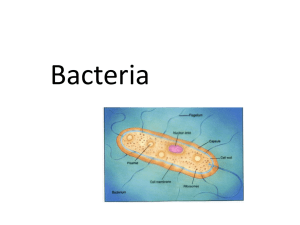
RAPID SPECTROPHOTOMETRIC DETECTION FOR ANALYSIS OF BACTERIAL CONTAMINATION IN WATER by
advertisement

RAPID SPECTROPHOTOMETRIC DETECTION FOR ANALYSIS OF BACTERIAL CONTAMINATION IN WATER by Sarah L. Spence c Copyright by Sarah L. Spence, 2011 All Rights Reserved A thesis submitted to the Faculty and the Board of Trustees of the Colorado School of Mines in partial fulfillment of the requirements for the degree of Master of Science (Applied Physics). Golden, Colorado Date Signed: Sarah L. Spence Signed: Lincoln D. Carr, PhD Thesis Advisor Signed: Cynthia Norrgran, MD Thesis Advisor Golden, Colorado Date Signed: Tom Furtak, PhD Department Head Department of Physics ii ABSTRACT Bacterial contamination in water is a hazard worldwide, from wells in third world countries to reclaimed water on the International Space Station. While traditional water testing techniques detect living bacteria in approximately 48 hours, we demonstrated that optical techniques can detect bacteria in as little as six hours. The Beer-Lambert Law, as applied to spectrophotometric turbidity studies, correlates the concentration of organismal growth in a solution to the absorption of visible light. By passing light through a sample of contaminated broth, we directly measure the intensity of the resulting light. We use this to calculate the transmittance and the absorption of light that passes through the solution. However, it is not entirely necessary to transform transmittance into absorption. A plot of transmittance over time tracks the inverse of the bacterial growth curve. Escherichia coli (E. coli ) was used as the contamination organism for this project. A sharp drop in transmittance is seen during the exponential growth phase of the bacteria being tested. We observed this change within six to twelve hours following the inoculation of the Escherichia coli into samples, using both a standard monochrometer and a device engineered specifically for this study. We employed cell counting algorithms to prove the consistency of the optical techniques with the confirmed presence of bacterial growth. The software algorithm utilizes a threshold to form a binary rendition of a full-color version of a sample slide. It then uses this binary data to calculate both an area fraction and the number of spherical or elliptical shapes present. This cell counting process indeed confirmed the growth of our intended contaminant. Our cultures were grown and maintained in a bioreactor for several months, in a full nutrient broth with glucose used as its limiting nutrient. The bioreactors serve to control the growth phase of the bacteria exhibited within the culture itself. By starving the bacteria, we were able to set them into a stationary growth phase, while supplying an abundant amount of glu- iii cose forced the population into an exponential growth phase. The individual growth patterns of each of these special cases were observed. We hypothesize that the length of the lag phase in the transplanted bacteria is affected by the difference in nutrients between the culture and the sample media. We anticipated seeing extremely short lag times in wild E. coli transplanted from water systems to a full nutrient broth. The Optical Bacteria Detector (OBD) was designed to be an effective and inexpensive device, with a limited use of consumables and minimum waste generation. It is a mobile, battery-operated field device that is in step with the spectrophotometer used in the laboratory. The OBD uses a phototransistor as a sensor and an LED with wavelength of approximately 500 nm. Data from the monochrometer shows that the sudden decrease in transmittance is most pronounced at this wavelength. The OBD can be tuned to test for other bacteria, such as Salmonella sps. and Vibrio fisheri, by changing the wavelength of the LED light source. Further work is being conducted on a second model of the OBD that will have the capability of testing the sample for scattering properties as well as absorption. The effects of Tyndall scattering will give the device the capability to suggest the size of the contaminant, which could be a life-saving determination of whether an infection is bacterial or parasitic. It is our hope that this research will be continued to detect the presence of many other lifethreatening pathogens, and continue to bridge the gap between the fields of biology and physics. iv TABLE OF CONTENTS ABSTRACT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . iii LIST OF FIGURES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . viii LIST OF TABLES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xii LIST OF SYMBOLS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xiii LIST OF ABBREVIATIONS . . . . . . . . . . . . . . . . . . . . . . . . . . . . xv ACKNOWLEDGEMENTS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xvi DEDICATION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xviii CHAPTER 1 INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 1.1 Motivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 1.2 Selection of the Bacterial Contamination . . . . . . . . . . . . . . . . . . 2 1.3 Escherichia coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 1.3.1 Anatomy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 1.3.2 Motility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6 1.3.3 Reproduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 1.3.4 Nutrient Usage . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 1.3.5 Growth Curve . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12 1.3.6 Physical Modeling of Biological Systems . . . . . . . . . . . . . 14 1.4 Previous Work . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 1.5 Biological Laboratory Techniques . . . . . . . . . . . . . . . . . . . . . 18 1.5.1 Media . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18 1.5.2 Inoculation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19 v 1.6 1.5.3 Gram Staining . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 1.5.4 Identification of Bacteria . . . . . . . . . . . . . . . . . . . . . . 23 Experiment Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23 1.6.1 Lab-grade Spectrophotometer . . . . . . . . . . . . . . . . . . . 26 1.6.2 Optical Bacteria Detector I . . . . . . . . . . . . . . . . . . . . 26 1.6.3 Optical Bacteria Detector II . . . . . . . . . . . . . . . . . . . . 26 CHAPTER 2 TURBIDITY STUDIES WITH SPECTROPHOTOMETRY . . . 27 2.1 Genesys 6 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28 2.2 Establishing Initial Conditions . . . . . . . . . . . . . . . . . . . . . . . 30 2.3 Assumptions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32 2.4 Testing Protocol 2.5 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 2.6 Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 2.6.1 Error Contributions . . . . . . . . . . . . . . . . . . . . . . . . . 39 2.6.2 Statistical Analysis . . . . . . . . . . . . . . . . . . . . . . . . . 43 2.6.3 Cell Counting . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45 CHAPTER 3 THE OPTICAL BACTERIA DETECTOR I: ABSORPTION ONLY . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51 3.1 Requirements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51 3.2 Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 3.3 Design . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 3.3.1 Circuit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 3.3.2 Housing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 vi 3.4 Testing Procedure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54 3.4.1 Lab Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 3.4.2 Field Testing . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 3.5 Difficulties . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57 3.6 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 3.7 Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 3.8 3.7.1 Error Contributions . . . . . . . . . . . . . . . . . . . . . . . . . 60 3.7.2 Statistical Analysis . . . . . . . . . . . . . . . . . . . . . . . . . 63 Linearity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65 CHAPTER 4 THE OPTICAL BACTERIA DETECTOR II: ABSORPTION AND SCATTERING . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 4.1 Theory . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 4.2 Engineering Design . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 4.3 Protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 4.4 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71 4.5 Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71 4.6 Future Work . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72 CHAPTER 5 CONCLUSION . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74 REFERENCES CITED . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79 APPENDIX A - VISUAL BASIC CODE FOR IMPORTING DATA . . . . . . . 84 APPENDIX B - GENESYS 6 AUTOMATION MACRO . . . . . . . . . . . Pocket APPENDIX C - VISUAL BASIC CODE FOR IMPORTING DATA . . . . . Pocket vii LIST OF FIGURES Figure 1.1 A representation of a bacterium with a single flagellum. The flagellum is powered by a proton turbine, and is produced by the bacterium after it has undergone approximately three divisions. Rotation of the flagellum in one direction will cause the bacterium to move forward, while rotation in the other direction will cause the bacterium to tumble and change directions. This results in random walk behavior. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8 Figure 1.2 The beginning of conjugation. One bacterium contains a plasmid with the gene encoding for a pilus while the other does not. . . . . . 10 Figure 1.3 A pilus projects from the bacterium containing the plasmid. This pilus is a temporary cytoplasmic connection between the two cells. . 10 Figure 1.4 Genetic material replicates and crosses into the adjacent cell via the pilus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 Figure 1.5 The pilus withdraws, resulting in two cells containing the same plasmid. The affected cell is now able to perform conjugation on other cells. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11 Figure 1.6 Schematic representation of the four phases of bacterial growth: lag phase, exponential phase, stationary phase and death phase. The lag phase occurs immediately after the bacteria is transplanted into a new environment, and no significant population growth occurs. The exponential phase is the result of each cell duplicating every 20-30 minutes. The stationary phase occurs when the population has reached its carrying capacity, and the death phase occurs when there is no longer enough nutrients to support the population. . . . 13 Figure 1.7 Our experimental representation of bacterial growth, created from data seen in Chapter 2. The lag, exponential and stationary phases are all clearly visible in this representation. . . . . . . . . . . . . . . 14 Figure 1.8 A stained sample slide photographed through a microscope camera. These images are used to automate the cell counting process. . . . . 16 Figure 1.9 A binary rendition of the slide above. The color image was first converted into a greyscale image, whereupon a threshold was used to convert it into a binary representation for processing. . . . . . . . 17 viii Figure 1.10 An inoculating loop used for the culturing of the bacteria. Can be used for both broth and agar media. . . . . . . . . . . . . . . . . . . 20 Figure 1.11 A positive gram-stain. Gram-positive bacteria retain the purple dye by way of a thick layer of peptidoglycan in their cell wall. . . . 22 Figure 1.12 Negative gram-stain. Gram-negative bacteria do not retain the purple dye of the gram-stain. . . . . . . . . . . . . . . . . . . . . . . 22 Figure 1.13 Spherical bacteria, referred to as cocci. . . . . . . . . . . . . . . . . 23 Figure 1.14 Rod-shaped bacteria, referred to as bacilli. . . . . . . . . . . . . . . 24 Figure 1.15 Spiral bacteria, referred to as spirilla. . . . . . . . . . . . . . . . . 24 Figure 1.16 General experimental setup. A light source is incident on a sample of bacteria. The cells suspended in this sample absorb fractions of the incident light. The transmitted light is then measured by a detector on the other side. . . . . . . . . . . . . . . . . . . . . . . . 25 Figure 2.1 Genesys 6 setup. The Genesys 6 uses tungsten halogen and deuterium bulbs as a light source and a photodiode as a sensor. It outputs data in percent transmittance. . . . . . . . . . . . . . . . . 27 Figure 2.2 Genesys 6 UV/Vis-Spectrophotometer. The Genesys 6 tests six cuvettes at once, and tests over a wide range of wavelengths. . . . . 29 Figure 2.3 A basic bioreactor setup. The bioreactor maintains a constant environment for the bacterial culture, by controlling temperature, pH and the amount of nutrient available. . . . . . . . . . . . . . . . . . 31 Figure 2.4 The bioreactor used in this experiment. A variable peristaltic pump was used to control the rate of glucose addition in order to manipulate initial growth phase of the culture. . . . . . . . . . . . . . . . 32 Figure 2.5 A labeled photograph of the setup. A second pump is used to collect culture solution from within the bioreactor. . . . . . . . . . . . . . . 35 Figure 2.6 Data taken by the Genesys 6 prior to implementation of new protocol. There is a wide spread in lag time for this data, however it was collected using several distinct batches of bacteria, without a stabilized protocol. . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 ix Figure 2.7 Data collected following the protocol revision. The spread of data is significantly narrower and follows two groupings corresponding to the two cultures used to collect the data itself. The transmittance values follow the inverse of an expected bacterial growth curve. . . . 37 Figure 2.8 Cuvettes tested with differing levels of initial bacterial concentration. The yellow lines, which display a significantly smaller lag phase, represent samples that were inoculated with five times as much bacteria as the other samples. . . . . . . . . . . . . . . . . . . 37 Figure 2.9 A comparison plot of bacteria prepared in stationary and exponential growth phases. The bacteria intially prepared in the stationary phase has a slightly shorter lag period than bacteria prepared in the exponential growth phase. . . . . . . . . . . . . . . . . . . . . . 38 Figure 2.10 Analysis in discrepancy between average values for bacteria prepared initially in the stationary phase and the exponential phase. The standardized difference between stationary and exponential phase increases at the end of the lag phase for the bacteria prepared in stationary phase. . . . . . . . . . . . . . . . . . . . . . . . 40 Figure 2.11 Curve fitted to the average values of the Genesys 6 data. Error-bars represent the variance in the Genesys 6 measurements over time for a sample of constant transmittance. . . . . . . . . . . . . . . . . . . 45 Figure 2.12 Cell counting in terms of colony forming units. This method counts individual cells, but does not account for cell clumping. . . . . . . . 46 Figure 2.13 Cell counting in terms of area fraction of cells. This is the preferred method, as it accounts more closely for the clumping of cells. . . . . 47 Figure 2.14 A mathematical fit of the cell count data shown above, in colony forming units. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 Figure 2.15 A mathematical fit of the area fraction data shown above. . . . . . 49 Figure 2.16 A normalized absorption curve as obtained from the Genesys 6 compared to corresponding area fraction data. This plot shows the correlation between actual cell growth and change in transmittance. 50 Figure 3.1 Basic setup for the OBD I. The OBD I uses a 560 nm LED as a light source, and a PNZ150 phototransistor as a detector. . . . . . . 51 x Figure 3.2 Circuit schematic for the OBD I. The output from the phototransistor is fed into an op-amp to amplify its signal, before being delivered to a voltmeter. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53 Figure 3.3 Top view of the housing for the OBD I. Features include an on/off switch, a cap for the sample container, and an LCD display. . . . . 54 Figure 3.4 Internal view of the housing for the OBD I. The circuit boards are mounted together, and the sample container is completely enclosed to prevent interference from ambient light. . . . . . . . . . . . . . . 55 Figure 3.5 Photograph of the OBD I. . . . . . . . . . . . . . . . . . . . . . . . 56 Figure 3.6 Complete data set taken by the OBD I. This data was taken over two separate runs using the same batch of bacteria. Though transmittance only falls to 0.60, these tests were performed using nutrient broth instead of LB. . . . . . . . . . . . . . . . . . . . . . . . . 59 Figure 3.7 Same data as above, a closer view, beginning at 12 hours. . . . . . . 60 Figure 3.8 Statistical analysis of the curve fitted to OBD data. Error-bars are calculated from the variation in measurements observed by the OBD I when testing a control sample. . . . . . . . . . . . . . . . . . 64 Figure 3.9 Experimental setup to test the linearity of the response of the OBD I. A variable lab power supply was used to supply voltage to the LED independent of the rest of the circuit. . . . . . . . . . . . . . . 66 Figure 3.10 Linearity analysis of the OBD I’s response. The initial jump at 2.0 V is the forward voltage drop necessary for the LED itself to turn on. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 Figure 4.1 Schematic for the scattering experiments with the OBD II. The light source in this case is a laser of wavelength 523 nm, and the detectors are provided by the same circuit as seen in Chapter 3. Detectors at 180◦ and 90◦ allow for us to calculate a ratio of scattered to transmitted light. . . . . . . . . . . . . . . . . . . . . . . . . . . 70 xi LIST OF TABLES Table 2.1 Error contributions for Genesys 6 testing . . . . . . . . . . . . . . . 42 Table 2.2 Parameter Values for Genesys Data . . . . . . . . . . . . . . . . . . 44 Table 2.3 Parameter Values for Cell Population . . . . . . . . . . . . . . . . . 48 Table 2.4 Parameter Values for Area Fraction . . . . . . . . . . . . . . . . . . 48 Table 3.1 Error contributions for OBD I testing Table 3.2 Parameter Values for OBD I . . . . . . . . . . . . . . . . 63 . . . . . . . . . . . . . . . . . . . . . 64 xii LIST OF SYMBOLS Transmittance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . T Absorption . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . A Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . c Intensity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . I Pathlength . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ` Molar absorptivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ε Time . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . t Population . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . P Diffusion Flux . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . J Diffusion Coefficient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . D Velocity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . v Frictional coefficient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ξ Density . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ρ Potential . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . U Reynolds Number . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Re Dynamic viscosity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . η Kinematic viscosity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ν Efficiency factor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Qext Extinction coefficient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . γ Particle Density . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . N Particle radius . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . a xiii Turbidity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . τ xiv LIST OF ABBREVIATIONS Optical Bacteria Detector . . . . . . . . . . . . . . . . . . . . . . . . . . . . OBD Optical Bacteria Detector I . . . . . . . . . . . . . . . . . . . . . . . . . . . OBD I Optical Bacteria Detector II . . . . . . . . . . . . . . . . . . . . . . . . . . OBD II Deoxyribonucleic Acid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . DNA Lysogeny Broth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . LB Colony forming unit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CFU National Institutes of Health . . . . . . . . . . . . . . . . . . . . . . . . . . . NIH Light-emitting diode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . LED Center for Disease Control . . . . . . . . . . . . . . . . . . . . . . . . . . . . . CDC Lipopolysaccharides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . LPS Ribonucleic Acid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . RNA Messenger Ribonucleic Acid . . . . . . . . . . . . . . . . . . . . . . . . . . mRNA Transfer Ribonucleic Acid . . . . . . . . . . . . . . . . . . . . . . . . . . . . tRNA Graphical User Interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . GUI Nephelometric Turbidity Units . . . . . . . . . . . . . . . . . . . . . . . . . xv NTU ACKNOWLEDGEMENTS This project has become such a large part of my life, that my life has become a large part of this project itself. My friends, colleagues and family have in many ways contributed to the success of the experiment and my perseverence to see it to completion. I must first thank my advisor and mentor, Dr. Cynthia Norrgran, not only for bringing me to this project, but for the many learning experiences since then. She has been there for every stumbling block and victory along the way, and I will never be able to fully express my gratitude for all she has done. I thank my other advisor, Dr. Lincoln Carr, for teaching me that being a biologist and a physicist need never be mutually exclusive, and Dr. Jeff Squier for his continuous input and support throughout the project. This project would have never advanced this far if not for the work of the students who came before me. Alyssa Cobb, who showed this experiment was possible at all, and Nick Hunter, who, in automating data collection, has saved me many long nights alone in the lab, have my special thanks. I thank Sean Bingel for the many hours spent staining and photographing slides. I must acknowledge Randy, Orlen, Mike and Justin, without whom the OBD would still be a poorly soldered circuit living inside a cookie tin. Erich Hoover has been an incredible resource for me as well; from circuit design to thesis writing and even telling me that perhaps it is time for a break, he has been both a friend and instructor throughout the course of this journey. My family and friends have perhaps the most of my gratitude. I thank my mother for her never-ending faith in me, as well as keeping me fed and caffeinated through the many long nights of work. I thank my father for both his technical advice and his moral support. I would never have gotten this far were it not for my parents’ love and encouragement. I am grateful for my friends, Ginger and Kim, who have somehow endured the constant ramblings of a project they know little about, and of xvi course Christine, who did not even hesitate to edit this thesis. I thank Michal for his ever-willingness to come to the rescue when I needed an extra set of hands, or just someone to talk to. Most importantly, I have to thank Shaun for never giving up on me, for keeping me awake at all hours of the night to record data, for not talking to me until I finished that last chapter, for spending Valentine’s Day weekend helping me write this thesis. He has been a bastion of support for me. No matter where the Air Force has sent him, he has always found a way to show his faith in me, and share in both victories and defeats. I will never be able to thank him enough for how much he has helped me this past year. xvii One of the advantages of being disorderly is that one is constantly making exciting discoveries. A. A. Milne xviii CHAPTER 1 INTRODUCTION Water contamination is a serious health concern for human populations around the world. In the developed world, bacterial contamination is detected through traditional laboratory techniques. The standard laboratory test for water contamination involves filtering the water and plating a sample of it in agar. The specimen is incubated in a heat-controlled environment for 24 to 48 hours, and examined for the formation of colonies. These processes are time consuming and therefore costly. [1] Developing countries often do not have access to laboratory facilities capable of performing these tests and they are too expensive for routine use. For these reasons, contaminated water sources are left unidentified and are still used for drinking water by the inhabitants. A simple, inexpensive method for detection of bacterial contamination is needed to protect these people from contaminated water sources. [2] The primary objective of this project is to develop a more effective means of detecting bacterial contamination in water. An ideal solution is a portable device that is small enough to fit in the glovebox of a car. The device must be powered by a replaceable, transportable power source (such as batteries), and be relatively inexpensive to produce (ideally less than $15). The device would be used in field conditions where testing in a laboratory would be unrealistic to impossible, and produce results in less than 24 hours. 1.1 Motivation To develop physical models of bacterial behavior, we must find a physical observ- able. This experiment uses the measurement of light intensity as a basis for modeling bacterial growth. In order to use this information, we employ a wide use of physical techniques. Automated data collection is required to record information at specific 1 intervals over a 24-hour period. A macro script was an efficient means to accomplish this. Additionally, new ways of collecting this information, such as portable field devices, require significant amounts of technical design. These analytical approaches supply a large amount of numeric data that is not characteristic of biological studies. As such, physics-based error analysis and plot-fitting are used to quantify and understand biological behavior. In addition to these physical techniques, biological methods must be used to conduct a clean experiment. This contribution from biology is limited to the basic bacteriology and laboratory techniques, including bacterial properties affecting population growth, cultures, inoculation, media use, staining and microscopy. 1.2 Selection of the Bacterial Contamination The contamination of water by human waste is one of the most lasting water purity problems in the world. Sites of natural disasters, such as the Haiti earthquake of 2010, are particularly vulnerable. [3] Human waste can contain enteric bacterial pathogens such as Salmonella sps., which causes widespread outbreaks, despite small concentrations of the contaminant in the water. Escherichia coli (E. coli ) acts as an indicator bacterium for enterobacteria species, because it can be detected more easily than other enteric bacteria. [4] It has been shown that E. coli is present in the gastrointestinal track of all humans in as little as 40 hours following birth. E. coli has been found on every continent and is hard to eradicate. [5] During the Vietnam War, it was found that 20% of wounds incurred during the war had been infected with E. coli due to the poor water quality. [6] This enforces the idea that water quality is not applicable only to drinking water, but water used for other purposes as well. Although E. coli is indigenous to the human gut, it can be a dangerous pathogen. The human immune system is accustomed to its own strain of E. coli present in a specific distribution. When this balance is disturbed, an immune response results. 2 The most common disease caused by E. coli is gastroenteritis, which can range in severity from mild to life-threatening. [5] The immediate reaction of the gastrointestinal system to E. coli, vomiting and diarrhea, is not itself particularly dangerous. The fluid loss caused by these symptoms, however, can result in severe dehydration. In the developed world, physicians are easily able to treat the dehydration with intravenous (IV) fluids, but in the regions of the world suffering most from these infections do not have the same capabilities. Dehydration is most dangerous to patients suffering from unrelated immunodeficiencies, such as geriatric and pediatric patients, as well as those subject to Human Immunodeficiency Virus (HIV) and Acquired Immune Deficiency Syndrome (AIDS). These latter groups are particularly prevalent in the developing world. Other common infections associated with E. coli include urinary tract infections (UTIs) and neonatal meningitis. [7] Rarely, E. coli infections can lead to more involved ailments. Hemolytic-uremic syndrome is caused by E. coli O157:H7, and presents with hemolytic anemia, thrombocytopenia and acute renal failure. This condition primarily affects children, and approximately one third of patients suffer permanent renal damage. [8] Peritonitis and mastitis are characterized by the inflammation of the peritoneum, the membrane encasing the abdominal cavity, and breast tissue respectively. They often must be treated with surgery. [9] [10] Septicemia, also called sepsis or “blood poisoning”, is a rare but serious complication of E. coli infection. Sepsis is an infection within the blood, which is often fatal (up to 60% of patients within 30 days). E. coli can also result in severe respiratory infections like Gram-negative pneumonia. [11] E. coli is also the standard bacteria for research purposes. More is known about E. coli than any other laboratory-grade bacterium. Because work with E. coli is standardized, research can continue without need for extraneous experiments to determine its basic physiology. This project is focused on the study of E. coli K12, because it is a safe, standardized lab strain. [12] 3 1.3 Escherichia coli In order to model the behavior of E. coli, we must first understand their structure and mechanisms. Bacterial structure, motility and metabolism shape the growth curve. 1.3.1 Anatomy By knowing the chemical composition of E. coli, we can make calculations regarding synthesis for reproduction and other intercellular pathways that affect their behavior. It is no surprise to find that 55.0% of the dry weight of a single cell of E. coli B/r is due to proteins, numbering approximately 2,350,000. It follows that for a cell to divide into two complete daughter cells, an additional two million proteins must be synthesized. The bacterium’s capacity to create these proteins acts as a determining factor in the reproduction interval of the culture. The nucleic material of the cell, RNA and DNA, follow at 20.5% and 3.1% total dry weight respectively. Lipids, small hydrophobic molecules important in energy storage and cell membrane structure, contribute 9.1% of the dry weight of the cell. Lipopolysaccharides (LPS), larger molecules composed of a polysaccharide bonded covalently to a lipid, are also key components of the cell wall and contribute an additional 3.4% of the dry weight. Glycogen and peptidoglycan each contribute 2.5% to the dry weight. Glycogen is an important molecule in energy storage for many organisms, while peptidoglycan is a molecule that forms a meshwork in the cell walls of all bacteria (and whose absence is a defining characteristic of similar lifeforms known as archaea). Polyamines, metabolites, cofactors and ions account for the remaining 3.9% [13] A bacterial cell wall consists of three separate membranes. The outermost membrane is a lipid bilayer. Lipid bilayers conforming to the fluid mosaic model are the basis of most membranes in biology. LPS molecules (phospholipids specifically) are composed of hydrophobic lipid tails, and a hydrophilic head. The bilayer is formed 4 when the lipid tails are drawn to one another, and the heads are pointed outward away from one another. Because the layers are not attached by any bonds, the molecules are free to shift with respect to one another. The layers are asymmetric, containing non-overlapping gaps filled with proteins. Lipid bilayers are also highly permeable to hydrophobic molecules. However, the outer membrane of E. coli is not particularly permeable to hydrophobic molecules, evidenced by the ineffectiveness of hydrophobic antibiotics. A second layer, the murein sacculus, lies beneath the lipid bilayer in the periplasm. This layer is composed of murein, also known as peptidoglycan. [14] This thin layer of meshwork acts as a filter between the cell and its environment, and is a crucial component of binary fission. Below the murein sacculus sits the cytoplasmic membrane. The cytoplasmic membrane is a second lipid bilayer; however, it is richer in transmembrane and other proteins. [15] [16] [17] [18] [19] The cell wall provides many functions for bacteria; among these are protection from the environment and, more importantly, protection against internal turgor pressure. [20] A single bacterium contains a high concentration of proteins and other materials, causing a large osmotic pressure to pull water from the environment into the cell. Without the structural integrity afforded the bacterium by its cell wall, water would rush pass the cell membrane, until the cell becomes so engorged with water it lyses. The rigidity of the cell wall prevents this occurrence, and operates as a physical barrier between the cell and its environment. [21] [22] [23] [24] [25] As prokaryotes, E. coli cells contain no membrane-bound organelles. They contain a single, circular strand of DNA, and the means by which to transcribe this DNA into RNA and translate that RNA into gene-expressing proteins. Ribosomes are the particles responsible for protein production. The two subunits of a ribosome encode proteins as prescribed by messenger RNA (mRNA) from amino acids delivered by transfer RNA (tRNA). E. coli cells also have external structures for motility called flagella, which will be discussed below. [26] 5 E. coli obtains nutrients from its environment through the process of diffusion (which will be discussed in greater depth in Section 1.3.2). This is the simplest and most efficient form of consumption in the biological world, but it comes at a cost. Diffusion is only a realistic means of consumption when the surface area to volume ratio is very high, which occurs at small radii. As such, the physical form of an E. coli bacterium is a rod of length 2 µm and diameter 1 µm. [27] [28] [29] [30] [31] [32] [33] 1.3.2 Motility Bacteria such as E. coli can grow specialized structures called flagella (see Figure 1.1). Flagella are comprised of an indeterminate number of fibers composed of a single protein, flagellin. These fibers are arranged in a helical pattern extending from the body of the bacterium to a length of 5-10 µm. An E. coli cell can contain any number of flagella, though the presence of flagella is most prevalent in wild strains. Flagella are powered by a proton turbine to propel the bacterium forward. The rotational element of the flagella is key to its motility, because reciprocal motion cannot move an object forward. An E. coli bacterium can use its flagella to swim at up to 70 cell lengths per second. However, flagella are costly to make, and a bacterium without any need to swim (such as one in a nutrient-rich environment) will stop the production of them. Bacteria without any means to swim are much less likely to survive harsh environments than those with flagella. [34] [23] [35] [36] [37] The path of a bacterium can be approximated using a random walk model. The flagellum only has the capability to propel the cell forwards. Reversing the direction of rotation of the flagellum does not put the bacterium “in reverse,” but rather causes it to tumble, turning it to an arbitrary orientation. [38] [39] Its flagellum can then be used to propel the cell forward in this secondary direction. [40] [41] [42] By using this random walk mechanism, a cell is able to significantly change its location to one 6 that is in a more nutrient-rich environment. This process, however, is completely random. A bacterium does not have the processing power to direct its flagellum up a concentration gradient. Random walks in three dimensions are complex fractals that lie out of the scope of this project; however, it is important to understand the driving force behind this form of bacterial motility. [43] [44] [45] [46] Fick’s Law (1.1) describes mass transfer that is dependent on a concentration gradient. [47] In this equation, J represents diffusion flux, D the diffusion coefficient, φ concentration, and x position. Diffusion is how E. coli obtains the nutrients needed to reproduce and thrive. [48] [49] [50] [51] J = −D ∂φ ∂x (1.1) Unfortunately, Fick’s Law only describes the simple diffusion of nutrients down a concentration gradient across the cell wall. Both Smoluchowski and Einstein formulated a diffusion model that accounts for the drift of particles. The Smoluchowski Equation (1.2) is a realistic insight into the mechanisms and speeds at which E. coli obtains its nutrition sources. It depends upon a density (ρ), time (t), spatial coordinates (r), a frictional coefficient (ξ), and a potential (U ). By considering the contribution from friction, this equation acts as a superior model for real world systems. [43] [52] ∂ρ(r, t) = D∇2 ρ(r, t) + ξ −1 ∇(ρ(r, t)∇U ) ∂t (1.2) An important physical quantity to consider when analyzing bacterial motility is the Reynolds number. The Reynolds number (Re) is a unitless comparison of the inertial forces and the viscous forces acting on a given object in a given medium, as is defined in (1.3), where U is the mean velocity of the object relative to the medium, L is a characteristic linear value, ρ is the density of the medium, η is the dynamic viscosity of the medium, and ν is the kinematic viscosity. [43] ρLU LU Re = = η ν 7 (1.3) This quantity allows us to make various approximations, given that the Reynolds number is either very large or very small. The Reynolds number for a single E. coli swimming at 10 µ m/s through water is about 10−5 , which lies within the realm of small Reynolds numbers. This is comparable to a human swimming through tar. Most significantly, the Navier-Stokes equation (1.4), which models the turbulent flow of Newtonian liquids, can be simplified and rewritten as the simpler Stokes equation (1.5).[43] 1 ∂v + (v · ∇)v = − ∇p + ν∇2 v ∂t ρ ∇p = η∇2 v (1.4) (1.5) This is an expression of laminar flow alone, where viscous forces outweigh inertial forces, which is suitable for systems in which the Reynolds number is much less than one. Flagellum Proton Turbine Figure 1.1: A representation of a bacterium with a single flagellum. The flagellum is powered by a proton turbine, and is produced by the bacterium after it has undergone approximately three divisions. Rotation of the flagellum in one direction will cause the bacterium to move forward, while rotation in the other direction will cause the bacterium to tumble and change directions. This results in random walk behavior. All considered, we can conclude that a bacterium must swim in order to acquire the correct amount of nutrients, unless the environment is nutrient rich. As the population grows, the relative amount of nutrients decreases. Non-flagellar bacteria are unable to survive, and only flagellar bacteria will continue to contribute to population growth. [43] 8 1.3.3 Reproduction The mechanism by which bacteria reproduce is vital to understanding its growth patterns. This section gives a brief overview of binary fission and the ways in which bacteria achieve variation in their genetic sequences. Bacteria, as prokaryotic cells, divide by way of binary fission. Binary fission is a reproduction process that results in two identical copies (or daughter cells) of the original mother cell. A bacterium contains a single, circular, double-stranded DNA molecule. Cell division begins with the replication of this DNA molecule, starting at a single point called the origin of replication, and moving both directions around the chromosome until they meet again at the terminus of replication, creating two separate molecules of DNA. During this time, the cell elongates, and begins to separate once the DNA replication is completed. Septation is the process by which the cells “pinch off;” a septum is formed by producing a new cell membrane and cell wall from the center of the elongated cell. Once the septum is complete, the reproduction cycle has come to an end, creating two identical cells. The amount of time between divisions of E. coli is between 20 and 30 minutes depending on the specific strain and the environment. [26] [53] [43] Though binary fission produces identical replicas of the original cell, bacteria can still exchange genetic information. This exchange of genetic information can occur through conjugation, transduction, and transformation. Conjugation involves the physical connection of cells by way of a pilus. A conjugative plasmid is necessary to create the pilus. A pilus is a small outgrowth of cytoplasm that reaches out and connects to another cell. This tubular pathway between cells allows segments of DNA to be exchanged with the gene that codes for the pilus. Many traits can be passed between cells in this manner, including antibiotic resistance. The process of conjugation is illustrated in Figure 1.2, Figure 1.3, Figure 1.4, and Figure 1.5. [26] Transduction transfers genetic information by way of a virus (also known as a 9 DNA Plasmid Figure 1.2: The beginning of conjugation. One bacterium contains a plasmid with the gene encoding for a pilus while the other does not. Pilus Figure 1.3: A pilus projects from the bacterium containing the plasmid. This pilus is a temporary cytoplasmic connection between the two cells. Figure 1.4: Genetic material replicates and crosses into the adjacent cell via the pilus. 10 Figure 1.5: The pilus withdraws, resulting in two cells containing the same plasmid. The affected cell is now able to perform conjugation on other cells. phage). Normally, when a phage attacks a cell, it injects its genetic information, which overtakes the cell machinery. The cell then produces copies of the phage, until the cell bursts, releasing new phages. [26] In transduction, the genetic information injected into the cell by the phage is incorporated into the existing genetic information of the cell. The cell then propagates this new DNA through either conjugation or binary fission. Transformation is the uptake by the cell of genetic information from its environment. The new genetic information is then incorporated in the same way as in transduction. Of course, genetic variation can also come about as a result of mutation. [26] 1.3.4 Nutrient Usage In order to survive, bacteria have evolved to have the ability to consume more than one source of energy. E. coli prefers to consume glucose over any other form of sugar. However, in environments that do not contain glucose, a bacterium can activate a gene that allows it to consume lactose instead. The lac operon is not induced in an environment that contains glucose. When lactose is present instead of glucose, the lac operon is turned on. The lac operon controls the genes that encode for the enzymes (β-glactosidase, lactose permease and lactose transacetylase) to metabolize lactose. 11 If the bacterium is introduced to an environment containing glucose, the lac operon is repressed. [54] [26] When the environment lacks the concentration of nutrients necessary to sustain the bacterial population, E. coli, like many species of bacteria, is able to enter a dormant state, where its metabolic needs are minimized, and population growth is stopped. This dormancy can be initiated by both low nutrient concentration in the environment and other factors affecting metabolism, such as temperature. Bacterial cultures in laboratories are kept at low temperatures in a refrigerator to induce dormancy. Different strains can survive different lengths of dormancy, though lab strains (such as E. coli K-12) must still be recultured regularly. [39] 1.3.5 Growth Curve Bacteria has a specific, sigmoidal curve that defines its growth, divided into four subsections: lag phase; exponential growth phase; stationary phase; death phase. The growth curve is charted as number of viable bacteria vs. time, where viable bacteria is defined to be bacteria that is living and capable of reproduction. The lag phase occurs immediately after the bacteria have been transplanted into a new location. The number of viable bacteria does not significantly increase during this time, because the bacteria are occupied healing any damage accumulated during transportation, as well as coming to terms with their new environment. The new environment may contain a different composition of nutrients, and the bacteria will change their metabolic processes to best make use of the nutrients now available to them. The exponential growth phase is then marked by a sudden increase in the number of viable bacteria. Because bacterial cell division involves essentially doubling the number of viable bacteria during each cell cycle, an exponential curve aptly represents rapid growth. However, this growth eventually evens out into a stationary phase, where the limited supply of food and the buildup of metabolic wastes make the environment unable to 12 support a larger colony of bacteria. In most population models, this is referred to as the carrying capacity. An expression describing carrying capacity (1.6), relates the population size (P ), the rate of population growth (r) and the carrying capacity itself (K). [26] P dP = rP (1 − ) dt K (1.6) The solution to this differential equation is then KP0 ert P (t) = . K + P0 (ert − 1) (1.7) When this buildup and lack of food become too great, the bacteria enter the death phase and gradually die off. Figure 1.6 and Figure 1.7 illustrate this growth trend. [26] Stationary Phase Exponential Phase Death Phase Bacterial Population Lag Phase Time Figure 1.6: Schematic representation of the four phases of bacterial growth: lag phase, exponential phase, stationary phase and death phase. The lag phase occurs immediately after the bacteria is transplanted into a new environment, and no significant population growth occurs. The exponential phase is the result of each cell duplicating every 20-30 minutes. The stationary phase occurs when the population has reached its carrying capacity, and the death phase occurs when there is no longer enough nutrients to support the population. 13 Figure 1.7: Our experimental representation of bacterial growth, created from data seen in Chapter 2. The lag, exponential and stationary phases are all clearly visible in this representation. 1.3.6 Physical Modeling of Biological Systems Physical models must be applied in order to fully understand bacterial behavior. The Beer-Lambert Law relates the concentration of a solute to the absorption of light by the solution, as shown by (1.8), where A is absorption, ε is the molar absorptivity, ` is the path-length of the light, and c is the concentration of the solute. [55] A = ε`c (1.8) This states that absorption is related to the changing concentration of the bacteria. However, this form is not very useful, as absorption is not a quantity that can be measured directly. Instead, transmittance can be measured and converted to absorption using (1.9). [55] A = − log10 (T ) (1.9) Transmittance, in turn, is defined by (1.10) in terms of the measured intensity, I, and the initial intensity, I0 . T = I I0 (1.10) Combining (1.9) and (1.10) yields (1.11). This is a simple, realistic way in which to indirectly measure absorption. 14 A = − log10 I I0 (1.11) (1.11) can then be combined with (1.8) to yield (1.12), which gives the concentration of the solute in terms of measurable quantities. − log10 (I/I0 ) c= ε` (1.12) The molar absorptivity ε is a constant that is intrinsic to the solute itself and is a measure of the degree to which a single particle of solute absorbs light at a particular wavelength. Because this property varies with the wavelength of light in question, it is most easily observed and calculated experimentally for the specifications in question. By way of the Beer-Lambert Law, one can determine that a change in the transmittance of light through a medium is consistent with the growth of some microbe in that medium. Whether or not the most prevalent microbe growing in the solution is the microbe in question must be determined by other means, such as the wavelength of light used and the medium itself. One of the major assumptions made in this project is that the change in transmittance of the sample is actually caused by growth of E. coli. To verify this, individual cells of E. coli were counted at one hour time intervals. In order to count individual cells, heat-sealed slides of the sample must be made and stained, as detailed in Section 1.5.3. Because the number of cells present on a slide can be too great to count by eye, another method must be employed. Images of the slides should be saved as image files by using a microscopic camera for further analysis. An example image of such a slide can be seen in Figure 1.8. ImageJ, an open source program made available by the National Institutes of Health (NIH) makes the cell counting process much less tedious. The software can define a threshold and create a binary analog to the original image (see Figure 1.9). A binary image is created from the high resolution jpeg image of the slide by first converting the image to greyscale. This greyscale image is then subject to a threshold 15 Figure 1.8: A stained sample slide photographed through a microscope camera. These images are used to automate the cell counting process. determined by isodata algorithm. This algorithm starts by taking an initial threshold to determine a basic distinction between objects and background. This is the initial value used in an iterative process that calculates the sum of the average values above and below the threshold. The threshold is incremented and this process continues until the value of the threshold is greater than the composite average value. (1.13) demonstrates the basis of this algorithm. [56] Background Average + Object Average Threshold = 2 (1.13) The threshold is then used to create a binary rendition of the original image, that is values higher than the threshold are replaced by 1 and values at or below the threshold are replaced by 0. This binary rendition allows the software to count the number of circular or elliptical shapes present. The software searches for a collection of pixels occupying a determined area that satisfy specific shape requirements. This is analogous to an approximate count of the number of cells present, without the bias introduced by manual counting. The software also is able to, instead of counting the number of cells present, 16 Figure 1.9: A binary rendition of the slide above. The color image was first converted into a greyscale image, whereupon a threshold was used to convert it into a binary representation for processing. calculate the area fraction, or the fraction of the total area of the image that is composed of spots representing cells. This latter method reduces the error introduced by the phenomenon of clumping - where many cells may be grouped together and counted as one “circle” by the former method. 1.4 Previous Work A. Cobb studied the growth of E. coli in detail using transmittance techniques by way of the Beer-Lambert Law. This research has led to a proof of concept that the presence and growth of bacteria can reliably be detected by the change in transmittance caused by increased turbity due to bacterial growth. Additionally, her research has indicated that E. coli responds best to and shows a significant change in transmittance when exposed to a light source of wavelength between 500nm and 600nm. Though previous experiments have indicated that 400nm may be a more optimal wavelength at which to perform these studies, we have chosen to use Cobb’s values because they were obtained with the same equipment and techniques as we are using 17 in this particular set of experiments. The ultraviolet (UV) spectrum was left specifically unexplored, because UV light causes bacterial death. In fact, UV lamps are used in operating rooms for sterilization. From this, efforts were made to create a device to perform the transmittance analysis of bacterial growth in a field setting. Because lab tools to track transmittance over time can be very costly (up to thousands of dollars), a field device would have to be a simplified version that only performed the necessary functions to reliably detect the presence of bacteria. This device is the OBD mentioned in Section 1.1. Several groups of undergraduate students have endeavored to design an OBD, which have ranged in sophistication from simplistic to a programmed Stamp chip, as well as a variety of physical designs. 1.5 Biological Laboratory Techniques Laboratory techniques for biological specimens differ greatly from physics and chemistry techniques. There is an increased need for clean techniques working with bacteria, not only to prevent the spread of an infection, but also to prevent contamination with other biologic organisms. These techniques are the basis of microbiological work in any laboratory. 1.5.1 Media The media in which bacteria are grown can greatly influence the growth speed of the organisms and the carrying capacity of the culture. There are two main categories of bacteria culture media, which differ mainly in physical state. Media can be either differential or selective. Most kinds of organisms can grow in differential media, but exhibit different growth behaviors. Selective media allows only organisms with certain properties (for example, gram-negative bacteria) to grow. [57] The most commonly used medium is called agar, which is a gel-type substance that is usually placed in 18 a petri dish, or occasionally a test tube. The sample is then streaked across the surface of the agar, generally with an inoculating loop. This process is called plating. After incubation, bacterial growth can be observed as visible colonies growing on the surface of the agar, and the amount of growth is measured in the number of colonies present (not the size of the colonies), thus the term colony forming unit or cfu. The agar can be prepared in such a way that it is nutrient rich. The types of nutrients included in the broth determine which species of bacteria are able to grow in this media. [57] This project, however, primarily uses a different form of bacteria media: broth. Broth is a thin, watery liquid that provides a complete, nutrient-rich environment for the intended organism. A given broth generally corresponds to an agar that is identical in nutrient content, differing only in physical form. E. coli is grown in LB (lysogeny broth) or its corresponding agar. LB contains peptides, casein peptones, vitamins, minerals and some trace elements. It can be made using three different formulas: LB-Miller, LB-Lennox, and LB-Luria. Unless otherwise stated, the broth used in the following experimentation was made according to the LB-Luria formula. Unlike an agar plating of bacteria, a bacteria culture grown in broth does not produce colonies. Bacterial growth in a broth media is evident by a change in turbidity; that is, the liquid becomes murkier and sometimes experiences a slight change in color. [57] 1.5.2 Inoculation In order to culture a new sample of E. coli, an inoculating loop is used. Inoculating loops utilize surface tension to collect the same, small volume of a given liquid consistently. An example of an inoculating loop can be seen in in Figure 1.10. An inoculating loop is sterilized by exposing it to an open flame until the metal of the loop turns red hot. The loop is then considered sterile, and should not be rested 19 Figure 1.10: An inoculating loop used for the culturing of the bacteria. Can be used for both broth and agar media. against any surface, as this will cause the loop to become contaminated. It should be noted that following sterilization, the loop must be allowed to cool for a short period of time (in the case of this experiment, 45-60 s) before exposure to the inoculum in order to ensure that no bacteria is damaged. [57] To collect the sample, the lip of the culture container must be briefly placed into the flame as well as the edge of the lid, in order to create an air current that prevents contamination of the culture by airborne pathogens. The lid should never be put down, as this can also cause the culture to become contaminated. The inoculating loop is then dipped into the broth and removed with the culture container held at an angle. The lip of the culture container as well as the lid should be flamed once more before recapping. [57] As with the culture container, the lip and lid of the sample container must be briefly flamed, and the lid must never be put down. The inoculating loop should be inserted into the sample broth and then removed. The lip and lid of the sample container must then be flamed once more before recapping. Following inoculation, the loop must not be put down until re-sterilized. This is done by again exposing it 20 to an open flame until the metal becomes red hot. [57] 1.5.3 Gram Staining There are two major divisions of bacteria – gram-negative and gram positive. Gram-positive (see Figure 1.11) bacteria have a thick outer coating of peptidoglycan, whereas gram-negative bacteria (see Figure 1.12) have a thin layer of peptidoglycan located between two other cell membranes. [58] Gram-staining shows whether a specific type of bacteria is gram positive (i.e. keeps the purple stain) or gram negative (where the purple stain is washed away). To perform a gram-stain, a heat-sealed slide of the sample must first be made. Heat-sealed slides are created by using an inoculating loop to place a small portion of the liquid sample on the center of the slide. The slide is then passed over a flame until the all water has evaporated from the slide. The heat-sealed slide is then dyed. Crystal Violet, the first dye, is applied for 30 seconds. The slide is then rinsed with water. This process is then repeated with the second dye, Iodine. Together, these dyes stain any cells on the slide purple. The slide is then rinsed with an alcohol (usually methanol) for three seconds, and is quickly rinsed with water. The alcohol dehydrates the cell membrane, so the gram-positive bacteria retains the stain, while the gram-negative bacteria does not. The slide is then counterstained with safranin. Safranin is a red/pink stain. When observed under a microscope, gram-positive bacteria appears purple, while any gram-negative bacteria will appear either pink or red depending on the intensity of the stain. Gram-staining is important in the medical world, as gram-negative and gram-positive bacteria behave differently in the presence of antibiotics such as penicillin. A gram-stain can direct a physician to choose the most effective antibiotic for the infection in question. In the research world, gram-staining is important for identification of bacteria under a microscope. [58] [57] 21 Figure 1.11: A positive gram-stain. Gram-positive bacteria retain the purple dye by way of a thick layer of peptidoglycan in their cell wall.[59] Figure 1.12: Negative gram-stain. Gram-negative bacteria do not retain the purple dye of the gram-stain.[59] 22 1.5.4 Identification of Bacteria Beyond the gram-staining divisions, bacteria can also be differentiated based on shape. There are spherical (cocci), rod-shaped (bacilli), and spiral (spirilla) bacteria. By determining the shape and gram-stain of a bacterium in a microscope, the species of the bacterium can be determined (or the possibilities narrowed down). Identifying bacteria is important in growth studies in order to confirm that the intended species of bacteria is indeed the most prevalent species in the test sample. E. coli is a gramnegative rod-shaped bacterium. Under a microscope, E. coli appears as either a pink or red rod, or sometimes spherical. It is important to note that in a culture of a rod-shaped bacteria viewed under a microscope, a spherical item does not necessarily mean the sample is contaminated, as rod-shaped bacteria can be viewed end-on and appear spherical in the “flattened” view from the microscope. [59] Figure 1.13: Spherical bacteria, referred to as cocci. [59] 1.6 Experiment Overview This experiment uses the principles of the Beer-Lambert Law to model bacterial growth. Monochromatic light is passed through a sample cuvette, and the intensity of the transmitted light is measured by a sensor (see Figure 1.16). The initial intensity 23 Figure 1.14: Rod-shaped bacteria, referred to as bacilli. [59] Figure 1.15: Spiral bacteria, referred to as spirilla. [59] 24 (immediately after the sample is created) is defined to be 100% transmittance. As this intensity measurement changes over time, the transmittance changes according to (1.10), which, by way of (1.12), indicates a change in bacterial concentration in the sample. Colloidal Solution Light Path Sensor Light Source Cuvette Figure 1.16: General experimental setup. A light source is incident on a sample of bacteria. The cells suspended in this sample absorb fractions of the incident light. The transmitted light is then measured by a detector on the other side. Because the Reynold’s number for an E. coli in water is so small, the cells are suspended in the broth, forming a colloidal solution. Colloidal solutions often result in the scattering of light, which can be seen in Figure 1.16, and is discussed in more detail in Chapter 4. The benefits of suspended bacteria resembling a colloidal solution include being able to introduce the light source at any height within the sample, as the suspension guarantees that the particles will not settle out to the bottom, thereby making particle density non-variable with respect to depth. Though this height is held constant throughout this experiment, any slight discrepancies can be assumed not to result in any additional systematic error. 25 1.6.1 Lab-grade Spectrophotometer The Genesys 6, a lab-grade spectrophotometer, was used in this experiment to obtain a baseline for the behavior of transmittance in response to bacterial growth, to which subsequent measurements were compared. The Genesys 6 is also capable of collecting transmittance data for all wavelengths of light in the visible spectrum. This capability was used to determine the optimum wavelength of light at which to observe the change in transmittance due to bacterial growth. At this wavelength, the drop in transmittance occurs most quickly and is the most sharp. 1.6.2 Optical Bacteria Detector I The Optical Bacteria Detector I (OBD I) accomplishes the same overall goals as the lab-grade spectrophotometer, with the exception of testing at multiple wavelengths. The OBD I uses the optimum wavelength determined by the use of the lab-grade spectrophotometer, and tests for changes in bacterial concentration in the same way. The notable difference with the OBD I is that it is a portable field device, manufactured in-house at a very low cost. 1.6.3 Optical Bacteria Detector II The Optical Bacteria Detector II (OBD II) is the second generation of the OBD I. The OBD II was also manufactured in-house with the goal of becoming a portable field device. It utilizes data collection of both light transmittance and light scattering. Differences in light scattering can determine the size of the organisms present in the sample. 26 CHAPTER 2 TURBIDITY STUDIES WITH SPECTROPHOTOMETRY In order to build the OBD, a complete set of data encompassing all visible wavelengths was needed to determine specifications, and to provide a reliable baseline for how readings from the OBD should behave. UV-Vis spectrophotometry is common amongst colorimetrists, but, unfortunately, not microbiologists. Turbidity is a quality of bacterial cultures that is often overlooked or at least taken for granted in microbiology. As such, there is no biological instrument yet designed for the purposes of this experiment. Instead, a standard UV-Vis spectrophotometer was used. Because this device was designed for the purpose of analyzing chemicals, not suspensions of microorganisms, we were forced to apply various alterations and additions to the standard protocol and software used. This chapter describes these experimental modifications, and the process of collecting and analyzing this data. Light Path Colloidal Solution Sensor Sample Cuvette n=0-5 Monochrometer Cuvette Figure 2.1: Genesys 6 setup. The Genesys 6 uses tungsten halogen and deuterium bulbs as a light source and a photodiode as a sensor. It outputs data in percent transmittance. 27 2.1 Genesys 6 In order to collect this data, a lab-quality spectrophotometer was used. The Genesys 6 is a UV-Vis spectrometer capable of running six samples (contained in standard cuvettes) at a time. The software associated with the Genesys 6 requires a baseline scan. This baseline scan is performed using the control sample of the group (the cuvette labeled “B” in the Genesys machine). The baseline scan is used to subtract out background noise, as well as to give an initial intensity measurement to calculate an initial transmittance value, which in turn is later used to calculate percent transmittance (see (1.10)). The wavelength range over which to test the sample can also be adjusted. In this experiment, the range is limited to the visible spectrum (390nm - 750nm). The Genesys 6 then outputs .dat files that report the percent transmittance (based on the baseline scan) resulting from each wavelength tested on the sample. [36] Unfortunately, the Genesys 6 is not automated or designed for time-dependent studies, which means data must be taken manually at each desired timestep. This becomes very time consuming and prevents the data from being as consistent, due to the inability to necessarily take data at the same time interval for each data set. Thanks to N. Hunter, there is now a macro program that collects data hourly from each sample, over a period of 22 hours with consistent specifications. The macro utilizes basic computer commands to cycle through a script that automatically selects the same options on the Genesys 6 graphical user interface (GUI) one would select manually. This GUI is available as open source software as well, and is called Datalyse. [60] The script cycles through this process each hour, having been set on a time delay counter. The code used for this macro can be seen in Appendix B. This improvement has allowed consistent data collection following the initial setup. 28 Figure 2.2: Genesys 6 UV/Vis-Spectrophotometer. The Genesys 6 tests six cuvettes at once, and tests over a wide range of wavelengths. 29 2.2 Establishing Initial Conditions Among the many difficulties in dealing with biological specimens is the inability to know which growth phase (see Section 1.3.5) a culture is in before testing begins. It was hypothesized that this initial condition could affect the lag time or the slope of the exponential growth rate after the bacteria is recultured. A bioreactor (see Figure 2.3) is the most efficient means for controlling initial conditions of living substances. A bioreactor operates by maintaining a constant environment for the organism (this method works extremely well for both bacteria and fungi). The bioreactor uses a temperature control system based on cycling heated/cooled water of the appropriate temperature around the bioreactor’s culture enclosure. The temperature is set at the optimum temperature for growth of the organism being maintained. The bioreactor chamber itself is filled with the desired broth in which to grow the organism. A limiting nutrient is then gradually added to the mixture. It is the rate at which this nutrient is added that can be used to control the growth phase of the culture. Limiting this nutrient (“starving” the bacteria), will force the bacteria into a stationary phase, while providing an excess of this nutrient will encourage the bacteria to enter the exponential growth phase. Additionally, the bioreactor can control foam and pH levels within the culture. A sensor for each is constantly in contact with the culture media, which is monitored by an external computing system that utilizes a negative feedback loop to pump a culture-friendly acid, base, or antifoaming agent into the enclosure. A motor and compressed air system keep the enclosure oxygenated (utilizing the same principles used in maintaining fishtanks) and properly mixed. This creates an consistent, controlled environment for the organisms. [38] In this project, LB was used as the broth and glucose was used for the limiting nutrient. The optimum temperature for growing E. coli is around 37◦ C (which, of course, is regular human body temperature). This setup eliminated systematic error occurring as a result of a culture being in a constant growth fluctuation. The 30 AntiFoam E. coli Acid Base Nutrient Temperature Control Temperature, Foam and pH Sensors Motorized Stirrer Culture Figure 2.3: A basic bioreactor setup. The bioreactor maintains a constant environment for the bacterial culture, by controlling temperature, pH and the amount of nutrient available. 31 particular setup used in this experiment can be seen in Figure 2.4. Figure 2.4: The bioreactor used in this experiment. A variable peristaltic pump was used to control the rate of glucose addition in order to manipulate initial growth phase of the culture. 2.3 Assumptions As with all experiments, certain assumptions had to be made in order to gather the necessary data. By far, the most important assumption was that the observed change in transmittance of a sample was due to bacterial growth, as opposed to other changes that could result in similar effects. Similarly, one must assume that the intended species of bacteria (E. coli) is growing in the sample, as opposed to another species that is able to subsist off of the same nutrients provided by the broth. Data supporting the validity of these assumptions can be seen in Section 1.3.6. It must also be assumed that the testing environment is clean. Though this would not be 32 the case in a field setting, it is necessary in order to gather the baseline data against which the field data is compared. To account for all of these assumptions, bacterial samples are kept isolated to the greatest degree possible, and standard precautions for handling biohazards are observed. 2.4 Testing Protocol A very specific procedure for testing samples in the Genesys 6 is necessary to acquire the proper data. 1. Arrange bacterial collection line (see Figure 2.5) in pump so it is in the pumpingout position. 2. Secure sterile retrieval bottle to the end of bacterial collection line. 3. Pump approximately 2cc of contaminated broth into retrieval bottle. 4. Reverse the position of the bacterial collection line to pump the remaining broth in the line back into the biostat. 5. Sterilize edges of broth bottle in alcohol flame. 6. Use sterile syringe to dispense 3 cc of broth into each of six cuvettes. 7. Sterilize edges of broth bottle and replace cap. 8. Sterilize inoculating loop. 9. Inoculate five cuvettes with bacteria from the retrieval bottle, sterilizing the inoculating loop between each. 10. Sterilize edges of retrieval bottle and empty into bleach. 11. Turn on Genesys and allow self-calibration. 33 12. Open Datalyse on the computer. 13. Place blank cuvette in the B-slot in the Genesys. 14. Choose the Genesys by selecting Choose Device, Other Devices, Genesys 6 UV/VIS. 15. Run baseline scan by choosing Genesys, Baseline Scan, confirm default settings. 16. Place the cuvettes in slots 1-5. 17. Set up macro by choosing the macro icon in the lower right hand corner. 18. Double click 2-19-09. 19. Change trigger to Word/Phrase. 20. Save. 21. Close. Do not shutdown. 22. Select Datalyse window. 23. Type “YEW”. 24. To clean up, place all cuvettes in a tub of dilute bleach. 25. In case of spill, dilute an equal amount of bleach into water. Spread bleach solution over affected area and wipe up using paper towels, while wearing gloves. 2.5 Results Data was taken with the Genesys 6 over many years, using many different cultures of bacteria, and our knowledge related to the experiment has increased, changed the way in which the experiment has been performed. Initially, specimens were not kept 34 Figure 2.5: A labeled photograph of the setup. A second pump is used to collect culture solution from within the bioreactor. in bioreactors, nor was the initial condition of the bacteria attempted to be controlled in any way. There was no data involving lab temperature, nor was there good recordkeeping concerning which data was taken with which bacteria subculture nor which batch media broth was used. Data sets taken under these conditions can be seen in Figure 2.6. Note that these data sets are widely spread, and even converge to different transmittance values. Though it is hypothesized that the above-mentioned systematic errors were responsible for these discrepancies, there is no means by which to prove this hypothesis either way. Following the disappointment encountered when analyzing the data seen in Figure 2.6, measures were taken to eliminate these systematic errors. Bioreactors were employed to control the initial state of the bacteria, and more diligent recordkeeping filled in the previously missing information regarding broth and temperature. Data collected following this revised protocol can be seen below in Figure 2.7. Note that the lag times were more consistent following the implementation of these procedural 35 Figure 2.6: Data taken by the Genesys 6 prior to implementation of new protocol. There is a wide spread in lag time for this data, however it was collected using several distinct batches of bacteria, without a stabilized protocol. corrections. Included in this new data collection is data taken with differing initial concentrations of bacteria in each cuvette. This experiment was performed by numbering the cuvettes to be put in the Genesys 6 by their corresponding label in the machine (0-5). A corresponding number of inoculating loops of bacteria were added to each cuvette; that is, the baseline sample had none, the first sample contained one loopful of bacteria, the second two loopfuls, and so on and so forth. As expected, the cuvettes containing more bacteria initially (in this case, those represented by the yellow lines) entered the exponential growth phase first. In order to observe the behavior of bacteria from one initial state transplanted into a new environment, the bioreactor was used to prepare cultures in both the exponential and the stationary phases of growth. Bacteria from each initial phase was tested with the Genesys 6, and can be viewed in Figure 2.9. Note the distinct differentiation between bacteria initially prepared in the stationary phase as opposed to bacteria prepared to be in the exponential growth phase. It is also important to address another revision in protocol that took place before these measurements. The 36 % Transmittance 100 80 60 40 20 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Time (hr) Figure 2.7: Data collected following the protocol revision. The spread of data is significantly narrower and follows two groupings corresponding to the two cultures used to collect the data itself. The transmittance values follow the inverse of an expected bacterial growth curve. Figure 2.8: Cuvettes tested with differing levels of initial bacterial concentration. The yellow lines, which display a significantly smaller lag phase, represent samples that were inoculated with five times as much bacteria as the other samples. 37 broth used, both for culturing and for testing was changed from LB to a nutrient broth. LB is very well suited for the growth of E. coli, while nutrient broth is geared toward growing a variety of specimens. This explains the change in slope of the growth curve and the increased difficulty in keeping samples uncontaminated. Figure 2.9: A comparison plot of bacteria prepared in stationary and exponential growth phases. The bacteria intially prepared in the stationary phase has a slightly shorter lag period than bacteria prepared in the exponential growth phase. Despite the wide spread of data sets present in this data collection, it is clear both that there is a definitive shape the plots conform to, and that this shape is consistent with what is accepted as the model of population growth for bacteria. 2.6 Analysis Though the curves seen in Figure 2.6 do follow the general trends of bacterial growth, they offer little to no insight regarding mathematical behavior of the organisms. However, the curves are concentrated in two distinct groupings, one centered about 7 hours, the other centered about 16 hours. It was this observation that led to the hypothesis that initial growth conditions were affecting the lag times in testing. 38 Taking care to eliminate systematic errors did well to decrease the spread of data from about 12 hours to about 4 hours (Figure 2.7). This data, however, also suffered the inconvenience of displaying two distinct groupings. Not only are there two points of high concentration for the exponential phase, but these two groups also diverge during the stationary phase - one group continues in a downward slope, whereas the other has a slight increase in transmittance, further supporting the hypothesis that some initial condition was causing the discrepancy. Comparing data taken from bacteria in these two initial growth phases, there are indeed two distinct groups, as the different cultures respond separately to a new environment. Interestingly enough, it is the stationary phase that begins to grow first. Figure 2.10 explores the difference between the transmittance values as Stationary(t) − Exponential(t) Inconsistency = Abs 1/2(Stationary(t) + Exponential(t)) (2.1) . We see a large spike in this value at eight hours, as the transmittance of the stationary phase bacteria begins to decrease. This shows that there is a significant difference between the lag time associated with bacteria prepared in the stationary and exponential phases. We hypothesize that the explanation for the extra lag in bacteria that had previously been in a constant state of exponential growth is that they suffer shock from being transplanted into an environment with a relatively low level of glucose. The bacteria may need time to adjust to the new nutrient concentrations. 2.6.1 Error Contributions Even with a meticulous, standardized protocol, there are many opportunities for error to contribute to our experimental results. The presence of biological specimens introduces an additional level of uncertainty that is very important to standardize. The possible sources of error, listed in order from most influential to least influential, 39 0.06 Inconsistency 0.05 0.04 0.03 0.02 0.01 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Time (hr) Figure 2.10: Analysis in discrepancy between average values for bacteria prepared initially in the stationary phase and the exponential phase. The standardized difference between stationary and exponential phase increases at the end of the lag phase for the bacteria prepared in stationary phase. are as follows: • Systematic Error – Biological 1. Genetic variation in culture ∗ A culture can evolve over a short period of time, due to the fast reproductive rate of E. coli. ∗ Re-culturing can introduce unknown elements to the culture, and any of the means of genetic variation discussed in Chapter One may or may not occur. ∗ Cultures started from various initial samples may not behave identically. 2. Contamination ∗ Equipment used is classified as clean, not sterile. 40 ∗ Protocol is clean, not sterile. Commensual bacteria from tester may contaminate samples. 3. Broth inconsistency ∗ No batch of broth is identical to another. Water may boil off during sterilization, affecting the broth concentration. ∗ Broth inside the bioreactor has a different nutrient composition than broth used in testing, due to the glucose added as limiting nutrient. 4. Culture concentration ∗ As in testing samples, the concentration of bacteria in a culture increases over time. This corresponds to a slight increase in initial inoculum concentration in test samples. – Procedural 1. Time discrepancies ∗ It takes the Genesys 6 four minutes to analyze a sample, while it only takes two minutes to prepare a sample. This leads to a two minute time discrepancy. ∗ This becomes very prevalent towards the end of a run, because the time discrepancy is cumulative. 2. Temperature of inoculating loop ∗ The inoculating loop is cooled before inoculation so as not to kill any bacteria. ∗ Inconsistent inoculating loop temperature at time of inoculation can change the number of living bacteria captured by the inoculating loop. ∗ Cooling time was standardized to 90 seconds to regulate this. 41 • Statistical Error 1. Tolerance of Genesys 6 – Directly affects raw measurements. – Characterized by Genesys 6 manual. – Represented as vertical error-bars in data analysis. 2. Cell Counting – Multiple methods yield different results. – Does not affect spectrophotometric studies. 3. Analytic Methods – Fit parameters accurate to one part in 100. Table 2.1 lists, in descending order, the quantitative values for these sources of error. An empty value field indicates a source of error that has been corrected following initial testing, and thus eliminated in data sets used for statistical analysis. Table 2.1: Error contributions for Genesys 6 testing Source of Error Type of Error Value Genetic Variability Biological ±0.73 hr Genesys 6 Tolerance Statistical Up to ±0.014 (transmittance) Time Discrepancies Procedural +2.0 ± 0.2 min Analytic Methods Statistical 1 part per 100 Contamination Biological Determined by Control Sample Broth Inconsistency Biological - Culture Concentration Biological - Inoculating Loop Temperature Procedural - Cell Counting Statistical - 42 2.6.2 Statistical Analysis The group of ‘revised’ data is composed of all standard experimental runs from 2009 onward. Though the data appears to suffer from a wide spread, closer inspection reveals that this is due to horizontal shifts. This would normally be very disconcerting, however further inspection reveals that data sets taken from the same culture have extremely small spreads. The greater the spread in time between the actual testing (for example, data observed in May compared to data observed in September), the greater the time shift observed when plotted. Taking this into consideration, the average time shift of these curves will correspond to the time shift present when all data sets are fitted mathematically to a single curve. Assuming this analysis is sufficient, we will proceed to analyze only the error in transmittance and absorption values. Like most population models, the bacterial growth curve is an S-shaped or sigmoid curve. A popular model for this curve is (2.2), where P is population, a, b, c and k are adjustable parameters, and t is time. This model stems from equation (1.7), having generalized the parameters. The parameters of this model allow us to account for different durations of the lag phase, various slopes of the exponential phase, and various carrying capacities. P = a b + ce−kt (2.2) However, the raw data we collect is not in a form that abides by this curve. Instead, we calculate percent absorption (a quantity that is directly correlated with the population of the culture) from percent transmittance by (2.3). A=1−T (2.3) Each data point was converted to percent absorption, and imported into Mathematica for data processing. The FindFit function was used to fit all of the data 43 points in the revised set to the model presented in (2.2). The resulting parameters (see Table 2.2) are expressed with two significant figures of accuracy. This model only accounts for the lag, exponential and stationary phases, however further modeling could include the death phase as well. The death phase is apparent in this data as a decrease in absorption around 16 hours. This new model would likely be of the same form with an additional exponential decay. Table 2.2: Parameter Values for Genesys Data Parameter Value a 1.35 b 0.023 c 10.8 k 0.77 All data points for each time step were then averaged to provide the points seen in Figure 2.11. The error contributed by the device itself was calculated by analyzing all of the control sets within the revised data collection. Theoretically, all values of uncontaminated controls should be precisely 100%, as the baseline function of the Genesys 6 software sets it as such. The standard deviation of these data points was calculated and set as the vertical errorbars seen in Figure 2.11. A reduced χ2 analysis of this data yields χ2red = 1.36. Ideally, χ2red = 1, and when χ2red >1, the fit has not fully expressed the data. As stated above, this discrepancy is likely due to this model not including the death phase, which is apparent in the plot itself. Figure 2.11 is very encouraging, as the fitted curve lies within the errorbars assigned from the error analysis. The point which is in least agreement with the curve is at the upper cusp of the S-curve, which, for bacterial detection, is not a point with which we are particularly concerned. These observations have helped develop a better understanding of the correlation 44 Figure 2.11: Curve fitted to the average values of the Genesys 6 data. Error-bars represent the variance in the Genesys 6 measurements over time for a sample of constant transmittance. between light transmittance and bacterial growth. It is with this knowledge that a smaller, more adaptable device can be created in order to apply this theory in the field. 2.6.3 Cell Counting The cell counting data can be seen in Figure 2.12 and Figure 2.13. Using the fitting methods described above, the area fractions and cell counts calculated by ImageJ were modeled mathematically. The parameters for cell population as a function of time (see Table 2.3) and area fraction as a function of time (see Table 2.4) are expressed below using two signficant figures. 45 Figure 2.12: Cell counting in terms of colony forming units. This method counts individual cells, but does not account for cell clumping. 46 Figure 2.13: Cell counting in terms of area fraction of cells. This is the preferred method, as it accounts more closely for the clumping of cells. 47 Table 2.3: Parameter Values for Cell Population Parameter Value a 13.0 b 0.025 c 0.43 k 0.74 Table 2.4: Parameter Values for Area Fraction Parameter Value a 0.87 b 0.22 c 8.98 k 0.79 Figure 2.14 and Figure 2.15 show the curves corresponding to Table 2.3 and Table 2.4 respectively, superimposed over their supporting data sets. Area Fraction 4 3 2 1 2 4 6 8 10 12 Time HhrL Figure 2.14: A mathematical fit of the cell count data shown above, in colony forming units. The same bacterial culture used for cell counting was then tested for transmittance 48 Population HcfuL 500 400 300 200 100 2 4 6 8 10 12 Time HhrL Figure 2.15: A mathematical fit of the area fraction data shown above. changing by the Genesys 6. This transmittance data can be seen in Figure 2.16. The correlation of these data sets support the assumption that bacterial growth is indeed causing the change in sample turbidity, as opposed to other factors. There is a certain amount of inherent error involved in the cell counting process. Most notably, using the same threshold for each image is only suitable when each slide is stained similarly. However, it is extremely difficult to achieve a consistent level of staining, particularly over a period of time. We have eliminated the human factor in pursuit of also eliminating bias; however, it has also decreased our ability to use human judgment. The other significant error incurred is the subject of clumping. There are many occurrences of cells growing extremely close to one another on the slide, so much so that the software is unable to count them as multiple entities. This is why we have chosen to rely on the area fraction data, as this method calculates the portion of the image contributed by cells. Also, cell counting is affected by the orientation of bacteria. This contribution is lessened when dealing with spherical bacteria, however E. coli is rod-shaped. A rod-shaped bacterium oriented on-end 49 Relative Growth 1.2 1 Area Fraction Absorption 0.8 0.6 0.4 0.2 0 1 2 3 4 5 6 7 8 9 10 11 Time (hr) Figure 2.16: A normalized absorption curve as obtained from the Genesys 6 compared to corresponding area fraction data. This plot shows the correlation between actual cell growth and change in transmittance. appears to be circular under a microscope. An algorithm constructed to determine the number of ellipses present would therefore have to be modified to deal with the elliptical eccentricity associated with bacterial orientation. Though area fraction still accounts for these particular bacteria, they contribute less to the actual value of area fraction than a bacterium on its side. By eliminating human bias, we also eliminate the ability to account for these inconsistencies. 50 CHAPTER 3 THE OPTICAL BACTERIA DETECTOR I: ABSORPTION ONLY This chapter details the development and testing of the Optical Bacteria Detector (OBD). Light Path Colloidal Solution Phototransistor LED Cuvette Figure 3.1: Basic setup for the OBD I. The OBD I uses a 560 nm LED as a light source, and a PNZ150 phototransistor as a detector. 3.1 Requirements In order to be effective as a field substitute to machines like the Genesys 6, the OBD must meet a variety of requirements. Most importantly, the change in transmittance due to bacterial growth must be clearly evident in the results gleaned from the OBD. These measurements will be taken with the same timestep (one hour) as in testing with the Genesys 6; however, the data will be taken and recorded manually. Ideally, when in use in the field, only an initial measurement and a followup measurement (observed after a predetermined period of time) will be taken and compared against one another to determine the presence of bacteria. The OBD will use cuvettes as sample collection containers. Cuvettes will be covered, and pre-filled with either powdered broth or a liquid broth concentrate. As discussed in Section 1.1, size and cost of the device are extremely important as well. Because this device will eventually 51 be used in remote areas and third world countries, it must be easily transportable and very affordable. These constraints have shaped the design and capabilities of the OBD itself. 3.2 Limitations As with all devices, the OBD I suffers from certain limitations. The device does not measure transmittance values for multiple wavelengths, nor is it automated. An automated device of this sort would perform tests at zero and twelve hours, and determine if the value has changed, and if it has, supply some sort of indicator, such as an LED or an alarm. The device is also unable to determine the specific species of bacteria growing in the water source. Though use of selective media could more closely determine certain qualities of the bacteria, such as response to a gram-stain, the device cannot determine these properties directly. With these limitations in mind, we can proceed to use the OBD I to detect contamination in water. 3.3 Design The design of the OBD can be divided into the physical design and the layout of the electronics. This section serves to describe and provide figures of both. 3.3.1 Circuit The circuit design for the OBD follows a very simple phototransistor circuit layout. A standard green LED of wavelength 560nm provides the light source. Though LEDs do not produce monochromatic light, the specificity they provide is close enough for the purposes of the OBD. A phototransistor (PNZ150) is used as the light sensor. The signal from the phototransistor is amplified by a LM358 op-amp, and delivered to a voltmeter. The voltage outputted by the op-amp is read in milliVolts via the voltmeter. A schematic of the circuitry of the OBD can be seen in Figure 3.2. 52 1 MΩ 1 kΩ 1.5 V 1.5 V LM358 + 1.5 V 1.5 V 1 kΩ 1 kΩ Vout Figure 3.2: Circuit schematic for the OBD I. The output from the phototransistor is fed into an op-amp to amplify its signal, before being delivered to a voltmeter. This circuit design can be improved by ensuring the linearity of the system. Instead of biasing the phototransistor by connecting it to ground through a resistor, it can be directly fed into the op-amp. Also, a voltage regulator, most likely a switching regulator, can be used between the battery supply and the circuit itself, to supply a more ideal voltage source. 3.3.2 Housing The housing for the device proved to be particularly challenging to design, as it must prevent external light from shining into the cuvette. The main part of the housing is built from a plastic container, with a metal screw-on lid. The metal sheet allows for openings for the electronic readout, an on/off switch, and the slot for the cuvette. The slot for the cuvette is a black plastic box, fitted to the size of the cuvette. Small holes are drilled through opposite sides to allow access for the LED and the phototransistor. A similar black, plastic cover blocks out the remainder of the 53 ambient light, and is attached to the metal lid by a length of fishingline. Schematics of this housing can be seen in Figure 3.3 and Figure 3.4, along with a photograph of the prototype in Figure 3.5. Sample Slot Cap Data Readout ON Power Switch OFF Figure 3.3: Top view of the housing for the OBD I. Features include an on/off switch, a cap for the sample container, and an LCD display. 3.4 Testing Procedure Two procedures apply for testing samples with the OBD: lab testing and field testing. The lab testing procedure was used to verify that the OBD was performing as anticipated, while the field testing procedure is that which would be used to test a potentially contaminated body of water for the presence of E. coli. 54 Digital Multimeter Cap Lid Circuit Board Power Supply LED Sensor Sample Slot Base Figure 3.4: Internal view of the housing for the OBD I. The circuit boards are mounted together, and the sample container is completely enclosed to prevent interference from ambient light. 55 Figure 3.5: Photograph of the OBD I. 56 3.4.1 Lab Testing The protocol for using the OBD to test bacterial samples is very similar to that detailed in Section 2.4. Following the inoculation of samples, an individual baseline of each sample cuvette must be taken. A reading for each cuvette must then be recorded for each sample every hour for the following 24 hours. Results can be normalized to one another by setting the initial values at 100%. Though data-taking by this method is more tedious than using the Genesys 6, the nature of the device and manual data acquisition allows more than five samples to be tested in a given 24 hour period. 3.4.2 Field Testing When using the OBD for field testing, different inoculation techniques must be used. Because the concentration of E. coli is much lower in a contaminated body of water, using an inoculating loop to culture bacteria from the water to a cuvette of sterile broth may not transfer enough viable bacteria to grow a healthy culture. Instead, water must be collected from the field source using a needle and syringe, and injected into a covered cuvette containing either highly concentrated liquid broth, or the base broth powder. The sample water will bring the broth to the desired concentration. A baseline measurement must be recorded for each sample at this initial time, and either subsequent measurements should be recorded every hour, or a single follow-up measurement must be recorded between 10 and 14 hours later. 3.5 Difficulties In the development of the OBD, many complications arose. These complications varied from difficulties involving the media, to inconsistencies in the OBD itself. In order to ensure that the OBD is a reliable means of detecting bacteria, these issues had to be addressed before significant amounts of data acquisition. Initially, field samples were to be collected by taking 3 cc of sample water and 57 injecting it into a covered cuvette containing a pre-measured amount of powdered broth. The cuvette would then be shaken to mix the broth. However, should the environmental temperature be significantly higher than room temperature, the broth powder tends to melt and congeal, making it very difficult to mix the broth media. To avoid this difficulty, prepared field testing cuvettes will contain 1 cc of triple concentrated liquid broth. The OBD itself suffered a few complications. The first of which was sensitivity to ambient light, particularly in direct sunlight. Initially, the readings from the OBD were dependent on whether or not the device was being used in direct sunlight or in the shade. In order to circumvent this issue, greater measures were taken to block light from the testing chamber, as well as to create a better seal on the removable cap. It is still recommended that the OBD be tested in approximately the same level of ambient light, to ensure that this will not affect the device’s performance. The OBD is also slightly sensitive to heat. The effect on readings is most easily observed when a sample is tested outside in the summer (anywhere from 90-100◦ F), followed by testing indoors at room temperature (approximately 70◦ F). At higher temperatures, the OBD can read up to 3 mV higher than at room temperature, as the electronic components themselves are sensitive to heat. This is only problematic when the OBD is expected to be taken into a different temperature environment during a test. For non-prototype versions of the OBD, the interior of the device will be filled with a thermally insulating foam that will prevent any electronic elements from being overly affected by the change in temperature. The prototype OBD will remain as is, in order to leave it open for adjustments to improve performance. Again, purposefully changing the OBD’s temperature environment is not recommended, in order to eliminate systematic error in testing. 58 3.6 Results Currently, the only results available from the OBD I are from lab testing. A plot showing the complete set of results obtained from lab testing of the OBD I can be seen in Figure 3.6, and a closer view in Figure 3.7. These results are normalized to 100%. Transmittance 1.0 0.8 0.6 0.4 0.2 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Time (hr) Figure 3.6: Complete data set taken by the OBD I. This data was taken over two separate runs using the same batch of bacteria. Though transmittance only falls to 0.60, these tests were performed using nutrient broth instead of LB. These results may appear different than those seen in Chapter 2, however they were obtained while using the nutrient broth instead of LB. We must compare this data to such plots as Figure 2.9. Both the data represented in Figure 2.9 and the data obtained with the OBD I reach a final transmittance value of approximately 0.60. This indicates that the OBD I is measuring the same properties as the Genesys 6, which was our intent. We hypothesize that the long lag time present in these samples is simply a characteristic of this culture of bacteria. 59 Transmittance 1.0 0.8 0.6 0.4 0.2 12 13 14 15 16 Time (hr) 17 18 19 Figure 3.7: Same data as above, a closer view, beginning at 12 hours. 3.7 Analysis Though much of the analysis of data taken with the OBD I resembles that of the data taken with the Genesys 6, there are more sources of error to consider, and a different baseline from which to calculate model parameters. 3.7.1 Error Contributions The biologically contributed error in testing with the OBD I is much the same as that described in the previous chapter. However, due to a new protocol, procedural error changes significantly. The sources of systematic and statistical error present in testing with the OBD is listed below: • Systematic Error – Biological 1. Genetic variation in culture 60 ∗ A culture can evolve over a short period of time, due to the fast reproductive rate of E. coli. ∗ Re-culturing can introduce unknown elements to the culture, and any of the means of genetic variation discussed in Chapter One may or may not occur. ∗ Cultures started from various initial samples may not behave identically. 2. Contamination ∗ Equipment used is classified as clean, not sterile. ∗ Protocol is clean, not sterile. Commensual bacteria from tester may contaminate samples. 3. Broth inconsistency ∗ No batch of broth is identical to another. Water may boil off during sterilization, affecting the broth concentration. ∗ Broth inside the bioreactor has a different nutrient composition than broth used in testing, due to the glucose added as limiting nutrient. 4. Culture concentration ∗ As in testing samples, the concentration of bacteria in a culture increases over time. This corresponds to a slight increase in initial inoculum concentration in test samples. – Procedural 1. Time discrepancies ∗ It takes only 30 seconds to analyze a sample with the OBD I, while it takes two minutes to prepare a sample. This leads to a 90 second time discrepancy in the opposite direction as when testing 61 with the Genesys 6. ∗ This becomes very prevalent towards the end of a run, because the time discrepancy is cumulative. 2. Temperature of inoculating loop ∗ The inoculating loop is cooled before inoculation so as not to kill any bacteria. ∗ Inconsistent inoculating loop temperature at time of inoculation can change the number of living bacteria captured by the inoculating loop. ∗ Cooling time was standardized to 90 seconds to regulate this. 3. Fluctuations in laboratory temperature ∗ Controlled by storing samples in a warm water bath set to 37.0◦ C. ∗ Though body temperature is optimal for growth of E. coli, it is not necessary. It is only necessary to standardize the temperature, though body temperature is the logical choice. 4. Handling of samples ∗ Cuvettes are capped, and handled only when wearing a fresh pair of nitrile gloves. ∗ It is easy to determine whether or not a sample has been subjected to smudging or contamination, both by direct observation and by observing the data itself. Only two samples were clearly affected (due to visible smudges on the cuvettes) and were not included in statistical analysis. • Statistical Error 1. Tolerance of OBD I 62 – Directly affects raw measurements. – Characterized by fluctuations in transmittance over time when analyzing a control sample. – Characterized in linearity of sensor. – Represented as vertical error-bars in data analysis. 2. Analytic Methods – Fit parameters accurate to one part in 100 Table 3.1 numerically describes the contributions of these errors, listed descending by numerical value. These should be taken into consideration when reviewing the contents of Section 3.7.2. An empty value field indicates that initial testing observed the contribution of error, and the source of error has been sufficiently corrected. Table 3.1: Error contributions for OBD I testing Source of Error Genetic Variability OBD I Tolerance Time Discrepancies Analytic Methods Contamination Broth Inconsistency Culture Concentration Inoculating Loop Temperature Laboratory Temperature Handling of Samples 3.7.2 Type of Error Value Biological ±0.73 hr Statistical Up to ±0.011 (transmittance) Procedural -1.5 ± 0.2 min Statistical 1 part per 100 Biological Determined by Control Sample Biological Biological Procedural Procedural Procedural - Statistical Analysis The FindFit process used in the previous chapter was then used to model the behavior of bacteria when measured the OBD I. The model described in (2.2) was again used to chart the behavior, and the parameters a, b, c, and k are again expressed to two significant figures in Table 3.2. 63 Table 3.2: Parameter Values for OBD I Parameter Value a 6.2 ∗ 10−3 b 4.7 ∗ 10−4 c 61.42 k 0.76 As done with the data in Chapter 2, an average absorption value was expressed for each of the timesteps involved. An error for each of these values was then calculated from the variance of the OBD I exposed to a stable sample that experiences no change in turbidity. This value is much higher than in the previous chapter, because the tolerance value for the OBD I is very low. This curve and errorbar analysis is shown in Figure 3.8. % Absorption 14 12 10 8 6 4 2 5 10 15 20 Time (hr) Figure 3.8: Statistical analysis of the curve fitted to OBD data. Error-bars are calculated from the variation in measurements observed by the OBD I when testing a control sample. This analysis demonstrates that the bacterial growth model applies to this data 64 very accurately. All but one of the average values calculated lie within the errorbar range of the fitted growth curve. Though this point is at an important timestep in our analysis, (the lower cusp of the S-curve), it is of greater value than the curve itself. This suggests that, on average, the sudden drops in transmittance values observed on the OBD are actually precursors to the drop predicted by the curve described by the parameters in Table 3.2. This data shows that the OBD has reliable capabilities, and that field testing will be the final trial to see whether or not it is a capable field device. The OBD accurately portrays the trend of bacterial growth, and follows predictable patterns. 3.8 Linearity A particular concern when constructing the OBD I was the linearity of its response. Phototransistors are easily saturated and this can affect the reliability of the data obtained through them. In order to determine the linearity of the system, I chose to use the voltage and current dependence of the light intensity from the LED to test the system’s response. It proved very difficult to use a filter to test linearity, because the geometry of testing conditions were too confined, and observing response outside of testing conditions is irrelevant. Instead, I wired the LED through the same resistance value as used in the OBD, and connected powered it via an adjustable lab power supply. This setup can be seen in Figure 3.9. I then proceeded to vary the supply voltage to the LED in 0.25 V increments, and observed the change in reading from the OBD I. This method was tested over a supply voltage range of about 2.0 V (the forward drop voltage of the LED) and 9.0 V. As seen in Figure 3.10, the sensor was not saturated, even beyond normal operating conditions. Having taken these results into consideration, it is reasonable to believe that the OBD I is operating in a linear regime. 65 Voltage Current (V) (A) Variable Power Supply Voltage PWR GND 1 kΩ ON Phototransistor (PNZ150) LED OFF Circuit Board Battery Supply Figure 3.9: Experimental setup to test the linearity of the response of the OBD I. A variable lab power supply was used to supply voltage to the LED independent of the rest of the circuit. 66 700 OBD Reading (mV) 600 500 400 300 200 100 0 0 2 4 6 8 10 Supply Voltage to LED (V) Figure 3.10: Linearity analysis of the OBD I’s response. The initial jump at 2.0 V is the forward voltage drop necessary for the LED itself to turn on. 67 CHAPTER 4 THE OPTICAL BACTERIA DETECTOR II: ABSORPTION AND SCATTERING The transmittance of light through a bacterial sample is a concept that has been explored very little by the scientific community. Though the information gained from testing such a delicate property on unreliable and everchanging samples may be limited, it is not without its benefits. This chapter explores the scattering properties of light through bacteria. 4.1 Theory Scattering of light off of particles is very dependent on the size of the particles in question. Ideally, the particles are spherical with a diameter on the order of the wavelength of light used. An E. coli bacterium measures one micron in diameter and two microns in length, and is not spherical. However, the spherical approximation is a good place to begin. The scattering of light off of spherical particles is called Mie scattering. Mie scattering is used in the medical field to detect abnormalities (usually cancer) by observing the interference patterns of the scattered light. [61] Similar to Mie scattering is Tyndall scattering, which negates the restrictions of shape that Mie scattering imposes. Tyndall scattering is used to determine the size of colloidal particles in a suspension, which is essentially the goal of this application of scattering. Tyndall scattering is most effective when shorter wavelengths of light are incident on the suspension. [61] Extinction measurements are highly important when analyzing colloidal solutions. Extinction can be expressed as a combination of the two quantities this study has been based on, absorption in scattering. Let us define extinction as in (4.1), where E is extinction and S is scattering. [61] E =S+A 68 (4.1) Considering this, we find that our earlier assumptions are not quite right. Assuming the intensity of light lost is due to absorption alone is incorrect and to accurately determine the absorption value we must observe both the transmitted intensity and the scattered intensity. This will be discussed further in Section 4.2. We can now use our definition of extinction mathematically. Let us define now I and I0 as measured intensity and initial intensity respectively. ` we will again define as pathlength and we will define a new variable, γ to be the extinction coefficient. The extinction coefficient is a function of an efficiency factor (Qext ) (4.2) for a medium containing N particles of radius a per unit volume (4.3). [61] 4 Qext = 2 Re[S(0)] x γ = N πa2 Qext (4.2) (4.3) We can now determine the intensity ratio to be defined as a function of ` and γ (4.4). I = e−γ` I0 (4.4) These supply a very elegant mathematical description of the extinction coefficient, but a simpler way to view it is simply the turbidity. Upon close inspection, we see that extinction indeed arises from turbidity itself. We can define τ to be the turbidity of the sample measured in Nephelometric Turbidity Units (NTU), which are the standard turbidity units for measuring the turbidity of drinking water, and rewrite (4.4) as (4.5). [61] I = e−τ ` I0 (4.5) A region of particular interest is the concept of higher order Tyndall spectra. La Mer and Johnson have done a good deal of work regarding this effect, and have classified the order of Tyndall spectra by particle radius. At its smallest, the radius of E. coli is 0.5 µm and at its largest it is 1 µm. La Mer and Johnson describe the existence of eight orders of Tyndall scattering for particles of radius 0.5 µm and ten 69 orders for particles of radius 0.6 µm, however the study did not include particles of larger radius. They observed these higher orders as resulting red bands. [61] 4.2 Engineering Design The circuitry for the OBD II is identical to that of the OBD I (seen in Figure 3.2), though doubled to accomodate two sensors. Currently, the OBD II is a benchtop device (unlike the OBD I field device), and thus lacks the more customized housing of the OBD I. In order to detect the effects of scattering, two sensors are arranged at 180◦ from the light source, and another arranged at 90◦ , as seen in Figure 4.1. The light source in this case is a pen laser of wavelength 523 nm , which allows for a more precise wavelength and orientation of incident light. Samples tested with the benchtop model were tested in a dark room to ensure there is no interference from ambient light. Sensors Pen Laser Sample Figure 4.1: Schematic for the scattering experiments with the OBD II. The light source in this case is a laser of wavelength 523 nm, and the detectors are provided by the same circuit as seen in Chapter 3. Detectors at 180◦ and 90◦ allow for us to calculate a ratio of scattered to transmitted light. 4.3 Protocol In order to obtain data with the OBD II, a sample must first be taken. Samples are prepared by dipping one end of the capillary tube into the desired culture. Capillary 70 action will draw the liquid up into the tube. The top of the tube must then be covered as the sample is moved to the OBD II and pressed into the clay block so that it stands erect. The light-blocking box should then be positioned over the device. After power is applied to the system, the voltage readings for both the transmittance sensor (the one positioned at 180◦ ) and the scattering sensor (the one positioned at 90◦ ) should be measured. These values represent the intensity of light both transmitted and scattered by the particles in the sample. 4.4 Results As initial testing of the scattering properties of a bacterial culture suspended in broth, the intensity of light at 180◦ and 90◦ to the incident beam was measured for each of 20 samples following 18 hours of growth. By comparing these measurements, we can develop a ratio of scattered light to transmitted light for E. coli in LB. The mean value for this ratio was 0.460 to three significant figures, with a standard deviation of 0.030 to three significant figures, when observed with the PNZ150 phototransistor and a pen laser of wavelength 523 nm. 4.5 Analysis Though these measurements are a starting point for further scattering studies, there was a great deal of error and inconsistency involved. The intensity of the pen laser is dependent on the charge of its batteries. This charge was low in testing, resulting in a lower, variable intensity. When new batteries were introduced, the intensity of the laser saturated the available reading from the OBD II. Additionally, cuvettes were used as the sample containers. For the purpose of measuring transmittance for studies invoking the Beer-Lambert law, cuvettes are ideal, because the calculation involves a concentration as opposed to the behavior of a single particle in that solution. A large cross-sectional area like that used here results 71 in interference of the different scattering behaviors of various particles. Also, because E. coli are rod-shaped, they will exhibit different scattering behavior depending on the angle at which light is incident, causing the combined behavior of many particles to be increasingly erratic. It is for this reason that analysis of scattering behavior when observed through cuvettes is limited to a few applications. By observing the scattering and transmittance intensities at each time interval, we can calculate a more precise value of light absorption than by measuring transmittance alone. With more knowledge and measurement of the constants in the Beer-Lambert law, we could find a value for the concentration of E. coli in the solution. Also, more detailed calculation of the ratio between transmittance and scattering could prove useful in microbial identification. We hypothesize that different contaminant organisms will exhibit a different scattering-to-transmittance ratio that falls within a range unique to their classification, for example bacterial vs. fungal. This would provide an additional option to selective media, as a means by which to determine contaminant source. 4.6 Future Work These scattering effects must be observed for other species in order to be truly use- ful. Because Tyndall scattering offers little to no information regarding the shape of the particle involved, it is the information about the size that presents great promise. Scattering effects do not require a large number of particles to be observed (as opposed to change in transmittance), so theoretically, a raw sample, not cultured over a length of time, will yield information. This information, whether corresponding directly to a size of particle, or presenting a sort of signature for that species, would suggest under which specifications a growth-transmittance study should be performed. For example, a scattering study that yields results suggesting that the particles present are larger (say, with a cross-sectional area of 50 microns) would lead one to perform a 72 growth-transmittance study for protozoa, not bacteria, using the corresponding media and wavelength appropriate for these organisms. The use of scattering would allow for quicker, more accurate detection of contamination in drinking water. [40] In order to decrease the effects of interference between too many particles, a capillary tube will be used to collect the sample instead of a cuvette. The smaller cross-sectional area of the capillary tube can accommodate no more than three cells of E. coli. The capillary tube will be held erect in a block of clay, between the laser and the sensors. A black, light-absorbent box must be used to block out any ambient light. For use of Tyndall scattering, it is important to use non-monochromatic light, in order to observe the colors of the visible light being scattered. To determine these colors through methods unbiased by human observation, filters should be used. Multiple sensors will be put in place, particularly for the detection of red and blue light, each at 90◦ from the incident beam-path. Each will have a filter that will only allow transmittance of that sensor’s prescribed wavelength range. The intensity measured at each of these sensors will provide a quantifiable measurement of which colors are being scattered most. It is our hypothesis that this experiment will yield additional information regarding the nature of bacterial growth and contamination in water. Though a benchtop model is described here, the design can easily be accomodated to fit in a portable system like the OBD I. 73 CHAPTER 5 CONCLUSION This thesis has explored the transmittance properties of light through bacteria, and demonstrated the correlation between transmittance plotted over time and the growth phases of the bacteria. Many of the challenges and assumptions in testing bacteria have been addressed, from the question of initial conditions to the various other unpredictabilities involved in dealing with biological specimens. We have employed the use of bioreactors in which to control culture environment and examine the effects on initial conditions. We have also prepared stained slides and utilized software from the NIH to count the number of actual cells present at a given time, and shown that the cell count corresponds directly to the change in absorption of light. Having controlled these various issues, the change in transmittance of light through bacteria was observed over time using the Genesys 6. This testing has given a baseline for how light transmittance responds to bacterial growth, and allowed for observation of the effects of other factors on this bacterial growth curve. The effects on lag time in the growth curve were confirmed to change with the initial concentration of bacteria in the sample container, as well as the effects on lag time of the initial growth phase of the bacterial culture. This has helped us to interpret the results observed from the OBD. We have developed a device capable of the timely detection of E. coli and performed initial testing with it. The OBD has demonstrated the ability to detect bacteria as the Genesys 6 has done, while staying within the confines of both budget and size. This design was expanded as well to include the capabilities of observing scattering effects of bacteria. The OBD, however, has many limitations. It is not a substitute for traditional laboratory tests, as specific properties of the bacteria can 74 be observed directly by these means. The OBD can only detect the presence of organisms, and, to an extent, the identity of these organisms. The selectivity of the broth media determines that only a certain subset of organisms can thrive off of it (those whose complete nutrient needs are met by the media), as well as the scattering properties of the organism. Even amongst single-celled organisms, size varies enough to change the scattering behavior of incident light. These properties make the OBD useful for confirming a suspected contamination, or identifying the presence of a contamination, though not the species. Though traditional lab methods take longer, they yield more useful information than the OBD ever will. By nature of dealing with biological specimens in this experiment, there are many errors involved. Through the course of experimentation, many separate cultures of bacteria have been used. Additionally, the same culture may change over the course of weeks or months, adapting to its environment. This can cause unpredictable changes in its behavior. Contamination also has a large effect on results. The materials used are assumed to be clean, though they are not sterile. Even still, the definition of clean does not ensure that there are not dormant species within the cuvettes or other equipment. Contamination can also occur due to errors in following laboratory technique, and human contribution of organisms from comensual bacteria or illness. Initially, we were concerned that the temperature of the inoculating loop was killing the bacteria in the course of inoculation. This concern was abated by standardizing the cooling time of the inoculating loop to 90 seconds. Inconsistency in broth can also cause the bacteria to behave differently. In particular, when using the bioreactor, the culture broth is extremely rich in nutrients while the sample broth is not. This can cause the bacteria to enter a state of shock and briefly go dormant to conserve nutrients. Additionally, the concentration of bacteria in the culture can ultimately affect the growth rate, by affecting the initial concentration of bacteria in the sample. This is particularly evident when using the bioreactors, as they have a relatively low 75 bacterial concentration compared to a standard culture vial. Errors in this system are not necessarily linked to the biological aspect. Any experiment incurs a certain amount of systematic and statistical error. One of particular concern is related to temperature. Bacterial growth is highly dependent on the temperature of the environment, particularly for E. coli, which is indigenous to the human gastrointestinal tract (body temperature). E. coli tested in the Genesys 6 is slightly more immune to this effect, as the electronic components of the device create their own incubator. Samples tested in the OBD, however, sit out in the lab with no temperature control. This was abated by suspending the samples in a fixedtemperature water-bath. Additionally, because the OBD I samples are changed out once an hour, there is an increased probability of smudging or contaminating the sample mid-run. There is also a good deal of time discrepancy caused by systematics. For example, there is approximately a two minute delay between the inoculation of each sample; however, there is a five minute delay between the testing of each sample. This could account for additional horizontal shifts in the various curves. Both models of the OBD are still in a development stage. Though the OBD I provides the data we had anticipated, there is much room for improvement. Currently, the sensor for the OBD I is not in a linear regime. That is, a fifty percent decrease in light intensity does not correspond to a fifty percent decrease in the voltage reading provided by the OBD. By changing the value of the resistor in line with the phototransistor, we could set the sensor to operating in a linear regime. With a linear response, we will see a more pronounced decrease in transmittance at the beginning of the exponential growth phase. The optimal resistance value can be found by using a potentiometer to vary the resistance, and observe the response when a filter of known transmittance is introduced to the system. This adjustment would optimize the effectiveness of the OBD I. The next logical step in this project is to perform field testing. While the OBD 76 functions under the stable conditions of a lab, and the fairly predictable growth and properties of cultured E. coli and clean broth, its real function is out in the field where conditions are not clean and stable (ambient light and temperature, for example, are constantly changing). Temperature changes, particularly those occuring between daytime and nighttime, can influence the growth of the bacterial samples, resulting in an atypical bacterial growth curve. In practical use, however, this is irrelevant. Ideally, the OBD will be used to test the initial reading of a sample, and a reading 12 hours later. If the transmittance has decreased by a significant amount, bacteria should be assumed to be present. In this way, temperature fluctuations do not affect the key result of a test with the OBD. Attempts at field testing were made during the OBD’s infancy, but these exact challenges called for a revision on the OBD’s physical design. No further attempts were made to field test following the OBD’s revision, as climate changes made the contamination site (the confluence of the South Platte River and Cherry Creek) more difficult to utilize. The OBD should be tested in a variety of environments with known and unknown levels of contamination. Should field testing prove the OBD sufficient under these conditions, efforts can be made to produce many OBD units and disperse them in parts of the world where laboratory testing is difficult or even impossible. The cost restrictions imposed on the OBD make this feasible. Following the implementation of an E. coli OBD, development of devices for other organisms should soon follow. E. coli was the most obvious place to start, as it is a standard in the world of bacteriology, but it is by far not the largest global threat. Research on other species of bacteria, such as cholera and salmonella, could help reduce the number of outbreaks in regions with lesser testing equipment. However, the technology used by the OBD should not be confined to use only in the detection of bacteria. Parasites must be considered also. Giardia is a protozoan parasite that has infected 50% of drinking water in the United States, and is considered by the 77 Center for Disease Control (CDC) to be serious enough as to be included on the list of bioterrorism threats. Fungi can be devastating as well. [41] Bret is a yeast used in making some forms of beer, but is devastating to the wine industry, because not only does it ruin the product, but it becomes embedded in wine barrels and building structure. [45] A rapid detection of Bret could save the industry millions of dollars. The potential applications of this technology are numerous, providing a rapid, inexpensive means for detecting the presence of biological contamination. Beyond the interest of scientific advancement is the human interest motivation for this research. The ability to test for the presence of these lifeforms without advanced laboratory equipment is the next big step in wiping out diseases that have ceased to exist in the developed world. While some strive to provide the underprivileged with the means to fight an ailment, this work provides a means by which to prevent infection from the beginning. With the advancement of this research, the worldwide detection of the presence of more and more pathogens will become possible. 78 REFERENCES CITED [1] Centers for Disease Control. Centers for disease control, January 2011. URL http://www.cdc.gov/. [2] World Health Organization. Water, health and ecosystems, January 2011. URL http://www.who.int/heli/risks/water/water/en/. [3] Doctors without Borders. Haiti: Water and sanitation, an essential role to fulfill, January 2011. URL http://www.doctorswithoutborders.org/news/ article.cfm?id=4960&cat=video. [4] United States Environmental Protection Agency. 5.11 fecal bacteria, January 2011. URL http://water.epa.gov/type/rsl/monitoring/vms511.cfm. [5] K. Todar. Pathogenic e. coli. http://www.textbookofbacteriology.net/e. coli.html, January 2011. [6] Max Sussman. Escherichia coli: Mechanisms of Virulence. Cambridge University Press, Cambridge, UK, 1997. [7] Kuby. Immunology. W.H. Freeman and Company, New York, NY, 2007. [8] Corrigan JJ and FG Boineau. Hemolytic-uremic syndrome. Pediatric Review, 22:11:365–9, 2001. [9] Mayo Clinic. Peritonitis, January 2011. URL http://www.mayoclinic.com/ health/peritonitis/DS00990. [10] PubMed Health. Breast infection, January 2011. URL http://www.ncbi.nlm. nih.gov/pubmedhealth/PMH0002460/. [11] Anevlavis S and Bouros D. Community acquired bacterial pneumonia. Expert Opin Pharmacother, 11:3:361–74, 2010. [12] Moselio Schaechter and Frederick C. Neidhardt. Escherichia coli and Salmonella Typhimurium. American Society for Microbiology, Washington, DC, 1987. [13] J. L. Ingraham, O. Maaloe, and F. C. Neidhardt. Growth of the bacterial cell. Sinauer Associates, Suderland, Mass., 1983. 79 [14] B. Lugtemberg and L. Von Alphen. Molecular architecture and functioning of the outer membrane of escherichia coli and other gram-negative bacteria. Biochim. Biophys. Acta, pages 737:51–115, 1983. [15] M. J. Osborn, J. E. Gander, E. Parisi, and J. Carson. Mechanism of assembly of the outer membrane of salmonella typhimurium. isolation and characterization of cytoplamic and outer membrane. J. Biol. Chem., pages 247:3962–3972, 1972. [16] H. Nikaido. Outer membrane of salmonella typhimurium: transmembrane diffusion of some hydrophobic substances. Biochim. Biophys., pages Act A 433:118– 132, 1976. [17] B. G. Cohen and A.D. Bangham. Diffusion of small nonelectolytes across liposome membranes. Nature (London), pages 236:173–174, 1972. [18] W. D. Stein. The movement of molecules across cell membranes. Acadenic Press, Inc., New York, 1967. [19] M. Inouye (ed.). Bacterial Outer Membranes. John Wiley & Sons, Inc., New York, 1979. [20] D.L. Melchoir and A. Carruthers. Studies of the relationship between bilayer water permeability and bilayer physical state. Biochemistry, pages 22:5797–5807, 1983. [21] L. Leive. The barrier function of the gram-negative envelope. Ann. N.Y. Acad. Sci., pages 235:109–127, 1974. [22] H. Nikaido and T. Nakae. The outer membrane of gram-negative bacteria. Adv. Microb. Physiol., pages 20:163–250, 1979. [23] L. G. Burman and J. T. Park. Molecular model for elongation of the murein sacculus of escherichia coli. Proc. Natl. Acad. Sci., pages 81:1844–1848, 1984. [24] T. Miura and S. Mizushima. Separation by density gradient centrifugation of two types of membranes from spheroplast membrane of escherichia coli k-12. Biochim. Piophys. Acta, pages 150:159–164, 1968. [25] C.A.. Schnaitman. Protein composition of the cell wall and cytoplasmic membrane of escherichia coli. J. Bacteriol., pages 104:890–901, 1970. [26] P. H. Raven. Biology. McGraw Hill, New York, NY, 2008. 80 [27] T. Nakae. Outer membrane of salmonella. isolation of protein complex that produces transmembrane channels. J. Biol Chem, pages 251:2176–2178, 1976. [28] T. Nakae. Identification of the outer membrane protein of e. coli that produces transmembrane channels in reconstituted vesicle membranes. Biochem Biophys Res. Commun., pages 71:877–884, 1976. [29] P. Bavoil, G. Nikaido, and K. von Meyenburg. Pleiotropic transport mutants of escherichia coli lack porn, a major outer membrane protein. Mol. Gen. Genet., pages 158:23–33, 1977. [30] I. R. Beacham, D. Haas, and E. Yagil. Mutants of escherichia coli ’cryptic’ for certain periplasmic enzymes: evidence for an alteration of the outer membrane. J. Bacteriol, pages 129:1034–1044, 1977. [31] J. F. Lutkenhaus. Role of a major outer membrane protein in escherichia coli. J. Bacteriol, pages 131:631–637, 1977. [32] H. Nikaido, S. A. Song, L. Shaltiel, and M. Nurminen. Outer membrane of salmonella. xiv. reduce transmembrane diffusion rates in porin-deficient mutants. Biochem. Biophys. Res. Commun., pages 76:324–330, 1977. [33] W. Weidel and H. Pelzer. Bagshaped macromolecules-a new outlook on bacterial cell walls. Adv. Enzymol, pages 26:193–232, 1964. [34] R. Hakenbeck, J.-V. Holtje, and H. Labischinski. The target of penicillin. Walter de Gruyter and Co., Berlin, Federal Republic of Germany, 1983. [35] H.C. Berg. Dynamic properties of bacterial flagellar motors. Nature (London), pages 249:77–79, 1974. [36] S.H. Larsen, R.W. Reader, E.N. Kort, W.-W. Tso, and J. Adler. Change in direction of flagellar rotation is the basis of the chemotactic response in escherichia coli. Nature (London), pages 249:74–77, 1974. [37] D. Abram, H. Koffler, and A.E. Vatter. Basal structure and attachment of flagella in cells of proteus vulgaris.. J. Bacteriol., pages 90:1337–1354, 1965. [38] M. Silverman and M.Simon. Flagellar rotation and the mechanism of bacterial motility. Nature (London), pages 249:73–74, 1974. [39] S.-I. Aizawa, G.E. Dean, C.J. Jones, R.M. Macnab, and S. Yamaguchi. Purification and characterization of the flagellar hook-basal body complex of salmonella typhimurium. J. Bacteriol., pages 161:836–849, 1985. 81 [40] G. Cohen-Bazire and J. London. Basal organelles of bacterial flagella.. J. Bacteriol., pages 94:458–465, 1967. [41] M. L. DePamphilis and J. Adler. Fine structure and isolation of the hook-basal body complex of flagella from escherichia coli and bacillus subtilis. J. Bacteriol., pages 105:384–395, 1971. [42] E.J. O’Brien and P.M. Bennett. Structure of straight flagella from a mutant salmonella. J. Mol. Biol., pages 70:133–152, 1972. [43] R. Phillips. Physical Biology of the Cell. Garland Science, New York, NY, 2009. [44] P. Nelson. Biological Physics: Energy, Information, Life. W. H. Freeman and Company, New York, 2008. [45] K. Dimmitt and M. I. Simon. Purification and partial characterization of bacillus subtilis flagellar hooks. J. Bacteriol., pages 108:282–286, 1971. [46] Y. Shirakihara and T. Wakabayashi. Three-dimensional image reconstruction of straight flagella from a mutant salmonella typhimurium. J. Mol. Biol., pages 131:485–507, 1979. [47] D.B. Kell. Diffusion of protein complex in prokaryotic membrane: fast, free, random or directed? Trends Biochem. Sci., pages 9:86–87, 1984. [48] P.B. Garland, K. Johnson, and G.R. Reid. The diffusional mobility of proteins in the cytoplasmic membrane of escherichia coli. Biochem. Soc. Trans.., pages 10:484–485, 1982. [49] D.B. Kell. Diffusion of protein complex in prokaryotic membrane: fast, free, random or directed? Trends Biochem. Sci., pages 9:86–87, 1984. [50] J. Konisky. Specific transport system and receptors for colicins and phages. John Wiley & Sons, Inc., New York, 1979. [51] S.and C. Ginsburgh Maloy, R. Simons, and W. Nunn. Transport of long and medium chain fatty acids by escherichia coli k-12. J. Biol. Chem., pages 356:3735–3742, 1981. [52] D.B. Kell. Diffusion of protein complex in prokaryotic membrane: fast, free, random or directed? Trends Biochem. Sci., pages 9:86–87, 1984. 82 [53] M.J. Osborn, J. E. Gander, E. Parisi, and J. Carson. Mechanism of assembly of the outer membrane of salmonella typhimurium. J. Biol. Chem., pages 247:3962– 3972, 1972. [54] Jr. M. H. Saier, H. Straud, L. S. Massman, J. J. Judice, M. J. Newman, and B. U. Feucht. Permease-specific mutations in salmonella typhimurium and escherichia coli that release the glycerol, maltose, melibiose, and lactose transport systems from regulation by the phosphoenolpyruvate: sugar phosphotransferase system. J. Bacteriol., 133:1358–67, 1978. [55] Marc Loudon. Organic Chemistry, 5th ed. Roberts and Company Publishers, Greenwood Village, CO, 2009. [56] National Institutes of Health. Imagej: Image processing and analysis in java, November 2010. URL http://rsbweb.nih.gov/ij/index.html. [57] B. Harisha. An Introduction to Practical Biotechnology. Laxmi Publications, Ltd., New Dehli, 2006. [58] J.W. Costerton, J.M. Ingram, and K.-J. Cheng. Structure and function of the cell envelope of gram-negative bacteria. Bacteriol.., pages 38:87–110, 1974. [59] Northwest College. Bacterial types, February 2011. URL http://biology. northwestcollege.edu/biology/b1010lab/bactypes.htm. [60] Carl Hemmingsen. Datalyse, August 2008. URL http://www.datalyse.dk/. [61] H. C. van de Hulst. Light Scattering by Small Particles. Dover Publications, New York, NY, 1957. 83 APPENDIX A - VISUAL BASIC CODE FOR IMPORTING DATA The following code is used to import individual data files into Excel 2003 in such a way that rows represent data from individual wavelengths and columns represent data from individual points in time. Hit Alt + F11 to open vb editor and go to [Insert] - [Module]. Paste the following code onto the right pane. Hit Alt + F11 again to return to Excel. Hit Alt + F8, choose “test” then click on [Run] to import data. Sub t e s t ( ) Dim myDir As String , f n As String , f f As I n t e g e r , t x t As String Dim d e l i m As String , n As Long , b ( ) , f l g As Boolean , x , t As Integer myDir = ‘ ‘ c : \ b a c k s l a s h t e s t ’ ’ d e l i m = vbTab f n = Dir ( myDir \& ‘ ‘ ∗ . dat ’ ’ ) Do While f n <> ‘ ‘ ’ ’ Redim b ( 1 To Rows . Count , 1 To 1 ) f f = FreeFile Open myDir \& ‘ ‘ \ b a c k s l a s h ’ ’ \& f n For I n p u t As \# f f Do While Not EOF( f f ) Li ne Input \# f f , t x t x = S p l i t ( txt , d e l i m ) I f Not f l g Then n = n + 1 : b(n , 1 ) = fn End I f I f UBound( x ) > 0 Then n = n + 1 b(n , 1 ) = x (1) End I f f l g = True Loop Close \# f f f l g = False t = t + 1 ThisWorkbook . S h e e t s ( 1 ) . C e l l s ( 1 , t ) . R e s i z e ( n ) . Value = b n = 0 f n = Dir ( ) Loop End Sub 84