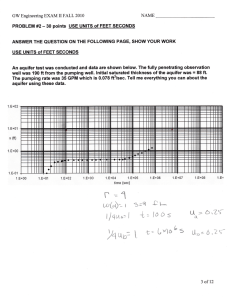
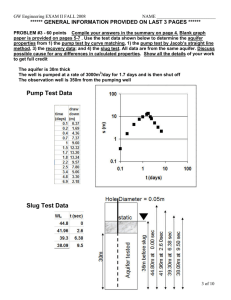
Natural organic matter in sedimentary basins and its
advertisement

Applied Geochemistry Applied Geochemistry 19 (2004) 1255–1293 www.elsevier.com/locate/apgeochem Natural organic matter in sedimentary basins and its relation to arsenic in anoxic ground water: the example of West Bengal and its worldwide implications J.M. McArthura,*, D.M. Banerjeeb, K.A. Hudson-Edwardsc, R. Mishrab, R. Purohitb, P. Ravenscroftd, A. Cronine, R.J. Howartha, A. Chatterjeef, T. Talukderf, D. Lowryg, S. Houghtona, D.K. Chadhah a Department of Earth Sciences, UCL, Gower St, London WC1E 6BT, UK Department of Geology, University of Delhi, Chattra Marg, Delhi 110007, India c School of Earth Science, Birkbeck, University of London, Malet St., London WC1E 7HX, UK d Arcadis Geraghty & Miller International Inc., 2 Craven Court, Newmarket CB8 7FA, UK e Robens Centre for Public and Environmental Health, University of Surrey, Guildford GU2 7XH, UK f Central Ground Water Authority, Bidhan Nagar, Salt Lake City, Kolkata 700091, India g Department of Geology, Royal Holloway University of London, Egham, Surrey TW20 0EX, UK h Central Ground Water Authority, 18/11 Jamnagar House, Mansingh Road, Delhi 110011, India b Received 16 September 2002; accepted 16 February 2004 Editorial handling is by Fuge Abstract In order to investigate the mechanism of As release to anoxic ground water in alluvial aquifers, the authors sampled ground waters from 3 piezometer nests, 79 shallow (<45 m) wells, and 6 deep (>80 m) wells, in an area 750 m by 450 m, just north of Barasat, near Kolkata (Calcutta), in southern West Bengal. High concentrations of As (200–1180 lg L1 ) are accompanied by high concentrations of Fe (3–13.7 mg L1 ) and PO4 (1–6.5 mg L1 ). Ground water that is rich in Mn (1–5.3 mg L1 ) contains <50 lg L1 of As. The composition of shallow ground water varies at the 100-m scale laterally and the metre-scale vertically, with vertical gradients in As concentration reaching 200 lg L1 m1 . The As is supplied by reductive dissolution of FeOOH and release of the sorbed As to solution. The process is driven by natural organic matter in peaty strata both within the aquifer sands and in the overlying confining unit. In well waters, thermotolerant coliforms, a proxy for faecal contamination, are not present in high numbers (<10 cfu/100 ml in 85% of wells) showing that faecally-derived organic matter does not enter the aquifer, does not drive reduction of FeOOH, and so does not release As to ground water. Arsenic concentrations are high (50 lg L1 ) where reduction of FeOOH is complete and its entire load of sorbed As is released to solution, at which point the aquifer sediments become grey in colour as FeOOH vanishes. Where reduction is incomplete, the sediments are brown in colour and resorption of As to residual FeOOH keeps As concentrations below 10 lg L1 in the presence of dissolved Fe. Sorbed As released by reduction of Mn oxides does not increase As in ground water because the As resorbs to FeOOH. High concentrations of As are common in alluvial aquifers of the Bengal Basin arise because Himalayan erosion supplies immature sediments, with low surface-loadings * Corresponding author. Fax: +44-20-7679-4166. E-mail address: j.mcarthur@ucl.ac.uk (J.M. McArthur). 0883-2927/$ - see front matter Ó 2004 Published by Elsevier Ltd. doi:10.1016/j.apgeochem.2004.02.001 1256 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 of FeOOH on mineral grains, to a depositional environment that is rich in organic mater so that complete reduction of FeOOH is common. Ó 2004 Published by Elsevier Ltd. 1. Introduction In the Bengal Basin, some 25% of wells that tap ground waters from alluvial aquifers contain high (>50 lg L1 ) concentrations of As, the consumption of which threatens the health of millions of people (e.g. Chatterjee et al., 1995; Chakraborti et al., 2002): by, for example, inducing cancers that may prove fatal (Smith et al., 2000; Yu et al., 2003). One model for the release of As to ground water, viz. reduction of sedimentary Fe oxides (FeOOH without mineralogical connotations hereinafter) and release of the sorbed As, was outlined by Gulens et al. (1979), adopted by Matisoff et al. (1982), and systematised by Korte and Fernando (1991). The process is driven by microbial reduction of organic matter (Nealson, 1997; Lovley, 1997; Banfield et al., 1998; Cummings et al., 1999; Chapelle, 2000; Lovely and Anderson, 2000). This mechanism was used by Nickson et al. (1998, 2000) to explain the occurrence of high concentrations of As in aquifers in the Bengal Basin: it was further developed by McArthur et al. (2001) and Ravenscroft et al. (2001), who proposed that organic carbon in buried peat was the driver of microbial reduction of FeOOH in the sediments, and that human organic waste in latrines might also locally contribute to As release by providing point-sources of organic matter to enhance local reduction of FeOOH (thereby confusing regional trends of As distribution by adding local signals). These authors also discussed, and dismissed, other mechanisms proposed to explain the release of As to ground water, such as oxidation of sedimentary pyrite, and competitive anion exchange of sorbed As with inorganic PO4 from fertiliser; they thereby broke the putative link between the development of irrigation with ground water and the presence of As poison in ground water. Although the Fe-reduction model is now widely accepted as valid for the Bengal Basin (Bandyopadhyay, 2002; Dowling et al., 2002; Harvey et al., 2002; Anawar et al., 2003; Tareq et al., 2003), the suggestion of McArthur et al. (2001) and Ravenscroft et al. (2001) that reduction is driven by buried peat has been disputed. For example, Pal et al. (2002) found peat in an As-free area, but did not find it in an As-affected area nearby. Harvey et al. (2002) suggest that reduction of FeOOH is driven by surface organic matter, from river beds, ponds, and soils, that is drawn into the aquifer by irrigation drawdown: this model seeks to re-link irrigation and high concentrations of As in ground water. Should such a linking lead to renewed calls to curb or ban irrigation with ground water, the livelihoods of the rural poor will be affected adversely. To better define the source of the organic matter driving reduction of FeOOH in the Bengal Basin and to further examine the behaviour of As and Fe in ground water, the authors installed 3 piezometer nests in a shallow (<45 m depth) aquifer near Barasat, in West Bengal (Fig. 1); measured vertical profiles of water composition in them; and took cores for study from the piezometer sites. One piezometer was sited where As poisoning of wells did not occur (site DP), one was where it was thought to be minor (site AP), and one where it was severe (site FP): the sites are less than 350 m apart (Fig. 1). Wells tapping the shallow (<45 m) and deep (>70 m) aquifers in the area were also sampled in order to ascertain the lateral extent, and degree, of changes in water quality, including As pollution. The content of thermotolerant coliform (TTC) bacteria in wells and surface ponds was measured in order to assess their content of faecally derived micro-organisms, the presence of which indicates the influence of human and animal organic waste. In order to investigate whether the wells changed their composition with time, they were analysed (TTC apart) on 3 occasions in 2 years. The results of these studies are presented here. 2. Location of area The study area is located at 22° 44.430 N, 88° 29.450 E (Indian/Bangladesh datum) in the contiguous villages of Joypur, Ardivok and Moyna (JAM hereinafter; Fig. 1) in southern West Bengal. The area is just north of the town of Barasat, Barasat-1 Block, 24 Parganas (North) District, some 20 km NNE of Calcutta. The climate of the area is tropical monsoonal with a dry season lasting from December to May. The Hugli River flows from north to south about 13 km to the west of the area, and the Sunti River (dry in most months) runs north-south 2 km to the east, joining the east-west draining Sunti Channel (also dry most months) at Mirathi, about 4 km to the north of the area. The area looks flat but has a regional slope to the north and east; i.e. towards the rivers. Superimposed on this is a microtopography that J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1257 Fig. 1. Location of wells and piezometers in the study area on the northern outskirts of Barasat town, West Bengal. Waved areas are ponds and rectangles represent some of the larger houses. Piezometer sites are double circles at AP, FP, DP. Arrowpoint in (b) shows field area. Roads in C are double lines, prominent tracks are single lines. Unfilled circles are deep wells (>70 m depth), filled circles are shallow wells (<45 m depth). N-34 is national highway 34. influences local hydrology: relative relief of 2–3 m exists where major roads are raised above ground level, and where ponds, perched above the local water table by several metres, have dry-season water levels up to 3 m below ground level. These numerous ponds range in size from <10 m diameter to around 100 m diameter, and are excavated to a depth that preserves natural sealing by the lower part of an aquitard that overlies the shallow aquifer. 3. Methods Piezometers consist of multiple, independent, wells drilled to different depths and spaced no more than 2 m apart. They were drilled using the reverse-circulation, hand-percussion, method (Ali, 2003). Using this method, mud and clay samples are recovered with undisturbed cores. In sands, the samples are disturbed. Separately at site AP (Fig. 1), percussive piston-sampling into plastic core-tubes was alternated with rotary wash-boring. The coring gave good recovery from precisely known depths in all strata. Cores were stored at room temperature until sampled for analysis, as the local groundwater temperature is 25 °C. A LECO C/S 125 Analyser was used to analyse the <0.5 mm fraction of sediments for total S (TS), total C (TC), and total organic C (TOC), the last after leaching samples with 10% v/v HCl. The difference between TC and TOC is reported as a percentage of calcite. To obtain values of d13 C for calcite, samples were analysed with a PrismÓ mass spectrometer using a common acid bath and NBS 19, a calcium carbonate reference material supplied by the National Bureau of Standards (now National Institute of Standards Technology). Ground waters were sampled, after purging, in February, 2002, November, 2002, and March 2003, from hand-pumped domestic and public wells. The depth of each well was recorded at the time of sampling by asking the owner how many pipes were used to install it: commonly, individuals buy 20-foot (6.1 m) lengths of pipe and employ a driller to install them. The depths of some wells are uncertain because of a change of owner, or some other circumstance. Water samples drawn from wells were not filtered: an accurate measurement was sought of the amount of As in water consumed by wellusers, who do not filter their water. All samples were crystal clear, i.e. without visible turbidity. Piezometers fitted with 1.8 m screens were installed in March 2003 in eastern JAM where As poisoning was absent (DP); in south-eastern JAM where it was believed to be minor (AP); and in northwestern JAM where it was severe (FP, 1258 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Fig. 1). The level above ground of each well in each piezometer nest, relative to an arbitrary level, was established by theodolite survey. Water levels were measured in order to establish hydraulic heads and the direction of ground-water flow. After extensive purging, piezometers were sampled via hand-pumps between 5 and 10 days after installation. Piezometer samples were filtered through 0.45 lm membrane filters. All samples for cation analysis were acidified to pH 2 with HNO3 . Samples taken for anion analysis and for stable isotope analysis were not acidified. Ammonium and NO 3 were determined within 2 weeks of collection, and mostly much sooner. Measurements of conductivity, and temperature were made at the well head. Measurements of dissolved O2 are not given as they are devalued by pump aeration. For piezometers and some wells, alkalinity was measured in the field by pH-titration with H2 SO4 and is reported as HCO3 : for wells where this was not done, HCO3 has been estimated by difference of charge balance. No wellsample contained H2 S that was detectable by smell after acidification of the sample with H2 SO4 and agitation. Analysis of waters for cations, total-S (reported as SO4 ) and total-P (reported as PO4 ) was by ICP-AES and analysis for anions and NH4 was by ion chromatography (including duplicate analysis for SO4 ). As a check on ICP determinations of P, PO4 was also determined spectophotometrically using the Mo-blue method. Concentrations of As were measured on acidified samples using graphite-furnace AAS. In order to assess particulate contributions to the dissolved load of well water, the concentration of Al in waters was measured and the turbidity of samples was recorded at collection. All samples were crystal clear, i.e. without visible turbidity, and concentrations of Al were <0.1 mg L1 , so it was assumed that the analysis is unaffected by particulate contributions. Measurement of d13 C of dissolved inorganic C (DIC) was done with a Micromass MultiflowÓ preparation system on-line to an IsoPrimeÓ mass spectrometer. After He flushing of samples, and addition of H3 PO4 , the isotopic composition of CO2 was measured after equilibration for 9 h at 30 °C. Drift corrections were made using a reference-gas pulse that bracketed each sample. Reference materials used were an in-house carbonate and NBS-19. Every third sample was accompanied by a standard, which was used to make corrections for drift, which was assumed to be linear; corrections were 6 0.4‰. Replicate analysis showed that repeatability was 6 ±0.1‰ for solid reference materials and 6 ±0.2‰ for solutions made from them. The average repeatability of the analysis was 0.06‰ for d13 C (n ¼ 134). Microbiological analysis for thermotolerant coliform bacteria was conducted in the field. The water was analysed, after purging, from 40 wells and 10 surface ponds in JAM, and 14 wells in the As-free area of Mirathi, some 4 km to the north of JAM. The isolation and enumeration of the TTC was carried out using membrane filtration and growth on membranes with lauryl SO4 broth (Oxoid, UK; Anon, 1994). The plates were incubated for 6 4 h at ambient temperature to aid bacterial resuscitation, before incubation at 44 0.5 °C for between 16 and 18 h. Yellow colonies were classified and counted as thermotolerant coliforms. Sanitary risks were quantified for each well in order to identify faecal sources, contaminant pathways and other factors that might contribute to contamination of wells (e.g. Lloyd and Bartram, 1991). Risk factors comprise observable faults in well construction and the proximity of sources of contamination, e.g. faecal material, and the distance between wells and latrines. 4. Results 4.1. Sedimentology A full description of the sediments and their composition will be presented elsewhere (Hudson-Edwards et al., 2004), so only an outline is given here (Table 1). Lithological logs of cored sediment were made on site and are reproduced in Fig. 2 without change. An upper aquitard of silts and silty clays (Fig. 2), of variable thickness (9–24 m), overlies an unconsolidated sand aquifer between 32 and 36 m in thickness (the shallow aquifer): the aquifer is underlain by a clay aquitard about 25 m thick, beneath which lies another sand aquifer (the deep aquifer) that is >60 m thick. The sediments were deposited between 22 and 1.2 ka ago (14 C and OSL ages from Hudson-Edwards et al., 2004) by the Ganges/Brahamaputra Rivers and their distributary channels. Dating of quartz in cores from the AP site by the OSL method showed that the aquifer sands are around 17 ka old below 32 m depth and <8 ka old above it (Hudson-Edwards et al., 2004), which explains why the lower part of the aquifer is twice as hard to core as the shallower levels, when judged by the number of hammer blows required to penetrate 0.5 m of sediment (around 90 below 32 m, around 50 above 32 m; McArthur et al., unpublished data). Correlations between the piezometer sites are shown on Fig. 2 and are based on the abundance of calcite and organic matter (TOC) in the sediments. The aquifer at site DP, and the lower 13 m of the aquifer at sites AP and FP, contains little or no calcite and almost no organic matter (<0.1%) and are therefore regarded as being of equivalent age (around 17 ka years). In the upper aquitard, the calcite-free level around 6–8 m depth in both AP and DP is assumed to correlate with the calcite-free level between 5 and 5.5 m in DP. Finally, in AP and FP, the upper few decimetres of the lower aquitard are brown, oxidised, and correlate (drilling at DP did not reach the lower aquitard). Table 1 Composition of sediment from piezometer sites in Joypur, Ardivok and Moyna (JAM), West Bengal, India Site DP Site AP Site FP TS % TOC % m bgl IC % as Cal TS % TOC % m bgl 1.5 3.0 4.6 5.2 6.1 7.6 9.1 10.7 12.2 13.7 14.3 15.2 15.2 15.2 16.8 16.8 18.3 19.8 19.8 21.3 21.3 22.9 24.4 25.9 27.4 29.0 30.5 32.0 33.5 35.1 36.6 38.1 39.6 41.1 42.7 44.2 45.7 47.2 48.8 50.3 51.8 57.9 59.4 3.17 5.67 9.50 0.08 < 0.05 26.9 2.00 1.17 1.58 2.25 1.17 1.25 0.25 < 0.05 0.67 5.00 0.67 6.50 7.75 0.08 0.33 0.08 < 0.05 0.17 0.08 0.08 0.17 < 0.05 < 0.05 0.08 0.08 < 0.05 0.08 < 0.05 < 0.05 < 0.05 < 0.05 0.50 0.83 < 0.05 0.08 2.67 0.08 0.425 0.027 0.014 0.052 0.023 0.001 0.023 0.044 0.051 0.430 0.122 0.221 1.27 2.24 0.036 0.332 0.010 0.006 0.000 0.000 0.001 0.007 0.007 0.007 0.012 0.007 0.006 0.005 0.006 0.007 0.005 0.008 0.007 0.008 0.008 0.006 0.005 0.009 0.007 0.006 0.007 0.011 0.006 1.46 0.66 0.36 4.87 2.32 0.27 0.38 2.25 0.56 1.51 1.52 4.52 7.61 13.3 1.90 33.7 0.52 0.29 0.20 0.16 0.13 0.28 0.10 0.07 0.10 0.09 0.08 0.09 0.09 0.11 0.07 0.10 0.07 0.08 0.08 0.07 0.05 0.11 0.06 0.11 0.08 0.11 0.10 2.10 2.50 2.70 3.00 3.05 3.05 3.20 3.50 3.64 3.80 4.00 4.50 4.57 4.57 4.80 5.03 5.20 5.50 5.57 5.70 5.86 5.92 6.00 6.00 6.03 6.10 6.10 6.25 6.50 6.50 6.75 7.00 7.62 7.62 7.75 8.00 8.50 8.77 9.14 9.14 9.14 9.95 11.35 12.40 13.25 3.83 3.83 3.08 3.00 0.33 3.17 1.67 5.00 3.58 5.83 5.67 6.83 0.08 7.33 5.50 0.08 5.67 4.25 6.58 6.08 0.25 3.33 0.25 0.42 0.25 0.33 0.42 0.75 0.25 1.50 0.67 0.58 0.42 0.42 1.25 10.3 1.58 4.67 0.17 0.75 5.17 3.75 4.42 4.00 3.83 0.009 0.006 0.006 0.005 0.014 0.028 0.003 0.007 0.005 0.007 0.008 0.009 0.041 0.020 0.008 0.016 0.012 0.021 0.007 0.011 0.010 0.009 0.029 0.119 0.015 0.018 0.039 0.014 0.010 0.015 0.020 0.040 0.016 0.033 0.012 0.010 0.028 0.054 0.023 0.065 0.154 0.013 0.014 0.011 0.009 0.44 0.24 0.39 0.27 0.33 0.44 0.36 0.31 0.35 0.28 0.35 0.35 0.78 0.15 0.38 1.12 0.32 0.39 0.39 0.39 0.78 0.67 0.61 0.72 0.60 0.70 1.58 0.37 0.35 0.25 0.53 0.44 0.25 0.33 0.45 0.21 0.35 0.24 0.33 0.78 0.65 0.28 0.22 0.40 0.25 0.5 1.4 1.5 1.5 2.3 3.0 3.0 3.2 4.6 5.0 5.9 6.1 6.1 7.4 7.6 7.6 7.8 9.1 9.1 9.1 9.5 10.7 11.3 12.2 12.2 13.3 14.2 14.3 17.0 18.8 20.8 22.6 24.4 26.1 27.9 28.8 29.5 30.5 31.6 33.6 35.3 37.0 39.0 40.8 42.5 IC % as Cal 4.98 5.49 2.25 4.33 5.46 1.75 4.92 6.52 6.33 7.00 5.34 1.08 1.75 0.58 0.08 0.00 5.52 5.67 3.83 6.17 4.99 5.33 5.57 3.67 3.83 4.75 5.73 5.31 4.91 4.81 3.18 3.78 3.85 3.57 3.45 2.02 2.71 1.87 0.87 0.65 0.73 0.79 0.79 0.60 1.08 TS % TOC % d13 C OM d13 C IC 0.04 0.00 0.00 0.00 0.02 0.00 0.01 0.04 0.02 0.06 0.01 0.00 0.01 0.00 0.04 0.08 0.01 0.06 0.01 0.03 0.00 0.05 0.00 0.01 0.01 0.00 0.01 0.01 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.01 0.62 0.04 0.00 0.00 0.00 0.01 0.02 0.01 0.01 0.53 0.24 0.49 0.40 0.45 0.42 0.40 0.53 0.56 0.44 0.66 0.86 0.79 0.89 2.92 2.63 0.20 0.62 0.58 0.42 0.72 0.58 0.35 0.60 0.78 0.17 0.17 0.17 0.18 0.33 0.14 0.13 0.14 0.17 0.23 0.10 7.20 0.17 0.18 0.12 0.10 0.09 0.08 0.09 0.08 )22.3 )21.2 )1.91 )1.61 )20.9 )0.69 )0.88 )22.0 )2.03 )21.4 )1.62 )20.2 )1.56 )23.6 )28.5 )25.0 )1.37 )9.77 )2.27 )1.80 )3.93 )2.55 )3.70 )2.99 )5.23 )4.57 )24.7 )24.3 1259 IC % as Cal J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 m bgl Site DP m bgl Site AP IC % as Cal TS % TOC % 1260 Table 1 (continued) Site FP IC % as Cal TS % TOC % m bgl IC % as Cal TS % TOC % d13 C OM d13 C IC 13.50 13.50 14.25 14.50 15.50 15.51 16.20 16.50 16.50 17.30 17.50 18.50 18.50 19.51 20.30 20.50 21.15 21.45 21.49 22.25 22.35 22.45 22.50 23.30 23.50 23.50 24.51 25.50 26.00 26.50 27.00 27.49 28.00 28.50 28.96 29.50 29.50 30.00 30.50 31.00 32.50 39.50 43.50 44.50 45.11 45.42 4.25 4.42 2.92 4.67 3.42 3.42 6.25 6.83 6.08 6.25 6.08 5.00 5.17 5.17 4.17 4.83 5.25 4.75 5.50 5.42 5.75 3.25 5.33 5.58 4.67 5.25 4.92 4.92 4.25 4.00 3.08 2.42 2.25 1.92 1.25 0.50 1.92 0.75 0.50 0.42 0.08 0.75 2.17 10.3 2.67 7.58 0.005 0.016 0.010 0.009 0.011 0.007 0.015 0.018 0.018 0.012 0.014 0.008 0.006 0.005 0.009 0.006 0.013 0.009 0.009 0.018 0.014 0.020 0.009 0.008 0.015 0.006 0.007 0.009 0.006 0.002 0.005 0.005 0.004 0.004 0.002 0.006 0.002 0.004 0.003 0.002 0.005 0.005 0.005 0.005 0.002 1.26 0.16 0.15 0.35 0.16 0.22 0.17 0.49 0.30 0.48 0.20 0.21 0.24 0.13 0.13 0.32 0.16 0.42 0.52 0.28 0.52 0.31 0.76 0.41 0.17 0.49 0.30 0.18 0.34 0.17 0.18 0.14 0.14 0.14 0.12 0.10 0.24 0.08 0.11 0.09 0.10 0.09 0.11 0.11 0.21 0.42 1.43 44.4 46.4 47.1 48.0 48.3 48.4 48.5 48.6 48.6 48.7 49.0 50.0 51.9 53.7 55.5 57.2 59.0 60.8 62.6 64.3 66.1 67.8 69.6 2.09 2.31 2.16 9.84 6.58 8.58 6.50 9.08 9.50 10.0 8.87 9.79 6.24 8.49 9.02 8.16 8.84 9.53 10.7 10.5 10.4 10.0 9.70 0.01 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.01 0.03 0.32 0.28 0.23 0.02 1.21 1.33 1.09 0.50 0.31 0.22 0.25 0.45 0.28 0.06 0.18 0.16 0.09 0.24 0.30 0.28 0.34 0.41 0.68 0.83 0.72 0.49 0.42 1.57 1.48 1.30 0.69 0.68 0.53 0.50 0.66 0.83 )23.5 )3.70 )2.63 )3.08 )4.13 )24.9 )20.7 )0.61 )22.9 )2.47 )24.9 )0.83 )0.40 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 m bgl J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 At site DP, in the east of the JAM area (Fig. 1), the upper confining layer is 24 m thick, with the lower half comprising impermeable clay (17–24 m). A calcite-free interval occurs around 5 m depth. Above 17 m depth, the confining layer contains abundant organic matter, especially at depths of around 5, 15 and 17 m: the deepest occurrence comprises peat sensu stricto with a concentration of total organic C (TOC) of 34% (Table 1); the peat contained well-preserved tree-leaves. Sulphur concentrations in the OM-rich units reach 2.2% at 15.2 m depth, but is only 0.33% at 17 m depth. Calcrete occurs at 19.8 m. Around 21 m depth, the clay is brown and oxidised and shows in Fig. 2 as a minimum concentration of total sulfur: the oxidised unit passes gradationally downward into the underlying unoxidised grey clay. There is a sharp transition at 24 m into the underlying aquifer sands, which are brown in colour through the full thickness of the aquifer. The mean concentration of TOC in the aquifer sands is 0.09%, with no sample containing more than 0.11%, and the concentration of total S in the aquifer sand does not exceed 0.012%. At the greatest depth reached (59.4 m), cuttings comprised brown sand mixed with grey silty clay, suggesting that the lower confining clay was nearby. However, the poor sample quality at this depth made it impossible to discover whether the underlying aquitard had an oxidised upper surface as is the case at the other piezometer sites. At AP, in southern JAM, the upper confining layer consists of silts and clayey silts. An interval free of calcite occurs between 6 and 8 m depth. Around 9 m depth, the silts grade downwards into fine sand in the upper part of the aquifer (Fig. 2). The aquifer sands are grey in colour above depths of 40 m, and brown in colour below it. Organic-rich units exist in the upper confining silts (1.6% TOC at 6 m depth; Table 1), and within the aquifer sands at 17 m (0.49% TOC) and 23 m (0.76% TOC). In aquifer sands, the mean concentration of TOC is 0.27%. Concentrations of TOC appear to decrease with depth below a depth of about 23 m, but an uncored interval from 32.5 to 38.5 m depth prevents certainty in this matter. Concentrations of total S reach 0.12% in the upper confining silts but do not exceed 0.02% in the aquifer sands. At a depth of 45 m, the aquifer sands give way abruptly to the underlying clay of the basal aquitard. The upper few decimetres of this aquitard are brown, oxidised, and contain <0.1% TOC and <0.001% of total S. The oxidised horizon grades downwards into grey clay in which the concentrations of these constituents are higher. A similar oxidised upper aquitard occurs at FP (see below): at both sites, the total S content of these intervals (Fig. 2) is low over several metres of the clay, suggesting that the oxidation is more pervasive that it appears. At FP, in central JAM, the sediments consist of silts and clayey silts in the upper confining layer (0–12 m) 1261 that grade into mostly medium sand in the main part of the aquifer, which is 32 m thick. The upper aquitard is free of calcite between depths of 6 and 8 m. The aquifer sands are grey in colour, except at the base of the aquifer (>40 m depth) where hints of brown occur in a general grey colouration. Organic-rich units exist towards the lower part of the upper confining silts (2.9% TOC at 8 m depth; Table 1), and within the aquifer sands at 29.5 m (7.2% TOC). A single sample at 22.9 m depth contains 0.71% TOC and may represent a lateral equivalent of the organic-rich horizon at 23 m depth in AP. Elsewhere in the sands, concentrations of TOC seldom exceed 0.3% and are lower than this in the basal part of the aquifer. Concentrations of total S reach 1.3% in the lower confining clay and 0.6% in the peaty unit at 29.5 m, but do not exceed 0.04% in the aquifer sands, nor do they exceed 0.06% in the upper aquitard. The underlying aquitard of grey clay has been proven by drilling to be 25 m thick at this site. The upper part of this clay is brown and oxidised, as it is at AP, and contains <0.1% TOC and <0.001% of total S; it grades downwards into grey clay in which concentrations of these constituents are higher. 4.2. Piezometer waters The composition of water from the 3 piezometers (Figs. 3–5), which tap only the shallow aquifer, is presented in the order of increasing concentration of As in the ground waters drawn from the aquifer (DP none; AP some; FP much), which is in the order of increasing concentration of organic matter in the aquifer sands (but not in the overlying aquitard; Fig. 2). The piezometer profiles reveal that the ground water is chemically stratified (Figs. 3–5), as expected (Parker et al., 1982; Karim et al., 1997; DPHE, 1999; McArthur et al., 2001). In the piezometer at DP, concentrations of aqueous As do not exceed 5 lg L1 (Table 2; Fig. 3). Concentrations of Mn are high and peak in the middle of the aquifer at 4.2 mg L1 , whilst concentrations of Fe show an inverse trend to those of Mn, being lowest in midaquifer (0.44 mg L1 at 43.7 m) and highest (2.4 mg L1 ) in the lowest piezometer. Concentrations of PO4 are 0.35 mg L1 at 25.8 m and decrease downwards. Concentrations of SO4 are higher than at the other sites and increase downward to be around 10 mg L1 in the bottom half of the aquifer. Values of d13 C(DIC) are around )2‰ in the upper part of the aquifer and )6‰ in the lower part and show no obvious relation to other parameters. In ground water at AP from depths <40 m (Fig. 4), concentrations of Fe (3.9–6.2 mg L1 ), PO4 (0.8–3.2 mg L1 ), and As (240–420 lg L1 ) are high. The maximum concentrations occur at depths of around 38 m (As), 26 m (Fe), and 21 m (P). The peak concentration for Fe occurs just below the peak concentration of 1262 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Fig. 2. Lithological logs and profiles of total organic C (TOC, bold line), total S (TS, light line) and CaCO3 in sediment at the piezometer sites DP, AP, FP, in the JAM area. Lithological logs are those made in the field and are reproduced without alteration. Lines of correlation are shown dotted. Note the logarithmic scale for TS and TOC. organic matter (0.72% TOC) in the aquifer sands at 23 m depth. At 26 m, the value of d13 C(DIC) also decreases by around 2‰, but overall values of between )2‰ and )10‰ are less negative than might be expected for a system so reducing. Towards the base of the aquifer (from the piezometer at 38.3 m depth downwards), concentrations of dissolved Fe, As, and PO4 decrease. Concentrations of SO4 decrease downwards from the uppermost piezometer, which is in the upper confining layers, and are <10 mg L1 below 32 m depth. In the piezometer waters at FP, concentrations of Fe are high (4–7 mg L1 , Fig. 5) over the entire aquifer thickness, but are not related to those of dissolved As nor to depth. Concentrations of As are around 430 lg L1 in the upper part of the aquifer, but are much higher in most of the lower part (>32 m depth), where they are >900 lg L1 in a considerable thickness of the aquifer. Near the base of the aquifer (lowermost piezometer at 43.4m depth), the As concentration (239 lg L1 ) is lower, but is still higher than it is in the base of the aquifer at AP (7 lg L1 ). Values of d13 C(DIC) range from +0.6‰ to )10.8‰ but show no strong relation to lithology, including the presence of units rich in organic matter. Concentrations of SO4 decrease downwards from the uppermost piezometer (in the upper confining unit) and are <1 mg L1 below 25 m depth. Concentrations of PO4 peak at 2.5 mg L1 around 20 m depth and decline downwards. 4.3. Ground water flow In the aquifers of the Bengal Basin, the natural flow regime combines 3 scales of flow-system, as described by Ravenscroft (2003). The first, at the kilometre scale in the shallow aquifer, is driven by local topography. On this are superimposed local systems, around microtopographic features such as levees, channels and ponds, that extend laterally for a few tens to perhaps a hundred metres, and vertically for 10–20 m. The third scale comprises natural flow to the south in the deeper aquifer. Water levels in the piezometers at the 3 sites in the shallow aquifer, recorded in March 2003, are shown in Fig. 6: each has a perched water table in the upper aquitard. The head differences between the aquifer and overlying aquitard are 0.32 m at site AP, 0.82 m at site FP, and 3.70 m at site DP, and reflect the degree of hydraulic connectivity between aquitard and aquifer. Within the aquifer sands, piezometric levels vary by less than 0.2 m and decrease downward, with the gradient being greatest (0.0013) at site AP and lowest (0.0007) at site DP. Ground water elevations in the lower part of the shallow aquifer indicate a hydraulic gradient of 0.0006 to the NNE at the time of measurement – in the dry season in early March, 2003. During the monsoon season, the direction reverses as local rivers to the north and east fill with water (McArthur et al., unpublished J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1263 Fig. 3. Lithologic log and piezometer profiles of groundwater composition in March, 2003, at site DP, Barasat, West Bengal. Horizontal dashed lines marks the boundary between the aquifer sands and the overlying confining unit: the base of the aquifer was not definitely reached, so no lower boundary is drawn. Note the very much thicker upper confining unit, and the very much lower concentrations of As, compared to other sites (Figs. 4,5). Alkalinity is reported as HCO3 . piezometric data). Time-series measurements of hydraulic heads, and the hydrogeology of the area, will be presented elsewhere (Hudson-Edwards et al., 2004). 4.4. Well waters In Table 3 are given the results of the chemical and microbiological analysis of the ground waters sampled from domestic and public wells. The compositions are typical of those found elsewhere in the Holocene sediments of the Bengal Basin (DPHE, 1999; McArthur et al., 2001; Harvey et al., 2002; Dowling et al., 2003). Shallow wells in the central and NW of JAM (Fig. 7) are rich in As (200–1180 lg L1 ), Fe (3–13.7 mg L1 ), and PO4 (1–6.5 mg L1 ), and lack Mn and SO4 . A group of shallow wells (14–18, 40–44) in the immediate vicinity of piezometer DP contain <10 lg L1 of As and more than 2 mg L1 of Mn. With few exceptions, high concentrations of As are not spatially coincident with high concentrations of Mn (Fig. 7) nor with high concentrations of SO4 (Fig. 7), and these parameters are not correlated (Figs. 8(a) and (b)). In cross-plots (Fig. 8) of species affected by redox reactions (Fe, SO4 ) or Fe reduction (As, PO4 , Fe), the data for shallow wells (excluding wells near the piezometer site DP, owing to their distinct sedimentological setting; Fig. 2) appear to fall into 3 groupings, on the basis of their As/Fe values, and their concentration of As: those with <17 lg L1 of As; those with >32 lg L1 of As and high As/Fe; and those with >32 lg L1 of As and low As/Fe. These 3 groups of wells appear to bear a relation to depth, a relation explained in a later section (Discussion). Wells with high As/Fe also have high As/PO4 (Fig. 8(d)) and distinctly less Fe and PO4 (Fig. 8(e)) than do wells with low As/PO4 (Figs. 8(c)–(e)). The ground waters are anoxic because they contain dissolved Fe. Concentrations of nitrate are generally < 1 mg L1 and the range in pH is small (6.76 to 7.32; Tables 2,3). Strong correlations exist between dissolved concentrations of Ca, Mg, and HCO3 (Figs. 9(a) and (b)). 1264 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Fig. 4. Lithologic log and piezometer profiles of groundwater composition in March, 2003, at Site AP, Barasat, West Bengal. Horizontal dashed lines mark the boundaries of the aquifer sands with confining units. The uppermost piezometer is screened within the silts of the upper confining unit. Alkalinity is reported as HCO3 . Shallow wells have Na/Cl ratios that scatter widely about the value for marine-salt (Fig. 9(c)) showing that some wells have gained Na and some lost it, relative to Cl. Many wells plot along a trend of increasing Ca and Na (Fig. 9(d)), as sodic feldspar and detrital calcite are weathered, but some plot to the right of this line, in a direction compatible with ion-exchange of Ca in solution for Na on mineral ion-exchange sites (trend of line B, Fig. 9(d)). The deep ground waters are fresher (mean EC 753 lS cm1 ) than are shallow ground waters (mean EC 957 lS cm1 ): they contain less Ca, Mg, Cl, and HCO3 , but more Na than most shallow wells (Fig. 9). Deep wells (>70 m) have higher Na/Cl and Na/Ca values than do shallow wells (Figs. 9(c) and (d)). Water from deep wells contains <17 lg L1 of As. Saturation indices, calculated for waters from shallow wells with Geochemist’s Workbench (v. 3.0; Bethke, 1998), show that most of the Joypur waters are oversaturated with calcite (SI +0.5 to +2.0), dolomite (SI +0.1 to +2.0) and aragonite (SI +0.1 to +0.5). Water samples with low concentrations of Fe are at equilibrium or undersaturated with siderite (SI +0.2 to )0.9) and vivianite (SI +0.3 to )3), and at equilibrium or oversaturated with rhodochrosite (SI +0.02 to +0.7). By contrast, samples high in Fe are saturated with vivianite (SI +0.8 to +3.7) and siderite (SI +0.3 to +1.5) and undersaturated with rhodochrosite (SI )0.3 to )1.3). Because Ba solubility is limited by the solubility of barite, concentrations of Ba in a few well waters (Tables 2, 3) approach the guideline value of 0.7 mg L1 (World Health Organization, 1993) where concentrations of SO4 are low. 4.5. Ground water abstraction Inhabitants draw water for domestic supply mostly from private wells in the shallow aquifer, typically with 12-foot (3.6 m) screens that have their screen-centres at depths around 25–45 m, but 6 wells in the study area tap the deep aquifer (>70 m depth). The deep aquifer is also exploited by 3 irrigation wells near the JAM area: one is 600 m to its east, a second is 1000 m to the north, and the third is 200 m to its SW. The 3 deep wells irrigate 34 ha of agricultural land that borders the villages: the shallow aquifer is not used much for irrigation. During J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1265 Fig. 5. Lithologic log and piezometer profiles of groundwater composition in March, 2003, at site FP, Barasat, West Bengal. Horizontal dashed lines mark the boundaries of the aquifer sands with confining units. The uppermost piezometer is screened in the silts of the upper confining unit. Alkalinity is reported as HCO3 . the development of both aquifers, abstractions must have modified the natural ground water flow regime. Nevertheless, natural flow at the kilometre scale appears still to dominate the shallow aquifer, as irrigation water is drawn from the deep aquifer only and shallow abstraction is but a small fraction of recharge. Deep abstraction for irrigation will induce regional down-flow through the clay aquitard between 45 and 70 m, but its thickness ensures that the effect is small. Human modifications have applied, at most, for 40 a, whereas the natural flow regime has existed in its present form for up to 1.2 ka, since this is the age of OM in the upper aquitards (Hudson-Edwards et al., 2004). The deep irrigation wells to the north and east of the JAM area are screened from 100 to 130 m and pump for 8 h a day in February and for 12 h a day in March and April to irrigate about 27 ha of rice and 7 ha of vegetables. The third, sited 200 m to the SW of the area, is 95 m deep and has a comparatively small abstraction. No abstraction volumes are available for these wells but, assuming typical crop water requirements (Ravenscroft, 2003) and that deep percolation comprises 33% of abstraction, the authors estimate that the annual abstraction from these wells is of the order of 400,000 m3 , with around 130,000 m3 being returned to the shallow aquifer as deep percolation losses. Because of the clay aquitard at 45–70 m, the cone of depression created by these wells must have spread widely in the deep aquifer as downward leakage balances withdrawal of water from confined storage. In JAM, there are about 100 hand tubewells taking water from the shallow aquifer. By assuming that each well is used by 10 people with an average water use of 20 L/d, the annual abstraction from the shallow aquifer is estimated to be of the order of 7300 m3 , which is 2% of the abstraction from the irrigation wells. 5. Discussion 5.1. Piezometer sediments The data and the correlations between the piezometer sites (Fig. 2) suggest that the sediments below a depth of 32 m in FP and AP are equivalent in age, and of the same age as the aquifer at DP; they also suggest that the sediment above 32 m in FP and AP is distinctly younger (<8 ka) than underlying sediment, which is at least 17 ka 1266 Table 2 Composition and hydraulic heads of ground waters from piezometers in Joypur, Ardivok and Moyna (JAM), West Bengal, India Depth mbgl Water level Temp. °C EC lS cm1 @ 25 °C pH As lg l1 Units are mg l1 unless specified otherwise Na K Mg Sr Ba Fe Mn NH4 HCO3 37.5 10.3 29.7 10.3 21.1 2.2 18.0 2.8 20.0 3.6 19.1 3.1 25.2 3.4 26.3 3.1 39.6 3.1 164 135 107 111 138 137 130 131 138 35.1 34.2 25.4 30.7 37.4 40.1 45.3 46.6 47.7 0.49 0.50 0.43 0.50 0.63 0.64 0.74 0.84 0.84 0.37 0.33 0.23 0.26 0.22 0.13 0.11 0.17 0.28 4.0 4.7 7.4 4.8 6.0 7.5 5.2 4.7 4.8 1.0 0.8 0.1 0.2 0.2 0.2 0.2 0.2 0.1 3.50 2.30 2.30 2.60 5.00 1.25 1.30 579 562 487 526 601 601 584 598 611 89.7 58.0 20.9 26.5 52.4 78.4 72.4 63.3 94.2 Cl SO4 Bal % NO3 PO4 H4 SiO4 65.5 33.2 0.7 1.5 0.4 0.6 0.4 0.4 0.1 0.23 0.53 0.33 0.40 0.18 0.49 0.56 0.17 0.47 0.54 0.72 2.50 1.57 1.84 1.21 1.05 1.08 0.88 52 47 70 66 61 59 51 57 68 )8.4 )10.8 0.6 )0.6 )3.5 )4.3 )5.0 )6.2 )4.7 )0.7 )1.0 1.7 )0.9 )0.2 )2.5 0.9 1.0 0.8 Site FP: location 22 deg, 44.500 6.9 94.45 26.7 14.5 93.62 19.4 93.58 26.7 22.4 93.57 26.7 27.9 26.7 31.2 93.58 26.8 37.0 93.56 26.9 39.8 93.57 26.8 43.4 93.57 26.7 min N, 88 deg, 29.430 min E 1210 6.54 35 1018 7.21 94 764 6.84 443 795 6.96 411 975 6.96 447 1022 6.96 448 1028 7.10 960 1024 7.08 958 1139 7.04 239 Site DP: location 22 deg, 44.511 14.3 97.22 25.8 93.52 26.1 31.9 93.54 26.3 37.6 93.52 43.7 93.52 26.3 50.1 93.53 26.3 55.6 93.51 26.4 min N, 88 deg, 29.561 min E 76.8 50.2 53.6 55.6 47.6 48.2 0.9 1.0 0.6 1.3 1.6 2.4 146 154 139 129 118 151 27.5 38.6 37.3 33.8 38.8 42.9 0.44 0.55 0.53 0.51 0.48 0.66 0.19 0.07 0.08 0.09 0.09 0.14 1.8 1.5 0.6 0.4 0.5 2.4 1.6 4.2 2.7 2.6 2.1 0.5 0.08 0.02 0.13 0.12 0.45 0.17 744 724 670 640 606 728 51.2 62.6 58.8 40.2 47.6 80.4 0.4 1.5 5.4 11.5 6.3 9.7 0.58 0.31 1.32 0.41 0.19 4.56 0.35 0.17 0.09 0.02 0.04 0.05 58 70 77 75 65 71 )2.1 )1.9 )6.8 )6.4 )6.4 )5.5 )2.2 )1.3 )1.2 )0.2 )0.5 )4.0 Site AP: location 22 deg, 44.347 6.9 94.13 26.7 14.3 93.81 26.5 20.6 93.72 26.3 25.9 93.71 26.1 31.9 93.70 26.3 38.3 26.5 41.0 26.5 41.8 26.4 42.8 93.69 26.5 min N, 88 deg, 29.505 min E 3152 6.70 135 250 1406 6.78 327 87.6 1307 7.04 258 58.8 1075 7.04 226 42.1 925 6.96 279 28.4 757 7.07 419 23.3 859 6.91 187 33.4 919 6.88 110 34.4 939 7.07 7 44.7 3.2 1.4 2.1 2.3 1.5 1.6 1.4 1.5 2.4 364 168 186 143 122 109 112 116 115 87.2 39.0 51.4 39.6 37.3 33.3 35.3 36.3 38.0 0.97 0.49 0.83 0.67 0.62 0.55 0.56 0.57 0.55 0.31 0.40 0.39 0.37 0.28 0.15 0.09 0.09 0.14 2.0 5.2 5.3 6.2 4.4 3.9 2.0 1.5 1.0 4.1 0.8 0.3 0.2 0.2 0.2 0.3 0.5 0.6 1.30 1.30 1.95 1.55 1.10 1.30 1.50 1.30 701 690 670 562 551 526 579 593 622 558 100 74.1 66.8 65.2 8.1 13.4 30.0 29.3 490.3 98.5 80.1 53.0 11.9 2.8 2.4 8.4 3.9 0.12 0.13 0.18 0.36 0.30 0.12 0.35 0.16 0.22 0.24 0.82 3.16 3.12 3.00 2.25 1.15 0.49 0.13 54 62 64 67 69 66 65 68 64 )6.8 )7.3 )7.4 )9.8 )6.6 )2.5 )1.2 )1.4 5.4 1.8 )1.9 2.8 1.3 )1.1 )0.5 8.8 11.7 16.1 11.2 4.3 1.0 18 29 12 6.1 9.4 2.2 0.07 0.11 0.08 0.04 0.04 0.04 0.3 0.0 0.24 0.16 10.1 16.9 4.0 6.0 4.6 6.0 0.35 0.35 1.5 0.5 0.7 30 25 5 Pond A 1 Pond A 2 Rainwater 1142 1175 1048 1031 999 1131 6.88 6.84 7.08 7.00 6.96 6.95 4 1 1 <1 1 4 20 17 12 Notes: depth mbgl is depth in metres to mid-screen of each piezometer, Water levels are m above an arbitrary datum. )3.6 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Ca d13 C DIC ‰ J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1267 0 10 Depth m 20 97.22 30 40 AP FP 50 DP 60 93.4 93.6 93.8 94.0 94.2 94.4 94.6 Water level (m above datum) Fig. 6. Water levels in March, 2003, piezometers at sites DP, AP, and FP, in metres relative to an arbitrary datum. At each site, the uppermost piezometer was completed close to the base of the upper confining unit. The higher head in the upper confining unit at Site DP arises from the impermeable clay unit that locally caps the aquifer at this site. At FP and AP, the aquifer sands are capped by more permeable silts. old. The lack of calcite in sediment at DP, and below 32 m depth at AP and FP, might be due to its removal during oxidative weathering for a period of around 9 ka prior to the deposition of overlying sediment, or to the deposition of the calcite-poor units by the Brahamaputra River: today, sediment of the Ganges River contains detrital calcite but sediment of the Brahamaputra River does not. This contrast is best seen by examining its reflection in the Sr content of ground water in Bangladesh (P. Ravenscroft; Maps of Ground-Water Composition for Bangladesh, http://www/es.ucl.ac.uk/research/ lag/as): where detrital calcite occurs, concentrations of Sr in ground water are high (>0.3 mg L1 ); where calcite is absent, concentrations of Sr are low (<0.3 mg L1 ). At DP, the aquifer sediments are brown over the entire depth of the aquifer. At AP, the aquifer sands are grey in colour (Fig. 10) above 40 m depth, and brown at greater depth. At FP, the sediments are grey throughout the full aquifer thickness (Fig. 10) but, at the very base of the aquifer (>40m depth), the grey colour is accompanied by hints of brown at some levels (44.5 m). The upper few decimetres of the underlying aquiclude at AP and FP is also brown in colour and oxidised. As others have done (van Geen et al., 2003a,b), the authors assume that the brown colour arises from the presence of FeOOH and that the grey colour occurs when these oxides have been destroyed (or given a sequestering surface coating) by reduction. Attempts to confirm this assumption, and to quantify the cause of the colour difference, using sequential chemical leaches were unsuccessful owing to uncertainty about the homogeneity, solubility, grain-size, and crystallinity, of likely reduced phases and of FeOOH. Further work on this issue will be reported elsewhere (Hudson-Edwards et al., 2004). Nevertheless, the colour difference does suggest an explanation for why concentrations of dissolved As and Fe seldom correlate well in ground water from alluvial aquifers where FeOOH-reduction is the dominant mechanism of As release: it is that most of the Fe(II) that is reduced is sequestered in solid phases, such as; siderite (Pal et al., 2002), Fe sulphide (Korte and Fernando, 1991; Rittle et al., 1995; Chakraborti et al., 2001) vivianite, magnetite (Harvey et al., 2002), and green rust (Frederickson et al., 1998). That not all reduced Fe stays in solution is clear from the fact that the highest concentration of Fe in the waters (13.7 mg L1 ; Table 3) requires the dissolution of only about 5 mg kg1 of solid FeOOH. Although the amount of FeOOH in the sediment is low (see later), it is not that low: the reduction of FeOOH must, therefore, result largely in the production of solid phases containing Fe(II), with only a small fraction of the Fe remaining in solution. The notable difficulty, then, in explaining the composition of ground water in alluvial basins is to account for those instances (as found in JAM; see later) where dissolved Fe and As do correlate: the expectation is that they should not. The brown colour occurs distally from the sources of organic matter, which are in the upper aquifer and overlying aquitard. At AP, only the base of the aquifer is brown (i.e. contains FeOOH), a level well below that where most of the organic matter is found. At site DP, the entire aquifer is brown, yet has abundant organic matter in the unit above it. An examination of that unit explains why: the upper confining layer is 24 m thick, but the lower half comprises impermeable clay (15–24 m) which, despite its high content of organic matter between depths of 15.2 and 16.8 m (Table 1, Fig. 2), prevents hydraulic connectivity being attained between 1268 Table 3 Chemical composition of, and content of thermotolerant coliform bacteria in, ground waters from water wells in Joypur, Ardivok and Moyna (JAM), West Bengal, India. Some TTC determinations were replicated, mostly on different days Well Lat. Lat. (deg.) (min.) Long. Long. San. (deg.) (min) Risk TTC a 2 b Depth Temp (cfu/100ml) (m) (˚C) EC pH (µS cm-1) Units are mg L–1 unless specified otherwise As Na K Ca Mg Sr Ba Fe Mn NH4 µg L-1 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 29.4413 29.4145 29.4295 29.4515 29.4515 29.3215 29.2765 29.2965 29.3205 29.4979 29.4745 29.4766 29.5605 29.5505 29.5538 29.5661 29.5615 29.5751 29.2745 29.3115 29.3085 29.3105 29.3095 29.4515 29.4465 29.4347 29.4245 29.4331 29.3953 29.3725 29.3875 29.3705 29.3805 29.3675 29.3460 29.3425 29.3405 29.5155 29.5776 29.5455 29.5538 29.5677 29.5924 29.4996 29.4594 29.4437 29.2764 29.3124 29.3436 8 11 12 0, 0, 1, 10 0 0 11 7 10 11 3 8 7 1 0 0 0 1 7 11 11 8 4 7 0 5 0 61, 29 0 1 6 7 0 0, 0 7 4 9 3 10 0 0 2 0 6 12 12 8 11 7 11 8 10 9 3 6 9 5 7 1, 5 0 6, 7, 6, 20 0 1 1 63, 0 4 8 1 1 0 33.5 39.6 18.3 45.7 30.5 39.6 45.7 39.6 39.6 42.7 26.3 26.5 26.4 26.6 26.2 26.5 26.6 26.9 27.7 26.2 26.0 26.1 26.0 26.1 26.2 26.0 1087 949 1045 909 1160 685 911 937 672 1009 1197 1107 1090 1011 951 1265 26.2 26.8 26.9 27.0 26.8 27.0 26.6 26.5 26.6 26.6 998 1051 1024 1003 1133 1110 791 1038 957 971 33.5 39.6 42.7 29.3 33.5 51.8 29.0 21.3 21.3 41.1 49.4 45.7 32.3 26.4 26.4 26.2 26.1 26.4 26.2 26.3 26.0 26.3 26.2 26.0 26.0 848 1212 991 932 1217 743 939 688 802 1090 1001 1226 7.18 7.01 7.12 6.98 7.03 7.12 7.02 7.04 7.10 6.99 6.88 7.00 6.91 6.95 6.93 6.84 7.32 7.02 6.96 7.03 7.04 7.08 6.99 7.07 7.18 7.01 7.15 7.07 7.16 6.99 7.04 7.04 6.99 7.08 7.09 7.08 7.13 7.22 7.02 7.02 43.3 26.2 867 7.10 909 908 988 1181 1195 800 6.94 7.08 6.78 6.88 7.01 6.97 33.5 44.5 45.7 48.8 33.5 43.9 44.2 30.5 33.5 33.5 33.5 39.6 39.6 39.6 41.1 45.7 33.5 33.5 27.4 16.8 44.5 361 1180 1090 131 626 96 317 310 163 <1 391 459 32 <1 <1 <1 <1 <1 233 245 330 415 370 4 <1 42 98 56 <1 167 135 174 380 270 590 390 673 6 5 <1 <1 <1 4 <1 145 152 138 310 142 30.3 25.3 26.4 21.6 36.5 13.4 47.6 47.5 17.2 25.4 45.7 46.8 38.2 38.1 30.2 49.5 43.4 35.4 23.8 50.3 43.7 43.9 41.3 21.8 25.6 22.3 33.1 35.5 21.3 39.8 22.3 39.2 30.6 19.2 25.2 18.3 21.6 31.8 36.6 58.7 41.9 35.0 44.1 22.8 29.9 26.1 45.4 56.7 23.2 3.3 3.3 3.5 2.1 2.9 1.4 3.5 4.5 2.4 2.3 2.5 1.7 1.3 1.6 1.3 1.1 1.0 1.1 3.2 3.6 3.6 3.8 3.8 1.8 2.7 2.5 2.3 2.0 1.9 3.1 1.9 1.9 2.6 1.7 2.2 7.4 2.3 1.9 1.1 0.9 1.1 0.9 1.0 1.7 1.8 2.9 4.0 3.6 3.3 133 120 128 114 144 91 100 105 86 127 160 132 143 131 127 181 145 134 129 108 120 136 134 105 128 124 117 109 111 143 118 101 141 91 113 81 100 141 127 163 136 114 130 125 118 130 147 134 100 45.1 36.1 40.1 31.5 42.5 28.6 34.5 33.8 24.1 37.9 35.0 37.5 37.5 34.5 34.5 35.1 40.4 33.6 43.4 36.9 37.2 43.2 42.7 30.1 43.5 39.2 36.4 32.5 34.0 48.1 41.2 36.8 45.9 31.8 38.3 31.6 30.7 40.4 32.8 32.5 36.1 31.3 33.3 33.3 29.8 36.4 41.2 42.9 30.4 0.72 0.66 0.72 0.58 0.71 0.44 0.55 0.54 0.41 0.57 0.60 0.59 0.69 0.56 0.63 0.35 0.55 0.52 0.72 0.58 0.60 0.68 0.67 0.46 0.66 0.60 0.58 0.54 0.54 0.78 0.60 0.57 0.70 0.49 0.63 0.49 0.50 0.59 0.50 0.50 0.53 0.44 0.46 0.48 0.56 0.64 0.64 0.63 0.51 0.04 0.12 0.16 0.42 0.31 0.32 0.35 0.32 0.10 0.34 0.10 0.05 0.05 0.05 0.06 0.04 0.03 0.58 0.33 0.41 0.58 0.60 0.03 0.14 0.05 0.08 0.06 0.08 0.14 0.52 0.42 0.18 0.25 0.09 0.38 0.10 0.05 0.04 0.24 0.03 0.03 0.05 0.10 0.06 0.14 0.56 0.30 0.39 8.8 6.3 7.0 3.4 11.3 4.7 3.4 4.6 7.1 0.2 8.1 13.7 0.1 0.6 1.1 0.6 0.2 0.1 10.9 3.8 7.2 8.3 7.7 0.1 0.2 0.2 2.2 1.6 0.1 3.1 7.9 7.9 5.1 5.7 5.7 4.8 6.1 0.1 0.2 0.1 0.4 0.1 2.1 0.2 3.3 7.2 10.8 7.4 7.7 0.15 0.06 0.05 0.52 0.06 0.05 0.02 0.14 0.07 2.25 0.05 0.14 1.76 2.14 2.94 5.29 2.80 2.36 0.07 0.04 0.04 0.06 0.06 0.94 0.56 0.74 0.64 0.76 1.17 0.41 0.07 0.08 0.14 0.05 0.07 0.04 0.08 1.40 2.35 2.97 2.36 2.07 3.01 2.1 0.8 0.2 0.1 0.1 0.1 2.9 2.8 0.5 1.0 0.3 0.2 1.5 0.9 3.5 0.1 0.0 0.1 0.0 0.1 0.4 0.5 1.4 0.6 1.4 1.0 1.0 0.7 1.8 1.8 0.1 0.7 1.8 0.6 1.2 1.5 0.1 0.0 SO 4 594 533 547 475 622 430 572 582 414 485 680 625 627 552 571 768 647 600 570 608 638 703 678 489 527 521 525 514 498 574 512 526 502 478 531 453 478 539 573 700 83.0 64.7 83.5 58.4 91.3 15.8 27.8 30.2 23.5 101 80.0 71.4 60.6 69.6 39.6 60.2 69.0 45.1 73.8 31.9 30.8 43.4 46.4 29.9 87.6 70.9 63.6 46.2 43.3 116 78.5 48.7 144 16.1 56.0 14.6 34.7 88.0 47.7 72.4 0.0 0.0 0.0 0.6 0.0 11.2 0.1 0.3 0.1 0.0 0.3 0.1 0.3 7.8 9.0 0.1 2.2 0.2 6.0 1.4 0.0 0.1 0.1 0.1 6.3 1.1 2.8 3.7 0.1 22.1 3.8 5.8 7.4 0.1 0.0 0.1 0.0 29.2 0.2 0.3 538 37.4 0.2 615 539 565 750 703 510 34.6 60.8 33.0 53.5 19.0 0.4 0.2 0.5 0.6 0.2 557 513 565 635 423 581 583 407 487 682 658 612 526 591 819 612 583 661 646 736 666 NO3 PO 4 H4 SiO 4 δ13C DIC Bal (‰) 0.3 0.2 0.3 0.1 0.1 0.5 0.4 0.2 0.1 0.2 0.4 0.3 0.3 0.3 0.3 0.2 0.1 0.1 0.1 0.3 0.1 0.2 0.1 0.3 0.1 0.5 0.1 0.6 0.4 0.4 0.2 0.3 0.7 0.3 0.3 0.1 0.3 0.5 0.3 0.0 0.3 0.0 1.7 1.5 1.3 0.7 2.3 2.3 2.1 3.1 0.3 1.8 0.9 0.2 0.0 0.1 0.3 0.0 3.5 2.1 1.1 3.3 3.7 0.1 0.1 0.1 0.3 0.0 0.2 0.2 3.0 3.4 1.1 2.9 1.1 3.0 1.7 0.1 0.1 0.2 0.1 0.0 0.2 0.2 1.2 1.5 5.8 4.9 6.5 48 59 58 71 65 67 59 60 98 72 67 53 77 68 81 67 68 76 68 59 67 67 67 75 68 71 71 67 66 68 66 68 66 63 59 67 59 72 78 63 -6.9 -10.1 -7.9 -3.6 -9.0 -6.6 0.9 -7.9 -8.2 -5.4 -7.4 -1.3 -6.4 -5.6 -6.6 -1.2 -0.3 0.3 -6.2 -7.7 -6.5 -4.0 -6.6 -7.8 -10.1 -5.1 -8.8 -9.0 -6.5 -10.3 -7.5 -7.8 -6.2 -7.5 -7.1 -0.5 71 -2.6 73 63 72 63 81 -7.6 2.5 -4.8 1.4 -5.3 -1.3 -0.9 0.6 -0.9 -0.2 0.6 -0.2 -0.2 -2.2 1.0 1.9 -1.6 -2.8 -0.9 -1.1 -3.9 -0.6 -2.2 0.6 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 JAM, Shallow Aquifer Ba 1b 22 44.5097 Ba 2 22 44.5430 Ba 3 22 44.5439 New Ba 4 22 44.4748 Old Ba 4 22 44.4748 Ba 6 22 44.4748 Ba 7 22 44.4830 Ba 8 22 44.5041 Ba 9 22 44.5067 Ba 10 22 44.4818 Ba 11 22 44.5007 Ba 12 22 44.5300 Ba 13 22 44.4673 Ba 14 22 44.4697 Ba 15 22 44.4777 Ba 16 22 44.4756 Ba 17 22 44.5284 Ba 18 22 44.5200 Ba 19 22 44.5178 Ba 20 22 44.5218 Ba 21 22 44.5262 Ba 22 22 44.5352 Ba 24 22 44.5469 Ba 25 22 44.4540 Ba 26 22 44.4334 Ba 27 22 44.4246 Ba 28 22 44.4316 Ba 29 22 44.4490 Ba 30 22 44.4248 Ba 31 22 44.4165 Ba 32 22 44.4158 Ba 33 22 44.4287 Ba 34 22 44.4321 Ba 35 22 44.4391 Ba 36 22 44.4297 Ba 37 22 44.4411 Ba 38 22 44.4604 Ba 39 22 44.4459 Ba 40 22 44.5003 Ba 41 22 44.5333 Ba 42 22 44.5080 Ba 43 22 44.5051 Ba 44 22 44.5100 Ba 45 22 44.4486 Ba 46 22 44.4918 Ba 47 22 44.5190 Ba 49 22 44.6118 Ba 50 22 44.6058 Ba 51 22 44.6238 HCO3c HCO3 Cl Field (diff) 29.4405 29.4645 29.3148 29.3082 29.2362 29.827 29.2260 29.2296 29.2662 29.5209 29.4807 29.4864 29.4516 29.4570 29.4210 29.3874 29.3646 29.3268 29.2986 29.2788 29.2458 29.4870 29.5314 29.5000 29.5030 29.5595 29.5572 29.5632 29.5948 29.4972 29.5940 29.6172 29.6736 29.7396 29.7510 29.3070 29.2296 9 7 7 0 0 6 137.2 115.8 125.0 88.4 115.8 121.9 32.0 35.1 44.2 43.6 34.4 44.5 45.1 41.1 36.6 25.9 33.5 29.0 39.6 44.5 44.5 39.6 40.2 44.5 46.0 39.6 28.0 38.4 38.4 26.2 38.4 44.5 40.8 42.1 33.5 32.0 26.7 26.8 27.7 26.5 26.5 26.5 26.4 26.4 26.3 26.4 26.3 26.2 26.4 25.9 26.0 25.9 26.1 Notes: a San. Risk is Sanitary Risk; see text for details. b Thermotolerant coliforms; colony-forming units per 100 ml. c Bicarbonate calculated from imbalance of ionic charge. Bal = ionic balance as 100*(sum cations - sum anions)/(sum cations + anions) as meq l 88 88 88 88 88 88 JAM, Deep Aquifer Ba 1a 22 44.5053 Ba 5 22 44.4735 Ba 23 22 44.5600 Ba 48 22 44.4324 Ba 65 22 44.5212 176-T 22 44.419 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 88 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 22 Ba 56 Ba 62 Ba 64 Ba 81 Ba 82 Ba 83 Ba 84 Ba 85 Ba 86 Ba 87 Ba 88 Ba 89 Ba 90 Ba 91 Ba 92 Ba 222 Ba 223 Ba 224 Ba 225 Ba 226 Ba 227 Ba 228 Ba 229 Ba 230 Ba 231 Ba 232 Ba 233 Ba 234 Ba 235 44.6124 44.6208 44.6118 44.5554 44.5536 44.3814 44.3664 44.3766 44.3796 44.3364 44.3250 44.3900 44.3988 44.3538 44.4174 44.4552 44.4408 44.2965 44.2907 44.3470 44.3508 44.3910 44.4054 44.4018 44.4450 44.4216 44.3604 44.3682 44.3604 44.3724 44.3700 22 22 Ba 52 Ba 53 -1 766 780 749 687 760 775 1126 872 1085 1120 1066 999 1069 1154 1039 1067 963 615 964 893 600 879 837 859 919 332 904 768 872 916 694 691 747 734 1070 1097 1205 6.78 7.15 7.28 7.20 6.76 7.30 7.01 6.98 7.00 6.91 6.98 6.93 7.06 7.03 7.03 6.89 7.10 7.05 6.99 6.80 7.16 7.18 7.22 6.91 6.88 7.00 7.03 7.03 7.00 6.99 7.13 7.04 6.98 7.02 7.01 7.03 7.08 6 17 3 7 7 3 192 405 444 4 34 1 49 1 <1 <1 <1 135 530 178 87 <1 189 167 105 8 16 <1 <1 <1 186 1130 5 <1 <1 237 151 47.9 44.3 51.8 44.9 54.0 56.5 56.1 41.5 47.5 36.7 33.6 34.9 31.9 34.1 25.2 32.7 27.3 13.0 28.5 25.1 12.8 35.5 27.6 33.9 35.6 27.1 22.6 18.2 23.5 34.3 18.8 16.3 30.6 19.9 29.1 30.8 59.1 4.4 3.6 3.8 3.7 3.3 4.6 4.1 3.5 3.9 2.1 2.1 2.1 2.3 2.6 2.3 2.5 2.2 4.3 2.5 2.0 4.0 1.6 2.1 1.4 1.5 1.9 1.8 1.7 1.2 2.6 1.9 1.5 1.5 1.2 1.4 3.8 4.0 86 90 82 72 82 87 128 102 124 139 136 128 140 147 142 132 123 83 121 118 80 112 113 112 118 122 123 108 120 126 103 105 106 103 146 137 135 24.3 26.7 22.6 31.4 22.4 24.2 41.1 31.5 38.9 42.8 40.7 38.8 40.1 44.5 39.0 39.7 37.3 22.1 37.7 33.1 20.8 35.3 33.3 34.9 36.5 35.9 37.1 30.5 33.3 35.7 29.7 29.4 29.4 31.7 42.9 42.1 44.3 0.51 0.52 0.51 0.61 0.50 0.54 0.60 0.49 0.62 0.69 0.66 0.63 0.66 0.69 0.68 0.64 0.61 0.26 0.59 0.54 0.31 0.58 0.54 0.56 0.58 0.58 0.58 0.60 0.51 0.54 0.50 0.45 0.51 0.41 0.57 0.62 0.67 0.16 0.16 0.15 0.18 0.15 0.16 0.44 0.34 0.53 0.15 0.13 0.12 0.09 0.14 0.07 0.08 0.04 0.19 0.14 0.38 0.25 0.06 0.12 0.09 0.09 0.04 0.04 0.04 0.04 0.08 0.36 0.03 0.05 0.02 0.05 0.8 1.4 1.0 0.6 0.8 0.8 6.2 4.8 5.8 0.3 0.3 0.2 0.8 0.3 0.2 1.9 0.4 5.7 6.0 8.2 3.7 0.3 6.7 2.0 1.6 0.1 1.1 0.1 0.1 0.0 5.7 0.6 0.1 0.0 2.7 0.42 10.7 0.46 5.8 0.15 0.19 0.15 0.1 0.1 0.13 0.1 0.0 0.1 1.5 0.6 0.8 0.7 0.6 0.8 0.7 0.5 0.1 0.1 0.1 0.2 0.77 0.22 0.35 0.50 2.10 1.04 1.84 2.16 0.88 0.11 0.74 1.46 2.69 4.33 0.1 0.1 0.1 1.3 0.0 0.0 0.0 1.0 2.4 2.9 2.5 2.5 2.6 2.6 2.5 1.9 2.4 2.0 2.3 1.7 1.7 2.0 0.0 0.8 0.9 0.2 0.0 0.2 0.0 492 516 477 477 482 526 766 595 653 537 573 685 568 547 580 699 523 394 570 615 345 606 606 598 591 570 586 530 544 588 505 513 507 519 582 652 699 523 498 534 474 546 583 485 526 495 513 615 604 579 593 559 579 0.7 0.2 5.8 1.5 7.8 2.6 4.3 0.9 0.1 1.9 8.4 7.3 4.9 0.2 4.8 3.2 0.6 0.1 9.7 0.1 9.4 27.9 11.0 3.6 13.4 30.4 36.6 27.0 8.3 33.7 45.2 16.5 6.8 26.0 11.1 98.3 16.1 13.5 14.6 13.9 13.2 12.2 2.5 0.4 0.8 24.2 44.6 0.4 70.9 95.3 69.8 65.5 13.2 46.0 0.3 41.9 30.6 0.9 0.3 37.6 91.4 61.2 51.1 64.8 0.1 0.3 0.4 0.4 0.4 0.2 0.4 0.2 0.2 0.2 0.1 0.4 0.2 0.3 0.2 0.5 0.3 0.4 0.0 0.0 0.0 0.0 0.0 0.1 3.0 2.9 3.3 0.2 0.2 0.1 0.4 0.0 0.2 0.1 0.4 2.9 1.6 2.2 2.2 0.2 1.7 1.2 0.6 0.2 0.3 0.1 0.2 0.1 2.3 1.0 0.3 0.0 0.1 6.1 2.2 81 75 76 64 71 72 52 69 62 65 68 69 74 73 72 69 63 85 71 73 74 66 67 58 69 67 69 62 61 74 66 58 0.3 -5.1 -0.5 -7.0 -1.6 -4.5 0.2 -0.1 0.3 1.8 -1.3 1.1 0.6 -0.7 0.1 1.5 -0.2 0.8 0.5 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1269 1270 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Fig. 7. Map of the distribution of As, Fe, Mn and SO4 in JAM wells. Symbols as on each figure. Wells near piezometer DP are rich in Mn and lack As: wells in northwestern JAM are rich in As and Fe. For a discussion of why, see text. the peat and the underlying aquifer sands. The best proof of the absence of downward transport is the presence of the brown clay at 21 m depth, which resulted from oxidative weathering; it has not been re-reduced during burial, and the resumption of reducing conditions, as would have happened had organic-rich water from the overlying peaty layers been percolating through it. The high water-level in the upper confining unit at DP (>3 m higher than that at AP or FP; Fig. 6) provides further proof of impermeability. 5.2. Piezometer waters The development of anoxia must be driven by consumption of O2 and NO3 in the units rich in organic matter, and by the reductive effect of small organic molecules (carboxylic acids, amines, sugars), derived by fermentative degradation of the organic matter, which are dispersed through the aquifer (Bergman et al., 1999). Where the sediments contain FeOOH (DP, base of AP), concentrations of dissolved As are low (<10 lg L1 ), despite the presence of dissolved Fe which attests to a degree of reduction of FeOOH. The authors attribute these low As concentrations to resorption, by residual FeOOH, of As released by partial reduction of FeOOH. For example, towards the base of the aquifer at AP (>38.3 m), concentrations of dissolved Fe, As, and PO4 decrease markedly (Fig. 4). The Fe concentration (0.97 mg L1 ) in the lowermost piezometer shows that reduction of FeOOH has occurred, but the brown colour of the sediment shows it has not gone to completion. The low concentration of As in the lowermost piezometer at AP (7 lg L1 ) is therefore attributed to resorption of As to residual FeOOH. Given sufficient time, organic matter migrating downwards will reduce all the Fe oxide in the lower part of the aquifer at AP and release all of its sorbed As to ground water, before continued recharge flushes the system clear of As and other constituents. A contrast between brown and grey coloration of sediment (particularly, between grey Holocene sediment and orange/brown Pleistocene sediment) and an association of brown coloration with low-As water has been noted in the area of Araihazar, Bangladesh (van Geen 1250 1250 1000 1000 As µg L-1 As µg L-1 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 750 500 0 1 2 (a) 3 Mn mg L 4 0 5 -1 10 20 1250 1250 1000 1000 750 500 250 30 SO4 mg L (b) As µg L-1 As µg L-1 500 0 0 40 50 -1 750 500 250 0 0 0 5 10 15 Fe mg L-1 (c) 0 2 4 6 8 PO4 mg L-1 (d) 50 7 SO4 mg L-1 6 PO4 mg L-1 750 250 250 5 4 3 40 30 20 2 10 1 0 0 0 (e) 1271 5 10 0 15 Fe mg L-1 (f) 5 10 15 Fe mg L-1 Fig. 8. Relation in March, 2003, in JAM wells between (a) As and Mn (b) As and SO4 , (c) As and Fe (d) As and PO4 (e) Fe and PO4 , (f) Fe and SO4 . Wells are categorized using As/Fe and As concentration as proxies for depth. Open squares are wells with >32 lg L1 of As and low As/Fe values; filled circles are wells with >32 lg L1 of As and high As/Fe values; small open circles are wells containing <16 lg L1 of As. See text for an explanation of the relation of these groupings to well depth. et al., 2003a), but in Araihazar, unlike in JAM, grey sediment does not always host As-rich water. That a grey Holocene aquifer overlies a Pleistocene aquifer of a ‘‘deep burnt orange’’ colour was also noted in Munshiganj, Bangladesh (Harvey et al., 2002) but, again, low concentrations of As prevailed over much of the thickness of the grey Holocene aquifer. The differences between these sites and JAM awaits explanation. The concentrations of As in most of the lower part (37.0–39.8 m) of the aquifer at FP are higher (>900 lg L1 ) than they are in the upper part of FP (94–448 lg L1 ) and are higher in both than they are at AP (maximum 420 lg L1 ). The authors attribute the differences to the fact that FP contains more organic matter than does AP, and higher peak concentrations of organic matter (e.g. 7.2% at 29.5 m in FP, 0.76% at 23 m in AP). This additional organic matter must be driving reduction of FeOOH in FP further than it has gone in AP, and results in ground water with higher concentrations of both Fe and As. This organic distribution 1272 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 800 HCO3 mg L-1 Ca mg L-1 200 150 100 700 600 500 400 300 50 15 25 35 45 Mg mg L-1 (a) 50 55 100 (b) 200 200 70 A Ca mg L-1 60 Na mg L -1 150 Ca mg L-1 50 40 30 150 100 B 20 50 10 0 0 0 (c) 50 100 Cl mg L 150 200 -1 0 (d) 20 40 Na mg L 60 -1 Fig. 9. Relation in March, 2003, in JAM wells between (a) Ca and Mg (b) Ca and HCO3 , (c) Cl and Na (d) Na and Ca. Line in (c) shows Na/Cl in marine salt. Arrow A in (d) represents a weathering trend: arrow B represents stoichiometric ion-exchange of Ca in solution with Na on mineral exchange-sites. Symbols as in Fig. 8, with the addition of large, open, circles for deep wells (>70 m), and crosses for wells in the vicinity of piezometer site DP, which, owing to their proximity to piezometer site DP, are inferred to be tapping an aquifer different sedimentologically from that tapped by other wells. does not, however, appear to explain the fact that the maximum As concentrations in AP and FP occur well below the maximum concentrations of PO4 and Fe: the control on dissolved P and Fe appears to be partially decoupled from the control on dissolved As. The different depths of the maxima in As, Fe, and PO4 is probably due simply to differences of the As/Fe/PO4 ratios in the aquifer sands above and below 32 m, given their different hardness and age (see Section 4): variable lithology is another reason why As and Fe seldom correlate well in ground water from alluvial basins. In DP, the brown colour of the aquifers sands at all depths (Fig. 10) attest to the presence of FeOOH throughout the aquifer: that the composition of ground water is quite different from those at sites AP and FP is therefore not unexpected. Concentrations of As are low (<5 lg L1 ), whilst concentrations of dissolved Mn and SO4 are high. The profiles and compositions at DP are best explained as resulting from the preservation of originally oxic connate water, which has subsequently been subject to creeping reduction as organic matter moves in laterally from surrounding sediments (e.g. site FP, which is 250 m to the west) to cause (presently) incomplete reduction of FeOOH and Mn(IV) oxides. Given the U-shaped profiles of As, Mn and Fe with depth (Fig. 3), organic matter must be diffusing upwards from the underlying organic-rich clay at this site (so its upper part cannot be oxidised?) as well as entering the aquifer laterally at shallow depths, so that the reduction potential decreases towards the mid-point of the aquifer: there, a peak in dissolved Mn (2–4 mg L1 , Fig. 3) coincides with a minimum in dissolved As (<2 lg L1 ) and Fe. Reduction of FeOOH dominates over reduction of Mn oxides towards the top and base of the aquifer. Concentrations of As are kept low (<5 lg L1 ) by resorption of As to residual-FeOOH. Oxides of Mn sorb As (Manning et al., 2002; Foster et al., 2003), so the As released by reduction of MnO2 must be re-sorbed to residual oxides of Mn and Fe, rather than remain in solution (as noted by St€ uben et al., 2003). The values of d13 C in dissolved inorganic C (DIC) are high for a redox-dominated system. They are kept from becoming more negative, and so from reflecting organic inputs more strongly, by the low pH of the ground water (6.76–7.32; Tables 2 and 3), which promotes rapid equilibration between DIC and detrital calcite, with a J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1273 Fig. 10. Colour of wet sediment from West Bengal, showing the grey colour of sediments in which reduction of FeOOH has gone to completion, and the brown colour of sediments that retain FeOOH. Groundwater from the former is enriched in As, that from the latter is not. (For interpretation of the reference to colour in this figure legend, the reader is referred to the web version of this article.) d13 C around )1‰ (Table 1; Hudson-Edwards et al., 2004). At AP, values of d13 C(DIC) decrease by around 2‰ at 26 m, a change that may reflect an organic contribution to DIC from the degradation of organic matter at 23m depth. At FP, the abundance of calcite approaches zero below 30 m depth; below this level, the more negative values of d13 C(DIC) reflect this diminished buffering capacity (Table 1). Surprisingly, the organic matter at 29.5 m in FP appears not to influence values of d13 C(DIC). In the upper part of the aquifer at DP, values of d13 C(DIC) are around )2‰: they are around )6‰ at greater depth. These high values must reflect equilibration with traces of calcite in the aquifer, although the analytical data suggests calcite is absent from DP. At sites AP and FP, concentrations of SO4 decrease downwards from the uppermost piezometers, which are in the upper confining layers (Figs. 4, 5), but it cannot be said with certainty whether these profiles represent downward passage (dilution and dispersion) of recent anthropogenic SO4 from the surface, infiltration of natural amounts of SO4 concentrated by evaporo-tran- spiration, or remobilisation of S in Fe sulphide from the organic-rich layers as oxic water passes through the uppermost part of the upper confining layers. It seems unlikely that they represent steady-state consumption of natural SO4 , at depth, by SO4 reduction, because the sites are <350 m apart and would have similar nearsurface concentrations were the profiles natural. In fact, concentrations at 7 m depth are 65 mg L1 at FP and 490 mg L1 at AP. The authors speculate that the high concentrations of SO4 at 7 m at AP result from surface loadings of gypsum as a result of house building, as it is used in plaster and the locality has several substantial houses. If this is the case, the profiles imply that SO4 from a surface source can penetrate to 30 m depth within a few tens of years. The chemical stratification revealed by the piezometers explains why wells may differ in composition despite being close to each other (<5 m) and of similar depth. Arsenic concentrations in the bottom 4 piezometers at the AP site decrease linearly with depth from 419 lg L1 at 38.3 m to 7 lg L1 at 42.8 m, a rate of change of 92 lg L1 m1 . The rate is 200 lg L1 m1 in the basal part 1274 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 of the aquifer at FP. Such high gradients will cause wells to differ substantially in As concentrations if they differ by no more than a metre in depth, a distance within the uncertainty with which a driller would record the depth of a well. Chemical stratification also compromises interpretions drawn from well-surveys of As concentrations, in the Ganges Plain and elsewhere: low concentrations of As in wells (e.g. as found near AP; Fig. 7) do not indicate that the aquifer they tap is free of As: using local knowledge of aquifer colour, a driller may emplace wells in the small thickness of good water in the brown sand at the base of polluted aquifers, if it exists (as it does at AP). Consequently, As in ground water would go undetected until breakthrough to wells, and its detection by analysis or the development of arsenocosis in consumers. Breakthrough and detection might be separated substantially in time and have serious consequences for health. Furthermore, the finding in an area of dangerous concentrations of As in only a small percentage of randomly distributed wells does not signal that the As is localized and the aquifer generally safe for development: it may show only that most wells tap water from above or beneath a poisoned part of the aquifer. 5.3. Well waters The chemical stratification of the ground water (Figs. 3–5), the differing depths of wells and the uncertainty about some well-depths, and the differing amounts of organic matter in the aquifer sands from place-to-place, conflate to obscure areal patterns in water quality (Fig. 7) and cross-plots of some dissolved constituents (Fig. 8): nevertheless, some patterns emerge. Wells near piezometer DP (14–18, 40–44) contain <10 lg L1 of As and more than 2 mg L1 of Mn, so the aquifer they tap must be sedimentologically and chemically similar to that seen in the DP piezometer, and be in a setting that is sedimentological distinct from those of other wells in the JAM area that lack Mn. The wells near DP must tap a part of the aquifer that is both free of organic matter and protected from direct infiltration of organic matter from above by a thick upper-aquiclude of clay. For wells near DP, the low concentrations of As and Fe, and the high concentration of Mn must be due to the fact that reduction of MnO2 dominates over reduction of FeOOH, and that where the latter occurs, it does not reduce all the FeOOH, so, in both cases, re-sorption keeps concentrations of As low. That concentrations of As are low when Mn is present is an observation not confined to the DP area: as Fig. 8(a) shows, As and Mn appear to be largely mutually exclusive in solution, a situation that extends to most shallow wells within 2 km of the JAM area (McArthur and Banerjee, unpublished data) and more widely in Bangladesh (data in DPHE, 1999). High concentrations of Mn might therefore indicate the presence of As-free connate water in an aquifer where reduction of FeOOH is incomplete. The exploitation of water at such sites will constitute mining of a finite resource and may draw in As-rich water. Such transport will be moderated by sorption to unreacted FeOOH. Regular monitoring of water quality is essential in such a complex situation. The composition of wells away from site DP can be interpreted if it is assumed that the vertical pattern of chemical stratification in the aquifer across the rest of the JAM area resembles that seen at AP and FP. The assumptions are: that As concentrations are low (<50 lg L1 ) where FeOOH is present to sorb As; that this occurs only at the base of the aquifer; that As concentrations reach a maximum at 35–40 m and decrease sharply below this depth; that ground water above this depth contains less As, although concentrations are still high; and that As concentrations reflect the amount of reduction that has occurred, and so the amount of organic matter in the sediments. A well-by-well analysis of As concentrations, however, does not reveal a clear relation between concentrations of As and well depth, probably because some well-depths are wrong. There is, however, some relation between well-depth and both As/ Fe and As concentrations: if the lowest concentrations of As (<20 lg L1 ) are assumed to represent wells emplaced in the base of the aquifer where FeOOH persists, as at AP, and if the remaining wells are divided into two groups on the basis of their As/Fe values, 3 divisions emerge (Fig. 8), viz. Group A; 19 wells with <1–16 lg L1 of As. Group B; 24 wells with >32 lg L1 of As and As/Fe between 13 and 56. Group C; 24 wells with >32 lg L1 of As and As/ Fe between 36 and 2025. The mean well-depths for each group are: 135 14 feet (41.1 ± 4.27 m) (A); 124 24 feet (37.8 ± 7.32 m) (B); and 108 20 feet (32.9 ± 6.1 m) (C), where all uncertainties are 1 s.d. Although these differences are not statistically significant, they are interesting because wells are constructed from 20-foot (6.1 m) lengths of plastic pipe, topped by 3–6 (0.91–1.82 m) feet of Fe pipe. The mean depths of these 3 groups suggest that they reflect groups of wells that are mostly made with 7, 6, or 5 pipes, with 7pipe wells (group A) being screened in brown sand at the base of the aquifer, 6-pipe wells (group C) being screened in the As maximum seen in the piezometer profiles to occur in the lower part of the aquifer, and 5-pipe wells (group B) being screened above the As maximum. The high standard deviation probably arises because some well depths are incorrect: nevertheless, enough are correct to allow the overall depth-control to be seen. The use of As/Fe, and As concentrations, as a proxy for depth seems reasonable because the As/Fe values in AP and FP (Fig. 11) change with depth, reflecting the fact that the maximum dissolved concentrations of As occur at a J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 depth greater than do those of Fe or PO4 , which are themselves not much separated in terms of depth (Figs. 4,5). The different As/Fe/PO4 values present at different depths (Fig. 11) probably arises because the younger sediment (<32 m depth) releases more Fe and PO4 on reduction, and less As, than the older sediment (>32 m depth): such lithological differences, reflected in ground water composition, must be one reason why Fe and As often correlate poorly in the well waters of the Ganges Plain. Dividing the wells onto groups A–C, however, yields correlations in the As-rich groups between As and Fe that are better than for the combined population, probably because the effect of mixing populations from different depths is reduced. The coefficient of correlation between As and Fe in the 3 populations (Fig. 8(c)) are: 0.01 for A (p P 0:1); 0.58 for B (p ¼ 0:01); and 0.86 for C (p ¼ 0:001, if one outlier, with 0.56 mg L1 of Fe and 1131 lg L1 of As, is excluded); the pooled population has a coefficient of 0.58 (p ¼ 0:001 excluding the outlier). In a pooled population of 213 wells within 2 km of JAM, the correlation coefficient between Fe and As is 0.39 (p ¼ 0:001; Fig. 12; Banerjee and McArthur, unpublished data). If the correlations have meaning, it can be only because the lower and intermediate concentrations of As, and the accompanying Fe, reflect a dynamic process that is proceeding extremely slowly, with the dissolved As and Fe concentrations representing a snapshot of slow, microbially mediated release, prior to sequestration of Fe into solid diagenetic phases, and resorption of As: the As and Fe are ‘kinetic species’ in the sense used in Fig. 13 (see next section). Higher concentrations of As may represent a state where all reactive FeOOH has been consumed, leaving the As in solution with no sorber to adhere to: this idea is also developed in a later section. For groups A–C, the correlation coefficients between PO4 and Fe (Fig. 8(e)) are 0.00 for A (p > 0:1); 0.50 for B (p ¼ 0:01); and 0.62 for C (p ¼ 0:001): the coefficient for the pooled population is 0.72 (p ¼ 0:001) and is enhanced by mixing populations B and C, one high in PO4 and Fe, the other lower in both. In a pooled population including data for 213 other wells within 2 km of JAM (Fig. 12; Banerjee and McArthur, unpublished data), the correlation coefficient between Fe and PO4 is 0.76 (n ¼ 213, p ¼ 0:001). Given the discussion above, the high concentrations of As and Fe in the northwestern part of JAM (Fig. 7) is probably due to two factors viz. that most wells there are emplaced at a depth near to the As maximum (are 6-pipe wells) and that organic matter around 30 m depth (as seen at FP) is more abundant in central and NW JAM than it is elsewhere. In much of southern and eastern parts of JAM, concentrations of As must be low (<50 lg L1 ) because wells there are screened at the base of the aquifer in the small thickness of brown sand revealed at AP. 1275 The considerations above show that real element associations can be disguised or enhanced by pooling data on too regional a scale, and that piezometer profiles are essential to fully understand the source, transport, and sinks, of As in the Bengal Basin. This discussion also serves to emphasise that the results of well-surveys must be evaluated in combination with a knowledge of the local geology, particularly aquifer colour, and well depth. 5.4. Diagenetic Iron sulphide Diagenetic Fe sulphide is a sink for dissolved As (Korte and Fernando, 1991; Rittle et al., 1995), and is present in the sediments in West Bengal (Fig. 2; Chakraborti et al., 2001). As much of the ground water in the JAM area lacks SO4 , as do most ground waters in surrounding areas (McArthur and Banerjee, unpublished data) and in the Ganges Plain generally (DPHE, 1999, 2001; Nickson et al., 2000; McArthur et al., 2001; Dowling et al., 2002), it must have been removed by SO4 reduction from most recharging water. As SO4 reduction is driven by microbial metabolism of organic matter, it is localised where organic matter is present. At all sites, total S is almost absent from aquifer sands but is relatively abundant in the organic-rich units (Fig. 2; Table 1), which are (or have been) the site of SO4 reduction and formation of diagenetic Fe sulphides. The traces of S in the sands probably represents Fe sulphide formed very early in the history of anoxia from reduction of SO4 in connate water. Reduction of SO4 is probably not occurring in the aquifer sands now, either because they lack SO4 (FP) or because SO4 -reducing bacteria are outcompeted by Fe-reducing bacteria (Chapelle and Lovley, 1992). The presence of SO4 in the upper aquifer at AP and FP (Figs. 4 and 5), where it has penetrated to levels below some organic-rich units, might show that the organic matter has lost much of its original reducing power through biodegradation. From these arguments, and the fact that most of the organic-rich units are stratigraphically above As-rich aquifers, the authors infer that little As is being removed now from ground water in the aquifer sands by sequestration in diagenetic sulphide. 5.5. Resorption as a control on aqueous As 5.5.1. The shallow aquifer Knapp et al. (2002) showed that aquifer sands from Virginia, USA, sorbed SO4 least where FeOOH in the sediments had been removed from grain surfaces by the establishment of reducing conditions. In JAM, As concentrations are low where the sediments are coloured brown by FeOOH, and are high where sediments are grey because all FeOOH has been reductively consumed (Figs. 10). Reduction is incomplete in those parts of the 1276 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 0 Depth, metres bgl 10 AP FP 20 30 40 50 60 0.00 DP 0.05 0.10 0.15 0.20 As / Fe 0 Depth, metres bgl 10 20 30 40 50 60 0.0 0.2 0.4 0.6 0.8 1.0 As / PO4 Fig. 11. Relation of As/Fe to depth at piezometer sites AP and FP in March, 2003. Values of As/Fe, and As concentration, are low where wells tap brown sediment at the base of the aquifer (more than about 40 m depth); are high where wells tap the aquifer between 35 and 40 m depth, and lower, but still high, where wells tap the aquifer at shallower depths. aquifer that are distal from sources of organic matter (e.g. the base of the aquifer at AP), or where no hydraulic continuity exists between organic-rich sediments and aquifer sands (at DP). Where reduction is incomplete, resorption to residual FeOOH will keep As concentrations low (as mooted by Ravenscroft et al., 2001), until all sorption sites become saturated by re-sorbed As, or all FeOOH has been consumed: this idea is implicit in the model of Welch et al. (2000) that explains the generation of high concentrations of As by progressive reduction of FeOOH. In Fig. 13, this model is shown more explicitly: As released to solution by reduction will exist in solution temporarily (this is termed ‘kinetic As’) until it is resorbed to residual FeOOH. Laboratory studies of microbial FeOOH-reduction show that As resorbs over a timescale of a few days to weeks (Figs. 3B and C in Dowdle et al., 1996; Figs. 6 and 7 in Jones et al., 2000). Resorption will decouple concentrations of As from those of Fe and leads to poor correlations between these constituents: this is one reason why these constituents seldom correlate well in ground waters in alluvial aquifers. Resorption of As will occur as ground water moves away from the influence of peaty sediment, where reduction is complete, to areas where it is not complete and FeOOH is available to sorb it (DPHE, 1999). 5.5.2. The deep aquifer Resorption may be one reason why the deep aquifers in JAM, and widely across the Bangladesh, are free of As (a very limited exceedance of 50 lg L1 of As in deep wells may be explained as contamination or mis-identification of wells). Sands of the deep aquifer in Bangladesh are typically described as ‘‘burnt-orange’’ (Harvey et al., 2002) or as ‘‘yellowish-brown to pale grey sands J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1277 1250 As µg L-1 1000 750 500 250 0 0 2 4 6 8 10 12 14 10 12 14 Fe mg L-1 7 PO4 mg L-1 6 5 4 3 2 1 0 0 2 4 6 8 Fe mg L-1 Fig. 12. Relation of As, PO4 , and Fe, in ground waters sampled from 213 wells in JAM in November, 2002, within 2 km of the study area (McArthur and Banerjee, unpublished data). By analogy with piezometer profiles, the poor correlation between As and Fe (r ¼ 0:39, n ¼ 213) arises because As/Fe values in ground water reflect As/Fe values in aquifer sediments, and are lithology-dependent. The better correlation between Fe and PO4 (r ¼ 0:76, n ¼ 213) reflects a more uniform distribution (less lithology-dependent concentration) of PO4 than of As in aquifer sands. Fig. 13. Schematic of dissolved concentrations of As as a function of the percentage of FeOOH reduced. Kinetic-As is that released by partial reduction of FeOOH but not immediately re-sorbed: its concentration represents a balance between the rates of FeOOH reduction and As sorption. Filled-arrow 1 shows position in the sequence of reduction of groundwater at site DP and the lowermost piezometer (42.8 m) at site AP; filled-arrow 2 shows the likely state of sediments and ground waters at site FP and the middle and upper aquifer at site AP. 1278 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 . . . cemented by secondary clay and Fe oxide’’ by Ravenscroft in DPHE (1999; Vol. S1-Main Report), because they were subjected to a long period of oxidative weathering during the lowering of sea-level that occurred between 125 and 18 ka. This weathering must have generated FeOOH by oxidation of biotite and other Fe-bearing minerals in the sediment. As a consequence, the higher content of FeOOH in Pleistocene sands of the deep aquifer distinguishes them (Stanley et al., 2000) from the Holocene sands of the shallow aquifer. The authors suggest that the establishment of anoxic conditions in the deep aquifer, as a consequence of the deposition of overlying, organic-rich, Holocene aquifers, has been sufficient to mobilise some Fe into ground water (Table 3; DPHE, 1999), but has been insufficient to reduce all its FeOOH, the remains of which may now act as a sorptive buffer to prevent As from accumulating in solution. An alternative explanation for the lack of As in waters from the deep aquifer, previously suggested by Ravenscroft (DPHE, 1999), McArthur et al. (2001), and Ravenscroft et al. (2001), and repeated in Smedley and Kinniburgh (2002), is that sorbed As was flushed from the deep aquifer during an extended period of oxidative weathering and flushing that occurred during the lowering of sea level between 125 and 18 ka. With available data, the authors cannot be certain which explanation is correct. In JAM, however, (where CH4 is not recorded) the DP profile shows that peat sensu stricto is encapsulated in impermeable clay: the deposition of the peat must have occurred in response to local waterlogging caused by the clay. The low permeability of this clay now prevents recharge from carrying soluble organic matter downwards into the underlying aquifer sands. The sediment profiles at FP and AP show that sites marginal to the peat depocentre (DP) developed lesser amounts of organic matter, in peaty layers that are lateral extensions of the main peat (i.e. are of a similar age; Hudson-Edwards et al., 2004). In these marginal environments, the thinner, perhaps intermittent, clay will allow greater hydraulic connectivity with underlying aquifer sands and so allow ingress of organic matter. During sedimentation, however, such sites would have been less waterlogged and so would have accumulated less organic matter than basin centres, and so would host less organic matter. Beyond the basin margin, permeability will be higher, but the amount of organic matter will be negligible. These ideas are illustrated in Fig. 14, in which a schematic cross-section through the JAM area shows the relation of permeability, organic matter, and sedimentary architecture. This model suggests that chemical reduction in the JAM aquifer (and by implication, elsewhere in the Bengal Basin) is driven by organic matter in peaty units that develop on the margins of peat basins where there is enough organic matter to drive redox, and sufficient permeability to allow organic matter to invade the aquifer in soluble form. 6. The driver for FeOOH reduction 6.1. Natural organic matter in sediments 6.1.1. The JAM Area The data for the JAM area suggest that reduction of FeOOH is driven by natural, subsurface, organic matter, in organic-rich units, formed at the periphery of local peat basins, that are in hydraulic continuity with the aquifer. The concentrations of organic matter in JAM sediments (low% levels) is more than adequate to account for the amount of reduction present: at 25 °C (the mean annual air temperature in JAM) infiltrating water contains around 8.3 mg L1 of dissolved O2 , an amount consumed by <0.001% of reactive organic matter in the sediment. That peaty sediment is connected with the presence of unwholesome water is confirmed by local experience in JAM: Ashraf Ali (Ardivok driller, pers. comm., 2003) has noted an association between bad water and ‘khalu mati’ (black soil, which the authors interpret to be peat, or peaty units) and between brown sediment and good water. The association suggests that connectivity (permeability) exists between aquifers and peat deposits in some parts of the Ganges Plain, perhaps where they are cut through by post-depositional channelling (H. Brammer, pers comm., 2001). 6.1.2. Other areas in the Bengal Basin The coincidence of high concentrations of As in groundwater and wetland environments, where plant productivity is high, was noted by Ravenscroft (DPHE, 1999) and has been reproduced in Smedley and Kinniburgh (2002), whilst both McArthur et al. (2001) and Ravenscroft et al. (2001) argued that peat, which forms in such wetlands, was the driver for reduction of FeOOH in the Ganges Plain. One reason for an emphasis on peat is that CH4 in combustible amounts is found at depth in some ground waters (Ahmed et al., 1998; Harvey et al., 2002) and the fact that CH4 in such amounts can be generated only by anoxic degradation of high concentrations of organic matter. If organic matter, accumulating marginally to peat depocentres, drives reduction of FeOOH (and so As release to ground water) it is crucial to establish the extent of peat deposition in the Bengal Basin. Peat is common beneath the Old Meghna Estuarine Floodplain in Comilla (Ahmed et al., 1998), Sylhet, and the Gopalganj-Khulna Peat Basins (Reimann, 1993; Brammer, 1996). Many wells in the area around Faridpur may be screened in waterlogged peat (Safiullah, 1998) and the shallow aquifer system in Lakshmipur contains peat (DPHE, 1999). Peat is often found in J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 geotechnical borings (piston samples), although it is rarely recorded during rotary drilling for water wells because such drilling masks its presence, unless the peat is very thick. Peat occurs extensively beneath the Asaffected areas of southern Samta village and Deulhi village in southwestern Bangladesh at a depth of about 10 m (AAN, 2000; Ishiga et al., 2000). Peat has been found in Holocene sediments around Mymensingh (Ishiga et al., 2000) and is well documented at Panigati, in SW Bangladesh (Islam and Tooley, 1999). Organicrich sediment has been reported to occur at 6 m and 25 m depth in Faridpur and at 28m depth in Sonargaon (Fig. 3 of Tareq et al., 2003). Sediment from a depth of 2.1 m at Gopalganj (100 km SW of Dhaka) contained 6% TOC (Nickson et al., 1998); sediment from a depth of 23m at Tepakhola (Faridpur municipality) contained 7.8% TOC (Safiullah, 1998). Peat is repeatedly mentioned by Umitsu (1987, 1993) and by Goodbred and Kuehl (2000) as being present in Bangladesh sediments; it has been reported to occur in southern West Bengal by Banerjee and Sen (1987), and also by Pal et al. (2002) who note that peat was present in the subsurface in an As-free area, but not in a nearby As-affected area. These authors do not state whether their peat was underlain by impermeable strata, as at the DP site, but it seems likely. It could be that the apparent lack of peat at their Asaffected site means that it was missed during coring. Harvey et al. (2002) also reported that no peat was found at an As-affected site in Bangladesh, but they report the presence of CH4 (marsh gas) in combustible amounts; this proves the presence of much organic matter in the aquifer close to their site. Thus, in the Bengal Basin, peat deposits are widespread, and so the margins of such deposits must be 1279 common. Despite the importance of peaty sediment as the redox driver in the Ganges Plain, there are two other potential drivers of redox: these are organic waste in latrines (McArthur et al., 2001; Ravenscroft et al., 2001), and organic matter leached from surface sources, such as soils, river beds, and ponds (Harvey et al., 2002). Each is considered in turn. 6.2. Other sources of organic matter 6.2.1. Organic matter in latrines Human organic waste in latrines does locally promote reduction of FeOOH in aquifers and contributes As to groundwater to locally enhance As concentrations, thereby confusing the regional signal of As derived from reduction driven by natural organic matter. The impact of point-sources can be seen in Singair, an As hot-spot in Holocene sediments some 10 km west of Dhaka, Bangladesh: of 15 shallow wells within 300 m of Singair mosque (Safiullah and McArthur, unpublished data), the 3 wells sited within 3 m of latrines contained 900 lg L1 of As, whilst the remaining 12 wells, all >15 m from latrines, contained between 100 and 300 lg L1 of As. Such an influence is not uncommon; new wells drilled through new concrete pit-latrines have been observed (McArthur, unpublished data). Point sources will be particularly strong where the polluting organic matter is in hydraulic continuity with the aquifer e.g. where the depth of a latrine is greater than the thickness of the upper confining layer and/or where wells and latrines are adjacent. But organic matter in latrines cannot be the main driver of As release to ground water because it is free of As in many areas of Bangladesh, despite the fact that the country is densely, and fairly evenly, populated. Fig. 14. Schematic of the relationship of peat basins to the occurrence of As enrichment in groundwater. Soluble organic matter to drive FeOOH-reduction can invade the aquifer only at the margins of peat basins, where the sediments are sufficiently permeable to allow its passage downwards in recharging ground water and the sedimentary source of OM is sufficiently abundant to supply it. Towards the basin centre, permeability decreases; away from the basin, the abundance of organic matter decreases: in both cases, enrichment of As decreases. 1280 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Where the depth of dug (pit) latrines is much less than the thickness of the upper confining layer, where wells are far from latrines, where sewage collection is engineered, or where the unsaturated zone is very thick (as it is on parts of the Madhupur and Barind Tracts), the impact of human sources of organic matter will be small, or negligible. Such is the case in JAM, where most dwellings have concrete-lined holding tanks with tops above ground level and where the depth of the upper confining layer is between 9 m (at FP and DP) and 24 m (at DP). Several considerations suggest that there is little connectivity between latrines and aquifer water in JAM. Firstly, thermotolerant (faecal) coliforms (presumptive Escherichia coli) are bacterial indicators of faecal pollution. Despite the very high risks of faecal pollution from poor well-completion, open access of many wells to animals, and the fact that there is generally a broad relationship between log(TTC) and sanitary risk scores (Cronin et al., 2003), the study wells mostly have a low TTC count (Table 3; summary statistics in Table 4): about 85% of the wells contained <10 cfu/100 ml of water. Furthermore, there is no correlation between TTC count and As concentrations in JAM wells. Given the high number of observable risks to JAM wells, their low TTC counts probably stem from the fact that most are >30 m deep, are sealed around the annulus by the clayey silt of the thick ( P 9 m thick) upper confining layer, and many have concrete completions of good quality, although the completions are mostly small in diameter. These factors reduce access of microbially polluted water into the aquifer via the annulus of the well. The implications of these results is that microbial contamination of the aquifer in the JAM area is on a small scale and so organic pollution of the aquifer by human waste is limited in volume and distribution. Hence, it is unlikely that faecal organics do much to drive reduction of FeOOH in JAM aquifers. sources of organic matter (rivers, ponds, and soils), drawn into the aquifer by irrigation drawdown, as has been suggested to occur in Bangladesh (Harvey et al., 2002). The authors note, however, that the occurrence of CH4 in combustible amounts around 30 m depth in Munshiganj, but in lessening amounts as depth diminishes (Harvey et al., 2002), and the presence of CH4 in ground water elsewhere in the Bengal Basin (Ahmed et al., 1998), can be due only to the presence of subsurface organic matter in high concentrations. Furthermore, if surface sources of organic matter in the Bengal Basin were of much importance in driving the release to ground water of large amounts of As, high As concentrations would not be confined to about 25% of wells (DPHE, 1999), or to Holocene sediments (it is largely absent from the Barind and Madhupur Tracts; DPHE, 1999; McArthur et al., 2001; Ravenscroft et al., 2001), nor would they be highest around 20 to 40 m depth across most of the Bengal Basin (Karim et al., 1997; McArthur et al., 2001; Ravenscroft et al., 2001; Harvey et al., 2002; van Geen et al., 2003a). Finally, although the 14 C ages of Harvey et al. (2002) for DIC show that very young water is penetrating to depths of around 20 m, the piezometer profiles of those authors show that concentrations of dissolved organic carbon (DOC) increases with depth to around 30 m depth, below which they scatter but remain high to 80 m depth, thereby proving the DOC has a deep source. The As maximum (and a maximum concentration of CH4 ) at 35–40 m depth cannot be caused by organic matter migrating downwards against a concentration gradient and then reacting at depth. The discrepancy noted by Harvey et al. (2002) between young 14 C ages for DIC and older 14 C ages for DOC is explicable in terms of a reservoir age for DOC and its derivation from older buried peat. 6.2.2. Surface sources of organic matter For reasons given above, it seems unlikely that reduction of FeOOH in the JAM area is driven by surface The results given here provide strong evidence that reduction of FeOOH across the Ganges Plain is driven overwhelmingly by buried, natural, organic matter. 6.3. The capacity to reduce FeOOH Table 4 Summary table of content of thermotolerant coliform bacteria in waters from wells, ponds, and piezometers, from Joypur, Ardivok and Moyna (JAM), and Mirathi, West Bengal, India, (March 2003). Colony forming units (cfu) per 100 ml. TNTC ¼ too numerous to count Summary statistics JAM wells Mirathi wells JAM ponds JAM piezos Numbers are cfu/100 ml Number, incl. repeats Maximum TTC Minimum TTC Non-detects (TTC ¼ 0) Mean TTC counts in wells with positive detects 49 63 0 23 10 15 65 0 1 14 11 TNTC 4400 0 – 21 TNTC 0 1 – Notes. cfu is colony-forming units. TTC is too numerous to count. J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 According to the equation for reduction of FeOOH, where FeOOH is used to represent Fe oxides 8FeOOH þ CH3 COOH þ 14H2 CO3 $ 8Fe2þ þ 16HCO 3 þ 12H2 O ð1Þ Given: the stoichiometry of this reaction; a density of 1 for buried peaty sediment; a C content of 7% in the sediment; a thickness of 50 cm for a peaty layer (as at 29.5 m depth in FP) with half the C bioavailable; and a porosity of 40%, then there will be about 1.1 g of bioreactive C under each cm2 of aquifer. The organic matter will consume 25 g of FeOOH which, given a porosity of 40%, a density of FeOOH of 2.4, and an aquifer containing 0.2% FeOOH, will be contained in an 8 m thickness of aquifer sand, which is half that of the aquifer thickness at FP below the peaty layer at 29.5 m depth, and is similar to the thickness in which concentrations of As exceed 900 lg L1 . Towards the base of the aquifer at FP, concentrations of As decrease whilst remaining high. Our calculation is very basic, but suggests that few aquifer volumes of water have passed vertically through the sands: had they done so, the organic matter, or the FeOOH, would be exhausted by now and the aquifer would be clear of both dissolved Fe and dissolved As. Although approximate, the calculations above suggest (and observation confirms) that there is enough organic matter in the aquifers in JAM for the entire aquifer thickness to be at risk: confusing the calculation are 3 factors. Firstly, the organic matter in the sediments at JAM is P 1.2 ka old (Hudson-Edwards et al., 2004), so it has been degrading for a substantial period of time. On deposition of the sediment, the amount of organic matter would have been greater and it would have been fresher, and more biodegradable, than it is now. Secondly, there is some uncertainty regarding the estimate of 0.2% for the amount of FeOOH in the aquifer. This figure represents the Fe, recalculated to FeOOH, that is leachable by ammonium oxalate at pH 3 (Table 5) from sands in mid-aquifer at site DP, which is the least reduced of the 3 piezometer sites (Figs. 3–5 and 10). The figure of 0.2% is a maximum because the leaches contained P, S, Al, Fe, Mg, (but not Ca, because Ca oxalate is insoluble) and it is not possible to apportion the leachable Fe between FeOOH and possible diagenetic phases formed by its partial reduction viz. magnetite, green rust, vivianite, siderite, Fe sulphide (Lovley et al., 1987; Frederickson et al., 1998; Chakraborti et al., 2001; Glasauer et al., 2003), or to possible detrital phases such as Fe-rich chlorite. Thirdly, the formation of magnetite (Fe2þ Fe3þ 2 O4 ) by reduction of sedimentary FeOOH reduces only one-third of the available pool of Fe(III) whilst sequestering the remaining two-thirds in the mineral structure (Frederickson et al., 1998) where it 1281 may be unavailable for reduction. Thus, the availability of FeOOH to sorb As in Bangladesh sediments will be some 70% less than supposed if magnetite is the main product of FeOOH reduction across the Ganges Plain. It can be inferred from the results of Harvey et al. (2002) that magnetite comprised most of the Fe that was extractable with oxalic acid solution from aquifer sediments from, Bangladesh, and so magnetite constituted up to 6% of total-Fe (although this inference is compromised by uncertainty about whether the mineral formed by oxidation of green rust during storage, or by reduction in the aquifer). 7. Other mechanisms of As release to ground water This study confirms that reduction of FeOOH is a good model for the release of high concentrations of As to anoxic ground water in the Ganges Plain and, by implication, worldwide, since reduction of FeOOH is not a site-specific process. Other mechanisms, such as reductive dissolution of Mn oxides (Smedley and Kinniburgh, 2002), competitive exchange with HCO3 (Appelo et al., 2002), and pH-dependent desorption from, or recrystallization of, FeOOH (Welch et al., 2000; Smedley and Kinniburgh, 2002), are not needed. The reasons why are discussed below. 7.1. Reduction of Mn oxides In JAM wells, concentrations of dissolved As are low (<33 lg L1 ) where concentrations of Mn exceed 1 mg L1 (Table 3; Fig. 8(a)). The reduction of Mn oxides is thermodynamically more favourable than is the reduction of FeOOH (Edmunds, 1986; St€ uben et al., 2003) and occurs before it. It follows that As, originally sorbed to Mn oxides but released by reduction, must be resorbed to FeOOH (or residual Mn oxides) rather than be released to ground water (St€ uben et al., 2003). This finding is contrary to that of Smedley and Kinniburgh (2002), who state that ‘‘Manganese oxides also undergo reductive desorption and dissolution and so could contribute to the As load of ground waters in the same way as Fe’’. 7.2. Exchange of sorbed As with HCO3 in ground water Potentially toxic concentrations of As in ground water are not common, and are particularly uncommon in potable water from oxic aquifers, in which HCO3 is the commonest and most abundant anion worldwide: it follows that the suggestion that As is released to solution by competitive exchange with HCO3 (Appelo et al., 2002) is unlikely to be correct. Ground waters from a variety of aquifer types in the UK contain HCO3 in concentrations of hundreds of m L1 , yet As concen- 1282 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Table 5 Provenance, and content of oxalate-extractable Fe, of sediment from aquifers in alluvial and deltaic settings worldwide Setting Source of sediment Nature of source % of oxalate-extractable Fe Min Max Data source Bangladesh West Bengal Himalayas Himalayas Ig, Met Ig, Met 0.1 0.05 0.5 0.16 West Bengal Himalayas Ig, Met 0.12 0.12 Campania, Italy Sarno & Latari Mountains Allegheny Mountains Ig, Sed (Lmst) 0.83 1.6 DPHE (1999, 2001) This work, site DP, oxidised This work, site AP, oxidised Adam et al. (2003) ? 0.4 41 Knapp et al. (2002) Coastal Virginia, USA Huhhot Basin, Inner Mongolia North Island, New Zealand NW Mississippi Lower Kissimmee Basin, Florida Da Qing & Man Han Mountains North Island uplands Met 0.67 3.4 Smedley et al. (2003) Ig, Sed. 3.7 8.1 Mississippi uplands Florida uplands Sed Sed Lmst? 1.2 0.07 1.9 0.25 Nguten and Sukias (2002) Rhoton et al. (2002) Reddy et al. (1995) trations are typically <10 lg L1 and natural concentrations above 50 lg L1 are rare (BGS, 1989). Moreover, concentrations of HCO3 in JAM are highest (744 mg L1 at site DP, Table 2; Fig. 3) where the ground water contains less than 5 lg L1 of As. Furthermore, the large range of As concentrations at AP and FP bear no relation to concentrations of HCO3 (Figs. 4, 5): this is best seen in the lower part of the aquifer at AP (Fig. 15), where concentrations of As decrease from 419 lg L1 (38.3 m) to 7 lg L1 (42.8 m) whilst pH varies by less than 0.2 units, HCO3 increases from 526 to 622 mg L1 , dissolved silica (as H4 SiO4 ) varies by only 4 mg L1 , dissolved PO4 decreases from 2.3 to 0.13 mg L1 , and dissolved Fe decreases from 3.9 to 0.97 mg L1 . These data emphasise the minimal role played by pH, HCO3 , and silica, in affecting concentrations of As in ground water in JAM and, by implication, in sediments elsewhere. Competitive exchange with natural organic matter (Redman et al., 2002), or other ligands (e.g. silica), may become important when considering mechanisms of As release that lead to concentrations of As of <50 lg L1 but, even at low concentrations, partial reduction of FeOOH and release of kinetic As (As released by reduction but soon resorbed to residual FeOOH, Fig. 13) may be adequate to explain low concentrations of As in anoxic environments. 7.3. Desorption of As induced by increasing pH Sorption of As to solids, notably sedimentary FeOOH, is generally held to be pH dependent, leading to the suggestion that desorption of As from mineral surfaces may contribute As to ground water as pH rises (Welch et al., 1988, 2000; Robertson, 1989; Smedley and Kinniburgh, 2002). A general observation that waters of higher pH tend to have higher concentrations of As (Welch et al., 1988; Robertson, 1989) is not in itself adequate to prove a causal connection between the two: other factors, such as residence time, evaporation, and weathering, may cause an increase in both. Whilst pHdependent desorption may release As to ground water in plumes of landfill leachate, or hydrocarbons where pH can exceed 10, there is little direct evidence that this process releases As to potable ground water, in which pH rarely exceeds 8.5. In waters from JAM, As concentrations range from <1 to 1180 lg L1 , despite the narrow range of pH (6.8–7.3; Table 3), a fact best illustrated by data from the base of the AP site (Fig. 15). It follows that pH is not a control on any sorption or desorption of As that may occur in JAM aquifers. Arsenic concentrations were linked to pH by Welch et al. (1988): the mechanism of pH-dependent desorption was used by Robertson (1989) to explain the presence of As in oxic potable ground water from 24 alluvial basins in Arizona, and has since been widely invoked. Robertson (1989) noted that concentrations of As increased as pH increased along flowlines in aquifers in several alluvial basins, and assumed the relation was causal. Many indicators in his data suggest otherwise: in Verde Valley ground waters, As concentration decreased with increasing pH (as he noted); the overall correlationcoefficient between pH and As concentration for his 24 sites was low (0.18, n ¼ 456; his Table 2); salinity increased along flowlines by a factor of between 4 and 8 (judged from his Fig. 3) suggesting either extensive evaporo-concentration, mixing with deeper more saline waters, or weathering, occurred as the water passed down the aquifer. It seems likely that As accumulated in J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 1283 Fig. 15. Detailed profiles of water composition in the basal aquifer at Site AP, showing the invariance in pH and in the concentrations of HCO3 , and H4 SiO4 , in contrast with the change in concentration of dissolved As, PO4 and Fe. solution along some flowlines in response to in-situ weathering of volcanic rocks, and that the accompanying increase in pH was a consequence of such weathering, not a cause of As accumulation. Robertson (1989) noted that ‘‘the basins containing the largest concentrations (of As) are bounded primarily by basalt and andesite’’ and so the basin-fill must be weathered materials from those rocks, which may have moderated the concentrations by resorption to weathered sediments. Robertson’s (1989) observation that concentrations of As correlated well with concentrations of Mo, F and V, at sites examined in detail, parallels the observation of Nicolli et al. (1989) that As, fluorine, and V, are associated in As-enriched ground water in central Argentina; both the association and the As enrichment were ascribed by Nicolli et al. (1989) to the weathering of dacitic volcanic ashfall layers in the loess aquifers. The relation of pH to As in solution can be further illustrated by reference to ground waters in aquifers of metamorphic bedrock in New England, USA (Ayotte et al., 2003). Higher concentrations of As (>10 lg L1 ) occur in waters with higher pH (7.5 to 9.3), but many samples, with pH ranging from 5 to 8.8, contain <2 lg L1 of As (Fig. 4 of Ayotte et al., 2003): this would not be the case if As concentrations were linked to pHrelated desorption. These authors note that the primary control on As concentrations is bedrock type. Furthermore, concentrations of As in UK aquifers are not related to pH (BGS, 1989): concentrations of <4 lgl occur in aquifers with a pH range from 7.0 to 9.5 (e.g. the Lincolnshire Limestone, Table 5.3 of BGS (1989)) and concentrations rarely exceeded 50 lg L1 , whatever the pH. Thus, the observation made by Welch et al. (1988) of a weak positive correlation between pH and concentrations of As in a large dataset of compositions of US ground waters may have explanations other than pH-induced desorption. Finally, Dixit and Hering (2003) show that sorption of both As(III) and As(V) to Fe oxides becomes less sensitive to pH as As concentrations, and Asaq /Fesediment ratios, decrease. At their lowest As concentration (750 lg L1 ), which approaches those found in the Bengal Basin, these authors showed that sorption is almost independent of pH over the range of pH common in most ground waters. Their data confirm that pH-dependent sorption does not contribute significantly to the release of As to anoxic potable ground waters of the composition, pH-range, and salinity typical of potable waters from alluvial aquifers. 8. The Himalayan connection 8.1. The Bengal Basin The authors postulate that the widespread occurrence of As concentrations above 100 lg L1 occurs only when sorption sites for As in an aquifer are saturated i.e. when reduction of FeOOH has gone largely to completion. Because high concentrations of As are common in Holocene sediments, they evidently have a particular susceptibility to undergo complete reduction of FeOOH. the authors explain this as follows. The Holocene sediment in the Bengal Basin derives from Himalayan rocks 1284 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 that are (mostly) igneous and metamorphic. Other than adjacent to the Indo-Burman ranges, surface coatings of FeOOH, with sorbed As, are not inherited from previous erosional cycles but must be created from crystalline rocks by weathering. The aquifer sands largely derive from erosion at a time of extended glaciation in the Himalayas when physical weathering dominated and the intensity of chemical weathering was limited by the low (glacial) temperatures during erosion. The immature sediment was transported rapidly to the lower reaches of the basin and buried in a waterlogged, anoxic environment and so had little opportunity to acquire much FeOOH through oxidative weathering. Further, much of the fine fraction was winnowed during transport and deposited offshore, thereby reducing the surface area of sediment available to host coatings of FeOOH. The abundance of fresh-looking muscovite and biotite in the Holocene sediments attests to the limited degree of chemical weathering they have undergone. Older sediments, such as the deep aquifer in Bangladesh, and sediment at DP, and below 32 m depth in JAM, has suffered more weathering. In Table 5, the content of diagenetically available Fe (as acid-oxalate leachable Fe) in oxidised brown sediments from JAM (from the base of the aquifer at AP, and from DP) is compared to that in sediments from elsewhere and shown to be low (<0.2%) in comparison. As the brown sediment at depth in JAM is of pre-Holocene age and has probably suffered a period of oxidative weathering (that would have increased its content of FeOOH), the original Holocene sediment may have contained even less. Admittedly, such comparisons are fraught with uncertainty: the leachable Fe may be in vivianite, green rust, magnetite, Fe sulphides, chlorite, and carbonate, as well as in oxides. Nevertheless, this data lends some support to the idea that ground water in the Bengal Basin is polluted by arsenic because its sediments contain little FeOOH (to sorb As) but have a superabundance of organic matter with which to reduce it, so total reduction is common. In this context, the comment of Harvey et al. (2002, p. 1604), that greycoloured sediment in an As-enriched profile in the Ganges Plain ‘‘contains little, if any, purely ferric [Fe(III)] oxyhydroxides’’, confirms that, where high concentrations of As are common, reduction of FeOOH has gone effectively to completion. 8.2. Implications for other areas In aquifers that contain much FeOOH and/or little organic matter, reduction of FeOOH will be incomplete and the release of As by reduction will be moderated by resorption to residual FeOOH. Such a process may be moderating As concentrations in groundwater in, for example, the alluvial aquifers of the Tualatin Basin, Oregon (Hinkle, 1997), where ground waters contain modest concentrations of As (<1–73 lg L1 ), and where those that exceed 50 lg L1 also contain some Fe (0.16– 1.9 mg L1 ) and PO4 (0.36–2.0 mg L1 ). Hinckle and Polette (1999) speculated that FeOOH reduction was one plausible mechanism of As release to ground water in reduced ground waters of the basin. The authors agree, but add that resorption of As to residual FeOOH has probably kept its concentration low. A more extreme example of resorption control may occur in alluvial aquifers of eastern Arkansas, where ground water contains up to 42 mg L1 of dissolved Fe, but less than 52 lg L1 of As (Kresse and Fazio, 2003). Although the aquifer sediments contain lignite and peat, the amount of organic matter available (i.e. biodegradable) appears to have been insufficient to reduce all of the sedimentary FeOOH, so concentrations of As are probably being moderated by resorption to unreduced oxides. Conversely, the Song Hong (Red River) Basin, in the vicinity of Hanoi, Vietnam, appears to be another basin where a superabundance of peat (up to 5 m in thickness; BGS, 1996) occurs in the area most poisoned by As (Fig. 16) and gives rise to ground water compositions similar from those in the Bengal Basin (Fig. 17): ground water in both areas is rich in As, Fe, NH4 and PO4 , and probably for the same reason – the organic matter in the Song Hong basis is abundant enough to drive FeOOH reduction to completion. Lesser amounts of peaty sediment occur elsewhere in the Song Hong delta (Tanabe et al., 2003) making it likely that toxic concentrations of As are widespread in the area’s ground water. 9. Temporal trends in As concentrations 9.1. Reasons why the composition of ground water may change with time All ground waters change their composition with time: the important question is, at what rate? In the Bengal Basin, some reasons for real change are clear from the chemical stratification of the ground water: water, and hence As, may move by bulk flow, both vertically and horizontally, and so redistribute As. With vertical gradients in As concentration of up to 200 lg L1 m1 (at FP in JAM), a small migration of the As front will severely poison aquifer levels that are presently safe to exploit. Additionally, seasonal fluctuations of the water table might affect the As concentration of wells screened near the water table, although this has not been observed over a monsoonal cycle elsewhere (van Geen et al., 2003a). In the longer term, redox reactions may deplete the store of As, or FeOOH, or redox driver, in the sediments and so cause the water composition to change with time. One thing that is unlikely to cause change is a change in the rate of ground water flow. Microbial reduction of FeOOH J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 occurs on a timescale of hours to days (e.g. Dowdle et al., 1996; Jones et al., 2000; Zobrist et al., 2000), as do in-situ redox reactions in the Ganges Plain (Harvey et al., 2002). Such rates are rapid in comparison to rates of ground water movement in the Ganges Plain, which are typically in the range of a few centimetres per day or less (Ravenscroft et al., 2001; Harvey et al., 2002), and so are likely to be de-coupled from rates of ground-water flow. Some reasons for apparent change with time in ground water composition include: (1) improving well-location as understanding of As pollution improves; (2) poor or inconsistent techniques for sampling, sample storage, or analysis; (3) inappropriate purging volume; (4) misidentification of the well; (5) transcription errors. 9.2. Long-term changes in the Bengal Basin There are insufficient time-series data to demonstrate or disprove a long-term change in water composition with time in the Bengal Basin, but tentative conclusions 1285 on long-term trends in composition can be inferred from data on the ages and As concentrations of wells documented in regional surveys (e.g. DPHE, 1999, 2001; van Geen et al., 2003a). For the shallow aquifer of Bangladesh, the proportion of wells falling above various threshold concentrations of As, plotted as a function of well age (Table 6; Fig. 18), suggests that, for the first ten years of existence, the exceedance for most threshold values of As increases as well-age increases, irrespective of region or threshold. A similar effect is not seen in data for Na (Fig. 18), an element that is largely conservative in solution, unaffected by redox transformations, and so is used as a comparator. For ages greater than 10 a, the large uncertainties in the proportion of wells exceeding a given threshold, which arise as a result of the paucity of older wells, makes uncertain the validity of an apparent increase in concentrations towards older ages. Similar deductions were made for a data set of 6 000 wells from the region of Araihazar (van Geen et al., 2003a), but were based on linear regression to highly skewed data (without log-transformation) and so may not be robust. The trend to 10 a seen in the DPHE data probably does not reflect the fact that an increasing awareness of the As problem might have lead drillers to site wells more Fig. 16. Comparison of the distribution of As in ground waters around Hanoi (after Berg et al., 2001) with the distribution of peat deposits (after BGS, 1996). The inner box shows the extent of the BGS map, and the lightly stippled area is where peat is >5 m thick. There is some indication that As enrichment is greatest where the peat is found, and further indication that the enrichment is greatest where the peat is thickest, to the immediate south of Hanoi. 1286 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 JAM Area, West Bengal Hanoi Area, Viet Nam Faridpur Area, Bangladesh Mn mg L -1 4 3 2 1 0 0 5 10 15 0 20 5 Fe mg L-1 10 15 20 0 5 Fe mg L-1 10 15 20 Fe mg L-1 PO4 mg L -1 12 10 8 6 4 2 0 0 5 10 15 SO4 mg L -1 NH4 mg L 0 20 5 -1 10 15 NH4 mg L 0 20 5 -1 10 15 NH4 mg L 20 -1 40 30 20 10 0 0 200 400 600 HCO3 mg L 800 0 200 -1 400 600 HCO3 mg L -1 800 0 200 400 600 HCO3 mg L 800 -1 Fig. 17. Comparison of the concentrations of Fe, Mn, PO4 , NH4 , SO4 , and HCO3 in wells tapping the As-affected shallow aquifers of Bangladesh (data from DPHE, 1999), West Bengal (this work) and the Song Hong (Red River) Basin around Hanoi (data from BGS, 1996, which did not report concentrations of As). All 3 areas contain waters rich in Fe, As, PO4 , and NH4 , which shows the influence of much reduction of FeOOH and organic-matter degradation. carefully than before – perhaps on the basis of sediment colour. Two considerations suggest that the trends may be real. Firstly, sampling was competed by 1998, when the As problem was not widely known about in rural areas. Secondly, there is a distinct difference in trend between redox-sensitive As and the more conservative tracer Na (Cl was not analysed for the national survey of Bangladesh, so could not be modelled). In the deep aquifers of the Bengal Basin, the establishment of anoxic conditions in the deep aquifer, as a consequence of the deposition of overlying, organic-rich, Holocene aquifers, has been sufficient to mobilise some Fe into groundwater in the deep aquifer (Table 3; DPHE, 1999), but has been insufficient to reduce the entire pool of FeOOH, which may now act as a sorptive buffer to prevent P or As from accumulating in solution. If FeOOH in the deep aquifer of the Bengal Basin does host sorbed As (i.e. the deep aquifer has not been flushed of it – see earlier), the potential exists for downwardmigrating organic matter to eventually reduce all its FeOOH and so mobilize As. The data in Table 7, compiled from DPHE (1999), suggest that this is not yet happening. In the most As-polluted enriched areas of Bangladesh, deep wells have remained free of As for up to 16 a. 9.3. Short-term trends in JAM In order to examine whether wells in the JAM area changed over the short time encompassed by the present study, the wells were sampled on 3 separate occasions over 18 months; in February, 2002, November, 2002, and March, 2003. In the most precise data viz. electrical conductivity, Ca, Mg, and Cl, some differences are seen in water composition between samplings. Changes are small and best illustrated by the correlation in relative J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 0.523 0.597 0.45 0.184 0.036 0.921 0.963 0.968 0.899 0.665 24 55 66 22 7 Depth <100 m: As 25 0.173 50 0.139 100 0.076 200 0.002 400 )0.019 0.587 1.406 0.645 0.204 0.041 0.944 0.985 0.982 0.921 0.736 34 143 120 28 9 Barisal: As 25 50 100 200 400 )0.135 )0.093 )0.044 )0.014 0.001 0.51 0.415 0.425 0.346 0.1 0.797 0.786 0.858 0.899 0.925 11 10 16 22 29 Khulna: As 25 50 100 )0.222 )0.333 )0.318 0.535 0.457 0.26 0.713 0.682 0.581 8 7 6 Chittagong: As 25 )0.199 100 )0.121 0.661 0.687 0.895 0.951 23 49 Rajshahi: As 25 0.103 0.772 9 0.416 0.329 0.233 0.15 0.568 0.505 0.391 0.451 4 4 3 4 change between these determinands (Fig. 19). The correlation between Ca and Mg might be an analytical artefact – despite the authors’ belief in its reality–because both were measured on the same solution, with the same ICP-AES, at the same time. The measurements of Cl and electrical conductivity were made independently of Ca; Cl by ion chromatography in the laboratory, and electrical conductivity by conductivity meter in the field. The correlation between Ca and these determinands must therefore be real. No changes were seen in con- 0.8 0.6 0 0.2 0.4 0.6 0.8 As > 50 µg L -1 a = 0.139 b = 0.146 c = 0.985 All wells L -1 0.4 0.6 0.8 Na > 60 mg a = - 0.199 b = 0.329 c = 0.505 0.2 Khulna: As ¼ 200, 400; no fit. Chittagong: As ¼ 50, 200 400; no fit. Rajshahi: As ¼ 50, 100, 200, 400; no fit. t90 ¼ time in years to reach 90% of the plateau value. Wells < 100m deep 0 1 All wells: Na mg L 40 )0.048 60 )0.199 100 )0.281 200 )0.182 0.4 All Wells: As lg L1 25 0.159 50 0.122 100 0.068 200 )0.005 400 )0.025 )0.001 t90 -1 0.2 c As > 50 µg L a = 0.122 b = 0.597 c = 0.963 0 b Proportion over threshold a Proportion over threshold Threshold value All wells: Proportion over threshold Table 6 Non-linear models for the proportion of wells exceeding given threshold concentration of As, as a function of age. The data are fitted with the function P ¼ b ðb aÞct , where P is the proportion exceeding a given threshold, a is the intercept, b is the plateau value, c is a fitted constant and t is time in years. Data from DPHE (2001) 1287 0 10 20 30 Well age in years Fig. 18. Trend of As concentration in wells with well age, represented by a model showing the proportion of wells above a threshold concentration as a function of time. Fitted numerically by the function [proportion over threshold] ¼ bðb aÞct , where a is intercept, b is plateau value, c is a constant and t is time in years. J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 centrations of As: the analytical repeatability for As analysis was around 10%, so the magnitude of any changes must be less than this amount; the data do not rule out changes in As concentration, but merely constrain the rate of change. Indeed, the piezometer profiles suggest that the depths at which ground water is enriched will increase as organic matter moves downwards through the aquifer, consuming FeOOH, although subsequent migration of the As released may be slowed by adsorption to such phases elsewhere in the aquifer. 4 Ca % difference 1288 2 0 -2 -4 -5 -4 -3 -2 -1 0 1 2 3 4 5 Mg % difference 9.4. Changes induced by irrigation pumping in the Bengal basin Ca % difference 4 2 0 -2 -4 -5 -4 -3 -2 -1 0 1 2 3 4 5 25 30 Measured EC % difference 4 Ca % difference An important issue is whether pumping for irrigation is spreading As through the shallow aquifers of the Bengal Basin, or is flushing them of As by inducing additional recharge. The authors believe the latter is the case. This contention may be best illustrated by site FP in the JAM area (Fig. 5). The piezometer profile shows that As concentration increases steeply between 31.2 and 37.0 m depth (or, given that there are only two control points, somewhere between). Given the large amount of organic matter at 29.5 m depth, the increase should occur across this unit and give higher concentrations of As at 31.2 m. That it does not suggests that the entire profile has been translocated downwards by between about 2 and 9 m by abstraction of water from shallow and deep aquifers. Such movement would also have lowered As concentrations above 19.2 m depth (Fig. 5) by drawing in new water to give the present profile. Water above 19.2 m depth has concentrations of Ca and Na quite different to those in deeper waters, adding support to this suggestion. Perhaps more convincingly, the depth profiles of Harvey et al. (2002), for groundwater in, Bangladesh, show that concentrations of dissolved constituents derived from differing sources (e.g. As from reduction of FeOOH; dissolved organic C, NH3 and CH4 from organic matter degradation; Ca from mineral weathering; HCO3 from multiple sources) all increase with depth to a peak around 35–40 m depth. Such a concordance of profiles chemically different species can be induced only by flushing, not by reactions in the aquifer. In this context, it is interesting to compare the profiles in Munshiganj and JAM. 2 0 -2 -4 -15 -10 -5 0 5 10 15 20 Cl % difference Fig. 19. Percentage difference in Ca concentration in wells, sampled in February 2002 and March 2003, plotted against the % change in Mg concentration (both measured by ICP-AES), the % change in EC (field measurement) and the % change in Cl concentration measured by ion chromatography. In JAM, As concentrations reach a maximum at depth in FP (Fig. 5) because of the OM-rich layer at 29.5 m depth. At AP, however (Fig. 4), the maximum is not Table 7 Summary of As concentrations, depth, and age, for deep wells in As-affected areas of Bangladesh. Data from DPHE (2001) Area No. of deep wells Age range in 1998 Range of As lg L1 Depth range (m) % of shallow wells with >50 g L1 As Chandpur Lakshmipur Noakhali 4 6 6 2–3 3–10 5–15 0–6 0–8 2–10 221–269 183–318 246–292 98 64 79 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 developed strongly (if data from the upper aquitard are excluded). The progression from a ‘flat’ profile, similar to that seen at AP, to the ‘peaked’ profile seen in Munshiganj can be achieved by depth-dependent flushing, which would lower the concentration of all species in the upper part of the aquifer (Fig. 20). Harvey et al. (2002) propose or imply, instead, that the shape of many element profiles results from redox reactions that are driven by organic matter from surface sources; this is unlikely, as dissolved species move down, not up, concentration gradients, and organic matter cannot move downwards and increase in concentration as it is being consumed by reaction. The suggestion of a surface source of organic matter is also at odds with the distribution of 14 C-ages, as discussed by van Geen et al. (2003b), who point out that Harvey et al. (2002) report measurable amounts of 14 C to 19 m depth but not below. A maximum in dissolved As concentration around 20–40 m depth is common across the Bengal Basin (Karim et al., 1997; DPHE, 1999; McArthur et al., 2001), but not universal: for example, in Araihazar, Bangladesh, it appears to be around 15 m depth (Fig. 6 of van Geen et al., 2003a), whilst in the region of Sylhet, it is around 60 m and may even be bi-modal, although as a result of differential subsidence, not pumping (Ravenscroft et al., 2001). The explanation that shallow maxima in As concentrations are caused by flushing of shallow levels in the aquifers seems reasonable because natural flow, and abstraction, particularly for irrigation, are confined to shallow depths – natural flow because of the lack of topography and the lack of natural outlets at deeper levels, the irrigation flow because shallow irrigation wells are at shallow depths in order to minimize the cost of installation. Ground water dating by Dowling et al. (2002) confirms that young water may be found to depths of 30 m in the Bengal Basin. If correct, the authors’ explanation for peaked profiles of As shows that the extensive development of ground water for irrigation is reducing the As problem in the ground water across the Bengal Basin. 1289 ated by resorption to residual FeOOH, frequent high concentrations of As (>100 lg L1 ) require the accumulation of organic matter in amounts sufficient to exhaust the entire store of FeOOH in a location that is in hydraulic continuity with aquifer sands. This condition is most easily met on the margins of buried peat basins, where organic matter accumulates in low percentage abundance in moderately permeable confining units or in the aquifer sands themselves, or where significant contributions from human and animal organic loading exist locally. As first proposed by P. Ravenscroft, as author of DPHE (1999, V1–3, 5, 6), by Ahmed and Ravenscroft (2000) and Ravenscroft et al. (2001), and since repeated by Smedley and Kinniburgh (2002), the global rise in sea-level between 18 and 6 ka, created settings worldwide for As-enriched aquifers to form. The large volume of accommodation space that resulted is now filled with recent sediments. The rates of sea level rise and sediment supply have been the primary controls on wetland formation, where organic-rich muds and alluvial or deltaic sands are closely juxtaposed. The problem of As in ground water will therefore be most prevalent in depositional areas where climate was hot and wet during the 10. Wider considerations The process of FeOOH-reduction is not site specific: it can explain the As problem in many sedimentary basins, both coastal and continental, because Fe oxides and sorbed As are ubiquitous components of clastic sediments. Whether the reduction of FeOOH will push As concentrations to hazardous levels depends on how near the process goes to completion. That, in turn, is governed by the balance between the amount of FeOOH and the amount and reactivity of organic matter available to reduce it. Whilst concentrations of up to a few tens of lg L1 of what the authors have called kinetic As may prove common where concentrations are moder- Fig. 20. Development of a peaked profile in As concentration as a result of abstraction for irrigation and public supply. Heavy pumping of the shallow aquifer for irrigation would enhance the rate of renewal of water in the aquifer to the depth of pumping. From an initial profile, A, similar to that seen in JAM at site AP, would develop a peaked profile, B, similar to that seen in Munshiganj by Harvey et al. (2002), by rapid renewal of water in the upper aquifer (shaded area between A and B) to a degree that diminishes with depth. Pumping from the deep aquifer (not shown) beneath the lower aquitard would induce slow translocation downward of the entire profile B. Localized OM-rich sediment shown as black stringers. 1290 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 climax of nearshore sediment deposition around 4–8 ka and where there was limited chemical weathering in the upper catchment. A set of globally applicable parameters–the depth of pre-Holocene incision, the relative rates of sea level rise and alluvial aggradation, the Holocene climatic record, and the geology of the upper catchment–therefore provide a general model for predicting the possible occurrence of As in coastal alluvial basins. Inland basins lack the drainage control imposed by global sea-level, so inland basins will be subject to local base-level controls and application of the outline model to such areas may be difficult. In a global context, the physical and demographic characteristics of the Bengal Basin represent an extreme, in which the extreme topography of the hinterland, its weathering under extremes of climate, and its crystalline geology, have conspired to exacerbate the pollution by minimising chemical weathering and production of FeOOH. Other areas that might be expected to suffer from the As problem, although perhaps to a lesser degree, include the deltas of tropical rivers such as the Mekong, Irrawaddy, and Chao Phraya rivers, and the coastal plains of Java and Sumatra. 11. Conclusions In the Bengal Basin, As is released to solution by reductive dissolution of FeOOH and release of its sorbed As to ground water. In the study area near Barasat in southern West Bengal, and by implication elsewhere in the Bengal Basin and worldwide in alluvial aquifers, the reduction is driven by natural organic matter buried in sediments. In JAM, the organic matter exists in moderate percentage abundance within the upper confining units of the aquifers and/or in discrete organic-rich units within aquifer sands. The organic matter accumulated in peat basins; the portion that drives reduction in the aquifer is that which accumulated on the margins of the basin where the sediments are sufficiently permeable to allow infiltration to the aquifer of the soluble organic products of fermentative degradation of the organic matter. Within the JAM area, which is typical of much of the southern Bengal Basin, surface sources of organic matter do not contribute to the reduction of FeOOH and so do not result in ground water containing high concentrations of As. In areas of the Bengal Basin where dug latrines penetrate the upper confining layer, sewage in hydraulic continuity with the aquifer may locally promote additional reduction of FeOOH and add to the As problem. Unless populations reach an urban density, however, this process will not exacerbate the regional As problem but will operate at the scale of individual wells. Where the reduction of FeOOH goes to completion, As concentrations often exceed 100 lg L1 . Where reduction of FeOOH is incomplete, concentrations of ki- netic As in solution are moderated (to <100 lg L1 ) by resorption to residual FeOOH. The extent of As mobilisation will be controlled by the balance between the quantity and biodegradability of organic matter, and the size of the FeOOH buffer available to sorb As. Reduction of FeOOH in the Bengal Basin commonly goes to completion because the sediments contain abundant natural organic matter to drive reduction but little FeOOH, owing to the immaturity of the sediment, which is produced and transported rapidly from a crystalline Himalayan source to its place of accumulation in the anoxic environment of the Bengal Basin. Acknowledgements We are indebted to the inhabitants of Joypur, Ardivok, and Moyna for allowing us to sample their wells. Particular thanks are due to Ali Ahmed Fakir, for his assistance over several years with logistics, and to him, Mrs. D. Chandra Ghatak, and Dual Ch. Shee, for allowing us to install piezometers on their land. We thank A. Osborn for assistance with analysis, much of which was done in the Wolfson Laboratory for Environmental Geochemistry (UCL-Birkbeck, University of London). Additional analysis was done using the NERC ICP-AES Facility at RHUL, with the permission of its Director, Dr. J.N. Walsh. Thanks are due Sam Tonkin for providing Fig. 1. This work was supported by the Royal Society, the British Council via a LINK Scheme grant to Delhi University and a new investigators grant to KHE (NERC Grant No. GR8/ 04292). A.C. thanks Drs. S. Peddle and R. Taylor for their support. We thank Laurent Charlet, Doris St€ uben, and an anonymous reviewer, for their helpful comments about our work. References AAN, 2000. Arsenic contamination in groundwater and hydrogeology, Samta village, western Bangladesh. Asian Arsenic Network. Report July 2000. Adam, P., Denaix, L., Terribile, F., Zampella, M., 2003. Characterization of heavy metals in contaminated volcanic soils of the Solofrana river valley (southern Italy). Geoderma (in press). Ahmed, K.M., Hoque, M., Hasan, M.K., Ravenscroft, P., Chowdhury, L.R., 1998. Occurrence and origin of water well CH4 gas in Bangladesh. J. Geol. Soc. India 51, 697–708. Ahmed, K.M., Ravenscroft, P., 2000. Extensive arsenic contamination of Bangladesh groundwater: curse of unmanaged development? In: Inyang, H.I., Ogunro, V.O. (Eds.), Proceedings of the 4th International Symposium on Environmental Geotechnology and Global Sustainable Development, vol. I. CEEST, University of Massachusetts, pp. 105–114. J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Ali, M., 2003. Review of drilling and tubewell technology for groundwater irrigation. In: Rahman, A.A., Ravenscroft, P. (Eds.), Groundwater Resources and Development in Bangladesh – Background to the Arsenic Crisis, Agricultural Potential and the Environment. University Press Ltd, Dhaka. Anawar, H.M., Akai, J., Komaki, K., Terao, H., Yoshioka, T., Ishizuka, T., Safiullah, S., Kata, K., 2003. Geochemical occurrence of arsenic in groundwater of Bangladesh: source and mobilization processes. J. Geochem. Explor. 77, 109– 131. Anon., 1994. The bacteriological examination of drinking water supplies 1994. Reports on Public Health and Medical Subjects No. 71: Methods for the examination of waters and associated materials. HMSO, London. Appelo, C.A.J., Van Der Weiden, M.J.J., Tournassat, C., Charlet, L., 2002. Carbonate ions and arsenic dissolution by groundwater. Environ. Sci. Technol. 36, 3096–3103. Ayotte, J.D., Montgomery, D.L., Flanagan, S.M., Robinson, K.W., 2003. Arsenic in groundwater in Eastern New England: occurrence, controls, and human health implications. Environ. Sci. Technol. 37, 2075–2083. Bandyopadhyay, R.K., 2002. Hydrochemistry of arsenic in Nadia District, West Bengal. J. Geol. Soc. India 59, 33–46. Banerjee, M., Sen, P.K., 1987. Palaeobiology in understanding the change of sea level and coastline in Bengal basin during Holocene period. Indian J. Earth Sci. 14, 307–320. Banfield, J.F., Nealson, K.H., Lovley, D.R., 1998. Geomicrobiology: interactions between microbes and minerals. Science 280, 54–55. Berg, M., Tran, H.C., Nguyen, T.C., Pham, H.V., Schertenleib, R., Giger, W., 2001. Arsenic contamination of ground and drinking water in Vietnam: a human health threat. Environ. Sci. Technol. 35, 2621–2626. Bergman, I., Lundberg, P., Nilsson, M., 1999. Microbial carbon mineralisation in an acid surface peat: effects of environmental factors in laboratory incubations. Soil Biol. Biochem. 31, 1867–1877. Bethke, C.M., 1998. The Geochemist’s Workbench Release 3.0. C.M. Bethke, Hydrogeology Program, University of Illinois. BGS, 1989. Trace element occurrence in British groundwaters. Research Report SD/89/3. BGS, 1996. NAFF Report WC/96/22. The effect of urbanization on the groundwater quality beneath the city of Hanoi, Vietnam. British Geological Survey. Brammer, H., 1996. The Geography of the Soils of Bangladesh. University Press Ltd, Dhaka. Chakraborti, D., Basu, G.K., Biswas, B.K.,, Chowdhury, U.K., Rahman, M.M., Paul, K., Roy, Chowdhury, T., Chanda, C.R., Lodh, D., 2001. Characterization of arsenic bearing sediments in Gangetic Delta of West Bengal, India. In: Chappell, W.R., Abernathy, C.O., Calderon, R.L. (Eds.), Arsenic Exposure and Health Effects IV. Elsevier, Oxford, pp. 53–77. Chakraborti, D., Rahman, M.M., Paul, K., Chowdhury, U.K., Sengupta, M.K., Lodh, D., Chanda, C.R., Saha, K.C., Mukherjee, S.C., 2002. Arsenic calamity in the Indian subcontinent – What lessons have been learned? Tantala 58, 3–22. 1291 Chapelle, F.H., 2000. The significance of microbial processes in hydrogeology and geochemistry. Hydrogeol. J. 8, 41–46. Chapelle, F.H., Lovley, D.R., 1992. Competitive-exclusion of sulphate reduction by Fe(III)-reducing bacteria – a mechanism for producing discrete zones of high-iron groundwater. Ground Water 30, 29–36. Chatterjee, A., Das, D., Mandal, B.K., Chowdhury, T.R., Samanta, G., Chakraborti, D., 1995. Arsenic in groundwater in 6 districts of West Bengal, India – the biggest arsenic calamity in the world 1. Arsenic species in drinkingwater and urine of the affected people. Analyst 120, 643– 650. Cronin, A.A., Breslin, N., Taylor, R., Pedley, S., 2003. Assessing the risks to groundwater quality from on-site sanitation and poor sanitary well completion, ‘Ecosan – closing the loop’. In: Proceedings of the 2nd International Conference on Ecological Sanitation, Lubeck, Germany, April. Cummings, D.E., Caccavo Jr., F., Fendorf, S., Rosensweig, R.F., 1999. Arsenic mobilization by the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY. Environ. Sci. Technol. 33, 723–729. Dixit, S., Hering, J.G., 2003. Comparison of arsenic (V) and arsenic (III) sorption to iron oxide minerals: implications for arsenic mobility. Environ. Sci. Technol. 37, 4182–4189. Dowdle, P.R., Laverman, A.M., Oremland, R.S., 1996. Bacterial dissimilatory reduction of arsenic (V) to arsenic (III) in anoxic sediments. Appl. Environ. Microbiol. 62, 1664–1669. Dowling, C.B., Poreda, R.J., Basu, A.R., Peters, S.L., Aggarwal, P.K., 2002. Geochemical study of arsenic release mechanisms in the Bengal Basin groundwater. Water Resour. Res. 38, 1173–1190. DPHE, 1999. Groundwater studies for arsenic contamination in Bangladesh. Final Report, Rapid Investigation Phase. Department of Public Health Engineering, Government of Bangladesh. Mott MacDonald (P. Ravenscroft; V1, 2, 3, 5, 6) and British Geological Survey (V4). DPHE, 2001. In: Kinniburgh, D.G., Smedley, P.L. (Eds). Arsenic Contamination of Groundwater in Bangladesh. BGS Technical Report, WC/00/19, British Geological Survey, Keyworth. Edmunds, W.M., 1986. In: Brandon, T.W. (Ed.), Groundwater Chemistry. Groundwater: Occurrence, Development and Protection. Inst. Water Eng. Sci., Water Practice Manual 5 (Chapter 3). Foster, A.L., Brown, G.E., Parks, G.A., 2003. X-ray absorption fine structure study of As(V) and Se(IV) sorption complexes on hydrous Mn oxides. Geochim. Cosmochim. Acta 67, 1937–1953. Frederickson, J.A., Zachara, J.M., Kennedy, D.W., Dong, H., Onstott, T.C., Hinman, N.W., Li, S.-M., 1998. Biogenic iron mineralization accompanying the dissimilatory reduction of hydrous ferric oxide by a groundwater bacterium. Geochim. Cosmochim. Acta 62, 3239–3257. Glasauer, S., Weidler, P.G., Langley, S., Beveridge, T.J., 2003. Control on Fe reduction and mineral formation by a subsurface bacterium. Geochim. Cosmochim. Acta 67, 1277–1288. Goodbred Jr., S.L., Kuehl, S.A., 2000. The significance of large sediment supply, active tectonism, and eustasy on margin sequence development: late Quaternary stratigraphy and 1292 J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 evolution of the Ganges–Brahmaputra delta. Sed. Geol. 133, 227–248. Gulens, J., Champ, D.R., Jackson, R.E., 1979. Influence of redox environments on the mobility of arsenic in groundwater. In: Jenne, E.A. (Ed.), Chemical Modeling in Aqueous Systems. American Chemical Society Symposium Series, vol. 93, pp. 81–95. Harvey, C.F., Swartz, C.H., Badruzzaman, A.B.M., KeonBlute, N., Yu, W., Ali, M.A., Jay, J., Beckie, R., Niedan, V., Brabander, D., Oates, P.M., Ashfaque, K.N., Islam, S., Hemond, H.F., Ahmed, M.F., 2002. Arsenic mobility and groundwater extraction in Bangladesh. Science 298, 1602– 1606. Hinkle, S.R., 1997. Quality of shallow ground water in alluvial aquifers of the Willamette Basin, Oregon, 1993–95. US Geol. Survey Water-Res. Investigations Report 97-4082–B. Hinckle, S.R., Polette, D.J., 1999. Arsenic in groundwater of the Willamette Basin, Oregon. USGS Water Resour. Invest. Rep. 98-4205. Hudson-Edwards, K., Banerjee, D., Ravenscroft, P., McArthur, J.M., Mishra, R., Purohit, R., Lowry, D., 2004. Sediments of the West Bengal and their relation to arsenic pollution (in preparation). Ishiga, H., Dozen, K., Yamazaki, C., Ahmed, F., Islam, M.B., Rahman, M.H., Sattar, M.A., Yamamoto, H., Itoh, K., 2000. Geological constrains on arsenic contamination of groundwater in Bangladesh. Abstract of 5th Forum on Asian arsenic contamination, Yokohama, Japan, RGAG, Saitama, pp. 53–62. Islam, M.S., Tooley, M.J., 1999. Coastal and sea level changes during the Holocene in Bangladesh. Quatern. Int. 55, 61–75. Jones, C.A., Langner, H.W., Anderson, K., McDermott, T.R., Inskeep, W.P., 2000. Rates of microbially mediated arsenate reduction and solubilization. Soil Sci. Soc. Am. 64, 600–608. Karim, M., Komori, Y., Alam, M., 1997. Subsurface As occurrence and depth of contamination in Bangladesh. J. Environ. Chem. 7, 783–792. Knapp, A.P., Herman, J.S., Mills, A.L., Hornberg, G.M., 2002. Changes in the sorption capacity of Coastal Plain sediments due to redox alteration of mineral surfaces. Appl. Geochem. 17, 387–398. Kresse, T., Fazio, J., 2003. Occurrence of arsenic in ground waters of Arkansas and implications for source and release mechanisms. Water Quality Report, Arkansas Ambient Ground-Water Monitoring Program, Arkansas Department of Environmental Quality. Korte, N.E., Fernando, Q., 1991. A review of arsenic(III) in groundwater. Crit. Rev. Environ. Contr. 21, 1–39. Lovley, D.R., 1997. Microbial Fe(III) reduction in subsurface environments. FEMS Microbiol. Rev. 30, 305–313. Lovely, D.R., Anderson, R.T., 2000. Influence of dissimilatory metal reduction on the fate of organic and metal contaminants in the subsurface. Hydrogeol. J. 8, 77–88. Lovley, D.R., Stolz, J.F., Nord, G.L., Phillips, E.J.P., 1987. Anaerobic production of magnetite by a dissimilatory ironreducing micro-organism. Nature 330, 252–254. Lloyd, B., Bartram, J., 1991. Surveillance solutions to microbiological problems in water quality control in developing countries. Water Sci. Technol. 24 (2), 61–75. Manning, B.A., Fendorf, S.E., Bostick, B., Suarez, D.L., 2002. arsenic (III) oxidation and Arsenic (V) adsorption reactions on synthetic birnessite. Environ. Sci. Technol. 36, 976–981. Matisoff, G., Khourey, C.J., Hall, J.F., Varnes, A.W., Strain, W., 1982. The nature and source of arsenic in Northeastern Ohio ground water. Ground Water 20, 446–455. McArthur, J.M., Ravenscroft, P., Safiullah, S., Thirlwall, M.F., 2001. Arsenic in groundwater: testing pollution mechanisms for sedimentary aquifers in Bangladesh. Water Resour. Res. 37, 109–117. Nealson, K.H., 1997. Sediment bacteria: Who’s there, what are they doing, and what’s new? Annu. Rev. Earth Planet. Sci. 25, 403–434. Nguten, L., Sukias, J., 2002. Phosphorus fractions and retention in drainage ditch sediments receiving surface runoff and subsurface drainage from agricultural catchments in the North Island, New Zealand. Agric., Ecosyst. Environ. 92, 49–69. Nickson, R., McArthur, J., Burgess, W., Ahmed, K.M., Ravenscroft, P., Rahman, M., 1998. Arsenic poisoning of Bangladesh groundwater. Nature 395, 338. Nickson, R.T., McArthur, J.M., Ravenscroft, P., Burgess, W.S., Ahmed, K.M., 2000. Mechanism of arsenic poisoning of groundwater in Bangladesh and West Bengal. Appl. Geochem. 15, 403–413. Nicolli, H.B., Suriano, J.M., Gomez Peral, M.A., Ferpozzi, L.H., Baleani, O.A., 1989. Groundwater contamination with arsenic and other trace elements in an area of the Pampa, Province of C ordoba, Argentina. Environ. Geol. Water Sci. 14, 3–16. Pal, T., Mukherjee, P.K., Sengupta, S., 2002. Nature of arsenic pollutants in groundwater of Bengal basin – a case study from Baruipur area, West Bengal, India. Curr. Sci. 82, 554– 561. Parker, J.W., Perkins, M.A., Foster, S.S.D., 1982. Groundwater quality stratification – its relevance to sampling strategy. ‘Commisie voor Hydrologisch Onderzoek’. TNO, Netherlands, Publication No. 31, pp. 43–54. Ravenscroft, P., 2003. Overview of the hydrogeology of Bangladesh. In: Rahman, A.A., Ravenscroft, P. (Eds.), Groundwater Resources and Development in Bangladesh – Background to the Arsenic Crisis, Agricultural Potential and the Environment. University Press Ltd, Dhaka. Ravenscroft, P., McArthur, J.M., Hoque, B.A., 2001. Geochemical and palaeohydrological controls on pollution of groundwater by arsenic. In: Chappell, W.R., Abernathy, C.O., Calderon, R.L. (Eds.), Arsenic Exposure and Health Effects IV. Elsevier, Oxford, pp. 53–77. Redman, A.D., Macalady, D.L., Ahmann, D., 2002. Natural organic matter affects arsenic speciation and sorption onto hematite. Environ. Sci. Technol. 36, 2889–2896. Reddy, K.R., Diaz, O.A., Scinto, L.J., Agami, M., 1995. Phosphorus dynamics in selected wetlands and streams of the lake Okeechobee Basin. Ecol. Eng. 5, 183–207. Reimann, K.-U., 1993. The Geology of Bangladesh. Gebruder Borntraeger, Berlin. Rhoton, F.E., Bigham, J.M., Lindbo, D.L., 2002. Properties of iron oxides in streams draining the Loess Uplands of Mississippi. Appl. Geochem. 17, 409–419. J.M. McArthur et al. / Applied Geochemistry 19 (2004) 1255–1293 Rittle, K.A., Drever, J.I., Colberg, P.J.S., 1995. Precipitation of arsenic during bacterial sulphate reduction. Geomicrobiol. J. 13, 1–11. Robertson, F.N., 1989. Arsenic in ground-water under oxidising conditions, south-western United States. Environ. Geochem. Health 11, 171–185. Safiullah, S., 1998. CIDA Arsenic Project Report: Monitoring and Mitigation of Arsenic in the Groundwater of Faridpur Municipality. Jahangirnagar University, Dhaka, Bangladesh. Smedley, P.L., Kinniburgh, D.G., 2002. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 17, 517–568. Smedley, P.L., Zhang, M., Zhang, G., Lou, Z., 2003. Mobilisation of arsenic and other trace elements in fluviolactustrine aquifers in the Huhhot Basin, Inner Mongolia. Appl. Geochem. 18, 1453–1477. Smith, A., Lingas, E., Rahman, M., 2000. Contamination of drinking-water by As in Bangladesh. Bull. WHO 78, 1093– 1103. Stanley, D.J., Hait, A.K., Jorstad, T.F., 2000. Iron-stained quartz to distinguish Holocene deltaic from Pleistocene alluvial deposits in small core samples. J. Coastal Res. 16, 357–367. St€ uben, D., Berner, Z., Chandrasekharam, D., Karmakar, J., 2003. Arsenic enrichment in groundwater of West Bengal, India: geochemical evidence for mobilization of As under reducing conditions. Appl. Geochem. 18, 1417–1434. Tanabe, S., Hori, K., Saito, Y., Haruyama, S., Vu, V.P., Kitamura, A., 2003. Song Hong (Red River) delta evolution related to millennium-scale Holocene sea-level changes. Quatern. Sci. Rev. 22, 2345–2361. 1293 Tareq, S.M., Safiullah, S., Anawar, H.M., Rahman, M.M., Ishazuka, T., 2003. Arsenic pollution in groundwater: a selforganizing complex geochemical process in the deltaic sedimentary environment, Bangladesh. Sci. Total Environ. 313, 213–226. Umitsu, M., 1987. Late quaternary environment and landform evolution in Bengal. Geog. Rev. Japan B 60, 164–178. Umitsu, M., 1993. Late quaternary environment and landforms in the Ganges Delta. Sed. Geol. 83, 177–186. van Geen, A., Zheng, Y., Versteeg, R., Stute, M., Horneman, A., Dhar, R., Steckler, M., Gelman, A., Small, C., Ahsan, H., Graziano, J.H., Hussain, I., Ahmed, K.M., 2003a. Spatial variability of arsenic in 6000 tube wells in a 25 km2 area of Bangladesh. Water Resour. Res. 39, art. no. 1140. van Geen, A., Zheng, Y., Stute, M., Ahmed, K.M., 2003b. Comment on ‘‘Arsenic mobility and groundwater extraction in Bangladesh’’. Science 300, 5619, 584C. Welch, A.H., Lico, S.M., Hughes, J.L., 1988. Arsenic in groundwater of the Western United States. Ground Water 26, 333–346. Welch, A.H., Westjohn, D.B., Helsel, D.R., Wanty, R.B., 2000. Arsenic in groundwater of the United States: occurrence and geochemistry. Ground Water, 38–589-604. WHO, 1993. Guidelines for Drinking Water Quality, vol. 1. World Health Organization, Geneva. Yu, W.H., Harvey, C.M., Harvey, C.F., 2003. Arsenic in groundwater in Bangladesh: a geostatistical and epidemiological framework for evaluating health effects, and potential remedies. Water Resour. Res. 39, 1146–1163. Zobrist, J., Dowdle, P.R., Davis, J.A., Oremland, R.S., 2000. Mobilization of arsenite by dissimilatory reduction of adsorbed arsenate. Environ. Sci. Technol. 34, 4747–4753.