Multipole Models Fit to 6-31G** Electric Potentials
advertisement

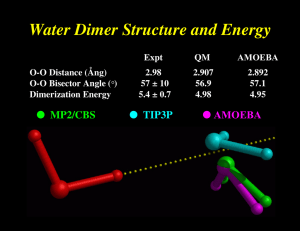
Multipole Models Fit to 6-31G** Electric Potentials Molecule Points Methane Water Ammonia Methanol Acetone Acetylene Formamide Me Acetate DiMe Amine NMA # Data 455 363 404 483 614 438 492 559 583 685 Relative RMS Error M only M+D M+D+Q 13.53 8.44 9.90 8.35 2.31 1.34 3.68 6.03 16.27 3.26 1.04 0.88 2.31 1.31 0.98 0.06 0.65 0.67 1.48 0.30 0.39 0.01 0.02 0.02 0.02 0.02 0.03 0.02 0.03 0.01 from D. E. Williams, J. Comput. Chem., 9, 745-763 (1988) Polytensor Formulation of Multipole Interactions Two Atoms A and B with Atom Centered Multipole Moments r A B Atom A t ∆ ∆ ∆ 3 1 () ● ● r 4 3 ∆ 2 ∆ = 2 2 ∆ Multipole Energy ∆ 1 ∆ Atom B ∆ ● Monopole (1 Component) ● Dipole (3 Components) ● Quadrupole (9 Components) ( n) i j k ∂ ∂ ∂ , i+j+k=n = ∂xi ∂yj ∂z k Dipole Polarizability All matter is polarized in direct proportion to the strength of an external field, where the proportionality constant is α , the polarizability: µinduced = α E (i.e., µ is linear, provided E is not too big!) Imagine a one-electron (e) atom with a radius of R placed in an electric field E. The electron’s orbit will be shifted away from the nucleus by a distance d. Then the induced dipole is given by: µinduced = αE = de At the equilibrium value of d , the external force on the electron due to the field must exactly counterbalance the internal force of displacement between the nucleus and the electron. These forces are: Fext = eE e2 sinθ , and Fint = 4πε0R2 e2d ≈ 4πε R3 0 = e 4πε0R3 µinduced Since Fext is equal to Fint , we obtain for the polarizability: α = 4π ε0 R3. Thus, neglecting the permittivity term, the polarizability should be roughly equal to the volume of the atom or molecule. For water, the experimental value of α = 1.48 Å3 suggests a radius of 1.14 Å, about 20% less than the standard water radius of 1.4 Å used in surface area calculations. The Importance of Polarization ● Inter-molecular polarization is necessary to describe gas-phase and condensed-phase properties within a single model ● Intra-molecular polarization is needed to treat the conformational dependence of electrostatics Water Dimer Structure and Energy O-O Distance (Ång) O-O Bisector Angle (°) Dimerization Energy ● MP2/CBS Expt QM AMOEBA 2.98 57 ± 10 5.4 ± 0.7 2.907 56.9 4.98 2.892 57.1 4.95 ● TIP3P ● AMOEBA π-Cation Interactions Catastrophic Failure of the Standard Model OPLS-AA CHARMM27 Amber ff94 Amber ff02 AMOEBA MP2/6-311+G(2d,2p) MP2/aVQZ CCSD(T)/CBS Expt (HPMS) Expt (CID) + ∆E0 K -Centroid -9.32 -11.06 -12.55 -15.87 -19.27 -18.4 -19.9 -20.6 2.90 2.81 2.74 2.63 2.81 2.81 2.79 2.79 ∆H298 -18.15 -20.1 -18.3 -17.7 Ion Selectivity by Benzene-Water + Na K + Molecular Dipole Polarizability E = [0.1, 0.1, 0.1] ● Based on Thole's modified dipole interaction model ● Isotropic atomic dipole polarizabilities are sufficient to reproduce experimental molecular polarizability tensors ● Induced dipoles further interactively induce each other within the molecule ● The field and interaction involved in induction are modified (damped) at short range Intermolecular Polarization for NMA # Dimer Monomer Monomer DMA/ QM Dipoles DMA DMA + Polarization ESP RRMS Total Dipole Dx Dy Dz 6.9% 7.88 7.73 0.09 1.51 16.6% 6.64 6.46 0.01 1.52 6.5% 7.83 7.69 0.03 1.48 ESP RRMS Total Dipole Dx Dy Dz 6.4% 8.85 -8.82 0.76 0.02 15.9% 7.49 -7.44 0.75 0.00 5.6% 8.85 -8.81 0.82 0.00 All calculations performed at MP2/6-311G++(2d,2p) interaction energy (kcal/mol) 12 H 2 - He 10 8 8 6 6 4 4 2 2 0 0 1.5 2.0 2.5 3.0 3.5 4.0 H 2 - Ne 1.5 2.0 2.5 3.0 3.5 4.0 distance (Å) distance (Å) rf = b / a a b 12 12 interaction energy (kcal/mol) Probing vdW Anisotropy 10 12 10 H 2 - He 10 8 8 6 6 4 4 2 2 0 0 H 2 - Ne 1.5 2.0 2.5 3.0 3.5 4.0 1.5 2.0 2.5 3.0 3.5 4.0 distance (Å) distance (Å) Small Molecule Database for Parametrization Neon Argon Krypton Graphite Dinitrogen Dioxygen Difluorine Methane Ethane Propane Butane Isobutane Pentane Cyclohexane Benzene Toluene/Xylenes Phenol/Cresols Ammonia Water Hydrogen Fluoride Hydrogen Sulfide Hydrogen Chloride Methanol Ethanol Isopropanol Methyl Amine Ethyl Amine n-Propyl Amine Dimethyl Amine Trimethyl Amine Formic Acid Acetic Acid Carboxylates Ureas Methyl Sulfide Ethyl Sulfide Dimethyl Sulfide Dimethyl Disulfide Formamide Acetamide N-Methyl Formamide Dimethyl Formamide N-Methyl Acetamide Dimethyl Acetamide Imidazoles Guanidine Indoles Tryptamine + various strained hydrocarbons, ions and monofunctionals Noble Gases and Homodiatomic Molecules H2 3.50 0.010 0.80 N2 3.72 0.076 1.04 O2 3.39 0.106 0.99 F2 3.22 0.109 0.96 Ne 3.15 0.073 Cl 2 3.925 0.340 0.935 Ar 3.82 0.260 Kr 4.09 0.359 T Ne Ar Kr Xe H2 N2 O2 F2 Cl 2 Density (g/cm3) (K) P (atm) expt 27.1 87.5 120.3 166.1 20.3 77.3 90.2 85.0 243.2 -4 -34 8 6 -3 -52 -5 -23 7 1.200 1.390 2.400 3.100 0.070 0.804 1.142 1.512 1.552 calc %error 1.203 1.394 2.405 3.100 0.069 0.830 1.145 1.511 1.564 0.3 0.3 0.2 0.0 -1.4 3.2 0.3 -0.1 0.8 H vap (kcal/mol) expt calc %error 0.420 1.554 2.161 3.018 0.218 1.332 1.628 1.554 4.857 0.421 1.554 2.163 3.018 0.216 1.346 1.617 1.565 4.851 0.2 0.0 0.1 0.0 -0.9 1.0 -0.6 0.7 0.1 Xe 4.37 0.498 AMOEBA Parameters for Water O-H Bond H-O-H Angle Urey-Bradley van der Waals O H Hreduction Polarizability O H b0 (Å) 0.9572 θ0 (deg) 108.50 l0 (Å) 1.5537 0 R (Å) 3.405 2.655 91% α (Å ) 0.837 0.496 3 Kb (kcal/Å2/mol) 529.6 Kθ (kcal/deg /mol) 34.05 Kl (kcal/Å /mol ) 38.25 2 2 ε (kcal/mol) 0.110 0.0135 O Multipoles Q dz Qxx Qyy Qzz (a.u.) -0.51966 0.14279 0.37928 -0.41809 0.03881 H Multipoles Q dx dy Qxx Qyy Qxz Qzz (a.u.) 0.25983 -0.03859 -0.05818 -0.03673 -0.10739 -0.00203 0.14412 Water Dimer Equilibrium Properties aAMOEBA De rO-O 4.96 2.892 4.18 57.2 2.02 2.54 α θ <µmol> µtot exp ab initio E tttt a 5.44 ± 0.7 f 2.976 f -1 ± 10 f 57 ± 10 b 4.98 a 2.907 a 4.2 a 56.9 5.02 b 2.912 b 5.5 b 55.6 e c 2.1 d 2.76 2.643 a f Based on calculations at CCSD(T)/TZ2P(f,d)+dif corrected for BSSE. Complete basis set estimate from correction of CCSD(T) calculations. c Derived from DMA calculation directly on water dimer minimum. d From MP2/TZ2P++ calculations. e Estimate after vibrational correction of experimental ∆H at 373 K. f Microwave spectra from molecular beam resonance experiments at 20 K. b H O α H O H θ H Liquid Water Properties 3.0 Soper ’00 Expt 2.0 AMOEBA Soper ’86 Heat Vaporization (kcal) 298K 10.51 10.48 Density (g/cc) 298K 0.997 323K 0.988 363K 0.962 1.0 0.0 AMOEBA 0 1 2 2.0 3 4 5 O - O Distance 6 7 8 1.000 0.992 0.964 Dielectric Constant 273K 87.7 86.8 298K 78.3 80.7 323K 69.9 66.5 -5 2 1.5 Diffusion (10 cm/s ) 298K 2.3 2.0 1.0 Cp (cal/mol K) 298K 18.0 0.5 0.0 20.9 / 27.6 Avg Mol Dipole (Debye) 2.78 2.6-3.0 0 1 2 3 4 5 O - H Distance 6 7 8 Epol / (Epol+Eperm) 30% Ammonia Monomer, Dimer and Liquid MONOMER Dipole Quadrupole Expt AMOEBA (unscaled) AMOEBA (60% Q) 1.471 1.528 1.528 -2.42, -2.45 -3.093 -2.491 DIMER ab Initio * AMOEBA Energy N..H 3.09 3.19 2.226 2.248 N..N 3.224 3.265 <HN..H 135 120 * aug-cc-pVQZ energy at 6-31+G* minimum LIQUID Hvap Pressure Expt AMOEBA 5.58 5.54 1 99 5 Dx10 T(K) 5.8 5.0 240 240 Methanol Dimer QM a # # AMOEBA CHARMM E 5.44 5.38 6.99 RO..O (Å) 2.87 2.91 2.80 a (deg) 44 44 23 b (deg) 179 174 178 (kcal/mol) MP2 Calculations from Mooij, et al., 1999 Self-Diffusion Coefficient (10 -5 cm2/s) 6 AMOEBA 5 Ammonia 4 3 NMA Methanol 2 Water 1 DMF 0 0 1 2 3 Expt 4 5 6 Heat of Vaporization (kcal/mol) 16 Alcohols Amines Amides Sulfides Aromatics 14 Expt 12 10 8 Pressure (NVT) 74±139 Atm 6 4 4 6 8 10 12 AMOEBA 14 16 Dielectric Constants: AMOEBA vs. Expt 100 AMOEBA Expt Dieletric Constant Formamide 80 Water 60 Acetamide 40 NH3 20 Methanol Ethanol 0 MeNH2 Formamide Dimer Energy Minima B C A E = -16.0 kcal/mol rms = 0.02 Å D E = -7.5 kcal/mol rms = 0.28 Å E = -10.3 kcal/mol rms = 0.04 Å E E = -7.3 kcal/mol rms = 0.03 Å E = -9.0 kcal/mol rms = 0.09 Å F E = -5.5 kcal/mol rms = 0.05 Å Formamide Dimer Association Energies A cyclic B side C D nonplan nonplan E side F head-tail MP2/6-31G** B3LYP/6-31G(d) -13.4 -13.4 -8.5 -8.3 -6.9 -6.7 -5.8 -6.0 -6.1 -6.7 -3.6 -3.2 MP2/aug-cc-pVTZ -16.1 -10.6 -8.2 -7.2 -6.9 -5.4 AMOEBA RMS -16.0 0.02 -10.3 0.04 -9.0 0.09 -7.5 0.28 -7.3 0.03 -5.5 0.05 OPLS-AA RMS -14.2 0.06 -7.8 0.24 -8.2 0.82 -8.2 1.03 -8.0 0.63 -2.7 0.16 AMBER RMS -16.8 0.06 -9.5 0.09 -9.6 0.22 -9.0 1.03 -8.9 0.67 -3.8 0.12 CHARMM RMS -13.0 0.05 -8.2 0.13 -8.0 0.21 -7.7 1.06 -7.6 0.74 -4.3 0.10 MM3 RMS -12.0 0.06 -6.5 0.24 -6.8 0.38 -6.8 1.37 -6.4 0.24 -1.5 0.25 Comparison of DMF Dimers: AMOEBA vs Dixon/Hay A C E = -4.97 kcal/mol rms = 0.08Å E = -7.80 kcal/mol rms = 0.24Å B D E = -5.37 kcal/mol rms = 0.09Å E = -8.79 kcal/mol rms = 0.15Å Dimethylformamide Dimer Structure and Energy A B C D BSSE Corrected -6.61 -4.07 -6.95 -5.35 -5.51 -3.31 -5.82 -4.14 -10.32 -5.86 -11.41 -8.34 -10.98 -6.36 -12.11 -8.90 AMOEBA (single) AMOEBA (opt) RMS -4.94 -4.97 0.08 -5.03 -5.37 0.09 -7.37 -7.80 0.24 -8.60 -8.79 0.15 OPLS-AA (single) OPLS-AA (opt) RMS -1.68 -3.45 0.25 -0.60 -3.54 0.64 -3.41 -5.42 0.41 -2.48 -5.18 0.69 MP2/DZP+diffuse # BSSE Corrected MP2/aug-cc-pVTZ # # QM Results from Vargas, et al., JACS, 122, 4750-4755 (2000) Parameterization for Polypeptides ● vdW parameters and atomic polarizabilities transfered from small molecules ● Atomic multipole parameters -> from small molecule fragments (?) -> from capped amino acids (?) conformational dependence via intramolecular polarization ● Torsional parameters obtained by fitting to conformational energy surfaces MP2/6-311+G(2d,2p) LMP2/cc-pVTZ(-f) 180 120 Psi 60 0 -60 -120 -180 -180 -120 -60 0 60 120 180 -180 -120 -60 1 2 3 60 Phi Phi 4 5 6 7 0 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 120 180 AMBER ff99 CHARMM27 180 180 120 120 3.2 2.8 2.4 2 1.6 1.2 0.8 0.4 60 0 -60 -120 -180 -180 -120 -60 0 60 120 60 0 -60 -120 -180 -180 180 -120 -60 0 60 120 OPLS-AA 180 120 Free Energy Surfaces 60 Psi Solvated Alanine Dipeptide 0 -60 -120 -180 -180 -120 -60 0 Phi 60 120 180 180 Torsional Energy Functional Forms ● Fourier series Etors = k1 [1+cos(f)] + k2 [1-cos(2f)] + k3 [1+cos(3f)] + .... • Bicubic spline Input: ●❷z ● ● ● Output: ● ● ● ¶z/¶x ¶z/¶y ¶z2/¶x¶y z(xi ,yi) Smooth first derivative Continuous second derivatives Comparison of QM/MM, PDB and AMOEBA Results AMOEBA QM/MM vs. PDB (Fixed Charge Water) -180 QM/MM, Jan Hermans, UNC PDB, Jane Richardson, Duke -120 -60 3.6 3.2 2.8 0 2.4 2 1.6 60 120 1.2 0.8 0.4 180 Conformational Populations Alpha Pass Beta Other Amber ff94 Amber ff99 CHARMM27 OPLS-AA OPLS-AA/L 68 77 46 13 23 5 10 2 9 8 26 13 52 75 65 1 1 0 3 4 SCCDFTB (Amber) SCCDFTB (CHARMM) SCCDFTB (CEDAR) 27 33 27 16 14 12 48 48 61 9 4 0 AMOEBA (Polar Water) AMOEBA (Fixed Water) 29 32 16 13 54 54 1 1 Valine Sidechain Energetics 12 LMP2/cc-pVTZ(-f) Energy (kcal/mol) 10 φ=-78 ψ=-32 AMOEBA 8 6 4 2 φ=-86 ψ=89 0 0 50 100 150 200 χ (degrees) 250 300 350 cis-N-Methylacetamide vs β-Sheet Model H CH3 CH3 C O N N CH3 CH3 C CH3 -20.5 -16.2 -18.7 -17.3 -11.3 -11.6 -11.5 CH3 N O cis-NMA MP2/(CEP)4-31G+(2d) BP/DZVP (BSSE) SIBFA TINKER AMBER94 CHARMM27 OPLS-AA O H O CH3 H O H H # N CH3 N O CH3 N CH3 H β -Sheet ∆E -17.5 -8.4 -17.1 -11.5 -14.8 -16.9 -16.9 +3.0 +7.8 +1.6 +5.8 -3.5 -5.3 -5.4 QM and SIBFA data from Gresh, et al., JACS, 121, 7885-7894 (1999)