HRA summary plans for Assessment and Approval
advertisement

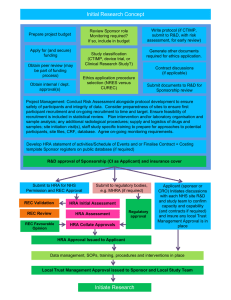
Summary of Plans for Health Research Authority Assessment and Approval Author: Janet Messer Date of Release: March 2014 Resources prepared by Debbie Corrigan Reviewed by: Janet Wisely Version No. & Status: Executive Summary v1 Summary of Plans for Health Research Authority (HRA) Assessment and Approval Contents Summary .............................................................................................................................. 1 1 Introduction ................................................................................................................... 4 2 Implementation model ................................................................................................... 5 3 Metrics and performance management ......................................................................... 7 4 Summary of investment and return ................................................................................ 8 Summary Following the establishment of the Health Research Authority (HRA), a multi-agency project group was set up to identify root causes of the issues described in a number of reports on research regulation and to propose solutions. The proposals have subsequently been tested and a wide range of projects is being taken forward to address multiple facets of research regulation. Details of these projects are available on the HRA website. The plans for HRA Assessment and Approval form a key component, providing a single, coherent and simple system for research approvals for the NHS in England. A typical large study requiring thousands of participants will need to be conducted across more than 20 NHS sites. The coordinating research team will need to get the study reviewed by a research ethics committee, and also seek permission from each NHS organisation. Currently researchers report that navigating these approvals and addressing varying and sometimes contradictory queries requires excessive effort from the research team and unnecessary costs for all concerned. The HRA will now streamline this complex process, so that researchers and NHS sites can rely on the HRA’s approval to address both legal and ethical aspects of the study in an integrated way. This will allow local research teams to work with their NHS site to set up and deliver the study. The HRA Approval will provide a single approval system for all studies in England, replacing the current separate systems for ethical review and NHS permissions with an integrated process and single approval that provides assurance to organisations hosting research. The HRA Approval incorporates assessments currently undertaken in study-wide review and multiple local R&D reviews and includes the Research Ethics Committee opinion. This will reduce the complexity of the approvals process for academic and industry research significantly. Site permissions will be replaced by straightforward decisions to deliver based on local capacity and capability. An options appraisal confirmed that implementation of selected components alone will not achieve the full potential of a single system. Although separate timelines for the individual review systems show improvement in average timelines, only implementation of the full HRA Approval will achieve improvements to the total elapsed time for applicants to complete the overall approvals process, and enable a single metric from one validation point to be captured. HRA Approval is also a necessary precursor to implementation of the clinical trials regulation from 2016-17. Establishing this radical new system will require an initial two year investment to ensure the HRA is properly resourced to deliver this important Government priority, which has been identified again in 1 1 the recent Select Committee report on Clinical Trials . An initial investment of £4.5 million in year 1 and £4.9 million in year 2 will be required, leading to operational costs in the third year of £4.1 million upon which efficiency savings could be made over time. The investment in one system managed by one organisation will realise the true benefit of the NHS in delivering research in the UK. These costs are less than the conservative estimate of annual savings, which include £3 million per year to sponsors of NHS research alone. In addition the new system maximises the potential economic benefits of the NHS in attracting and delivering research in the UK, and represents a safe investment to deliver an important Government priority. However, the key saving of the HRA Approval is anticipated to be in the total elapsed time from submission to HRA to last site set up, and in actual effort in navigating the process, due to the radically simplified system. Currently, unanticipated time spent in gaining approvals, and resultant difficulties in completing studies result in grant extensions and increased costs to funders and commercial sponsors. Where studies fail to complete due to delays in set-up of sites, not only is the whole grant wasted but the potential results of the research are also wasted. For commercial research, delays in overall time to initiation of sites have an overall impact on recruitment of participants in the UK, with significant implications for budgets allocated from global companies to the UK. The success of the HRA Approval will be measured through metrics that provide a holistic overview, rather than measuring individual components of the research process in isolation. Perceptions of researchers and the life sciences industry will primarily be influenced by the predictability and proportionality of the new system. 2 The House of Lords Science and Technology Committee report on clinical trials states: We welcome moves by the HRA to streamline NHS governance arrangements and stress the importance of this initiative, which, in our view, should be given the highest priority. Following completion of the feasibility study, we recommend that a timeline detailing the next steps be published as part of the HRA’s response to this Report. The Government should assist the HRA in its efforts to meet this priority, including making additional resources available if necessary. The HRA believes the HRA Approval is a key activity to protect and promote the interests of patients and the public in health research. 1 2 http://www.parliament.uk/science/ http://www.parliament.uk/science/ 2 The new process Sites HRA Site feasibility and selection Single application • Application to HRA • Single validation capacity & capability ready to deliver capacity & capability ready to deliver HRA approval • Assessment • REC opinion 3 1 Introduction The aim of HRA Approval for research is to address the issues with current processes, and the practical operation of these processes, that result in unnecessary duplication, inefficiency and complexity for researchers in both academia and industry. A feasibility study, including small-scale piloting, was conducted by the HRA between January and June 2013. This study demonstrated that an HRA Approval based on one application and consisting of an integrated assessment addressing legal and management aspects of research applications, plus the Research Ethics Committee (REC) opinion was feasible and would streamline and simplify processes, to achieve the government’s ambition of unifying the approvals system for health research. The process would involve one application to HRA and an assessment, alongside the REC opinion to form an HRA Approval. The implementation of the process would be supported by mechanisms to ensure that this approval is accepted by others (including clarifying that responsibility for audit and inspection findings relating to the approval rests with the HRA rather than local Trusts). This would eliminate duplication of assessment across individual sites, requirements for extra documentation or further checking. It will provide a basis for unifying the approval system for health research with other regulators and review bodies. The options are all proposed on the basis that the HRA Approval would be available to all studies requiring REC review with NHS participants, and all studies not requiring REC review but requiring NHS permission, according to current criteria (for example research involving NHS staff). The process would be available to single-centre and multi-centre studies in England. The system would be available for all studies and would not be restricted to NIHR Portfolio studies, which are currently the only studies eligible to use the NIHR Coordinated System for gaining NHS Permission (CSP). The options for implementation are all costed, although the details remain confidential. Cost effectiveness has been considered for the wider NHS and other stakeholders, including funders, sponsors and industry. 4 2 Implementation model The HRA Assessment will consider the following components, which will sit alongside the independent REC opinion: Participant interests Contracting Compliance and delivery Clinical support service technical elements Research passport requirements The components draw on the study-wide criteria agreed across all UK permission systems, which have been tested over time. The Assessment will include additional opportunities for a single review of technical elements that are currently duplicated across the NHS, particularly radiation, information governance and pharmacy. The HRA Assessment will confirm compliance with standard contracts, agreements and templates to improve consistency and avoid delays resulting from negotiation of sponsor-specific or site-specific preferences. The REC opinion will itself draw on and take assurance from the assessment. For example the review of indemnity and insurance arrangements will no longer be undertaken by the committee. This will allow the committee to focus on reviewing ethical considerations to maximise the value of the independent committee. Some areas will be assessed and approved by NHS staff within the HRA. Clinical service reviews will be managed through the HRA using expert assessors in NHS organisations e.g. pharmacy and radiation. The assessments that are relevant to the REC opinion will be provided to the REC to enable RECs to focus on ethical aspects of the study. It is envisaged that the delivery of the HRA Approval will be through a regional service, building on the existing HRA Centres and aligning with the new boundaries for NIHR Local Clinical Research Networks, which are also aligned with Academic Health Science Networks. There are currently five HRA Centres: a model that has been developed to achieve maximum efficiencies and geographical coverage. Although the actual premises of individual centres remain open to future contractual negotiation, the general geographical coverage is consistent with a model of each HRA Centre serving a cluster of networks. A proposed arrangement is set out below. Implementation through such a model would allow close integration between HRA Assessment and REC services, along with geographical alignment for provision of advice and support. Current premises would need to be reviewed in order to provide the necessary infrastructure. Initially recruitment of staff would be primarily to project posts to develop the plans and implementation timetable; some of these will be fixed term posts or secondments. Operational staff will be employed across the HRA Centres, with some posts delivering early phases of implementation, and further posts over the course of the implementation timetable. 5 North Centre North East & Cumbria Yorkshire & Humber North West Coast East Centre East Midlands Eastern North Thames North West & Midlands Centre Greater Manchester West Midlands Thames Valley & S. Midlands South West Centre South West Peninsula Wessex West of England South East Centre Kent, Surrey and Sussex South London North West London 6 3 Metrics and performance management Significant changes to the processes in the approvals process will have implications for the current metrics used in performance management, including the NIHR 70 day benchmark, Clinical Research Network (CRN) High Level Objectives and internal CRN targets. Although these targets and benchmarks have been successful in driving reductions in timelines, the metrics for the REC and R&D systems are measured in isolation, and analysis now shows that without integration there is a risk that any further downward pressure on timelines will simply move delays around the system. There is already feedback from applicants that some sites are preventing local submissions from being made until targets can be achieved. Currently applicants are recommended to apply for REC review and CSP study-wide review in parallel. In fact practice is variable, particularly for non-commercial studies where submission to CSP is not permitted until funding has been confirmed, whereas some applicants apply to REC whilst funding is being finalised. Sixty percent of studies are given a provisional opinion at first review so the elapsed time from REC submission to final opinion may be longer than the target time for R&D review. The overall impact for the applicant is that the time from REC submission to completion of study-wide review can be considerably longer than the individual performance data from HRA or CRN would indicate. Analysis has also shown that completion of study-wide review is often prevented by delays in the final REC opinion letter getting passed on to the relevant person to complete study-wide review (due to the different models for study-wide review in networks). Integrated information systems where R&D staff at sites are able to directly access information about REC and other regulatory processes will avoid unnecessary delays. Integration of REC and R&D processes in the HRA Approval will require different metrics as the current metrics based around timing of R&D submission and SSI submission will no longer be relevant. The HRA Approval may result in changes to the way in which research teams set up sites, as feedback indicates that some applicants deliberately spread out application to sites because of the work currently involved in negotiation with each site. Simpler processes for setting up sites should allow more sites to be set up in parallel, allowing more time for recruitment of participants. As noted elsewhere, a key measure of performance will also be the time to HRA Approval against the predicted time given at the start of the HRA Approval process. This takes into account the importance of proportionality and predictability to support good planning, with well-managed studies being expected to proceed rapidly, whereas studies with complex requirements or novel considerations would be recognised as needing longer. 7 4 Summary of investment and return Initially HRA will establish a programme team. Once plans and timetables are agreed with relevant stakeholders, HRA Approval will be rolled out, on an application type basis. The HRA will start with studies that have minimal impact on patient care and clinical services. Roll out will then move to studies in primary care where, increasingly, support for research is being offered through large geographical clusters. The HRA will also look to include other specific study types where current processes present particular difficulties e.g. rare diseases in the early phases of implementation. In the meantime, work will continue on standardising the requirements for technical reviews for pharmacy, radiation and contracting. Roll out is anticipated to be completed by the end of 2015 and will be for all studies in England within a UK-wide coordinated framework. Early HRA action will include appointing application managers to address issues within the approvals systems for studies not in the early phase of roll out. In addition staff will be recruited to work in geographic clusters to work collaboratively with stakeholders in enabling any necessary local changes to systems, structures and behaviours to support a seamless transition to HRA Approval. These staff will identify existing areas of good practice to share and work with existing teams who are actively involved in improvement programmes. The costs of implementation are therefore composed of the following staff roles: project teams, information systems, guidance and policy, engagement and operations. Summary of investment required for additional functions Investment area for additional resources HRA Assessment and Approval IS support HRA associated organisational change REC Operations Totals Total costs 2014/15 (£’k) 3012 395 942 166 4515 Total costs 2015/16 (£’k) 3438 282 1003 166 4889 Total costs 2016/7 (£’k) 2920 114 1003 166 4203 Workforce assumptions Staff by location type London Outside London 2014/15 Headcount WTE 31 25 69 54.85 100 79.85 2015/16 Headcount WTE 26 23.1 67 66.1 93 89.2 2016/17 Headcount WTE 23 14.85 62 55.1 85 69.95 On average it is conservatively estimated that there would be a direct saving to HRA, NHS, or CRN of about 10 - 20 hours per study for hospital-based studies depending on the type of study, and about 10 hours per study for primary care studies. The grades of staff currently involved in these tasks are highly variable. There are no data available on the costs of R&D review for non-portfolio studies. Many of these studies that involve the NHS are likely to be single-centre studies. For multi-centre studies, there would be savings for all additional sites where study-wide and local reviews are undertaken at each 8 site. The costs are difficult to determine as they are difficult to separate out from the costs of undertaking feasibility and the decision to deliver, and include staff outside R&D offices. In those areas where staff are working in full accordance with guidelines for pragmatic and proportionate review the cost savings will be less than in those areas where staff continue to duplicate reviews or have not harmonised arrangements locally. The savings related to staff outside R&D offices are of particular note as the proposed process has the potential to release clinical and non-clinical service staff who are involved to varying extents in duplicating review of studies, eg radiation, data protection, finance, and Human Resources. Without factoring in any savings to sponsors or research teams, the yearly costs of the HRA Approval for all studies in England, are lower than the total yearly costs of one part of the current system for NIHR portfolio studies. It is not intended to suggest that individuals currently undertaking activities that are indicated as a saving would no longer have a role, only that these activities would no longer be required. The savings in costs to sponsors will vary considerably depending on the staff currently allocated to obtaining approvals and permissions, but can be illustrated by a non-commercial case study supplied to the HRA in which 0.8WTE were required for a period of over six months to support the set up and initiation of a study with 23 sites, approximating to 20 hours per site. A CRO similarly reported allowing 20 hours per site to obtain permission, including contracting, for commercial studies. Taking into account the continuing need for sponsors to conduct feasibility, site assessment, site preparation and training and initiation, a conservative estimated saving of five hours per site for a study of five sites would save 25 hours. Given that some elements of the interaction with both RECs and NHS R&D are undertaken by Chief Investigators and Principal Investigators who are often consultants, and other roles are undertaken by coordinators and research nurses, a conservative mid-salary of £40,000 is assumed. The saving of 25 hours per study for sponsors across 4000 studies involving the NHS would be in the region of £3,000,000 in a year. However, it is also anticipated that the streamlined, simpler system will enable researchers to complete the overall approvals process in a shorter time. This is where greatest savings will be found in reality. Staff costs for non-commercial research are often funded for a specified period through grants. Unanticipated time spent in gaining approvals, and resultant difficulties in completing studies, requires grant extensions with increasing costs to funders. In a large flagship non-commercial study setting up sites during 2013, despite very few issues being raised during the permissions process, a 9 contract research organisation has had to be brought in to dedicate time to chasing up local research teams and R&D staff. Where studies fail to complete due to delays in set-up of sites, not only is the whole grant wasted but the potential results of the research are also wasted. For commercial research, delays in overall time to initiation of sites has an overall impact on recruitment of participants in the UK, with significant implications for budgets allocated from global companies to the UK. For smaller bioscience companies, delays in commencing trials have a significant impact, as milestone payments from investors are often triggered by completion of stages of the research process such as site set-up and recruitment. Analysis of a sub-set of studies in one region found that the median time from REC submission to completion of study-wide review was 146 days. This time encompassed a median time to final REC opinion of 65 days (with no clock stopping), and a median time to complete study-wide review of 86 days (including time waiting for the REC opinion). The data showed a median delay from REC submission to R&D submission of 46 days. Feedback from researchers indicates that applicants stagger the applications processes because of the complexity of navigating the separate processes at the same time. Similarly researchers, particularly for non-commercial research, stagger the submissions for site permissions due to the time required to negotiate individually with each site. In an example provided to HRA, for 11 sites where permission had been obtained (further sites had not issued permission at the time the example was shared and were not included in the data), the elapsed time from the first submission to the last permission was 118 days, and the elapsed time from initial contact with the first site to the last permission was 155 days. In this example the median time from site submission to permission was 37.5 days, a little over the target time for CSP of 30 days, although some sites were still in process and would exceed target times. However, the median time from initial contact with the site to submission to the site was 57.5 days, with much of that time reported to be spent in liaison with local research teams and preparing local applications. Given that the initial contact with sites was not started in this study until eight months after REC approval, the total elapsed time from submission to REC, even before all sites were set up, was over eighteen months. It is difficult to accurately quantify the savings and efficiencies for sponsors and researchers as they found it difficult to assess the impact in detail of such a radically different system. However, there was virtually unanimous support for the proposals as a much simpler system comparing favourably with systems in other European countries. Measurement of impact across a range of parties will form a key part of the implementation plan. The key saving of the HRA Approval is anticipated to be in the total elapsed time from submission to HRA to last permission at a site and in actual effort in navigating the process, due to the simplified system. The HRA Approval will replace the separate REC and study-wide processes, and site permissions will be replaced by straightforward decisions to deliver based on local capacity and capability. 10