Lecture 34: Prions. PRION Prion diseases can occur through: Infectious
advertisement

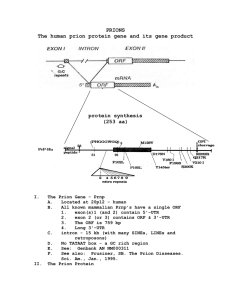
Lecture 34: Prions. PRION = Proteinaceous Infectious particle Prion diseases can occur through: 1. Infection: Infectious 2. As a dominantly inherited genetic disorder: Familial 3. Consequent to a spontaneous mutation: Sporadic Mammalian Prion diseases. Characterized as Spongiform Encephalopathies. Invariably fatal. Characterized by amyloid plaques or spongy (spongiform) appearance of the affected areas of the brain, due to accumulation of vesicular structures in the brains of infected organisms. Vacuoles are seen in one neuron and in the neuropiles. Astrocytes with small nucleus proliferate. No inflammatory cells infiltrae in the brain. •For many years, prion diseases were thought to be caused by slow-acting viruses, and were referred to as ‘slow virus diseases’, ‘transmissible spongiform encephalopathies’, or ‘unconventional viral diseases’. •First discovered by showing that injection of a brain extract from a patient into a chimpanzee could result in infection. Incubation times to clinical disease are long (years). •Inherited forms tend to appear in mid-life. Human Prion Diseases • Human prion disease should be considered in any patient who develops a progressive subacute or chronic decline in cognitive or motor function. Typically adults between 40 and 80 years of age. Creutzfeldt-Jakob disease (CJD). Characterized by a progressive dementia • iCJD (infectious) – injection of brain matter from a CJD patient into chimps suggested that CJD was a result of an infectious agent. – Patients have been infected iatrogenically from injections of human growth hormone derived from human pituitary gland extracts. • fCJD: (familial). • sCJD (spontaneous) – Dominantly inherited trait, i.e. heterozygous individuals develop fCJD. – Penetrance is 100%, i.e. if the carriers live long enough, they will all eventually develop prion disease. – Inherited forms have been demonstrated in a number of families. Genealogic investigations from 4 southern English families show that they are related and argue for the existence of a single founder born more than 200 years ago. – Lybian jews. Had been thought to be due to the consumption of lightly cooked/raw sheep brain and eyeballs. Later shown to be due to a specific mutation. – Similar mutation found in people originating from Orava in North Central Slovakia, in a cluster of families from Chile, and a large German family living in the US. – CJD can occur in the absence of a familial history or infection. Can be due to a spontaneous mutation on the PrP gene, or even in the presence of only wild-type genes: a hint into the nature of the process. Human Prion Diseases • Gerstmann-Steussler-Scheinker disease (GSS) – The first of the spongiform encephalopathies that was described as a genetically inherited trait. – Linkage analysis from GSS families demonstrated that PrP was the responsible gene. – GSS may also arise spontaneously. • Fatal Familial Insommnia – FFI: characterized by adults generally over age 50 who present with a progressive sleep disorder who die within about a year of onset. – More than 30 families worldwide have been identified. • FSI (fatal sporadic insomnia). The sporadic form of FFI. • Kuru – Affects the Fore people of New Guniea. Transmitted through ritual cannabalism. Regional distribution of PrPSc in transgenic mice inoculated with brain extracts from humans who died of prion disease Mouse inoculated with brain extract from FFI patient Mouse inoculated with brain extract from fCJD (E200K) patient Hippocampus Ventral posterior lateral thalamic nucleus Neocortex Habenula Hypothalamus Thalamus Amygdala Animal Prion Diseases Scrapie • The classic spongiform encephalopathy of sheep • Characterized by progressive loss of motor control • Propensity of infected animals to obsessively rub or scrape themselves against things (fenceposts, sides of enclosures, etc) to the point of scraping off all their hair and rubbing their skin raw. • Horizontally transmissible through the herd. Animal Prion Diseases Mad Cow disease (Bovine Spongiform Encephalopathy or BSE) • Characterized by progressive loss of motor control. • Transmissible. • Long incubation period is dose dependent: 2-10 years with a mean period of 5 years. • Route of infection has been linked infective sheep and cattle tissue in meat and bone meal, a component of feed for cattle and other domestic livestock. • A change in the traditional feed-rendering process was linked to the rise in BSE. The traditional includes an extraction step with organic solvents, which is thought to have extracted out the prion protein. In Great Britian in the early 1980's a new cost effective method was implemented that omitted the organic extraction step. The result was the BSE epidemic of the mid- to late-80's (thanks to Maggie Thacher). Animal Prion Diseases BSE (Con’t) • Following the introduction of the ruminant feed ban in July1988, and the wholesale slaughter of infected herds, BSE is declining in Great Britian. • Small epidemic in France linked to human growth hormone derived from cadavers • BSE cow found in Canada in 2003 • BSE cow found in USA in 2004 – came from Canadian herd. Risks to human health • New variant of CJD (vCJD) began to appear in humans 8 years after the first known case of BSE in cattle. Although there have only been 21 confirmed cases of vCJD to date, the young ages of the patients and the lack of mutant PrP alleles suggests that they developed disease via an infectious route. Animal Prion Diseases Other mammalian prion diseases • Transmissible Mink Encephalopathy (TME): Mink are carnivores, raised on farms, infection results from being fed prion-containing meat and bone meal. • Feline Spongiform Encephalopathy (FSE): Infection results from being fed prion-containing beef. • Exotic Ungulate Encephalopathy (EUE): occurs in the greater kudu, nyala, oryx, i.e. ungulates housed in zoos. Infection results from being fed prioncontaining meat and bone meal. • Chronic Wasting Disease (CWD). Occurs in mule deer and elk. Unknown etiology, perhaps due to contact with infected farm animals. Concern in Wyoming that it will spread to cattle. Etiology, molecular biology & biochemistry The Prion Protein (PrP). •Discovery of the prion protein used a mouse scrapie model. •Pruisner and co-workers infected mice by intracerebellar injection with brain extracts from Scrapie infected sheep. •Provided a bioassay that could be used to identify the prion-causing agent. •The infectious fraction is: •Nuclease resistant. •Resistant to UV irradiation at 254 nm. •Sensitive to proteases. Suggests that the agent is a protein. •Strongly suggests that the agent is not a nucleic acid. The Prion Protein (PrP). Amyloid plaque stained with Congo Red Bifefringence under polarized light The Prion Protein •Infectious agent purified to a single protein species •Named the Prion Protein (PrP). •Infectious dose: ~105 PrPSc molecules correspond to one ID50 unit of prions. •This leaves open the formal possibility of some nucleic acid contamination, and that the infectious agent is a small protein-associated nucleic acid. •No difference in the quantity of PrP between normal and diseased brains. •Constitutively expressed in brains of adult animals. •Highly regulated during development. •Cellular functions •Long term survival of Purkinje neurons?? •Circadian activity rhythms and patterns?? Molecular characterization of the PrP gene. • Protein sequence analysis was used to clone the PrP gene. • Highly conserved gene among all vertebrates. Prion protein gene structure. • PrP is encoded by a single open reading frame (ORF) encoding 253 amino acids in all known mammalian and avian PrP genes. Rules out the possibility that the infective form arises from alternative RNA splicing. Molecular characterization of the PrP gene. Alignment of the 44 known PrP sequences shows a striking degree of consevation between the mammalian sequences, suggestion the retention of an important function through evolution. • However, PrP-null mice are fine. Therefore, PrP is not an essential protein. PrP protein structure • PrP post-translationally processed to remove: – a 22 amino acid NH2-terminal signal peptide • at the COOH-terminus, 23 residues are removed during the addition of a glycophosphatidyl inositol (GPI) moiety that anchors the protein to the cell membrane. • NH2-Terminal sequence repeats • Amino terminal domain of mammalian PrP contains 5 copies of a P(H/Q)GGG(G)WGQ octarepeat sequence • Insertions of octarepeats have been linked to Human disease • Deletions of octarepeats do not appear to cause disease, however, deletions do not prevent disease. PrP protein structure • Conserved Ala-Gly region • Between A113 to Y128 is a highly conserved Glyand Ala-rich region. • Structurally, the sequence appears to have been selected for its properties of flexibility, and perhaps for the ability to undergo conformational change. • Closest protein sequence is spider dragline silk, a highly β-sheet rich protein that is capable of forming filamentous polymers. • A single point mutation A117V is linked to GSS. • Two β-sheet (S12 and S2) and three α-helix (A, B & C) forming regions are conserved PrP protein structure fCJD Human mutations •extra copies of the octarepeat •D178N •E200K sCJD •Homozygosity for M129V fGSS FFI •P102L •A117V (the sequence polymorphism that was used to demonstrate the inherited nature of GSS and prion disease). •Requires double mutation: D178N + M129V Structural studies • PrPc is protease-sensitive; • PrPsc is protease-resistant • PrPc is α-helix rich: 40% a-helix, little β-sheet; • PrPsc is β-sheet rich: 30% a-helix, 45% β-sheet • No differences in postranslational chemical modifications were found between the two forms of the protein. • Rules out this as a possible cause of prion diseases. Structural studies •PrPc is soluble •PrPsc forms insoluble filaments 45,000X magnification electron micrograph of yeast prion protein fibers formed in the test tube. The rigid fibers are similar to those observed in amyloid diseases of mammals. Allosteric model of Prion propagation Prion form is “enciphered” thru protein conformation Prion form Normal form + Heterodimer Homodimer Subunit interaction model for prion formation Normal form Prion form + Heterodimer Homodimer Crystal seed model Propagation of the prion form of the proteins 104 104 104 104 35 35 35 Polymerization 35 35 35 35 Prion formation in vitro (yeast prion system) PSI 35 35 35 35 Prion formation in vitro (Human prion system) A major technical hurdle was that nobody had been able to establish an in vitro prion propagation system using PrP. This was accomplished (Jackson, G.S.. et al., 1998. Science 283: 1935 – 37). Overcoming the species barrier…implications for hBSE Advantages of the prion model •Self-encoded protein explains the lack of lymphocytic infiltration •Epidemiology is best explained by the prion model: •Spontaneous forms are due to the rare, stochastic conversion of native, wild-type protein to prion form. •Inherited form is due to the PrP gene mutations that make the conformational conversion more energetically favorable. •Infectious because infectious form also makes the conformational conversion of endogenous PrPc more energetically favorable. •Templating/Enciphering •Incubation times. Once the species barrier has been crossed, of the host species PrPc to PrPsc becomes more energetically favorable. •Strain differences, e.g. FFI vs CJD, and deposition patterns are explained by the prion hypothesis. Problems with the prion model •Cannot make or isolate completely pure PrPsc . •Therefore, the argument can be made that infectious innoculum contains a small amount of an infectious nucleic acid. Q: Does requirement for transmissability limit our conceptual understanding of prion disease? By the allosteric model, any protein that can template a conformational change should also fit the definition. Opens up new possibilities, e.g. Altzheimers, Sickle Cell disease Early-Onset Familial Alzheimer Disease With Coexisting-Amyloid and Prion Pathology (JAMA. 2000;283:1689-1691) Figure. Double Immunostaining for -Amyloid and Prion Protein (PrP) in the Frontal Cortex Senile plaques immunopositive for -amyloid40 (arrowheads in panels A and C), PrP106-126(arrowheads in B), and for -amyloid40 plus PrP106-126 (double arrowheads in C) are shown. Twodifferent chromogens were used, first diaminobenzidine dihydrochloride to reveal the -amyloid peptide (reddish-brown) and second, benzidine dihydrochloride to reveal the PrP106-126 (blue). When both signals are superposed, the blue signal appears dark and is localized in the center of the plaque. Scale bar is 50 µm for panels A and B, and 75 µm for panel C. Fundamental significance of viroids • Molecular signals that induce host polymerase to accept viroids as templates for transcription (missing from cellular RNAs?) • Molecular mechanisms of viroid replication (operative in uninfected cells?) • How do viroids induce disease? • Why are viroids restricted to higher plants? • How did viroids originate? T.O. Diener (1987) First report of viroid disease - Schultz and Folsom (1923) Viroid disease symptoms Potato spindle tuber viroid chrysanthemum stunt viroid Apple viroid disease Healthy Infected Avocado Sunblotch Viroid Coconut cadang-cadang viroid Viroids •Smallest known agents of infectious disease •Discovered by T.O. Diener (1971) •Small, covalently closed circular, single stranded RNAs •No protein coding capacity (246399 nt) •Autonomous replication (no helper virus) •29 species (8 genera, 2 families) Viroid classification • Host range may be broad (HSVd) or narrow (CCCVd) • Mechanically transmissable • Not insect transmitted • Most important vector = man Viroid pathogenicity Host contribution • Induction of “pathogenesis related” proteins • Protein kinases / signaling cascades Viroid contribution • Role of pathogenicity domain • Induction of RNA silencing Viroid Movement, i.e. spread within infected plant • Nuclear / chloroplast import • Cell-to-cell movement • Long distance via phloem Movement of viroids and viruses Infected leaf Source-to-sink movement Moves from infected leaf to roots, up phloem to actively photosynthesizing leaves, then back down the plant Developmental controls on movement as well Origin of viroids Three pieces of evidence that viroids are relics of a precellular RNA world: •Structural periodicity / polyploid genomes: Suggests that they are constructed from very ancient functional RNA domains. • Minimal ribozymes / genomic tags: represent ancient enzymes • Viroids / introns / transposons: all ancient and phylogenetically related to one another Phylogeny of subviral RNAs: They’re genetically related to one another ASBVd Viroids LTSV ASSVd HDV: hepatitis delta virus AGVd SCMoV GYSVd Plant Satellite viruses SNMV GVd1B VTMoV HLVd CTiVd SatTRSV CCCVd HSVd CLVd CEVd PSTVd TASVd 100 nt TPMVd CSVd Elena et al. 1991. Proc. Natl. Acad. Sci. USA 88, 5631-5634. SatArMV Hepatitis delta virus: a human viroid? • Rod-like secondary structure • Replicates in nucleus • RNA polymerase II • Ribozyme-mediated cleavage • Protein coding region /RNA editing Some interesting musings… •Viroids can only be propagated horizontally/vegetatively. •Cannot be propagated vertically, do not make it through meiosis. •Origins of sex? •Implications for animal cloning?