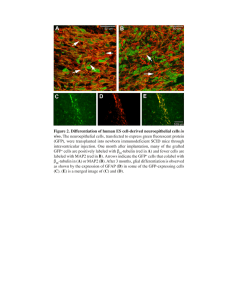
The aggregation-prone intracellular serpin SRP-2 fails to
advertisement

Genetics: Early Online, published on March 18, 2015 as 10.1534/genetics.115.176180 The aggregation-prone intracellular serpin SRP-2 fails to transit the ER in C. elegans Richard M. Silverman1, Erin E. Cummings1, Linda P. O’Reilly1, Mark T. Miedel1, Gary A. Silverman1, Cliff J. Luke1, David H. Perlmutter1 and Stephen C. Pak1,2 1 Departments of Pediatrics and Cell Biology, University of Pittsburgh School of Medicine, Children's Hospital of Pittsburgh of UPMC and Magee-Womens Hospital Research Institute, 4401 Penn Avenue, Pittsburgh, PA 15224, USA 2 Correspondence should be addressed to: paksc@upmc.edu (SCP) ACKNOWLEDGMENTS This study was supported by grants from the National Institutes of Health (DK079806 and DK081422). Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Thanks to Dan Lawrence, University of Michigan for providing the mouse neuroserpin cDNA. AUTHOR CONTRIBUTIONS RMS, EEC, LPO, MTM and SCP conducted the experiments and performed the image analysis, CJL performed the computational analysis; RMS, DHP, GAS and SCP analyzed the data and wrote the manuscript. COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests. 1 Copyright 2015. ABSTRACT Familial Encephalopathy with Neuroserpin Inclusions Bodies (FENIB) is a serpinopathy that induces a rare form of presenile dementia. Neuroserpin contains a classical signal peptide and like all extracellular serpins is secreted via the endoplasmic reticulum (ER)-Golgi pathway. The disease phenotype is due to gain-of-function missense mutations that cause neuroserpin to misfold and aggregate within the ER. In a previous study, nematodes expressing a homologous mutation in the endogenous C. elegans serpin, srp-2, was reported to model the ER proteotoxicity induced by an allele of mutant neuroserpin. Our results suggest that SRP-2 lacks a classical N-terminal signal peptide and is a member of the intracellular serpin family. Using confocal imaging and an ER co-localization marker, we confirmed that GFP tagged wild-type SRP-2 localized to the cytosol and not the ER. Similarly, the aggregation-prone SRP-2 mutant formed intracellular inclusions that localized to the cytosol. Interestingly, wild-type SRP2, targeted to the ER by fusion to a cleavable N-terminal signal peptide, failed to be secreted and accumulated within the ER lumen. This ER retention phenotype is typical of other obligate intracellular serpins forced to translocate across the ER membrane. Neuroserpin is a secreted protein that inhibits trypsin-like proteinase. SRP-2 is a cytosolic serpin that inhibits lysosomal cysteine peptidases. We concluded that SRP-2 is neither an orthologue nor a functional homologue of neuroserpin. Furthermore, animals expressing an aggregation-prone mutation in SRP-2 do not model the ER proteotoxicity associated with FENIB. INTRODUCTION Members of the serine proteinase inhibitor (serpin) superfamily are best known as physiological regulators of proteolytic cascades associated with coagulation, thrombolysis, inflammation and cell death (SILVERMAN et al. 2010; WHISSTOCK et al. 2010). To accomplish this task serpins fold into a highly conserved metastable structure consisting of three - sheets, 8-9 helices and an exposed reactive center loop (RCL), which serves as bait for target proteinases (HUBER AND CARRELL 1989; IRVING et al. 2000). After a target proteinase binds and cleaves its cognate RCL, strain on the serpin scaffold is relieved, thereby triggering a conformational change that traps the serpin and proteinase in a covalent complex (HUNTINGTON et al. 2000). Due to the metastability of the native serpin fold, which is crucial for its inhibitory activity, even single nonsynonymous amino acid changes make these proteins highly susceptible to misfolding and aggregation (HUNTINGTON 2006). This aggregation-prone phenotype is most evident in patients with 1-antitrypsin (1AT)/SERPINA1 deficiency (ATD). The most common mutation, Z (E342K), impairs the latter stages of serpin folding and facilitates domain swaps between monomers, yielding oligomers and higher order polymers (YAMASAKI et al. 2011). Although a small percentage of the monomers of the mutant protein (ATZ) are constitutively secreted via the endoplasmic reticulum (ER)-Golgi pathway, toxic monomers and higher order species accumulate within the ER of hepatocytes (PERLMUTTER 2002; PERLMUTTER 2011; SILVERMAN et al. 2013). Ultimately, these ERretained ATZ species, which appear as periodic-acid Schiff positive (PAS+), diastase 2 resistant inclusions in histological liver specimens, lead to cirrhosis and in some cases hepatocellular carcinoma (PERLMUTTER 2006; PERLMUTTER 2007; SILVERMAN et al. 2013). Similar types of mutations have resulted in the intracellular accumulation of other extracellular serpins such as antithrombin (SERPINC1), C1 esterase inhibitor (SERPING1), 1-antichymotrypsin (SERPINA3) and neuroserpin (NS/SERPINI1) (ROUSSEL et al. 2011). Collectively, these disorders, which all involve proteins secreted via the classical ER-Golgi pathway, have been designated serpinopathies and are characterized by: 1) a loss-of-function phenotype due to decreased circulating levels of the inhibitor and 2) a gain-of-function phenotype due to cellular proteotoxicity associated with retention of misfolded proteins within ER of the synthesizing cells (CARRELL AND LOMAS 1997; CARRELL AND LOMAS 2002; CARRELL 2005). In 2001, the serpin nomenclature committee divided the 36 human serpins into 9 clades (A-I) (SILVERMAN et al. 2001). Unique among this group is the clade B serpins, which belong to the larger evolutionarily well-conserved group of intracellular serpins (REMOLD-O'DONNELL 1993; SILVERMAN et al. 2004). In comparison to the other human serpins (clades A and C-I), which are all secreted proteins containing cleavable Nterminal signal peptides, the intracellular serpins (clade B) lack a recognizable secretion signal as well as N- and C-terminal extensions. Generally, these serpins possess a nucleo-cytosolic subcellular distribution (BIRD et al. 2001; SILVERMAN et al. 2004). However, one member of the intracellular serpin family, chicken ovalbumin, is secreted by the chicken oviduct (PALMITER et al. 1978). Also, but to a much lesser extent and only under certain conditions, plasminogen-activator type 2 (PAI2/SERPINB2) appears to be inefficiently secreted (BELIN 1993; BELIN et al. 2004). Neither ovalbumin nor PAI2/SERPINB2 possess a cleavable N-terminal signal peptide (PALMITER et al. 1978). Mutagenesis studies suggest that embedded hydrophobic motifs in the N-terminus facilitate secretion with that of PAI2/SERPINB2 being very inefficient (TABE et al. 1984; BELIN et al. 2004). Although some of these secreted intracellular serpins possess complex N-linked carbohydrates suggestive of Golgi processing, secretion still occurs in the presence of tunicamycin (KELLER AND SWANK 1978) and may involve unconventional signal pathway(s) similar to those used by FGF-2 and IL-1 (NICKEL AND RABOUILLE 2009; GIULIANI et al. 2011; MALHOTRA 2013). Indeed, forced expression of wild-type protease inhibitor 6 (PI6)/SERPINB6 and MASPIN/SERPINB5 into the ER-Golgi pathway by fusing a N-terminal signal peptide leads to overt polymerization and ER retention (SCOTT et al. 1996; TEOH et al. 2010). None of the 12 other human clade B serpins appear to be secreted. Although, for example, high levels of the squamous cell carcinoma antigen 1 (SCCA1)/SERPINB3 and SCCA2/SERPINB4 are detected in the circulation of some patients with advanced stages of malignancy and other conditions, these proteins appear to be passively released concomitant with cell death (UEMURA et al. 2000). To gain better insight into the biologic function of the intracellular serpin family, we examined their role in C. elegans, which only encodes clade B-like/intracellular serpins. The C. elegans genome encodes nine intracellular serpins. However, only SRP-1, SRP-2, SRP-3, SRP-6 and SRP-7 are synthesized as full length proteins and serve as bona fide proteinase inhibitors, with the rest being transcribed pseudogenes or non-inhibitory variants (PAK et al. 2004; LUKE et al. 2006; PAK et al. 2006; LUKE et al. 3 2007). Similar to the clade B serpins, these nematode serpins lacked a signal peptide and the N- and C-terminal extensions typical of serpins transiting the conventional ERGolgi secretory pathway. Green fluorescent protein (GFP)-nematode serpin fusions show a diffuse cytoplasmic appearance and no evidence of secretion into the intestinal cell lumen, the pseudocoelomic space or the cuticle (PAK et al. 2004; LUKE et al. 2006; PAK et al. 2006; LUKE et al. 2007). Recently, one of the C. elegans serpin genes, srp-2, was cloned, mutagenized and reintroduced in the nematode germline by biolistic transformation (SCHIPANSKI et al. 2013). This mutation was introduced into a conserved residue within the serpin scaffold and is homologous to a highly polymerogenic mutation found in some patients with the serpinopathy, Familial Encephalopathy with Neuroserpin Inclusion Bodies (FENIB) (DAVIS et al. 2002; GOOPTU AND LOMAS 2009). The authors conclude that this transgenic strain accumulates mutant SRP-2 in the ER and therefore serves as an invertebrate model of FENIB. Moreover, other investigators have used these transgenic animals to study the relationship between a luminal misfolded ER protein and the unfolded protein response (UPR) and ER-associated degradation (ERAD) pathways under different experimental conditions (DENZEL et al. 2014; HOU et al. 2014). However, using colocalization studies and comparisons to the canonical serpinopathy caused by ATZ, we show unequivocally that wild-type and mutant SRP-2 resided in the cytosol and not in the ER. While these animals expressing mutant SRP-2 could be used to study the effects of cytosolic serpin polymerization, the markedly different expression patterns, inhibitory profiles and subcellular localization of SRP-2 as compared to those of human NS/SERPINI1, confounds the ability to use these animals to model accurately the interaction between aging and ER overload in patients with the serpinopathy, FENIB. Furthermore, interesting experimental results regarding the effects of mutant SRP-2 on ER proteostasis pathways should be viewed from the perspective that this proteotoxic stress originated from within the cytosol and not the ER (DENZEL et al. 2014; HOU et al. 2014). MATERIALS AND METHODS Multiple sequence alignments and phylogenetic comparison of C. elegans SRP-2 with human serpins. Human and C .elegans serpin protein sequences were downloaded from the NCBI database (http://www.ncbi.nlm.nih.gov). Accession numbers for each of the serpins are given in supplemental Table 1. Sequence alignment was performed using the ClustalW algorithm (THOMPSON et al. 1994) using MacVector software (v 13.5.0, MacVector, Inc.), which also includes a similarity and identity matrix. Phylogenetic analysis of the multiple sequence alignment was performed using the uncorrected neighbor-joining algorithm and bootstrapped 1000 times using the same MacVector software. Construction of SRP-2 transgene fusions. SRP-2 expression constructs were generated using the plasmid, p2332. p2332 was generated by inserting a 4 kb nhx-2 promoter fragment (NEHRKE AND MELVIN 2002; NEHRKE 2003) into the HindIII/XbaI sites of from the promoter-less C. elegans expression vector containing GFP with an N4 terminal signal peptide (sGFP), pPD95.85 (kind gift from Dr. Andrew Fire, Stanford University School of Medicine). The Pnhx-2ssrp-2::GFP fusion construct was generated by inserting the full-length (1.6kb) srp-2 gene (minus the stop codon) into the KpnI site of the plasmid, p2332. The KpnI site lies immediately downstream of the N-terminal signal peptide and allows targeting of the sSRP-2::GFP fusion protein to the ER-Golgi secretory pathway. The Pnhx-2srp-2::GFP (lacking the synthetic signal peptide sequence) was generated by inserting the full-length srp-2 gene (minus the stop codon) into the NheI/KpnI sites of the plasmid, p2332. The double digest removes the synthetic signal peptide and allows assessment of endogenous SRP-2 N-terminus in protein trafficking. Pnhx-2ssrp-2H302R::GFP and Pnhx-2srp-2H302R::GFP constructs were generated by sitedirected mutagenesis using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies/Stratagene, Santa Clara, CA). A Psrp-2srp-2::GFP expression construct was generated by ligating a 5.5 kb srp-2 promoter region (forward primer: TTTAATAAGCTTTAGTTTCAGATGGTGG, reverse primer: TATATAAAGCTTGTCGGAAAATTATGACACTTTTGG), the full-length srp-2 gene (minus the stop codon) and the 0.85 kb GFP fragment into the canonical expression vector pPD49.26 (kind gift from Dr. Andrew Fire) as previously described (PAK et al. 2004). A Psrp-2sGFP::ATM expression construct was generated by replacing the nhx-2 promoter in the plasmid Pnhx-2sGFP::ATM with the srp-2 promoter as described (GOSAI et al. 2010). C. elegans strains and culture conditions. Animals were routinely cultured at 22 ˚C on nematode growth medium (NGM) plates seeded with E. coli strain, OP50, unless otherwise specified in the text. The C. elegans strain, N2, was obtained from the Caenorhabditis Genetics Center ((CGC), http://www.cbs.umn.edu/CGC/). Transgenic strains were generated by injecting the respective plasmids into the gonad of young adult N2 hermaphrodites at a final concentration 100 ng/µl. The Pnhx-2sGFP::ATZ expression vector was generated as previously described (GOSAI et al. 2010; LONG et al. 2014). Pnhx-2GFP::ATZ (cytoplasmic) was generated by mutating the ATG of the signal peptide to a KpnI restriction enzyme recognition site causing translation to begin at the start of GFP. Lines expressing Pnhx-2srp-2::GFP, Pnhx-2ssrp-2::GFP, Pnhx-2srp2H302R::GFP and Pnhx-2ssrp-2H302R::GFP were generated by co-injecting the plasmid with Pnhx-2sDsRed::KDEL at a final DNA concentration of 100 ng/µl. A complete list of worm strains and their genotypes is shown in Table S2. Microscopic imaging. For microscopic image acquisition, approximately 12 animals were transferred to a 35 mm MatTek glass bottom culture dish (MatTek, Ashland, MA) containing 6 µl of 50 mM sodium azide. Confocal images were collected using a Leica TCS SP8 microscope. GFP fluorescence was excited using a 488 nm argon laser and red fluorophores with a 561 nm solid-state laser with either a 20x 0.6NA Apochromat air objective or a 40x 1.3NA oil Apochromat CS2 objective. Fluorescence images were captured using a spectral HyD detector over ~100 Z-planes. DIC images were collected using a transmitted light detector on the 488 nm Argon laser line. Confocal images were acquired using LAS AF software (Leica Microsystems, Mannheim, Germany) and visualized, rendered and analyzed using Volocity Software 5 (v6.11, Perkin Elmer, Waltham, MA). Co-localization analyses were performed using the co-localization module on the Volocity Software (BARLOW et al. 2010). RESULTS SRP-2 is an improbable orthologue of NS/SERPINI1. A comparison of the amino acid sequence of NS/SERPINI1 to those from the C. elegans serpin family, showed SRP-2 to have the greatest similarity to NS/SERPINI1 (SCHIPANSKI et al. 2013). However, the “best hit” criteria can be misleading as the highest degree of amino acid similarity/identity relationship may still not yield a true orthologous relationship, but rather a similarity only slightly higher than that of the baseline among all serpin superfamily members. Indeed, the degree of amino acid similarity (~30%) between SRP-2 and NS/SERPINI1 is in the general range for serpin homologues even between evolutionarily divergent species (vide infra, scaffold conservation). Thus, amino acid similarity analysis alone is insufficient evidence to conclude that SRP-2 and NS/SERPINI1 are functional homologues or orthologues. In the exhaustive phylogenic analysis by Irving et al., the human serpins are divided into 9 distinct clades (A-I) (IRVING et al. 2000). If SRP-2 was most homologous to NS/SERPINI1, then comparisons back to the major vertebrate serpin clades (A-I) should yield the reciprocal result (i.e., SRP-2 should show highest homology with NS/SERPINI1). Using ClustalW (THOMPSON et al. 1994), we performed a multiple sequence amino acid alignment between SRP-2 and the entire human serpin family (n=36, clades A-I). SRP-2 showed greatest amino acid sequence similarity (47.7%) and identity (26.8%) with SERPINB7 (Fig. 1A). Moreover, SRP-2 showed greater homology with most of the clade B/intracellular serpins than with NS/SERPINI1, which ranked 15 of 36 on the list. Phylogenetic analysis using the uncorrected neighbor-joining algorithm and bootstrapped 1000 times also showed that SRP-2 formed its own orphan clade, distinct from that containing NS/SERPINI1 (Fig. 1B). This result was similar to that of a previous study showing that the C. elegans serpins, which like the clade B serpins, form their own clade (L) (IRVING et al. 2000). Thus, the C. elegans serpins are closer in homology to each other and to the clade B serpins than they are to NS/SERPINI1. Although overall amino acid similarity does not appear to make SRP-2 more homologous to NS/SERPINI1 than any of the other C. elegans serpins, SRP-2 may show structural motifs that make it a better candidate. We re-examined the primary amino acid sequence of the proteins to determine if this was the case. Like most of the human serpins (except those in clade B), NS/SERPINI1 contains an N-terminal extension with a classical signal peptide. This signal peptide was detected by three of the most common signal peptide prediction programs, Signal P 4.1 (PETERSEN et al. 2011), Phobius (KALL et al. 2007) and PrediSi (HILLER et al. 2004) (Table 1). SRP-2, like the clade B/intracellular serpins has no N-terminal extension, and analysis of its Nterminus by Signal P 4.1, Phobius and PrediSi failed to detect a cleavable signal peptide (Table 1). PrediSi did predict a cleavable signal peptide only when the Nterminal signal sequence length was extended to >50 amino acids. Since, cleavage of the first 50 amino acids of SRP-2 would eliminate key structural elements of the serpin 6 (including helix A, strand 6B and helix B), and prohibit proper folding, we concluded that SRP-2 does not contain a cleavable signal peptide. NS/SERPINI1, like many of the extracellular serpins, contains a C-terminal extension. This extension is 10 amino acids longer than that of the canonical serpin, 1AT/SERPINA1. In neurons, NS/SERPINI1 undergoes regulated secretion from dense core vesicles. The terminal 13 amino acids, which ends in an FEEL ER-retention-like motif is critical for targeting NS/SERPINI1 to dense core vesicles (ISHIGAMI et al. 2007). In contrast, SRP-2, like all intracellular serpins, lacks a C-terminal extension. In fact, all of the C. elegans serpins harbor a truncated C-terminus that is about 1-2 amino acids shorter than those in clade B (LUKE et al. 2006). We next compared the RCL (amino acids P17-P4’) of NS/SERPINI1 with that of SRP-2. The proximal hinge of region (P17-P9) of inhibitory serpins is typically identified by the motif: P17E, P16E/K/R, P15G, P14T/S and P12-P9(A/G/S)4 (IRVING et al. 2000). This motif is well-conserved since this portion of s4A must fold back and insert into sheet A for the serpin to form an inhibitory complex with the proteinase. Since both serpins are bona fide protease inhibitors, we expected that the corresponding residues for NS/SERPINI1 and SRP-2 conformed to this consensus (EEGSEAAAV and EDGTTAAAA, respectively). However, the exposed loop region (P8-P5’) of inhibitory serpins, which also contains the reactive center (P1-P1’) defines serpin specificity. Thus, this region is more variable and has evolved to be preferentially bound and cleaved (between P1 and P1’ using Schechter and Berger numbering (SCHECHTER AND BERGER 1967)) by different target proteases. These regions, especially at the crucial P1 positions (preceding the cleavage site ) are divergent, with a basic residue (SGMIAISRMAVLY) for NS/SERPINI1 versus an acidic residue (SAFKVQLEMMIMA) residue for SRP-2. This difference accounts for the biochemical studies showing that NS/SERPINI1 and SRP-2 inhibit profoundly different types of proteases. NS/SERPINI1 inhibits trypsin-like proteases (prefer cleavage after R residues), such as the plasminogen activators (t-PA and u-PA) and plasmin (HASTINGS et al. 1997; OSTERWALDER et al. 1998). In contrast, SRP-2 inhibits granzyme B-like serine proteinases (prefer cleavage after acidic residues) (PAK et al. 2004). Moreover, SRP-2, unlike NS/SERPINI1, harbors a hydrophobic residue at the P2 position, which signals a preference for lysosomal cysteine proteinases. Indeed SRP-2 is also a potent inhibitor of the lysosomal cysteine proteinases, cathepsins K, L and S (PAK et al. 2004). Thus, while NS/SERPINI1 and SRP-2 are both inhibitory serpins, they have evolved distinct reactive centers and inhibitory profiles. Interestingly, the only C. elegans serpin with a basic residue at either the canonical P1 or P1’ position (serpins can use alternative P1 sites) is SRP-7A, B and C (LUKE et al. 2006). srp-7 encodes at least three isoforms by alternative splicing of three terminal exons with different RCLs (LUKE et al. 2006). However, it has yet to be determined whether SRP-7 inhibits any trypsin-like proteases. All inhibitory-type serpins show a high degree of similarity within the serpin scaffold and only diverge, as described above, in the distal portion of the RCL that defines specificity for the proteinase active site (WHISSTOCK et al. 1998). In the analysis by Irving et al., 51 positions showed a high degree of amino acid conservation among the entire superfamily (72-95%), which included an evolutionary diverse set of 219 7 unique serpin amino acid sequences (IRVING et al. 2000). If the average serpin molecule contains ~ 400 residues, then ~25% of the total sequence should be highly conserved among random serpin family members. This high degree of amino acid similarity reflects the strict folding requirements required for the formation of the metastable state and accounts for the ranges of identity (24-31%) observed between NS/SERPINI1 and any of the five C. elegans inhibitory-type serpins (SRP-1, -2, -3, -6 and -7). Consistent with the knowledge that pathological missense mutations generally reside within evolutionarily conserved regions of proteins, the positions of 4 of the 5 NS/SERPINI1 mutations that induce FENIB target the set of 51 conserved serpin residues (DAVIS et al. 2002; GOOPTU AND LOMAS 2009). Moreover, the five wild-type amino acid residues, which are mutated in the six NS/SERPINI1 mutations described to date, are conserved in SRP-2, as well as all of the C. elegans inhibitory-type serpins (Table 2). Taken together, there does not appear to be any distinguishing primary structural or functional motif to suggest that NS/SERPINI1 was more related to SRP-2 than to any other C. elegans serpin. Wild-type and aggregation-prone SRP-2 with an FENIB-like mutation failed to transit the ER-Golgi pathway. Six different FENIB mutations have been described, with the H338R mutation inducing the greatest degree of polymerization and generating the earliest clinical symptoms (DAVIS et al. 2002; GOOPTU AND LOMAS 2009). The His 338 position in strand 5A is highly conserved (>70%) among members of the serpin superfamily and is present in all of the functional C. elegans serpins (Table 2). Transgenes containing either the wild-type or H302R-containing (the H338R equivalent position on the SRP-2 scaffold) srp-2 gene fused to the N-terminus of YFP and driven by either the endogenous srp-2 or unc-54 (muscle specific) promoters were used to generate several transgenic lines (SCHIPANSKI et al. 2013). Animals expressing the PsrpH302R ::YFP transgene accumulate polymerized material in a perinuclear location 2srp-2 as shown by native acrylamide gel electrophoresis and fluorescence microscopy, respectively (SCHIPANSKI et al. 2013). While a perinuclear distribution can be consistent with ER accumulation, aggregation-prone proteins in the cytosol also can accumulate in the perinuclear regions as electron-dense deposits (LINK et al. 2006) or as more complex structures, such as aggresomes and juxta-nuclear quality control compartments (JUNQs) (AMEN AND KAGANOVICH 2014). To help differentiate among these possibilities, we generated SRP-2 wild-type and mutant transgenes as described above. However, we directed expression to the intestine using the nhx-2 promoter so we could compare SRP-2 subcellular localization to that of well-characterized transgenic animals expressing either the wild-type (sGFP::ATM) or Z mutant (sGFP::ATZ) forms of the canonical secreted serpin, 1AT/SERPINA1 (GOSAI et al. 2010; LONG et al. 2014; O'REILLY et al. 2014). As stated earlier, the Z mutation impairs folding and is retained in the ER as monomers, oligomers and higher order polymers (PERLMUTTER 2002; PERLMUTTER 2011; SILVERMAN et al. 2013). A functional signal peptide (s) fused to GFP was used to replace the wild-type signal of the two 1AT/SERPINA1 isoforms. Several validation studies, including electron and confocal microscopy show that sGFP directed these proteins into the ER-Golgi secretory pathway (GOSAI et al. 2010; LONG et al. 2014; O'REILLY et al. 2014). To confirm ERlocalization by fluorescence confocal imaging, all of transgenic animals used in this study were co-injected also with a second transgene expressing sDsRed fused to the 8 ER retrieval signal, KDEL (sDsRed::KDEL) (PELHAM 1995). Of note, SRP-2 is also expressed in many tissues, including the intestine, so ectopic expression was not a concern (PAK et al. 2004). Also, we have fused GFP to either the N- or C-terminus of different serpins and neither has been shown to impair folding or inhibitory activity (UEMURA et al. 2000). All of our 1AT/SERPINA1 transgenes have GFP fused to the Nterminus, but all of the srp-2-containing transgenes contain GFP fused to the Cterminus so we did not interfere with the function of the natural N-terminus of the protein. The C-terminal location of GFP would also permit a more direct phenotypic comparison between our SRP-2 transgenes with those previously described (SCHIPANSKI et al. 2013). As shown by Long et al., and repeated here for a direct comparison (LONG et al. 2014), sGFP::ATZ forms large intracellular inclusions within dilated ER cisterna that colocalize with the sDsRed::KDEL marker (Fig. 2A-E). In contrast, a new transgene lacking the signal peptide directed GFP::ATZ into the cytosol and failed to co-localize with sDsRed::KDEL (Fig. 2F-J). As expected, SRP-2::GFP (i.e., the wild-type protein) also showed a diffuse cytoplasmic distribution, which did not co-localize with sDsRed::KDEL (Fig. 2K-O). To determine whether SRP-2::GFP could be directed into the classical secretory pathway, we fused the same functional N-terminal signal peptide used with the 1AT/SERPINA1 transgenes to SRP-2::GFP. The signal peptide directed sSRP-2::GFP to the ER where it appeared to form large inclusions, much like sGFP::ATZ, that also co-localized with the sDsRed::KDEL marker (Fig. 2P-T). This result was not surprising, as at least two members of the clade B serpin family, MASPIN/SERPINB5 and SERPINB6, appear to be obligate intracellular proteins and accumulate in the ER as endo-H sensitive aggregates when they are forced into the secretory pathway by a synthetic signal peptide (SCOTT et al. 1996; TEOH et al. 2010). Moreover, SERPINB6 no longer maintains its inhibitory activity when extracted from the ER. However, this result is not always the case for clade B serpins. Forced translocation of sSCCA1/SERPINB3::GFP or sSCCA2/SERPINB4::GFP into the ER-Golgi pathway of mammalian cells leads to secretion of active inhibitors as confirmed by pulse-chase analysis and proteinase inhibitor assays, respectively (UEMURA et al. 2000). In this regard, we concluded that sSRP-2::GFP behaved more like sSERPINB6 and sMASPIN/SERPINB5 and is likely an obligate intracellular serpin. Next, we examined the subcellular localization of SRP-2H302R::GFP. As expected, this mutant fusion protein formed intracellular aggregates (Fig. 2U-Y). However, these aggregates did not co-localize with the sDsRed::KDEL marker and thus did not appear to be within the ER. SRP-2H302R::GFP co-localized with sDsRed::KDEL only when the synthetic signal peptide was attached to its N-terminus (Fig. 2Z-DD). Taken together, these imaging studies confirmed that the H302R mutation caused SRP-2 to aggregate, however, these aggregates localized to the cytosol and not the ER. Wild-type SRP-2 was not secreted via a conventional or unconventional pathway. While the previous co-localization studies suggested that SRP-2 was not secreted via the ER-Golgi pathway, we sought to determine whether SRP-2 was secreted by any other means. Previous work using the srp-2 promoter showed that this element drives robust expression in hypodermal and seam cells throughout development and in early embryos (PAK et al. 2004). Since the hypodermal and seam 9 cells secrete collagen, we reasoned that heterologous protein secretion by these cells would be more easily visualized microscopically by the latter becoming trapped within or beneath the extracellular cuticle. Indeed, a transgene containing the srp-2 promoter driving sGFP::ATM expression previously showed that ATM was secreted and trapped within the cuticle (LONG et al. 2014). We repeated this analysis on adult animals for comparison to similarly staged transgenic animals expressing SRP-2::GFP driven by its endogenous promoter. Unlike the sGFP::ATM expressing animals (Fig. 3A), SRP2::GFP was only detected within the hypodermal cells and none was detected within or beneath the extracellular cuticle (Fig. 3B). Using these same transgenic lines, we also examined embryos at the comma stage, as they exhibit a small, but well delineated extracellular region located between the embryo and the eggshell, the perivitelline space. As expected, sGFP::ATM (Fig. 3C and D), but not SRP-2::GFP (Fig. 3E and F), was secreted into this space. Indeed, sGFP::ATM was secreted so efficiently, that the protein was difficult to detect within the cells of the embryo. In contrast, SRP-2::GFP was only detectable within the cells. Taken together these imaging studies suggested that SRP-2 was not secreted and behaved as an obligate intracellular serpin. Since srp-2 and NS/SERPINI1 were not orthologous and animals expressing mutated SRP-2 cannot accurately model FENIB, we considered whether mutant NS/SERPINI1 might be expressed in C. elegans in a fashion analogous to that which we had done with sGFP::ATZ. We generated transgenes containing either a wild-type (sGFP::NS) or mutant (sGFP::NSG392E) form of murine NS/Serpini1, and using the xnp-1 promoter to drive expression predominantly in neuronal cells. Preliminary imaging studies showed that sGFP::NS was expressed diffusely in the cell body and axons, whereas sGFP::NSG392E formed large intracellular aggregates (Supplemental Fig. 1). Thus, mutant NS was retained within the neuronal cell body in a manner similar to that observed with the canonical extracellular serpin mutant, ATZ. Further characterization of these animals will be required to determine if the aggregates localized to the ER and whether they can serve as an invertebrate model of FENIB. DISCUSSION The serpinopathy, FENIB, is a rare autosomal dominant neurodegenerative disease characterized by progressive deterioration of cognition, memory and visuospatial skills (DAVIS et al. 1999a; DAVIS et al. 1999b). Some patients also develop progressive myoclonus epilepsy (PME) (TAKAO et al. 2000). Histopathological examination shows the intracellular inclusions, Collins bodies, within neurons located in the deeper layers of the cerebral cortex and subcortical nuclei, including the substantia nigra (DAVIS et al. 1999a; DAVIS et al. 1999b). Collins bodies are comprised almost exclusively of misfolded and/or polymerized NS/SERPINI1 and are located within dilated cisternae of the rough ER (DAVIS et al. 1999a; DAVIS et al. 1999b). At least 6 different missense NS/SERPINI1 mutations have been identified, and based on their location within the serpin scaffold, confer different degrees of instability relative to the native fold (DAVIS et al. 2002; HAGEN et al. 2011). The more destabilizing mutations (G392R > G392E > H338R > S52R/L47P > S49P) are associated with a 10 greater number and more pervasive distribution of Collins bodies, and an earlier onset and more severe progression of dementia and PME (DAVIS et al. 2002; GOOPTU AND LOMAS 2009). Transient transfection of the mutant genes into cell lines also show that the more destabilizing mutations result in the formation of longer polymers and a greater degree of ER retention (MIRANDA et al. 2004; MIRANDA et al. 2008). Taken together, these data show that FENIB is a excellent model for conformational-dependent proteotoxicity because the disease is due to highly penetrant autosomal dominant missense mutations within a single gene; the phenotype is ageing-dependent, with more destabilizing mutations presenting earlier and with more severe neuronal loss; and severity is correlated with the degree of misfolded protein accumulation within the ER, which places strain directly on ER proteostasis pathways. Since deterioration of these pathways has been implicated in the pathogenesis of neurodegeneration and ageing (YOSHIDA 2007; GUERRIERO AND BRODSKY 2012), new model systems are needed to enhance our understanding of disease mechanisms and to develop novel therapeutic strategies. Thus, modeling aspects of complex serpinopathies in a facile genetic system like C. elegans is one approach to gain molecular genetic insight into these disorders (SILVERMAN et al. 2009; GOSAI et al. 2010; LONG et al. 2014; O'REILLY et al. 2014). However, we suggest that using SRP-2 to model the protein misfolding aspects of FENIB is misleading for at least 5 reasons: 1) SRP-2 is most homologous by amino acid similarity to the intracellular clade B serpins in mammals rather than the secreted/extracellular NS/SERPINI1, 2) SRP-2, like the clade B serpins, does not contain a classical signal peptide, an N- or C-terminal extension, or a specialized 13 amino acid C-terminal targeting signal (to dense core vesicles) present in NS/SERPINI1 (PAK et al. 2004; SILVERMAN et al. 2004; ISHIGAMI et al. 2007), 3) SRP-2 and NS/SERPINI1 have different reactive center portions of their RCLs and display distinct protease inhibitory profiles (HASTINGS et al. 1997; OSTERWALDER et al. 1998; PAK et al. 2004), 4) SRP-2, in contrast to NS/SERPINI1, is expressed predominately in nonneuronal tissues except for the phasmid neurons (HASTINGS et al. 1997; PAK et al. 2004), and 5) SRP-2 (cytosol) and NS/SERPINI1 (ER-Golgi secretory pathway, dense core vesicles) occupy distinct subcellular compartments (OSTERWALDER et al. 1996; PAK et al. 2004; ISHIGAMI et al. 2007). Many of the serpinopathy-inducing missense mutations cluster in several highly conserved structural elements within the serpin scaffold: the shutter region, the proximal and distal hinge regions and strands 5A and 5B (HUBER AND CARRELL 1989; STEIN AND CARRELL 1995; IRVING et al. 2000). The clustering of missense mutations in these sites holds true for many of the type 1 serpin deficiencies (decreased circulating levels of the protein) associated with serpinopathies involving1AT/SERPINA1, 1antichymotrypsin/SERPINA3, antithrombin/SERPINC1, C1 esterase inhibitor/SERPING1 and heparin-cofactor II/SERPIND1 (HUBER AND CARRELL 1989; STEIN AND CARRELL 1995; IRVING et al. 2000). Based on this degree of conservation, it was not surprising that the polymerizing H338R mutation located on s5A in NS/SERPINI1 (DAVIS et al. 2002; GOOPTU AND LOMAS 2009; HAGEN et al. 2011) resulted in the intracellular aggregation and accumulation of the SRP-2 when introduced into the homologous position on the nematode scaffold (H302R) (this study and (SCHIPANSKI et al. 2013)). Indeed, the positions of all the known FENIB mutations fall on conserved amino acid positions located in all the C. elegans inhibitory-type serpins, and would be 11 expected to give similar aggregation-prone phenotypes if introduced into any inhibitorytype serpin genes. Thus, the ability of SRP-2 to partially model the polymerogenic effects of the H338R mutation cannot in itself be used to confer a higher degree of functional homology to NS/SERPINI1 as compared to any of the other C. elegans serpins. The lack of highly homologous NS-like gene in C elegans (i.e., an orthologue) is supported further by a study by Kumar and Ragg (KUMAR AND RAGG 2008). They used microsynteny and gene structure to analyze the evolutionary of origins of neuroserpinlike genes. Dating back to the emergence of Deuterostomes, neuroserpin like genes are closely linked to PDCD10 (programmed cell death protein 10/TFAR15/cerebral cavernous malformation type 3). These serpins are distinguished by several common features and include: a classical signal peptide; an RCL containing one or more basic residues in the reactive center, including an arginine residing at the canonical P1 position; and a variable length C-terminal extension (4-13 amino acids), ending with a KDEL or FEEL motif. As described above, these latter targeting motifs lead to ER-Golgi recycling, and in the case of mammalian neuroserpins, targeting to dense core vesicles (ISHIGAMI et al. 2007). Interestingly, both C. elegans and D. melanogaster, both contain a PDCD10 orthologue, but neither is closely linked to a serpin gene. In C. elegans for example, ccm-3/C14A4.1 and srp-2 map to chromosomes II and V, respectively (http://www.wormbase.org). This observation suggests the neuroserpin evolved from an ancestral serpin gene after Ecdysozoa (C. elegans) and Deuterostomia (vertebrates) diverged from Bilateria. Taken together, these studies suggest that intracellular serpins, but not a neuroserpin-like gene, evolved with the C. elegans and D. melanogaster lineages. Conceivably, the most challenging aspect of using the SRP-2H302R::GFP C. elegans strain to model FENIB is that SRP-2 appears to be an obligate intracellular serpin that resides in the cytosol, while NS/SERPINI1 is facultatively or constitutively secreted (depending on the cell type) via the classical ER-Golgi secretory pathway. This difference in subcellular localization is important. Eukaryotic cells are compartmentalized, and the proteostasis machinery in these compartments has become highly specialized to facilitate the proper folding, post-translational modification and disposition of different protein species (BUCHBERGER et al. 2010; BENYAIR et al. 2014). For example, the types of chaperones, co-chaperones and carbohydrate processing enzymes used to assist proper protein folding differ between the ER and the cytosol (BUCHBERGER et al. 2010; BENYAIR et al. 2014). Moreover, the elimination of misfolded proteins located within these compartments differs to a certain extent. For example, the ER associated degradation (ERAD) pathway extracts misfolded ER luminal and transmembrane proteins and delivers them to the cytosol for degradation (MEUSSER et al. 2005; KINCAID AND COOPER 2007; HEBERT et al. 2010). Although misfolded and aggregated proteins in either the ER or cytosol may be degraded by ubiquitin-proteasomal system (UPS) or autophagy, the pathways for arriving to these terminal sites may involve different E3 ligases, ubiquitin binding proteins and shuttling factors (RAASI AND WOLF 2007; KROEGER et al. 2009; YOSHIDA AND TANAKA 2010; HOUCK et al. 2014). Also, the surveillance and signaling systems that monitor and respond to proteotoxicity differ significantly between the ER (the unfolded protein response, (UPR)) 12 and the cytosol (the heat shock/stress response (HSR)) (RICHTER et al. 2010). These compartment-specific stress responses explain a paradoxical observation in the study of Schipanski et al. (SCHIPANSKI et al. 2013). They show that genetic loss of heat shock factor-1 (hsf-1), one of the master transcriptional regulators of the cytosolic HSR, resulted in a substantially greater increase in SRP-2H302R::GFP aggregation (increase ~6.5 fold) than after the loss of any of the three UPR sensors/effectors: ire-1, atf-6 or pek-1 (increase ~3-4 fold) (SCHIPANSKI et al. 2013). While down-regulation of the UPR sensors had a similar effect on SRP-2H302R::GFP aggregation, it was not to the same magnitude as that observed with loss of HSF-1. This result is more easily reconciled knowing that SRP-2H302R::GFP aggregation occurred in the cytosol and not the ER. Moreover, this result emphasizes that compartmentalization of stress-response pathways does not preclude extensive cross-talk between sensors and that this communication may make it difficult at times to determine the actual location of the proteotoxic species (LIU AND CHANG 2008; BUCHBERGER et al. 2010; RICHTER et al. 2010; HELDENS et al. 2011; ROTH et al. 2014). There are two other studies that make experimental inferences based on the notion that SRP-2H302R::YFP was aggregating within the ER instead of the cytosol (DENZEL et al. 2014; HOU et al. 2014). First, Hou et al. show that loss-of-function of the Mediator subunit, mdt-15, causes an increase in ER membrane phospholipid saturation and activation of the UPR (HOU et al. 2014). To determine whether activation of the UPR is due to a change in ER lipids or secondary to some untoward effect of the mutation on ER proteostasis, they examined the effects of the mdt-15 mutation in animals expressing the “ER folding sensor”, SRP-2H302R::YFP. Since depletion of mdt15 had no effect on the degree of SRP-2H302R::YFP aggregation, they conclude that this mutation activates the UPR directly by altering ER membrane lipid content and not secondarily by impairing protein folding capability within the ER and stimulating the UPR by increasing the accumulation of misfolded species. Further, evidence for this direct effect is supported by no increase in a second ER folding sensor, the luminal ERAD substrate, CPL-1W32A,Y35A::YFP (MIEDEL et al. 2012; HOU et al. 2014). While the overall conclusions of this study are supported by the CPL-1 misfolding mutant, we would be hesitant to draw this conclusion based on the experiments using SRP-2H302R::YFP per se. Second, Denzel et al., show that enhancing the activity of the hexosamine pathway (HP) leads to an increase in N-glycan precursors and improved ER protein homeostasis as shown by decreased aggregation of the mutant “ER” protein, SRP-2H302R::YFP (DENZEL et al. 2014). Since SRP-2H302R::YFP actually localized to the cytosol, it was not surprising that the investigators were able to demonstrate the beneficial effects of enhanced HP activity is extended to the cytosol, as the proteotoxicity of two welldescribed cytosolic aggregation-prone proteins, polyglutamine repeats (polyQ40) and synuclein is reduced (DENZEL et al. 2014). Transgenic C. elegans strains expressing these proteins have served as disease models for Huntington’s and Parkinson’s disease, respectively (MORLEY et al. 2002; VAN HAM et al. 2008). Interestingly, the only other purported ER-specific folding sensor assayed in this study employed a C. elegans strain expressing human sA1-42 peptide driven by the muscle-specific unc-54 promoter (LINK 1995; LINK 2001). While the signal peptide of the A1-42 fragment is cleaved, the peptide is directed out of the ER by an unknown mechanism and deposited in the cytosol where it forms fibrils and aggregates that impair muscle movement (LINK et al. 2001). While it 13 is likely that enhanced HP decreases protein misfolding in the ER, all of the folding sensors used in this study, especially SRP-2H302R::YFP reside in the cytosol and suggest that the highly beneficial effects of the HP pathway on misfolded proteins was only confirmed for species residing within this compartment and not the ER. Moreover, by recognizing that SRP-2 localizes to the cytosol instead of the ER, the results of the two latter publications provide compelling evidence that compartmental protein quality control pathways are highly interdependent. Thus, these model systems should prove valuable in determining the currently ill-defined signaling mechanism(s) that coordinate the UPR or HSR to misfolded and/or aggregation prone proteins primarily localizing to the opposing compartment (LIU AND CHANG 2008; HELDENS et al. 2011; ROTH et al. 2014). We concluded that animals expressing aggregation-prone SRP-2H302R do not appropriately model serpinopathies induced by classically secreted serpins. Moreover, they fail to capture the effects of ER overload and Collins body formation on cellular demise occurring in patients with FENIB (SCHIPANSKI et al. 2013). Since Collins bodies are PAS+ and diastase resistant ER inclusions, and analogous to the dilated rough ER cisternae containing ATZ in livers of patients with ATD (LOMAS et al. 1992; DAVIS et al. 1999a; DAVIS et al. 1999b; YAZAKI et al. 2001; PERLMUTTER 2007), we suggest that signal peptide containing, aggregation-prone mutant serpins, such as NS/SERPINI1 itself or AT/SERPINA1 (GOSAI et al. 2010; LONG et al. 2014; O'REILLY et al. 2014) might prove to be better candidates to investigate the relationship between ER overload, ageing and dementia. Taken together, studies using mutated intracellular and secreted serpins underscore the differential effects that the subcellular microenvironment (e.g., redox state, chaperone complement and carbohydrate modifying enzymes) has on protein folding, misfolding, aggregation and degradation. Since C. elegans strains expressing SRP-2 mutants successfully model protein aggregation in the cytosol, they represent a unique tool to study compartment-specific proteostasis mechanisms. Moreover, we are aware of only two human diseases associated with the homozygous loss of clade B/intracellular serpins. In both cases, nucleotide changes lead to premature stop codons. These loss-of-function mutations in SERPINB6 and SERPINB7 are associated with non-syndromic hearing loss (SIRMACI et al. 2010) and Nagashima-type palmoplantar keratosis (KUBO et al. 2013; MIZUNO et al. 2014; YIN et al. 2014), respectively. As the use of whole genome and exome sequencing for the identification of disease related alleles for undiagnosed and rare diseases increases, we expect that gain-of-function missense mutations also will be detected in clade B/intracellular serpin family members. In this context, the SRP-2H302R::YFP mutants described by Schipanski et al. (SCHIPANSKI et al. 2013) may be valuable in discerning the pathogenesis of these new disease phenotypes associated with intracellular serpinopathies. 14 REFERENCES Amen, T., and D. Kaganovich, 2014 Dynamic droplets: the role of cytoplasmic inclusions in stress, function, and disease. Cell Mol Life Sci: in press. Barlow, A. L., A. Macleod, S. Noppen, J. Sanderson and C. J. Guerin, 2010 Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of pearson's correlation coefficient. Microsc Microanal 16: 710-724. Belin, D., 1993 Biology and facultative secretion of plasminogen activator inhibitor-2. Thromb Haemost 70: 144-147. Belin, D., L. M. Guzman, S. Bost, M. Konakova, F. Silva et al., 2004 Functional activity of eukaryotic signal sequences in Escherichia coli: the ovalbumin family of serine protease inhibitors. J Mol Biol 335: 437-453. Benyair, R., N. Ogen-Shtern and G. Z. Lederkremer, 2014 Glycan regulation of ERassociated degradation through compartmentalization. Semin Cell Dev Biol: in press. Bird, C. H., E. J. Blink, C. E. Hirst, M. S. Buzza, P. M. Steele et al., 2001 Nucleocytoplasmic distribution of the ovalbumin serpin PI-9 requires a nonconventional nuclear import pathway and the export factor crm1. Mol Cell Biol 21: 5396-5407. Buchberger, A., B. Bukau and T. Sommer, 2010 Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40: 238-252. Carrell, R. W., 2005 Cell toxicity and conformational disease. Trends Cell Biol 15: 574580. Carrell, R. W., and D. A. Lomas, 1997 Conformational disease. Lancet 350: 134-138. Carrell, R. W., and D. A. Lomas, 2002 Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med 346: 45-53. Davis, R. L., P. D. Holohan, A. E. Shrimpton, A. H. Tatum, J. Daucher et al., 1999a Familial encephalopathy with neuroserpin inclusion bodies. Am J Pathol 155: 1901-1913. Davis, R. L., A. E. Shrimpton, R. W. Carrell, D. A. Lomas, L. Gerhard et al., 2002 Association between conformational mutations in neuroserpin and onset and severity of dementia. Lancet 359: 2242-2247. Davis, R. L., A. E. Shrimpton, P. D. Holohan, C. Bradshaw, D. Feiglin et al., 1999b Familial dementia caused by polymerization of mutant neuroserpin. Nature 401: 376-379. Denzel, M. S., N. J. Storm, A. Gutschmidt, R. Baddi, Y. Hinze et al., 2014 Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell 156: 1167-1178. Giuliani, F., A. Grieve and C. Rabouille, 2011 Unconventional secretion: a stress on GRASP. Curr Opin Cell Biol 23: 498-504. Gooptu, B., and D. A. Lomas, 2009 Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem 78: 147-176. 15 Gosai, S. J., J. H. Kwak, C. J. Luke, O. S. Long, D. E. King et al., 2010 Automated highcontent live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One 5: e15460. Guerriero, C. J., and J. L. Brodsky, 2012 The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92: 537-576. Hagen, M. C., J. R. Murrell, M. B. Delisle, E. Andermann, F. Andermann et al., 2011 Encephalopathy with neuroserpin inclusion bodies presenting as progressive myoclonus epilepsy and associated with a novel mutation in the Proteinase Inhibitor 12 gene. Brain Pathol 21: 575-582. Hastings, G. A., T. A. Coleman, C. C. Haudenschild, S. Stefansson, E. P. Smith et al., 1997 Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J Biol Chem 272: 33062-33067. Hebert, D. N., R. Bernasconi and M. Molinari, 2010 ERAD substrates: which way out? Semin Cell Dev Biol 21: 526-532. Heldens, L., S. M. Hensen, C. Onnekink, S. T. van Genesen, R. P. Dirks et al., 2011 An atypical unfolded protein response in heat shocked cells. PLoS One 6: e23512. Hiller, K., A. Grote, M. Scheer, R. Munch and D. Jahn, 2004 PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res 32: W375-379. Hou, N. S., A. Gutschmidt, D. Y. Choi, K. Pather, X. Shi et al., 2014 Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci U S A 111: E2271-2280. Houck, S. A., H. Y. Ren, V. J. Madden, J. N. Bonner, M. P. Conlin et al., 2014 Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol Cell 54: 166-179. Huber, R., and R. W. Carrell, 1989 Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry 28: 89518966. Huntington, J. A., 2006 Shape-shifting serpins--advantages of a mobile mechanism. Trends Biochem Sci 31: 427-435. Huntington, J. A., R. J. Read and R. W. Carrell, 2000 Structure of a serpin-protease complex shows inhibition by deformation. Nature 407: 923-926. Irving, J. A., R. N. Pike, A. M. Lesk and J. C. Whisstock, 2000 Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res 10: 1845-1864. Ishigami, S., M. Sandkvist, F. Tsui, E. Moore, T. A. Coleman et al., 2007 Identification of a novel targeting sequence for regulated secretion in the serine protease inhibitor neuroserpin. Biochem J 402: 25-34. Kall, L., A. Krogh and E. L. Sonnhammer, 2007 Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res 35: W429-W432. Keller, R. K., and G. D. Swank, 1978 Tunicamycin does not block ovalbumin secretion in the oviduct. Biochem Biophys Res Commun 85: 762-768. Kincaid, M. M., and A. A. Cooper, 2007 ERADicate ER stress or die trying. Antioxid Redox Signal 9: 2373-2387. 16 Kroeger, H., E. Miranda, I. MacLeod, J. Perez, D. C. Crowther et al., 2009 Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem 284: 22793-22802. Kubo, A., A. Shiohama, T. Sasaki, K. Nakabayashi, H. Kawasaki et al., 2013 Mutations in SERPINB7, encoding a member of the serine protease inhibitor superfamily, cause Nagashima-type palmoplantar keratosis. Am J Hum Genet 93: 945-956. Kumar, A., and H. Ragg, 2008 Ancestry and evolution of a secretory pathway serpin. BMC Evol Biol 8: 250. Link, C. D., 1995 Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A 92: 9368-9372. Link, C. D., 2001 Transgenic invertebrate models of age-associated neurodegenerative diseases. Mech Ageing Dev 122: 1639-1649. Link, C. D., V. Fonte, B. Hiester, J. Yerg, J. Ferguson et al., 2006 Conversion of green fluorescent protein into a toxic, aggregation-prone protein by C-terminal addition of a short peptide. J Biol Chem 281: 1808-1816. Link, C. D., C. J. Johnson, V. Fonte, M. Paupard, D. H. Hall et al., 2001 Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging 22: 217-226. Liu, Y., and A. Chang, 2008 Heat shock response relieves ER stress. EMBO J 27: 1049-1059. Lomas, D. A., D. L. Evans, J. T. Finch and R. W. Carrell, 1992 The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357: 605-607. Long, O. S., J. A. Benson, J. H. Kwak, C. J. Luke, S. J. Gosai et al., 2014 A C. elegans model of human alpha1-antitrypsin deficiency links components of the RNAi pathway to misfolded protein turnover. Hum Mol Genet 23: 5109-5122. Luke, C. J., S. C. Pak, D. J. Askew, Y. S. Askew, J. E. Smith et al., 2006 Selective conservation of the RSL-encoding, proteinase inhibitory-type, clade l serpins in Caenorhabditis species. Front Biosci 11: 581-594. Luke, C. J., S. C. Pak, Y. S. Askew, T. L. Naviglia, D. J. Askew et al., 2007 An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell 130: 1108-1119. Malhotra, V., 2013 Unconventional protein secretion: an evolving mechanism. EMBO J 32: 1660-1664. Meusser, B., C. Hirsch, E. Jarosch and T. Sommer, 2005 ERAD: the long road to destruction. Nat Cell Biol 7: 766-772. Miedel, M. T., N. J. Graf, K. E. Stephen, O. S. Long, S. C. Pak et al., 2012 A ProCathepsin L Mutant Is a Luminal Substrate for Endoplasmic-ReticulumAssociated Degradation in C. elegans. PLoS One 7: e40145. Miranda, E., I. MacLeod, M. J. Davies, J. Perez, K. Romisch et al., 2008 The intracellular accumulation of polymeric neuroserpin explains the severity of the dementia FENIB. Hum Mol Genet 17: 1527-1539. Miranda, E., K. Romisch and D. A. Lomas, 2004 Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J Biol Chem 279: 28283-28291. 17 Mizuno, O., T. Nomura, S. Suzuki, M. Takeda, Y. Ohguchi et al., 2014 Highly prevalent SERPINB7 founder mutation causes pseudodominant inheritance pattern in Nagashima-type palmoplantar keratosis. Br J Dermatol 171: 847-853. Morley, J. F., H. R. Brignull, J. J. Weyers and R. I. Morimoto, 2002 The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99: 10417-10422. Nehrke, K., 2003 A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. J Biol Chem 278: 4465744566. Nehrke, K., and J. E. Melvin, 2002 The NHX family of Na+-H+ exchangers in Caenorhabditis elegans. J Biol Chem 277: 29036-29044. Nickel, W., and C. Rabouille, 2009 Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol 10: 148-155. O'Reilly, L. P., O. S. Long, M. C. Cobanoglu, J. A. Benson, C. J. Luke et al., 2014 A genome-wide RNAi screen identifies potential drug targets in a C. elegans model of alpha1-antitrypsin deficiency. Hum Mol Genet 23: 5123-5132. Osterwalder, T., P. Cinelli, A. Baici, A. Pennella, S. R. Krueger et al., 1998 The axonally secreted serine proteinase inhibitor, neuroserpin, inhibits plasminogen activators and plasmin but not thrombin. J Biol Chem 273: 2312-2321. Osterwalder, T., J. Contartese, E. T. Stoeckli, T. B. Kuhn and P. Sonderegger, 1996 Neuroserpin, an axonally secreted serine protease inhibitor. Embo J. 15: 29442953. Pak, S. C., V. Kumar, C. Tsu, C. J. Luke, Y. S. Askew et al., 2004 Srp-2 is a cross-class inhibitor that participates in post-embryonic development of the nematode Caenorhabditis elegans: Initial characterization of the Clade L serpins. J Biol Chem 279: 15448-15459. Pak, S. C., C. Tsu, C. J. Luke, Y. S. Askew and G. A. Silverman, 2006 The Caenorhabditis elegans muscle specific serpin, SRP-3 neutralizes chymotrypsinlike serine peptidases. Biochemistry 45: 4474-4480. Palmiter, R. D., J. Gagnon and K. A. Walsh, 1978 Ovalbumin: a secreted protein without a transient hydrophobic leader sequence. Proc Natl Acad Sci U S A 75: 94-98. Pelham, H. R., 1995 Sorting and retrieval between the endoplasmic reticulum and Golgi apparatus. Curr Opin Cell Biol 7: 530-535. Perlmutter, D. H., 2002 The cellular response to aggregated proteins associated with human disease. J Clin Invest 110: 1219-1220. Perlmutter, D. H., 2006 Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res 60: 233-238. Perlmutter, D. H., 2007 Liver disease in alpha1-antitrypsin deficiency, pp. 483-508 in Molecular and cellular aspects of the serpinopathies and disorders in serpin activity, edited by G. K. Silverman and D. A. Lomas. World Scientific Publishing Co. Perlmutter, D. H., 2011 Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annual review of medicine 62: 333-345. 18 Petersen, T. N., S. Brunak, G. von Heijne and H. Nielsen, 2011 SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785786. Raasi, S., and D. H. Wolf, 2007 Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol 18: 780-791. Remold-O'Donnell, E., 1993 The ovalbumin family of serpin proteins. FEBS Lett. 315: 105-108. Richter, K., M. Haslbeck and J. Buchner, 2010 The heat shock response: life on the verge of death. Mol Cell 40: 253-266. Roth, D. M., D. M. Hutt, J. Tong, M. Bouchecareilh, N. Wang et al., 2014 Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol 12: e1001998. Roussel, B. D., J. A. Irving, U. I. Ekeowa, D. Belorgey, I. Haq et al., 2011 Unravelling the twists and turns of the serpinopathies. FEBS J 278: 3859-3867. Schechter, I., and A. Berger, 1967 On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: 157-162. Schipanski, A., S. Lange, A. Segref, A. Gutschmidt, D. A. Lomas et al., 2013 A novel interaction between aging and ER overload in a protein conformational dementia. Genetics 193: 865-876. Scott, F. L., P. B. Coughlin, C. Bird, L. Cerruti, J. A. Hayman et al., 1996 Proteinase inhibitor 6 cannot be secreted, which suggests it is a new type of cellular serpin. J. Biol. Chem. 271: 1605-1612. Silverman, G. A., P. I. Bird, R. W. Carrell, F. C. Church, P. B. Coughlin et al., 2001 The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem 276: 33293-33296. Silverman, G. A., C. J. Luke, S. R. Bhatia, O. S. Long, A. C. Vetica et al., 2009 Modeling molecular and cellular aspects of human disease using the nematode Caenorhabditis elegans. Pediatr Res 65: 10-18. Silverman, G. A., S. C. Pak and D. H. Perlmutter, 2013 Disorders of protein misfolding: alpha-1-antitrypsin deficiency as prototype. J Pediatr 163: 320-326. Silverman, G. A., J. C. Whisstock, D. J. Askew, S. C. Pak, C. J. Luke et al., 2004 Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell Mol Life Sci 61: 301-325. Silverman, G. A., J. C. Whisstock, S. P. Bottomley, J. A. Huntington, D. Kaiserman et al., 2010 Serpins flex their muscle: I. Putting the clamps on proteolysis in diverse biological systems. J Biol Chem 285: 24299-24305. Sirmaci, A., S. Erbek, J. Price, M. Huang, D. Duman et al., 2010 A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet 86: 797-804. Stein, P. E., and R. W. Carrell, 1995 What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 2: 96-113. Tabe, L., P. Krieg, R. Strachan, D. Jackson, E. Wallis et al., 1984 Segregation of mutant ovalbumins and ovalbumin-globin fusion proteins in Xenopus oocytes. Identification of an ovalbumin signal sequence. J Mol Biol 180: 645-666. 19 Takao, M., M. D. Benson, J. R. Murrell, M. Yazaki, P. Piccardo et al., 2000 Neuroserpin mutation S52R causes neuroserpin accumulation in neurons and is associated with progressive myoclonus epilepsy. J Neuropathol Exp Neurol 59: 1070-1086. Teoh, S. S., J. C. Whisstock and P. I. Bird, 2010 Maspin (SERPINB5) Is an Obligate Intracellular Serpin. J Biol Chem 285: 10862-10869. Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994 CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673-4680. Uemura, Y., S. C. Pak, C. Luke, S. Cataltepe, C. Tsu et al., 2000 Circulating serpin tumor markers SCCA1 and SCCA2 are not actively secreted but reside in the cytosol of squamous carcinoma cells. Int J Cancer 89: 368-377. van Ham, T. J., K. L. Thijssen, R. Breitling, R. M. Hofstra, R. H. Plasterk et al., 2008 C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet 4: e1000027. Whisstock, J., R. Skinner and A. M. Lesk, 1998 An atlas of serpin conformations. Trends Biochem Sci 23: 63-67. Whisstock, J. C., G. A. Silverman, P. I. Bird, S. P. Bottomley, D. Kaiserman et al., 2010 Serpins flex their muscle: II. Structural insights into target peptidase recognition, polymerization, and transport functions. J Biol Chem 285: 24307-24312. Yamasaki, M., T. J. Sendall, M. C. Pearce, J. C. Whisstock and J. A. Huntington, 2011 Molecular basis of alpha1-antitrypsin deficiency revealed by the structure of a domain-swapped trimer. EMBO Rep 12: 1011-1017. Yazaki, M., J. J. Liepnieks, J. R. Murrell, M. Takao, B. Guenther et al., 2001 Biochemical characterization of a neuroserpin variant associated with hereditary dementia. Am J Pathol 158: 227-233. Yin, J., G. Xu, H. Wang, J. Zhao, L. Duo et al., 2014 New and Recurrent SERPINB7 Mutations in Seven Chinese Patients with Nagashima-Type Palmoplantar Keratosis. J Invest Dermatol 134: 2269-2272. Yoshida, H., 2007 ER stress and diseases. FEBS J 274: 630-658. Yoshida, Y., and K. Tanaka, 2010 Lectin-like ERAD players in ER and cytosol. Biochim Biophys Acta 1800: 172-180. 20 TABLES Table 1. Signal peptide prediction analysis Protein N-terminal sequence SignalP 4.1 NS/SERPINI1 MAFLGLFSLLVLQSMATG-ATF YES Signal Peptide Prediction Prob Phobius Prob PrediSi 0.91 YES 1.00 Prob YES 0.81 α1AT/SERPINA1 MPSSVSWGILLLAGLCCLVPVSL-AED YES 0.87 YES 1.00 YES PAI-2/SERPINB2 MEDLCVANTLFALNLFKHLAKASPTQN NO 0.20 YES 0.77 NO MASPIN/SERPINB5 MDALQLANSAFAVDLFKQLCEKEPLGN NO 0.14 NO 0.01 NO C. elegans SRP-2 MSDNATSQTDFALKLLATLPHSGSVVL NO 0.20 NO 0.00 NO1 C. elegans SRP-6 MSDSSSDEKMGLLLHSETDFGLSLLRQ NO 0.12 NO 0.00 NO sGFP MHKVLLALFFIFLAPASALA-VSE YES 0.95 YES 1.00 YES 1 PrediSi predicted a signal peptide for SRP-2 only if the N-terminal sequence length was extended to >50 amino acids. 1.00 0.33 0.00 0.36 0.02 1.00 Table 2. NS/SERPINI1/neuroserpin mutations occur at highly conserved residues within serpin scaffold that are also retained in all C. elegans inhibitory serpins serpin structural motif1 2 s6B hB hB2 s5A2 s5B2 NS/SERPINI13 L47P S49P S52R H338R G392R/E SERPINA14 F51 S53 S56 H334 G386 5 SRP-1 F32 S34 S37 H307 G363 L26 S28 S31 H302 G356 SRP-25 SRP-35 F25 S27 S30 H304 G358 SRP-65 F35 S37 S40 H316 G372 F26 S28 S31 H307 G363 SRP-7A5 1 s, strand; b, helix. 2 Amino acid is conserved in >70% of serpin family members (IRVING et al. 2000). 3 Amino acid position and change for NS/SERPINI1 mutations based on its own numbering. 4 Corresponding amino acid position based on canonical SERPINA1 numbering. 5 Corresponding amino acid position based the initiator methionine for each intracellular C. elegans serpin. 21 Table 3. Comparison between NS/SERPINI1 and SRP-2 Feature NS/SERPINI1 SRP-2 number of amino acids N- and C-terminal extensions cleavable signal peptide secreted subcellular localization RCL P4-P1’ inhibitory profile 410 yes 359 no yes yes ER, dense core vesicles AISRM t-PA, uPA, plasmin tissue distribution neurons, pancreas, testes no no cytosol VQLEM lysosomal cysteine peptidases (cathepsin L, S, K); granzyme B hypoderm, intestine 22 FIGURE LEGENDS Figure 1. C. elegans SRP-2 protein sequence compared to human serpin family members. (A) Percent similarity and identity scores of the primary amino acid sequence of SRP-2 compared to the 36 human (Hsa) members. Human serpins are listed in order of increasing to decreasing percent similarity and have the prefix SERPIN before the clade and member number omitted. (B) An unrooted uncorrected phylogram of the multiple sequence alignment performed using the neighbor-joining method and bootstrapped 1000 times. Numbers at branch point indicate the percent those branch points occurred in the 1000 generated trees. Figure 2. Confocal image analysis of transgenic animals expressing wild-type and mutant (± s)SRP-2::GFP transgenes. GFP and DsRed fluorescence images of animals co-expressing sDsRed::KDEL and sGFP::ATZ (A-D), GFP::ATZ (F-I), SRP2::GFP (K-N), sSRP-2::GFP (P-S), SRP-2H302R::GFP (U-X) and sSRP-2H302R::GFP (ZCC) under the control of the intestinal-specific promoter from nhx-2. White arrowheads highlight regions that co-localize in the GFP (A, P, Z) and DsRed (B, Q, AA) channels. White arrows highlight regions that do not co-localize in the GFP (U) and DsRed (W) channels. Regions highlighted by white boxes are shown at higher magnifications as merged images (D, I, N, S, X and CC). Co-localization plots were generated using the co-localization feature in the Volocity package. Images were likely co-localized if the colocalization plot showed a linear relationship and the Pearson’s correlation coefficient was greater than 0.750 (BARLOW et al. 2010). Figure 3. Comparison of confocal images from transgenic animals expressing a classical secreted serpin, sGFP::ATM versus the intracellular serpin, SRP-2::GFP. Maximum intensity projection fluorescence images of the posterior region of animals expressing sGFP::ATM (A) and SRP-2::GFP (B) under the control of the srp-2 promoter. The srp-2 promoter is primarily active in hypodermal cells throughout development (PAK et al. 2004). The hypodermis is responsible for synthesizing the cuticle and secreted products become incorporated or trapped within this structure. As shown previously (LONG et al. 2014) and re-imaged here for comparison, sGFP::ATM expressed under the control of the srp-2 promoter showed little hypodermal retention and instead accumulated within the cuticle of the worm (A, arrowheads). These results suggested that sGFP::ATM was efficiently secreted by the hypodermal cells. Unlike sGFP::ATM however, SRP-2::GFP was detected within hypodermal cells (B, arrows), but no protein was detected beneath or within the cuticle. These results further support our findings suggesting that SRP-2 was an obligate intracellular serpin. We also examined comma stage embryos from transgenic animals expressing sGFP::ATM (C and D (DIC merge)) or SRP-2::GFP (E and F (DIC merge)). sGFP::ATM was rapidly secreted and mostly visible within the extracellular perivitelline space between the embryo and the egg shell (C and D, asterisk and arrowheads). In contrast, SRP-2::GFP was only visible within cells. These images confirmed that wild-type SRP-2 localized to the cytosol and there was no detectable secretion. 23 SUPPLEMENTARY FIGURE LEGENDS Figure S1. Confocal images of transgenic animals expressing NS/SERPINI1 transgenes. GFP fluorescence images of the head region of animals expressing wildtype (A) and mutant (B) NS/SERPINI1 under the control of the xnp-1 promoter. Animals expressing sGFP::NSwt show diffuse GFP expression in most neuronal cell bodies and axons (A, inset, yellow arrow). In contrast, animals expressing the sGFP::NSG392E mutant show little to no GFP expression in the axons and instead accumulate GFP in the perinuclear region (B, inset, red arrows). Mo, mouth. TB, terminal bulb. N, nucleus. Ax, axon. 24 SUPPLEMENTARY TABLES Table S1. Human serpin accession numbers Name Accession # Common Name SERPINA1 P01009.3 α1-antitrypsin SERPINA2 P20848.1 SERPINA3 P01011.2 SERPINA4 AAH14992.1 Antichymotrypsin SERPINA5 P05154.3 PAI-3 SERPINA6 P08185.1 CBG SERPINA7 P05543.2 TBG SERPINA8 P01019.1 Angiotensin SERPINA9 Q86WD7.3 Centerin SERPINA10 Q9UK55.1 PZI SERPINA11 Q86U17.2 SERPINA12 Q8IW75.1 Vaspin SERPINB1 P30740.1 MNEI SERPINB13 Q9UIV8.2 Headpin SERPINB2 P05120.2 PAI-2 SERPINB3 P29508.2 SCCA1 SERPINB4 P48594.2 SCCA2 SERPINB5 P36952.2 Maspin SERPINB6 P35237.3 PI-6 SERPINB7 O75635.1 Megsin SERPINB8 P50452.2 SERPINB9 P50453.1 PI-9 PI-10 SERPINB10 P48595.1 SERPINB11 Q96P15.1 SERPINB12 Q96P63.1 Yukopin SERPINC1 NP_000479.1 Antithrombin III SERPIND1 AAH35028.1 Heparin Cofactor II SERPINE1 Q8NC51.2 PAI-1 SERPINE2 AAH15663.1 Nexin SERPINE3 A8MV23.2 SERPINF1 P36955.4 PEDF SERPINF2 P08697.3 α2-antiplasmin SERPING1 P05155.2 C1 Inhibitor SERPINH1 P50454.2 HSP47 SERPINI1 Q99574.1 Neuroserpin SERPINI2 O75830.1 Pancpin 25 Table S2. Transgenic lines used in this study Strain name VK2460 VK2461 VK2462 VK2463 VK2464 VK2465 VK1483 VK11 VK2261 VK2262 Genotype vkEx2460[nhx-2p::srp-2::GFP;nhx-2p::sDsRed::KDEL] vkEx2461[nhx-2p::ssrp-2::GFP;nhx-2p::sDsRed::KDEL] vkEx2462[nhx-2p::srp-2(H302R)::GFP;nhx-2p::sDsRed::KDEL] vkEx2463[nhx-2p::ssrp-2(H302R)::GFP;nhx-2p::sDsRed::KDEL] vkEx2464[nhx-2p::sGFP::ATZ;nhx-2p::sDsRed::KDEL] vkEx2465[nhx-2p::GFP::ATZ;nhx-2p::sDsRed::KDEL] vkEx1483[srp-2p::sGFP::ATM] vkls8[srp-2p::srp-2::GFP] vkEx2261[xnp-1p::sGFP::mNS;myo-2p::mCherry] vkEx2262[xnp-1p::sGFP::mNS(G392E);myo-2p::mCherry] 26