Aplastic anemia Deepa Jeyakumar, MD Assistant Clinical Professor of Medicine
advertisement

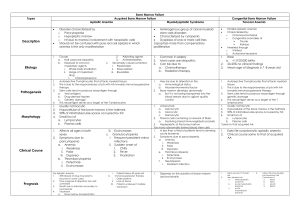
Aplastic anemia Deepa Jeyakumar, MD Assistant Clinical Professor of Medicine 10/15/14 ST Pt is a 43 year old woman who was in her usual state of health until 03/16/11 when she presented to outside ER with left flank pain and dark urine for two days. Found to have hct of 26 with platelet count of 26k. No hemolysis on labs. Peripheral smear reveals no atypical cells with few large platelets. Initially thought to have ITPgiven IVIG x 1. Did not response to IVIG. Required daily platelet transfusions. Bone marrow biopsy markedly hypocellular marrow with 80% fat. Cellularity is composed of entirely maturing erythroid elements. Myeloid elements are markedly decreased. Occasional segmented forms noted. Megakaryocytes rare. Stainable iron increased. No ringed sideroblasts. Reticulin focally increased. No features of parvovirus seen. Several blast forms seen. Diagnosis of Aplastic Anemia Marrow is profoundly hypocellular with decrease in all elements. Residual hematopoietic cells are morphologically normal. Malignant infiltrates and fibrosis is absent. Hematopoiesis is non-megaloblastic. Severity Moderate aplastic anemia Marrow cellularity <30% Absence of severe pancytopenia Depression of at least two of three blood elements below normal. Severe o o Bone marrow cellularity <25% or marrow showing <50% normal with two of three peripheral blood count criteria: ANC <500 Plt <20k Retic count <40k Very Severe All of above plus ANC less than 200. Classification Inherited Fanconi’s anemia, dyskeratosis congenita, Shwachman-Diamond Syndrome, Reticular dysgenesis, Amegakaryocytic thrombocytopenia, familial aplastic anemia, preleukemia (monosomy 7) and nonhematologic disease (Down, Dubowitz, Seckel) Acquired Irradiation drugs and chemicals: cytotoxic agents, benzene, idiosyncratic reaction, chloramphenicol, NSAIDS, antiepileptics, Gold viruses: EBV, Hepatitis virus (non-A,non-B, non-C, non-G), Parvovirus (transient aplastic crisis or pure red cell aplasia), HIV Immune diseases: eosinophilic fasciitis, hyperimmunoglobulinemia, thymoma and thymic carcinoma, GvHD in immunodeficiency PNH Pregnancy Idiopathic Differential Diagnosis Pancytopenia with hypocellular bone marrow - Inherited aplastic anemia - Hypoplastic AML Pancytopenia with cellular bone marrow Acquired aplastic anemia Hypoplastic MDS Primary bone marrow diseases PNH Myelophthisis Hairy cell leukemia Hypersplenism Overwhelming infection Brucellosis Sarcoidosis - -MDS Myelofibrosis Bone marrow lymphoma SLE, Sjogren’s disease Vitamin B12 and folate deficiency - Alcoholism Ehrlichiosis tuberculosis Hypocellular bone marrow with or without cytopenia Q fever Mycobacteria Hypothyroidism - Legionaires disease - Tuberculosis - Anorexia nervosa Hypocellular AML & hypocellular MDS Epidemiology International Aplastic Anemia and Agranulocytosis Study (IAAAS) found 2 confirmed cases per one million people (two PNH units) Thailand- 4 cases per million Cumulative survival has increased over the past few decades Etiology and Pathogenesis Genetic predisposition found in HLADR2. This correlates to response to immunosuppressants. Similar results found in hypoplastic MDS. Pathogenesis Immune-mediated T-cell destruction of marrow Young demonstrated that removal of lymphocytes from aplastic bone marrow improved colony number in tissue culture and addition of lymphocytes to normal marrow inhibited hematopoiesis in vitro. Immune Destruction of Hematopoiesis Telomere shortening Originally thought to be due to stem-cell exhaustion. Telomere shortening also found in X-linked form of dyskeratosis congenita due to mutations in DKCI. Telomere shortening also found in mutations in TERC found in AD patients with constitutional Subsequent analysis of patients with acquired aplastic anemia found mutations in TERC and TERT. Interestingly, family members of patients who share these mutations can have normal blood counts but hypocellular marrows, reduced CD34 counts and poor hematopoietic colony formations and short telomeres. Therefore, 1/3 to ½ of patients with aplastic anemia have short telomeres but mutations are only found in 10% of patients. Treatment Treatment ATG: Lymphocyte numbers decreased within the first few days of therapy and then return to pretreatment levels within a week or so. Appears to be immunomodulatory as well as lymphocytotoxic- producing a state of tolerance by preferential depletion of activated T cells. Rabbit appears to be more potent that the horse formulation. Cyclosporine: its selective effects on T-cell function is due to direct inhibition on the expression of nuclear regulatory proteins, resulting in decreased T-cell proliferation and activation. Intensive Immunosuppression Clinical Endpoints Response defined as transfusion independence. Relapse defined as requirement of additional immunosuppresants. About 50% response rate with horse ATG. Happens in 30-40% of patients. Clonal evolution occurs in 15% of cases. Into MDS, AML, PNH Improving on ATG & cyclosporine for first line management of AA? Addition of high dose steroids did not improve outcomes and just added to toxicity. Addition of G-CSF and GM-CSF did not improve outcomes Addition of mycophenolate did not improve response rates or outcomes. Sirolimus was equally ineffective. Cyclophosphamide was associated with a higher death rate due to prolonged neutropenia. Relapsed/Refractory Aplastic Anemia Rabbit ATG- if patient has not seen it before and had a decent response to initial treatment. Alemtuzumab has been shown in the relapsed setting to be effective. Cyclophosphamide has a 50% response rate in relapsed setting. Moderate Aplastic Anemia Role for duclizumab, humanized monoclonal antibody to IL-2 receptor Role for androgen therapy HLA-matched sibling allotransplantation Risk factors for graft failure Heavily transfused Long time between diagnosis and transplantation Alternative Donor Transplant A 24-year-old man undergoes follow-up evaluation for treatment of aplastic anemia. Two of his siblings are HLA-identical matches. Hemoglobin Leukocyte count Platelet count Reticulocyte count 8.3 g/dL (83 g/L) (following transfusion of 1 unit of irradiated packed erythrocytes last week) 500/µL (0.5 × 109/L) with 23% neutrophils, 3% band forms, and 71% lymphocytes 26,000/µL (26 × 109/L) 0.2% Review of the bone marrow biopsy done 2 weeks ago confirms the diagnosis of aplastic anemia, demonstrating an aplastic bone marrow with normal cytogenetics. Which of the following is the most appropriate treatment? A: Allogeneic hematopoietic stem cell transplantation B: Antithymocyte globulin, corticosteroids, and cyclosporine C: Autologous hematopoietic stem cell transplantation D: Corticosteroids E: Granulocyte colony-stimulating factor Thank You!