ADVANCED UNDERGRADUATE LABORATORY EXPERIMENT 28 MASS SPECTROMETER
advertisement

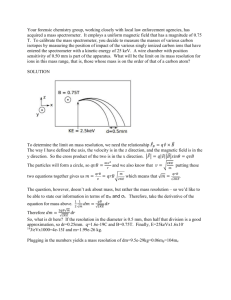
ADVANCED UNDERGRADUATE LABORATORY EXPERIMENT 28 MASS SPECTROMETER Revised: February 2005 by Jason Harlow June 1997 by Joe Vise March 1988 by John Pitre Introduction In nuclear physics, spectrometers are used to measure relative isotopic masses and to determine the relative abundances of isotopes as they are produced by nuclear events such as fission. Mass spectrometers are also used to study ionization processes in gases, the results of ion-molecular interactions and the structure of complex organic compounds. In the field of geophysics and geology, spectrometers are applied to the measurement of geological time, and the variation in isotopic abundances in rocks and minerals produced by changes in physical conditions at the time of deposition. Mass spectrometers have also served as analytical tools for gas analysis and as vacuum system leak detectors. In this experiment students learn how to operate a mass spectrometer and the vacuum system associated with it. Students plot a spectrum of the near-vacuum in the chamber, and investigate the ionization process for two different pure gases. Theory The most common type of mass spectrometer employs a transverse magnetic field to separate the components of a narrow beam of monoenergetic ions. The magnitude of the force acting on an ion of charge q and velocity v during its passage through a magnetic field B which is perpendicular to v is F = qvB (1) The trajectory of ions of mass M in the magnetic field is along an arc of a circle with radius R as is demonstrated in Figure 1. Since the centripetal force F is given by Mv 2 (2) F R then the radius of the circular trajectory is given by Mv (3) R qB In the simplest type of spectrometers, ion beams are produced by accelerating ions through a potential difference V in an electrostatic lens system. On leaving this system the ions have kinetic energy 1 Mv 2 qV (4) 2 The radius of curvature in the magnetic field is therefore 1 2 MV R (5) B q 2 Figure 1. Ion paths in a mass spectrometer. The simplest example of this type of magnetic deflection spectrometer is one in which the entire trajectory, from source to detector, is in the transverse magnetic field. Under this condition, the maximum separation, or resolution, of adjacent masses occurs after a deflection of 180°. The geometrical refocussing of slightly divergent ion beams of a given mass is also best at a deflection of 180°. This arrangement is used in the small mass spectrometer (MS-10) manufactured by Associated Electrical Industries Ltd which is used in this experiment. Figure 2 shows a schematic arrangement of the MS10 mass spectrometer. Assembly details and mechanical drawings are given in the instruction manual and are available from the technician if required. 3 Figure 2. Schematic arrangement of the MS10 mass spectrometer. The ions are accelerated by the accelerating voltage +V with respect to ground applied to the ion source cage. The ions travel perpendicular to the uniform magnetic field, B, and describe circular orbits. The radius of orbit varies for each particular type of ion and is determined by its mass and the accelerating voltage. Thus the total ion beam is separated, by the action of the fixed magnetic field, into individual beams containing ions corresponding to one particular mass. By varying the value of accelerating voltage, individual beams are brought in turn to focus on the collector where the ions give up their charge. The ion current at the collector is detected by an electrometer amplifier whose output is displayed on a meter or recorder. 4 The Ionization Process Within The Source When a gas sample is introduced into the spectrometer, ions will be formed in the ion source, shown in Figure 3, by electron bombardment. The electrons for this purpose are emitted from a hot rhenium wire filament and are accelerated parallel to the external magnetic field by the electron voltage Ve between the filament and the ion source cage. Ve can be varied from 0 to 150 V and these energetic electrons drift across the ion source chamber producing ionization along the electron beam path. Figure 3. Schematic of details of the ion source. NOTE: The ion cage is a solid box with holes in it. The electron beam is incident on a plate or “trap” on the opposite side of the source, and the current to this plate is used as a convenient measure for the purpose of stabilizing the electron beam intensity. The trap voltage Vt, of about 35 V with respect to the cage, simply allows the beam to be collected and has no other effect on the operation of the spectrometer. Ions produced by ionizing collisions are pushed out of the ion source cage by a voltage Vr on the repeller plate which is typically 1 V. The penetration of the ion accelerating voltage V into the ion source cage also helps to extract the ions. The mass spectrometer detects ion beams of different mass to charge ratio extracted from the ion source. The ion current I is given by I K (V , B) N 0 Qi (Ve ) d I e (6) where No is the gas density in the ion source, Qi(Ve) is the ionization cross section of the atoms for bombardment by electrons of energy eVe, d is the effective path length of the electrons in the source and Ie is the electron current traversing the source. K(V,B) is a quantity dependent on ion accelerating voltage V and the magnetic field B, and represents the efficiency of extraction of ions from the source. For a complete discussion of equation (6) see Barnard, p. 85, Duckworth, Chap. 3 and Field, p. 41. Ions will not be formed in a particular gas until the energy of the bombarding electrons exceeds the first ionization potential of the gas. The electron energy at which ions first are detected should thus be a measure of the ionization potential of the gas in the source. In a simple mass spectrometer such as the MS10, this is a very rough measure indeed, because the energy spectrum of the bombarding electrons is broadened by their inherent thermal energy spread and the voltage drop along the length of the filament. The energy spectrum is also shifted by space charges and contact potentials in the source as well as the drawing out field of the ion accelerating potential. 5 Figure 4. Ion current I versus electron accelerating voltage Ve. Special sources have been designed to avoid these difficulties, but in simple spectrometers the result of these effects is to superimpose a tail on the leading edge of the ion current versus electron energy curve as is illustrated in Figure 4. Morrison (1953) suggested the following approach for determining the ionization potential from this curve. 6 Figure 5. I and its first two derivatives versus Ve (Idealized). For ionization close to the ionization potential Vi, the probability of ionization is proportional to the energy excess of the ionizing electrons above the ionization potential. Thus, the yield of ions, or the ion current will be proportional to Ve−Vi. This has been proven experimentally by Fox et al. The graph of ion current as a function of electron energy is therefore a straight line, at least in a small range of energies above the ionization potential. This is true in Figure 4 if we were to ignore the tail near the onset of ionization. Figure 5(a) is an idealized version of Figure 4. The first derivative of ion current with respect to electron energy per unit charge is a step function at Vi as in Figure 5(b) and the second derivative will be a delta function at Vi as in Figure 5(c). As mentioned above, these functions are shifted by potential biases in the ion source and broadened, in particular by electron thermal energy spread, so that Figures 5(b) and ( c ) become Figures 6 (a) and (b), respectively. 7 Figure 6. More realistic versions of the second and third graphs in Figure 5. Morrison suggested that the peak of the second derivative plot would be a less subjective measure of the “appearance potential” than the first appearance of ions. This method has the advantage of illustrating the thermal energy spectrum of the electrons in the ion source, and the linear nature of the ionization probability above the appearance potential. Measurement of Ionization Cross Sections Equation (6) along with the ideal gas equation pV = nRT (7) may be used to determine the ionization cross section Qi(Ve) if one assumes an effective path length d = 0.1 cm and an efficiency of ion extraction from the source K(V,B)=1 given by the manufacturers of the mass spectrometer. The electron trap current Ie may be measured directly. The ion current I is determined by knowing that a full scale reading on the 1000 range of the amplifier corresponds to 10−10 A. In fact, the numbers on the amplifier range factor all refer to the ion current of a full scale needle deflection in units of 10−13 A. The ionization gauge is calibrated for N2 and the pressure reading must be multiplied by a factor F if the pressure of other gases are being measured. In particular, F = 0.83 for Ar and F = 4.35 for Ne. The readings for P will be suspect if the mass spectrometer is used in the dynamic mode since there will be pressure differentials throughout the system and the pressure in the ionization gauge head will not be the pressure in the ionization chamber of the mass spectrometer. One can convert changes in the measured ion current into estimates of changes in partial gas pressure in the ionization chamber if one knows the sensitivity S of the mass spectrometer. Sensitivity is defined as the change in the ion current I at a given mass, resulting from a change in pressure P of this isotopic species. I S (8) P At pressures below about 5 × 10−5 torr, this sensitivity is constant for a given gas and given operating conditions in the source. For a trap current Ie of 50 µA, an electron accelerating voltage of 70 V and an ion repeller voltage of +1.0 V, the manufacturer quotes a sensitivity of 5.6 × 10−5 A/torr for Ar and 1.0 × 10−5 A/torr for Ne. These values are typical and some variation is expected from instrument to instrument. Experiment The main goals of this experiment are: become familiar with the operation of the mass spectrometer analyse the mass spectrum of the near-vacuum present in the mass spectrometer pump out the spectrometer and re-analyse the spectrum of remaining gas find the first ionization energies of Argon and Neon 8 determine the ionization cross-section of Argon and Neon A schematic diagram of the mass spectrometer, vacuum and gas handling systems is given in Figure 7. Figure 7. Schematic Diagram of the Apparatus. In general, mass spectrometer systems are configured so that the mass spectrometer may be isolated from the rest of the system by ultra high bakeable metal vacuum valves. When the mass spectrometer is isolated from the pumps and the rest of the system, the pressure in the spectrometer will gradually rise as adsorbed gasses (mainly N2 and O2) are released from the interior metal surfaces but sufficiently slowly so that the total pressure is still less than 10−5 torr after 30 minutes. Under these circumstances the mass spectrometer is said to operate in the STATIC mode and very small samples of gas can be analysed. The mass spectrometer in the undergraduate laboratory is not configured for high vacuum operation since large amounts of sample gas are available. Also, “high” background pressures of about 10−5 torr are useful as a multi-component sample when learning the basics of mass spectrometer operation. When working with sample gasses the mass spectrometer is operated in the DYNAMIC mode. Gas is allowed to leak into the spectrometer from the sample line via a controlled-leak valve and is pumped away by the spectrometer pumps. 9 Initial Startup The electronics should be turned on immediately upon arriving since the amplifier requires an hour to reach its final stabilized level. Also, after the filament has been turned on, the accelerator voltage and the electron source require a half-hour to reach final stability. The boldface steps should be repeated or checked each time the mass spectrometer is turned on. When you are done for the day or wish to leave the equipment for more than an hour or so, please follow the “Shutting Down the System” procedures which begin on page 14. 1. Turn AMPLIFIER RANGE FACTOR to 1000, its least sensitive setting. Note that one must multiply the number on the amplifier range factor by 10−13 Amps to get the reading for the maximum deflection of the needle. For example, at setting 1000, the needle pointing to 1 (full scale) would indicate a current of 10−10 Amps. 2. Turn MAINS to ON. 3. Turn the THERMOCOUPLE GAUGE CONTROL to ON and set the TC SELECTOR to monitor the pressure in the mass spectrometer chamber as indicated on the diagram on the THERMOCOUPLE GAUGE CONTROL. This step is necessary because the THERMOCOUPLE GAUGE CONTROL contains a relay which prevents the FILAMENT of the mass spectrometer from being turned on until the pressure in the mass spectrometer as measured by the thermocouple gauge is less than 1 millitorr. 4. Turn the FILAMENT switch to ON. If the pressure in the mass spectrometer should rise above 1 millitorr or if the TC SELECTOR switch is changed to a different position, the FILAMENT will automatically shut off. 5. Monitor the ELECTRON VOLTS output with a multimeter and adjust the ELECTRON BEAM CONTROL until Ve is 70 V. Record the actual value an uncertainty. 6. Turn TRAP µA to 50. 7. Zero the AMPLIFIER using the COARSE and FINE controls. When this is being done, the MASS dial should be set in between integer mass readings, so that the expected current is zero. Progressively increase the AMPLIFIER sensitivity while adjusting the zero on each range. Return the AMPLIFIER setting to 1000, its least sensitive range. As the electronics stabilize, you may need to repeat this zeroing step. 8. Turn the MASS SELECTOR switch to 12-45. 9. Turn the SCAN switch to MANUAL. 10. Turn the MASS dial slowly and look for peaks from residual gasses in the mass spectrometer. The most prominent peaks should be at 18 (water) and 28 (nitrogen). Progressively increase the AMPLIFIER sensitivity until these peaks are found. 11. Set the MASS dial exactly to 28 and then adjust the FINE TUNE CONTROL until the meter reading is a maximum. After the completion of this step the MASS dial has now been calibrated throughout its entire range. Notice that there is considerable play in the MASS dial and one should always turn the dial in a clockwise sense when searching for various isotopes. 12. Turn on the RG1000 digital ion gauge. (PWR button) 13. Connect a recorder to the AMPLIFIER RECORDER jack. Turn the SCAN switch to AUTO. Use high sensitivity and record and identify the residual gasses using the cracking patterns given in Appendix I. Make sure that you can explain the relative heights of the peaks in simple cracking patterns. What are the components for the hydrocarbons that give rise to the various peaks? 10 14. Refer to the next section on “Starting Up the Vacuum System” and evacuate the mass spectrometer. Again, record and identify the residual gasses using Appendix I. Make an attempt to explain the differences between this run and the previous run before the mass spectrometer was evacuated using the diffusion pump. 15. Remember that the AMPLIFIER zero may continue to slowly drift for an hour after initial startup. Starting Up the Vacuum System Examine the layout of the system and find the: ROTARY PUMP ATMOS LEAK or leak to atmosphere value (NEVER TOUCH THIS) ROTARY PUMP COLD TRAP, an open liquid N2 (LN2) dewar DIFFUSION PUMP DIFFUSION PUMP COLD TRAP, with plastic funnel for filling with LN2 BAFFLE VALVE ION PUMP PERMANENT MAGNET Mass spectrometer body All three thermocouple gauges (TC1, TC2, TC3) Gas bottles and regulators GAS LEAK VALVE Trace your way through the pumping system to see which valves need to be opened or closed in succession to pump the system out. Trace you way through the gas handling system to see which valves need to be opened or closed in succession to admit gas to the system. 1. Turn on the THERMOCOUPLE GAUGE CONTROL and set the TC SELECTOR switch to the appropriate position to monitor the pressure at the ROTARY PUMP as indicated on the diagram on the THERMOCOUPLE GAUGE CONTROL. Remember that the FILAMENT, if it is ON, will shut off. 2. Switch on the ROTARY PUMP. The switch is on the motor. 3. Check the ROTARY PUMP COLD TRAP to see if water has condensed in the bottom. If there is water in the bottom, carefully insert a Kimwipe to absorb it. Remove the Kimwipe with the pickup tool provided. 4. When the pressure at the ROTARY PUMP is less than about 10 millitorr, fill the trap between the ROTARY PUMP and the DIFFUSION PUMP with liquid nitrogen. The purpose of this trap is to prevent high pressure ROTARY PUMP oils from reaching the MASS SPECTROMETER either through the DIFFUSION PUMP or through the gas handling lines. In most vacuum systems, a cold trap is not placed between the ROTARY PUMP and the DIFFUSION PUMP. Note if the pressure changes upon adding the liquid nitrogen. 5. Open the valve between the ROTARY PUMP and the DIFFUSION PUMP. 6. Plug in the DIFFUSION PUMP. The cooling fan should start immediately and the base of the pump should start to get hot after about 3 minutes. 11 7. Allow about 20 minutes for the DIFFUSION PUMP oil to boil and then fill the DIFFUSION PUMP COLD TRAP with liquid nitrogen. 8. Set the TC SELECTOR back to monitor the pressure in the mass spectrometer chamber so that the filament may come back on. 9. The mass spectrometer body is still isolated from the DIFFUSION PUMP by the BAFFLE VALVE and it is being pumped by the ION PUMP. Record the pressure using the “torr” setting on the on the ION PUMP CONTROL unit. 10. Open the BAFFLE VALVE and monitor the pressure on the ION PUMP CONTROL. The pressure should rise as the valve is opened because gas is released from the O-ring to the metal seal. The pressure should fall to less than the pressure in the previous step within a minute. If this does not happen, close the BAFFLE VALVE immediately and ask for help. Admitting Sample Gas to the System 1. After the BAFFLE VALVE has been opened in the section on “Starting Up the Vacuum System”, the pressure should be allowed to fall to less than 10−7 range as measured on the ION PUMP CONTROL unit. 2. The pressure of sample gas in the spectrometer will be monitored by the digital IONIZATION GAUGE. The pressure will momentarily rise as the filament (FIL) is turned on. This is due to outgassing of the filament as it becomes hot. Note that the pressure registered by the IONIZATION GAUGE may be a factor of ten greater than that at the flange to the ION PUMP which is the pressure measured by the ION PUMP CONTROL. This is because the hot IONIZATION GAUGE is a source of gas and there is a large pressure gradient between the IONIZATION GAUGE and the diffusion pump. 3. If the pressure is above 10−7 torr, turn on the degassing filament (DEG) in the IONIZATION GAUGE. Again the pressure will rise sharply as adsorbed gasses escape from the hot degassing filament. The pressure may rise as the glass envelope of the IONIZATION GAUGE becomes hot. 4. When the pressure falls to less than 10−7 torr, or after about ten minutes, turn off the degassing filament (DEG). Wait about 10 minutes as the IONIZATION GAUGE cools and the pressure falls. Please do not leave the degassing filament on for more than about 10 or 15 minutes, or it can damage the system. 5. Turn off the ION PUMP! This pump is almost always left on, but for this section only you may ignore the big “DON’T SHUT THIS POWER SUPPLY OFF” warning. The reason we are temporarily turning it off is that the ion pump should not be used to pump on large amounts of Neon or Argon. 6. Turn the TC SELECTOR to the position to monitor the pressure in the gas handling lines. 7. Before pumping out the lines to the gas handling system close the valve between the DIFFUSION PUMP and the ROTARY PUMP. Note that the pressure in the spectrometer will gradually rise when the DIFFUSION PUMP is isolated from the ROTARY PUMP. Whenever the pressure in the spectrometer rises above 10−6 torr, open the valve between the DIFFUSION PUMP and the ROTARY PUMP and reduce this pressure before proceeding. 12 8. With the valve between the DIFFUSION PUMP and the ROTARY PUMP closed, open the valve between the ROTARY PUMP and the MANIFOLD to the gas samples, if indeed it is closed. 9. Wait until the pressure in the gas handling lines is less than 10 millitorr before opening any valves. In the next steps refer to figure 8. Figure 8. Schematic diagram of the gas handling system. 10. Open the valve A at the MANIFOLD between the MANIFOLD and the REGULATOR (turn counterclockwise). Remember to monitor the pressure on the IONIZATION GAUGE and recall that the valve between the DIFFUSION PUMP and the ROTAY PUMP must be closed as the valves in the gas handling lines are opened. 11. Check that the valve D on the gas bottle is closed (fully clockwise) and the silver gas reducing valve C on the REGULATOR is closed (full counterclockwise). Note that valve C opens by pushing. Once it turns freely counterclockwise, it is completely closed. If you continue unscrewing it, the silver handle will fall off, and then you can screw it back in. 12. Open the valve B at the REGULATOR between the REGULATOR and the MANIFOLD (this valve is in fact redundant). The gas handling lines will now be pumped out. 13. Close the valves B and A. The pressure should fall again to less than 10 millitorr. 14. Close the valve between the MANIFOLD and the ROTARY PUMP. 15. Open the valve between the DIFFUSION PUMP and the ROTARY PUMP. 16. The next steps will admit gas to the gas handling lines. Open the valve D on the top of the gas bottle (counterclockwise). 17. Turn the silver reducing valve C slowly clockwise until the pressure reads about 50 kPa or about one half an atmosphere. 18. Open the valve B. The pressure on the regulator will momentarily fall and then rise again to 50 kPa. 19. Open the valve A. There will now be about one half an atmosphere of gas in the gas handling lines. 20. There now should be enough gas in the lines to perform your experiment. In order to prevent your gas bottle from leaking out, you should now close valves A, B, C and D. 13 21. Turn the THERMOCOUPLE GAUGE selector to monitor the pressure in the mass spectrometer and turn on the FILAMENT in the mass spectrometer. 22. Open the black main valve on the GAS LEAK VALVE. There will be an initial burst of gas into the system as the valve is opened but the pressure will fall immediately to the 10−8 range. 23. Start off by admitting about 10−6 torr of gas to the mass spectrometer. Half-close the baffle valve. Gradually open the silver leak valve on the GAS LEAK VALVE and monitor the pressure rise in the mass spectrometer with the IONIZATION GAUGE. Do not exceed 10−5 torr. Measuring the Ionization Potential for the Gas Sample 1. Measure the REPELLER VOLTAGE. Its magnitude should be 1.00 ± 0.05 Volts. You may adjust it with a flat-head screwdriver. 2. Set the MASS dial to observe the highest peak of the gas that you have admitted to the system. (i.e. 40 for Argon, 20 for Neon.) 3. Reduce the ELECTRON VOLTS to the region of onset of ionization where the ion current falls to zero. 4. Make a rough plot at half volt intervals of ion current versus ELECTRON VOLTS over a five volt interval to determine the region where the onset of ionization occurs. 5. Now select a smaller two to three volt region which includes the ionization potential Vi (see figures 4 and 6). Adjust the gas pressure so that readings can be taken when the AMPLIFIER is on the 100 or higher setting. Do not exceed 10−5 torr. 6. Monitor the RECORDER OUTPUT with a digital voltmeter and plot ion current versus ELECTRON VOLTS at 0.1 volt intervals. For each ELECTRON VOLT setting use the FINE TUNE control to obtain the maximum reading which means that the ion beam is focussed on the collector. Repeat this measurement since small fluctuations in the ion ACCELERATOR VOLTS causes the ion current peak to shift. 7. Use your data to find the “appearance potential” for the gas you are using. This provides a measurement of the first ionization energy of the gas. 8. Repeat these measurements for at least one more gas. Be sure to close the valve between the diffusion pump and the gas handling lines when you pump out the gas from the gas handling lines. Measuring Ionization Cross Sections 1. Set the ELECTRON VOLTS TO 70, the TRAP current 50, and the ION REPELLER to 1.0 V. 2. Measure the ION CURRENT and record the pressure. 3. Calculate the ionization cross-section and compare your value to that of others (see for example Massey or McDaniel). 4. Repeat this measurement for another gas. Shutting Down the System 1. If the BAFFLE VALVE has not been opened, proceed to step 13. 14 2. If no gas has been admitted to the system, proceed to step 10. 3. Close the silver leak valve on the GAS LEAK VALVE (clockwise=closed). 4. Close the black main valve on the GAS LEAK VALVE. 5. Close the valve between the DIFFUSION PUMP and the ROTARY PUMP and pump out the gas handling lines. The same precautions should be taken in monitoring the pressure in the mass spectrometer when pumping out the gas handling lines as was done in the section on “Admitting Gas To The System”. 6. Turn off the FILAMENT in the mass spectrometer. 7. Turn the THERMOCOUPLE GAUGE selector to monitor the pressure in the gas handling lines. 8. Open the valve between the gas handling lines and the ROTARY PUMP. This will pump out your sample gas so that future users do not find the lines contaminated. 9. After the pressure has fallen to less than 10 millitorr, close the valve between the gas handling lines and the ROTARY PUMP, and open the valve between the DIFFUSION PUMP and the ROTARY PUMP. 10. Turn on the ION PUMP! 11. Turn off the IONIZATION GAUGE (FIL) and wait a few minutes for it to cool. 12. Close and open the BAFFLE VALVE a couple of times, while monitoring the pressure on the ION PUMP CONTROL dial. The pressure should stabilize at about 10−6 torr initially. If it is much greater than this, open the BAFFLE VALVE and allow the IONIZATION GAUGE to cool further and repeat this procedure. 13. With the BAFFLE VALVE closed, unplug the DIFFUSION PUMP. Be careful you pull the correct plug! 14. Close the valve between the DIFFUSION PUMP and the ROTARY PUMP. 15. THE ION PUMP MUST BE LEFT ON. 16. Turn off the ROTARY PUMP. 17. Turn off the other various instruments, including the PWR on the digital ion gauge, the TC gauge, the Filament and Mains on the electronics box, the multimeters and the chart recorder. References 1. G.P. Barnard, Modern Mass Spectrometry, The Institute of Physics, 1953. (QC 451 B3 T) 2. H.E. Duckworth, Mass Spectroscopy, Cambridge University Press, 1958. (QC 451 D8) 3. F.H. Field and J.L. Franklin, Instrumental Factors Affecting Electron Energy, Academic Press, 1970. (QC 702 F5) 4. R.E. Fox et al, Phys. Rev. 84, 859, 1951. 5. H.S.W. Massey and E.H.S. Burhop, Electronic and Ionic Impact Phenomena, Clarendon Press, 1952. (QC 794.6C6M38 1969) 6. E.W. McDaniel, Collision Phenomena in Ionized Gases, J. Wiley and Sons, 1964. (QC 721 M24) 7. J.D. Morrison, J. Chem. Phys. 19, 1305, 1951. 8. J.D. Morrison, J. Chem. Phys. 21, 1767, 1953. 9. J.D. Morrison and A.J.C. Nicholson, J. Chem. Phys. 20, 1021, 1952. 15 Appendix I. Cracking Patterns on the MS10 This appendix gives relative intensities of the outputs of various molecules. The different peaks are due to molecular dissociation (breaking up of molecules during the ionization process), different ionization states and isotopes. 16 17 18