Chapter 2. X-Ray Photoelectron and Auger Electron Spectroscopy
advertisement

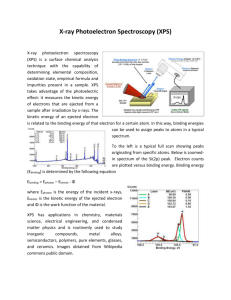
Chapter 2. X-Ray Photoelectron and Auger Electron Spectroscopy 2.1 Introduction In X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES), electrons emitted after the interaction between primary X-rays or electrons and a sample are detected. The interaction is illustrated in Fig. 2.1 for AES. The amount of electrons having escaped from the sample without energy loss is typically measured in the range of 20 to 2000 eV. The data is represented as a graph of intensity versus electron energy. Due to the impact of the primary beam, the atoms in the sample are ionised, and electrons are liberated from the surface, either as a result of the photoemission process (XPS), or of the radiationless de-excitation in the Auger electron emission process (AES). Prim ary Electron Beam Secondary Electrons Auger Electrons 0.4-5 nm Backscattered Electrons Characteristic X-rays Sam ple Surface Volum e of Prim ary Excitation 1-3 µm Fig. 2.1: Schematic representation of the primary beam – sample interaction in the case of an AES analysis. 1 Although XPS and AES are comparable methods in the sense that both are based on the use of a spectrometer to measure electrons of relatively low energy, the main difference between the two methods consists in the source of the primary radiation, which is necessary to provoke ionisation of the atoms. AES makes use of an electron gun while XPS relies on soft X-rays. As a consequence of that, one of the main differences is the lateral resolution of the two methods. Since there is a continuous evolution in the design of the equipment and the performance of the techniques, it is difficult to express an absolute value for it, but for AES the lateral resolution typically is situated in the 10 to 100 nm range, while by means of XPS only a lateral resolution of a few to 100 µm can be reached. In both methods low energy electrons are measured, giving rise to comparable depth and sensitivity values, which are respectively in the order of nanometers (see Fig. 2.1) and of about 0.1 % atomic concentration. This type of measurements is necessarily performed under high vacuum conditions, and only samples restricted in size can be analysed. From this point of view, XPS and AES cannot be considered as non-destructive techniques, although the analyses themselves are not destructive in nature. On the other hand, thanks to their spatial resolution, a small amount of material suffices for the analysis. Moreover, samples can be analysed with XPS and AES in the as-received condition, and can in many cases be put back in their original position afterwards. In this chapter, we try to give practical information on XPS and AES, so that the reader can understand the basic principles, the characteristics and the potential of the methods and their instrumentation. More detailed information may be found in dedicated books. Strongly recommended are e.g. the books of D. Briggs and M.P. Seah [1], T.A. Carlson [2], J.F. Watts [3], B. Agius et al. [4], M. Thompson et al. [5], and V.I. Nefedov [6]. More practical information, e.g. spectra and data, is given in [7-9]. We can also refer to the 2 proceedings of important international conferences such as ECASIA "European Conference for Application of Surface and Interface Analysis" and QSA "Quantitative Surface Analysis". Many papers on XPS and AES can be found in journals. “Surface Science” and “Applied Surface Science”, for example, mostly communicate about the fundamental physical background of the XPS and AES analysis, while e.g. “Surface and Interface Analysis” and “Journal of the Vacuum Science and Technology” report more on the applications. At the end of this chapter, a paragraph is dedicated to a literature overview of the use of XPS and AES for the analysis of cultural heritage materials. 2.2 Basic concepts of XPS and AES 2.2.1 Principle of X-ray photoelectron spectroscopy In the case of XPS, electrons are liberated from the specimen as a result of a photoemission process. An electron is ejected from an atomic energy level by an X-ray photon, mostly from an Al-Kα or Mg-Kα primary source, and its energy is analysed by the spectrometer. The XPS process is schematically represented in Fig. 2.2 for the emission of an electron from the 1s shell of an atom. Photoelectron Efermi level 2p X-ray (photon) Binding Energy 2s 1s Fig. 2.2 : Schematic representation of the XPS process. 3 The experimental quantity that is measured is the kinetic energy of the electron, which depends on the energy hν of the primary X-ray source. The characteristic parameter for the electron is its binding energy. The relation between these parameters is given by Eq. (2.1) : EB = hν – EK – W (2.1) where EB and EK are respectively the binding and the kinetic energy of the emitted photoelectron, hν is the photon energy, and W is the spectrometer work function. In a first approximation, the work function is the difference between the energy of the Fermi level EF and the energy of the vacuum level EV, which is the zero point of the electron energy scale : W = EF – E V (2.2) This quantity is to be determined by calibration for the spectrometer used. From Eq. (2.1) it is clear that only binding energies lower than the exciting radiation (1486.6 eV for Al-Kα and 1253.6 eV for Mg-Kα) are probed. Each element has a characteristic electronic structure and thus a characteristic XPS spectrum. Fig. 2.3 shows the XPS spectrum of Ag. x 10 4 Ag3d5/2 3.5 Ag3d3/2 3 2.5 2 0 1200 1000 800 600 400 Binding Energy (eV) 200 -Ag4p -Ag4d -Ag4s 0.5 Ag3p1/2 -Ag3s 1 -AgMNN 1.5 Ag3p3/2 -AgMNN c/s 0 Fig. 2.3 : XPS spectrum of Ag. Conditions : Al-Kα at 350 W, pass energy = 58.7 eV. In the spectrum, a number of peaks appears on a background. The 4 background originates from photoelectrons which undergo energy changes between photoemission from the atom and detection in the spectrometer. The observed peaks can be grouped into three types : peaks originating from photoemission (i) from core levels, (ii) from valence levels at low binding energies (0 to 20 eV) and (iii) from X-ray excited Auger emission (between 1100 and 1200 eV). Although valence level spectra have their analytical value (e.g. in the study of the electronic structure of materials), they contribute little to the analysis of cultural heritage materials, and will not be discussed here. The main information comes from the core level peaks and the Auger peaks. As can be seen in Fig. 2.3, the notation for XPS and AES peaks is different. The nomenclature employed to describe XPS and AES features is based on the momenta associated with the orbiting paths of electrons around atomic nuclei, indicated by the quantum numbers n, l, j. And yet the translation into the notation is different for both techniques. XPS uses the spectroscopic notation : first the principal quantum number (n = 1, 2, 3, …), then l = 0, 1, 2, … indicated as s, p, d, …respectively, and finally the j value given as a suffix (1/2, 3/2, 5/2, …). The AES nomenclature on the contrary usually follows the X-ray notation : the states with n = 1, 2, 3, … are designated K, L, M, …, while the combinations of l and j are given the suffixes 1, 2, 3, …. Both notations are listed in Table 2.1. In the case of Ag, the Al-Kα radiation is only energetic enough to probe the core levels up to the 3s shell. The non s-level peaks are doublets. This is related to the fact that when l > 0, two possible states characterised by j arise (see Table 2.1). More details on the difference in energy between the two states and on their relative intensities can be found in [1]. It is also noted that the core level peaks have different intensities and widths. The relative intensities are governed by the ionization efficiencies of the different core shells, designated by ionization cross section. The line width, defined as the 5 full width at half-maximum intensity (FWHM), is a convolution of several contributions : the natural width of the core level, the width of the X-ray line and the resolution of the analyser. Table 2.1 : XPS and X-ray notation, respectively used for XPS and AES peaks. quantum number notation N l j XPS X-ray 1 0 1/2 1s1/2 K 2 0 1/2 2s1/2 L1 2 1 1/2 2p1/2 L2 2 1 3/2 2p3/2 L3 3 0 1/2 3s1/2 M1 3 1 1/2 3p1/2 M2 3 1 3/2 3p3/2 M3 3 2 3/2 3d3/2 M4 2 5/2 3d5/2 M5 etc. etc. 3 etc. The Auger peaks in the spectrum, MNN peaks in the case of Ag, originate from the decay of the ionized atoms to their ground state. The principle of the Auger emission is discussed in the next paragraph. 2.2.2 Principle of Auger electron spectroscopy AES uses, in contrast to XPS, electrons as primary radiation. The analyzed electrons are not the emitted core electrons, but the Auger electrons that are ejected as a consequence of the return of the ionized atom to its ground state. Fig. 2.4 shows a schematic representation of the processes involved in the emission of an Auger electron. 6 (a) (b) Auger Electron X-ray Photon Incident Beam Fig. 2.4 : Schematic representation of the X-ray fluorescence (a) and Auger electron emission (b) processes. For the example shown here, a hole is created on the K level in the initial ionization step. This requires a primary energy greater than the binding energy of the electron in that shell. For the ionization to be efficient, a primary energy of about 5 times the binding energy is taken. In practice, typical primary energies are 5 and 10 keV. The hole can be produced by either the primary beam, or the backscattered secondary electrons The atom relaxes by filling the hole with an electron coming from an outer level, in the example shown as L1. As a result, the energy difference EK – EL1 becomes available as excess energy, which can be used in two ways. The emission of an X-ray at that energy may occur or the energy may be given to another electron, either in the same level or in a more shallow one, as is the case in the example, to be ejected. The first of the two competing processes is X-ray fluorescence, the second Auger emission. Fortunately, the probability for Auger emission is much higher for core levels with binding energies below about 2 keV, as illustrated in Fig. 2.5. 7 Fig. 2.5 : Relative probabilities of X-ray fluorescence and Auger emission. An AES transition is written as ABC, where A indicates the level of ionization, B the level where the second electron involved in the transition comes from, and C the level from which the Auger electron is emitted. For A, B and C the X-ray notation (see Table 2.1) is used. The Auger transition represented in Fig. 2.4 is called KL1L2,3. Electrons originating from the valence band, are often denoted by V. For example in the case of Ag, the M4,5N4,5N4,5 doublet (see Fig. 2.6) is an MVV transition. More details on the notations and the corresponding electronic configurations can be found in [1]. The kinetic energy EABC of an Auger transition ABC in an atom of atomic number Z is given by Eq. (2.3) : EABC(Z) = EA(Z) – EB(Z) – EC(Z)* - W (2.3) where EI are the binding energies on the Ith atomic level. The star in EC* indicates that it is the binding energy on C in the presence of a hole. In good approximation Eq. (2.4) applies [10] : EABC(Z) = EA(Z) – ½ [EB(Z) + EB(Z+1)] – ½ [EC(Z) + EC(Z+1)] – W (2.4) Eq. (2.4) shows that the kinetic energy of an Auger electron is independent of the type of primary beam (i.e. electrons or X-rays) and its energy. For this reason, AES spectra are always plotted on a kinetic energy scale. Since the kinetic energy is only a function of the atomic energy levels, all elements of 8 the periodic table have a unique spectrum. Fig. 2.6 shows, as an example, the Auger spectrum of Ag. (b) (a) 4 5 6 x 10 6 5.5 x 10 4 5 2 4.5 c/s c/s 0 4 -2 3.5 -4 3 -6 2.5 2 220 240 260 280 300 320 340 Kinetic Energy (eV) 360 380 400 -8 220 240 260 280 300 320 340 Kinetic Energy (eV) 360 380 400 Fig. 2.6 : AES spectrum of Ag, (a) direct spectrum, (b) differentiated spectrum. Conditions : Ep = 5 keV, ∆E/E = 0.25%. The AES peaks are superimposed on an important background of different types of secondary electrons. This is the reason why, in many cases, AES spectra are represented in the differentiated form. Note that the MNN peaks of Ag in the AES and in the XPS spectrum are at different energies. This is because both techniques do not use the same energy scale : kinetic energies for AES spectra against binding energies for XPS spectra. The energy positions of X-ray and electron induced Auger transitions differ, as shown by Eq. (2.1), by the value hν, the energy of the X-ray radiation. If in XPS the Xray source is changed from Mg-Kα to Al-Kα, the position of the XPS peaks remains, while the AES peaks move by 233 eV. This allows the two types of peaks to be easily distinguished. As is the case for XPS peaks, the relative intensities of AES transitions are governed by their respective core shell ionization efficiencies due to the primary electron beam. Yet, for AES the situation is more complex, since there is additional ionization due to back-scattered electrons (see Fig. 2.1). 9 (a) (b) Fig. 2.7 : Influence of back-scattering on intensity and spatial distribution [11]. Ip is peak intensity due to primary ionization, Ib to back-scattered induced ionization. Primary beam at (a) 5 keV, (b) 20 keV and 40° angle of incidence. The back-scattering factors depend both on the energy and the angle of incidence of the primary beam, and they influence the intensities as well as the spatial distribution of the detected Auger electrons as illustrated in Fig. 2.7. The sample consists of a 40 nm thick Al layer on Au. The Al KLL peak is shown. In general, AES peaks are broader than XPS peaks. This is related to the complex multiplet splitting due to the number of final states after the transition that are permitted (see Table 2.1). The KLL series, for example, consists of five components, i.e. KL1L1 (1 transitions), KL1L2,3 (2 transitions) and KL2,3L2,3 (3 transitions, of which one is forbidden). Two elements contribute to the peak broadening in solid species : peak overlap in the multiplet structure, and solid state peak broadening. In what follows, the analytical possibilities and the strengths and weaknesses of XPS and AES are discussed. This is however impossible without considering first the instrumentation 10 2.3 XPS and AES instruments 2.3.1 General set-up The techniques are comparable in configuration and contain mainly the following parts: (i) a primary beam source, for AES an electron gun and for XPS an X-ray source, (ii) an electron energy analyser, combined with a detection system and (iii) a sample stage, all contained within a vacuum chamber. As for most techniques, the system is operated and controlled by a computer, usually provided with software allowing mathematical treatment of the An example of a full set-up is given in Fig. 2.8, while Fig. 2.9 shows a schematic diagram of an XPS configuration with an X-ray source, a monochromator and an hemispherical sector analyser (HSA). In Fig. 2.9 a different configuration, used in AES, containing a cylindrical mirror analyser (CMA) with a central electron gun is depicted. data. Fig. 2.8: Picture of a full set-up: 5800 Multitechnique system of Physical Electronics combining XPS and AES. 11 X-ray Source Energy Analyzer Hemispherical sector analyzer Electron Source Quartz Crystal Monochromator Al kα x-rays Input Lens 15 kV electrons Rowland Circle Aluminum Anode Multi-channel Detector Photoelectrons Sample Fig. 2.9: Schematic representation of an XPS set-up with an hemisperical sector analyser. Electron Source Multi-Channel Detector Cylindrical Mirror Analyzer Sample Ion Gun Fig. 2.10: Schematic representation of a cylindrical mirror analyser used in AES. 12 XPS and AES apparatus is in a continuous evolution, but with both techniques the most radical progress has been the improvement of the lateral resolution. By the introduction of field Emission AES, the level of 10 nm can now be reached. The lateral resolution of XPS currently reaches the level of a few micrometers, and XPS mapping facilities are becoming more frequently available than in the past. In the following paragraphs, the most essential parts of the instrumentation will be discussed in more detail. 2.3.2 The vacuum system The electron spectrometer and sample room must be operating under ultra high vacuum (UHV), typically in the range of 10-8 to 10-10 torr. The reason for this is twofold. The low energy electrons are elastically and non elastically scattered by residual gas molecules leading to a loss of intensity and of energy so that not only the intensity of the peaks is affected but also the noise in the spectrum increases. The second reason is that lowering the vacuum level to e.g. 10-6 torr, would immediately result in the formation of a monolayer of residual gas absorbed on the sample surface in less than a second. A vacuum of 10-10 torr allows measurements to be carried out for about an hour before a monofilm is formed. The fact that AES and XPS are methods achieving a depth resolution of a few nanometer with a detection limit lower than 1 % of a monolayer, clearly establishes the high requirements on the vacuum. Even in the case of a vacuum of 10-10 torr, typically carbon peaks are found as a result of surface contamination. Therefore, often the sample is cleaned by a slight sputtering, prior to the analysis. The sputtering technique will be discussed in paragraph 2.5.5, but at this stage we already want to stress that sputtering may induce variations in the surface composition and should be handled with care, especially for a non-destructive analysis of cultural heritage material . The way in which such a vacuum is obtained depends on the specific design, 13 but is generally based on diffusion, sputter ion and turbo molecular pumps, combined with auxiliary tools such as Ti sublimation pumps. The high vacuum level imposes high requirements on the used materials. Stainless steel is often selected for the fabrication of the analysis chamber and the joints are usually made of Cu rings. The trajectory of the electrons is strongly influenced by the earth's magnetic and electric fields and consequently a screening is placed around the system. All UHV systems need occasionally to be baked out to remove contaminants from the chamber walls, the stage and other contact points. 2.3.3 The X-ray source for XPS In contrast to the electron source, the X-ray source energy depends on the choice of the anode material, resulting in the availability of a number of discrete energies rather than a continuous variation of the energy, as exists for electron and ion guns. The photon energy must be sufficiently high to excite intense photoelectron peaks from all elements of the periodic table (see paragraph 2.2.1). For XPS analysis, it is very important to consider the energy resolution of the primary X-rays. As explained in paragraph 2.2.1, the width of the XPS line depends, among other factors, on the line width of the primary X-ray line, and this strongly affects the energy resolution obtained in the spectra. The most commonly applied configuration consists of a twin anode, providing Al-Kα and Mg-Kα photons with line energies/line widths of respectively 1486.6 eV/0.85 eV and 1253.6 eV/0.70 eV respectively. In rare cases, higher energy anodes such Si-Kα (1739.5 eV/1eV), Zr-Lα (2042.4 eV/1.7 eV) and Ti-Kα (4510.0 eV/2 eV) are used for the excitation of higher energy electron levels such as the 1s level of heavy elements. The X-ray line width can be reduced from 0.85 eV to 0.4 eV by using a monochromator. Its action is based on Bragg reflection of the X-rays on a single crystal, e.g. natural quartz, as shown in Fig. 2.9. The quartz crystal is placed on the surface of a Rowland or focussing sphere, together with the anode and the sample. The X-rays are dispersed by diffraction on the crystal 14 and refocussed on the sample surface. Other benefits are the removal of contributions to the XPS spectrum of satellite peaks and of the Bremsstrahlung continuum coming from the X-ray spectrum of the anode. The drawback of the use of a monochromator is a severe loss in intensity of the primary X-rays, e.g. up to 40 % for Al-Kα radiation, and thus of the resulting XPS peaks. Acquisition times are drastically increased, and detecting low elemental concentrations becomes impossible. As a result, in practice a monochromator is only used in cases where a high energy resolution, typically 0.4 eV, is required (see paragraph 2.5.4). Focusing of X-rays is much more complicated compared to electron beams, and it is only recently that focussed X-ray systems enabling to reach a lateral resolution of a few micrometers, are introduced in XPS. Depending on the constructor of the equipment, two trends may be noted. The first possibility is to apply an aperture system in order to select only a fraction of the emitted photoelectrons for detection. The newest development is the so-called microfocus monochromator, where focusing of the X-rays is realized by intermediate of an electron gun. 2.3.4 The electron gun for AES An AES system employs an electron gun, either based on a thermionic or on a field emitter source. A thermionic source uses thermal energy to give electrons sufficient energy to escape over the work function of the source, while in a field emission source the work function barrier is reduced. The primary beam source in an AES system is comparable to that in scanning electron microscopes (SEM). For both techniques, there is an increasing trend of using a field emitter type electron source. In the case of a field emission (FE) source, the techniques of AES and SEM are respectively denoted FEAES and FESEM. In conventional AES, the only function of the incident beam is to produce ionization in the core levels of the atoms in order to initiate the Auger transitions (see paragraph 2.2.2). From that point of view, the energy level 15 and energy dispersion of the primary electrons are less important than for XPS. The main characteristic of the electron gun is its brightness. It is defined as the number of electrons emitted per unit area and unit solid angle. It determines the number of primary electrons impinging on the surface of the analysed material and therefore directly determines the data acquisition time and sensitivity of the AES system. The simplest form of a thermionic system is a W wire in the shape of a hairpin. A small electric current provokes heating in the wire, so that the electrons achieve energies higher than the work function of about 4.5 eV and are able to escape from the wire. Yet, most AES systems with a thermionic source use a LaB6 filament because its brightness is higher compared to of W filament sources. A real breakthrough on the level of brightness was the introduction of field emission sources [12]. Field emission is achieved by applying a strong electrostatic field between a filament, in the shape of a needle with a tip radius of about 50 nm, and an extraction electrode. The filament is usually a wire of a W single crystal fashioned into a sharp point. The significance of the small tip radius is that the electric field can be concentrated to extreme levels, up to 109 V/m or more. Applying a high field results in narrowing the barrier as well in width as in height, allowing the electrons to tunnel directly through the barrier and omitting the requirement of thermal energy. The small area of emission from the tip into a small solid angle provides a high brightness compared to thermionic sources. Moreover, a more chromatic (i.e., mono-energetic) beam is obtained, reducing for example chromatic aberrations in the electron lenses, so that smaller spot sizes can be achieved. Cleanliness of the W single crystal is extremely important, since adsorption of impurities will increase the work function. Therefore the gun compartment of the instrument is differentially pumped. The final spot size on the sample surface with a particular FE electron gun is a function of the lens system, the beam current and the energy (see Fig. 2.11). 16 Beam Diameter (nm) 1000 20 kV 10 kV 100 5 kV 3 kV 10 1 10 Beam Current (nA) 50 Fig. 2.11: Relation between spot size, beam current and energy for a FE gun. Fig. 2.12 shows an example of an FEAES analysis, resolving Cu particles with a dimension smaller than 100 nm proving that it is one of best methods to combine high lateral resolution (10 nm range) with high depth resolution (nm range). (a) (b) Fig. 2.12: FEAES analysis of small Cu particles. (a) SEM image, (b) AES image based on the Cu mapping. Note however that the obtained lateral resolution depends also on the characteristics of the sample under investigation. 17 2.3.5 Detection of electron energy Mainly two types of detectors are used in AES and XPS systems: the cylindrical mirror analyser (CMA) and the hemispherical sector analyser (HSA). In the past, the CMA was preferred for AES systems merely for geometrical reasons and for its high analyser transmission function, and the HSA analyser for XPS for its superior energy resolution. More and more, however, depending on the constructor and on the requirements of the customer, the HSA detector is also used in AES systems. A multifunctional AES-XPS system is evidently operated by only one electron detector, and mostly a HSA type is chosen. Yet, it is important to mention that not all of the requirements imposed by the type of analysis coincide for XPS and AES . 2.3.5.1 The cylindrical mirror analyser The CMA, shown in Fig. 2.10, consists of two concentric cylinders, the inner cylinder at ground potential while the potential of the outer cylinder is ramped negatively. A proportion of the emitted Auger electrons will pass through the defining aperture in the inner cylinder. Depending on the potential applied on the outer cylinder, electrons of the desired energy pass through the detector aperture and are refocused on the electron detector and measured by a channel electron multiplier. By scanning the potential, a signal proportional to the number of emitted electrons is obtained as a function of the kinetic energy. Unfortunately, the measured spectrum not only contains Auger electrons, but also low energy (typically between 0 and 50 eV) secondary electrons and elastically and inelastically backsacttered electrons, with their energies depending on the primary energy that is used (usually between 1 and 10 keV). The AES peaks are superimposed as weak features on a relatively intense background. Therefore, very often the spectrum is recorded while applying a small ac potential modulation to the analyser, so that an analogue derivative spectrum is obtained. Nowadays, with the use of more sensitive multiple electron detection systems and powerful computer systems, most of the spectra are recorded in the direct mode. The signal to 18 noise ratio is improved by scanning the energy over the cylinders several times so that counts are accumulated. After recording the spectrum in the direct mode, it can be differentiated numerically as is shown in Fig. 2.6. The sensitivity of the CMA is related to the analyser transmission function, giving the effective number of electrons that are measured by the analyser for a particular energy, and is superior compared to the HSA. The energy resolution of a CMA is relative to the energy of the peak. Its best relative energy resolution (about 0.25 % ∆E/E) is clearly inferior compared to that of the HSA, as will be discussed below. Therefore, the CMA is not often used for XPS analysis, except maybe in the past where a kind of double CMA was introduced. Yet this configuration is no longer considered in new systems. The major advantage of the CMA is that there are less shadowing effects when analysing rough surfaces and small particles, which is often the case for cultural heritage materials (see also paragraph 2.7). This is due to the coaxial position of the electron gun and the detector, as noted in Fig. 2.10, ensuring that the collection of the electrons emitted from the surface is done coaxially around the electron gun. This effect is shown in Fig. 2.13, where the SEM picture and the AES images (see paragraph 2.5.7) of Ni spheres on an In substrate are shown for a coaxial geometry. The SEM picture is taken by the secondary electron detector incorporated in the AES system. The AES image is obtained by scanning of the primary lens system over the surface. At each position, the intensity of a number of AES peaks is measured, providing information on the lateral distribution of elements (see also paragraph 2.5.7). Operated in this mode, the technique is also called scanning Auger microscopy (SAM). A drawback of the use of an electron gun and an analyser operating under the same angle is that angle resolved depth profiling (see paragraph 2.5.5) is not possible. 19 (a) (b) Fig. 2.13: Images of Ni spheres on an In substrate, (a) SEM and (b) coaxial AES imaging based on the Ni mapping. 2.3.5.2 The hemispherical sector analyser All constructors use HSA detectors for electron dispersion analysis in XPS. A typical configuration is shown in Fig. 2.9. The HSA is designed to have a constant and as high as possible energy resolution for the detection of photoelectrons. The best energy resolution in XPS is 0.4 eV, corresponding to the line width of the monochromator. In order to reduce the size of the analyser, it is standard practice to retard the kinetic energies of the photoelectrons either to a user-selected analyser energy, called pass energy, or by a user-selected ratio. The first mode is called fixed analyser transmission mode (FAT), also known as the constant analyser energy mode (CAE). In this mode of operation, which is applied for the detection of photoelectrons, a constant voltage is applied across the hemispheres allowing electrons of a particular energy to pass between them. The most important characteristic in this case is a constant energy resolution in the spectrum as a function of the energy, in contrast to the CMA analyser where a relative energy resolution is obtained. The electrons are emitted from the specimen and transferred to the focal point of the analyser by the lens assembly. At this point they are retarded electrostatically before entering the analyser itself. Those electrons with energies matching the pass energy of the analyser are transmitted, detected and counted by the electron detector. The retarding field potential is then 20 ramped, and so the electrons are counted as function of energy. Since Auger electrons have a higher kinetic energy than photoelectrons, they need to be more retarted. When recording XAES (X-ray induced AES) or AES spectra, the second mode is applied. In the so-called constant retard ratio (CRR), also known as fixed retard ratio (FRR), the voltages of the hemispheres of the analyser are changed with the energy of the spectrum so that the ratio of the electron kinetic energy to the pass energy is constant. In this mode, the transmission function is optimized at the expense of the energy resolution. To improve the sensitivity of the HSA, the electron detection is done by a multichannel detector system. Depending on the type of system, the number of electron multipliers may go up to 16. This parallel electron detection is especially useful when a monochromator is used due to the loss of intensity of the primary X-rays. 2.3.6 The ion gun In AES and XPS instruments, sample sputtering can be performed by means of beams of energetic primary ions. Sputtering is useful for two reasons. One is to clean the sample prior to the analysis, because often the surface is contaminated with dirt or residual gas from the atmosphere. The second reason to sputter a sample is to record depth profiles, where the composition is probed in depth by the collection of AES or XPS spectra as a function of the sputtering time. In that case, the ion bombardment is carried out in a sequential manner with the ion gun switched off when the spectrum is recorded. The depth profile is built up by the measurement of the intensity of the recorded peaks versus etching or sputtering time. The main parameters for sputtering are the energy and the current of the ion beam, the distribution of the current in the sputtered zone and finally the spot size. Three different types of ion guns are in use in XPS and AES systems: the cold static spot gun, the electron impact source and the duoplasmatron type. It is important to note that their characteristics are very distinct, resulting in large 21 differences in obtained sputter profiles. The cold static spot gun has usually a large beam size of 5 to 10 mm and is therefore only used for large spot XPS depth profiling or for precleaning of the sample. The latter operation is often done in a configuration where the ion gun is mounted in a separate chamber, called preparation vacuum chamber. The gun is back-filled with an inert gas such as Ar having a pressure of about 10-6 torr, with a variable potential between 1 and 10 kV. A discharge to form Ar+ is realised by an external magnet and the positively ions are accelerated and extracted by a simple focus electrode. In the electron impact source, the ions are created by electrons emitted by a heated filament, accelerated into a cylindrical grid, where they collide with Ar gas atoms. The ion energy is controlled by the potential applied to the grid and ion energies up to 5 kV can be achieved. This ion source produces an ion beam with a narrow energy spread and spot sizes from 2 mm to 50 µm. It can be operated in static mode or it can be rastered over the surface to produce a larger and more uniform crater. For the rapid removal of material and etching of large areas, the duoplasmatron design of ion source is sometimes preferred. A magnetically constricted arc is used to produce a dense plasma from which the ion beam is extracted, focused and rastered across the specimen by a set of deflector plates. This type of gun provides intense ion beams with a narrow energy spread, very suitable for small spot focusing. However, due to its high price, it is not so much used in AES or XPS systems in contrast to secondary ion microscopy (SIMS) instruments. 2.3.7 The sample holder and stage The mounting of the samples on the sample holder should be done in such a way that electrical conduction is guaranteed. This is achieved by using metallic clips or bolt-down assemblies. Alternatively a metal loaded tape may also be used. In the case of powders, the particles can be pressed into an indium foil. 22 The sample holder is mounted on a sample stage which allows for high resolution positioning in the x,y,z and θ directions. To an increasing extent, especially in new systems (and highly appreciated in industrial laboratories) remote control stages are introduced. In this manner, in an automated fashion, different sample areas may be analysed, and if a parking stage is available, an automatic exchange of samples is possible. Triggered by the growing interest of the semiconductor industry, the vacuum systems are made accessible for large samples, allowing to analyse semi-conductor wafers without preceding sample preparation. In most research instruments in use in university laboratories, the sample size is limited in area to a few cm2 and in thickness to a few mm. In some systems additional sample manipulation facilities are present such as e.g. cooling or heating of the sample holder in situ during the analysis. A valuable tool in materials characterisation is a separate preparation chamber connected to the analysis system, enabling sample treatments such as heating or tensile testing. After pretreatment, the sample is introduced into the vacuum chamber for analysis through a lock gate without coming in contact with the air. The angle between the ion beam, the primary beam and the detected electron beam may induce shadowing and redeposition effects when sputtering rough surfaces. To optimize the sputtering conditions, a sample stage with tilt and rotation capabilities is useful. In [13] the Zalar rotation is applied to this purpose. 2.4 Sample Requirements Sample requirements are different for AES and XPS. Although it is also possible to detect and analyse Auger peaks in an XPS facility, the AES method is based on the use of an primary beam of electrons. This means that in principle the sample has to be conductive. In general, a sample can be analysed in AES if one can obtain good SEM pictures from it without coating. For AES investigations, isolating samples cannot be coated with a conductive 23 C or Au layer as is commonly done in SEM-EDX analysis since the escape depth of the Auger electrons is only a few nm. Thus, AES is especially appreciated for the analysis of metals and passive films on metals, but there is a growing tendency to analyse semi conducting and ceramic materials with it. Our own experience shows that e.g. Al covered with a non conducting oxide film of about 0.1 µm is measurable [13]. In other cases, special attributes, such as a conductive grid to be placed on the sample, are necessary to limit charging effects. Another possibility is to adapt the working conditions. This can be done by varying the angle of incidence of the primary electrons, or, more complicated, by performing the analysis under conditions, i.e. low primary electron energy and low current, where the impinging primary current is balanced by escaping electrons. In principle the requirement of a conductive sample is less stringent with a primary X-ray beam. However, practice shows that also in XPS charging effects may occur when analysing non-conducting samples, especially when using a monochromator. Therefore the constructors of XPS equipment add in their newest configurations a neutralisation system which is able to compensate the charge automatically. Both for XPS and AES, the samples should be stable in a UHV system of 10-10 torr. Polymers with low vapour pressure, wet samples and porous materials need to be handled with care. Many cultural heritage materials belong to this category, as will be shown in paragraph 2.7. Most operators prefer flat samples, firstly because the interpretation of the data is more straightforward, and secondly because roughness deteriorates the depth resolution in sputtering mode (see paragraph 2.5.5). 2.5 Information in XPS and AES spectra 2.5.1 Surface analysis As well for XPS as for AES, the primary beam has a penetration depth of a few micrometers. The photo electron (XPS) or Auger electron (AES) can only travel a limited distance, called attenuation length λ, before being inelastically 24 scattered. The characteristic depth d from which photoelectrons and Auger electrons are emitted, called the escape depth, is given by: d = λ(E) cos θ (2.5) where θ is the angle of emission from the surface normal. The attenuation length varies according to the element which is emitting the electron and to the matrix, and depends on the energy of the emission. Typical values of λ are, as shown in Fig. 2.14, in the range of 1 to 10 atom layers [14]. This is the basis for the surface sensitivity of XPS and AES spectroscopy. Fig. 2.14 : Dependence of attenuation length λ on the emitted electron energy [14]. 2.5.2 Qualitative analysis The qualitative analysis of a specimen consists in identifying the elements that are present. For this purpose, a survey or wide energy scan spectrum is recorded. As outlined in paragraph 2.2, each element has a characteristic XPS and AES spectrum. In [7,8], the spectra of all elements can be found. Nowadays instrumentation, both for XPS and AES, is equipped with data treatment systems running automatic peak identification tools. The identification of the composing elements of a sample under investigation is in most cases straightforward, except if peaks are overlapping. Which of both techniques to choose for a qualitative analysis depends mainly on two factors. 25 (a) x 10 2 (b) x 10 4 -Cr2p3/2 4 1 1.8 0.5 1.6 1.4 -0.5 O1 -O KLL 0.8 -1 Cr2 450 -O1s 0.4 Cr1 400 -Cr3p 0.6 -1.5 -2 1 -Cr2s Cr c/s Cr 1.2 c/s -Cr LMM 0 0.2 500 550 Kinetic Energy (eV) 600 0 1200 1000 800 600 400 Binding Energy (eV) 200 0 Fig. 2.15 : Complementary information in the AES (a), and XPS (b) spectra of an oxidized Cr sample. Conditions : Ep = 5 keV for AES, Al-Kα at 350 W, pass energy = 58.7 eV for XPS. First, the nature of the specimen is important: conductive specimens can be analysed by both techniques, while for non-conductive specimens XPS is more appropriate, although some remedies against charging effects exist (see paragraph 2.4). Secondly, the lateral resolution required for the analysis should be considered. If surface distributions on the micrometer scale are expected, AES is the proper method. The combined use of both techniques can be very useful for the analysis of complex spectra. This is illustrated in Fig. 2.15 for a Cr sample with an oxide layer at the surface. In the AES spectrum, the Cr and O peaks overlap, while in the XPS spectrum they are clearly separated. 2.5.3 Quantitative analysis The determination of the surface composition of a specimen is more complicated. To this purpose, data is collected in the multiplex mode. The relevant energy windows are selected, and the peak intensities are measured with high energy resolution and signal-to-noise ratio. The calculation of the composition based on first principles is in practice hardly ever done. The proportion between the peak intensity and the concentration of the emitting element depends in a complex manner on the 26 intensity of the primary beam, the ionization cross section, on the probability of Auger transition in the case of AES, on the attenuation length, on the spectrometer transmission efficiency and on the detector efficiency. A more practical approach consists in incorporating those parameters into sensitivity factors. The intensity of a signal from an element A, IA, in a solid is proportional to its molar fraction xA: xA = IA / IA0 (2.6) where IA0 is the intensity from a pure A sample, and may be considered as a sensitivity factor. In general what is used in the commercial data treatment software is a set of relative sensitivity factors, normalized to a reference element. For this reference, in most cases Cu or Ag, the sensitivity factor is set to unity. The molar fraction xA in a homogeneous specimen composed of i elements, is then given by : xA = NA / Σ Ni = (IA / SA) / Σ (Ii / Si) (2.7) where Ii is the area of the peak generated by constituent i, Ni its number of moles and Si its relative sensitivity factor. Since the sensitivity factors include instrumental parameters, they need to be determined on each spectrometer, and for each primary beam intensity. Moreover, since the escape depth is matrix and surface roughness dependent, using the values of sensitivity factors included in the data treatment software introduces errors. A more correct quantification can be obtained by using sensitivity factors that are determined on an appropriate set of reference samples with representative matrix effects, as shown in [15]. This may however be very difficult for cultural heritage materials. Anyway, the technique cannot be applied rigorously on heterogeneous samples, since in that case the assumption of constant sensitivity factors is not valid. The problem is more severe in AES analysis, due to the contribution of back-scattered electrons induced 27 ionization. An additional difficulty encountered in quantitative analysis is the determination of the peak areas Ii (see Eq. (2.7)). XPS peaks, and even more pronounced AES peaks, appear on a background. background correction is not straightforward. In some cases, the In most commercial data handling systems different possibilities exist. The simplest one is the straight line between two suitably chosen points. More complex methods are the Shirley and the Tougaard background correction procedures [1]. 2.5.4 Chemical analysis XPS and AES not only allow to identify and quantify the constituting elements of a sample, but make it also possible to obtain information on their chemical state. In this respect, XPS is favoured with respect to AES, and the reason for it is to be found in the nature of the respective transitions that are used in both methods. The core level peaks in XPS show a clear shift in binding energy, the so-called chemical shift, related with differences in the chemical environment of the emitting element. This is shown in Fig. 2.16 for the C peak of a polyethylene terephtelate (PET) sample. The capability to distinguish between different chemical states is the main characteristic of XPS. Due to this, another acronym is in use for this technique: ESCA, which stands for ‘Electron Spectroscopy for Chemical Analysis’. 28 1800 Peak FWHM (eV) CH C-O O=C-O 0.99 0.98 0.76 CH C 1s c/s O=C-O C-O 0 298 288 278 Binding Energy (eV) Fig. 2.16 : Chemical shift of the C 1s peak as a function of its bonding with O. The shifts are typically a few to 10 eV or more, and therefore detectors with a high energy resolution are used (see paragraph 2.3.5.2). To subtract chemical information , it is imperative to determine peak positions as accurately as possible. The line of interest is preferentially evoked by means of a monochromatic X-ray source, and recorded with the highest possible energy resolution. When dealing with small chemical shifts, overlapping peaks may anyway occur in the spectra. Peak deconvolution and peak fitting tools are available in the commercial data handling systems. An illustration is seen in Fig. 2.16, where the C 1s peak is deconvoluted in its components. In AES, the situation is less promising. Firstly, most AES lines are by nature broader than XPS lines. Secondly, the Auger transition involves three electrons and the overall chemical shift is influenced by the three energy levels concerned. In general, similar shifts of a few eV as for XPS are to be expected, especially when core electrons are involved in the transition. Since in AES-specific equipment, detectors with lower energy resolution are used (see paragraph 2.3.5.1) it is clear that chemical shifts are not often measured 29 accurately. Moreover, peaks corresponding to transitions with valence electrons are broad and poorly defined, which makes the assignment of chemical shifts nearly impossible. In that case, the identification of the chemical state may be done by a peak shape analysis. In fact the shape of a CVV or CCV peak, C being a core level, is related to the density of states (DOS) in the valence band. Since the DOS varies from one chemical environment to another, a variation in peak shape may be observed. This effect is commonly noticed in Auger spectra of non-metallic elements such as C, S, O, N. Fig. 2.17 shows a few examples of chemical effect in differentiated AES spectra. Al KLL Al LMM dE(E)/EdN Elemental Al Elemental Al Al Oxide Al Oxide C KLL 40 50 60 70 Kinetic Energy (eV) 80 90 1280 1300 1320 1340 1360 1380 1400 1420 Kinetic Energy (eV) Graphitic C Si KLL Si LMM Elemental Si W Carbide 230 240 250 260 / 270 280 290 Elemental Si 300 Si Oxide Kinetic Energy (eV) Si Oxide 50 60 70 80 90 Kinetic Energy (eV) 100 110 1500 1520 1540 1560 1580 1600 1620 1640 Kinetic Energy (eV) Fig. 2.17 : Chemical shifts and line shape effects in AES spectra of Al, Si and C. An additional difficulty that occurs when determining chemical shifts is the electrostatic charging of the specimen. In case of metallic specimens, this problem can be easily avoided by using a proper mounting procedure, but for poorly conducting materials a ‘charging’ shift can be observed, especially in 30 AES where a an electron beam is used as primary radiation. However, also in XPS the phenomenon is encountered. Although an internal standard, e.g. the C 1s for XPS or the C KVV line for AES, may be used to estimate the value of the charging shift, it is not very accurate. In order to solve this problem, the Auger parameter was introduced [16]. The Auger parameter α is the difference between the kinetic energies of a photoelectron line and an Auger line in the XPS spectrum . Taking Eq. (2.1) into account, the expression for α becomes : α = EK + EB – hν (2.8) where EK is the kinetic energy of the sharpest Auger line and EB the binding energy of the most intense photoelectron line. The modified Auger parameter α’ is introduced to ensure a positive value : α’ = α + hν = EK + EB (2.9) Since α and α’ are related to a difference in energy measured on the same sample in the same spectrometer, any static charge effects cancel out. The Auger parameter has a unique value for each chemical state and can be used as a fingerprint. Tabulations of the Auger parameter can be found in handbooks (see, e.g., [7]). 2.5.5 In-depth analysis XPS and AES can be used to provide compositional information as a function of depth. It can be obtained by non-destructive and destructive techniques. The two most commonly applied methods, i.e. angular resolved measurements and ion sputtering, are discussed below. 2.5.5.1 Angular resolved measurements This method is almost exclusively used in XPS and is non-destructive since no material is removed. It is called angle-resolved XPS (ARXPS). The principle of this method is represented in Fig. 2.18. 31 High take-off angle Low take-off angle e- e- Analyzer X-rays α α d d = λ sin α d 500eV Ar+ Ion Beam Fig. 2.18 : Principle of ARXPS. d = sampling depth, l = attenuation length. The intensity I of electrons emitted from a depth d is given by the BeerLambert relationship : I = I0 exp (-d / λ sin α) (2.10) where I0 is the intensity from an infinitely thick clean substrate, α is the electron take-off angle relative to the sample surface and λ and d are as defined above. At 90°, 95% of the signal intensity emerges from a distance 3λ, while at 15°, this is reduced to a distance of 0.8 λ. In the newest equipment, the measurements are performed in such a way that the analysed area remains constant throughout the tilt range. For example, in case of a metal surface M covered by a thin organic overlayer containing C, the ratio of the peak intensities IM and IC, when measured as a function of α, will vary. Valuable information on the thickness of the overlayer is gained, but the technique is limited to thin layers (of a few nm). 2.5.5.2 Sputter profiles Sputtering is a destructive method. The sample is bombarded with highly 32 energetic ions (mostly Ar+ ions with an energy of 1 to 5 keV) , the surface atoms are sputtered away and the residual surface is analysed. By this technique, layers up to 1 to 2 µm are accessible. AES or XPS spectra are recorded, either discontinuously after the subsequent sputter steps or simultaneously with the sputtering. The original data consist of signal intensities of the detected elements, mostly peak-to-peak heights in AES and peak areas in XPS, as a function of sputtering time. A typical example is shown in Fig. 2.19. Fig. 2.19 : XPS sputter profile of a Ti oxide layer on top of a TiNb alloy. Sputter conditions : 4 kV Ar+ on a 2mm x 2mm area. Analysis conditions : Al-Kα at 350W. To obtain the original concentration distribution of the elements and/or their chemical states two transformation steps are required. Depth scale calibration is the first step. The sputter rate, which relates sputter time and depth, depends on instrumental parameters, but is also affected by the specimen. The instrumental effects can be determined by a calibration measurement on a layer of known thickness. In practice, very often a standard reference sample of Ta2O5/Ta with 30 and 100 nm thickness is used for calibration. In general, however, the sputter rate varies with the composition, and is by consequence sample dependent. Even for a constant 33 composition, sputtering induced effects, e.g. where a component is preferentially sputtered, may cause non-linearities in the sputter time and depth relationship. In the second step, peak intensities are translated into concentrations of elements taking into account their respective sensitivity coefficients. The obtained concentration profile deviates from the real one because differences in escape depth of Auger or photoelectrons originating from different elements are neglected. A number of effects disturbs the sputtering profiles. This is expressed by the depth resolution. The most common definition of the depth resolution is the difference in depth coordinate between 84 and 16% of the intensity change at an interface. Besides the differences in escape depth, other parameters contribute to the broadening of profiles. The most important are instrumental parameters, sample characteristics and radiation-induced effects. The influence of the instrumentation is on the level of the quality of the vacuum (risk of reaction of the sputtered surface with residual gases), the purity of the ion beam (importance of a differentially pumped ion gun) and of the etch crater. The etch crater must coincide with the analysis area, and analysis of the crater walls must be avoided. In this perspective, XPS depth profiling is unfavourable compared to AES due to its larger spot size. The main sample characteristics limiting depth resolution are (a) surface roughness, possibly causing shadowing effects between the ion beam and the primary radiation and non-uniform sputter yields, and (b) preferential sputtering of one component in the study of alloys or compounds. Finally radiation-induced effects comprise among others implantation of primary ions, ion-induced reactions, e.g. reduction of Cu2+ to Cu+, and atomic mixing through knock-on effects (displacement of atoms to deeper layers). 2.5.6 Data analysis Data analysis is a technique that may be helpful in interpreting 34 multidimensional data sets with overlapping spectral features. They are generated either in chemical analysis problems where the same spectral region is studied on different samples (e.g. metal oxide, hydroxide, sulphide, …), or either in depth profiling studies where the same sample is measured under different conditions (e.g., a paint layer on top of a substrate). The two most commonly applied multivariate techniques are LLSF (linear least squares fitting) and FA (factor analysis). Both assume that each spectrum can be represented by a linear combination of component spectra : [D] = [R] . [C] (2.11) where [D] is the data matrix of recorded XPS or AES spectra, [R] is the matrix of spectra of independent components and [C] is a matrix of weighting factors or concentrations. In LLSF, [C] is determined by a linear least squares minimalisation. Data analysis by means of FA is more complex, and several techniques exist, but the basic idea is first to identify the number of independent components and then to determine the true component spectra together with the concentrations. Illustrations of data analysis can e.g. be found in [17,18]. 2.5.7 Imaging Distributions of elements and chemical states over the surface are measurable using rastering techniques. Obviously, for this application AES is in favour compared to XPS in view of its smaller analysed area (see paragraph 2.3). Imaging in AES is also named SAM, Scanning Auger Microscopy. A well focused incident electron beam is rastered over the surface, and the relevant spectra are collected. Peak intensities are translated into gray scale values. Examples are shown in Fig. 2.12 and 2.13. The smaller the spot size, the smaller the influence of the back-scattering [1]. For spot sizes larger than 100 nm, the back-scattering phenomena may modify the images in a complex manner. With the developments in small spot XPS, imaging in XPS is nowadays also feasible, as illustrated in Fig. 2.20. 35 2.6 Comparison of XPS, AES and other surface analytical techniques XPS and AES are often combined in one experimental set-up because they are complementary. Sometimes, the system is also equipped with other sources and detectors, so that other techniques can be applied simultaneously. A few examples are: energy dispersive X-ray analysis (EDX), secondary ion mass spectroscopy (SIMS), scanning tunneling microscopy (STM) or ellipsometry. Making a list of all the available surface analytical methods is even for the most experienced surface scientist an extremely difficult job as the number of techniques continuously increases, especially when newer techniques such as scanning probe or light reflection methods are considered. Each of the many techniques has its advantages, and it is beyond the scope of this chapter to compare them all. The main common characteristic of XPS and AES is their high surface sensitivity. Another technique often used for the same reason is SIMS. In Table 2.2 these techniques are compared. The characteristic values that are listed here are only indicative since all equipment is in continuous evolution. Moreover, some differences in performance exist between the commercially available systems. 36 (a) Polyethylene Mapping Area Substrate Adhesion Layer Base Coat Clear Coat 695 x 320µm 1072 x 812µm (b) C O Cl S Fig. 2.20 : (a) cross section image and (b) C, O, Cl and Si XPS images of a painted substrate. 37 Table 2.2 : Comparison of the main characteristics of XPS, AES and SIMS. characteristics XPS AES SIMS primary beam X-rays electrons ions analyzed beam electrons (energy) electrons (energy) ions (mass) type of sample all, charging possible conductive all, charging possible area of analysis 10 µm 10 nm 100 nm surface selectivity 1 to 5 nm 1 to 5 nm 0.1 to 1 nm elemental all except H, He all except H, He all 0.1 % 0.1 % < 1 ppm (dynamic) identification sensitivity 100 ppm (static) quantification requiring standards requiring standards requiring close standards molecular not possible not possible mostly possible shift, straightforward shift and shape not possible identification nature of chemical bonding requiring data analysis depth profiling elemental, chemical elemental, chemical elemental destructive nature none if not sputtered none if not sputtered always 38 REFERENCES [1] D. Briggs and M.P. Seah, ‘Practical Surface Analysis. Volume 1 – Auger and X-ray Photoelectron Spectroscopy’, Second Edition, John Wiley and Sons, Chichester, 1990 [2] T.A. Carlson, ‘Photoelectron and Auger Spectroscopy’, Plenum Press, New York, 1975 [3] J.F. Watts, ‘An Introduction to Surface Analysis by Electron Spectroscopy’, Oxford University Press, Oxford, 1990 [4] G. Hollinger, P. Pertosa, Chapter 3 in ‘Surfaces interfaces et films minces – Observation et analyse’, Edited by B. Agius et al., Dunod, Paris, 1990 [5] M. Thompson, M.D. Baker, A. Christie, J.F. Tyson, ‘Auger Electron Spectroscopy’, John Wiley and Sons, New York, 1985 [6] V.I. Nefedov, ‘X-ray Photoelectron Spectroscopy of Solid Surfaces’, English Edition, VSP BV, Utrecht, 1988 [7] J.F. Moulder, W.F. Stickle, P.E. Sobol, K.D. Bomben, ‘Handbook of X-ray Photoelectron Spectroscopy’, Edited by J. Chastain, R.C. King Jr., Physical Electronics, USA, 1995 [8] K.D. Childs, B.A. Carlson, L.A. LaVanier, J.F. Moulder, D.F. Paul, W.F. Stickle, D.G. Watson, ‘Handbook of Auger Electron Spectroscopy’, Third Edition by C.L. Hedberg, Physical Electronics, USA, 1995 [9] G. Beamson, D. Briggs, ‘High Resolution XPS of Organic Polymers.The Scienta ESCA300 Database’, John Wiley and Sons, Chichester, 1982 [10] M.F. Chung, L.H. Jenkins, Surf. Sci., 21 (1970) 253 [11] N.M. glezos, A.G. Nassiopoulow, Surf. Sci., 254 (1991) 314 [12] J.I. Goldstein, D.E. Newbury, P.Echlin, D.C. Joy, A.D. Romig Jr., C.E. Lyman, C. Fiori, E. Lifshin, Chapter 2 in ‘Scanning Electron Microscopy and X-ray Microanalysis’, Second Edition, Plenum Press, New York, 1992 [13] J. De Laet, K. De Boeck, H. Terryn, J. Vereecken, Surf. Interface Anal., 22 (1994) 175 [14] M.P. Seah, W.A. Dench, Surf. Interface Anal., 1 (1979) 2 [15] M. Detroye, F. Reniers, C. Buess-Herman, J. Vereecken, Proceedings ECASIA 97, 7th European Conference on Applications of Surface and Interface Analysis, 16-20/06/97, Göteborg, Sweden, Edited by I. Olefjord, L. Nyborg, D. Briggs, John Wiley, 1997, 995 [16] C.D. Wagner, L.H. Gale, R.H. Raymond, Anal. Chem., 51 (1979) 466 and references cited herein [17] F. Reniers, A. Hubin, H. Terryn, J. Vereecken, Surf. Interface Anal., 21 (1994) 483 [18] G. Treiger, I. Bondarenko, P. Van Espen, G. Goeminne, N. Roose, H. Terryn, Proceedings ECASIA 95, 6th European Conference on Applications of Surface and Interface Analysis, 913/10/95, Montreux, Switzerland, Edited by H.J. Mathieu, B. Reihl, D. Briggs, John Wiley, 1996, 775 39 40