene expression values were adjusted to the mean value of the control group
(untreated cells) set at 1 for each gene. * for p < 0.05, ** for p < 0.01 and *** for p < 0.0001 using
Student’s test
Figure 2
Figure 2. Effect of JDP2 knockdown on the expression of preeclamptic plasma deregulated
genes. To inhibit JDP2 expression, HUVECs (A) were transfected with 30 pmol of siRNA specific for
JDP2 (siJDP2). The same amount of nonspecific double-stranded RNA was used as a negative control
(siCO). Two days after the transfection, the cells were harvested and total RNA was subjected to
quantitative RT-PCR using primers specific for JDP2 and for several genes found to be modified by
the preeclamptic plasma. Levels of expression were normalized to that of GAPDH and SDHA. Data
are shown as relative gene expression values adjusted to the mean value of the control group (siCO),
which was set at 1 for each gene. Error bars represent the standard deviation (±SD).
117
Results
Figure 3
Figure 3. Effects of TNF-α inhibition on the expression of a selection of genes modified by
preeclampic plasma. The HUVECs were cultured for 24 hours in quadruplicates, in the presence of
preeclamptic plasma with/or without 10 µg/ml of the TNF-α inhibitor Etanercept. Subsequently, the
expression of several genes found to be modified by the preeclamptic plasma in the previous
experiments was analyzed using quantitative RT-PCR. Expression ratio with or without Etanercept
are shown for the 12 genes analyzed (a ratio of 1 meaning no effect of TNF-α inhibition). Inhibition of
TNF-α signaling had a significant effect only on NOX4 expression (* p = 0.02). A trend was however
observed for BCL2A1 ( # p = 0.07) and VEGFA ( ## p = 0.08). Error bars represent the standard
deviations.
118
Results
References
1.
Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R: Pre-eclampsia, Lancet 2010,
376:631-644
2.
James JL, Whitley GS, Cartwright JE: Pre-eclampsia: fitting together the placental, immune
and cardiovascular pieces, J Pathol 2010, 221:363-378
3.
VanWijk MJ, Kublickiene K, Boer K, VanBavel E: Vascular function in preeclampsia,
Cardiovasc Res 2000, 47:38-48
4.
Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP: Pathophysiology of
hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction,
Am J Physiol Heart Circ Physiol 2008, 294:H541-550
5.
Baker PN, Stranko CP, Davidge ST, Davies PS, Roberts JM: Mechanical stress eliminates the
effects of plasma from patients with preeclampsia on endothelial cells, Am J Obstet Gynecol
1996, 174:730-736
6.
Taylor RN, Crombleholme WR, Friedman SA, Jones LA, Casal DC, Roberts JM: High plasma
cellular fibronectin levels correlate with biochemical and clinical features of preeclampsia
but cannot be attributed to hypertension alone, Am J Obstet Gynecol 1991, 165:895-901
7.
Baker PN, Davidge ST, Roberts JM: Plasma from women with preeclampsia increases
endothelial cell nitric oxide production, Hypertension 1995, 26:244-248
8.
Endresen MJ, Morris JM, Nobrega AC, Buckley D, Linton EA, Redman CW: Serum from
preeclamptic women induces vascular cell adhesion molecule-1 expression on human
endothelial cells in vitro: a possible role of increased circulating levels of free fatty acids, Am
J Obstet Gynecol 1998, 179:665-670
9.
Hayman R, Brockelsby J, Kenny L, Baker P: Preeclampsia: the endothelium, circulating
factor(s) and vascular endothelial growth factor, J Soc Gynecol Investig 1999, 6:3-10
10. Matsubara K, Matsubara Y, Hyodo S, Katayama T, Ito M: Role of nitric oxide and reactive
oxygen species in the pathogenesis of preeclampsia, J Obstet Gynaecol Res 2010, 36:239247
11. Taylor RN, de Groot CJ, Cho YK, Lim KH: Circulating factors as markers and mediators of
endothelial cell dysfunction in preeclampsia, Semin Reprod Endocrinol 1998, 16:17-31
12. Baker PN, Krasnow J, Roberts JM, Yeo KT: Elevated serum levels of vascular endothelial
growth factor in patients with preeclampsia, Obstet Gynecol 1995, 86:815-821
13. Davidge ST, Stranko CP, Roberts JM: Urine but not plasma nitric oxide metabolites are
decreased in women with preeclampsia, Am J Obstet Gynecol 1996, 174:1008-1013
14. Sankaralingam S, Xu Y, Sawamura T, Davidge ST: Increased lectin-like oxidized low-density
lipoprotein receptor-1 expression in the maternal vasculature of women with preeclampsia:
role for peroxynitrite, Hypertension 2009, 53:270-277
15. Donker RB, Asgeirsdottir SA, Gerbens F, van Pampus MG, Kallenberg CG, te Meerman GJ,
Aarnoudse JG, Molema G: Plasma factors in severe early-onset preeclampsia do not
substantially alter endothelial gene expression in vitro, J Soc Gynecol Investig 2005, 12:98106
119
Results
16. Bian C, Zhao K, Tong GX, Zhu YL, Chen P: Immortalization of human umbilical vein
endothelial cells with telomerase reverse transcriptase and simian virus 40 large T antigen,
J Zhejiang Univ Sci B 2005, 6:631-636
17. Gavard J, Gutkind JS: VEGF controls endothelial-cell permeability by promoting the betaarrestin-dependent endocytosis of VE-cadherin, Nat Cell Biol 2006, 8:1223-1234
18. Huang da W, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene
lists using DAVID bioinformatics resources, Nat Protoc 2009, 4:44-57
19. Huang da W, Sherman BT, Lempicki RA: Bioinformatics enrichment tools: paths toward the
comprehensive functional analysis of large gene lists, Nucleic Acids Res 2009, 37:1-13
20. Pan J, Nakade K, Huang YC, Zhu ZW, Masuzaki S, Hasegawa H, Murata T, Yoshiki A,
Yamaguchi N, Lee CH, Yang WC, Tsai EM, Obata Y, Yokoyama KK: Suppression of cell-cycle
progression by Jun dimerization protein-2 (JDP2) involves downregulation of cyclin-A2,
Oncogene 2010, 29:6245-6256
21. Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA: The interplay between endoplasmic
reticulum stress and inflammation, Immunol Cell Biol 2012, 90:260-270
22. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D: Dynamic interaction of BiP and
ER stress transducers in the unfolded-protein response, Nat Cell Biol 2000, 2:326-332
23. Schiffrin EL: Role of endothelin-1 in hypertension and vascular disease, Am J Hypertens
2001, 14:83S-89S
24. George EM, Palei AC, Granger JP: Endothelin as a final common pathway in the
pathophysiology of preeclampsia: therapeutic implications, Curr Opin Nephrol Hypertens
2012, 21:157-162
25. Barnes G, Japp AG, Newby DE: Translational promise of the apelin--APJ system, Heart 2010,
96:1011-1016
26. Quazi R, Palaniswamy C, Frishman WH: The emerging role of apelin in cardiovascular
disease and health, Cardiol Rev 2009, 17:283-286
27. Cobellis L, De Falco M, Mastrogiacomo A, Giraldi D, Dattilo D, Scaffa C, Colacurci N, De Luca
A: Modulation of apelin and APJ receptor in normal and preeclampsia-complicated
placentas, Histol Histopathol 2007, 22:1-8
28. Simsek Y, Celik O, Yilmaz E, Karaer A, Dogan C, Aydin S, Ozer A: Serum levels of apelin,
salusin-alpha, and salusin-beta in normal pregnancy and preeclampsia, J Matern Fetal
Neonatal Med 2012,
29. Szabo C: Hydrogen sulphide and its therapeutic potential, Nat Rev Drug Discov 2007, 6:917935
30. Eberhardt RT, Forgione MA, Cap A, Leopold JA, Rudd MA, Trolliet M, Heydrick S, Stark R,
Klings ES, Moldovan NI, Yaghoubi M, Goldschmidt-Clermont PJ, Farber HW, Cohen R,
Loscalzo J: Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia, J
Clin Invest 2000, 106:483-491
31. Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J: Cellular redox state and
endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthasedeficient mice, Arterioscler Thromb Vasc Biol 2002, 22:34-41
32. Holwerda KM, Bos EM, Rajakumar A, Ris-Stalpers C, van Pampus MG, Timmer A, Erwich JJ,
Faas MM, van Goor H, Lely AT: Hydrogen sulfide producing enzymes in pregnancy and
preeclampsia, Placenta 2012, 33:518-521
120
Results
33. Bedard K, Krause KH: The NOX family of ROS-generating NADPH oxidases: physiology and
pathophysiology, Physiol Rev 2007, 87:245-313
34. Frey RS, Ushio-Fukai M, Malik AB: NADPH oxidase-dependent signaling in endothelial cells:
role in physiology and pathophysiology, Antioxid Redox Signal 2009, 11:791-810
35. Lener B, Koziel R, Pircher H, Hutter E, Greussing R, Herndler-Brandstetter D, Hermann M,
Unterluggauer H, Jansen-Durr P: The NADPH oxidase Nox4 restricts the replicative lifespan
of human endothelial cells, Biochem J 2009, 423:363-374
36. Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F: Important role of
Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial
cells in vitro, Arterioscler Thromb Vasc Biol 2007, 27:2319-2324
37. Jaulmes A, Sansilvestri-Morel P, Rolland-Valognes G, Bernhardt F, Gaertner R, Lockhart BP,
Cordi A, Wierzbicki M, Rupin A, Verbeuren TJ: Nox4 mediates the expression of plasminogen
activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells, Thromb
Res 2009, 124:439-446
38. E G, Cao Y, Bhattacharya S, Dutta S, Wang E, Mukhopadhyay D: Endogenous vascular
endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating
VEGF receptor-2 transcription, J Biol Chem 2012, 287:3029-3041
39. Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP,
Iruela-Arispe ML: Autocrine VEGF signaling is required for vascular homeostasis, Cell 2007,
130:691-703
40. Rowe J, Campbell S, Gallery ED: Plasma from preeclamptic women stimulates decidual
endothelial cell growth and prostacyclin but not nitric oxide production: close correlation of
prostacyclin and thromboxane production, J Soc Gynecol Investig 2001, 8:32-38
41. Rajakumar A, Chu T, Handley DE, Bunce KD, Burke B, Hubel CA, Jeyabalan A, Peters DG:
Maternal gene expression profiling during pregnancy and preeclampsia in human
peripheral blood mononuclear cells, Placenta 2011, 32:70-78
42. Karsan A, Yee E, Kaushansky K, Harlan JM: Cloning of human Bcl-2 homologue:
inflammatory cytokines induce human A1 in cultured endothelial cells, Blood 1996,
87:3089-3096
43. Vogler M: BCL2A1: the underdog in the BCL2 family, Cell Death Differ 2012, 19:67-74
44. Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP: Altered global
gene expression in first trimester placentas of women destined to develop preeclampsia,
Placenta 2009, 30:15-24
45. Lasorella A, Uo T, Iavarone A: Id proteins at the cross-road of development and cancer,
Oncogene 2001, 20:8326-8333
46. Benezra R, Rafii S, Lyden D: The Id proteins and angiogenesis, Oncogene 2001, 20:83348341
47. Zhao Q, Beck AJ, Vitale JM, Schneider JS, Gao S, Chang C, Elson G, Leibovich SJ, Park JY, Tian
B, Nam HS, Fraidenraich D: Developmental ablation of Id1 and Id3 genes in the vasculature
leads to postnatal cardiac phenotypes, Dev Biol 2011, 349:53-64
48. Cooley SM, Donnelly JC, Geary MP, Rodeck CH, Hindmarsh PC: Maternal insulin-like growth
factors 1 and 2 (IGF-1, IGF-2) and IGF BP-3 and the hypertensive disorders of pregnancy, J
Matern Fetal Neonatal Med 2010, 23:658-661
121
Results
49. Rutkowski DT, Kaufman RJ: That which does not kill me makes me stronger: adapting to
chronic ER stress, Trends Biochem Sci 2007, 32:469-476
50. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH: Autocrine tumor necrosis factor alpha
links endoplasmic reticulum stress to the membrane death receptor pathway through
IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression, Mol
Cell Biol 2006, 26:3071-3084
51. Cullinan SB, Diehl JA: Coordination of ER and oxidative stress signaling: the PERK/Nrf2
signaling pathway, Int J Biochem Cell Biol 2006, 38:317-332
52. Gotoh T, Mori M: Nitric oxide and endoplasmic reticulum stress, Arterioscler Thromb Vasc
Biol 2006, 26:1439-1446
53. Outinen PA, Sood SK, Pfeifer SI, Pamidi S, Podor TJ, Li J, Weitz JI, Austin RC: Homocysteineinduced endoplasmic reticulum stress and growth arrest leads to specific changes in gene
expression in human vascular endothelial cells, Blood 1999, 94:959-967
54. Kokame K, Kato H, Miyata T: Homocysteine-respondent genes in vascular endothelial cells
identified by differential display analysis. GRP78/BiP and novel genes, J Biol Chem 1996,
271:29659-29665
55. Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK,
Malinow MR, Austin RC: Homocysteine-induced endoplasmic reticulum stress causes
dysregulation of the cholesterol and triglyceride biosynthetic pathways, J Clin Invest 2001,
107:1263-1273
56. Dong XY, Tang SQ: Insulin-induced gene: a new regulator in lipid metabolism, Peptides
2010, 31:2145-2150
57. Yeh M, Cole AL, Choi J, Liu Y, Tulchinsky D, Qiao JH, Fishbein MC, Dooley AN, Hovnanian T,
Mouilleseaux K, Vora DK, Yang WP, Gargalovic P, Kirchgessner T, Shyy JY, Berliner JA: Role
for sterol regulatory element-binding protein in activation of endothelial cells by
phospholipid oxidation products, Circ Res 2004, 95:780-788
58. Hubel CA, Lyall F, Weissfeld L, Gandley RE, Roberts JM: Small low-density lipoproteins and
vascular cell adhesion molecule-1 are increased in association with hyperlipidemia in
preeclampsia, Metabolism 1998, 47:1281-1288
59. Potter JM, Nestel PJ: The hyperlipidemia of pregnancy in normal and complicated
pregnancies, Am J Obstet Gynecol 1979, 133:165-170
60. Aronheim A, Zandi E, Hennemann H, Elledge SJ, Karin M: Isolation of an AP-1 repressor by a
novel method for detecting protein-protein interactions, Mol Cell Biol 1997, 17:3094-3102
61. Jin C, Ugai H, Song J, Murata T, Nili F, Sun K, Horikoshi M, Yokoyama KK: Identification of
mouse Jun dimerization protein 2 as a novel repressor of ATF-2, FEBS Lett 2001, 489:34-41
62. Wardell SE, Boonyaratanakornkit V, Adelman JS, Aronheim A, Edwards DP: Jun dimerization
protein 2 functions as a progesterone receptor N-terminal domain coactivator, Mol Cell Biol
2002, 22:5451-5466
63. Weidenfeld-Baranboim K, Bitton-Worms K, Aronheim A: TRE-dependent transcription
activation by JDP2-CHOP10 association, Nucleic Acids Res 2008, 36:3608-3619
122
Results
Supplementary Figures
Figure S1
Figure S1. Plasma levels of TNF-α and sVCAM-1 measured in nine preeclamptic (PE) and eleven
matched pregnant control (CO) women. Bars represent mean values, and error bars the standard
deviation (SD). Differences between the two groups were determined using a student's test.
123
Results
Figure S2
Figure S2. Endothelial gene expression in HUVECs exposed to TNF-α. Expression of IL-1β, IL-8,
ICAM-1, VCAM-1, and E-Selectin after 5 hours incubation with TNF-αafter 5 hours incubation. Gene
expression was determined by quantitative real time RT-PCR. Relative gene expression values are
adjusted to the mean value of the control group (untreated cells) which was set at 1 for each gene.
Bars represent mean values, and error bars the standard deviation (SD). Differences between the two
groups were determined using a student's test.
124
Results
Figure S3
Figure S3. Effect of preeclamptic plasma on HUVEC monolayers permeability. HUVECs were
cultured on collagen coated Transwells (Costar) and allowed to form monolayers. Cells were then
stimulated with 10% preeclamptic or control plasma and the endothelial monolayer permeability was
assessed by the passage of FITC-conjugated dextran. The values express the ratio between sample
(preeclamptic or control plasma) and non-stimulated cells on three independent experiments: * for p <
0.05, ** for p < 0.01 and *** for p < 0.001 using Student’s test.
Figure S4
Figure S4. Comparison of the respective effects of early and late onset preeclampsia plasma on the
expression of genes found modified in the microarray experiment. The expression was evaluated by
quantitative RT-PCR. Data from the control group were obtained from 10 patients. In grey, mild cases
(late onset) preeclampsia are presented (n=7), and in black more severe (early onset) preeclampsia
(n=5) are presented. In most cases, no significant difference between the two types of preeclamptic
125
Results
plasma could be observed relative to controls, except for Endothelin1 which was more induced when
the plasma from severe cases was used (see text). Error bars are SEM.
Figure S5
Figure S5. Effect of JDP2 knockdown on the expression of preeclamptic plasma deregulated
genes in the HMEC-1. To inhibit JDP2 expression, HMEC-1 were transfected with 30 pmol of
siRNA specific for JDP2 (siJDP2). The same amount of nonspecific double-stranded RNA was used
as a negative control (siCO). Two days after the transfection, the cells were harvested and total RNA
was subjected to quantitative RT-PCR using primers specific for JDP2 and for several genes found to
be modified by the preeclamptic plasma. Levels of expression were normalized to that of GAPDH
and SDHA. Data are shown as relative gene expression values adjusted to the mean value of the
control group (siCO), which was set at 1 for each gene. Error bars represent the standard deviation
(±SD).
126
Results
2. Paper 2 (in preparation)
A complete hypoxic response in endothelial cells depends on the
transcription factor Jun-Dimerization Protein 2
Summary
The gene expression profile of HUVEC line after a short time treatment with preeclamptic
plasma allowed us to pinpoint JDP2 as a factor playing a role in managing cellular behavior
under the stress condition induced by preeclamptic plasma. In this regard, the knock-down of
JDP2 in HUVEC cells mimics the expression profile of cells treated with preeclamptic plasma
at least for the genes VEGF-A, IGFBP3, and BCL2A1 (Calicchio et al. 2013).
In the last 15 years 32 papers only deal with the characterization of JDP2 function, following
the discovery of its interaction with the proto-oncogene c-Jun in 1997. JDP2 appears to play a
remarkable role in important physiological processes like differentiation, cell cycle regulation,
cellular senescence and in pathological outcomes as well, in particular cancer progression and
stress conditions, like infections and UV irradiation.
Its implication in the regulation of VEGF, one of the master genes involved in endothelial cell
growth and more generally in angiogenesis and vasculogenesis, prompted us to go deeper in
the characterization of its implication in a specific stress condition : hypoxia. Hypoxia is one
of the main drivers of a correct feto-placental development, cancer progression and
vascularization in growing tumors, in particular through its action on VEGF expression.
The aim of this work was to characterize the role of JDP2 in endothelial response in the
context of hypoxia. For this, a protocol to cultivate HUVECs in normoxic and hypoxic
conditions was set up. We evaluated the impact of JDP2 knock-down on the expression of
VEGF under hypoxic conditions. Then, we focused specifically on the contribution of the
HRE binding site present in the VEGF promoter, on VEGF expression in case of JDP2 downregulation.
We show that JDP2 expression is necessary for achieving complete VEGF up-regulation
under-hypoxic conditions and in particular that JDP2 contributes to VEGF expression by
127
Results
modulating HIF-1 activating properties under hypoxic conditions. This work has been done in
collaboration with Dr. Jacques Mathieu and Dr. Carole Peyssonnaux (Team “Gènes,
nutriments et fer”, Institut Cochin, Paris).
128
Results
A complete hypoxic response in endothelial cells depends on the
transcription factor Jun-Dimerization Protein 2
Rosamaria Calicchio1, Jacques R.R. Mathieu1, Francisco Miralles1, Carole Peyssonnaux1,
Daniel Vaiman1,
1
INSERM U1016-CNRS UMR8104, Université Paris Descartes, Institut Cochin, 24, rue du
Faubourg Saint-Jacques, 75014 Paris, France.
Abstract
Hypoxia refers to a condition of oxygen shortage that drives important cellular processes like
differentiation, proliferation and angiogenesis in physiological and pathological conditions,
like feto-placental development and tumor progression. Its action is mediated by the well
conserved Hypoxia inducible factors (HIF) family. Under hypoxic conditions, HIF-1α is
stabilized, forms a heterodimer with HIF-1β and regulates the expression of genes presenting
a Hypoxia responsive element (HRE) in their promoter. Here we investigated the role of Jun
Dimerization Protein 2, a member of AP-1 family, on the regulation of Vascular Endothelial
Growth factor (VEGF) under hypoxic conditions in the HUVEC cell line. We showed that,
under hypoxic conditions JDP2 depletion reduced VEGF expression by interfering with HIF1 activating properties. In parallel we evaluated the effects of JDP2 knock-down on AP-1
family members expression, because of their known implication in HIF-1 regulation of gene
expression. We showed for the first time that JDP2 is necessary for a full cellular response to
hypoxia in HUVECs by modulating HIF-1 effects on gene expression, emphasizing the
function of JDP2 as an ancillary factor of the hypoxic response. Further studies are needed to
clarify its function in the process of angiogenesis in normal and pathological situations.
129
Results
Introduction
Vasculogenesis refers to the process, typical of the first stages of embryonic development, which leads
to the formation of the vascular network of the newborn. In adult tissues, evidences of vasculogenesis
come from the organization of new vessels after an ischemic injury, and it involves progenitor cells
from the bone marrow “niche”
1,2
. During embryonic development the process of vasculogenesis
implies the migration and differentiation of hemangiogenic progenitor cells into angioblastic and
hematopoietic cell population and results in the formation of the heart and the primitive vascular
plexus in the growing embryo 3,4. The vascular tree is then completed by angiogenesis, which refers to
the migration of endothelial cells and circulating progenitor cells, the proliferation and the formation
of new vessels from the preexisting vascular system through vascular sprouting and intussusception 5,6.
Angiogenesis occurs in adults as well, in physiological conditions, like menstrual cycle, or in
pathological situations like tumor progression 7–9
Angiogenesis and vasculogenesis are also the main drivers of the formation of the placental vascular
network, which supports feto-placental circulation. It implies the formation of new vessels and the
adaptation of maternal vasculature to the needs of the growing fetus, in the process of placentation.
Organization of the placental vasculature starts very early during placental development,
approximately 21 days post-conception, with the differentiation of hemangiogenic progenitor cells
which populate chorionic villi
10,11
. At this step of placentation two main forces, strongly
interconnected, drive a correct vascularization: the presence of growth factors, in particular VEGF and
PlGF, and an hypoxic environment, which persists until 8-10 weeks of gestation 12.
The VEGF family includes VEGF-A, -B, C, -D, -E, -F, PlGF, and three receptors VEGFR-1, -2, -3
13,14
. VEGF-A and PlGF are highly expressed in early placenta
11
. VEGF-A can specifically interact
with VEGFR-1 and VEGFR-2 in order to trigger the gene expression cascade necessary for blood
vessels formation 15.
VEGF expression is augmented by low oxygen tension
16
, which enhances vascularization and
trophoblast differentiation during early stages of placentation. Hypoxia action is mediated by the
Hypoxia Inducible Factors (HIFs) family, which includes two types of factors, type α and type β
subunits.
HIF-α subunits are sensitive to oxygen tension and under normoxic conditions undergo rapidly
proteasomal degradation. Under low oxygen tension, HIF1-α is stabilized, forms heterodimers with
130
Results
HIF1-β and activates transcription of genes having a HRE consensus sequence in their proximal or
distal promoter 17.
In pathological situations, impaired vascular remodeling occurs and may be associated with
preeclampsia, a human pregnancy disorder which affects 5-8% of human pregnancies. While disease
is presumably caused by early defects, late maternal complications associated with hypertension,
proteinuria, exacerbated pro-inflammatory and pro-coagulant state, occur from the second trimester
and affect the whole maternal organism 18.
In a previous work we found that Jun Dimerization protein 2 (JDP2) mRNA level, was strongly
decreased in the HUVEC cell line exposed to preeclamptic plasma. We also showed that it was
directly involved in the down-regulation of VEGF expression (Calicchio et al. 2013). Consistently,
microarray analysis showed a strong decrease in the amount of JDP2 mRNA in preeclamptic placentas
compared with placentas from normal pregnancies 19.
JDP2 is a member of the AP-1 family involved in proliferation, differentiation, and cancer
progression. As other members of AP-1 family, it can form heterodimers with other AP-1 proteins or
CEBP proteins and binds the TPA consensus sequence (TRE) or the c-AMP consensus sequences
(CRE). It can act as a transcription factor with inhibiting properties, as described in the case of the cJun and ATF3 promoters, or as an activator, by interacting with the partners CHOP-10 and
progesterone receptor 20,21.
Several studies concur to suggest that the AP-1 pathway participates in the hypoxia-induced cellular
response. According to the cell type, it has been shown that hypoxia induces the expression of some
members of AP-1 family, like c-jun, c-fos, and junB 22; moreover phosphorylation and transcriptional
activation of AP-1 members are also ensured by the increased activity of Mitogen-activated protein
kinases (MAPK), such as Jun NH2-terminal kinase (JNK) and extracellular signal-regulated kinase 1
and 2 (ERK1/2) under hypoxic conditions
23,24
. Activated AP-1 dimers, in turn, activate several
hypoxia target genes, like VEGF 25, IL-8 26, eNOS 27, MMP-2 28 and endothelin-1 29.
All these findings prompted us to evaluate the impact of JDP2 on hypoxia-induced gene expression,
by analyzing the effect of JDP2 knock–down on VEGF transcription and the contribution of the AP-1
pathway and the HIF machinery to the regulation of the VEGF promoter which encompasses both
HRE and AP-1 family binding sites. We demonstrate that JDP2 is necessary for the expression of
VEGF under hypoxic condition through the regulation of HIF transcriptional activity. These results
identify JDP2 as a novel regulator of VEGF, one of the master genes involved in a correct vascular
131
Results
development, and, in the case of preeclampsia, the guilty party of the impaired placental vascular
adaptation and the later maternal dysfunction. In addition, the effect of JDP2 on the isolated HRE
element suggests that JDP2 could play a very general function in achieving a complete transcriptional
response to hypoxia.
132
Results
Materials and methods
Endothelial cells culture
The endothelial cell line used in our study was the Human Umbilical Vein Endothelial Cell (HUVEC)
line immortalized with the large T-antigen of SV40 and ectopic expression of the hTERT (human
Telomerase Reverse Transcriptase). Since the establishment of this cell line, its phenotype has been
maintained over passages. Cells were routinely cultured in uncoated 75 cm2 tissue culture flasks at
37°C and 5% CO2. Culture medium consisted of DMEM GlutaMAX supplemented with 10% (vol/vol)
heat-inactivated fetal calf serum (FCS), 100 IE/ml penicillin, and 100 mg/mL streptomycin.
To analyze the effect of hypoxia on gene expression, cells were seeded in six-well plates, placed in a
InVivo2Hypoxia Workstation 500 (Ruskin) chamber at 37°C and exposed to an oxygen-depleted
atmosphere (0.5% O2, 5% CO2) or maintained as control at 37°C in a humidified normal atmosphere
(20% O2–5% CO2).
JDP2 gene silencing and hypoxic cell culture
HUVECs were cultivated in DMEM/Glutamax and 10% FCS on day 0, and were transfected on day 1,
in serum-free medium with 10 pmol of siRNA oligonucleotides targeting human JDP2 (Qiagen) or
non-targeting controls using Lipofectamine RNAiMAX Qiagen. The control siRNA sequence of the
sense strand is UAGCAAUGACGAAUGCGUAdTdT. The sense and antisense sequences of the
duplex specific for human JDP2 are 5’-GCCAUGAGUUGCAACCAAATT-3’ and 5’UUUGGUUGCAACUCAUGGCTT -3’. After 6 hours, the medium was changed to complete
medium. Total RNA was extracted after 48 hours and processed for quantitative RT-PCR. JDP2
siRNA silencing was performed also in association with hypoxic cell culture. In this case, 6 hours after
the transfection, the medium was changed to complete medium and after 24 hours cells were incubated
at 0.5% oxygen in the hypoxic chamber InVivo2Hypoxia Workstation 500 from Ruskin for 24 hours
before RNA or protein extraction.
RNA isolation
Total RNA was isolated from HUVECs with Trizol (Invitrogen Life Technology) according to the
manufacturer’s instructions and treated with DNase I (Invitrogen Life Technology) to eliminate
genomic DNA contamination. The quality of the RNA was analyzed using the Agilent Bioanalyser
2100.
Quantitative RT-PCR
Quantitative RT-PCR was used to validate the expression level of a subset of genes that showed
differential expression in the HUVECs treated with preeclamptic plasma. Twelve genes were selected
based upon their fold differences and biological relevance. The RNA samples were reversetranscripted according to a standardized protocol. Briefly, 2 µg of total DNase-treated RNA were
reverse transcribed in a volume of 25 µL at 39°C using the Superscript reverse transcriptase
133
Results
(Invitrogen) during 1 hour. Quantitative PCR was carried out in duplicates on 8 controls, 9
preeclamptic samples individually using the amplification kit LC480 SYBR Green Master Kit (Roche)
and the reaction was performed in a Light Cycler 480 Thermocycler (Roche). Primers (Table 1) were
designed for the coding sequences (GENBANK) of the different genes to be analyzed, using the
PRIMER3 software (http://frodo.wi.mit.edu/cgi-bin/primer3). The different pairs were chosen to cover
all of the previously described isoforms and aligned with basic local alignment search tool software
(BLAST) to avoid nonspecific annealing. 35 cycles were performed with the following 3 temperature
steps (95°C for 10 s, 55°C for 15 s, and 72°C for 15s). Finally, samples were submitted to a
progressive temperature elevation (from 65 to 99°C at 0.1°C/s), resulting in a fusion curve, enabling to
check the PCR products homogeneity. The threshold cycle number (Ct) values were collected with the
LightCycler software (Roche) and analyzed through a second derivative maximum method. These Cts
were normalized by the Ct values obtained for 3 reporter genes, succinate dehydrogenase subunit A
(SDHA), and glyceraldehydes-3-phsphate dehydroenase (GAPDH), and Cyclophilin A.
Table 1. Primers used for RT-qPCR analysis
Western blot
Cells were washed three times in cold PBS. Then cells were scraped and resuspended in histidinesucrose buffer (Sucrose 0.25 M L-Histidine 0.03 M) with an EDTA-free protease inhibitor cocktail
(Roche inhibitors 1X, PMSF 0,2 mM) and left for 30 min at 4°C on a rotating platform. The lysate
was centrifuged at 5000 rpm for 8 min at 4°C to pellet nuclear fraction. The pellet was then
resuspended in Hepes buffer (Hepes KOH 20 mM pH 7.9, glycerol 25%, NaCl 420 mM, MgCl 2
1.5mM, EDTA 0.2 mM, PMSF 0.2 mM, DTT 0.5 mM) supplemented with a protease inhibitor
cocktail (Roche inhibitors 1X). After 30 min on ice, lysates were spun down at 14000 rpm, 8 min at
4°C, to obtain the nuclear protein fraction (supernatant). Proteins amount was quantified with the BCA
protein assay kit (Thermo Scientific, IL 61101, USA) according to manufacturer’s instructions.
Nuclear proteins were resolved by SDS-PAGE by sodium dodecyl sulphate polyacrylamide gel
electrophoresis (5% gels). Proteins in the gel were transferred to a polyvinylidene difluoride (PVDF)
membrane (Hybond-P; GE Healthcare Ltd., U.K) in a transfer Buffer (Transfer Buffer Biorad 10X,
0,1% SDS, 10% Methanol) using a Mini-Trans-Blot electrophoretic transfer cell (Bio-Rad Life
134
Results
Science Group, Hercules, SA) overnight at 35 V. Membranes were blocked for 1 hour at 4°C with
PBS buffer supplemented with 5% nonfat dry milk. Immunoblotting detection was performed using a
1:100 dilution anti-HIF1α (BD, 610958 Clone 54), and 1:1000 anti- HSC-70 (sc-7298, Santa Cruz
Biotechnology) in a PBS buffer supplemented with 0.1% Tween-20 and 5% nonfat dry milk at 4°C
overnight. Incubation with the secondary antibody was performed at RT for 45 minutes using a 1:3000
dilution of anti-mouse antibodies (Dako A/S, DK 2600 Glostrup) conjugated to horseradish peroxidase
(HRP) in a PBS buffer supplemented with 0.05% Tween-20 and 1% nonfat dry milk. Immunoreactive
bands were detected using the enhanced chemiluminescent HRP Substrate Immobilon Western
(Millipore Corporation, USA). Proteins were visualized using Image Quant Las4000 mini (GE
Healthcare).
Plasmids
pGL3-VEGF-Luc and pGL3-HRE-Luc contain respectively the VEGF promoter (2400bp) and six
HRE elements upstream of the firefly Luciferase reporter gene in a pGL3 vector.
Luciferase assays
HUVECs were seeded 24 h before transfection in 24-well plates at 50% of confluence. Cells were then
transfected using the Lipofectamine 2000 reagent (Invitrogen) (1 µL/well) and rinsed 5 hours later. In
order to assess transfection efficiency, the Renilla luciferase vector (pRL-RSV, Promega) was
systematically co-transfected in all experiments. Cells were transfected with 500 ng of reporter
luciferase (pGL3-VEGF-Luc or pGL3-HRE-Luc), 10 pmol of siRNA (siRNA control or siJDP2) and
10 ng of pRL-RSV. Cells were harvested after 48 h. The day following transfection, the wells were
rinsed in order to remove dead cells and fresh medium was added. 24h after transfection cells were
incubated at 0.5% oxygen in the hypoxic chamber for 24 hours or left at the control oxygen level (20%
oxygen). The cells were rinsed before the cell lysis that preceded the luciferase measurement. The
cellular viability was checked under light microscope, without clear differences between the different
transfection conditions. In addition, the amount of proteins was quantified and not strongly altered by
transfections. By all means, the use of pRL-RSV as an internal control made it possible to take into
consideration exclusively the viable transfected cells. Transcriptional activity was assessed by the
Dual-Luciferase Reporter Assay System (Promega). Luminescence was measured using a FLUOstar
OPTIMA Microplate Reader (BMG labtech). The experiments were performed three times
independently with 6 replicates per experiment for each condition. The observed firefly activity was
divided by the activity recorded from the Renilla luciferase vector, and the mean values of the
replicates were calculated.
Statistical analysis
Statistical significance was assessed by performing one-way analysis of variance (ANOVA) followed
by Bonferroni post-hoc test. Values of P< 0.05 were considered significant. (* and # for p ≤ 0.05, **
and ## for p ≤ 0.01, *** and ### for p ≤ 0.0001). * refers to statistical analysis between normoxic and
hypoxic conditions, # refers to statistical analysis between siRNA control and siJDP2 under hypoxic
conditions.
135
Results
Results
1. JDP2 regulates VEGF expression under hypoxic conditions
In our previous work we showed that JDP2 knock-down decreases VEGF-A expression in
HUVEC cells cultivated in the presence of human plasma (Calicchio et al. 2013). In order to better
define the role of JDP2 on VEGF expression we tested here the effects of JDP2 on VEGF expression
when this expression is induced by hypoxia. For this, we cultivated untransfected HUVECs in
normoxia and hypoxia (20% and 0.5% oxygen respectively). In parallel, HUVECs transfected with a
control siRNA (siCtl) or a siRNA against JDP2 (siJDP2) were cultivated in the same conditions, in
normoxia and hypoxia.
We could show that transfection itself had no effect on VEGF expression, neither in normoxia
nor in hypoxia, in the presence or absence of control siRNA. Then we analyzed VEGF expression in
three separate conditions: in normoxia (20% O2), hypoxia (0.5% O2), and hypoxia +siJDP2 (referring
to HUVEC transfected with siJDP2 and cultivated for 24 hours at 0.5% O2). VEGF expression was
increased 8 fold after 24 hours of culture under hypoxic conditions, compared to a normoxic
environment; this induction falls down to 5 fold in cells transfected with siJDP2 and cultivated for 24
hours under hypoxic conditions. Statistical analysis showed that VEGF was significantly up-regulated
by hypoxia but that this increase was significantly lower when the siJDP2 was added (Figure 1.A).
Hypoxia by itself has no effect on JDP2 expression as shown in Figure 1.B. The knock-down
of JDP2 was efficient in normoxia as well as in hypoxia. This implies that JDP2 is necessary to
achieve a full level of VEGF transcriptional induction under hypoxia. In normoxia, the basal level of
VEGF-A is not affected by JDP2 extinction.
136
Results
Figure 1: A) Effect of siJDP2 VEGF (A) and JDP2 (B) expression in normoxic and hypoxic
conditions. HUVEC cells were cultivated 24 hours in normoxia (20% O2) or hypoxia (0.5 % O2).
Equivalent conditions were performed for HUVECs previously transfected with a control siRNA
(siCtl) or siJDP2. After 24 hours of normoxia or hypoxia, total RNA was extracted and VEGF and
JDP2 gene expression was measured by qRT-PCR. Results are the mean value of two independent
experiments, with 2 biological replicates per experiment for each condition. Relative gene expression
was normalized on gene expression of HUVECs transfected with siCtl in normoxia. Error bars
represent the SEM.
Since VEGF expression can be regulated at many levels, including transcription, mRNA stability,
and translation we evaluated by mean of Luciferase/Renilla assays, the effect of JDP2 on VEGF
transcriptional activity through ~2400 bp of its promoter. The pGL3-VEGF-Luc containing the VEGF
promoter cloned upstream of the firefly Luciferase reporter gene was transfected in cells exposed to
normoxia or hypoxia during 24 hours. We measured the relative luminescence in normoxic and
hypoxic conditions in cells co-transfected with siCtl or siJDP2 or without additional transfection
(Figure 2). Under hypoxic conditions, the VEGF promoter activity was increased up to 2.5 fold
compared to normoxia. HUVECs transfected with siJDP2 and cultivated in normoxia showed no
modification of luciferase activity; on the contrary, cells transfected with siJDP2, after 24 hours under
hypoxic conditions, showed a significant increase of 1.8 fold in luciferase activity compared to
normoxia; nevertheless this significant increase of luciferase activity was itself significantly less
pronounced than in the absence of the siJDP2, reproducing the observations on the endogenous VEGF.
This suggests again that the full transcriptional induction of VEGF by hypoxia requests the presence
of JDP2.
137
Results
Figure 2: Effect of siJDP2 on VEGF promoter activity in normoxia and hypoxia. The biological
activity of the VEGF promoter, following transient transfection into the HUVECs in presence of siCtl
or siJDP2, was measured by luciferase assays. Data are shown as mean fold ± SEM of three
independent experiments (including four replicates per condition). HUVEC cells were transfected with
pGL3-VEGF-Luc plus either siCtl or siJDP2. 24h after transfection cells were cultivated for 24h in
normoxia or hypoxia. Results are represented as fold induction of Relative Luminescence Unit (RLU)
compared to HUVECs transfected with siCtl under normoxic condition. Error bars represent the SEM.
2. JDP2 impacts HRE promoter activity
The hypothesis that JDP2 could impact the HIF-1 contribution to VEGF expression under
hypoxic conditions prompted us to evaluate the role of JDP2 on HRE promoter activity by Luciferase
assay. HUVECs were cultivated in 24-well plates and co-transfected with pGL3-HRE-Luc, containing
six HRE elements upstream of the firefly Luciferase reporter gene, supplemented either with siCtl or
siJDP2, in normoxia or hypoxia. Hypoxia induced HRE-mediated luciferase activity 30 fold compared
to normoxic conditions; by contrast, HUVEC cells co-transfected with siJDP2 after 24 hours at 0.5%
oxygen, displayed an increase of only 15-fold of HRE activity compared to normoxic conditions.
Therefore, siJDP2 transfection reduced by 2-fold the hypoxia-dependent induction of the HRE
promoter activity (Figure 3).
138
Results
Figure 3: Effect of siJDP2 on HRE promoter activity. The biological activity of HRE sequences,
following transient transfection into the HUVEC cell line in presence of control siCtl or siJDP2 was
measured by luciferase assays. Data are shown as mean fold ± SEM of three independent experiments
(including four replicates per condition). HUVECs were transfected with the pGL3-HRE-Luc plus
either siCtl or siJDP2. 24h after transfection HUVECs were cultivated for 24h in normoxia or hypoxia.
Results are represented as fold induction of Relative Luminescence Unit (RLU) compared to HUVECs
transfected with siCtl under normoxic conditions.
3. Effects of siJDP2 on HIF-1α and HIF-2α mRNA levels and HIF-1α protein expression
under hypoxic conditions
Luciferase assays pointed out the capacity of JDP2 to interfere with hypoxia–induced
activation of VEGF expression, at least in part by reducing HRE promoter activity. In order to
evaluate whether the effect of JDP2 was mediated by deregulating HIF-1α or HIF-2α, we quantified
the gene expression and protein levels of HIF-1α and HIF-2α in normoxia and hypoxia with or without
JDP2. After 24h at 0.5% oxygen, the level of HIF-1α was strongly decreased in hypoxia,
independently of JDP2: no significant difference in HIF-1α gene expression was detected between
untransfected cells and cells transfected with siJDP2 (Figure 4A). HIF-2α expression does not vary
significantly between normoxia and hypoxia (Figure 4B).
139
Results
Figure 4: Effect of siJDP2 and hypoxia on HIF-1α (A) and HFI-2α (B) gene expression. HUVEC
cells were cultivated 24 hours in normoxia or hypoxia. Equivalent conditions were performed for
HUVECs previously transfected with siCtl or siJDP2. 24 hours after normoxia or hypoxia, total RNA
was extracted and HIF-1α and HIF-2α gene expression was measured by qRT-PCR. Results are the
mean value of two independent experiments, with two biological replicates per experiment for each
condition. Relative gene expression was normalized on gene expression of HUVECs transfected with
siCtl under normoxic conditions. Error bars represent SEM.
A Western blot analysis of HIF-1α protein level in cells transfected with siCtl or siJDP2 in
normoxic or hypoxic conditions was performed as well, in order to evaluate the possible impact of
JDP2 on HIF-α stabilization or degradation (Figure 5). Protein quantification revealed a strongly
increased HIF-1α protein level under hypoxic conditions, but no effect of JDP2 extinction. We were
not able to detect HIF-2α protein level, probably because of a limited amount of this factor in nuclear
protein extracts or because of the weak sensibility of the antibody.
140
Results
Figure 5: Effect of siJDP2 on HIF-1α protein level under hypoxic conditions. HUVEC cells were
cultivated 24 hours in normoxia or hypoxia. Equivalent conditions were performed for HUVECs
previously transfected with siCtl or siJDP2. Western blots show nuclear extracts revealed for HIF-1α
and HSC-70, the latter one being used as loading control.
4. Effects of siJDP2 on the expression of AP-1 family members in the HUVEC line
under normoxic and hypoxic conditions
Since JDP2 interacts with several members of this family, we decided to evaluate the
expression level of c-jun, junB, junD, fos, cebpβ, atf3 and atf4 in normoxia, hypoxia and in cells where
JDP2 has been down-regulated (Figure 6). The expression level was measured by qRT-PCR. Hypoxia
enhanced the expression of C-JUN, and ATF3, while no significant changes was detected for JUNB,
JUND, CEBP,CHOP-10 and FOS mRNAs. Inhibition of JDP2 under normoxic conditions reduced
ATF3 expression in normoxia and hypoxia, leading to an ATF3 expression level in hypoxia, similar to
the expression in normoxia.
141
Results
142
Results
Figure 6: Effect of siJDP2 and hypoxia on AP-1 family members expression. HUVEC cells were
cultivated 24 hours in normoxia or hypoxia. Equivalent conditions were performed for HUVECs
previously transfected with siCtl or siJDP2 24 hours after normoxia or hypoxia, total RNA was
extracted and AP-1 proteins gene expression was measured by qRT-PCR. Results are the mean value
of two independent experiments, with 2 biological replicates per experiment for each condition.
Relative gene expression was normalized on gene expression of HUVECs transfected with siCtl in
normoxic conditions. Error bars represent SEM.
143
Results
Discussion
We show in this study that JDP2 is a novel actor of the hypoxia cascade, at least in endothelial
cells. Its action was independent of an effect on HIF, and reciprocally, hypoxia and therefore HIF
induction was not able to modulate JDP2. Under hypoxic condition, the regulation of VEGF in these
cells was clearly dependent on two cascades of transcription regulators, one involving the HIF
pathway and one involving the AP-1 pathway.
Hypoxia refers to the stress condition in which an oxygen shortage induces a cellular response
mediated by the expression of a battery of genes presenting a Hypoxia Responsive Element (HRE)
within their promoters or enhancers. Hypoxia-induced gene expression is mediated by the HIF
transcription factors family. The better characterized actor is HIF-1, composed by heterodimers of
HIF-1α and HIF-1β (ARNT). Under hypoxic conditions HIF-1α is stabilized, translocated into the
nucleus, and in association with HIF-1β and p300, binds HRE, thereby activating gene expression.
Oxygen tension is one of the main regulators of cellular processes like differentiation, proliferation
and migration, and more generally, of all physiological and pathological events in which these cellular
adaptations are implicated, like feto-placental development and cancer progression. Hence the
importance to finely define the possible actors involved in cellular response to hypoxia.
Impact of JDP2 on HIF1α dependent activation of VEGF under hypoxic condition. Here, we show
that JDP2 plays a crucial role in orchestrating a complete response to oxygen shortage, since JDP2
knock-down reduces VEGF expression in the HUVEC cell line after 24 hours at 0.5% oxygen. VEGF
is a mitogenic factor involved in proliferation, permeability and angiogenesis. Under hypoxic
conditions, VEGF induction is enhanced by the activating factor HIF-1, which binds specifically the
HRE consensus sequence present in the VEGF promoter.
16
. The inhibiting effect of JDP2 affects
specifically the capacity of HIF-1 to fully activate VEGF expression under hypoxic conditions since
HUVECs co-transfected with pGL3-HRE-Luc and siJDP2 showed a reduced HRE promoter activity in
luciferase assays. All our experiments showed that in the absence of JDP2, the activation of the VEGF
promoter (as well as HRE binding sites alone), reached only half the level attained in the presence of
JDP2.
In HUVECs, we found that HIF-1α mRNA was decreased in hypoxic conditions, suggesting that
either directly or not, a negative feedback loop of the hypoxia-stabilized HIF-1α protein may affect
HIF-1α gene expression, since the protein was detectable only in cells cultured at 0.5% oxygen.
144
Results
We showed that JDP2 does not impact HIF-1α neither at the mRNA or protein levels. Thus it is more
likely that JDP2 interferes on HIF-1α capability to activate hypoxia target genes without perturbing its
stability. We cannot conclude whether JDP2 interacts directly or not with HIF-1α, but it could
interfere with co-activators and cofactors. Indeed, HIF-1α activation depends not only on oxygendependent stabilization, but also on its acetylation level and on its interaction with the co-activator
p300. Acetylation at specific lysines has opposite effects on protein stabilization, and it is influenced
by the cooperation of proteins with HAT activity and HDACs. HDACs implication in HIF-1α stability
has been demonstrated by the use of specific HDACs inhibitors in vivo and in vitro, which results in
HIF-1α destabilization, impaired expression of its target genes and reduced tumor mass and
angiogenesis in a mouse model for xenograft tumor growth
30–33
. Therefore, JDP2 could participate as
a HAT inhibitor or a HDAC recruiter. In fact JDP2 interacts with HDAC1, 2, 3, 5, 6 and HDAC10
through the bZIP domain at the N-terminal region 34. JDP2 interacts and recruits specifically HDAC3
on gene promoters thus modulating the epigenetic landscape and gene expression: in particular this
mechanism has been described in details concerning the JDP2-dependent inactivation of c-Jun
expression via the recruitment of HDAC3 on c-Jun promoter 35. Interestingly, HDAC3 is also a target
of HIF-1α: it is increased under hypoxic conditions and participates to HIF-1α stabilization and
transactivation through the interaction with the oxygen sensing domain ODDD
36,37
, which makes
more seductive the possible role of JDP2 in HDAC3 recruitment and HIF-1 activation.
JDP2 modulation of AP-1 and HIF-1 interaction. There are more and more evidences that hypoxiainduced gene expression is the result of a complex interplay between HIF factors and other
transcription factors that cooperate to the cellular response. Concerning endothelial specific
modulation, it has been shown that endothelin-1 (ET-1) expression is driven by GATA-2 and AP-1 38,
and the presence of consensus sequences for these both factors is necessary, in addition to HRE, to
enhance ET-1 expression under hypoxic conditions
39
. Similarly, hypoxia dependent Plasminogen
Activator Inhibitor-1 (PAI-1 aka SERPINE1) expression is the result of the collaboration of Egr-1,
HIF-1α, and C/EBPα
40
. More recently high-throughput approaches based on HIF-1α chromatin
immunoprecipitation coupled to genome-wide expression profiles and whole genome sequencing
confirm the high specificity of HIF factors for the HRE binding sequence and consequent activation of
a hundred of genes induced by hypoxia. Nevertheless it seems that HIF-1α binds only a weak amount
of potential or predictive binding sites
. Additionally, HIF-1α had preferentially an activating
41–43
effect on genes expression, while the mechanism required for the repression under hypoxic condition
could be regulated indirectly by HIF or completely bypass HIF machinery
44,45
. All these findings
show that the response to hypoxia is more complex than expected, and that HIF could be at the head of
an integrative multiprotein system aiming at discriminating responsive genes according to cell type,
chromatin context, and promoter composition 42,45. A recent study based on computational predictions
145
Results
of transcription factor binding sites highlights the over-representation of specific transcription factors
binding sites near HRE consensus sequence which could participate to the activation of hypoxia
responsive genes in different tissues and cell lines, in particular AP-1 binding site (TRE) and c-Amp
responsive element (CRE) binding sites 46. In parallel, AP-1, ATF3, CEBP, CREB are stress response
genes, induced under hypoxic conditions
47
, which have been identified as enriched transcription
factors in core HIF binding regions. Interestingly JDP2 seems to be the common partner of the
transcription factors families mentioned above, since it can bind several members of AP-1 and CEBP
family. Moreover, it recognizes and binds also CRE consensus sequence, together with CREB
transcription factors, a part TRE element (AP-1 binding site).
The AP-1 family participates to the regulation of important cellular processes like proliferation,
survival, differentiation and cellular transformation, and response to environmental stress. The
implication of the AP-1 family in stress response and specifically hypoxia depends strictly on the cell
type, the experimental conditions, the type of stress, and the subunits that form AP-1 dimers.
Different members of the AP-1 family have been found activated under hypoxic conditions, like c-Jun,
c-Fos and JunB 22, but the well characterized mechanism of AP-1 implication in hypoxic cellular stress
is concentrated on c-Jun action. c-Jun has been described as an early response gene immediately
expressed under stress condition
48,49
. Like all the members of the same family, it forms dimers with
several partners and modulates gene expression. Its transcriptional activity and protein stability are
enhanced by the phosphorylation of serine 63 and 73 by Jun NH2-terminal kinase (JNK)
50,51
; c-Jun
phosphorylation can also be induced by other mitogen–activated protein kinases (MAPK), like
extracellular signal regulated kinase 1 and 2 (ERK1/2) and p38 MAPK 52,53.
A complex interplay involves c-Jun and HIF-1 regulation under hypoxia, consisting of a mutual
regulation at the transcriptional and post-translational levels. The model proposed is that c-jun
expression, as an early response gene, is quickly induced under hypoxia by pathways which do not
depend on HIF-1α stabilization; later on, its expression is maintained by HIF-1 expression and concurs
to participate with HIF factors to manage cellular response to hypoxia 54. Furthermore an increase in
activation of JNK and ERK1/2 under hypoxia
23,24
is correlated with an increased phosphorylation
status of c-Jun and consequent increase of c-Jun DNA binding activity 55,56. Once activated, c-Jun can
directly activate hypoxia responsive genes, like VEGF, by acting as a transcription factor 57,58, or it can
act in a multi-proteic complex in association with HIF-1 55,59. In fact it has been shown that c-Jun can
directly bind HIF-1α at the level of its ODDD domain, thus stabilizing protein and preventing HIF-1α
proteasomal dependent degradation. Its action as oxygen defender is independent of its transctiptional
activity, since mutations in Ser 63 or 73, or inactivation of JNK, do not interfere with HIF-1α
146
Results
interaction
59
. Another study points out the necessity of the phosphorylated status of cJun for the
interaction with HIF-1α 55.
Our study brings a consolidated link between the AP-1 family (through JDP2 modulation, for instance
in pathological states such as preeclampsia) and the hypoxic response, which led us to investigate
direct putative JDP2 effects on AP-1 members under hypoxic and normoxic conditions. Indeed, our
experiments confirmed a hypoxia induced expression of c-Jun and ATF3 in HUVEC cells, as already
described in the literature 22,60.
Overall, we did not observe numerous significant effects of JDP2 knock-down on the expression of
AP-1 members neither in normoxia nor in hypoxia, except in the case of ATF3: in normoxia JDP2
knock-down was associated with down-regulation of ATF3, and under hypoxic conditions it blocks
the activating effect of hypoxia on ATF3 expression.
ATF3, a member of the ATF/CREB (CRE-binding protein) family, which is a subfamily of the AP-1
group, is a stress response gene induced by different cellular stresses, such as oxidative stress, ER
stress, toxic agents, pro-apoptotic agents
61–64
, and tumor progression
described as a regulator of HIF-2α in UV mediated cellular apoptosis
65–67
68
. ATF-3 has also been
and ChIP-on-chip analysis
identifies VEGF signaling pathway as associated to stress-induced ATF3 69. During hypoxia or anoxia
ATF3 mRNA level is increased in a HIF and p53 independent fashion
60
and its increased
transcriptional activity is enhanced by activation of c-Jun-NH(2)-terminal kinase (JNK) 70.
ATF3 displays a strong similarity with JDP2 71 which may explain somehow their partially redundant
action: in fact they share the same function with the same protein partners, both are transcriptional
activators by forming dimers with the common partner CHOP-10 21, and transcriptional repressors on
the ATF3 promoter 34. Both can interact and recruit several members of the HDAC family on gene
promoter thereby regulating gene expression
34
; both have a double nature of oncogene and tumor
suppressor 72–74, according to the cellular background and protein partners.
JDP2 is classically described as a transcriptional repressor, like for example in the case of c-jun and
ATF3 expression
35,75
. It has also been shown that JDP2 over-expression is associated with the up-
regulation of c-Jun and ATF3 expression in vivo in a model of hepatocellular carcinoma in mice 72. In
this model, JDP2 over-expression in the liver seems to produce no significant phenotype in mice, apart
from a weak up-regulation of c-jun. But when mice were treated with the genotoxic agent
diethylnitrosamine, it potentiates cell proliferation and tumor formation in transgenic mice compared
to wild type. So it is possible that perturbation of JDP2 expression according to cell type, stress
147
Results
condition, and molecular partner(s) have opposite effects on gene expression and this could explain
why in our system the down-regulation of JDP2 does not translate into a massive up-regulation of its
putative target genes.
JDP2 janus nature of activator and inhibitor is common to ATF3 as well. It has been show that ATF3
binding to cyclin D1 promoter has a double effect of activator or inhibitor whether it binds AP-1 site
76
or cyclic AMP response element (CRE) on cyclin D1 promoter 77.
In our system, JDP2 implication could lie in maintaining or perturbing the equilibrium among AP-1
members. In fact more and more data in the literature agree with the fact that AP-1 function is strictly
cell type specific, and within this specificity the dimer composition is the key point to regulate gene
expression in term of activation or repression. To that, if we add that a stress condition does perturb
this equilibrium, different co-operations and interactions among AP-1 family members could drive the
AP-1 involvement in stress cellular response.
Concerning the hypoxia response, the contribution of individual AP-1 subunits to hypoxia response
genes expression is still lacking, like the mechanism of activation of AP-1 proteins under hypoxic
conditions. Some studies concentrate on the composition of AP-1 dimers involved in the hypoxic
response, which lead to the identification of JunB/cFos 78, cJun/cFos 79, JunB/Fra-1 and JunB/FosB
80
.
Thus, we could speculate that JDP2 and ATF3 reduced expression perturbs the interactions between
AP-1 members under hypoxic conditions and their effects on gene expression. Further studies need to
clarify the protein composition of AP-1 dimers under hypoxic conditions in our model and a possible
link with the reduction of AP-1 subunits availability, like JDP2 and ATF3, and response to hypoxia.
In summary the present work pinpoints JDP2 as a novel actor of VEGF regulation and more generally
of hypoxia sensing in endothelial cells. More and more evidences point out the different possible axes
in which JDP2 could be implicated in such regulation, in particular as a transcription factor, by
binding AP-1 consensus sequence, as a regulator of AP-1 signaling pathway and as a HIF-1
modulator. Moreover it helps clarifying its involvement in important processes like angiogenesis in
physiological and pathological conditions. Interestingly JDP2 has already been described as oncogene
and tumor suppressor, and maybe its implication in the regulation of VEGF could better define its
field of action during tumor progression.
Our previous study links JDP2 down-regulation to endothelial dysfunction during preeclampsia and a
recent study found JDP2 strongly decreased in third trimester preeclamptic placentas
19
. It could be
interesting to analyze whether this transcriptional effect has repercussions on the protein content in the
148
Results
preeclamptic placenta, and then to understand how this deregulation affects hypoxia sensing and
response in this organ, as well as adaptive capabilities of the placenta to react to stressful conditions,
such as encountered even in normal pregnancies.
References
1.
Ii, M. et al. Concurrent vasculogenesis and neurogenesis from adult neural stem cells. Circ.
Res. 105, 860–868 (2009).
2.
Tepper, O. M. et al. Adult vasculogenesis occurs through in situ recruitment,
proliferation, and tubulization of circulating bone marrow-derived cells. Blood 105, 1068–1077
(2005).
3.
Demir, R., Seval, Y. & Huppertz, B. Vasculogenesis and angiogenesis in the early human
placenta. Acta Histochem. 109, 257–265 (2007).
4.
Patan, S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 117, 3–32 (2004).
5.
Adams, R. H. & Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis.
Nat. Rev. Mol. Cell Biol. 8, 464–478 (2007).
6.
Gerhardt, H. et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell
filopodia. J. Cell Biol. 161, 1163–1177 (2003).
7.
Maas, J. W. et al. Endometrial angiogenesis throughout the human menstrual cycle. Hum.
Reprod. Oxf. Engl. 16, 1557–1561 (2001).
8.
Rl, B., K, F., Ma, L., Jr, M. M. & B, H. Angiogenesis and tumor progression of melanoma.
Quantification of vascularity in melanocytic nevi and cutaneous malignant melanoma. Lab.
Investig. J. Tech. Methods Pathol. 67, 331–337 (1992).
9.
Ugurel, S., Rappl, G., Tilgen, W. & Reinhold, U. Increased Serum Concentration of
Angiogenic Factors in Malignant Melanoma Patients Correlates With Tumor Progression and
Survival. J. Clin. Oncol. 19, 577–583 (2001).
10.
Demir, R., Kaufmann, P., Castellucci, M., Erbengi, T. & Kotowski, A. Fetal vasculogenesis
and angiogenesis in human placental villi. Acta Anat. (Basel) 136, 190–203 (1989).
11.
Kaufmann, P., Mayhew, T. M. & Charnock-Jones, D. S. Aspects of human fetoplacental
vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 25, 114–126
(2004).
12.
Hustin, J. & Schaaps, J. P. Echographic [corrected] and anatomic studies of the
maternotrophoblastic border during the first trimester of pregnancy. Am. J. Obstet. Gynecol. 157,
162–168 (1987).
13.
Huppertz, B. et al. Placental angiogenesis, maternal and fetal vessels--a workshop report.
Placenta 28 Suppl A, S94–96 (2007).
14.
Jakeman, L. B., Armanini, M., Phillips, H. S. & Ferrara, N. Developmental expression of
binding sites and messenger ribonucleic acid for vascular endothelial growth factor suggests a
role for this protein in vasculogenesis and angiogenesis. Endocrinology 133, 848–859 (1993).
15.
Charnock-Jones, D. S. & Burton, G. J. Placental vascular morphogenesis. Baillières Best Pr.
Res. Clin. Obstet. Gynaecol. 14, 953–968 (2000).
149
Results
16.
Wang, G. L. & Semenza, G. L. Purification and characterization of hypoxia-inducible factor
1. J. Biol. Chem. 270, 1230–1237 (1995).
17.
Semenza, G. L. & Wang, G. L. A nuclear factor induced by hypoxia via de novo protein
synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional
activation. Mol. Cell. Biol. 12, 5447–5454 (1992).
18.
Roberts, J. M. & Gammill, H. S. Preeclampsia: recent insights. Hypertension 46, 1243–
1249 (2005).
19.
Nishizawa, H. et al. Microarray analysis of differentially expressed fetal genes in placental
tissue derived from early and late onset severe pre-eclampsia. Placenta 28, 487–497 (2007).
20.
Wardell, S. E., Boonyaratanakornkit, V., Adelman, J. S., Aronheim, A. & Edwards, D. P. Jun
dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol.
Cell. Biol. 22, 5451–5466 (2002).
21.
Weidenfeld-Baranboim, K., Bitton-Worms, K. & Aronheim, A. TRE-dependent
transcription activation by JDP2-CHOP10 association. Nucleic Acids Res. 36, 3608–3619 (2008).
22.
Ausserer, W. A., Bourrat-Floeck, B., Green, C. J., Laderoute, K. R. & Sutherland, R. M.
Regulation of c-jun expression during hypoxic and low-glucose stress. Mol. Cell. Biol. 14, 5032–
5042 (1994).
23.
Laderoute, K. R. et al. Mitogen-activated protein kinase phosphatase-1 (MKP-1)
expression is induced by low oxygen conditions found in solid tumor microenvironments. A
candidate MKP for the inactivation of hypoxia-inducible stress-activated protein kinase/c-Jun Nterminal protein kinase activity. J. Biol. Chem. 274, 12890–12897 (1999).
24.
Müller, J. M., Krauss, B., Kaltschmidt, C., Baeuerle, P. A. & Rupec, R. A. Hypoxia induces cfos transcription via a mitogen-activated protein kinase-dependent pathway. J. Biol. Chem. 272,
23435–23439 (1997).
25.
Salnikow, K. et al. The Regulation of Hypoxic Genes by Calcium Involves c-Jun/AP-1,
Which Cooperates with Hypoxia-Inducible Factor 1 in Response to Hypoxia. Mol. Cell. Biol. 22,
1734–1741 (2002).
26.
Shi, Q. et al. Cooperation between transcription factor AP-1 and NF-kappaB in the
induction of interleukin-8 in human pancreatic adenocarcinoma cells by hypoxia. J. Interf.
Cytokine Res. Off. J. Int. Soc. Interf. Cytokine Res. 19, 1363–1371 (1999).
27.
Hoffmann, A., Gloe, T. & Pohl, U. Hypoxia-induced upregulation of eNOS gene expression
is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. J. Cell.
Physiol. 188, 33–44 (2001).
28.
Riches, K. et al. Chronic hypoxia inhibits MMP-2 activation and cellular invasion in
human cardiac myofibroblasts. J. Mol. Cell. Cardiol. 47, 391–399 (2009).
29.
Yamashita, K., Discher, D. J., Hu, J., Bishopric, N. H. & Webster, K. A. Molecular regulation
of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator
protein-1, GATA-2, AND p300/CBP. J. Biol. Chem. 276, 12645–12653 (2001).
30.
Ellis, L., Hammers, H. & Pili, R. Targeting tumor angiogenesis with histone deacetylase
inhibitors. Cancer Lett. 280, 145–153 (2009).
31.
Kato, H., Tamamizu-Kato, S. & Shibasaki, F. Histone deacetylase 7 associates with
hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 279, 41966–
41974 (2004).
150
Results
32.
Kong, X. et al. Histone deacetylase inhibitors induce VHL and ubiquitin-independent
proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 26, 2019–2028
(2006).
33.
Qian, D. Z. et al. Class II histone deacetylases are associated with VHL-independent
regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 66, 8814–8821 (2006).
34.
Darlyuk-Saadon, I., Weidenfeld-Baranboim, K., Yokoyama, K. K., Hai, T. & Aronheim, A.
The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3,
recruit multiple HDAC members to the ATF3 promoter. Biochim. Biophys. Acta 1819, 1142–1153
(2012).
35.
Jin, C. et al. JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit
the retinoic acid-induced differentiation of F9 cells. Mol. Cell. Biol. 22, 4815–4826 (2002).
36.
Kim, M. S. et al. Histone deacetylases induce angiogenesis by negative regulation of
tumor suppressor genes. Nat. Med. 7, 437–443 (2001).
37.
Kim, S.-H. et al. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep.
17, 647–651 (2007).
38.
Kawana, M., Lee, M. E., Quertermous, E. E. & Quertermous, T. Cooperative interaction of
GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol. Cell. Biol. 15, 4225–4231
(1995).
39.
Yamashita, K., Discher, D. J., Hu, J., Bishopric, N. H. & Webster, K. A. Molecular regulation
of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator
protein-1, GATA-2, AND p300/CBP. J. Biol. Chem. 276, 12645–12653 (2001).
40.
Liao, H., Hyman, M. C., Lawrence, D. A. & Pinsky, D. J. Molecular regulation of the PAI-1
gene by hypoxia: contributions of Egr-1, HIF-1α, and C/EBPα. FASEB J. 21, 935–949 (2007).
41.
Mole, D. R. et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and
HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem.
284, 16767–16775 (2009).
42.
Xia, X. & Kung, A. L. Preferential binding of HIF-1 to transcriptionally active loci
determines cell-type specific response to hypoxia. Genome Biol. 10, R113 (2009).
43.
Xia, X. et al. Integrative analysis of HIF binding and transactivation reveals its role in
maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. U. S. A. 106, 4260–4265
(2009).
44.
Ortiz-Barahona, A., Villar, D., Pescador, N., Amigo, J. & del Peso, L. Genome-wide
identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model
integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res.
38, 2332–2345 (2010).
45.
Schödel, J. et al. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq.
Blood 117, e207–217 (2011).
46.
Villar, D. et al. Cooperativity of Stress-Responsive Transcription Factors in Core HypoxiaInducible Factor Binding Regions. PLoS ONE 7, e45708 (2012).
47.
Cummins, E. P. & Taylor, C. T. Hypoxia-responsive transcription factors. Pflügers Arch.
Eur. J. Physiol. 450, 363–371 (2005).
48.
Lau, L. F. & Nathans, D. Expression of a set of growth-related immediate early genes in
BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc. Natl. Acad. Sci. U. S. A. 84,
1182–1186 (1987).
151
Results
49.
Ryder, K. & Nathans, D. Induction of protooncogene c-jun by serum growth factors. Proc.
Natl. Acad. Sci. U. S. A. 85, 8464–8467 (1988).
50.
Hibi, M., Lin, A., Smeal, T., Minden, A. & Karin, M. Identification of an oncoprotein- and
UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev.
7, 2135–2148 (1993).
51.
Musti, A. M., Treier, M. & Bohmann, D. Reduced ubiquitin-dependent degradation of c-Jun
after phosphorylation by MAP kinases. Science 275, 400–402 (1997).
52.
Chen, K.-C., Chiou, Y.-L. & Chang, L.-S. JNK1/c-Jun and p38 alpha MAPK/ATF-2 pathways
are responsible for upregulation of Fas/FasL in human chronic myeloid leukemia K562 cells
upon exposure to Taiwan cobra phospholipase A2. J. Cell. Biochem. 108, 612–620 (2009).
53.
Morton, S., Davis, R. J., McLaren, A. & Cohen, P. A reinvestigation of the multisite
phosphorylation of the transcription factor c-Jun. EMBO J. 22, 3876–3886 (2003).
54.
Laderoute, K. R. et al. The response of c-jun/AP-1 to chronic hypoxia is hypoxia-inducible
factor 1 alpha dependent. Mol. Cell. Biol. 22, 2515–2523 (2002).
55.
Alfranca, A. et al. c-Jun and Hypoxia-Inducible Factor 1 Functionally Cooperate in
Hypoxia-Induced Gene Transcription. Mol. Cell. Biol. 22, 12–22 (2002).
56.
Yao, K. S., Xanthoudakis, S., Curran, T. & O’Dwyer, P. J. Activation of AP-1 and of a nuclear
redox factor, Ref-1, in the response of HT29 colon cancer cells to hypoxia. Mol. Cell. Biol. 14,
5997–6003 (1994).
57.
Guma, M. et al. Genetic and pharmacological inhibition of JNK ameliorates hypoxiainduced retinopathy through interference with VEGF expression. Proc. Natl. Acad. Sci. U. S. A.
106, 8760–8765 (2009).
58.
Salnikow, K. et al. The Regulation of Hypoxic Genes by Calcium Involves c-Jun/AP-1,
Which Cooperates with Hypoxia-Inducible Factor 1 in Response to Hypoxia. Mol. Cell. Biol. 22,
1734–1741 (2002).
59.
Yu, B. et al. c-Jun protects hypoxia-inducible factor-1alpha from degradation via its
oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res. 69, 7704–
7712 (2009).
60.
Ameri, K. et al. Induction of activating transcription factor 3 by anoxia is independent of
p53 and the hypoxic HIF signalling pathway. Oncogene 26, 284–289 (2006).
61.
Chen, B. P., Wolfgang, C. D. & Hai, T. Analysis of ATF3, a transcription factor induced by
physiological stresses and modulated by gadd153/Chop10. Mol. Cell. Biol. 16, 1157–1168
(1996).
62.
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of
Toll-like receptor 4. Nature 441, 173–178 (2006).
63.
Hai, T., Wolfgang, C. D., Marsee, D. K., Allen, A. E. & Sivaprasad, U. ATF3 and stress
responses. Gene Expr. 7, 321–335 (1999).
64.
Suganami, T. et al. Activating transcription factor 3 constitutes a negative feedback
mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage
activation in obese adipose tissue. Circ. Res. 105, 25–32 (2009).
65.
Janz, M. et al. Classical Hodgkin lymphoma is characterized by high constitutive
expression of activating transcription factor 3 (ATF3), which promotes viability of
Hodgkin/Reed-Sternberg cells. Blood 107, 2536–2539 (2006).
152
Results
66.
Pelzer, A. E. et al. The expression of transcription factor activating transcription factor 3
in the human prostate and its regulation by androgen in prostate cancer. J. Urol. 175, 1517–1522
(2006).
67.
Yin, X., Dewille, J. W. & Hai, T. A potential dichotomous role of ATF3, an adaptiveresponse gene, in cancer development. Oncogene 27, 2118–2127 (2008).
68.
L Turchi, E. A. Hif-2alpha mediates UV-induced apoptosis through a novel ATF3dependent death pathway. Cell Death Differ. 15, 1472–80 (2008).
69.
Tanaka, Y. et al. Systems Analysis of ATF3 in Stress Response and Cancer Reveals
Opposing Effects on Pro-Apoptotic Genes in p53 Pathway. PLoS ONE 6, e26848 (2011).
70.
Chen, S.-C., Liu, Y.-C., Shyu, K.-G. & Wang, D. L. Acute hypoxia to endothelial cells induces
activating transcription factor 3 (ATF3) expression that is mediated via nitric oxide.
Atherosclerosis 201, 281–288 (2008).
71.
Aronheim, A., Zandi, E., Hennemann, H., Elledge, S. J. & Karin, M. Isolation of an AP-1
repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17, 3094–
3102 (1997).
72.
Bitton-Worms, K., Pikarsky, E. & Aronheim, A. The AP-1 repressor protein, JDP2,
potentiates hepatocellular carcinoma in mice. Mol. Cancer 9, 54 (2010).
73.
Heinrich, R., Livne, E., Ben-Izhak, O. & Aronheim, A. The c-Jun dimerization protein 2
inhibits cell transformation and acts as a tumor suppressor gene. J. Biol. Chem. 279, 5708–5715
(2004).
74.
Yin, X., Dewille, J. W. & Hai, T. A potential dichotomous role of ATF3, an adaptiveresponse gene, in cancer development. Oncogene 27, 2118–2127 (2008).
75.
Weidenfeld-Baranboim, K. et al. The ubiquitously expressed bZIP inhibitor, JDP2,
suppresses the transcription of its homologue immediate early gene counterpart, ATF3. Nucleic
Acids Res. 37, 2194–2203 (2009).
76.
Allan, A. L., Albanese, C., Pestell, R. G. & LaMarre, J. Activating transcription factor 3
induces DNA synthesis and expression of cyclin D1 in hepatocytes. J. Biol. Chem. 276, 27272–
27280 (2001).
77.
Lu, D., Wolfgang, C. D. & Hai, T. Activating transcription factor 3, a stress-inducible gene,
suppresses Ras-stimulated tumorigenesis. J. Biol. Chem. 281, 10473–10481 (2006).
78.
Norris, M. L. & Millhorn, D. E. Hypoxia-induced protein binding to O2-responsive
sequences on the tyrosine hydroxylase gene. J. Biol. Chem. 270, 23774–23779 (1995).
79.
Song, M. S., Park, Y. K., Lee, J. H. & Park, K. Induction of glucose-regulated protein 78 by
chronic hypoxia in human gastric tumor cells through a protein kinase C-epsilon/ERK/AP-1
signaling cascade. Cancer Res. 61, 8322–8330 (2001).
80.
Bergman, M. R. et al. A functional activating protein 1 (AP-1) site regulates matrix
metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1
and JunB-FosB heterodimers. Biochem. J. 369, 485–496 (2003)
153
Discussion
and
perspectives
Discussion & Perspectives
Discussion and perspectives
1. Preeclampsia: a vascular perspective
The organization of the vascular network is the key point of a correct tissue growth or more
largely organism development, and its maintenance is of primary importance for the nutrient
supply and gas exchange in the whole organism.
Preeclampsia is a pregnancy disorder, which develops spontaneously only in the human
species and is responsible for maternal morbidity and mortality. The root cause, as well as the
later symptoms, involves alterations of the vascular system. Indeed, the triggering event rest
on a defect in vascular remodeling during the process of placentation; the perturbed blood
flow affecting the feto-placental unit leads to oxidative and nitrative stress, apoptotic and
necrotic events and the release of placental particles in the maternal circulation which activate
the endothelial system and impair vascular function, thereby affecting the whole maternal
organism during pregnancy and later maternal health. The perturbed endothelial status is
linked to the symptomatic phase of the disease, which occurs after 20 weeks of gestation and
includes hypertension and proteinuria, both defining clinically preeclampsia. In some cases
HELLP syndrome, oedema and eclampsia can complicate the clinical picture.
Despite efforts and findings of the last decades many questions about preeclampsia still
animate the scientific community and push research to go deeper into the understanding of the
causes of the disease, of its development and its repercussions on the future maternal status.
Concerning the symptomatic step of the disease, the so-called maternal syndrome, researches
were mainly concentrated on the identification of plasma factors which impair endothelial
barrier, and on the characterization of the endothelial damage.
Several studies, including ours, show that endothelial cells are responsive to plasma factors of
preeclamptic pregnancies: cultures of primary endothelial cells with preeclamptic plasma
result in increased permeability, increased expression of markers of cellular activation,
increased oxidative stress, and release of pro-inflammatory and activating molecules.
154
Discussion & Perspectives
In our project HUVEC were cultivated with plasma coming either from healthy pregnant
women or preeclamptic patients. This project was based on the idea that endothelial cells
could be used as living biosensors “receptive” to factors released in the maternal circulation
during preeclamptic pregnancies and “reactive” through modulation of the gene expression
profile.
The first step of our work was to verify whether our cellular model was responsive to
preeclamptic plasma factors: culture of HUVECs with preeclamptic plasma indeed induced an
increased permeability and the expression of markers of cellular activation, such as IL-8, IL6, ICAM, VCAM and E-Selectin in the same way as pro-inflammatory cytokines. While
working on immortalized cells may somehow temper the applicability of the findings to in
vivo situation, it presents also some clear advantages, not the least being the avoidance of
genetic variability which could influence cellular response, in a patient-dependent fashion.
Indeed some data in the literature are discordant about endothelial response to preeclamptic
plasma. It could be plausible that part of this discrepancy in results is influenced by primary
cells genetic background. And it could also be noticed that the composition of human plasma
coming from preeclamptic or normotensive patients is very variable in terms of circulating
factors concentration, and adds complexity to the experimental system. Hence the importance
to simplify the experimental system as much as possible, in order to improve the
reproducibility and reduce the variability.
Agenome wide-approach allows depicting the expressions status of endothelial cells and
tracing the pathways involved in endothelial response to preeclamptic plasma. The innovation
of this kind of approach consists in investigating the endothelial reaction in its complexity
and try to infer how the change in gene expression could participate to endothelial altered
functions.
The first finding of this study was to point out that preeclamptic plasma imposes a change in
gene expression in endothelial cells, in contrast to a previous more ancient study failing to
show strong global expressional changes in primary endothelial cells treated with
preeclamptic plasma (Donker et al., 2005). The reason of this discrepancy could lie in the
technical limitation of microarrays in 2005, or as described before, in the fact that a different
cellular model has been used for the genome wide study (HUVEC primary cells in this case).
155
Discussion & Perspectives
We found a total of 116 genes significantly modified in HUVECs treated with preeclamptic
plasma.
We showed that preeclamptic plasma acts on cell survival and on functions specific of
endothelial cells, like angiogenesis and vaso- regulation.
It was known from the literature that preeclamptic plasma stimulates cellular proliferation
(Rowe et al., 2001). We showed that this could be the result of up-regulation of genes
involved in mitosis and cell cycle progression (CCNB1, CCNB2, CDC20, CDCA8, NEK2, and
CASC5) and anti-apoptotic genes (BCL2A1, BIRC5 and ID3) and concomitant downregulation of apoptotic genes (IGFBP3, CHAC1, DDIT3). Interestingly down-regulation of
the pro-apoptotic branch of the Unfolded Protein Response (UPR) pathway (ATF4, CEBP,
CHAC1, TRIB3, XBP-1 and DDIT4) could also participate in the endothelial struggle for
survival. UPR is activated in response to overload of misfolded proteins and aim to promote
protein correct folding. Failure to this response leads to an increase of not functional proteins,
reduced protein synthesis and finally cell death.
Amongst our novel results, vaso-regulation was equally impaired by the up-regulation of
vasoactive molecules (ET-1, APLN, CBS), and the down-regulation of VEGF-A, one of the
most important mediator of proliferation, angiogenesis, permeability in endothelial cells. At
the same time endothelial cells participate to increased oxidative stress and a pro-coagulation
state through the up-regulation of NOX4, one of the main sources of reactive oxygen species
(ROS) in endothelial cells.
This study defines the response of endothelial cells after a short treatment of preeclamptic
plasma and demonstrates that endothelial cells actively participate to the maintenance of
oxidative stress, increased pro-inflammatory and pro-coagulant state and vasoconstrinction all
along pregnancy complicated by preeclampsia. Defining transcriptomic endothelial response
to preeclamptic plasma could help progressing towards the knowledge of the vascular biology
of preeclampsia, but also stimulates some reflections about endothelial damage during
pregnancy.
In particular two main questions about preeclampsia are debated by the scientific community:
when and how!
156
Discussion & Perspectives
When? At which step of the disease, does the vascular system start to suffer of an impaired
function? Is it affected even before the apparition of the clinical symptoms of hypertension
and proteinuria?
Preeclamptic symptoms only worsen all along pregnancy, and according to our data,
endothelial cells respond to third trimester plasma with a positive feedback in order to
maintain and aggravate the symptoms depicted before. In this context, it could be interesting
to set a retrospective study in order to evaluate how endothelial cells respond to the plasma
coming from early stages of pregnancy. This could offer some trails to the very first triggers
of endothelial activation and maternal symptoms and maybe help finding new early
biomarkers of the disease.
In fact one of the most challenging goals in research on preeclampsia is to define a set of
biomarkers to ensure an early diagnosis and treatment even before the clinical manifestations
of the disease. Some circulating factors have already been found modified since the first
trimester (Table 3, chapter 1), but the definition of a well characterized set of biomarkers that
would improuve the early diagnosis is still an open and stimulating field of research.
How? Our study depicted the whole genome expression profile of HUVECs exposed to
preeclamptic plasma and the functional clustering analysis helps inferring the principal
pathways involved in the endothelial response. This could be the starting point of a more
exhaustive study focused on protein level, localization and function, in order to characterize
deeper the pathways involved in endothelial dysfunction.
In fact some of the genes identified by our study are very attractive candidates with respect to
their function in the pathophysiology of preeclampsia and deserve a deeper characterization. It
is the case of VEGF-A regulation: during preeclampsia circulating VEGF-A is at least
partially inactivated by the increased expression of its soluble receptor sFlt-1, thus competing
for the binding of VEGF-A to its cognate membrane receptor, VEGFR (Maynard et al., 2003).
Here we show that endothelial cells actively participate to the lack of VEGF signaling cascade
through down-regulation of endogenous VEGF.
Interestingly VEGF reduction could be
linked to the down-regulation of the UPR pathway and specifically ATF4. In fact it has been
shown that activation of UPR pathway by oxidized phospholipids induced angiogenesis
157
Discussion & Perspectives
through the up-regulation of VEGF, mediated by ATF4 (Oskolkova et al., 2008). It could be
plausible that in our model system VEGF-shrinkage, results partly of decreased activating
properties of ATF4, albeit further studies are requested to define ATF4 possible implication in
VEGF-A down-regulation during preeclampsia.
Another interesting finding which needs a deeper investigation is the role of NOX4 in
endothelial dysfunction. Indeed we showed that it is up-regulated in endothelial cells treated
with preeclamptic plasma and this could be the result of increased plasmatic level of TNF-α.
Since NOX4 is directly involved in increased levels of reactive oxygen species and fosters
indirectly exaggerate pro-coagulant state through the regulation of expression of the
plasminogen activator -1 (SERPINE1, aka PAI-1) (Jaulmes et al., 2009), a better
understanding of its regulation and down-stream implication could help determining if NOX4
is a suitable target for therapeutic approaches.
2. Preeclampsia and beyond: the future maternal health
The importance to accurately define endothelial damage during preeclampsia is of primary
importance in order to evaluate the risks for the future maternal health. Preeclamptic
symptoms usually disappear after the delivery of the feto-placental unit. Nevertheless the
maternal vascular system seems to be inevitably compromised even several years after a
pregnancy complicated by preeclampsia.
Reduced endothelial response to vasodilators
persists even 15-25 years after such pregnancies (Lampinen et al., 2006; Ramsay et al., 2003)
and increased level of circulating TNF-α, sFLt-1 and C-reactive protein were detected in
women affected by preeclampsia even eight years after parturition (Kvehaugen et al., 2011).
Impact on future maternal health has been confirmed also by epidemiological studies showing
the high risk of hypertension and end-stage renal diseases in women previously affected by
preeclampsia several years after the delivery (Carty et al., 2010; Vikse et al., 2008). It should
be noticed that pathological conditions that increase the risk of preeclampsia, like chronic
hypertension, diabetes and chronic inflammation, are also predisposing factors for
cardiovascular disease. In this pathological context, the preeclamptic syndrome could
invalidate vascular system in a more permanent way, which could explain the higher risk of
complications for the future maternal health.
158
Discussion & Perspectives
During preeclampsia circulating damaging factors barrage continuously endothelial cells and
perturb the function of the vascular system during pregnancy. But in addition they could
impress a sort of “memory” of this prolonged stress condition through epigenetic
modifications.
Epigenetics refers to heritable changes in gene expression throughout mitosis or even meiosis,
which do not affect the cellular genetic code. Epigenetic regulation is perpetuated by three
main mechanisms: microRNA, methylation of CG rich domains (CpG islands) in DNA
sequence (usually the promoter regions) and chemical modifications of the histones tails.
The analysis of the DNA methylation status of preeclamptic placentas highlights a global
level of hypermethylation of CpG islands compared to the methylation status of normal
placentas (Kulkarni et al., 2011); the main goal is to investigate whether changes in CpG
islands methylation have an impact on gene expression, and in some cases it has been
confirmed: for example, hypomethylation of TIMP3 promoter, a gene involved in trophoblast
invasion, is associated to gene upregulation in preeclamptic placentas (Xiang et al., 2013).
Concerning the vascular system, a recent study analyzed the pattern of CpG methylation of
omental arteries from normal pregnant women and preeclamptic patients: 236 genes have
been found differentially methylated with a false discovery rate of <5%. Interestingly
functional clustering attribute these genes to important vascular functions linked to
preeclampsia, like smooth muscle contraction, thrombosis, inflammation, redox homeostasis,
sugar metabolism, and amino acid metabolism (Mousa et al., 2012). It remains to be
confirmed whether the DNA methylation rate leads to changes in gene expression, but the
most striking finding of this work is that preeclampsia could affect the epigenetic status of
endothelial cells in a deeper way than translational or transcriptional level, and that these
changes could persist and impact the future maternal health.
159
Discussion & Perspectives
3. JDP2: role in the endothelial response to preeclampsia and in
hypoxia sensing
A genome wide approach allows investigating a scientific issue without any preconception.
Results often help to confirm previous findings, but moreover it can open new paths of
research completely unsuspected at the beginning.
In our case, microarray approach confirmed that endothelial cells are responsive to factors
present in preeclamptic plasma and participates actively to vaso- regulation and oxidative
stress through the transcriptional modification of important genes involved in these functions,
like EDN1, APLN, CBS, NOX4. In parallel we showed for the first time that endothelial cells
participate to the lack of VEGF signaling cascade by down-regulation of endogenous VEGF.
Microarray also allows to identify a novel actor of endothelial response, Jun Dimerization
Protein 2 (JDP2), the gene on which we decided to focus our research. JDP2 down regulation
has been reported also in a genome-wide study on the third trimester preeclamptic placentas
(Nishizawa et al., 2007). No changes in JDP2 expression has been detected in the first
trimester placentas of women who later develop preeclampsia (Founds et al., 2009), thus
suggesting that its deregulation maybe needs a stress condition and that its implication may
play a role in the later steps of the disease.
JDP2 belongs to AP-1 family and is implicated in the regulation of important cellular
processes like proliferation, differentiation, cell cycle regulation and tumor progression. It can
acts through three main mechanisms: as transcription factor, as modulator of AP-1
transcription factors family, or as architect of chromatin folding through the recruitment of
HDACs, the inhibition of HAT activity of some proteins, and the regulation of the
methylation status of histone tails.
In our first work we showed that down-regulation of JDP2 in HUVECs cultivated with
plasma coming from normotensive pregnant women is sufficient to reproduce the
transcriptomic modifications of HUVECs cultivated with preeclamptic plasma at least for
three genes, VEGF-A, IGFBP3 and BCL2A1.
160
Discussion & Perspectives
Even though the role of IGFBP3 has not been completely characterized its expression seems
to play an important role for the placental development: in fact it inhibits trophoblast
proliferation (Karen et al., 2007) and its depletion, together with the increase in caspase-10
and death receptor 3 (DR-3) is associated to the apoptosis in preeclamptic placentas (Han et
al., 2006). Interestingly IGFBP3 decrease has been detected not only in preeclamptic
placentas and maternal circulation of women affected by preeclampsia, but it is also
associated with other pregnancy disorders such as Intrauterine Growth Restriction (IUGR)
(Verkauskiene et al., 2007) and multiple pregnancies (Langford et al., 1995); this suggests
that all those pregnancy complications share, at least in part, the alterations of some common
pathway cascade. We showed that IGFBP3 follows faithfully JDP2 expression profile and
that down-regulation of JDP2 is sufficient to down-regulate IGFBP3 as much as preeclamptic
plasma, thus suggesting a possible involvement of JDP2 in its regulation in other pregnancy
disorders as well.
BCL2A1 belongs to the BCL2 antiapoptotic family, and it has a pro-survival function by
reducing the release of cytochrome C from mitochondrial compartments and blocking caspase
activation and apoptotic signaling cascade (Vogler, 2012). It has been found modified also in
patients suffering of vascular disorders like acute coronary syndrome (Silbiger et al., 2013)
and pulmonary arterial hypertension (Pendergrass et al., 2010). It is highly expressed in the
hematopoietic system; its transcriptional activation is enhanced by pro-inflammatory stimuli
and activation of NF-κB pathway. BCL2A1 expression is inversely correlated to that of
JDP2, thus pointing out to the possibility that AP-1 signaling pathway may play an important
role in its regulation. BCL2A1 up- regulation is linked to the development of hematological
malignancies, like lymphoblastic and chronic lymphocytic leukemia and B-cell lymphoma
(Nagy et al., 2003), but also solid tumors like breast, colon, ovarian and prostate cancer (Choi
et al., 1995; Vogler, 2012); moreover its expression increased with the progression of tumor
stage, suggesting a possible implication with the more aggressive stages of the disease (Yoon
et al., 2003). Interestingly high incidence of loss of JDP2 has been detected in the later and
severe stage of prostate cancer patient and JDP2 over-expression in a PC-3 cells, a cellular
model of the advanced stage of prostate cancer, reduces cellular proliferation and tumor
formation in mice (Heinrich et al., 2004).The correlation between JDP2 and BCL2A1
expression could be a new field of investigation on the role of JDP2 in tumor progression and
161
Discussion & Perspectives
maybe clarify the role of JDP2 in cancer biology, still fluctuating between the role of
oncogene and the role of tumor suppressor.
A. JDP2: a new supervisor of endothelial hypoxic response
In the second part of my PhD work we moved from a whole genome to a targeted gene
approach in order to better characterize the role of JDP2 on VEGF regulation and the link, if
any, with hypoxia. The possible role of JDP2 on the hypoxia –dependent VEGF expression
could circumscribe JDP2 action on the process of angiogenesis during placentation, which
need both growth factors (VEGF and PlGF) and a hypoxic environment to be fully
accomplished. Moreover it can give new insights concerning the perturbed placental vascular
remodeling in the pathological context of preeclampsia, in which JDP2 was found strongly
down-regulated in third trimester preeclamptic placentas (Nishizawa et al., 2007) and
HUVEC line treated with preeclamptic plasma (Calicchio et al. 2013).
We found that JDP2 is necessary for the full hypoxia induced expression of VEGF; which
without JDP2 is roughly halved. We demonstrated that this VEGF partial induction is partly
due to reduced HIF-1 dependent activation of VEGF transcription, as shown by the reduced
HRE transcriptional activity in HUVEC lacking JDP2 expression.
Globally our study raises a lot of questions and interesting scientific insights, probably much
more than answers. Our work repositions JDP2 as a transcriptional activator (direct or
indirect?) under hypoxic conditions but not itself modified by hypoxia, and in general, as a
new molecular actor necessary for the fulfillment of cellular response to hypoxic condition.
We demonstrated that JDP2 does not impact directly HIF-1 protein stability, leaving to
speculate that its action is targeted to HIF-1 binding to HRE consensus sequence or HIF-1
transactivation. Whether JDP2 interacts directly or not with HIF-1 with some consequences
on HIF-1 DNA binding activity is one of the main questions arising from this work.
HIF-1 transactivation depends on its acetylation status and on its interaction with the coactivator p300. In the active state, acetylation at Lys- 674 by PCAF reinforces the interaction
between HIF-1α and p300 (Lim et al., 2010), which in turns, acetylates the residue Lys- 709
on HIF-1α, thus increasing protein stability. On the opposite, acetylated Lys- 532 within the
162
Discussion & Perspectives
N-terminal tail perturbs HIF-1α stability (Geng et al., 2012). Acetylation/de-acetylation
balance can also be influenced by HDAC recruitments. It is known from the literature that
JDP2 can interact with p300 and PCAF and blocks their HAT activity in vitro (Pan et al.,
2003): following this scenario we would expect mostly an increased HIF-1 activity in the case
of JDP2 down-regulation, but it is not the case. Nevertheless it could be suitable to verify the
putative interaction between JDP2 and its known partners with HAT activity involved in
hypoxia response, in order to assess if it interferes or not in the acetylation rate of the active
HIF-1 complex.
Several HDACs have been identified in the literature as regulators of HIF-1α acetylation
status, and consequently as stabilizers of HIF-1α status, like Sirtuin1 (Lim et al., 2010),
HDAC3 (Kim et al., 2007) HDAC4 (Geng et al., 2011), HDAC6 (Qian et al., 2006) and
HDAC7 (Kato et al., 2004). Intriguingly HDAC3 is a common partner of HIF-1 and JDP2
(Kim et al., 2001, 2007), thus stimulating the interest to go deeper in a possible role of JDP2
as HDAC3 recruiter and indirectly HIF-1 trans-activator.
So far, HDACs implication in angiogenesis during tumor development stimulates the interest
to go further on a possible link between HDACs, hypoxia and JDP2. Hypoxia is a driving
force for angiogenesis since it stimulates VEGF expression and its receptor VEGFR-1
(Forsythe et al., 1996; Gerber et al., 1997). Recently it has been shown that other proteins,
which equally promotes angiogenesis, are targeted by HIF-1α, like angiopoietin 2 (Simon et
al., 2008), Stem Cell Factor, (Han et al., 2008) and semaphoring 4D (Sun et al., 2009). The
implication of hypoxia in tumor angiogenesis is the subjects of interest of many scientific
works: more than 3000 papers deals with hypoxia, angiogenesis and tumor development.
Nevertheless it should be taken into account that hypoxia-induced angiogenesis occurs even
in important physiological processes like placentation, in which the field of research seems to
be less developed.
Tumor growth is usually associated with a hypoxic environment and needs the
accomplishment of an angiogenic program to develop. In this context, HIF-1α has been
found increased in several tumors together with VEGF (Brahimi-Horn and Pouysségur, 2005;
Zhong et al., 1998), and is an important stimulator of vascular plasticity during tumor
progression. HDAC1 induces the expression of VEGF and HIF-1α through down-regulation
163
Discussion & Perspectives
of two important tumor suppressors, p53 and pVHL (Kim et al., 2001); HDACs inhibitor, like
Trichostatin (TSA), blocks the VEGF expression and its receptors and counterbalance the
pro-angiogenic activity of VEGF by the expression of its competitor semaphorin II (Deroanne
et al., 2002). All these findings guide research through the development of HDACs inhibitors
as anti-cancer therapy by targeting specifically tumor angiogenesis.
HDACs implication in angiogenesis and placentation is still a virgin field of research: at now
it has been shown that class II HDACs (HDAC 4, 5, 6, 7, 9 and 10) together with HIF-1α and
HIF-1β are necessary to the correct trophoblast differentiation, but their role all along
placentation has still not been established. Interestingly HIF-1β deficient trophoblast stem
cells show impaired differentiation (almost restricted to spongiotrophoblasts and giant cells),
HDACs mislocalization, and hyperacetylated histones. The same altered profile could be
reproduced by blocking HDACs, thus suggesting an active cooperation between HDACs and
HIF factors in order to have the best differentiation performance (Maltepe et al., 2005).
Since JDP2 is implicated in tumor progression, VEGF regulation, and HDAC recruitment, it
could be a suitable candidate as an active actor in hypoxia induced angiogenesis in
physiological and pathological outcomes.
B. JDP2 involvement in the transcriptional modification of AP-1
members under hypoxic condition
More and more evidences point to the possibility that the cellular response to hypoxia results
from the formation of a multi-protein complex which acts as an enhanceosome headed by HIF
dimers which aims to organize cellular response (Schödel et al., 2011; Xia and Kung, 2009).
JDP2 is an important regulator of AP-1 members and AP-1 protein family is involved in the
cellular response to hypoxia, we evaluated the potential transcriptional modifications of AP-1
members in HUVECs under hypoxic conditions, with and without JDP2: we demonstrated
that lack of JDP2 interferes with the hypoxia-dependent expression of ATF3. It has been
shown that both JDP2 and ATF3 could bind ATF3 promoter at the level of a CRE element
and a non canonical ATF/CRE elements, and that they inhibit ATF3 expression through the
recruitment of multiple HDACs on ATF3 promoter (Darlyuk-Saadon et al., 2012). Here we
164
Discussion & Perspectives
showed for the first time that JDP2 could have some role other than transcriptional repressor
and that its expression is necessary for ATF3 induction under hypoxic condition. Moreover it
has been shown that under anoxic condition ATF3 is more strongly expressed, and activation
of transcription does not depend on HIF-1 but needs the activation of c-Jun NH2-terminal
kinase (Ameri et al., 2006). This suggests that AP-1 family is necessary to ATF3 response to
hypoxia, and we demonstrated that JDP2 could be the AP-1 member involved in ATF3
transcriptional induction. Further studies are needed to define JDP2 action as direct or indirect
and its molecular partners.
The high degree of homology in the bZIP domain between JDP2 and ATF3 is probably linked
to their redundant action in term of their protein partners, their function as transcriptional
activator or repressor and their function of HDACs recruiters. Similarity refers also to their
double nature of repressor or activatior : ATF3 may play as inhibitor of transcription by
forming homodimers, or activator by forming heterodimers with c-Jun (Hai and Curran,
1991). Because of c-Jun implication in HIF-1 and VEGF regulation, ATF3 depletion may
impair c-Jun activating property an probably participate to the partial loss of
VEGF
expression in HUVECs lacking JDP2 under the hypoxic stress. All these findings led to
speculate about a co-participation of JDP2 and ATF3 in the hypoxia response regulation via
AP-1 members interaction and stimulate the interest on a further characterization of their
possible regulation of HIF-1 transcriptional activity through a direct or indirect interaction.
C. A Possible role of JDP2 on VEGF expression mediated by histone
modifications
Gene expression is the result of interaction of transcription factors and DNA, organized in a
permissive chromatin structure. Accessible chromatin results from post- translational
modifications in histone tails which are associated to activation of transcription.
Hypoxia imposes a modification in gene expression which ensures the cellular survival in
presence of a stress condition. A recent work has shown that hypoxic conditions are
responsible for a global reduction in transcription (Johnson et al., 2008). Globally hypoxic
condition induces also changes in histone modifications associated with either transcriptional
activation or inhibition. In fact hypoxia stimulates the increase in H3K9 methylation and a
165
Discussion & Perspectives
decrease in H3K9acetylation which could be linked to the observed inhibition of
transcription. In parallel other marks of activation of transcription are increased as well, like
increased di- and tri-methylation of H3K4 and increased acetylation of H3K14 (Chen et al.,
2006; Johnson et al., 2008; Tausendschön et al., 2011; Xia and Kung, 2009; Xia et al., 2009).
If we specifically consider VEGF epigenetic marks induced by the hypoxic environment, we
could find a correlation between increased gene expression under hypoxic condition and
“permissive” histone modifications, like increase in tri-metyhylation of H3K4, increased
histone H3 acetylation, and decreased tri-methylation of H3K27 (Johnson et al., 2008; Jung
et al., 2005).
Decreased of H3K27 tri-methylation has been correlated with induced expression of p16 and
p19 during replicative senescence. In this case JDP2 plays a fundamental role in chromatin
remodeling and gene expression: in fact under stress condition JDP2 blocks the interaction of
Polycomb repressive complex (PRC) on p16 and p19 promoter thus preventing methylation of
H3K27 and allowing the expression of p16 and p19, provoking cellular growth arrest and
cellular senescence (Nakade et al., 2009). Blockade of JDP2 in mouse embryonic fibloblasts
(MEFs) results in reduced p16 expression and escape from replicative senescence. According
to the model proposed by Nakade and colleagues JDP2 protects histone tails from methylation
by masking methylation sites and participates in maintaining an open chromatin state,
permissive to transcription.
In our system, we could imagine that JDP2 has a similar effect on VEGF promoter, since it is
known from the literature that increased VEGF level under hypoxic condition is associated
with reduce methylation level of H3K27. Further confirmations will be required to evaluate
the methylation status of H3K27 on VEGF promoter in our cellular model.
166
Conclusion
Conclusion
This PhD work aimed to characterize the transcriptional endothelial response induced by
preeclamptic plasma. We demonstrate that endothelial cells react to the stress induced by
preeclamptic plasma through a change in gene expression which produces important vascular
functions like vasoregulation, oxidative stress and coagulant state.
Among modified genes, we decided to focus on JDP2 and characterize its role in hypoxia
induced VEGF expression. We demonstrate that JDP2 expression is necessary for a full
cellular response to oxygen penury, and in particular that JDP2 endorses HIF-1 activating
properties under hypoxic condition in endothelial cells.
These findings open new fields of investigation on the molecular mechanism through which
JDP2 may participate to cellular adaptation in case of oxygen shortage and moreover on its
implication in all physiological and pathological conditions in which oxygen tension is an
important stimulator, such as cellular differentiation and cellular proliferation. It also
encourages revisiting the mechanisms by which hypoxia influence gene expression and
suggests that novel actors can be identified in this well-studied cascade.
167
Bibliography
Bibliography
Bibliography
Abrahams, V.M., Kim, Y.M., Straszewski, S.L., Romero, R., and Mor, G. (2004). Macrophages
and apoptotic cell clearance during pregnancy. Am. J. Reprod. Immunol. New York N 1989 51, 275–
282.
ACOG Committee on Practice Bulletins--Obstetrics (2002). ACOG practice bulletin. Diagnosis
and management of preeclampsia and eclampsia. Number 33, January 2002. Obstet. Gynecol. 99,
159–167.
Ahmed, A., Singh, J., Khan, Y., Seshan, S.V., and Girardi, G. (2010). A new mouse model to
explore therapies for preeclampsia. PloS One 5, e13663.
Ahringer, J. (2000). NuRD and SIN3 histone deacetylase complexes in development. Trends
Genet. TIG 16, 351–356.
Akolekar, R., de Cruz, J., Foidart, J.-M., Munaut, C., and Nicolaides, K.H. (2010). Maternal
plasma soluble fms-like tyrosine kinase-1 and free vascular endothelial growth factor at 11 to 13
weeks of gestation in preeclampsia. Prenat. Diagn. 30, 191–197.
Albertsson, P., Kim, M.H., Jonges, L.E., Kitson, R.P., Kuppen, P.J., Johansson, B.R., Nannmark,
U., and Goldfarb, R.H. (2000). Matrix metalloproteinases of human NK cells. Vivo Athens Greece 14,
269–276.
Alexander, B.T., Kassab, S.E., Miller, M.T., Abram, S.R., Reckelhoff, J.F., Bennett, W.A., and
Granger, J.P. (2001). Reduced uterine perfusion pressure during pregnancy in the rat is associated with
increases in arterial pressure and changes in renal nitric oxide. Hypertension 37, 1191–1195.
Alfranca, A., Gutiérrez, M.D., Vara, A., Aragonés, J., Vidal, F., and Landázuri, M.O. (2002). cJun and Hypoxia-Inducible Factor 1 Functionally Cooperate in Hypoxia-Induced Gene Transcription.
Mol. Cell. Biol. 22, 12–22.
Alroy, I., Soussan, L., Seger, R., and Yarden, Y. (1999). Neu differentiation factor stimulates
phosphorylation and activation of the Sp1 transcription factor. Mol. Cell. Biol. 19, 1961–1972.
Amburgey, O.A., Chapman, A.C., May, V., Bernstein, I.M., and Cipolla, M.J. (2010). Plasma
from preeclamptic women increases blood-brain barrier permeability: role of vascular endothelial
growth factor signaling. Hypertension 56, 1003–1008.
Ameri, K., Hammond, E.M., Culmsee, C., Raida, M., Katschinski, D.M., Wenger, R.H., Wagner,
E., Davis, R.J., Hai, T., Denko, N., et al. (2006). Induction of activating transcription factor 3 by
anoxia is independent of p53 and the hypoxic HIF signalling pathway. Oncogene 26, 284–289.
Andrews, P.M. (1981). Investigations of cytoplasmic contractile and cytoskeletal elements in the
kidney glomerulus. Kidney Int. 20, 549–562.
Angel, P., and Karin, M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation
and transformation. Biochim. Biophys. Acta 1072, 129–157.
168
Bibliography
Anim-Nyame, N., Gamble, J., Sooranna, S.R., Johnson, M.R., and Steer, P.J. (2003).
Microvascular permeability is related to circulating levels of tumour necrosis factor-alpha in preeclampsia. Cardiovasc. Res. 58, 162–169.
Arany, Z., Huang, L.E., Eckner, R., Bhattacharya, S., Jiang, C., Goldberg, M.A., Bunn, H.F., and
Livingston, D.M. (1996). An essential role for p300/CBP in the cellular response to hypoxia. Proc.
Natl. Acad. Sci. U. S. A. 93, 12969–12973.
Ark, M., Yilmaz, N., Yazici, G., Kubat, H., and Aktaş, S. (2005). Rho-associated protein kinase II
(rock II) expression in normal and preeclamptic human placentas. Placenta 26, 81–84.
Arngrímsson, R., Sigurõardóttir, S., Frigge, M.L., Bjarnadóttir, R.I., Jónsson, T., Stefánsson, H.,
Baldursdóttir, A., Einarsdóttir, A.S., Palsson, B., Snorradóttir, S., et al. (1999). A genome-wide scan
reveals a maternal susceptibility locus for pre-eclampsia on chromosome 2p13. Hum. Mol. Genet. 8,
1799–1805.
Aronheim, A., Zandi, E., Hennemann, H., Elledge, S.J., and Karin, M. (1997). Isolation of an AP1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17, 3094–
3102.
Arribas, S.M., Hinek, A., and González, M.C. (2006). Elastic fibres and vascular structure in
hypertension. Pharmacol. Ther. 111, 771–791.
Ashur-Fabian, O., Yerushalmi, G.M., Mazaki-Tovi, S., Steinberg, D.M., Goldshtein, I.,
Yackobovitch-Gavan, M., Schiff, E., Amariglio, N., and Rechavi, G. (2012). Cell free expression of
hif1α and p21 in maternal peripheral blood as a marker for preeclampsia and fetal growth restriction.
PloS One 7, e37273.
Auer, J., Camoin, L., Guillonneau, F., Rigourd, V., Chelbi, S.T., Leduc, M., Laparre, J., Mignot,
T.-M., and Vaiman, D. (2010). Serum profile in preeclampsia and intra-uterine growth restriction
revealed by iTRAQ technology. J. Proteomics 73, 1004–1017.
August, P., Lenz, T., Ales, K.L., Druzin, M.L., Edersheim, T.G., Hutson, J.M., Müller, F.B.,
Laragh, J.H., and Sealey, J.E. (1990). Longitudinal study of the renin-angiotensin-aldosterone system
in hypertensive pregnant women: deviations related to the development of superimposed
preeclampsia. Am. J. Obstet. Gynecol. 163, 1612–1621.
Ayala, D.E., Ucieda, R., and Hermida, R.C. (2012). Chronotherapy With Low-Dose Aspirin for
Prevention of Complications in Pregnancy. Chronobiol. Int.
Bachour, A., Teramo, K., Hiilesmaa, V., and Maasilta, P. (2008). Increased plasma levels of
inflammatory markers and upper airway resistance during sleep in pre-eclampsia. Sleep Med. 9, 667–
674.
Bakhti, A., and Vaiman, D. (2011). Prevention of gravidic endothelial hypertension by aspirin
treatment administered from the 8th week of gestation. Hypertens. Res. Off. J. Jpn. Soc. Hypertens.
34, 1116–1120.
169
Bibliography
Ballegeer, V.C., Spitz, B., De Baene, L.A., Van Assche, A.F., Hidajat, M., and Criel, A.M.
(1992). Platelet activation and vascular damage in gestational hypertension. Am. J. Obstet. Gynecol.
166, 629–633.
Ballegeer, V.C., Spitz, B., De Baene, L.A., Van Assche, A.F., Hidajat, M., and Criel, A.M.
(1992). Platelet activation and vascular damage in gestational hypertension. Am. J. Obstet. Gynecol.
166, 629–633.
Banerjee, S., Smallwood, A., Nargund, G., and Campbell, S. (2002). Placental morphogenesis in
pregnancies with Down’s syndrome might provide a clue to pre-eclampsia. Placenta 23, 172–174.
Banzola, I., Farina, A., Concu, M., Sekizawa, A., Purwosunu, Y., Strada, I., Arcelli, D.,
Simonazzi, G., Caramelli, E., and Rizzo, N. (2007). Performance of a panel of maternal serum markers
in predicting preeclampsia at 11-15 weeks’ gestation. Prenat. Diagn. 27, 1005–1010.
Bartoli, M., Platt, D., Lemtalsi, T., Gu, X., Brooks, S.E., Marrero, M.B., and Caldwell, R.B.
(2003). VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 17, 1562–
1564.
Barton, J.R., and Sibai, B.M. (2008). Prediction and prevention of recurrent preeclampsia. Obstet.
Gynecol. 112, 359–372.
Baumann, M.U., Bersinger, N.A., Mohaupt, M.G., Raio, L., Gerber, S., and Surbek, D.V. (2008).
First-trimester serum levels of soluble endoglin and soluble fms-like tyrosine kinase-1 as firsttrimester markers for late-onset preeclampsia. Am. J. Obstet. Gynecol. 199, 266.e1–6.
Beckman, J.S., and Koppenol, W.H. (1996). Nitric oxide, superoxide, and peroxynitrite: the good,
the bad, and ugly. Am. J. Physiol. 271, C1424–1437.
Beckmann, I., Visser, W., Struijk, P.C., van Dooren, M., Glavimans, J., and Wallenburg, H.C.
(1997). Circulating bioactive tumor necrosis factor-alpha, tumor necrosis factor-alpha receptors,
fibronectin, and tumor necrosis factor-alpha inducible cell adhesion molecule VCAM-1 in
uncomplicated pregnancy. Am. J. Obstet. Gynecol. 177, 1247–1252.
Beitelshees, A.L., Navare, H., Wang, D., Gong, Y., Wessel, J., Moss, J.I., Langaee, T.Y., CooperDeHoff, R.M., Sadee, W., Pepine, C.J., et al. (2009). CACNA1C gene polymorphisms, cardiovascular
disease outcomes, and treatment response. Circ. Cardiovasc. Genet. 2, 362–370.
Belfort, M.A. (1992). The effect of magnesium sulphate on blood flow velocity in the maternal
retina in mild pre-eclampsia: a preliminary colour flow Doppler study. Br. J. Obstet. Gynaecol. 99,
641–645.
Bell, E.L., and Guarente, L. (2011). The SirT3 divining rod points to oxidative stress. Mol. Cell
42, 561–568.
Belo, L., Santos-Silva, A., Caslake, M., Pereira-Leite, L., Quintanilha, A., and Rebelo, I. (2005).
Oxidized-LDL levels in normal and pre-eclamptic pregnancies: contribution of LDL particle size.
Atherosclerosis 183, 185–186.
Benckert, C., Jonas, S., Cramer, T., Marschall, Z. von, Schäfer, G., Peters, M., Wagner, K.,
Radke, C., Wiedenmann, B., Neuhaus, P., et al. (2003). Transforming Growth Factor β1 Stimulates
170
Bibliography
Vascular Endothelial Growth Factor Gene Transcription in Human Cholangiocellular Carcinoma
Cells. Cancer Res. 63, 1083–1092.
Benirschke, K. (1973). The human placenta. J. D. Boyd and W. J. Hamilton. Heffer, Cambridge,
365 pp. 1970. Teratology 8, 77–78.
Bhattacharya, S., Michels, C.L., Leung, M.K., Arany, Z.P., Kung, A.L., and Livingston, D.M.
(1999). Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1.
Genes Dev. 13, 64–75.
Bhaumik, S.R., Smith, E., and Shilatifard, A. (2007). Covalent modifications of histones during
development and disease pathogenesis. Nat. Struct. Mol. Biol. 14, 1008–1016.
Bills, V.L., Salmon, A.H., Harper, S.J., Overton, T.G., Neal, C.R., Jeffery, B., Soothill, P.W., and
Bates, D.O. (2011). Impaired vascular permeability regulation caused by the VEGF₁₆₅b splice variant
in pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 118, 1253–1261.
Blagosklonny, M.V., An, W.G., Romanova, L.Y., Trepel, J., Fojo, T., and Neckers, L. (1998).
p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 273, 11995–11998.
Blazek, E., Wasmer, S., Kruse, U., Aronheim, A., Aoki, M., and Vogt, P.K. (2003). Partial
oncogenic transformation of chicken embryo fibroblasts by Jun dimerization protein 2, a negative
regulator of TRE- and CRE-dependent transcription. Oncogene 22, 2151–2159.
Bosco, C., González, J., Gutiérrez, R., Parra-Cordero, M., Barja, P., and Rodrigo, R. (2012).
Oxidative damage to pre-eclamptic placenta: immunohistochemical expression of VEGF, nitrotyrosine
residues and von Willebrand factor. J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med.
Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 25, 2339–2345.
Bosio, P.M., Cannon, S., McKenna, P.J., O’Herlihy, C., Conroy, R., and Brady, H. (2001).
Plasma P-selectin is elevated in the first trimester in women who subsequently develop pre-eclampsia.
BJOG Int. J. Obstet. Gynaecol. 108, 709–715.
Bossy-Wetzel, E., Bakiri, L., and Yaniv, M. (1997). Induction of apoptosis by the transcription
factor c-Jun. EMBO J. 16, 1695–1709.
Boulanger, C., and Lüscher, T.F. (1990). Release of endothelin from the porcine aorta. Inhibition
by endothelium-derived nitric oxide. J. Clin. Invest. 85, 587–590.
Brahimi-Horn, M.C., and Pouysségur, J. (2005). The hypoxia-inducible factor and tumor
progression along the angiogenic pathway. Int. Rev. Cytol. 242, 157–213.
Brosens, I., Robertson, W.B., and Dixon, H.G. (1967). The physiological response of the vessels
of the placental bed to normal pregnancy. J. Pathol. Bacteriol. 93, 569–579.
Brown, C.D., Barnes, K., and Turner, A.J. (2000). Functional significance of the isoforms of
endothelin-converting enzyme-1. J. Cardiovasc. Pharmacol. 36, S26–27.
Brown, M.A., Lindheimer, M.D., de Swiet, M., Van Assche, A., and Moutquin, J.M. (2001). The
classification and diagnosis of the hypertensive disorders of pregnancy: statement from the
International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertens. Pregnancy Off.
J. Int. Soc. Study Hypertens. Pregnancy 20, IX–XIV.
171
Bibliography
Bruick, R.K., and McKnight, S.L. (2001). A conserved family of prolyl-4-hydroxylases that
modify HIF. Science 294, 1337–1340.
Buhimschi, I.A., Saade, G.R., Chwalisz, K., and Garfield, R.E. (1998). The nitric oxide pathway
in pre-eclampsia: pathophysiological implications. Hum. Reprod. Update 4, 25–42.
Burton, G.J., and Caniggia, I. (2001). Hypoxia: implications for implantation to delivery-a
workshop report. Placenta 22 Suppl A, S63–65.
Burton, G.J., Hempstock, J., and Jauniaux, E. (2003). Oxygen, early embryonic metabolism and
free radical-mediated embryopathies. Reprod. Biomed. Online 6, 84–96.
Burton, G.J., Jauniaux, E., and Watson, A.L. (1999). Maternal arterial connections to the
placental intervillous space during the first trimester of human pregnancy: the Boyd collection
revisited. Am. J. Obstet. Gynecol. 181, 718–724.
Buurma, A., Cohen, D., Veraar, K., Schonkeren, D., Claas, F.H., Bruijn, J.A., Bloemenkamp,
K.W., and Baelde, H.J. (2012). Preeclampsia is characterized by placental complement dysregulation.
Hypertension 60, 1332–1337.
Buurma, A.J., Turner, R.J., Driessen, J.H.M., Mooyaart, A.L., Schoones, J.W., Bruijn, J.A.,
Bloemenkamp, K.W.M., Dekkers, O.M., and Baelde, H.J. (2013). Genetic variants in pre-eclampsia: a
meta-analysis. Hum. Reprod. Update 19, 289–303.
Caniggia, I., Grisaru-Gravnosky, S., Kuliszewsky, M., Post, M., and Lye, S.J. (1999). Inhibition
of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic
pregnancies. J. Clin. Invest. 103, 1641–1650.
Caniggia, I., Grisaru-Gravnosky, S., Kuliszewsky, M., Post, M., and Lye, S.J. (1999). Inhibition
of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic
pregnancies. J. Clin. Invest. 103, 1641–1650.
Caniggia, I., Mostachfi, H., Winter, J., Gassmann, M., Lye, S.J., Kuliszewski, M., and Post, M.
(2000). Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast
differentiation through TGFbeta(3). J. Clin. Invest. 105, 577–587.
Caniggia, I., Mostachfi, H., Winter, J., Gassmann, M., Lye, S.J., Kuliszewski, M., and Post, M.
(2000a). Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast
differentiation through TGFbeta(3). J. Clin. Invest. 105, 577–587.
Caniggia, I., Winter, J., Lye, S.J., and Post, M. (2000b). Oxygen and placental development
during the first trimester: implications for the pathophysiology of pre-eclampsia. Placenta 21 Suppl A,
S25–30.
Carbia-Nagashima, A., Gerez, J., Perez-Castro, C., Paez-Pereda, M., Silberstein, S., Stalla, G.K.,
Holsboer, F., and Arzt, E. (2007). RSUME, a small RWD-containing protein, enhances SUMO
conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131, 309–323.
Carbillon, L. (2012). First trimester uterine artery Doppler for the prediction of preeclampsia and
foetal growth restriction. J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia
Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 25, 877–883.
172
Bibliography
Carbillon, L. (2012). First trimester uterine artery Doppler for the prediction of preeclampsia and
foetal growth restriction. J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia
Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 25, 877–883.
Carlino, C., Stabile, H., Morrone, S., Bulla, R., Soriani, A., Agostinis, C., Bossi, F., Mocci, C.,
Sarazani, F., Tedesco, F., et al. (2008). Recruitment of circulating NK cells through decidual tissues: a
possible mechanism controlling NK cell accumulation in the uterus during early pregnancy. Blood
111, 3108–3115.
Carmeliet, P., Moons, L., Luttun, A., Vincenti, V., Compernolle, V., De Mol, M., Wu, Y., Bono,
F., Devy, L., Beck, H., et al. (2001). Synergism between vascular endothelial growth factor and
placental growth factor contributes to angiogenesis and plasma extravasation in pathological
conditions. Nat. Med. 7, 575–583.
Carr, D.B., Epplein, M., Johnson, C.O., Easterling, T.R., and Critchlow, C.W. (2005). A sister’s
risk: family history as a predictor of preeclampsia. Am. J. Obstet. Gynecol. 193, 965–972.
Carson, D.D., Bagchi, I., Dey, S.K., Enders, A.C., Fazleabas, A.T., Lessey, B.A., and Yoshinaga,
K. (2000). Embryo implantation. Dev. Biol. 223, 217–237.
Carty, D.M., Delles, C., and Dominiczak, A.F. (2010). Preeclampsia and future maternal health.
J. Hypertens. 28, 1349–1355.
Cascio, S., D’Andrea, A., Ferla, R., Surmacz, E., Gulotta, E., Amodeo, V., Bazan, V., Gebbia, N.,
and Russo, A. (2010). miR-20b modulates VEGF expression by targeting HIF-1α and STAT3 in
MCF-7 breast cancer cells. J. Cell. Physiol. 224, 242–249.
Castellucci, M., Scheper, M., Scheffen, I., Celona, A., and Kaufmann, P. (1990). The
development of the human placental villous tree. Anat. Embryol. (Berl.) 181, 117–128.
Caughey, A.B., Stotland, N.E., Washington, A.E., and Escobar, G.J. (2005). Maternal ethnicity,
paternal ethnicity, and parental ethnic discordance: predictors of preeclampsia. Obstet. Gynecol. 106,
156–161.
Cerón-Mireles, P., Harlow, S.D., Sánchez-Carrillo, C.I., and Núñez, R.M. (2001). Risk factors for
pre-eclampsia/eclampsia among working women in Mexico City. Paediatr. Perinat. Epidemiol. 15, 40–
46.
Chafetz, I., Kuhnreich, I., Sammar, M., Tal, Y., Gibor, Y., Meiri, H., Cuckle, H., and Wolf, M.
(2007). First-trimester placental protein 13 screening for preeclampsia and intrauterine growth
restriction. Am. J. Obstet. Gynecol. 197, 35.e1–7.
Chaiworapongsa, T., Romero, R., Espinoza, J., Bujold, E., Mee Kim, Y., Gonçalves, L.F.,
Gomez, R., and Edwin, S. (2004). Evidence supporting a role for blockade of the vascular endothelial
growth factor system in the pathophysiology of preeclampsia. Young Investigator Award. Am. J.
Obstet. Gynecol. 190, 1541–1547; discussion 1547–1550.
Chaiworapongsa, T., Romero, R., Kim, Y.M., Kim, G.J., Kim, M.R., Espinoza, J., Bujold, E.,
Gonçalves, L., Gomez, R., Edwin, S., et al. (2005). Plasma soluble vascular endothelial growth factor
receptor-1 concentration is elevated prior to the clinical diagnosis of pre-eclampsia. J. Matern.-Fetal
173
Bibliography
Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat.
Obstet. 17, 3–18.
Chaiworapongsa, T., Romero, R., Yoshimatsu, J., Espinoza, J., Kim, Y.M., Park, K., Kalache, K.,
Edwin, S., Bujold, E., and Gomez, R. (2002). Soluble adhesion molecule profile in normal pregnancy
and pre-eclampsia. J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean.
Perinat. Soc. Int. Soc. Perinat. Obstet. 12, 19–27.
Chan, J.C.Y., Knudson, O., Wu, F., Morser, J., Dole, W.P., and Wu, Q. (2005). Hypertension in
mice lacking the proatrial natriuretic peptide convertase corin. Proc. Natl. Acad. Sci. U. S. A. 102,
785–790.
Chatterjee, P., Chiasson, V.L., Kopriva, S.E., Young, K.J., Chatterjee, V., Jones, K.A., and
Mitchell, B.M. (2011). Interleukin 10 deficiency exacerbates toll-like receptor 3-induced
preeclampsia-like symptoms in mice. Hypertension 58, 489–496.
Chavarría, M.E., Lara-González, L., García-Paleta, Y., Vital-Reyes, V.S., and Reyes, A. (2008).
Adhesion molecules changes at 20 gestation weeks in pregnancies complicated by preeclampsia. Eur.
J. Obstet. Gynecol. Reprod. Biol. 137, 157–164.
Chavarría, M.E., Lara-González, L., González-Gleason, A., Sojo, I., and Reyes, A. (2002).
Maternal plasma cellular fibronectin concentrations in normal and preeclamptic pregnancies: a
longitudinal study for early prediction of preeclampsia. Am. J. Obstet. Gynecol. 187, 595–601.
Chavez, J.C., and LaManna, J.C. (2002). Activation of hypoxia-inducible factor-1 in the rat
cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J.
Neurosci. Off. J. Soc. Neurosci. 22, 8922–8931.
Chelbi, S.T., Veitia, R.A., and Vaiman, D. (sous presse). Why Preeclampsia still exists? Med.
Hypotheses.
Chelbi, S.T., Wilson, M.L., Veillard, A.-C., Ingles, S.A., Zhang, J., Mondon, F., GascoinLachambre, G., Doridot, L., Mignot, T.-M., Rebourcet, R., et al. (2012). Genetic and epigenetic
mechanisms collaborate to control SERPINA3 expression and its association with placental diseases.
Hum. Mol. Genet. 21, 1968–1978.
Chen, H., Yan, Y., Davidson, T.L., Shinkai, Y., and Costa, M. (2006). Hypoxic stress induces
dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer
Res. 66, 9009–9016.
Chen, R., Dioum, E.M., Hogg, R.T., Gerard, R.D., and Garcia, J.A. (2011). Hypoxia increases
sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 286, 13869–
13878.
Chen, T.-H., Wang, J.-F., Chan, P., and Lee, H.-M. (2005). Angiotensin II stimulates hypoxiainducible factor 1alpha accumulation in glomerular mesangial cells. Ann. N. Y. Acad. Sci. 1042, 286–
293.
Choi, S.S., Park, I.C., Yun, J.W., Sung, Y.C., Hong, S.I., and Shin, H.S. (1995). A novel Bcl-2
related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow.
Oncogene 11, 1693–1698.
174
Bibliography
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M.L., Rehman, M., Walther, T.C., Olsen, J.V., and
Mann, M. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular
functions. Science 325, 834–840.
Chowdhury, R., Hardy, A., and Schofield, C.J. (2008). The human oxygen sensing machinery and
its manipulation. Chem. Soc. Rev. 37, 1308–1319.
Churchill, D., and Duley, L. (2002). Interventionist versus expectant care for severe preeclampsia before term. Cochrane Database Syst. Rev. CD003106.
Cipolla, M.J. (2007). Cerebrovascular Function in Pregnancy and Eclampsia. Hypertension 50,
14–24.
Clark, B.A., Halvorson, L., Sachs, B., and Epstein, F.H. (1992). Plasma endothelin levels in
preeclampsia: elevation and correlation with uric acid levels and renal impairment. Am. J. Obstet.
Gynecol. 166, 962–968.
Clowse, M.E.B., Jamison, M., Myers, E., and James, A.H. (2008). A national study of the
complications of lupus in pregnancy. Am. J. Obstet. Gynecol. 199, 127.e1–127.e6.
Compernolle, V., Brusselmans, K., Acker, T., Hoet, P., Tjwa, M., Beck, H., Plaisance, S., Dor,
Y., Keshet, E., Lupu, F., et al. (2002). Loss of HIF-2alpha and inhibition of VEGF impair fetal lung
maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat.
Med. 8, 702–710.
Conde-Agudelo, A., Althabe, F., Belizán, J.M., and Kafury-Goeta, A.C. (1999). Cigarette
smoking during pregnancy and risk of preeclampsia: a systematic review. Am. J. Obstet. Gynecol.
181, 1026–1035.
Conrad, K.P., Gandley, R.E., Ogawa, T., Nakanishi, S., and Danielson, L.A. (1999). Endothelin
mediates renal vasodilation and hyperfiltration during pregnancy in chronically instrumented
conscious rats. Am. J. Physiol. 276, F767–776.
Coonrod, D.V., Hickok, D.E., Zhu, K., Easterling, T.R., and Daling, J.R. (1995). Risk factors for
preeclampsia in twin pregnancies: a population-based cohort study. Obstet. Gynecol. 85, 645–650.
Cooper, J.C., Sharkey, A.M., McLaren, J., Charnock-Jones, D.S., and Smith, S.K. (1995).
Localization of vascular endothelial growth factor and its receptor, flt, in human placenta and decidua
by immunohistochemistry. J. Reprod. Fertil. 105, 205–213.
Cowden Dahl, K.D., Fryer, B.H., Mack, F.A., Compernolle, V., Maltepe, E., Adelman, D.M.,
Carmeliet, P., and Simon, M.C. (2005). Hypoxia-inducible factors 1alpha and 2alpha regulate
trophoblast differentiation. Mol. Cell. Biol. 25, 10479–10491.
Craven, C.M., Morgan, T., and Ward, K. (1998). Decidual spiral artery remodelling begins before
cellular interaction with cytotrophoblasts. Placenta 19, 241–252.
Crews, J.K., Herrington, J.N., Granger, J.P., and Khalil, R.A. (2000). Decreased EndotheliumDependent Vascular Relaxation During Reduction of Uterine Perfusion Pressure in Pregnant Rat.
Hypertension 35, 367–372.
175
Bibliography
Croken, M.M., Nardelli, S.C., and Kim, K. (2012). Chromatin modifications, epigenetics, and
how protozoan parasites regulate their lives. Trends Parasitol. 28, 202–213.
Cruickshank, J.K., and Beevers, D.G. (1982). Epidemiology of hypertension: blood pressure in
blacks and whites. Clin. Sci. Lond. Engl. 1979 62, 1–6.
Cui, X.-L., Brockman, D., Campos, B., and Myatt, L. (2006). Expression of NADPH oxidase
isoform 1 (Nox1) in human placenta: involvement in preeclampsia. Placenta 27, 422–431.
Cui, Y., Wang, W., Dong, N., Lou, J., Srinivasan, D.K., Cheng, W., Huang, X., Liu, M., Fang, C.,
Peng, J., et al. (2012). Role of corin in trophoblast invasion and uterine spiral artery remodelling in
pregnancy. Nature 484, 246–250.
Dai, B., Liu, T., Zhang, B., Zhang, X., and Wang, Z. (2013). The polymorphism for endothelial
nitric oxide synthase gene, the level of nitric oxide and the risk for pre-eclampsia: a meta-analysis.
Gene 519, 187–193.
Daikoku, T., Cha, J., Sun, X., Tranguch, S., Xie, H., Fujita, T., Hirota, Y., Lydon, J., DeMayo, F.,
Maxson, R., et al. (2011). Conditional deletion of Msx homeobox genes in the uterus inhibits
blastocyst implantation by altering uterine receptivity. Dev. Cell 21, 1014–1025.
Daikoku, T., Matsumoto, H., Gupta, R.A., Das, S.K., Gassmann, M., DuBois, R.N., and Dey,
S.K. (2003). Expression of Hypoxia-inducible Factors in the Peri-implantation Mouse Uterus Is
Regulated in a Cell-specific and Ovarian Steroid Hormone-dependent Manner EVIDENCE FOR
DIFFERENTIAL FUNCTION OF HIFs DURING EARLY PREGNANCY. J. Biol. Chem. 278, 7683–
7691.
Dalgard, C.L., Lu, H., Mohyeldin, A., and Verma, A. (2004). Endogenous 2-oxoacids
differentially regulate expression of oxygen sensors. Biochem. J. 380, 419–424.
Darlyuk-Saadon, I., Weidenfeld-Baranboim, K., Yokoyama, K.K., Hai, T., and Aronheim, A.
(2012). The bZIP repressor proteins, c-Jun dimerization protein 2 and activating transcription factor 3,
recruit multiple HDAC members to the ATF3 promoter. Biochim. Biophys. Acta 1819, 1142–1153.
Darmochwal-Kolarz, D., Leszczynska-Gorzelak, B., Rolinski, J., and Oleszczuk, J. (1999). T
helper 1- and T helper 2-type cytokine imbalance in pregnant women with pre-eclampsia. Eur. J.
Obstet. Gynecol. Reprod. Biol. 86, 165–170.
Davidge, S.T., Stranko, C.P., and Roberts, J.M. (1996). Urine but not plasma nitric oxide
metabolites are decreased in women with preeclampsia. Am. J. Obstet. Gynecol. 174, 1008–1013.
Davisson, R.L., Hoffmann, D.S., Butz, G.M., Aldape, G., Schlager, G., Merrill, D.C., Sethi, S.,
Weiss, R.M., and Bates, J.N. (2002). Discovery of a spontaneous genetic mouse model of
preeclampsia. Hypertension 39, 337–342.
De Vries, J.I.P., van Pampus, M.G., Hague, W.M., Bezemer, P.D., and Joosten, J.H. (2012). Lowmolecular-weight heparin added to aspirin in the prevention of recurrent early-onset pre-eclampsia in
women with inheritable thrombophilia: the FRUIT-RCT. J. Thromb. Haemost. JTH 10, 64–72.
176
Bibliography
Dechend, R., Viedt, C., Müller, D.N., Ugele, B., Brandes, R.P., Wallukat, G., Park, J.-K., Janke,
J., Barta, P., Theuer, J., et al. (2003). AT1 receptor agonistic antibodies from preeclamptic patients
stimulate NADPH oxidase. Circulation 107, 1632–1639.
Dekel, N., Gnainsky, Y., Granot, I., and Mor, G. (2010). Inflammation and implantation. Am. J.
Reprod. Immunol. New York N 1989 63, 17–21.
Dekker, G., and Robillard, P.-Y. (2007). Pre-eclampsia: Is the immune maladaptation hypothesis
still standing? An epidemiological update. J. Reprod. Immunol. 76, 8–16.
Dekker, G.A., Kraayenbrink, A.A., Zeeman, G.G., and van Kamp, G.J. (1991). Increased plasma
levels of the novel vasoconstrictor peptide endothelin in severe pre-eclampsia. Eur. J. Obstet. Gynecol.
Reprod. Biol. 40, 215–220.
Dekker, G.A., Robillard, P.Y., and Hulsey, T.C. (1998). Immune maladaptation in the etiology of
preeclampsia: a review of corroborative epidemiologic studies. Obstet. Gynecol. Surv. 53, 377–382.
Demir, R., and Erbengi, T. (1984). Some new findings about Hofbauer cells in the chorionic villi
of the human placenta. Acta Anat. (Basel) 119, 18–26.
Demir, R., Kayisli, U.A., Seval, Y., Celik-Ozenci, C., Korgun, E.T., Demir-Weusten, A.Y., and
Huppertz, B. (2004). Sequential expression of VEGF and its receptors in human placental villi during
very early pregnancy: differences between placental vasculogenesis and angiogenesis. Placenta 25,
560–572.
Deroanne, C.F., Bonjean, K., Servotte, S., Devy, L., Colige, A., Clausse, N., Blacher, S., Verdin,
E., Foidart, J.-M., Nusgens, B.V., et al. (2002). Histone deacetylases inhibitors as anti-angiogenic
agents altering vascular endothelial growth factor signaling. Oncogene 21, 427–436.
Dey, S.K., Lim, H., Das, S.K., Reese, J., Paria, B.C., Daikoku, T., and Wang, H. (2004).
Molecular cues to implantation. Endocr. Rev. 25, 341–373.
Dhanabal, M., Ramchandran, R., Waterman, M.J., Lu, H., Knebelmann, B., Segal, M., and
Sukhatme, V.P. (1999). Endostatin induces endothelial cell apoptosis. J. Biol. Chem. 274, 11721–
11726.
Dioum, E.M., Chen, R., Alexander, M.S., Zhang, Q., Hogg, R.T., Gerard, R.D., and Garcia, J.A.
(2009). Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase
sirtuin 1. Science 324, 1289–1293.
Discher, D.J., Bishopric, N.H., Wu, X., Peterson, C.A., and Webster, K.A. (1998). Hypoxia
regulates beta-enolase and pyruvate kinase-M promoters by modulating Sp1/Sp3 binding to a
conserved GC element. J. Biol. Chem. 273, 26087–26093.
Dong, W., Li, Y., Gao, M., Hu, M., Li, X., Mai, S., Guo, N., Yuan, S., and Song, L. (2012). IKKα
contributes to UVB-induced VEGF expression by regulating AP-1 transactivation. Nucleic Acids Res.
40, 2940–2955.
Donker, R.B., Asgeirsdóttir, S.A., Gerbens, F., van Pampus, M.G., Kallenberg, C.G.M., te
Meerman, G.J., Aarnoudse, J.G., and Molema, G. (2005). Plasma factors in severe early-onset
177
Bibliography
preeclampsia do not substantially alter endothelial gene expression in vitro. J. Soc. Gynecol. Investig.
12, 98–106.
Doridot, L., Passet, B., Méhats, C., Rigourd, V., Barbaux, S., Ducat, A., Mondon, F., Vilotte, M.,
Castille, J., Breuiller-Fouché, M., et al. (2013). Preeclampsia-Like Symptoms Induced in Mice by
Fetoplacental Expression of STOX1 Are Reversed by Aspirin Treatment. Hypertension 61, 662–668.
Du, H., Liu, H., Zhao, J., Wu, Y., Gong, X., Zhou, Q., Xu, J., Li, Y., Shi, X., and Qiao, F. (2011).
Effects of maternal serum on permeability of glomerular endothelial cell membrane. J. Huazhong
Univ. Sci. Technol. Med. Sci. Hua Zhong Ke Ji Xue Xue Bao Yi Xue Ying Wen Ban Huazhong Keji
Daxue Xuebao Yixue Yingdewen Ban 31, 17–20.
Duckitt, K., and Harrington, D. (2005). Risk factors for pre-eclampsia at antenatal booking:
systematic review of controlled studies. BMJ 330, 565.
Duley, L., Gülmezoglu, A.M., and Henderson-Smart, D.J. (2003). Magnesium sulphate and other
anticonvulsants for women with pre-eclampsia. Cochrane Database Syst. Rev. CD000025.
Duley, L., Henderson-Smart, D.J., and Meher, S. (2006). Drugs for treatment of very high blood
pressure during pregnancy. Cochrane Database Syst. Rev. CD001449.
Dunn, C.L., Kelly, R.W., and Critchley, H.O.D. (2003). Decidualization of the human
endometrial stromal cell: an enigmatic transformation. Reprod. Biomed. Online 7, 151–161.
Ema, M., Taya, S., Yokotani, N., Sogawa, K., Matsuda, Y., and Fujii-Kuriyama, Y. (1997). A
novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates
the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad.
Sci. U. S. A. 94, 4273–4278.
Enders, A.C. (1976). Cytology of human early implantation. Res. Reprod. 8, 1–2.
Enders, A.C. (1989). Trophoblast differentiation during the transition from trophoblastic plate to
lacunar stage of implantation in the rhesus monkey and human. Am. J. Anat. 186, 85–98.
Epstein, A.C., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O’Rourke, J., Mole, D.R., Mukherji,
M., Metzen, E., Wilson, M.I., Dhanda, A., et al. (2001). C. elegans EGL-9 and mammalian homologs
define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54.
Eremina, V., Sood, M., Haigh, J., Nagy, A., Lajoie, G., Ferrara, N., Gerber, H.-P., Kikkawa, Y.,
Miner, J.H., and Quaggin, S.E. (2003). Glomerular-specific alterations of VEGF-A expression lead to
distinct congenital and acquired renal diseases. J. Clin. Invest. 111, 707–716.
Eriksson, A., Cao, R., Roy, J., Tritsaris, K., Wahlestedt, C., Dissing, S., Thyberg, J., and Cao, Y.
(2003). Small GTP-binding protein Rac is an essential mediator of vascular endothelial growth factorinduced endothelial fenestrations and vascular permeability. Circulation 107, 1532–1538.
Esplin, M.S., Fausett, M.B., Fraser, A., Kerber, R., Mineau, G., Carrillo, J., and Varner, M.W.
(2001). Paternal and Maternal Components of the Predisposition to Preeclampsia. N. Engl. J. Med.
344, 867–872.
178
Bibliography
Estellés, A., Gilabert, J., Aznar, J., Loskutoff, D.J., and Schleef, R.R. (1989). Changes in the
plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in
patients with severe preeclampsia. Blood 74, 1332–1338.
Estellés, A., Gilabert, J., Aznar, J., Loskutoff, D.J., and Schleef, R.R. (1989). Changes in the
plasma levels of type 1 and type 2 plasminogen activator inhibitors in normal pregnancy and in
patients with severe preeclampsia. Blood 74, 1332–1338.
Even-Ram, S., Uziely, B., Cohen, P., Grisaru-Granovsky, S., Maoz, M., Ginzburg, Y., Reich, R.,
Vlodavsky, I., and Bar-Shavit, R. (1998). Thrombin receptor overexpression in malignant and
physiological invasion processes. Nat. Med. 4, 909–914.
Falcao, S., Stoyanova, E., Cloutier, G., Maurice, R.L., Gutkowska, J., and Lavoie, J.L. (2009).
Mice overexpressing both human angiotensinogen and human renin as a model of superimposed
preeclampsia on chronic hypertension. Hypertension 54, 1401–1407.
Farina, A., Sekizawa, A., De Sanctis, P., Purwosunu, Y., Okai, T., Cha, D.H., Kang, J.H.,
Vicenzi, C., Tempesta, A., Wibowo, N., et al. (2008). Gene expression in chorionic villous samples at
11 weeks’ gestation from women destined to develop preeclampsia. Prenat. Diagn. 28, 956–961.
Fauque, P., Jouannet, P., Lesaffre, C., Ripoche, M.-A., Dandolo, L., Vaiman, D., and Jammes, H.
(2007). Assisted Reproductive Technology affects developmental kinetics, H19 Imprinting Control
Region methylation and H19 gene expression in individual mouse embryos. BMC Dev. Biol. 7, 116.
Feinberg, R.F., Kliman, H.J., and Wang, C.L. (1994). Transforming growth factor-beta stimulates
trophoblast oncofetal fibronectin synthesis in vitro: implications for trophoblast implantation in vivo.
J. Clin. Endocrinol. Metab. 78, 1241–1248.
Feldser, D., Agani, F., Iyer, N.V., Pak, B., Ferreira, G., and Semenza, G.L. (1999). Reciprocal
positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res.
59, 3915–3918.
Feng (2009). Estrogen and progestin regulate HIF-1α expression in ovarian cancer cell lines via
the activation of Akt signaling transduction pathway. Oncol. Rep. 21.
Fest, S., Aldo, P.B., Abrahams, V.M., Visintin, I., Alvero, A., Chen, R., Chavez, S.L., Romero,
R., and Mor, G. (2007). Trophoblast-macrophage interactions: a regulatory network for the protection
of pregnancy. Am. J. Reprod. Immunol. New York N 1989 57, 55–66.
Fickling, S.., Williams, D., Vallance, P., Nussey, S.., and Whitley, G.S.. (1993). Plasma
concentrations of endogenous inhibitor of nitric oxide synthesis in normal pregnancy and preeclampsia. The Lancet 342, 242–243.
Finley, L.W.S., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., Teruya-Feldstein, J.,
Moreira, P.I., Cardoso, S.M., Clish, C.B., et al. (2011). SIRT3 opposes reprogramming of cancer cell
metabolism through HIF1α destabilization. Cancer Cell 19, 416–428.
Fitzgerald, D.J., Entman, S.S., Mulloy, K., and FitzGerald, G.A. (1987). Decreased prostacyclin
biosynthesis preceding the clinical manifestation of pregnancy-induced hypertension. Circulation 75,
956–963.
179
Bibliography
Fitzgerald, D.J., Rocki, W., Murray, R., Mayo, G., and FitzGerald, G.A. (1990). Thromboxane
A2 synthesis in pregnancy-induced hypertension. Lancet 335, 751–754.
Fitzpatrick, E., Johnson, M.P., Dyer, T.D., Forrest, S., Elliott, K., Blangero, J., Brennecke, S.P.,
and Moses, E.K. (2009). Genetic association of the activin A receptor gene (ACVR2A) and preeclampsia. Mol. Hum. Reprod. 15, 195–204.
Fitzpatrick, T.E., and Graham, C.H. (1998). Stimulation of plasminogen activator inhibitor-1
expression in immortalized human trophoblast cells cultured under low levels of oxygen. Exp. Cell
Res. 245, 155–162.
Flamme, I., Fröhlich, T., von Reutern, M., Kappel, A., Damert, A., and Risau, W. (1997). HRF, a
putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible
factor-1 alpha and developmentally expressed in blood vessels. Mech. Dev. 63, 51–60.
Foidart, J.-M., Munaut, C., Chantraine, F., Akolekar, R., and Nicolaides, K.H. (2010). Maternal
plasma soluble endoglin at 11-13 weeks’ gestation in pre-eclampsia. Ultrasound Obstet. Gynecol. Off.
J. Int. Soc. Ultrasound Obstet. Gynecol. 35, 680–687.
Forbes, K., Westwood, M., Baker, P.N., and Aplin, J.D. (2008). Insulin-like growth factor I and II
regulate the life cycle of trophoblast in the developing human placenta. Am. J. Physiol. Cell Physiol.
294, C1313–1322.
Forsythe, J.A., Jiang, B.H., Iyer, N.V., Agani, F., Leung, S.W., Koos, R.D., and Semenza, G.L.
(1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor
1. Mol. Cell. Biol. 16, 4604–4613.
Founds, S.A., Conley, Y.P., Lyons-Weiler, J.F., Jeyabalan, A., Hogge, W.A., and Conrad, K.P.
(2009). Altered global gene expression in first trimester placentas of women destined to develop
preeclampsia. Placenta 30, 15–24.
Founds, S.A., Conley, Y.P., Lyons-Weiler, J.F., Jeyabalan, A., Hogge, W.A., and Conrad, K.P.
(2009). Altered global gene expression in first trimester placentas of women destined to develop
preeclampsia. Placenta 30, 15–24.
Freedman, S.J., Sun, Z.-Y.J., Poy, F., Kung, A.L., Livingston, D.M., Wagner, G., and Eck, M.J.
(2002). Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc. Natl.
Acad. Sci. U. S. A. 99, 5367–5372.
Friedman, A., Kaufer, D., and Heinemann, U. (2009). Blood-brain barrier breakdown-inducing
astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 85, 142–149.
Friedman, S.A. (1988). Preeclampsia: a review of the role of prostaglandins. Obstet. Gynecol. 71,
122–137.
Friedman, S.A., Schiff, E., Emeis, J.J., Dekker, G.A., and Sibai, B.M. (1995). Biochemical
corroboration of endothelial involvement in severe preeclampsia. Am. J. Obstet. Gynecol. 172, 202–
203.
180
Bibliography
Funai, E.F., Paltiel, O.B., Malaspina, D., Friedlander, Y., Deutsch, L., and Harlap, S. (2005). Risk
factors for pre-eclampsia in nulliparous and parous women: the Jerusalem perinatal study. Paediatr.
Perinat. Epidemiol. 19, 59–68.
Furuse, M., Hirase, T., Itoh, M., Nagafuchi, A., Yonemura, S., Tsukita, S., and Tsukita, S. (1993).
Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol. 123, 1777–
1788.
Furuse, M., Itoh, M., Hirase, T., Nagafuchi, A., Yonemura, S., Tsukita, S., and Tsukita, S. (1994).
Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin
at tight junctions. J. Cell Biol. 127, 1617–1626.
Gandley, R.E., Conrad, K.P., and McLaughlin, M.K. (2001). Endothelin and nitric oxide mediate
reduced myogenic reactivity of small renal arteries from pregnant rats. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 280, R1–7.
Garmendia, J.V., Gutiérrez, Y., Blanca, I., Bianco, N.E., and De Sanctis, J.B. (1997). Nitric oxide
in different types of hypertension during pregnancy. Clin. Sci. Lond. Engl. 1979 93, 413–421.
Gaugler-Senden, I.P.M., Huijssoon, A.G., Visser, W., Steegers, E.A.P., and de Groot, C.J.M.
(2006). Maternal and perinatal outcome of preeclampsia with an onset before 24 weeks’ gestation.
Audit in a tertiary referral center. Eur. J. Obstet. Gynecol. Reprod. Biol. 128, 216–221.
Genbacev, O. (2001). To proliferate or to divide - to be or not to be. Early Pregnancy Online 5,
63–64.
Genbacev, O., Joslin, R., Damsky, C.H., Polliotti, B.M., and Fisher, S.J. (1996). Hypoxia alters
early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental
defects that occur in preeclampsia. J. Clin. Invest. 97, 540–550.
Genbacev, O., Krtolica, A., Kaelin, W., and Fisher, S.J. (2001). Human cytotrophoblast
expression of the von Hippel-Lindau protein is downregulated during uterine invasion in situ and
upregulated by hypoxia in vitro. Dev. Biol. 233, 526–536.
Genbacev, O., Zhou, Y., Ludlow, J.W., and Fisher, S.J. (1997). Regulation of human placental
development by oxygen tension. Science 277, 1669–1672.
Geng, H., Harvey, C.T., Pittsenbarger, J., Liu, Q., Beer, T.M., Xue, C., and Qian, D.Z. (2011).
HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J.
Biol. Chem. 286, 38095–38102.
Geng, H., Harvey, C.T., Pittsenbarger, J., Liu, Q., Beer, T.M., Xue, C., and Qian, D.Z. (2011).
HDAC4 protein regulates HIF1α protein lysine acetylation and cancer cell response to hypoxia. J.
Biol. Chem. 286, 38095–38102.
Geng, H., Liu, Q., Xue, C., David, L.L., Beer, T.M., Thomas, G.V., Dai, M.-S., and Qian, D.Z.
(2012). HIF1α protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 287, 35496–
35505.
181
Bibliography
Geng, H., Liu, Q., Xue, C., David, L.L., Beer, T.M., Thomas, G.V., Dai, M.-S., and Qian, D.Z.
(2012). HIF1α protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 287, 35496–
35505.
Gerald, D., Berra, E., Frapart, Y.M., Chan, D.A., Giaccia, A.J., Mansuy, D., Pouysségur, J.,
Yaniv, M., and Mechta-Grigoriou, F. (2004). JunD reduces tumor angiogenesis by protecting cells
from oxidative stress. Cell 118, 781–794.
Gerber, H.P., Condorelli, F., Park, J., and Ferrara, N. (1997). Differential transcriptional
regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is
up-regulated by hypoxia. J. Biol. Chem. 272, 23659–23667.
Gerhardt, A., Goecke, T.W., Beckmann, M.W., Wagner, K.J., Tutschek, B., Willers, R., Bender,
H.-G., Scharf, R.E., and Zotz, R.B. (2005). The G20210A prothrombin-gene mutation and the
plasminogen activator inhibitor (PAI-1) 5G/5G genotype are associated with early onset of severe
preeclampsia. J. Thromb. Haemost. JTH 3, 686–691.
Getsios, S., Chen, G.T., Huang, D.T., and MacCalman, C.D. (1998). Regulated expression of
cadherin-11 in human extravillous cytotrophoblasts undergoing aggregation and fusion in response to
transforming growth factor beta 1. J. Reprod. Fertil. 114, 357–363.
Giardina, J.B., Green, G.M., Rinewalt, A.N., Granger, J.P., and Khalil, R.A. (2001). Role of
endothelin B receptors in enhancing endothelium-dependent nitric oxide-mediated vascular relaxation
during high salt diet. Hypertension 37, 516–523.
Gille, J., Reisinger, K., Asbe-Vollkopf, A., Hardt-Weinelt, K., and Kaufmann, R. (2000).
Ultraviolet-A-induced transactivation of the vascular endothelial growth factor gene in HaCaT
keratinocytes is conveyed by activator protein-2 transcription factor. J. Invest. Dermatol. 115, 30–36.
Gille, J., Swerlick, R.A., and Caughman, S.W. (1997). Transforming growth factor-α-induced
transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2dependent DNA binding and transactivation. EMBO J. 16, 750–759.
Giudice, L.C. (1999). Potential biochemical markers of uterine receptivity. Hum. Reprod. Oxf.
Engl. 14 Suppl 2, 3–16.
Goldberg, M.A., Dunning, S.P., and Bunn, H.F. (1988). Regulation of the erythropoietin gene:
evidence that the oxygen sensor is a heme protein. Science 242, 1412–1415.
Goldenberg, R.L., Culhane, J.F., Iams, J.D., and Romero, R. (2008). Epidemiology and causes of
preterm birth. Lancet 371, 75–84.
Görlach, A., and Bonello, S. (2008). The cross-talk between NF-kappaB and HIF-1: further
evidence for a significant liaison. Biochem. J. 412, e17–19.
Görlach, A., Diebold, I., Schini-Kerth, V.B., Berchner-Pfannschmidt, U., Roth, U., Brandes, R.P.,
Kietzmann, T., and Busse, R. (2001). Thrombin activates the hypoxia-inducible factor-1 signaling
pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ.
Res. 89, 47–54.
182
Bibliography
Gothié, E., Richard, D.E., Berra, E., Pagès, G., and Pouysségur, J. (2000). Identification of
alternative spliced variants of human hypoxia-inducible factor-1alpha. J. Biol. Chem. 275, 6922–6927.
Gould, P.S., Gu, M., Liao, J., Ahmad, S., Cudmore, M.J., Ahmed, A., and Vatish, M. (2010).
Upregulation of urotensin II receptor in preeclampsia causes in vitro placental release of soluble
vascular endothelial growth factor receptor 1 in hypoxia. Hypertension 56, 172–178.
Granger, J.P., Alexander, B.T., Llinas, M.T., Bennett, W.A., and Khalil, R.A. (2002).
Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction.
Microcirc. New York N 1994 9, 147–160.
Gray, M.J., Zhang, J., Ellis, L.M., Semenza, G.L., Evans, D.B., Watowich, S.S., and Gallick, G.E.
(2005). HIF-1α, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that
regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas.
Oncogene 24, 3110–3120.
Greenberg, S.G., Baker, R.S., Yang, D., and Clark, K.E. (1997). Effects of continuous infusion of
endothelin-1 in pregnant sheep. Hypertension 30, 1585–1590.
Gris, J.-C., Chauleur, C., Molinari, N., Marès, P., Fabbro-Peray, P., Quéré, I., Lefrant, J.-Y.,
Haddad, B., and Dauzat, M. (2011). Addition of enoxaparin to aspirin for the secondary prevention of
placental vascular complications in women with severe pre-eclampsia. The pilot randomised
controlled NOH-PE trial. Thromb. Haemost. 106, 1053–1061.
Groten, T., Kreienberg, R., Fialka, I., Huber, L., and Wedlich, D. (2000). Altered subcellular
distribution of cadherin-5 in endothelial cells caused by the serum of pre-eclamptic patients. Mol.
Hum. Reprod. 6, 1027–1032.
Gu, Y., Groome, L.J., Alexander, J.S., and Wang, Y. (2012). PAR-2 triggers placenta-derived
protease-induced altered VE-cadherin reorganization at endothelial junctions in preeclampsia. Placenta
33, 803–809.
Gu, Y., Lewis, D.F., Alexander, J.S., and Wang, Y. (2009). Placenta-derived chymotrypsin-like
protease (CLP) disturbs endothelial junctional structure in preeclampsia. Reprod. Sci. Thousand Oaks
Calif 16, 479–488.
Gu, Y.Z., Moran, S.M., Hogenesch, J.B., Wartman, L., and Bradfield, C.A. (1998). Molecular
characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit,
HIF3alpha. Gene Expr. 7, 205–213.
Guilbert, L., Robertson, S.A., and Wegmann, T.G. (1993). The trophoblast as an integral
component of a macrophage-cytokine network. Immunol. Cell Biol. 71 ( Pt 1), 49–57.
Gupta, S., Agarwal, A., and Sharma, R.K. (2005). The role of placental oxidative stress and lipid
peroxidation in preeclampsia. Obstet. Gynecol. Surv. 60, 807–816.
Gurdol, F., Yurdum, L.M., Ozturk, U., Isbilen, E., and Cakmakoglu, B. (2012). Association of the
CC chemokine receptor 5 (CCR5) polymorphisms with preeclampsia in Turkish women. Arch.
Gynecol. Obstet. 286, 51–54.
183
Bibliography
H Schröcksnadel, G.D. (1993). Tumor markers in hypertensive disorders of pregnancy. Gynecol.
Obstet. Invest. 35, 204–208.
Hai, T., and Curran, T. (1991). Cross-family dimerization of transcription factors Fos/Jun and
ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. U. S. A. 88, 3720–3724.
Halder, J.B., Zhao, X., Soker, S., Paria, B.C., Klagsbrun, M., Das, S.K., and Dey, S.K. (2000).
Differential expression of VEGF isoforms and VEGF(164)-specific receptor neuropilin-1 in the mouse
uterus suggests a role for VEGF(164) in vascular permeability and angiogenesis during implantation.
Genes. New York N 2000 26, 213–224.
Haller, H., Hempel, A., Homuth, V., Mandelkow, A., Busjahn, A., Maasch, C., Drab, M.,
Lindschau, C., Jüpner, A., Vetter, K., et al. (1998). Endothelial-cell permeability and protein kinase C
in pre-eclampsia. Lancet 351, 945–949.
Hammer, A., and Dohr, G. (2000). Expression of Fas-ligand in first trimester and term human
placental villi. J. Reprod. Immunol. 46, 83–90.
Han, J.Y., Kim, Y.S., Cho, G.J., Roh, G.S., Kim, H.J., Choi, W.J., Paik, W.Y., Rho, G.J., Kang,
S.S., and Choi, W.S. (2006). Altered gene expression of caspase-10, death receptor-3 and IGFBP-3 in
preeclamptic placentas. Mol. Cells 22, 168–174.
Han, V.K., Bassett, N., Walton, J., and Challis, J.R. (1996). The expression of insulin-like growth
factor (IGF) and IGF-binding protein (IGFBP) genes in the human placenta and membranes: evidence
for IGF-IGFBP interactions at the feto-maternal interface. J. Clin. Endocrinol. Metab. 81, 2680–2693.
Han, Z.-B., Ren, H., Zhao, H., Chi, Y., Chen, K., Zhou, B., Liu, Y., Zhang, L., Xu, B., Liu, B., et
al. (2008). Hypoxia-inducible factor (HIF)-1 alpha directly enhances the transcriptional activity of
stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis 29,
1853–1861.
Haram, K., Svendsen, E., and Abildgaard, U. (2009). The HELLP syndrome: Clinical issues and
management. A Review. BMC Pregnancy Childbirth 9, 8.
Harris, A.L. (2002). HYPOXIA — A KEY REGULATORY FACTOR IN TUMOUR GROWTH.
Nat. Rev. Cancer 2, 38–47.
Harris, L.K. (2010). Review: Trophoblast-vascular cell interactions in early pregnancy: how to
remodel a vessel. Placenta 31 Suppl, S93–98.
Harris, L.K., and Aplin, J.D. (2007). Vascular remodeling and extracellular matrix breakdown in
the uterine spiral arteries during pregnancy. Reprod. Sci. Thousand Oaks Calif 14, 28–34.
Hata, Y., Duh, E., Zhang, K., Robinson, G.S., and Aiello, L.P. (1998). Transcription factors Sp1
and Sp3 alter vascular endothelial growth factor receptor expression through a novel recognition
sequence. J. Biol. Chem. 273, 19294–19303.
Hayashi, T., Yamada, K., Esaki, T., Kuzuya, M., Satake, S., Ishikawa, T., Hidaka, H., and Iguchi,
A. (1995). Estrogen increases endothelial nitric oxide by a receptor-mediated system. Biochem.
Biophys. Res. Commun. 214, 847–855.
184
Bibliography
He, H., Venema, V.J., Gu, X., Venema, R.C., Marrero, M.B., and Caldwell, R.B. (1999).
Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin
through flk-1/KDR activation of c-Src. J. Biol. Chem. 274, 25130–25135.
Healy, D.L. (1991). Endometrial prolactin and implantation. Baillières Clin. Obstet. Gynaecol. 5,
95–105.
Heidbreder, M., Qadri, F., Jöhren, O., Dendorfer, A., Depping, R., Fröhlich, F., Wagner, K.F.,
and Dominiak, P. (2007). Non-hypoxic induction of HIF-3alpha by 2-deoxy-D-glucose and insulin.
Biochem. Biophys. Res. Commun. 352, 437–443.
Heilmann, L., Schorsch, M., Hahn, T., and Fareed, J. (2011). Antiphospholipid syndrome and
pre-eclampsia. Semin. Thromb. Hemost. 37, 141–145.
Heinrich, R., Livne, E., Ben-Izhak, O., and Aronheim, A. (2004). The c-Jun dimerization protein
2 inhibits cell transformation and acts as a tumor suppressor gene. J. Biol. Chem. 279, 5708–5715.
Heinrich, R., Livne, E., Ben-Izhak, O., and Aronheim, A. (2004). The c-Jun dimerization protein
2 inhibits cell transformation and acts as a tumor suppressor gene. J. Biol. Chem. 279, 5708–5715.
Hennessy, A., Thornton, C.E., Makris, A., Ogle, R.F., Henderson-Smart, D.J., Gillin, A.G., and
Child, A. (2007). A randomised comparison of hydralazine and mini-bolus diazoxide for hypertensive
emergencies in pregnancy: the PIVOT trial. Aust. N. Z. J. Obstet. Gynaecol. 47, 279–285.
Herrmann, J., and Lerman, A. (2001). The endothelium: dysfunction and beyond. J. Nucl.
Cardiol. Off. Publ. Am. Soc. Nucl. Cardiol. 8, 197–206.
Hertig, A., Berkane, N., Lefevre, G., Toumi, K., Marti, H.-P., Capeau, J., Uzan, S., and Rondeau,
E. (2004). Maternal serum sFlt1 concentration is an early and reliable predictive marker of
preeclampsia. Clin. Chem. 50, 1702–1703.
Herzog, M. (1909). A contribution to our knowledge of the earliest known stages of placentation
and embryonic development in man. Am. J. Anat. 9, 361–400.
Hess, J., Angel, P., and Schorpp-Kistner, M. (2004). AP-1 subunits: quarrel and harmony among
siblings. J. Cell Sci. 117, 5965–5973.
Hess, J., Angel, P., and Schorpp-Kistner, M. (2004). AP-1 subunits: quarrel and harmony among
siblings. J. Cell Sci. 117, 5965–5973.
Hiby, S.E., Apps, R., Sharkey, A.M., Farrell, L.E., Gardner, L., Mulder, A., Claas, F.H., Walker,
J.J., Redman, C.W., Redman, C.C., et al. (2010). Maternal activating KIRs protect against human
reproductive failure mediated by fetal HLA-C2. J. Clin. Invest. 120, 4102–4110.
Hiby, S.E., Walker, J.J., O’Shaughnessy, K.M., Redman, C.W.G., Carrington, M., Trowsdale, J.,
and Moffett, A. (2004). Combinations of Maternal KIR and Fetal HLA-C Genes Influence the Risk of
Preeclampsia and Reproductive Success. J. Exp. Med. 200, 957–965.
Hill, C., Würfel, A., Heger, J., Meyering, B., Schlüter, K.-D., Weber, M., Ferdinandy, P.,
Aronheim, A., Schulz, R., and Euler, G. (2013). Inhibition of AP-1 signaling by JDP2 overexpression
protects cardiomyocytes against hypertrophy and apoptosis induction. Cardiovasc. Res. 99, 121–128.
185
Bibliography
Hills, F.A., Elder, M.G., Chard, T., and Sullivan, M.H.F. (2004). Regulation of human villous
trophoblast by insulin-like growth factors and insulin-like growth factor-binding protein-1. J.
Endocrinol. 183, 487–496.
Hirano, H., Imai, Y., and Ito, H. (2002). Spiral artery of placenta: development and pathologyimmunohistochemical, microscopical, and electron-microscopic study. Kobe J. Med. Sci. 48, 13–23.
Holden, D.P., Fickling, S.A., Whitley, G.S., and Nussey, S.S. (1998). Plasma concentrations of
asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy and
preeclampsia. Am. J. Obstet. Gynecol. 178, 551–556.
Hsu, C.D., Iriye, B., Johnson, T.R., Witter, F.R., Hong, S.F., and Chan, D.W. (1993). Elevated
circulating thrombomodulin in severe preeclampsia. Am. J. Obstet. Gynecol. 169, 148–149.
Hsu, C.D., Iriye, B., Johnson, T.R., Witter, F.R., Hong, S.F., and Chan, D.W. (1993). Elevated
circulating thrombomodulin in severe preeclampsia. Am. J. Obstet. Gynecol. 169, 148–149.
Hua, Z., Lv, Q., Ye, W., Wong, C.-K.A., Cai, G., Gu, D., Ji, Y., Zhao, C., Wang, J., Yang, B.B.,
et al. (2006). MiRNA-Directed Regulation of VEGF and Other Angiogenic Factors under Hypoxia.
PLoS ONE 1, e116.
Huang, C., Han, Y., Wang, Y., Sun, X., Yan, S., Yeh, E.T.H., Chen, Y., Cang, H., Li, H., Shi, G.,
et al. (2009). SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 deSUMOylation. EMBO J. 28, 2748–2762.
Huang, X., Le, Q.-T., and Giaccia, A.J. (2010). MiR-210 – micromanager of the hypoxia
pathway. Trends Mol. Med. 16, 230–237.
Hubel, C.A., Lyall, F., Weissfeld, L., Gandley, R.E., and Roberts, J.M. (1998). Small low-density
lipoproteins and vascular cell adhesion molecule-1 are increased in association with hyperlipidemia in
preeclampsia. Metabolism. 47, 1281–1288.
Hubel, C.A., Wallukat, G., Wolf, M., Herse, F., Rajakumar, A., Roberts, J.M., Markovic, N.,
Thadhani, R., Luft, F.C., and Dechend, R. (2007). Agonistic angiotensin II type 1 receptor
autoantibodies in postpartum women with a history of preeclampsia. Hypertension 49, 612–617.
Hung, T.-H., and Burton, G.J. (2006). Hypoxia and reoxygenation: a possible mechanism for
placental oxidative stress in preeclampsia. Taiwan. J. Obstet. Gynecol. 45, 189–200.
Huppertz, B., Kingdom, J., Caniggia, I., Desoye, G., Black, S., Korr, H., and Kaufmann, P.
(2003). Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the
maternal circulation. Placenta 24, 181–190.
Huppertz, B., Sammar, M., Chefetz, I., Neumaier-Wagner, P., Bartz, C., and Meiri, H. (2008).
Longitudinal determination of serum placental protein 13 during development of preeclampsia. Fetal
Diagn. Ther. 24, 230–236.
Hustin, J., and Schaaps, J.P. (1987). Echographic [corrected] and anatomic studies of the
maternotrophoblastic border during the first trimester of pregnancy. Am. J. Obstet. Gynecol. 157,
162–168.
186
Bibliography
Hwang, H.C., Martins, C.P., Bronkhorst, Y., Randel, E., Berns, A., Fero, M., and Clurman, B.E.
(2002). Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and
high-throughput insertion site analysis. Proc. Natl. Acad. Sci. U. S. A. 99, 11293–11298.
Ietta, F., Wu, Y., Winter, J., Xu, J., Wang, J., Post, M., and Caniggia, I. (2006). Dynamic HIF1A
regulation during human placental development. Biol. Reprod. 75, 112–121.
Ignarro, L.J., Buga, G.M., Wood, K.S., Byrns, R.E., and Chaudhuri, G. (1987). Endotheliumderived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad.
Sci. 84, 9265–9269.
Imperatore, A., Rolfo, A., Petraglia, F., Challis, J.R.G., and Caniggia, I. (2010). Hypoxia and
preeclampsia: increased expression of urocortin 2 and urocortin 3. Reprod. Sci. Thousand Oaks Calif
17, 833–843.
Inoue, A., Yanagisawa, M., Kimura, S., Kasuya, Y., Miyauchi, T., Goto, K., and Masaki, T.
(1989). The human endothelin family: three structurally and pharmacologically distinct isopeptides
predicted by three separate genes. Proc. Natl. Acad. Sci. U. S. A. 86, 2863–2867.
Ioannidou, S., Deinhardt, K., Miotla, J., Bradley, J., Cheung, E., Samuelsson, S., Ng, Y.-S., and
Shima, D.T. (2006). An in vitro assay reveals a role for the diaphragm protein PV-1 in endothelial
fenestra morphogenesis. Proc. Natl. Acad. Sci. 103, 16770–16775.
Irving, J.A., and Lala, P.K. (1995). Functional role of cell surface integrins on human trophoblast
cell migration: regulation by TGF-beta, IGF-II, and IGFBP-1. Exp. Cell Res. 217, 419–427.
Isaacs, J.S., Jung, Y.-J., Mimnaugh, E.G., Martinez, A., Cuttitta, F., and Neckers, L.M. (2002).
Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative
pathway. J. Biol. Chem. 277, 29936–29944.
Isaacs, J.S., Jung, Y.J., Mole, D.R., Lee, S., Torres-Cabala, C., Chung, Y.-L., Merino, M., Trepel,
J., Zbar, B., Toro, J., et al. (2005). HIF overexpression correlates with biallelic loss of fumarate
hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–
153.
Ishihara, N., Matsuo, H., Murakoshi, H., Laoag-Fernandez, J.B., Samoto, T., and Maruo, T.
(2002). Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either
preeclampsia or intrauterine growth retardation. Am. J. Obstet. Gynecol. 186, 158–166.
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J.M., Lane,
W.S., and Kaelin, W.G., Jr (2001). HIFalpha targeted for VHL-mediated destruction by proline
hydroxylation: implications for O2 sensing. Science 292, 464–468.
Iwahashi, M., Muragaki, Y., Ooshima, A., Yamoto, M., and Nakano, R. (1996). Alterations in
distribution and composition of the extracellular matrix during decidualization of the human
endometrium. J. Reprod. Fertil. 108, 147–155.
Iyer, N.V., Kotch, L.E., Agani, F., Leung, S.W., Laughner, E., Wenger, R.H., Gassmann, M.,
Gearhart, J.D., Lawler, A.M., Yu, A.Y., et al. (1998). Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12, 149–162.
187
Bibliography
Jaakkola, P., Mole, D.R., Tian, Y.-M., Wilson, M.I., Gielbert, J., Gaskell, S.J., Kriegsheim, A.
von, Hebestreit, H.F., Mukherji, M., Schofield, C.J., et al. (2001). Targeting of HIF-α to the von
Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 292, 468–
472.
Jabbour, H., Gubbay, O., and Critchley, H. (2002). Prolactin action and signalling in the human
endometrium. Reprod. Med. Rev. 10, 117–132.
Jackson, P., Barrowclough, I.W., France, J.T., and Phillips, L.I. (1980). A successful pregnancy
following total hysterectomy. Br. J. Obstet. Gynaecol. 87, 353–355.
Jacobs, M., Nassar, N., Roberts, C.L., Hadfield, R., Morris, J.M., and Ashton, A.W. (2011).
Levels of soluble fms-like tyrosine kinase one in first trimester and outcomes of pregnancy: a
systematic review. Reprod. Biol. Endocrinol. RBE 9, 77.
Jaulmes, A., Sansilvestri-Morel, P., Rolland-Valognes, G., Bernhardt, F., Gaertner, R., Lockhart,
B.P., Cordi, A., Wierzbicki, M., Rupin, A., and Verbeuren, T.J. (2009). Nox4 mediates the expression
of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells.
Thromb. Res. 124, 439–446.
Jauniaux, E., Gulbis, B., and Burton, G.J. (2003). The human first trimester gestational sac limits
rather than facilitates oxygen transfer to the foetus--a review. Placenta 24 Suppl A, S86–93.
Jauniaux, E., Hempstock, J., Greenwold, N., and Burton, G.J. (2003). Trophoblastic Oxidative
Stress in Relation to Temporal and Regional Differences in Maternal Placental Blood Flow in Normal
and Abnormal Early Pregnancies. Am. J. Pathol. 162, 115–125.
Jauniaux, E., Watson, A.L., Hempstock, J., Bao, Y.P., Skepper, J.N., and Burton, G.J. (2000).
Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early
pregnancy failure. Am. J. Pathol. 157, 2111–2122.
Jauniaux, E., Watson, A.L., Hempstock, J., Bao, Y.P., Skepper, J.N., and Burton, G.J. (2000).
Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early
pregnancy failure. Am. J. Pathol. 157, 2111–2122.
Jensen, D.E., Black, A.R., Swick, A.G., and Azizkhan, J.C. (1997). Distinct roles for Sp1 and
E2F sites in the growth/cell cycle regulation of the DHFR promoter. J. Cell. Biochem. 67, 24–31.
Jeong, J.W., Bae, M.K., Ahn, M.Y., Kim, S.H., Sohn, T.K., Bae, M.H., Yoo, M.A., Song, E.J.,
Lee, K.J., and Kim, K.W. (2002). Regulation and destabilization of HIF-1alpha by ARD1-mediated
acetylation. Cell 111, 709–720.
Jiang, B.H., Jiang, G., Zheng, J.Z., Lu, Z., Hunter, T., and Vogt, P.K. (2001).
Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth
Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 12, 363–369.
Jiang, B.H., Zheng, J.Z., Leung, S.W., Roe, R., and Semenza, G.L. (1997). Transactivation and
inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by
oxygen tension. J. Biol. Chem. 272, 19253–19260.
188
Bibliography
Jin, C., Kato, K., Chimura, T., Yamasaki, T., Nakade, K., Murata, T., Li, H., Pan, J., Zhao, M.,
Sun, K., et al. (2006). Regulation of histone acetylation and nucleosome assembly by transcription
factor JDP2. Nat. Struct. Mol. Biol. 13, 331–338.
Jin, C., Li, H., Murata, T., Sun, K., Horikoshi, M., Chiu, R., and Yokoyama, K.K. (2002). JDP2, a
repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced
differentiation of F9 cells. Mol. Cell. Biol. 22, 4815–4826.
Jin, C., Ugai, H., Song, J., Murata, T., Nili, F., Sun, K., Horikoshi, M., and Yokoyama, K.K.
(2001). Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett.
489, 34–41.
Joberty, G., Petersen, C., Gao, L., and Macara, I.G. (2000). The cell-polarity protein Par6 links
Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2, 531–539.
Johansen, M., Redman, C.W., Wilkins, T., and Sargent, I.L. (1999). Trophoblast deportation in
human pregnancy--its relevance for pre-eclampsia. Placenta 20, 531–539.
Johnson, A.B., Denko, N., and Barton, M.C. (2008). Hypoxia induces a novel signature of
chromatin modifications and global repression of transcription. Mutat. Res. 640, 174–179.
Jung, J.E., Kim, H.S., Lee, C.S., Park, D.-H., Kim, Y.-N., Lee, M.-J., Lee, J.W., Park, J.-W.,
Kim, M.-S., Ye, S.K., et al. (2007). Caffeic acid and its synthetic derivative CADPE suppress tumor
angiogenesis by blocking STAT3-mediated VEGF expression in human renal carcinoma cells.
Carcinogenesis 28, 1780–1787.
Jung, J.E., Lee, H.G., Cho, I.H., Chung, D.H., Yoon, S.-H., Yang, Y.M., Lee, J.W., Choi, S.,
Park, J.-W., Ye, S.-K., et al. (2005). STAT3 is a potential modulator of HIF-1-mediated VEGF
expression in human renal carcinoma cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 19, 1296–
1298.
Jung, Y.-J., Isaacs, J.S., Lee, S., Trepel, J., and Neckers, L. (2003). IL-1beta-mediated upregulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between
inflammation and oncogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 17, 2115–2117.
Käär, K., Jouppila, P., Kuikka, J., Luotola, H., Toivanen, J., and Rekonen, A. (1980). Intervillous
blood flow in normal and complicated late pregnancy measured by means of an intravenous 133Xe
method. Acta Obstet. Gynecol. Scand. 59, 7–10.
Kabawat, S.E., Mostoufi-Zadeh, M., Driscoll, S.G., and Bhan, A.K. (1985). Implantation site in
normal pregnancy. A study with monoclonal antibodies. Am. J. Pathol. 118, 76–84.
Kajino, T., Torry, D.S., McIntyre, J.A., and Faulk, W.P. (1988). Trophoblast antigens in human
seminal plasma. Am. J. Reprod. Immunol. Microbiol. AJRIM 17, 91–95.
Kallio, P.J., Okamoto, K., O’Brien, S., Carrero, P., Makino, Y., Tanaka, H., and Poellinger, L.
(1998). Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the
CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 17, 6573–6586.
189
Bibliography
Kaluz, S., Kaluzová, M., Liao, S.-Y., Lerman, M., and Stanbridge, E.J. (2009). Transcriptional
control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1)
show? Biochim. Biophys. Acta BBA - Rev. Cancer 1795, 162–172.
Kam, E.P., Gardner, L., Loke, Y.W., and King, A. (1999). The role of trophoblast in the
physiological change in decidual spiral arteries. Hum. Reprod. Oxf. Engl. 14, 2131–2138.
Kametas, N.A., Krampl, E., McAuliffe, F., Rampling, M.W., and Nicolaides, K.H. (2004).
Pregnancy at high altitude: a hyperviscosity state. Acta Obstet. Gynecol. Scand. 83, 627–633.
Kanasaki, K., Palmsten, K., Sugimoto, H., Ahmad, S., Hamano, Y., Xie, L., Parry, S., Augustin,
H.G., Gattone, V.H., Folkman, J., et al. (2008). Deficiency in catechol-O-methyltransferase and 2methoxyoestradiol is associated with pre-eclampsia. Nature 453, 1117–1121.
Kanasaki, K., Palmsten, K., Sugimoto, H., Ahmad, S., Hamano, Y., Xie, L., Parry, S., Augustin,
H.G., Gattone, V.H., Folkman, J., et al. (2008). Deficiency in catechol-O-methyltransferase and 2methoxyoestradiol is associated with pre-eclampsia. Nature 453, 1117–1121.
Karen, F., D, A.J., and Melissa, W. (2007). IGFBP-3 has both IGF-dependent and
&#150;independent effects on cytotrophoblast proliferation in the human placenta.
Karin, M., Liu, Z. g, and Zandi, E. (1997). AP-1 function and regulation. Curr. Opin. Cell Biol. 9,
240–246.
Karmakar, S., and Das, C. (2002). Regulation of trophoblast invasion by IL-1beta and TGF-beta1.
Am. J. Reprod. Immunol. New York N 1989 48, 210–219.
Karmakar, S., and Das, C. (2004). Modulation of ezrin and E-cadherin expression by IL-1beta
and TGF-beta1 in human trophoblasts. J. Reprod. Immunol. 64, 9–29.
Karumanchi, S.A., Maynard, S.E., Stillman, I.E., Epstein, F.H., and Sukhatme, V.P. (2005).
Preeclampsia: a renal perspective. Kidney Int. 67, 2101–2113.
Kato, H., Tamamizu-Kato, S., and Shibasaki, F. (2004). Histone deacetylase 7 associates with
hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 279, 41966–
41974.
Kato, H., Tamamizu-Kato, S., and Shibasaki, F. (2004). Histone deacetylase 7 associates with
hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 279, 41966–
41974.
Kaufmann, P., Mayhew, T.M., and Charnock-Jones, D.S. (2004). Aspects of human fetoplacental
vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 25, 114–126.
Keith, B., Adelman, D.M., and Simon, M.C. (2001). Targeted mutation of the murine
arylhydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt.
Proc. Natl. Acad. Sci. U. S. A. 98, 6692–6697.
Keogh, R.J., Harris, L.K., Freeman, A., Baker, P.N., Aplin, J.D., Whitley, G.S., and Cartwright,
J.E. (2007). Fetal-derived trophoblast use the apoptotic cytokine tumor necrosis factor-alpha-related
apoptosis-inducing ligand to induce smooth muscle cell death. Circ. Res. 100, 834–841.
190
Bibliography
Keyes, L.E., Armaza, J.F., Niermeyer, S., Vargas, E., Young, D.A., and Moore, L.G. (2003).
Intrauterine growth restriction, preeclampsia, and intrauterine mortality at high altitude in Bolivia.
Pediatr. Res. 54, 20–25.
Khalil, A., Cowans, N.J., Spencer, K., Goichman, S., Meiri, H., and Harrington, K. (2009). First
trimester maternal serum placental protein 13 for the prediction of pre-eclampsia in women with a
priori high risk. Prenat. Diagn. 29, 781–789.
Khalil, R.A., and Granger, J.P. (2002). Vascular mechanisms of increased arterial pressure in
preeclampsia: lessons from animal models. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 283, R29–
R45.
Kiely, D.G., Cargill, R.I., Struthers, A.D., and Lipworth, B.J. (1997). Cardiopulmonary effects of
endothelin-1 in man. Cardiovasc. Res. 33, 378–386.
Kim, M.S., Kwon, H.J., Lee, Y.M., Baek, J.H., Jang, J.E., Lee, S.W., Moon, E.J., Kim, H.S., Lee,
S.K., Chung, H.Y., et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of
tumor suppressor genes. Nat. Med. 7, 437–443.
Kim, S.-H., Jeong, J.-W., Park, J.A., Lee, J.-W., Seo, J.H., Jung, B.-K., Bae, M.-K., and Kim, K.W. (2007). Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 17, 647–651.
Kim, S.Y., Park, S.Y., Lim, J.H., Lee, B.Y., Yang, J.H., and Ryu, H.M. (2012). Hypoxia
inducible factor-1α gene polymorphisms in Korean patients with pre-eclampsia. J. Endocrinol. Invest.
35, 670–675.
Kim, W., Bennett, E.J., Huttlin, E.L., Guo, A., Li, J., Possemato, A., Sowa, M.E., Rad, R., Rush,
J., Comb, M.J., et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified
proteome. Mol. Cell 44, 325–340.
Kimura, M. (2008). IRF2-binding protein-1 is a JDP2 ubiquitin ligase and an inhibitor of ATF2dependent transcription. FEBS Lett. 582, 2833–2837.
Kitamoto, Y., Takeya, M., Tokunaga, H., and Tomita, K. (2001). Glomerular endothelial cells are
maintained by vascular endothelial growth factor in the adult kidney. Tohoku J. Exp. Med. 195, 43–
54.
Kjøller, L., Kanse, S.M., Kirkegaard, T., Rodenburg, K.W., Rønne, E., Goodman, S.L., Preissner,
K.T., Ossowski, L., and Andreasen, P.A. (1997). Plasminogen activator inhibitor-1 represses integrinand vitronectin-mediated cell migration independently of its function as an inhibitor of plasminogen
activation. Exp. Cell Res. 232, 420–429.
Klockenbusch, W., Goecke, T.W., Krüssel, J.S., Tutschek, B.A., Crombach, G., and Schrör, K.
(2000). Prostacyclin deficiency and reduced fetoplacental blood flow in pregnancy-induced
hypertension and preeclampsia. Gynecol. Obstet. Invest. 50, 103–107.
Klonoff-Cohen, H.S., Savitz, D.A., Cefalo, R.C., and McCann, M.F. (1989). An epidemiologic
study of contraception and preeclampsia. JAMA J. Am. Med. Assoc. 262, 3143–3147.
191
Bibliography
Klungsøyr, K., Morken, N.H., Irgens, L., Vollset, S.E., and Skjaerven, R. (2012). Secular trends
in the epidemiology of pre-eclampsia throughout 40 years in Norway: prevalence, risk factors and
perinatal survival. Paediatr. Perinat. Epidemiol. 26, 190–198.
Knight, M., and UKOSS (2007). Eclampsia in the United Kingdom 2005. BJOG Int. J. Obstet.
Gynaecol. 114, 1072–1078.
Koch, S., Rabinstein, A., Falcone, S., and Forteza, A. (2001). Diffusion-weighted imaging shows
cytotoxic and vasogenic edema in eclampsia. AJNR Am. J. Neuroradiol. 22, 1068–1070.
Koelman, C.A., Coumans, A.B., Nijman, H.W., Doxiadis, I.I., Dekker, G.A., and Claas, F.H.
(2000). Correlation between oral sex and a low incidence of preeclampsia: a role for soluble HLA in
seminal fluid? J. Reprod. Immunol. 46, 155–166.
Koga, K., Osuga, Y., Tajima, T., Hirota, Y., Igarashi, T., Fujii, T., Yano, T., and Taketani, Y.
(2010). Elevated serum soluble fms-like tyrosine kinase 1 (sFlt1) level in women with hydatidiform
mole. Fertil. Steril. 94, 305–308.
Kondo, K., and Kaelin, W.G., Jr (2001). The von Hippel-Lindau tumor suppressor gene. Exp.
Cell Res. 264, 117–125.
Kozak, K.R., Abbott, B., and Hankinson, O. (1997). ARNT-Deficient Mice and Placental
Differentiation. Dev. Biol. 191, 297–305.
Kubes, P., Suzuki, M., and Granger, D.N. (1991). Nitric oxide: an endogenous modulator of
leukocyte adhesion. Proc. Natl. Acad. Sci. U. S. A. 88, 4651–4655.
Kulkarni, A., Chavan-Gautam, P., Mehendale, S., Yadav, H., and Joshi, S. (2011). Global DNA
methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA
Cell Biol. 30, 79–84.
Kumasawa, K., Ikawa, M., Kidoya, H., Hasuwa, H., Saito-Fujita, T., Morioka, Y., Takakura, N.,
Kimura, T., and Okabe, M. (2011). Pravastatin induces placental growth factor (PGF) and ameliorates
preeclampsia in a mouse model. Proc. Natl. Acad. Sci. U. S. A. 108, 1451–1455.
Kupferminc, M.J., Rimon, E., Many, A., Sharon, M., Lessing, J.B., and Gamzu, R. (2011). Low
molecular weight heparin treatment during subsequent pregnancies of women with inherited
thrombophilia and previous severe pregnancy complications. J. Matern.-Fetal Neonatal Med. Off. J.
Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 24, 1042–1045.
Kvehaugen, A.S., Dechend, R., Ramstad, H.B., Troisi, R., Fugelseth, D., and Staff, A.C. (2011).
Endothelial function and circulating biomarkers are disturbed in women and children after
preeclampsia. Hypertension 58, 63–69.
Kwon, Y.-W., Kwon, K.-S., Moon, H.-E., Park, J.A., Choi, K.-S., Kim, Y.-S., Jang, H.-S., Oh,
C.-K., Lee, Y.-M., Kwon, Y.-G., et al. (2004). Insulin-like growth factor-II regulates the expression of
vascular endothelial growth factor by the human keratinocyte cell line HaCaT. J. Invest. Dermatol.
123, 152–158.
192
Bibliography
La Ferla, K., Reimann, C., Jelkmann, W., and Hellwig-Bürgel, T. (2002). Inhibition of
erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB.
FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 16, 1811–1813.
Lachmeijer, A.M., Arngrímsson, R., Bastiaans, E.J., Frigge, M.L., Pals, G., Sigurdardóttir, S.,
Stéfansson, H., Pálsson, B., Nicolae, D., Kong, A., et al. (2001). A genome-wide scan for
preeclampsia in the Netherlands. Eur. J. Hum. Genet. EJHG 9, 758–764.
Lafayette, R.A., Druzin, M., Sibley, R., Derby, G., Malik, T., Huie, P., Polhemus, C., Deen,
W.M., and Myers, B.D. (1998). Nature of glomerular dysfunction in pre-eclampsia. Kidney Int. 54,
1240–1249.
Laivuori, H., Lahermo, P., Ollikainen, V., Widen, E., Haiva-Mallinen, L., Sundstrom, H.,
Laitinen, T., Kaaja, R., Ylikorkala, O., and Kere, J. (2003). Susceptibility Loci for Preeclampsia on
Chromosomes 2p25 and 9p13 in Finnish Families. Am. J. Hum. Genet. 72, 168–177.
Lala, P.K., and Chakraborty, C. (2003). Factors regulating trophoblast migration and
invasiveness: possible derangements contributing to pre-eclampsia and fetal injury. Placenta 24, 575–
587.
Lampinen, K.H., Rönnback, M., Kaaja, R.J., and Groop, P.-H. (2006). Impaired vascular
dilatation in women with a history of pre-eclampsia. J. Hypertens. 24, 751–756.
Land, S.C., and Tee, A.R. (2007). Hypoxia-inducible factor 1alpha is regulated by the
mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 282, 20534–
20543.
Lando, D., Peet, D.J., Gorman, J.J., Whelan, D.A., Whitelaw, M.L., and Bruick, R.K. (2002b).
FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxiainducible factor. Genes Dev. 16, 1466–1471.
Lando, D., Peet, D.J., Whelan, D.A., Gorman, J.J., and Whitelaw, M.L. (2002a). Asparagine
hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861.
Langford, K.S., Nicolaides, K.H., Jones, J., Abbas, A., McGregor, A.M., and Miell, J.P. (1995).
Serum insulin-like growth factor-binding protein-3 (IGFBP-3) levels and IGFBP-3 protease activity in
normal, abnormal, and multiple human pregnancy. J. Clin. Endocrinol. Metab. 80, 21–27.
Lash, G.E., Otun, H.A., Innes, B.A., Bulmer, J.N., Searle, R.F., and Robson, S.C. (2005).
Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active
proteases. Biol. Reprod. 73, 374–381.
Lash, G.E., Otun, H.A., Innes, B.A., Bulmer, J.N., Searle, R.F., and Robson, S.C. (2006). Low
oxygen concentrations inhibit trophoblast cell invasion from early gestation placental explants via
alterations in levels of the urokinase plasminogen activator system. Biol. Reprod. 74, 403–409.
Lash, G.E., Schiessl, B., Kirkley, M., Innes, B.A., Cooper, A., Searle, R.F., Robson, S.C., and
Bulmer, J.N. (2006). Expression of angiogenic growth factors by uterine natural killer cells during
early pregnancy. J. Leukoc. Biol. 80, 572–580.
193
Bibliography
Lawlor, D., Morton, S., Nitsch, D., and Leon, D. (2005). Association between childhood and
adulthood socioeconomic position and pregnancy induced hypertension: results from the Aberdeen
children of the 1950s cohort study. J. Epidemiol. Community Health 59, 49–55.
Le Bouteiller, P., and Piccinni, M.-P. (2008). Human NK cells in pregnant uterus: why there?
Am. J. Reprod. Immunol. New York N 1989 59, 401–406.
Lee, H., Park, H., Kim, Y.J., Kim, H.J., Ahn, Y.M., Park, B., Park, J.H., and Lee, B.E. (2005).
Expression of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in human preeclamptic
placenta: possible implications in the process of trophoblast apoptosis. Placenta 26, 226–233.
Lee, J.-W., Bae, S.-H., Jeong, J.-W., Kim, S.-H., and Kim, K.-W. (2004). Hypoxia-inducible
factor (HIF-1)alpha: its protein stability and biological functions. Exp. Mol. Med. 36, 1–12.
Lee, S.B., Wong, A.P., Kanasaki, K., Xu, Y., Shenoy, V.K., McElrath, T.F., Whitesides, G.M.,
and Kalluri, R. (2010). Preeclampsia: 2-methoxyestradiol induces cytotrophoblast invasion and
vascular development specifically under hypoxic conditions. Am. J. Pathol. 176, 710–720.
Lejeune, B., Van Hoeck, J., and Leroy, F. (1982). Satellite versus total DNA replication in
relation to endopolyploidy of decidual cells in the mouse. Chromosoma 84, 511–516.
Leung, D.N., Smith, S.C., To, K.F., Sahota, D.S., and Baker, P.N. (2001). Increased placental
apoptosis in pregnancies complicated by preeclampsia. Am. J. Obstet. Gynecol. 184, 1249–1250.
Levine, R.J., Lam, C., Qian, C., Yu, K.F., Maynard, S.E., Sachs, B.P., Sibai, B.M., Epstein, F.H.,
Romero, R., Thadhani, R., et al. (2006). Soluble Endoglin and Other Circulating Antiangiogenic
Factors in Preeclampsia. N. Engl. J. Med. 355, 992–1005.
Levine, R.J., Maynard, S.E., Qian, C., Lim, K.-H., England, L.J., Yu, K.F., Schisterman, E.F.,
Thadhani, R., Sachs, B.P., Epstein, F.H., et al. (2004a). Circulating angiogenic factors and the risk of
preeclampsia. N. Engl. J. Med. 350, 672–683.
Levine, R.J., Qian, C., Leshane, E.S., Yu, K.F., England, L.J., Schisterman, E.F., Wataganara, T.,
Romero, R., and Bianchi, D.W. (2004b). Two-stage elevation of cell-free fetal DNA in maternal sera
before onset of preeclampsia. Am. J. Obstet. Gynecol. 190, 707–713.
Levy, R., Smith, S.D., Chandler, K., Sadovsky, Y., and Nelson, D.M. (2000). Apoptosis in human
cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am. J.
Physiol. Cell Physiol. 278, C982–988.
Lewis, C.E., and Pollard, J.W. (2006). Distinct role of macrophages in different tumor
microenvironments. Cancer Res. 66, 605–612.
Li, Y., Fanning, A.S., Anderson, J.M., and Lavie, A. (2005). Structure of the conserved
cytoplasmic C-terminal domain of occludin: identification of the ZO-1 binding surface. J. Mol. Biol.
352, 151–164.
Liljedahl, U., Karlsson, J., Melhus, H., Kurland, L., Lindersson, M., Kahan, T., Nyström, F.,
Lind, L., and Syvänen, A.-C. (2003). A microarray minisequencing system for pharmacogenetic
profiling of antihypertensive drug response. Pharmacogenetics 13, 7–17.
194
Bibliography
Lim, J.H., Kim, S.Y., Kim, D.J., Park, S.Y., Han, H.W., Han, J.Y., Lee, S.W., Yang, J.H., and
Ryu, H.M. (2010). Genetic polymorphism of catechol-O-methyltransferase and cytochrome P450c17α
in preeclampsia. Pharmacogenet. Genomics 20, 605–610.
Lim, J.-H., Lee, Y.-M., Chun, Y.-S., Chen, J., Kim, J.-E., and Park, J.-W. (2010). Sirtuin 1
modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell
38, 864–878.
Lim, J.-H., Lee, Y.-M., Chun, Y.-S., Chen, J., Kim, J.-E., and Park, J.-W. (2010). Sirtuin 1
modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell
38, 864–878.
Lin, D., Edwards, A.S., Fawcett, J.P., Mbamalu, G., Scott, J.D., and Pawson, T. (2000). A
mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity.
Nat. Cell Biol. 2, 540–547.
Lin, J., and August, P. (2005). Genetic thrombophilias and preeclampsia: a meta-analysis. Obstet.
Gynecol. 105, 182–192.
Lipinski, M.J., Frias, J.C., and Fayad, Z.A. (2006). Advances in detection and characterization of
atherosclerosis using contrast agents targeting the macrophage. J. Nucl. Cardiol. Off. Publ. Am. Soc.
Nucl. Cardiol. 13, 699–709.
Lipsitz, P.J. (1971). The clinical and biochemical effects of excess magnesium in the newborn.
Pediatrics 47, 501–509.
Lisy, K., and Peet, D.J. (2008). Turn me on: regulating HIF transcriptional activity. Cell Death
Differ. 15, 642–649.
Liu, B., Peng, X.-C., Zheng, X.-L., Wang, J., and Qin, Y.-W. (2009). MiR-126 restoration downregulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 66,
169–175.
Liu, C.-J., Tsai, M.-M., Hung, P.-S., Kao, S.-Y., Liu, T.-Y., Wu, K.-J., Chiou, S.-H., Lin, S.-C.,
and Chang, K.-W. (2010). miR-31 Ablates Expression of the HIF Regulatory Factor FIH to Activate
the HIF Pathway in Head and Neck Carcinoma. Cancer Res. 70, 1635–1644.
Liu, J., Chakraborty, C., Graham, C.H., Barbin, Y.P., Dixon, S.J., and Lala, P.K. (2003).
Noncatalytic domain of uPA stimulates human extravillous trophoblast migration by using
phospholipase C, phosphatidylinositol 3-kinase and mitogen-activated protein kinase. Exp. Cell Res.
286, 138–151.
Liu, Y.V., Baek, J.H., Zhang, H., Diez, R., Cole, R.N., and Semenza, G.L. (2007). RACK1
competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90
inhibitor-induced degradation of HIF-1alpha. Mol. Cell 25, 207–217.
Llurba, E., Sánchez, O., Domínguez, C., Soro, G., Goya, M., Alijotas-Reig, J., and Cabero, L.
(2013). Smoking during pregnancy: changes in mid-gestation angiogenic factors in women at risk of
developing preeclampsia according to uterine artery Doppler findings. Hypertens. Pregnancy Off. J.
Int. Soc. Study Hypertens. Pregnancy 32, 50–59.
195
Bibliography
Lo, Y.M., Leung, T.N., Tein, M.S., Sargent, I.L., Zhang, J., Lau, T.K., Haines, C.J., and Redman,
C.W. (1999). Quantitative abnormalities of fetal DNA in maternal serum in preeclampsia. Clin. Chem.
45, 184–188.
Lockwood, C.J., Yen, C.-F., Basar, M., Kayisli, U.A., Martel, M., Buhimschi, I., Buhimschi, C.,
Huang, S.J., Krikun, G., and Schatz, F. (2008). Preeclampsia-related inflammatory cytokines regulate
interleukin-6 expression in human decidual cells. Am. J. Pathol. 172, 1571–1579.
Loke, Y.W., King, A., and Burrows, T.D. (1995). Decidua in human implantation. Hum. Reprod.
10, 14–21.
López-Jaramillo, P., Casas, J.P., and Serrano, N. (2001). Preeclampsia: from epidemiological
observations to molecular mechanisms. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Médicas E
Biológicas Soc. Bras. Biofísica Al 34, 1227–1235.
López-Jaramillo, P., Herrera, J.A., Arenas-Mantilla, M., Jáuregui, I.E., and Mendoza, M.A.
(2008). Subclinical infection as a cause of inflammation in preeclampsia. Am. J. Ther. 15, 373–376.
López-Jaramillo, P., Herrera, J.A., Arenas-Mantilla, M., Jáuregui, I.E., and Mendoza, M.A.
(2008). Subclinical infection as a cause of inflammation in preeclampsia. Am. J. Ther. 15, 373–376.
Luo, J.C., and Shibuya, M. (2001). A variant of nuclear localization signal of bipartite-type is
required for the nuclear translocation of hypoxia inducible factors (1alpha, 2alpha and 3alpha).
Oncogene 20, 1435–1444.
Lyall, F. (2005). Priming and remodelling of human placental bed spiral arteries during
pregnancy--a review. Placenta 26 Suppl A, S31–36.
Lyall, F., Greer, I.A., Boswell, F., Young, A., Macara, L.M., and Jeffers, M.D. (1995).
Expression of cell adhesion molecules in placentae from pregnancies complicated by pre-eclampsia
and intrauterine growth retardation. Placenta 16, 579–587.
Lysiak, J.J., Hunt, J., Pringle, G.A., and Lala, P.K. (1995). Localization of transforming growth
factor beta and its natural inhibitor decorin in the human placenta and decidua throughout gestation.
Placenta 16, 221–231.
Mabjeesh, N.J., Escuin, D., LaVallee, T.M., Pribluda, V.S., Swartz, G.M., Johnson, M.S.,
Willard, M.T., Zhong, H., Simons, J.W., and Giannakakou, P. (2003). 2ME2 inhibits tumor growth
and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3, 363–375.
Madazli, R., Benian, A., Aydin, S., Uzun, H., and Tolun, N. (2002). The plasma and placental
levels of malondialdehyde, glutathione and superoxide dismutase in pre-eclampsia. J. Obstet.
Gynaecol. J. Inst. Obstet. Gynaecol. 22, 477–480.
Magee, L.A. (2003). Hydralazine for treatment of severe hypertension in pregnancy: metaanalysis. BMJ 327, 955–0.
Magee, L.A., Abalos, E., von Dadelszen, P., Sibai, B., Easterling, T., Walkinshaw, S., and CHIPS
Study Group (2011). How to manage hypertension in pregnancy effectively. Br. J. Clin. Pharmacol.
72, 394–401.
196
Bibliography
Magee, L.A., Yong, P.J., Espinosa, V., Côté, A.M., Chen, I., and von Dadelszen, P. (2009).
Expectant management of severe preeclampsia remote from term: a structured systematic review.
Hypertens. Pregnancy Off. J. Int. Soc. Study Hypertens. Pregnancy 28, 312–347.
Maharaj, A.S.R., Saint-Geniez, M., Maldonado, A.E., and D’Amore, P.A. (2006). Vascular
endothelial growth factor localization in the adult. Am. J. Pathol. 168, 639–648.
Makino, Y., Cao, R., Svensson, K., Bertilsson, G., Asman, M., Tanaka, H., Cao, Y., Berkenstam,
A., and Poellinger, L. (2001). Inhibitory PAS domain protein is a negative regulator of hypoxiainducible gene expression. Nature 414, 550–554.
Makino, Y., Uenishi, R., Okamoto, K., Isoe, T., Hosono, O., Tanaka, H., Kanopka, A., Poellinger,
L., Haneda, M., and Morimoto, C. (2007). Transcriptional up-regulation of inhibitory PAS domain
protein gene expression by hypoxia-inducible factor 1 (HIF-1): a negative feedback regulatory circuit
in HIF-1-mediated signaling in hypoxic cells. J. Biol. Chem. 282, 14073–14082.
Maltepe, E., Keith, B., Arsham, A.M., Brorson, J.R., and Simon, M.C. (2000). The role of
ARNT2 in tumor angiogenesis and the neural response to hypoxia. Biochem. Biophys. Res. Commun.
273, 231–238.
Maltepe, E., Krampitz, G.W., Okazaki, K.M., Red-Horse, K., Mak, W., Simon, M.C., and Fisher,
S.J. (2005). Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate
in the placenta. Dev. Camb. Engl. 132, 3393–3403.
Manaster, I., and Mandelboim, O. (2010). The unique properties of uterine NK cells. Am. J.
Reprod. Immunol. New York N 1989 63, 434–444.
Many, A., Hubel, C.A., Fisher, S.J., Roberts, J.M., and Zhou, Y. (2000). Invasive
cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am. J. Pathol. 156, 321–331.
Marks, D.S., Vita, J.A., Folts, J.D., Keaney, J.F., Welch, G.N., and Loscalzo, J. (1995). Inhibition
of neointimal proliferation in rabbits after vascular injury by a single treatment with a protein adduct
of nitric oxide. J. Clin. Invest. 96, 2630–2638.
Maruyama, K., Fukasaka, M., Vandenbon, A., Saitoh, T., Kawasaki, T., Kondo, T., Yokoyama,
K.K., Kidoya, H., Takakura, N., Standley, D., et al. (2012). The transcription factor Jdp2 controls bone
homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation.
Immunity 37, 1024–1036.
Masson, N., Singleton, R.S., Sekirnik, R., Trudgian, D.C., Ambrose, L.J., Miranda, M.X., Tian,
Y.-M., Kessler, B.M., Schofield, C.J., and Ratcliffe, P.J. (2012). The FIH hydroxylase is a cellular
peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 13, 251–257.
Masson, N., Willam, C., Maxwell, P.H., Pugh, C.W., and Ratcliffe, P.J. (2001). Independent
function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl
hydroxylation. EMBO J. 20, 5197–5206.
Matsuo, K., Kooshesh, S., Dinc, M., Sun, C.-C.J., Kimura, T., and Baschat, A.A. (2007). Late
postpartum eclampsia: report of two cases managed by uterine curettage and review of the literature.
Am. J. Perinatol. 24, 257–266.
197
Bibliography
Mattila, P., Majuri, M.L., Mattila, P.S., and Renkonen, R. (1992). TNF alpha-induced expression
of endothelial adhesion molecules, ICAM-1 and VCAM-1, is linked to protein kinase C activation.
Scand. J. Immunol. 36, 159–165.
Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E.,
Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. (1999). The tumour suppressor protein
VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275.
Maynard, M.A., Evans, A.J., Shi, W., Kim, W.Y., Liu, F.-F., and Ohh, M. (2007). Dominantnegative HIF-3 alpha 4 suppresses VHL-null renal cell carcinoma progression. Cell Cycle Georget.
Tex 6, 2810–2816.
Maynard, S.E., Min, J.-Y., Merchan, J., Lim, K.-H., Li, J., Mondal, S., Libermann, T.A., Morgan,
J.P., Sellke, F.W., Stillman, I.E., et al. (2003). Excess placental soluble fms-like tyrosine kinase 1
(sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J.
Clin. Invest. 111, 649–658.
Maynard, S.E., Min, J.-Y., Merchan, J., Lim, K.-H., Li, J., Mondal, S., Libermann, T.A., Morgan,
J.P., Sellke, F.W., Stillman, I.E., et al. (2003). Excess placental soluble fms-like tyrosine kinase 1
(sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J.
Clin. Invest. 111, 649–658.
Maynard, S.E., Min, J.-Y., Merchan, J., Lim, K.-H., Li, J., Mondal, S., Libermann, T.A., Morgan,
J.P., Sellke, F.W., Stillman, I.E., et al. (2003). Excess placental soluble fms-like tyrosine kinase 1
(sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J.
Clin. Invest. 111, 649–658.
McGrath, J.C., Deighan, C., Briones, A.M., Shafaroudi, M.M., McBride, M., Adler, J., Arribas,
S.M., Vila, E., and Daly, C.J. (2005). New aspects of vascular remodelling: the involvement of all
vascular cell types. Exp. Physiol. 90, 469–475.
McKeeman, G.C., Ardill, J.E.S., Caldwell, C.M., Hunter, A.J., and McClure, N. (2004). Soluble
vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who
have preeclampsia develop. Am. J. Obstet. Gynecol. 191, 1240–1246.
McKinnon, T., Chakraborty, C., Gleeson, L.M., Chidiac, P., and Lala, P.K. (2001). Stimulation of
human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving
inhibitory G protein(s) and phosphorylation of MAPK. J. Clin. Endocrinol. Metab. 86, 3665–3674.
McMahon, S., Charbonneau, M., Grandmont, S., Richard, D.E., and Dubois, C.M. (2006).
Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective
inhibition of PHD2 expression. J. Biol. Chem. 281, 24171–24181.
Meade, E.S., Ma, Y.Y., and Guller, S. (2007). Role of hypoxia-inducible transcription factors
1alpha and 2alpha in the regulation of plasminogen activator inhibitor-1 expression in a human
trophoblast cell line. Placenta 28, 1012–1019.
Medica, I., Kastrin, A., and Peterlin, B. (2007). Genetic polymorphisms in vasoactive genes and
preeclampsia: a meta-analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 131, 115–126.
198
Bibliography
Meekins, J.W., Pijnenborg, R., Hanssens, M., McFadyen, I.R., and van Asshe, A. (1994). A study
of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic
pregnancies. Br. J. Obstet. Gynaecol. 101, 669–674.
Meher, S., and Duley, L. (2007). Nitric oxide for preventing pre-eclampsia and its complications.
Cochrane Database Syst. Rev. Online CD006490.
Mello, G., Parretti, E., Fatini, C., Riviello, C., Gensini, F., Marchionni, M., Scarselli, G.F.,
Gensini, G.F., and Abbate, R. (2005). Low-molecular-weight heparin lowers the recurrence rate of
preeclampsia and restores the physiological vascular changes in angiotensin-converting enzyme DD
women. Hypertension 45, 86–91.
Mendelsohn, M.E., O’Neill, S., George, D., and Loscalzo, J. (1990). Inhibition of fibrinogen
binding to human platelets by S-nitroso-N-acetylcysteine. J. Biol. Chem. 265, 19028–19034.
Meseguer, M., Aplin, J.D., Caballero-Campo, P., O’Connor, J.E., Martín, J.C., Remohí, J.,
Pellicer, A., and Simón, C. (2001). Human endometrial mucin MUC1 is up-regulated by progesterone
and down-regulated in vitro by the human blastocyst. Biol. Reprod. 64, 590–601.
Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J., and Brüne, B. (2003). Nitric oxide impairs
normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol. Biol. Cell 14, 3470–
3481.
Michell, B.J., Chen, Z., Tiganis, T., Stapleton, D., Katsis, F., Power, D.A., Sim, A.T., and Kemp,
B.E. (2001). Coordinated Control of Endothelial Nitric-oxide Synthase Phosphorylation by Protein
Kinase C and the cAMP-dependent Protein Kinase. J. Biol. Chem. 276, 17625–17628.
Michiels, C., Minet, E., Michel, G., Mottet, D., Piret, J.-P., and Raes, M. (2001). HIF-1 and AP-1
Cooperate to Increase Gene Expression in Hypoxia: Role of MAP Kinases. IUBMB Life 52, 49–53.
Milanini-Mongiat, J., Pouysségur, J., and Pagès, G. (2002). Identification of two Sp1
phosphorylation sites for p42/p44 mitogen-activated protein kinases: their implication in vascular
endothelial growth factor gene transcription. J. Biol. Chem. 277, 20631–20639.
Mills JL, DerSimonian R, Raymond E, and et al (1999). Prostacyclin and thromboxane changes
predating clinical onset of preeclampsia: A multicenter prospective study. JAMA 282, 356–362.
Minchenko, A., and Caro, J. (2000). Regulation of endothelin-1 gene expression in human
microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Mol. Cell.
Biochem. 208, 53–62.
Minet, E., Michel, G., Mottet, D., Piret, J.P., Barbieux, A., Raes, M., and Michiels, C. (2001). cJUN gene induction and AP-1 activity is regulated by a JNK-dependent pathway in hypoxic HepG2
cells. Exp. Cell Res. 265, 114–124.
Mitra, S., Goyal, T., and Mehta, J.L. (2011). Oxidized LDL, LOX-1 and atherosclerosis.
Cardiovasc. Drugs Ther. Spons. Int. Soc. Cardiovasc. Pharmacother. 25, 419–429.
Moin, S.M., Chandra, V., Arya, R., and Jameel, S. (2009). The hepatitis E virus ORF3 protein
stabilizes HIF-1α and enhances HIF-1-mediated transcriptional activity through p300/CBP. Cell.
Microbiol. 11, 1409–1421.
199
Bibliography
Molnár, M., and Hertelendy, F. (1995). Signal transduction in rat myometrial cells: comparison of
the actions of endothelin-1, oxytocin and prostaglandin F2 alpha. Eur. J. Endocrinol. Eur. Fed. Endocr.
Soc. 133, 467–474.
Moncada, S., and Vane, J.R. (1978). Pharmacology and endogenous roles of prostaglandin
endoperoxides, thromboxane A2, and prostacyclin. Pharmacol. Rev. 30, 293–331.
Moncada, S., Palmer, R.M., and Higgs, E.A. (1991). Nitric oxide: physiology, pathophysiology,
and pharmacology. Pharmacol. Rev. 43, 109–142.
Mondon, F., Mignot, T.-M., Rebourcet, R., Jammes, H., Danan, J.-L., Ferré, F., and Vaiman, D.
(2005). Profiling of oxygen-modulated gene expression in early human placenta by systematic
sequencing of suppressive subtractive hybridization products. Physiol. Genomics 22, 99–107.
Mor, G., Romero, R., Aldo, P.B., and Abrahams, V.M. (2005). Is the trophoblast an immune
regulator? The role of Toll-like receptors during pregnancy. Crit. Rev. Immunol. 25, 375–388.
Moses, E.K., Lade, J.A., Guo, G., Wilton, A.N., Grehan, M., Freed, K., Borg, A., Terwilliger,
J.D., North, R., Cooper, D.W., et al. (2000). A Genome Scan in Families from Australia and New
Zealand Confirms the Presence of a Maternal Susceptibility Locus for Pre-Eclampsia, on
Chromosome 2. Am. J. Hum. Genet. 67, 1581–1585.
Mount, P.F., Kemp, B.E., and Power, D.A. (2007). Regulation of endothelial and myocardial NO
synthesis by multi-site eNOS phosphorylation. J. Mol. Cell. Cardiol. 42, 271–279.
Mousa, A.A., Archer, K.J., Cappello, R., Estrada-Gutierrez, G., Isaacs, C.R., Strauss, J.F., and
Walsh, S.W. (2012). DNA Methylation is Altered in Maternal Blood Vessels of Women With
Preeclampsia. Reprod. Sci. 1933719112450336.
Mütze, S., Rudnik-Schöneborn, S., Zerres, K., and Rath, W. (2008). Genes and the preeclampsia
syndrome. J. Perinat. Med. 36, 38–58.
Myatt, L., Rosenfield, R.B., Eis, A.L., Brockman, D.E., Greer, I., and Lyall, F. (1996).
Nitrotyrosine residues in placenta. Evidence of peroxynitrite formation and action. Hypertension 28,
488–493.
Nadar, S.K., Al Yemeni, E., Blann, A.D., and Lip, G.Y.H. (2004). Thrombomodulin, von
Willebrand factor and E-selectin as plasma markers of endothelial damage/dysfunction and activation
in pregnancy induced hypertension. Thromb. Res. 113, 123–128.
Nagamatsu, T., and Schust, D.J. (2010). The contribution of macrophages to normal and
pathological pregnancies. Am. J. Reprod. Immunol. New York N 1989 63, 460–471.
Nagy, B., Lundán, T., Larramendy, M.L., Aalto, Y., Zhu, Y., Niini, T., Edgren, H., Ferrer, A.,
Vilpo, J., Elonen, E., et al. (2003). Abnormal expression of apoptosis-related genes in haematological
malignancies: overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br. J.
Haematol. 120, 434–441.
Nakade, K., Pan, J., Yamasaki, T., Murata, T., Wasylyk, B., and Yokoyama, K.K. (2009). JDP2
(Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative
senescence. J. Biol. Chem. 284, 10808–10817.
200
Bibliography
Nakade, K., Pan, J., Yamasaki, T., Murata, T., Wasylyk, B., and Yokoyama, K.K. (2009). JDP2
(Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative
senescence. J. Biol. Chem. 284, 10808–10817.
Nakade, K., Pan, J., Yoshiki, A., Ugai, H., Kimura, M., Liu, B., Li, H., Obata, Y., Iwama, M.,
Itohara, S., et al. (2007). JDP2 suppresses adipocyte differentiation by regulating histone acetylation.
Cell Death Differ. 14, 1398–1405.
Naruse, K., Lash, G.E., Bulmer, J.N., Innes, B.A., Otun, H.A., Searle, R.F., and Robson, S.C.
(2009). The urokinase plasminogen activator (uPA) system in uterine natural killer cells in the
placental bed during early pregnancy. Placenta 30, 398–404.
National Collaborating Centre for Women’s and Children’s Health (UK) (2010). Hypertension in
Pregnancy: The Management of Hypertensive Disorders During Pregnancy (London: RCOG Press).
Nava-Salazar, S., Sanchez-Rodriguez, E.N., Mendoza-Rodriguez, C.A., Moran, C., RomeroArauz, J.F., and Cerbon, M.A. (2011). Polymorphisms in the hypoxia-inducible factor 1 alpha gene in
Mexican patients with preeclampsia: A case-control study. BMC Res. Notes 4, 68.
Neal, C.R., Hunter, A.J., Harper, S.J., Soothill, P.W., and Bates, D.O. (2004). Plasma from
women with severe pre-eclampsia increases microvascular permeability in an animal model in vivo.
Clin. Sci. Lond. Engl. 1979 107, 399–405.
Neumann, P., Gertzberg, N., Vaughan, E., Weisbrot, J., Woodburn, R., Lambert, W., and
Johnson, A. (2006). Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and
nitration of actin. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L674–L684.
Nevo, O., Soleymanlou, N., Wu, Y., Xu, J., Kingdom, J., Many, A., Zamudio, S., and Caniggia, I.
(2006). Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is
mediated by HIF-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1085–1093.
Nicol, C.J., Zielenski, J., Tsui, L.C., and Wells, P.G. (2000). An embryoprotective role for
glucose-6-phosphate dehydrogenase in developmental oxidative stress and chemical teratogenesis.
FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 14, 111–127.
Nicolaides, K.H., Bindra, R., Turan, O.M., Chefetz, I., Sammar, M., Meiri, H., Tal, J., and
Cuckle, H.S. (2006). A novel approach to first-trimester screening for early pre-eclampsia combining
serum PP-13 and Doppler ultrasound. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet.
Gynecol. 27, 13–17.
Nishi, H., Nakada, T., Hokamura, M., Osakabe, Y., Itokazu, O., Huang, L.E., and Isaka, K.
(2004). Hypoxia-inducible factor-1 transactivates transforming growth factor-beta3 in trophoblast.
Endocrinology 145, 4113–4118.
Nishizawa, H., Pryor-Koishi, K., Kato, T., Kowa, H., Kurahashi, H., and Udagawa, Y. (2007).
Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and
late onset severe pre-eclampsia. Placenta 28, 487–497.
Nishizawa, H., Pryor-Koishi, K., Kato, T., Kowa, H., Kurahashi, H., and Udagawa, Y. (2007).
Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and
late onset severe pre-eclampsia. Placenta 28, 487–497.
201
Bibliography
Niu, G., Wright, K.L., Huang, M., Song, L., Haura, E., Turkson, J., Zhang, S., Wang, T.,
Sinibaldi, D., Coppola, D., et al. (2002). Constitutive Stat3 activity up-regulates VEGF expression and
tumor angiogenesis. Oncogene 21, 2000–2008.
Nochy, D., Birembaut, P., Hinglais, N., Freund, M., Idatte, J.M., Jacquot, C., Chartier, M., and
Bariety, J. (1980). Renal lesions in the hypertensive syndromes of pregnancy: immunomorphological
and ultrastructural studies in 114 cases. Clin. Nephrol. 13, 155–162.
Noris, M., Perico, N., and Remuzzi, G. (2005). Mechanisms of disease: Pre-eclampsia. Nat. Clin.
Pract. Nephrol. 1, 98–114; quiz 120.
Noris, M., Todeschini, M., Cassis, P., Pasta, F., Cappellini, A., Bonazzola, S., Macconi, D.,
Maucci, R., Porrati, F., Benigni, A., et al. (2004). L-arginine depletion in preeclampsia orients nitric
oxide synthase toward oxidant species. Hypertension 43, 614–622.
North, R.A., Ferrier, C., Long, D., Townend, K., and Kincaid-Smith, P. (1994). Uterine artery
Doppler flow velocity waveforms in the second trimester for the prediction of preeclampsia and fetal
growth retardation. Obstet. Gynecol. 83, 378–386.
Nova, A., Sibai, B.M., Barton, J.R., Mercer, B.M., and Mitchell, M.D. (1991). Maternal plasma
level of endothelin is increased in preeclampsia. Am. J. Obstet. Gynecol. 165, 724–727.
Novaro, V., Colman-Lerner, A., Ortega, F.V., Jawerbaum, A., Paz, D., Lo Nostro, F., Pustovrh,
C., Gimeno, M.F., and González, E. (2001). Regulation of metalloproteinases by nitric oxide in human
trophoblast cells in culture. Reprod. Fertil. Dev. 13, 411–420.
Nunbhakdi-Craig, V., Machleidt, T., Ogris, E., Bellotto, D., White, C.L., and Sontag, E. (2002).
Protein phosphatase 2A associates with and regulates atypical PKC and the epithelial tight junction
complex. J. Cell Biol. 158, 967–978.
Oskolkova, O.V., Afonyushkin, T., Leitner, A., von Schlieffen, E., Gargalovic, P.S., Lusis, A.J.,
Binder, B.R., and Bochkov, V.N. (2008). ATF4-dependent transcription is a key mechanism in VEGF
up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of
unfolded protein response. Blood 112, 330–339.
Osungbade, K.O., and Ige, O.K. (2011). Public Health Perspectives of Preeclampsia in
Developing Countries: Implication for Health System Strengthening. J. Pregnancy 2011.
Paarlberg, K.M., de Jong, C.L., van Geijn, H.P., van Kamp, G.J., Heinen, A.G., and Dekker, G.A.
(1998). Vasoactive mediators in pregnancy-induced hypertensive disorders: a longitudinal study. Am.
J. Obstet. Gynecol. 179, 1559–1564.
Pagé, E.L., Robitaille, G.A., Pouysségur, J., and Richard, D.E. (2002). Induction of hypoxiainducible factor-1alpha by transcriptional and translational mechanisms. J. Biol. Chem. 277, 48403–
48409.
Pagès, G. (2007). Sp3-mediated VEGF regulation is dependent on phosphorylation by extracellular signals regulated kinases (Erk). J. Cell. Physiol. 213, 454–463.
Palmer, R.M., Ashton, D.S., and Moncada, S. (1988). Vascular endothelial cells synthesize nitric
oxide from L-arginine. Nature 333, 664–666.
202
Bibliography
Palmer, S.K., Moore, L.G., Young, D., Cregger, B., Berman, J.C., and Zamudio, S. (1999).
Altered blood pressure course during normal pregnancy and increased preeclampsia at high altitude
(3100 meters) in Colorado. Am. J. Obstet. Gynecol. 180, 1161–1168.
Pan, J., Jin, C., Murata, T., and Yokoyama, K.K. (2003). Sequence specific transcription factor,
JDP2 interacts with histone and inhibits p300-mediated histone acetylation. Nucleic Acids Res. Suppl.
2001 305–306.
Pan, Y., Mansfield, K.D., Bertozzi, C.C., Rudenko, V., Chan, D.A., Giaccia, A.J., and Simon,
M.C. (2007). Multiple factors affecting cellular redox status and energy metabolism modulate
hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell. Biol. 27, 912–925.
Panza, J.A., Quyyumi, A.A., Callahan, T.S., and Epstein, S.E. (1993). Effect of antihypertensive
treatment on endothelium-dependent vascular relaxation in patients with essential hypertension. J. Am.
Coll. Cardiol. 21, 1145–1151.
Papageorghiou, A.T., Yu, C.K.H., Cicero, S., Bower, S., and Nicolaides, K.H. (2002). Secondtrimester uterine artery Doppler screening in unselected populations: a review. J. Matern.-Fetal
Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat.
Obstet. 12, 78–88.
Papapetropoulos, A., García-Cardeña, G., Madri, J.A., and Sessa, W.C. (1997). Nitric oxide
production contributes to the angiogenic properties of vascular endothelial growth factor in human
endothelial cells. J. Clin. Invest. 100, 3131–3139.
Parrish, M.R., Martin, J.N., Jr, Lamarca, B.B., Ellis, B., Parrish, S.A., Owens, M.Y., and May,
W.L. (2013). Randomized, placebo controlled, double blind trial evaluating early pregnancy
phytonutrient supplementation in the prevention of preeclampsia. J. Perinatol. Off. J. Calif. Perinat.
Assoc.
Parrish, M.R., Murphy, S.R., Rutland, S., Wallace, K., Wenzel, K., Wallukat, G., Keiser, S., Ray,
L.F., Dechend, R., Martin, J.N., et al. (2010). The effect of immune factors, tumor necrosis factoralpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1
and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 23,
911–916.
Patel, J., Landers, K., Mortimer, R.H., and Richard, K. (2010). Regulation of Hypoxia Inducible
Factors (HIF) in Hypoxia and Normoxia During Placental Development. Placenta 31, 951–957.
Peluffo, G., and Radi, R. (2007). Biochemistry of protein tyrosine nitration in cardiovascular
pathology. Cardiovasc. Res. 75, 291–302.
Pendergrass, S.A., Hayes, E., Farina, G., Lemaire, R., Farber, H.W., Whitfield, M.L., and
Lafyatis, R. (2010). Limited Systemic Sclerosis Patients with Pulmonary Arterial Hypertension Show
Biomarkers of Inflammation and Vascular Injury. PLoS ONE 5, e12106.
Peng, J., Zhang, L., Drysdale, L., and Fong, G.-H. (2000). The transcription factor EPAS1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci.
97, 8386–8391.
203
Bibliography
Pernow, J., Kaijser, L., Lundberg, J.M., and Ahlborg, G. (1996). Comparable potent coronary
constrictor effects of endothelin-1 and big endothelin-1 in humans. Circulation 94, 2077–2082.
Perrotta, S., Nobili, B., Ferraro, M., Migliaccio, C., Borriello, A., Cucciolla, V., Martinelli, V.,
Rossi, F., Punzo, F., Cirillo, P., et al. (2006). Von Hippel-Lindau-dependent polycythemia is endemic
on the island of Ischia: identification of a novel cluster. Blood 107, 514–519.
Perry, K.G., Jr, and Martin, J.N., Jr (1992). Abnormal hemostasis and coagulopathy in
preeclampsia and eclampsia. Clin. Obstet. Gynecol. 35, 338–350.
Peterson, H., Laivuori, H., Kerkelä, E., Jiao, H., Hiltunen, L., Heino, S., Tiala, I., Knuutila, S.,
Rasi, V., Kere, J., et al. (2009). ROCK2 allelic variants are not associated with pre-eclampsia
susceptibility in the Finnish population. Mol. Hum. Reprod. 15, 443–449.
Petit, P., Top, M., Chantraine, F., Brichant, J.F., Dewandre, P.Y., and Foidart, J.M. (2009).
[Treatment of severe preeclampsia: until when and for what risks/benefits?]. Rev. Médicale Liège 64,
620–625.
Pijnenborg, R., McLaughlin, P.J., Vercruysse, L., Hanssens, M., Johnson, P.M., Keith, J.C., Jr,
and Van Assche, F.A. (1998). Immunolocalization of tumour necrosis factor-alpha (TNF-alpha) in the
placental bed of normotensive and hypertensive human pregnancies. Placenta 19, 231–239.
Piu, F., Aronheim, A., Katz, S., and Karin, M. (2001). AP-1 Repressor Protein JDP-2: Inhibition
of UV-Mediated Apoptosis through p53 Down-Regulation. Mol. Cell. Biol. 21, 3012–3024.
Platt, D.H., Bartoli, M., El-Remessy, A.B., Al-Shabrawey, M., Lemtalsi, T., Fulton, D., and
Caldwell, R.B. (2005). Peroxynitrite increases VEGF expression in vascular endothelial cells via
STAT3. Free Radic. Biol. Med. 39, 1353–1361.
Pohl, U., Holtz, J., Busse, R., and Bassenge, E. (1986). Crucial role of endothelium in the
vasodilator response to increased flow in vivo. Hypertension 8, 37–44.
Pongcharoen, S., Somran, J., Sritippayawan, S., Niumsup, P., Chanchan, P., Butkhamchot, P.,
Tatiwat, P., Kunngurn, S., and Searle, R.F. (2007). Interleukin-17 expression in the human placenta.
Placenta 28, 59–63.
Poon, L.C.Y., Akolekar, R., Lachmann, R., Beta, J., and Nicolaides, K.H. (2010). Hypertensive
disorders in pregnancy: screening by biophysical and biochemical markers at 11-13 weeks. Ultrasound
Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 35, 662–670.
Poon, L.C.Y., Syngelaki, A., Akolekar, R., Lai, J., and Nicolaides, K.H. (2013). Combined
screening for preeclampsia and small for gestational age at 11-13 weeks. Fetal Diagn. Ther. 33, 16–27.
Popovici, R.M., Kao, L.C., and Giudice, L.C. (2000). Discovery of new inducible genes in in
vitro decidualized human endometrial stromal cells using microarray technology. Endocrinology 141,
3510–3513.
Pottecher, T., Luton, D., Zupan, V., and Collet, M. (2009). Prise en charge multidisciplinaire des
formes graves de prééclampsie (SFAR : Société française d’anesthésie et de réanimation CNGOF :
Collège national des gynécologues et obstétriciens français SFNN : Société française de néonatologie
SFMP : Société française de médecine périnatale).
204
Bibliography
Potter, M.D., Barbero, S., and Cheresh, D.A. (2005). Tyrosine phosphorylation of VE-cadherin
prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J. Biol.
Chem. 280, 31906–31912.
Pringle, K.G., Kind, K.L., Thompson, J.G., and Roberts, C.T. (2007). Complex interactions
between hypoxia inducible factors, insulin-like growth factor-II and oxygen in early murine
trophoblasts. Placenta 28, 1147–1157.
Purwosunu, Y., Sekizawa, A., Okazaki, S., Farina, A., Wibowo, N., Nakamura, M., Rizzo, N.,
Saito, H., and Okai, T. (2009). Prediction of preeclampsia by analysis of cell-free messenger RNA in
maternal plasma. Am. J. Obstet. Gynecol. 200, 386.e1–7.
Qian, D., Lin, H.-Y., Wang, H.-M., Zhang, X., Liu, D.-L., Li, Q.-L., and Zhu, C. (2004).
Normoxic induction of the hypoxic-inducible factor-1 alpha by interleukin-1 beta involves the
extracellular signal-regulated kinase 1/2 pathway in normal human cytotrophoblast cells. Biol. Reprod.
70, 1822–1827.
Qian, D.Z., Kachhap, S.K., Collis, S.J., Verheul, H.M.W., Carducci, M.A., Atadja, P., and Pili, R.
(2006). Class II histone deacetylases are associated with VHL-independent regulation of hypoxiainducible factor 1 alpha. Cancer Res. 66, 8814–8821.
Qian, D.Z., Kachhap, S.K., Collis, S.J., Verheul, H.M.W., Carducci, M.A., Atadja, P., and Pili, R.
(2006). Class II histone deacetylases are associated with VHL-independent regulation of hypoxiainducible factor 1 alpha. Cancer Res. 66, 8814–8821.
Qing, X., Redecha, P.B., Burmeister, M.A., Tomlinson, S., D’Agati, V.D., Davisson, R.L., and
Salmon, J.E. (2011). Targeted inhibition of complement activation prevents features of preeclampsia
in mice. Kidney Int. 79, 331–339.
Qiu, C., Phung, T.T.T., Vadachkoria, S., Muy-Rivera, M., Sanchez, S.E., and Williams, M.A.
(2006). Oxidized low-density lipoprotein (Oxidized LDL) and the risk of preeclampsia. Physiol. Res.
Acad. Sci. Bohemoslov. 55, 491–500.
Queenan, J.T., Jr, Kao, L.C., Arboleda, C.E., Ulloa-Aguirre, A., Golos, T.G., Cines, D.B., and
Strauss, J.F., 3rd (1987). Regulation of urokinase-type plasminogen activator production by cultured
human cytotrophoblasts. J. Biol. Chem. 262, 10903–10906.
Quero, L., Dubois, L., Lieuwes, N.G., Hennequin, C., and Lambin, P. (2011). miR-210 as a
marker of chronic hypoxia, but not a therapeutic target in prostate cancer. Radiother. Oncol. 101, 203–
208.
Raijmakers, M.T.M., Dechend, R., and Poston, L. (2004). Oxidative stress and preeclampsia:
rationale for antioxidant clinical trials. Hypertension 44, 374–380.
Rajakumar, A., and Conrad, K.P. (2000). Expression, ontogeny, and regulation of hypoxiainducible transcription factors in the human placenta. Biol. Reprod. 63, 559–569.
Rajakumar, A., Brandon, H.M., Daftary, A., Ness, R., and Conrad, K.P. (2004). Evidence for the
functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae.
Placenta 25, 763–769.
205
Bibliography
Rajakumar, A., Cerdeira, A.S., Rana, S., Zsengeller, Z., Edmunds, L., Jeyabalan, A., Hubel, C.A.,
Stillman, I.E., Parikh, S.M., and Karumanchi, S.A. (2012). Transcriptionally active syncytial
aggregates in the maternal circulation may contribute to circulating soluble fms-like tyrosine kinase 1
in preeclampsia. Hypertension 59, 256–264.
Rajakumar, A., Michael, H.M., Daftary, A., Jeyabalan, A., Gilmour, C., and Conrad, K.P. (2008).
Proteasomal activity in placentas from women with preeclampsia and intrauterine growth restriction:
implications for expression of HIF-alpha proteins. Placenta 29, 290–299.
Rajakumar, A., Whitelock, K.A., Weissfeld, L.A., Daftary, A.R., Markovic, N., and Conrad, K.P.
(2001). Selective overexpression of the hypoxia-inducible transcription factor, HIF-2alpha, in
placentas from women with preeclampsia. Biol. Reprod. 64, 499–506.
Rajakumar, A., Whitelock, K.A., Weissfeld, L.A., Daftary, A.R., Markovic, N., and Conrad, K.P.
(2001). Selective overexpression of the hypoxia-inducible transcription factor, HIF-2alpha, in
placentas from women with preeclampsia. Biol. Reprod. 64, 499–506.
Ramsay, J.E., Stewart, F., Greer, I.A., and Sattar, N. (2003). Microvascular dysfunction: a link
between pre-eclampsia and maternal coronary heart disease. BJOG Int. J. Obstet. Gynaecol. 110,
1029–1031.
Rana, S., Powe, C.E., Salahuddin, S., Verlohren, S., Perschel, F.H., Levine, R.J., Lim, K.-H.,
Wenger, J.B., Thadhani, R., and Karumanchi, S.A. (2012). Angiogenic factors and the risk of adverse
outcomes in women with suspected preeclampsia. Circulation 125, 911–919.
Ranta, V., Viinikka, L., Halmesmäki, E., and Ylikorkala, O. (1999). Nitric oxide production with
preeclampsia. Obstet. Gynecol. 93, 442–445.
Redman, C.W., and Sargent, I.L. (2000). Placental debris, oxidative stress and pre-eclampsia.
Placenta 21, 597–602.
Redman, C.W.G., and Sargent, I.L. (2004). Preeclampsia and the systemic inflammatory
response. Semin. Nephrol. 24, 565–570.
Rein, D.T., Schondorf, T., Gohring, U.J., Kurbacher, C.M., Pinto, I., Breidenbach, M., Mallmann,
P., Kolhagen, H., and Engel, H. (2002). Cytokine expression in peripheral blood lymphocytes
indicates a switch to T(HELPER) cells in patients with preeclampsia. J. Reprod. Immunol. 54, 133–
142.
Reisinger, K., Kaufmann, R., and Gille, J. (2003). Increased Sp1 phosphorylation as a mechanism
of hepatocyte growth factor (HGF/SF)-induced vascular endothelial growth factor (VEGF/VPF)
transcription. J. Cell Sci. 116, 225–238.
Repovic, P., Fears, C.Y., Gladson, C.L., and Benveniste, E.N. (2003). Oncostatin-M induction of
vascular endothelial growth factor expression in astroglioma cells. Oncogene 22, 8117–8124.
Richard, D.E., Berra, E., and Pouyssegur, J. (2000). Nonhypoxic pathway mediates the induction
of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J. Biol. Chem. 275, 26765–26771.
206
Bibliography
Rigourd, V., Chauvet, C., Chelbi, S.T., Rebourcet, R., Mondon, F., Letourneur, F., Mignot, T.M.,
Barbaux, S., and Vaiman, D. (2008). STOX1 overexpression in choriocarcinoma cells mimics
transcriptional alterations observed in preeclamptic placentas. PloS One 3, e3905.
Ristimäki, A., Ylikorkala, O., and Viinikka, L. (1990). Effect of growth factors on human
vascular endothelial cell prostacyclin production. Arter. Dallas Tex 10, 653–657.
Roberge, S., Giguère, Y., Villa, P., Nicolaides, K., Vainio, M., Forest, J.-C., von Dadelzen, P.,
Vaiman, D., Tapp, S., and Bujold, E. (2012a). Early administration of low-dose aspirin for the
prevention of severe and mild preeclampsia: a systematic review and meta-analysis. Am. J. Perinatol.
29, 551–556.
Roberge, S., Villa, P., Nicolaides, K., Giguère, Y., Vainio, M., Bakthi, A., Ebrashy, A., and
Bujold, E. (2012b). Early administration of low-dose aspirin for the prevention of preterm and term
preeclampsia: a systematic review and meta-analysis. Fetal Diagn. Ther. 31, 141–146.
Roberts, J.M., and Gammill, H.S. (2005). Preeclampsia: recent insights. Hypertension 46, 1243–
1249.
Roberts, J.M., Taylor, R.N., Musci, T.J., Rodgers, G.M., Hubel, C.A., and McLaughlin, M.K.
(1989). Preeclampsia: an endothelial cell disorder. Am. J. Obstet. Gynecol. 161, 1200–1204.
Roberts, W.G., and Palade, G.E. (1997). Neovasculature induced by vascular endothelial growth
factor is fenestrated. Cancer Res. 57, 765–772.
Rodesch, F., Simon, P., Donner, C., and Jauniaux, E. (1992). Oxygen measurements in
endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 80, 283–285.
Rodesch, F., Simon, P., Donner, C., and Jauniaux, E. (1992). Oxygen measurements in
endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 80, 283–285.
Rodger, M.A., Betancourt, M.T., Clark, P., Lindqvist, P.G., Dizon-Townson, D., Said, J.,
Seligsohn, U., Carrier, M., Salomon, O., and Greer, I.A. (2010). The Association of Factor V Leiden
and Prothrombin Gene Mutation and Placenta-Mediated Pregnancy Complications: A Systematic
Review and Meta-analysis of Prospective Cohort Studies. PLoS Med 7, e1000292.
Roggensack, A.M., Zhang, Y., and Davidge, S.T. (1999). Evidence for peroxynitrite formation in
the vasculature of women with preeclampsia. Hypertension 33, 83–89.
Rolfo, A., Many, A., Racano, A., Tal, R., Tagliaferro, A., Ietta, F., Wang, J., Post, M., and
Caniggia, I. (2010). Abnormalities in Oxygen Sensing Define Early and Late Onset Preeclampsia as
Distinct Pathologies. PLoS ONE 5, e13288.
Romero, R., Nien, J.K., Espinoza, J., Todem, D., Fu, W., Chung, H., Kusanovic, J.P., Gotsch, F.,
Erez, O., Mazaki-Tovi, S., et al. (2008). A longitudinal study of angiogenic (placental growth factor)
and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1)
factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for
gestational age neonate. J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia
Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 21, 9–23.
207
Bibliography
Rossi, A.C., and Mullin, P.M. (2011). Prevention of pre-eclampsia with low-dose aspirin or
vitamins C and E in women at high or low risk: a systematic review with meta-analysis. Eur. J. Obstet.
Gynecol. Reprod. Biol. 158, 9–16.
Roten, L.T., Johnson, M.P., Forsmo, S., Fitzpatrick, E., Dyer, T.D., Brennecke, S.P., Blangero, J.,
Moses, E.K., and Austgulen, R. (2009). Association between the candidate susceptibility gene
ACVR2A on chromosome 2q22 and pre-eclampsia in a large Norwegian population-based study (the
HUNT study). Eur. J. Hum. Genet. EJHG 17, 250–257.
Rowe, J., Campbell, S., and Gallery, E.D. (2001). Plasma from preeclamptic women stimulates
decidual endothelial cell growth and prostacyclin but not nitric oxide production: close correlation of
prostacyclin and thromboxane production. J. Soc. Gynecol. Investig. 8, 32–38.
Ruiz, M., Pettaway, C., Song, R., Stoeltzing, O., Ellis, L., and Bar-Eli, M. (2004). Activator
Protein 2α Inhibits Tumorigenicity and Represses Vascular Endothelial Growth Factor Transcription
in Prostate Cancer Cells. Cancer Res. 64, 631–638.
Ryan, H.E., Lo, J., and Johnson, R.S. (1998). HIF-1 alpha is required for solid tumor formation
and embryonic vascularization. EMBO J. 17, 3005–3015.
Saito, S., Umekage, H., Sakamoto, Y., Sakai, M., Tanebe, K., Sasaki, Y., and Morikawa, H.
(1999). Increased T-helper-1-type immunity and decreased T-helper-2-type immunity in patients with
preeclampsia. Am. J. Reprod. Immunol. New York N 1989 41, 297–306.
Sakai, M., Tsuda, H., Tanebe, K., Sasaki, Y., and Saito, S. (2002). Interleukin-12 secretion by
peripheral blood mononuclear cells is decreased in normal pregnant subjects and increased in
preeclamptic patients. Am. J. Reprod. Immunol. New York N 1989 47, 91–97.
Sakakibara, A., Furuse, M., Saitou, M., Ando-Akatsuka, Y., and Tsukita, S. (1997). Possible
involvement of phosphorylation of occludin in tight junction formation. J. Cell Biol. 137, 1393–1401.
Salafia, C.M., Pezzullo, J.C., Ghidini, A., Lopèz-Zeno, J.A., and Whittington, S.S. (1998).
Clinical correlations of patterns of placental pathology in preterm pre-eclampsia. Placenta 19, 67–72.
Salamonsen, L.A., Nie, G., Hannan, N.J., and Dimitriadis, E. (2009). Society for Reproductive
Biology Founders’ Lecture 2009. Preparing fertile soil: the importance of endometrial receptivity.
Reprod. Fertil. Dev. 21, 923–934.
Salmon, J.E., Heuser, C., Triebwasser, M., Liszewski, M.K., Kavanagh, D., Roumenina, L.,
Branch, D.W., Goodship, T., Fremeaux-Bacchi, V., and Atkinson, J.P. (2011). Mutations in
complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE
cohort. PLoS Med. 8, e1001013.
Salmon, J.E., Heuser, C., Triebwasser, M., Liszewski, M.K., Kavanagh, D., Roumenina, L.,
Branch, D.W., Goodship, T., Fremeaux-Bacchi, V., and Atkinson, J.P. (2011). Mutations in
complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE
cohort. PLoS Med. 8, e1001013.
Salnikow, K., Kluz, T., Costa, M., Piquemal, D., Demidenko, Z.N., Xie, K., and Blagosklonny,
M.V. (2002). The Regulation of Hypoxic Genes by Calcium Involves c-Jun/AP-1, Which Cooperates
with Hypoxia-Inducible Factor 1 in Response to Hypoxia. Mol. Cell. Biol. 22, 1734–1741.
208
Bibliography
Sandrim, V.C., Palei, A.C.T., Metzger, I.F., Gomes, V.A., Cavalli, R.C., and Tanus-Santos, J.E.
(2008). Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble
fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension 52, 402–407.
Sankaralingam, S., Xu, Y., Sawamura, T., and Davidge, S.T. (2009). Increased lectin-like
oxidized low-density lipoprotein receptor-1 expression in the maternal vasculature of women with
preeclampsia: role for peroxynitrite. Hypertension 53, 270–277.
Sargent, I.L., Borzychowski, A.M., and Redman, C.W.G. (2006). Immunoregulation in normal
pregnancy and pre-eclampsia: an overview. Reprod. Biomed. Online 13, 680–686.
Savvidou, M.D., Hingorani, A.D., Tsikas, D., Frölich, J.C., Vallance, P., and Nicolaides, K.H.
(2003). Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in
pregnant women who subsequently develop pre-eclampsia. Lancet 361, 1511–1517.
Scazzocchio, E., Figueras, F., Crispi, F., Meler, E., Masoller, N., Mula, R., and Gratacos, E.
(2013). Performance of a first-trimester screening of preeclampsia in a routine care low-risk setting.
Am. J. Obstet. Gynecol. 208, 203.e1–203.e10.
Schäffer, L., Scheid, A., Spielmann, P., Breymann, C., Zimmermann, R., Meuli, M., Gassmann,
M., Marti, H.H., and Wenger, R.H. (2003). Oxygen-regulated expression of TGF-beta 3, a growth
factor involved in trophoblast differentiation. Placenta 24, 941–950.
Schlafke, S., and Enders, A.C. (1975). Cellular basis of interaction between trophoblast and
uterus at implantation. Biol. Reprod. 12, 41–65.
Schmoranzer, J., Fawcett, J.P., Segura, M., Tan, S., Vallee, R.B., Pawson, T., and Gundersen,
G.G. (2009). Par3 and dynein associate to regulate local microtubule dynamics and centrosome
orientation during migration. Curr. Biol. CB 19, 1065–1074.
Schödel, J., Oikonomopoulos, S., Ragoussis, J., Pugh, C.W., Ratcliffe, P.J., and Mole, D.R.
(2011). High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–
217.
Schreiber, M., Kolbus, A., Piu, F., Szabowski, A., Möhle-Steinlein, U., Tian, J., Karin, M.,
Angel, P., and Wagner, E.F. (1999). Control of cell cycle progression by c-Jun is p53 dependent.
Genes Dev. 13, 607–619.
Schreurs, M.P.H., Hubel, C.A., Bernstein, I.M., Jeyabalan, A., and Cipolla, M.J. (2013).
Increased oxidized low-density lipoprotein causes blood-brain barrier disruption in early-onset
preeclampsia through LOX-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 27, 1254–1263.
Scortegagna, M., Ding, K., Oktay, Y., Gaur, A., Thurmond, F., Yan, L.-J., Marck, B.T.,
Matsumoto, A.M., Shelton, J.M., Richardson, J.A., et al. (2003). Multiple organ pathology, metabolic
abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat. Genet. 35,
331–340.
Sebire, N.J., Roberts, L., Noble, P., Wallace, E., and Nicolaides, K.H. (2000). Raised maternal
serum inhibin A concentration at 10 to 14 weeks of gestation is associated with pre-eclampsia. BJOG
Int. J. Obstet. Gynaecol. 107, 795–797.
209
Bibliography
See, J.R., Marlon, A.M., Feikes, H.L., and Cosby, R.S. (1975). Effect of direct revascularization
surgery on coronary collateral circulation in man. Am. J. Cardiol. 36, 734–738.
Selak, M.A., Armour, S.M., MacKenzie, E.D., Boulahbel, H., Watson, D.G., Mansfield, K.D.,
Pan, Y., Simon, M.C., Thompson, C.B., and Gottlieb, E. (2005). Succinate links TCA cycle
dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85.
Seligman, S.P., Buyon, J.P., Clancy, R.M., Young, B.K., and Abramson, S.B. (1994). The role of
nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 171, 944–948.
Semenza, G.L. (2004). Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiol.
Bethesda Md 19, 176–182.
Semenza, G.L., and Wang, G.L. (1992). A nuclear factor induced by hypoxia via de novo protein
synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional
activation. Mol. Cell. Biol. 12, 5447–5454.
Seo, S.B., McNamara, P., Heo, S., Turner, A., Lane, W.S., and Chakravarti, D. (2001).
Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing
the set oncoprotein. Cell 104, 119–130.
Seoud, M.A.-F., Nassar, A.H., Usta, I.M., Melhem, Z., Kazma, A., and Khalil, A.M. (2002).
Impact of advanced maternal age on pregnancy outcome. Am. J. Perinatol. 19, 1–8.
Serdar, Z., Gür, E., Develioğlu, O., Colakoğullari, M., and Dirican, M. (2002). Placental and
decidual lipid peroxidation and antioxidant defenses in preeclampsia. Lipid peroxidation in
preeclampsia. Pathophysiol. Off. J. Int. Soc. Pathophysiol. ISP 9, 21.
Seth, A., Sheth, P., Elias, B.C., and Rao, R. (2007). Protein phosphatases 2A and 1 interact with
occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J.
Biol. Chem. 282, 11487–11498.
Shaamash, A.H., Elsnosy, E.D., Makhlouf, A.M., Zakhari, M.M., Ibrahim, O.A., and EL-dien,
H.M. (2000). Maternal and fetal serum nitric oxide (NO) concentrations in normal pregnancy, preeclampsia and eclampsia. Int. J. Gynaecol. Obstet. Off. Organ Int. Fed. Gynaecol. Obstet. 68, 207–
214.
Shembrey, M.A., and Noble, A.D. (1995). An instructive case of abdominal pregnancy. Aust. N.
Z. J. Obstet. Gynaecol. 35, 220–221.
Shimokawa, T., and Smith, W.L. (1992). Prostaglandin endoperoxide synthase. The aspirin
acetylation region. J. Biol. Chem. 267, 12387–12392.
Sibai, B.M. (2003). Diagnosis and management of gestational hypertension and preeclampsia.
Obstet. Gynecol. 102, 181–192.
Sibai, B.M. (2005). Diagnosis, prevention, and management of eclampsia. Obstet. Gynecol. 105,
402–410.
Sibai, B.M., and Stella, C.L. (2009). Diagnosis and management of atypical preeclampsiaeclampsia. Am. J. Obstet. Gynecol. 200, 481.e1–7.
210
Bibliography
Sibai, B.M., Ramadan, M.K., Usta, I., Salama, M., Mercer, B.M., and Friedman, S.A. (1993).
Maternal morbidity and mortality in 442 pregnancies with hemolysis, elevated liver enzymes, and low
platelets (HELLP syndrome). Am. J. Obstet. Gynecol. 169, 1000–1006.
Sikkema, J.M., van Rijn, B.B., Franx, A., Bruinse, H.W., de Roos, R., Stroes, E.S., and van
Faassen, E.E. (2001). Placental superoxide is increased in pre-eclampsia. Placenta 22, 304–308.
Silbiger, V.N., Luchessi, A.D., Hirata, R.D.C., Lima-Neto, L.G., Cavichioli, D., Carracedo, A.,
Brión, M., Dopazo, J., García-García, F., dos Santos, E.S., et al. (2013). Novel genes detected by
transcriptional profiling from whole-blood cells in patients with early onset of acute coronary
syndrome. Clin. Chim. Acta Int. J. Clin. Chem. 421, 184–190.
Silver, R.K., Kupferminc, M.J., Russell, T.L., Adler, L., Mullen, T.A., and Caplan, M.S. (1996).
Evaluation of nitric oxide as a mediator of severe preeclampsia. Am. J. Obstet. Gynecol. 175, 1013–
1017.
Simionescu, N., Simionescu, M., and Palade, G.E. (1978). Open junctions in the endothelium of
the postcapillary venules of the diaphragm. J. Cell Biol. 79, 27–44.
Simón, C., Martin, J.C., Meseguer, M., Caballero-Campo, P., Valbuena, D., and Pellicer, A.
(2000). Embryonic regulation of endometrial molecules in human implantation. J. Reprod. Fertil.
Suppl. 55, 43–53.
Simon, M.-P., Tournaire, R., and Pouyssegur, J. (2008). The angiopoietin-2 gene of endothelial
cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central
factors GATA-2 and Ets-1. J. Cell. Physiol. 217, 809–818.
Singh, J., Ahmed, A., and Girardi, G. (2011). Role of complement component C1q in the onset of
preeclampsia in mice. Hypertension 58, 716–724.
Siristatidis, C., Nissotakis, C., Chrelias, C., Iacovidou, H., and Salamalekis, E. (2006).
Immunological factors and their role in the genesis and development of endometriosis. J. Obstet.
Gynaecol. Res. 32, 162–170.
Skjaerven, R., Wilcox, A.J., and Lie, R.T. (2002). The interval between pregnancies and the risk
of preeclampsia. N. Engl. J. Med. 346, 33–38.
Sladek, S.M., Magness, R.R., and Conrad, K.P. (1997). Nitric oxide and pregnancy. Am. J.
Physiol. 272, R441–463.
Slomiany, M.G., and Rosenzweig, S.A. (2004). IGF-1–Induced VEGF and IGFBP-3 Secretion
Correlates with Increased HIF-1α Expression and Activity in Retinal Pigment Epithelial Cell Line
D407. Invest. Ophthalmol. Vis. Sci. 45, 2838–2847.
Smith, S.D., Dunk, C.E., Aplin, J.D., Harris, L.K., and Jones, R.L. (2009). Evidence for immune
cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 174,
1959–1971.
Song, G., Kim, J., Bazer, F.W., and Spencer, T.E. (2008). Progesterone and interferon tau
regulate hypoxia-inducible factors in the endometrium of the ovine uterus. Endocrinology 149, 1926–
1934.
211
Bibliography
Sontag, J.-M., and Sontag, E. (2006). Regulation of cell adhesion by PP2A and SV40 small tumor
antigen: an important link to cell transformation. Cell. Mol. Life Sci. CMLS 63, 2979–2991.
SPARGO, B., McCARTNEY, C.P., and WINEMILLER, R. (1959). Glomerular capillary
endotheliosis in toxemia of pregnancy. Arch. Pathol. 68, 593–599.
Spinella, F., Rosanò, L., Del Duca, M., Di Castro, V., Nicotra, M.R., Natali, P.G., and Bagnato,
A. (2010). Endothelin-1 Inhibits Prolyl Hydroxylase Domain 2 to Activate Hypoxia-Inducible Factor1α in Melanoma Cells. PLoS ONE 5, e11241.
Sroga, J.M., Ma, X., and Das, S.K. (2012). Developmental regulation of decidual cell polyploidy
at the site of implantation. Front. Biosci. Sch. Ed. 4, 1475–1486.
Stan, R.V., Tkachenko, E., and Niesman, I.R. (2004). PV1 Is a Key Structural Component for the
Formation of the Stomatal and Fenestral Diaphragms. Mol. Biol. Cell 15, 3615–3630.
Stark, J.M. (1993). Pre-eclampsia and cytokine induced oxidative stress. BJOG Int. J. Obstet.
Gynaecol. 100, 105–109.
Staun-Ram, E., and Shalev, E. (2005). Human trophoblast function during the implantation
process. Reprod. Biol. Endocrinol. RBE 3, 56.
Staun-Ram, E., Goldman, S., Gabarin, D., and Shalev, E. (2004). Expression and importance of
matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod. Biol.
Endocrinol. 2, 59.
Steinbrech, D.S., Mehrara, B.J., Saadeh, P.B., Greenwald, J.A., Spector, J.A., Gittes, G.K., and
Longaker, M.T. (2000). Hypoxia increases insulinlike growth factor gene expression in rat osteoblasts.
Ann. Plast. Surg. 44, 529–534; discussion 534–535.
Stolze, I.P., Tian, Y.-M., Appelhoff, R.J., Turley, H., Wykoff, C.C., Gleadle, J.M., and Ratcliffe,
P.J. (2004). Genetic Analysis of the Role of the Asparaginyl Hydroxylase Factor Inhibiting Hypoxiainducible Factor (HIF) in Regulating HIF Transcriptional Target Genes. J. Biol. Chem. 279, 42719–
42725.
Strickland, L.A., Jubb, A.M., Hongo, J.-A., Zhong, F., Burwick, J., Fu, L., Frantz, G.D., and
Koeppen, H. (2005). Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour
endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J. Pathol. 206, 466–
475.
Sugimoto, H., Hamano, Y., Charytan, D., Cosgrove, D., Kieran, M., Sudhakar, A., and Kalluri, R.
(2003). Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF
antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J. Biol. Chem. 278, 12605–
12608.
Sun, Q., Zhou, H., Binmadi, N.O., and Basile, J.R. (2009). Hypoxia-inducible factor-1-mediated
regulation of semaphorin 4D affects tumor growth and vascularity. J. Biol. Chem. 284, 32066–32074.
Taddei, S., Virdis, A., Mattei, P., Ghiadoni, L., Sudano, I., and Salvetti, A. (1996). Defective Larginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation 94, 1298–
1303.
212
Bibliography
Takimoto, E., Ishida, J., Sugiyama, F., Horiguchi, H., Murakami, K., and Fukamizu, A. (1996).
Hypertension induced in pregnant mice by placental renin and maternal angiotensinogen. Science 274,
995–998.
Tal, R., Shaish, A., Barshack, I., Polak-Charcon, S., Afek, A., Volkov, A., Feldman, B., Avivi,
C., and Harats, D. (2010). Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice:
possible implications for preeclampsia and intrauterine growth restriction. Am. J. Pathol. 177, 2950–
2962.
Tang, M., Wang, G., Lu, P., Karas, R.H., Aronovitz, M., Heximer, S.P., Kaltenbronn, K.M.,
Blumer, K.J., Siderovski, D.P., Zhu, Y., et al. (2003). Regulator of G-protein signaling-2 mediates
vascular smooth muscle relaxation and blood pressure. Nat. Med. 9, 1506–1512.
Tannetta, D.S., Dragovic, R.A., Gardiner, C., Redman, C.W., and Sargent, I.L. (2013).
Characterisation of syncytiotrophoblast vesicles in normal pregnancy and pre-eclampsia: expression of
flt-1 and endoglin. PloS One 8, e56754.
Tausendschön, M., Dehne, N., and Brüne, B. (2011). Hypoxia causes epigenetic gene regulation
in macrophages by attenuating Jumonji histone demethylase activity. Cytokine 53, 256–262.
Taylor, R.N., Crombleholme, W.R., Friedman, S.A., Jones, L.A., Casal, D.C., and Roberts, J.M.
(1991). High plasma cellular fibronectin levels correlate with biochemical and clinical features of
preeclampsia but cannot be attributed to hypertension alone. Am. J. Obstet. Gynecol. 165, 895–901.
Taylor, R.N., Crombleholme, W.R., Friedman, S.A., Jones, L.A., Casal, D.C., and Roberts, J.M.
(1991). High plasma cellular fibronectin levels correlate with biochemical and clinical features of
preeclampsia but cannot be attributed to hypertension alone. Am. J. Obstet. Gynecol. 165, 895–901.
Taylor, R.N., Varma, M., Teng, N.N., and Roberts, J.M. (1990). Women with preeclampsia have
higher plasma endothelin levels than women with normal pregnancies. J. Clin. Endocrinol. Metab. 71,
1675–1677.
Tazuke, S.I., Mazure, N.M., Sugawara, J., Carland, G., Faessen, G.H., Suen, L.-F., Irwin, J.C.,
Powell, D.R., Giaccia, A.J., and Giudice, L.C. (1998). Hypoxia stimulates insulin-like growth factor
binding protein 1 (IGFBP-1) gene expression in HepG2 cells: A possible model for IGFBP-1
expression in fetal hypoxia. Proc. Natl. Acad. Sci. 95, 10188–10193.
Textor, B., Sator-Schmitt, M., Richter, K.H., Angel, P., and Schorpp-Kistner, M. (2006). c-Jun
and JunB Are Essential for Hypoglycemia-Mediated VEGF Induction. Ann. N. Y. Acad. Sci. 1091,
310–318.
Thomopoulos, C., Tsioufis, C., Michalopoulou, H., Makris, T., Papademetriou, V., and
Stefanadis, C. (2013). Assisted reproductive technology and pregnancy-related hypertensive
complications: a systematic review. J. Hum. Hypertens. 27, 148–157.
Thorne-Lyman, A., and Fawzi, W.W. (2012). Vitamin D during pregnancy and maternal,
neonatal and infant health outcomes: a systematic review and meta-analysis. Paediatr. Perinat.
Epidemiol. 26 Suppl 1, 75–90.
213
Bibliography
Tian, H., Hammer, R.E., Matsumoto, A.M., Russell, D.W., and McKnight, S.L. (1998). The
hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and
protection against heart failure during embryonic development. Genes Dev. 12, 3320–3324.
Treins, C., Giorgetti-Peraldi, S., Murdaca, J., Semenza, G.L., and Obberghen, E.V. (2002).
Insulin Stimulates Hypoxia-inducible Factor 1 through a Phosphatidylinositol 3-Kinase/Target of
Rapamycin-dependent Signaling Pathway. J. Biol. Chem. 277, 27975–27981.
Tsatsaris, V., Goffin, F., Munaut, C., Brichant, J.-F., Pignon, M.-R., Noel, A., Schaaps, J.-P.,
Cabrol, D., Frankenne, F., and Foidart, J.-M. (2003). Overexpression of the soluble vascular
endothelial growth factor receptor in preeclamptic patients: pathophysiological consequences. J. Clin.
Endocrinol. Metab. 88, 5555–5563.
Tse, W.K., Whitley, G.S.J., and Cartwright, J.E. (2002). Transforming growth factor-beta1
regulates hepatocyte growth factor-induced trophoblast motility and invasion. Placenta 23, 699–705.
Tsikouras, P., Dafopoulos, A., Trypsianis, G., Vrachnis, N., Bouchlariotou, S., Liatsikos, S.A.,
Dafopoulos, K., Maroulis, G., Galazios, G., Teichmann, A.T., et al. (2012). Pregnancies and their
obstetric outcome in two selected age groups of teenage women in Greece. J. Matern.-Fetal Neonatal
Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet. 25,
1606–1611.
Tuffnell, D.J., Jankowicz, D., Lindow, S.W., Lyons, G., Mason, G.C., Russell, I.F., Walker, J.J.,
and Yorkshire Obstetric Critical Care Group (2005). Outcomes of severe pre-eclampsia/eclampsia in
Yorkshire 1999/2003. BJOG Int. J. Obstet. Gynaecol. 112, 875–880.
Tuohy, J.F., and James, D.K. (1992). Pre-eclampsia and trisomy 13. Br. J. Obstet. Gynaecol. 99,
891–894.
Turko, I.V., and Murad, F. (2002). Protein nitration in cardiovascular diseases. Pharmacol. Rev.
54, 619–634.
Van Dijk, M., Drewlo, S., and Oudejans, C.B.M. (2010). Differential methylation of STOX1 in
human placenta. Epigenetics 5, 736–742.
Van Dijk, M., Mulders, J., Poutsma, A., Könst, A.A.M., Lachmeijer, A.M.A., Dekker, G.A.,
Blankenstein, M.A., and Oudejans, C.B.M. (2005). Maternal segregation of the Dutch preeclampsia
locus at 10q22 with a new member of the winged helix gene family. Nat. Genet. 37, 514–519.
Vanderlelie, J., Venardos, K., Clifton, V.L., Gude, N.M., Clarke, F.M., and Perkins, A.V. (2005).
Increased biological oxidation and reduced anti-oxidant enzyme activity in pre-eclamptic placentae.
Placenta 26, 53–58.
Vanhoutte, P.M. (2003). Endothelial control of vasomotor function: from health to coronary
disease. Circ. J. Off. J. Jpn. Circ. Soc. 67, 572–575.
Vassilopoulos, A., and Papazafiri, P. (2005). Attenuation of oxidative stress in HL-1
cardiomyocytes improves mitochondrial function and stabilizes Hif-1alpha. Free Radic. Res. 39,
1273–1284.
214
Bibliography
Venkatesha, S., Toporsian, M., Lam, C., Hanai, J., Mammoto, T., Kim, Y.M., Bdolah, Y., Lim,
K.-H., Yuan, H.-T., Libermann, T.A., et al. (2006). Soluble endoglin contributes to the pathogenesis of
preeclampsia. Nat. Med. 12, 642–649.
Verkauskiene, R., Beltrand, J., Claris, O., Chevenne, D., Deghmoun, S., Dorgeret, S., Alison, M.,
Gaucherand, P., Sibony, O., and Lévy-Marchal, C. (2007). Impact of fetal growth restriction on body
composition and hormonal status at birth in infants of small and appropriate weight for gestational age.
Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 157, 605–612.
Vikse, B.E., Irgens, L.M., Leivestad, T., Skjaerven, R., and Iversen, B.M. (2008). Preeclampsia
and the risk of end-stage renal disease. N. Engl. J. Med. 359, 800–809.
Vogler, M. (2012). BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 19, 67–74.
Wang, A., Rana, S., and Karumanchi, S.A. (2009). Preeclampsia: The Role of Angiogenic
Factors in Its Pathogenesis. Physiology 24, 147–158.
Wang, G.L., and Semenza, G.L. (1995). Purification and characterization of hypoxia-inducible
factor 1. J. Biol. Chem. 270, 1230–1237.
Wang, G.L., Jiang, B.H., Rue, E.A., and Semenza, G.L. (1995). Hypoxia-inducible factor 1 is a
basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S.
A. 92, 5510–5514.
Wang, X., Matsumoto, H., Zhao, X., Das, S.K., and Paria, B.C. (2004). Embryonic signals direct
the formation of tight junctional permeability barrier in the decidualizing stroma during embryo
implantation. J. Cell Sci. 117, 53–62.
Wang, Y., and Walsh, S.W. (1996). Antioxidant activities and mRNA expression of superoxide
dismutase, catalase, and glutathione peroxidase in normal and preeclamptic placentas. J. Soc. Gynecol.
Investig. 3, 179–184.
Wang, Y., and Walsh, S.W. (2001). Increased superoxide generation is associated with decreased
superoxide dismutase activity and mRNA expression in placental trophoblast cells in pre-eclampsia.
Placenta 22, 206–212.
Wang, Y., Gu, Y., Granger, D.N., Roberts, J.M., and Alexander, J.S. (2002). Endothelial
junctional protein redistribution and increased monolayer permeability in human umbilical vein
endothelial cells isolated during preeclampsia. Am. J. Obstet. Gynecol. 186, 214–220.
Wang, Y., Walsh, S.W., and Kay, H.H. (1992). Placental lipid peroxides and thromboxane are
increased and prostacyclin is decreased in women with preeclampsia. Am. J. Obstet. Gynecol. 167,
946–949.
Wang, Y., Walsh, S.W., and Kay, H.H. (1992). Placental lipid peroxides and thromboxane are
increased and prostacyclin is decreased in women with preeclampsia. Am. J. Obstet. Gynecol. 167,
946–949.
Wardell, S.E., Boonyaratanakornkit, V., Adelman, J.S., Aronheim, A., and Edwards, D.P. (2002).
Jun dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol.
Cell. Biol. 22, 5451–5466.
215
Bibliography
Watson, A.L., Palmer, M.E., Jauniaux, E., and Burton, G.J. (1997). Variations in expression of
copper/zinc superoxide dismutase in villous trophoblast of the human placenta with gestational age.
Placenta 18, 295–299.
Watson, A.L., Skepper, J.N., Jauniaux, E., and Burton, G.J. (1998). Changes in concentration,
localization and activity of catalase within the human placenta during early gestation. Placenta 19, 27–
34.
Wei, D., Le, X., Zheng, L., Wang, L., Frey, J.A., Gao, A.C., Peng, Z., Huang, S., Xiong, H.Q.,
Abbruzzese, J.L., et al. (2003a). Stat3 activation regulates the expression of vascular endothelial
growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 22, 319–329.
Wei, L.-H., Kuo, M.-L., Chen, C.-A., Chou, C.-H., Lai, K.-B., Lee, C.-N., and Hsieh, C.-Y.
(2003b). Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a
STAT3 pathway. Oncogene 22, 1517–1527.
Weidenfeld-Baranboim, K., Bitton-Worms, K., and Aronheim, A. (2008). TRE-dependent
transcription activation by JDP2-CHOP10 association. Nucleic Acids Res. 36, 3608–3619.
Weidenfeld-Baranboim, K., Hasin, T., Darlyuk, I., Heinrich, R., Elhanani, O., Pan, J., Yokoyama,
K.K., and Aronheim, A. (2009). The ubiquitously expressed bZIP inhibitor, JDP2, suppresses the
transcription of its homologue immediate early gene counterpart, ATF3. Nucleic Acids Res. 37, 2194–
2203.
Weidenfeld-Baranboim, K., Koren, L., and Aronheim, A. (2011). Phosphorylation of JDP2 on
threonine-148 by the c-Jun N-terminal kinase targets it for proteosomal degradation. Biochem. J. 436,
661–669.
Weiner, C.P. (1991). Preeclampsia-eclampsia syndrome and coagulation. Clin. Perinatol. 18,
713–726.
Weinstein, L. (1982). Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a
severe consequence of hypertension in pregnancy. Am. J. Obstet. Gynecol. 142, 159–167.
Wenger, R.H., Rolfs, A., Marti, H.H., Guénet, J.L., and Gassmann, M. (1996). Nucleotide
sequence, chromosomal assignment and mRNA expression of mouse hypoxia-inducible factor-1
alpha. Biochem. Biophys. Res. Commun. 223, 54–59.
Whitley, G.S.J., and Cartwright, J.E. (2010). Cellular and Molecular Regulation of Spiral Artery
Remodelling: Lessons from the Cardiovascular Field. Placenta 31, 465–474.
WHO | WHO recommendations for prevention and treatment of pre-eclampsia and eclampsia.
Wiener, C.M., Booth, G., and Semenza, G.L. (1996). In vivo expression of mRNAs encoding
hypoxia-inducible factor 1. Biochem. Biophys. Res. Commun. 225, 485–488.
Wiesener, M.S., Jürgensen, J.S., Rosenberger, C., Scholze, C.K., Hörstrup, J.H., Warnecke, C.,
Mandriota, S., Bechmann, I., Frei, U.A., Pugh, C.W., et al. (2003). Widespread hypoxia-inducible
expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. Off. Publ. Fed.
Am. Soc. Exp. Biol. 17, 271–273.
Williams, Z. (2012). Inducing tolerance to pregnancy. N. Engl. J. Med. 367, 1159–1161.
216
Bibliography
Witlin, A.G., Friedman, S.A., and Sibai, B.M. (1997). The effect of magnesium sulfate therapy on
the duration of labor in women with mild preeclampsia at term: a randomized, double-blind, placebocontrolled trial. Am. J. Obstet. Gynecol. 176, 623–627.
Wong, V. (1997). Phosphorylation of occludin correlates with occludin localization and function
at the tight junction. Am. J. Physiol. - Cell Physiol. 273, C1859–C1867.
Woods, L.L. (1989). Importance of prostaglandins in hypertension during reduced uteroplacental
perfusion pressure. Am. J. Physiol. 257, R1558–1561.
Worley, K.C., Hnat, M.D., and Cunningham, F.G. (2008). Advanced extrauterine pregnancy:
diagnostic and therapeutic challenges. Am. J. Obstet. Gynecol. 198, 297.e1–7.
Wu, F., and Wilson, J.X. (2009). Peroxynitrite-dependent activation of protein phosphatase type
2A mediates microvascular endothelial barrier dysfunction. Cardiovasc. Res. 81, 38–45.
Xia, X., and Kung, A.L. (2009). Preferential binding of HIF-1 to transcriptionally active loci
determines cell-type specific response to hypoxia. Genome Biol. 10, R113.
Xia, X., Lemieux, M.E., Li, W., Carroll, J.S., Brown, M., Liu, X.S., and Kung, A.L. (2009).
Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone
methylation homeostasis. Proc. Natl. Acad. Sci. U. S. A. 106, 4260–4265.
Xiang, Y., Zhang, X., Li, Q., Xu, J., Zhou, X., Wang, T., Xing, Q., Liu, Y., Wang, L., He, L., et
al. (2013). Promoter hypomethylation of TIMP3 is associated with pre-eclampsia in a Chinese
population. Mol. Hum. Reprod. 19, 153–159.
Xu, Y., Liu, Z., and Guo, K. (2012). The effect of JDP2 and ATF2 on the epithelial-mesenchymal
transition of human pancreatic cancer cell lines. Pathol. Oncol. Res. POR 18, 571–577.
Yamada, S., Pokutta, S., Drees, F., Weis, W.I., and Nelson, W.J. (2005). Deconstructing the
cadherin-catenin-actin complex. Cell 123, 889–901.
Yamashita, K., Discher, D.J., Hu, J., Bishopric, N.H., and Webster, K.A. (2001). Molecular
regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator
protein-1, GATA-2, AND p300/CBP. J. Biol. Chem. 276, 12645–12653.
Yang, X.-J., and Seto, E. (2003). Collaborative spirit of histone deacetylases in regulating
chromatin structure and gene expression. Curr. Opin. Genet. Dev. 13, 143–153.
Yinon, Y., Nevo, O., Xu, J., Many, A., Rolfo, A., Todros, T., Post, M., and Caniggia, I. (2008).
Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: hypoxic
regulation via transforming growth factor-beta 3. Am. J. Pathol. 172, 77–85.
Ylikorkala, O., Pekonen, F., and Viinikka, L. (1986). Renal prostacyclin and thromboxane in
normotensive and preeclamptic pregnant women and their infants. J. Clin. Endocrinol. Metab. 63,
1307–1312.
Yoon, H.-S., Hong, S.-H., Kang, H.-J., Ko, B.K., Ahn, S.-H., and Huh, J.-R. (2003). Bfl-1 gene
expression in breast cancer: its relationship with other prognostic factors. J. Korean Med. Sci. 18, 225–
230.
217
Bibliography
Young, M.R., Li, J.J., Rincón, M., Flavell, R.A., Sathyanarayana, B.K., Hunziker, R., and
Colburn, N. (1999). Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is
required for tumor promotion. Proc. Natl. Acad. Sci. U. S. A. 96, 9827–9832.
Yu, J., Shixia, C.Z., Wu, Y., and Duan, T. (2011). Inhibin A, activin A, placental growth factor
and uterine artery Doppler pulsatility index in the prediction of pre-eclampsia. Ultrasound Obstet.
Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 37, 528–533.
Yuanhong, X., Feng, X., Qingchang, L., Jianpeng, F., Zhe, L., and Kejian, G. (2010).
Downregulation of AP-1 repressor JDP2 is associated with tumor metastasis and poor prognosis in
patients with pancreatic carcinoma. Int. J. Biol. Markers 25, 136–140.
Zafarmand, M.H., Nijdam, M.-E., Franx, A., Grobbee, D.E., and Bots, M.L. (2008). The
angiotensinogen gene M235T polymorphism and development of preeclampsia/eclampsia: a metaanalysis and meta-regression of observational studies. J. Hypertens. 26, 1726–1734.
Zamudio, S., Wu, Y., Ietta, F., Rolfo, A., Cross, A., Wheeler, T., Post, M., Illsley, N.P., and
Caniggia, I. (2007). Human placental hypoxia-inducible factor-1alpha expression correlates with
clinical outcomes in chronic hypoxia in vivo. Am. J. Pathol. 170, 2171–2179.
Zelzer, E., Levy, Y., Kahana, C., Shilo, B.Z., Rubinstein, M., and Cohen, B. (1998). Insulin
induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO
J. 17, 5085–5094.
Zhang, E.G., Smith, S.K., Baker, P.N., and Charnock-Jones, D.S. (2001). The regulation and
localization of angiopoietin-1, -2, and their receptor Tie2 in normal and pathologic human placentae.
Mol. Med. Camb. Mass 7, 624–635.
Zhang, H., Zhang, Y., Yang, F., Li, L., Liu, S., Xu, Z., Wang, J., and Sun, S. (2011). Complement
component C4A and apolipoprotein A-I in plasmas as biomarkers of the severe, early-onset
preeclampsia. Mol. Biosyst. 7, 2470–2479.
Zhang, Y., Gu, Y., Li, H., Lucas, M.J., and Wang, Y. (2003). Increased endothelial monolayer
permeability is induced by serum from women with preeclampsia but not by serum from women with
normal pregnancy or that are not pregnant. Hypertens. Pregnancy Off. J. Int. Soc. Study Hypertens.
Pregnancy 22, 99–108.
Zhang, Y., Zhao, S., Gu, Y., Lewis, D.F., Alexander, J.S., and Wang, Y. (2005). Effects of
peroxynitrite and superoxide radicals on endothelial monolayer permeability: potential role of
peroxynitrite in preeclampsia. J. Soc. Gynecol. Investig. 12, 586–592.
Zhang, Z., Li, Y., Zhang, L.L., Jia, L.T., and Yang, X.Q. (2012). Association of 14 bp
insertion/deletion polymorphism of the HLA-G gene in father with severe preeclampsia in Chinese.
Tissue Antigens 80, 158–164.
Zhao, J., Gu, Y., Fan, R., Groome, L.J., and Wang, Y. (2011). Factors derived from preeclamptic
placentas perturb polarity protein PARD-3 expression and distribution in endothelial cells. Reprod.
Sci. Thousand Oaks Calif 18, 164–171.
218
Bibliography
Zhong, H., Agani, F., Baccala, A.A., Laughner, E., Rioseco-Camacho, N., Isaacs, W.B., Simons,
J.W., and Semenza, G.L. (1998). Increased expression of hypoxia inducible factor-1alpha in rat and
human prostate cancer. Cancer Res. 58, 5280–5284.
Zhong, L., D’Urso, A., Toiber, D., Sebastian, C., Henry, R.E., Vadysirisack, D.D., Guimaraes,
A., Marinelli, B., Wikstrom, J.D., Nir, T., et al. (2010). The histone deacetylase Sirt6 regulates glucose
homeostasis via Hif1alpha. Cell 140, 280–293.
Zhong, X.Y., Laivuori, H., Livingston, J.C., Ylikorkala, O., Sibai, B.M., Holzgreve, W., and
Hahn, S. (2001). Elevation of both maternal and fetal extracellular circulating deoxyribonucleic acid
concentrations in the plasma of pregnant women with preeclampsia. Am. J. Obstet. Gynecol. 184,
414–419.
Zhou, C.C., Ahmad, S., Mi, T., Abbasi, S., Xia, L., Day, M.-C., Ramin, S.M., Ahmed, A.,
Kellems, R.E., and Xia, Y. (2008a). Autoantibody from women with preeclampsia induces soluble
Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of
activated T-cells signaling. Hypertension 51, 1010–1019.
Zhou, C.C., Zhang, Y., Irani, R.A., Zhang, H., Mi, T., Popek, E.J., Hicks, M.J., Ramin, S.M.,
Kellems, R.E., and Xia, Y. (2008b). Angiotensin receptor agonistic autoantibodies induce preeclampsia in pregnant mice. Nat. Med. 14, 855–862.
Zhou, Y., Fisher, S.J., Janatpour, M., Genbacev, O., Dejana, E., Wheelock, M., and Damsky,
C.H. (1997). Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for
successful endovascular invasion? J. Clin. Invest. 99, 2139–2151.
Zhou, Y., McMaster, M., Woo, K., Janatpour, M., Perry, J., Karpanen, T., Alitalo, K., Damsky,
C., and Fisher, S.J. (2002). Vascular endothelial growth factor ligands and receptors that regulate
human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver
enzymes, and low platelets syndrome. Am. J. Pathol. 160, 1405–1423.
Zhou, Y., McMaster, M., Woo, K., Janatpour, M., Perry, J., Karpanen, T., Alitalo, K., Damsky,
C., and Fisher, S.J. (2002). Vascular endothelial growth factor ligands and receptors that regulate
human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver
enzymes, and low platelets syndrome. Am. J. Pathol. 160, 1405–1423.
Zhu, X., Wu, S., Dahut, W.L., and Parikh, C.R. (2007). Risks of proteinuria and hypertension
with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and
meta-analysis. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 49, 186–193.
Zundel, W., Schindler, C., Haas-Kogan, D., Koong, A., Kaper, F., Chen, E., Gottschalk, A.R.,
Ryan, H.E., Johnson, R.S., Jefferson, A.B., et al. (2000). Loss of PTEN facilitates HIF-1-mediated
gene expression. Genes Dev. 14, 391–396.
219
Supplemental
Papers
Supplemental papers
220
Landscape of Transcriptional Deregulations in the
Preeclamptic Placenta
Daniel Vaiman, Rosamaria Calicchio, Francisco Miralles*
INSERM U1016-CNRS UMR8104, Université Paris Descartes, Institut Cochin, Paris, France
Abstract
Preeclampsia is a pregnancy disease affecting 5 to 8% of pregnant women and a leading cause of both maternal and fetal
mortality and morbidity. Because of a default in the process of implantation, the placenta of preeclamptic women
undergoes insufficient vascularization. This results in placental ischemia, inflammation and subsequent release of placental
debris and vasoactive factors in the maternal circulation causing a systemic endothelial activation. Several microarray
studies have analyzed the transcriptome of the preeclamptic placentas to identify genes which could be involved in
placental dysfunction. In this study, we compared the data from publicly available microarray analyses to obtain a
consensus list of modified genes. This allowed to identify consistently modified genes in the preeclamptic placenta. Of
these, 67 were up-regulated and 31 down-regulated. Assuming that changes in the transcription level of co-expressed
genes may result from the coordinated action of a limited number of transcription factors, we looked for over-represented
putative transcription factor binding sites in the promoters of these genes. Indeed, we found that the promoters of upregulated genes are enriched in putative binding sites for NFkB, CREB, ANRT, REEB1, SP1, and AP-2. In the promoters of
down-regulated genes, the most prevalent putative binding sites are those of MZF-1, NFYA, E2F1 and MEF2A. These
transcriptions factors are known to regulate specific biological pathways such as cell responses to inflammation, hypoxia,
DNA damage and proliferation. We discuss here the molecular mechanisms of action of these transcription factors and how
they can be related to the placental dysfunction in the context of preeclampsia.
Citation: Vaiman D, Calicchio R, Miralles F (2013) Landscape of Transcriptional Deregulations in the Preeclamptic Placenta. PLoS ONE 8(6): e65498. doi:10.1371/
journal.pone.0065498
Editor: Ana Claudia Zenclussen, Otto-von-Guericke University Magdeburg, Germany
Received March 12, 2013; Accepted April 26, 2013; Published June 13, 2013
Copyright: ß 2013 Vaiman et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits
unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was conducted in a laboratory supported by the CNRS and INSERM. RC is a recipient of a fellowship from the French Research Ministry (Paris
XI University). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist.
* E-mail: francisco.miralles@inserm.fr
oxidative stress and inflammation, with subsequent release of
placental factors and debris into the maternal circulation. These
circulating factors are supposed to cause a widespread ECs
activation leading to the multisystem dysfunction characteristic of
the maternal syndrome of PE [3,4]. Since the placenta plays a
central role in the development of the disease, identifying the
molecular mechanisms altered in the preeclamptic placenta
comparatively to the non-pathologic placenta is fundamental to
understand the initiation and evolution of this disease. In this
context microarray-based genome-wide transcriptional profiling
was used in several studies based on the comparison of the
preeclamptic and non-pathologic placenta as reviewed by Louwen
and collaborators [5]. In general, similar categories of differentially-expressed genes were reported including genes involved in:
vascular regulation, inflammation, cell proliferation, apoptosis,
differentiation, and cellular metabolism. However, in some cases
the results appeared controversial in respect to some of the genes
of interest. These differences may originate from the type of PE,
the sampling of the placenta, the gestational age, ethnicity, mode
of delivery, the microarray platforms and the filtering and
statistical analysis. To overcome these differences we compared
the lists of modified genes extracted from the publicly available
datasets on microarray experiments concerning the preeclamptic
placenta. The intersection of these gene-expression data sets,
considering both up- and down-regulated genes, allowed obtaining
Introduction
Preeclampsia (PE) is a pregnancy complication affecting
approximately 5–8% of pregnant women and capable of causing
both maternal and fetal morbidity and mortality. The disease
develops after 20 weeks of gestational age and is characterized by
elevated maternal blood pressure (140 mmHg/90 mmHg) and
proteinuria (.300 mg/24h), endothelial cells (ECs) dysfunction
and systemic inflammation [1]. In addition, PE can lead to
eclampsia (when convulsions develop), and may be associated with
the HELLP syndrome (Hemolysis, Elevated Liver enzymes and
Low Platelet count). Both conditions may induce severe complications such as cerebral hemorrhage, lung edema or liver
hemorrhage and rupture. PE symptoms appear after 20 weeks
of gestational age, but sometimes much later by the end of
pregnancy, and even, quite surprisingly, post-partum [2]. Those
PEs who initiate early are generally more severe (blood pressure
.160 mmHg/110 mmHg and proteinuria .300 mg/24h) and
associated to a greater rate of intrauterine growth retardation and
of iatrogenic prematurity.
Defective placentation is generally described as being at the root
of the disease. Several studies have established that in the
developing preeclamptic placenta, the normal process of trophoblast invasion and remodeling of the uterine maternal spiral
arteries is impaired. This default in placental development in early
pregnancy results in reduced placental perfusion, placental
PLOS ONE | www.plosone.org
1
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
a minimal list of genes which are consistently modified in PE.
Then, we have used this consensus list to explore the transcriptional mechanisms involved in preeclampsia-specific placental
dysfunction. This strategy has been used recently by Tapia and
coworkers to identify with success transcription factors (TFs)
involved in endometrial receptivity [6]. Transcriptional mechanisms control the expression of genes mainly through the action of
TFs. These proteins bind to the DNA regulatory sequences of the
genes at specific sites known as transcription factor binding sites
(TFBS). Usually, the transcriptional activity of a gene requires the
binding of several TFs, which act cooperatively to activate or
repress transcription [7]. Therefore, we have used several
bioinformatic tools allowing detecting over-representation of
TFBS and of sets of TFBS in the promoters of genes. This way
we identified a number of TFs which are likely involved in the
regulation of the set of consistently modified genes in PE. These
TFs may be instrumental in the transcriptomic modifications
undergone by the preeclamptic placenta and their involvement in
this disease can now be tested in the wet laboratory.
Functional Clustering
The list of genes consistently up- and down-regulated within the
microarray datasets was submitted to the GENOMATIX GeneRanker tool for functional annotation and pathway analysis. This
allowed gaining information on the biological significance of these
genes.
Identification of Over-represented TFBS in the Proximal
Promoter of the Genes Consistently Modified in the
Preeclamptic Placenta
The sequences of the proximal promoter of the genes associated
with the preeclamptic placenta were retrieved from the Data Base
of Transcriptional Start Sites (DBTSS), [14]. For the purposes of
this study the proximal promoter was defined as the region
comprised within 1000 base pairs (bp) upstream and 200 bp
downstream of the transcriptional start site (TSS). These sequences
were used to search for potential TFBS using the following free
softwares: CREMAG, a web tool that searches over-represented
TFBS in a set of sequences using the TRANSFAC and JASPAR
vertebrate position-weight matrices [15]. The analysis was
performed with the default parameters. We used a 70%
conservation threshold and a maximum number of 20 most
conserved TFBSs in non-coding regions between 1000 bp
upstream and 200 bp downstream of the TSS. TELIS (Transcription Element Listening System) is a Java server-side application which identifies transcription-factor binding motifs (TFBMs)
that are over-represented among the promoters [16]. It consists of
two parts: PromoterScan and PromoterStats. PromoterScan finds
the number of occurrences of specific TFBMs in promoters and
stores the results in MySQL database. PromoterStats uses zstatistics to find matrices which are over-represented (or underrepresented) on the specific differentially expressed promoter set.
The transcription factor affinity prediction (TRAP) method
calculates the affinity of transcription factors for DNA sequences
on the basis of a biophysical model [17]. This method has proven
to be useful for several applications, including for determining
which transcription factors have the highest affinity in a set of
sequences [18]. TFM-explorer is a program for analyzing
regulatory regions of eukaryotic genomes. It takes a set of coregulated gene sequences, and search for locally over-represented
TFBS [19]. The algorithm proceeds in two steps: (i) it scans
sequences for detecting all potential transcription factor binding
sites, using weight matrices from JASPAR or TRANSFAC. (ii) it
extracts significant clusters (region of the input sequences
associated with a factor) by calculating a score function. The
web tool TOUCAN uses the MotifScanner algorithm to search for
Materials and Methods
Data Sets used in this Study
We searched the public DataSets assembled from the Gene
Expression Omnibus (GEO) repository, to identify expression
microarray datasets that compared the expression of preeclamptic
versus normal placentas. The keywords: preeclampsia, placenta,
microarrays and gene-expression, were used for this search. To be
included in our study the microarray experiments had to be done
with placental biopsies collected at delivery and at relatively
comparable gestational ages (30–39 weeks). This allowed to
identify six datasets (Table 1). The GEO accession numbers of
the studies are: GSE10588, GSE4707, GSE30186, GSE25906,
GSE24129 and GSE14722, [8,9,10,11,12,13]. The data from
each study were analyzed with Geo2R to identify genes
significantly modified (P-value #0.05 and Log2 Fold
Change = 60.2). This generated a list of modified genes (upand down-regulated) for each study. Subsequently the lists of
modified genes were confronted using the GENOMATIX list
comparison tool (Genomatix Software GmbH, Munich, Germany)
to identify those genes which were consistently modified (up- or
down-regulated genes). Those showing similar modification in at
least 4 studies were considered relevant and included in two final
lists (consistently up-regulated and down-regulated genes).
Table 1. Preeclamptic placenta microarrays analyzed in this study.
Study
GEO accession
PE/Co Gest. Age
PE samples Co samples (Weeks)
Delivery
Microarrays plataform
Sitras et al., 2009 [9]
GSE10588
17
26
34/39
CS
ABI HGSM Version 2
Nishizawa et al., 2007 [12]
GSE4707
13
8
32/32
CS
Agilent-012391 Whole Human
Genome Oligo Microarray G4112A
Meng et al., 2011 [10]
GSE30186
6
6
36/39
CS
Illumina HumanHT-12 V4.0
Tsai et al., 2011 [11]
GSE25906
23
37
33/37
Labor
Illumina human-6 v2.0
Nishizawa et al., 2011 [13]
GSE24129
8
8
34/38
CS
Affymetrix Human Gene 1.0 ST Array
Win et al., 2009 [14]
GSE14722
12
11
32/31
CS/Labor
Affymetrix Human Genome U133
Plus 2.0
*Gestational age (weeks).
doi:10.1371/journal.pone.0065498.t001
PLOS ONE | www.plosone.org
2
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
potential TFBS in a set of sequences using the TRANSFAC or
JASPAR vertebrate databases. The information obtained from the
MotifScanner is subsequently processed by the statistics function of
TOUCAN to identify over-represented TFBS [20,21]. We used
several different TFBS prediction software’s because these
bioinformatics tools usually generate a number of false positives.
Thus, only TFBS predicted by more than one tool were
considered as true positives.
highest scores include the peroxisome proliferative activated
receptor alpha, lipid, hypoxia inducible factor 1, FMS like
receptor tyrosine kinase 3 and vascular endothelial growth factor
pathways. In addition, we noticed that in at least three out of the
six microarray studies some of the consistently modified genes in
the preeclamptic placenta encode TFs. Among the up-regulated
genes we found: LIMD1 (LIM domain-containing protein 1),
BHLHE40 (Basic helix-loop-helix family member e40), VDR
(Vitamin D 1,25-dihydroxyvitamin D3 receptor), CEBPA
(CCAAT/enhancer binding protein, alpha), BCL6 (B-cell CLL/
lymphoma 6), ARID3A (AT rich interactive domain 3A) and
NRIP1 (Nuclear receptor interacting protein 1). Among the downregulated genes: TFDP2 (Transcription factor Dp-2), ZFAND5
(Zinc finger, AN1-type domain 5), BHLHE41 (Basic helix-loophelix family, member e41), and NR2F1 (Nuclear receptor
subfamily 2, group F, member 1). These TFs were also included
in further analyses.
Identification of Regulatory Modules
To identify common regulatory modules in a set of promoter
sequences we used the Genomatix FrameWorker software.
FrameWorker identifies significant complex models of TFBS
present in the promoter sequences of a set of co-regulated genes.
The models/FrameWorkers are defined as all the TFBs that occur
in the same order and in a certain distance range in all (or a subset)
of the input sequences. To determine the P-value of the models, a
background promoter sequence set of 5000 human promoters is
scanned with the models generated by the software. This allows
calculating the probability to found the same models in a set of
randomly selected promoters.
Identification of Over-represented TFBS among the
Consistently Modified Genes
Co-expressed groups of genes are expected to share regulatory
elements which are responsible of the co-regulation. Thus, to
identify the putative common regulatory elements the lists of upand down-regulated genes were analyzed with bioinformatics
tools. First the proximal promoter sequences of the genes (1000 bp
up-stream and 200 bp downstream of the TSS) were retrieved
from the DBTSS data base, and subsequently analyzed with
several public TFBS detection tools: CREMAG, TELIS, TRAP,
TFM-Explorer, and TOUCAN. Only those TFBS showing a P
value #0.05 for their observed frequency versus their predicted
frequency were considered. The results of these analysis are listed
in Table 7 and Table 8. The most significant over-represented
TFBS found in the up-regulated genes list correspond to NFkB
(Nuclear factor kappa B), RREB1 (Ras responsive element binding
protein 1), SP1 (Specificity protein 1), ARNT (Aryl hydrocarbon
receptor nuclear translocator), CREB1 (cAMP responsive element
binding protein 1) and AP-2 (Activating enhancer-binding protein
2). In the down-regulated list the most significant over-represented
TFBS are MZF1 (Myeloid zinc finger 1), E2F1 (E2F transcription
factor 1), MEF2A (Myocyte enhancer factor 2A) and NFYA
(Nuclear transcription factor Y, alpha). Some TFBS, such as SP1,
E2F1, ARNT, and MZF1 appear over-represented in both upand down-regulated genes.
Transcription Factors Interaction
TFs interactions were identified through the Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING) database v9.0.
This database contains known and predicted physical and
functional protein-protein interactions [22]. STRING was used
in the protein mode, and only interactions based in experimental
protein-protein interaction and curated databases with confidence
levels over 0.5 were considered.
Results
Identification of Genes Consistently Associated with the
Preeclamptic Placenta
The intersection of the lists of modified genes extracted from the
microarray studies of the preeclamptic placenta yielded a short list
of genes being consistently modified in the different studies. We
identified a total 98 modified genes of which 67 were up-regulated
and 31 down-regulated. Table 2 (up-regulated) and Table 3
(down-regulated) show a selection of consistently modified genes in
PE (Complete lists are provided as Tables S1 and S2). The most
consistently up-regulated genes were LEP and FLT1 (present in
the totality of studies), followed by QPCT, SIGLEC6, ENG,
BCL6, INHA, EBI3, PAPP2 and HTRA1 (found modified in five
studies). The most consistently down-regulated gene modified in
all the studies was CLDN1. Followed by genes present at least in
four out of six studies including among others ABAT, SOD1,
GCLM, APLN, ABCG2, and NR2F1.
Search for Regulatory Modules in TFs Consistently
Modified in Preeclampsia
The intersection of the microarrays of preeclamptic placentas
indicates that a few TFs appear consistently modified at the
transcriptional level (either up- or down-regulated). Thus, these
transcriptionally co-regulated TFs could share common regulatory
elements in their promoters. These elements are often organized
into defined motifs (frameworks) of two or more TFBs which are
located in the promoter of the genes in a specific orientation,
separated by a given distance and working in concert. We used the
Genomatix FrameWorker software to identify putative regulatory
modules among the TFs consistently modified in the preeclamptic
placenta. Among the promoter sequences of the TFs consistently
up-regulated we got seven significant models (i.e. modules) of three
elements present in the promoter of five genes out of seven. The
most significant model (P#7.82610211) was composed of TFBs
for the Zinc finger transcription factors EGRF (Early growth
response family), E2FF (E2F-myc activator/cell cycle regulator)
and ZF5F (binding site for the transcription factor Zfp161);
Functional Clustering Analysis
We then used the GENOMATIX Gene Ranker software to
perform functional and network analysis of the consistently
modified genes. This made it possible to identify functional gene
classifiers (Table 4 and 5) and pathways (Table 6) that are
significantly enriched in the preeclamptic placenta. Among the upregulated genes the most significant functional categories were
signaling and signal transduction, the regulation of biological
quality, interferon-gamma biosynthetic process, the regulation of B
cell differentiation and cell proliferation. The list of downregulated genes was enriched in transcripts involved in the
response to regulation of sulfur metabolism, blood vessel size
and blood circulation, cellular homeostasis, and the responses to
chemical stimulus and oxidative stress. The pathways with the
PLOS ONE | www.plosone.org
3
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Table 2. Partial list of consistently up-regulated genes in the preeclamptic placenta.
Gene
EntrezGene ID
GSE4707
GSE24129
GSE10588
GSE25906
GSE14722
GSE30186
LEP
3952
1
1
1
1
1
1
FLT1
2321
1
1
1
1
1
1
HTRA1
5654
1
1
1
1
1
-
QPCT
25797
1
1
1
1
1
1
SPAG4
6676
1
1
1
1
1
-
INHA
3623
1
1
1
1
1
-
PAPPA2
60676
1
1
1
1
1
-
SIGLEC6
946
1
1
1
1
1
-
ENG
2022
1
1
1
1
1
-
INHBA
3624
1
1
1
1
1
-
BCL6
604
-
1
1
1
1
-
SLC26A6
65010
-
1
1
1
-
1
GREM2
64388
1
1
1
1
1
-
EBI3
10148
1
1
1
1
1
-
HTRA4
203100
1
1
1
1
-
-
FSTL3
10272
1
1
1
1
1
-
BHLHE40
8553
1
1
1
1
1
-
The table shows some of the consistently up-regulated genes in the six preeclamptic placenta microarray studies analyzed. The microarrays are identified by their GEO
data set accession number (GSE). (1) Indicates modified in the microarrays, (-) Indicates not-modified. *Complete consensus list of up-regulated genes in the
preeclamptic placenta is provided as Table S1.
doi:10.1371/journal.pone.0065498.t002
to TFBS for E2FF, RXRF (Retinoid 6 receptor heterodimerbinding sites), KLFS (Kruppel-like factors) and ZF02 (C2H2 zinc
finger transcription factors 2). This module was present in the
promoter of three out of four genes (Figure 1B).
(Figure 1A). In addition, an alternative regulatory module of two
elements (EGRF and E2FF) was found present in the promoter of
six out of the seven TFs consistently up-regulated in the
preeclamptic placenta (P#8.7661028). In the case of the
consistently down-regulated TFs we found one highly significant
model (P#1.99610210) composed of six elements corresponding
Table 3. Partial list of consistently down-regulated genes in the preeclamptic placenta.
Gene
EntrezGene ID
GSE4707
GSE24129
GSE10588
GSE25906
GSE14722
GSE30186
CLDN1
9076
1
1
1
1
1
1
ABAT
18
1
1
1
-
1
1
MFF
56947
-
1
1
1
1
-
GCLM
2730
-
1
1
1
1
1
F13A1
2162
-
1
1
1
1
-
SOD1
6647
-
1
1
1
1
-
APLN
8862
1
1
-
1
-
1
ABCG2
9429
-
1
1
1
1
-
GOT1
2805
-
1
1
1
1
-
SLC23A2
9962
-
1
1
1
-
1
OLFML3
56944
-
1
-
1
1
1
LEPREL1
55214
1
1
1
-
1
-
BHLHE41
79365
-
1
1
-
1
-
FAM101B
359845
1
1
1
1
-
-
NR2F1
7025
-
1
1
1
1
-
The table shows some of the consistently down-regulated genes in the six preeclamptic placenta microarray studies analyzed. The microarrays are identified by their
GEO data set accession number (GSE). (1) Indicates modified in the microarrays, (-) Indicates not-modified. *Complete consensus list of down-regulated genes in the
preeclamptic placenta is provided as Table S2.
doi:10.1371/journal.pone.0065498.t003
PLOS ONE | www.plosone.org
4
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Table 4. Biological processes annotation clusters for up-regulated genes as reported by the GENOMATIX webtool.
Database
Functional annotation
N6 of genes
Genes
P-Value
GO:0023052
Signaling
36
HTRA1, HEXB, CEBPA, DDR1, LYN, CYP26A1, ENG, APLP2, EBI3, KIT,
HLPDA, SYDE1, RASEF, PREX1, BAD, VDR, INSIG1, FLT1, LHB, FSTL3,
SCARB1, LIMD1, MIF, BHLHE40, SIGLEC6, TREM1, LEP, GREM2, ERRFI1,
NRIP1, INHBA, CORO2A, ERO1L, INHA, SH3BP5, NEK111
1.73E-06
GO:0065008
Regulation of biological quality
25
HTRA1, HEXB, DDR1, LYN, CYP26A1, ENG, APLP2, BCL6, KIT, GAPDH,
EZR, BAD, VDR, KIF2A, LHB, HTRA4, SCARB1, BHLHE40, PAPPA2,
TREM1, LEP, INHBA, INHA, HK2, PROCR
3.42E-06
GO:0030099
Myeloid cell differentiation
7
CEBPA, LYN, KIT, FSTL3, LEP, INHBA, INHA7
7.47E-06
GO:0007165
signal transduction
32
tHTRA1, CEBPA, DDR1, LYN, CYP26A1, ENG, APLP2, EBI3, KIT, SYDE1, RASEF,
PREX1, BAD, VDR, INSIG1, FLT1, LHB, FSTL3, SCARB1, LIMD1, MIF, TREM1,
LEP, GREM2, ERRFI1, NRIP1, INHBA, CORO2A, ERO1L, INHA, SH3BP5, NEK111
1.72E-05
GO:0045577
B cell differentiation
3
BAD, INHBA, INHA1
1.81E-05
GO:0022414
Reproductive process
15
GPX3, HEXB, DDR1, APLP2, KIT, IGSF8, VDR, FLT1, LHB, LEP, NRIP1,
INHBA, SPAG4, INHA, HK2
2.74E-05
GO:0045072
Interferon-gamma biosynthesis
3
EBI3, INHBA, INHA2
2.98E-05
GO:0008283
Cell proliferation
16
CEBPA, DDR1, LYN, ENG, EBI3, KIT, HLPDA, IGSF8, BAD, VDR, INSIG1,
FLT1, SCARB1, MIF, INHBA, INHA
5.99E-05
doi:10.1371/journal.pone.0065498.t004
functional analysis identified several categories including: signaling, biological quality regulation, myeloid cell regulation, and cell
proliferation among the up-regulated genes. Blood vessel regulation, blood circulation, cellular homeostasis and response to
oxidative stress were the functional categories identified as
enriched in the down-regulated genes. Consistently with preeclampsia pathophysiology, pathway analysis showed an overrepresentation of genes involved in peroxisome proliferative
activated receptor alpha, lipid biosynthesis, hypoxia, and VEGF
response. Subsequently, we extended our analysis by searching
common TFs possibly involved in gene regulation in preeclamptic
women’s placentas. Several bioinformatics tools detected overrepresented TFBSs in the promoters of the PE-associated genes.
Inside up-regulated genes promoters we found an over-representation of TFBSs for NFKB, SP1, RREB1, ARNT, CREB1 and
AP-2. Conversely, among the down-regulated genes we found a
prevalence of TFBSs for MZF-1, NFYA, E2F1 and MEF2A.
Interestingly several transcriptionnally modified genes were
themselves transcription factors. Below, we discuss the molecular
mechanisms of action of all these TFs, and how they might be
related to the placental dysfunction in the context of PE.
NFkB. Belongs to the REL family of TFs which in mammals
is composed of five members: RelA/p65, RelB, c-Rel, p50(NFkB1)
Transcription Factors Interactome
We used the STRING database to search for known
interactions among the TFs identified as consistently modified in
the preeclamptic placenta and also with those identified through
our TFBS analysis. Subsequently, we used the STRING functions
to extend the network and display close interacting factors. As
shown in Figure 2, the majority of the TFs modified in the
preeclamptic placenta including those inferred from the TFBS
analysis present a close functional association. In addition, we
identified that the transcription factor EP300 (E1A binding protein
p300) is connected with the largest number of preeclampsiaassociated TFs in an extended interaction network.
Discussion
The molecular basis of transcriptional alterations in the
preeclamptic placenta remains elusive. Herein, we identified
several TFs which are putatively involved in the regulation of
genes that are consistently associated with PE. We started our
analysis by intersecting publicly available datasets from microarrays analysis of preeclamptic placentas. This allowed building a
consensus list of modified genes in the preeclamptic placenta. Of
these, 67 were up-regulated and 31 down-regulated. The
Table 5. Biological processes annotation clusters for down-regulated genes as reported by the GENOMATIX webtool.
P-Value
Database
Functional annotation
N6 of genes
Genes
GO:0006790
Sulfur compound metabolism
4
GCLM, SOD1, ENPP1, GOT1
1.41E-04
GO:0050880
Regulation of blood vessel size
3
GCLM, SOD1, APLN
5.71E-04
GO:0006536
Glutamate metabolic process
2
GCLM, GOT1
4.03E-04
GO:0006979
Response to oxidative stress
4
GCLM, SOD1, SLC23A2, SEPP1
4.85E-04
GO:0008015
Blood circulation
4
GCLM, ABAT, SOD1, APLN
1.34E-03
GO:0042311
Vasodilation
2
APLN, SOD1
2.17E-03
GO:0065008
Regulation of biological quality
10
HSD17B1, GCLM, SOD1, APLN, ABCG2, F13A1, ABAT, NRCAM, ENPP1,
GOT1
3.15E-03
doi:10.1371/journal.pone.0065498.t005
PLOS ONE | www.plosone.org
5
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Table 6. Signal transduction pathways as reported by the GENOMATIX web tool.
Pathway
N6 of genes
Observed genes
P-value
Peroxisome Proliferative Activated Receptor Alpha
5
VDR, LHB, SCARB1, LEP, NRIP1
5.04E-05
Lipid
8
LYN, PREX1, EZR, SCARB1, LEP, ARID3A, HK2, PROCR
2.97E-04
Hypoxia inducible Factor 1, alpha subunit
4
NDRG1, FLT1, MIF, ERO1L
2.02E-03
FSM Like Receptor Tyrosine Kinase 3
3
CEBPA, LYN, KIT
2.71E-03
Vascular Endothelial Growth Factor
5
ENG, KIT, PREX1, FLT1, ERO1L
3.08E-03
Nuclear Receptor Subfamily 1, Group H
2
VDR, SCARB1
4.60E-03
BCL2 Associated Athanogene
2
BAD, VDR
8.15E-03
Chemokine (CXC Motif) Receptor 4
3
KIT, PREX1, MIF
9.09E-03
TEK Tyrosine Kinase
2
ENG, FLT1
9.83E-03
Nuclear Receptor Subfamily 2, GroupF
1
NR2F1
4.56E-03
doi:10.1371/journal.pone.0065498.t006
and p52(NFkB2). NFkB proteins bind to kB sites as dimers, either
homodimers or heterodimers, and can exert both positive and
negative effects on gene transcription. Signaling mediated by
NFkB stimulates inflammation, invasion, angiogenesis, and cell
proliferation and it is also associated with apoptosis regulation
[23]. NFkB is known to be involved in PE at several levels and in
different cell types. Placental NFkB has been found activated
nearly 10-fold in PE [24]. In vitro experiments show that oxidative
Table 7. Transcription factor binding sites over-represented in the consensus list of up-regulated genes.
TFBS detection tools
CREMAG (1)
CREMAG (2)
TELIS (2)
TRAP (1)
TFM-explorer (1)
TFM-explorer (2)
TOUCAN (2)
CREB1
CEBPA
REST
GC
INSM1
KLF4
SP1
SP1
OLF1
AP2
AP2
SP1
GC
AP2
NFIL3
CEBPA
MZF1
KLF4
ARNT
MZF1
PAX5
HLF
ATF2
NFKB
EBF1
MYC
AP2
E2F1
ELK1
CREB1
ARNT
PAX5
RREB1
NFKB
NFKB
PAX5
E2F1
CREL
RREB1
EGR1
RREB1
MAZR
PAX4
E4BP4
IK2
TP53
MAX
TP53
NFYA
ATF
SP1
Tcfcp2l1
MYC
MYCMAX
ATF2
MZF1
ESR1
HIF1A
USF
NRF2
SRY
EGR1
SPZ1
HOX13
NGFIC
STAT3
NFKB
CAP
E2F1
OCT1B
MYCMAX
BARBIE
SPZ1
SP1
MZF1
ARNT
AP4
AP1
REST
EGR1
MYF
RREB1
AP1
GC
ESRRB
CREB1
PLAG1
STAF
ARNT
HNF4A
ZFX
E2F1
CREB1
SPZ1
ESR2
TFBS with a prevalence P value #0.05 are shown. (1) and (2) indicate that the TFBS weight matrices used for the analysis were respectively JASPAR or TRANSFAC. TFBS
predicted by more than one analysis tool appear in bold.
doi:10.1371/journal.pone.0065498.t007
PLOS ONE | www.plosone.org
6
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Table 8. Transcription binding sites over-represented in the consensus list of down-regulated genes.
TFBS detection tools
CREMAG (1)
CREMAG (2)
TELIS(2)
TRAP (1)
TFM-explorer (1)
TFM-explorer (2)
TOUCAN
NFYA
AP1
COUP
MZF1
NFYA
CEBP
TAXCREB
MZF1
SRF
MEF2A
E2F1
KLF4
AP4
NFYA
ARNT
PAX4
NRSF
MZF1
CTCF
HLTF
NFYA
E2F1
GC
IRF2
NFATC2
PAX6
FOXF1
COUP
Evi1
OCT-1B
AP4
MYF
FOXL1
OCT-6B
OCT-1B
SP1
SP1
Zfp423
MAFB
Gfi-1
CREBP1
MZF1
AP2
EGR1
TBP
ISRE
E2F1
ARNT
NKX25
FOXD1
Evi1
MEF2A
MEF2A
E2F1
E2F1
SP1
FOXD1
TATA
HNF4
SPI1
MEF2A
OCT-1B
SREBP1
MEF2A
FOXA2
GATA-1
PAX5
NR3C1
EGR1
ARNT
XFD2
PDX1
HNF3B
AP4
NFE2L2
En-1
FOXF1
MYB
NFYA
TBP
TFBS with a prevalence P value #0.05 are shown. (1) and (2) indicate that the TFBS weight matrices used for the analysis were respectively JASPAR or TRANSFAC. TFBS
predicted by more than one analysis tool appear in bold.
doi:10.1371/journal.pone.0065498.t008
stability of SP1 [32]. In the placenta, SP1 is involved in the
differentiation of the cytotrophoblast and regulates the expression
of several proteins including ID-1, Syncytin, the AT1 receptor, the
11beta-hydroxysteroid dehydrogenase type 2 (11b-HSD2) and the
pregnancy-specific glycoprotein 5, as well as several cullin genes
involved in the dynamics of protein recycling [33]. Moreover,
compound Sp1/Sp3 heterozygous mice show severely reduced
spongiotrophoblast layer and a disorganized labyrinth layer.
Within the spongiotrophoblast layer both spongiotrophoblast cells
and trophoblast glycogen cells are reduced. Haploinsufficiency of
both Sp1 and Sp3 also leads to a severe disruption of the normal
labyrinth layer architecture [34]. In response to oxidative-stress
induced by hypoxia, SP1 becomes activated and induces the
expression of several factors including VEGFA, b-enolase,
cyclooxygenase 2, and carbonic anhydrase 9. SP1 is also involved
in the inflammatory response and, together with NFkB and AP-1,
up-regulates the expression of VCAM1 and ICAM1 adhesion
molecules, tumor growth factor (TGF-b) and platelet-derived
growth factor (PDGFb), and, finally, monocytes chemotactic
protein-1 (MCP1) and osteopontin cytokines (28).
AP2. The activator protein-2 (AP-2) family consists of five
members, AP-2a, AP-2b, AP-2c, AP-2d, and AP-2e, encoded by
different genes. These isoforms can directly transactivate their
target genes by binding the same GC-rich consensus sequence
[35]. AP-2a and AP-2c are expressed in the placenta, and they
control syncytiotrophoblast-specific gene expression [36,37]. In
AP-2c-deficient mice all derivatives of the throphoblast cells are
formed, however both the embryo and the extraembryonic tissues
are severely growth retarded. This growth retardation is based on
a reduced proliferation of the cells of the ectoplacental cone and a
reduced number of giant cells [38]. In addition, AP-2c has been
shown to regulate the genes for adenosine deaminase (ADA),
human placental lactogen, and human chorionic gonadotropin-b
stress, a hallmark of preeclamptic placenta, causes NFkB
activation in a trophoblast-like cell line, which is enhanced by
TNF-a [24]. In addition, trophoblast cells respond to TLR3
activation by signaling through both NFkB and IRF pathways
resulting in expression of inflammatory mediators and, in
particular, the PE-related anti-angiogenic factor sFLT-1 [25]. In
endothelial cells (ECs) preeclamptic plasma up-regulates NFkB
activity by 2.5-fold compared with normal plasma [26]. This
results in ECs activation. Several factors in the preeclamptic
plasma induce endothelial NFkB activation, including cytokines,
lipid peroxides, peroxinitirites, and shed membrane microparticles
(mainly derived from apoptotic cytotrophoblasts, leukocytes and
platelets), [26,27]. Increased endogenous activation of NFkB
associated with TNF-a and IL-1b release has been detected in
PBMC in PE as compared to normal pregnancies [28]. Several
factors associated with PE have been shown to be able to induce
NFkB activation including adiponectin, leptin, cytokines (TNF-a,
IL-6), lipid peroxides, and agonistic auto-antibodies to the
angiotensin II receptor type I (AT1-AA); [29,30]. Moreover
experiments studying placental ischemia-reperfusion in vitro and
in vivo provide strong evidence indicating that oxidative stress and
ROS production can activate the NFkB signalling pathway [31].
Activation of the NFkB pathway in the placenta, together with
other stress signaling pathways (p38, MAPK, JNK), results in the
placental production of inflammatory mediators, apoptotic debris,
and anti-angiogenic mediators.
SP1. is a ubiquitously expressed Zinc Finger TF that regulates
the expression of thousands of genes implicated in the control of
cellular processes. SP1 is also involved in chromatin remodeling
through interactions with chromatin-modifying factors such as
EP300 and histone deacetylases (HDACs). Although constitutively
expressed, phosphorylation, acetylation, sumoylation, ubiquitylation, and glycosylation influence the transcriptional activity and
PLOS ONE | www.plosone.org
7
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Figure 1. Transcription factors modules in the promoter sequences of TFs consistently modified in PE. The TFs consistently modified
(either up- or down-regulated ) in PE were analyzed with the Genomatix FrameWorker software to identify common regulatory modules. (A) We
identified seven common significant four-element modules in the promoters of five out of seven consistently up-regulated TFs. Here we show the
most significant (P#7.8610211) regulatory module composed of TFBs for EGRF, E2FF and ZF5F. (B) We identified one significant 6-element regulatory
module (P#1.9610210) in the promoters of tree out of four TFs down-regulated in the preeclamptic placenta. This module is composed of TFBS for
E2FF, RXRF, KLFs and ZF02.
doi:10.1371/journal.pone.0065498.g001
consistent with the reported increased expression of AP-2 in PE
and its known role in trophoblasts genes regulation.
CREB1. The cAMP responsive element binding protein 1
(CREB1), a member of the leucine zipper family of DNA-binding
proteins, is ubiquitously expressed and binds as a homodimer to
the cAMP response element (CRE). In the placenta, CREB
contributes to the regulation of PLGF gene expression [42].
Moreover in cytotrophoblast cells CREB, modulates human
chorionic gonadotropin (hCG) gene-expression by a direct
protein-protein interaction with AP-2a [43]. Also, a recent study
has shown that hCG added to cytotrophoblast cells lines (JEG-3,
BeWo) or to placental explants induces endogenous leptin
expression. This induction appears to be mediated by CREB [44].
[37,39,40]. The expressions of AP-2a and AP-2c have been found
elevated in the preeclamptic placentas in comparison with the
gestational age-matched control placentas [41]. Moreover, the
over expression of AP-2a or AP-2c in an extravillous trophoblast
(EVT) cell line, decreased its migratory and invasive abilities [41].
This was associated with reduced expression of protease activated
receptor-1 and matrix metalloproteinases and a significant
induction of plasminogen activator inhibitor-1 and the tissue
inhibitor of metalloproteinase-1. The same study has shown that in
this EVT cell line TNF-a (which is present at higher levels in PE)
induces both AP-2a and AP-2c expression. Thus, the overrepresentation of genes containing TFBS for AP-2 in our study is
PLOS ONE | www.plosone.org
8
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Figure 2. Transcription factors interaction network in the preeclamptic placenta. TFs modified in the preeclamptic placenta were analyzed
with STRING v9.0 to identify putative interactions. Blue lines represent the evidence for the association. The thickness of the line is proportional to the
confidence level of the interaction. TFS found to be modified at the transcriptional level in the preeclamptic placenta appear in red (up-regulated)
and green (down-regulated).
doi:10.1371/journal.pone.0065498.g002
ARNT (HIF-1b). ARNT (aryl hydrocarbon receptor nuclear
translocator) is the beta subunit (HIF-1b) of the heterodimeric
transcription factor, hypoxia-inducible factor 1 (HIF-1). HIF-1 is a
ubiquitous TF complex involved in the regulation of the cellular
responses to oxygen deprivation (hypoxia). Under normoxic
conditions the HIF-1a subunit is constitutively transcribed,
translated and hydroxylated at multiple proline residues. This
hydroxylation targets HIF-1a for proteasomal degradation. In
PLOS ONE | www.plosone.org
hypoxia, mitochondria-derived ROS inhibits HIF-1a hydroxylation, enabling nuclear translocation, heterodimerization with the
constitutively expressed ARNT (HIF-1b), binding to DNA,
interaction with the co-activators p300/CBP and subsequent
activation of hypoxia–responsive genes. In the developing placenta
ARNT (HIF-1b) plays a critical role in cell differentiation [45].
Moreover, as a component of the HIF-1 complex ARNT (HIF-1b)
regulates the expression of placental genes responsive to hypoxia.
9
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
Studies in both preeclamptic patients and animal models have
revealed the existence of hypoxia in the preeclamptic placenta
[46,47,48]. Hypoxia in PE, is believed to be the consequence of
shallow invasion of the decidua by the cytotrophoblasts resulting in
impaired remodeling of the spiral arteries. This leads to reduced
uteroplacental blood flow causing placental hypoxia, oxidative
stress, and inflammation. The analysis of placental explants and
in vitro studies on cytotrophoblasts have shown that several factors
involved in the maternal manifestations of the preeclamptic
syndrome are transcriptionally regulated by the HIF-1 complex
including: Endothelin 1 [49,50], Endoglin [51], the antiangiogenic
factor sFLT-1 [52], Leptin [53], and the vasoconstrictors
Urotensin II [54], Urocortin-2 and Urocortin-3 [55]. Therefore,
the fact that the analysis of the promoters of consistently modified
genes in PE reveals and over-representation of HIF-ANRT
binding sites is consistent with the central role played by hypoxia
in the development of PE.
RREB1. is a zinc finger TF that binds to RAS-responsive
elements (RREs) of gene promoters [56]. In the placenta, RREB1
is expressed in the extravillous cytotrophoblasts were it could be
involved in pathological repression of the human leukocyte antigen
G (HLAG). HLA-G is expressed in the human placenta and
amnios, and plays an essential role in the maternal tolerance
toward the fetus through the inhibition of the NK and T
lymphocyte-mediated direct cytotoxicity. Both circulating HLA-G
and HLA-G protein expressed in the extravillous cytototrophoblasts are reduced in PE [57,58], possibly trough oxidative stress
[59]. RREB1 can inhibit expression of HLA-G by binding to
RREs within the HLA-G promoter [60]. RREB1 is also involved
in the response to cellular stress as it binds to the p53 gene core
promoter and up-regulates p53 transcription. One known effect of
the oxidative stress in PE is to cause oxidative DNA damage [61].
Thus, it is tempting to speculate that RREB1 could activate p53
gene expression in the preeclamptic placenta. However, at present
there are contradictory studies concerning the up-regulation of
p53 in the preeclamptic placenta [62].
MZF1. Myeloid zinc finger 1 (MZF1) is a member of the
SCAN domain family of TFs. MZF1 is preferentially expressed in
hematopoietic cells, and may be involved in the transcriptional
regulation of hematopoietic-specific genes [63]. A putative role in
placental physiology or pathology is currently unknown. However,
the human placenta has been recognized to work as a
hematopoietic organ during the embryonic and fetal development
[64]. Increased hematopoietic activity in the preeclamptic placenta
has been suggested [65]. On the other hand MZF1, together with
SP1 and ZBTB7B has been involved in the regulation of the
SERPINA3 in the cytotrophoblastic cell line JEG3 [66].
SERPINA3 is a serine protease inhibitor known to be upregulated in human placental diseases (including PE) in association
with a hypomethylation of the 5’ region of the gene [67]. Over
expression of SERPINA3 in JEG-3 cells, decreased cell adhesion
to the extracellular matrix and to neighboring cells, but protects
them from apoptosis.
E2F1. The E2F family of TFs controls the expression of genes
involved in cell proliferation, differentiation, apoptosis, and DNA
repair. In the context of PE, a recent study has reported the upregulation of E2F1, together with several genes involved in cell
cycle progression, in peripheral blood mononuclear cells (PBMCs)
isolated from severe preeclamptic women [68]. In our analysis we
did not detect E2F1 among the consensus up-regulated genes in
PE. However, we found that its partner, TFDP2, is downregulated in the preeclamptic placentas. Thus down-regulation of
TFDP2 might result in impaired DNA-binding of E2F1, and lead
to the deregulation of genes controlled by the E2F1-TFDP2
PLOS ONE | www.plosone.org
complex. On the other hand, it has been reported that under
hypoxic conditions E2F1 and p53 are up-regulated, and are able
to down-regulate expression from the VEGF promoter [69]. The
minimum VEGF promoter mediating transcriptional repression
by E2F1, was found to be composed of an E2F1-binding site with
four SP1 sites in close proximity. Of note, it is known that E2F1
and Sp1 proteins physically and functionally interact and show
functional synergism in promoters having binding sites for both
[70]. In ECs, E2F1 can induce the expression of FLT-1, KDR,
and ANGPT2, through a mechanism involving VEGF stimulation, and both Histones and E2F1 acetylation [71]. Previous
studies had shown that the expression of FLT-1 and KDR is
regulated by Sp1 proteins. [72]. Thus, we find again the
association between E2F1 and SP1 binding sites in the regulation
of this antiangiogenic genes.
MEF2A. (Myocyte enhancer factor 2A) belongs to the MADS
(MCM1, agamous, deficiens, SRF) family of TFs and plays a
pivotal role in the development of various organ systems, including
the cardiovascular system [73]. The implication of this TF in
placental development or in preeclampsia has not been studied.
However, its role in the control of gene expression in smooth
muscle cells (SMCs) and ECs suggests that it might be involved in
the vascularization of the placenta. In vascular SMCs, MEF2A has
been shown to be activated via reactive oxygen species and p38
mitogen-activated protein kinase. This leads to the induction of the
transcription factor KLF5 in response to angiotensin II [74].
KLF5 has been found consistently up-regulated in cardiovascular
diseases [75]. Within ECs, shear stress stimulates induction of
KLF2 via the MEK5/ERK5/MEF2 pathway, which ultimately
leads to MEF2A binding to and transactivating the KLF2
promoter [76]. KLF2, has been reported to be essential for the
anti-inflammatory and antithrombotic functions of the endothelium [77]. The mechanisms by which KLF2 achieve its antiinflammatory function are multiple and include inhibition of
NFkB, activator protein-1 (AP-1), and activating transcription
factor 2 (AP-2). Thus, the ROS produced in preeclamptic placenta
could be involved in the activation of MEF2A in SMCs. On the
other hand in the ECs, MEF2A activation could be part of an
adaptive response seeking to protect the cells against inflammation
and thrombosis (two characteristics of PE).
NFYA. associates with a dimer composed of NF-YB, and NFYC subunits, forming a trimer that binds to DNA. The complex
recognizes the pentanucleotide CCAAT, a motif present in the
promoter regions of many genes [78]. The DNA interaction of the
complex occurs through NFYA, suggesting a role as the regulatory
subunit. ROS play also an important role in NFY regulation [79].
When oxidized, NFYB forms homodimers remaining localized in
the cytoplasms, as a consequence the formation of the trimer and
subsequent DNA binding is impaired. NF-Y is known to interact
with several TFs to mediate the synergistic activation of specific
classes of promoters. The most frequent TFs partners of NFY
include: SREBP, SP1, KLFs, OCT-1 and E2F1. NFY seems to be
also involved in the response to cell stress. Thus, NFY directly
controls the expression of TFs genes such as P53 (DNA-damage),
XBP1, CHOP/DDIT3 (ER stress), and HSF1 (Heat shock), [78].
The role of NFY in the regulation of genes involved in the
response to cell stress could represent a link between this TF and
PE. In this sense, NFYA and OCT-1 (another TF which appears
over-represented in our analysis) synergistically regulate a P53independent induction of GADD45 subsequently to DNA-damage
[80]. The GADD45 stress sensor protein has been suggested to be
the link between placental stress and the pathogenesis of PE
through the induction of FLt-1. Thus in stressed placental explants
10
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
GADD45a initiated a signaling cascade culminating in FLt-1
induction [81].
In addition to the TFs identified by our bioinformatic TFBS
analysis, some of the genes consistently modified in the
preeclamptic placenta encode TFs. Among the up-regulated genes
we found: LIMD1, BHLHE40, VDR, CEBPA, BCL6, ARID3A
and NRIP1. Among the down-regulated genes: TFDP2,
ZFAND5, BHLHE41, and NR2F1.
LIMD1 inhibits E2F-mediated transcription, and suppresses the
expression of the majority of genes with E2F1-responsive elements
[82]. The up-regulation of this TF in the preeclamptic placenta
seems coherent with the detection of an over-representation of
TFBS for E2F1 among the down-regulated genes. On the other
hand, LIMD1 has been recently involved in the regulation of the
hypoxia response through a mechanism involving HIF1-a
degradation [83]. LIMD1 up-regulation in the preeclamptic
placenta might result from a feed-back mechanism aiming to
regulate the transcriptional activity of the HIF complex.
BHLHE40 (DEC1/STRA13) is another TF up-regulated in PE,
known to be expressed in the cytotrophoblasts and fibroblast cells
of the placenta [84]. Its gene expression is regulated by various
extracellular stimuli, such as growth factors, serum starvation,
hormones, nutrients, cytokines, and hypoxia through HIF-1a
activation. CEBPA (CCAAT/enhancer-binding protein alpha)
coordinates proliferation arrest and the differentiation of trophoblastic cells [85]. CEBPA is known to activate the expression of the
leptin gene [86]. Thus, the up-regulation of CEBPA is probably
related to the increased expression of leptin (one of the most
consistently modified genes in the PE placenta). BCL6 mediates
transcriptional repression and interacts with components of
histone deacetylase co-repressor complexes including N-CoR
and SMRT [87]. It is involved in a multiple biological processes
including: regulation of inflammatory response; negative regulation of cell growth; negative regulation of transcription, response
to DNA damage stimulus, negative regulation of B cell apoptosis.
It has been speculated that up-regulation of BCL6 in the
preeclamptic placenta could be related to deregulated DNAdamage response, cell cycle arrest, cell survival and immune
response in trophoblast cells [5]. ARID3A is a nuclear matrixassociated TF that stimulates immunoglobulin heavy chain (IgH)
expression and Cyclin E1/E2F-dependent cell cycle progression.
[88,89]. NRIP1 (also known as RIP140) has been shown to bind
and repress the transcriptional activity of several nuclear receptors
including the estrogen receptors, the peroxisome proliferatoractivated receptors, the vitamin D receptor, thyroid hormone
receptors, and estrogen-related receptors [90]. NRIP1 has a major
role as co-regulator of genes involved in lipid and glucose
metabolism, in heart, skeletal muscle, and liver. Its biological role
in the placenta is currently unknown. However, we found in our
study that the most significantly up-regulated pathways concern
the peroxisome proliferators-activated receptor and lipids biosynthesis. Its implication in placental inflammation through its
cooperation with NFkB is also possible. TFDP2 is a member of
the E2F/DP family [91]. As mentioned above, it binds DNA
cooperatively with E2F family members. The down-regulation of
TFDP2 implies impaired E2F1 driven transcription, and seems to
be coherent with the fact that TFBS for E2F1 are over-represented
among the down-regulated genes in the PE placenta. ZFAND5
plays a role in the regulation of NFkB activation and apoptosis.
Over-expression of ZFAND5 sensitizes cells to TNF-induced
apoptosis [92]. BHLHE41 (DEC2/SHARP) is associated with the
regulation of apoptosis, circadian rhythm and the response to
hypoxia [93]. This TF binds to HIFs and promotes HIF
proteasomal degradation by serving as the HIF-presenting factor
PLOS ONE | www.plosone.org
to the proteasome independently from pVHL (von Hippel-Lindau
tumor suppressor), hypoxia and the ubiquitination machinery.
BHLHE41 therefore determines the intrinsic instability of HIF
proteins to act in parallel to, and cooperate with, oxygen levels
[94]. Therefore down-regulation of BHLHE41, is probably related
to the up-regulation of hypoxia responsive genes in the PE
placenta. NR2F1 (COUP-TFI) is a member of the orphan
subfamily of nuclear receptors required for multiple physiologic
and biologic functions, including heart and vascular system
function and cholesterol/lipid homeostasis [95]. Little is known
about a putative role of NR2F1 in the placenta. A study identified
NR2F1 as a repressor of the hLHR (Luteinizing hormone
receptor) gene transcription in JAR cells (issued from a human
placental choriocarcinoma), [96]. In the placenta LH mediates
gonadotropin signals and triggers intracellular responses that
participate in maturation and function of the gonads as well as the
regulation of steroidogenesis and gametogenesis. Nevertheless, we
observe that TFBS for COUP are over-represented in the list of
down-regulated genes in the PE placenta.
Another TF worth mentioning here is STOX1 (storkhead box
1). To date only two PE susceptibility genes have been identified
(ACVR2A and STOX1). Of these, STOX1 encodes a wingedhelix TF showing great similarity with the FOX family of TFs
[97]. STOX1 has been found to be involved in trophoblast
dysfunction in PE. Over-expression of STOX1 in the JEG-3
choriocarcinoma cell line (as a model for trophoblasts), deregulates
many genes which are also modified in the preeclamptic placenta
[98]. Transgenic mice over-expressing the human version of
STOX1 develop a syndrome similar to severe human PE. During
pregnancy, the mice undergo a steep increase in blood pressure,
develop proteinuria and renal histology reveals accumulation of
fibrin [99]. Here, we have compared the transcriptome of the
JEG-3 cells over-expressing STOX1 and the list of consistently
modified genes in PE and found a significant correlation (data not
shown). Genes such as LEP, ENG, EBI3, FSTL3, SPAG4, LHB,
TMEM45A, GCLM, TFDP2, or TSPAN12 that we find
consistently modified in PE, are also transcriptionally modified
in the JEG-3 over-expressing STOX1. The microarrays analyzed
in the present study do not reveal any significant modifications in
the transcriptional levels of STOX1. However, STOX1 is known
to be post-transcriptionally regulated. When phosphorylated by
Akt, the STOX1 protein is inhibited from entering the nucleus
and subsequently degraded by ubiquitination. In the absence of
phosphorylation STOX1 is addressed to the nucleus [97]. As the
STOX1 DNA-binding domain shows great similarity to FOX
transcription factors it has been proposed that STOX1 binds to
the FOX binding sites in the promoters of target genes. In our
analysis FOX binding sites are detected as over-represented
among the consistently down-regulated genes.
Having identified a set of TFs which are likely involved in the
transcriptional modifications of the preeclamptic placenta, we
investigated the putative interactions within them. The interactomics analysis using the STRING software showed that most of
these TFs present close interactions. Moreover, by extending the
interaction network we found that many of them were strongly
connected with a pivotal TF: EP300. This protein is ubiquitously
expressed and functions as a scaffolding actor between the TFs
and the RNA polymerase II. It functions also as a histone
acetyltransferase that regulates transcription via chromatin
remodeling [100]. Among others, it mediates cAMP-gene
regulation by binding specifically to phosphorylated CREB
protein [101]. EP300 has been also identified as a co-activator
of HIF-1a, and, thus, plays a role in the stimulation of hypoxiainduced genes such as VEGF [102]. The loss of one functional
11
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
copy of the gene causes a rare disease in infants, the RubinsteinTaybi syndrome. This disease is characterized by growth
retardation, dysmorphic features, skeletal abnormalities and
mental retardation [103]. Interestingly, three of the babies out of
the seven reported cases, were born from women who developed
preeclampsia during the pregnancy [104]. This suggests that there
could be an association between EP300 heterozygotic deleterious
mutations and PE. The interaction of EP300 with most of the TFs
identified in our study enhances its possible implication in PE.
In summary, our study has identified a number of TFs which
could be key regulators of the changes in gene expression observed
in the preeclamptic placenta. This allows developing hypothesis
about the molecular mechanisms at work in the diseased placenta.
However, there are a number of limitations of our study which
must be taken into consideration. We have drawn a list of
consistently modified genes in PE from the publicly available
microarray data sets. That corresponds to only six studies from a
total of 20 published microarray studies on preeclampsia.
Unfortunately, the datasets corresponding to the majority of
studies have not been deposited in public databases. Moreover, the
authors do not provide in their manuscripts complete lists of
modified genes. The access to more datasets would have increased
the statistical power of the study, and presumably identified even
more striking commonalities. Another aspect to consider is that
these microarray experiments were done on placental samples
which are composed of different cell types. This heterogeneity can
cause noise that disturbs the correct prediction of a co-regulated
gene set, and hence of the TFs involved in their regulation. Finally,
we arbitrarily chose to limit the size of the promoters to be
analyzed to 1200 bp. We postulated that the TFs regulating the
activity of the modified genes would bind TFBS close to the TSS (1000/+200 bases). If we had chosen other promoter lengths we
might get different results. In a previous study published in 2006,
Vasarhelyi et al analyzed the promoters of genes found to be
modified in preeclamptic placentas [105]. They reported an overrepresentation of TFBS corresponding to NFkB(p50), SREBP and
E47. Except for NFkB, the TFs identified in their study are
different to those reported here, these differences being probably
due to the data used for the studies. Vasarhelyi et al extracted data
from a number of studies performed between years 2002 to 2005
[47,106,107,108]. At that time, microarrays offered only a partial
covering of the human genome. Thus, we used more recent data
corresponding to microarrays with full coverage of the human
genome.
Despite all this caveats our study allowed to identify a number of
TFs involved in PE. Although a few of them are found to be
consistently modified in the preeclamptic placenta at the
transcriptional level, many of the TFs identified by our study
(NFkB, CREB, ARNT, SP1, E2F1, NFYA…) are regulated by
post-transcriptional mechanisms. These post-transcriptional modifications (acetylation, methylation, phosphorylation, sumoylation,
etc… ), can be triggered by cellular stresses which are known to be
associated with PE such as hypoxia, inflammation, oxidative stress,
DNA-damage, etc… The validity of the hypothesis raised by our
bioinformatic study need to be confirmed by experimental studies
analyzing the implication of these TFs (including their posttranscriptional modifications) in both, in vitro models and in vivo in
preeclamptic placentas.
Supporting Information
Table S1 Complete list of consistently up-regulated genes in the
preeclamptic placenta. The lists of up-regulated genes for each of
the six preeclamptic placenta microarrays analyzed in this study
were confronted using the GENOMATIX list comparison tool
(Genomatix Software GmbH, Munich, Germany). This allowed to
identify those genes which were consistently up-regulated. Those
showing similar modification in at least 4 studies were considered
relevant and included in a final list of consistently up-regulated
genes.
(XLSX)
Table S2 Complete list of consistently down-regulated genes in
the preeclamptic placenta. The lists of down-regulated genes for
each of the six preeclamptic placenta microarrays analyzed in this
study were confronted using the GENOMATIX list comparison
tool (Genomatix Software GmbH, Munich, Germany). This
allowed to identify those genes which were consistently downregulated. Those showing similar modification in at least 4 studies
were considered relevant and included in a final list of consistently
down-regulated genes.
(XLSX)
Author Contributions
Conceived and designed the experiments: DV FM. Performed the
experiments: DV RC FM. Analyzed the data: DV RC FM. Wrote the
paper: DV FM.
References
9. Meng T, Chen H, Sun M, Wang H, Zhao G, et al. (2012) Identification of
differential gene expression profiles in placentas from preeclamptic pregnancies
versus normal pregnancies by DNA microarrays. OMICS 16: 301–311.
10. Tsai S, Hardison NE, James AH, Motsinger-Reif AA, Bischoff SR, et al. (2011)
Transcriptional profiling of human placentas from pregnancies complicated by
preeclampsia reveals disregulation of sialic acid acetylesterase and immune
signalling pathways. Placenta 32: 175–182.
11. Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, et al. (2007)
Microarray analysis of differentially expressed fetal genes in placental tissue
derived from early and late onset severe pre-eclampsia. Placenta 28: 487–497.
12. Nishizawa H, Ota S, Suzuki M, Kato T, Sekiya T, et al. (2011) Comparative
gene expression profiling of placentas from patients with severe pre-eclampsia
and unexplained fetal growth restriction. Reprod Biol Endocrinol 9: 107.
13. Winn VD, Gormley M, Paquet AC, Kjaer-Sorensen K, Kramer A, et al. (2009)
Severe preeclampsia-related changes in gene expression at the maternal-fetal
interface include sialic acid-binding immunoglobulin-like lectin-6 and pappalysin-2. Endocrinology 150: 452–462.
14. Suzuki Y, Yamashita R, Sugano S, Nakai K (2004) DBTSS, DataBase of
Transcriptional Start Sites: progress report 2004. Nucleic Acids Res 32: D78–
81.
15. Piechota M, Korostynski M, Przewlocki R (2010) Identification of cisregulatory elements in the mammalian genome: the cREMaG database. PLoS
One 5: e12465.
1. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R (2010) Pre-eclampsia.
Lancet 376: 631–644.
2. Sibai BM (2012) Etiology and management of postpartum hypertensionpreeclampsia. Am J Obstet Gynecol 206: 470–475.
3. James JL, Whitley GS, Cartwright JE (2010) Pre-eclampsia: fitting together the
placental, immune and cardiovascular pieces. J Pathol 221: 363–378.
4. Wang A, Rana S, Karumanchi SA (2009) Preeclampsia: the role of angiogenic
factors in its pathogenesis. Physiology (Bethesda) 24: 147–158.
5. Louwen F, Muschol-Steinmetz C, Reinhard J, Reitter A, Yuan J (2012) A
lesson for cancer research: placental microarray gene analysis in preeclampsia.
Oncotarget 3: 759–773.
6. Tapia A, Vilos C, Marin JC, Croxatto HB, Devoto L (2011) Bioinformatic
detection of E47, E2F1 and SREBP1 transcription factors as potential
regulators of genes associated to acquisition of endometrial receptivity. Reprod
Biol Endocrinol 9: 14.
7. Davidson EH (2006) The Regulatory Genome: Gene Regulatory Networks in
Development and Evolution; Burlington M, editor. San Diego, USA: Academic
Press.
8. Sitras V, Paulssen RH, Gronaas H, Leirvik J, Hanssen TA, et al. (2009)
Differential placental gene expression in severe preeclampsia. Placenta 30:
424–433.
PLOS ONE | www.plosone.org
12
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
44. Maymo JL, Perez Perez A, Maskin B, Duenas JL, Calvo JC, et al. (2012) The
Alternative Epac/cAMP pathway and the MAPK pathway mediate hCG
induction of leptin in placental cells. PLoS One 7: e46216.
45. Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E (2000)
Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses.
Genes Dev 14: 3191–3203.
46. Lunell NO, Nylund LE, Lewander R, Sarby B (1982) Uteroplacental blood
flow in pre-eclampsia measurements with indium-113m and a computer-linked
gamma camera. Clin Exp Hypertens B 1: 105–117.
47. Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, et al. (2005) Molecular
evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 90:
4299–4308.
48. Combs CA, Katz MA, Kitzmiller JL, Brescia RJ (1993) Experimental
preeclampsia produced by chronic constriction of the lower aorta: validation
with longitudinal blood pressure measurements in conscious rhesus monkeys.
Am J Obstet Gynecol 169: 215–223.
49. Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA (2001) Molecular
regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxiainducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol
Chem 276: 12645–12653.
50. Minchenko A, Caro J (2000) Regulation of endothelin-1 gene expression in
human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia
responsive element. Mol Cell Biochem 208: 53–62.
51. Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C (2002)
Endoglin expression is regulated by transcriptional cooperation between the
hypoxia and transforming growth factor-beta pathways. J Biol Chem 277:
43799–43808.
52. Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, et al. (2006) Increased
expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia
is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 291: R1085–
1093.
53. Grosfeld A, Andre J, Hauguel-De Mouzon S, Berra E, Pouyssegur J, et al.
(2002) Hypoxia-inducible factor 1 transactivates the human leptin gene
promoter. J Biol Chem 277: 42953–42957.
54. Gould PS, Gu M, Liao J, Ahmad S, Cudmore MJ, et al. (2010) Upregulation of
urotensin II receptor in preeclampsia causes in vitro placental release of soluble
vascular endothelial growth factor receptor 1 in hypoxia. Hypertension 56:
172–178.
55. Imperatore A, Rolfo A, Petraglia F, Challis JR, Caniggia I (2010) Hypoxia and
preeclampsia: increased expression of urocortin 2 and urocortin 3. Reprod Sci
17: 833–843.
56. Thiagalingam A, De Bustros A, Borges M, Jasti R, Compton D, et al. (1996)
RREB-1, a novel zinc finger protein, is involved in the differentiation response
to Ras in human medullary thyroid carcinomas. Mol Cell Biol 16: 5335–5345.
57. Steinborn A, Varkonyi T, Scharf A, Bahlmann F, Klee A, et al. (2007) Early
detection of decreased soluble HLA-G levels in the maternal circulation
predicts the occurrence of preeclampsia and intrauterine growth retardation
during further course of pregnancy. Am J Reprod Immunol 57: 277–286.
58. Zhu X, Han T, Yin G, Wang X, Yao Y (2012) Expression of human leukocyte
antigen-G during normal placentation and in preeclamptic pregnancies.
Hypertens Pregnancy 31: 252–260.
59. Zhou X, Zhang GY, Wang J, Lu SL, Cao J, et al. (2012) A novel bridge
between oxidative stress and immunity: the interaction between hydrogen
peroxide and human leukocyte antigen G in placental trophoblasts during
preeclampsia. Am J Obstet Gynecol 206: 447 e447–416.
60. Flajollet S, Poras I, Carosella ED, Moreau P (2009) RREB-1 is a transcriptional
repressor of HLA-G. J Immunol 183: 6948–6959.
61. Kimura C, Watanabe K, Iwasaki A, Mori T, Matsushita H, et al. (2012) The
severity of hypoxic changes and oxidative DNA damage in the placenta of
early-onset preeclamptic women and fetal growth restriction. J Matern Fetal
Neonatal Med.
62. Crocker I (2007) Gabor Than Award Lecture 2006: pre-eclampsia and villous
trophoblast turnover: perspectives and possibilities. Placenta 28 Suppl A: S4–
13.
63. Bavisotto L, Kaushansky K, Lin N, Hromas R (1991) Antisense oligonucleotides from the stage-specific myeloid zinc finger gene MZF-1 inhibit
granulopoiesis in vitro. J Exp Med 174: 1097–1101.
64. Barcena A, Kapidzic M, Muench MO, Gormley M, Scott MA, et al. (2009)
The human placenta is a hematopoietic organ during the embryonic and fetal
periods of development. Dev Biol 327: 24–33.
65. Centlow M, Carninci P, Nemeth K, Mezey E, Brownstein M, et al. (2008)
Placental expression profiling in preeclampsia: local overproduction of
hemoglobin may drive pathological changes. Fertil Steril 90: 1834–1843.
66. Chelbi ST, Wilson ML, Veillard AC, Ingles SA, Zhang J, et al. (2012) Genetic
and epigenetic mechanisms collaborate to control SERPINA3 expression and
its association with placental diseases. Hum Mol Genet 21: 1968–1978.
67. Auer J, Camoin L, Guillonneau F, Rigourd V, Chelbi ST, et al. (2010) Serum
profile in preeclampsia and intra-uterine growth restriction revealed by iTRAQ
technology. J Proteomics 73: 1004–1017.
68. Rajakumar A, Chu T, Handley DE, Bunce KD, Burke B, et al. (2011)
Maternal gene expression profiling during pregnancy and preeclampsia in
human peripheral blood mononuclear cells. Placenta 32: 70–78.
69. O’Connor DJ, Lu X (2000) Stress signals induce transcriptionally inactive E2F1 independently of p53 and Rb. Oncogene 19: 2369–2376.
16. Cole SW, Yan W, Galic Z, Arevalo J, Zack JA (2005) Expression-based
monitoring of transcription factor activity: the TELiS database. Bioinformatics
21: 803–810.
17. Roider HG, Kanhere A, Manke T, Vingron M (2007) Predicting transcription
factor affinities to DNA from a biophysical model. Bioinformatics 23: 134–141.
18. Roider HG, Manke T, O’Keeffe S, Vingron M, Haas SA (2009) PASTAA:
identifying transcription factors associated with sets of co-regulated genes.
Bioinformatics 25: 435–442.
19. Tonon L, Touzet H, Varre JS (2010) TFM-Explorer: mining cis-regulatory
regions in genomes. Nucleic Acids Res 38: W286–292.
20. Aerts S, Van Loo P, Thijs G, Mayer H, de Martin R, et al. (2005) TOUCAN 2:
the all-inclusive open source workbench for regulatory sequence analysis.
Nucleic Acids Res 33: W393–396.
21. Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, et al. (2003) Toucan:
deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res 31:
1753–1764.
22. Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. (2011)
The STRING database in 2011: functional interaction networks of proteins,
globally integrated and scored. Nucleic Acids Res 39: D561–568.
23. Hayden MS, Ghosh S (2012) NF-kappaB, the first quarter-century: remarkable
progress and outstanding questions. Genes Dev 26: 203–234.
24. Vaughan JE, Walsh SW (2012) Activation of NF-kappaB in placentas of
women with preeclampsia. Hypertens Pregnancy 31: 243–251.
25. Nakada E, Walley KR, Nakada T, Hu Y, von Dadelszen P, et al. (2009) Tolllike receptor-3 stimulation upregulates sFLT-1 production by trophoblast cells.
Placenta 30: 774–779.
26. Takacs P, Kauma SW, Sholley MM, Walsh SW, Dinsmoor MJ, et al. (2001)
Increased circulating lipid peroxides in severe preeclampsia activate NFkappaB and upregulate ICAM-1 in vascular endothelial cells. FASEB J 15:
279–281.
27. Cooke CL, Davidge ST (2002) Peroxynitrite increases iNOS through NFkappaB and decreases prostacyclin synthase in endothelial cells. Am J Physiol
Cell Physiol 282: C395–402.
28. Luppi P, Tse H, Lain KY, Markovic N, Piganelli JD, et al. (2006) Preeclampsia
activates circulating immune cells with engagement of the NF-kappaB
pathway. Am J Reprod Immunol 56: 135–144.
29. Haugen F, Drevon CA (2007) Activation of nuclear factor-kappaB by high
molecular weight and globular adiponectin. Endocrinology 148: 5478–5486.
30. Dechend R, Viedt C, Muller DN, Ugele B, Brandes RP, et al. (2003) AT1
receptor agonistic antibodies from preeclamptic patients stimulate NADPH
oxidase. Circulation 107: 1632–1639.
31. Cindrova-Davies T, Spasic-Boskovic O, Jauniaux E, Charnock-Jones DS,
Burton GJ (2007) Nuclear factor-kappa B, p38, and stress-activated protein
kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to
oxidative stress: effects of antioxidant vitamins. Am J Pathol 170: 1511–1520.
32. Tan NY, Khachigian LM (2009) Sp1 phosphorylation and its regulation of
gene transcription. Mol Cell Biol 29: 2483–2488.
33. Gascoin-Lachambre G, Buffat C, Rebourcet R, Chelbi ST, Rigourd V, et al.
(2010) Cullins in human intra-uterine growth restriction: expressional and
epigenetic alterations. Placenta 31: 151–157.
34. Kruger I, Vollmer M, Simmons DG, Elsasser HP, Philipsen S, et al. (2007)
Sp1/Sp3 compound heterozygous mice are not viable: impaired erythropoiesis
and severe placental defects. Dev Dyn 236: 2235–2244.
35. Mohibullah N, Donner A, Ippolito JA, Williams T (1999) SELEX and missing
phosphate contact analyses reveal flexibility within the AP-2[alpha] protein:
DNA binding complex. Nucleic Acids Res 27: 2760–2769.
36. Ito T, Nomura S, Okada M, Katsumata Y, Kikkawa F, et al. (2002) Ap-2 and
Ikaros regulate transcription of human placental leucine aminopeptidase/
oxytocinase gene. Biochem Biophys Res Commun 290: 1048–1053.
37. Richardson BD, Langland RA, Bachurski CJ, Richards RG, Kessler CA, et al.
(2000) Activator protein-2 regulates human placental lactogen gene expression.
Mol Cell Endocrinol 160: 183–192.
38. Werling U, Schorle H (2002) Transcription factor gene AP-2 gamma essential
for early murine development. Mol Cell Biol 22: 3149–3156.
39. Johnson W, Jameson JL (1999) AP-2 (activating protein 2) and Sp1 (selective
promoter factor 1) regulatory elements play distinct roles in the control of basal
activity and cyclic adenosine 3’,5’-monophosphate responsiveness of the human
chorionic gonadotropin-beta promoter. Mol Endocrinol 13: 1963–1975.
40. Shi D, Kellems RE (1998) Transcription factor AP-2gamma regulates murine
adenosine deaminase gene expression during placental development. J Biol
Chem 273: 27331–27338.
41. Kotani T, Iwase A, Ino K, Sumigama S, Yamamoto E, et al. (2009) Activator
protein-2 impairs the invasion of a human extravillous trophoblast cell line.
Endocrinology 150: 4376–4385.
42. Depoix C, Tee MK, Taylor RN (2011) Molecular regulation of human
placental growth factor (PlGF) gene expression in placental villi and trophoblast
cells is mediated via the protein kinase a pathway. Reprod Sci 18: 219–228.
43. Cheng HC, Shih HM, Chern Y (2002) Essential role of cAMP-response
element-binding protein activation by A2A adenosine receptors in rescuing the
nerve growth factor-induced neurite outgrowth impaired by blockage of the
MAPK cascade. J Biol Chem 277: 33930–33942.
PLOS ONE | www.plosone.org
13
June 2013 | Volume 8 | Issue 6 | e65498
Transcription Factors in the Preeclamptic Placenta
89. Webb CF, Bryant J, Popowski M, Allred L, Kim D, et al. (2011) The ARID
family transcription factor bright is required for both hematopoietic stem cell
and B lineage development. Mol Cell Biol 31: 1041–1053.
90. Fritah A, Christian M, Parker MG (2010) The metabolic coregulator RIP140:
an update. Am J Physiol Endocrinol Metab 299: E335–340.
91. Hitchens MR, Robbins PD (2003) The role of the transcription factor DP in
apoptosis. Apoptosis 8: 461–468.
92. Huang J, Teng L, Li L, Liu T, Chen D, et al. (2004) ZNF216 Is an A20-like
and IkappaB kinase gamma-interacting inhibitor of NFkappaB activation. J Biol
Chem 279: 16847–16853.
93. Fujimoto K, Shen M, Noshiro M, Matsubara K, Shingu S, et al. (2001)
Molecular cloning and characterization of DEC2, a new member of basic
helix-loop-helix proteins. Biochem Biophys Res Commun 280: 164–171.
94. Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, et al. (2012)
SHARP1 suppresses breast cancer metastasis by promoting degradation of
hypoxia-inducible factors. Nature 487: 380–384.
95. Zhang Y, Dufau ML (2004) Gene silencing by nuclear orphan receptors. Vitam
Horm 68: 1–48.
96. Zhang Y, Dufau ML (2000) Nuclear orphan receptors regulate transcription of
the gene for the human luteinizing hormone receptor. J Biol Chem 275: 2763–
2770.
97. van Dijk M, Oudejans CB (2011) STOX1: Key Player in Trophoblast
Dysfunction Underlying Early Onset Preeclampsia with Growth Retardation.
J Pregnancy 2011: 521826.
98. Rigourd V, Chauvet C, Chelbi ST, Rebourcet R, Mondon F, et al. (2008)
STOX1 overexpression in choriocarcinoma cells mimics transcriptional
alterations observed in preeclamptic placentas. PLoS One 3: e3905.
99. Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, et al. (2013)
Preeclampsia-Like Symptoms Induced in Mice by Fetoplacental Expression of
STOX1 Are Reversed by Aspirin Treatment. Hypertension 61: 662–668.
100. Eckner R, Arany Z, Ewen M, Sellers W, Livingston DM (1994) The adenovirus
E1A-associated 300-kD protein exhibits properties of a transcriptional
coactivator and belongs to an evolutionarily conserved family. Cold Spring
Harb Symp Quant Biol 59: 85–95.
101. Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, et al. (2010) CBP/p300 double
null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J 29: 3660–3672.
102. Kwon HS, Kim DR, Yang EG, Park YK, Ahn HC, et al. (2012) Inhibition of
VEGF transcription through blockade of the hypoxia inducible factor-1alphap300 interaction by a small molecule. Bioorg Med Chem Lett 22: 5249–5252.
103. Rubinstein JH, Taybi H (1963) Broad thumbs and toes and facial
abnormalities. A possible mental retardation syndrome. Am J Dis Child 105:
588–608.
104. Tsai AC, Dossett CJ, Walton CS, Cramer AE, Eng PA, et al. (2011) Exon
deletions of the EP300 and CREBBP genes in two children with RubinsteinTaybi syndrome detected by aCGH. Eur J Hum Genet 19: 43–49.
105. Vasarhelyi B, Cseh A, Kocsis I, Treszl A, Gyorffy B, et al. (2006) Three
mechanisms in the pathogenesis of pre-eclampsia suggested by overrepresented transcription factor-binding sites detected with comparative
promoter analysis. Mol Hum Reprod 12: 31–34.
106. Tsoi SC, Cale JM, Bird IM, Kay HH (2003) cDNA microarray analysis of gene
expression profiles in human placenta: up-regulation of the transcript encoding
muscle subunit of glycogen phosphorylase in preeclampsia. J Soc Gynecol
Investig 10: 496–502.
107. Reimer T, Koczan D, Gerber B, Richter D, Thiesen HJ, et al. (2002)
Microarray analysis of differentially expressed genes in placental tissue of preeclampsia: up-regulation of obesity-related genes. Mol Hum Reprod 8: 674–
680.
108. Pang ZJ, Xing FQ (2004) DNA microarrays detect the expression of apoptosisrelated genes in preeclamptic placentas. J Perinat Med 32: 25–30.
70. Lin SY, Black AR, Kostic D, Pajovic S, Hoover CN, et al. (1996) Cell cycleregulated association of E2F1 and Sp1 is related to their functional interaction.
Mol Cell Biol 16: 1668–1675.
71. Pillai S, Kovacs M, Chellappan S (2010) Regulation of vascular endothelial
growth factor receptors by Rb and E2F1: role of acetylation. Cancer Res 70:
4931–4940.
72. Higgins KJ, Abdelrahim M, Liu S, Yoon K, Safe S (2006) Regulation of
vascular endothelial growth factor receptor-2 expression in pancreatic cancer
cells by Sp proteins. Biochem Biophys Res Commun 345: 292–301.
73. Potthoff MJ, Olson EN (2007) MEF2: a central regulator of diverse
developmental programs. Development 134: 4131–4140.
74. Oishi Y, Manabe I, Imai Y, Hara K, Horikoshi M, et al. (2010) Regulatory
polymorphism in transcription factor KLF5 at the MEF2 element alters the
response to angiotensin II and is associated with human hypertension. FASEB J
24: 1780–1788.
75. Dong JT, Chen C (2009) Essential role of KLF5 transcription factor in cell
proliferation and differentiation and its implications for human diseases. Cell
Mol Life Sci 66: 2691–2706.
76. van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, et
al. (2006) Shear stress sustains atheroprotective endothelial KLF2 expression
more potently than statins through mRNA stabilization. Cardiovasc Res 72:
231–240.
77. Atkins GB, Jain MK (2007) Role of Kruppel-like transcription factors in
endothelial biology. Circ Res 100: 1686–1695.
78. Dolfini D, Gatta R, Mantovani R (2012) NF-Y and the transcriptional
activation of CCAAT promoters. Crit Rev Biochem Mol Biol 47: 29–49.
79. Nakshatri H, Bhat-Nakshatri P, Currie RA (1996) Subunit association and
DNA binding activity of the heterotrimeric transcription factor NF-Y is
regulated by cellular redox. J Biol Chem 271: 28784–28791.
80. Jin S, Fan F, Fan W, Zhao H, Tong T, et al. (2001) Transcription factors Oct-1
and NF-YA regulate the p53-independent induction of the GADD45 following
DNA damage. Oncogene 20: 2683–2690.
81. Xiong Y, Liebermann DA, Holtzman EJ, Jeronis S, Hoffman B, et al. (2013)
Preeclampsia-associated stresses activate Gadd45a signaling and sFlt-1 in
placental explants. J Cell Physiol 228: 362–370.
82. Sharp TV, Munoz F, Bourboulia D, Presneau N, Darai E, et al. (2004) LIM
domains-containing protein 1 (LIMD1), a tumor suppressor encoded at
chromosome 3p21.3, binds pRB and represses E2F-driven transcription. Proc
Natl Acad Sci U S A 101: 16531–16536.
83. Foxler DE, Bridge KS, James V, Webb TM, Mee M, et al. (2012) The LIMD1
protein bridges an association between the prolyl hydroxylases and VHL to
repress HIF-1 activity. Nat Cell Biol 14: 201–208.
84. Meinhardt G, Husslein P, Knofler M (2005) Tissue-specific and ubiquitous
basic helix-loop-helix transcription factors in human placental trophoblasts.
Placenta 26: 527–539.
85. Bamberger AM, Makrigiannakis A, Schroder M, Bamberger CM, Relakis C, et
al. (2004) Expression pattern of the CCAAT/enhancer-binding proteins C/
EBP-alpha, C/EBP-beta and C/EBP-delta in the human placenta. Virchows
Arch 444: 149–152.
86. He Y, Chen H, Quon MJ, Reitman M (1995) The mouse obese gene. Genomic
organization, promoter activity, and activation by CCAAT/enhancer-binding
protein alpha. J Biol Chem 270: 28887–28891.
87. Basso K, Dalla-Favera R (2010) BCL6: master regulator of the germinal center
reaction and key oncogene in B cell lymphomagenesis. Adv Immunol 105:
193–210.
88. Herrscher RF, Kaplan MH, Lelsz DL, Das C, Scheuermann R, et al. (1995)
The immunoglobulin heavy-chain matrix-associating regions are bound by
Bright: a B cell-specific trans-activator that describes a new DNA-binding
protein family. Genes Dev 9: 3067–3082.
PLOS ONE | www.plosone.org
14
June 2013 | Volume 8 | Issue 6 | e65498
Send Orders for Print-Reprints and e-Reprints to reprints@benthamscience.net
Current Pharmaceutical Design, 2014, 20, 000-000
1
DNA Methylation, An Epigenetic Mode of Gene Expression Regulation in Reproductive Science
Rosamaria Calicchio1,2,3*, Ludivine Doridot1,2,3*, Francisco Miralles1,2,3, Céline Méhats1,2,3 and
Daniel Vaiman1,2,3,#
1
Inserm, U1016, Institut Cochin, Paris, France; 2CNRS, UMR8104, Paris, France; 3Université Paris Descartes, Paris, France
Abstract: DNA methylation is an important part of the epigenetic code governing gene expression. In human reproductive diseases, recent studies have shown the existence of deviations from the normal methylation profile at various genome loci. In this review, this type
of epigenetic alterations is explored in pathological spermatogenesis, ovarian diseases, placental syndromes, such as preeclampsia and Intra-Uterine Growth Restriction, uterine diseases such as endometriosis, and putative pathophysiological effects of Assisted Reproductive
Technologies. We review the notion of epigenetics, the technical methods available to analyze methylation, and the known associations
between reproductive diseases and DNA methylation, focusing on human pathologies and on animal models when available. We show
that imprinted genes control regions (ICRs) are a prominent and frequent target of methylation anomalies in reproductive disorders, but
such alterations also affect non-imprinted genes. The mechanistic aspects of gene regulation in response to methylation anomalies are
also discussed in this review when they have been investigated.
Keywords: DNA methylation, epigenetics, reproduction, placenta, sperm, infertility, medically assisted reproduction, imprinted genes.
1. GENERAL INTRODUCTION ON DNA METHYLATION
In human (and other mammalian) cell nuclei, the distance separating two consecutive base pairs of DNA is estimated at 0.3 nm
[1]. Since there are 3 billion base pairs in one haploid genome, the
total size of the haploid genome is around one meter. The nuclei of
most cells are ~ 5-10 m Therefore organism biology is confronted
to two major challenges: (i) how to store this huge molecule in such
a small volume? and (ii) given that this first challenge is solved,
how can chromatin work after condensation in such a small volume?
Condensation of the chromatin in the nucleus is achieved
through a series of multi-layered gene condensations involving
wrapping around the histone core in nucleosomes, and the constitution of fibers of 11 nm, themselves packed in 30 nm fibers, then in
300 nm looped domains, 700 nm condensed coiled that constitute
the arms of a metaphase chromosome [2]. Once this ‘library’ is
constituted, access to the pertinent information (materialized by the
expression of a given gene at a controlled and specific moment in a
tissue, or during development) seems to constitute a conundrum.
Nevertheless, cells and organisms thrive. Epigenetics is the reading
rule of the genetic material, acting as a gatekeeper authorizing an
exquisitely precise regulation of gene expression. It is characterized
as heritable through mitosis and sometimes through meiosis, and
not based on modifications of the DNA primary sequence. Three
major mechanisms of epigenetic regulation have been described,
DNA methylation, modification of the ‘histone code’ and synthesis
of non-coding small RNA, such as micro RNA, that are stored in
the cells and are generally operative to inhibit gene expression.
These three mechanisms are in fact acting as a network [3].
In this review, we will focus on DNA methylation and its
known impact in human reproductive diseases (Fig. 1). In mammals, methylation occurs mainly at the C of CpG dinucleotides. The
palindromic structure of CpG dinucleotides allows methylation on
both strands. DNA methylation is achieved by the action of DNA
#Address correspondence to this author at the Institut Cochin, Genetics and
Development Department, 24 rue du Faubourg St Jacques, 75014, Paris,
France; Tel: 00 33 1 44412301; Fax: 00 33 1 44412302;
E-mail: daniel.vaiman@inserm.fr
*These two authors contributed equally to the text.
1381-6128/14 $58.00+.00
methyl transferases (DNMT1, DNMT2, DNMT3A, DNMT3B and
DNMT3L) encoded by related genes and their action and targets
have been recently reviewed [4]. A developing field of research is
linked to the existence of active mechanisms of demethylation, or to
the specific excision of DNA fragments encompassing methylated
CpG dinucleotides, via the action of TET (1-3), AID and GADD45
proteins [5-7]. A novel modification of CpG islands, hydroxymethylation, is also emerging as a growing field in the understanding of DNA methylation in mammalian cells. In reproductive science, the known reprogramming of the embryo necessitates an active demethylation of the paternal genome, and may involve hydroxy-methylation as an intermediate [8].
The central question raised by DNA methylation is its function
in regulating gene expression. While the current dogma assimilates
gene methylation with transcriptional repression, numerous exceptions are known and global correlations between expression and
methylation are low. In fact, the number of studies attempting to
associate DNA methylation to gene expression in a mechanistic
way are few. So, while DNA methylation is probably technically
speaking the easiest modification to analyze, it is also paradoxically
the least studied in terms of its direct or indirect impact on gene
expression.
Analyses of gene methylation were in fact carried out initially
in the context of parental imprinting. Genomic imprinting refers to
a system of gene expression regulation which ensures the establishment and maintenance of allele-specific gene expression,
strictly linked to parental origin. Imprinted genes have been discovered in plants, insects and mammals. In mammals, that are the focus
of this review, about 70 imprinted genes have been discovered in
humans and more than 100 in mice (see for instance
http://igc.otago.ac.nz/home.html). Theoretical [9] and experimental
considerations [10] strongly suggest, however, that these genes may
be much more numerous. Imprinting is established in the gonad and
implemented in a sex-dependent fashion on the germ cells. Part of
this programmation at least is associated with differential methylation at specific loci, called Imprinting Control Regions, or ICR. The
most extensively described controls in a mirror fashion the expression of two paradigmatic imprinted genes, IGF2 and H19, whose
imprinted status seems conserved in all Therian mammals [11].
IGF2 and H19 are located at the telomeric part of chromosome 11
© 2014 Bentham Science Publishers
2 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
Fig. (1). An overview of the four reproductive organs studied in this review and of the diseases that are evoked. In the center is represented a DNA molecule,
methyl residues being positioned on the red strand. DNMT1 (in blue) is the DNA Methyl Transferase that transmits the methylation from the existing strand to
the neo-synthesized strand. DNMT3A (in yellow) and DNMT3L (in green) form a heterodimer that dimerizes and associates at specific positions to the chromatin to proceed to a de novo DNA methylation. This vision does not present the complex of proteins that drives this methylation which is clearly site-specific
in response to various external stimuli.
in humans (11p15). The ICR that controls the expression of the two
genes (generally called ICR1) is located between the two genes and
~3kb 5’ of H19 (Fig. 2). In the classical model, when this ICR is
demethylated, the Zinc Finger Transcription Factor protein CTCF is
able to bind and to trigger the activation of H19 from a distal (telomeric) enhancer. This is the situation on the maternal allele. In the
reciprocal situation encountered on the male allele, the ICR is
methylated, the enhancer activates the IGF2 promoter (~90 kb upstream). It is clear however that this model is oversimplified since
the regulation on large genomic elements cannot be accounted for
only by the linear order of genes and regulatory elements, but rather
by a 3-D structure [12, 13].
In studies on reproductive diseases, altered patterns of expression of imprinted genes are recurrently found, as shown all along
this review. Amongst the most known reproductive defects linked
with imprinting, and thus emphasizing the dissymmetry between
the two parental genomes are hydatiform moles [14], where two
male genomes initiate the development without any female contribution. The loss of DNA methylation specific of different maternal
DMRs in the female germ line, leads to a total failure of fetal tissues development and overgrowth of placental structures. Predisposition to this epigenetic disease has been linked to mutations in
NLRP7[15], setting light on the possible interaction between epigenetic and genetic mechanisms [16].
2. UPDATES ON METHODOLOGIES TO IDENTIFY DIFFERENTIALLY METHYLATED REGIONS
In a test tube, DNA methylation is erased during amplification
of the newly synthesized strands that rapidly become an over-
whelming proportion of the targeted DNA molecules. Hence identification of differentially methylated regions relies on a methylationdependent treatment and/or recognition. Three methods are commonly used to discriminate methylated DNA:
•
endonuclease digestion with methylation sensitive/insensitive
enzymes
•
affinity enrichment using antibodies specific to methylated
cytosines (5mC) or for hydroxymethylatedcytosines(5hmC) or
specific to proteins interacting with methylated DNA
•
bisulfite treatment that drives chemical conversion of DNA
(changing all the cytosines in thymidines except if they were
methylated.
The three methods (described in more details thereafter, and
charted as Fig. 3) have firstly been validated along targeted approaches and more recently served as bases for genome-wide studies.
2.a. Targeted Approaches
Historically, Riggs and Holliday independently proposed that
chemical modifications of the DNA such as methylation could influence gene expression [17, 18]. The observations were made after
two-dimensional gel electrophoresis, chromatography and/or radioisotope incorporation. The discovery of endonucleases, the enzymes that cut DNA, capable of discriminate methylated from unmethylated DNA in the late seventies boosted the research in this
domain and isoschizomer pairs such as MspI/HpaII have been
shown to access up to 98,5% of the CpG Islands or 91,1% of the
RefSeq promoters [19, 20]. Both recognize and cut at the same
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
3
Fig. (2). A schematic representation of the two alleles (maternal and paternal) present at the paradigmatic IGF2/H19 imprinted locus. On the maternal chromosome, the imprinting control region (ICR) is demethylated (white lollipops). This allows the binding of the CTCF Zinc Finger Transcription factor, leading to
insulation of the left part of the ICR. In this case, the enhancer activates H19 expression, while IGF2 (in grey) is not expressed. On the paternal allele, methylation of the ICR (black lollipops) prevents CTCF binding; then the enhancer activates the IGF2 promoter, leading to expression from the paternal allele, while
H19 (in grey) is not expressed. The actual distance between H19 and the ICR is ~3 kb, while the actual distance between H19 and IGF2 is ~90 kb.
sequence, CCGG, but methylation of the second C in this motif
prevents digestion by HpaII. Other couples have been described
such as DpnI/DpnII or SmaI/XmaI, but enzyme-based methods are
confined to specific recognition elements and can therefore interrogate only a subset of all sites of methylation. Indeed, not all CpG
are located with CCGG sequences. Moreover the resolution remains
modest; it depends on the sequence, myriads of bands may be generated from one region and polyclonal or mixed methylation patterns may render the results not interpretable.
The next revolution in DNA methylation studies arose from the
discovery of the fact that methyl-cytosines can be recognized by
specific antibodies [21]. Later on, antibodies specific to proteins
bound to methylated cytosines (methyl binding domains) were developed [22]. These approaches, called MeDIP (for Methyl DNA
ImmunoPrecipitation) and MBDIP (for Methyl Binding protein
DNA) respectively, made it possible to generate libraries enriched
in CpG with high levels of cytosine methylation, thus large and
dense CpGs islands. These libraries have nevertheless a low resolution and are unable to distinguish methylation at the single-base
level. Moreover these methods require relatively large amounts of
input genomic DNA; they discriminate similar sequences with difficulty and false positive results may be obtained due to capture of
unmethylated DNA. One advantage of the affinity enrichment approach is the possibility of specifically pulling down hydroxymethylated DNA using antibodies raised against 5hmC.
The discovery in 1970 that uracil, thymidine, and deoxycytidine
were subjected to sulfonation at position six of their pyrimidine
rings upon bisulfite treatment [23, 24] prepared the launch of the
bisulfite conversion era. Ten years later, Wang and coworkersdemonstrated that 5mC is also sensitive to bisulfite treatment but at
a much slower rate than cytosine [25] and Frommer and coworkersestablished the nowadays classical method to identify methylation
patterns in individual strands of particular genomic sequences: they
utilized bisulfite-induced modification of genomic DNA, under
conditions whereby cytosine is converted to uracil, while5mC remains nonreactive[26]. Next, PCR performed with two sets of
strand-specific primers yield a pair of fragments, one from each
strand, in which all uracil and thymine residues have been amplified
as thymine and only 5-methylcytosine residues have been amplified
as cytosine. Then PCR products are sequenced. Targeting the bisulfite induced sequence changes to specifically amplify either methylated or unmethylated alleles made it possible for the first time to
easily map methylated DNA at the single-base resolution. There are
however several limitations to this technique: i) bias and measurement errors may be introduced by incomplete bisulfite conversion,
ii) DNA may be damaged by the treatment, iii) bisulfite treated
DNA shows a reduced sequence complexity; with the exception of
5
mC, only three different bases instead of four are present, meaning
that more than 90% of Cytidines have been changed into Uracils.
This decreases hybridization specificity and renders PCR primer
design difficult, iv) PCR efficiency may be different for methylated
versus unmethylated version of the same sequence; true quantification may then not be easily obtained, v) bisulfite treatment makes
no distinction between 5mC and 5hmC. Anyway the "gold standard"
for methylation studies remains sodium bisulfite conversion of
DNA followed by cloning and sequencing. However the scaling up
of bisulfite conversion sequencing approaches to analysis of the
entire genome is impractical, in that it requires synthesis of a vast
numbers of primers and a priori knowledge or assumption of the
sequences in specific loci; the use of degenerate primer sequences
only adds difficulties, reducing additionally amplification specificity.
One should keep in mind that there are dynamic changes in
response to the cellular environment and various other stimuli and
that methylated DNA patterns are specific to tissue and developmental stages. Inadequate genome coverage or sample size may
introduce bias in the analysis due to inter-individual epigenomic
variation. Single-base pair resolution only is able to give a comprehensive read-out of methylation patterns in a specific cell type.
To circumvent limitations to interrogate the methylation pattern
in a genomic locus of interest and determine the context of DNA
methylation sites, genome-wide DNA methylation at single-base
pair resolution profiling techniques have been developed this last
decade.
2.b. Genome-wide Approaches
Microarray hybridization adaptation and next generation sequencing have considerably enriched the genome-wide DNA methylation panel of techniques, firstly in organisms with small genome,
but recently, these approaches have been applied to much complex
organisms, including mammals. Two main approaches have been
developed: array- and sequencing-based. These approaches need a
good balance between genome-wide coverage, resolution, and
throughput costs and still require the recognition of methylated
DNA (enzymatically, by affinity enrichment, or chemical conversion).
2.b.1. Array-based
Several techniques have been developed using enzymatic methods such as methylated CpG island amplification, differential methylation amplification which relies on the digestion of one pool of
4 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
Fig. (3). An overview of the major technical possibilities available to analyze DNA methylation. Some approaches are adequate to analyze a specific locus
(left box), some are of interest to analyze the whole genome (right box). The genomic DNA is processed along the three possibilities represented in the upper
part. The arrows correspond to possibilities of genome-wide analyses, and to technical destinations of the DNA, which can be PCR-amplified or ligated. The
large arrows correspond to issues where data mining is necessary, essentially because it correspond to genome-wide analyses, where bioinformatics plays a
pivotal position in order to be able to interpret the data.
genomic DNA with a methylation sensitive-enzyme and a mock
digestion of another pool, or the use of the methylation-dependent
endonuclease McrBC to cut randomly the genomic DNA. HpaII
tiny fragment enrichment by ligation-mediated PCR (HELP)
method where the fragments may be hybridized on dedicated arrays
or detected by high-throughput sequencing represents also an enzyme-based approach of choice. Coupled enzymatic methods to
array-based analysis provide low resolution although improvements
derive from use of pools of restriction enzymes. Dedicated arrays
are required.
Affinity enrichment methods permit rapid and efficient genome-wide assessment of DNA methylation, but as mentioned
above, they do not interrogate individual CpG dinucleotides. Typically, these approaches comprises Chromatin ImmunoPrecipitation
(ChIP) followed by microarray hybridization (ChIP-chip) on a tiling array or to a feature microarray, such as a CpG island array,
where the input DNA and the enriched DNA are labeled with different fluorescent dyes. Dedicated arrays are also required. Nowa-
days, next generation sequencing (ChIP-seq) methods are preferred
to the array hybridization after the ChIP.
In this context, the prevailing view that DNA methylation occurs predominantly at CpG dinucleotides islands in the human genome has been challenged with new findings in studies using next
generation sequencing technologies. For instance, in human stem
cells 25% of cytosine methylation are found in a non-CpG context,
in contrast with human fibroblasts, where almost all methylation are
in a CG context [27]. Moreover the assumption that DNA methylation regulates gene expression mainly through its effects at 5' promoters has been also challenged, since less than 3% CpG islands
have been found methylated in 5' promoters, the majority of 5mC
being located in intragenic and intergenic regions. To interrogate
not only CpGs but a maximum of 5mC, microarrays have been designed with probes specific of methylated and unmethylated sequences for previously annotated loci. Recently developed, the
Infinium Human Methylation 450 BeadChip of Illumina interrogates over 450,000 CpG sites out of the ~28 106CpG sites in the
DNA Methylation
human genome (0.02%). The Illumima method requires a wholegenome amplification after bisulphite conversion, followed by
fragmentation and hybridization of the sample to methylationspecific DNA oligomers that are linked to individual bead types.
Each bead type corresponds to a specific DNA CpG site and methylation state. This method offers a good balance between genomewide coverage, resolution (at the single-base level), and throughput
(possibility to run 12 samples at the same time).
In addition to the limitations brought by the required step to
discriminate enzymatically, by affinity, or chemically the methylated DNA, array-based methods have some other drawbacks: i)
they necessitate an appropriately designed microarray, ii) hybridization artefacts exist, iii) they often require a whole genome amplification step that introduces technical biases, iv) analyses of the relative fluorescent signal intensity to extract DNA methylation information often require bioinformatics adjustements and one has to
deal with choices of threshold necessary to interpret the raw data.
2.b.2. Sequencing-based
Again, these approaches require being able to distinguish methylated from unmethylated DNA, hence, they rely on affinityenrichment, enzymatic or chemical conversion with bisulfite. Once
the DNA is obtained, high-throughput sequencing is performed
using the Roche or Illumina platforms. One has to decide the number and the depths of the reads, knowing that a minimum number of
reads is required to reliably detect differential methylation among a
given pair of samples. Low sequencing depths are often sufficient
to detect strong differences such as global loss of DNA methylation.
Several methods have been developed these last decades. Two
seminal papers in Nature Biotechnologies in 2010 [28, 29] compared the different approaches, affinity-enrichment, enzymatic or
chemical conversion with bisulfite and high-throughput sequencing:
•
MeDIP-seq and MDBIP-seq provide a relative enrichment of
methylated DNA rather than absolute DNA methylation levels.
But the affinity enrichment approaches do not interrogate
CpG-poor genomic regions and combination with an enzymatic digestion of the unmethylated DNA with a methylationsensitive enzyme (MRE-seq) increased greatly genomic coverage. These methods are accessible at reduced costs. However,
these two enrichment-based methods poorly detected repetitive
DNA, where numerous CpGs can be found, or differential
methylated regions (DMR), for which deep sequencing appearsoften required. Futhermore, allele-specific epigenetic
status cannot be interrogated by these approaches.
•
Whole-genome Bisulphite-seq, MethylC-seq, a shotgun sequencing of bisulphite DNA, provides single-base resolution
and has the highest level of coverage and resolution. However
it does not discriminate 5mC and 5hmC and is very expensive.
•
Reduced Representation Bisulfite sequencing (RRBS) has a
reduced genomic coverage compared to the two enrichmentbased methods; RRBS reads cover less than 10% of the 28
106CpGs. However RRBS has the lowest cost per CpG covered in CpG islands, gives a single-based resolution and allows
interrogation of epigenetic status on an allele-specific basis.
Targeted bisulfite sequencing using padlock capture is also used
to interrogate the methylome [30]. This decreases costs since only
genomic regions of interest are analyzed, but may generate complex
cross-hybridization structures, resulting in a possible increase of
false discoveries.
If a single locus validation is important for targeted work on a
specific locus, rather than obtaining a global methylome vision,
with an acceptable False Discovery Rate, all these approaches need
a validation step that can be performed either by pyrosequencing of
bisulfite DNA or clonal bisulfite sequencing.
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
5
The future in genome-wide epigenetic study will be to be able
to directly assess the methylation status of a cytosine at the singlebase resolution, such as nanopore or single-molecule real-time sequencing.
3. EXAMPLES OF GENOMIC METHYLATION ALTERATIONS IN REPRODUCTIVE SCIENCES AND HUMAN REPRODUCTIVE DISEASES
A summary of the literature analyzed in this review in relation
with DNA methylation and reproductive diseases is presented as
Table 1.
3.a. Sperm Methylation Anomalies – Spermatogenesis
One couple out of six seeks medical assistance for procreation
due to fertility defects. While most of these defects are considered
as idiopathic [31], it is generally admitted that one third originates
from female infertility, one third to male infertility and one third to
cases where the origin cannot be elucidated. Clearly in male infertilities, defects in spermatogenesis are the cause of most cases, and
are induced by mutations in genes constituting a highly complex
and multifactorial network [32], as shown by the wealth of data
provided by mouse gene-invalidation models. In fact, at least 1000
genes are estimated to be involved in spermatogenesis defects[33].
Outside these strictly genetic considerations, it is interesting to note
that the sperm chromatin is very different from the chromatin of
other cells; specifically, the genome is compacted to an extreme
point and packed around specific highly basic (lysine-rich) proteins
called protamines [34]. During spermatogenesis, classical histones
are substituted progressively by transition histones and ultimately
by protamines [35]. It implies that the epigenetic structure of the
sperm is considerably changed and may suggest that epigenetic
marks could be altered in the sperm of infertile men [36]. Amongst
these epigenetic marks, it has been shown that DNA methylation at
a genome-wide scale through examination of 2600 loci by restriction landmark genomic scanning, is quite different between male
germ cells and somatic cells[37].
While the abnormal methylation may be widespread in the genome of infertile men, the targeted approach has focused on a quite
limited number of genes up to now, most presumably because researchers concentrated their attention on them. It is quite probable
than in the years to come, the use of high throughput technologies
will strongly increase the number of loci potentially affected at the
methylation level in the convoluted processes of male gametogenesis. In 2003, Benchaib and coworkers interrogated the global methylation pattern of the sperm after immunostaining of the sperm
5
mC. Despite this somewhat imprecise method, this early study was
able to reveal that overall sperm DNA methylation was correlated
with pregnancy rate[38].
Many studies focused on the methylation status of DAZ and
DAZL (Deleted in azoospermia and Deleted in Azoospermia-like,
respectively). DAZ is located on the Y chromosome, belongs to the
AZF region, partly or totally deleted in 10% of the male infertilities
[39, 40]. DAZL is located on chromosome 3. Alike DAZ, it is testis
specific, and homologous to the Boule infertility locus of Drosophila. In 1997, Chai et al, demonstrated that the 5’ region of DAZ
and DAZL are hypomethylated in sperm, but not in other tissues.
Navarro-Costa showed that patients with OAT (Oligo-AzooTeratozoospermia, meaning sperm too few or absent and/or malformed) displayed increased methylation at the CpG island located
in the promoter of DAZL, while the promoter of DAZ remained
unmethylated in both groups[39]. This result is consistent with the
study of Wu (2010) that did not see methylation alterations of DAZ
after the comparison of 174 idiopathic infertile patients with 58
fertile controls [41].
The only mechanistic insight on the function of the altered
methylation of DAZL today was studied in the pig model [42]. In
6 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Table 1.
Calicchio et al.
Recapitulation of the phenotypes and observations described in this review.
Species
Tissue
Disease
Human
Umbilical cord
IUGR spon-
Inducedphenotype
Genes analyzed: Ex-
Genes analyzed:
pression
Methylation
genome-wide
genome-wide
Gene Activity/Outcome
nt
taneous
Yucatan
Liver
minipigs
IUGR spon-
Betaine-
taneous
homcysteinemethyltran
lower
sferase (BHMT)
Cystathionine-g-lyase
(CGL) : lower
Rat
Hippocampus
IUGR in-
Changes in behav-
duced
iour and IUGR
methyl-
(aged offspring are
donor defi-
more anxious and
cient
have better learning
lower
GR
GR
nt
HSD11b2
HSD11b2
nt
NNAT
NNAT
nt
Reelin : lower
Reelin
nt
ability)
TGFb signaling(smad7,
Smurf2, Smad2 and
Smad3)
Human
Umbilical cord
LINE1
spontaneous
low and high
LINE-1 methylation decreased
birthweight,
premature
infants
Human
Fetaladrenal cortex
IUGR
Nicotine treatment
StAR
StAR promoter one CpG (377) sensitive to nicotine
treatment target of PAX6
Rat
Sheep
Hippocampus
Heart
IUGR
IUGR
See paper
Placental function
DUSP5 decreased at
sex-specific alterations of
Protein decreased at day 21
Day 0 et Day 21 (den-
CpG methylation and histone
only, ERK phosphorylation
tate gyrus)
code
increased
IGF2 increased
IGF2/H19 unchanged
Greater relative left ventricle
restriction
Human
Placenta
FetalGrowth
weight
IGF1R increased
IGF1R unchanged
IGF2R increased
IGF2R unchanged
IGF2 decreased
IGF2 not significant at two
Restriction
Rat
Pancreas
IUGR
putative DMR
Bilateral uterine
Pdx1 induced by Ex-
prevention of Dnmt1 binding
Phosphorylation of USF1,
artery ligation
endin4 treatment, afetr
induce demethylation
Association with PCAF at
followed by Ex-
being extinguished
the proximal promoter of
endine-4 treatment
trhough IUGR induc-
Pdx1, increase of HAT
tion
activity, prevention of
Dnmt1 binding
Human
Placenta
IUGR
Small for gesta-
genome-wide DNA
Genome-wide analysis identi-
tional age
methylation
fies profiles specific of IUGR
patterns of DNA methylation
in human placenta are relia-
in placental methylation
bly and significantly associated with infant growth
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
7
(Table 1) Contd….
Inducedphenotype
Genes analyzed: Expression
Genes analyzed:
Species
Tissue
Disease
Human
Placenta
Severe
DNA methylation of placenta
Genome-wide (MedIP on
Mild overall hypermethy-
IUGR
imprinted genes
Chip) focused on all
lation of the DMRs
Methylation
Gene Activity/Outcome
imprinted genes
Mouse
Neural tube
IUGR in-
Alcool treat-
5-MeC, MBD1, DNMT1
Altered DNA methylation
duced
ment/AZA treat-
expression
following drug(s) treat-
ment
Human
Placenta
ment
H19 was increased in IUGR
FetalGrowth
H19 methylation lower
Restriction
increased H19 transcription in the FGR group of
placentas.
Mouse
Lungs
Induced
Antenatal mater-
ACE1 mRNA increased but
Methylation unchnaged in
IUGR
nalhypoxia
protein decreased
the promoter
Ace2 increased mRNA and
protein
At-1b increased
Renin increased protein
mmir 199b, 27b, 200b, 468
decreased
Human
Placenta
IUGR
TBX15 expression regulated
Promoter induction is
by PDX1, repressed in IUGR
methylation and PDX1dependent
IGF2 no significant
Human
Blood cells
IUGR
differences
GNASAS no significantdifferences
INSIGF no significant
differences
LEP no significant differences
Mouse
All tissues
IUGR in-
substitution of the
duced
CTCF binding site
IGF2
with Chicken b
globin insulator x2
Human
SRS
Uniparental disomy chromo-
44% of the patients with
some 7
hypomethylation on
chromosome 11p15
Mouse
embryo and placenta
IUGR
Alcohol induced,
Igf2 decreased ~1,5 fold
correction by
Moderate decrease at 4
CpG of Igf2
methyl donors for
malformation in
paricular
Human
Placenta/Blood/fetal
Preeclampsia
TIMP3 expression inversely
Illumina 1505 CpG, 34
and IUGR
correlated with methylation
loci hypomethylated in
EOPET, 5 in IUGR;
TIMP3
CAPG
GLI2
8 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
(Table 1) Contd….
Species
Tissue
Disease
Inducedphenotype
Genes analyzed: Expression
Genes analyzed:
Methylation
Gene Activity/Outcome
KRT13
Human
cord blood, placenta and umbili-
Idiopathic
cal cord
IUGR
IGF2/H19
LINE-1 methylation,
ICR1 of IGF2/H19
DMR2 hypomethylation
Diagnostic/prognosticseems possi-
Human
Fetal DNA in maternal plasma
IUGR/preeclampsia
SRY
ble?
hypermethylated
RASSF1A
RASSF1A
Beta-globin
Rat
Pancreas
IUGR
Bilateral uterine
Change in mRNA expression
artery ligation
of genes associated with cell
1400 loci identified by
the HELP technique at
death, vascularisation, beta-
conserved intergenic loci
cell proliferation and insulin
secretion
Human
CD34+ hematopoietic stem cells
IUGR
near
Silver-
PLAGL1
from cord blood
Human
Russell
syndrome,
IUGR
IGF2R several (~10%)
patients with a complete
gain of methylation
PEG10
MEST1
GRB10
KCNQ1OT1
H19 loss of methylation
IGF2P0
DLK1
PEG3
NESPAS
Human
Placenta
IUGR,
IGF2/H19 ICR1
Illumina Golden Gate/
Preeclamp-
Bisulfite Pyrosequencing
sia,
11p15 ICR1 demethy-
PE+IUGR
lated in normotensive
IUGR placentas only.
ICR2 not modified,
LINE-1 not modified
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
9
(Table 1) Contd….
Species
Tissue
Rats
Hypothalamus
Disease
Induced phenotype
Genes analyzed: Expression
Genes analyzed:
Methylation
IUGR,
isocaloric low
Cell differentia-
POMC promoter overall
induced
protein; (Restricted
tion/Cytoskeleton, catch-up
demethylated at 7 posi-
and control fetuses
growth corrects the phenotype
tions at PND12
adopted randomly
delayed placental leptin
by restricted or
surge; expression modifiedin
control mothers,
IUGR Bmp2,4,7, cyclin ,
for estimating
double cortin, Dnmt1,
separately the
Dnmt3a
Gene Activity/Outcome
effects of gestation
and lactation)
Mouse
early neurulation
IUGR in-
IUGR
duced by
Links methylation expression
Genome-wide MedIP-
for 84 genes
chip chromosomes 7, 10
alcohol
and X more frequent;
consomption
imprinted genes, cell
cycle, growth, apoptosis,
cancer and olfaction
Human
Leukocytes
BWS/SRS
IUGR/overgrowth
IGF2/H19
Genetic defects affecting
methylation of ICR1 in
21 BWS and16 SRS
patients outside the CTCF
binding sites
Human
Placenta
IUGR/PE
IUGR
Cullin4B, Cullin7
Cullin7 promoter hy-
SP1 may act as a regula-
pomethylated in IUGR
tor induced by IUGR of
Cullin genes
Human
Leukocytes
SRS/ BWS
five maternally and two
167 patients, Loss of
paternally methylated loci
methylation at 11p15
concern RSS patients,
SRS and BWS patients,
LOM can involve both
outsied ICR1 and 2, LOM
paternally and maternally
at other loci
methylated loci in the
(DLK1/GTL2 DMR
same patient
(>2/3 patients). Multilocus LOM for SRS patients.
Human
SRS IUGR
Exclusion of LOT1
and postnatal
(ZAC1/PLAGL1 in SRS)
growth
retardation
Impact of epigenetic
mechanisms in development of the CVS
Human
Leukocytes
SRS
UPD (chr7)and 11p15
epimutation are not
always associated assessed for a group of 188
patients
multilocus LOM can also
10 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
(Table 1) Contd….
Genes analyzed:
Species
Tissue
Disease
Induced phenotype
Genes analyzed: Expression
Rat
Liver
IUGR
Bilateral uterine
IGF1: Days 0 and 21 IUGR
Methylation analyzed > at
artery ligation
decreases the hematic and
several positions among
seric expression
12 CpG sites. Several
Methylation
Gene Activity/Outcome
modifications in both
directions at D0 and D21.
Strong hypermethylation
at D0 At Prom2 (6 CpG
analyzed) rather normalized at P21. Histone (6
modif, P1 taken as reference.
Mouse
Placenta
IUGR in-
IUGR
H19 DMR less methy-
duced by
lated (paternal alleles).
alcohol
Imprinting unaffected
exposure
periimplantatory
Human
DNA (blood cells?)
SRS
201 patients at IGF2/H19
40% of the patients with
epimutation at the locus
amongst 201
Human
Placenta and neonatal blood
IUGR
IGF2 expression decreased,
KvDMR and H19 DMR,
one case of H19 biallelic
20 controls and 24 SGA,
expression
alterations leading to
abiallelic expression of
H19 in one case
Human
Blood cells
BWS/SRS
technique by Highresolution Melting curves
Rat
Pancreas
IUGR
IUGR induced by
PDX1 low levels in beta cells.
bilateral arteries
Inactivation reversible by
Evaluation of DNA from
islets of 5 IUGR and 5
ligation
HDAC inhibition.
control animals revealed
that at age 2 weeks none
of the 14 CpG sites in
the Pdx1 promoter were
methylated in islets from
either IUGR or control
animals. At 6 months of
age (n = 5 animals per
group), Pdx1 DNA methylation levels across the
CpG island averaged
51.3% ± 10.3% compared
with no CpG methylation
in controls (P < 0.05 vs.
controls). No single CpG
site was consistently
methylated.
Human
DNA (blood cells?)
SRS
Human
DNA (blood cells?)
SRS
IGF2/H19
ICR1 hypomethylation
Analysis of several regions: 14q32, 6q24 ICR2.
Only 11p15.5 and UPD7
seem important.
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
11
(Table 1) Contd….
Species
Tissue
Disease
Rat
Skeletal muscle (proxy C2C12)
IUGR
Human
Induced phenotype
Genes analyzed:
Methylation
Glut4 Promoter, three
SRS
Human
Genes analyzed: Expression
IGF2/H19
SRS
Gene Activity/Outcome
IUGR induce epigentics
CpG clusters affecting the
alterations that modifies
binding of MEF2A,
GLUT4 transcriptioni in
MEF2D and MYOD
skeletal muscle.
Epimutations at 11p15
IGFBP3 and IGF1 modi-
(19/44), UPD7 (5/44)
fied.
11p15
Technical approach to
screen by multiplex ligation the 11p15 region
Human
SRS
Evaluation of the prevalence of epimetuationsa at
11p15 in SRS
Human
DNA (leukocytes)
SRS
After IVF
31 Cytosines at
In this case abnormal
PEG/MEST (7q31) and
methylation at the
23 at a the H19 DMR at
PEG/MEST locus
11p15
Mouse
Placenta
IUGR
induced by oral
Igf2 down-regulated
infection with
hypermethylation of the
Igf2 P0 promoter
Campylobacter
Rectus
Human
DNA
SRS
11p15 epimutations are
associated to SRS
Rat
Liver
IUGR
Bilateral uterine
DUSP5 mRNA reduced
artery ligation
methylation of exon 2 of
dephosphorylates ERk1
DUSP5
and 2 inducing phosphorylation of p612 IRS-1,
insulin resistance
SRS and
fetal and postnatal
BWS
growth retardation
Imprinted domains in 11p5
Loss of DNA methylation
fetal and postnatal growth
is epigenetically controlled by different ICRs,
at 11p15 and other chromosomal regions
Human
DNA
SRS and 11p15 epimutations,
no in IUGR idiopathic
Rat
brain
IUGR
Bilateral uterine
Cerebral DNA methyla-
artery ligation
tion reduced at D0, decrease of DNMT1,
MECP2, HDAC1, remain
low at d21
Human
Placenta
IUGR
PHLDA2 increased, MEST3,
DNA methylation at
MEG, GATM, GNAS
PHLDA2 and MEST
PLAGL1 decreased
non imprinted: LEP, CRH,
HPGD, INHBAn IGF1,
INDAO, PSGs GLRX,
AGTR1, DSCR1, SLC
Human
DNA
SRS
Epimutations at 11p15
12 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
(Table 1) Contd….
Genes analyzed: Expression
Genes analyzed:
Species
Tissue
Disease
Induced phenotype
Rats
Liver
IUGR
Bilateral uterinear-
S-adenosylhomocystein,
consequences on DNA
tery ligation
decrease methioninadenosyl-
methylation
Methylation
Gene Activity/Outcome
transferase and cystathionebeta synthase
Mouse
Mouse embryos
Growth alterations
imprinted loci in Chrs 11 and
7p
Human
Placenta
IUGR
Growth retardation
ESX1L
imprinted in mice, not in
humans, no variation in
human IUGR (methylation and expression)
Rat
Kidney
IUGR
Bilateral uterinear-
p53
tery ligation
Human
Mouse
Liver of 1 day old mice
hypomethylations exons
5-8
IUGR
Growth retardation
duplication of 11p15 maternal
IUGR
Growth retardation
IGF2
Deletion of a 54 bp
intragenic methylationc an
methylated core region in
increases levels of tran-
DMR2 on the paternal
scription
allele reduced Igf2
mRNA levels
Human
Placenta
Preeclampsia
CAPN2, EPHX2,
Microarray analysis
This study demonstrated
ADORA2B, SOX7, CXCL1
identified 296 genes that
aberrant patterns of DNA
showed significantly
methylation in PE
& CDX1
aberrant DNA methylation in preeclampsia (PE)
Human
Placenta
Preeclampsia
TIMP3, CAPG, MEST
and Early
The promoter of TIMP3
gene-specific
was confirmed to be
hypomethylation may be a
onset
significantly hypomethy-
common phenomenon in
Preeclampsia
lated in EOPET placentas
EOPET placentas, and that
(EOPET)
TIMP3 could serve as a
potential prenatal diagnostic marker for EOPET.
Human
maternal plasma
Preeclampsia
RASSF1A
Hypermethylation of
utility of hypermethylated
RASSF1A in PE
RASSF1A sequences in
maternal plasma as a
gender- and polymorphism-independent marker
for pre-eclampsia.
Human
maternal plasma, Placenta
Preeclampsia
RASSF1A, SERPINB5
Human
Placenta
Preeclampsia
MMP9
hypomethylated in PE
Reduced synthesis of
placentas
MMP9 in PE placentas
may result from epigenetic
changes of the methylation status of CpG sites in
the promoter region.
Human
Placenta
Preeclampsia/IUGR
SERPINA3, A5, A8, B2, B5,
Hypomethylation of
and B7
SERPINA3 promoter in
PE and IUGR
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
13
(Table 1) Contd….
Species
Tissue
Disease
Placenta
Human
JEG-3 cells
Human
Placenta
Induced phenotype
Preeclampsia/IUGR
Preeclampsia/IUGR
Preeclampsia
Genes analyzed: Expression
TBX15, PDX1
Genes analyzed:
Methylation
Gene Activity/Outcome
Hypomethylation of
Reduced expression of
TBX15 promoter
TBX15 in IUGR
Increased global DNA
A positive association
SERPINA3
Global DNA methylation
methylation levels were
between global DNA
seen in the PE group
methylation and systolic
and diastolic blood pressure was seen in the term
PE group
Human
Sperm
ART
The proportion of sperm
with DNA fragmentation
appears to be potentially
useful as a predictor of
ICSI outcome, whereas
embryo quality based on
morphological criteria,
appeared unaffected by
DNA fragmentation
Human/Mouse
medakafish
germ cells and testis somatic cells
sperm, ovary
DAZ and DAZL
DAZ and DAZL
gametogene-
the odazl or its protein is a
sis
marker for germ cells
during embryogenesis and
at critical stages of gametogenesis in both sexes of
medaka
Human
spermatozoa, leukocytes, pla-
ART
centa
Inheritable male
DAZ and DAZL
The 5' end of both genes
infertility
are hypomethylated in
spermatozoa but not in
leukocytes or placenta,
consistent with the expression pattern of the genes
Human
sperm
oligoasthenozoospermic
DAZL
increased methylation
defects in the DAZL
Incorrect DNA methylation of the DAZL promoter CpGisland associates with defective human
sperm.
Pig
somatic and germcells
Azoospermia
Male infetility
DAZL
Methylation supresses
DNA methylation may
DAZL promoteractivity
suppress DAZL expression in somatic cells by
interfering with Sp1
binding
Human
testicular biopsies
idiopathic
VASA
azoospermic
or severely
oligospermic
Human
Sperm
oligospermic
high VASA TDMR
methylation rates in MA
MEST and H19
14 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Calicchio et al.
(Table 1) Contd….
Species
Tissue
Disease
Human
Sperm
azoospermia,
Induced phenotype
Genes analyzed: Expression
H19, IG-GTL2 and MEST
vasectomy
Genes analyzed:
Methylation
Gene Activity/Outcome
Hypomethylation of H19
DMR
reversal
Human
Sperm
H19 and IGF2
oligosper-
Perturbations of the 6th
mic, terato-
CTCF site of the H19
zoospermic
DMR
and OAT
Human
Sperm
Human
Sperm
low sperm motility
HDAC1, SURT3, DNMT3A
abnormal methylation
severe OZ
and 67
moderate OZ
Human
Sperm
oligospermic
LIT1, MEST, SNRPN,
significantlyincreased-
patients and
PLAGL1, PEG3, H19 and
methylation
patients with
IGF2
known
anomalies of
the protamines
Human
Sperm
ART, Oligospermia
XIST, DNMT3L
Mouse
Blastocysts
ART Superovulation
H19
Superovulation clearly
H19 could be used as a
disrupted H19
sensor of the invasiveness
methylation gene
of the ART
expression in individual
Female rat
Spermatogonia
vinclozolin,
Inheritable male
methoxy-
infertility
Global DNA methylation
blastocysts
altered DNA methylation
patterns in the germ line
chlor
Human
Endometrium biopsies
Endometri-
84 nuclear receptor genes
DNA methylation and
osis
transcriptional repression
signaling as the most
affected pathway in endometrium in women with
endometriosis
Human/Mouse
Eutopic endometrium
Endometri-
Hoxa10, IGFFBP1, Pgr-AB
osis
hypermethylation of
Hoxa10 in the endometriosis
Human
Endometrium biopsies
Endometri-
Hoxa10
Hypermethylation
osis
Reduced expression of
Hoxa10 in endometrion is
associated with hypermethylation
Human
cystic endometriosis lesions of
Endometri-
the ovary
osis
SF1
Methylation reduces SF1
increased methylation of
expression
SF1 promoter in endometrial cells
Human
Endometrial cells
Endometriosis
PR-B
partial methylation of PR-
prolonged exposure of
B promoter associated
endometrial cells to TNF-
with a reduction in its
alpha induces partial
expression
methylation of PR-B
promoter
DNA Methylation
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
15
(Table 1) Contd….
Species
Tissue
Human
Endometrium biopsies
Disease
Induced phenotype
Genes analyzed: Expression
Genes analyzed:
Methylation
Gene Activity/Outcome
APC, CDKN2A, PYCARD,
RARB, RASSF1 and ESR1
Human
Endometrium biopsies
OMA, DIE and SUP
Global DNA methylation
35 genes alterations of
methylation associated to
expression modifications
Mouse
Ovary
PCOS
LHR, AR, FSHR and H19
LHR gene was demethy-
evidence for close linkage
lated in PCOS
between DNA demethylation of LHR and PCOS
Mouse
Oocytes, blastocytes
ART
DNA methylation of im-
maternal as well as pater-
printed genes
nal H19 methylation was
perturbed by superovulation
Mouse
blastocystes
H19
Culture medium affects
H19 methylation and
expression
Human
placenta and cord blood
ART
DNA methylation at more
ART may have an effect
than 700 genes (1536 CpG
on global patterns of
sites)
DNA methylation and
gene expression
Human
Lymphocytes and buccal cells
ART
KvDMR and CTCF binding
differences in methylation
sites in H19, IGF2 DMR0 and
in IGF2 and IGF2R
IGF2R
this important article, the authors cloned a minimal promoter region
of 149 bp in front of the luciferase reporter gene, and showed that
in vitro methylation leads to a complete extinction of the promoter
activity in primordial germ cells, accessible in their pig model.
Then, observing the presence of several putative binding sites for
the SP1 Zinc Finger Transcription Factor, the authors demonstrated
actual binding to the promoter element using gel-shift assays, unless the promoter element was methylated. These observations were
confirmed by Chromatin Immunoprecipitation (ChIP). Overall,
these data suggest that from a mechanistic point of view, SP1 may
contribute to the regulated expression of DAZLA in germ cells. To
note, this study is one of the very scarce that attempts to correlate
experimentally DNA methylation, Transcription Factor tethering
and gene expression.
Among the other studies on specific genes, Sugimoto and coworkers focused their work on the human VASA gene [43]. VASA is
a member of the DEAD-box protein family with ATP-dependent
RNA helicase catalytic activity, which alike DAZL has RNA binding activity and is involved in the early steps of spermatogenesis.
Potentially, this study reflects the idea that any so-called ‘spermatogenetic’ gene with a CpG island nearby is a potential candidate
to analyze a methylation status in patients with abnormal spermatogenesis. In this study, the authors analyzed a CpG island by
MALDI-TOF on a sample of 131 idiopathic azoospermic or severely oligospermic patients. In this case, the analysis was carried
out on a complex tissue, testicular biopsies. The authors chose on
their samples the ones where a sperm maturation arrest was detected histologically (17 out of 131), it is known then, that VASA is
silenced, since this gene is necessary for progression through meio-
sis [44]). In six patients of the maturation arrest group (n = 17), the
methylation rate of each CpG unit within an amplicon was especially high; the average rate was 77.4±3.07%. The methylation rate
was low in the remaining 11 patients, with an average of 29.3±
2.68%(P<0.0001), the controls being at 16.3 ± 4.8 %.
An even larger number of studies on spermatogenetic defects
concentrated on imprinted genes.
Probably one of the first evidence that defective spermatogenesis was associated with methylation in imprinted genes came from
the seminal study of Marques and coworkers [45]. This study focused on MEST and H19, and showed significant anomalies for the
latter. This prompted an international research effort aiming at correlating abnormal epigenetic marks on imprinted genes and abnormal spermatogenesis. Kobayashi and coworkers studied H19,
GTL2, PEG1, LIT1, ZAC, PEG3 and SNRPN using a combined
bisulfite-PCR restriction analysis (COBRA) and sequencing technique, in parallel with non-imprinted repetitive sequences (LINE
and Alu). The authors analyzed the seven Differentially Methylated
Regions (DMRs) in the sperm of 97 men from infertile couples, and
discovered 14 samples with abnormal paternal methylation at H19
and GTL2 DMRs, and 20 with abnormal maternal methylation at
PEG1, LIT1, ZAC, PEG3 and SNRPN
Marques and coworkers studied H19 and MEST in oligospermic
and control patients by bisulfite treatment followed by cloning and
sequencing [46]. Similarly, Minor and coworkers analyzed
Oligoazoospermic and control patients at three imprinted genes,
H19, IG-GTL2 and MEST, by the same approach. They revealed
that H19 DMR methylation was decreased in azoospermic men.
The most important idea is that alterations also exist in Oligoazoo-
16 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
important idea is that alterations also exist in Oligoazoospermia,
showing that even without known gametogenesis defects, imprinting anomalies exist [47]. They show that in azoospermic patients,
the 6th binding site of CTCF near H19 where complete methylation
was expected was significantly reduced especially in cases of
obstructive azoospermia due to congenital bilateral absence of vas
deferens and in secretory azoospermia due to hypospermatogenesis.
MEST was analyzed in the same study, and was found demethylated as expected for an imprinted gene in the sperm. The study by
Boissonnas and coworkers in 2010 was probably the first to use
pyrosequencing on oligospermic, teratozoospermic and OAT patient sperm. This approach allowed giving a precise measure of the
degree of methylation alterations in H19 and IGF2 in sperm cells,
where the DMR is supposed to be set to a methylation level of
100%. There was an overall demethylation in the patient sperm,
proportional to the severity of the sperm anomalies and strongly
marked at the 6thCTCF binding site of H19. The bisulfite analysis is
based on billions of molecules while in bisulfite/cloning/sequencing, it is generally less than 20 clones that are analyzed. However, this approach gives no information on the phase of the methylation i.e. if a molecule is methylated at all the consecutive CpG
(see methods).
The development of genome-wide approaches to studying genomic methylation will undoubtedly rejuvenate and enrich the
growing database of spermatogenic-associated methylation anomalies. Recently Pacheco and coworkers [48] used such genome-wide
analysis of methylation with the Human BeadChipIllumina array
that allowed the simultaneous analysis of 27,578 CpG dinucleotides. Interestingly, clustering of the methylation data from 21 patients yielded a strong correlation with the motility parameters of
the sperm cells. 9,189 CpG were significantly different, 80% of
which were hypomethylated in low-motility samples, and 194 were
associated with imprinted genes. The authors performed a simultaneous transcriptome analysis that revealed modifications of genes
potentially involved in regulating the epigenetic structure of the
chromatin, such as histone deacetylase HDAC1, sirtuin SIRT3 and
the de novo methylation enzyme DNMT3A. This recent study emphasizes the importance of high-throughput analysis in the future.
Using another technical approach (Bisulfite PCR-Luminex
methylation analysis), Sato and coworkers examined 8 imprinted
loci (ZDBF2, H19, GTL2, PEG1, LIT1, ZAC, PEG3, SNRPN) in
337 patients (209 normal, 61 severe Oligozoospermia and 67 moderate Oligozoospermia). 13.9% of the patients showed an abnormal
methylation at one or more of these imprinted loci.
Similarly, in 2010, Hammoud and coworkers analyzed CpG
methylation at 7 imprinted loci by bisulfite sequencing: LIT1,
MEST, SNRPN, PLAGL1, PEG3, H19 and IGF2 on three male
populations, normal donors (n=5), oligospermic patients (n=10) and
patients with known anomalies of the protamines (n=10). MEST,
KCNQ1 (overlapping LIT1) as well as SNRPN and H19, were affected in oligospermic patients.
In 2009, Kobayashi and coworkers studied the DNA methylation of seven imprinted genes and of XIST, the non-coding RNA
initiating X chromosome inactivation [49], in 78 paired DNA samples (paternal DNA and embryo DNA) in order to evaluate whether
these errors were due to the use of Assisted Reproductive Technologies (ART) or directly transmitted by the sperm [49]. ART and
especially ICSI is of course often used for patients that suffer from
oligo/astheno/teratozoospermia, and as discussed here, these alterations are often associated to abnormal methylation in the sperm at
imprinted loci [50]. Our group was one of the first to report anomalies of the methylation of imprinted genes (especially H19) in mice
embryos at the blastocyst stage, following the use of ART [51],
these anomalies inducing long-term consequences on gene expression in the placenta [52]. Recently, the same type of anomalies has
been reported in human embryos, accessible to research purposes in
some countries [53]. It is therefore a quite relevant question to
Calicchio et al.
know whether the alterations in the embryo are a consequence of
ART or an inheritance of an abnormal methylation profile from the
paternal sperm. The study of Kobayashi showed that amongst the
17 cases of abnormal methylation in the ART sample, 7 cases(41%)
presented with the similar anomaly in the parental sperm. In the
same study, the authors correlated DNMT3L variants with abnormal
paternal methylation. DNMT3L, discovered in 2001 [54], is an
atypical DNA methyl transferase that does not possess any catalytic
moiety. However, this enzyme is a cofactor of DNMT3A [55], and
seems required for the correct apposition of imprinted marks during
the early development of germ cells. In mice, the KO of this enzyme induces a ‘meiotic catastrophe’, where numerous mis-pairings
between the parental chromosomes in the germ cells during meiosis
trigger the abortion of the whole gametogenetic process. DNMT3L
seems largely involved in the methylation of repetitive sequences
(especially LINEs), which appears indispensable for the correct
ongoing of meiosis [54]. Some data that our group published between the links of DNMT3L and the methylation of specific loci on
the chromosomes in the case of endometriosis (see § 3c), substantiate the pivotal role of this factor as agitating a flag where a DNA
sequence should be methylated by an active DNA methyltransferase.
Another recent issue in the epigenetic ‘game’ in male gametogenesis is the possible involvement of environmental influences,
and in particular of endocrine disrupters, with the additional idea
that such alterations could be transmitted epigenetically through
meiosis and therefore on several consecutive generations. The
seminal work for such ideas was probably published by Michael
Skinner group in 2005 [56]. Rats exposed to high doses of the fungicide vinclozolin, presented anomalies of the genital tract, spermatogenesis and several parameters of the sperm biology that seem
heritable during several generations. As reviewed by Price [57], it
appears that the data were less obvious in humans, at least in 2007,
certainly because the control of the observational data is much more
complex and the genetic background much more heterogeneous
than on experimental rodent models.
3.b. Ovary
DNA methylation in the ovarian context has less been studied
per se, except in relation with ovarian cancer which is beyond the
scope of this review. Here we will concentrate on diseases that are
of ovarian origin but are rather direct causes of infertility, Polycystic Ovary Syndrome (PCOS) and Premature Ovarian Failure (POF).
3.b.1. Polycystic Ovary
PCOS is one of the most frequent ovary disorder (5 – 15% of
women of reproductive age), strongly correlated to endocrine disequilibrium [58]. Ovary is normally synthesizing androgens that are
aromatized in estrogens by the CYP19 – aromatase enzyme. Imbalance in this regulation leads to excessive androgenic activity, defects in ovulation and the appearance of cysts on the ovaries that are
detectable by ultrasound. Genetic analysis showed that polymorphisms of the gene encoding the androgen receptor (AR, located on
the X chromosome) are associated with PCOS [59]. This gene encompasses a highly polymorphic (CAG) repeat encoding a glutamine tract, the shorter alleles being generally considered as the
most active, albeit this is sometimes debated; indeed the study of
Hockey and coworkers showed a higher frequency of alleles > 22
repeats in the PCOS population. A more recent study in Indian
women failed to find a clear association between PCOS and AR
short allele expression [60]. The expression of androgen receptor
appears regulated by methylation of a CpG island in the 5’ region,
as shown in prostate cancers [61]. In 2010, Laisk and coworkers
studied epigenetic variations at this locus using HpaII-amplification
from heterozygous patients, but failed to associate repeat length, X
chromosome inactivation patterns and susceptibility to PCOS. In
2010, Xu and coworkers failed to show differences in the methylation profile of circulating DNA on 20 controls and 20 PCOS
DNA Methylation
women using a 5-methyl CpG antibody followed by a ELISA-like
test [62]. In a targeted study, Zhu and coworkers used postbisulphite approaches to analyze the methylation status of LHR, AR,
FSHR and H19 in a mouse model where PCOS was induced by
DHEA (Dehydroepiandrosterone) treatment, and showed that the
LHR gene was demethylated in PCOS [63]. Finally very recently, a
systematic analysis was performed in a rhesus monkey model prenatally androgenized, using the InfiniumHumanMethylation27
BeadChip plat form [64]. However the authors focus on omental
tissues near the gastrointestinal tract, and not directly on the ovarian
tissue. The authors suggest that visceral adipose tissue is particularly interesting due to its potential links with metabolic disease,
itself frequently shown as a risk factor for PCOS. In infants, 163
regions were found modified and 325 in adults. The analysis of
genes nearby showed significantly enriched pathways in infants
(anti-proliferative role of TOB (Transducer of ErbB2) in T cell
signaling, VDR/RXR activation, Methionine metabolism, Complement system, Nucleotide excision repair pathway) and adult s
(TGF signaling, Axonal guidance signaling, Polyamine regulation in Colon Cancer, Wnt/B-catenin signaling). These differences (interestingly not common between infants and adults) are
nevertheless probably linked to the injection of testosterone propionate to the females, and may not be specific to the disease, strictly
speaking.
3.b.2. Premature Ovarian Failure
Premature Ovarian Failure (POF) may be defined as an ovarian
failure occurring before menopause, and according to the clinical
definition before the age of 40. Genetically speaking, mapping studies of POF revealed two important regions on chromosome X [65],
one of them having recently been shown to belong to a heterochromatic domain [66]. In 2010, Laisk and coworkers could show that
short variants in the Androgen receptor CAG repeat are associated
with POF [67]. These is presumably linked to skewed X chromosome inactivation, reported in 2007 for AR[68]. The detail of the
epigenetic mechanisms at work in this ‘X-choice’ is however not
elucidated. Another gene that has been investigated for the disease
is PGRMC1 (Progesterone Receptor Membrane Component-1), also
located on the X chromosome [69].
3.c. Endometriosis
Endometriosis is a frequent invalidating disease of reproduction, characterized by the implantation of endometrium tissue outside the uterine cavity. In the classical vision of this disease, regurgitation of the menses is at the origin of the lesions [70]. It may
affect utero-sacred ligaments, the peritoneal part of the rectum or
the vagina, and often the ovary. Endometriosis is associated with
pain [71, 72], and is considered a major cause of female infertility,
even though a recent study suggests that the eutopic uterine tissue
of endometric patients is similar to that of uteri from control patients, free of endometriosis lesions, as revealed by transcriptome
analyses [73]. More recently, Zelenko and coworkers suggested by
analyzing the expression of Nuclear Receptor genes the existence of
differences at the epigenetic level. More precisely, they started by
analyzing the expression of 84 nuclear receptor genes [74]. Nuclear
receptors such as members of the RAR/RXR/PPAR family are
known to interact with enzymes that modify histones and methylate/demethylate DNA. The modified genes were functionally clustered using Ingenuity Pathway Analysis (IPA), and epigenetic
pathways were identified as the most strongly modified in the endometrium of endometriotic women compared to controls.
The idea that endometriosis may be accompanied by epigenetic
modifications is also substantiated by mouse models where uterine
tissue is grafted ectopically in the peritoneum. In this situation,
hypermethylation of Hoxa10 was observed [75]. This is in accordance with observations in humans where several studies indicated
that the promoter of HOXA10 is modified by methylation in the
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
17
lesions [76], this being clearly confirmed by various studies [77],
and clearly associated with expression alterations [78].
Accordingly, several recent studies focused on specific genes to
attempt identifying specific differences in the methylation profile at
various CpG islands, essentially but not exclusively focusing on
genes involved in the regulation of steroidogenesis, the other being
rather potentially involved in cancer. Namely, PTGS2, HOXA10,
HOXA11, PAX2, SF1, PGRB, E-cadherin, ESR1 (=ERA), CYP19,
MLH1, p16 were analyzed individually to date [77, 79-88]. ERA
was reported as not being modified by methylation more than 10
years ago [89]. It may be that at the head of this steroidogenic cascade, SF1 plays the role of a bandmaster since it is a key gene to
activate estrogen synthesis. The transcription of this gene was
found induced more than 100 fold in endometriomas[78, 90], albeit
this increase may be partly due to the proximity of ovarian (non
endometriotic) tissue as revealed by immunohistochemistry [91]. 5aza-2’ deoxycytidine treatment increases SF1 mRNA more than 50
fold in endometrial cells, this induction being regulated through a
CpG island located between position -85/239. This site is a conditional binding site for MeCP2, an important factor to mediate
chromatin opening or locking [90]. The methylation status of SF1
in endometriosis remains however to be directly studied.
In a recent review, Bulun and coworkers speculated that in endometriosis the promoter of ERB is not adequately methylated leading to its overexpression [92]. This overexpression relative to ERA
would be the initial event indirectly triggering a down-regulation of
the progesterone receptor, consistently with the reduction described
by Borghese and co-workers [78]. Progesterone resistance is part of
the pathogenesis in this disease at least for a defined category of
patients for whom the symptoms are not improved by progesterone
treatment. The regulation of progesterone response may also be
driven by methylation alterations of the PR promoter, especially the
B isoform. This has been substantiated in a cellular model by Wu
and coworkers that stimulate by TNF an immortalized line of
epithelial endometriotic cells during 30 days, thus triggering a partial methylation of PR-B promoter associated with a reductionin its
expression [93].
In line with the putative links between cancer and endometriosis, Vestergaard and coworkers analyzed biopsies from ectopic
lesions from 23 patients, and analyzed the promoter methylation of
APC, CDKN2A, PYCARD, RARB, RASSF1 and ESR1, without identifying significant alterations from this epigenetic point of view
[94].
Systematic analysis of genome methylation in endometriosis
was recently performed [95], and revealed by ChIP on chip following immunoprecipitation with an anti-methylC antibody, and hybridation to anAffymetrix promoter CpG arrays. In fine, 25,000 promoters were analyzed after IP starting from pools of three different
subtypes of endometriosis (ovarian, deep infiltrating and superficial). In 35 genes alterations of methylation were associated to expression modifications. Quite interestingly, demethylations were
randomly distributed, but hypermetyhlation was biased towards the
extremity of chromosomes [95]. This observation was correlated
with a recent paper [96], that showed that specific isoforms of
DNMT3L were linked to abnormal hypermethylation of chromosome ends in the general populations. This observation prompted to
analyze DNMT3L variants with a high density of SNPs in endometriotic and control patients [97]. Following this study, a specific
DNMT3L haplotype covering intron9-exon10-intron10 was very
strongly associated with the risk of endometrioma (RR= 7.21).
Since DNMT3L is not catalytically active, this article suggests that
it could drive the methylation activity of cofactors interacting with
it such as DNMT3A [55]. Several studies showed that DNMT are
not expressed appropriately in endometriotic lesions [78, 98]. In the
last study, interestingly, DNMT1 was found reduced more than
twice at the expression level. This would be quite consistent with
the random demethylation observed along chromosomes in endo-
18 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
metrioma lesions, since the down regulation of DNMT1 could lead
to a loss of the methylation maintenance through cell divisions.
3.d. Medically Assisted Reproduction
Assisted Reproductive Technologies (ART) group the complete
set of artificial methods involving the handling of eggs and sperm
in order to achieve a pregnancy. Thus it goes from oocyte and
sperm donation, in vitro Fertilization (IVF), to ‘ultra-invasive’
techniques such as intracytoplasmic sperm injection (ICSI), when a
sperm cell, from a highly deteriorated spermogram or even starting
from a sperm cell with DNA damage, is injected into an ovocyte
[99]. Nowadays ART conceived children represent 1-4 % of the
newborn population in industrialized countries [100]. More than 4
million babies have been conceived through ART worldwide since
the beginning of IVF about 30 years ago [101]. Moreover, the use
of ART increases steadily, with more than a doubling between 1996
and 2007 in the USA [102]. The technical expertise of these approaches is fully recognized since children are quite efficiently
obtained. Notwithstanding, since a few years, questions started to
emerge about the long term evaluation of health risks for these children, especially with the development of epigenetics. Not exhaustively, it seems that ART increases the risk of placental diseases,
Intra-Uterine Growth Restriction, and of course twin pregnancies
[103]. Later in life, it seems possible that ART is associated with an
increased risk of developing the metabolic syndrome, a plague for
modern societies associating type II diabetes, obesity and cardiovascular disorders [104].
Concrete ideas suggesting increased risks for the health of
ART-conceived compared to naturally conceived children, originate
from two kinds of studies: those based on epidemiological data, and
those based on studies on animal models, especially mice.
Mouse studies revealed modifications on DNA methylation of
imprinted genes in oocytes after superovulation [105], and in blastocystes after embryo culture [51, 106]. Transcriptome analysis
showed that in vitro culture before transplantation considerably
modifies the expression profile of placental genes, and in particular
imprinted genes [52, 107]. Such defects were also observed in human oocytes [108]. These modifications could have long-term effect and explain in part the increased risks observed after the use of
ART.
From epidemiological studies, it was observed an increased risk
of very preterm (<32 weeks) and preterm (<37) delivery, very low
(<1,5 kg) and low (<2,5 kg) birth weight and perinatal mortality
when ART was used [109, 110]. There is also an increased risk for
preeclampsia or gestational hypertension (x 2.7) in pregnancies
achieved thanks to IVF [111, 112]. Nevertheless, these increased
risks could be overestimated because the comparison was made
with naturally conceived children from fertile parents, which may
not be the best control population. Indeed, when ART conceived
children are compared to children naturally conceived from subfertile parents (defined as those who need more than 12 months to
initiate a pregnancy), the associations between ART and prematurity or low birth weight are not found any more [113]. Again, however, when all the confounding factors are taken into account, the
risk seems to be real [114].
Associations between ART and imprinted genes disorders are
recurrently found despite their relative rarity making the analysis
difficult. In fact, after ART, the risks for Beckwith-Wiedemann
Syndrome (BWS) are estimated at 1/4000, more than 3 times the
risk in the general population [115-117]. Interestingly, a large majority of BWS cases after ART are due to imprinting defect in the
H19-IGF2 locus, while it’s only the cause of around 50 % of BWS
in the general population. Several studies [118-121]also suggest an
association between ART and Angelman Syndrome (AS), but due
to the rarity of this syndrome, it is premature to give an accurate
estimation of the increased risk. What seems certain is that, similarly to the BWS situation, the part of the syndrome caused by im-
Calicchio et al.
printing defects is increased by the use of ART: 5% in the general
AS cases, and around 70 % in the ART AS cases. However,
Ludwig and colleagues suggest that the subfertility of the parents
could be partly responsible, as 20% of AS patients are born from
subfertile couples [120]. Other reports suggest that the prevalence
of Silver-Russel Syndrome and retinoblastoma could also be increased after the use of ART [122-124].
In definitive, to obtain a comprehensive vision of ART effects,
ART-conceived children have to be followed in large studies, since
it seems clear that genomic imprinting is impacted by ART, even if
the responsible mechanisms involved are not understood. However,
it is reassuring to see that the vast majority of ART-conceived children is overall in good health. This may be due to modulation of the
consequences of DNA methylation defects by the placenta, as was
suggested by Fauque and colleagues [51] in mice, and seem to be
confirmed in human with DNA methylation differences in placenta
and cord blood [102], which are not found anymore later in life
[125], except maybe for IGF2 and IGF2R(generally not imprinted
in humans) in mouth epithelium cell samples. Finally, the future
will inevitably be based upon the new high-throughput methods to
study DNA methylation that will allow addressing genome-wide
DNA methylation defects.
3.e. Placental and Fetal Membranes Diseases
The placenta in Therian mammals is an organ of incredible
importance. Its dysfunction will inevitably trigger pregnancy losses.
At full development, the human placenta presents as a ‘pie’ of ~ 20
cm in diameter and two quite different sides morphologically as
well as functionally. One side faces the mother endometrium and
one faces the baby. Near the center of this latter, the umbilical cord
roots. The placenta plays at least three crucial roles in the development of the baby: endocrine, immunological and nutritional. The
embryo-placental unit is surrounded by two membranes, the amnion
and the chorion. These membranes are of extreme importance at the
time of delivery. Recent results suggest that alterations in DNA
methylation of specific cell types of these membranes could be
related to Premature Rupture of fetal Membrane (PROM) and thus
involved in prematurity.
3.e.1. Preeclampsia
Preeclampsia is a quite mysterious disease of human pregnancy,
frequent (~5% of pregnancy), potentially letal (up to 16% of maternal death if unattended), generally drifted towards the first gestation, and whose severity decreases with successive pregnancies
with the same partner. It is clinically characterized by a de novo
gestational hypertension and by proteinuria, developing from midgestation [126]. It is classically admitted that the placenta plays a
pivotal role in the development of the symptoms. It has been demonstrated that the external layer of the placenta, the syncytiotrophoblast sheds debris towards the general circulation (in higher
amounts in the case of placental diseases), thus bringing vasoactive
molecules (such as endothelin 1, ET1) either directly or carried by
micro/nanoparticles [127, 128] to the maternal organs and affect the
endothelium in a generalized fashion, thus probably participating to
the hypertensive state of the mother.
The preeclamptic placenta presents with a high level of expression deregulation compared to normal placentas, as revealed by
numerous transcriptomic studies on the comparison of placental
gene expression between normal and pathological pregnancies
[129-134]. Part of these deregulations may be due to epigenetic
modulation, especially by differential CpG methylation between
normal and pathological placentas [135]. Using targeted and whole
genome approaches, variations in methylation has been found in
numerous genes in the preeclamptic placenta: 296 loci in [136] that
used MedIP on chip with placental DNA from severe preeclampsias, of which some were validated by bisulfite sequencing, and
were close to the following genes:CAPN, EPHX2, ADORA2B,
SOX7, CXCL1 and CDX1. Yuen and coworkers revealed methyla-
DNA Methylation
tion variations in 34 loci that were hypomethylated in early onset
preeclamptic placentas, albeit these variations were mild (~10-15%
max) [137]. Four of these loci were validated by bisulphite sequencing. TIMP3, able to limit the actions of metalloproteinases
important for trophoblast invasion, was shown to be hypomethylated and to vary at the expression level in this study. Amongst the
first variations found, RASSF1A and SERPINB5 (maspin), were
identified [138, 139], as well as SERPINA3 [140], MMP9
[141],Cullin7 and Cullin 4B [142]. The links between methylation
alterations and gene expression have seldom been explored from a
mechanistic point of view. In vitro methylation of promoter elements in SERPINA3 and in TBX15 has nevertheless revealed that
DNA polymorphisms may or may not generate variations in gene
expression, and that some transcriptional factors may be sensitive to
these modifications. More specifically, Chelbi and coworkers
showed that the transcription factor ZBTB7B, regulated differentially the expression of SERPINA3, according to its methylation
status, but only for a specific allele, the regulation of the other allele
being methylation-insensitive [143]. Along the same line, PDX1
regulates differentially the promoter of the lipid-metabolism related
transcription factor TBX15[144]. These fine-tuned adaptations present in placental tissue, may constitute the basis of adaptation of
this organ to external threats (hypoxia/ hyperoxia/ xenohormones or
other endocrine disrupters, viruses, etc.)
In addition, the level of whole genome methylation has been
addressed [145] and revealed an increased methylation level in the
preeclampsia group. Whether this abnormal methylation concerns
mainly repetitive elements (LINEs for instance) was not specifically analyzed by the authors.
3.e.2. Intra Uterine Growth Restriction (IUGR)
There is an abundant literature on methylation alterations accompanying Intra-Uterine Growth Restriction. It is a disease
loosely defined as the incapacity of a fetus to grow to its normal
(optimal or genetically defined) potential. Often, it is confounded
with ‘SGA’ (Small for Gestational Age, which can be not linked to
a pathological status) as opposed to ‘AGA’ (Adequate for Gestational Age). We will treat SGA and IUGR indifferently in the rest
of this review. In humans, the genetic/ethnic variability, the modification of human size throughout generations, makes necessary to
use reference curves that are evolving during the years. Often, a
proxy of IUGR/SGA, birth weight is used, which, while easy to
collect, does not indicate the rupture in the growth curve that is
characteristic of IUGR. In about one third of the cases, preeclampsia (PE) is accompanied by IUGR. Contrary to PE that is a human
disease, IUGR can be induced in various animal models and by
various procedures as extensively reviewed in 2005 [146].
In particular, the rat model has been extensively studied. Two
major procedures were implemented to induce IUGR in this species, isocaloric hypoproteic alimentation of the females during gestation, and bilateral ligation of the uterine arteries. In this latter
model one of the first alterations of DNA methylation was observed
on the gene p53 in the fetal kidney, where the arteries ligation triggers hypomethylation of the exons 5-8 of the gene [147], this being
associated with an increased apoptosis in this organ, in adults. In
the liver, following the same procedure (rats were analyzed at birth
– D0- and 21 days later -D21), it was shown that enzymes involved
in methyl-radicals metabolism such as methionine adenosyltransferase and cystathione synthase are perturbed, [148]. The author
speculate that uteroplacental insufficiency affects hepatic onecarbon metabolism and subsequent DNA methylation; global analysis of the methylation level showed an overall hypomethylation in
the liver. In accordance with this observation we showed in another
model of growth restricted rats where the expression of genes was
analyzed simultaneously in 5 organs near birth, that DNMTs are
essentially reduced in the liver [149]. In the liver and in the same
model, Fu and co-workers showed that the expression of the phosphatase Dusp5 is reduced, along with a methylation of its second
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
19
exon [150]. Dusp5 dephosphorylates enzymes of the MAP Kinase
pathway, responsible in particular of the phosphorylation of IRS-1
at P 612, suggesting an action of this deregulation on the insulin
pathway and therefore on growth. In 2009, the same team showed
that the promoter P2 of Igf-1 is hypermethylated at D0 in the liver
(6 CpG analyzed), albeit this alteration comes back to normal at
D21 [151]. As in the liver, methylation-modifying enzymes Dnmt1
and Mecp2 were down-regulated from D0 and remain low at D21 in
the brain [152]. This observation was substantiated on another
IUGR-induced model in the rat, where it was shown that Dnmt1
and Dnmt3a are modified in neural tissue (hypothalamus). In this
study, the authors focused on the POMC gene and showed a demethylation at 12 CpG at postnatal D12[153]. In the IUGR rats,
behavior was also recently studied, together with alterations of gene
expression in the hippocampus. Methylation and expression was
studied for GR, HSD11b2, NNAT and reelin. Only reelin was found
down-regulated, none of the genes showed differences in their
methylation status [154]. In the same tissue (hippocampus), after
induction of IUGR by a methyl deficient diet, Dusp5 was shown
decreased at d0 and d21, this being accompanied by sex-specific
alterations of CpG methylation and of the histone code [155]. In
skeletal muscle, it was shown as well that three CpG in the Glut4
promoter affect the binding of Mef2A, Mef2D and MyoD, three
factors crucial for muscle growth. This suggested that glucose
transport could be a target of epigenetic mechanisms inducing a
defective growth in IUGR through epigenetic mechanisms. Always
in a mechanistic vision of the consequences of IUGR, the interesting work of Park and coworkers, focused on the pancreas, where
the level of Pdx1, the major transcription factor for pancreatic betacell development is reduced by IUGR. More precisely, in young
pups (2 weeks), the 14 CpG islands of the Pdx1 promoter were not
methylated in control and IUGR rats. Later, at 6 months of age,
IUGR rats showed a 51.3 % methylation level at these positions
associated to diabetes in adulthood, while the controls showed no
evidence of methylation. This (together with alterations of the histone code in this model) argues in favor of anin utero programming
of adult disease [156]. The central role of Pdx1 was further studied
byPinney and coworkers (2011), where the author showed that
Pdx1 expression can be restored by Exendin4 treatment (a longacting glucagon-like peptide, administered to the newborn from D0
to D6). In this study, Dnmt1 binding to the Pdx1 promoter was
studied and shown to be prevented through the recruitment of the
transcription factor Usf1 and the histone acetyl transferase Pcaf,
thus protecting against methylation and inactivation [157]. Global
methylation analysis in the rat pancreas was also performed [158],
by the HELP technique, whichmade it possible identifying 1400
differentially methylated loci, nearby genes involved in cell death,
vascularization, and beta cell proliferation.
Mice were also used to study the effects of IUGR, often in relation with imprinted genes. In 2001, Murrell showed that deletion of
the IGF2/H19 DMR2, does not affect the imprinted status of the
locus, but induces IUGR through reduction of Igf2 expression. Essentially mice were used to study gene methylation in relation with
IUGR in two contexts: the impact of and on imprinted genes, and
the effects of environmental perturbations, such as alcohol exposure. In the context of imprinted genes, Monk and coworkers studied in 2003 the proximal mouse chromosome 11 region which
shares similarities with the chromosome 7 of humans in the region
associated with the Silver-Russel Syndrome (SRS), [159]. SRS is
an imprinting syndrome that associates ante and post-natal growth
retardation, macrocephaly and body asymmetries (~1/50,000
births). In 10% of the cases, SRS is linked to a uniparental disomy
of chromosome 7 [160]. SRS is often associated with demethylation
of the IGF2/H19 locus (description and function of the locus is
depicted in (Fig. 2) and paragraph b). Recently, manipulations of
the mouse genome allowed altering the imprinting code at the locus
[161]. The authors substituted elegantly one of the important CTCF
binding sites separating H19/IGF2 (Fig. 2 and § 3a) with the
20 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
chicken -globin insulator element, exhibiting enhancer blocking
by CTCF and chromatin barrier functions by USF1 and VEZF1.
When the normal ICR1 is inherited by the father, it is completely
methylated, allowing Igf2 expression. When the chicken globin
insulator is used, it is unmethylated on both alleles, allowing CTCF
binding. This triggers a quasi-extinction of Igf2 (~10% residual
expression) and overexpression of H19, with a very severe IUGR
(carriers are half the size of their littermates). Mice were also used
to analyze IUGR induced by environmental causes. In 2011, Goyal
and coworkers induced IUGR in mice by exposing the mothers to
hypoxia [162]. Expression of Ace1, Ace2, At-1b was followed. The
methylation of the ACE promoter was analyzed but found unchanged, despite the variations in gene expression. In several studies, the impact of alcohol exposure was evaluated on the neural
development in mice [163-166]. Overall in all the conditions studied, IUGR was induced by this exposure; the H19 DMR was not
found altered in terms of methylation of the imprinting control region [166], although there was a correlation between methylation
and placental weight (but not with embryo weight). In 2011, some
of the effects of alcohol exposure (rather malformations than
IUGR) could be corrected by methyl donors, may be through a
decreased methylation at 4 CpG in Igf2[164]. It has been reported
that in the brain, alcohol exposure altered the timely developmental
DNA methylation, in a way similar to that obtained by administration of the anti-methylation agent 5-azacytidine [167]. Liu and coworkers investigated genome-wide alterations of DNA methylation
[168], and detected anomalies in imprinted genes, genes involved in
cell cycle, cancer, apoptosis, growth and olfaction (olfactory receptors are generally mono-allelically expressed, which may explain
why they are found in this category, of primary targets of environmental stress acting on DNA methylation). Interestingly, these
genes were found enriched on chromosome 7, 10 and X. The methylation alterations were associated with gene expression deregulations in 84 genes. Among environmental exposure, it has been
shown in an isolated study [169] in the mouse model that IUGR
may be induced by bacterial infection with Campylobacter Rectus
(an oral micro-organism) and triggers methylation of the placental
promoter of Igf2, P0 and the down-regulation of its expression.
In humans there is an important corpus of reports linking imprinting defects and IUGR associated with the SRS. Many studies
confirmed the presence of epimutations at 11p15 that are linked
with SRS [170-180]. Overall, it seems that ~40 - 60% of the SRS
are associated with such epimutations, while around 10% are associated with UniparentalDisomies of chromosome 7. The location of
the epimutations at 11p15 appear as patient-specific. In 2010, genetic defects affecting the methylation of ICR1 at the IGF2/H19
locus was found in 16 patients outside the CTCF binding site [181].
The imprinted disease that plays a reciprocal role to the SRS is the
BWS, characterized by a double expression of IGF2 and fetal overgrowth. In BWS, ICR1 has gained methylation, while it has often
lost methylation in SRS. BWS was shown associated to genetic
anomalies in ICR1 in the same study. Concerning SRS, a part of
research is now focused on the unexplained cases. In 2010 Turner
and coworkers showed that SRS could be associated with a gain of
methylation at the IGF2R locus [182], the imprinting status of
which seems to differ in humans according to individuals. A recent
study of SRS patients [123] detected an imprinting defect in the
PEG/MEST locus, in a sample of patients that followed IVF. As
mentioned before (§3.d), this could enter in the growing corpus of
evidences showing an increased risk of methylation disorders following the use of assisted reproduction techniques. Many studies on
IUGR kept a focus on imprinted genes, often in placental tissue.
Among them McMinn and coworkers studied the methylation and
expression of numerous genes in IUGR [183]. They found 6 imprinted genes that were modified (PHLDA2 increased, MEST,
MEG3, GATM, GNAS and PLAGL1 decreased), and showed that
PHLDA2 and MEST were also presenting changes in DNA methylation. In idiopathic IUGR, biallelic expression of H19 was found
Calicchio et al.
once [93]. Expression of H19 was found increased in IUGR placent
a while its methylation level was lower [184]. Using the first Illumina platform for methylation analysis (Golden Gate), Bourque and
co-workers analyzed the 11p15.5 ICR1 (H19/IGF2) and showed
that it was decreased in IUGR [185]. This was confirmed independently [186]. Always focusing on imprinted genes, Lambertini and
co-workers used MeDIP on chip, and showed a slight tendency
towards hypermethylation in differentially methylated regions of
the imprinted genes [187]. Few studies focused on non-imprinted
genes. In 2010, the Golden Gate platform (ancient version, with
1536 CpG only) allowed to identify 5 loci differentially (up) methylated in IUGR placentas [137]. Other genome-wide approaches
were used using the novel generation 27KBeadChipplateform and
allowed to detect specific methylation signatures in IUGR placentas
[188]. Consistently, Michels and coworkers found an association
between LINE-1 methylation in the placenta and birth weight [189].
MSAP-PCR allowed to show that the promoter of the transcription
factor TBX15 was differentially methylated in IUGR placentas
[144]. TBX15 is involved in lipid metabolism [190]. In this case
methylation of the TBX15 promoter is positively correlated with
baby weight and the promoter is regulated by the transcription factor PDX1in a methylation-dependent fashion. In 2010, GascoinLachambre and coworkers showed that DNA methylation is modified in the CUL7 promoter, this gene being strongly up-regulated in
IUGR [142]. Several teams focused on epigenetic marks present in
cord blood cells. The last study published [191] did not identify
significant differences, as in the study of Tobi (2011), that focused
on imprinted genes [192]. To this respect it is important to note the
interesting study of Einstein and coworkers (2010) that isolated
CD34+ hematopoietic stem cells from cord blood and analyzed the
methylation level genome-wide by the HELP technique. 56 loci
were identified near 35 genes, several of which point to a network
where the transcription factor HNF4A occupies a central position.
This suggests that genome-wide approaches could be very efficient
to discover differences if they are applied to purified cell populations, where differences are not blurred by the tissue heterogeneity.
3.e.3. Preterm labor-Preterm Premature Rupture of Membranes
(PPROM)
To date, only one publication describes epigenetic variation in
the promoter of one gene and an increased risk to preterm premature rupture of membranes and preterm labor [193]. J Strauss III
and coll. demonstrated that a single-nucleotide polymorphism in the
promoter of the metalloprotease MMP1 increases its methylation
status and increases the expression of MMP1 in amnion fibroblastsin a population of Afro-American women. These women are
more prone to PPROM. It has also been postulated that nutrition
and environmental exposure would induce epigenetic modifications
in the gestational tissues and increase odds to a preterm delivery,
but no sound study demonstrates this postulate to date.
4. CONCLUSIONS
Technical access to the epigenetic code is a key towards understanding the rules that define gene expression patterns, in physiological as well as pathological situations. Reproductive diseases in
humans presented in this review are all prone to alterations in DNA
methylation, with a recurrent finding of alterations in imprinted
gene profile, putting them in front as indicators of disorders. In the
future, genome-wide approaches will be implemented and should
allow developing integrative databases, ultimately at the tissue type
level of genome polymorphism, epigenome marks and gene expression.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of
interest.
ACKNOWLEDGEMENTS
DNA Methylation
Declared none.
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
[28]
REFERENCES
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
[13]
[14]
[15]
[16]
[17]
[18]
[19]
[20]
[21]
[22]
[23]
[24]
[25]
[26]
[27]
Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953; 171: 737-8.
Eickbush TH, Moudrianakis EN. The compaction of DNA helices
into either continuous supercoils or folded-fiber rods and toroids.
Cell 1978; 13: 295-306.
Guil S, Esteller M. DNA methylomes, histone codes and miRNAs:
tying it all together. Int J Biochem Cell Biol 2009; 41: 87-95.
Kinney SR, Pradhan S. Regulation of expression and activity of
DNA (cytosine-5) methyltransferases in mammalian cells. Prog
Mol Biol Transl Sci 2011; 101: 311-33.
De Carvalho DD, You JS, Jones PA. DNA methylation and cellular
reprogramming. Trends Cell Biol 2010; 20: 609-17.
Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5methylcytosine by TET1 promotes active DNA demethylation in
the adult brain. Cell 2011; 145: 423-34.
Niehrs C, Schafer A. Active DNA demethylation by Gadd45 and
DNA repair. Trends Cell Biol 2012; 22: 220-7.
Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the
paternal genome upon fertilization involves genome-wide oxidation
of 5-methylcytosine. Proc Natl Acad Sci USA 2011; 108: 3642-7.
Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL,
Hartemink AJ. Computational and experimental identification of
novel human imprinted genes. Genome Res 2007; 17: 1723-30.
Barbaux S, Gascoin-Lachambre G, Buffat C, et al. A genome-wide
approach reveals novel imprinted genes expressed in the human
placenta. Epigenetics 2012: in press.
Smits G, Mungall AJ, Griffiths-Jones S, et al. Conservation of the
H19 noncoding RNA and H19-IGF2 imprinting mechanism in therians. Nat Genet 2008; 40: 971-6.
Kato Y, Sasaki H. Imprinting and looping: epigenetic marks control interactions between regulatory elements. Bioessays 2005; 27:
1-4.
Krueger C, Osborne CS. Raising the curtains on interchromosomal
interactions. Trends Genet 2006; 22: 637-9.
Kajii T, Ohama K. Androgenetic origin of hydatidiform mole.
Nature 1977; 268: 633-4.
Murdoch S, Djuric U, Mazhar B, et al. Mutations in NALP7 cause
recurrent hydatidiform moles and reproductive wastage in humans.
Nat Genet 2006; 38: 300-2.
Bestor TH, Bourc'his D. Genetics and epigenetics of hydatidiform
moles. Nat Genet 2006; 38: 274-6.
Holliday R, Pugh JE. DNA modification mechanisms and gene
activity during development. Science 1975; 187: 226-32.
Riggs AD. X inactivation, differentiation, and DNA methylation.
Cytogenet Cell Genet 1975; 14: 9-25.
Bird AP, Southern EM. Use of restriction enzymes to study eukaryotic DNA methylation: I. The methylation pattern in ribosomal
DNA from Xenopus laevis. J Mol Biol 1978; 118: 27-47.
Cedar H, Solage A, Glaser G, Razin A. Direct detection of methylated cytosine in DNA by use of the restriction enzyme MspI. Nucleic Acids Res 1979; 6: 2125-32.
Adouard V, Dante R, Niveleau A, Delain E, Revet B, Ehrlich M.
The accessibility of 5-methylcytosine to specific antibodies in double-stranded DNA of Xanthomonas phage XP12. Eur J Biochem
1985; 152: 115-21.
Hendrich B, Bird A. Identification and characterization of a family
of mammalian methyl-CpG binding proteins. Mol Cell Biol 1998;
18: 6538-47.
Hayatsu H, Wataya Y, Kai K, Iida S. Reaction of sodium bisulfite
with uracil, cytosine, and their derivatives. Biochemistry 1970; 9:
2858-65.
Hayatsu H, Wataya Y, Kazushige K. The addition of sodium bisulfite to uracil and to cytosine. J Am Chem Soc 1970; 92: 724-6.
Wang RY, Gehrke CW, Ehrlich M. Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res 1980; 8: 4777-90.
Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 1992; 89:
1827-31.
Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res 2009; 19: 959-66.
[29]
[30]
[31]
[32]
[33]
[34]
[35]
[36]
[37]
[38]
[39]
[40]
[41]
[42]
[43]
[44]
[45]
[46]
[47]
[48]
[49]
[50]
[51]
21
Bock C, Tomazou EM, Brinkman AB, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat
Biotechnol 2010; 28: 1106-14.
Harris RA, Wang T, Coarfa C, et al. Comparison of sequencingbased methods to profile DNA methylation and identification of
monoallelic epigenetic modifications. Nat Biotechnol 2010; 28:
1097-105.
Deng J, Shoemaker R, Xie B, et al. Targeted bisulfite sequencing
reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol 2009; 27: 353-60.
Layman LC. Human gene mutations causing infertility. J Med
Genet 2002; 39: 153-61.
Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med 2008; 14: 1197-213.
Schultz N, Hamra FK, Garbers DL. A multitude of genes expressed
solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA 2003; 100:
12201-6.
Rousseaux S, Reynoird N, Escoffier E, Thevenon J, Caron C,
Khochbin S. Epigenetic reprogramming of the male genome during
gametogenesis and in the zygote. Reprod Biomed Online 2008; 16:
492-503.
Wykes SM, Krawetz SA. The structural organization of sperm
chromatin. J Biol Chem 2003; 278: 29471-7.
Ooi SL, Henikoff S. Germline histone dynamics and epigenetics.
Curr Opin Cell Biol 2007; 19: 257-65.
Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs
prior to meiosis in male germ cells. Dev Biol 2007; 307: 368-79.
Benchaib M, Ajina M, Lornage J, Niveleau A, Durand P, Guerin
JF. Quantitation by image analysis of global DNA methylation in
human spermatozoa and its prognostic value in in vitro fertilization: a preliminary study. Fertil Steril 2003; 80: 947-53.
Navarro-Costa P, Nogueira P, Carvalho M, et al. Incorrect DNA
methylation of the DAZL promoter CpG island associates with defective human sperm. Hum Reprod 2010; 25: 2647-54.
Ghorbian S. Routine diagnostic testing of Y chromosome deletions
in male infertile and subfertile. Gene 2012; 503: 160-4.
Wu W, Lu C, Xia Y, et al. Lack of association between DAZ gene
methylation patterns and spermatogenic failure. Clin Chem Lab
Med 2010; 48: 355-60.
Linher K, Cheung Q, Baker P, Bedecarrats G, Shiota K, Li J. An
epigenetic mechanism regulates germ cell-specific expression of
the porcine Deleted in Azoospermia-Like (DAZL) gene. Differentiation 2009; 77: 335-49.
Sugimoto K, Koh E, Sin HS, et al. Tissue-specific differentially
methylated regions of the human VASA gene are potentially associated with maturation arrest phenotype in the testis. J Hum Genet
2009; 54: 450-6.
Medrano JV, Ramathal C, Nguyen HN, Simon C, Reijo Pera RA.
Divergent RNA-binding proteins, DAZL and VASA, induce meiotic progression in human germ cells derived in vitro. Stem Cells
2012; 30: 441-51.
Marques CJ, Carvalho F, Sousa M, Barros A. Genomic imprinting
in disruptive spermatogenesis. Lancet 2004; 363: 1700-2.
Marques CJ, Costa P, Vaz B, et al. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia.
Mol Hum Reprod 2008; 14: 67-74.
Marques CJ, Francisco T, Sousa S, Carvalho F, Barros A, Sousa M.
Methylation defects of imprinted genes in human testicular spermatozoa. Fertil Steril 2010; 94: 585-94.
Pacheco SE, Houseman EA, Christensen BC, et al. Integrative
DNA methylation and gene expression analyses identify DNA
packaging and epigenetic regulatory genes associated with low motility sperm. PLoS One 2011; 6: e20280.
Kobayashi H, Hiura H, John RM, et al. DNA methylation errors at
imprinted loci after assisted conception originate in the parental
sperm. Eur J Hum Genet 2009; 17: 1582-91.
Boissonnas CC, Abdalaoui HE, Haelewyn V, et al. Specific epigenetic alterations of IGF2-H19 locus in spermatozoa from infertile
men. Eur J Hum Genet 2010; 18: 73-80.
Fauque P, Jouannet P, Lesaffre C, et al. Assisted Reproductive
Technology affects developmental kinetics, H19 Imprinting Control Region methylation and H19 gene expression in individual
mouse embryos. BMC Dev Biol 2007; 7: 116.
22 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
[52]
[53]
[54]
[55]
[56]
[57]
[58]
[59]
[60]
[61]
[62]
[63]
[64]
[65]
[66]
[67]
[68]
[69]
[70]
[71]
[72]
[73]
[74]
Fauque P, Mondon F, Letourneur F, et al. In vitro fertilization and
embryo culture strongly impact the placental transcriptome in the
mouse model. PLoS One 2010; 5: e9218.
Chen SL, Shi XY, Zheng HY, Wu FR, Luo C. Aberrant DNA
methylation of imprinted H19 gene in human preimplantation embryos. Fertil Steril 2010; 94: 2356-8, 2358 e1.
Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and
the establishment of maternal genomic imprints. Science 2001;
294: 2536-9.
Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of
Dnmt3a bound to Dnmt3L suggests a model for de novo DNA
methylation. Nature 2007; 449: 248-51.
Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 2005; 308: 1466-9.
Germain AM, Romanik MC, Guerra I, et al. Endothelial dysfunction: a link among preeclampsia, recurrent pregnancy loss, and future cardiovascular events? Hypertension 2007; 49: 90-5.
ESHRE/ASRM. Revised 2003 consensus on diagnostic criteria and
long-term health risks related to polycystic ovary syndrome
(PCOS). Hum Reprod 2004; 19: 41-7.
Hickey T, Chandy A, Norman RJ. The androgen receptor CAG
repeat polymorphism and X-chromosome inactivation in Australian
Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab 2002; 87: 161-5.
Dasgupta S, Sirisha PV, Neelaveni K, et al. Androgen receptor
CAG repeat polymorphism and epigenetic influence among the
south Indian women with Polycystic Ovary Syndrome. PLoS One
2010; 5: e12401.
Nakayama T, Watanabe M, Suzuki H, et al. Epigenetic regulation
of androgen receptor gene expression in human prostate cancers.
Lab Invest 2000; 80: 1789-96.
Xu N, Azziz R, Goodarzi MO. Epigenetics in polycystic ovary
syndrome: a pilot study of global DNA methylation. Fertil Steril
2010; 94: 781-3 e1.
Zhu JQ, Zhu L, Liang XW, Xing FQ, Schatten H, Sun QY. Demethylation of LHR in dehydroepiandrosterone-induced mouse
model of polycystic ovary syndrome. Mol Hum Reprod 2010; 16:
260-6.
Xu N, Kwon S, Abbott DH, et al. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like
phenotypes in prenatally androgenized rhesus monkeys. PLoS One
2011; 6: e27286.
Vaiman D. Fertility, sex determination, and the X chromosome.
Cytogenet Genome Res 2002; 99: 224-8.
Rizzolio F, Pramparo T, Sala C, et al. Epigenetic analysis of the
critical region I for premature ovarian failure: demonstration of a
highly heterochromatic domain on the long arm of the mammalian
X chromosome. J Med Genet 2009; 46: 585-92.
Laisk T, Haller-Kikkatalo K, Laanpere M, et al. Androgen receptor
epigenetic variations influence early follicular phase gonadotropin
levels. Acta Obstet Gynecol Scand 2010; 89: 1557-63.
Bretherick KL, Metzger DL, Chanoine JP, et al. Skewed Xchromosome inactivation is associated with primary but not secondary ovarian failure. Am J Med Genet A 2007; 143A: 945-51.
Mansouri MR, Schuster J, Badhai J, et al. Alterations in the expression, structure and function of progesterone receptor membrane
component-1 (PGRMC1) in premature ovarian failure. Hum Mol
Genet 2008; 17: 3776-83.
Sampson J. Peritoneal endometriosis due to menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet
Gynecol 1927; 14: 422-469.
Noel JC, Anaf V, Borghese B, Vaiman D, Fayt I, Chapron C. The
steroidogenic factor-1 protein is not expressed in various forms of
endometriosis but is strongly present in ovarian cortical or medullary mesenchymatous cells adjacent to endometriotic foci. Fertil
Steril 2011: in press.
de Ziegler D, Borghese B, Chapron C. Endometriosis and infertility: pathophysiology and management. Lancet 2010; 376: 730-8.
Fassbender A, Verbeeck N, Bornigen D, et al. Combined mRNA
microarray and proteomic analysis of eutopic endometrium of
women with and without endometriosis. Hum Reprod 2012; 27:
2020-9.
Zelenko Z, Aghajanova L, Irwin JC, Giudice LC. Nuclear receptor,
coregulator signaling, and chromatin remodeling pathways suggest
involvement of the epigenome in the steroid hormone response of
Calicchio et al.
[75]
[76]
[77]
[78]
[79]
[80]
[81]
[82]
[83]
[84]
[85]
[86]
[87]
[88]
[89]
[90]
[91]
[92]
[93]
[94]
[95]
[96]
endometrium and abnormalities in endometriosis. Reprod Sci 2012;
19: 152-62.
Lee B, Du H, Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod 2009; 80: 79-85.
Wu Y, Halverson G, Basir Z, Strawn E, Yan P, Guo SW. Aberrant
methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am J
Obstet Gynecol 2005; 193: 371-80.
Szczepanska M, Wirstlein P, Luczak M, Jagodzinski PP, Skrzypczak J. Reduced expression of HOXA10 in the midluteal endometrium from infertile women with minimal endometriosis. Biomed Pharmacother 2010; 64: 697-705.
Borghese B, Mondon F, Noel JC, et al. Gene expression profile for
ectopic versus eutopic endometrium provides new insights into endometriosis oncogenic potential. Mol Endocrinol 2008; 22: 255762.
Wang D, Chen Q, Zhang C, Ren F, Li T. DNA hypomethylation of
the COX-2 gene promoter is associated with up-regulation of its
mRNA expression in eutopic endometrium of endometriosis. Eur J
Med Res 2012; 17: 12.
Szczepanska M, Wirstlein P, Skrzypczak J, Jagodzinski PP. Expression of HOXA11 in the mid-luteal endometrium from women
with endometriosis-associated infertility. Reprod Biol Endocrinol
2012; 10: 1.
de Graaff AA, Delvoux B, Van de Vijver KK, Kyama CM,
D'Hooghe TM, Dunselman GA, Romano A. Paired-box gene 2 is
down-regulated in endometriosis and correlates with low epidermal
growth factor receptor expression. Hum Reprod 2012; 27: 1676-84.
Jichan N, Xishi L, Guo SW. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in adenomyosis and its rectification by a histone deacetylase inhibitor and a demethylation agent.
Reprod Sci 2010; 17: 995-1005.
Xue Q, Lin Z, Cheng YH, et al. Promoter methylation regulates
estrogen receptor 2 in human endometrium and endometriosis. Biol
Reprod 2007; 77: 681-7.
Xue Q, Zhou YF, Zhu SN, Bulun SE. Hypermethylation of the
CpG island spanning from exon II to intron III is associated with
steroidogenic factor 1 expression in stromal cells of endometriosis.
Reprod Sci 2011; 18: 1080-4.
Izawa M, Taniguchi F, Uegaki T, et al. Demethylation of a nonpromoter cytosine-phosphate-guanine island in the aromatase gene
may cause the aberrant up-regulation in endometriotic tissues. Fertil Steril 2010; 95: 33-9.
Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Promoter hypermethylation of progesterone receptor isoform B (PR-B) in endometriosis. Epigenetics 2006; 1: 106-11.
Ren F, Wang D, Jiang Y. Epigenetic inactivation of hMLH1 in the
malignant transformation of ovarian endometriosis. Arch Gynecol
Obstet 2012; 285: 215-21.
Martini M, Ciccarone M, Garganese G, et al. Possible involvement
of hMLH1 p16(INK4a) and PTEN in the malignant transformation
of endometriosis. Int J Cancer 2002; 102: 398-406.
Lelievre-Pegorier M, Merlet-Benichou C. The number of nephrons
in the mammalian kidney: environmental influences play a determining role. Exp Nephrol 2000; 8: 63-5.
Xue Q, Lin Z, Yin P, et al. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5' CpG island in endometriosis. J Clin Endocrinol Metab 2007; 92: 3261-7.
Noel JC, Borghese B, Vaiman D, Fayt I, Anaf V, Chapron C. Steroidogenic factor-1 expression in ovarian endometriosis. Appl Immunohistochem Mol Morphol 2010; 18: 258-61.
Bulun SE. Endometriosis. N Engl J Med 2009; 360: 268-79.
Wu Y, Starzinski-Powitz A, Guo SW. Prolonged stimulation with
tumor necrosis factor-alpha induced partial methylation at PR-B
promoter in immortalized epithelial-like endometriotic cells. Fertil
Steril 2008; 90: 234-7.
Vestergaard AL, Thorup K, Knudsen UB, et al. Oncogenic events
associated with endometrial and ovarian cancers are rare in endometriosis. Mol Hum Reprod 2011; 17: 758-61.
Borghese B, Barbaux S, Mondon F, et al. Research resource: genome-wide profiling of methylated promoters in endometriosis reveals a subtelomeric location of hypermethylation. Mol Endocrinol
2010; 24: 1872-85.
El-Maarri O, Kareta MS, Mikeska T, et al. A systematic search for
DNA methyltransferase polymorphisms reveals a rare DNMT3L
DNA Methylation
[97]
[98]
[99]
[100]
[101]
[102]
[103]
[104]
[105]
[106]
[107]
[108]
[109]
[110]
[111]
[112]
[113]
[114]
[115]
[116]
[117]
[118]
[119]
variant associated with subtelomeric hypomethylation. Hum Mol
Genet 2009; 18: 1755-68.
Borghese B, Santulli P, Hequet D, et al. Genetic polymorphisms of
DNMT3L involved in hypermethylation of chromosomal ends are
associated with greater risk of developing ovarian endometriosis.
Am J Pathol 2012; 180: 1781-6.
Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1,
DNMT3A, and DNMT3B in women with endometriosis. Fertil
Steril 2007; 87: 24-32.
Ahmadi A, Ng SC. Fertilizing ability of DNA-damaged spermatozoa. J Exp Zool 1999; 284: 696-704.
de Mouzon J, Goossens V, Bhattacharya S, et al. Assisted reproductive technology in Europe, 2006: results generated from European registers by ESHRE. Hum Reprod 2010; 25: 1851-62.
Halliday JL, Ukoumunne OC, Baker HW, et al. Increased risk of
blastogenesis birth defects, arising in the first 4 weeks of pregnancy, after assisted reproductive technologies. Hum Reprod 2010;
25: 59-65.
Katari S, Turan N, Bibikova M, et al. DNA methylation and gene
expression differences in children conceived in vitro or in vivo.
Hum Mol Genet 2009; 18: 3769-78.
de Mouzon J, Goossens V, Bhattacharya S, et al. Assisted reproductive technology in Europe, 2007: results generated from European registers by ESHRE. Hum Reprod 2012; 27: 954-66.
Cardozo E, Pavone ME, Hirshfeld-Cytron JE. Metabolic syndrome
and oocyte quality. Trends Endocrinol Metab 2011; 22: 103-9.
Market-Velker BA, Zhang L, Magri LS, Bonvissuto AC, Mann
MR. Dual effects of superovulation: loss of maternal and paternal
imprinted methylation in a dose-dependent manner. Hum Mol
Genet 2010; 19: 36-51.
Doherty AS, Mann MR, Tremblay KD, Bartolomei MS, Schultz
RM. Differential effects of culture on imprinted H19 expression in
the preimplantation mouse embryo. Biol Reprod 2000; 62: 152635.
Fauque P, Ripoche MA, Tost J, et al. Modulation of imprinted gene
network in placenta results in normal development of in vitro manipulated mouse embryos. Hum Mol Genet 2010; 19: 1779-90.
Sato A, Otsu E, Negishi H, Utsunomiya T, Arima T. Aberrant
DNA methylation of imprinted loci in superovulated oocytes. Hum
Reprod 2007; 22: 26-35.
Helmerhorst FM, Perquin DA, Donker D, Keirse MJ. Perinatal
outcome of singletons and twins after assisted conception: a systematic review of controlled studies. BMJ 2004; 328: 261.
Jackson RA, Gibson KA, Wu YW, Croughan MS. Perinatal outcomes in singletons following in vitro fertilization: a meta-analysis.
Obstet Gynecol 2004; 103: 551-63.
Shevell T, Malone FD, Vidaver J, et al. Assisted reproductive
technology and pregnancy outcome. Obstet Gynecol 2005; 106:
1039-45.
Chen XK, Wen SW, Bottomley J, Smith GN, Leader A, Walker
MC. In vitro fertilization is associated with an increased risk for
preeclampsia. Hypertens Pregnancy 2009; 28: 1-12.
Romundstad LB, Romundstad PR, Sunde A, et al. Effects of technology or maternal factors on perinatal outcome after assisted fertilisation: a population-based cohort study. Lancet 2008; 372: 73743.
Thomopoulos C, Tsioufis C, Michalopoulou H, Makris T, Papademetriou V, Stefanadis C. Assisted reproductive technology and
pregnancy-related hypertensive complications: a systematic review.
J Hum Hypertens 2013; 27(3): 148-57.
DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro
fertilization with Beckwith-Wiedemann syndrome and epigenetic
alterations of LIT1 and H19. Am J Hum Genet 2003; 72: 156-60.
Maher ER, Afnan M, Barratt CL. Epigenetic risks related to assisted reproductive technologies: epigenetics, imprinting, ART and
icebergs? Hum Reprod 2003; 18: 2508-11.
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le
Bouc Y. In vitro fertilization may increase the risk of BeckwithWiedemann syndrome related to the abnormal imprinting of the
KCN1OT gene. Am J Hum Genet 2003; 72: 1338-41.
Cox GF, Burger J, Lip V, et al. Intracytoplasmic sperm injection
may increase the risk of imprinting defects. Am J Hum Genet 2002;
71: 162-4.
Orstavik KH, Eiklid K, van der Hagen CB, et al. Another case of
imprinting defect in a girl with Angelman syndrome who was con-
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
[120]
[121]
[122]
[123]
[124]
[125]
[126]
[127]
[128]
[129]
[130]
[131]
[132]
[133]
[134]
[135]
[136]
[137]
[138]
[139]
[140]
23
ceived by intracytoplasmic semen injection. Am J Hum Genet
2003; 72: 218-9.
Ludwig M, Katalinic A, Gross S, Sutcliffe A, Varon R, Horsthemke B. Increased prevalence of imprinting defects in patients
with Angelman syndrome born to subfertile couples. J Med Genet
2005; 42: 289-91.
Sutcliffe AG, Peters CJ, Bowdin S, et al. Assisted reproductive
therapies and imprinting disorders--a preliminary British survey.
Hum Reprod 2006; 21: 1009-11.
Bliek J, Terhal P, van den Bogaard MJ, et al. Hypomethylation of
the H19 gene causes not only Silver-Russell syndrome (SRS) but
also isolated asymmetry or an SRS-like phenotype. Am J Hum
Genet 2006; 78: 604-14.
Kagami M, Nagai T, Fukami M, Yamazawa K, Ogata T. SilverRussell syndrome in a girl born after in vitro fertilization: partial
hypermethylation at the differentially methylated region of
PEG1/MEST. J Assist Reprod Genet 2007; 24: 131-6.
Moll AC, Imhof SM, Cruysberg JR, et al. Incidence of retinoblastoma in children born after in-vitro fertilisation. Lancet 2003; 361:
309-10.
Puumala SE, Nelson HH, Ross JA, Nguyen RH, Damario MA,
Spector LG. Similar DNA methylation levels in specific imprinting
control regions in children conceived with and without assisted reproductive technology: a cross-sectional study. BMC Pediatr 2012;
12: 33.
Redman CW, Sargent IL. Latest advances in understanding
preeclampsia. Science 2005; 308: 1592-4.
Goswami D, Tannetta DS, Magee LA, et al. Excess syncytiotrophoblast microparticle shedding is a feature of early-onset preeclampsia, but not normotensive intrauterine growth restriction.
Placenta 2006; 27: 56-61.
Lok CA, Van Der Post JA, Sargent IL, et al. Changes in microparticle numbers and cellular origin during pregnancy and preeclampsia. Hypertens Pregnancy 2008; 27: 344-60.
Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H,
Udagawa Y. Microarray analysis of differentially expressed fetal
genes in placental tissue derived from early and late onset severe
pre-eclampsia. Placenta 2007; 28: 487-97.
Toft JH, Lian IA, Tarca AL, et al. Whole-genome microarray and
targeted analysis of angiogenesis-regulating gene expression (ENG,
FLT1, VEGF, PlGF) in placentas from pre-eclamptic and smallfor-gestational-age pregnancies. J Matern Fetal Neonatal Med
2008; 21: 267-73.
Varkonyi T, Nagy B, Fule T, et al. Microarray profiling reveals
that placental transcriptomes of early-onset HELLP syndrome and
preeclampsia are similar. Placenta 2011; 32 Suppl: S21-9.
Centlow M, Wingren C, Borrebaeck C, Brownstein MJ, Hansson
SR. Differential gene expression analysis of placentas with increased vascular resistance and pre-eclampsia using whole-genome
microarrays. J Pregnancy 2011; 2011: 472354.
Tsai S, Hardison NE, James AH, et al. Transcriptional profiling of
human placentas from pregnancies complicated by preeclampsia
reveals disregulation of sialic acid acetylesterase and immune signalling pathways. Placenta 2011; 32: 175-82.
Loset M, Mundal SB, Johnson MP, et al. A transcriptional profile
of the decidua in preeclampsia. Am J Obstet Gynecol 2011; 204: 84
e1-27.
Chelbi ST, Vaiman D. Genetic and epigenetic factors contribute to
the onset of preeclampsia. Mol Cell Endocrinol 2008; 282: 120129.
Jia RZ, Zhang X, Hu P, et al. Screening for differential methylation
status in human placenta in preeclampsia using a CpG island plus
promoter microarray. Int J Mol Med 2012; 30: 133-41.
Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE,
Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset
preeclampsia. Eur J Hum Genet 2010; 18: 1006-12.
Tsui DW, Chan KC, Chim SS, et al. Quantitative aberrations of
hypermethylated RASSF1A gene sequences in maternal plasma in
pre-eclampsia. Prenat Diagn 2007; 27: 1212-8.
Bellido ML, Radpour R, Lapaire O, et al. MALDI-TOF mass array
analysis of RASSF1A and SERPINB5 methylation patterns in human placenta and plasma. Biol Reprod 2010; 82: 745-50.
Chelbi ST, Mondon F, Jammes H, et al. Expressional and epigenetic alterations of placental serine protease inhibitors: SERPINA3
24 Current Pharmaceutical Design, 2014, Vol. 20, No. 00
[141]
[142]
[143]
[144]
[145]
[146]
[147]
[148]
[149]
[150]
[151]
[152]
[153]
[154]
[155]
[156]
[157]
[158]
[159]
[160]
[161]
is a potential marker of preeclampsia. Hypertension 2007; 49: 7683.
Wang Z, Lu S, Liu C, Zhao B, Pei K, Tian L, Ma X. Expressional
and epigenetic alterations of placental matrix metalloproteinase 9 in
preeclampsia. Gynecol Endocrinol 2010; 26: 96-102.
Gascoin-Lachambre G, Buffat C, Rebourcet R, et al. Cullins in
human intra-uterine growth restriction: expressional and epigenetic
alterations. Placenta 2010; 31: 151-7.
Chelbi ST, Wilson ML, Veillard AC, et al. Genetic and epigenetic
mechanisms collaborate to control SERPINA3 expression and its
association with placental diseases. Hum Mol Genet 2012; 21:
1968-78.
Chelbi ST, Doridot L, Mondon F, et al. Combination of promoter
hypomethylation and PDX1 overexpression leads to TBX15 decrease in vascular IUGR placentas. Epigenetics 2011; 6: 247-55.
Kulkarni A, Chavan-Gautam P, Mehendale S, Yadav H, Joshi S.
Global DNA methylation patterns in placenta and its association
with maternal hypertension in pre-eclampsia. DNA Cell Biol 2011;
30: 79-84.
McMillen IC, Robinson JS. Developmental origins of the metabolic
syndrome: prediction, plasticity, and programming. Physiol Rev
2005; 85: 571-633.
Pham TD, MacLennan NK, Chiu CT, Laksana GS, Hsu JL, Lane
RH. Uteroplacental insufficiency increases apoptosis and alters p53
gene methylation in the full-term IUGR rat kidney. Am J Physiol
Regul Integr Comp Physiol 2003; 285: R962-70.
MacLennan NK, James SJ, Melnyk S, et al. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol Genomics 2004; 18: 43-50.
Vaiman D, Gascoin-Lachambre G, Boubred F, et al. The Intensity
of IUGR-Induced Transcriptome Deregulations Is Inversely Correlated with the Onset of Organ Function in a Rat Model. PLoS One
2011; 6: e21222.
Fu Q, McKnight RA, Yu X, Callaway CW, Lane RH. Growth
retardation alters the epigenetic characteristics of hepatic dual
specificity phosphatase 5. FASEB J 2006; 20: 2127-9.
Eustache F, Mondon F, Canivenc-Lavier MC, et al. Chronic dietary
exposure to a low-dose mixture of genistein and vinclozolin modifies the reproductive axis, testis transcriptome, and fertility. Environ Health Perspect 2009; 117: 1272-9.
Ke X, Lei Q, James SJ, et al. Uteroplacental insufficiency affects
epigenetic determinants of chromatin structure in brains of neonatal
and juvenile IUGR rats. Physiol Genomics 2006; 25: 16-28.
Coupe B, Amarger V, Grit I, Benani A, Parnet P. Nutritional programming affects hypothalamic organization and early response to
leptin. Endocrinology 2010; 151: 702-13.
Konycheva G, Dziadek MA, Ferguson LR, et al. Dietary methyl
donor deficiency during pregnancy in rats shapes learning and
anxiety in offspring. Nutr Res 2011; 31: 790-804.
Ke X, McKnight RA, Caprau D, et al. Intrauterine growth restriction affects hippocampal dual specificity phosphatase 5 gene expression and epigenetic characteristics. Physiol Genomics 2011;
43: 1160-9.
Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of
type 2 diabetes following intrauterine growth retardation in rats is
associated with progressive epigenetic silencing of Pdx1. J Clin Invest 2008; 118: 2316-24.
Pinney SE, Jaeckle Santos LJ, Han Y, Stoffers DA, Simmons RA.
Exendin-4 increases histone acetylase activity and reverses epigenetic modifications that silence Pdx1 in the intrauterine growth retarded rat. Diabetologia 2011; 54: 2606-14.
Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem 2010; 285: 15111-8.
Monk D, Smith R, Arnaud P, et al. Imprinted methylation profiles
for proximal mouse chromosomes 11 and 7 as revealed by methylation-sensitive representational difference analysis. Mamm Genome
2003; 14: 805-16.
Eggerding FA, Schonberg SA, Chehab FF, Norton ME, Cox VA,
Epstein CJ. Uniparental isodisomy for paternal 7p and maternal 7q
in a child with growth retardation. Am J Hum Genet 1994; 55: 25365.
Lee DH, Singh P, Tsark WM, Szabo PE. Complete biallelic insulation at the H19/Igf2 imprinting control region position results in fe-
Calicchio et al.
[162]
[163]
[164]
[165]
[166]
[167]
[168]
[169]
[170]
[171]
[172]
[173]
[174]
[175]
[176]
[177]
[178]
[179]
[180]
[181]
[182]
[183]
tal growth retardation and perinatal lethality. PLoS One 2010; 5:
e12630.
Goyal R, Leitzke A, Goyal D, Gheorghe CP, Longo LD. Antenatal
maternal hypoxic stress: adaptations in fetal lung ReninAngiotensin system. Reprod Sci 2011; 18: 180-9.
Zhou FC, Chen Y, Love A. Cellular DNA methylation program
during neurulation and its alteration by alcohol exposure. Birth Defects Res A Clin Mol Teratol 2011; 91: 703-15.
Downing C, Johnson TE, Larson C, et al. Subtle decreases in DNA
methylation and gene expression at the mouse Igf2 locus following
prenatal alcohol exposure: effects of a methyl-supplemented diet.
Alcohol 2011; 45: 65-71.
Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol
exposure alters DNA methylation profiles in mouse embryos at
early neurulation. Epigenetics 2009; 4: 500-11.
Haycock PC, Ramsay M. Exposure of mouse embryos to ethanol
during preimplantation development: effect on DNA methylation in
the h19 imprinting control region. Biol Reprod 2009; 81: 618-27.
Woods AK, Hoffmann DS, Weydert CJ, et al. Adenoviral delivery
of VEGF121 early in pregnancy prevents spontaneous development
of preeclampsia in BPH/5 mice. Hypertension 2011; 57: 94-102.
Zhao L, Zhou H, Lu L, et al. Identification of quantitative trait loci
controlling rice mature seed culturability using chromosomal segment substitution lines. Plant Cell Rep 2009; 28: 247-56.
Bobetsis YA, Barros SP, Lin DM, et al. Bacterial infection promotes DNA hypermethylation. J Dent Res 2007; 86: 169-74.
Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in SilverRussell syndrome. Nat Genet 2005; 37: 1003-7.
Eggermann T, Schonherr N, Meyer E, et al. Epigenetic mutations
in 11p15 in Silver-Russell syndrome are restricted to the telomeric
imprinting domain. J Med Genet 2006; 43: 615-6.
Schonherr N, Meyer E, Roos A, Schmidt A, Wollmann HA, Eggermann T. The centromeric 11p15 imprinting centre is also involved in Silver-Russell syndrome. J Med Genet 2007; 44: 59-63.
Netchine I, Rossignol S, Dufourg MN, et al. 11p15 imprinting
center region 1 loss of methylation is a common and specific cause
of typical Russell-Silver syndrome: clinical scoring system and
epigenetic-phenotypic correlations. J Clin Endocrinol Metab 2007;
92: 3148-54.
Eggermann T, Meyer E, Caglayan AO, Dundar M, Schonherr N.
ICR1 epimutations in llp15 are restricted to patients with SilverRussell syndrome features. J Pediatr Endocrinol Metab 2008; 21:
59-62.
Binder G, Seidel AK, Martin DD, et al. The endocrine phenotype
in silver-russell syndrome is defined by the underlying epigenetic
alteration. J Clin Endocrinol Metab 2008; 93: 1402-7.
Eggermann T, Schonherr N, Jager S, et al. Segmental maternal
UPD(7q) in Silver-Russell syndrome. Clin Genet 2008; 74: 486-9.
Wojdacz TK, Dobrovic A, Algar EM. Rapid detection of methylation change at H19 in human imprinting disorders using methylation-sensitive high-resolution melting. Hum Mutat 2008; 29: 125560.
Bartholdi D, Krajewska-Walasek M, Ounap K, et al. Epigenetic
mutations of the imprinted IGF2-H19 domain in Silver-Russell
syndrome (SRS): results from a large cohort of patients with SRS
and SRS-like phenotypes. J Med Genet 2009; 46: 192-7.
Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical
pictures. Horm Res 2009; 71 Suppl 2: 30-5.
Eggermann T, Spengler S, Bachmann N, et al. Chromosome 11p15
duplication in Silver-Russell syndrome due to a maternally inherited translocation t(11;15). Am J Med Genet A 2010; 152A: 14847.
Demars J, Shmela ME, Rossignol S, et al. Analysis of the
IGF2/H19 imprinting control region uncovers new genetic defects,
including mutations of OCT-binding sequences, in patients with
11p15 fetal growth disorders. Hum Mol Genet 2010; 19: 803-14.
Turner CL, Mackay DM, Callaway JL, et al. Methylation analysis
of 79 patients with growth restriction reveals novel patterns of
methylation change at imprinted loci. Eur J Hum Genet 2010; 18:
648-55.
McMinn J, Wei M, Schupf N, Cusmai J, Johnson EB, Smith AC,
Weksberg R, Thaker HM, Tycko B. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction.
Placenta 2006; 27: 540-9.
DNA Methylation
[184]
[185]
[186]
[187]
[188]
Current Pharmaceutical Design, 2014, Vol. 20, No. 00
Koukoura O, Sifakis S, Zaravinos A, et al. Hypomethylation along
with increased H19 expression in placentas from pregnancies complicated with fetal growth restriction. Placenta 2011; 32: 51-7.
Bourque DK, Avila L, Penaherrera M, von Dadelszen P, Robinson
WP. Decreased placental methylation at the H19/IGF2 imprinting
control region is associated with normotensive intrauterine growth
restriction but not preeclampsia. Placenta 2010; 31: 197-202.
Tabano S, Colapietro P, Cetin I, et al. Epigenetic modulation of the
IGF2/H19 imprinted domain in human embryonic and extraembryonic compartments and its possible role in fetal growth restriction. Epigenetics 2010; 5: 313-24.
Lambertini L, Lee TL, Chan WY, et al. Differential methylation of
imprinted genes in growth-restricted placentas. Reprod Sci 2011;
18: 1111-7.
Banister CE, Koestler DC, Maccani MA, Padbury JF, Houseman
EA, Marsit CJ. Infant growth restriction is associated with distinct
patterns of DNA methylation in human placentas. Epigenetics
2011; 6: 920-7.
Received: April 20, 2013
Accepted: July 18, 2013
[189]
[190]
[191]
[192]
[193]
25
Michels KB, Harris HR, Barault L. Birthweight, maternal weight
trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS One 2011; 6: e25254.
Gesta S, Bluher M, Yamamoto Y, et al. Evidence for a role of
developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci USA 2006; 103: 6676-81.
Adkins RM, Tylavsky FA, Krushkal J. Newborn umbilical cord
blood DNA methylation and gene expression levels exhibit limited
association with birth weight. Chem Biodivers 2012; 9: 888-99.
Tobi EW, Heijmans BT, Kremer D, et al. DNA methylation of
IGF2, GNASAS, INSIGF and LEP and being born small for gestational age. Epigenetics 2011; 6: 171-6.
Wang H, Ogawa M, Wood JR, et al. Genetic and epigenetic
mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum Mol
Genet 2008; 17: 1087-96.
Modele +
ANCAAN-762; No. of Pages 6
ARTICLE IN PRESS
Disponible en ligne sur
www.sciencedirect.com
Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
Mise au point
Dysfonction endothéliale : rôle dans le syndrome maternel de la
prééclampsie et conséquences à long terme pour le système cardiovasculaire
Endothelial dysfunction: Role in the maternal syndrome of preeclampsia and long-term
consequences for the cardiovascular system
R. Calicchio a,∗ , C. Buffat b , D. Vaiman a , F. Miralles a,∗∗
b
a Inserm U1016-CNRS UMR8104, université Paris Descartes, institut Cochin, 24, rue du Faubourg-Saint-Jacques, 75014 Paris, France
Laboratoire de biologie moléculaire, génétique oncologique et endocrinienne, hôpital de la Conception, AP–HM, 147, boulevard Baille, 13385 Marseille, France
Reçu le 28 janvier 2013 ; accepté le 8 mars 2013
Résumé
La prééclampsie (PE) est une maladie de la grossesse et une cause majeure de mortalité et de morbidité maternelle et fœtale. Il s’agit d’une
pathologie complexe caractérisée par une hypertension et une protéinurie maternelles. Le placenta prééclamptique libère dans la circulation
maternelle des facteurs qui induisent une dysfonction endothéliale systémique. Nous révisons ici les études montrant le rôle central de l’endothélium
dans le développement du syndrome maternel de la PE. Nous présentons aussi des résultats originaux montrant comment des facteurs présents dans
le plasma prééclamptique modifient le transcriptome des cellules endothéliales. Ainsi, des gènes impliqués dans des fonctions essentielles telles
que la vasorégulation, le stress oxydatif, l’apoptose et la prolifération cellulaire sont differentiellement exprimés dans les cellules endothéliales
exposées à du plasma provenant de grossesses prééclamptiques ou normales. Finalement nous discutons les évidences qui lient la dysfonction
endothéliale du syndrome prééclamptique à un risque accru de pathologie cardiovasculaire sur le long terme. Une compréhension approfondie des
modifications subies par la cellule endothéliale dans la PE est essentielle afin de développer des nouvelles thérapies permettant de mieux traiter la
maladie et de prévenir ses séquelles sur le système cardiovasculaire des femmes.
© 2013 Elsevier Masson SAS. Tous droits réservés.
Mots clés : Prééclampsie ; Endothélium ; Pathologies cardiovasculaires ; Transcriptomes ; Épigénétique
Abstract
Preeclampsia is a pregnancy disorder being a leading cause of maternal and fetal mortality and morbidity. It is a complex multisystem disease
characterized by hypertension and proteinuria. In preeclampsia the placenta releases factors into the maternal circulation which cause a systemic
endothelial dysfunction. Here, we review data demonstrating the central role played by the endothelium in the development of the maternal
syndrome of preeclampsia. We present also original data showing how circulating factors present in the plasma of preeclamptic women can alter
the transcriptome of endothelial cells. The expression of genes involved in essential functions such as vasoregulation, oxidative stress, apoptosis
and cell proliferation show differential expression when endothelial cells are exposed to preeclamptic or normal pregnancy plasma. We conclude
by discussing the growing evidences that the alterations of the endothelium during preeclampsia are linked to an increased risk of cardiovascular
diseases latter on life. Therefore, a better understanding of the modifications undergone by the endothelial cells during preeclampsia is essential to
develop new therapeutic approaches to both, manage preeclampsia and to prevent the long-term sequelae of the disease on women cardiovascular
system.
© 2013 Elsevier Masson SAS. All rights reserved.
Keywords: Preeclampsia; Endothelium; Cardiovascular diseases; Transcriptomics; Epigenetics
∗
∗∗
Auteur correspondant.
Co-auteur correspondant.
Adresses e-mail : rosamaria.calicchio@inserm.fr (R. Calicchio), francisco.miralles@inserm.fr (F. Miralles).
0003-3928/$ – see front matter © 2013 Elsevier Masson SAS. Tous droits réservés.
http://dx.doi.org/10.1016/j.ancard.2013.03.002
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002
Modele +
ANCAAN-762; No. of Pages 6
2
ARTICLE IN PRESS
R. Calicchio et al. / Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
1. La prééclampsie : définition et physiopathologie
La prééclampsie (PE) est une maladie de la gestation qui se
manifeste généralement au début du dernier trimestre de la grossesse et qui se caractérise par une hypertension (> 140/90 mmHg
pour la systole et la diastole respectivement) et une protéinurie maternelles (> 300 mg/24 h). Cette pathologie affecte 5 %
à 7 % des grossesses et demeure une cause majeure de morbidité et de mortalité à la fois chez la mère et le fœtus [1].
La PE concerne l’ensemble de l’organisme de la femme, avec
toutefois une incidence particulière sur les systèmes cardiovasculaire, hépatique, rénal, cérébral ainsi que sur les mécanismes
de contrôle de la coagulation. La PE est caractérisée par une
vasoconstriction généralisée qui résulte d’une augmentation de
la résistance périphérique et d’une augmentation de la pression
artérielle moyenne. Un œdème hépatique est présent chez la
plupart des femmes prééclamptiques. Dans la PE, des degrés
variables d’insuffisance rénale sont associés à une lésion glomérulaire caractéristique, l’endothéliose glomérulaire due à la
formation de dépôts de fibrine. Finalement, la PE est accompagnée d’une activation locale ou disséminée de la cascade de
coagulation. Les complications de la PE sont multiples. Les plus
communes étant l’éclampsie (une crise convulsive tonicoclonique) et le syndrome Hemolysis, Elevated Liver enzymes, Low
Platelet count (HELLP) constitué par l’association hémolyse,
cytolyse hépatique et thrombopénie. Une autre complication
grave de la PE est l’insuffisance rénale aiguë qui dans les cas
extrêmes aboutit à une nécrose corticale.
La physiopathologie de la PE reste encore mal connue. Toutefois, le placenta semble être à l’origine de la pathologie [1,2].
Dans une grossesse non pathologique, lors de la formation du
placenta, les cytotrophoblastes fœtaux envahissent et remanient
profondément la structure des artères spiralées jusqu’au niveau
du premier tiers du myomètre. Ces modifications provoquent
une augmentation importante du débit sanguin en direction du
placenta. Dans les grossesses prééclamptiques, la placentation
est perturbée car les cytotrophoblastes ne parviennent pas à
envahir correctement le myomètre et à transformer les artères
spiralées. Cet échec morphogénétique provoque une hypoxie
placentaire, entraînant un stress oxydatif et une inflammation
locale qui dans un second temps déclenchent une dysfonction
endothéliale maternelle systémique. Plusieurs études ont montré
qu’en situation de PE le placenta ischémique libère dans la circulation sanguine des facteurs responsables de l’apparition de la
maladie maternelle (débris syncitiaux, radicaux libres, facteurs
angiogéniques et anti-angiogéniques, hormones placentaires,
cytokines pro-inflammatoires, etc.). Ces molécules perturbent
le fonctionnement de l’endothélium maternel et provoquent
l’apparition du syndrome. Ainsi, le plasma des patientes
atteintes de PE présente des concentrations anormales de facteurs angiogéniques et anti-angiogéniques tels le VEGF, PLGF,
sFlt-1, et sEng [2–4]. Le Vascular endothelial growth factor
(VEGF) et Placental growth factor (PLGF) sont des facteurs
angiogéniques jouant un rôle fondamental pour le maintien de
l’homéostasie vasculaire. De plus, le VEGF possède un effet
vasodilatateur par le biais de l’induction de la production endothéliale de NO et PGL2 . Le VEGF et PLGF se fixent tous les
deux au récepteur VEGFR-1 ou Fms-like tyrosine kinase (Flt1).
Toutefois, un épissage alternatif génère une forme soluble du
récepteur au VEGF, le sFlt-1, capable elle aussi de fixer le VEGF
et le PLGF. sFlt-1 empêche ainsi l’interaction de ces deux facteurs angiogéniques avec leurs récepteurs situés essentiellement
à la surface des cellules endothéliales. Ainsi des taux élevés
de sFlt-1 circulant privent le système vasculaire de signaux
nécessaire à sa survie et/ou maintenance. Un deuxième facteur
soluble, sEng, est retrouvé à des taux anormalement élevés dans
le plasma des femmes prééclamptiques. La sEng est produite par
clivage protéolytique du récepteur à l’endogline. Ce récepteur
est exprimé par les syncytiotrophoblaste et les cellules endothéliales et agit comme un corécepteur du facteur pro-angiogénique
TGF-1. Comme pour sFlt-1, l’augmentation du taux circulant
de sEng prive l’endothélium d’un signal essentiel à son fonctionnement. Il a été montré que l’augmentation de sFlt-1 et sEng
et la diminution des concentrations plasmatiques et urinaires de
VEGF et du PLGF précèdent l’apparition du syndrome clinique
de la PE et sont corrélées avec la sévérité de la maladie [5].
2. La prééclampsie, une maladie de l’endothélium
maternel
De nombreuses preuves montrent que le syndrome maternel de la PE est la conséquence d’un dysfonctionnement de
l’endothélium vasculaire. En plus de l’endothéliose glomérulaire du rein, des modifications structurales de l’endothélium
de la veine ombilicale et des vaisseaux utéroplacentaires
ont été décrites [6,7]. Les modifications fonctionnelles
de l’endothélium maternel sont mises en évidence par
l’augmentation du taux sérique de différents marqueurs
de l’activation endothéliale : Von Willebrand factor (VWF),
endothéline-1, fibronectine, formes solubles des molécules
d’adhésion (E-selectin, VCAM, and ICAM), interleukine-6 (IL6) et interleukin-8 (IL-8), [8]. Aussi, les niveaux plasmatiques
des facteurs anti-angiogéniques sFlt-1 et sEng sont augmentés
en PE [5,9]. L’implication de l’endothélium dans l’activation
de la cascade de coagulation dans le plasma prééclamptique
est révélé par des taux élevés de fibronectine, thrombomoduline et par la modification de la balance entre l’activateur et
inhibiteur du plasminogène et entre la prostacycline (PGI2 ) et
le thromboxane (TxA2 ) [10]. Le tonus vasculaire et la résistance périphérique sont sous contrôle de facteurs dérivés de
l’endothélium. Par conséquent, la modification de la fonction
endothéliale pendant la PE a un impact important sur la régulation du tonus vasculaire. Une diminution de la vasodilatation
dépendante de l’endothélium a été démontrée par des techniques
non invasives chez les femmes prééclamptiques [11–13].
Ces données prouvent que la PE est une maladie de
l’endothélium. La glomérulose endothéliale est probablement
la cause de la protéinurie. Le défaut dans la vasodilatation endothélium-dépendante est la cause de l’hypertension et
déclenche une intense vasoconstriction dans différents organes
provoquant une hypoperfusion. L’activation des cellules endothéliales est une cause probable d’inflammation systémique.
Finalement, l’augmentation de la perméabilité des cellules endothéliales peut expliquer l’œdème caractéristique de la PE.
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002
Modele +
ARTICLE IN PRESS
ANCAAN-762; No. of Pages 6
R. Calicchio et al. / Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
3. Effets du plasma prééclamptique sur les cellules
endothéliales
Des études in vitro ont montré que le plasma des femmes
prééclamptiques induit des réponses spécifiques dans les cellules endothéliales. L’incubation de cellules endothéliales avec
le plasma de patientes prééclamptiques augmente le niveau
d’expression de marqueurs de l’activation endothéliale tels
l’endothéline-1, PDGF, VCAM-1, ICAM-1, fibronectine, iNOS
et NOX2, [14–18]. De plus la production de prostacyclines, NO
et radicaux libres est incrémentée après exposition au plasma
prééclamptique [8,14,19]. Finalement, le plasma prééclamptique augmente la sécrétion par les cellules endothéliales de
chemokines (MCP-1) et des cytokines IL-6 et Il-8 [20–22]. Ces
travaux montrent que le plasma prééclamptique contient des facteurs capables d’agir directement sur les cellules endothéliales
et de déclencher leur activation.
Une seule étude a essayé de déterminer les effets de la PE
sur l’expression globale des gènes dans l’endothélium maternel.
À l’aide de puces ADN, Donker et al. ont analysé le transcriptome de deux lignées de cellules endothéliales cultivées pendant
24 heures avec du plasma issu de femmes avec une grossesse normale ou prééclamptique [23]. De façon surprenante, cette étude
n’a pas montré de différences significatives dans l’expression
des gènes. Les auteurs ont donc conclu que le plasma prééclamptique ne contient pas de facteurs capables d’agir directement sur
les cellules endothéliales. Ce résultat semble en contradiction
3
avec les études in vitro mentionnées plus haut. La présence dans
le plasma prééclamptique de facteurs capables de modifier l’état
de transcription des gènes de l’endothélium est une question de
grande importance. La réponse à cette question est fondamentale, aussi bien, afin de mieux connaître l’état physiologique
de la cellule endothéliale dans le contexte de cette pathologie,
que dans le cadre de la recherche de biomarqueurs ou de nouvelles approches thérapeutiques. C’est pourquoi, considérant les
avances techniques dans le domaine de l’analyse du transcriptome depuis l’étude de Donker et al., nous avons conduit une
nouvelle étude afin d’évaluer les effets du plasma prééclamptique sur le transcriptome des cellules endothéliales.
4. Analyse du transcriptome de cellules endothéliales
exposées au plasma prééclamptique
Nous avons comparé le transcriptome d’une lignée de
cellules endothéliales issues du cordon ombilical (HUVEC)
après une exposition de 24 heures à 10 % de plasma provenant de grossesses prééclamptiques ou normales. Cette
analyse a mis en évidence 116 gènes montrant un profil
d’expression modifié dans les cellules HUVEC traitées
avec du plasma prééclamptique. Une partie de ces gènes
joue un rôle majeur dans la physiopathologie des cellules
endothéliales. L’analyse non supervisée a mis en évidence
cinq catégories de gènes selon leur fonction : vasorégulation,
mitose et cycle cellulaire, régulation de l’apoptose, biosynthèse
Tableau 1
Analyse bio-informatique du transcriptome des cellules Huvec traitées avec du plasma prééclamptique.
DAVID
Catégorie fonctionnelle
Nombre de gènes
Symbole de gène
Valeur de p
Cycle cellulaire
13
1,30 × 10–5
Apoptose
11
CKAP2, TXNIP, TRNP1, CASC5, CDC20, BIRC5, PTTG1 SMC4,
CCNB1, CDCA8, SPAG5, PLK1, CCNA2
PHLPP1, CKAP2, TSC22D3, CHAC1, BCL2A1, TRIB3, BIRC5, PMAIP1,
RTEL1, PHLDA1, DDIT4
CEBPB, VEGFA, SPHK1, BCL2A1, BIRC5, HSPA5, RTEL1, CITED2
VEGFA, EDN1, SPHK1, ITGB3, IGFBP3, INSR, CITED2
CEBPB, VEGFA, SPHK1, BCL2A1, RAG1, BIRC5, HSPA5
RTEL1, CITED2
PTTG1, CCNB1, CCNA2, PLK1, PSRC1, CDC20, CDCA8, SPAG5,
SMC4, KIF20A, EDN1, INSR, BIRC5, CASC5, CCNB2
PTTG1, CCNB1, CCNA2, TXNIP, PLK1, PSRC1, CDC20, KPNA2,
CKAP2, CDCA8, SPHK1, SPAG5, GAS2L3, CITED2, SMC4, TRNP1,
KIF20A, EDN1, ID3, INSR, BIRC5, CASC5, CCNB2
AGMO, GGT5, INSIG1, FASN, EDN1, TRIB3
SLIT3, CHAC1, PHLDA1, PMAIP1, TXNIP, SLIT2, IGFBP3, GARS,
HSPA5, VEGFA, CKAP2, SPHK1, PHLPP1, DDIT4, RAG1, CITED2,
CLN5, CEBPB, ID3, TRIB3, BCL2A1, BIRC5
CCNB1, VEGFA, DDIT4, STC2, CITED2, EDN1
ITGB3, SLIT2, VEGFA, SPHK1, EDN1, INSR
ADAMTS1, CCNB1, ITGB3, SLIT2, VEGFA, SPHK1, CITED2, ERRFI1,
EDN1, ID3, INSR
HSPA5, SPHK1, CITED2, CEBPB, BCL2A1, BIRC5
PTTG1, CCNB1, CCNA2, ZSWIM7, PMAIP1, PLK1, ATF4, HSPA5,
VEGFA, INSIG1, CBS, STC2, RTEL1, EDN1
8,45 × 10–9
Anti-apoptose
Régulation de la migration cellulaire
Régulation négative de l’apoptose
8
7
9
6,67 × 10–5
4,70 × 10–4
9,65 × 10–4
2,61 × 10–3
GENOMATIX
Phase M du cycle cellulaire
15
Cycle cellulaire
23
Biosynthèse des acides gras
Régulation de l’apoptose
6
22
Réponse à l’hypoxie
Régulation positive de la motilité
Développement du système cardiovasculaire
6
6
11
Anti-apoptose
Réponse cellulaire au stress
6
14
2,99 × 10–6
1,48 × 10–4
2,38 × 10–4
6,50 × 10–4
6,50 × 10–4
8,98 × 10–4
6,41 × 10–3
2,00 × 10–3
Liste des principaux gènes modifiés dans les cellules Huvec exposées à du plasma prééclamptique et association avec les catégories fonctionnelles alterées, suite à
l’analyse bio-informatique sur David et Genomatix. Les gènes surexprimés dans les cellules HUVEC traitées avec du plasma prééclamptique sont montrés en gras.
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002
Modele +
ANCAAN-762; No. of Pages 6
4
ARTICLE IN PRESS
R. Calicchio et al. / Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
des acides gras, réponse au stress du réticulum endoplasmique (Tableau 1). Ces modifications transcriptomiques
sont cohérentes avec les études publiées sur la dysfonction
endothéliale. En effet, la dérégulation de facteurs impliqués
dans les fonctions de vasorégulation (EDN1, APLN, et CBS)
pourrait être liée au développement de l’hypertension suite à
l’augmentation de la vasoconstriction. La balance observée
entre l’augmentation de gènes anti-apoptotiques et la diminution
des facteurs pro-apoptotiques (y compris les gènes impliqués
dans le contrôle du stress du réticulum endoplasmique) pourrait
résulter d’une réponse adaptative des cellules endothéliales à
des facteurs présents dans le plasma prééclamptique (cytokines
pro-inflammatoires et facteurs anti-angiogéniques). Cette
réponse adaptative permettrait aux cellules endothéliales de
survivre au stress provoqué par le plasma prééclamptique.
Notre analyse montrait aussi une augmentation de
l’expression du gène NOX4 dans les cellules traitées avec du
plasma prééclamptique. NOX4 code une NADPH oxidase qui
est, dans l’endothélium, une source majeure de radicaux libres et
d’H2 O2 [24]. Les radicaux libres, par l’activation de différentes
voies de signalisation (Akt, Src, mAPK), régulent l’expression
des gènes impliqués dans la croissance, l’apoptose, la migration,
l’angiogenèse, la perméabilité et l’inflammation [24].
5. Conséquences à long terme de la prééclampsie sur le
système cardiovasculaire maternel
Les symptômes de la PE disparaissent complètement après
l’accouchement, mais des études épidémiologiques indiquent
que sur le long terme, il y a des conséquences pour le système cardiovasculaire maternel. Environ 20 % des femmes
ayant développé une grossesse prééclamptique présentent de
l’hypertension ou de la microalbuminurie dans un délai de sept
ans après l’accouchement, comparé à seulement 2 % des femmes
avec des grossesses normales [25]. De plus, le risque à long
terme de maladie cardiovasculaire et cérébrovasculaire est doublé chez les femmes avec une PE associée à une hypertension
gestationnelle comparée aux femmes d’un même âge. Finalement, les femmes ayant subi une PE présentent un risque accru
de développer une pathologie rénale terminale [26]. La PE et les
maladies cardiovasculaires partagent beaucoup de facteurs de
risque communs, y compris l’hypertension chronique, le diabète,
l’obésité, la maladie rénale, et le syndrome métabolique [27].
L’augmentation à long terme, des pathologies cardiovasculaires
chez les femmes ayant subi une PE peut être le résultat de facteurs de risque partagés avec les autres pathologies vasculaires
ou le résultat de subtils dommages vasculaires provoqués par la
PE, persistant après la grossesse et jouant un rôle nocif avec le
vieillissement du système vasculaire. La persistance d’une dysfonction vasculaire après la PE a fait l’objet de plusieurs études et
semble être maintenant un fait établi. L’étude la plus importante
a porté sur une période couvrant trois années post-partum et a
examiné 78 femmes ayant subi une PE, 35 femmes avec des PE
récurrentes et 48 femmes avec des grossesses normales [28]. Les
femmes ayant subi une PE présentaient une diminution significative de la réponse aux vasodilatateurs. Cette anomalie apparaissait plus marquée chez celles qui ont fait des PE récurrentes.
Cela a été confirmé par d’autres études montrant la persistance
d’anomalies de la réponse endothéliale aux vasodilatateurs des
nombreuses années (15 à 25) après une PE [29,30]. Par ailleurs,
il a été constaté que huit années après la survenue d’une PE les
concentrations de TNF-a, sFlt-1 et protéine C-réactive circulant
dans le sang maternel sont anormalement élevées [31].
6. Implication des mécanismes épigénétiques dans la
prééclampsie
Plusieurs études ont suggéré un rôle de la génétique ou
de mutations de gènes spécifiques dans la susceptibilité à la
PE [32]. Toutefois, peu d’études ont analysé l’implication des
mécanismes épigénétiques. L’épigénétique est définie comme
l’étude des changements héritables de l’état de transcription
des gènes, n’impliquant aucune altération de la séquence ADN.
Ces changements sont en général réversibles et sont portés par
des modifications dites épigénétiques. Ces modifications sont
transmissibles lors de la mitose et/ou la méiose. Il existe trois
mécanismes majeurs de régulation épigénétique : la méthylation
de l’ADN, diverses modifications chimiques des histones et la
régulation de l’expression des gènes par des petits ARN non
codants (tels les micro-ARN). Ces trois mécanismes agissent de
manière coordonnée.
Concernant le rôle de l’épigénétique dans la PE, la majorité
des études a porté sur la méthylation de l’ADN. Chez les mammifères, la méthylation de l’ADN a lieu sur le C des dinucléotides
CpG. Ces dinucléotides sont préférentiellement localisés au
niveau du promoteur des gènes (formant des îlots CpG). Cette
méthylation est effectuée par les méthyltrasférases de l’ADN
(DNMT1, DNMT2, DNMT3A, DNMT3B et DNMT3L). Ces
enzymes transfèrent un groupement méthyl (CH3 ) sur le carbone 5 de la cytosine d’un dinucléotide CpG. De manière
générale l’hypométhylation des îlots CpG est associée à une
augmentation de l’expression des gènes et l’hyperméthylation
à une diminution de leur expression. Les études de transcriptomique ont montré que le placenta prééclamptique présente
d’importantes modifications d’expression de gènes [33–36].
Cela a conduit à postuler que certaines de ces modifications
peuvent être dues à des changements de méthylation de l’ADN
dans les îlots CpG. Diverses études globales ont montré des
variations de méthylation des régions régulatrices de plusieurs
gènes [37,38]. TIMP3, un gène impliqué dans le contrôle de
l’invasion trophoblastique a été trouvé hypométhylé dans le
placenta prééclamptique, et il a été démontré que cette hypométhylation affectait réellement l’expression du gène [39]. D’autres
gènes pressentant des modifications de méthylation dans le
placenta prééclamptique sont RASSF1A et SERPINB5 [40,41],
MMP9 [42], Cullin7 et Cullin 4B [43] et SERPINA3 [44]. Par
ailleurs, le niveau de méthylation global a été analysé indiquant
une hyperméthylation de l’ADN dans le placenta prééclamptique [45].
Dans le contexte du système vasculaire, une étude très récente
a comparé, à l’aide de puces spécialement conçues pour ce
propos (Illumina Human Methylation27 BeadChip), l’état de
méthylation de quelque 27 000 îlots CpG (correspondant à près
de 14 000 gènes) dans des échantillons d’artères isolées de
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002
Modele +
ANCAAN-762; No. of Pages 6
ARTICLE IN PRESS
R. Calicchio et al. / Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
l’omentum de femmes avec une grossesse prééclamptique ou
normale [46]. L’étude a révélé une diminution significative de
la méthylation des îlots CpG correspondant à 65 gènes. Ces
gènes sont impliqués dans des fonctions aussi diverses que le
contrôle de la contraction des muscles lisses, la coagulation,
l’inflammation, l’homéostasie redox, ou le métabolisme des
sucres et des acides aminés. Cela montre que, l’hypométhylation
des séquences régulatrices de certains gènes impliqués dans la
régulation du système vasculaire est associée à la PE. Cependant, cette étude ne permet pas de conclure si ces différences de
méthylation sont préexistantes à la survenue de la PE ou bien
la conséquence de la PE. Il est toutefois intéressant de noter ici
que le stress oxydatif, qui est une des conséquences de la PE, est
connu comme étant un facteur capable de modifier la méthylation de l’ADN [47,48]. Cette étude, bien que limitée aux artères
de l’omentum, montre qu’il est tout à fait possible qu’un épisode de PE puisse induire des modifications de la méthylation de
l’ADN maternel pouvant altérer l’expression des gènes impliqués dans la fonction vasculaire. Avec l’âge ces modifications
pourraient jouer un rôle dans la plus grande susceptibilité aux
pathologies cardiovasculaires observée chez les femmes ayant
subies une PE.
7. Conclusion
Le rôle de l’endothélium dans le développement du syndrome
maternel de la PE est démontré par une multitude d’études.
Cependant, une compréhension approfondie des modifications
subies par la cellule endothéliale dans la PE pourra contribuer à une meilleure prise en charge de cette pathologie. Cela
devrait aussi permettre de mieux appréhender les conséquences
de la PE pour la future santé cardiovasculaire des patientes et
d’orienter les recherches afin de proposer des nouvelles thérapeutiques. Étant donné les difficultés éthiques et techniques que
suppose l’accès à l’endothélium maternel, le développement de
modèles cellulaires et animaux permettant d’analyser au niveau
moléculaire l’impact de la PE sur le système vasculaire semble
fondamental.
Déclaration d’intérêts
Les auteurs déclarent ne pas avoir de conflits d’intérêts en
relation avec cet article.
Références
[1] Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia.
Lancet 2010;376:631–44.
[2] Mutter WP, Karumanchi SA. Molecular mechanisms of preeclampsia.
Microvasc Res 2008;75:1–8.
[3] Wang A, Rana S, Karumanchi SA. Preeclampsia: the role of angiogenic
factors in its pathogenesis. Physiology (Bethesda) 2009;24:147–58.
[4] Noris M, Perico N, Remuzzi G. Mechanisms of disease: pre-eclampsia.
Nat Clin Pract Nephrol 2005;1:98–114, quiz 120.
[5] Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for
later cardiovascular disease. Circulation 2011;123:2856–69.
5
[6] Shanklin DR, Sibai BM. Ultrastructural aspects of preeclampsia. I.
Placental bed and uterine boundary vessels. Am J Obstet Gynecol
1989;161:735–41.
[7] Wang Y, Gu Y, Granger DN, Roberts JM, Alexander JS. Endothelial junctional protein redistribution and increased monolayer permeability in human
umbilical vein endothelial cells isolated during preeclampsia. Am J Obstet
Gynecol 2002;186:214–20.
[8] Sankaralingam S, Xu Y, Sawamura T, Davidge ST. Increased lectin-like
oxidized low-density lipoprotein receptor-1 expression in the maternal vasculature of women with preeclampsia: role for peroxynitrite. Hypertension
2009;53:270–7.
[9] Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, et al. Soluble
endoglin and other circulating antiangiogenic factors in preeclampsia. N
Engl J Med 2006;355:992–1005.
[10] Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP.
Pathophysiology of hypertension during preeclampsia: Linking placental
ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol
2008;294:H541–50.
[11] VanWijk MJ, Kublickiene K, Boer K, VanBavel E. Vascular function in
preeclampsia. Cardiovasc Res 2000;47:38–48.
[12] Yoshida A, Nakao S, Kobayashi M, Kobayashi H. Flow-mediated vasodilation and plasma fibronectin levels in preeclampsia. Hypertension
2000;36:400–4.
[13] Yamamoto T, Suzuki Y, Kojima K, Suzumori K. Reduced flow-mediated
vasodilation is not due to a decrease in production of nitric oxide in preeclampsia. Am J Obstet Gynecol 2005;192:558–63.
[14] Baker PN, Davidge ST, Roberts JM. Plasma from women with preeclampsia increases endothelial cell nitric oxide production. Hypertension
1995;26:244–8.
[15] Endresen MJ, Morris JM, Nobrega AC, Buckley D, Linton EA, Redman CW. Serum from preeclamptic women induces vascular cell adhesion
molecule-1 expression on human endothelial cells in vitro: a possible role
of increased circulating levels of free fatty acids. Am J Obstet Gynecol
1998;179:665–70.
[16] Hayman R, Brockelsby J, Kenny L, Baker P. Preeclampsia: the endothelium, circulating factor(s) and vascular endothelial growth factor. J Soc
Gynecol Invest 1999;6:3–10.
[17] Matsubara K, Matsubara Y, Hyodo S, Katayama T, Ito M. Role of nitric
oxide and reactive oxygen species in the pathogenesis of preeclampsia. J
Obstet Gynaecol Res 2010;36:239–47.
[18] Taylor RN, de Groot CJ, Cho YK, Lim KH. Circulating factors as markers and mediators of endothelial cell dysfunction in preeclampsia. Semin
Reprod Endocrinol 1998;16:17–31.
[19] Davidge ST, Stranko CP, Roberts JM. Urine but not plasma nitric oxide
metabolites are decreased in women with preeclampsia. Am J Obstet Gynecol 1996;174:1008–13.
[20] Kauma S, Takacs P, Scordalakes C, Walsh S, Green K, Peng T. Increased endothelial monocyte chemoattractant protein-1 and interleukin-8 in
preeclampsia. Obstet Gynecol 2002;100:706–14.
[21] Takacs P, Kauma SW, Sholley MM, Walsh SW, Dinsmoor MJ, Green
K. Increased circulating lipid peroxides in severe preeclampsia activate
nf-kappab and upregulate icam-1 in vascular endothelial cells. FASEB J
2001;15:279–81.
[22] Takacs P, Green KL, Nikaeo A, Kauma SW. Increased vascular endothelial cell production of interleukin-6 in severe preeclampsia. Am J Obstet
Gynecol 2003;188:740–4.
[23] Donker RB, Asgeirsdottir SA, Gerbens F, van Pampus MG, Kallenberg CG,
te Meerman GJ, et al. Plasma factors in severe early-onset preeclampsia do
not substantially alter endothelial gene expression in vitro. J Soc Gynecol
Invest 2005;12:98–106.
[24] Frey RS, Ushio-Fukai M, Malik AB. Nadph oxidase-dependent signaling in
endothelial cells: Role in physiology and pathophysiology. Antioxid Redox
Signal 2009;11:791–810.
[25] Carty DM, Delles C, Dominiczak AF. Preeclampsia and future maternal
health. J Hypertens 2010;28:1349–55.
[26] Vikse BE, Irgens LM, Leivestad T, Skjaerven R, Iversen BM. Preeclampsia and the risk of end-stage renal disease. N Engl J Med 2008;359:
800–9.
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002
Modele +
ANCAAN-762; No. of Pages 6
6
ARTICLE IN PRESS
R. Calicchio et al. / Annales de Cardiologie et d’Angéiologie xxx (2013) xxx–xxx
[27] Yinon Y, Kingdom JC, Odutayo A, Moineddin R, Drewlo S, Lai V, et al.
Vascular dysfunction in women with a history of preeclampsia and intrauterine growth restriction: insights into future vascular risk. Circulation
2010;122:1846–53.
[28] Chambers JC, Fusi L, Malik IS, Haskard DO, De Swiet M, Kooner JS.
Association of maternal endothelial dysfunction with preeclampsia. JAMA
2001;285:1607–12.
[29] Ramsay JE, Stewart F, Greer IA, Sattar N. Microvascular dysfunction: a
link between pre-eclampsia and maternal coronary heart disease. BJOG
2003;110:1029–31.
[30] Lampinen KH, Ronnback M, Kaaja RJ, Groop PH. Impaired vascular dilatation in women with a history of pre-eclampsia. J Hypertens
2006;24:751–6.
[31] Kvehaugen AS, Dechend R, Ramstad HB, Troisi R, Fugelseth D, Staff AC.
Endothelial function and circulating biomarkers are disturbed in women
and children after preeclampsia. Hypertension 2011;58:63–9.
[32] Pridjian G, Puschett JB. Preeclampsia. Part 2: experimental and genetic
considerations. Obstet Gynecol Surv 2002;57:619–40.
[33] Nishizawa H, Ota S, Suzuki M, Kato T, Sekiya T, Kurahashi H, et al.
Comparative gene expression profiling of placentas from patients with
severe pre-eclampsia and unexplained fetal growth restriction. Reprod Biol
Endocrinol 2011;9:107.
[34] Varkonyi T, Nagy B, Fule T, Tarca AL, Karaszi K, Schonleber J,
et al. Microarray profiling reveals that placental transcriptomes of earlyonset hellp syndrome and preeclampsia are similar. Placenta 2011;
32 Suppl.:S21–9.
[35] Centlow M, Wingren C, Borrebaeck C, Brownstein MJ, Hansson SR.
Differential gene expression analysis of placentas with increased vascular resistance and pre-eclampsia using whole-genome microarrays. J
Pregnancy 2011;2011:472354.
[36] Tsai S, Hardison NE, James AH, Motsinger-Reif AA, Bischoff SR, Thames
BH, et al. Transcriptional profiling of human placentas from pregnancies
complicated by preeclampsia reveals disregulation of sialic acid acetylesterase and immune signalling pathways. Placenta 2011;32:175–82.
[37] Jia RZ, Zhang X, Hu P, Liu XM, Hua XD, Wang X, et al. Screening
for differential methylation status in human placenta in preeclampsia
[38]
[39]
[40]
[41]
[42]
[43]
[44]
[45]
[46]
[47]
[48]
using a cpg island plus promoter microarray. Int J Mol Med 2012;30:
133–41.
Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson WP.
DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet
2010;18:1006–12.
Xiang Y, Zhang X, Li Q, Xu J, Zhou X, Wang T, et al. Promoter hypomethylation of timp3 is associated with pre-eclampsia in a chinese population.
Mol Hum Reprod 2013;19(3):153–9.
Tsui DW, Chan KC, Chim SS, Chan LW, Leung TY, Lau TK, et al. Quantitative aberrations of hypermethylated rassf1a gene sequences in maternal
plasma in pre-eclampsia. Prenat Diagn 2007;27:1212–8.
Bellido ML, Radpour R, Lapaire O, De Bie I, Hosli I, Bitzer J, et al.
Maldi-tof mass array analysis of rassf1a and serpinb5 methylation patterns
in human placenta and plasma. Biol Reprod 2010;82:745–50.
Wang Z, Lu S, Liu C, Zhao B, Pei K, Tian L, et al. Expressional and epigenetic alterations of placental matrix metalloproteinase 9 in preeclampsia.
Gynecol Endocrinol 2010;26:96–102.
Gascoin-Lachambre G, Buffat C, Rebourcet R, Chelbi ST, Rigourd V, Mondon F, et al. Cullins in human intra-uterine growth restriction: Expressional
and epigenetic alterations. Placenta 2010;31:151–7.
Chelbi ST, Mondon F, Jammes H, Buffat C, Mignot TM, Tost J, et al.
Expressional and epigenetic alterations of placental serine protease inhibitors: serpina3 is a potential marker of preeclampsia. Hypertension
2007;49:76–83.
Kulkarni A, Chavan-Gautam P, Mehendale S, Yadav H, Joshi S. Global
DNA methylation patterns in placenta and its association with maternal
hypertension in pre-eclampsia. DNA Cell Biol 2011;30:79–84.
Mousa AA, Strauss 3rd JF, Walsh SW. Reduced methylation of the thromboxane synthase gene is correlated with its increased vascular expression
in preeclampsia. Hypertension 2012;59:1249–55.
Hitchler MJ, Domann FE. An epigenetic perspective on the free radical
theory of development. Free Radic Biol Med 2007;43:1023–36.
Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts
induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S
A 1994;91:1261–4.
Pour citer cet article : Calicchio R, et al. Dysfonction endothéliale : rôle dans le syndrome maternel de la prééclampsie et conséquences à long
terme pour le système cardiovasculaire. Ann Cardiol Angeiol (Paris) (2013), http://dx.doi.org/10.1016/j.ancard.2013.03.002