Quantum hydrodynamic model for the enhanced moments of inertia
advertisement

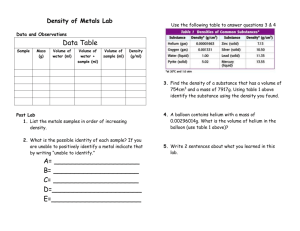
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 4 22 JULY 2002 Quantum hydrodynamic model for the enhanced moments of inertia of molecules in helium nanodroplets: Application to SF6 Kevin K. Lehmanna) and Carlo Callegarib) Department of Chemistry, Princeton University, Princeton, New Jersey 08544 共Received 28 August 2001; accepted 29 April 2002兲 The increase in moment of inertia, ⌬I, of SF6 in helium nanodroplets is calculated using the quantum hydrodynamic approach 关Callegari et al., Phys. Rev. Lett. 83, 5058 共1999兲; 84, 1848 共2000兲兴, which we extend here to an explicit three-dimensional treatment. Three plausible helium densities are reconstructed by interpolation of previously published ‘‘density cuts’’ in terms of an expansion into cubic harmonics 共several interpolation strategies are presented兲. This allows us to predict a value of ⌬I that ranges from as low as 30 u•Å2 to as high as 318 u•Å2 . The lower limit reproduces the prediction of Kwon et al. 关J. Chem. Phys. 113, 6469 共2000兲兴, who use the same hydrodynamic model and an unpublished density based upon a Path Integral Monte Carlo calculation. These values can be compared with the experimentally measured ⌬I 共310⫾10 u•Å2 ) for large (N⭓103 He atoms兲, and with Fixed Node, Diffusion Monte Carlo calculations by Lee, Farrelly, and Whaley 关Phys. Rev. Lett. 83, 3812 共1999兲兴, which found ⌬I⫽290–305 u•Å2 for N ⫽8 –20 helium atoms. The present results show that the value of ⌬I obtained from the hydrodynamic model is quite sensitive to physically reasonable variations in the helium density; therefore one has to be careful as to which density to use. Because the model is based upon the assumption that the helium is in the ground ‘‘quasienergy’’ state of the helium-molecule time-dependent potential, we propose that calculations should be done using densities calculated at 0 K rather than at finite temperature. We have extended our original algorithm to also handle irregular boundaries. We find that in the present case the calculated value of ⌬I only changes by a few percent. © 2002 American Institute of Physics. 关DOI: 10.1063/1.1486443兴 there is consensus that the additional moment of inertia, ⌬I, arises from the kinetic energy of helium motion that is correlated with the rotation of the solute. In the case of SF6 – HeN 共Ref. 7兲 a fixed frame, fixed node, diffusion Monte Carlo 共DMC兲 calculation on small clusters with N⫽8 –20 has recovered rotational level spacings (⬀B eff) in excellent agreement with those observed for SF6 in a 共much larger兲 He nanodroplet. Such calculations give limited dynamical information, however, and thus leave open the question of how to physically characterize the helium motion. Two quantitatively predictive dynamical models have been put forth that invoke very distinct types of helium motion.8 –10 In one,8,9 a proposed ‘‘nonsuperfluid fraction’’ of helium is calculated using Path Integral Monte Carlo 共PIMC兲 methods, and is assumed to rotate rigidly with the molecule, provided that the molecule–He potential is sufficiently anisotropic relative to the induced rotational energy. To date, this ‘‘two-fluid model’’ has been applied to SF6 and OCS.8,9 For SF6 it gives a result in excellent agreement with experiment: the predicted increase in the effective moment of inertia, ⌬I two-fluid ⫽327 u•Å2 almost exactly matches the experimental value ⌬I exp⫽310⫾10 u•Å2 共Ref. 4兲 derived from the rovibrational spectrum.11 The second approach, introduced by our group,10 is based upon a hydrodynamic treatment for the helium flow. Its key assumptions are: 共1兲 a continuum fluid; 共2兲 constant solvation density in the frame rotating with the molecule 共adiabatic following兲; 共3兲 ideal fluid flow 共aviscous and irro- I. INTRODUCTION There is considerable current interest in the spectroscopy of atoms and molecules solvated in liquid helium. In particular, 4 He nanodroplets1,2 are currently the best system to study, on a microscopic scale, some unique physical properties: He is the sole substance known to remain a liquid as T→0 K, and is also the most easily accessible superfluid. These properties are also relevant to chemistry, as they make 4 He droplets an almost ideal ‘‘matrix’’ for the production and characterization of novel species.3 Notably, even large and highly anisotropic solute molecules exhibit rotationally resolved spectra, though with rotational constants, B eff , often considerably smaller than for the same molecule in the gas phase. Physically this means that the effective moments of inertia for rotation of molecules in liquid helium, I eff , are several times larger than those of the isolated 共gas phase兲 molecules.1,4 – 6 The number of molecules and aggregates being formed and detected in He droplets is steadily increasing, as is their complexity. A theory capable of quantitative predictions for I eff will be valuable inasmuch as the rotational constants of an unknown spectral band can be used to identify the chemical carrier of the transition strength, as is often done in gas phase spectroscopy. Although the exact dynamics still has to be elucidated, a兲 Electronic mail: lehmann@princeton.edu Current address: Condensed Matter Physics, M/C 114-36, Caltech, Pasadena, CA 91125. Electronic mail: cal@caltech.edu b兲 0021-9606/2002/117(4)/1595/9/$19.00 1595 © 2002 American Institute of Physics Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp 1596 J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 K. K. Lehmann and C. Callegari tational兲. The ‘‘classical look’’ of condition 共1兲 might give the impression that this approach is no longer valid when the variation of helium density is large on an atomic length scale, which is exactly our situation. In reality the hydrodynamic equation used, Eq. 共13兲, can be derived from the variational optimization of a many-body helium wave function of the form, 冋 ⌿⫽ 共 R 兲 exp ⫺ im ប 兺i 共 r i 兲 册 , 共1兲 where (R) is the highly correlated ground state wave function, m the atomic mass of 4 He, and 兺 i (r i ) a phase function written as a sum of one-body terms. Condition 共2兲 follows from the assumption that the rotational frequency of the molecule is much less than the excitation frequencies of the helium that are coupled to the rotation. The hydrodynamic approach has been applied to a number of linear molecules; its estimates of the additional moment of inertia, ⌬I h , were found to be in good agreement with ⌬I exp for the heavier molecules, including OCS. Conjusteau et al.13 have experimentally established that for lighter molecules, specifically HCN and DCN, adiabatic following breaks down, and that the sign of the observed deviations is consistent with the approximations of the hydrodynamic theory in these cases. Lee, Farrelly, and Whaley7 had previously demonstrated numerically that the adiabatic following approximation breaks down for SF6 if the free molecule rotational constant is increased by a factor of 10 over its known value, making it similar in size to that of HCN and DCN. The hydrodynamic model has been used by Kwon et al.9 to calculate ⌬I h for SF6 ; the reported value, ⌬I h⫽31 u•Å2 , is taken by Kwon et al. as evidence of the failure of the hydrodynamic approach since it is only about 10% of ⌬I exp . This result would indeed suggest that the hydrodynamic model has failed for this molecular system, however no discussion was given as to the robustness of this result with respect to changes in the density used. In the present work, we present the results of calculations which use several different density functions, based upon both thermally averaged and zero-temperature helium densities around SF6 . We demonstrate a large sensitivity of the calculated ⌬I h to reasonable variations of the density, and we present reasons why the zero-temperature density should be used in hydrodynamic calculations of the helium contribution to the effective moment of inertia. Because of the different symmetry of the system 共O h) compared to the linear molecules we treated in Ref. 10 (C ⬁ v), it is no longer possible to remove one degree of freedom by separation of variables. As a result, both the helium density and the velocity potential have to be calculated in three dimensions; this requires several changes to our original approach; in addition we extend our original algorithm so that irregular boundaries could be used. The computational procedures used in the present case are described in Sec. II B for the He density, and in Sec. II C for the velocity potential; the results are presented in Sec. III and discussed in Sec. IV. II. CALCULATIONS A. Notation We use spherical coordinates (r, , )⬟(r,⍀); explicit dependence on coordinates may be omitted when obvious. The SF6 共solute兲 molecule is oriented with the S–F bonds lying along the Cartesian axes which instantaneously coincide for the laboratory-fixed and molecule-fixed frames of reference; (r,⍀) is the three-dimensional helium density around the solute molecule; (r,⍀) is the velocity potential 共see Sec. II C兲; v(r,⍀) the Eulerian velocity of the fluid in the laboratory frame. O h is the point group of the SF6 molecule, which also determines the symmetry of the helium density; C 2 , C 3 , and C 4 are the associated symmetry axes; 2 (r), 3 (r), 4 (r) are radial cuts of the density along those axes, e.g., along ⍀ 2 ⫽( /4,0), ⍀ 3 ⫽ 关 arccos(1/冑3), /4兴 , ⍀ 4 ⫽( /2,0), respectively; 0 (r) is the spherically averaged density at each r, defined as (1/4 ) 兰 (r,⍀)d⍀. T L (L ⫽0, 4, 6, 8, 10, . . . 兲 are cubic harmonics,16 i.e., the linear combinations of the spherical harmonics, Y LM , that transform as A 1g in the O h point group. In this work we will only use harmonics up to L⫽8, for which the explicit expressions are T 0 ⫽1, 共2兲 T 4⫽ 冑 7 Y ⫹ 3 40 T 6⫽ 冑 冑7 Y 60⫺ 共 Y 64⫹Y 6,⫺4 兲 , 2 2 T 8⫽ 冑33 4 ⫹ 1 4 Y 80⫹ 冑 冑 1 2 5 共 Y 44⫹Y 4,⫺4 兲 , 6 冑 共3兲 共4兲 7 共 Y 84⫹Y 8,⫺4 兲 6 65 共 Y 88⫹Y 8,⫺8 兲 . 6 共5兲 Several papers present a procedure to calculate these and higher harmonics. We cite among these the original work of von der Lage and Bethe,16 where the term ‘‘Kubic Harmonics’’ is first introduced, that of Fox and Ozier,17 and the works of Fehlner and Vosko, who give powerful quadrature formulas for integration of functions with octahedral symmetry.18,19 We use here the popular normalization 兰 (T L ) 2 d⍀⫽4 , i.e., the T L have unit rms value on the sphere. B. Reconstructing the helium density For the cylindrically symmetric molecules dealt with in our earlier hydrodynamics work10 it was convenient to use helium density functional 共DF兲 theory14 to calculate the helium density around a static solute molecule. For systems with cylindrical or higher symmetry, DF is computationally many orders of magnitude less expensive than Quantum Monte Carlo 共QMC兲 methods. Speed comes at the price of a more approximate treatment, compared to QMC, but the DF Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 Moments of inertia in helium nanodroplets treatment has been shown to accurately reproduce the static properties of pure and doped He clusters,20 at least when the dopant–helium interaction is not too large. Currently, DF has not been implemented in three dimensions; because of this, and of the fact that we want to compare our results to those of Kwon et al.,9 we chose to use published densities that have been calculated by QMC methods. Two of those exist: one calculated by Barnett and Whaley15 and based upon a DMC calculation with a second order estimator 共hereafter, density I兲; the other calculated by Kwon et al.9 by path integral quantum Monte Carlo 共hereafter, density II兲. We will describe our procedure as applied to density I; further details that only pertain to density II are postponed to a later section. We were unable to obtain the three dimensional densities from the authors of the respective papers. We then decided to focus on density I, for which a more extensive set of data samples has been published. We extract the radial isotropic density 0 (r), and the radial cuts 2 (r), 3 (r), 4 (r) by digitizing the plots in Figs. 7 and 8 of Ref. 15, for a cluster of 69 He atoms, the largest for which density cuts were reported. The standard approach to interpolate a sparse set of data is to write the interpolating function as a series expansion in terms of complete set of functions that are a basis for the given symmetry, and to use the available data to determine the coefficients of the lowest terms of the expansion. More explicitly we write 共 r,⍀ 兲 ⫽ 0 共 r 兲 ⫹a 4 共 r 兲 T 4 共 ⍀ 兲 ⫹a 6 共 r 兲 T 6 共 ⍀ 兲 ⫹a 8 共 r 兲 T 8 共 ⍀ 兲 ⫹•••. 共6兲 Formally, it would be straightforward to obtain the expansion coefficients, from a L (r)⫽(4 ) ⫺1 兰 (r,⍀)T L d⍀, if enough sampling directions were available that the integral could be reliably calculated. Instead we assume that the density contains only the terms explicitly given in Eq. 共6兲. We then evaluate Eq. 共6兲 along ⍀ 2 , ⍀ 3 , ⍀ 4 , thus obtaining a system of linear equations which, when inverted, gives the equations for a 4 (r),a 6 (r), and a 8 (r), a 4共 r 兲 ⫽ 1 143冑21 关 189 0 共 r 兲 ⫺128 2 共 r 兲 ⫺189 3 共 r 兲 ⫹128 4 共 r 兲兴 , a 6 共 r 兲 ⫽⫺ 冑 1 55 2 关 5 0 共 r 兲 ⫹32 2 共 r 兲 ⫺27 3 共 r 兲 13 ⫺10 4 共 r 兲兴 , a 8 共 r 兲 ⫽⫺ 冑 8 65 共7兲 共8兲 3 关 35 0 共 r 兲 ⫺16 2 共 r 兲 ⫺9 3 共 r 兲 187 ⫺10 4 共 r 兲兴 . 共9兲 Figure 1 shows the expansion coefficients thus calculated. We refer to this scheme as ‘‘direct expansion.’’ It is apparent from this figure that the density in the first solvation shell 共the most important for determination of the moment of 1597 FIG. 1. The expansion coefficients, 0 (r),a 4 (r),a 6 (r), and a 8 (r) as a function of the He–SF6 radial distance. inertia兲 is not fully converged with the present truncation of the density expansion. One of the consequences is that there are regions where the density calculated by Eq. 共6兲 becomes negative; this is not a serious problem, however, as it only occurs in regions that can be considered as occupied by the SF6 molecule, where the density can be safely set to the threshold value min required by the hydrodynamic equations 共see below兲. It is a more serious problem that even in regions where it is positive, such a density may be a poor representation of the true density. We later implemented a much better approach than the direct expansion of the helium density by Eq. 共6兲. Nevertheless the results obtained by this rather crude method are presented for completeness since they help demonstrate the sensitivity of the results to changes in the assumed density function. This lack of convergence arises from the large variation of the density 共at fixed r) in the first solvation shell, in particular near the SF6 repulsive wall. The same problem has plagued the series expansion of the intermolecular potential between a spherical atom and a nonspherical molecule; expansions of such potentials into a series of orthogonal functions are notorious for their terrible convergence properties at short distances, due to the fact that near the repulsive wall, at fixed r, the potential at different ⍀ can vary by several orders of magnitude. It has been recognized that a superior approach is an indirect expansion, where one first expresses the radial cuts of the potential 共at fixed ⍀) as an analytic function of r and of a set of parameters, and subsequently expand each of these parameters into a series of spherical harmonics. With a judicious choice, the parameters are slowly varying with angle, and therefore much better convergence can be achieved. The method has been successfully applied to numerous potentials, e.g., to He–CH4 共Ref. 21兲 and He–SF6 共Ref. 12兲 共incidentally, the latter potential is the one that was used to calculate the helium densities we utilize here兲. A functional form f (r) that was found to closely reproduced the radial density cuts is what we call a sum of ‘‘modified half Gaussians,’’ explicitly 共see also Fig. 2兲, Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp 1598 J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 K. K. Lehmann and C. Callegari FIG. 2. Radial density cuts, 0 (r), 2 (r), 3 (r), and 4 (r) as a function of the He–SF6 radial distance, and their fits to ‘‘modified half Gaussians’’ 共see text兲. Also shown is the physical interpretation of the fitting parameters. 2 w i⫽ 再 冉 冏 冏冊 兺 i⫽1 A i exp ⫺ w⫺ i 共 r⬍c i 兲 f 共 r 兲⫽ w⫹ i bi r⫺c i wi 共 r⭓c i 兲 ; b i⫽ 再 ⫹c 1,6T 6 (⍀)⫹c 1,8T 8 (⍀) 其 兴. Note that for convenience in tabulating the expansion coefficients, this form is slightly different than that used for the density, Eq. 共6兲. Finally we write 共10兲 , b⫺ i 共 r⬍c i 兲 b⫹ i 共 r⭓c i 兲 . 2 共 r,⍀ 兲 ⫽ Note that the exponential functions are Gaussians only for b i ⫽2 共hence the term ‘‘modified’’兲, and that f (r) is continuous and has a continuous first derivative as long as b ⫾ i ⬎1. We fitted 0 (r), 2 (r), 3 (r), 4 (r) with the functional form of Eq. 共10兲 by least squares minimization, obtaining the parameters reported in Table I. The density cuts and the best fitting curves are shown in Fig. 2; in all cases we found that the fitting function would reproduce the original data within the limit of their noise. Therefore we find it convenient, and justified, to use the best fitting analytic function as a smooth interpolation of the original data. An expansion into cubic harmonics was then calculated by use of Eqs. 共6兲–共9兲, with replaced by the parameter to be expanded 关for example, c 1 (⍀)⫽c 1,0兵 1⫹c 1,4T 4 (⍀) 兺 i⫽1 冋冏 A i 共 ⍀ 兲 exp ⫺ r⫺c i 共 ⍀ 兲 w i共 ⍀ 兲 冏 册 b i (⍀) . 共11兲 We will refer to this scheme as ‘‘indirect expansion.’’ Note that for an arbitrary function f, f (x)⫽ f (x), therefore c 1,0 in the example above 共and likewise any other parameter兲 is not the entry in row 0 of Table I. Rather the isotropic part of each parameter has to be calculated selfconsistently, by constraining the spherical average of the density to match 0 (r). We call these ‘‘spherically averaged parameters,’’ and report them in row L⫽0 of Table I. In the same table, we also report the other cubic harmonic expansion coefficients for each parameter. TABLE I. Best fit parameters for radial cuts of density I to Eq. 共10兲 are given in rows 0 through 4 . The row labeled L⫽0 contains the ‘‘spherically averaged parameters’’ for which Eq. 共12兲 best fits the 0 curve; the last three rows contain the dimensionless coefficients of the higher cubic harmonics for the expansion of each parameter in the ‘‘indirect expansion’’ scheme 共see text兲. 0 2 3 4 L⫽0 L⫽4 L⫽6 L⫽8 a A1 (Å⫺3) c1 共Å兲 0.0715 0.093 0.205 0.0506 4.518 4.493 4.170 4.801 0.0786 ⫺0.569 0.242 0.216 4.721 0.046 ⫺0.010 ⫺0.026 w⫺ 1 共Å兲 w⫹ 1 共Å兲 b⫺ 1 b⫹ 1 A2 (Å⫺3) c2 共Å兲 w⫺ 2 共Å兲 Parameters for fits to radial density and density cuts along C n axes 0.619 0.674 2.499 1.493 0.0268 7.557 1.190 0.489 0.685 2.374 1.642 0.0178 8.029 1.779 0.418 0.592 2.400 1.649 0.0444 7.561 1.129 0.559 0.856 2.857 1.692 0.0298 7.64a 1.275 Cubic harmonic expansion coefficients for each line shape parameter 0.690 0.564 2.530 1.230 0.0268 7.664 1.268 0.134 0.045 0.052 ⫺0.090 ⫺0.103 ⫺0.006 ⫺0.046 ⫺0.023 ⫺0.002 0.013 0.016 0.211 ⫺0.014 ⫺0.113 ⫺0.158 0.141 ⫺0.002 0.190 0.027 0.010 0.086 w⫹ 2 共Å兲 b⫺ 2 b⫹ 2 1.295 1.730 0.927 1.578 2.403 2.782 2.692 2.157 1.387 1.867 1.535 1.576 1.220 0.045 ⫺0.121 0.116 2.675 ⫺0.047 ⫺0.022 ⫺0.019 1.360 ⫺0.079 ⫺0.049 0.136 Fixed. Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 Moments of inertia in helium nanodroplets 1599 TABLE II. Fitting parameters for density II. See Table I for details. 0 2 3 4 L⫽0 L⫽4 L⫽6 L⫽8 A1 (Å⫺3) c1 共Å兲 0.0863 0.0919 0.165 0.0677 4.384 4.382 4.112 4.741 0.0944 ⫺0.266 0.130 0.054 4.430 0.037 ⫺0.006 ⫺0.002 w⫺ 1 共Å兲 w⫹ 1 共Å兲 b⫺ 1 b⫹ 1 冕 c2 共Å兲 w⫺ 2 共Å兲 Parameters for fits to radial density and density cuts along C n axes 0.500 0.635 2.682 1.480 0.0266 7.350 1.075 0.455 0.586 2.648 1.474 0.0269 7.329 1.080 0.396 0.501 2.458 1.489 0.0313 7.346 0.949 0.551 0.659 2.972 1.553 0.0205 7.635 1.436 Cubic harmonic expansion coefficients for each line shape parameter 0.473 0.586 2.758 1.443 0.0266 7.376 1.105 0.086 0.066 0.054 0.001 ⫺0.097 0.009 0.104 ⫺0.011 ⫺0.019 ⫺0.006 0.007 0.016 0.003 ⫺0.001 ⫺0.006 ⫺0.001 ⫺0.013 0.022 ⫺0.009 0.003 0.021 ⫹ 兲 d⍀ 共 r,⍀,A 1,0 ,c 1,0 , . . . ,b 2,0 w⫹ 2 共Å兲 b⫺ 2 b⫹ 2 1.282 1.282 1.070 1.347 2.054 2.088 2.497 1.969 1.442 1.442 1.295 1.527 1.260 0.054 ⫺0.028 ⫺0.006 2.094 ⫺0.067 0.033 0.017 1.428 0.039 ⫺0.015 0.000 of He atoms in a cluster is increased the density of complete shells increases, but not significantly. Figure 12 of Ref. 9, which reports the isotropic density for a cluster of 100 He atoms, then gives us an upper limit for the density of a 64 He atoms cluster calculated at 0.3125 K. As a higher temperature will have, if any, the effect of delocalizing the He density, i.e., of smoothing the first solvation shell, we think that the above proves that our replacement for the spherically averaged density is acceptable. A posteriori the agreement between the value of ⌬I h we obtain with this density and the value calculated by Kwon et al. also justifies our approximations. In summary, density II represents a cluster of 64 He atoms and is constructed using the radial density cuts from a PIMC calculation at 0.3125 K from Ref. 9, and the spherically averaged density from a PIMC calculation at 0.625 K from Ref. 8. The fitting parameters, and their expansion into cubic harmonics, that reproduce density II are reported in Table II. Finally we also fitted the superfluid fraction of density II, reconstructed from Figs. 2 and 3 of Ref. 8. The fitting parameters, and their expansion into cubic harmonics, that reproduce this ‘‘superfluid density’’ are reported in Table III. The self-consistent calculation was performed by fitting 0 (r), as recreated from its analytic expression, to the function, 共 4 兲 ⫺1 A2 (Å⫺3) 共12兲 ⫹ as free by least-square minimization, with A 1,0 ,c 1,0 , . . . ,b 2,0 parameters of the fit. The integral in Eq. 共12兲 was numerically evaluated at each iteration by 6-point quadrature, with the formula provided in Ref. 18. While, as we will see, density I produces satisfactory results which are consistent with ⌬I exp , it does not agree with the much lower value found by Kwon et al.9 Upon visual inspection, it is evident that the PIMC density used by Kwon et al., calculated for 64 He atoms, is considerably more isotropic than the DMC density we use 共compare Fig. 3 of Ref. 9 with Fig. 8 of Ref. 15兲. The question then arises: How sensitive to density anisotropy is the prediction of the hydrodynamic treatment? Unfortunately Kwon et al. only report the radial density cuts 2 (r), 3 (r), and 4 (r), but not the spherically averaged density, 0 (r), which is an essential ingredient of our approach. Nevertheless, the importance of this test grants, in our opinion, the extra approximations we’ll make to bypass this problem. Lacking an isotropic density consistent with the above PIMC density cuts, we replace it with that obtained from the data 共for 64 He atoms兲 in Figs. 2共a兲 and 2共b兲 of Ref. 8. The latter density is calculated at 0.625 K, as opposed to 0.3125 K, but we do not expect the difference to be substantial. Our opinion is based on the following facts: Figure 7 of Ref. 15 tells us that as the number C. Hydrodynamic calculations 1. Hydrodynamic equations Because the helium motion is assumed to be irrotational (“⫻v⫽0), the Eulerian velocity of the fluid in the laboratory frame, v, can be written as the gradient of a scalar func- TABLE III. Fitting parameters for the superfluid fraction of density II. See Table I for details. 0 2 3 4 L⫽0 L⫽4 L⫽6 L⫽8 A1 (Å⫺3) c1 共Å兲 0.0439 0.0408 0.0681 0.0306 4.401 4.321 4.143 4.698 0.0478 ⫺0.164 0.090 ⫺0.034 4.419 0.035 ⫺0.002 ⫺0.004 w⫺ 1 共Å兲 w⫹ 1 共Å兲 b⫺ 1 b⫹ 1 A2 (Å⫺3) c2 共Å兲 w⫺ 2 共Å兲 Parameters for fits to radial density and density cuts along C n axes 0.512 0.652 2.719 1.462 0.0162 7.339 1.083 0.372 0.651 1.964 1.628 0.0179 7.224 1.035 0.432 0.461 3.163 1.441 0.0204 7.264 0.866 0.488 0.720 2.758 1.645 0.0136 7.466 1.427 Cubic harmonic expansion coefficients for each line shape parameter 0.468 0.613 2.773 1.480 0.0162 7.367 1.125 0.070 0.093 0.015 0.010 ⫺0.126 0.010 0.135 0.035 ⫺0.050 0.093 ⫺0.020 0.014 0.003 ⫺0.007 ⫺0.056 0.008 ⫺0.054 0.039 0.036 ⫺0.005 ⫺0.010 w⫹ 2 共Å兲 b⫺ 2 1.278 1.277 1.111 1.403 2.029 1.857 2.197 1.779 1.450 1.459 1.403 1.622 1.261 0.054 ⫺0.018 0.004 2.046 ⫺0.029 0.026 ⫺0.033 1.439 0.029 0.001 0.020 b⫹ 2 Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp 1600 J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 K. K. Lehmann and C. Callegari tion, v⫽⫺“ , where is termed the velocity potential. In order to calculate we need to specify the motion of the SF6 , which we will assume to be a classical rotation with angular velocity around the z axis. Because of the octahedral symmetry of the helium solvation density around SF6 , the net rotational kinetic energy is independent of the axis about which rotation takes place. Imposing continuity gives the following equation: “• 共 “ 兲 ⫽ 冉 冊 t ⫽⫺ 共 “ 兲 • 共 ⫻r兲 , 共13兲 LF where the second equality comes from the assumption that the helium density remains time-independent in the molecule-fixed frame. LF means that the partial derivative is taken with respect to a point fixed in the laboratory reference frame; the corresponding partial with respect to a point fixed in the molecular frame is zero by construction. The boundary conditions on are that in the direction normal to the boundary the fluid velocity matches the velocity of the boundary, i.e., / n⫽“ •n̂⫽⫺( ⫻r)•n̂, with n̂ the unit vector normal to the boundary surface and pointing out of the fluid. The solution to Eq. 共13兲 is linear in and thus is calculated for unit . Equating the helium kinetic energy with a rotational kinetic energy defines ⌬I h : 冕 冋 冕 冉 冊 冕 1 1 ⌬I h 2 ⫽ m He 2 2 1 ⫽ m He ⫺ 2 兩 “ 兩 2 dV t dV⫹ 共14兲 册 共 “ 兲 •dS . 共15兲 Equation 14 holds for any velocity potential; Eq. 共15兲 has been derived from Eq. 共14兲 using vector identities and Eq. 共13兲. As such, the two estimates for ⌬I h need only be equal for a that is solution to Eq. 共13兲. It can also be shown that the net orbital angular momentum produced by the helium flow is, for the solution of Eq. 共13兲, JHe⫽⌬I h . Note that, consistent with common convention, dS in the second integral of Eq. 共15兲 is defined to point out of the fluid region. With our choice of coordinates 共SF6 centered at r⫽0 and oriented with its S–F bonds lying on the Cartesian axes兲 must be invariant to reflection in the xy plane, therefore its domain can be restricted to the interval 关 0, /2兴 and the condition / ⫽0 imposed in the plane ⫽ /2 共the xy plane兲. Reflection in each of the four symmetry planes containing the z axis is equivalent to inverting the direction of rotation, i.e., to a change in the sign of . Therefore the domain can be restricted to the interval 关 0, /4兴 and ⫽0 imposed in the planes ⫽0 and ⫽ /4. In most of our calculations, the inner and outer boundaries in the radial direction are spherical surfaces at r⫽r min and r⫽r max 共this condition can be relaxed, see Sec. II C 3兲. On both boundaries the condition / n⫽0 was imposed, which implies that no helium flux out of the domain of the solution was allowed. Since either or / n are zero on all the boundary surfaces, the surface integral in Eq. 共15兲 is zero. 2. Numeric solution The inhomogeneous partial differential equation, Eq. 共13兲 was solved numerically on a uniformly-spaced grid of points. The radial grid consists of up to 201 points; r min is always 3.5 Å, for r max we tried the values 10 and 15 Å. Grids with up to 161 points were used for each angular coordinate, in the restricted domains specified above. Equation 共13兲 was cast into the more convenient form, ⵜ 2 ⫹(“ ln )•(“⫹⫻r)⫽0, converted to a 7-point finite-difference equation in the chosen grid of points, and solved by Gauss-Seidel iteration with successive over-relaxation.22 This equation becomes singular when ⫽0, therefore we clip the density to a threshold value, min , i.e., all grid points where ⬍ min are assigned the value ⫽ min . Solutions were iterated until the rms change in on the grid points was less than a fixed fraction, ⑀ (⫽10⫺6 in this work兲 of the rms value of on the grid points. In the Gauss–Seidel method, the change in the value of each grid point is proportional to the error in the finite difference equation evaluated at that point, so this convergence criteria is equivalent to one based on the residuals in this equation. In a test calculation with the convergence criteria tightened to ⑀ ⫽10⫺10, the norm of was approximately 0.01% lower and ⌬I h approximately 0.04% higher than when ⑀ ⫽10⫺6 was used. After an initial transient, the convergence estimator, the norm of , and the predicted values of ⌬I h all appear to converge exponentially, approximately with the same rate, with the number of Gauss–Seidel iteration steps. For the largest grid used, with ⬇5.1 million points, a typical run required about 1100 iteration cycles 共several hours of computation time on a personal computer兲 with an overrelaxation parameter of 1.9 ( in the notation used in Ref. 22兲. We tested min values in the range 10⫺7 –10⫺5 Å , and found that the estimate for ⌬I h changed by ⬃1% 共with higher values of min giving lower values of ⌬I h ). Refining the grid from 101⫻81⫻81 to 201⫻161⫻161 points changed the estimate for ⌬I h by ⱗ0.5%. 3. Nonspherical boundaries The use of a rigid, spherical inner boundary and a lower bound on the density are mathematical artifices introduced to accelerate convergence with respect to grid size and GaussSeidel iteration. It is natural to ask whether these rather ad hoc choices significantly affect the final value of the kinetic energy. The velocity of the fluid, ⫺“ , need not approach zero in the region where the density goes to zero; rather, it will vary considerably upon small changes of the boundary conditions. While the effect of the choice of boundary condition will propagate into a region of higher density, by conservation of flux it should damp as the density gets larger. It is clear on physical and mathematical grounds that in the limit that min→0 and “ bounded, the kinetic energy of the solution will be independent of the exact boundary conditions. However, since the equation for depends only logarithmically on the density 关through the term “(ln )兴, convergence may be legitimately expected to be slow. At the urging of the referee, we have modified our code to include a Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 FIG. 3. The first order estimator procedure of Fox 共Ref. 23兲. In a normal 5-point finite-difference scheme, 0 ⫽g( 1 , 2 , 3 , 4 ), where g is the finite-difference operator for the equation being solved. If one or more points lie outside of the boundary 共here, left of the thick line兲, their values have to be estimated at the beginning of each Gauss–Seidel iteration by use of the boundary conditions and the values from the previous iteration. In the example shown, this is done by solving for 1 by using the set of equations: ( / n) B⫽( 1 ⫺ C)/k and C⫽ 0 ⫹( 2 ⫺ 0 )h, with k⫽C1 and h⫽0C/02. The procedure is easily generalized to three dimensions. The normal to the surface is along ⫺“(ln ), which is estimated by numerical derivatives of the function ln, which is tabulated on each grid point. curved inner boundary surface, defined as the loci of (r,⍀)⫽ min , and we did not clip the density to min at large r. To overcome the problem that the inner boundary no longer falls exactly on grid points, we use the first order estimator discussed by Fox23 共see Fig. 3兲, generalized from two to three dimensions. In general, programming this procedure involves treatment of many special cases, but in the present problem is simplified by the fact that at the inner boundary, “ remains nearly parallel to r̂, and thus the boundary always moves by less than one radial grid point for a unit change in the or grid. Calculations were performed for the ‘‘indirect expansion’’ of density I, using min values of 10⫺5 and 10⫺7 Å⫺3 . Also, r min was reduced to a value of 2.5 Å so that the surface defined by ⫽ min is contained inside the grid for all ⍀. The ⌬I h thus calculated differed by less than 1% from those calculated with a spherical inner boundary and with the density clipped to min in the outer region. We consider this to be a convincing demonstration that the kinetic energy is not significantly affected 共to be precise, no more than by the choice of grid size兲 by the use of a computationally easier boundary condition in a region where the density is very small. Moments of inertia in helium nanodroplets 1601 FIG. 4. Radial kinetic energy density, calculated at unit , for DMC and PIMC He densities. Plotted values includes the r 2 Jacobian factor. We repeated the calculation using the indirect expansion form of the DMC density 关Eq. 共11兲 and Table I兴. With r max ⫽10 Å, the resulting values for ⌬I h were 281.1–281.7 u•Å2 . At this radius, however, the He density is still significant. The calculation was repeated with r max⫽15 Å, at which point all the calculated densities are on the order of 10⫺5 Å⫺3 or less. This gave values of 316.4 –317.5 u•Å2 for ⌬I h . Figure 4 shows the radial kinetic energy density 共including the r 2 weighting兲. About 49% of the kinetic energy is carried by helium in the first solvation shell, while the rest by helium in the second solvation shell. Calculations were also performed using the indirect expansion form of the PIMC density 关Eq. 共11兲 and Table II兴. This PIMC density gave ⌬I h ⫽29.47– 29.56 u•Å2 , far below any value from the DMC densities. About 85% of the kinetic energy is carried by helium in the first solvation shell. Our results can be compared with a value of 31 u•Å2 for ⌬I h reported by Kwon et al., computed with the total PIMC helium density. We take the close agreement between our results and those of Kwon et al. 共when referred to the same densities兲 as support for the adequacy of our parameter-based interpolation to reproduce the full three dimensional helium density distribution. Kwon and Whaley8 also reported the ‘‘superfluid’’ densities along the same symmetry axes, as calculated using PIMC and their proposed superfluid estimator. Repeating the hydrodynamic calculation using the interpolating form of these densities gave ⌬I h ⫽ 14.58 –14.64 u•Å2 . This can be compared to the ‘‘superfluid’’ contribution to ⌬I h of 22 u•Å2 reported by Kwon et al.9 III. NUMERICAL RESULTS The first calculations were performed using the direct expansion into cubic harmonics of the DMC density. The two integral estimates for ⌬I h are found to be 171 and 168 u•Å2 , respectively. Convergence with respect to grid size, density cut off and convergence criteria appear to be within a few percent. Given the radial range of the data available for cubic harmonic expansion, the calculations were performed for r min⫽3.5 Å and r max⫽10 Å. The calculated ⌬I h are ⬇55% of the value of ⌬I exp⫽310⫾10 u•Å2 extracted from the observed effective rotational constant of SF6 in 4 He nanodroplets.4 IV. DISCUSSION Our results show that the calculated value of ⌬I h for SF6 in helium is extremely sensitive to the details of the helium density, particularly the angular anisotropy. Values of ⌬I h calculated using DMC densities span the range from 55% to 102% of ⌬I exp . A calculation using more recently reported PIMC densities gave a much lower ⌬I h of only 10% of ⌬I exp . This can be contrasted with the results reported in Ref. 10, where for heavier rotors, the theory appeared to systematically overestimate the size of ⌬I h . Similarly, the hydrodynamic approach was found to overestimate the in- Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp 1602 J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 creased moment of inertia when applied to the model problem of a planar rotor interacting with a rigid ring of He atoms.30 The values of ⌬I h calculated with the PIMC and DMC densities differ by an order of magnitude. This was quite surprising to us, based upon the qualitative similarity of the two densities. It is easily seen that, if the density is scaled by a uniform factor, will not change and ⌬I h will scale by the same factor. Qualitatively, one can compare the peak values for the PIMC and DMC densities. For the raw densities the peak lies along the C 3 and is about 20% higher for the PIMC density. For the angular average of the densities, the PIMC one still has a higher peak value, by about 25%. In all cases large differences are observed in the first solvation shell and much smaller ones in the second solvation shell. Both of these facts would, in themselves, suggest that the kinetic energy should be higher for the PIMC density. The PIMC based density does have a significantly reduced angular anisotropy compared to the DMC density. For example, the maximum value for 兩 d /d 兩 共which is the source term in the hydrodynamic equation兲 is 0.46 Å⫺3 for the DMC density and 0.28 Å⫺3 for the PIMC density. This would suggest that the kinetic energy of the PIMC density should be less than that of the DMC density, as is calculated. While these differences are significant, they are far below the resulting difference in kinetic energy. A principal result of this work is the demonstration of this high sensitivity, which was far higher than we had expected at the outset. Given the sensitivity of the results reported above to the density 共and thus to the angular anisotropy of the potential, which is poorly characterized experimentally兲 we do not believe it is possible at this time to reach any definitive conclusions about the accuracy of the hydrodynamic model for this particular molecule. Even given a fixed potential, there appears to be a substantial variation in the density calculated by the DMC and PIMC methods. While DMC calculations have ‘‘trial function bias,’’ the differences are much larger than the remaining bias as estimated by Barnett and Whaley.15 The PIMC calculations suffer possible bias based upon the use of the ‘‘primitive action’’ for the He–SF6 interaction potential.24 No results have yet been published that would allow the reader to assess the convergence of the published PIMC densities with respect to this bias, nor with respect to length of simulation run. It has been recently reported25 that for the case of a Na⫹ cation in helium, a similar PIMC calculation26 gave an even qualitatively incorrect radial helium density, which was blamed on metastable trapping of the PIMC simulations. If we take the reported DMC and PIMC results as accurate, we can ask which should be used in the hydrodynamic calculations. Since experiments are done at a finite temperature of 0.38 K, one might expect that PIMC calculations at 0.3125 K would be superior to the ground state 共i.e., zero temperature兲 DMC results. However, as discussed above, the hydrodynamic model can be derived from the variational minimization of a trial wave function 关Eq. 共1兲兴 that consists of the helium ground state wavefunction multiplied by a phase function written as a sum of one body terms. This ‘‘Feynman-type’’ of approximation is widely used in the K. K. Lehmann and C. Callegari theory of liquid helium, and should provide a rigorous bound on the zero temperature effective moment of inertia.27,28 We plan to present a detailed derivation of this statement in a later publication. The relevance to the present discussion is that the quantum theory we have for the use of the hydrodynamic approach is based upon excitations of the ground state of the system. As such, this approach is only theoretically consistent if we us as input the ground state helium density for the system, i.e., that which is directly estimated by DMC calculations. Of course, a PIMC density can be used as well if it is calculated at a temperature low enough that the density is sufficiently close to that of the ground state. Based upon the liquid drop model estimates of the excitation energies of liquid helium,29 this would be expected to be the case for calculations done at 0.3125 K, but if it is not, then the hydrodynamic calculations should be done with the ground state helium density. Of course, such a zero temperature theory cannot predict the temperature dependence of the effective rotational constants, but experiments on SF6 have shown that there is a negligible temperature dependence of this quantity over the range T⫽0.15–0.5 K. The fixed node, Diffusion Monte Carlo calculations of Lee et al.7 were in excellent agreement with the experimental rotational excitation energies. From the effective rotational constants reported in that work, ⌬I⫽290–305 u•Å2 can be calculated for clusters with N⫽8 –20 helium atoms. This suggest that, barring cancellation of errors, the nodal structure of the wave function assumed in that work 共which had the nodal properties of the rigid rotor wave function for the SF6 ) should be a reasonable description of the true many body wave function for this system. It is useful, therefore, to examine how consistent the hydrodynamic model is with the functional form assumed in that work. The many body wave function of Eq. 共1兲 gives the amplitude in the frame rotating with the SF6 . Quantization of the SF6 rotational motion in the adiabatic approximation results in a full wave function that is the product of the helium wave function, Eq. 共1兲, times the rigid rotor wave function for the SF6 . Since the helium ground state wave function is nodeless, this wave function will have, in the limit that the one-body phase terms m /បⰆ1, the same exact nodes as those assumed in the DMC calculation of Lee et al. The maximum value for for our solution is ⬃5 Å2 , and for the J⫽1 level ⬇4 B eff , where B eff is the effective moment of inertia of SF6 in helium, 1.04 GHz. This gives a maximum hydrodynamic phase of ⬇0.02 over the entire solution space, and thus the above inequality is everywhere satisfied. We would like to point out that for the model problem of a planar rotor coupled to a ring of helium atoms 共which can be solved exactly兲, the errors in the equivalent fixed node approximation was of this same size and yet the DMC estimate of the rotational excitation energy had errors of at most a few percent.30 We can conclude, in contrast with unsubstantiated claims made by Kwon et al.,9 that the fixed nodes assumed in the DMC calculation of Lee et al. are entirely consistent with the results of the present quantum hydrodynamic treatment. Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp J. Chem. Phys., Vol. 117, No. 4, 22 July 2002 ACKNOWLEDGMENTS The authors wish to acknowledge Professor Giacinto Scoles for many helpful discussions. This work was supported by the National Science Foundation and the Air Force Office of Scientific Research. J. P. Toennies and A. F. Vilesov, Annu. Rev. Phys. Chem. 49, 1 共1998兲. A special issue on helium nanodroplets was published by J. Chem. Phys. 115共22兲 共2001兲. 3 K. K. Lehmann and G. Scoles, Science 279, 2065 共1998兲. 4 M. Hartmann, R. E. Miller, J. P. Toennies, and A. F. Vilesov, Phys. Rev. Lett. 95, 1566 共1995兲. 5 C. Callegari, R. Schmied, K. K. Lehmann, and G. Scoles, in Ref. 2, p. 10090. 6 C. Callegari, A. Conjusteau, I. Reinhard, K. K. Lehmann, and G. Scoles, J. Chem. Phys. 113, 10535 共2000兲. 7 E. Lee, D. Farrelly, and K. B. Whaley, Phys. Rev. Lett. 83, 3812 共1999兲. 8 Y. Kwon and K. B. Whaley, Phys. Rev. Lett. 83, 4108 共1999兲. 9 Y. Kwon, P. Huang, M. V. Patel, D. Blume, and K. B. Whaley, J. Chem. Phys. 113, 6469 共2000兲. 10 C. Callegari, A. Conjusteau, I. Reinhard, K. K. Lehmann, G. Scoles, and F. Dalfovo, Phys. Rev. Lett. 83, 5058 共1999兲; 84, 1848 共2000兲. 11 We should point out that this level of agreement is far better than what should be expected given the considerable uncertainty in the anisotropy of the He–SF6 interaction potential used 共the two parameters that determine the anisotropy of the He–SF6 potential of Ref. 12 have reported values of ⫺0.6⫾0.3 and 0.14⫾0.14). To be conservative, one should therefore only compare theoretical values 共which have so far all been obtained with the same potential兲, although we take the agreement between models that use 1 2 Moments of inertia in helium nanodroplets 1603 rather different assumptions as an indication that the potential of Ref. 12 might be significantly more accurate than the error estimates would suggest. 12 R. T Pack, E. Piper, G. A. Pfeffer, and J. P. Toennies, J. Chem. Phys. 80, 4940 共1983兲. 13 A. Conjusteau, C. Callegari, I. Reinhard, K. K. Lehmann, and G. Scoles, J. Chem. Phys. 113, 4840 共2000兲. 14 M. Casas, F. Dalfovo, A. Lastri L Serra, and S. Stringari, Z. Phys. D: At., Mol. Clusters 35, 67 共1995兲. 15 R. N. Barnett and K. B. Whaley, J. Chem. Phys. 99, 9730 共1993兲. 16 F. C. von der Lage and H. A. Bethe, Phys. Rev. 71, 612 共1947兲. 17 K. Fox and I. Ozier, J. Chem. Phys. 52, 5044 共1970兲. 18 W. R. Fehlner, S. B. Nickerson, and S. H. Vosko, Solid State Commun. 19, 83 共1976兲. 19 W. R. Fehlner and S. H. Vosko, Can. J. Phys. 54, 2159 共1976兲. 20 F. Dalfovo, Z. Phys. D: At., Mol. Clusters 29, 61 共1994兲. 21 U. Buck, K. H. Kohl, A. Kohlhase, M. Faubel, and V. Staemmler, Mol. Phys. 55, 1255 共1985兲. 22 W. H. Press, B. P. Flannery, S. A. Teukolsky, and W. Vetterling, Numerical Recipes 共Cambridge University Press, Cambridge, 1986兲. 23 L. Fox, Numerical Solution of Ordinary and Partial Differential Equations 共Pergamon Press, New York, 1962兲, Chap. 21. 24 D. M. Ceperley, Rev. Mod. Phys. 67, 279 共1995兲. 25 M. Buzzacchi, D. E. Galli, and L. Reatto, Phys. Rev. B 64, 094512 共2001兲. 26 A. Nakayama and K. Yamashita, J. Chem. Phys. 112, 10966 共2000兲. 27 A. J. Leggett, Physica Fennica 8, 125 共1973兲. 28 K. Kim and W. F. Saam, Phys. Rev. B 48, 13735 共1993兲. 29 D. M. Brink and S. Stringari, Z. Phys. D: At., Mol. Clusters 15, 257 共1990兲. 30 K. K. Lehmann, J. Chem. Phys. 114, 4643 共2001兲. Downloaded 24 Jun 2003 to 128.112.71.198. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp