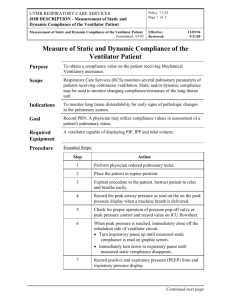
Ventilatory Failure, Ventilator Support, and Ventilator Weaning
advertisement

P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Martin J. Tobin,*1 Franco Laghi,1 and Amal Jubran1 ABSTRACT The development of acute ventilatory failure represents an inability of the respiratory control system to maintain a level of respiratory motor output to cope with the metabolic demands of the body. The level of respiratory motor output is also the main determinant of the degree of respiratory distress experienced by such patients. As ventilatory failure progresses and patient distress increases, mechanical ventilation is instituted to help the respiratory muscles cope with the heightened workload. While a patient is connected to a ventilator, a physician’s ability to align the rhythm of the machine with the rhythm of the patient’s respiratory centers becomes the primary determinant of the level of rest accorded to the respiratory muscles. Problems of alignment are manifested as failure to trigger, double triggering, an inflationary gas-flow that fails to match inspiratory demands, and an inflation phase that persists after a patient’s respiratory centers have switched to expiration. With recovery from disorders that precipitated the initial bout of acute ventilatory failure, attempts are made to discontinue the ventilator (weaning). About 20% of weaning attempts fail, ultimately, because the respiratory controller is unable to sustain ventilation and this failure is signaled by development of rapid shallow breathing. Substantial advances in the medical management of acute ventilatory failure that requires ventilator assistance are most likely to result from research yielding novel insights into the operation of the respiratory control C 2012 American Physiological Society. Compr Physiol 2:2871-2921, 2012. system. Introduction This review encompasses aspects of control of breathing that pertain to three states sequentially experienced by a patient with respiratory disease of sufficient severity to require admission to an intensive care unit. Firstly, the patient may have such severe derangements of respiratory function that he or she develops ventilatory failure. Secondly, the ventilatory failure may be of sufficient severity to require the institution of mechanical ventilation. Thirdly, after the precipitating respiratory disorder has resolved to a reasonable degree, the patient is weaned from the ventilator; these weaning attempts, however, may be followed by recurrences of respiratory distress of such severity as to require the temporary reinstitution of mechanical ventilation. Ventilatory Failure Relationship between ventilation and PaCO2 Ventilatory failure is conventionally defined as an increase in arterial carbon dioxide tension (PaCO2 ) that exceeds the normal range. When PaCO2 is higher than 45 mmHg, the respiratory system is deemed to have failed in maintaining an adequate level of ventilation. As such, the physiological determinants of PaCO2 constitute the framework for consideration of ventilatory failure (306). In healthy subjects, PaCO2 remains remarkably stable despite wide variations in minute ventilation (54). Such Volume 2, October 2012 stability depends on exquisite control of breathing achieved primarily through negative feedback. According to this model, the controlled variable is PaCO2 , the controlled system is the pulmonary gas-exchange organ, and the controller is the entire system of respiratory centers, neurons, and muscles that achieve alveolar ventilation (269). The system is designed to maintain a stable PaCO2 despite fluctuations in CO2 production by the body (V̇ CO2 ), and a reference PCO2 , or “set point,” is maintained in the brainstem. Under steady-state conditions, the relationship between alveolar CO2 tension (PACO2 ), alveolar ventilation (V̇ A ) and V̇ CO2 is given by the alveolar-gas equation: PACO2 = K V̇ CO2 V̇A where K is a constant that converts measurement of V̇ CO2 from standard conditions to body temperature conditions. Because PaCO2 is in equilibrium with PACO2 and because V̇ A equals minute ventilation (V̇ E ) minus dead-space * Correspondence to mtobin2@lumc.edu of Pulmonary and Critical Care Medicine, Edward Hines Jr. Veterans Affairs Hospital and Loyola University of Chicago Stritch School of Medicine, Hines, Illinois Published online, October 2012 (comprehensivephysiology.com) 1 Division DOI: 10.1002/cphy.c110030 C American Physiological Society Copyright 2871 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology Ventilation (L/min) 40 30 20 10 0 40 20 0 60 PaCO2 (torr) Figure 1 The relationship between minute ventilation (V̇ E ) and arterial carbon dioxide tension (PaCO2 ) in a healthy subject with a constant level of CO2 production (200 mL/min) and a dead space to tidal volume ratio of 0.40; this relationship is termed the metabolic hyperbola. Also shown is a normal normoxic ventilatory response to CO2 of 5 L/min/mmHg, the controller curve (dotted straight line). The set point of the system is determined by the intersection of the two relationships (269). (V D /V T ) ventilation, PaCO2 = K V̇ CO2 V̇E (1 − VD /VT ) For any given values of V D /V T and V̇ CO2 , this equation describes a curve, the metabolic hyperbola, on a plot of V̇ E against PaCO2 . Figure 1 shows the relationship between PaCO2 and V̇ E for a constant V̇ CO2 of 200 mL/min and a constant V D /V T of 0.40 in the absence of normal feedback control between V̇ E and PaCO2 . This relationship implies that at a constant V̇ CO2 , an increase in V̇ A must be accompanied by a reciprocal fall of PaCO2 . In other words, the metabolic hyperbola represents the simple response of the controlled system (the organ of pulmonary gas exchange) to changes in ventilation per se. Increases in V D /V T or V̇ CO2 will produce different hyperbolae, shifted upward and to the right. The sensitivity of the controller system (the entire system of respiratory centers, respiratory neurons and respiratory muscles) is evaluated by forcing an increase in PaCO2 and measuring the consequent increase in V̇ E (213, 269). Increases in PaCO2 to test the sensitivity of the controller system can be achieved with the steady-state method, in which the subject breathes air enriched with different concentrations of CO2 , or with the rebreathing method. The resulting V̇ E PaCO2 relationship, termed the controller curve, is usually described as a straight line (Fig. 1). The greater the sensitivity of the controller system to an increase in V̇ CO2 , the steeper will be the controller curve. Sensitivity to CO2 is reported as the slope of the controller curve (V̇ E per mmHg), and the normal range in healthy adults is 0.5 to 8.0 L/min/mmHg (1.5-5.0 in 80% of subjects) (269). 2872 In a steady state, the values of PaCO2 and V̇ E must simultaneously satisfy the equations of both the controller curve and the metabolic hyperbola. That is, the subject must lie at the intersection between the metabolic hyperbola (controlled system) and the controller curve (controller system); this unique point is the operating point or set point of the system, which specifies the normal resting levels of V̇ E and PaCO2 (269). According to the alveolar-gas equation, an increase in metabolic rate or an increase in V D /V T can theoretically produce an increase in PaCO2 . In reality, a sole increase in V̇ CO2 or a sole increase in V D /V T is never sufficient to constitute a primary mechanism of hypercapnia (306). Substantial increases in either V̇ CO2 or V D /V T are readily compensated for by a normal ventilatory apparatus. Accordingly, an increase in PaCO2 always signifies an abnormality in the mechanical or control components of the respiratory system. An increase in CO2 or V D /V T can, however, aggravate hypercapnia when the respiratory system is already compromised. Patients with chronic obstructive pulmonary disease (COPD) are especially vulnerable to the development of hypercapnia when treated with supplemental oxygen (13, 65, 70, 225). Several mechanisms may contribute to the development of hypercapnia: a decrease in hypoxic ventilatory response consequent to the administration of oxygen; an increase in dead space consequent to worsening of ventilation-perfusion relationships secondary to release of hypoxic vasoconstriction; and the Haldane effect (for any given amount of CO2 bound to hemoglobin, PaCO2 is considerably higher in the presence of a high vs. a low oxygen saturation). Figure 2 provides an example of why some patients develop marked hypercapnia after administration of oxygen and others do not. Clinical disorders producing ventilatory failure Table 1 lists a large number of clinical disorders that can lead to ventilatory failure. The classification is based on anatomical site, extending from conditions that affect the central nervous system to those affecting the neuromuscular apparatus, the airways, or lung tissue. Problems with control of breathing for four of these disorders are discussed later: central alveolar hypoventilation (briefly), neuromuscular disorders, COPD (in greater detail), and the acute respiratory distress syndrome (ARDS). Central alveolar hypoventilation Several structural and functional disorders of the central nervous system can produce hypoventilation and ventilatory failure. Among these is central alveolar hypoventilation, which is caused by a failure of the respiratory centers to provide sufficient activation to the lower motor neurons. Ambulatory patients with central alveolar hypoventilation exhibit hypercapnia despite normal spirometric measurements of lung function. In mechanically ventilated patients, the disorder is suspected when patients develop marked increases in PCO2 despite relatively normal measurements of resistance Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Comprehensive Physiology Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Table 1 Examples of Clinical Disorders that Produce 20 Ventilation (L/min) Ventilatory Failure A 10 B 0 10 30 50 70 PaCO2 (torr) Figure 2 Schematic representation of mechanisms that contribute to variable degrees of hypercapnia in patients with COPD who receive supplemental oxygen. Patient A has a baseline PaCO2 of 30 mmHg (green symbol) and a V D /V T of 0.40 (the lower black metabolic hyperbola). Administration of supplemental oxygen results in a 2-L decrease in minute ventilation (blue symbol; represented as a vertical drop for the sake of simplicity). This patient has a normal CO2 controller response (slope of the red line, 1.0 L/min/mmHg), which brings his minute ventilation back up to the black hyperbola (red arrowhead). Consequently, administration of supplemental oxygen produces an increase in PCO2 of only 1.5 torr. Patient B also has a baseline V D /V T of 0.40, but his PCO2 is 50 mmHg (green symbol). Administration of supplemental oxygen again results in a 2-L decrease in minute ventilation (blue symbol). This patient has a depressed CO2 controller response (slope of the red line, 0.2 L/min/mmHg), and administration of oxygen produces an increase in dead space from 0.40 to 0.60 secondary to worsening of ventilation-perfusion relationships (consequent to release of hypoxic vasoconstriction and the development of CO2 -induced bronchodilation); the patient accordingly moves from the lower black metabolic hyperbola to the upper blue hyperbola. Consequently, administration of supplemental oxygen produces an increase in PCO2 of 15 torr (10fold greater than in Patient A). and elastance and no apparent evidence of lower motor neuron disease (127). Specific features of various hypoventilation syndromes are discussed in detail in other articles of the series. Neuromuscular disorders Respiratory muscle weakness frequently goes undetected in patients with neuromuscular disease until ventilatory failure is precipitated by aspiration pneumonia or cor pulmonale. Diagnosis is delayed because limb muscle weakness prevents patients from exceeding their limited ventilatory capacity (39). A few patients develop severe respiratory muscle weakness despite little or no peripheral muscle weakness (112). Severe weakness of the inspiratory muscles produces a restrictive pattern: decreases in vital capacity, total lung capacity (TLC), and functional residual capacity (FRC), with a relatively normal ratio of forced expiratory volume in one second to forced vital capacity (FEV1 /FVC). Whether expiratory muscles are weak or not, residual volume remains relatively normal. Diffusing capacity, corrected for alveolar volume, is not only normal in patients with respiratory muscle weak- Volume 2, October 2012 A. Central disorders Central nervous system: structural Primarily (upper or lower) neuronal disorders Central (primary) alveolar hypoventilation Cerebrovascular accidents Congenital diseases (Arnold-Chiari II malformation) Degenerative diseases (Parkinson’s disease) Infections (polio virus infection) Spinal cord injury Amyotrophic lateral sclerosis Paraneoplastic disorder (anti-Hu antibodies) Primarily myelin disorders Multiple sclerosis Central nervous system: functional Drugs (narcotics, sedatives, anesthetics) Metabolic alkalosis Resetting of CO2 set point Sleep-related hypoventilation B. Neuromuscular disorders Peripheral nerve Metabolic disorders (diabetes mellitus, porphyria) Vasculitis Guillain-Barre syndrome Neuralgic amyotrophy Trauma Neuromuscular junction Myasthenia gravis Lambert-Eaton syndrome Botulism Tick paralysis Drugs (neuromuscular blocking agents) Muscle disorders Critical illness myopathy Inflammatory/Autoimmune disorders Endocrinopathies Congenital myopathies Drugs (colchicine), electrolyte disturbances C. Mechanical derangements Obstructive diseases COPD Asthma Cystic fibrosis Restrictive diseases Pulmonary parenchyma Idiopathic pulmonary fibrosis and congeners Acute respiratory distress syndrome (ARDS) Pulmonary resection Chest wall deformity Trauma and flail chest Scoliosis Ankylosing spondylitis Thoracoplasty Fibrothorax Abdominal distension Obesity Ascites ness, but it can also be normal in patients with pulmonary fibrosis (135) and is of limited value in differentiating the two conditions. Vital capacity is normal, or only minimally reduced, if respiratory muscle strength is more than 50% of predicted (61). This finding results from the sigmoid shape of the pressure-volume relationship of the respiratory system. When strength is less than 50% of predicted, however, the loss in vital capacity is greater than expected (61,78). The decrease 2873 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning is secondary to the associated decrease in compliance of the chest wall and lungs (78, 172). The latter probably results from fibrosis consequent to recurrent aspirations, arrested lung growth in early-onset abnormalities of the chest wall, and diffuse microatelectasis (306). The decreased chest-wall compliance probably results from stiffening of tendons and ligaments of the rib cage, activity of antagonistic muscles (expiratory muscle activity during inspiration in myotonic dystrophy), thoracic deformities accompanying muscle weakness (kyphoscoliosis in poliomyelitis), fibrotic changes or spasticity of involved muscles, and ankylosis of the costosternal and thoracovertebral joints (306). Patients with respiratory muscle weakness take rapid shallow breaths (172), possibly as a result of afferent signals in weakened respiratory muscles, intrapulmonary receptors, or both (30, 172). PaCO2 may be reduced early in the disease (217), but hypercapnia is likely when respiratory muscle strength falls to 25% of predicted (31). Reduction in strength, however, does not consistently predict alveolar hypoventilation because factors such as elastic load and breathing pattern also contribute (172). Abnormalities in respiratory muscle performance may initially be evident only during sleep (10,293), during immersion in water (176,231) or, in patients who are able to exercise, during physical activity (241). When inspiratory strength and vital capacity are 50% of predicted, hypoventilation can occur with minor upper respiratory tract infections. Patients with neuromuscular diseases, irrespective of the primary pathology (10, 113), commonly develop abnormalities during sleep: frequent arousals, increased Stage 1 sleep, decreased rapid-eye movement sleep, hypoventilation, and hypoxemia (27). Patients with diaphragmatic weakness or diaphragmatic paralysis are at particular risk of developing hypoventilation during rapid-eye movement sleep (27, 242, 254, 293). To decrease this likelihood, the central nervous system can adopt two strategies: phasic recruitment of inspiratory muscles other than the diaphragm during rapideye movement sleep (10) or suppression of rapid-eye movement sleep (10, 27, 293). The failure of all patients to develop these adaptive strategies may explain discrepancies among reports on oxygenation during sleep in patients with isolated diaphragmatic paralysis (147, 254, 293). Chronic obstructive pulmonary disease COPD is estimated to affect more than 200 million people worldwide, and now constitutes the fourth most common cause of death. Suspicion for the presence of COPD is aroused by taking a patient’s history and performing physical examination. The diagnosis, however, hinges largely on spirometric measurements, specifically the demonstration of a forced expiratory volume in 1 s (FEV1 ) below the normal range combined with a decrease in the FEV1 /FVC ratio of less than 70% or so (230). Being defined in this manner, the development of ventilatory failure in COPD is commonly perceived to have resulted from a mechanical impediment, primarily from 2874 Comprehensive Physiology Flow Normal Vmax COPD Vmax Pleural pressure Inspiration Expiration Figure 3 Schematic representation of the isovolume pressure-flow relationship. Patients with COPD exhibit an initial diagonal segment, where increases in pressure produce increases in airflow (the effortdependent region on the left), followed by a flat portion, where increases in pressure do not produce increases in flow (the effortindependent region on the right). Compared with a healthy subject, the slope of the initial diagonal segment is decreased, indicating an increase in airway resistance, and maximum flow is much reduced in the remaining portion as a result of expiratory flow limitation. [Modified, with permission, from Pride and Macklem (206).] an increase in the resistance to air flow during expiration. In reality, the pathophysiology of ventilatory embarrassment and ventilatory failure in COPD involves many factors other than airway resistance. Even when COPD is severe, the increase in resistance in the effort-dependent range is only 5 to 10 cmH2 O/L/s above normal (37) (Fig. 3). Subjects facing inspiratory and expiratory resistances of this magnitude can achieve a normal level of ventilation with very minor adjustments in respiratory motor output (306). Studies using modeling techniques reveal that uncomplicated obstructive disease imposes virtually no stress on the respiratory control system despite reductions in FEV1 to 50% of predicted (306). Even when FEV1 is reduced to 20% of predicted, the increase in respiratory motor output needed to maintain normocapnia is fairly small. The main problem in COPD is not the increase in airway resistance per se, but expiratory airflow limitation that results from decreased tethering of the airways secondary to rupture of elastic fibers (206). At rest, expiratory airflow limitation is present in about 60% of patients with COPD (72). Figure 3 provides a schematic representation of the pressure-flow relationship in COPD (304). In the initial diagonal segment (on the left), the slope of the relationship is less than in healthy subjects, indicating that airway resistance is increased. In this region, airflow in patients with COPD is sensitive to applied pressure during inspiration and also over a finite range of expiration (just as in healthy subjects). Over the remaining portion of the relationship, expiratory flow is much lower than in healthy subjects and it is also independent of applied pressure. This region represents flow limitation, which, by definition, cannot be compensated for by greater expiratory muscle action. Expiratory airflow limitation Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology delays lung emptying, with the result that inspiration begins before the respiratory system has returned to its relaxation volume—a state known as dynamic hyperinflation (206). In COPD, dynamic hyperinflation can also develop or worsen when ventilatory demands are increased. When healthy subjects experience increased ventilatory demands, they increase minute ventilation through increases in both tidal volume and respiratory frequency. At the end of a tidal inspiration, a finite time is required for lung volume to return to passive FRC. The minimum time is determined by maximum expiratory flow (3). When patients with severe COPD and expiratory flow limitation develop tachypnea, they necessarily shorten the time available for expiration. The decrease in expiratory time means that the next inspiration will commence before lung volume has returned to passive FRC, thus producing dynamic hyperinflation (143). Although flow limitation is the main instigator of the dynamic hyperinflation, the increased airway resistance is a contributing factor. The time constant (resistance × compliance) of the normal passive respiratory system is about 0.3 s (3 cmH2 O/L/s × 0.1 L/cmH2 O = 0.3 s). Substantial increases in resistance will impinge on the time needed for expiration. In a patient with a respiratory frequency of 40 breaths per minute and a threefold increase increase in resistance, this factor will contribute to the development of dynamic hyperinflation (309). In patients with severe obstruction, this built-in response can be counterproductive and may exaggerate hypercapnia rather than relieve it (304). Specifically, the development of dynamic hyperinflation decreases the effectiveness of the respiratory system through at least four mechanisms. First, dynamic hyperinflation forces the respiratory muscles to operate on the upper less compliant portion of the pressure-volume curve (206). In this region, greater inspiratory effort is required to achieve increases in tidal volume, reflecting the distinctly nonlinear relationship between the intensity of inspiratory activation and resulting tidal volume (304). Consequently, increases in ventilatory demands that are accompanied by increases in respiratory frequency may lead to a decrease, rather than an increase, in tidal volume. In this way, the ventilatory challenge resembles that typically associated with restrictive lung disease. The decrease in tidal volume may be sufficiently severe as to completely offset the increase in minute ventilation anticipated with the increase in frequency. As tidal volume decreases, V D /V T necessarily increases and thus hypercapnia can accompany the tachypneic response (277). Thus, increased inspiratory muscle activation is highly inefficient from an energetic perspective in meeting increased ventilatory demands that occur in patients with severe COPD (304). Second, dynamic hyperinflation, which develops or worsens in response to tachypnea, leads to an increase in the oxygen cost of breathing because the next inspiration typically commences before the respiratory system has returned to its relaxation volume. Consequently, a threshold load is imposed on the inspiratory muscles, termed auto or intrinsic positive end-expiratory pressure (PEEP) (Fig. 4) (143). Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Third, end-expiratory lung volume is usually determined by the static equilibrium between inwardly directed elastic recoil of the lungs and outwardly directly recoil of the thoracic cage (relaxation volume). The outwardly directed forces help the inspiratory muscles to inflate the lungs. When endexpiratory lung volume lies above 70% of predicted total lung capacity (218), however, thoracic elastic recoil is directed inward and the inspiratory muscles have to work not only against the elastic recoil of the lungs but also against that of the thoracic cage. The fourth mechanism whereby dynamic hyperinflation decreases the effectiveness of the respiratory system in the expulsion of CO2 is through a decrease in the resting length of the diaphragm primarily as a result of a decrease in the length of the zone of apposition (143). The zone of apposition normally constitutes 60% of the diaphragm’s total area, but only 40% in patients with COPD (44). The smaller zone of apposition means that a small portion of the rib cage is exposed to the positive abdominal pressure produced by diaphragmatic contraction, and this further limits the capacity of the diaphragm to produce rib-cage expansion (140, 143). Other mechanisms through which hyperinflation decreases the capacity of the respiratory muscles to generate negative intrathoracic pressure include worsening of the length-tension relationship, decrease in the curvature of the diaphragm, and change in the mechanical arrangement of costal and crural components of the diaphragm (143). In addition to functional weakness secondary to hyperinflation, respiratory muscle weakness in COPD may also result from reductions in specific force (defined as the maximum tetanic force per cross-sectional area)—reported by some (153, 190) but not all investigators (256)—and muscle injury especially in patients with acute-on-chronic COPD (232). The reduced pressure output of the inspiratory muscles in some patients with COPD can be completely explained by muscle shortening consequent to hyperinflation (44, 180, 237, 279). Similowski and co-workers (237) found that some patients with COPD had greater transdiaphragmatic pressure (in response to phrenic nerve stimulation) than did healthy subjects at equivalent lung volumes. The finding suggests that the inspiratory muscles had adapted to hyperinflation. The adaptation is probably secondary to shortening of diaphragmatic sarcomeres (reported in patients with mild-tomoderate COPD (187) and in a hamster model of emphysema (80)), which cause a leftward shift of the length-tension relationship. Whether these adaptations to hyperinflation actually occur in patients with COPD remains highly controversial (46). Hyperinflation is not only detrimental but also protective. First, it optimizes expiratory flow by limiting dynamic airway closure during exhalation. Second, hyperinflation may protect the diaphragm against fatigue insofar as susceptibility to fatigue is greater when a muscle is made to contract at optimum muscle length rather than when it is shortened (88). Third, the increased mechanical load associated with increased hyperinflation and airway obstruction may produce diaphragmatic 2875 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology Onset inspir effort Flow (L/s) Onset inspir flow 2 0 –2 Paw cmH2O 25 15 5 –5 Pes cmH2O 10 0 –10 0 2 4 6 8 10 0 1 2 3 4 Time (s) Figure 4 Differing degrees of intrinsic positive end-expiratory pressure (PEEP) in patients with COPD. Tracings are flow, airway pressure (Paw ), and esophageal pressure (Pes ) in two patients with COPD receiving assisted ventilation with pressure support set at 20 cmH2 O (no external PEEP is applied). The patient on the left had a myocardial infarction complicated by congestive heart failure, and the patient on the right had sepsis. Intrinsic PEEP, estimated as the difference in esophageal pressure between the onset of inspiratory effort (vertical blue line) and the onset of inspiratory flow (vertical red line), was 0.5 cmH2 O in the patient on the left and 10.6 cmH2 O in the patient on the right. This method of estimating intrinsic PEEP is based on the assumption that the change in esophageal pressure reflects the inspiratory muscle pressure required to counterbalance the end-expiratory elastic recoil of the respiratory system. remodeling consisting of fast-to-slow fiber shift (153), where slow fibers are more fatigue resistant than are the fast fibers (143). Patients with COPD have an increased risk of coronary artery disease, and 20% to 30% exhibit chronic heart failure (150). Increased stress on the myocardium during an exacerbation of COPD or an episode of weaning failure could overwhelm an already compromised cardiac reserve and cause acute left-ventricular failure (125,151). An increased respiratory load resulting from interstitial edema secondary to acute left-ventricular failure together with decreased blood flow to the respiratory muscles may markedly impair respiratory muscle performance and reduce the time to task failure (14). Healthy subjects recruit their expiratory muscles when coping with increased ventilatory demands. Activity of the expiratory muscles helps inspiration by moving end-expiratory lung volume down to below passive FRC, and thus leads to the storage of elastic energy in the respiratory system. At the completion of exhalation, the transversus abdominis relaxes and the release of stored energy causes intrathoracic pressure to fall and inspiratory flow to start before the diaphragm has begun to contract (143). In this way, expiratory muscle activity during expiration can assist the inspiratory muscles 2876 in expanding the respiratory system during inspiration. The effectiveness of the expiratory muscles in sparing inspiratory muscle work can be substantial. Phasic activation of the expiratory muscles of the order of 10% of maximum can produce an increase in tidal volume equivalent to that achieved by a doubling of inspiratory muscle activity (305). Recruitment of the expiratory muscles is probably part of the constrained response of the respiratory centers to increased ventilatory demands (143). In patients with COPD who have expiratory flow limitation, the associated dynamic airway collapse makes it impossible for the expiratory muscles to increase expiratory flow, and thus these patients are unable to lower endexpiratory lung volume below passive FRC. Consequently, the entire burden of breathing must be borne by the inspiratory muscles. The level of PaCO2 in patients with respiratory disorders is usually maintained within the normal range until the disease has reached a very advanced stage (143). The preservation of ventilation despite an increase in mechanical load is achieved through greater respiratory muscle recruitment than in healthy subjects (62). That is, load compensation must be taking place (309). Immediate (first breath) load compensation to maintain normocapnia occurs only in wakefulness; during sleep, Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology application of resistive, or elastic loads does not produce immediate load compensation, with the result that tidal volume falls and PaCO2 increases (114, 296). Evidence for the occurrence of load compensation has led to considerable interest in studying the response to externally applied loads. When resistive loads are applied in healthy subjects, the steady-state response typically consists of slow deep breathing. In contrast, patients with COPD exhibit an increase in respiratory frequency, and when they develop progressive ventilatory failure, tidal volume becomes less than normal (268, 277). The discrepancy between the response of healthy subjects to externally applied loads and patients with COPD results from the fact that external resistive loads do not accurately simulate the mechanical load of COPD. As already discussed, an increase in airway resistance is only one component of the mechanical challenge in COPD (309). Patients also face an elastic load as a result of breathing on the upper flat portion of the pressure-volume curve, muscle weakness consequent to hyperinflation, and an inspiratory threshold load consequent to the auto-PEEP imposed by dynamic hyperinflation (143). In addition, patients with COPD are exposed to numerous other stimuli including airway inflammation, parenchymal inflammation, hormonal and electrolyte disturbances, high ventilatory requirements (increase in dead space, metabolic rate, or both), and pulmonary vascular disease. No external device that simulates this complex combination of mechanical loads and other perturbations has been developed and tested. That patients breathe rapidly rather than slowly suggests that the influence of increased resistance per se on the breathing pattern is fairly small, and less than the effect of other stimuli that promote tachypnea (268). In patients with COPD, as in many patients with ventilatory failure (306), the combination of an increased load on the respiratory muscles and their decreased capacity to generate flow and pressure (143) raises the question of whether contractile fatigue might contribute to the development of ventilatory failure and hypercapnia. Contractile fatigue can be short lasting or long lasting (143). Short-lasting fatigue results from accumulation of inorganic phosphate, failure of the membrane electrical potential to propagate beyond T-tubules, and to a much lesser extent intramuscular acidosis (143). Shortlasting fatigue appears to have a protective function because it can prevent injury to the sarcolemma caused by forceful muscle contractions (318). Long-lasting fatigue (139) is consistent with the development of, and recovery from, muscle injury (122, 318). The topic of respiratory muscle fatigue is complex and the interested reader is referred to a detailed review of the topic (143). Modest intermittent resistive loading (2 h a day over 4 days) can disrupt sarcomeres and the sarcolemma of diaphragmatic fibers in dogs (319). This mechanism may also occur in patients. In a study of 21 patients with FEV1 ranging from 16% to 122% of predicted, the proportion of abnormal fibers in the diaphragm was correlated with airflow obstruction (160). Abnormalities consisted of myofibers with internally located nuclei, lipofuscin pigmentation (sign of Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning oxidative stress), small angulated fibers, inflammation, and necrosis; these abnormalities occupied 4% to 34% of the diaphragm (160). Sarcomere disruption has also been reported in 18 patients with COPD (188). The density and area of disruptions in the patients was twice that seen in 11 control subjects, and it was correlated with FEV1 and hyperinflation (188). Diaphragmatic damage has been reported in patients dying of COPD (232) and also in those dying of status asthmaticus (67) and in infants dying of the sudden infant death syndrome (236). Circumstantial evidence suggestive of contractile fatigue in patients experiencing respiratory distress has also been reported (32, 48, 71, 101, 127). One of the major puzzles in COPD is why do some patients develop hypercapnia and others do not (269). A variety of factors have been put forward, including respiratory muscle function (17), configuration of the diaphragm (200), respiratory mechanics (17), abnormalities in ventilation-perfusion relationships and increased dead-space ventilation (13, 287). The wide scatter together with the fact that the normal response to added loads in conscious humans is normocapnic suggests that some abnormality in the control of breathing contributes to the hypercapnia. It has been suggested that the development of hypercapnia results from innate or premorbid differences in the chemical control of breathing. The hypothesis is that patients who exhibit hypercapnia have depressed chemical responsiveness even before they develop COPD (130, 175). This proposition, however, is impossible to test once hypercapnia has become established because the presence of a mechanical abnormality inevitably alters the ventilatory response to CO2 and hypoxia; moreover, chronic hypercapnia and hypoxia can also depress chemical responsiveness even in the absence of mechanical abnormalities. It had been thought that patients presenting with a blue-bloater profile have hypercapnia as a result of decreased respiratory motor output and that patients presenting with a pink-puffer profile maintain eucapnia through increases in respiratory motor output. Repeated attempts to generate data to support this simplistic picture have been to no avail. Indeed, even when patients develop acute increases in PaCO2 as a result of severe progressive respiratory failure they develop increases in respiratory drive (173, 227, 277). In addition to its detrimental effects, hypercapnia can be advantageous in patients with abnormal respiratory mechanics because it can make breathing more efficient. When alveolar PCO2 is increased, more CO2 is expelled for a given exhaled volume. Thus, the oxygen cost to the respiratory muscles in excreting CO2 is more efficient in hypercapnic patients. In patients experiencing an acute exacerbation of COPD, a further mechanism that may contribute to the development of hypercapnia is impaired voluntary activation of the diaphragm. Topeli and co-workers (280) investigated this possibility using the twitch-interpolation technique in 15 patients with stable COPD. When the phrenic nerves are stimulated during a voluntary contraction, the increase in transdiaphragmatic pressure reflects the proportion of muscle 2877 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning result of hyperinflation), a threshold load (as a result of autoPEEP), and functional weakness of the inspiratory muscles (as a result of hyperinflation) with only a small increase in inspiratory airway obstruction. 15 0 Transdiaphragmatic pressure (cmH2O) Comprehensive Physiology 100 Acute respiratory distress syndrome 50 0 1s Figure 5 Tracings of transdiaphragmatic pressure (Pdi ) during bilat- eral phrenic nerve stimulation in a patient with COPD. During a forceful Mueller maneuver, stimulation produced a superimposed twitch pressure (arrow). On the right is a twitch pressure achieved by stimulation during resting breathing just after the Mueller maneuver. The ratio of the amplitude of superimposed twitch pressure to resting twitch pressure (expressed as a percentage) measures the extent that muscle is not recruited by the central nervous system during the Mueller maneuver. The extent of muscle recruitment is usually expressed as the voluntary activation index, which is calculated as: 100 minus the superimposed twitch pressure to resting twitch pressure ratio. In the displayed example, the amplitude of the superimposed twitch pressure is 19% of the amplitude of resting twitch pressure, yielding a voluntary activation index of 81%; if the superimposed stimulus had evoked no increase in pressure, the activation index would have been 100% (143). Of note, a twitch pressure recorded shortly after forceful contractions (potentiated twitch) is greater than a twitch pressure recorded after 15 to 20 min of rest (nonpotentiated twitch) (140). Nonpotentiated twitches are usually used to quantify diaphragmatic force output at rest, and changes in force following fatiguing protocols (140, 143). ARDS is a form of noncardiogenic pulmonary edema that results from severe acute alveolar injury. The main physiological features are marked increase in respiratory elastance and resistance, a decrease in the number of functional lung units, decrease in FRC, severe hypoxemia secondary to intrapulmonary shunt, and pulmonary hypertension. Despite the overall reduction in FRC (79), patients with ARDS commonly exhibit dynamic hyperinflation and intrinsic PEEP (137) as a result of loss of lung recoil, increased peripheral airway resistance, or both. Breathing at a low lung volume also promotes airway closure and gas trapping, which is further aggravated by the increase in closing volume that results with alveolar edema and the deficiency of surfactant in ARDS (137). While the abnormalities in respiratory mechanics, gas exchange and pulmonary vascular resistance impose stresses on the respiratory controller, systematic studies of the effect of ARDS on respiratory controller function have not been conducted. Ventilator Support Goal of mechanical ventilation not recruited by voluntary activation (Fig. 5). Voluntary activation was higher in six patients who had hypercapnia than in nine patients with normocapnia, 95% versus 89%; the value in normocapnic patients was equivalent to that reported in healthy subjects (88%; (5)). The extent of voluntary activation of the diaphragm and PaCO2 were both positively correlated with inspiratory muscle load (280). The results suggest that, contrary to expectations, development of hypercapnia is not the result of impaired voluntary activation of the diaphragm and that patients with a high load may have learned to fully activate their diaphragm on an intermittent basis (280). The latter explanation is supported by data that suggest that suprapontine compensatory mechanisms are active in defending ventilation in awake human subjects challenged with an inspiratory load (212). The ability to mount an increase in voluntary drive to the diaphragm may be especially important during an acute exacerbation of COPD. Moreover, patients with COPD who are capable of lowering PaCO2 upon acute voluntary increases in ventilation exhibit significant correction of chronic hypercapnia with the chronic administration of respiratory stimulants such as medroxyprogesterone or acetazolamide (244-246). In summary, the pathophysiology of COPD presents a curious paradox in that the name of the disease focuses on airway obstruction and the diagnosis is based on decreased airflow during expiration, yet the mechanical consequences occur during inspiration and consist of an elastic load (as a 2878 Many patients who require mechanical ventilation have relatively normal arterial blood gases but have clinical signs of increased work of breathing: nasal flaring, vigorous activity of the sternomastoid muscles, tracheal tug, recession of the suprasternal, supraclavicular and intercostal spaces, paradoxical motion of the abdomen, and pulsus paradoxus (144). The oxygen cost of breathing of patients in acute respiratory distress is increased to as much as 50% of total oxygen consumption (85). By decreasing respiratory work, mechanical ventilation allows precious oxygen stores to be rerouted from the respiratory muscles to other vulnerable tissue beds. The institution of mechanical ventilation is most commonly based on a physician’s clinical gestalt, formed through assessing a patient’s signs and symptoms, rather than because a patient satisfies a certain set of criteria on a checklist. Indeed, the most common—and honest—reason that mechanical ventilation is instituted is a tautology: a physician thinks that “the patient looks like he (or she) needs to be placed on the ventilator” (144). Modes of mechanical ventilation The most commonly used ventilator modes are assist-control ventilation, pressure support, and intermittent mandatory ventilation (IMV). Figure 6 shows the typical appearance of tidal volume and airway pressure in patients receiving ventilator assistance with each mode. Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning VT, L ACV Paw (cmH2O) Printer Name: Yet to Come SIMV PSV 1.0 1.0 1.5 0.5 0.5 0.75 0.0 0.0 0.0 35 35 15 15 –5 –5 30 0 4 8 12 20 10 0 0 2 Time (s) 4 6 Time (s) 8 0 4 8 12 Time (s) Figure 6 Recordings of tidal volume (V T ) and airway pressure (Paw ) in patients receiving ventilator assistance with assist-control ventilation (ACV), synchronized intermittent mandatory ventilation (SIMV), and pressure support ventilation (PSV). The ordinates and abscissae differ from one panel to the next because the tracings were recorded in three different patients. See text for details. Assist-control ventilation With assist-control ventilation, the ventilator delivers a breath either when triggered by the patient’s inspiratory effort (pressure or flow triggered) or, independently, if such an effort does not occur within a preselected time period (162). When the patient’s inspiratory effort successfully triggers the machine, the total respiratory rate can exceed the preset rate. All breaths are delivered under positive pressure by the machine. The breath delivered by the ventilator can have a predetermined volume (volume-cycled form of assist-control ventilation) or a predetermined inflation pressure that is applied for a predetermined inflation time (pressure-control form of assistcontrol ventilation). When delivering the breath, the ventilator adjusts pressure output such that total applied pressure (Paw + Pmus ) remains in excess of elastic recoil until the target volume (volume-cycled ventilation) or target inflation time (pressurecontrol ventilation) is reached (308). With this mode, there is no inherent coupling between the end of a patient’s inspiratory effort and the onset of expiratory flow. If the patient stops making an inspiratory effort before a set tidal volume or the set target inflation time is delivered, the ventilator simply continues to deliver gas until the target volume or the target inflation time is reached. Conversely, when the set tidal volume (or the set inflation time) is reached before the end of a patient’s inspiratory effort, the ventilator will cycle off while patient effort is ongoing and this may cause expiratory flow to begin prematurely (308). By design, the volume of gas delivered during the volumecycled form of assist-control ventilation is independent of patient effort, except where the set tidal volume is less than that which a patient would generate based on his or her own level of effort. The greater the inspiratory effort made by the patient, the lower will be the airway pressure during the volume-cycled form of assist-control ventilation, such that the overall airway pressure on the ventilator monitor bears an inverse relationship to patient-generated pressure. During the Volume 2, October 2012 pressure-control form of assist-control ventilation, the more the patient contributes to inspiration, the less volume of gas is provided directly by the machine, and ventilator-generated volume bears an inverse relationship to patient-generated effort. The amount of active work performed by a patient ventilated in volume-cycled assist-control is critically dependent on the trigger sensitivity and inspiratory flow settings. Even when these settings are selected appropriately, patients actively perform about a third of the work performed by the ventilator during passive conditions (165). When employing the volume-cycled form of assist-control ventilation, the clinician selects some tidal volume and an inspiratory flow; indirectly, the clinician is also selecting a mechanical inspiratory time despite having no idea of the duration of the patient’s own neural inspiratory time. Likewise, when employing the pressure-cycled form of assist-control ventilation, the clinician selects a mechanical inspiratory time despite having no idea of the duration of the patient’s own neural inspiratory time. Given that neural inspiratory time varies widely in duration both within patients and among patients, it is very unlikely that machine inspiratory time will coincide with that of a patient. Pressure-support ventilation Pressure-support ventilation is a patient-triggered, pressuretargeted, and flow-cycled mode of mechanical ventilation (33). The physician sets a level of pressure that assists every spontaneous effort, and the patient can alter respiratory frequency, inspiratory time, and tidal volume. Tidal volume is determined by the pressure setting, the patient’s effort and pulmonary mechanics, in contrast to volume-targeted ventilation such as volume-cycled assist-control ventilation, where a guaranteed volume is delivered. With volume-targeted ventilation, the inspiratory flow setting is a crucial determinant of patient work. There is no flow setting with pressure 2879 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology 2 Flow (L/s) PS 5 PS 0 1 0 –1 Paw (cmH2O) –2 30 20 10 0 Pes (cmH2O) –10 10 0 –10 –20 2 PS 10 Flow (L/s) 1 PS 20 0 –1 Paw (cmH2O) –2 30 20 10 0 Pes (cmH2O) –10 10 0 –10 –20 0 2 4 6 8 10 0 2 4 6 8 10 Time (s) Figure 7 Increase in elastic recoil and expiratory muscle recruitment during pressure support. Flow (inspiration directed upward), airway pressure (Paw ), and esophageal pressure (Pes ) in a patient receiving pressure-support of 0 (upper left panel), 5 cmH2 O (upper right panel), 10 cmH2 O (lower left panel), and 20 cmH2 O (lower right panel). As pressure support was increased from 0 to 20 cmH2 O, respiratory frequency decreased from 20 to 13 breaths/min and tidal swings in esophageal pressure decreased from 20 to 10 cmH2 O. End-inspiratory esophageal pressure returned to preinspiratory values at pressure support of 0, whereas the end-inspiratory value was higher than preinspiratory esophageal pressure at pressure support of 20, suggesting recruitment of expiratory muscles and increased elastic recoil. The increase in airway pressure above the preset level (best seen at pressure support of 5 and 10) probably resulted from relaxation of the inspiratory muscles while mechanical inflation was still active; the extent of expiratory muscle contribution to the increase in airway pressure cannot be determined in the absence of measurements of chest-wall recoil pressure. The flow signal at the start of inhalation demonstrates an initial spike that increases as the level of support increases; this phenomenon reflects the need for higher flows to achieve the higher target airway pressures. support, although the initial peak flow determines the speed of pressurization and the initial pressure ramp profile. Pressure support can be very effective in decreasing the work of inspiration. The degree of inspiratory muscle unloading, however, is variable, with a coefficient of variation of up to 96% among patients (129). The level of pressure delivered by the ventilator is usually adjusted in accordance with changes in the patient’s respiratory frequency. The frequency, how- 2880 ever, which signals a satisfactory level of respiratory muscle rest has never been well defined, and recommendations range from 16 to 30 breaths per minute (129). Cycling to exhalation is triggered by a decrease in inspiratory flow to a preset level, such as 5 L/min or 25% of peak inspiratory flow, depending on the manufacturer’s algorithm. The algorithm for “cycling-off” of mechanical inflation can cause problems in patients with obstructive airway diseases, Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology Flow (L/s) Paw (cmH2O) –2 40 Transversus abdominis EMG (arbitrary units) –10 10 10 3 20 Time (s) Figure 8 Recordings of flow, airway pressure (Paw ), and transversus abdominis electromyography (EMG) in a critically ill patient with COPD receiving pressure support of 20 cmH2 O. The onset of expiratory muscle activity (vertical dotted line) occurred when mechanical inflation was only partly completed. [From Parthasarathy S, Jubran A, Tobin MJ. Cycling of inspiratory- and expiratory-muscle groups with the ventilator in airflow limitation. Am J Respir Crit Care Med 1998; 158: 1471-1478 (192).] because increases in resistance and compliance produce a slow time constant (of the respiratory system) (Fig. 7). The longer time needed for flow to fall to the threshold value can cause mechanical inflation to persist into neural expiration. In 12 patients with COPD receiving pressure support of 20 cmH2 O, Jubran et al. (129) found that five recruited their expiratory muscles while the machine was still inflating the thorax (Fig. 8). The patients who recruited their expiratory muscles during mechanical inflation had an average time constant of 0.54 s, as compared with an average of 0.38 s in the patients who did not exhibit expiratory muscle activity. The persistence of mechanical inflation into neural expiration is very uncomfortable, as well recognized with use of inverse-ratio ventilation (where, contrary to the normal pattern, inflation lasts longer than exhalation) (7). Intermittent mandatory ventilation With IMV, the patient receives periodic positive-pressure breaths from the ventilator at a preset flow, volume, and frequency, but the patient can also breathe spontaneously between these mandatory breaths (221). A problem not raised at the time IMV was introduced is the ability of a patient to adapt to the intermittent nature of ventilator assistance. It had been assumed that the degree of respiratory muscle rest achieved by IMV would be proportional to the number of mandatory breaths delivered. Studies, however, have demonstrated that Volume 2, October 2012 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 2 0 9:35 that is not the case; instead, inspiratory effort is equivalent for spontaneous and assisted breaths during IMV (117, 166). Indeed, the tension-time index (the product of two fractions: [mean pressure per breath/maximum inspiratory pressure] × [fractional inspiratory time, T I /T TOT ]) for both the spontaneous and assisted breaths was above the threshold associated with task failure of the respiratory muscles at IMV rates of 14 breaths per minute or less (166). Similarly, at a moderate level of machine assistance (where the ventilator accounts for 20%-50% of total ventilation), Imsand et al. (117) reported that electromyographic activity of the diaphragm and sternomastoid muscles was equivalent for assisted and the adjacent unassisted breaths. That inspiratory muscle activation is equivalent during assisted and unassisted breaths has led to the notion that activation is “preprogrammed” before the onset of the breath and that it is not altered by inter-breath changes in the level of assistance. Younes (312) considers this explanation untenable because several investigators have revealed immediate changes in inspiratory output with changes in ventilator settings (such as with changes in inspiratory flow). It is more likely, he argues (312), that the lack of differences in electrical activation between assisted and unassisted breaths in the study of Imsand et al. (117) were related to factors that minimized the development of responses. For example, differences in flow between assisted and unassisted breaths in IMV are not always large (98). All of the patients in the study of Imsand et al. (117) had severe COPD, and reflexes mediated by mechanoreceptors are attenuated in such patients (202,282,314). Moreover, patients with COPD have dynamic hyperinflation and a substantial proportion of neural inspiratory time will have elapsed before the commencement of inspiratory flow. Under these conditions, there is no reason to expect any difference between neural output in assisted and unassisted breaths because in both cases virtually all of neural inspiratory time will take place without receiving ventilator assistance (312). The studies on IMV emphasize how alterations in respiratory load from one breath to the next are not sufficient to arouse reflex compensation. In the past, it had been believed that an increase in the mechanical load produced reflex increases in respiratory drive (41). Despite extensive research, no load-related source of respiratory drive has been identified and it is thought that none exists (309). An additional finding pertinent to this issue is that occlusion of the airway during inspiration in anesthetized animals does not produce an increase in inspiratory muscle activity during the first occluded breath other than what can be explained by prolongation of inspiration (secondary to loss of vagal feedback) (309). As such, relief of respiratory distress by mechanical ventilation is unlikely to be secondary to simple unloading of respiratory muscles. It is more likely that relief is related to a decrease in drive achieved by other means, such as improvement in gas exchange, improvement in blood pressure (in hemodynamically compromised patients), or decrease in metabolic rate secondary to relief of anxiety and agitation (313). 2881 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning Control of breathing during mechanical ventilation The respiratory control system consists of an automatic (metabolic) control system that integrates peripheral information in the brainstem and a behavioral or voluntary controller in supramedullary and cortical structures (161, 286). In healthy subjects, the automatic drive to breathe has three main sources (313). First, chemical drive, which is related to changes in arterial PO2 and to arterial and brain PCO2 and pH, and is mediated by peripheral and central chemoreceptors. The second is metabolic drive, which is related to the metabolic rate and mediated by unknown mechanisms. Third is wakefulness drive, which is manifested through its disappearance during sleep. Metabolic rate does not appear to be sufficiently high to produce oscillation of the respiratory centers during sleep; instead, respiratory rhythm is almost completely dependent on chemical factors (PaCO2 , pH, PaO2 ) (313). Consequently, a minor decrease in PaCO2 in the presence of normoxia can produce central apnea during sleep (243) and anesthesia (86). In addition to the automatic controller, the voluntary controller (forebrain) transmits signals to the respiratory system along neural pathways, some of which can bypass the automatic metabolic controller in the brainstem (58,185,186,216). These observations raise two points. First, within some range, awake human subjects can develop a wide variety of breathing patterns at will. Second, when mechanically ventilated, the manner in which a patient responds to changes in ventilator settings is heavily influenced by how the respiratory control system responds in different circumstances, including state of wakefulness or sleep. Wakefulness drive and behavioral factors influencing ventilated patients The pattern of breathing during hypocapnia highlights the importance of cortical-subcortical interactions in the control of ventilation. Hypocapnia induces recurrent apneas during sleep and anesthesia (86, 243), but it fails to induce apneas during wakefulness (56, 207, 235, 311). These observations provide circumstantial evidence for the existence of a ventilatory oscillator that is sensitive to PCO2 (83) and for a “wakefulness drive to breathe.” The suprapontine origin of the wakefulness stimulus is suggested by its disappearance in patients with forebrain lesions (201). The magnitude of the wakefulness stimulus is quite variable among healthy subjects (207). Various forms of mental activity can alter the overall output of the respiratory controller and can influence breath-tobreath variability of breathing pattern (161, 275). In general, mental activity causes greater alterations of breath-to-breath variability in tidal volume and expiratory time than in inspiratory time (161). The behavioral response to a given perturbation is also highly varied: the same mechanical stimulus may elicit respiratory acceleration in some subjects and respiratory slowing in others (15). 2882 Printer Name: Yet to Come Comprehensive Physiology Behavioral responses to mechanical perturbations are important determinants of patient-ventilator interactions. Changes in the mechanical state of the respiratory system during mechanical ventilation are promptly sensed by mechanoreceptors and relayed to higher brain centers. Alterations in lung volume, flow, and airway pressure are readily perceived by awake patients and may evoke behavioral respiratory responses, with an intention of preserving a tidal volume to which one is accustomed, attempting to minimize unpleasant sensations, purposeless panic, and the startle response—in short, to enhance comfort (313). Chemical control of breathing during mechanical ventilation Changes in PaO2 , PaCO2 , and pH influence respiratory motor output through altered activity of chemoreceptors (54). In turn, changes in respiratory motor output produce alterations in blood gas tensions. In general, the chemical feedback system tends to moderate the changes in blood gas tensions that would otherwise occur as a result of changes in metabolic rate or the gas-exchanging properties of the respiratory system (308). Changes in PaO2 are sensed by the peripheral chemoreceptors, located close to the bifurcation of the common carotid arteries (54), and also in the rostral ventrolateral medulla, which responds by increasing ventilation and sympathetic efferent activity (55, 184, 258). The peripheral chemoreceptors respond to reductions in PaO2 (but not oxygen saturation) and arterial pH, and an increase in PaCO2 —the latter probably mediated via intracellular acidification. The peripheral chemoreceptors increase ventilation much more rapidly than the central chemoreceptors; a lag equal to the circulation time from the lungs to the carotid body elapses before a change in PO2 at the lungs begins to influence breathing, and the half-time response is about 10 s (93). The time constant of the central chemoreceptors is 75 s (124). Thus, the carotid bodies offer faster fine tuning of respiratory control than the central chemoreceptors. Recent research emphasizes the role of the retrotrapezoid nucleus, located in close proximity to the ventral surface of the medulla oblongata, which appears to function generally along the lines previously suggested for the hypothetical ventral surface chemoreceptors (104). This nucleus has a dual function: to drive respiration and to stabilize PCO2 . Neuroanatomical and physiologic findings from recent research have elucidated how sensory input from the peripheral chemoreceptors influence the gain of the central chemoreceptors in response to changes in central CO2 (24, 104). The ventilatory response to PaO2 is hyperbolic, being almost flat in the high PaO2 range and the slope increases progressively as PaO2 decreases (313). The aortic bodies play a minor role in modulating spontaneous respiratory activity, although they have a discernible effect when their gain is increased by hypercapnia (292). In conscious healthy humans, resting ventilation falls about 15% in the absence of carotid body activity. The precise Volume 2, October 2012 P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology role of the carotid bodies, however, during natural breathing is uncertain because hypoxia is rare in healthy subjects and people survive with few problems when the carotid bodies are removed (158, 289). Peripheral chemoreceptors are likely to be more important in hypoxic patients, as demonstrated by worsening hypercapnia when patients with respiratory failure breathe a high concentration of oxygen (13,65,70,225). There is a multiplicative interaction between PCO2 and PO2 at the carotid bodies, in that the sensitivity to hypoxia is greater in the presence of hypercapnia and almost disappears during hypocapnia (89). An increase in PaCO2 and/or hydrogen ion (with or without hypercapnia) results in an increase in the respiratory motor output (313). The response is mediated both by peripheral and central chemoreceptors (54). Studies using carotid body denervation in unanesthetized animals show that the slope of the steady-state ventilatory response to inhaled CO2 is determined by a 60% to 80% contribution from central chemoreception and 20% to 40% contribution from peripheral chemoreception (63). The ventilatory response to CO2 in healthy subjects exhibits a very wide range, 0.47 to 8.16 L/min/mmHg (110,118,214), although approximately 80% of subjects have a response between 1.5 and 5 L/min/mmHg (118). The ventilatory response to CO2 is enhanced in the presence of hypoxia and/or metabolic acidosis (313). Alterations in ventilatory demand are met through a combination of alterations in tidal volume and respiratory frequency. Hey et al. (107) compared the responses to hypoxia, hypercapnia, metabolic acidosis, elevated body temperature, and exercise in healthy subjects. The responses to pyrexia differed from those to other stimuli in that they were primarily tachypneic in nature. For all other stimuli, increases in ventilatory demand were met by initial increases in tidal volume. Respiratory frequency changed little until a much higher level of respiratory stimulation. Typically a fivefold increase in minute ventilation (from 8 to 40 L/min) is accomplished through a threefold increase in tidal volume (for example, from 0.65 to 2.0 L) and only a 50% to 60% increase in respiratory frequency (307). Greater increases in ventilatory demand are achieved mainly through increases in respiratory frequency rather than tidal volume. This pattern of response should be borne in mind when employing different ventilator modes. With assist-control, if an increase in ventilatory demand causes a patient to predominantly increase effort per breath, as opposed to respiratory frequency, the ventilator will be unresponsive: tidal volume is pre-set with this mode, and an increase in patient effort per breath will not produce an increase in minute ventilation (313). With pressure support, an increase in patient effort per breath will result in increases in tidal volume (33). In contrast, when an increase in ventilatory demand causes a patient to predominantly increase respiratory frequency, minute ventilation will increase similarly with both ventilator modes provided the changes in a patient’s true neural respiratory rate are reflected faithfully in the ventilator rate. Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning The aforementioned responses to hypercapnia and hypoxemia occur in mechanically ventilated patients irrespective of whether they are awake or asleep (313). Responses to hypocapnia, however, occur only during sleep (and anesthesia) (86, 243) and are not present during wakefulness (56, 207, 235, 311) (see below). As already stated, hyperoxia can decrease minute ventilation during wakefulness in hypoxic patients (13, 65, 70, 225). Control of breathing during hypocapnia Patients receiving mechanical ventilation are commonly exposed to levels of PaCO2 that extend well into the hypocapnic range. The ventilatory response to CO2 below resting PCO2 has been the subject of considerable interest for several decades, and it is also particularly important in the management of ventilated patients (54, 63). The behavior of the respiratory controller in the presence of hypocapnia differs between wakefulness and sleep, and thus these states will be discussed separately. Control of breathing during hypocapnia while awake Based on limited data, it was widely believed in the past that the ventilatory response to CO2 was discontinuous, and made up of two segments. Above eupnea, the response was known to be steep, producing a linear segment. Below eupnea, the response was viewed as gradually decreasing and then essentially flat. At the lowest PCO2 , a finite level of rhythmic ventilation is still observed in awake subjects, which is related to the wakefulness drive to breathe (313). The terms “hockey-stick” and “dogleg” were used to describe the configuration of the two segments, and the intersection was viewed as the normal resting set point (90) (Fig. 9). 40 30 Minute ventilation (L/min) P1: OTA/XYZ 20 10 0 20 30 40 50 PACO2 (mmHg) Figure 9 Schematic representation of the “dogleg” or “hockey-stick” configuration of the ventilatory response to CO2 during wakefulness. At alveolar carbon dioxide tension (PACO2 ) levels above eupnea, minute ventilation increases linearly in proportion to increases in PACO2 . At PACO2 levels below eupnea, the ventilatory response gradually decreases and becomes essentially flat. [Based on data presented by Nielsen M, Smith H. Studies on the regulation of respiration in acute hypoxia. Acta Physiol Scand 1952; 24: 293-313 (182).] 2883 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning Change in frequency (b/m) dP/dt (cmH2O.s) Among investigators who argued for the presence of a discontinuous “hockey-stick” configuration, some estimated the apneic threshold by extrapolating the hypercapnic ventilatory response to the zero V̇E intercept (54). Such extrapolation, however, is highly uncertain because of the unknown shape of the CO2 response near and below eupnea (63, 132). Other proponents of the “hockey-stick” configuration employed alveolar hypoxia to induce hypocapnia (107, 182)—a highly problematic approach. The presence of a “hockey-stick” configuration has been challenged by several investigators (196, 207, 313). Awake subjects exhibit CO2 responsiveness over a wide range, extending down to PaCO2 levels in the low 20s. At the lowest PCO2 , there remains a finite rhythmic output, and the magnitude of this “minimum” output varies considerably among subjects (207,313). In 16 healthy awake subjects, Patrick et al. (196) used assist-control ventilation to induce hypocapnia; hyperoxia was added to avoid the problem with the older methodology. All subjects continued rhythmic breathing despite high tidal volumes and severe hypocapnia (alveolar PCO2 of approximately 25 mmHg). As inspired CO2 was increased in steps to increase end-tidal carbon dioxide tension (PET CO2 ) from about 25 mmHg to about 40 mmHg, respiratory motor output (quantified as muscle pressure) increased progressively and the slope of the motor response increased. In contrast, the response of respiratory frequency to increases in PCO2 was very weak or absent in the hypocapnic range, and increased significantly only above 36 mmHg (196) (Fig. 10). These observations have particular relevance in the management of ventilated patients. Contrary to common assumptions, respiratory frequency did not fall as a result of negative chemical feedback: decreases in PCO2 down to 25 mmHg had no effect on respiratory frequency (196). 12 8 4 0 1 0 26 28 30 32 34 36 38 40 42 PET CO2 (mmHg) Figure 10 Contrasting response of respiratory motor output and respiratory frequency over a 15 mmHg variation in PCO2 . Average responses of change in respiratory motor output, quantified as change in pressure over time (dP/dt), and change in respiratory frequency in response to changes in end-tidal carbon dioxide tension (PET CO2 ) from about 26 to 41 mmHg. Respiratory motor output increased progressively in response to increases in PET CO2 , even at hypocapnic levels, whereas the response of respiratory frequency to change in PET CO2 was very weak or absent in the hypocapnic range and increased significantly only when PET CO2 exceeded 36 mmHg. [Adapted, with permission, from Patrick et al. (196).] 2884 Printer Name: Yet to Come Comprehensive Physiology In further research from the same group, Puddy et al. (207) studied 18 healthy awake subjects receiving assist-control ventilation. Ventilator tidal volume was increased in steps from the minimum tolerable level up to 80% of the subject’s inspiratory capacity or the ventilator’s maximum tidal volume. Increases in tidal volume were accompanied by little change in respiratory frequency (Fig. 11). The decrease in respiratory frequency was statistically significant but very small relative to the increase in tidal volume, approximately 12% versus 100%; consequently, ventilation increased substantially and PET CO2 dropped precipitously (Fig. 11). The findings of Puddy et al. (207) and Patrick et al. (196) have been corroborated by other investigators. Lofaso et al. (155), Morrell et al. (174), and Scheid et al. (229) observed no significant decrease in respiratory frequency with the addition of pressure support of 10 cmH2 O in awake subjects. Also using pressure support of approximately 10 cmH2 O, Georgopoulos et al. (95) found that respiratory frequency remained relatively stable over an even broader range of PET CO2 , 23 to 45 mmHg; frequency increased significantly only when PET CO2 approached 50 mmHg. These observations (95, 155, 174, 196, 207, 229) have particular relevance in the management of ventilated patients. Most patients are ventilated with some assisted mode, where the patient’s respiratory center output determines the rate of ventilator triggering. The delivered volume is largely preset, either by a selected volume [assist control (162), IMV (221)] or by a set level of pressure [pressure support (33)]. If the volume is excessive in relation to a patient’s CO2 stores, contrary to the assumption of many clinicians, respiratory frequency will not fall as a result of negative chemical feedback. Hypocapnia will follow and the associated respiratory alkalemia can cause cardiac arrhythmias and seizures (16,133,219). Because tidal volume is preset by the ventilator, a decrease in respiratory frequency becomes the only means of guarding against hypocapnia. Many clinicians assume that a fall in PaCO2 consequent to an increase in delivered tidal volume will lead to a fall in respiratory frequency as a result of negative chemical feedback. An additional factor of considerable clinical importance is the ventilatory response to hypoxia during acute stable hypocapnia, which has been investigated by Corne et al. (52). Stable levels of PET CO2 below eucapnia of 6 mmHg (mild hypocapnia) and 12 mmHg (moderate hypocapnia) were achieved in eight healthy subjects using volume-cycled ventilation. The respiratory motor output response, assessed in terms of change in muscle pressure (Pmus ), was depressed during mild hypocapnia versus eucapnia: 0.26 ± 0.33 versus 0.53 ± 0.59 cmH2 O per percentage oxygen saturation (SaO2 %). During moderate hypocapnia, the Pmus response was negligible: 0.003 ± 0.09 cmH2 O per SaO2 %—a value that is not significantly different from zero. Mild and moderate hypocapnia caused proportional reductions in the response of respiratory frequency to hypoxia. In all subjects, including those who displayed a vigorous response to hypoxia at eucapnia, the hypoxic response was completely ablated Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 40 20 Minute ventilation (L/min) Respiratory frequency (breaths/min) Comprehensive Physiology 16 12 8 32 24 16 8 4 0 0 0.4 0.8 1.2 1.6 2.0 2.4 Tidal volume (L) f at VT min 14.1 ± 3.9 f at VT max 12.4 ± 4.0 0 0 . 0.4 0.8 1.2 1.6 2.0 2.4 Tidal volume (L) VE at VT min 13.0 ± 4.2 . VE at VT max 23.0 ± 7.5 Figure 11 Limited decreases in respiratory frequency in response to increases in ventilator tidal volume. Individual changes in respiratory frequency (left panel) and minute ventilation (right panel) in healthy subjects as tidal volume was increased from a minimum to a maximum setting during assist-control ventilation, resulting in tidal volumes of 944 ± 198 and 1867 ± 277 (SD) mL, respectively. The average respiratory frequency and minute ventilation at the minimum and maximum tidal volume settings are listed below the horizontal axes. Doubling of delivered tidal volumes produced minimal decreases (∼12%) in respiratory frequency. [Adapted, with permission, from Puddy et al. (207).] when PET CO2 was reduced by an average of 11 mmHg below eucapnia. The progressive depression of the hypoxic response with decreases in PCO2 is of special concern in the management of ventilated patients because it may prevent the respiratory motor output from increasing vigorously in response to a ventilator malfunction, such as a disconnected circuit, and thus lead to hypoxic brain injury. Control of breathing during hypocapnia while asleep In anaesthetized cats, stepwise reductions in PCO2 in the perfusate of the pontomedullary region produced progressive decreases in tidal volume with no change in breathing stability (19). These data suggest that hypocapnia at the carotid or central chemoreceptors is not in itself sufficient to cause apnea (63). Despite these observations, most if not all studies performed in sleeping animals (177) and sleeping humans (170, 243, 298) support the presence of an apnea-hypopnea threshold for CO2 . These data (56, 87, 243) indicate that rhythmic breathing during sleep is critically dependent on the level of PCO2 in arterial blood. Reductions in PaCO2 of only a few torr below the awake, eupneic value consistently produce central apnea and periodic breathing in sleeping human subjects (157,178). Moreover, susceptibility to periodic breathing during hypoxia (146) or in patients with Cheyne-Stokes respiration in congestive heart failure (120, 248, 294) have been shown to coincide with a relatively high degree of ventilatory responsiveness to Volume 2, October 2012 hypoxia and/or CO2 . Paradoxically, however, driving ventilation higher and reducing steady-state background PaCO2 can also alleviate central apnea and periodic breathing during sleep (22, 59, 121, 259). These two sets of observations are compatible only if the apneic CO2 threshold changes in response to sustained hyperventilation. To investigate this issue, Nakayama et al. (177) used pressure support to increase tidal volume, reduce PaCO2 , and produce apnea and periodic breathing in sleeping dogs, and then examined the effects of several respiratory stimuli and inhibitors on the apnea threshold. They used the difference between PET CO2 during eupnea and the PET CO2 that resulted in apnea (apnea threshold), PET CO2 , as an index of the susceptibility to apnea. During normoxia, apnea occurred when PET CO2 was decreased from 39 to 34 mmHg (PET CO2 of −5 mmHg). Changes in background ventilatory drive were found to alter the apnea threshold and, thus, also the proximity of the spontaneous PCO2 to the apnea threshold. Increased respiratory motor output induced by metabolic acidosis and nonhypoxic peripheral chemoreceptor stimulation (almitrine) produced stable hyperventilation (PET CO2 of 28-29 mmHg). Apnea occurred when PET CO2 was decreased to 22 mmHg during metabolic acidosis and to 23 mmHg during almitrine infusion (PET CO2 −6 mmHg). In other words, increased respiratory motor output induced by metabolic acidosis or almitrine produced widening of PET CO2 from −5 mmHg at baseline to −7 and −6 mmHg, respectively. The exception to 2885 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning this pattern was hypoxia that produced stable hyperventilation (PET CO2 of 31 mmHg). With this background of increased respiratory motor output, apnea occurred when the PET CO2 was decreased to 27 mmHg, which corresponds to a PET CO2 of −4 mmHg. That is, despite a substantial increase in ventilatory drive and an attendant decrease in spontaneous PaCO2 , hypoxia increased the ventilatory sensitivity to CO2 below eupnea and significantly reduced PET CO2 . To further define the role of hypoxia in producing central apnea and periodic breathing, Xie et al. (297) used pressure support to lower PaCO2 in human subjects during sleep. Pressure support induced hypopneas (failure to trigger the ventilator) and apneas (no breathing effort) in all subjects during both normoxia and hypoxia (SaO2 80%). Eupneic PET CO2 was 45.0 ± 1.1 mmHg during normoxia and it fell to 42.1 ± 1.4 mmHg during hypoxia. The apnea threshold was equivalent with hypoxia and normoxia (∼41 mmHg), as was the hypopnea threshold (∼42.5 mmHg). The main determinant of the development of central apneas and periodic breathing was PET CO2 , which was 3.4 ± 0.3 mmHg during normoxia and 1.1 ± 0 mmHg during hypoxia (the pattern for the hypopnea threshold was similar). Periodic breathing and central apneas are especially common during sleep in patients with congestive heart failure (120, 121), and studies conducted in these patients have yielded fundamental insights into the effects of mechanical ventilation on control of breathing. To investigate the mechanism of breathing instability in patients with heart failure, Xie et al. (298) recorded PET CO2 during spontaneous breathing and used pressure support to estimate the apnea threshold in 19 stable patients with congestive heart failure, 12 of whom exhibited and 7 of whom did not exhibit central sleep apnea. The patients without central sleep apnea experienced a significant increase in PET CO2 at the onset of sleep (of approximately 2.5 mmHg), whereas patients with central sleep apnea did not. As a result, the difference in PET CO2 between eupnea and the apnea threshold was smaller in the patients with central sleep apnea than in those without central sleep apnea: −2.8 ± 0.3 versus −5.1 ± 0.7 mmHg. The ventilatory response to CO2 below eupnea was higher in patients with central sleep apnea than in those without central sleep apnea: 3.86 ± 0.79 versus 1.47 ± 0.18 L/min/mmHg. The close proximity of PET CO2 during eupnea to the apnea threshold makes a transient reduction of PCO2 much more likely to fall below the apnea threshold, and the increased ventilatory response to CO2 below eupnea makes the respiratory system more sensitive to any reduction of CO2 —both factors predisposing to breathing instability. The failure of PET CO2 to increase during sleep in the patients with central sleep apnea signifies the presence of some additional ventilatory drive that offsets the removal of “the wakefulness drive to breathe.” Possibilities include an increase in interstitial pressure that stimulates pulmonary irritant and juxtacapillary receptors (96). In contrast with marked alterations in respiratory motor output that result from slight reductions in chemoreceptor 2886 Printer Name: Yet to Come Comprehensive Physiology stimulation consequent to very small decreases in PCO2 , large increases in PO2 have no comparable effects on respiratory motor output in most patients (63). Induction of a state of extreme hyperoxia (PaO2 > 500 mmHg) using extracorpeal perfusion of the isolated carotid body does not produce apnea (247). Effect of mechanical ventilation on sleep quality in critically ill patients Behavioral factors and the wakefulness drive to breathe can interfere with patient-ventilator interaction and thus lead to dysynchrony between the cycling of a ventilator and a patient’s respiratory rhythm. By ablating these stimuli, sleep might be expected to enhance respiratory muscle rest during mechanical ventilation (195). Moreover, ventilated patients experience numerous derangements, including sepsis, abnormal gas exchange, and increased work of breathing, which may disturb sleep (21, 99). To characterize the extent and nature of sleep disruption in critically ill patients, Cooper et al. (50) performed polysomnography in 20 mechanically ventilated patients. Eight patients exhibited electrophysiological features of sleep, and these experienced 42 arousals and awakenings per hour of sleep. This level of sleep fragmentation is equivalent to that experienced by patients with obstructive sleep apnea who have excessive day-time sleepiness and impaired cognition (103). Gabor et al. (91) investigated the factors that might lead to disrupted sleep in ventilated patients and found that 21% of disrupted sleep resulted from noise and a further 7% resulted from all medical care. While much of the unexplained sleep disruption is likely to result from the underlying illness, operation of the ventilator, and especially the choice of ventilator mode, may be another contributor. To address this possibility, Parthasarathy and Tobin (194) compared the effects of pressure support and assist-control ventilation on sleep in 11 critically ill patients. Six patients developed central apneas during pressure support, but not during assist-control by virtue of the backup rate (Fig. 12). The occurrence of apneas during pressure support was not related to tidal volume, as the pressure level was titrated to achieve a tidal volume equivalent to that during assist-control ventilation (8 mL/kg). Sleep fragmentation, measured as the sum of arousals and awakenings, was greater during pressure support than during assist-control: 79 ± 7 versus 54 ± 7 events per hour. Disturbed sleep during pressure support was related to the development of central apneas (r = 0.57), which in turn was significantly related to PET CO2 (r = −0.83) (Fig. 13). PET CO2 was the most important determinant for the development of apneas: as PET CO2 grew wider, the number of central apneas decreased. The addition of 100 mL of dead space to the ventilator circuit in the six patients who developed apneas produced a 4.3 mmHg increase in end-tidal CO2 , decreased the frequency of central apneas, from 53 to 4 apneas/h, and the frequency of arousals and awakenings, from 83 to 44 events/h. This study shows that while critically ill patients have a background level of sleep disturbance, Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Assist control Pressure support C -A1 4 O3-A2 ROC LOC Chin Leg VT RC AB 1 min Figure 12 Polysomnographic tracings during assist-control ventilation and pressure support in a representative patient. Electroencephalogram (C4 -A1 , O3 A2 ), electrooculogram (ROC, LOC), electromyograms (Chin and Leg), integrated tidal volume (V T ), rib-cage (RC), and abdominal (AB) excursions on respiratory inductive plethysmography are shown. Arousals and awakenings, indicated by horizontal bars, were more numerous during pressure support than during assistcontrol ventilation. [Adapted, with permission, from Parthasarathy S and Tobin MJ. Effect of ventilator mode on sleep quality in critically ill patients. Am J Respir Crit Care Med 166: 1423-1429, 2002 (194).] Volume 2, October 2012 No dead space Dead space 8 PETCO2 minus apnea threshold (mmHg) secondary to factors such as pain, medications, staff interruptions, noise and light, the mode of mechanical ventilation can further aggravate sleep disruption. The observation that ventilator mode can aggravate sleep disruption was confirmed by Bosma et al. (26). These investigators performed polysomnography in 13 ventilated patients who received pressure-support ventilation or proportional assist ventilation (PAV) according to a randomized crossover design. (PAV is a form of synchronized ventilator support in which the ventilator generates pressure in proportion to instantaneous patient effort while correcting for respiratory system elastance and resistance.) The two modes were adjusted to achieve a similar degree of inspiratory muscle unloading. Pressure support of 9.2 ± 2.8 cmH2 O achieved a 54 ± 3% decrease in pressure-time product per minute. The resistive and elastic proportionality factors were adjusted during PAV to obtain a 53 ± 5% decrease in pressure-time product per minute. At these settings, tidal volume was higher with pressure support than with PAV: 0.63 ± 0.13 versus 0.59±0.13 L (SD). End-tidal CO2 for the entire night was lower with pressure support than with PAV: 37.3 ± 5.3 versus 39.4 ± 6.8 mmHg (P < 0.05). Overall sleep quality was significantly inferior with pressure support. More arousals occurred with pressure support than with PAV: median (range) of 16 (2–74) versus 9 (1–41) per hour of sleep. Awakenings were greater with pressure support than with PAV: 5.5 (1–24) versus 3.5 (0–24) per hour. Patient-ventilator asynchronies per hour were also greater with pressure support than with PAV: 53 ± 59 versus 24 ± 15. Moreover, the number of patient-ventilator asynchronies per hour correlated significantly with the number of 6 r = –0.83 P < 0.001 4 2 0 –2 –4 –6 0 40 80 Central apneas per hour Figure 13 The difference between the average end-tidal CO2 and the apnea threshold plotted against the number of central apneas per hour of pressure support alone (closed symbols) and pressure support with added dead space (open symbols) in six patients. The average end-tidal CO2 was measured during both sleep and wakefulness. The mean number of central apneas per hour was strongly correlated with the end-tidal CO2 during a mixture of both sleep and wakefulness (including the transitions between sleep and wakefulness) (r = −0.83, P < 0.001). [Adapted, with permission, from Parthasarathy S and Tobin MJ. Effect of ventilator mode on sleep quality in critically ill patients. Am J Respir Crit Care Med 166: 1423-1429, 2002 (194).] 2887 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning arousals per hour (r2 = 0.71). In brief, for an equivalent degree of inspiratory muscle unloading, sleep was more fragmented with pressure support than with PAV, and the poorer quality of sleep was related to increased patient-ventilator dysynchrony with pressure support. Cabello et al. (40) undertook polysomnography in 15 mechanically ventilated patients and compared sleep quality while patients were randomized between assist-control ventilation, pressure support set by the patient’s attending physician, and pressure support continuously adjusted by a closed-loop knowledge-based system. Sleep architecture, sleep quantity, and sleep fragmentation were equivalent with the three ventilator modes. The findings differ from those reported by Parthasarathy and Tobin (194) in part because of the method of selecting tidal volume in the study of Cabello et al. (40). A tidal volume of 8 mL/kg was employed during assist-control ventilation, whereas a tidal volume between 6 and 8 mL/kg was targeted during pressure support (40). The median (25-75th percentile) tidal volumes were 500 mL (380-500) during assist-control ventilation, 450 mL (357-521) during clinician-adjusted pressure support, and 390 mL (330-492) during automated pressure support. The findings of these three studies of mechanical ventilation during sleep in critically ill patients suggest that pressure support, when carefully adjusted to avoid hyperventilation, does not increase the amount of sleep fragmentation over that experienced with assist-control ventilation. If, however, pressure support is set in the manner of everyday clinical practice, it is likely to lead to sleep fragmentation (26, 194). The findings of Parthasarathy and Tobin (194) carry important implications for how physicians set the ventilator. Compared to wakefulness, sleep lowered respiratory rate by 32.6 ± 6.1% during pressure support and by 14.9 ± 3.2% during assist-control. Likewise, sleep raised end-tidal CO2 by 11.0 ± 2.3% during pressure support and by 4.6 ± 1.8% during assist-control ventilation. In clinical practice, the level of pressure support is typically adjusted in accordance with a patient’s respiratory rate, which provides reasonable guidance as to a patient’s inspiratory effort (129). Pressure is commonly titrated during the daytime without the clinician being sure whether the patient is asleep or awake. If the patient is asleep at the time of the adjustment, a point at which respiratory rate will be relatively low, then, on awakening, the increase in a patient’s rate may cause a considerable increase in effort. Ventilator-induced neuromechanical inhibition When assisted ventilation is used to progressively increase tidal volume, apnea results during sleep when PaCO2 is lowered by as little as 2 mmHg over 2-3 breaths (63). Even in the absence of hypocapnia, Dempsey and colleagues have demonstrated that positive-pressure ventilation can induce inhibition of the respiratory centers and even apnea in animals and human subjects during nonrapid eye movement (NREM) sleep (106, 148, 163, 228, 295). These investigators argue that mechanical ventilation can markedly inhibit or even elimi- 2888 Printer Name: Yet to Come Comprehensive Physiology nate respiratory motor output solely as a result of alterations in mechanoreceptors in the lungs or chest wall—so-called neuromechanical inhibition (64). Neuromechanical inhibition is manifested as changes in the amplitude of respiratory motor output, 20% to 60% decreases in the diaphragmatic electromyogram (Edi ), transdiaphragmatic pressure (Pdi ), and respiratory timing, and prolonged expiratory time and apnea (64). In sleeping (228) or lightly anesthetized animals (136), a single ventilator breath can produce a prolonged expiratory time when the breath is delivered during the “inflationsensitive” phase of expiration. When a single breath is applied in the first 25% to 65% of expiration, the resumption of diaphragmatic electromyographic (EMG) activity is delayed and expiratory time is prolonged; the effect on expiratory prolongation is increased when the tidal volume of the single breath is 75% to 150% of eupneic tidal volume—the greater the tidal volume, the greater the expiratory time prolongation (228). A ventilator breath that is about half eupneic tidal volume does not significantly prolong expiratory time (228). When a single ventilator breath of any volume is applied in the last 25% of expiration, expiratory time does not change (228). Application of a ventilator breath in the 65% to 75% range of expiratory time has a variable effect—expiratory time may be prolonged, shortened or unchanged (228). A single ventilator breath alters only the expiratory time of the perturbed breath and does not affect the next respiratory cycle (228). In sleeping humans during normocapnic conditions, Rice et al. (215) studied the effect of increased tidal volume, with and without increases in respiratory rate, through the respective use of controlled and assist-control ventilation. During and after assist-control ventilation, an increase in tidal volume to 135% to 220% of the eupneic value never completely eliminated inspiratory motor output and expiratory time was only slightly (although significantly) prolonged. During controlled mechanical ventilation, in contrast, an increase in respiratory rate by 1-3 breaths/min above the mean eupneic value and an increase in tidal volume to 165% to 200% of the baseline eupneic value eliminated inspiratory motor output at the cessation of mechanical ventilation, and expiratory time was prolonged by two to four times the control value (215). (The prolongation of expiratory time with assist-control ventilation was less than 20%-25% of the prolongation observed after controlled ventilation at comparable increases in tidal volume.) These results suggest that the combination of increases in respiratory rate in combination with (moderate) elevations in tidal volume during mechanical ventilation can reset respiratory rhythm and inhibit respiratory motor output more than that achieved by increases in tidal volume alone. The duration of apnea following the cessation of normocapnic controlled ventilation is influenced by several factors. One, apnea duration is proportional to the increase in ventilator tidal volume (once inspiratory motor output has been silenced by an increase in ventilator frequency) (228, 295). Two, apnea duration increases in proportion to the duration of the passive controlled ventilation, at least up to about 1 min of Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology controlled ventilation (228). The latter response is similar to the prolongation of expiratory time that occurs after repeated electrical stimulation of vagal or superior laryngeal nerves, where expiratory duration is proportional to the duration of the phasic sensory input (315). Three, controlled ventilation at an increased frequency can eliminate respiratory motor output even when PaCO2 is as much as 5 mmHg above normocapnia (228); postventilator apneas occur in the presence of such hypercapnia, however, only when the ventilator tidal volume is much higher than eupneic tidal volume (64). Why does a constant ventilator frequency (above the average eupneic frequency) cause silencing of the diaphragmatic EMG? A single ventilator breath affects the respiratory controller only if it is delivered during the “inflation-sensitive” phase of the respiratory cycle (136). For each repeated ventilator breath to be delivered precisely during the inflation-sensitive phase is unlikely unless the inflation-sensitive phase becomes prolonged. Dempsey and Skatrud postulated that such prolongation might result from a cumulative carryover of an inhibitory influence, which in effect would widen the inflation-sensitive phase as controlled ventilation is continued beyond the first delivered breath (64). In support of such a cumulative effect are the observations that the duration of postventilator apneas during normocapnic mechanical ventilation is dependent on the total number of ventilator cycles and the amplitude of their tidal volume (148, 228). The mechanisms responsible for the inhibitory aftereffects of normocapnic mechanical ventilation are not precisely known. It has been reasoned that the means of informing the respiratory controller of the adequacy of ventilation must involve neurally mediated sensory information (concerning pressure, volume, and/or muscle tension) from one or more sites of mechanoreception (136, 148, 156, 239). The sustained inhibition of respiratory motor output in animals and humans does not appear to involve vagal or intercostal afferents (148, 238). When a sensory stimulus is delivered, several investigators have documented that changes in synaptic efficacy persist beyond the duration of the stimulus (321). Recordings from the nucleus of the solitary tract show that potentiation of synaptic input to these neurons enhances inhibitory outflow and reduces the neuronal response to subsequent afferent chemoreceptor input (171). Accordingly, the apneas that follow mechanical ventilation may reflect an imbalance between a dominant continued short-term suppression of inspiratory motor output and a rise in chemoreceptor input as asphyxia intensifies during the postventilator apnea (64). Dempsey has argued that the resumption of normal rhythm following the apneas must represent the eventual dominance of the excitatory chemoreceptor input over the continued “inertia” of central respiratory motor neurons; even at these high levels of PaCO2 (and reduced PO2 ), however, a central inhibitory effect is still present as indicated by the reduced amplitude and gradual recovery of successive spontaneous breaths (64). The occurrence of apnea following the cessation of (normocapnic) controlled ventilation supports the concept of Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning strong central “inhibitory memory” effects imposed by repetitive mechanoreceptor feedback (228). Satoh et al. (228) commented that the apnea induced by controlled ventilation resembles the prolonged apneas in patients with central sleep apnea, which often last well beyond the normal threshold for chemoreceptor excitation. Once the medullary rhythm generator is silenced by mechanical or chemical mechanisms, the respiratory rhythm is not easily restored (228). It is possible that mechanical feedback linked to ventilator frequency plays an important role in the resetting of the natural breathing cycle. If resetting of respiratory rhythm is sufficiently strong, Puddy and co-workers (207) postulated that increased ventilator frequency may cause apnea by “superseding” the inherent rhythm—a case of where the tidal volume and rate set on the ventilator exceed the tidal volume and frequency demands of the patient—as opposed to reflecting the action of a built-in inhibitory neural mechanical feedback that suppresses ventilatory drive. It should be noted, however, that use of an augmented tidal volume can in itself inhibit respiratory motor output. Studies demonstrating neuromechanical inhibition have been largely, although not exclusively (149), conducted during sleep to eliminate the confounding influence of behavioral responses. When healthy subjects received controlled mechanical ventilation during wakefulness, Puddy et al. (207) found little evidence of neuromechanical inhibition. The investigators maintain that the apneas were confined to circumstances where the subject remained passive and both the delivered tidal volume and mandatory ventilator frequency exceeded the subject’s tidal volume and frequency demands at that moment. It should be noted, however, that Puddy et al. (207) did not control for end-tidal PCO2 in their experiments. Despite the presence of hypocapnia, awake subjects did not experience postventilator apnea, which suggests that behavioral influences played an important role. Neuromechanical inhibition has been mainly documented in instances where hyperventilation has been passively induced; apneas, however, have also been observed during sleep when a transient ventilatory overshoot and hypocapnia were elicited through the release of a brief airway occlusion (mimicking obstructed apnea) (115) and with the use of pressure support to increase tidal volume and reduce PaCO2 (297). In six healthy subjects, Sharshar et al. (234) investigated excitability of the diaphragmatic motor cortex by measuring diaphragmatic motor-evoked potentials elicited by transcranial magnetic stimulation during isocapnic volume-cycled ventilation. Isocapnic volume-cycled ventilation produced a decrease in the motor-evoked potentials elicited by transcranial magnetic stimulation, but not in those elicited by phrenic-nerve stimulation. These results suggest that isocapnic volume-cycled ventilation causes depression of the diaphragmatic motor cortex. Whether or not the observed depression is another manifestation of the neuromechanical inhibition reported by Dempsey and colleagues remains to be determined. 2889 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning Flow delivery Paw cmH2O I-E switchover 5 0 Triggering 250 ms Figure 14 Three points at which patient-ventilator dysynchrony may arise: point of triggering (cycling-on function), inspiratory flow delivery (posttrigger inflation), and the point of switchover between inspiration and expiration (cycling-off function). Each is discussed in the text. Patient-ventilator dysynchrony For effective unloading of the inspiratory muscles, the ventilator should cycle in synchrony with the patient’s central respiratory rhythm (266). Dysynchrony may originate at several points in the cycling of the two pumps (192) (Fig. 14). If the onset of machine assistance lags significantly behind the commencement of patient inspiratory muscle activity (trigger delay), the ventilator’s ability to produce respiratory muscle rest is markedly impaired (152, 220). Inadequate respiratory muscle rest may also result from failure of the gas-delivery system to meet a patient’s inspiratory flow demands (165, 220, 288). Most studies of patient-ventilator dysynchrony have focused on these two inspiratory phenomena, and scant attention has been paid to expiratory events during ventilation. Depending on the algorithm used for “cycling off” of the ventilator, recruitment of the expiratory muscles can occur during mechanical inflation (129). A final source of dysynchrony is a delay in the termination of a patient’s expiratory muscle activity before the onset of the next mechanical inflation. All of the foregoing considerations are further complicated by the recognition that the inspiratory and expiratory muscle groups can contract simultaneously (66). Framework for evaluating patient-ventilator interaction The interplay between a patient and a ventilator depends on a number of factors, and, most importantly, the physiologic characteristics of a given patient. During mechanical ventilation, the total pressure applied to the respiratory system (Ptotal ) is the sum of the pressure provided by the ventilator (Paw ) and the pressure generated by the patient’s respiratory muscles (Pmus ) (264). Thus, at any time, (t), from the onset of inspiratory flow, Ptotal(t) = Paw(t) + Pmus(t) 2890 Printer Name: Yet to Come Comprehensive Physiology The fundamental relationship between the patient and the ventilator is contained in the equation of motion. At a given moment, the total pressure applied to the respiratory system is balanced by (or dissipated against) the opposing forces produced by the resistive and elastic properties of the respiratory system—assuming that inertia is negligible. The pressure dissipated against resistance (Pres ) is a function of the instantaneous rate of flow [Pres = f (V̇ )] (308). The pressure dissipated against the elastic properties (Pel ) at a given moment is a function of how far respiratory volume is from the relaxation volume of the respiratory system (FRC) as opposed to the volume inhaled. For ease of convenience, the function of volume that defines the instantaneous elastic recoil [Pel = f (V)] is commonly considered as a constant (elastance), describing recoil pressure per unit of volume above FRC. This approach may be reasonable in a healthy subject during resting ventilation, because the usual tidal volume of 400 mL constitutes a small fraction of vital capacity, and it is positioned on the linear portion of the sigmoid pressure-volume relationship of the respiratory system. The approach can be very misleading in ventilated patients, where set tidal volume (600-800 mL) may take up half or more of vital capacity and the delivered volume may be located close to TLC, consequent to dynamic hyperinflation, or be located close to residual volume, consequent to abdominal distention or obesity. At both extremes, the pressure-volume relationship is grossly nonlinear. As a result of these influences, the total pressure applied to the respiratory system (Ptotal (t) ) can be calculated as: Ptotal(t) = V̇(t) · Rrs + V(t) · E rs where Rrs is the resistance and Ers the elastance of the respiratory system, and V̇ (t) and V (t) are, respectively, instantaneous inspiratory flow and volume above passive FRC at time t from the onset of inspiration. The values of Rrs and Ers are readily measured during passive mechanical ventilation using the end-inspiratory occlusion technique (38). The response of a patient to the institution of mechanical ventilation depends on the patient’s breathing pattern, which in turn depends on the degree of chemoreceptor stimulation secondary to hypoxia or hypercapnia, the degree of agitation secondary to mechanical derangements of respiratory function, the degree of respiratory muscle weakness, systemic disorders, medications that have been administered (such as sedatives), and many other factors. Point of triggering (cycling-on function) Most ventilators contain a demand valve and deliver positivepressure assistance whenever a patient’s inspiratory effort decreases pressure in the ventilator circuit by 1-2 cmH2 O (Fig. 15). The latter value, selected by a clinician, is termed the set sensitivity (223). Patients reach the set sensitivity by activating their inspiratory muscles. For a given trigger sensitivity, the delay between onset of patient inspiratory effort and onset of ventilator assistance is a function of a patient’s Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 20 Onset inspiratory effort r = 0.78 Valve opening Patient effort during ventilator assistance (cmH2O.s) I:E flow switch Flow (L/s) 1 0 Pes cmH2O Paw cmH2O –1 20 10 0 –5 15 10 5 0 10 0 –5 0 0 1 2 3 4 Time (s) Figure 15 Patient effort related to ventilator triggering. Tracings of flow, airway pressure (Paw ), and esophageal pressure (Pes ) in a patient receiving the volume-cycled form of assist-control ventilation. The vertical blue line represents the onset of patient inspiratory effort, the vertical red line represents the onset of flow delivery (opening of the ventilator valve), and the vertical green line represents the transition from inspiratory flow to expiratory flow; the dashed tracing above the Pes recording on the bottom panel is an estimate of the patient’s chest-wall recoil pressure, measured during passive ventilation. The effort of triggering is divided into two phases. First, the effort of the trigger phase (single hatched area). Second, the effort of the posttrigger phase (double hatched area) is the area enclosed between esophageal pressure and estimated chest-wall recoil pressure between the onset of flow delivery and the switch from inspiratory flow to expiratory flow. respiratory motor output. When a patient’s respiratory drive is low, assistance may not begin until well into the patient’s inspiratory time, thereby causing the ventilator to cycle almost completely out of phase with the patient’s respiratory cycle. If respiratory motor output is extremely low, no triggering may take place and the ventilator will operate at the backup rate (controlled ventilation) much or all the time. When the threshold to open the demand valve is reached and the ventilator starts to provide positive-pressure assistance, the inspiratory neurons do not simply switch off, and a patient may expend considerable inspiratory effort throughout the machine-cycled inflation (165). The level of patient effort during this posttrigger phase is closely related to a patient’s respiratory motor output at the point of triggering (152) (Fig. 16). If respiratory motor output at the point of triggering is important, one might intuitively expect that effort during the time of triggering would determine patient effort during the remainder of inspiration. Such an association was suggested by Giuliani and co-workers (98). To investigate this issue, Leung and co-workers (152) applied graded levels of pressure support in 11 critically ill patients. They achieved a fourfold reduction in overall patient effort as compared with unas- Volume 2, October 2012 20 40 60 Respiratory motor output (cmH2O.s) Figure 16 Patient effort during the time that the ventilator is delivering a breath (measured as inspiratory pressure-time product per breath in cmH2 O.s) is closely related to a patient’s respiratory motor output (measured as dP/dt in cmH2 O.s) at the moment that a patient triggers the ventilator (r = 0.78). The inspiratory muscles of a patient who has a low respiratory drive at the time of triggering the ventilator will perform very little work during the remainder of inspiration when the ventilator is providing assistance. Conversely, the inspiratory muscles of a patient who has a high respiratory drive will expend considerable effort throughout the period of inspiration even though the mechanical ventilator is providing assistance. [Based on data published in Leung P, Jubran A and Tobin MJ. Comparison of assisted ventilator modes on triggering, patient effort, and dyspnea. Am J Respir Crit Care Med 155: 1940-1948, 1997 (152).] sisted breathing. Yet patient effort during the time of triggering did not change. The constancy of effort during the trigger phase was probably secondary to different factors becoming operational as the level of ventilator assistance was varied (Fig. 17). When patients were receiving a low level of pressure support, their respiratory motor output was high and the time to trigger the machine was short; as a result, they generated large swings in intrathoracic pressure over a short time. When patients received a high level of pressure support, their respiratory motor output was lower but more time was needed to trigger the machine; as a result, they generated much smaller swings in intrathoracic pressure, but over a longer time. Thus, increases in the level of ventilator assistance do not substantially decrease patient effort during the time of triggering. Opening a demand valve during pressure triggering can impose substantial effort (223). In an attempt to overcome this problem, flow triggering was introduced. With this approach, the ventilator is triggered when a patient’s effort creates a difference between the inspiratory and expiratory base flow in the circuit. Aslanian and co-workers (12) found that the time required for triggering was 43% less with flow triggering than with pressure triggering, and effort during the time of triggering was decreased by 62%. Patient effort during the posttriggering phase, however, was equivalent for pressure and flow triggering. As a result, the overall effect on total 2891 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology 15 cmH2O.s 40 Total PTP dP/ dt 20 0 cmH2O.s 10 Seconds 0.6 5 Trigger PTP 0 0.3 Trigger time 0 0 20 40 60 80 100 0 20 40 60 80 100 Pressure support (%) Figure 17 Graded increases in pressure support produced a decrease in total pressure-time product (PTP) per breath (closed symbols), whereas PTP during the trigger phase (open symbols) did not change (left panel). The constancy of PTP during triggering probably resulted from different factors becoming operational at different levels of assistance (right panel). At low levels of pressure support, respiratory motor output (dP/dt) and intrinsic positive end-expiratory pressure (PEEPi ) were high but triggering time was short, resulting in a large change in pleural pressure over a brief interval. At high levels of pressure support, dP/dt and PEEPi were low but triggering time was long, resulting in a smaller change in pleural pressure over a longer time. [Adapted, with permission, from Leung P, Jubran A and Tobin MJ. Comparison of assisted ventilator modes on triggering, patient effort, and dyspnea. Am J Respir Crit Care Med 155: 1940-1948, 1997 (152).] patient effort was small, and the clinical benefit of flow triggering appears to be much less than commonly stated. Most studies of patient-ventilator interaction have been based on indirect measurements, where the onset and offset of respiratory muscle activity have been estimated from recordings of airflow combined with airway, esophageal pressure, and transdiaphragmatic pressures (32, 129, 165, 223, 262). Parthasarathy et al. systematically evaluated the concordance between such indirect estimates and a more direct measurement of neural activity, namely, the crural Edi (193). Estimates of the duration of inspiration based on flow, esophageal pressure, and transdiaphragmatic pressures revealed substantial differences from the time durations measured with the Edi . The average differences (bias error) ranged from −52 to 714 ms. The standard deviation of these differences ranged from 74 to 221 ms. When inspiratory time measured on the recording of the Edi was taken as the reference standard, the inspiratory time estimated from the transdiaphragmatic pressure (from the initial deflection of the signal until the signal returns to baseline) had a mean difference of 57% from the reference value and a scatter of 87% (Fig. 18). Such discrepancies can adversely affect the operation of the ventilator. Because a ventilator algorithm is based on recordings of flow and airway pressure, errors in estimating the onset of inspiratory time may give rise to delay in triggering, and errors in estimating the duration of inspiratory time that may cause mechanical inflation to persist into expiration. A delay in opening of the inspiratory valve may arise from a decreased respiratory motor output (152, 262) or increased 2892 intrinsic PEEP (PEEPi ) (9, 193). In five critically ill patients, significant delays were noted between the onset of patient inspiratory effort (measured by crural Edi ) and the onset of inspiratory flow (193) (Fig. 19). The delay between the onset of inspiratory effort and the time the ventilator was triggered was correlated with the level of PEEPi of the breaths (r = 0.59). This observation suggests that when elastic recoil pressure at the end of expiration is high, the subsequent inspiratory effort also needs to be proportionally increased if the ventilator is to be successfully triggered. A ventilator that is designed to sense the patient’s effort at a neural level (Edi ) instead of sensing the final result of patient effort (changes in airway pressure or flow) should achieve better patient-ventilator synchrony (240). A new mode, neurally adjusted ventilator assist (NAVA) (240), shows promise in that regard. The machine has yet to undergo rigorous evaluation in the ICU. Failure to trigger When receiving high levels of pressure support or assistcontrol ventilation, a quarter to a third of a patient’s inspiratory efforts may fail to trigger the machine (152) (Fig. 20). The number of ineffective triggering attempts increases in direct proportion to the level of ventilator assistance (152). In a study of factors contributing to ineffective triggering, a decrease in the magnitude of inspiratory effort at a given level of assistance was not the cause; indeed, effort was 38% higher during nontriggering attempts than during the triggering phase of attempts that successfully opened the Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning 0.5 360 Raw Edi (arb. units) Phase angle (degrees) 240 –0.5 0.5 Processed Edi (arb. units) –0.5 0.5 –120 –360 Pes (arb. units) Flow Pdi Figure 19 The phase angle between the indirect estimate of the on- –0.5 –90 0 180 360 degrees 25 0 –25 2 Flow (L/s) 0 –240 MA Edi Pes (cmH2O) 120 set of neural inspiratory time and its reference measurement in five patients during mechanical ventilation; estimates from the esophageal pressure (Pes ) tracings are shown as closed squares, estimates from flow tracings are shown as closed circles, and estimates from transdiaphragmatic pressure (Pdi ) tracings are shown as closed triangles. The closed symbols represent the mean difference (bias) in phase angle; the open symbols to the right of each closed symbol represent the mean difference between the two measurements noted during the reproducibility testing of the reference measurement. The error bars represent ±2 SD (twice the precision). A positive phase angle indicates a delay in the onset of neural inspiratory time for the flow-based measurements. [Modified, with permission, from Parthasarathy S, Jubran A and Tobin MJ. Assessment of neural inspiratory time in ventilatorsupported patients. Am J Respir Crit Care Med 162: 546-552, 2000 (193).] 0 –2 Figure 18 Estimations of the duration of neural inspiratory time. Representative tracings of the raw crural diaphragmatic electromyogram (Edi ), the processed Edi achieved by removing electrocardiography (EKG) artifacts by computer, the moving average (MA) of the processed Edi , esophageal pressure (Pes ), and flow in a patient breathing spontaneously. The relationship between an indirect estimate of the onset of neural inspiratory time and its onset on the diaphragmatic EMG signal was assessed by calculation of the phase angle, expressed in degrees. In this example, the onset of inspiratory time is estimated as occurring earlier (negative phase angle of 15◦ ) by esophageal pressure-based measurements and later (positive phase angle of 110◦ ) by flow-based measurements. The duration of inspiratory time as estimated by esophageal pressure (hatched horizontal bar) is longer than the true inspiratory time measured by diaphragmatic electromyogram (note that the hatched bar is wider than 0-360◦ on the solid black bar of the reference measurement). The duration of inspiratory time as estimated by flow-based measurements is shorter (clear horizontal bar) than the true inspiratory time measured by diaphragmatic electromyogram (note that the open white bar is narrower than 0-360◦ on the solid black bar of the reference measurement). [Adapted, with permission, from Parthasarathy S, Jubran A and Tobin MJ. Assessment of neural inspiratory time in ventilator-supported patients. Am J Respir Crit Care Med 162: 546-552, 2000 (193).] Volume 2, October 2012 ventilator valve (152). Significant differences, however, were noted in the characteristics of the breaths before the triggering and nontriggering attempts. Breaths before nontriggering attempts had a higher tidal volume than did the breaths before triggering attempts, 486 ± 19 and 444 ± 16 mL, respectively, and a shorter expiratory time, 1.02 ± 0.04 and 1.24 ± 0.03 s, respectively. An abbreviated expiratory time does not allow the lung to return to its relaxation volume, leading to an increase in elastic recoil pressure. Indeed, PEEPi was higher at the onset of nontriggering attempts than at triggering attempts: 4.22 ± 0.26 and 3.25 ± 0.23 cmH2 O, respectively. Thus, nontriggering results from premature inspiratory efforts that are not sufficient to overcome the increased elastic recoil associated with dynamic hyperinflation (152). When triggering fails as a result of dynamic hyperinflation, the nontriggering on that breath allows the lungs to more completely empty in preparation for the next breath. Occasionally, two or three failed triggering attempts take place before triggering becomes successful. Because there is no lung inflation during a failed triggering attempt, mechanical exhalation continues for a longer time and end-expiratory volume continues to fall until triggering becomes successful. This pattern may occur repeatedly such that it resembles the 2893 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology Flow (L/s) 1.5 I 0 Flow Paw cmH2O –1.5 50 E 0 Radians –50 20 Pes cmH2O 0 –90 0 90 180 270 360 Mechanical 0 Subject 1, 60° –20 0 5 10 15 20 25 Time (s) Transversus abdominis EMG Subject 2, –45° Figure 20 Failure to trigger the ventilator. Flow, airway pressure (Paw ) and esophageal pressure (Pes ) in a patient with COPD who is receiving assist-control ventilation at the following settings: tidal volume 600 mL, inspiratory flow 60 L/min, trigger sensitivity −2 cmH2 O and positive end-expiratory pressure 0 cmH2 O. The patient’s intrinsic respiratory rate is 28 breaths/min, whereas the number of breaths delivered by the ventilator is 16 breaths/min. That is, 43% of the patient’s inspiratory efforts fail to trigger ventilator assistance. Contractions of the inspiratory muscles during the failed triggering attempts cause a temporary deceleration of expiratory flow and much less obvious decreases in airway pressure. The temporary decelerations in expiratory flow are followed by temporary accelerations of expiratory flow that coincide with the termination of the unsuccessful inspiratory effort. Wenckebach pattern of atrioventricular block on an electrocardiograph (EKG) (313). In addition to increase in elastic recoil pressure (152), an elevated PEEPi can also result from an increase in expiratory muscle activity. Parthasarathy et al. (192) investigated the relative contributions of these two factors to ineffective triggering in healthy subjects receiving pressure support, in whom they induced airflow limitation with a Starling resistor. The phase relationship of the “on-switch” and “off-switch” of pertinent muscles with cycling of the ventilator was defined using wire electrodes to obtain electromyographic recordings of the diaphragmatic crura and transversus abdominis muscles. The degree of nonsynchronization between the machine and the patient was expressed in terms of phase angle (θ ) (198). When the start of the transversus abdominis EMG signal coincides with the end of mechanical inflation, phase angle is zero; when the transversus abdominis EMG activity starts after the end of mechanical inflation, phase angle has a positive value; and when the transversus abdominis EMG activity starts before the end of mechanical inflation, phase angle has negative units (Fig. 21). All nine subjects exhibited nontriggering efforts, and these were more frequent at a pressure support of 10 and 20 cmH2 O than at 0 cmH2 O (Fig. 22). The phase angle of the nontriggering attempts was more negative than that of the attempts that successfully triggered the ventilator: −32.6 ± 3.1 versus −12.6 ± 1.8◦ (P = 0.0002) at a pressure support of 10 cmH2 O, and −50 ± 9.4◦ versus −19.1 ± 3.4◦ (P = 0.01) 2894 Figure 21 The concordance between neural expiratory time and mechanical expiratory time can be quantified in terms of the phase angle, expressed in degrees. If neural activity begins simultaneously with the machine, the phase angle (θ ) is zero. Neural activity beginning after the offset of mechanical inflation results in a positive phase angle (60◦ for Subject 1). Neural activity beginning before the onset of mechanical inflation results in a negative phase angle (−45◦ for Subject 2). [Adapted, with permission, from Parthasarathy S, Jubran A and Tobin MJ. Cycling of inspiratory and expiratory muscle groups with the ventilator in airflow limitation. Am J Respir Crit Care Med 158: 1471-1478, 1998 (192).] at a pressure support of 20 cmH2 O. That is, the length of time that the expiratory muscles had been active before the cycling-off of mechanical inflation was longer for nontriggering attempts than for triggering attempts. In other words, mechanical inflation continued into neural expiration for a longer time before the failed triggering attempts than before the successful triggering attempts. Such continuation of mechanical inflation into neural expiration not only directly counters expiratory flow but also decreases the time available for unopposed expiratory flow. This leads to an increase in elastic recoil at the start of the ensuing inspiration, which in turn necessitates a greater inspiratory effort to achieve effective triggering of the ventilator (105). In this way, the time that a patient commences an expiratory effort (in relation to cycling-off of mechanical inflation) partly determines the success of the ensuing inspiratory effort in triggering the ventilator. Double triggering Some patients exhibit two mechanical inflations within a single neural inspiration, a phenomenon known as double triggering (Fig. 23). With assist-control ventilation, double triggering is likely when the set mechanical inspiratory time is substantially less than a patient’s neural inspiratory time. In this situation, mechanical inflation terminates while the patient is still making an inspiratory effort. After a brief period, the ventilator may trigger again, resulting in a second Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning Non triggering attempts Phase angle (degrees) 0 –30 –60 –90 PS 0 PS 10 PS 20 Figure 22 Phase angle between neural and mechanical expiratory times before triggering (closed circles) and nontriggering (open circles) attempts. At pressure support (PS) of 10 and 20 cmH2 O, the phase angle before nontriggering attempts exceeded that before triggering attempts, indicating that neural expiratory time during late mechanical inflation was longer before nontriggering attempts than before triggering attempts (for pressure support 10, P = 0.0002; for pressure support 20, P = 0.01). [Adapted, with permission, from Parthasarathy S, Jubran A and Tobin MJ. Cycling of inspiratory and expiratory muscle groups with the ventilator in airflow limitation. Am J Respir Crit Care Med 158: 1471-1478, 1998 (192).] inflation within the same neural inspiration and, thus, greater alveolar distension than with the delivery of a single tidal volume (313). With pressure-support ventilation, double triggering is likely when the time constant is short (low resistance, high elastance) and patient neural inspiratory time is relatively long (a slow, spontaneous respiratory rate) (308). Following the loss of ventilator pressure after a first triggering attempt, volume decreases because Pmus alone is not sufficient to sustain elastic recoil. During this phase, the persistence of neural inhalation will cause a progressive increase in Pmus , while elastic recoil continues to decrease. If the patient’s neural inspiratory time is sufficiently long, a point is reached where Pmus will exceed elastic recoil and flow will become inspiratory and double triggering will occur (307). The mechanical and cellular derangements seen in patients with ARDS have been demonstrated experimentally to pose major risks for the development of ventilator-induced lung injury (68, 290). Randomized clinical trials have also shown an increased mortality in patients ventilated with a high tidal volume (12 mL/Kg) (1, 6). Consequently, it has become standard practice to lower the delivered tidal volume to minimize alveolar overdistension—for example, to keep the inspiratory pressure during a pause at the end of inspiration (plateau pressure) to 32 cmH2 O or lower (265). A low tidal volume is typically accompanied by a short mechanical inspiratory time and thus these patients are especially susceptible to double triggering. Accordingly, conscious attempts to lower tidal volume can paradoxically result in greater alveolar distension than occurs with conventional tidal volume settings (Fig. 24). An additional (and often unrecognized) factor that may produce alveolar distension is the tachypnea-associated increase in intrinsic PEEP that accompanies lowering of tidal volume (60). 0 –1 –2 1.0 Liter 50 Paw cmH2O Flow 1 L/s Triggering attempts 20 Volume 0.5 0 Pes cmH2O 10 Pes cmH2O –10 20 0 –10 5 0 2 4 6 8 Time (s) –10 0 10 20 30 Time (s) Figure 23 Four incidents of double triggering, each indicated by an arrowhead symbol (↑). Airway pressure (Paw ) and esophageal pressure (Pes ) in a patient with COPD and pneumonia who was receiving assist-control ventilation at the following settings: tidal volume 600 mL, inspiratory flow 60 L/min, trigger sensitivity −2 cmH2 O and positive end-expiratory pressure 5 cmH2 O. The duration of neural inhalation of the double-triggered breaths, roughly equivalent to the width of the associated swings in esophageal pressure, was substantially longer than the neural inhalation of the normally triggered breaths. Volume 2, October 2012 Figure 24 Volume stacking caused by double triggering. Flow (top panel), volume (middle panel), and esophageal pressure (lower panel) in a patient with COPD receiving assist-control ventilation. During first breath, esophageal pressure remains positive indicating that the patient did not trigger the inflation. During the second breath, esophageal pressure becomes negative indicating active inspiratory effort, which lasts more than 1 s; the duration of mechanical inflation is 0.6 s. The longer duration of neural inspiration as compared with mechanical inflation causes the ventilator to deliver a second breath before there is time for exhalation. As a result, end-inspiratory lung volume increases (breath stacking) with a consequent increase in elastic recoil. The increase in elastic recoil is responsible for the higher peak expiratory flow on the second breath as compared with the first breath. 2895 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology Flow 60 L/s Flow 90 L/s Flow (L/s) 2.0 0 –2.0 Paw cmH2O 60 0 –60 Pes cmH2O 30 0 –30 0 4 8 0 4 8 Time (s) Figure 25 Influence of ventilator flow setting on patient effort. Flow (inspiration directed upward), airway pressure (Paw ), and esophageal pressure (Pes ) in a patient with respiratory failure who is receiving assist-control ventilation; inspiratory flow is set at 60 L/min in the left panel and at 90 L/min in the right panel. At an inspiratory flow of 60 L/min (left panel), the pronounced negative deflection in airway pressure (patient effort to trigger the ventilator) together with subsequent extensive scalloping signifies that the inspiratory flow delivered by the ventilator is insufficient to meet the high demand. At an inspiratory flow of 90 L/min (right panel), the small negative deflection in airway pressure together with the subsequent smooth convex contour signifies that the delivered flow satisfies the patient’s respiratory drive. Accordingly, the flow of 90 L/min achieved greater unloading of the respiratory muscles, as signaled by the shorter duration of inspiratory effort and the smaller swings in esophageal pressure. Setting of inspiratory flow and tidal volume When a patient is first connected to a ventilator, inspiratory flow is set at some default value, such as 60 L/min. Many critically ill patients, however, have an elevated respiratory drive and the initial setting of flow may be insufficient to meet flow demands. As a result, patients will struggle against their own respiratory impedance and that of the ventilator, with consequent increase in the work of breathing (Fig. 25). Clinicians sometimes increase flow with a goal of shortening inspiratory time and thus increasing expiratory time. But an increase in flow causes immediate and persistent tachypnea; as a result, expiratory time may be shortened (209). In healthy subjects, Puddy and Younes (209) found that an increase in inspiratory flow from 30 to 90 L/min caused respiratory frequency to increase from 8.8 to 14.1 breaths/min. The increase in frequency was nearly complete within the first two breaths and did not change thereafter despite the development of respiratory alkalosis. This flow-associated tachypnea has been shown to be independent of breathing route, temperature of inspired gas, and delivered volume; it also persists after anesthesia of the airway, denervation of receptors in the lungs and chest wall, and during sleep (94). In addition to the effect 2896 of delivered flow on frequency, investigators have shown that frequency is influenced by delivered tidal volume—an effect seen under isocapnic and hypocapnic conditions, and during wakefulness and sleep (262). When studying the effects of a change in a ventilator’s flow or tidal volume, the results may be influenced unwittingly by simultaneous changes in ventilator inspiratory time. When inspiratory flow is increased and tidal volume kept constant, ventilator inspiratory time must decrease. When tidal volume is increased and inspiratory flow kept constant, ventilator inspiratory time must increase. Laghi et al. (141) undertook a series of experiments in healthy volunteers to delineate the separate influences of these three settings—flow, tidal volume, and ventilator inspiratory time—in mediating flow-associated tachypnea. For inspiratory flow of 30, 60, and 90 L/min, the respective frequencies were 12.9, 15.5, and 18.2 breaths/min. Because tidal volume was kept constant, an increase in flow was accompanied by a proportional decrease in ventilator inspiratory time. The increase in frequency was proportional to the decrease in ventilator inspiratory time (r = 0.69). When flow and tidal volume were held constant and ventilator inspiratory pauses of as much as 2 s were imposed, frequency Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning again changed as a function of ventilator inspiratory time (r = 0.86). When flow was increased from 30 to 60 L/min, and tidal volume adjusted to maintain a constant ventilator inspiratory time, frequency decreased from 17.9 to 16.0 breaths/min. This series of observations shows that imposed inspiratory time can determine frequency independently of delivered inspiratory flow and tidal volume (141). The findings have clinical implications. Physicians often increase tidal volume to lower PaCO2 . An increase in tidal volume without change in inspiratory flow, however, must be accompanied by a longer inspiratory time, which is likely to decrease frequency. The consequent change in minute ventilation and PaCO2 will fall short of that intended. (The opposite scenario holds when a lower tidal volume is selected.) The effects of delivered flow, tidal volume, and inspiratory time on frequency can be interpreted according to current understanding of the Hering-Breuer reflex. First, for a fixed inspiratory time, an increase in inspiratory flow will necessarily increase end-inspiratory lung volume. An increase in delivered volume causes a progressive increase in the activity of the slowly adapting receptors (stretch receptors). This activity is transmitted by the vagus to the brainstem where it inhibits inspiration (negative feedback). Any decrease in neural inspiratory time is also likely to be accompanied by a decrease in neural expiratory time, because the two phases are closely linked (47). Accordingly, decreases in neural inspiratory time and expiratory time secondary to lung inflation are likely to be major factors in explaining flow-associated tachypnea. Second, a decrease in neural inspiratory time fosters the extension of mechanical inflation into neural expiratory time, which, in turn, causes prolongation of neural expiratory time (316,322). A mechanical expiratory time that exceeds neural inspiration, as can occur with imposed inspiratory pauses, higher tidal volume, or slower flow rates, is thus an important modulator of frequency. In summary, the response of frequency to a change in ventilator settings is complex, because it depends on the relative dominance of physiologic mechanisms acting in different directions. High flows are often used with the intent of lowering PEEPi by achieving a shorter inspiratory time and, thus, allowing more time for exhalation. The associated tachypnea could cause expiratory time to shorten, and thus increase PEEPi , or to lengthen, secondary to continuation of mechanical inflation into neural expiration; which of these opposing scenarios prevails has yet to be determined. One of the main reasons that clinicians increase inspiratory flow is to decrease inspiratory time in the hope of allowing more time for expiration and thus decrease PEEPi , especially in patients with COPD. Because flow usually leads to an increase in rate, the expected shortening of expiratory time might actually produce an increase in PEEPi . Laghi et al. (142) studied this phenomenon in ten patients with COPD (Fig. 26). As with healthy subjects, an increase in flow from 30 to 90 L/min caused respiratory rate to increase from 16.1 ± 1.0 to 20.8 ± 1.5 breaths/min. Despite the increase in rate, PEEPi fell from 7.0 ± 1.3 to 6.4 ± 1.1 cmH2 O. The decrease in PEEPi arose because of an increase in expiratory time, 2.1 ± 0.2 to 2.3 ± 0.2 s, which allowed more time for lung deflation. Why did expiratory time increase? An increase in inspiratory 90 L/min 60 L/min 30 L/min Swing 16.8 19.5 21.5 PEEPi 13.3 14.4 15.6 Flow (L/s) 2 0 Pes cmH2O –2 40 0 Chest volume (a.u.) –40 1 0 –1 0 10 20 30 Time (s) Figure 26 Continuous recordings of flow, esophageal pressure (Pes ), and the sum of rib-cage and abdominal motion in a patient with COPD receiving assist-control ventilation at a constant tidal volume. As flow increased from 30 to 60 and 90 L/min (from right to left; i.e., the opposite of the usual presentation), frequency increased (from 18 to 23 and 26 breaths/min, respectively), PEEPi decreased (from 15.6 to 14.4 and 13.3 cmH2 O, respectively), and end-expiratory lung volume also fell. Increases in flow from 30 L/ min to 60 and 90 L/min also led to decreases in the swings in Pes from 21.5 to 19.5 and 16.8 cmH2 O, respectively. [Adapted, with permission, from Laghi F, Segal J, Choe WK and Tobin MJ. Effect of imposed inflation time on respiratory frequency and hyperinflation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 1365-1370, 2001 (142).] Volume 2, October 2012 2897 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology PTP/min PTP/breath 14 300 200 100 9 0 20 40 60 80 100 25 15 4 0 IMV PS 35 Breaths/min cmH2O.s/breath 400 cmH2O.s/min Frequency 0 20 40 60 80 100 0 20 40 60 80 100 Ventilator assistance (%) Figure 27 Changes in pressure-time product (PTP) per minute (left panel), PTP per breath (middle panel), and frequency (right panel) as the level of intermittent mandatory ventilation (IMV) and pressure support (PS) were progressively increased in 11 ventilator-dependent patients. Ventilator assistance of 0% represents unassisted breathing; PS of 100% represents the level necessary to achieve a tidal volume equivalent to that during assist-control ventilation (10 mL/kg); IMV of 100% is the same ventilator rate as during assist-control ventilation. The left panel shows that the rate of change in PTP per minute over the entire span of assistance did not differ between PS and IMV. The middle panel shows that the rate of change in PTP per breath decreased linearly as the rate of IMV was increased; the decrease with PS was linear only up a medium level and decreased little thereafter. The right panel shows that respiratory frequency decreased linearly as PS was increased, whereas it changed little with IMV until a high level of assistance was provided. See text for interpretation. [Based on data from Leung P, Jubran A and Tobin MJ. Comparison of assisted ventilator modes on triggering, patient effort, and dyspnea. Am J Respir Crit Care Med 155: 1940-1948, 1997 (152).] flow is usually achieved by shortening of mechanical inspiratory time. The shortened inspiratory time combined with time-constant inhomogeneity of COPD will cause overinflation of some lung units to persist into neural expiration. In anesthetized animal models (316,322) and in sleeping human subjects (116), continued inflation during neural expiration causes stimulation of the vagus, which prolongs expiratory time. Whether vagal stimulation is responsible for prolongation of expiratory time during wakefulness in humans remains to be determined. Mode-specific effects of inspiratory unloading Leung and co-workers (152) undertook a head-to-head comparison of the relative effectiveness of progressive increases in the number of ventilator-assisted breaths during IMV and in the level of pressure support in unloading the inspiratory muscles. Over the entire span of assistance, the rate of change in effort per minute did not differ between the modes (analyzed in terms of slopes). The efficacy of unloading, however, did differ according to the stages of assistance. From zero assistance up to a medium level of assistance (60% of maximum), the decrease in patient effort per minute was greater with pressure support than with IMV; the converse was observed as ventilator assistance was increased further to a maximal level (Fig. 27). The changes in effort per breath and frequency help with understanding the different responses (152). Effort per breath decreased linearly as the rate of IMV was increased. Effort per breath also decreased linearly as pressure support was increased to a medium level, but little thereafter, presumably because pressure support provides assistance only if a patient makes some effort. Frequency decreased linearly as 2898 pressure support was increased; with IMV, frequency changed little until a high level of assistance was provided. As IMV is increased from a medium to a high level, all of a patient’s ventilatory requirements will be met and frequency decreases dramatically. Thus, a greater rate of respiratory unloading occurred between medium and high levels of assistance with IMV than with pressure support. Because these two modes are used primarily to provide partial, as opposed to complete, assistance, the greater decrease in patient effort per minute with pressure support at 20% to 60% of assistance makes it a clinically more useful mode than IMV. Pressure support and IMV are commonly combined in a given patient. In an international survey of mechanical ventilation (74), this combination tied with assist-control ventilation as the most commonly used mode of ventilation in North America (34% for each). The rationale for combining the modes is unclear, but presumably clinicians use pressure support to overcome the work imposed by the endotracheal tube and demand valve during the nonmandatory breaths. Examining the response of the respiratory centers to this combination of modes provides useful insight into patient-ventilator interaction. A decrease in the number of mandatory breaths produces a decrease in the average tidal volume (152), with inevitable increase in dead space to tidal volume ratio. To avoid a decrease in alveolar ventilation, the patients increased respiratory motor output, inspiratory effort, and frequency. Adding pressure support of 10 cmH2 O caused a decrease in effort at any given IMV rate. Interestingly, the decrease in effort during the mandatory ventilator breaths was related to the decrease in respiratory motor output during the intervening breaths (r = 0.67) (152). In other words, the reduction in motor output during the intervening breaths achieved by adding Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning pressure support was carried over to the mandatory breaths, facilitating greater unloading. Onset inspir effort I:E flow switch Inspiration-expiration switching (cycling-off function) Volume 2, October 2012 Flow (L/s) 0 Paw H2O –1.5 70 30 –10 70 Pes H2O Patients are commonly ventilated with a volume-cycled mode, such as the volume-cycled form of assist-control ventilation or IMV. This system should more precisely be termed timecycled ventilation because inspiratory flow is preset and the ventilator adjusts inspiratory time to achieve a given tidal volume. Although inflation time is constant with a time-cycled machine, patients invariably display considerable breath-tobreath variability in neural inspiratory time (278). Accordingly, if the machine delivers the set tidal volume before the end of a patient’s neural inspiratory time, ventilator assistance will cease while the patient continues to make an inspiratory effort and double triggering (two ventilator breaths for a single effort) will be a likely consequence. During assist-control ventilation, when a patient’s neural inspiratory time is short, ventilator inflation may continue into neural expiration and thus decrease the time available for lung emptying. The sense of being unable to empty the lungs may cause patients to activate the expiratory muscles with the result that the patient appears to fight or buck the ventilator (Fig. 28). The decrease in time for emptying also increases the likelihood for dynamic hyperinflation and thus inspiratory efforts that fail to trigger the ventilator. The algorithm for cycling-off of mechanical inflation during pressure support varies among brands (33). On most ventilators, the termination of inspiratory assistance during pressure support is set at a default threshold, such as 25% of the peak inspiratory flow or inspiratory flow of 5 L/min. That is, when inspiratory flow falls to 25% of the peak value or below 5 L/min, inspiratory pressurization switches off. Such algorithms can be problematic in patients with COPD because increases in resistance and compliance produce a slow timeconstant of the respiratory system (see Fig. 8). The longer time needed for flow to fall to the threshold value can cause mechanical inflation to persist into neural expiration. In 12 patients with COPD receiving pressure support of 20 cmH2 O, five recruited their expiratory muscles while the machine was still inflating the thorax (129). The patients who recruited their expiratory muscles during mechanical inflation had an average time constant of 0.54 s, compared with an average of 0.38 s in the patients who did not exhibit expiratory muscle activity. The persistence of mechanical inflation into neural expiration is very uncomfortable, as is well recognized with use of inverse-ratio ventilation. Algorithms that achieve better coordination between the end of mechanical inflation and the onset of a patient’s expiration may lessen this form of patient-ventilator asynchrony (299). In ten critically ill patients, Tassaux et al. (260) investigated the influence of variation in the cycling-off threshold on patient-ventilator interaction. Compared with the usual default setting (25% of the peak inspiratory flow), a threshold of 1.5 30 –10 0 2 4 Time (s) 6 Figure 28 Expiratory activity during mechanical inflation. Flow, airway pressure (Paw ) and esophageal pressure (Pes ) in a patient with COPD who is receiving assist-control ventilation. The patient’s first two inspiratory efforts (red vertical lines) trigger the ventilator successfully. The third and fourth inspiratory efforts are very weak (arrows) and do not trigger a mechanical inflation. The last inspiratory effort triggers the ventilator, and at the end of this mechanical inflation (blue vertical line) the patient recruits his expiratory muscles, producing an increase in esophageal pressure above the expected elastic recoil of the lung (dashed tracing) before the termination of mechanical inflation. Expiratory muscle recruitment produces a transient cessation of expiratory flow, which corresponds to a transient increase in esophageal pressure to approximately 20 cmH2 O. 75% of peak inspiratory flow produced a predictable decrease in inspiratory time (from 0.72 ± 0.25 to 0.68 ± 0.23 s), a decrease in PEEPi (from 5.4 ± 2 to 4.8 ± 1.9 cmH2 O), and fewer nontriggering efforts. When the threshold for the termination of inspiratory pressurization was set at 10% of peak inspiratory flow, inspiratory time increased marginally as did the number of nontriggering efforts. While the threshold of 75% of peak inspiratory flow resulted in fewer nontriggering efforts, it also resulted in a lower tidal volume as compared with the default threshold (25% of peak inspiratory flow): 0.46 ± 0.19 versus 0.62 ± 0.2 L (SD). The best trade-off between number of nontriggering efforts and the size of tidal volume needs to be balanced for each individual patient. Younes and co-workers (314) studied 50 patients ventilated with proportional-assist ventilation (PAV), most of whom did not have COPD. Exhalation was delayed intermittently by briefly delaying the opening of the exhalation valve. In response to a delay in the opening of the expiratory valve, all but 5 of 50 patients developed an increase in the duration of neural expiratory time. The increase in duration of neural expiratory time, however, offset only 36 ± 20% of the delay in expiration. Consequently, dynamic hyperinflation worsened. When delays in onset of expiration were 2899 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning introduced in a computer-simulated model, similar increases in dynamic hyperinflation were noted (69). Although the magnitude of expiratory effort does not appear to influence the success of triggering attempts, the time that expiratory efforts commence in relation to the cycling of the ventilator is an important factor. Parthasarathy and co-workers quantified the relationship between the onset of expiratory muscle activity, measured with a wire electrode in the subject’s transversus abdominis, and the termination of mechanical inflation by the ventilator (192). When mechanical inflation extended into the early part of the patient’s (neural) expiration, resulting in a negative phase angle, the time available for unopposed exhalation was shortened. The inadequate time for exhalation leads to hyperinflation with an associated increase in elastic recoil. As a result, patients need to generate greater inspiratory effort to successfully trigger the ventilator. In this way, the time that a patient commences an expiratory effort (in relation to cycling-off of mechanical inflation) partly determines the success of the ensuing inspiratory effort in triggering the ventilator. A further factor that confounds the delineation of mechanisms of patient-ventilator dysynchrony is that inspiratory and expiratory muscle groups can contract simultaneously (66). In the study of Parthasarathy et al. (192), the time of overlap was defined as the period during which late expiratory (transversus abdominis) muscle activity overlapped with early inspiratory muscle (diaphragmatic) activity (see Fig. 18). The average duration of inspiratory and expiratory muscle overlap was on the order of 60 to 90 ms in the subjects, with a range of 0 to 200 ms. Although this period is comparable to the time taken to trigger a ventilator of 100 to 200 ms (220), it did not appear to have any consistent effect on the success of triggering attempts—the period of overlap was not different for triggering and nontriggering attempts at pressure support of 0, 10, or 20 cmH2 O. Variability of breathing The central respiratory pattern generator can be viewed as a homeostatic system influenced by various feedback loops. The confluence of these feedback signals is processed by a controller that produces a homeostatic output exhibiting considerable breath-to-breath variability (35, 278). Studies employing measures of coefficients of variation (standard deviation/mean) of breath components have revealed a wide range of variability in healthy subjects (161,274,283). Breath components that reflect volume and respiratory motor output display greater variability than time components in a variety of experimental conditions, with the exception of slow-wave sleep (161). During slow-wave sleep, breathing is controlled primarily by the metabolic control system located in the brainstem, whereas higher neural centers exert a greater influence on breathing during wakefulness and REM sleep (199). Thus, the greater breath-to-breath variability of respiratory volume compared with respiratory timing does not appear to be an intrinsic property of the central pattern generator, but instead 2900 Printer Name: Yet to Come Comprehensive Physiology reflects the modulating action of higher neural centers on respiratory output (161). Coefficients of variation provide a measure of gross variability, but they do not define the structure within a time series (263). Data sets may have the same coefficient of variation yet differ in the sequential ordering in a time series. A typical breath series looks quite erratic and unpredictable, yet signal-analysis techniques reveal significant structured behavior. Gross variability can be divided into a fixed part, namely the mean of a time series, and a variable part, which is the deviation of the magnitude of each breath from the mean. The variable deviation can in turn be divided into a random uncorrelated, or white noise, fraction and a nonrandom structured fraction (28, 29, 124, 128). The random fraction of breath variability, which can be viewed as the freedom to vary the respiratory cycle, constitutes more than 90% of breath-component activity in awake healthy subjects. The converse of unpredictable random behavior is predictable oscillatory behavior, which accounts for less than 1% of normal breath-component activity. Correlated behavior falls between the two extremes and is partly predictable in that the volume and timing of one respiratory cycle is significantly related to that of the preceding respiratory cycle. Correlated behavior is usually about 3%-10% of normal breath-component activity, and it together with the tiny amount of oscillatory behavior can be viewed as the constraint on respiratory freedom (28, 29, 124, 128). That breath-to-breath variability is mainly random in nature makes it possible for the respiratory system to engage in tasks other than gas exchange, such as speaking (28, 29, 36, 124, 128, 205, 278). A lack of “tightness” in the control of breathing may also be an important buffer against the development of dyspnea (45,108). When mechanical loads are imposed on the respiratory system or when the metabolic needs of the body are increased, it becomes increasingly difficult for suprapontine ventilatory commands to compete against the automatic control system (36, 252). Such challenges are generally accompanied by significant decreases in the random fraction (28, 29, 124). This decrease in random variability may lessen the freedom of the respiratory system to undertake behavioral tasks (28, 29, 100). Abnormalities in the rhythmicity of breathing can have devastating consequences and be responsible for sudden death in patients with the sleep apnea syndrome, sudden infant death syndrome, Cheyne-Stokes syndrome, and other disorders (269). Information on how breath-to-breath variation in controller output contributes to the clinical manifestations of lung disease is scant, and limited to one disease state. Patients with restrictive lung disease exhibit distinctive alterations in the variability of breathing (Fig. 29). The random fraction of breath variability was as much as 27 times smaller in patients with restrictive lung disease than in healthy subjects (30). Conversely, correlated behavior was up to three times greater in the patients. Random variability in tidal volume and expiratory time exhibited the most striking changes, which were also the fraction and breath component most affected Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 1500 Tidal volume (mL) 1000 500 0 2000 1500 1000 500 0 20 40 60 Time (min) Figure 29 Breath-to-breath values of tidal volume during 1 h of resting breathing in a patient with restrictive lung disease (top panel) and in a healthy control subject (bottom panel). The coefficient of variation for tidal volume in the patient (0.13) was more than five times smaller than that in the healthy subject (0.72). [Adapted, with permission, from Brack T, Jubran A and Tobin MJ. Dyspnea and decreased variability of breathing in patients with restrictive lung disease. Am J Respir Crit Care Med 165: 1260-1264, 2002 (30).] when healthy subjects breathed against an external elastic load (28). When ventilation is measured nonobtrusively and without the use of a mouthpiece, as by means of inductive plethysmography, considerable variation in the average level of ventilation is evident among healthy subjects. For example, a 15-min nonobtrusive recording of resting breathing in 65 healthy subjects revealed a tidal volume of 383 ± 91 (SD) ml (268). Equivalent nonobtrusive recordings revealed higher tidal volumes in patients with stable COPD: 447 ± 139 mL in 16 eucapnic patients and 476 ± 158 mL in 12 hypercapnic patients (268). The highest tidal volumes among patients with various respiratory disorders were observed in 15 subjects with symptomatic but stable asthma: 679 ± 275 mL (268). In addition to the substantial variation in average tidal volume among subjects, each individual subject exhibited considerable breath-to-breath variability in all breath components. Gross within-day, within-subject, breath-to-breath variability in tidal volume, as reflected by coefficient of variation, was 33.0 ± 14.9% (SD) in 47 young healthy subjects (21-50 years) and significantly higher in 18 healthy older subjects (60-81 years), 44.0 ± 14.7% (274). The substantial variability in breathing pattern between subjects, further magnified by the considerable breath-to-breath variability within subjects, poses a problem in selecting settings during mechanical ventilation. If tidal volume is set during volume-cycled assist-control ventilation at the average value during unassisted breathing, 5-7 mL/kg, the wide breath-to-breath variation in ventilatory demand means that around half of the delivered tidal volumes Volume 2, October 2012 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 2000 0 9:35 will be lower than the patient’s demand, which may lead to dyspnea (308). It is possible that comfort will be attained only when the set tidal volume approaches or exceeds the greatest demand within the range of spontaneous breathing. If a higher tidal volume were selected, such as 12-15 mL/kg, it is likely that the breath-to-breath variation in demand will be satisfied in virtually all patients. Most of the demand of the ventilatory control system, however, would be satisfied with a lower volume setting. Younes, however, has speculated that the wide variation in breathing pattern between subjects and also the large variability on a breath-to-breath basis may explain why a high tidal volume setting (12-15 mL/kg, as opposed a normal value of 5-6 mL/kg) has been traditionally recommended during assist-control ventilation (308) (Fig. 30). More recently, much lower tidal volumes have been employed, especially in patients with ARDS, as randomized trials have revealed a significantly lower mortality with a tidal volume of 6 mL/kg as compared with a tidal volume of 12 mL/kg (1, 6). PAV is a form of ventilator assistance in which the ventilator generates pressure in proportion to patient effort. While receiving PAV, patients are able to achieve greater variation in breath components than with conventional ventilator modes. In 11 patients being ventilated for a variety of reasons, Marantz et al. (164) altered the level of PAV from near-maximal levels to the lowest tolerable level of assistance. In this way, the constraint of abnormal respiratory mechanics on the respiratory control system was progressively lessened as the level of PAV was gradually increased. The most consistent finding was that variation in the level of PAV produced no consistent effect on respiratory frequency, tidal volume, or minute ventilation. At the highest and lowest level of assistance, respiratory frequency was respectively 25.0 ± 4.2 and 25.7 ± 3.9 breaths/min. Likewise, minute ventilation did not change consistently between the highest and lowest level of assistance: 12.8 ± 5.4 (SD) versus 11.6 ± 4.3 L/min. These findings suggest that within an individual patient and for a particular state there exists a unique set of values for tidal volume, respiratory frequency and minute ventilation that are largely independent of the mechanical load; if the level of PAV is increased, patient effort is decreased to maintain the desired ventilatory targets. While the values of a breath component may be relatively constant and independent of load within a given patient, Marantz et al. (164) observed considerable variation in the values of a breath component among the patients. Minute ventilation, which reflects ventilatory demand, varied from 5.6 to 18.7 L/min among patients. At the highest level of PAV, tidal volume ranged from 203 mL (3.4 mL/kg) to 844 mL (9.4 mL/kg) among patients. The average tidal volume was 517 ± 217 mL (7.46 ± 3.25 mL/kg) at the highest level of PAV, which was considerably less than the delivered tidal volume while the patients had been receiving IMV or pressure support before entry into the study. The patients’ tidal volume during PAV was not correlated with any measure of respiratory mechanics or with the underlying disease. The only variable that correlated with tidal volume was minute ventilation: r = 0.91. 2901 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology 2.0 Flow (L/s) 1.0 0.0 –1.0 –2.0 Esophageal pressure (mmHg) 12 8 4 0 –4 –8 0 25 50 75 Time (s) Figure 30 Variability of breathing. Time series of airflow and esophageal pressure in a patient with acute respiratory failure secondary to an acute exacerbation of COPD who is being ventilated with pressure support of 5 cmH2 O (PEEP, 0 cmH2 O). The esophageal-pressure tracing exhibits wandering values at end-expiration and varying inspiratory excursions; these features are somewhat less evident on the flow tracing. The strength of this correlation suggests that more than 80% of the variability in tidal volume in ventilated patients is determined by alterations in ventilatory demand, presumably arising from interindividual differences in ventilatory drive. Because factors other than ventilatory demand accounted for less than 20% of the variability in tidal volume, Marantz et al. (164) went on to argue that receptor stimulation or malfunction secondary to a given underlying disease plays little role in the regulation of tidal volume in ventilated patients; whether this possibility, based on correlative data, can be generalized remains to be determined. As witnessed with PAV, another new ventilator mode also achieves greater breath-to-breath variation in breath components than allowed with conventional modes. NAVA, which delivers positive pressure in relation to a patient’s diaphragmatic electrical activity, is still in the early stages of clinical application. In 12 patients with a moderate lung injury who were being weaned from the ventilator, Schmidt et al. (230) found that switching from pressure support to NAVA resulted in no change in the mean values of breath components but there were significant increases in the coefficients of variation of tidal volume and mean inspiratory flow (V T /T I ). Similar trends were observed with autocorrelation analysis: the lag of V T /T I was lower at the highest setting of NAVA compared with both pressure support and the lowest level of NAVA; moreover, the autocorrelation coefficient of V T /T I and of respiratory frequency was lower at the highest setting of NAVA than at the lowest level of NAVA. The observation that progressive unloading of the respiratory system with increasing levels of NAVA produced an increase in breath-to-breath vari- 2902 ability of breathing and a decrease in the correlated fraction is consistent with studies in healthy subjects during external loading (28, 29) and in patients with restrictive lung disease (30). At the present time, it is not known whether induced changes in variability of breathing in ventilator-supported patients lead to improvements in patient outcome. PAV and NAVA are not the only methods of increasing the variability of breathing during mechanical ventilation. The variation in breathing during mechanical ventilation with PAV and NAVA is generated by the patient’s respiratory centers. More recently, investigators have examined the effects of variation in breathing imposed by a ventilator, so-called “noisy ventilation” (257, 261). Studies conducted in experimental animals have revealed a number of benefits in response to the deliberate imposition of some variability during mechanical ventilation (257, 261). Noisy ventilation involves the deliberate imposition of a coefficient of variation in tidal volume of up to 40%. In animal models of lung injury, noisy ventilation has been shown to improve oxygenation, reduce peak inspiratory airway pressure, reduce elastance, and decrease histological damage even when compared with the use of a low tidal volume (11, 25, 92, 169, 250, 251). To date, noisy ventilation has not been studied in human subjects. Ventilator Weaning When patients have recovered sufficiently from the illness that precipitated the need for mechanical ventilation, attempts are made to disconnect them from the machine. In the early days Volume 2, October 2012 P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology of intensive care units and mechanical ventilation (from late 1950s to mid 1960s), physicians were worried about discontinuing the ventilator too abruptly; it was believed that patients needed to slowly learn how to breathe on their own again. Consequently, the approach to the ventilator withdrawal was very slow—just a minute or two of spontaneous breathing once or twice a day (267). This gradualist approach was appropriately called “weaning.” Today, it is recognized that the majority of patients who demonstrate reasonable recovery of respiratory function after a period of mechanical ventilation (patients who perform well on weaning-predictor tests) will tolerate the first trial of spontaneous breathing without a gradualist weaning approach. Nevertheless, the short descriptor weaning is used in place of the more accurate but cumbersome term “discontinuation of mechanical ventilation,” and attempts to introduce other descriptors have not been successful. Of patients who are deemed ready for a weaning trial, about 20% to 30% will develop severe distress during the first trial of spontaneous breathing and require the resumption of mechanical ventilation. If the trial is extended, these weaning-failure patients will develop hypercapnia unless severe hypoxemia first intervenes. Because these patients develop acute respiratory failure over a relatively short period of time (usually 15-90 min), they have become the most employed experiment of nature for studying the pathophysiology of acute respiratory failure. Moreover, the patients who tolerate a weaning trial, and successfully sustain spontaneous ventilation following extubation, provide a control group for comparison. The mechanisms that cause weaning failure can be divided into those occurring at the level of control of breathing, mechanics of the lung and chest wall, the respiratory muscles, the cardiovascular system, and gas-exchange properties of the lung. Control of breathing Patients who fail a trial of spontaneous breathing commonly develop hypercapnia. This observation led many investigators to suspect that depression of respiratory motor output was responsible. When respiratory motor output was directly measured, however, it was found to increase between the onset and end of a failed weaning trial as the patients were developing progressive hypercapnia. In the study of Tobin et al. (277), patients developed acute severe distress accompanied by an increase in PaCO2 , from 42 to 56 mmHg, and a fall in pH from 7.43 to 7.35. Between the beginning and end of the trial, which lasted 40 ± 11 min, the patients developed an increase in mean inspiratory flow (V T /T I ): 265 ± 27 to 328 ± 32 mL/s (Fig. 31). No patient had a value of V T /T I below the 95% confidence limits of normal subjects. Subsequent studies using the airway occlusion method (P0.1) as a measure of respiratory motor output revealed that respiratory motor output is higher in weaning-failure patients than in weaning-success patients (43, 173, 226, 227). The major mechanism for acute hypercapnia in weaningfailure patients is a change in the pattern of breathing rather Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 500 400 Success Failure 300 VT P1: OTA/XYZ 200 100 0 0 1 2 3 Time (s) Figure 31 The mean respiratory cycle during spontaneous breathing in seven weaning-failure and ten weaning-success patients. The early termination of inspiratory time in the weaning-failure patients leads to a decrease in tidal volume. The decrease in inspiratory time, coupled with a decrease in expiratory time, results in a faster respiratory frequency. Bars represent 1 SE. [Adapted, with permission, from Tobin MJ, et al. The pattern of breathing during successful and unsuccessful trials of weaning from mechanical ventilation. Am Rev Respir Dis 134: 11111118, 1986 (277).] than respiratory center depression. Patients who go on to fail a weaning trial develop an almost immediate change in breathing pattern as soon as they are disconnected from a ventilator (277) (Fig. 32). The dominant finding is a shortening of inspiratory time. For example, in the study of Tobin et al. (277), inspiratory time was 0.8 ± 0.1 s in weaning-failure patients versus 1.4 ± 0.3 s in weaning-success patients. At the level of the respiratory centers, expiratory time is strongly coupled to inspiratory time (47). Consequently, expiratory time was also shorter in the weaning-failure patients than in the weaning-success patients: 1.2 ± 0.3 versus 2.5 ± 0.5 s. The combined changes in inspiratory time and expiratory time led to a marked increase in respiratory frequency: 33 ± 2 versus 21 ± 3 breaths/min. Because the rate of inspiratory flow (V T /T I ) was equivalent in the two patient groups, the short inspiratory time resulted in a lower tidal volume in the failure patients: 194 ± 23 versus 398 ± 56 mL. The decrease in tidal volume was balanced by the increase in frequency, and thus minute ventilation was equivalent in the two groups. A decrease in tidal volume without an increase in minute ventilation must result in higher overall dead-space ventilation (V D /V T ); indeed, the combined changes in tidal volume and respiratory frequency accounted for 81% of increase in PaCO2 observed in the weaning-failure patients. An attempt to compensate for the low tidal volume through an increase in frequency will produce an increase in work of breathing. According to the model of Otis et al. (189), the increase in work with rapid shallow breathing is exponential: work = 4035e (0.0017*f /V T ) , r = 0.90 (272) (Fig. 33). 2903 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology 0.6 100 Start of trial PaCO2 = 48 End of trial PaCO2 = 73 Tidal volume (L) Respiratory frequency (breaths/min) 0.5 50 0.4 0.3 0.2 0.1 Tidal volume (mL) 0 0.0 800 –20 400 –5 0 5 10 15 20 Figure 34 Two superimposed Campbell diagrams of work of 0 8 4 12 Minutes 16 20 Figure 32 A time-series, breath-by-breath plot of respiratory frequency and tidal volume in a patient who failed a weaning trial. The arrow indicates the point of resuming spontaneous breathing. Rapid, shallow breathing developed almost immediately after discontinuation of the ventilator. [Adapted, with permission, from Tobin MJ, et al. The pattern of breathing during successful and unsuccessful trials of weaning from mechanical ventilation. Am Rev Respir Dis 134: 1111-1118, 1986 (277).] 3.0 Work (Joules/min) –10 Pes (cmH2O) 0 2.5 2.0 Work = 4035e (0.0017* f/VT) r = 0.90 1.5 0 50 150 100 200 Frequency/tidal volume (b/min/L) 250 Figure 33 The relationship between work of breathing and the frequency-tidal volume (f/V T ) ratio for a constant alveolar ventilation of 4 L/min based on the model of Otis and collaborators (189). Work of breathing is least for f/V T ratios of 37 to 59; it increases exponentially as f/V T increases from 59 to 234/min/L. [Adapted, with permission, from Tobin MJ, Laghi F and Brochard L. Role of the respiratory muscles in acute respiratory failure of COPD: lessons from weaning failure. J Appl Physiol 107: 962-970, 2009 (272).] 2904 –15 breathing in a patient at the start and end of a trial of spontaneous breathing. Over the course of the weaning trial, the patient developed an increase in PaCO2 from 48 to 73 mmHg, but minimal change in overall work of breathing, suggesting that the hypoventilation resulted from either failure of respiratory motor output to increase during the weaning trial or an inability of the respiratory controller to sense the rise in PaCO2 . Several groups of investigators have shown that rapid shallow breathing (high frequency, low tidal volume) is a characteristic finding in weaning-failure patients (227, 271, 285) (Fig. 32). The degree of rapid shallow breathing can be quantified by calculating the ratio of respiratory frequency to tidal volume (f /V T ) (302). The higher the ratio, the more severe is the rapid, shallow breathing. An f /V T ratio of 100 breaths/min/L has been shown to be the best predictor of weaning outcome (302), and has become part of routine diagnostic testing in ventilated patients. While most patients who fail a weaning trial exhibit an elevated respiratory motor output and also excessive mechanical load or insufficient respiratory muscle strength (or both), a small proportion of weaning-failure patients display none of these features. For example, Jubran and Tobin (127) observed that 2 of 17 (12%) weaning-failure patients developed PaCO2 values greater than 70 mmHg during a weaning trial, and yet detailed measurements of their lung mechanics and respiratory muscle function were within the range of the weaning-success patients (Fig. 34). On the basis of exclusion, depression of respiratory motor output was judged to be the dominant reason for acute hypercapnia in these patients. Respiratory mechanics Physiological variables for quantifying lung mechanics can be grouped under three major headings: resistance, elastance, and gas trapping. The most detailed data on respiratory mechanics in patients being weaned from mechanical ventilation comes from a study by Jubran and Tobin of 31 patients with Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 R insp,L (cmH2O/L/s) 15 Edyn,L (cmH2O/L) 40 PEEPi (cmH2O) Comprehensive Physiology 5 10 Failure Success *P < 0.003 5 0 * 0 2.5 * 0 0 Start End Figure 35 Inspiratory resistance of the lung (Rinsp,L ), dynamic lung elastance (Edyn,L ), and intrinsic positive end-expiratory pressure (PEEPi ) in 17 weaning-failure patients and 14 weaning-success patients. Data displayed were obtained during the second and last minute of a T-tube trial, and at one third and two-thirds of the trial duration. Between the onset and end of the trial, the failure group developed increases in Rinsp,L (P < 0.009), Edyn,L (P < 0.0001), and PEEPi (P < 0.0001) and the success group developed increases in Edyn,L (P < 0.006) and PEEPi (P < 0.02). Over the course of the trial, the failure group had higher values of Rinsp,L (P < 0.003), Edyn,L (P < 0.006), and PEEPi (P < 0.009) than the success group [Adapted, with permission, from Jubran A and Tobin MJ. Pathophysiologic basis of acute respiratory distress in patients who fail a trial of weaning from mechanical ventilation. Am J Respir Crit Care Med 155: 906-915, 1997 (127).] COPD undergoing a weaning trial (127). Over the course of a trial of spontaneous breathing lasting 45 ± 8 min, 17 patients developed acute distress and an increase in PaCO2 (from 45 to 58 mmHg), requiring the reinstitution of mechanical ventilation. The remaining 14 patients tolerated the trial and were successfully extubated; these served as a control group. At the start of the weaning trial, inspiratory lung resistance was markedly elevated, but not significantly different, in the failure and success patients: 9.0 ± 1.7 versus 5.3 ± 1.1 cmH2 O/L/s (127) (Fig. 35). By the end of the trial, resistance increased to 14.8 ± 2.0 cmH2 O/L/s in the failure patients, but it did not change in the success patients. The mechanism of the progressive increase in inspiratory resistance in the failure patients is not known, although bronchoconstriction consequent to heightened airway reactivity is a possible candidate. Dynamic lung elastance was higher in failure patients than in success patients at the start of the trial: 21.2 ± 3.4 versus 9.9 ± 1.7 cmH2 O/L (127) (Fig. 35). At the end of the trial, elastance increased to 34.1 ± 4.0 cmH2 O/L in the failure patients and to 14.0 ± 2.0 cmH2 O/L in the success patients. The elevated elastance at the start of the trial was Volume 2, October 2012 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning * 20 9:35 probably secondary to frequency dependence of elastance. The mechanism of the progressive increase in elastance over the course of the trial is uncertain, but it may be related to progressive dynamic hyperinflation (285) as discussed later. PEEPi , an indirect measure of gas trapping, was higher in the failure patients than in the success patients at the onset of the trial: 2.0 ± 0.5 versus 0.7 ± 0.1 cmH2 O. By the end of the trial, PEEPi increased to 4.1 ± 0.8 cmH2 O in the failure patients and to 1.1 ± 0.2 cmH2 O in the success patients (127) (Fig. 35). Jubran and Tobin did not partition total PEEPi into the component resulting from expiratory muscle contraction and that resulting from an increase in end-expiratory lung volume. This information was subsequently obtained by the same group of investigators (191), who partitioned total PEEPi into that resulting from expiratory muscle contraction (abdominal muscles, expiratory rib-cage muscles, or both) by calculating the rise in gastric pressure (Pga ) between the onset of expiratory flow and the point of rapid decline in esophageal pressure (Pes ), and the remaining portion, suggesting an increase in end-expiratory lung volume. After correcting for expiratory-muscle contribution, the remaining portion of total PEEPi increased between the start and end of the weaning trial in seven of the ten patients (191). These data suggest that many weaning-failure patients develop dynamic hyperinflation. Expiratory flow limitation (134) and tachypnea, through a decrease in time available for exhalation (277), are the most likely determinants of dynamic hyperinflation. It should be recognized that it has not been possible to obtain direct measurements of end-expiratory lung volume in patients experiencing acute respiratory failure, and the use of esophageal pressure to estimate this entity is based on many assumptions (310). Other investigators have also reported a worsening of lung mechanics in weaning-failure patients. An innovative approach was employed by Vassilakopoulos et al. (285). They first studied patients at the end of a T-tube trial. Then they reinstituted ventilation in the assist-control mode, sedated the patients, and hyperventilated them to abolish spontaneous respiratory muscle activity. Later, they adjusted the ventilator settings to simulate a patient’s pattern of spontaneous breathing, and measured lung mechanics under passive conditions. The investigators studied patients at two points in time: shortly after they first failed a T-tube trial, and about 9 days later, shortly before they were successfully extubated. Between the time of weaning failure and weaning success, airway resistance decreased from 9.6 ± 3.4 to 7.9 ± 3.3 cmH2 O/L/s, static PEEPi decreased from 6.1 ± 2.5 to 3.8 ± 2.7 cmH2 O, and static respiratory compliance did not change. The investigators also disconnected the patients from the ventilator (after first delivering some breaths simulating spontaneous breathing), and allowed them to exhale freely until zero expiratory flow was reached. This point was taken as the elastic equilibrium volume of the respiratory system, and the increase in FRC secondary to gas trapping (PEEPi ) was taken as the difference between inspired and expired volume. This 2905 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Patient effort The deterioration in lung mechanics during a failed weaning trial leads to increased work of breathing. The increased respiratory work is made manifest by greater swings in esophageal pressure (Fig. 36). In the study of Jubran and Tobin, pressuretime product was not different in the weaning-failure and weaning-success patients at the onset of a spontaneous breathing trial: 255 ± 59 and 158 ± 23 cmH2 O.s/min (normal, 94 ± 12) (127). At the end of the trial, pressure-time product increased more in the failure patients than in the success patients: 388 ± 68 versus 205 ± 25 cmH2 O.s/min. The increase in effort in the failure patients resulted from worsening of all elements of respiratory mechanics. Partitioning of the increase in pressure-time product at end of the trial revealed that the fraction caused by PEEPi increased by 111%, that caused by the non-PEEPi elastic component increased by 33%, and the fraction caused by the resistive component increased by 42% (127). In 60 patients being weaned from mechanical ventilation, Jubran et al. (123) characterized the changes in esophagealpressure swings over the course of a trial of spontaneous breathing (Fig. 37). The median time (plus interquartile range) to reach ±10% of the average value of esophageal pressure during the last minute of the trial was 7.5 (4.2–14.8) min in the weaning-failure patients and 5 (2–8.5) min in the weaning-success patients (Fig. 38). In contrast, frequency-totidal volume ratio (f /V T ), a measure of rapid shallow breathing, reached ±10% of its final average value at 2 (1–2) min in both the weaning-success and weaning-failure patients. The 2906 Flow (L/s) 1.25 0 –1.25 50 Pes cmH2O volume was 327 ± 180 mL during weaning failure, and it fell to 213 ± 175 mL at the time of weaning success. The observation that weaning-failure patients display more severely deranged lung mechanics than do weaningsuccess patients raises the question of whether the derangements might be detectable even before patients reattempt spontaneous breathing (that is, while patients are still receiving full ventilator support). Jubran and Tobin (126) studied lung and chest wall mechanics of patients before the onset of a weaning trial. Inspiratory resistance was about 14 times higher than that in healthy subjects but it was equivalent in the failure and success patients: 13.9 and 13.0 cmH2 O/L/s, respectively. Static elastance of the lung and the chest wall were similar in the two groups (126). Dynamic elastance of the lung was higher in the failure patients than in the success patients, 28 ± 3 versus 17.8 ± 2 cmH2 O/L, and this was the only measurement of passive mechanics that differentiated the two groups. Dynamic PEEPi during passive ventilation was equivalent in the groups. This picture contrasts with the more severely deranged mechanics in failure patients than in success patients during a weaning trial (127). The difference indicates that something in the act of spontaneous breathing, rather than an intrinsic abnormality in respiratory mechanics, is responsible for the marked difference between failure and success patients during a weaning trial. Comprehensive Physiology 0 –50 0 2 4 0 Time (s) 2 4 6 Figure 36 Respiratory effort during a weaning trial. Recordings of flow (inspiration directed upward) and esophageal pressure (Pes ) in a patient with severe COPD who failed a weaning trial (left panel) and in a weaning-success patient, who had been intubated because of an opiate overdose (and had no lung disease) (right panel), 10 min into a trial of spontaneous breathing. The weaning-failure patient exhibits a steeper fall and greater excursion in esophageal pressure than did the weaning-success patient—features that signify greater respiratory motor output. Despite the threefold larger excursion in esophageal pressure in the weaning-failure patient, peak inspiratory flow in this patient is only twice as great as in the weaning-success patient, signifying more abnormal mechanics in the weaning-failure patient. The duration of respiratory cycle was shorter in weaning-failure patient signifying tachypnea. The expiratory flow in the weaning-failure patient demonstrates a supramaximal flow transient at the beginning of exhalation that is typical of expiratory flow limitation. more gradual and progressive increase in esophageal pressure over time may have resulted from a slow increase in PCO2 , as commonly occurs in weaning-failure patients (127, 277). The relative constancy in f /V T may be related to the increases in both inspiratory resistive and inspiratory elastic loads that occur in weaning-failure patients. These loads have opposing effects on breathing pattern. A resistive load slows respiratory frequency while preserving tidal volume, thereby producing a decrease in f /V T (29, 309). In contrast, an elastic load increases respiratory frequency accompanied by a decrease in tidal volume, both of which will produce an increase in f /V T (28, 208). Respiratory muscles Research into the mechanisms whereby abnormalities of the respiratory muscles might contribute to weaning failure has focused on inspiratory muscle strength and respiratory muscle fatigue. Muscle strength has been assessed by measuring the pressure generated during a maximal inspiratory effort against an occluded airway (143). Early investigators reported that maximal inspiratory pressure (PI max) was lower in weaning-failure patients than in weaning-success patients, but later investigators reported no difference between the two groups (84, 138, 226, 278, 317) The pattern of reporting suggests the possibility of test-referral bias, whereby patients Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning Flow (L/s) 0.8 0.4 0.0 –0.4 Pes (cmH2O) 10 0 –10 Paw (cmH2O) –20 8 4 0 –4 10 0 30 20 Time (s) 25 250 20 200 f /VT, (breaths/min/L) Pes swings (cmH2O) Figure 37 Progressive increase in inspiratory effort in a weaning-failure patient. Flow (top panel), esophageal pressure (Pes , middle panel), and airway pressure (Paw , lower panel) in a patient who developed severe respiratory distress while receiving continuous positive airway pressure (CPAP) of 5 cmH2 O. The patient had developed respiratory failure (requiring mechanical ventilation) after developing a pulmonary embolus subsequent to undergoing lobectomy for lung cancer. The swings in esophageal pressure became progressively more negative over the first 30 s of the trial. 15 10 5 150 100 50 0 0 10 20 30 0 0 40 Time (min) 10 20 30 40 Figure 38 Time-series plot of swings in esophageal pressure (Pes ; left panel) and frequencyto-tidal volume ratio (f/V T ; right panel) during a trial of spontaneous breathing in a weaningfailure patient. Black dots represent 1-min averages. The solid line indicates the average value of Pes swings and f/V T of the final minute of the trial. The dashed lines indicate ±10% of the final minute values of Pes swings and f/V T . The time taken to reach ±10% of the final value was 14 min for Pes swings and 2 min for f/V T . [Adapted, with permission, from Jubran A, et al. Weaning prediction: esophageal pressure monitoring complements readiness testing. Am J Respir Crit Care Med 171: 1252-1259, 2005 (123).] Volume 2, October 2012 2907 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning with the lowest values of PI max were deliberately excluded from the subsequently conducted studies (271). Another consideration is the well-recognized difficulty of ensuring reliable measurements of PI max, given its total dependence on patient motivation and cooperation—an even greater challenge in critically ill patients (143). Studies using phrenic nerve stimulation, and specifically the technique of twitch interpolation, revealed that patients being weaned from mechanical ventilation typically make submaximal efforts when PI max is being measured. Moreover, Laghi et al. (138) found that six of nine weaning-failure patients had twitch Pdi values below 10 cmH2 O. Healthy subjects have twitch Pdi values of 35 to 39 cmH2 O, and stable patients with COPD have values of 17 to 20 cmH2 O. Contrary to previous thinking, these data indicate that weaning-failure patents may have considerable muscle weakness. Evidence has been accumulating for the presence of ventilator-induced respiratory muscle injury as a mechanism responsible for respiratory muscle weakness (273, 284). A seminal study showed that 11 days of controlled mechanical ventilation produced a 46% decrease in respiratory muscle strength in baboons (8). Subsequent studies have revealed that complete cessation of diaphragmatic activity with controlled mechanical ventilation—alone (222) or in combination with neuromuscular blocking agents (303)—results in injury and atrophy of diaphragmatic fibers. Muscle fibers generate less force in response to stimulation, not simply because of their decreased bulk but even when normalized for cross-sectional area. The decrease in diaphragmatic force ranges from 20% to more than 50%. The alterations in muscle function occur rapidly, within 12 h of instituting mechanical ventilation in rats (168) and within one day in patients (119). These alterations increase as ventilator duration is prolonged (119, 204). Oxidative stress appears to be the most proximal mechanism in the biochemical cascade that leads to ventilatorinduced muscle injury (203, 291). Oxidative stress decreases contractility by causing protein oxidation and by promoting protein catabolism (203). Other mechanisms that contribute to muscle-protein loss include apoptosis (168) and decreased protein synthesis (233). Levine et al. (154) reported human data that support the findings of the animal studies. They obtained biopsies of the costal diaphragms from 14 brain-dead organ donors. These patients exhibited diaphragmatic inactivity and had received mechanical ventilation for 18 to 69 h. They also obtained intraoperative biopsies of the diaphragms of eight patients undergoing thoracic surgery for suspected lung cancer; these control patients had experienced diaphragmatic inactivity and mechanical ventilation for 2 to 3 h. Histologic measurements revealed marked diaphragmatic atrophy in the brain-dead patients. Compared with the control group, the mean crosssectional areas of muscle fibers were significantly decreased by more than 50%. Many of these findings have been corroborated by Jaber et al. (119), who also reported a progressive decrease in diaphragmatic contractility of mechanically ventilated patients—the extent of which was correlated with 2908 Printer Name: Yet to Come Comprehensive Physiology the duration of ventilator support. In the study of Levine et al. (154), the cross-sectional area of fibers of the pectoralis major, a muscle not affected by mechanical ventilation, was equivalent in the two groups. This finding indicates that the diaphragmatic atrophy experienced by the brain-dead patients was not part of some generalized muscle-wasting disorder. Biochemical and gene-expression studies suggest that the atrophy results from oxidative stress leading to muscle protein degradation. Evidence of oxidative stress is indicated by a 23% lower concentration of glutathione in the diaphragms of brain-dead patients than in controls (154). Evidence of enhanced muscle protein degradation is indicated by a 100% to 400% greater expression of calpain-1, 2, and 3 (119) and 150% greater expression of active caspase-3 in the braindead patients than in the controls (154). Calpains are calciumactivated proteases implicated in several aspects of skeletal muscle injury and atrophy (119), and caspase-3 is an enzyme that can dissociate proteins from the myofibrillar lattice, a critical step in muscle proteolysis. Muscle proteolysis typically involves the ubiquitinproteasome pathway, a cytosolic ATP-dependent protease system (143). In this system, proteins catabolized by the proteasome are first “tagged” with a small chain of ubiquitin molecules. Tagging with ubiquitin is an ATP-requiring process that involves specific “ubiquitin ligases” such as atrogin1 and muscle ring finger-1 (MuRF-1) (143). In the study of Levine et al. (154), the number of messenger RNA transcripts for atrogin-1 and MuRF-1 were 200% and 590% higher, respectively, in the brain-dead patients than in the controls. In the study of Jaber et al. (119), ubiquitinated proteins were 19% higher in the brain-dead patients than in the controls. For years, researchers had believed that most if not all weaning-failure patients develop respiratory muscle fatigue by the completion of a failed weaning trial. This belief was largely based on observations made by Cohen et al. (49) in a study of 12 patients who exhibited difficulties during weaning. Seven patients developed a shift in the power spectrum of the EMG signal recorded from the diaphragm, a finding judged to signify muscle fatigue (32). In the study of Cohen et al. (49), six of the seven patients also exhibited paradoxical motion of abdomen (inward displacement of abdomen during inspiration) and four exhibited respiratory alternans (phasic alternation between the contribution of the rib-cage and abdominal compartments to tidal volume). The changes in rib cage-abdominal motion were not observed in the five patients who did not develop EMG changes. The investigators concluded that respiratory muscle fatigue was a common cause of weaning failure and its presence could be detected by finding paradoxical motion of abdomen. Subsequent detailed recordings of rib cage-abdominal motion revealed that when paradoxical motion of the abdomen occurs in weaning-failure patients, it occurs immediately upon disconnection from the ventilator and it displays no progression over time (270). In addition, mathematical computations of the extent of abdominal paradox were no greater in failure patients than in success patients. In studies of healthy Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology volunteers, fatigue was found to be neither a necessary nor a sufficient condition for the development of abnormal rib cage-abdominal motion (276). These data indicate that rib cage-abdominal motion cannot be used for detecting respiratory muscle fatigue. The studies, however, did not exclude the possibility that fatigue is common in weaning-failure patients. Investigators have evaluated tension-time index as an index of fatigability. Studies in healthy volunteers have shown that task failure becomes inevitable when subjects breathe against an inspiratory load that causes the tension-time index to rise above a threshold of 0.15 (18). In a number of studies (42, 127, 285), weaning-failure patients more commonly had tension-time index values above 0.15 than did weaningsuccess patients. At completion of a weaning trial, other measures of inspiratory effort, such as pressure-time product, were also markedly higher in weaning-failure patients, 388 ± 68 (SE) cmH2 O.s/min, than in weaning-success patients, 205 ± 25 cmH2 O.s/min (normal, 94 ± 12 cmH2 O.s/min) (127). Given that the high pressure-time product occurred in conjunction with marked elevations in respiratory frequency, 37.1 ± 2.5 breaths/min, it is evident weaning-failure patients experience workloads that are sufficient to induce task failure and, possibly, respiratory muscle fatigue. EMG power spectrum and tension-time index provide only indirect evidence of fatigue, and do not provide direct proof of its occurrence. The most direct method for detecting fatigue in patients is to stimulate the phrenic nerves and measure the resulting change in Pdi .When employing this technique in critically ill patients, it is especially challenging to ensure that successive twitches are all generated at the same end-expiratory lung volume, to ensure a constant degree of neural depolarization by the stimulator, and ensure that twitch potentiation (the transient increase in pressure that occurs with a recent forceful contraction) does not occur (139, 140). Controlling for these factors, Laghi et al. (138) studied 11 weaning-failure patients and eight weaningsuccess patients before and after a T-tube trial. Twitch Pdi was 8.9 ± 2.2 H2 O before and 9.4 ± 2.4 H2 O after the trial in the weaning-failure patients (Fig. 39). The respective values in the weaning-success patients were 10.3 ± 1.5 and 11.2 ± 1.8 cmH2 O. Not a single patient developed a decrease in twitch Pdi . The absence of fatigue was surprising because seven of the nine weaning-failure patients had a tension-time index above the threshold reported to lead to task failure and fatigue (0.15) (18). One likely reason that patients did not develop fatigue is because physicians reinstituted mechanical ventilation before there was sufficient time for its development. The relationship between the tension-time index and the length of time that a load can be sustained until task failure follows an inversepower function. Bellemare and Grassino (18) expressed the relationship as: time to task failure = 0.1 * (tension-time index)−3.6 . The increase in the tension-time index over the course of the weaning trial (138) and predicted time to task failure (18) are shown in Figure 40. At the point that the physician reinstituted mechanical ventilation, patients were Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning After Before 15 Pes (cmH2O) Pga (cmH2O) 0 –15 40 20 0 Pdi (cmH2O) 40 20 0 10 R-CMAP (a.u.) 0 –10 10 L-CMAP (a.u.) 0 –10 0 0.2 0.4 0 Time (s) 0.2 0.4 Figure 39 Esophageal pressure (Pes ), gastric pressure (Pga ), transdiaphragmatic pressure (Pdi ), and compound motor action potential (CAMP) of the right and left hemidiaphragm after phrenic nerve stimulation before (left) and after (right) a T-tube trial in a weaning failure patient. The end-expiratory value of Pes and the amplitude of the right and left CAMPs were the same before and after the trial, indicating that the stimulations were delivered at the same lung volume and that the stimulations achieved the same extent of diaphragmatic recruitment. The amplitude of twitch Pdi elicited by phrenic nerve stimulation was the same before and after weaning. [Adapted, with permission, from Laghi F, et al. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120-127, 2003 (138).] predicted to be an average of 13 minutes away from task failure. Moreover, the time to task failure was underestimated because diaphragmatic recruitment during maximal voluntary contractions was incomplete (138). In other words, patients display clinical manifestations of severe respiratory distress for a substantial time before they develop fatigue. In an intensive care setting, these clinical signs will lead clinicians to reinstitute mechanical ventilation before fatigue has time to develop. Other factors that might have protected the respiratory muscles from contractile fatigue include development of dynamic hyperinflation because susceptibility to fatigue is greater when a fatiguing protocol is conducted at optimum muscle length rather than when a muscle is shortened (88), and activation of extradiaphragmatic muscles of respiration (191). In a study of 19 patients being weaned from mechanical ventilation (191), all but one of the 11 weaning-failure patients exhibited expiratory muscle activity (the exception being a patient with paraplegia). Expiratory muscle activity—as quantified by the expiratory rise in Pga —was absent in all but three of eight weaning-success patients, and its magnitude was trivial in the remainder (Fig. 41). At the onset of the trial, the expiratory rise in Pga was equivalent in the failure and success groups, 0.9 ± 0.5 and 0.1 ± 0.1 cmH2 O, respectively (P = 0.3). At the end of the trial, the expiratory rise in 2909 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology 6 50 Expiratory rise in Pga (cmH2O) Predicted time to task failure (min) 60 40 30 20 10 0.3 0.2 Tension-time 0.1 index 0.0 2 0 0 10 20 30 40 50 Duration of spontaneous breathing trial (min) Figure 40 Interrelationship between the duration of a spontaneous breathing trial, tension-time index of the diaphragm, and predicted time to task failure in nine patients who failed a trial of weaning from mechanical ventilation. The patients breathed spontaneously for an average of 44 min before a physician terminated the trial. At the start of the trial, the tension-time index was 0.17, and the formula of Bellemare and Grassino (18) (see text for details) predicted that patients could sustain spontaneous breathing for another 59 min before developing task failure. As the trial progressed, the tension-time index increased and the predicted time to development of task failure decreased. At the end of the trial, the tension-time index reached 0.26. That patients were predicted to sustain spontaneous breathing for another 13 min before developing task failure clarifies why patients did not develop a decrease in diaphragmatic twitch pressure. In other words, physicians interrupted the trial on the basis of clinical manifestations of respiratory distress, before patients had sufficient time to develop contractile fatigue. [Adapted, with permission, from Laghi F, Tobin MJ: Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003; 168:10 (143).] Pga increased to 4.4 ± 1.1 cmH2 O in the failure group (P = 0.0005), whereas it did not change, 0.1 ± 0.1 cmH2 O, in the success group (P = 0.4). Compared with the success group, the failure group exhibited larger increases in expiratory rise in Pga (P = 0.004). In the failure group, expiratory muscle activity accounted for 53 ± 4% of total PEEPi throughout the weaning trial. In patients with COPD experiencing acute respiratory failure, heightened activation of the expiratory muscles represents an automatic component of the response of the respiratory system to very high levels of ventilatory stimulation (183,301,305). Consistent with this viewpoint is the observed correlation between expiratory muscle activation and respiratory motor output, estimated as the rate of change in Pes : r = 0.57 (0.12-0.82), P = 0.02 (191). Thus, irrespective of the underlying ventilatory defect (77), expiratory muscle recruitment appears to be part of the normal response of the respiratory centers to increased ventilatory demands (77,143). In healthy subjects, expiratory muscle recruitment helps inspiration because the active reduction in end-expiratory lung 2910 4 Last minute First minute Spontaneous breathing trial, time sextiles Figure 41 Expiratory rise in gastric pressure (Pga ) during the course of a weaning trial in failure ( ) and success patients ( ). Between the onset and the end of the trial, increases in expiratory rise in Pga (P = 0.0005) occurred in failure patients but not in the success patients. Over the course of the trial, failure patients had higher values of expiratory rise in Pga (P = 0.004) than success patients. Bars represent ± SE. [Modified, with permission, from Parthasarathy S, Jubran A, Laghi F and Tobin MJ. Sternomastoid, rib-cage and expiratory muscle activity during weaning failure. J Appl Physiol 103: 140-147, 2007 (191).] volume stores elastic energy in the diaphragm and abdomen. At the end of exhalation, the expiratory muscles relax and the release of stored energy causes intrathoracic pressure to fall and inspiratory flow to start before the diaphragm begins to contract (143). When stable, about 60% of patients with COPD have expiratory flow limitation (72), which hinders the expiratory muscles from lowering lung volume and, thus, patients cannot benefit from this action of the expiratory muscles. In the patients of Parthasarathy et al. (191), expiratory muscle contraction was, if anything, associated with an increase, rather than a decrease, in end-expiratory lung volume. The expiratory muscles are effective in assisting inspiration in the upright posture, but they may be less effective in semirecumbent patients. Even when it is beneficial, expiratory muscle contraction naturally imposes a cost in energy expenditure (4). When expiratory muscle contraction is of no potential benefit, as in patients with expiratory flow limitation, it may be detrimental and contribute to the development of acute respiratory failure. In the study of Parthasarathy et al. (191), sternomastoid EMG activity, measured with fine-wire electrodes, was evident in 83 ± 9% of all the breaths in the weaning-failure group and in 19 ± 10% of all breaths in the weaning-success group (P = 0.002) (Fig. 42). Sternomastoid activity became evident within the first minute of the trial in 8 of the 11 failure patients and 1 of the 8 success patients. By the end of the trial, sternomastoid activity was noted in all failure patients but in only three of the success patients, and this activity was modest. The immediate increase in sternomastoid activity in Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Comprehensive Physiology Ventilatory Failure, Ventilator Support, and Ventilator Weaning First minute 2 Flow (L/s) Last minute 40% of trial duration 1 0 –1 Pes (cmH2O) –2 10 0 –10 EMGscm (arb units) –20 2 1 0 –1 –2 0 2 4 6 8 0 2 4 6 8 0 2 4 6 8 Time (s) Figure 42 Representative tracings of flow, Pes , and EMGscm in a weaning-failure patient. Recordings were obtained during the first minute of the weaning trial, 40% of trial duration, and last minute of the trial. Phasic inspiratory activity of the sternomastoid muscle was evident within the first minute of the trial, and it increased progressively over the course of the trial. Note that phasic activity of the sternomastoids persists into expiration. [Adapted, with permission, from Parthasarathy S, Jubran A, Laghi F and Tobin MJ. Sternomastoid, rib-cage and expiratory muscle activity during weaning failure. J Appl Physiol 103: 140-147, 2007 (191).] the failure patients probably results from increased respiratory motor output in response to a combination of decreased capacity of the respiratory muscles to generate pressure (138) and an increase in mechanical load that occurs early during the weaning trial. In addition to increased sternomastoid activity, weaning-failure patients displayed greater inspiratory rib-cage muscle contribution to tidal breathing throughout the trial than did the success patients (Fig. 43). A striking feature of the weaning-failure patients is the timing at which different muscle groups become active (191). The sequence begins with activity of the diaphragm and with greater activity of inspiratory rib-cage muscles than is the case in the success patients; recruitment of sternomastoids and rib-cage muscles is near maximum within 4 min of trial commencement, whereas the expiratory muscles are not recruited until quite late in the trial [at 17-20 min (191)]. The existence of a hierarchy of respiratory muscle activation is supported by the known delayed activation of the sternomastoid muscles (111) and expiratory muscles in healthy volunteers (145,300) and in ambulatory patients with COPD (53). In addition to extradiaphragmatic muscle recruitment, another factor that may protect the diaphragm of weaning-failure patients from long-lasting fatigue is the activation of neural pathways designed to inhibit inspiratory muscle recruitment in the face of potentially fatiguing loads. This possibility is supported by studies conducted in laboratory animals wherein inspiratory loading causes respiratory failure and acidosis be- Volume 2, October 2012 fore force output decreases or substrate is depleted in the diaphragm (2, 181, 211, 224). These observations suggest that central (57, 255) and reflex mechanisms (109, 249) affect the breathing pattern (210) and α-motoneuron firing rates (167) in response to loading. Two neural pathways may convey information from the respiratory muscles to the central nervous system (57, 197). One pathway transmits information from mechanoreceptors [Golgi tendon organs and muscle spindles (109, 159)] in the dorsal column, and relays it to the brainstem and thalamus before reaching the sensimotor cortex (159, 320). This pathway may participate in proprioceptive control of the respiratory muscles, integrating movements originating in the motor cortex (255). The second pathway consists of vagal (2,159) and possibly phrenic nerve afferents [group IV phrenic afferent fibers (109)] that reach the amygdala after relaying in the brainstem and then projecting to the mesocortex [cingulated gyrus (159)]. This pathway may deal with respiratory nociception (51, 255), such as dyspnea [through the relay in the amygdala (20)], and the ventilatory response to carbon dioxide [through the relay in the brainstem, ventral cerebellum, and limbic system (102)] (51,255). In addition to these two pathways projecting to the central nervous system, there is evidence for the existence of a spinal pathway responsible for phrenic-to-phrenic reflex inhibition (249). The net effect of these reflex pathways may be to inhibit the inspiratory muscles in the face of potentially fatiguing loads, thereby protecting them from irreversible damage at the cost 2911 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning Comprehensive Physiology Weaning success –30 Start End –20 ΔPes (cmH2O) –10 0 Weaning failure –40 –30 –20 –10 0 –30 –15 0 15 –30 –15 0 15 ΔPga (cmH2O) Figure 43 Plots of tidal changes in esophageal pressure (Pes ) against tidal changes in gastric pressure (Pga ) in a weaning-success patient and a weaningfailure patient. At the start of a weaning trial, the success patient (top left panel) exhibited swings in esophageal pressure that became markedly more negative between the onset (closed symbol) and the end of inspiration (open symbol); in contrast, gastric pressure increased only slightly. Therefore, the slope of the Pes Pga plot at the onset of weaning (top left panel) was much greater than the slope recorded in healthy subjects during resting breathing, where the tidal change in gastric pressure is often greater than the tidal change in esophageal pressure. The steep Pes -Pga plot (top left panel) indicates a greater-than-usual contribution of the rib-cage muscles to tidal breathing than that of the diaphragm. Between the onset (top left panel) and the end of weaning (top right panel), the slope of the Pes -Pga plot changed very little, indicating a constant contribution of the diaphragm and rib-cage muscles to tidal breathing over the course of the weaning trial. In the case of the weaning-failure patient, the inspiratory swings in esophageal pressure and gastric pressure had a similar pattern at the start of the trial to that in the success patient (bottom left panel). At the end of the trial, the failure patient exhibited a markedly negative slope in the Pes -Pga plot, signifying a further increase in inspiratory rib-cage muscle recruitment that was out of proportion to diaphragmatic recruitment. of CO2 retention. Finally, an increase in CO2 during loading may also protect the respiratory muscles by decreasing production of reactive oxygen species (253). Cardiovascular performance Although the respiratory muscles may not develop fatigue, they have to overcome a huge workload. Thus, they depend on an efficient transport of oxygen by the cardiovascular system. Aware of this fact, several researchers have examined cardiovascular performance during weaning. Lemaire et al. (151) studied 15 patients with severe COPD who were difficult to wean (seven of whom had documented ischemic heart disease). During unassisted breathing the patients developed evidence of cardiovascular stress including increases in transmural pulmonary artery wedge pressure, cardiac in- 2912 dex, and left- and right-ventricular end-diastolic volume. The investigators attributed the increase in left-ventricular end-diastolic volume to augmentation of venous return (secondary to low pleural pressure during spontaneous breathing and central translocation of blood volume secondary to peripheral vasoconstriction) and increased left-ventricular afterload (secondary to markedly negative pleural pressure swings and increased catecholamine release). Jubran et al. (125) continuously recorded cardiovascular performance and mixed venous oxygen saturation in eight weaning-failure and 11 weaning-success patients over the course of trials of spontaneous breathing that lasted about 40 min. Immediately before the trial, mixed venous oxygen saturation was equivalent in the two groups. On discontinuation of the ventilator, saturation fell progressively in the failure patients (to 51.5 ± 7.9% at end of trial), whereas Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology it did not change in the success patients. Right- and leftventricular afterload increased in the failure patients—most likely because the negative swings in intrathoracic pressure were more extreme during the course of spontaneous respiration. At the completion of the trial, the level of oxygen consumption was equivalent in patients who could be weaned and in those who could not. How the cardiovascular system met oxygen demands differed in the two groups (125). In the weaning-success patients, oxygen demands were met through an increase in oxygen delivery, mediated by the expected increase in cardiac output on discontinuation of positivepressure ventilation. In the weaning-failure patients, oxygen demands were met through an increase in oxygen extraction, and these patients had a relative decrease in oxygen delivery (125). The greater oxygen extraction caused a substantial decrease in mixed venous O2 saturation, contributing to the arterial hypoxemia that occurred in some patients (125). In an investigation of similar design, Zakynthinos et al. (317) studied 12 weaning-success and 18 weaning-failure patients during a spontaneous breathing trial. Half of the failure patients increased their oxygen consumption and this increase was met mainly by an increase in oxygen extraction. The remaining half of the failure patients did not exhibit an increase in oxygen consumption; instead, increase in oxygen delivery was accompanied by a decrease in oxygen extraction. These studies (125, 151, 317) demonstrate variability in circulatory and global tissue oxygenation responses during weaning failure. In these investigations (125,151,317), all weaning-failure patients developed respiratory distress, suggesting that they developed increases in respiratory motor output. The response of the respiratory centers, however, was not uniform. During a weaning trial, the patients of Lemaire et al. (151) experienced a decrease in the negative swings in intrathoracic pressure. In contrast, Jubran et al. (125) recorded an increase in the negative swings in intrathoracic pressure. Many patients in these investigations developed acute or acute-on-chronic alveolar hypoventilation. It is not known how the respiratory centers integrate afferent information from the cardiovascular system, respiratory system and peripheral muscles in generating neural volleys to the respiratory muscles. Gas exchange A primary goal of mechanical ventilation is to improve gas exchange, and accordingly one expects some deterioration in gas exchange with the resumption of spontaneous breathing. The most detailed study of gas exchange during weaning is that conducted by Beydon et al. (23). They studied eight patients with COPD who were considered ventilator dependent (although the patients were able to sustain at least 1-3 h of spontaneous breathing). When switched from controlled ventilation, patients developed an increase in frequency, fall in tidal volume (without change in minute ventilation), and an increase in PaCO2 (41 to 49 mmHg). Using the multiple inert gas technique, the investigators found that the distribution Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning of ventilation to regions of ventilation-perfusion (V̇A / Q̇) relationships above 100 (i.e., dead space) increased from 39 ± 8% during controlled ventilation to 46 ± 7% during spontaneous breathing. Perfusion of low V̇A / Q̇ regions was higher during spontaneous breathing than during controlled ventilation (15 ± 11 vs. 6 ± 8%). The investigators also performed isotope scans, which revealed a decrease in V̇A / Q̇ ratios between the apex and the base of the lungs. This observation indicated that the low V̇A / Q̇ units identified by the inert-gas technique were located at the lung bases. The major determinant of the V̇A / Q̇ abnormalities was the size of tidal volume: it correlated with perfusion in the low V̇A / Q̇ range, the decrease of V̇A / Q̇ ratios in the bases, and widening of the isotopic craniocaudal gradient. The maldistribution of V̇A / Q̇ ratios during spontaneous breathing was improved by controlled ventilation but not by pressure support of 10 cmH2 O. Torres et al. (281) used the multiple inert-gas technique to study eight patients with COPD who were apparently successfully weaned. Measurements were first obtained during assist-control ventilation (tidal volume 700 mL, frequency 12 breaths/min). On discontinuation of the ventilator, the patients developed rapid shallow breathing (relative to ventilator settings) and acute respiratory acidosis (increase in PaCO2 from 49 to 59 mmHg, decrease in pH from 7.42 to 7.36). Spontaneous breathing caused an overall worsening of ventilationperfusion inequality: the fraction of cardiac output distributed to low V̇A / Q̇ (< 0.1) areas increased from 9.4 to 19.6% and the dispersion of ventilation distribution increased. Despite the deterioration in V̇A / Q̇ relationships, the expected fall in PO2 was prevented by an increase in cardiac output (4.7 to 6.7 L/min) and increase in mixed venous PO2 (37 to 42 mmHg). A largely similar pattern of gas exchange was reported by Ferrer et al. (82), who studied seven patients with COPD that were not yet ready to tolerate complete discontinuation of mechanical ventilation. Extubation Virtually no pathophysiologic studies have been conducted on patients who develop such severe respiratory distress following the removal of an endotracheal tube that they require reintubation and the reinstitution of mechanical ventilation. Two factors make it especially challenging to conduct research in this setting. The first is instrumentation. The recording of swings in intrathoracic pressure, as reflected by esophageal pressure, is relatively easy, but of limited value on its own. The derivation of most physiologic indices, such as pulmonary resistance, compliance and intrinsic PEEP, requires a simultaneous measurement of airflow. The use of a mouthpiece or face mask for recording airflow is particularly difficult in a recently extubated dyspneic patient, and is likely to distort the very variables an investigator wishes to measure. The second challenge is the timing. Weaning failure almost invariably occurs within the first hour of attempted spontaneous breathing. The time course for the development of postextubation 2913 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning cardiorespiratory distress extends over a longer span, and many patients do not develop distress until many hours after extubation. Consequently, investigators and their instruments have typically long left the scene by the time the problem becomes manifest. Management of weaning Discontinuation of mechanical ventilation needs to be carefully timed. Premature discontinuation places severe stress on the respiratory and cardiovascular systems, which can impede a patient’s recovery. Unnecessary delays in ventilator discontinuation can lead to a host of complications and several measurements are used to aid decision making. The level of oxygenation must be satisfactory before one attempts to discontinue mechanical ventilation. Yet in many patients with satisfactory oxygenation, such attempts fail. The use of traditional predictors of the success or failure of attempts—maximal inspiratory pressure, vital capacity, and minute ventilation—frequently yield false-positive or falsenegative results (266). A more reliable predictor is the ratio of respiratory frequency to tidal volume (f /V T ) (302). The higher the ratio, the more severe the rapid, shallow breathing and the greater the likelihood of unsuccessful weaning. The reliability of f /V T ratio as a predictor of weaning outcome has been evaluated by at least 29 groups of investigators (271). When data from the studies were entered into a Bayesian model with pretest probability as the operating point, the reported positive-predictive values were significantly correlated with the values predicted by the original report on f /V T (302), r = 0.86 ( P < 0.0001); likewise, reported negative-predictive values were correlated with the values predicted, r = 0.82 (P < 0.0001) (271). The average sensitivity of 0.87 indicates that f /V T is a reliable screening test for successful weaning. Once the recorded values on weaning-predictor tests suggest that a patient has a reasonably good chance of tolerating the discontinuation of mechanical ventilation, the next step is to undertake a formal weaning trial. Two approaches involve the use of a T-tube circuit, whereby the patient either proceeds immediately to a trial of spontaneous breathing lasting for up to two hours or trials begin briefly (lasting only 5-10 min) and the duration is gradually extended (weaning in the literal sense of the work). Two other approaches involve a gradual reduction in the level of ventilator assistance. When IMV is used for this purpose, the number of ventilator-assisted breaths is gradually decreased in response to patient tolerance. When pressure support is used, the level of pressure is gradually decreased (266). Until the early 1990s, it was widely believed that all weaning methods were equally effective, and physician judgment was regarded as the critical determinant. Results of randomized controlled trials, however, indicate that the period of weaning is as much as three times as long with IMV as with trials of spontaneous breathing (34, 75). In a study involving patients who experience respiratory difficulties during wean- 2914 Comprehensive Physiology ing, trials of spontaneous breathing halved the weaning time as compared with pressure support (75); in another study, the weaning time was similar with the two methods (34). Performing trials of spontaneous breathing once a day is as effective as performing such trials several times a day (75)—but much simpler. In a subsequent study, half-hour trials of spontaneous breathing were as effective as 2-h trials in patients undergoing their first weaning attempt (73). When patients are able to sustain spontaneous ventilation without undue discomfort, they are extubated. About 10% to 20% of such patients require reintubation (34, 75). Mortality among patients who require reintubation is more than six times as high as mortality among patients who can tolerate extubation (73). The reason for the higher mortality is unknown; it is not clearly related to the development of new problems after extubation or to complications of reinserting the tube. Indeed, the need for reintubation may simply be a marker of a more severe underlying illness. Several investigators have studied the usefulness of noninvasive ventilation as a means of making the process of weaning and extubation more expeditious and effective. At least three groups of investigators have reported that the institution of noninvasive ventilation in certain patients (in particular those with COPD) is beneficial if instituted at the point when patients have just failed a trial of spontaneous breathing (81, 97, 179). In contrast, two groups of investigators have reported that noninvasive ventilation is not beneficial in postextubated patients when instituted after they already have clinical manifestations of respiratory distress. In the latter two studies, however, the level of ventilator assistance, pressure support of 5 cmH2 O (131) or delivered tidal volume of as little as 5 mL/kg (76), may have been inadequate to truly test the efficacy of noninvasive ventilation in this circumstance. Coda The respiratory control system plays a fundamental role in the three topics covered in this article: ventilatory failure, mechanical ventilation, and ventilator weaning. Abnormalities in respiratory mechanics and pulmonary gas exchange contribute to the development of ventilatory failure, but it is the performance of the respiratory controller that determines whether or not a patient will be able to sustain spontaneous ventilation without requiring ventilator assistance. While a patient is being supported by mechanical ventilation, abnormalities in respiratory mechanics and gas exchange again influence the selection of ventilator settings, but the most challenging component of ventilator management—achieving satisfactory interaction between the patient and the machine—depends crucially on knowledge of the respiratory control system. Indeed, new ventilator modes introduced with the specific intent of improving patient-ventilator synchronization, such as PAV and NAVA, operate through modulation of respiratory motor output on a breath-by-breath basis. Finally, when patients fail a trial of weaning from mechanical ventilation, abnormalities Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology of respiratory muscle function, cardiovascular performance and respiratory mechanics are all involved, but the physiological hallmark of weaning failure—rapid shallow breathing—is a manifestation of altered respiratory motor output. Despite the fundamental importance of control of breathing in every aspect of acute respiratory failure and ventilator management, it is the component of pulmonary physiology that poses the greatest challenge to clinicians and with which they feel the greatest unease. At least two factors contribute to this unsatisfactory state of affairs. One is the lack of direct methods for monitoring the major sensory inputs to the respiratory control system (mechanoreceptor and vagal stimuli, and so on) and the absence of a simple measure of respiratory motor output, in contrast with the ready availability of methods for monitoring gas exchange and respiratory mechanics at a patient’s bedside. The second, and more fundamental, factor is the complexity of the physiological mechanisms involved in the respiratory control system and the likelihood that our knowledge has progressed considerably less in this field than in other areas of respiratory pathophysiology. References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 342: 1301-1308, 2000. Adams JM, Farkas GA, Rochester DF. Vagal afferents, diaphragm fatigue, and inspiratory resistance in anesthetized dogs. J Appl Physiol 64: 2279-2286, 1988. Agostoni E, Hyatt RE. Static behavior of the respiratory system. In: Macklem PT, Mead J, editors. Handbook of Physiology: The Respiratory System, Vol. II, Bethesda: American Physiological Society, 1986, pp. 113-130. Aliverti A, Macklem PT. The major limitation to exercise performance in COPD is inadequate energy supply to the respiratory and locomotor muscles. J Appl Physiol 105: 749-751, 2008. Allen GM, McKenzie DK, Gandevia SC, Bass S. Reduced voluntary drive to breathe in asthmatic subjects. Respir Physiol 93: 29-40, 1993. Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 338: 347-354, 1998. Amato MBP, Marini JJ. Pressure-controlled and inverse-ratio ventilation. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill, Inc., 2006, pp. 251-272. Anzueto A, Peters JI, Tobin MJ, de los SR, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ. Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med 25: 1187-1190, 1997. Appendini L, Patessio A, Zanaboni S, Carone M, Gukov B, Donner CF, Rossi A. Physiologic effects of positive end-expiratory pressure and mask pressure support during exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 149: 1069-1076, 1994. Arnulf I, Similowski T, Salachas F, Garma L, Mehiri S, Attali V, BehinBellhesen V, Meininger V, Derenne JP. Sleep disorders and diaphragmatic function in patients with amyotrophic lateral sclerosis. Am J Respir Crit Care Med 161: 849-856, 2000. Arold SP, Mora R, Lutchen KR, Ingenito EP, Suki B. Variable tidal volume ventilation improves lung mechanics and gas exchange in a rodent model of acute lung injury. Am J Respir Crit Care Med 165: 366-371, 2002. Aslanian P, El Atrous S, Isabey D, Valente E, Corsi D, Harf A, Lemaire F, Brochard L. Effects of flow triggering on breathing effort during partial ventilatory support. Am J Respir Crit Care Med 157: 135-143, 1998. Aubier M, Murciano D, Milic-Emili J, Touaty E, Daghfous J, Pariente R, Derenne JP. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis 122: 747-754, 1980. Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 14. Aubier M, Trippenbach T, Roussos C. Respiratory muscle fatigue during cardiogenic shock. J Appl Physiol 51: 499-508, 1981. 15. Axen K, Haas SS. Range of first-breath ventilatory responses to added mechanical loads in naive men. J Appl Physiol 46: 743-751, 1979. 16. Ayres SM, Grace WJ. Inappropriate ventilation and hypoxemia as causes of cardiac arrhythmias. Am J Med 46: 495-505, 1969. 17. Begin P, Grassino A. Inspiratory muscle dysfunction and chronic hypercapnia in chronic obstructive pulmonary disease. Am Rev Respir Dis 143: 905-912, 1991. 18. Bellemare F, Grassino A. Effect of pressure and timing of contraction on human diaphragm fatigue. J Appl Physiol 53: 1190-1195, 1982. 19. Berkenbosch A, van Beek JH, Olievier CN, De Goede J, Quanjer PH. Central respiratory CO2 sensitivity at extreme hypocapnia. Respir Physiol 55: 95-102, 1984. 20. Bernard JF, Huang GF, Besson JM. The parabrachial area: Electrophysiological evidence for an involvement in visceral nociceptive processes. J Neurophysiol 71: 1646-1660, 1994. 21. Berry RB, Mahutte CK, Light RW. Effect of hypercapnia on the arousal response to airway occlusion during sleep in normal subjects. J Appl Physiol 74: 2269-2275, 1993. 22. Berssenbrugge A, Dempsey J, Iber C, Skatrud J, Wilson P. Mechanisms of hypoxia-induced periodic breathing during sleep in humans. J Physiol 343: 507-524, 1983. 23. Beydon L, Cinotti L, Rekik N, Radermacher P, Adnot S, Meignan M, Harf A, Lemaire F. Changes in the distribution of ventilation and perfusion associated with separation from mechanical ventilation in patients with obstructive pulmonary disease. Anesthesiology 75: 730738, 1991. 24. Blain GM, Smith CA, Henderson KS, Dempsey JA. Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO(2). J Physiol 588: 2455-2471, 2010. 25. Boker A, Graham MR, Walley KR, McManus BM, Girling LG, Walker E, Lefevre GR, Mutch WA. Improved arterial oxygenation with biologically variable or fractal ventilation using low tidal volumes in a porcine model of acute respiratory distress syndrome. Am J Respir Crit Care Med 165: 456-462, 2002. 26. Bosma K, Ferreyra G, Ambrogio C, Pasero D, Mirabella L, Braghiroli A, Appendini L, Mascia L, Ranieri VM. Patient-ventilator interaction and sleep in mechanically ventilated patients: Pressure support versus proportional assist ventilation. Crit Care Med 35: 1048-1054, 2007. 27. Bourke SC, Gibson GJ. Sleep and breathing in neuromuscular disease. Eur Respir J 19: 1194-1201, 2002. 28. Brack T, Jubran A, Tobin MJ. Effect of elastic loading on variational activity of breathing. Am J Respir Crit Care Med 155: 1341-1348, 1997. 29. Brack T, Jubran A, Tobin MJ. Effect of resistive loading on variational activity of breathing. Am J Respir Crit Care Med 157: 1756-1763, 1998. 30. Brack T, Jubran A, Tobin MJ. Dyspnea and decreased variability of breathing in patients with restrictive lung disease. Am J Respir Crit Care Med 165: 1260-1264, 2002. 31. Braun NM, Arora NS, Rochester DF. Respiratory muscle and pulmonary function in polymyositis and other proximal myopathies. Thorax 38: 616-623, 1983. 32. Brochard L, Harf A, Lorino H, Lemaire F. Inspiratory pressure support prevents diaphragmatic fatigue during weaning from mechanical ventilation. Am Rev Respir Dis 139: 513-521, 1989. 33. Brochard L, Lellouche F. Pressure-support ventilation. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill Inc., 2011, pp. 221-250. 34. Brochard L, Rauss A, Benito S, Conti G, Mancebo J, Rekik N, Gasparetto A, Lemaire F. Comparison of three methods of gradual withdrawal from ventilatory support during weaning from mechanical ventilation. Am J Respir Crit Care Med 150: 896-903, 1994. 35. Brown DR, Forster HV, Greene AS, Lowry TF. Breathing periodicity in intact and carotid body-denervated ponies during normoxia and chronic hypoxia. J Appl Physiol 74: 1073-1082, 1993. 36. Bunn JC, Mead J. Control of ventilation during speech. J Appl Physiol 31: 870-872, 1971. 37. Burrows B, Saksena FB, Diener CF. Carbon dioxide tension and ventilatory mechanics in chronic obstructie lung disease. Ann Intern Med 65: 685-700, 1966. 38. Burton SL, Hubmayr RD. Determinants of patient-ventilator interactions: Bedside waveform analysis. In: Principles and Practice of Intensive Care Monitoring. New York: McGraw-Hill, Inc., 1998, pp. 655-666. 39. Buyse B, Demedts M, Meekers J, Vandegaer L, Rochette F, Kerkhofs L. Respiratory dysfunction in multiple sclerosis: A prospective analysis of 60 patients. Eur Respir J 10: 139-145, 1997. 40. Cabello B, Thille AW, Drouot X, Galia F, Mancebo J, d’Ortho MP, Brochard L. Sleep quality in mechanically ventilated patients: Comparison of three ventilatory modes. Crit Care Med 36: 1749-1755, 2008. 41. Campbell EJM, Howell JBL. Proprioceptive control of breathing. In: de Reuck AVS, O’Connor M, editors. Ciba Foundation Symposium on Pulmonary Structure and Function, Boston: Little, Brown, 1962, pp. 2952. 2915 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning 42. Capdevila X, Perrigault PF, Ramonatxo M, Roustan JP, Peray P, d’Athis F, Prefaut C. Changes in breathing pattern and respiratory muscle performance parameters during difficult weaning. Crit Care Med 26: 79-87, 1998. 43. Capdevila XJ, Perrigault PF, Perey PJ, Roustan JP, d’Athis F. Occlusion pressure and its ratio to maximum inspiratory pressure are useful predictors for successful extubation following T-piece weaning trial. Chest 108: 482-489, 1995. 44. Cassart M, Pettiaux N, Gevenois PA, Paiva M, Estenne M. Effect of chronic hyperinflation on diaphragm length and surface area. Am J Respir Crit Care Med 156: 504-508, 1997. 45. Chonan T, Mulholland MB, Cherniack NS, Altose MD. Effects of voluntary constraining of thoracic displacement during hypercapnia. J Appl Physiol 63: 1822-1828, 1987. 46. Clanton TL, Levine S. Respiratory muscle fiber remodeling in chronic hyperinflation: Dysfunction or adaptation? J Appl Physiol 107: 324-335, 2009. 47. Clark FJ, Von Euler C. On the regulation of depth and rate of breathing. J Physiol 222: 267-295, 1972. 48. Cohen CA, Zagelbaum G, Gross D, Roussos C, Macklem PT. Clinical manifestations of inspiratory muscle fatigue. Am J Med 73: 308-316, 1982a. 49. Cohen CA, Zagelbaum G, Gross D, Roussos C, Macklem PT. Clinical manifestations of inspiratory muscle fatigue. Am J Med 73: 308-316, 1982b. 50. Cooper AB, Thornley KS, Young GB, Slutsky AS, Stewart TE, Hanly PJ. Sleep in critically ill patients requiring mechanical ventilation. Chest 117: 809-818, 2000. 51. Corfield DR, Fink GR, Ramsay SC, Murphy K, Harty HR, Watson JD, Adams L, Frackowiak RS, Guz A. Evidence for limbic system activation during CO2 -stimulated breathing in man. J Physiol 488 (Pt 1): 77-84, 1995. 52. Corne S, Webster K, Younes M. Hypoxic respiratory response during acute stable hypocapnia. Am J Respir Crit Care Med 167: 1193-1199, 2003. 53. Criner GJ, Celli BR. Ventilatory muscle recruitment in exercise with O2 in obstructed patients with mild hypoxemia. J Appl Physiol 63: 195-200, 1987. 54. Cunningham DJC, Robbins PA, Wolff CB. Integration of respiratory responses to changes in alveolar partial pressures of CO2 and O2 and in arterial pH. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, Section 3. The Respiratory System. Volume II: Control of Breathing, part 2, Bethesda, MD: Am Physiol Soc, 1986, pp. 475-528. 55. Curran AK, Rodman JR, Eastwood PR, Henderson KS, Dempsey JA, Smith CA. Ventilatory responses to specific CNS hypoxia in sleeping dogs. J Appl Physiol 88: 1840-1852, 2000. 56. Datta AK, Shea SA, Horner RL, Guz A. The influence of induced hypocapnia and sleep on the endogenous respiratory rhythm in humans. J Physiol 440: 17-33, 1991. 57. Davenport PW, Reep RRL. Cerebral cortex and respiration. In: Dempsey J, Pack AL, editors. Regulation of Breathing. New York: Marcel Dekker, 1995, pp. 365-388. 58. Davis JN, Plum F. Separation of descending spinal pathways to respiratory motoneurons. Exp Neurol 34: 78-94, 1972. 59. DeBacker WA, Verbraecken J, Willemen M, Wittesaele W, DeCock W, Van deHeyning P. Central apnea index decreases after prolonged treatment with acetazolamide. Am J Respir Crit Care Med 151: 87-91, 1995. 60. de Durante G, del Turco M, Rustichini L, Cosimini P, Giunta F, Hudson LD, Slutsky AS, Ranieri VM. ARDSNet lower tidal volume ventilatory strategy may generate intrinsic positive end-expiratory pressure in patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 165: 1271-1274, 2002. 61. De Troyer A, Borenstein S, Cordier R. Analysis of lung volume restriction in patients with respiratory muscle weakness. Thorax 35: 603-610, 1980. 62. De Troyer A, Leeper JB, McKenzie DK, Gandevia SC. Neural drive to the diaphragm in patients with severe COPD. Am J Respir Crit Care Med 155: 1335-1340, 1997. 63. Dempsey JA. Crossing the apnoeic threshold: Causes and consequences. Exp Physiol 90: 13-24, 2005. 64. Dempsey JA, Skatrud JB. Apnea following mechanical ventilation may be caused by nonchemical neuromechanical influences. Am J Respir Crit Care Med 163: 1297-1298, 2001. 65. Dick CR, Liu Z, Sassoon CS, Berry RB, Mahutte CK. O2 -induced change in ventilation and ventilatory drive in COPD. Am J Respir Crit Care Med 155: 609-614, 1997. 66. Dodd DS, Brancatisano T, Engel LA. Chest wall mechanics during exercise in patients with severe chronic air-flow obstruction. Am Rev Respir Dis 129: 33-38, 1984. 67. Douglass JA, Tuxen DV, Horne M, Scheinkestel CD, Weinmann M, Czarny D, Bowes G. Myopathy in severe asthma. Am Rev Respir Dis 146: 517-519, 1992. 2916 Printer Name: Yet to Come Comprehensive Physiology 68. Dreyfuss D, Saumon G. Ventilator-induced lung injury: Lessons from experimental studies. Am J Respir Crit Care Med 157: 294-323, 1998. 69. Du HL, Ohtsuji M, Shigeta M, Chao DC, Sasaki K, Usuda Y, Yamada Y. Expiratory asynchrony in proportional assist ventilation. Am J Respir Crit Care Med 165: 972-977, 2002. 70. Dunn WF, Nelson SB, Hubmayr RD. Oxygen-induced hypercarbia in obstructive pulmonary disease. Am Rev Respir Dis 144: 526-530, 1991. 71. Efthimiou J, Fleming J, Spiro SG. Sternomastoid muscle function and fatigue in breathless patients with severe respiratory disease. Am Rev Respir Dis 136: 1099-1105, 1987. 72. Eltayara L, Becklake MR, Volta CA, Milic-Emili J. Relationship between chronic dyspnea and expiratory flow limitation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 154: 1726-1734, 1996. 73. Esteban A, Alia I, Tobin MJ, Gil A, Gordo F, Vallverdu I, Blanch L, Bonet A, Vazquez A, de Pablo R, Torres A, de La Cal MA, Macias S. Effect of spontaneous breathing trial duration on outcome of attempts to discontinue mechanical ventilation. Spanish Lung Failure Collaborative Group. Am J Respir Crit Care Med 159: 512-518, 1999. 74. Esteban A, Anzueto A, Alia I, Gordo F, Apezteguia C, Palizas F, Cide D, Goldwaser R, Soto L, Bugedo G, Rodrigo C, Pimentel J, Raimondi G, Tobin MJ. How is mechanical ventilation employed in the intensive care unit? An international utilization review. Am J Respir Crit Care Med 161: 1450-1458, 2000. 75. Esteban A, Frutos F, Tobin MJ, Alia I, Solsona JF, Valverdu I, Fernandez R, de La Cal MA, Benito S, Tomas R. A comparison of four methods of weaning patients from mechanical ventilation. Spanish Lung Failure Collaborative Group. N Engl J Med 332: 345-350, 1995. 76. Esteban A, Frutos-Vivar F, Ferguson ND, Arabi Y, Apezteguia C, Gonzalez M, Epstein SK, Hill NS, Nava S, Soares MA, D’Empaire G, Alia I, Anzueto A. Noninvasive positive-pressure ventilation for respiratory failure after extubation. N Engl J Med 350: 2452-2460, 2004. 77. Estenne M, Derom E, De Troyer A. Neck and abdominal muscle activity in patients with severe thoracic scoliosis. Am J Respir Crit Care Med 158: 452-457, 1998. 78. Estenne M, Gevenois PA, Kinnear W, Soudon P, Heilporn A, De Troyer A. Lung volume restriction in patients with chronic respiratory muscle weakness: The role of microatelectasis. Thorax 48: 698-701, 1993. 79. Falke KJ, Pontoppidan H, Kumar A, Leith DE, Geffin B, Laver MB. Ventilation with end-expiratory pressure in acute lung disease. J Clin Invest 51: 2315-2323, 1972. 80. Farkas GA, Roussos C. Diaphragm in emphysematous hamsters: Sarcomere adaptability. J Appl Physiol 54: 1635-1640, 1983. 81. Ferrer M, Esquinas A, Arancibia F, Bauer TT, Gonzalez G, Carrillo A, Rodriguez-Roisin R, Torres A. Noninvasive ventilation during persistent weaning failure: A randomized controlled trial. Am J Respir Crit Care Med 168: 70-76, 2003. 82. Ferrer M, Iglesia R, Roca J, Burgos F, Torres A, Rodriguez-Roisin R. Pulmonary gas exchange response to weaning with pressure-support ventilation in exacerbated chronic obstructive pulmonary disease patients. Intensive Care Med 28: 1595-1599, 2002. 83. Fiamma MN, Straus C, Thibault S, Wysocki M, Baconnier P, Similowski T. Effects of hypercapnia and hypocapnia on ventilatory variability and the chaotic dynamics of ventilatory flow in humans. Am J Physiol Regul Integr Comp Physiol 292: R1985-R1993, 2007. 84. Fiastro JF, Habib MP, Shon BY, Campbell SC. Comparison of standard weaning parameters and the mechanical work of breathing in mechanically ventilated patients. Chest 94: 232-238, 1988. 85. Field S, Sanci S, Grassino A. Respiratory muscle oxygen consumption estimated by the diaphragm pressure-time index. J Appl Physiol 57: 44-51, 1984. 86. Fink B, Hanks E, Ngai S, Papper E. Central regulation of respiration during anesthesia and wakefulness. Ann N Y Acad Sci U S A 109: 892900, 1963. 87. Fink BR. Influence of cerebral activity in wakefulness on regulation of breathing. J Appl Physiol 16: 15-29, 1961. 88. Fitch S, McComas A. Influence of human muscle length on fatigue. J Physiol 362: 205-213, 1985. 89. Fitzgerald RS, Parks DC. Effect of hypoxia on carotid chemoreceptor response to carbon dioxide in cats. Respir Physiol 12: 218-229, 1971. 90. Forster HV, Klein JP, Hamilton LH, Kampine JP. Regulation of PaCO2 and ventilation in humans inspiring low levels of CO2. J Appl Physiol 52: 287-294, 1982. 91. Gabor JY, Cooper AB, Crombach SA, Lee B, Kadikar N, Bettger HE, Hanly PJ. Contribution of the intensive care unit environment to sleep disruption in mechanically ventilated patients and healthy subjects. Am J Respir Crit Care Med 167: 708-715, 2003. 92. Gama dA, Spieth PM, Pelosi P, Carvalho AR, Walter C, SchreiberFerstl A, Aikele P, Neykova B, Hubler M, Koch T. Noisy pressure support ventilation: A pilot study on a new assisted ventilation mode in experimental lung injury. Crit Care Med 36: 818-827, 2008. Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 93. Gardner WN. The pattern of breathing following step changes of alveolar partial pressures of carbon dioxide and oxygen in man. J Physiol 300: 55-73, 1980. 94. Georgopoulos D, Mitrouska I, Bshouty Z, Webster K, Anthonisen NR, Younes M. Effects of breathing route, temperature and volume of inspired gas, and airway anesthesia on the response of respiratory output to varying inspiratory flow. Am J Respir Crit Care Med 153: 168-175, 1996. 95. Georgopoulos D, Mitrouska I, Bshouty Z, Webster K, Patakas D, Younes M. Respiratory response to CO2 during pressure-support ventilation in conscious normal humans. Am J Respir Crit Care Med 156: 146-154, 1997. 96. Giesbrecht GG, Younes M. Respiratory response to pulmonary vascular congestion in intact conscious dogs. J Appl Physiol 74: 345-353, 1993. 97. Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: A prospective, randomized controlled study. Am J Respir Crit Care Med 160: 86-92, 1999. 98. Giuliani R, Mascia L, Recchia F, Caracciolo A, Fiore T, Ranieri VM. Patient-ventilator interaction during synchronized intermittent mandatory ventilation. Effects of flow triggering. Am J Respir Crit Care Med 151: 1-9, 1995. 99. Gleeson K, Zwillich CW, White DP. The influence of increasing ventilatory effort on arousal from sleep. Am Rev Respir Dis 142: 295-300, 1990. 100. Goldberger AL. Non-linear dynamics for clinicians: Chaos theory, fractals, and complexity at the bedside. Lancet 347: 1312-1314, 1996. 101. Goldstone JC, Green M, Moxham J. Maximum relaxation rate of the diaphragm during weaning from mechanical ventilation. Thorax 49: 54-60, 1994. 102. Gozal D, Hathout GM, Kirlew KA, Tang H, Woo MS, Zhang J, Lufkin RB, Harper RM. Localization of putative neural respiratory regions in the human by functional magnetic resonance imaging. J Appl Physiol 76: 2076-2083, 1994. 103. Guilleminault C, Partinen M, Quera-Salva MA, Hayes B, Dement WC, Nino-Murcia G. Determinants of daytime sleepiness in obstructive sleep apnea. Chest 94: 32-37, 1988. 104. Guyenet PG. The 2008 Carl Ludwig Lecture: Retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol 105: 404416, 2008. 105. Haluszka J, Chartrand DA, Grassino AE, Milic-Emili J. Intrinsic PEEP and arterial PCO2 in stable patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 141: 1194-1197, 1990. 106. Henke KG, Arias A, Skatrud JB, Dempsey JA. Inhibition of inspiratory muscle activity during sleep. Chemical and nonchemical influences. Am Rev Respir Dis 138: 8-15, 1988. 107. Hey EN, Lloyd BB, Cunningham DJ, Jukes MG, Bolton DP. Effects of various respiratory stimuli on the depth and frequency of breathing in man. Respir Physiol 1: 193-205, 1966. 108. Heywood P, Murphy K, Corfield DR, Morrell MJ, Howard RS, Guz A. Control of breathing in man; insights from the ‘locked-in’ syndrome. Respir Physiol 106: 13-20, 1996. 109. Hill JM. Increase in the discharge of muscle spindles during diaphragm fatigue. Brain Res 918: 166-170, 2001. 110. Hirshman CA, McCullough RE, Weil JV. Normal values for hypoxic and hypercapnic ventilaroty drives in man. J Appl Physiol 38: 1095-1098, 1975. 111. Hudson AL, Gandevia SC, Butler JE. The effect of lung volume on the co-ordinated recruitment of scalene and sternomastoid muscles in humans. J Physiol 584: 261-270, 2007. 112. Hughes PD, Polkey MI, Moxham J, Green M. Long-term recovery of diaphragm strength in neuralgic amyotrophy. Eur Respir J 13: 379-384, 1999. 113. Hukins CA, Hillman DR. Daytime predictors of sleep hypoventilation in Duchenne muscular dystrophy. Am J Respir Crit Care Med 161: 166-170, 2000. 114. Iber C, Berssenbrugge A, Skatrud JB, Dempsey JA. Ventilatory adaptations to resistive loading during wakefulness and non-REM sleep. J Appl Physiol 52: 607-614, 1982. 115. Iber C, Davies SF, Chapman RC, Mahowald MM. A possible mechanism for mixed apnea in obstructive sleep apnea. Chest 89: 800-805, 1986. 116. Iber C, Simon P, Skatrud JB, Mahowald MW, Dempsey JA. The BreuerHering reflex in humans. Effects of pulmonary denervation and hypocapnia. Am J Respir Crit Care Med 152: 217-224, 1995. 117. Imsand C, Feihl F, Perret C, Fitting JW. Regulation of inspiratory neuromuscular output during synchronized intermittent mechanical ventilation. Anesthesiology 80: 13-22, 1994. 118. Irsigler GB. Carbon dioxide response lines in young adults: The limits of the normal response. Am Rev Respir Dis 114: 529-536, 1976. 119. Jaber S, Petrof BJ, Jung B, Chanques G, Berthet JP, Rabuel C, Bouyabrine H, Courouble P, Koechlin-Ramonatxo C, Sebbane M, Similowski T, Scheuermann V, Mebazaa A, Capdevila X, Mornet D, Mercier Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning J, Lacampagne A, Philips A, Matecki S. Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med 183: 364-371, 2011. 120. Javaheri S. A mechanism of central sleep apnea in patients with heart failure. N Engl J Med 341: 949-954, 1999. 121. Javaheri S, Parker TJ, Wexler L, Liming JD, Lindower P, Roselle GA. Effect of theophylline on sleep-disordered breathing in heart failure. N Engl J Med 335: 562-567, 1996. 122. Jiang TX, Reid WD, Road JD. Delayed diaphragm injury and diaphragm force production. Am J Respir Crit Care Med 157: 736-742, 1998. 123. Jubran A, Grant BJ, Laghi F, Parthasarathy S, Tobin MJ. Weaning prediction: Esophageal pressure monitoring complements readiness testing. Am J Respir Crit Care Med 171: 1252-1259, 2005. 124. Jubran A, Grant BJ, Tobin MJ. Effect of hyperoxic hypercapnia on variational activity of breathing. Am J Respir Crit Care Med 156: 11291139, 1997. 125. Jubran A, Mathru M, Dries D, Tobin MJ. Continuous recordings of mixed venous oxygen saturation during weaning from mechanical ventilation and the ramifications thereof. Am J Respir Crit Care Med 158: 1763-1769, 1998. 126. Jubran A, Tobin MJ. Passive mechanics of lung and chest wall in patients who failed or succeeded in trials of weaning. Am J Respir Crit Care Med 155: 916-921, 1997. 127. Jubran A, Tobin MJ. Pathophysiologic basis of acute respiratory distress in patients who fail a trial of weaning from mechanical ventilation. Am J Respir Crit Care Med 155: 906-915, 1997. 128. Jubran A, Tobin MJ. Effect of isocapnic hypoxia on variational activity of breathing. Am J Respir Crit Care Med 162: 1202-1209, 2000. 129. Jubran A, Van de Graaff WB, Tobin MJ. Variability of patient-ventilator interaction with pressure support ventilation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 152: 129136, 1995. 130. Kawakami Y, Irie T, Shida A, Yoshikawa T. Familial factors affecting arterial blood gas values and respiratory chemosensitivity in chronic obstructive pulmonary disease. Am Rev Respir Dis 125: 420-425, 1982. 131. Keenan SP, Powers C, McCormack DG, Block G. Noninvasive positivepressure ventilation for postextubation respiratory distress: A randomized controlled trial. JAMA 287: 3238-3244, 2002. 132. Khoo MC. Determinants of ventilatory instability and variability. Respir Physiol 122: 167-182, 2000. 133. Kilbourn KH. Shock, seizures, and coma with alkalosis during mechanical ventilation. Ann Intern Med 65: 977-984, 1966. 134. Kimball WR, Leith DE, Robins AG. Dynamic hyperinflation and ventilator dependence in chronic obstructive pulmonary disease. Am Rev Respir Dis 126: 991-995, 1982. 135. King TE, Jr., Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: Scoring system and survival model. Am J Respir Crit Care Med 164: 1171-1181, 2001. 136. Knox CK. Characteristics of inflation and deflation reflexes during expiration of the cat. J Neurophysiol 36: 284-295, 1973. 137. Koutsoukou A, Armaganidis A, Stavrakaki-Kallergi C, Vassilakopoulos T, Lymberis A, Roussos C, Milic-Emili J. Expiratory flow limitation and intrinsic positive end-expiratory pressure at zero positive endexpiratory pressure in patients with adult respiratory distress syndrome. Am J Respir Crit Care Med 161: 1590-1596, 2000. 138. Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ. Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 167: 120-127, 2003. 139. Laghi F, D’Alfonso N, Tobin MJ. Pattern of recovery from diaphragmatic fatigue over 24 hours. J Appl Physiol 79: 539-546, 1995. 140. Laghi F, Harrison MJ, Tobin MJ. Comparison of magnetic and electrical phrenic nerve stimulation in assessment of diaphragmatic contractility. J Appl Physiol 80: 1731-1742, 1996. 141. Laghi F, Karamchandani K, Tobin MJ. Influence of ventilator settings in determining respiratory frequency during mechanical ventilation. Am J Respir Crit Care Med 160: 1766-1770, 1999. 142. Laghi F, Segal J, Choe WK, Tobin MJ. Effect of imposed inflation time on respiratory frequency and hyperinflation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 163: 13651370, 2001. 143. Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med 168: 10-48, 2003. 144. Laghi F, Tobin MJ. Indications for mechanical ventilation. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill Inc., 2006, pp. 129-162. 145. Laghi F, Topeli A, Tobin MJ. Does resistive loading decrease diaphragmatic contractility before task failure? J Appl Physiol 85: 1103-1112, 1998. 146. Lahiri S, Maret K, Sherpa MG. Dependence of high altitude sleep apnea on ventilatory sensitivity to hypoxia. Respir Physiol 52: 281-301, 1983. 147. Laroche CM, Carroll N, Moxham J, Green M. Clinical significance of severe isolated diaphragm weakness. Am Rev Respir Dis 138: 862-866, 1988. 2917 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning 148. Leevers AM, Simon PM, Dempsey JA. Apnea after normocapnic mechanical ventilation during NREM sleep. J Appl Physiol 77: 2079-2085, 1994. 149. Leevers AM, Simon PM, Xi L, Dempsey JA. Apnoea following normocapnic mechanical ventilation in awake mammals: A demonstration of control system inertia. J Physiol 472: 749-768, 1993. 150. Le Jemtel TH, Padeletti M, Jelic S. Diagnostic and therapeutic challenges in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol 49: 171-180, 2007. 151. Lemaire F, Teboul JL, Cinotti L, Giotto G, Abrouk F, Steg G, MacquinMavier I, Zapol WM. Acute left ventricular dysfunction during unsuccessful weaning from mechanical ventilation. Anesthesiology 69: 171-179, 1988. 152. Leung P, Jubran A, Tobin MJ. Comparison of assisted ventilator modes on triggering, patient effort, and dyspnea. Am J Respir Crit Care Med 155: 1940-1948, 1997. 153. Levine S, Nguyen T, Kaiser LR, Rubinstein NA, Maislin G, Gregory C, Rome LC, Dudley GA, Sieck GC, Shrager JB. Human diaphragm remodeling associated with chronic obstructive pulmonary disease: Clinical implications. Am J Respir Crit Care Med 168: 706-713, 2003. 154. Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358: 1327-1335, 2008. 155. Lofaso F, Isabey D, Lorino H, Harf A, Scheid P. Respiratory response to positive and negative inspiratory pressure in humans. Respir Physiol 89: 75-88, 1992. 156. Lofaso F, Simonneau G, Ladurie FL, Cerrina J, Chapelier A, Brenot F, Dartevelle P, Herve P. Frequency of mechanical ventilation and respiratory activity after double lung transplantation. Respir Physiol 92: 319-327, 1993. 157. Lorenzi-Filho G, Rankin F, Bies I, Douglas BT. Effects of inhaled carbon dioxide and oxygen on cheyne-stokes respiration in patients with heart failure. Am J Respir Crit Care Med 159: 1490-1498, 1999. 158. Lugliani R, Whipp BJ, Seard C, Wasserman K. Effect of bilateral carotidbody resection on ventilatory control at rest and during exercise in man. N Engl J Med 285: 1105-1111, 1971. 159. Macefield G, Hagbarth KE, Gorman R, Gandevia SC, Burke D. Decline in spindle support to alpha-motoneurones during sustained voluntary contractions. J Physiol 440: 497-512, 1991. 160. Macgowan NA, Evans KG, Road JD, Reid WD. Diaphragm injury in individuals with airflow obstruction. Am J Respir Crit Care Med 163: 1654-1659, 2001. 161. Mador MJ, Tobin MJ. Effect of alterations in mental activity on the breathing pattern in healthy subjects. Am Rev Respir Dis 144: 481-487, 1991. 162. Mancebo J. Assist-control ventilation. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill Inc., 2006, pp. 183-200. 163. Manchanda S, Leevers AM, Wilson CR, Simon PM, Skatrud JB, Dempsey JA. Frequency and volume thresholds for inhibition of inspiratory motor output during mechanical ventilation. Respir Physiol 105: 1-16, 1996. 164. Marantz S, Patrick W, Webster K, Roberts D, Oppenheimer L, Younes M. Response of ventilator-dependent patients to different levels of proportional assist. J Appl Physiol 80: 397-403, 1996. 165. Marini JJ, Capps JS, Culver BH. The inspiratory work of breathing during assisted mechanical ventilation. Chest 87: 612-618, 1985. 166. Marini JJ, Smith TC, Lamb VJ. External work output and force generation during synchronized intermittent mechanical ventilation. Effect of machine assistance on breathing effort. Am Rev Respir Dis 138: 11691179, 1988. 167. Marsden CD, Meadows JC, Merton PA. Isolated single motor units in human muscle and their rate of discharge during maximal voluntary effort. J Physiol 217: 12P-13P, 1971. 168. McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK. Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med 175: 150-159, 2007. 169. McMullen MC, Girling LG, Graham MR, Mutch WA. Biologically variable ventilation improves oxygenation and respiratory mechanics during one-lung ventilation. Anesthesiology 105: 91-97, 2006. 170. Meza S, Mendez M, Ostrowski M, Younes M. Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. J Appl Physiol 85: 1929-1940, 1998. 171. Mifflin SW. Convergent carotid sinus nerve and superior laryngeal nerve afferent inputs to neurons in the NTS. Am J Physiol 271: R870-R880, 1996. 172. Misuri G, Lanini B, Gigliotti F, Iandelli I, Pizzi A, Bertolini MG, Scano G. Mechanism of CO(2) retention in patients with neuromuscular disease. Chest 117: 447-453, 2000. 2918 Printer Name: Yet to Come Comprehensive Physiology 173. Montgomery AB, Holle RH, Neagley SR, Pierson DJ, Schoene RB. Prediction of successful ventilator weaning using airway occlusion pressure and hypercapnic challenge. Chest 91: 496-499, 1987. 174. Morrell MJ, Shea SA, Adams L, Guz A. Effects of inspiratory support upon breathing in humans during wakefulness and sleep. Respir Physiol 93: 57-70, 1993. 175. Mountain R, Zwillich C, Weil J. Hypoventilation in obstructive lung disease. The role of familial factors. N Engl J Med 298: 521-525, 1978. 176. Mulvey DA, Aquilina RJ, Elliott MW, Moxham J, Green M. Diaphragmatic dysfunction in neuralgic amyotrophy: An electrophysiologic evaluation of 16 patients presenting with dyspnea. Am Rev Respir Dis 147: 66-71, 1993. 177. Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Effect of ventilatory drive on carbon dioxide sensitivity below eupnea during sleep. Am J Respir Crit Care Med 165: 1251-1260, 2002. 178. Naughton M, Benard D, Tam A, Rutherford R, Bradley TD. Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure. Am Rev Respir Dis 148: 330-338, 1993. 179. Nava S, Ambrosino N, Clini E, Prato M, Orlando G, Vitacca M, Brigada P, Fracchia C, Rubini F. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease. A randomized, controlled trial. Ann Intern Med 128: 721-728, 1998. 180. Newell SZ, McKenzie DK, Gandevia SC. Inspiratory and skeletal muscle strength and endurance and diaphragmatic activation in patients with chronic airflow limitation. Thorax 44: 903-912, 1989. 181. Nichols DG, Buck JR, Eleff SM, Shungu DC, Robotham JL, Koehler RC, Traystman RJ. Diaphragmatic fatigue assessed by 31P-magnetic resonance spectroscopy in vivo. Am J Physiol 264: C1111-C1118, 1993. 182. Nielsen M, Smith H. Studies on the regulation of respiration in acute hypoxia. Acta Physiol Scand 24: 293-313, 1952. 183. Ninane V, Rypens F, Yernault JC, De Troyer A. Abdominal muscle use during breathing in patients with chronic airflow obstruction. Am Rev Respir Dis 146: 16-21, 1992. 184. Nolan PC, Dillon GH, Waldrop TG. Central hypoxic chemoreceptors in the ventrolateral medulla and caudal hypothalamus. Adv Exp Med Biol 393: 261-266, 1995. 185. Orem JM. Central neural interactions between sleep and breathing. In: Saunders NA, Sullivan CE, editors. Sleep and Breathing. New York: Marcel Dekker, 1984, pp. 91-135. 186. Orem JM. The wakefulness stimulus for breathing. In: Saunders NA, Sullivan CE, editors. Sleep and Breathing. New York: Marcel Dekker, Inc., 1994, pp. 113-154. 187. Orozco-Levi M, Gea J, Lloreta JL, Felez M, Minguella J, Serrano S, Broquetas JM. Subcellular adaptation of the human diaphragm in chronic obstructive pulmonary disease. Eur Respir J 13: 371-378, 1999. 188. Orozco-Levi M, Lloreta J, Minguella J, Serrano S, Broquetas JM, Gea J. Injury of the human diaphragm associated with exertion and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 164: 17341739, 2001. 189. Otis AB, Fenn WO, Rahn H. Mechanics of breathing in man. J Appl Physiol 2: 592-607, 1950. 190. Ottenheijm CA, Heunks LM, Sieck GC, Zhan WZ, Jansen SM, Degens H, de Boo T, Dekhuijzen PN. Diaphragm dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 172: 200-205, 2005. 191. Parthasarathy S, Jubran A, Laghi F, Tobin MJ. Sternomastoid, rib cage, and expiratory muscle activity during weaning failure. J Appl Physiol 103: 140-147, 2007. 192. Parthasarathy S, Jubran A, Tobin MJ. Cycling of inspiratory and expiratory muscle groups with the ventilator in airflow limitation. Am J Respir Crit Care Med 158: 1471-1478, 1998. 193. Parthasarathy S, Jubran A, Tobin MJ. Assessment of neural inspiratory time in ventilator-supported patients. Am J Respir Crit Care Med 162: 546-552, 2000. 194. Parthasarathy S, Tobin MJ. Effect of ventilator mode on sleep quality in critically ill patients. Am J Respir Crit Care Med 166: 1423-1429, 2002. 195. Parthasarathy S, Tobin MJ. Sleep in the intensive care unit. Intensive Care Med 30: 197-206, 2004. 196. Patrick W, Webster K, Puddy A, Sanii R, Younes M. Respiratory response to CO2 in the hypocapnic range in awake humans. J Appl Physiol 79: 2058-2068, 1995. 197. Peiffer C, Poline JB, Thivard L, Aubier M, Samson Y. Neural substrates for the perception of acutely induced dyspnea. Am J Respir Crit Care Med 163: 951-957, 2001. 198. Petrillo GA, Glass L, Trippenbach T. Phase locking of the respiratory rhythm in cats to a mechanical ventilator. Can J Physiol Pharmacol 61: 599-607, 1983. 199. Phillipson EA, Bowes G. Control of breathing during sleep. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, The Respiratory System. Control of breathing, Volume II, Bethesda: Am Physiol Soc, 1986, sect. 3, pp. 649-689. Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 200. Pitcher WD, Cunningham HS. Oxygen cost of increasing tidal volume and diaphragm flattening in obstructive pulmonary disease. J Appl Physiol 74: 2750-2756, 1993. 201. Plum F, Brown HW, Snoep E. Neurologic significance of posthyperventilation apnea. JAMA 181: 1050-1055, 1962. 202. Polacheck J, Strong R, Arens J, Davies C, Metcalf I, Younes M. Phasic vagal influence on inspiratory motor output in anesthetized human subjects. J Appl Physiol 49: 609-619, 1980. 203. Powers SK, Kavazis AN, Levine S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med 37: S347S353, 2009. 204. Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 92: 1851-1858, 2002. 205. Preas HL, Jubran A, Vandivier RW, Reda D, Godin PJ, Banks SM, Tobin MJ, Suffredini AF. Effect of endotoxin on ventilation and breath variability: Role of cyclooxygenase pathway. Am J Respir Crit Care Med 164: 620-626, 2001. 206. Pride NB, Macklem PT. Lung mechanics in disease. In: Handbook of Physiology: The Respiratory System, Vol. III. Bethesda: American Physiological Society, 1986, pp. 659-692. 207. Puddy A, Patrick W, Webster K, Younes M. Respiratory control during volume-cycled ventilation in normal humans. J Appl Physiol 80: 17491758, 1996. 208. Puddy A, Younes M. Effect of slowly increasing elastic load on breathing in conscious humans. J Appl Physiol 70: 1277-1283, 1991. 209. Puddy A, Younes M. Effect of inspiratory flow rate on respiratory output in normal subjects. Am Rev Respir Dis 146: 787-789, 1992. 210. Radell PJ, Eleff SM, Nichols DG. Effects of loaded breathing and hypoxia on diaphragm metabolism as measured by (31)P-NMR spectroscopy. J Appl Physiol 88: 933-938, 2000. 211. Radell PJ, Eleff SM, Traystman RJ, Nichols DG. In vivo diaphragm metabolism: Comparison of paced and inspiratory resistive loaded breathing in piglets. Crit Care Med 25: 339-345, 1997. 212. Raux M, Straus C, Redolfi S, Morelot-Panzini C, Couturier A, Hug F, Similowski T. Electroencephalographic evidence for pre-motor cortex activation during inspiratory loading in humans. J Physiol 578: 569-578, 2007. 213. Read DJC. A clinical method for assessing the ventilatory response to carbon dioxide. Australas Ann Med 16: 20-32, 1967. 214. Rebuck AS, Read J. Patterns of ventilatory response to carbon dioxide during recovery from severe asthma. Clin Sci 41: 13-21, 1971. 215. Rice AJ, Nakayama HC, Haverkamp HC, Pegelow DF, Skatrud JB, Dempsey JA. Controlled versus assisted mechanical ventilation effects on respiratory motor output in sleeping humans. Am J Respir Crit Care Med 168: 92-101, 2003. 216. Rikard-Bell GC, Bystrzycka EK, Nail BS. Cells of origin of corticospinal projections to phrenic and thoracic respiratory motoneurones in the cat as shown by retrograde transport of HRP. Brain Res Bull 14: 39-47, 1985. 217. Rimmer KP, Golar SD, Lee MA, Whitelaw WA. Myotonia of the respiratory muscles in myotonic dystrophy. Am Rev Respir Dis 148: 1018-1022, 1993. 218. Rochester DF, Braun NM. Determinants of maximal inspiratory pressure in chronic obstructive pulmonary disease. Am Rev Respir Dis 132: 42-47, 1985. 219. Rotheram EB, Safar P, Robin E. CNS disorder during mechanical ventilation in chronic pulmonary disease. JAMA 189: 993-996, 1964. 220. Sassoon CS. Mechanical ventilator design and function: The trigger variable. Respir Care 37: 1056-1069, 1992. 221. Sassoon CS. Intermittent mandatory ventilation. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. New York: McGrawHill Inc., 2006, pp. 201-220. 222. Sassoon CS, Caiozzo VJ, Manka A, Sieck GC. Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol 92: 2585-2595, 2002. 223. Sassoon CS, Gruer SE. Characteristics of the ventilator pressure- and flow-trigger variables. Intensive Care Med 21: 159-168, 1995. 224. Sassoon CS, Gruer SE, Sieck GC. Temporal relationships of ventilatory failure, pump failure, and diaphragm fatigue. J Appl Physiol 81: 238245, 1996. 225. Sassoon CS, Hassell KT, Mahutte CK. Hyperoxic-induced hypercapnia in stable chronic obstructive pulmonary disease. Am Rev Respir Dis 135: 907-911, 1987. 226. Sassoon CS, Mahutte CK. Airway occlusion pressure and breathing pattern as predictors of weaning outcome. Am Rev Respir Dis 148: 860-866, 1993. 227. Sassoon CS, Te TT, Mahutte CK, Light RW. Airway occlusion pressure. An important indicator for successful weaning in patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 135: 107-113, 1987. Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning 228. Satoh M, Eastwood PR, Smith CA, Dempsey JA. Nonchemical elimination of inspiratory motor output via mechanical ventilation in sleep. Am J Respir Crit Care Med 163: 1356-1364, 2001. 229. Scheid P, Lofaso F, Isabey D, Harf A. Respiratory response to inhaled CO2 during positive inspiratory pressure in humans. J Appl Physiol 77: 876-882, 1994. 230. Schmidt M, Demoule A, Cracco C, Gharbi A, Fiamma MN, Straus C, et al. Neurally adjusted ventilatory assist increases respiratory variability and complexity in acute respiratory failure. Anesthesiology 112(3): 670681, 2010. 231. Schoenhofer B, Koehler D, Polkey MI. Influence of immersion in water on muscle function and breathing pattern in patients with severe diaphragm weakness. Chest 125: 2069-2074, 2004. 232. Scott A, Wang X, Road JD, Reid WD. Increased injury and intramuscular collagen of the diaphragm in COPD: Autopsy observations. Eur Respir J 27: 51-59, 2006. 233. Shanely RA, Van Gammeren D, DeRuisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, Powers SK. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med 170: 994-999, 2004. 234. Sharshar T, Ross ET, Hopkinson NS, Porcher R, Nickol AH, Jonville S, Dayer MJ, Hart N, Moxham J, Lofaso F, Polkey MI. Depression of diaphragm motor cortex excitability during mechanical ventilation. J Appl Physiol 97: 3-10, 2004. 235. Shea SA. Behavioural and arousal-related influences on breathing in humans. Exp Physiol 81: 1-26, 1996. 236. Silver MM, Smith CR. Diaphragmatic contraction band necrosis in a perinatal and infantile autopsy population. Hum Pathol 23: 817-827, 1992. 237. Similowski T, Yan S, Gauthier AP, Macklem PT, Bellemare F. Contractile properties of the human diaphragm during chronic hyperinflation. N Engl J Med 325: 917-923, 1991. 238. Simon PM, Griffin DM, Landry DM, Skatrud JB. Inhibition of respiratory activity during passive ventilation: A role for intercostal afferents? Respir Physiol 92: 53-64, 1993. 239. Simon PM, Habel AM, Daubenspeck JA, Leiter JC. Vagal feedback in the entrainment of respiration to mechanical ventilation in sleeping humans. J Appl Physiol 89: 760-769, 2000. 240. Sinderby C, Navalesi P, Beck J, Skrobik Y, Comtois N, Friberg S, Gottfried SB, Lindstrom L. Neural control of mechanical ventilation in respiratory failure. Nat Med 5: 1433-1436, 1999. 241. Sinderby C, Weinberg J, Sullivan L, Lindstrom L, Grassino A. Electromyographical evidence for exercise-induced diaphragm fatigue in patients with chronic cervical cord injury or prior poliomyelitis infection. Spinal Cord 34: 594-601, 1996. 242. Skatrud J, Iber C, McHugh W, Rasmussen H, Nichols D. Determinants of hypoventilation during wakefulness and sleep in diaphragmatic paralysis. Am Rev Respir Dis 121: 587-593, 1980. 243. Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol 55: 813-822, 1983a. 244. Skatrud JB, Dempsey JA. Relative effectiveness of acetazolamide versus medroxyprogesterone acetate in correction of chronic carbon dioxide retention. Am Rev Respir Dis 127: 405-412, 1983b. 245. Skatrud JB, Dempsey JA, Bhansali P, Irvin C. Determinants of chronic carbon dioxide retention and its correction in humans. J Clin Invest 65: 813-821, 1980. 246. Skatrud JB, Dempsey JA, Iber C, Berssenbrugge A. Correction of CO2 retention during sleep in patients with chronic obstructive pulmonary diseases. Am Rev Respir Dis 124: 260-268, 1981. 247. Smith CA, Saupe KW, Henderson KS, Dempsey JA. Ventilatory effects of specific carotid body hypocapnia in dogs during wakefulness and sleep. J Appl Physiol 79: 689-699, 1995. 248. Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med 162: 2194-2200, 2000. 249. Speck DF, Revelette WR. Attenuation of phrenic motor discharge by phrenic nerve afferents. J Appl Physiol 62: 941-945, 1987. 250. Spieth PM, Carvalho AR, Guldner A, Pelosi P, Kirichuk O, Koch T, de Abreu MG. Effects of different levels of pressure support variability in experimental lung injury. Anesthesiology 110: 342-350, 2009. 251. Spieth PM, Carvalho AR, Pelosi P, Hoehn C, Meissner C, Kasper M, Hubler M, von Neindorff M, Dassow C, Barrenschee M, Uhlig S, Koch T, de Abreu MG. Variable tidal volumes improve lung protective ventilation strategies in experimental lung injury. Am J Respir Crit Care Med 179: 684-693, 2009. 252. Stanley NN, Cunningham EL, Altose MD, Kelsen SG, Levinson RS, Cherniack NS. Evaluation of breath holding in hypercapnia as a simple clinical test of respiratory chemosensitivity. Thorax 30: 337-343, 1975. 253. Stofan DA, Callahan LA, DiMarco AF, Nethery DE, Supinski GS. Modulation of release of reactive oxygen species by the contracting diaphragm. Am J Respir Crit Care Med 161: 891-898, 2000. 2919 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 9:35 Ventilatory Failure, Ventilator Support, and Ventilator Weaning 254. Stradling JR, Warley AR. Bilateral diaphragm paralysis and sleep apnoea without diurnal respiratory failure. Thorax 43: 75-77, 1988. 255. Straus C, Zelter M, Derenne JP, Pidoux B, Willer JC, Similowski T. Putative projection of phrenic afferents to the limbic cortex in humans studied with cerebral-evoked potentials. J Appl Physiol 82: 480-490, 1997. 256. Stubbings AK, Moore AJ, Dusmet M, Goldstraw P, West TG, Polkey MI, Ferenczi MA. Physiological properties of human diaphragm muscle fibres and the effect of chronic obstructive pulmonary disease. J Physiol 586: 2637-2650, 2008. 257. Suki B, Alencar AM, Sujeer MK, Lutchen KR, Collins JJ, Andrade JS, Jr., Ingenito EP, Zapperi S, Stanley HE. Life-support system benefits from noise. Nature 393: 127-128, 1998. 258. Sun MK, Reis DJ. Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol 266: R245-R256, 1994. 259. Sutton JR, Houston CS, Mansell AL, McFadden MD, Hackett PM, Rigg JR, Powles AC. Effect of acetazolamide on hypoxemia during sleep at high altitude. N Engl J Med 301: 1329-1331, 1979. 260. Tassaux D, Gainnier M, Battisti A, Jolliet P. Impact of expiratory trigger setting on delayed cycling and inspiratory muscle workload. Am J Respir Crit Care Med 172: 1283-1289, 2005. 261. Thammanomai A, Hueser LE, Majumdar A, Bartolak-Suki E, Suki B. Design of a new variable-ventilation method optimized for lung recruitment in mice. J Appl Physiol 104: 1329-1340, 2008. 262. Tobert DG, Simon PM, Stroetz RW, Hubmayr RD. The determinants of respiratory rate during mechanical ventilation. Am J Respir Crit Care Med 155: 485-492, 1997. 263. Tobin MJ. Non-invasive monitoring of ventilation. In: Tobin MJ, editor. Principles and Practice of Intensive Care Monitoring. New York: McGraw-Hill, Inc., 1998a, pp. 465-495. 264. Tobin MJ. Respiratory mechanics in spontaneously-breathing patients. In: Tobin MJ, editor. Principles and Practice of Intensive Care Monitoring. New York: McGraw-Hill, Inc., 1998b, pp. 617-654. 265. Tobin MJ. Culmination of an era in research on the acute respiratory distress syndrome. N Engl J Med 342: 1360-1361, 2000. 266. Tobin MJ. Advances in mechanical ventilation. N Engl J Med 344: 1986-1996, 2001. 267. Tobin MJ. Remembrance of weaning past: The seminal papers. Intensive Care Med 32: 1485-1493, 2006. 268. Tobin MJ, Chadha TS, Jenouri G, Birch SJ, Gazeroglu HB, Sackner MA. Breathing patterns. 2. Diseased subjects. Chest 84: 286-294, 1983. 269. Tobin MJ, Gardner WN. Monitoring of the control of ventilation. In: Tobin MJ, editor. Principles and Practice of Intensive Care Monitoring. New York: McGraw-Hill, Inc., 1998, pp. 415-464. 270. Tobin MJ, Guenther SM, Perez W, Lodato RF, Mador MJ, Allen SJ, Dantzker DR. Konno-Mead analysis of ribcage-abdominal motion during successful and unsuccessful trials of weaning from mechanical ventilation. Am Rev Respir Dis 135: 1320-1328, 1987. 271. Tobin MJ, Jubran A. Variable performance of weaning-predictor tests: Role of Bayes’ theorem and spectrum and test-referral bias. Intensive Care Med 32: 2002-2012, 2006. 272. Tobin MJ, Laghi F, Brochard L. Role of the respiratory muscles in acute respiratory failure of COPD: Lessons from weaning failure. J Appl Physiol 107: 962-970, 2009. 273. Tobin MJ, Laghi F, Jubran A. Narrative review: Ventilator-induced respiratory muscle weakness. Ann Intern Med 153: 240-245, 2010. 274. Tobin MJ, Mador MJ, Guenther SM, Lodato RF, Sackner MA. Variability of resting respiratory drive and timing in healthy subjects. J Appl Physiol 65: 309-317, 1988. 275. Tobin MJ, Perez W, Guenther SM, D’Alonzo G, Dantzker DR. Breathing pattern and metabolic behavior during anticipation of exercise. J Appl Physiol 60: 1306-1312, 1986. 276. Tobin MJ, Perez W, Guenther SM, Lodato RF, Dantzker DR. Does rib cage-abdominal paradox signify respiratory muscle fatigue? J Appl Physiol 63: 851-860, 1987. 277. Tobin MJ, Perez W, Guenther SM, Semmes BJ, Mador MJ, Allen SJ, Lodato RF, Dantzker DR. The pattern of breathing during successful and unsuccessful trials of weaning from mechanical ventilation. Am Rev Respir Dis 134: 1111-1118, 1986. 278. Tobin MJ, Yang KL, Jubran A, Lodato RF. Interrelationship of breath components in neighboring breaths of normal eupneic subjects. Am J Respir Crit Care Med 152: 1967-1976, 1995. 279. Topeli A, Laghi F, Tobin MJ. Can diaphragmatic contractility be assessed by twitch airway pressures in patients with chronic obstructive pulmonary disease? Am J Respir Crit Care Med 160: 1369-1374, 1999. 280. Topeli A, Laghi F, Tobin MJ. The voluntary drive to breathe is not decreased in hypercapnic patients with severe COPD. Eur Respir J 18: 53-60, 2001. 281. Torres A, Reyes A, Roca J, Wagner PD, Rodriguez-Roisin R. Ventilation-perfusion mismatching in chronic obstructive pulmonary disease during ventilator weaning. Am Rev Respir Dis 140: 1246-1250, 1989. 2920 Printer Name: Yet to Come Comprehensive Physiology 282. Tryfon S, Kontakiotis T, Mavrofridis E, Patakas D. Hering-Breuer reflex in normal adults and in patients with chronic obstructive pulmonary disease and interstitial fibrosis. Respiration 68: 140-144, 2001. 283. Tulla H, Takala J, Alhava E, Hendolin H, Manninen H, Kari A. Breathing pattern and gas exchange in emergency and elective abdominal surgical patients. Intensive Care Med 21: 319-325, 1995. 284. Vassilakopoulos T. Ventilator-induced diaphragmatic dysfunction. In: Tobin MJ, editor. Principles and Practice of Mechanical Ventilation. Third edition. New York: McGraw-Hill Inc, 2012 (in press). 285. Vassilakopoulos T, Zakynthinos S, Roussos C. The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med 158: 378-385, 1998. 286. Von Euler C. Brain stem mechanisms for generation and control of breathing pattern. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, The Respiratory System. Control of Breathing, Volume II, Bethesda: Am Physiol Soc, 1986, sect. 3, pp. 1-67. 287. Wagner PD, Dantzker DR, Dueck R, Clausen JL, West JB. Ventilationperfusion inequality in chronic obstructive pulmonary disease. J Clin Invest 59: 203-216, 1977. 288. Ward ME, Corbeil C, Gibbons W, Newman S, Macklem PT. Optimization of respiratory muscle relaxation during mechanical ventilation. Anesthesiology 69: 29-35, 1988. 289. Wasserman K, Whipp BJ, Koyal SN, Cleary MG. Effect of carotid body resection on ventilatory and acid-base control during exercise. J Appl Physiol 39: 354-358, 1975. 290. Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 110: 556-565, 1974. 291. Whidden MA, Smuder AJ, Wu M, Hudson MB, Nelson WB, Powers SK. Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol 108: 1376-1382, 2010. 292. Whipp BJ. Carotid bodies and breathing in humans. Thorax 49: 10811084, 1994. 293. White JE, Drinnan MJ, Smithson AJ, Griffiths CJ, Gibson GJ. Respiratory muscle activity and oxygenation during sleep in patients with muscle weakness. Eur Respir J 8: 807-814, 1995. 294. Wilcox I, McNamara SG, Dodd MJ, Sullivan CE. Ventilatory control in patients with sleep apnoea and left ventricular dysfunction: Comparison of obstructive and central sleep apnoea. Eur Respir J 11: 7-13, 1998. 295. Wilson CR, Satoh M, Skatrud JB, Dempsey JA. Non-chemical inhibition of respiratory motor output during mechanical ventilation in sleeping humans. J Physiol 518 (Pt 2): 605-618, 1999. 296. Wilson PA, Skatrud JB, Dempsey JA. Effects of slow wave sleep on ventilatory compensation to inspiratory elastic loading. Respir Physiol 55: 103-120, 1984. 297. Xie A, Skatrud JB, Dempsey JA. Effect of hypoxia on the hypopnoeic and apnoeic threshold for CO(2) in sleeping humans. J Physiol 535: 269-278, 2001. 298. Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med 165: 1245-1250, 2002. 299. Yamada Y, Du HL. Analysis of the mechanisms of expiratory asynchrony in pressure support ventilation: A mathematical approach. J Appl Physiol 88: 2143-2150, 2000. 300. Yan S, Lichros I, Zakynthinos S, Macklem PT. Effect of diaphragmatic fatigue on control of respiratory muscles and ventilation during CO2 rebreathing. J Appl Physiol 75: 1364-1370, 1993. 301. Yan S, Sliwinski P, Gauthier AP, Lichros I, Zakynthinos S, Macklem PT. Effect of global inspiratory muscle fatigue on ventilatory and respiratory muscle responses to CO2. J Appl Physiol 75: 1371-1377, 1993. 302. Yang KL, Tobin MJ. A prospective study of indexes predicting the outcome of trials of weaning from mechanical ventilation. N Engl J Med 324: 1445-1450, 1991. 303. Yang L, Luo J, Bourdon J, Lin MC, Gottfried SB, Petrof BJ. Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med 166: 1135-1140, 2002. 304. Younes M. Load responses, dyspnea, and respiratory failure. Chest 97: 59S-68S, 1990. 305. Younes M. Determinants of thoracic excursions during exercise. In: Whipp BJ, Wasserman K, editors. Exercise: Pulmonary Physiology and Pathophysiology. . New York: Marcel Dekker, Inc., 1991, pp. 1-65. 306. Younes M. Mechanisms of ventilatory failure. Current Pulmonology 14: 243-292, 1993a. 307. Younes M. Patient-ventilator interaction with pressure-assisted modalities of ventilator support. Semin Resp Med 15: 299-322, 1993b. 308. Younes M. Interactions between patients and ventilators. In: Roussos C, editor. The Thorax. New York: Marcel Dekker, 1995a, pp. 23672420. 309. Younes M. Mechanisms of respiratory load compensation. In: Demsey JA, Pack AL, editors. Regulation of Breathing. New York: Marcel Dekker, Inc., 1995b, pp. 867-922. Volume 2, October 2012 P1: OTA/XYZ P2: ABC JWBT335-c110030 JWBT335/Comprehensive Physiology July 31, 2012 Comprehensive Physiology 310. Younes M. Dynamic intrinsic PEEP (PEEP(i),dyn): Is it worth saving? Am J Respir Crit Care Med 162: 1608-1609, 2000. 311. Younes M. Apnea following mechanical ventilation may not be caused by neuromechanical influences. Am J Respir Crit Care Med 163: 12981301, 2001. 312. Younes M. Control of breathing during mechanical ventilation. In: Slutsky AS, Brochard L, editors. Mechanical Ventilation. Berlin: Springer, 2004, pp. 63-82. 313. Younes M, Georgopoulos D. Control of breathing relevant to mechanical ventilation. In: Marini JJ, Slutsky AS, editors. Physiological Basis of Ventilator Support. New York: Marcel Dekker, Inc., 1998, pp. 1-73. 314. Younes M, Kun J, Webster K, Roberts D. Response of ventilatordependent patients to delayed opening of exhalation valve. Am J Respir Crit Care Med 166: 21-30, 2002. 315. Younes M, Polacheck J. Central adaptation to inspiratory-inhibiting expiratory-prolonging vagal input. J Appl Physiol 59: 1072-1084, 1985. 316. Younes M, Vaillancourt P, Milic-Emili J. Interaction between chemical Volume 2, October 2012 9:35 Printer Name: Yet to Come Ventilatory Failure, Ventilator Support, and Ventilator Weaning factors and duration of apnea following lung inflation. J Appl Physiol 36: 190-201, 1974. 317. Zakynthinos S, Routsi C, Vassilakopoulos T, Kaltsas P, Zakynthinos E, Kazi D, Roussos C. Differential cardiovascular responses during weaning failure: Effects on tissue oxygenation and lactate. Intensive Care Med 31: 1634-1642, 2005. 318. Zhu E, Comtois AS, Fang L, Comtois NR, Grassino AE. Influence of tension time on muscle fiber sarcolemmal injury in rat diaphragm. J Appl Physiol 88: 135-141, 2000. 319. Zhu E, Petrof BJ, Gea J, Comtois N, Grassino AE. Diaphragm muscle fiber injury after inspiratory resistive breathing. Am J Respir Crit Care Med 155: 1110-1116, 1997. 320. Zifko UA, Young BG, Remtulla H, Bolton CF. Somatosensory evoked potentials of the phrenic nerve. Muscle Nerve 18: 1487-1489, 1995. 321. Zucker S. Short-term synaptic plasticity. Ann Rev Neurosci 12: 13-31, 1989. 322. Zuperku EJ, Hopp FA, Kampine JP. Central integration of pulmonary stretch receptor input in the control of expiration. J Appl Physiol 52: 1296-1315, 1982. 2921