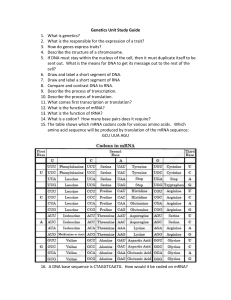
Lincoln University Digital Thesis
advertisement

Lincoln University Digital Thesis Copyright Statement The digital copy of this thesis is protected by the Copyright Act 1994 (New Zealand). This thesis may be consulted by you, provided you comply with the provisions of the Act and the following conditions of use: you will use the copy only for the purposes of research or private study you will recognise the author's right to be identified as the author of the thesis and due acknowledgement will be made to the author where appropriate you will obtain the author's permission before publishing any material from the thesis. Population and diet of the New Zealand fur seal (Arctocephalus forsteri): molecular approaches A thesis submitted in partial fulfilment of the requirements for the Degree of PhD of Ecology at Lincoln University by Arsalan Emami Khoyi Lincoln University 2015 Abstract of a thesis submitted in partial fulfilment of the requirements for the Degree of PhD of Ecology Abstract Population and diet of the New Zealand fur seal (Arctocephalus forsteri): molecular approaches by Arsalan Emami Khoyi The recent increase in the New Zealand fur seal (Arctocephalus forsteri) population has given rise to socio-economic concerns regarding the potential conflicts with human interests. Elaboration of a comprehensive management strategy has been hindered by the paucity of solid information concerning New Zealand fur seal ecology. Recent developments in massive parallel DNA sequencing and computational infrastructures were used to address some of the major areas of conflict with human commercial interests. The first focus of the current study was to test a series of non-destructive methods for collecting biological samples for high- throughput DNA analysis. A second focus of the study was application of whole mitochondrial genomes in conjunction with Y chromosome Zinc fingers (ZFY) from New Zealand fur seals throughout the whole range of the species distribution in an Approximate Bayesian Computation framework to reconstruct the recent demographic history of the species. The pristine population size (pre-human colonisation), historical population size after human first arrival and the bottleneck population size were estimated. There was enough variability left in the mitochondrial genomes to detect the 18 th -19thcentury’s population bottleneck in the species. The pattern observed in ZFY data set was more complicated indicating more subtle population genetics dynamics. Mitochondrial DNA were ii uniform in its distribution with few distant haplotypes that could represents the presence of old lineages or potential introgression from other sympatric species. The intriguing pattern observed in ZFY data also resulted in the discovery of a rare genomic event called ‘’ectopic gene conversion” between non- recombining parts of Y and X chromosomes in the New Zealand fur seal genome The third focus of the study is on the fine scale population structure of NZ fur seals at a local scale around Banks Peninsula, -South Island, and New Zealand. No evidence of local population structure was found in the area suggesting the presence of substantial gene flow among colonies at a local scale. Moreover, the “spill over“ colony expansion dynamics, suggested previously as a pattern for recolonizing new habitat, was supported at the local scale using genetic data. Most of the newly-established colonies in the area showed the highest degree of genetic structure similarities with older colonies in their vicinity emphasizing the important role of “spill over” dynamics of older colonies in formation of new colonies. The data significantly support multi recolonization events with occasional local recruitment of immigrating individuals. There is a short mitogenomic announcement in chapter five where I used the complete mitogenomes of New Zealand fur seals in addition to three mustelid species (all de novo sequenced in the current research) to re-examine the origin of pinnipeds in the light of new available mitogenomes. The final focus of the study used molecular-based methods to identify the prey and parasite items of the New Zealand fur seal from massive parallel sequencing of faeces and regurgitates. The overlap between the diet of the seals and commercial fisheries were also estimated. iii Data supported a generalist pattern of feeding behaviour of the New Zealand fur seal. As many as 64 prey species were identified from faecal samples and/or regurgitates in a single colony. Surprisingly, only 10% of species in fur seals diet were species of commercial interest. The population and diet data will provide marine ecosystem managers with an increased knowledge necessary for elaborating any long-term conservation plan for the New Zealand fur seal. Keywords: New Zealand fur seals, Banks Peninsula, mitochondrial, zinc finger, ABC, population genetics, diet iv Acknowledgements I would like to express my sincere gratitude to my supervisors, Dr Adrian Paterson, Dr Robert Cruickshank and advisor Dr Laura Boren, for their patience, excellent guidance and immense support during my PhD. I would like also to thank Dr James Ross and Dr Elaine Murphy for their exemplary generosity and encouragement during my research. I am sincerely thankful of Lincoln University and New Zealand Department of Conservation and its rangers for providing me with the unique opportunity of pursuing current research and consolidating my knowledge. A special thanks to Dr David Hartley for his excellent ideas and perseverance in laboratory work and to Dr Andrea Betancourt for patiently walking me through the fascinating world of population genetics. I would like to express my appreciation to all other people at Lincoln University and elsewhere who contributed to my research. I thank Rhmaty family for supporting me during all of my education in the past quarter of a century. I also express my respects to Dr Masoud Afzali for his inspiring presence in the hardest period of my life and his encouragements to strive towards reaching my goals. I am also grateful to my mother and father for their sacrifices to help me finish my education. v Table of Contents Once you have edited this document you will need to update the Table of Contents to reflect the changes, and to remove any error messages. To update the Table of Contents, right-click anywhere on the grey area below, select ‘Update Field’ (and select ‘Update Entire Table’ if prompted). Delete this note when you are familiar with how to update this table of contents. Abstract ....................................................................................................................................... ii Acknowledgements ...................................................................................................................... v Table of Contents ........................................................................................................................ vi List of Tables ............................................................................................................................. viii List of Figures .............................................................................................................................. ix Chapter 1 Introduction ............................................................................................................ 1 1.1 Evolution of seals ......................................................................................................................5 1.2 Otaridae ....................................................................................................................................6 1.3 Fur seals ....................................................................................................................................7 1.3.1 New Zealand fur seal....................................................................................................7 1.3.2 Exploitation history ......................................................................................................9 Chapter 2 A research diary of the specimen collection methods ...............................................12 2.1 Flipper notching ..................................................................................................................... 12 13 2.2 Non-invasive methods of DNA collection .............................................................................. 14 2.2.1 Buccal Swabs ..............................................................................................................14 2.2.2 Environmental DNA....................................................................................................17 2.2.3 Dart Biopsy .................................................................................................................19 Chapter 3 New Zealand fur seal population genetics using complete mitochondrial DNA and evidence for X to Y gene conversion ........................................................................................26 3.1 Introduction ........................................................................................................................... 27 3.2 Material and methods ........................................................................................................... 32 3.2.1 Sample collection .......................................................................................................32 3.2.2 Justification for number of sequenced individuals ....................................................32 3.2.3 Whole mitochondrial genome sequencing and assembly ........................................36 3.2.4 Y Chromosome zinc finger sequencing and assembly ...............................................38 3.2.5 An overview of the analysis .......................................................................................39 3.3 Results .................................................................................................................................... 52 3.3.1 Mitochondrial DNA ....................................................................................................52 3.3.2 Estimating posterior distribution of the parameters .................................................56 3.3.3 ZFY data ......................................................................................................................56 3.4 Discussion............................................................................................................................... 71 3.4.1 Y chromosome zinc finger ..........................................................................................76 vi Chapter 4 Fine scale population structure and colony expansion dynamics of New Zealand fur seals (Arctocephalus forestri) around Bank Peninsula ..............................................................84 4.1 Introduction ........................................................................................................................... 85 4.2 Material and Methods ........................................................................................................... 89 4.2.1 Tissue collection .........................................................................................................89 4.2.2 Control region amplification/ analysis .......................................................................91 4.3 Results .................................................................................................................................... 93 4.4 Discussion............................................................................................................................. 103 Chapter 5 A mitogenomic announcement: complete mitochondrial genome of the stoat (Mustela erminae) and New Zealand fur seal (Arctocephalus forsteri) and their significance for mammalian phylogeny ......................................................................................................... 107 Chapter 6 Identifying prey and parasite items from New Zealand fur seal faeces using massive parallel sequencing ............................................................................................................... 118 6.1 Introduction ......................................................................................................................... 118 6.2 Material and Methods ......................................................................................................... 123 6.3 Results .................................................................................................................................. 125 6.3.1 Stewart Island ..........................................................................................................126 6.3.2 Fiordland ..................................................................................................................126 6.3.3 Kaikoura ...................................................................................................................127 6.3.4 Cape Palliser .............................................................................................................127 6.3.5 Bay of Plenty ............................................................................................................127 6.4 Discussion............................................................................................................................. 128 6.4.1 New Zealand fur seal parasites ................................................................................128 6.4.2 New Zealand fur seal prey Items .............................................................................129 Chapter 7 Conclusion ............................................................................................................ 137 Appendix A Brief history of Zinc finger markers ............................................................................143 References ........................................................................................................................... 146 vii List of Tables Table 3-1. Long PCR primer combinations designed to amplify New Zealand fur seal whole mitochondrial genomes. ...............................................................................................38 Table 3-2 Basic statistics calculated from 50 New Zealand fur seals mitogenomes .........................53 Table 3-3 the summary statistics (mode, median and mode) calculated from posterior distribution of the estimated parameters. The bottleneck population size (Nbot), the size of the fur seal population after first arrival of humans in to New Zealand (Nmao) and pristine population size (Npris).....................................................................................61 Table 3-4 Bias and precision of ABC analysis of bottleneck and historical population size. The number are calculated from posterior distribution of the parameter. (NBOT), the size of population after first arrival of humans in to New Zealand (NMAO and pristine population size (NPRIS). The true values correspond to the known value based up on them the pseudo data were created. ...........................................................................62 Table 3-5 Basic statistics calculated from the first group of ZFY sequences.....................................63 Table 3-6 Basic statistics calculated the second group of ZFY sequences. .......................................64 Table 3-7 BLAST results for the first consensus sequence calculated from New Zealand fur seal ZFY data. ..............................................................................................................................67 Table 3-8BLAST results of the second group of New Zealand fur seal ZFY consensus sequence. ....68 Table 3-9 Position and length of putative gene conversion sites among New Zealand fur seals group 1 consensus ZFY sequence and Californian sea lion ZFX (Next page). .............69 Table 4-1 Absolute pairwise shared haplotypes among different colonies in Banks Peninsula. Number inside parenthesis represent the number of samples taken from each colony. ..........................................................................................................................95 Table 5-1 Annotation of the protein coding genes in New Zealand fur seal complete mitogenome. ..............................................................................................................115 Table 5-2 Annotation of the tRNA genes in New Zealand fur seal complete mitogenome. ...........116 Table 5-3 Annotation of the rRNA genes in New Zealand fur seal complete mitogenome............117 Table 6-1 Fish and squid species identified from New Zealand fur seal faecal samples across 5 different colonies across New Zealand.......................................................................133 Table 6-2 Parasite species identified from New Zealand fur seal faecal samples across 5 different colonies across New Zealand.......................................................................134 viii List of Figures Figure 2-1 A New Zealand fur seal neutralized for biopsy collection by researchers at Banks Peninsula. Picture from author.....................................................................................13 Figure 2-2 A standard Schulz mouth gag. Picture kindly provided by medical- Tool Company (http://www.medical-tools.com/ .................................................................................16 Figure 2-3 A Pneu dart DNA collection device. Picture from the company web site (http://www.pneudart.com) ........................................................................................22 Figure 3-1 New Zealand colonies where tissue biopsies were collected. .........................................33 Figure 3-2 Variation is the resolution power of different 100 bp long windows of New Zealand fur seal mitochondrial DNA in identifying correct number of haplotypes. X axis is position in the fur seal genome and Y axis is the number of identified haplotypes for that window. Zero means that those fragments don’t have resolution power to differentiate between sequences at all. In other word the sequence it is not variable. ........................................................................................................................36 Figure 3-3 A median- joining network constructed from whole mitochondrial genomes. The different coloured lines (black, green, yellow and red) demonstrate mutation steps between neighbouring haplotypes. Empty circles show median haplotypes while the full circles show the observed haplotypes. The yellow, magenta and green haplotypes were considerably separated from the rest of network. ............................................................................54 Figure 3-4 A Bayesian phylogenetic tree reconstructed from complete mitochondrial DNA of seven different species of Otaridae. Numbers on each node represent posterior probabilities. The scale bar represents base substitutions in the mitogenomes. ........55 Figure 3-5 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a population that passed through a bottleneck 250 years ago (above) and a population with constant size in the past 250 years (below). The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 80% of the variation in posterior distributions. .............................................................................................57 Figure 3-6 Posterior probability of the bottleneck model (scenario1) and a constant size population (scenario2) calculated from a subset of 500 and 2000 closest simulated data set in a direct estimation and logistic regression method. X axis is subset of closest data set and Y axis is the posterior probability. ...............................................58 Figure 3-7 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a population that passed through a bottleneck 150 years ago The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 70% of the variation in posterior distributions ...............................................................................59 Figure 3-8 Posterior distributions of the bottleneck population size , the size of population after first arrival of humans in to New Zealand, historical size and pristine population size of New Zealand fur seals. Note that the numbers on the Y axis are direct counts. .....60 Figure 3-9 A Bayesian phylogenetic tree for ZFY sequences. The (a) and (b) phylogenetic tree correspond separately to the each clade in the main tree in more detail. Scale bar represent generation in New Zealand fur seals calculated based on Y chromosome mutation rate of 1 x 𝟏𝟎𝟖 (Xue, Wang et al. 2009) .......................................................63 Figure 3-10 A Bayesian phylogenetic tree constructed using consensus sequences from each zfy group and zfx – zfy sequences from a closely related species, Californian sea lions. Scale bar represents base substitution in zfy sequences. ............................................69 Figure 4-1 Locations of the colonies around Bank Peninsula from where samples were sourced. .90 Figure 4-2 the haplotype structure in each location in the Banks Peninsula. H1-H56 corresponds to the different haplotypes identified in the area. ...........................................................95 ix Figure 4-3 A Bayesian Phylogenetic tree of mitochondrial DNA control region from Bank Peninsula. The scale bar show the substitution in mitochondrial DNA control region (Black and grey represent different clades in the area ..................................................................97 Figure 4-4 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a model depicting multisource nature of recolonization of the Banks Peninsula.The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 50% of the variation in posterior distributions. .......................................98 Figure 4-5 Posterior probability of multisource pattern of recolonization of Banks Peninsula (scenario1) and a single massive recolonization (scenario2) and gradual increase from a small population of size 100 (scenario3) calculated from a subset of 2000 closest simulated data set in a logistic regression estimation method. X axis is subset of closest data set and Y axis is the posterior probability. .........................................100 Figure 4-6 the mantel test on mitochondrial DNA control region from Banks Peninsula. X axis is geographical distance and Y Axis is genetic distance. ................................................101 Figure 4-7 Spatial autocorrelation of the mitochondrial DNA control region from Banks Peninsula. X axis is distance classes and Y axis (Ay) is genetic distance. .....................................102 Figure 4-8 Genetic landscape of mitochondrial DNA control region from Banks Peninsula. X and Y axis are geographic coordinates of the sampled colonies and Z axis is genetic distance among haplotypes. .....................................................................................................103 Figure 5-1 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent posterior distribution and scale bar is time in millions of years. ...............................113 Figure 5-2 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent estimated divergence date (Millions years ) and scale bar is time in millions years.Figure 5-1 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent posterior distribution and scale bar is time in millions of years............................................................................................................................114 x Chapter 1 Introduction Applications of DNA-based methods to wildlife ecology issues is a fast developing domain for ecological studies. The advent of new sequencing technologies has opened a new horizon for wildlife managers to answer many questions of ecological relevance. Examples of DNA-based methods applications to the major areas of wildlife science are numerous. Molecular approaches have been successfully tested in wildlife forensics (Alacs, Georges et al. 2010), estimating census and effective population size (Gardner, Royle et al. 2010, Harris, Winnie et al. 2010, Brinkman, Person et al. 2011), diet and trophic dynamics (Carreon-Martinez and Heath 2010, Deagle, Chiaradia et al. 2010), fine scale population structure (Sellas, Wells et al. 2005, Warnock, Rasmussen et al. 2010), tracking of individuals (Ernest, Penedo et al. 2000), diversity (Nybom 2004) and population genomics (Hohenlohe, Bassham et al. 2010). The population of fur seals (Arctocephalus forsteri) in New Zealand is increasing (Taylor 1982, Dix 1993, Lalas and Harcourt 1995, Taylor, Barton et al. 1995, Bradshaw, Lalas et al. 2000) and escalating conflict with fisheries and coastal region inhabitants seems inevitable (Lalas and Bradshaw 2001). Density-dependent factors, such as habitat quality, are not strong enough to regulate population expansion at this stage (Crawley 1990). The future of this species will be a major area of concern for all New Zealand marine ecosystem managers in coming years. A solid understanding of fur seal ecology is needed for managers to develop management plans as well as clarification to the extent of potential conflict with human interests. One of the most important sources of conflict between fur seals and human interests is competition for food resources (Trites, Christensen et al. 1997, Bradshaw, Lalas et al. 2000). 1 The expansion of fishing industries and the application of the modern processing and storage technologies have changed the traditional concept of commercial species. Species classified as non-commercial are now of increasing economic value which, in turn, expands the potential degree of conflict between humans and fur seals. Understanding the extent of competition between seals and humans is not possible without precise knowledge of fur seal diets. There have been several valuable studies concerning New Zealand fur seal diets in New Zealand (see Chapter 5 on diet). Several issues prevent the large-scale application of these studies. First, the fur seal diet is dynamic, it changes according to time and space and it is difficult to generalize findings from one study to the whole populations in all of their distribution areas. Each study is no more than a snapshot which, at best, represents characteristics of a particular population at a specific time. Second, none of the methods used to determine fur seal diet are free of biases. Comparisons of data obtained using different methods are not always valid. Each method of diet reconstruction performs well in identifying some elements of the diet but does not re-construct other contributors to an individual diet with equal precision (Tollit, Steward et al. 1997). A combination of different methods can provide the most precise estimation and that is why any new methods of diet estimation is worth investigating. Major contributors to the fur seal diet have been identified using traditional hard-parts methods (see Chapter 5). Uncertainty concerning the contribution of both small-sized species (with easily degradable hard-parts) and large-sized species (that cannot be consumed completely and, therefore, usually lack diagnostic hard parts) still persists. Furthermore, traditional methods are inappropriate for analysing the contribution of soft-bodied species (i.e. sharks, rays and skates). 2 Molecular techniques can be used as a complementary method for more accurate diet estimation and the new method has the potential to provide scientists with a range of new data, otherwise unobtainable by traditional methods. The current study is the first application of molecular methods to study fur seal diet in New Zealand. A molecular approach may validate (or invalidate) earlier traditional studies of diet and provide ecosystem managers with more solid knowledge of New Zealand fur seal diet for any future decision making. By targeting the diversity and complexity of fur seal diet, the more accurate role of this animal in marine food webs will be clarified and the potential fields of conflict with human interests will be established. These results will help decision makers, and especially conservation authorities, to better understand the dynamics of fur seal diets and, therefore, to construct more precise ecosystem management plans. A second focus of the current research will address the issue of genetic diversity in New Zealand fur seal populations. What effect has the severe population bottleneck and subsequent re-colonisation of New Zealand by fur seals had on genetic structure and variation in the species? Is it necessary to continue with existing conservation plans? At what point will we need to intervene (if at all)? Current knowledge of fur seal population biology does not allow us to answer these questions confidently. It is expected that, due to a history of severe over-harvesting during the last few centuries, the current fur seal population in New Zealand does not show a high degree of genetic diversity. A thorough evaluation of this hypothesis has yet to be done. The actual effect of this possible bottleneck on the present day population has yet to be identified. Furthermore, there are several populations of this species in the southern, eastern, western and northern parts of the South Island. The degree of genetic diversity within and among these populations needs 3 further investigation. Without exact answers to these questions we cannot decide on the future of current management programs. This thesis follows four different objectives: In chapter two the advantages and disadvantages of different methods of biological sample collection are discussed and the rationale for why dart biopsy and/or flipper –notching were chosen as the preferred method for sample collection is expressed in detail. Finally, some concerns regarding the use of these methods are reviewed. In chapter three two models of evolutionary history (constant size versus a bottleneck model) in the evolutionary history of New Zealand fur seals is evaluated simultaneously. The timing of the bottleneck, its size and the size of the fur seal population before human large-scale colonisation of New Zealand are estimated. Consistency between two markers with different inheritance pattern (mitochondrial DNA versus Zinc Finger on Y chromosome) in reconstructing the evolutionary history of New Zealand fur seals are tested. In chapter four, the fine-scale population genetic structure of New Zealand fur seals in Banks Peninsula is studied. Female philopatry and stability of matrilineal societies are investigated. The founder effect and its severity in the recolonization of Banks Peninsula is tested in more detail. In chapter five the evolutionary history of pinnipeds were re-examined using few new mitogenomes sequenced in this research. In chapter six the main contributors to the fur seal diet are identified using a massive parallel sequencing method on faecal DNA. The potential overlaps with commercial or recreational fisheries are investigated. 4 1.1 Evolution of seals The first mammalian species appeared on land approximately 200 million years ago. The existence of mammals adapted to life in the sea represents “a return to the ancestral home” rather than direct descent from marine ancestors (Reeves and Stewart 2003). Several independent lineages re-entered the sea (Reeves and Stewart 2003). The small, primitive, hoofed, probably hippopotamus-like, ancestors of cetaceans (whales, dolphins, and porpoises) and the sub-ungulate ancestors of the sirenia (manatees, dugongs, and sea cows) were the first mammals to re-invade the seas. Mammals similar to modern day cetaceans and sirenians flourished in early Eocene waters more than 50 million years ago (Doming 2002, Fordyce 2002 ). Later, possibly in the late Oligocene or early Miocene (15-30 million years ago), the bear- or weasel-like ancestors of the Otariidae (fur seals and sea lions), Odobenidae (walrus) and Phocidae (true seals) began re-invading the aquatic habitats (Berta 2002). The latest waves of water re-invasions resulted in modern species like the polar bears, sea otters, and marine otters (Heyning and Lento 2002). This study focuses on pinnipeds in general and otariids in particular. The earliest known otariid is Pithanotaria, with remains found from several localities in California dating from 12-13 million year ago. Pithanotaria was a small animal, 1.5 m in length, the size of a Galapagos fur seal (Arctocephalus galapagoensis), the smallest living otariid (Macdonald 2001). Around 8 million years ago there were otariids in the North Pacific that demonstrated an increase in body size and were clearly sexually dimorphic, similar to modern sea lions except in minor differences in their limb bones and dentition. First to diverge from the main otariid stem was the Northern fur seal Callorhinus sp. whose separation has been dated as at least 6 million years ago (Macdonald 2001). Soon after the 5 split the otariids dispersion through southern hemisphere (South America, South Africa, and Australasia) started. Geological records from 6 million to about 2-3 million years ago showed limited diversification in this linage which remained similar to the modern southern hemisphere fur seals Arctocephalus spp. However, in the past two million years, the southern linage radiated into many species. This radiation was accompanied by a change in body size and development of singe rooted check teeth (Macdonald 2001). Today’s marine mammals are a diverse group of more than 120 different species (Martin and Reeves 2009) that spend the whole or a major part of their life cycle in the water, mainly in the oceans and open seas. The degree of water dependency varies among members of this group. Some species, like cetaceans and sirenians, spend all of their life cycle in the water unable to survive outside water for very long, while others, including members of the families Otariidae, Odobenidae, and Phocidae, are still dependent on land for part of their life cycle, mainly breeding. Given the importance of otariids in the current research, the remaining part of this chapter will focus on this group of pinnipeds. 1.2 Otaridae Members of the family Otariidae (eared seals, fur seals and sea lions) are discerned from members of the Phocidae by their external ear pinnae, a fact that gives rise to their common name “eared seals” (Otariidae means “little ear”). Compared to Phocidae, members of the Otaridae family have larger fore flippers and smaller hind flippers. They are distributed in both hemispheres. They are found along the coast of north eastern Asia, western North America, South America, southern Africa, southern Australia, New Zealand and many, predominantly southern, oceanic islands (Novak 2003). Otariids have stream-lined, sleek bodies that provide them with manoeuvrability needed to swim underwater. They are relatively fast on land and 6 can move up to a speed of 20 km/h. The strong fore flippers not only support their body mass on land but also provide them with necessary steering power in the water. They can also easily scull with the rear end of their body as members of phobias lineage do. To reach higher speeds, they usually porpoise, projecting themselves in shallow arcs out of the water. (Crawley 1990). Total length in otarriids ranges from 120 to 350 cm and the weight varies between 271100 kg (Novak 2003). Extreme sexual dimorphism is obvious in this group of animals, males are usually much larger than females. 1.3 Fur seals Fur seals are a group of eared seals, including nine different species belonging to two separate genera (Crawley 1990). A dense, soft hair under their outer covering of stiff, waterproof hair led to a specific appearances that give rise to their common name “fur seals”. Eight species of fur seals belong to the genus Arctocephalus (the generic name means ‘bear head‘ (Berta, Sumich et al. 2006)) and the one remaining species, to the genus Callorhinus (the generic name means ‘beautiful nose’ (Berta, Sumich et al. 2006)). Based on fossil records and anatomical evidence, Repenning (1977) suggests that divergence between the Callorhincus - Arctocephalus lineages was probably in the late Miocene, whereas the radiation of sea lions from the Arctocephalus lineage occurred in the late Pliocene or early Pleistocene. 1.3.1 New Zealand fur seal The New Zealand fur seal (Arctocephalus forsteri), sometimes known as the Antipodean fur seal, Australasian fur seal, Black fur seal or South Australian fur seal (Saundry 2009), is New Zealand’s most common native marine mammal (Saundry 2009). Arctocephalus forsteri are 7 found in suitable rocky habitat throughout New Zealand, from the Three King’s islands to Macquarie Island. Even though their distribution is spread over 2700 km their presence has been reported to be discontinuous and seasonally variable (Singleton , 1972; Csordas et al, 1965, as cited in Crawley 1990). The botanist George Forster in 1773 was the first scientist to describe “sea bears” in his account of the second voyage of Cook to New Zealand. A scientific description of this species was published later in 1828 (Todd 2002). Crawley and Wilson (1976) described the species as “dark grey-brown in appearance; the coat colour of adults merges from a dark grey-brown dorsally to a lighter grey-brown ventrally. The thick under fur is a rich chestnut colour; the guard hairs are coarse and dark grey, often with white tips which impart a silvery sheen to the dry fur. Sexual dimorphism is obvious in this species with males reaching up to185 kg in weight and 2.5 m in length, whereas females seldom exceed 50 kg and 1.5 m length” (Crawley and Warneke 1979). As in other pinnipeds, New Zealand fur seals are active both day and night and possess acute vision and good hearing under water (Novak 2003). New Zealand fur seals usually use their olfactory sense to discern other individual seals (King 1983). The brain size in relation to body size is similar to terrestrial, non-diving carnivores (Armstrong 1982). (Miller 1975) observed continuous presence of individual fur seals in a colony throughout the year. However, the dynamics of the colony in terms of numbers, ages and sex classes differs substantially between seasons. First to arrive on shore are breeding males. Soon after their arrival, males establish territories which they might hold for as long as nine weeks. Within a few weeks pregnant females arrive to give birth to the previous year pups. Although the exact time of harem formation is unknown, Stirling (1970) suggests that the serious harem 8 formation might start in mid-November. Females usually mate with the first male they meet, which is usually the harem holder of the territory where their last year pup was born (Miller 1975). Mating can take anywhere between 5-38 minutes (Miller, 1974). The whole gestation period including delayed implementation takes up to a year. At birth, pups (which are usually a single ) are about 55 cm long and weigh 3.5 kg (Crawley and Wilson 1976). Male sexual maturity has been reported between 5-9 years (Dickie and Dawson 2006) although fur seals probably do not form their own harem until they reach age 10 (Mattlin, 1978, as cited in Dickie and Dawson 2006). Female sexual maturity is estimated at between 4 and 6 years of age (Dickie and Dawson 2006). New Zealand fur seals are among the deepest diving fur seals and have the longest dive duration (Mattlin, Gales et al. 1998). Maximum dive duration and depth were 9.3 min and 312 m for females and 14.8 min and >380 m in males (Page, McKenzie et al. 2005). New Zealand fur seals used different habitat as their prime foraging areas. While most of females choose continental shelf waters, males usually prefer deeper continental shelf breaks or pelagic waters (Page, McKenzie et al. 2005). 1.3.2 Exploitation history Pinnipeds have been hunted by humans living in coastal areas for centuries (Todd 2002). Maori hunted fur seals for food and clothing and their teeth were used to make composite fishhooks (Trotter 1972). It has been suggested that Maori subsistence hunting may have caused near elimination of fur seals in mainland New Zealand (Lalas and Bradshaw 2001). Commercial sealing probably started when Australian settlers carried out large scale hunting in Bass Strait in the 18th century. Soon after they turned their attention to the coast of New Zealand and its 9 neighbouring islands (Hutton and Drummond 1923). Capitan Cook observed fur seals in New Zealand only on the coast of the South Island in his first voyage. On his second voyage, “the first fresh meat eaten by him at Dusky Bay was obtained from the first seal killed there. During his stay of nearly two months at the southern Sounds, Cook’s crew killed many seals, using the flesh for food, the skin for repairs to the rigging and the blubber for oil for his lamps” (Hutton and Drummond 1923). Small scale commercial sealing began in the New Zealand waters in 1792-3, when around 4500 skins were taken in Dusky Sound. Systematic large scale exploitation probably started in the early 1800s (Crawley 1990). Commercial hunting was carried out so vigorously that the New Zealand population came close to extinction in many localities. The actual number of seals killed and skins taken was never published mainly due to the secrecy of the industry. The magnitude of the harvest is indicated by official cargo reports, 60,000 skins from the Antipodes Islands in 1804-1805 and 53 000 in 1906-1907 (Macnab, 1907 , as cited in Crawley 1990) and a total of more than 180,000 skins from Macquarie Island between 1810-1813 (Csordas, 1958, as cited in Crawley 1990) show the scale of the hunting. Hutton and Drummond (1923) wrote “as early as 1810 the effects of reckless slaughter began to be felt, and it was the discovery of Campbell Island and Macquarie Island in that year which gave fresh life to the trade “. By the 1830s, the seal population had been eliminated on the Antipodes and Macquarie Islands, and only a few fragmented populations survived on the Snares, Bounty, Chatham, Auckland and Campbell Islands (Macnab 1907 , as cited in Crawley 1990). The eventual poor economics of sealing and an imposition of a system of a closed season and hunting permits from 1875 to 1916 eventually saved this species from extinction. Excepting 10 Campbell Island in 1924-1926, and parts of southern New Zealand in 1946, no killing of fur seals, other than for scientific proposes, has been allowed over the last hundred years. Protection is now guaranteed under the New Zealand Protection Act (1978), except in cases where the Act allows for commercial fisherman to kill seals actually caught in the act of robbing nets and damaging fishing gear. The severe decline of the seal population during the last two centuries likely resulted in a bottleneck that had severe impacts on the genetic diversity of the modern population. Local populations are likely descended from a few dozen individuals that found refuge in the remote areas during large-scale harvesting and then recolonized mainland areas. The current number of New Zealand fur seals is unknown. Barton (1996) estimated the population size as over 100,000 individuals. Taking an annual 5% increases in population size in to account (Taylor 1982), the current population is assumed to be about 200,000 individuals (Laura Boren, Department on Conservation personal communication). 11 Chapter 2 A research diary of the specimen collection methods Finding an adequate source of biological sample for DNA-based studies is the first step in initiating any large scale genetic studies. In this chapter I detail the process by which I tested collecting methodology to minimise impact on individual seals while maintaining high integrity of data. ifferent potential sources of biological sample collection from New Zealand fur seals were evaluated thoroughly. Based on the observed efficiency and disruptiveness (Lefort, Boyer et al. 2014) of the different methods, the most optimal method was chosen for the rest of the study. 2.1 Flipper notching Flipper notching is the method of choice in biological sample collection from pinnipeds (Worthington Wilmer, Allen et al. 1999, Hoffman and Amos 2005, Robertson and Gemmell 2005, Hoffman, Forcada et al. 2006, de Oliveira, Hoffman et al. 2008). The method involves the collection of biopsy samples from the inter-digital margin of fore-flippers by restraining animals, mainly females and pups rather than aggressive males. In this method, the individual pups to be sampled are chosen based on a strategy that minimises panicking other pups which could result in injuries. Two trained researchers encircle the targeted individual. One person captures the seal. While the first person is neutralizing the animal by slightly pressing against its head and hind flipper(Majluf and Goebel 1992), the second person performs the biopsy operation and wound disinfection (Figure 2.1). The success rate of the flipper notched biological samples in this research was extremely high. No samples failed to provide mitochondrial DNA or zinc finger amplifications, justifying the large –scale use of the method in pinnipeds studies. From more than 100 ear notches samples 12 collected throughout New Zealand, 100% of them showed enough concentration and purification for amplification of mitochondrial DNA and Zinc finger. Whether the whole mitochondrial DNA was considered or a short fragment of the control region no sign of DNA degradation or external contamination was detected. Despite being an excellent source of biological sample for DNA studies, this method involved some minor degrees of damage to the animal physical integrity which could result in external bleeding. This method is widely used for biological sample collection from suckling pups which are more vulnerable to excessive bleeding and external stress. To alleviate the potential stress caused by ear notching I assessed other less intrusive methods. Figure 2-1 A New Zealand fur seal neutralized for biopsy collection by researchers at Banks Peninsula. Picture from author. 13 2.2 Non-invasive methods of DNA collection Non-invasive tissue sampling for DNA analysis has been used in many conservation genetic studies, especially on rare and illusive species. Despite being a preferred method for scientists, the poor quality and low quantity of DNA generally recovered using non-invasive methods has hampered their large-scale use in studies of wild populations (Taberlet, Waits et al. 1999). Access to an adequate source of DNA is an important consideration in genomic analysis, which in turn will considerably facilitate comprehensive study of wild animals (Brooks, Williamson et al. 2003). Priority is given to methods of DNA collection that do not jeopardise the physical integrity of individuals, although some minor disturbance in the behaviour of subject animals is often unavoidable. There are several non-invasive approaches that have been tested in the current research to sample DNA. 2.2.1 Buccal Swabs Despite wide use of saliva as a source of DNA in human studies, its application in wild populations studies remains scarce (Williams, Blejwas et al. 2003). Saliva as an alternative to other non-invasive source of DNA (faecal samples, hairs , etc.) would substantially reduce the effort needed to collect samples in the field (Inoue, Inoue-Murayama et al. 2007). Brooks, Williamson et al. (2003) first tested saliva as a source of DNA in a comparative genomic study of non-human mammals, including cows and pigs. Oberbauer, Grossman et al. (2003) used DNA recovered from saliva in a genome linkage study in canine species, and Meldgaard, Bollen et al. (2004) successfully PCR-amplified mouse DNA from buccal swabs. In wild populations, salivary DNA has been successfully recovered from amphibians (Pidancier, Miquel et al. 2003) 14 and reptiles (Williams, Blejwas et al. 2003). To test the feasibility of saliva as a source of DNA, the issue of capturing animals, and then keeping their mouth open long enough to take an effective sample, needs to be considered, especially for non-domesticated wild animals. In New Zealand fur seals, capturing seals pups definitely involved some degree of stress and unease. It is up to the person collecting the buccal swabs to choose the least stressful capture and handling methods for the target individual. (Pidancier, Miquel et al. 2003) . Capturing seals pups for this research was not difficult since many pups in the early stages of their life have not developed a natural fear of humans, and are easily approachable to very close distances. The rocky nature of their colony facilitated their capture, even when they were hiding in the spaces between rocks. However, opening the mouth, and keeping it open during operation, was not a trivial task. To keep the mouth cavity open during swabbing, a modified smaller version of a Schulze mouth gag (medical-tools. Pakistan) (Figure 2-2) was designed to fit inside the mouth cavity of a fur seal pup. The gag is placed in the mouth cavity and a handle on upper part of the gag provides the operator with the necessary grip to keep the month open. To test the method, three researchers approached pups cautiously to minimise unnecessary stress. While the first person had captured the pup and neutralized it with slight pressure against its neck and hind flipper, the second researcher fitted the mouth gag inside the pup’s mouth. The third researcher swabbed gently inside the mouth cavity for 30-45 seconds using a swab brush provided in the Master Amp DNA extraction kit (Catalogue number MB7901S). The whole process took no more than one minute. If this time was exceeded then the sampling was terminated and the animal released. 15 The swabs were air-dried for few minutes before putting them into a plastic cover and kept cold until they could be analysed in the Molecular Ecology Lab at Lincoln University, usually within few days. Figure 2-2 A standard Schulz mouth gag. Picture kindly provided by medical- Tool Company (http://www.medical-tools.com/ DNA was extracted using a protocol recommended in the manufacturer’s instructions for the Master Amp DNA extraction kit. The DNA concentration was measured using a Qubit quantitation kit (Life Technologies, Catalogue number Q33217). A 1kb fragment of the control region of the mitochondrial genome was amplified (for details of PCR amplification and 16 thermal cycles see the chapter 4). Amplicons were screened on a 2% agarose gel to assess the success rate of the process. 2.2.2 Environmental DNA Humans shed approximately 400,000 skin cells each day, from all parts of the body (Wickenheiser 2002). Trace amounts of DNA may be deposited on objects coming into contact with the body. Depending on the nature, frequency, and duration of contact, as much as several hundred nanograms of DNA can be present on touched objects (Van Oorschot, Weston et al. 1998, Castella and Mangin 2008, Sewell, Quinones et al. 2008, Raymond, van Oorschot et al. 2009). Assuming that all mammals shed similar amount of skin cells, and hence DNA, as humans, the method was evaluated on New Zealand fur seals. Two different methods for collecting DNA from fur seal habitats were tested: (1) rock surface swabbing, and (2) tape lifting. In both approaches, an adult animal was chosen based on a strategy that caused minimal disturbance to the rest of colony. The focal animal was approached carefully and encouraged to move from their haul-out rock. The surface of the rock where the seal had been resting was thoroughly swabbed with a slightly wet sterile swab or, alternatively, a small piece of water-soluble tape was stuck to the surface and then lifted repeatedly 30-40 times. Swabs and/or tapes were immediately placed inside a sterile vial and kept cold until DNA could be extracted in the Molecular Ecology Lab at Lincoln University. For DNA extraction, a Qiagen DNA Investigator extraction kit (Catalogue number 56504) was used, according to the manufacturer’s instructions. The DNA concentration was measured using a Qubit quantitation kit. A 1kb fragment of the mitochondrial control region was amplified as explained in the chapter 4. The resulting amplicons were run on a 2% agarose gel to assess the amplification success rate. 17 Ten samples for each different source of DNA were tested. The success rate was different for different DNA sources. 80% of the buccal swabs gave positive amplicons of the expected size for this fragment of mitochondrial DNA. The BLAST results matched perfectly with New Zealand fur seals, or closely related species. Buccal swabs consistently gave a DNA yield of less than 3 µg, which is consistent with results of (Walker, Najarian et al. 1999). The DNA quantity from buccal swabs is at least two orders of magnitude less than peripheral blood samples, which have an mean yield in excess of 100 µg (Walker, Najarian et al. 1999).. Only one sample from ten rock surface swabs and tape lifts gave a very faint band at the expected size of approximately 1kb. Despite repeating the amplification several times, the DNA concentration and/purity was not suitable for Sanger sequencing. None of the BLAST results matched a mammalian species in their first five hits. Leforte (Lincoln University, personal communication) introduced the new term “non-disruptive” DNA sampling to emphasize that it is not only adverse effects of sampling on the physical integrity of an individual animal that needs to be taken into account, but also fitness and long-term psychological effects of sample collection should be considered. The results of this study are a classic example of a situation where a “non-invasive” method is in fact seriously disruptive to an animal’s psychological condition. Mouth swabbing, despite being apparently non-invasive, and without any obvious damage to the animals’ physical integrity, involved substantial amounts of stress for the target individuals. Observations show that extra sensibility of the nasal area, accompanied by the need to neutralise individuals for relatively long period of time, are the main factors causing discomfort in the focal individuals. The swabbing process itself does not seem to disturb the animals significantly as the few individuals that were easily handled without a need for a mouth gag did not show any signs of discomfort and continued to play with swab even after the operation had ended. 18 Experience with seal pups has shown that, apart from the capturing of individuals by researchers itself, the most stressful situation for a pup is probably touching its mouth and nasal areas, which could provoke a sense of asphyxiation. Based on these results, mouth swabbing was considered more disruptive than traditional, relatively invasive, flipper notching. Buccal swabs were not used in the rest of study, although the possibility of saliva as a source of DNA in other animals (amphibians, birds), with different sensibilities, remains open. The reason that environmental DNA was not successful in the current research is yet to be clarified. Forensic studies have shown that shedding of epithelial cells from human hands is varies among individuals, with some individuals shedding cells more easily than others (Bright and Petricevic 2004). Actions such as hand-washing could also decrease the amount of epithelial cells shed by an individual (Van Oorschot, Weston et al. 1998). It is possible that dense fur covering the body, along with constant submersion in sea water could have some impacts on epithelial cell shedding resulting in a low recovery rate. In conclusion the low amplification success rate, coinciding with highly degraded nature of the extracted DNA, implied that environmental DNA was not a reliable source for this study. 2.2.3 Dart Biopsy The remote collection of tissue samples from wild, marine mammals has historically been the most effective, and least invasive, method of tissue collection for ecological studies (Brown, Kraus et al. 1991). Remote collection, if carried out according to proper instructions, has less adverse impacts on animals than other tissue collection methods (Karesh, Smith et al. 1987, Brown, Corkeron et al. 1994, Gauthier and Sears 1999, BearzI 2000, Krützen, Barré et al. 2002, Gorgone, Haase et al. 2008, Jefferson and Hung 2008), although some adverse effects have been noted (e.g. BearzI 2000). 19 A minor degree of disturbance to target individuals is inevitable in most sampling operations. Caudron (2006) summarised three factors that influence the degree of invasiveness of any sample collecting operation in wild populations as (1) the duration of sampling, (2) the number of humans involved in the operation and (3) the sampling distance (i.e. the distance between a target animal and the person responsible for collecting the biopsy). Although these factors influence all the individuals in the colony, it is known that different individuals may react differently to similar environmental stressors (BearzI 2000, Kiszka, Simon-Bouhet et al. 2010). The eventual response of individuals may depend on individual physiological and psychological factors (Barrett Lennard, Smith et al. 1996), age and size (Peters 1986, Gauthier and Sears 1999), gender (Brown, Kraus et al. 1991, Gauthier and Sears 1999), reproductive and hormonal conditions, illness or concurrent pathologies, behaviour at the time of sampling (Barrett Lennard, Smith et al. 1996), and previous experience (Barrett Lennard, Smith et al. 1996). Examples of the use of remote biopsies in the studies of wild populations of pinnipeds are numerous (e.g. Hoberecht, Vos et al. 2006, Hoffman, Forcada et al. 2006, Curtis, Stewart et al. 2007, Hoffman, Forcada et al. 2007, Hoffman and Forcada 2009, Nino-Torres, Gardner et al. 2009, Hoffman and Nichols 2011). Although it is not always necessary to determine the individual from which the biopsy sample originates, in some studies involving parentage assignment or fine-scale population social structure, determining the origin of DNA is of pivotal importance (Krützen, Barré et al. 2002). One method to correctly determine the origin of a DNA sample is to capture target animals before collecting the sample. Capturing wild animals is always a risky operation for both humans and animals. Although the development of tranquilizers has facilitated the capture and handling of target animals, it is still one of the most challenging parts of an ecological study. The use of tranquilizers needs considerable expertise in both animal anaesthesiology and constant monitoring of target animals for any signs of breathing irregularities and/or 20 thermal stress (Gemmell and Majluf 1997). In general, chemical capture and anaesthesia of wild mammals usually involves some degree of uncertainty regarding the animal’s recovery, even in healthy individuals. The results of a survey published in the British Journal of Anaesthesia shows that approximately 0.01-0.05% of deaths in humans, 0.1% in dogs and cats, and 1% in horses, are directly or indirectly attributable to anaesthesia (Jones 2001). Several studies show that immobilisation of pinnipeds and other marine and terrestrial mammals, both with and without anaesthetic, may result in a mortality rate of around 3-20% (Loughlin and Spraker 1989, Baker, Fedak et al. 1990, Work, DeLong et al. 1993, Heath, Calkins et al. 1996). As an alternative, which allows for high quality tissue samples without the need for chemical and/or physical immobilisation, a biopsy sample can be obtained by shooting a biopsy dart projectile with an appropriate propelling device. This method permits selective, non-destructive sampling of fresh, uncontaminated tissue samples for genetic and ecotoxicological studies (Gemmell and Majluf 1997). In the current research the applicability of a new remote biopsy collection system developed by (Pneu –Dart. Pennsylvania-USA) on New Zealand fur seals was tested. A purpose made DNA collection dart system was constructed. The dart was a 120mm cylindrical shape projectile consisting of a 100 mm body and a 20 mm barbed biopsy needle encased within a 25 mm tubular cutter (Fig 2.3). The barb needle protrudes approximately 4mm from the tip of the tubular cutter. A X-2 Gauged CO2 Projector pistol (Pneu –Dart. Pennsylvania-USA) was used to propel the biopsy dart towards the target animals. The biopsy system were tested on four male New Zealand fur seals in Kaikoura, New Zealand. The DNA dart successfully penetrated into the male fur seal bodies. All four attempts successfully collected blubber tissue. The reaction of the males to the biopsy operation was a brief search in the direction of assailant usually accompanied by a sudden loud noise which 21 tip length will cause excessive injury and/or unsuccessful sampling. Gemmell (1997) reported that careful biopsy sampling should not cause any tissue damage, or bone fracturing or chipping at the point of impact. Hoberecht (2006), in his study on sea lions, observed a small circular wound at the point of impact that generally bled for about 10 minutes. After 30-60 minutes it became difficult to discern the dart penetration site from other abrasions on the animal’s skin. He also reported that the amount of bleeding was affected by the ambient temperature. Bleeding continues for longer on hot sunny days compared to cold rainy days. Another cause of concern is the complication of the wound healing process in sea water. This mysterious and rapid process in marine mammals has attracted a considerable amount of attention (e.g. WÜRsig and WÜRsig 1977, Bertilsson‐Friedman 2006, Van Den Hoff and Morrice 2008). Marine mammals are prone to many different causes of skin trauma in their daily life. Territorial disputes, the rocky nature of their habitat, and living in a challenging environment with regular encounters with carnivorous predators, leave their mark on an animal’s body. For example, one study in Monteron Bay showed that 36% of dolphins bear the scars of an encounter with sharks (Corkeron, Morris et al. 1987). Surprisingly, most of these wounds heal very quickly. Wursig (1977) reported that pigment spots and bite marks made by conspecific bottlenose dolphins disappeared in less than one year. Another study showed that dart wounds on dolphins normally heal in less than two months (Krützen, Barré et al. 2002). Efficient healing of soft tissue trauma in marine mammals is further evidenced by the survival of southern elephant seals (Van Den Hoff and Morrice 2008) and Hawaiian monk seals (Bertilsson‐Friedman 2006) after severe shark attack injuries. The process of wound healing in marine mammals can be studied from two different perspectives: (1) the impact of sea water on the wound healing process and (2) specific biochemical adaptations. Corkeron (1987) suggested that exposure to the osmotic pressure 23 of sea water causes the formation of a buffer layer of degenerating cells that prevents the trauma from spreading to the deeper tissues. Corkeron (1987) also proposed that constant and gentle irrigation of the wound by sea water will facilitate the wound healing process. Another important aspect of wound healing in marine mammals is the biochemical structure of blubber tissue. Blubber accumulates many toxic materials of anthropogenic origin, such as organohalogenes and heavy metals from their food resource. In addition to these pollutant organohalogenes, blubber can also accumulate organohalogenes of natural origin (Vetter, Scholz et al. 2001, Vetter and Jun 2003, Vetter, Jun et al. 2003). Although the origins of these non-human derived organohalogene compounds are unknown, several similar substances are produced by marine organisms such as algae and plankton (Gribble 2003). These organohalogenes are known to have antimicrobial properties, such as inhibition of biofilm formation (Janssens, Steenackers et al. 2008), and antibiotic activity (Ezaki, Koyama et al. 1983). The extent to which these substances are released in damaged blubber tissue and help to prevent the spread of wounds to the adjacent tissues, interfering in the wound healing process, is yet to be studied (Zasloff 2011). In conclusion, the complication of the biopsy wound seems a very remote possibility. Remote biopsy however appears to involve some degree of unease and pain in target individuals. Gemmell (1997) observed that South American fur seals recoiled from the impact of the dart biopsy and searched briefly for the assailant, which indicates the painfulness of dart penetration. The sea lion, with its larger body size, also turned in the direction of the dart and escaped to sea (Hoberecht, Vos et al. 2006). It does not seem that this brief, painful experience has any long-term impact on animal behaviour. At least in the latter study, the sea lion returned to land in less than five minutes. Krutzen (2002), in a study on dolphins, observed that dolphins reacted equally to being hit or missed by a dart. It was probably a sudden disturbance in their environment that caused their reaction and actual dart penetration seems 24 to have less impact. He also observed that target animals did not change their long-term behaviour towards humans because they were still easily approachable by researchers. It is suggested that, at least with regards to male New Zealand fur seals, that the relatively invasive remote collection method is less disruptive that all other methods involving the live capture and anaesthesia of animals. Individuals do not have to tolerate stressful physical restraint and the disturbance in the colony is minimal. Using advanced propellers and retriever systems, individual may be targeted from a safe, for both seal and researcher, distance. Targeted individuals are usually mature males that are used to wounds and bruises in their life cycle. When the method is compared to the flipper notching of young pups in early month of their life the advantages of taking biopsy from adult males is more obvious. The blubber tissue collected using this method is also suitable for other studies, such as food web dynamics using radioactive isotopes which traditionally required capture and anaesthesia of target individuals. Respecting the sensibilities of the public opinions, which understandably oppose the shooting of the New Zealand fur seals even by a dart gun, and after a thorough and detailed consultations with New Zealand Department of Conservation, it was decided to choose the seal pups flipper notching method for the rest of the this research. However, the application of this methods in future studies after answering public concerns remain a feasible nondisruptive alternative for future studies in this species and other similar species. 25 Chapter 3 New Zealand fur seal population genetics using complete mitochondrial DNA and evidence for X to Y gene conversion Summary New Zealand fur seals are one of the many pinniped species that have passed through a population bottleneck due to historical commercial sealing. Investigating sex-specific demographic dynamics during and after a population bottleneck requires reconstructing population dynamic history using genetic markers with different inheritance patterns. I reconstructed the demographic history of New Zealand fur seals in a Bayesian framework using maternally inherited whole mitochondrial DNA sequences and sequences from an intron of the male-specific zinc-finger Y chromosomal protein gene (ZFY). Both mitogenomic and Y chromosomal data suggest the presence of two separate population refuges for New Zealand fur seals surviving large-scale human harvest. Approximate Bayesian Computation (ABC) analysis of the New Zealand fur seal mitogenome strongly supports the hypothesis of a population bottleneck around 35 generations ago, coinciding with the peak of commercial sealing in the Southern hemisphere. There appears to have been a catastrophic decline in effective population size of the New Zealand fur seal to approximately less than 1% of its historical size during the commercial sealing period. Mitogenomic data are consistent with a pristine effective population size of 62700 that first declined to 20600 after the establishment of Polynesian populations into New Zealand and then dropped to an extreme low of 95 breeding individuals during commercial sealing. Y chromosome data identify more subtle population demographic dynamics. The ZFY data are consistent with a series of gene conversion and/or recombination events in this region of the 26 Y chromosome with its X chromosome counterpart. The ZFY region of the Y chromosome is situated outside the pseudo autosomal region. Chromosomal rearrangement in this region is rare and might indicate an adaptive mechanism to counteract gradual degeneration of the Y chromosome due to a high mutation rate. 3.1 Introduction The study of genetic variability is of great relevance in conservation ecology and evolutionary biology (Lehman 1998, Hoelzel, Fleischer et al. 2002, Valqui, Hartl et al. 2010). Genetic diversity provides natural selection with the necessary variation to act on populations. Individual fitness and population viability are equally influenced by genetic variation. Natural populations can pass through episodes of substantially low densities, often termed ‘bottlenecks’, due to a variety of natural and human-influenced factors. These episodes of population bottlenecks usually result in the reduction of effective population size (Wright 1938), loss of heterozygosity (Nei, Maruyama et al. 1975), loss of alleles (Maruyama and Fuerst 1985), and greater frequency of linkage disequilibrium (Hartl and Clark 1997). Populations that pass through population bottlenecks are more prone to developmental instability, decline in competitiveness, loss of disease and/or parasite resistance, and a reduced survival rate (Hoelzel, Fleischer et al. 2002). Genetic drift can further threaten the viability of these small populations, not only by random elimination of whatever diversity has been left, but also by acting as the predominant mode of evolution, replacing natural selection (Lacy 1997). There is a substantial risk that deleterious alleles can become fixed in small populations due to the stochastic action of genetic drift replacing more favourable alleles (Avise 1994, Keller, Arcese et al. 1994, Soule 2005). 27 Burger and Lynch (1995) reported that fluctuation in genetic variation in small populations is of sufficient magnitude to limit adaptations of these populations to changing environments, and eventually causes extinction. Larger populations are more resilient to environmental stochastisity. Each case of extinction is an instance of a population failing to adapt rapidly enough to natural, or human-induced, environmental changes (Lacy 1997). Spielman, Brook et al. (2004), in a meta-analysis of many different taxa, found that genetic diversity is on average 35% lower in endangered species compared to non-endangered organisms. The impact of bottlenecks on genetic diversity depends mainly on demographic factors. The effects of a severe bottleneck may be minimized if recovery is rapid (Nei, Maruyama et al. 1975, Miller and Hedrick 2001) and in some cases, a bottleneck can even be beneficial in purging deleterious alleles from populations (Weber, Stewart et al. 2004). The role of genetic variation in species viability is the subject of contentious debate. Decline in genetic variation may have negative impacts on mate acquisition, infant mortality, resistance to disease , physiological responses to stress, social dominance, longevity, and other components of fitness in wild populations (Lacy 1997). Packer, Pusey et al. (1991) found that, in an isolated population of African lions (Panthera leo) that had passed through a recent population bottleneck of no more than 10 individuals in the Ngorongoro Crater of Tanzania, the frequency of sperm abnormalities was higher and sperm motilities were lower, than in the neighbouring large populations of the Serengeti. Asian lions (Panthera leo persica) reduced to a single population in the Gir forest in western India displayed a similarly low degree of genetic variation, low sperm counts, and an exceptionally high rate of spermatozoa deformity (Obrien, Martenson et al. 1987, Wildt, Bush et al. 1987). 28 Despite the essential role of genetic diversity in species’ evolutionary potentials there are several examples of animals with low levels of genetic variation that survive well, at least in short term, in nature (Simberloff 1988, Caro and Laurenson 1994). Northern elephant seals (Mirounga angustirostris) are recovering rapidly to very large numbers despite experiencing a severe population bottleneck size of an estimated 10-30 individuals (Hoelzel, Halley et al. 1993). African cheetahs (Acinonyx jubatus) have survived even with a low level of genetic variation due to several population bottlenecks in the past (O'brien, Wildt et al. 1983, O'brien, Roelke et al. 1985, Obrien 1994). Pinnipeds are a group of animals whose genetic diversity is of pivotal importance for their long term survival (Hoffman, Matson et al. 2006). The 2008 IUCN (International Union for Conservation of Nature) review of the status of the mammals of the world classified pinnipeds, alongside other marine mammals, as disproportionately threatened and of particular concern. One in three species of pinnipeds is considered threatened compared to one in five species for other mammals (Kovacs, Aguilar et al. 2012). The long history of commercial harvesting in the 18th and 19th centuries pushed many pinniped species out of their historical ranges and decreased their population densities close to extinction. The remaining populations were highly fragmented, and reduced in their distribution range, with subsequent reductions in genetic diversity (Weber, Stewart et al. 2000, Wynen, Goldsworthy et al. 2000, Pastor, Garza et al. 2004, Schultz, Baker et al. 2009). All extant fur seal and sea lion species have survived population bottlenecks, with different degrees of severity (Reeves, Stewart et al. 1992, Stewart, Yochem et al. 1993, Wynen, Goldsworthy et al. 2001, Nowak 2003). Several studies have investigated the effects of genetic variation on marine mammal fitness. Coltman, Bowen et al. (1998) found a positive correlation between birth weight, neonatal survival rate and genetic variation in harbour seals (Phoca vitulina). Amos, Wilmer et al. (2001) 29 reported a similar trend between life-time success and heterozygosity in long-fin pilot whales (Globicephala melas) and grey seals (Halichoerus grypus). Acevedo-Whitehouse, Gulland et al. (2003) showed that California sea lions (Zalophus californianus) stranded due to illness had less genetic diversity than control animals stranded due to trauma. Similarly, Bean, Amos et al. (2004) demonstrated a higher genetic diversity in healthy versus dead grey seals pups. A strong correlation between genetic diversity and fitness has also been suggested for harp seals (Pagophilus groenlandicus) (Kretzmann, Mentzer et al. 2006) and bottle nose dolphins (Tursiops sp.) in Shark Bay Australia (Frère, Krützen et al. 2010). New Zealand fur seals (Arctocephalus forsteri) survived commercial sealing of the 18th and 19th centuries in dramatically reduced populations probably in a few remote refuges. The reduction in population size resulted in a considerable loss of genetic diversity in the species (Lento, Mattlin et al. 1994). Fur seal populations in many southern areas may all descend from small groups that survived the massive human harvest in regional refuges. Since the enforcement of a series of protective measures in the early 20th century inhibiting commercial sealing, the population of New Zealand fur seal seals has recovered rapidly from the brink of extinction (Taylor 1982, Dix 1993, Lalas and Harcourt 1995, Taylor, Barton et al. 1995, Bradshaw, Lalas et al. 2000). Taylor (1982) estimated that the New Zealand fur seal population was growing at least 2% per year in the 1980s and this may have reached 5% in the Bounty Islands, and possibly elsewhere. Assuming a steady increase of 5% per year, the current estimate for the New Zealand fur seal population in 2015 is around 200,000 (Laura Boren, Department of Conservation, personal communication). It is believed that the current population size is still considerably smaller than it was before humans arrived in New Zealand, although historical data about New Zealand fur seal population size is scarce. 30 Even though the census number of fur seals in New Zealand is undoubtedly increasing rapidly, a solid knowledge of genetic diversity is required to make any conclusions about the implications of current population trends for the continued survival and long-term management of the species. There is a clear distinction between the current prospect of survival and the evolutionary potential for survival. While the low level of genetic diversity might not affect the former in the short-term, the latter will be more significantly affected. Lento, Mattlin et al. (1994), in a study of mitochondrial cytochrome b sequences, suggested that a high level of genetic diversity may still exist in the species as a whole, even when there is limited gene flow between broad geographical regions. Robertson and Gemmell (2005) used microsatellite data to study genetic variation and relationships among seven different fur seal colonies around New Zealand. They found that, despite a history of severe population bottlenecks, fur seals still possess enough microsatellite variation for this to be informative in population genetics studies. However, a rigorous test for the bottleneck that the seals passed through, and its amplitude, is still missing from the scientific literatures. In this study, the genetic diversity among New Zealand fur seal populations is examined using two genetic markers: (1) the maternally inherited complete mitochondrial DNA and (2) an intron of the paternally inherited ZFY gene on the Y-chromosome. The application of complete mitochondrial DNA markers for detecting a bottleneck signal was tested and the mitochondrial marker was used in a Bayesian framework to reconstruct the recent dynamic demographic history of the species. 31 3.2 Material and methods 3.2.1 Sample collection Up to 10 tissue samples were collected from most of the major breeding colonies throughout New Zealand, including Cape Palliser, Taranaki, Kaikoura, Banks Peninsula, Otago Peninsula, Stewart Island, Fiordland, Open Bay Island, Cape Foulwind, Kahurangi National Park, and the Bounty Islands (Figure3.1). Tissue biopsies were taken from the interdigital web of the left fore-flipper using a medium-sized piglet ear-notcher (Majluf and Goebel 1992). The earnotcher was thoroughly disinfected after each use with 70% ethanol to prevent contamination. The samples were transferred to 99% ethanol immediately and kept cold until DNA could be extracted in the Molecular Ecology Laboratory at Lincoln University, usually within a few weeks. The whole process was done in accordance with permit 35069-MAR issued by the New Zealand Department of Conservation. DNA was extracted from tissue samples using the Qiagen DNAeasy Blood &Tissue Kit (Catalogue number 69506) according to the manufacturer’s instructions, and DNA was stored at -20C. 3.2.2 Justification for number of sequenced individuals For two gene copies the rate of coalescence when scaling by a unit of 2N will be one. The reason is that the probability of a single coalescent event is 1 2𝑁 which means the rate will be 2N and when re-scaled by unit of 2N it becomes 1. When considering the limit of large populations (N ⟶∞) we can ignore the possibility that two, or even more coalescence 32 Figure 3-1 New Zealand colonies where tissue biopsies were collected. 33 events, happen in a single generation (Kingman 1982) which will facilitate understanding the process substantially. We also know that there are n (n-1)/2 possible distinct combinations of n pairs of any objects in a set of n objects (In our case the number of sampled haploid fur seal genomes). Looking backward in the time the expected time until the first coalescent event in more than two samples will be exponentially distributed with a mean: 1 𝑛(𝑛−1)/2 = 2/[𝑛(𝑛 − 1)]. After the first coalescent event the remaining (n-1) lineages will coalesce with the same probability but now there are (n-1) lineages instead of n. Replacing n with n-1 in the above formula 2/ [(n-1) (n-2)]. The whole process continues until the last two haplotypes coalesce into a most recent common ancestor (MRCA) of all lineages. This is important because we can calculate expected time to the MRCA of all sampled haplotypes according to following equation: E [TMRCA] = ∑𝑛2 2 𝑘(𝑘−1) . This formula is the sum of all intervals in a coalescence tree. K represents the number of DNA haplotypes in the sample. This formula predicts that for example the expected time for the MRCA of the 5 samples is 1.6 (multiply by 2N) while we increase the number of samples to 50 it is higher at 1.96. With further increases of the number of samples the expected time will only slightly increase and approach the theoretical value of 2. These results mean sampling more than 50 individuals, in addition to the logistical difficulties and financial burdens, will 34 not add substantial extra information to the downstream analysis. I decided to sequence the entire length of the mitogenome for 50 individuals since this number will be enough to capture most of the variation in mt DNA at the level of populations. Closely related to the notion of expected time until MRCA is the total tree length of the sampled data: 2𝐾 E [Total length of tree] = ∑𝑛𝑘=2 𝐾𝐸[𝑡𝑘] = ∑𝑛2 𝑘(𝑘−1) =2 ∑𝑛−1 𝑘=1 1/𝑘 . Where K represents the number of sampled haplotypes. The reason that expected time until MRCA of the linages is multiplied by a factor K is due to the fact that for a sample consisting of k lineages the total length becomes K times𝐸[𝑡𝑘]. Et represents the expected coalescence time for k lineages. The importance of this finding will be clear in the next section where I explain my choice of summary statistics. Given the apparent absence of recombination in the mitogenome, how much extra diversity can be obtained by sequencing whole length of the mitochondrial DNA which is still costly and tedious? To answer this question a Python script was composed that takes sequences from successive windows of an arbitrary 100bp long from the first sequence of the current data set of 50 seal mitochondrial DNA. The script compares each 100bp windows of the first sequence to the corresponding region of the remaining 49 fur seal mitogenomes and counts the total number of haplotypes that could be identified from only that particular 100 bp windows and moves on until the end of sequence. Different segments of mitochondrial DNA were found to not be the same in their resolution power. While some segments identified the 49 haplotypes, even in short 100 bp windows, 35 some segments were conservative and performed poorly in haplotype identification (Figure 3.2) which is usually the first desired statistics in any simulation. To detect the maximum variability, especially in species with limited a priori knowledge of the mitochondrial DNA diversity, it is highly recommended to sequence the whole length of mitochondria for more reliable population inference. Figure 3-2 Variation is the resolution power of different 100 bp long windows of New Zealand fur seal mitochondrial DNA in identifying correct number of haplotypes. X axis is position in the fur seal genome and Y axis is the number of identified haplotypes for that window. Zero means that those fragments don’t have resolution power to differentiate between sequences at all. In other word the sequence it is not variable. 3.2.3 Whole mitochondrial genome sequencing and assembly The mitochondrial DNA sequence for New Zealand fur seals (National Centre for Biotechnology Information (NCBI) accession number NC004023.1) was used to design a series 36 of primers to amplify the entire genome in fragments of 3-6 kb in length (Table 3.1). Seven amplicons were chosen that had approximately 500bp overlap. All seven fragments were amplified from 50 samples chosen from throughout New Zealand. Long PCRs were performed using 5µl of template DNA as a starting material. The thermal cycle contained 25 cycles of denaturing steps at 95 C for 30 seconds, followed by an annealing temperature of 55°C for 45 seconds, and a 330 second extension at 72 °C. The same primers were also used to amplify the entire mitochondrial genome using Phi29 (NEB, catalogue number M0269S) in a rolling circle isothermal (30°C) amplification for 10 hours. Amplified DNA was then prepared for sequencing using an Illumina Nextera XT library preparation kit and subsequently sequenced on an Illumina MiSeq machine following the manufacturer’s instructions. Whole mitochondrial DNA sequences were assembled using CLC Genomics software package v7.5 (CLC Bio Inc.). The New Zealand fur seal mitochondrial genome previously submitted to the NCBI (Lin, McLenachan et al. 2002), despite being labelled as complete, was found to be incomplete, as it does not include approximately 1kb of the genome . To overcome this problem, data available for a closely related species, Guadalupe fur seal (Arctocephalus townsendi), were used as a reference guide to assemble the genome. 20% of data had one or few segment(s) with a coverage of 10X. Given that these positions were conserved in all high coverage assemblies, they were included in the downstream data analysis. 37 20 (0.01% error rates) were discarded using Codon Code Aligner software (Codon Code Corporation, Dedham, MA). Under this criterion, only high quality segments were analysed. The downside of this rigorous filtering were slightly larger numbers of deleted nucleotides in the middle of some sequences that are interpreted as missing data in downstream analysis. The resulting filtered sequences were aligned using the Clustal W2 web service (McWilliam, Li et al. 2013) setting parameters to their default value. 3.2.5 An overview of the analysis Haplotype analyses were performed using Arlequin v3.5 (Excoffier, Laval et al. 2005), with default settings except where explicitly stated otherwise. A median-joining haplotype network was estimated from mitogenomics data using the program PopArt (Bandelt, Forster et al. 1999). A Bayesian skyline plot was also reconstructed from mitochondrial genomes using BEAST v1.8.1 (Drummond and Rambaut 2007) and Tracer v1.5 (Rambaut and Drummond 2003) to estimate relative population size. A series of Bayesian phylogenetic tree were constructed for the mitogenomics and ZFY data using the software package BEAST v1.8.1 (Drummond and Rambaut 2007). Two Approximate Bayesian Computations (ABCs) were performed on the assembled New Zealand fur seal mitochondrial genomes to test for evidence of the population bottleneck and also to estimate the bottleneck population size, historical population size, and pristine population size using a methodology implemented in the software package DIYABC v2.0.4 (Cornuet, Pudlo et al. 2014). The aim of the first analysis was to compare two models of population evolution: a constant population size model against a model with a bottleneck. In the second analysis, the pristine population size, historical population size and bottleneck population size were estimated in a population bottleneck model. 39 In the following section I take a closer look at methods applied in the analysis section of this chapter and next chapter. Bayesian skyline plot Bayesian skyline plot is one of the commonly used methods to estimate the demographic dynamics of the populations. An exact term search of the word in google scholar conducted in 2th of July 2015 returned 1540 exact matches! Estimating a skyline plot consists of two equally important steps (Ho and Shapiro 2011): 1-Estimating genealogy from aligned sequences 2-Inferring the population history from genealogy The first step involves estimating a tree topology from sequence alignments. Branch length of the tree should be proportional to the time, calculated either in the units of mutations rate or generations. Phylogenetic errors especially in species with long generation times or loci with low mutation rates is considered in this step. However, estimating the average consensus tree over a large number of trees, particularly in a Bayesian framework, alleviates the uncertainty associated with this step (Drummond, Rambaut et al. 2005). The second step depends on the timing of the coalescent method rather than an exact topology of aligned sequenced. In a sample consisting of n haplotypes the expected time until first coalescence is simply ϒ 2𝑁 = 1 𝑛(𝑛−1)/2 Which with a rearrangement we get: 40 ϒ * n (n-1) /2 = 2N Where ϒ represent the expected coalescence time. These calculations are correct for all intervals (𝑡𝑛 , 𝑡𝑛−1, , … . 𝑡2 ) from leaf nodes until the MRCA. Note that 𝑡𝑛 is time until the first coalescent event occurs in a sample of n sequences. After this event only n-1 sequences will be left and 𝑡𝑛−1 is the time needed until the second coalescent event and so on. Associated with this step is a coalescent error. Coalescence is a stochastic process, any single estimated genealogy represents only a single realization of the event and might not accurately represent the real history of sampled sequences. It has been shown that variance in the height of a coalescent tree is calculated as: 1 𝑉𝑎𝑟𝑇(𝐼) = (Variance in coalescent tree length consisting of K sequences) = 4 ∑𝑘𝑖=2 𝑖 2 (𝑖−1)2 This error is sometimes so considerable that obtaining an inference about the real history of a population may be completely obscured by the stochasticity of the coalescence process, especially in case of short single locus. ABC (Approximate Bayesian Computation) Bayesian statistics is a powerful tool in scientific inference and has been applied extensively in modern population genetics (Marjoram and Tavaré 2006). Coinciding with unprecedented developments in computer power, Bayesian frameworks (Acevedo-Whitehouse, Gulland et al. 2003) provide scientists with unparalleled opportunities to answer many population genetics questions which were difficult or impossible otherwise (Cornuet, Pudlo et al. 2014). Approximate Bayesian methods are based on the fundamental Bayes rule formula: 41 ᴘ (ᴓ|Data) = C. (Data|ᴓ) ᴘ (ᴓ) Where ᴓ is the parameter (or model) of interest and ᴘ (ᴓ) is our prior belief in the parameter of interest (or model). C is a normalizing constant that we ignore for the sake of simplicity. The F (Data|ᴓ) term, the likelihood of the data, is relatively difficult to calculate. A great advantage of ABC method is to replace complex likelihood functions with a stochastic simulation (Tavaré, Balding et al. 1997, Cornuet, Pudlo et al. 2014). A random vector of prior distribution of ᴓ are then sampled. This vector will be accepted if the summary statistics of the sampled vector £ = ( £1 , £2 , £3 , …,£𝑛 ) are close enough to the observed summary statistics: £-£𝑂𝑏𝑠𝑒𝑟𝑣𝑒𝑑< ℰ ℰ is called the acceptance tolerance level. The distribution of the accepted parameter values is an estimate of the posterior distribution of the calculated parameter : £- £𝑂𝑏𝑠𝑒𝑟𝑣𝑒𝑑= =√∑𝑛𝑠𝑡𝑎𝑡 𝑗=1 (𝑆𝑖𝑗−𝑆𝑗(𝑂𝐵𝑆𝐸𝑅𝑉𝐸𝐷))12 𝑉𝑗 Sij is the jth summary statistics calculated from the ith set of simulated data. Si(Observed) is observed summary statistics and Vj is the variance of jth summary statistics across all simulated data set. A linear regression analysis is performed using only the nearest simulated data set to the observed value where the desired parameter is the dependent variable and summary statistics are the independent variables. This regression will be used to calculate the posterior distribution of each parameter. 42 A more simple likelihood equation can also be calculated around observed data which might mirror the distribution of the unknown likelihood formula. Some precision will definitely be lost by using this method but it is still very constructive in the case of complicated likelihood equations. This method assumes that the likelihood is constant for all accepted values which might not be the case. Recently, a new variants of ABC emerged that take the variation in the likelihood of accepted value into consideration (Wegmann, Leuenberger et al. 2010). Excoffier, Estoup et al. (2005) suggests three different steps for initiating any ABC analysis: 1- Generate a large amount of simulated data. 2- Select only part of the simulated data whose differences with the observed data are less than an arbitrary threshold (see above) 3- Estimate the posterior distribution of the desired parameter using a Linear Regression Model. The next step in initiating an ABC analysis is the correct choice of summary statistics that fairly represent the information contained within the genetic data. Choice of summary statistics: A common method of summarizing genetic data is representing them in the terms of mean number of pairwise differences, which is usually labelled π. To calculate π, the number of sites in which each pair of sequences differ are calculated and then divided by the number of such comparisons: π= ∑𝑖<𝑗 𝑑𝑖𝑗 𝑛(𝑛−1)/2 43 Under the infinite sites model, the nucleotide differences between two sequences are exactly equal to the number of mutations in their respective ancestral lineage Ө: Ө = E [𝑑𝑖𝑗 ] Therefore: E[π] = E [ Which is equal to: ∑𝑖<𝑗 𝑑𝑖𝑗 𝑛(𝑛−1)/2 E[π] = ] E [𝑑𝑖𝑗 ] 𝑛(𝑛−1)/2 = ө𝑛(𝑛−1)/2 𝑛(𝑛−1)/2 E[π] = ө This equation shows that the average number of pairwise differences is an unbiased estimator of ө under the neutral coalescence model. This estimator of the ө, whose exact value might never be known, is sometimes called Tajima’s ө ( ө̂ 𝑇 ) after the Japanese population geneticist Fumio Tajima. For more details please see Tajima (1983). Another common summary statistic used in this research was the number of segregating sites (S), which is defined as the count of number of sites which are different in a sample of DNA sequences. The expected number of such a sites can be easily derived from a neutral coalescence model. We have already showed above that the total length of a neutral coalescent tree is equal to: 44 E [Total length of tree] = ∑𝑛𝑘=2 𝐾𝐸[𝑡𝑘] = ∑𝑛2 2𝐾 𝑘(𝑘−1) =2 ∑𝑛−1 𝑘=1 1/𝑘 The infinite site model predicts that the total number of mutation on a lineage of length ᴦ is ө simply r 2. Therefore, the mean number of mutation on a tree with total length of 2 ∑𝑛−1 𝑘=1 1/𝑘 will be: ө E[S] =[2 ∑𝑛−1 𝑘=1 1/𝑘 ] [ ] 2 Rearranging the equation above for ө gives another estimator of ө named after population geneticist G.A Watterson: 𝐸[𝑆] Ө = ∑𝑛−1 𝑘=1 1/𝑘 This equation is summarized as: 𝑆 ө̂𝑠 = ∑𝑛−1 𝑘=1 1/𝑘 Calculating the expected value (E) from both sides of above equation gives: 𝑆 𝐸[ө̂𝑠 ]=𝐸[∑𝑛−1 𝑘=1 1/𝑘 ]=ө 45 It is noteworthy that both of above-mentioned estimators are the best attempt to estimate the real value of the parameter ө, whose exact value might never be definitively known. Both of these estimators are closely related to the mutation rate (µ) and the effective population size 𝑁𝑒 . The reason that I emphasize these two estimators here is that I use these summary statistics to calculate a third summary statistic, which is highly important in population genetics: ̂ . In a neutrally evolving population both estimators of ө should lead to an Tajima’s𝐷 approximately similar estimate of the parameter ө. Any statistically significant differences between these two ө estimators represent the possibility that something complex has happened in the studied population evolutionary history. ̂ as a measure of differences in ө calculated from Fumio Tajima initially suggested Tajima′s 𝐷 any of the above mentioned estimators: ̂=ө ̂𝑇 - ө Tajima′𝑠 𝐷 ̂𝑠 ̂ is more than trivial and depends on factors like selection, population Interpreting Tajima′s 𝐷 demographic history, and mutation rate. Studying selection is not in the scope of this research. ̂ and population demographic history represents However, the correlation between Tajima′s 𝐷 substantial information regarding evolutionary history of the focal populations. It is known that in populations that have gone through a bottleneck, S recovers more rapidly ̂ . We can express this negative number than π, which results in an average negative Tajima’s𝐷 as the result of an excess of haplotypes in very low frequency, which will impact π more than S. 46 ̂ test is more complicated and out of Detailed calculation of the significance level of Tajima𝐷 the scope of current research. Tajima (1989) found a similarity between test statistics and a beta distribution with mean zero and variance 1. He suggested a method based on this similarity for calculating significance level for the test. Later Simonsen, Churchill et al. (1995) developed a more detailed method based on confidence interval and grid search application which has largely been used in a Bayesian frame work to define significance level for test statistics. Several others summary statistics has also been used in current ABC calculations including: the number of haplotypes, the number of segregating sites, the variance of pairwise genetic distances, the number of private segregating sites, the mean of number of the rarest nucleotide at segregating sites, and the variance of the number of the rarest nucleotide at segregating sites. The mean number of the rarest nucleotide at segregating sites has been chosen here because it has been shown that this summary statistic is more sensitive to recent demographic variation (Cornuet, Pudlo et al. 2014). The reason is simple: a recent population expansion will results in many singletons which are sequences that happen only once in the data set. High number of singletons will results in a low value for test statistics. We expect to see opposite trends in the case of recent decline in the population demographic history. Effective population size in Approximate Bayesian framework The effective population size is the number of individuals in the Wright- Fisher model that would produce the same amount of genetic drift as is observed in the real population (Nielsen 47 and Slatkin 2013). Here we emphasize the long term coalescence effective population size, which has largely been used in the current research. To explain I will start with a simple example. Imagine a fur seal population that has a population increase from 2N to 2Nc (c > 0). The coalescence probability would be: 1 2𝑁 + 1 2𝑁𝑐 1 .We know that effective population size is the inverse of the coalescence probability of2𝑁𝑒. P and q are the intervals of time spent in each period. If 1 1 1 𝑝 2𝑁 + 𝑞 2𝑁𝑐 = 2𝑁𝑒 Then Ne= 1 𝑝 𝑞 + 2𝑁 2𝑁 In this case the result is nothing but a harmonic mean of population size! The conclusion is that in rapidly fluctuating population size the long-term effective population size is simply the harmonic mean of the actual population size during the period. Choice of priors in the ABC analysis ABC, as in all Bayesian computational pipelines, is extremely sensitive to the selection of the appropriate priors for estimated parameters Excoffier, Estoup et al. (2005). The famous rule of computational science “garbage in – garbage out” is nowhere more obvious than in Approximate Bayesian statistics. Five demographic priors were carefully selected in the current research: (1) the actual effective population size of the current population, (2) the time of the bottleneck, (3) the size 48 of bottleneck, (4) the size of population before the bottleneck but after arrival of first New Zealand inhabitants; and (5) the pristine population size (pre-human arrival). To estimate the effective population size of the current population an approximately 5x 108 simulations were performed in Bayesian mode of the software package Lamarc (Kuhner 2006). The average Ɵ, was estimated at 0.0297. Assuming a mutation rate from 2.7 x 10−5 to 3 x 10−6, according to Schneider and Excoffier (1999), for complete mitochondrial DNA, the estimated Ɵ could correspond to an effective population size between 1100 to 9900 breeding individuals. We designed the prior for effective population size as a normal distribution with a minimum equal to 1100, a maximum equal to 9900, a mean of 5500, and a standard deviation of 1000. The size of the population bottleneck was considered to be between 2 and 1000 breeding individual’s .The historical population size and pristine population size was assumed to be a uniform distribution with a maximum of 100000 and 200000 individuals respectively A second step, confirmative ABC analysis, was also performed where the posterior distribution of the above parameters were used to select a series of normally distributed priors with a mean set to the medians of the posterior parameters estimated in the first phase. The hypothetical time for the bottleneck was set to 35 generations ago. This prior has some inherent uncertainty. The exact generation time in New Zealand fur seal is poorly known and is between 4 and 7 (see introduction) years in females. Assuming an average 4-5 years generation time for females fur seal (Matthee, Fourie et al. 2006) the prior range coincides with the intensive phase of commercial sealing in New Zealand around 140-175 years ago. 49 The historical size of the population was chosen as approximately 70 generation ago (corresponding to approximately 350 years ago) to coincide before the arrival of the European settlers to the New Zealand to show the impact of early subsistence hunting on this species before onset of commercial sealing. The timing of historical population size was also consistent with earlier suggestion by McGlone (1989) that the most severe modification of the New Zealand wild fauna happened between 750 – 500 years ago when increasing human populations over-exploited many animal species. The pristine population size pre-dates the first reported human settlement in New Zealand (200 generation ago in fur seals, which is approximately 1000 years ago) (Wilmshurst, Anderson et al. 2008, McWethy, Whitlock et al. 2010). The mitochondrial DNA mutation rate was assumed to be between 2.7 x 10−5 and 3 x 10−6, according to the Schneider and Excoffier (1999) double methodology estimates for human mitochondrial DNA genomes, which are the most reliable for whole mitochondrial DNA of a mammalian species. Bottleneck test A model depicting a constant population size with natural fluctuation in size over the past 175 years was evaluated against a model suggesting a recent population bottleneck using mitogenomic data. Each evolutionary scenario was simulated 1 x 106 times and summary statistics were calculated for each model separately. First, a graphical model-checking analysis involving principal component analysis was performed to verify the degree of consistency between selected evolutionary scenarios, designed priors, estimated posteriors and actual observed data. The aim of the principal component analysis was to reduce the original variable space of eight summary statistics to a 50 lower number of non-correlated, synthesized variables and to subsequently better visualize the proximity and correlations of statistical units. To check the posterior probability of each evolutionary scenario two separate methods were used: 1 – A direct estimate 2- A logistic regression estimate. In the direct method the number of times a specific scenario summary statistics was found in a set of n nearest data sets in ABC simulations was assumed to be the probability of that specific evolutionary scenario (Cornuet, Pudlo et al. 2014). In the logistic regression method a polychotomic weighted logistic regression was performed on a set of n nearest data sets in simulations with proportions of each scenario as the dependent variable and the difference between simulated and observed data set as an independent variable (Beaumont 2008). The intercept of the regression was taken as a point estimate for the probability of each scenario. To calculate the confidence in the choice of evolutionary scenario, explicitly the chance of a type I error, which is the probability of rejecting the correct evolutionary hypothesis, a new data set for each scenario was simulated and subjected to the same ABC analysis to calculate their respective posterior probability. The proportion of time that ABC analysis wrongly identified the alternative model (e.g., a constant population model in this case) as having higher posterior value over a bottleneck model was considered as the probability of type I error (rejecting a correct hypothesis in favour of alternative false hypothesis). 51 ABC analysis offers an efficient way to thoroughly assess its performances on the data that have been generated during simulations. To calculate the precision and bias of ABC analysis, pseudo observed data of known parameter values were simulated using the software package DIYABC v2.0.4. The simulated data set mimics the exact configurations of the observed data set. The comparison of real (true) parameter values with those estimated from pseudo observed data subjected to the same ABC analysis can indicate the level of precision in the ABC analysis process. 3.3 Results 3.3.1 Mitochondrial DNA A total of 49 haplotypes were identified from 50 individuals. From 16616 bases, 1288 sites (7.8%) were polymorphic. The mean number of pairwise differences was 244.03 ± 106.14. The mean nucleotide diversity was 0.014 ± 0.007. Tajima ө̂(𝑇) (Tajima 1983) and Waterson ө̂(𝑊) (Watterson 1975) were estimated as 244.03 ± 117.79 and 267.68 ± 73.64, respectively. ̂ ) was slightly, but not significantly, negative (-0.52, p-value= 0.33) (Table3.2). The Tajima’s D (𝐷 sequences are likely evolving according to the Hasegawa, Kishino and Yano (HKY) substitution model. A median-joining network showed two separate clades with a substantial number of missing intermediate haplotypes. A few haplotypes were substantially distinct (>100bp) from others, suggesting the presence of very old lineages and/or possible introgression from another sympatric fur seal species (Figure 3.2). 52 A Bayesian Skyline plot for the mitochondrial genomes calibrated using the mitochondrial mutation rate calculated by Schneider and Excoffier (1999), despite showing a recent population perturbation around 50 generations ago, reconstructed an effective population size of fur seal population that was unrealistically low, implying possible violations of some of the basic conditions necessary for performing Bayesian skyline. A consensus sequence was built using all New Zealand mitogenomes. When the consensus sequence was used in a Table 3-2 Basic statistics calculated from 50 New Zealand fur seals mitogenomes Parameter Statistic No. of transitions 908 No. of transversions 317 No. of substitutions 1225 No. of indels 94 Bayesian phylogenetic tree, including all other otarid species whose complete mitochondrial DNA is available in the public databases, it strongly groups New Zealand fur seals with the Guadeloupe fur seal (Arctocephalus towsendi), rather than the geographically closer South African fur seal (Arctocephalus pusilus ) (Figure 3.3). The genetic distance between A. forsteri and A. towsendi was 5% while the genetic distance between A. forsteri and A. pusilus was 7%. The distance between A. forsteri and Australian and New Zealand sea lions were respectively 6.9% and 6.8%. 53 Figure 3-3 A median- joining network constructed from whole mitochondrial genomes. The different coloured lines (black, green, yellow and red) demonstrate mutation steps between neighbouring haplotypes. Empty circles show median haplotypes while the full circles show the observed haplotypes. The yellow, magenta and green haplotypes were considerably separated from the rest of network. 54 Figure 3-4 A Bayesian phylogenetic tree reconstructed from complete mitochondrial DNA of seven different species of Otaridae. Numbers on each node represent posterior probabilities. The scale bar represents base substitutions in the mitogenomes. The consistency between the selected model, priors, posteriors and observed data was visualised using a graphical model check (Figure 3.4). While the combinations of models and parameters are consistent, the mitogenomic data support a population bottleneck model over a constant size population which incorporates natural fluctuation in the population size up to 10% of its actual size. A direct (see above) estimate of the model posterior probabilities 55 supports a population bottleneck model (P =0.01). The regression model also supports a population bottleneck model (P = 0.007) (Figure 3.5 and 3.6). 3.3.2 Estimating posterior distribution of the parameters A graphical model check was performed on the bottleneck model and showed that the combination of the chosen model and parameters were consistent (Figure 3.7). The ABC simulation phase estimated a population bottleneck size of 95 breeding individuals, a historical population size of 20600 individuals and a pristine population size of 62700. To estimate posterior probability of desired parameters in the ABC analysis phase, 10763 simulated data sets, nearest to the observed value ( approximately 1% of total simulation), were chosen and the posterior distributions for each parameter were estimated. The second phase ABC parameter estimates were consistent with an exploratory phase and show some minor improvements in parameter estimations. Bias and precision were estimated only for the main ABC analysis (Table 3.4). The ABC analysis performed well and captured the true values in the bias and precision analysis (see above). 3.3.3 ZFY data The ZFY Bayesian phylogenetic tree separated the sequences into two completely distinct groups. More surprisingly, the time for coalescence was substantial indicating that more complex evolutionary scenarios are involved in this fragment of Y chromosome evolutionary history (Figure 3.10). Each group showed a high level of genetic variation with its own group. However, the diversity within each group was an order of magnitude lower than between the groups (Table 3.5 – 3.6). This is despite the lack of intra-species genetic variation reported for the same regions in several other pinniped species (Curtis, Stewart et al. 2007). 56 Figure 3-5 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a population that passed through a bottleneck 250 years ago (above) and a population with constant size in the past 250 years (below). The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 80% of the variation in posterior distributions. 57 Figure 3-6 Posterior probability of the bottleneck model (scenario1) and a constant size population (scenario2) calculated from a subset of 500 and 2000 closest simulated data set in a direct estimation and logistic regression method. X axis is subset of closest data set and Y axis is the posterior probability. 58 Figure 3-7 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a population that passed through a bottleneck 150 years ago The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 70% of the variation in posterior distributions 59 Figure 3-8 Posterior distributions of the bottleneck population size , the size of population after first arrival of humans in to New Zealand, historical size and pristine population size of New Zealand fur seals. Note that the numbers on the Y axis are direct counts. 60 Figure 3-8-B Posterior distributions of the second ABC analysis for bottleneck population size , the size of population after first arrival of humans in to New Zealand, historical size and pristine population size of New Zealand fur seals. Note that the numbers on the Y axis are direct counts. 61 Table 3-3 the summary statistics (mode, median and mode) calculated from posterior distribution of the estimated parameters. The bottleneck population size (Nbot), the size of the fur seal population after first arrival of humans in to New Zealand (Nmao) and pristine population size (Npris). Parameter mean median mode nbot 1.62E+02 9.53E+01 4.25E+01 nhis 2.77E+04 2.06E+04 8.64E+03 npris 7.25E+04 6.27E+04 3.40E+04 Table 3-4 Bias and precision of ABC analysis of bottleneck and historical population sizes. N is current population effective size, nbot is the population bottleneck size, nhis is the size of population after first arrival of humans in to New Zealand and pristine population size (npris) correspond to the size of population before human arrival. The true values correspond to the known hypothetical value based up on them the pseudo-simulated data were created. Parameter N nbot nhis npris useq_1 True values Means Medians 5.28E+03 5.31E+03 5.30E+03 5.27E+03 -5.27E+03 5.01E+02 5.82E+02 5.70E+02 5.01E+02 -4.99E+02 5.00E+04 4.79E+04 4.59E+04 4.76E+04 -4.52E+04 1.00E+05 1.22E+05 1.20E+05 1.24E+05 -1.26E+05 1.50E-05 1.56E-05 1.52E-05 -1.50E05 -1.49E-05 Modes 5.29E+03 5.43E+02 2.26E+04 1.12E+05 1.37E-05 62 Figure 3-9 A Bayesian phylogenetic tree for ZFY sequences. The (a) and (b) phylogenetic tree correspond separately to the each clade in the main tree in more detail. Scale bar represent generation in New Zealand fur seals calculated based on Y chromosome mutation rate of 1 x 𝟏𝟎𝟖 (Xue, Wang et al. 2009) 63 Table 3-5 Basic statistics calculated from the first group of ZFY sequences Parameter Number of individuals Number of haplotypes No. of transitions No. of transversions No. of substitutions No. of indels Theta_S s.d. Theta_S Theta_pi s.d. Theta_pi Mean number of pairwise differences s.d.Mean number of pairwise differences Nucleotide diversity s.d.Nucleotide diversity Value 28 26 67 45 112 0 25.6973 8.32936 17.3915 8.87055 17.3915 7.96739 0.02058 0.0105 Table 3-6 Basic statistics calculated the second group of ZFY sequences. Parameter Number of individuals Number of haplotypes No. of transitions No. of transversions No. of substitutions Theta_S s.d. Theta_S Theta_pi s.d. Theta_pi Mean number of pairwise differences s.d.Mean number of pairwise differences Nucleotide diversity s.d.Nucleotide diversity Value 36 28 31 27 58 12.5399 4.03461 6.69841 3.5951 6.69841 3.23443 0.00776 0.00417 64 The BLAST results of all members of these separate groups identified the Californian sea lions ZFY last intron as the best match (Table 3.7 – 3.8). The Californian sea lion (Zalophus californianus) is the closest relative to the New Zealand fur seal for which data is available in Gen bank. A second more precise BLAST were performed using only Californian sea lion ZFX and ZFY and restricting the E-value to zero. The best match for all sequences remained the Californian sea lion ZFY. A Bayesian phylogenetic tree built, using only Californian sea lion ZFX and ZFY and two consensus sequences estimated for each New Zealand fur seal ZFY group, strongly grouped one consensus sequence with Californian sea lion ZFY and, surprisingly, the other consensus sequences with Californian sea lion ZFX (Figure 3.11 ). This pattern was also observed in the neighbour joining tree and reflected in the pairwise distances between four sequences. To compensate for the lack of ZFX data from New Zealand fur seal, all the ZFX data from all other pinnipeds species were collected from public DNA repositories and used as a hypothetical replacement for the missing New Zealand fur seal ZFX data. Almost all of these data were submitted to Genbank by Curtis et al. (2007a) from a single study in which the primer pairs used in this research were first developed. When the New Zealand fur seal ZFY sequences were tested for potential gene conversion between ZFY and ZFX one clade showed several statistically significant putative gene conversion events involving almost all of the pinnipeds from which ZFX data are available, including Californian sea lions. The pattern remained the same when only two consensus sequences from each New Zealand fur seal ZFY group were compared against Californian sea lion ZFX and ZFY. At least four statistically significant putative gene conversion were detected in the sequence length between one of the New Zealand fur seal ZFY groups and the ZFX homologue of Californian sea lion (Table 3.9). 65 The GENECONV program pipeline allows the verification of the results(Sawyer 1999). The software applies a filter to the data by excluding sites that show no variation among them. These constant sites are most likely under high selection pressure (Sawyer 1999). In the next step the algorithm searches for pairwise identical sequences of considerable size that show an unusually high match score (+1 value for each matches and a user defined penalty for each mismatch). It is noteworthy that mismatches could have occurred due to two separate mechanisms: occurrence of mutation after potential gene conversion, and incomplete mismatch repair following a gene conversion event. GENECONV uses an innovative method to calculate significance level for such fragments: first it transfers the alignments in twodimensional array where rows and columns represent the sequences and polymorphic sites, respectively. The highest scoring fragment for both global and pairwise alignment is chosen. An approximately 10,000 permutation is performed on each column containing polymorphic sites. GENECONV selects the pairwise P value as the proportion of selected permuted alignments that the maximal fragment scores as higher than what was originally calculated for these fragments. In addition, the global permutations P value has a built in multiple comparison correction factor according to (Bonferroni 1936) for all sequences in the data set which considerably improves the reliability of data. 66 Table 3-7 BLAST results for the first consensus sequence calculated from New Zealand fur seal ZFY data. 67 Table 3-8BLAST results of the second group of New Zealand fur seal ZFY consensus sequence. 68 Figure 3-10 A Bayesian phylogenetic tree constructed using consensus sequences from each zfy group and zfx – zfy sequences from a closely related species, Californian sea lions. Scale bar represents base substitution in zfy sequences. Table 3-9 Position and length of putative gene conversion sites among New Zealand fur seals group 1 consensus ZFY sequence and Californian sea lion ZFX (Next page). * BC = Bonferroni-corrected KA (BLAST-like) Values 69 Putative gene conversion Zalophus_californianus_zinc_finger_protein_(ZFX) / A. P value BC* P Start End Total number of number of differences value position position Length polymorphic sites in with putative the fragment conversion candidate 0.0002 0.00111 181 232 52 27 0 0.0005 0.00312 39 80 42 25 0 0.0013 0.00522 82 125 44 24 0 0.0062 0.02456 142 178 37 21 0 forsteri ZFY(1) Zalophus_californianus_zinc_finger_protein_(ZFX) / A. forsteri ZFY(1) Zalophus_californianus_zinc_finger_protein_(ZFX) / A. forsteri ZFY(1) Zalophus_californianus_zinc_finger_protein_(ZFX) / A. forsteri ZFY(1) 70 3.4 Discussion Beebee and Rowe (2004) described populations as dynamic entities showing constant changes in their densities and distributions, which have long-term impacts on their viability through continuous changes in their genetic structures. Many pinniped species have experienced bottlenecks of different degrees of severity over the past few centuries, which may have depleted large parts of their historical genetic diversity. New Zealand fur seals mitochondrial data in the current study show high haplotype diversity and low nucleotide diversity, which mirrors the pattern observed in other fur seal species (Lento, Haddon et al. 1997, Baker, Loughlin et al. 2005, Matthee, Fourie et al. 2006, Coltman, Stenson et al. 2007) Mitogenomics data, as found earlier by (Lento, Mattlin et al. 1994, Lento, Haddon et al. 1997), clearly group New Zealand fur seal into two separate clades throughout the range of this species. The two clades that have been identified may correspond to the presence of two population refuges for New Zealand fur seals that escaped commercial sealing and potential post-sealing secondary contacts. It is also possible that the ancient pattern of New Zealand colonization is responsible for the observed structure (Weber, Stewart et al. 2004, Matthee, Fourie et al. 2006). The homogenous distribution of mitochondrial haplotypes throughout New Zealand could be interpreted as a sign that individuals from both of these clades gradually recolonized New Zealand and are regularly found in the same breeding colonies, where they interbreed. The historical extent of gene flow between these two ancient clades is unknown, and was probably only limited by geographical distance separating their distribution areas. 71 Lento, Haddon et al. (1997), using mitochondrial cytochrome b, suggested an evolutionary scenario where two ancient sub species of New Zealand fur seals, one inhabiting mainland New Zealand and Western Australia (A. forsteri forsteri) and other living in the Sub Antarctic islands (A. forsteri snarensis) met and hybridized after cessation of commercial sealing. Our mitogenomic data are consistent with this evolutionary scenario. Few haplotypes in one clade were substantially different from others as found in a study of a shorter region of mitochondrial DNA Lento, Mattlin et al. (1994). It is possible that these different haplotypes are very old lineages that escaped the bottleneck coalescent ‘blackhole’ where other lineages coalesced in their common ancestor. This pattern is observed in other mammals. For example, Wayne, Meyer et al. (1990) found a similar pattern in population of eastern African black-backed jackals where he suggested evolution rate differentiation among individulas. There is also the possibility that these distant haplotypes are the result of hybridization with other fur seal species. Until complete mitogenome are available for all of other Arctocephalus species, especially Sub-Antarctic fur seal (A. tropicalis) and Antarctic fur seal (A. gazella), we can not definitely investigate this hypothesis. The Bayesian phylogenetic tree identified Guadelupe fur seal as a closest relative to the New Zealand fur seal among all other Otaridea species whose complete mitogenomes are available. Both species are sister taxa to a clade including New Zealand sea lion (Phocarctos hookeri) and Australian sea lions (Neophoca cinerea). The geographically closer South African fur seals (A. pusilus) were more distantly related. The Guadelupe fur seal, or extinct close relative are a potential candidate for previous hybridization with the New Zealand fur seal as Lento, Mattlin et al. (1994) speculate. 72 Tajima’s D and the ABC analysis both consistently suggest a population that passed through a sever recent bottleneck. A Bayesian skyline plot failed to reconstruct a coherent demographic history, despite identifying a putative population perturbation coinciding with commercial sealing, as the estimated effective population sizes were unacceptably low. To understand why the skyline plats performed poorly with the current data we need to return to the basic of Bayesian skyline plot methodology. As I explain above Bayesian skyline take advantage of simple relation between the coalescent interval size and the effective population size which is summarized in form of: ϒ * n (n-1) /2 = 2Ne. ϒ is the coalescent interval size and n is the number of sampled genealogies. That the estimated Ne was unrealistically small may be explained as a result of ϒ becoming very small since the number of sampled genealogies n is constant (start from 50 and end at 2, when the last two genealogies finally coalesce into their MRCA). An evolutionary scenario that may give rise to this pattern is when coalescent events happen in short successive intervals that usually occur when the size of population becomes very small (Ho and Shapiro 2011). A visual inspection of the genealogies in the phylogenetic package BEAST tree log file indicated that for the majority of trees the external branches were extremely short with most of the genealogies coalescing almost immediately after or during the bottleneck period, giving a signal of a population with a small effective size of 1000 individuals. The mitogenome data in our research are probably not informative enough for a Bayesian skyline approach and other methodologies that apply combinations of random coalescence simulations and actual genealogy of the sequences. Drummond, Rambaut et al. (2005) has 73 highlighted the requirements for a set of highly informative genetic data for a correct demographic history reconstruction. More importantly, most of the coalescence-based methods, including Bayesian skyline, assume a single, large, interbreeding and panmictic population. The violation of this assumption could have major implications in reconstructing population demographic history (Städler, Haubold et al. 2009, Chikhi, Sousa et al. 2010, Heller, Chikhi et al. 2013). The effect of violating the panmixia assumption and sampling individuals from different populations (demes) is usually similar to a panmictic population that is decreasing in size towards the present (Wakeley 1999, Beaumont 2004, Nielsen and Beaumont 2009, Peter, Wegmann et al. 2010). Wakeley (1999) has identified two major phases for such lineages when one traces them backward in the time. First, there is a “scattering phase “as one starts from time t=0 (present), this phase continues until all sampled lineages either coalesced or migrated in two separate demes. Following the “scattering phase” there is the “collecting phase” where lineages based in different demes migrate to a single deme to be able to finally coalesce. Based on effective migration rate between different demes this phase could result in a very long coalescence interval. This transition between two phases could be interpreted as a decline in effective population size as Heller, Chikhi et al. (2013) call this phenomenon the “structure effect”. Fur seals are known to have structured maternal population dynamics in large geographical scales which are the result of highly philopatric female behaviour (Lento, Mattlin et al. 1994, Baker, ANTONELIS et al. 1995). This population structure will impact on the skyline analysis, possibly making it an inappropriate method to use in this study. The abovementioned structure effect which is the result of assuming an inappropriate panmictic model to the structured population rather than an issue with Bayesian Skyline algorithm per se, made interpretation of the recent population decline in species with highly structured population 74 dynamic, as it is the case of New Zealand fur seals, controversial. The fact that the assumption of panmixia has largely been ignored in the many of researches using the skyline approach may require a re-evaluation of many of studies applying this method (Chikhi, Sousa et al. 2010, Heller, Chikhi et al. 2013). The ABC algorithm is seemingly less sensitive to the violation of the panmixia assumption probably because it is at least partly independent of the true genealogy of sequences. Although the exact nature of the impacts of violating the panmixia assumption on ABC estimates are yet to be determined. Now I take a closer look at the effect of population structure in coalescence genealogy simulation. Start with a simple case of two alleles, each in a separate deme. Genes cannot coalesce until they are both in a single deme. Looking backward in time, there are two independent scenarios possible for these pair of genes: either they coalesce or migrate to another deme. Assume that the rate of mutation from one deme to another is M. Since there are two independent demes in this case the total mutual rate of migration is M+M=2M which means expected time until an immigration event happens is 1 2𝑀 . E [Dif] and E [Sim] represent expected coalescence times when two genes are in different and similar demes respectively. Coalescent and immigration are two independent event and we can add them to get the probability of immigration + coalescent together: 1 1+2𝑀 Note 1 in the dominator in expected coalescence time scaled in unit of 2N which is 1 and 2M is expected immigration rate. When two individuals are in two different demes: 75 1 E [Dif] = 2𝑀 + E [sim] This equation express that in this case first two genes have to immigrate into one deme with 1 rate 2𝑀 plus the standard probability of coalescence when they are in the same deme E [sim]. In the case where both of the genes are in the same deme, looking backward either a coalescence or an immigration could happen which is represented in the form E [sim] = 1 1+2𝑀 2𝑀 + 1+2𝑀 E [Dif]. Solving these equations we get: E [sim] = 2 1 E [Dif] = 2𝑀 +2 For more complicated cases of multi coalescence event with d demes see (Hamilton 2011). It is an interesting finding. it shows that in the case where individuals are in different demes with a low migration rate M (moderate gene flow among demes ) then E [Dif] will become far larger than E [Sim] and therefore result in a longer waiting time until the coalescent event. 3.4.1 Y chromosome zinc finger Genes outside the pseudo-autosomal region (PAR) of the Y chromosome are believed not to recombine with their X chromosome counterparts (Pamilo and Bianchi 1993). These genes are thought to have been genetically isolated for up to 350 million years (Rosser, Balaresque et al. 2009). Coinciding with a higher mutation rate in the Y chromosome, the fate of genes situated 76 in non-recombining regions depends on many inter- and intra-chromosomal processes. These genes might become highly specialized for male function, and, therefore, subjected to strong selection pressure, but it is equally likely that the higher rate of male-driven mutation could degenerate many of these genes through processes such as Muller’s ratchet or hitchhiking. Slattery, Sanner-Wachter et al. (2000) suggested another mechanism, called ectopic gene conversion, through which the Y chromosome might maintain its function in eutherian mammals by the exchange of genetic material with its X chromosome homologues. This ectopic gene conversion is the result of inadvertent homology searches preceding and essential to synapsis of homologous chromosomes (Steele, Morris et al. 1991). Since ectopic gene conversion does not involve the formation of a synaptonemal complex, and reciprocal exchange (Jinks-Robertson, Sayeed et al. 1997, McKim, Green-Marroquin et al. 1998), it is relevant to non-recombining regions of the Y chromosome. This form of recombination could present an adaptive editing mechanism that compensates for gradual degeneration of functional genes situated in non-recombining regions. Ectopic gene conversion seems to be more common in mammals than initially thought. Instances have been reported in pallas cats (Otocolobus manul), ocelots (Leopardus pardalis), tigrinas (L.tigrinas), margay (L. weidii), pampas cats (Lynchailurus colocolo), Geoffroy’s cats (Oncifelis geoffroyi), crab-eating foxes (Cerdocyon thous), and mice (Mus musculus) (Pamilo and Bianchi 1993, Slattery, SannerWachter et al. 2000). Slattery, Sanner-Wachter et al. (2000) reported that the rate of conversion between X and Y chromosomes is approximately 7.4 X 10−8 in Felidae. Similarly, Rosser, Balaresque et al. (2009) found that the rate of X to Y conversion calibrated in units of bases per generation is four to five orders of magnitude faster than the Y substitution rate in humans. The ZFY data in 77 the current research could indicate the presence of a similar mechanism in New Zealand fur seals. To explain the evolutionary pattern observed in ZFY data I suggest the following hypothesis. First , Even though the primer used here has already been shown to be sequence specific (Curtis, Stewart et al. 2007) we cannot completely rule out the possibility of co-amplification of ZFX in some samples. However, co-amplification is unlikely as no single instance of a double bands pattern was observed in the electrophoresis gels. One will expect that in case of coamplification of ZFX that at least some samples would display a double band. Between 40-50 % of all samples failed to show any bands, which expect in the case of co-amplification of ZFX be less since every individuals has an X that should have been co-amplified in that case . Also, ten known females (all lactating, Laureline Meynier, Personal communication) were amplified for ZFY. As expected, in case of specificity of primers, all of these individuals failed to amplify for the ZFY sequence. Even if a group of sequences are in fact co-amplified zfx, the high similarity of these sequences BLAST search to Californian sea lion ZFY, rather than to ZFX, is still surprising and could indicate more complex evolutionary patterns in the history of the New Zealand fur seal ZFX. The second hypothesis is the occurrence of a gene duplication event in the past that resulted in two paralogous copy of the same gene. Each copy subsequently has evolved independently. One copy might later have experienced a conversion event with its homologous ZFX which results in the observed pattern. An instance of ZFX gene duplication has already been documented in the member of the family Muridae (Pamilo and Bianchi 1993). The possibility of the same event in pinnipeds evolutionary history is yet to be explored. 78 A third hypothesis is that both of ZFY groups observed in this research are in fact genuine ZFY sequences. One lineages have had a gene conversion event with its homologous ZFX in the past while the other lineages has preserved the old non-recombined version. Unfortunately, current data do not allow exact determination of the nature of the causal mechanism. A more detailed study of the phenomenon is currently underway in our laboratory using massive parallel sequencing of whole ZFX and ZFY genes (40-60kb). An extensive comparative study of mammalian sex chromosomes suggests instances where all of these mechanism are in action. For example, the Y chromosome may exchange genetic material with its X counterpart through recombination in the PAR. These genes could then migrate to non-recombining segments of the Y chromosome through mechanisms, such as inversion and/or intra-chromosomal translocation (Ellison, Li et al. 1996, Lahn and Page 1997). Impact of first Polynesian subsistence hunting The current data show that subsistence hunting of New Zealand fur seals by Maori, despite having a considerable impacts on fur seals population and reducing the population by about one third, was not detrimental to the existence of the species. This finding is consistent with Lalas and Bradshaw (2001) who suggest subsistence hunting may have resulted in near elimination of fur seals from mainland New Zealand. We also suggest that the impact of Polynesian subsistence hunting was probably highest in the first 100 years of the establishments in the New Zealand. A similar trend has been reported in the dynamics of the extinction of 11 species of the extinct birds Moa (Holdaway and Jacomb 2000) and other species in New Zealand (McGlone 1989). 79 Assuming the same number of breeding males to breeding females might be an over estimation for a species with polygamous mating system. In addition, the polygamous mating system in fur seals could result in a phenomenon where small numbers of breeding males could show an amount of genetic variation usually observed in a population with larger effective population size than actual small census population size. The relationship between variance in the number of offspring and effective population size is known Losos, Baum et al. (2013) and described by the equation: 𝑁𝑒 = 𝑁− ɕ2 1 2 In fact understanding this formula in the light of coalescence theory and basic life story observations is not very difficult. I return to two basic equations of the coalescence theory. First: For the sake of simplicity I start with a sample with two haplotypes: The probability of coalescence is simply: Probability of coalescence = 1 2𝑁𝑒 We ca re-arrange the equation in term of𝑁𝑒 : 1 2 Ne = 𝑝𝑟𝑜𝑏𝑎𝑏𝑖𝑙𝑖𝑡𝑦 𝑜𝑓 𝑐𝑜𝑎𝑙𝑒𝑠𝑐𝑒𝑛𝑐𝑒 80 Now we turn to the model: We can calculate the number of offspring for an individual I with expression∑𝑚𝑖 = 2𝑁 . The total probability that the individual in parent generation sired two 𝑚𝑖(𝑚𝑖−1) random individuals from offspring generations is ∑ =2𝑁(2𝑁−1). We need to consider two other calculations: E [𝑚𝑖 ] = 1 and E [𝑚𝑖2 ] = ɕ2 +1 By slight modification of above formula while taking into account the fact that this formula is realization of original coalescence 1 2 Ne = 𝑝𝑟𝑜𝑏𝑎𝑏𝑖𝑙𝑖𝑡𝑦 𝑜𝑓 𝑐𝑜𝑎𝑙𝑒𝑠𝑐𝑒𝑛𝑐𝑒 We obtain the formula: 𝑁𝑒 = 𝑁− ɕ2 1 2 . If ɕ2 , which is variance in the number of offspring, is less than one, then the relationship 𝑁 between the two parameters will approximate to 𝑁𝑒 ≈ɕ2. This relationship illustrates cases where effective population sizes could be larger than actual census sizes. The correlation between variance in number of offspring and effective populations size has traditionally been used in genetic management of domestic and wild populations (for more details see Guerrant, Havens et al. 2004). I will explain this idea further with a simple scenario. Imagine a fur seal population consisting of 1010 adult males of equal strength, such that no individual could monopolize mating access to females, either because the male was not strong or fast enough to do so or because of the large dispersion of females over a territory, making encounters between rival males 81 uncommon. If each member of the hypothetical male population randomly sire a single pup or by chance no pups at all we will have a variance in offspring sire by each males equal to 0.25 implying that 0.25 Ne = N ! However, if alpha males form harems of up to 10 females and deprive other males from mating access then the variance will be 100 times higher, around 25, implying that 25Ne= N. Of course, this is an extreme case which assumes no limitation in the number of receptive females but less extreme scenarios are likely to have occurred in actual fur seal populations. Male New Zealand fur seals are highly polygamous, such that only a few dominant males around the age of ten form large harems of up to 9 females that exclude other subordinate and recently matured males from the colony (Carey 1989). Excluded males usually form satellite aggregations in the vicinity of the main colony, waiting for rare opportunities to sneak mattings when the dominant males are too occupied to guard all the females. Dominant males have substantially larger body sizes, with high fat content which could have made these individuals attractive targets for commercial and trophy seal hunters. We suggest that eliminating dominant males by hunting provided other subordinate males with more mating opportunities. This pattern results in more sub-ordinate males siring next generation progeny, which in turn decreases the variance in the number of progeny sired by males. Lower density of competing males, as well as elimination of powerful dominant individuals who usually monopolize mating opportunities, could distribute mating opportunities more evenly among the other males. Moreover, lower female population density and their dispersion over a vast area throughout New Zealand could potentially decrease the risk of territorial encounters among males that usually result in the exclusion of subordinate males. 82 A combination of all of these factors could result in a male population that displays an effective population size which is larger than its actual census size. These data show that population demographic dynamics are also subject to the pressures imposed by natural selection, although in a less conspicuous manner, and highlight the adaptive benefits of resilient demographic dynamics in times of environmental stress which help New Zealand fur seals to successfully re colonize New Zealand in around 100 years. This research confirms that the New Zealand fur seal has moved through a severe bottleneck, less than 1% of pre-human arrival population, due to sealing. Variation is not geographically constrained, and two very distinct clades are found throughout the current fur seal range. Haplotype diversity is high consisting with a population that recovering from a bottleneck. Few haplotypes are distantly related to the other haplotypes which could indicate relict lineages or hybridizations with other species. The zfy evolutionary history is more complex and involves more subtle mechanism involved in this part of the genome evolutionary dynamics. 83 Chapter 4 Fine scale population structure and colony expansion dynamics of New Zealand fur seals (Arctocephalus forestri) around Bank Peninsula Summary New Zealand fur seals are one of many pinniped species that survived the 18th and 19th century’s commercial sealing in dangerously low numbers. After the enforcement of a series of protection measurements in the early 20th New Zealand fur seals began to recover from the brink of extinction. Here I report a fine scale population structure of New Zealand fur seals at Banks Peninsula South Island New Zealand using mitochondrial DNA control region. I identify two separate clades in Banks Peninsula. Although the dispersion of genetic variation in the area is uniform, one colony in particular (Otanerito Bay) showed significant Fst value compared to several other colonies in the area. The most abundant haplotype in the area showed a slightly aggregated structure. The Horseshoe colony showed the least number of shared haplotypes with other colonies suggesting a different origin of re colonization of this specific colony. The genetic data support the spill-over dynamics of colony expansion already suggested for this species. Approximate Bayesian analysis suggests recolonization of the area from two main clades across New Zealand with a most likely admixture coefficient of 0.57 to form Banks Peninsula population. The population of fur seals in the area are probably in the early phase of maturity in the colony expansion dynamic. 84 4.1 Introduction The traditional view of a species as a single large, random mating unit is usually challenged when facing actual field observations (Coltman, Pilkington et al. 2003). Many species consist of several sub-populations with different degrees of co-ancestry and relatedness (Sugg, Chesser et al. 1996). In most mammalian species, males are the main dispersing sex. Female philopatric behaviour usually gives rise to matrilineal social groups (Greenwood 1980). Malemediated gene flow, along with female philopatry and polygynous mating systems, are strategies that have evolved to avoid the devastating impacts of inbreeding that can arise when both sexes exhibit philopatric behaviour (Chesser 1991). A high degree of co-ancestry among members of a matrilineal group may also facilitate kin selection and local adaptation with long term beneficial evolutionary consequences (Coltman, Pilkington et al. 2003). Marine environments provide animals with immense dispersal potential, with relatively few barriers to gene flow (Mirimin, Miller et al. 2011). Defining distinct populations in marine ecosystems has a major role to play in species management and conservation. Hoelzel (1998) suggested that in many marine mammals, fine-scale population genetic structure arises from specialization of behaviour, which in turn could facilitate resource partitioning among individuals (Skulason and Smith 1995). Fine-scale population-structure is more pronounced in species with socially structured or breeding-group population dynamics, where matrilocal females in stable social groups mate with males whose associations with females varies from permanent bond to semi-permanent 85 association. This non-random dispersal of individuals in social groups will result in groups that are genetically similar due to co-ancestry, and differentiated from neighbouring groups (Sugg, Chesser et al. 1996, Storz 2005). Such a population structure has been reported for numerous mammalian species including black-tailed prairie dogs (Cynomys ludovicianus) (Chesser 1983), red howler monkeys (Pope 1992), several cercophithecine primates (Schwartz and Armitage 1980, Turner 1981, Dracopoli, Brett et al. 1983, Melnick 1987, Melnick 1987, Kawamoto 1996), bats (Desmodus rotundus) (Wilkinson 1985), Richardson’s ground squirrels ( Urocitellus richardsonii) (van Staaden, Chesser et al. 1994), rabbits (Oryctolagus cuniculus) (Surridge, Ibrahim et al. 1999), lions (Panthera leo) (Spong, Stone et al. 2002), bottlenose dolphins (Tursiops truncates) (Urian, Hofmann et al. 2009), and ungulates (Coltman, Pilkington et al. 2003). Few studies have investigated the pattern of recolonization of an area after an initial disappearance by a mammalian species. Carr, Bowman et al. (2007) found that mustelid fishers (Martes pennati) in southern Ontario, Canada recolonized the area from multiple source populations after initial removal in the 1950s. The multi-source nature of recolonization results in homogenization of genetic variation at local scales. Recently, Bonin, Goebel et al. (2013) in a study using microsatellite data reported the multi-source nature of recolonization of Livingston Island by Antarctic fur seals (Arctocephalus gazella), with obvious population differentiation between Livingston Island and the larger nearby South Georgia Island populations, indicating the contributions of several source populations to the reinvasion history of Livingston Island, rather than a single spill-over event from larger South Georgia colonies to smaller Livingston Island colonies in their vicinities. New Zealand fur seals, Arctocephalus forsteri (Lesson,1828), are a group of pinnipeds that show an extreme polygamous mating system in which just a few males sire most of the 86 offspring in next generation(Crawley and Wilson 1976). This mating system has important impacts on species demographic dynamics and colony expansion patterns. Since end of the large scale commercial sealing of the 18th and 19th centuries, populations of many pinniped species, including New Zealand fur seals, have started to recover from the brink of extinction (Macdonald 2001). Roux (1987) suggested four successive phases for the process of fur seal recolonization: (1) the survival phase, (2) the establishment phase, (3) recolonization, and (4) maturity. Individuals surviving the large scale human harvest initially form a few founding colonies during the establishment phase. This stage is immediately followed by a recolonization phase when shortage of space in the older colonies accelerates the formation of new colonies, primarily in the vicinity of the founder colonies. The process of spilling over of older colonies into new colonies continues until density dependent factors, such as absolute shortage of space on land or food resources at sea, limit the establishment of new colonies and subsequently the colonies enter the maturity phase. Intra-regional fine-scale population genetics of New Zealand fur seals is poorly known. Two studies have previously investigated the broader population structure in this species. Lento, Mattlin et al. (1994) found limited gene flow across the broad geographical range of the New Zealand population. Similarly, Robertson and Gemmell (2005) reported moderate gene flow (an average FST value = 0.017 for all pairwise comparisons) between seven colonies throughout New Zealand. Despite providing valuable information at a broad scale, neither of these studies examined population structure at a local scale, or the consequences of fine-scale population structure for the dynamics of colony expansion. Banks Peninsula is situated in the middle of the east coast of New Zealand’s South Island. Originally formed from two extinct volcanos, Banks Peninsula was isolated by ocean as 87 recently as 15,000 years ago, at the end of last glaciation period, and remained isolated from the mainland until the alluvial plains of Canterbury reached its base and reconnected it to the mainland (McLintock 1966). Different species have colonized the area from different source populations throughout the New Zealand mainland (Banks, Mitchell et al. 2002). Banks Peninsula offers an opportunity to study the fine-scale population genetics of New Zealand fur seals. This area has been re-colonized in the past 40 years (Wilson 1981). Many colonies are even younger and have established in the last decade (Baird 2011). The local bays provide suitable habitat for New Zealand fur seals and the relative timings of recolonizations for major colonies in the area are known. Wilson (1981) was the first researcher to report the re-appearance of four New Zealand fur seals pups, and one female, in haul-outs on the eastern side of Horseshoe Bay on Banks Peninsula in 1973. Despite observing seals at many other locations throughout the area, no pups or females were reported from any of these colonies, which were mainly occupied by immature, sub-adult males. Ryan, Hickling et al. (1997) found that many of the previously identified haul-out sites had become breeding colonies. None of the study sites in our research were reported as breeding colonies or haul-out sites by Ryan, Hickling et al. (1997) except the Horseshoe Bay and Island Bay colonies, which at that time was reported as a non-breeding sites. Baird (2011) was the first to suggest all of the colonies included in our research were wellestablished breeding colonies. Records suggest that many of these colonies were established between 1997 and 2010. The recent establishment history of these colonies could provide an insight into the dynamics of colony expansion in New Zealand fur seals at a local scale . 88 We report here a study of matrilineal population structure and expansion dynamics in New Zealand fur seals on a local scale at Banks Peninsula, in the South Island of New Zealand. Alternative hypotheses of multiple source recolonization events versus a single recolonization event, and a spill-over pattern of colony expansion, previously reported as a potential model for colony expansion dynamics Bradshaw, Lalas et al. (2000), were thoroughly evaluated using genetic data from the mitochondrial DNA control region. 4.2 Material and Methods 4.2.1 Tissue collection Tissue samples were collected from the interdigital web of a fur seal pup’s left fore-flipper using a medium-size piglet ear-notcher (Majluf and Goebel 1992). Between 9 and 10 individuals from all major breeding colonies with more than 50 individuals from around Banks Peninsula were sampled (Figures 4.1 – 4.2). The samples were transferred to 99% ethanol immediately and kept cold until DNA could be extracted in the Molecular Ecology Laboratory at Lincoln University, usually within a few weeks. The whole process was done in accordance with permit 35069-MAR issued by the New Zealand Department of Conservation (DOC). DNA was extracted from tissue samples using a Qiagen DNAeasy Blood &Tissue Kit (Catalogue number 69506) according to the manufacturer’s instructions, and DNA was stored at -20C. 89 Figure 4-1 Locations of the colonies around Bank Peninsula from where samples were sourced. 90 4.2.2 Control region amplification/ analysis A fragment of the mitochondrial control region, approximately 1kb long, was amplified and sequenced using T-Thr, and T-Phe primers as explained in Goldsworthy, Francis et al. (2000). These primers bind to tRNA gene regions on either side of the control region. Sequences were aligned using Clustal W2 (Larkin, Blackshields et al. 2007). Haplotype analyses were performed in Arlequin v.3.5 (Excoffier, Laval et al. 2005) using default settings except where explicitly stated otherwise. A Bayesian phylogenetic tree was constructed using default parameters in the package BEAST v.1.8 (Drummond and Rambaut 2007). Three separate Approximate Bayesian Computation (ABC) analyses were performed using DIYABC v2 (Cornuet, Pudlo et al. 2014). In the first model, it was assumed that the current Banks Peninsula population is descended from a hypothetical founder population of up to 1500 individuals that gradually increased to the current population size. In the second model, the probability of a scenario with a single massive recolonization event was investigated. In the third, and final model, a multi-source origin of recolonization with population expansion in the area was evaluated. To test this model, a homologous region was sequenced from 50 additional fur seals randomly chosen from around New Zealand and the Sub-Antarctic Islands and was subjected to the same Bayesian phylogenetic analysis. Two clades identified by the analysis were considered as representative of the putative source populations for the recolonization of Banks Peninsula. To calculate exponential growth rate(g), the phylogenetic package Lamarc v.2.1.10 (Kuhner 2006) was used in the Bayesian mode. Eight summary statistics were calculated from a single population: (1) number of haplotypes, (2) number of segregating sites, (3) mean pairwise genetic distance, (4) variance of pairwise genetic distances, (5) Tjima’s D, (6) number of private segregating sites, (7) mean frequency of the rarest nucleotide at segregating sites, and (8) the variance of the frequency of the rarest 91 nucleotide at segregating sites. In addition five other summary statistics were calculated from pool of samples: (1) number of distinct haplotypes in the pool sample, (2) number of segregating sites in the pool sample, (3) mean of within sample pairwise differences, (4) mean of between sample pairwise differences, and FST between two samples as explained in (Cornuet, Pudlo et al. 2014). A maximum likelihood analysis adapted from (Choisy, Franck et al. 2004) were performed to estimate the coefficient of admixture of two putative populations that have probably given rise to the current Bank Peninsula population. Principle Component Analyses were performed to test the consistency of simulated and observed data in different models. A mantel test and spatial analyses of intercolony genetic variation was performed on the data set using software package AIS v1 (Miller 2005) to test for possible non-random patterns of genetic variation around the edge of Banks Peninsula. Two separate statistics are of special interest: spatial autocorrelation index and index of allelic aggregation (non-uniform dispersion of the alleles in the area). The program AIS that was used in the current study uses an unconventional algorithm to perform a spatial autocorrelation study. The program estimates the statistic Ay for a user defined distance class y as: Ay = ∑𝑛𝑖 ∑𝑛𝑗>𝑖 𝐷𝑖𝑗𝑤𝑖𝑗 / ∑𝑛𝑖 ∑𝑛𝑗>1 𝑤𝑖𝑗 Where n is number of sampled individuals, Dij is genetic distance between haplotype i and j. wij is a binary matrix which take value of 1 if geographical distance between haplotype i and j fall within distance class y and 0 otherwise. The P value for the test was calculated using a 92 randomization methodology. Allelic aggregation index (Rj) was calculated for a set of N copies of allele j ( 𝑁𝑗 ) sampled from different localities across landscape as: 𝑅𝐽 = 𝑑𝑗 /𝑙 𝑗 Where 𝑑𝑗 is observed average nearest neighbour distance for observation of allele j and 𝑙 𝑗 is calculated as = 1/(2 2√𝑁𝑗 /𝐴). Where A is the size of landscape area. If 𝑅𝐽 = 1 the samples are randomly distributed across the area. When 𝑅𝐽 < 1 samples shows a clumped distribution across the area. Where 𝑅𝐽 > 1 samples show a uniform distribution across the landscape. The uniform distribution is different from a random distribution. P value for each allele will be calculated by randomly re-shuffling genotypes across landscape. 4.3 Results The genetic data identified 56 control region haplotypes from 86 Banks Peninsula individuals. These haplotypes were polymorphic at 100 sites. 46 of the 56 haplotypes occurred only once. A haplotype with 14 counts (16% of the total) was the most abundant haplotype in the region. The mean number of pairwise differences between haplotypes was estimated as 23.53 (SD ± 10.45). Mean nucleotide diversity over all loci was 0.0426 (SD ± 0.021). Tajima ө̂(𝑇) (Tajima 93 1983) and Watterson ө̂(𝑊) (Watterson 1975) were calculated as 23.53 (SD ± 11.58) and 16.12 (SD ± 4.33), respectively. Colonies throughout Banks Peninsula showed different degrees of shared haplotypes. Most of the colonies showed the highest number of shared haplotypes with their adjacent colonies, rather than colonies farther apart. For example, the Damons Bay colony at the entrance of Akaroa Harbour, a busy recreational port, showed the highest number of shared haplotypes with the Otanerito and Island Bay colonies, which are in its immediate vicinity (Table 4.1). Analysis of molecular variance (AMOVA) (Excoffier, Smouse et al. 1992) found that almost all variation was within populations. The null hypothesis of lack of population structure around Banks Peninsula (FST = 0) could not be rejected (P = 0.675 ± 0.014). However, the pairwise FST was significant between Otanerito-Teoka (0.13), Otanerito-Le Bons (0.18), and Otanerito– Horseshoe (0.12) (Table 4.2). 94 Figure 4-2 the haplotype structure in each location in the Banks Peninsula. H1-H56 corresponds to the different haplotypes identified in the area. 95 Table 4-1 Absolute pairwise shared haplotypes among different colonies in Banks Peninsula. Number inside parenthesis represent the number of samples taken from each colony. Teoka Horseshoe Island Damons Otanerito Goughs LeBons Teoka(9) - Horseshoe (9) 1 Island (10) 3 2 Damons (9) 5 1 9 Otanerito (10) 3 0 6 12 Goughs (9) 2 1 5 4 3 Le Bons (10) 3 1 4 4 3 2 West head (10) 4 2 3 4 3 2 5 Long lookout (10) 1 0 2 4 3 1 1 WestHead 1 Spatial analysis of molecular variance (SAMOVA) (Dupanloup, Schneider et al. 2002) also identified the Otanerito Bay colony as maximally differentiated from the remaining eight 96 colonies (FST =0.08) , although the result was not statistically significant when performing a permutation test ( P value = 0.6). Horseshoe Bay colony had the fewest number of shared haplotypes with any other colonies in the area. Two obvious clades within Banks Peninsula were observed in the Bayesian phylogenetic analysis (Figure 4.2). Approximate Bayesian computations analysis was consistent with a population that has formed from admixture of two separate clades from New Zealand mainland and/or Sub Antarctic Island populations. The most likely admixture coefficient from the above mentioned clades to form the Bank Peninsula population was estimated as 0.57. Figure 4-3 A Bayesian Phylogenetic tree of mitochondrial DNA control region from Bank Peninsula. The scale bar show the substitution in mitochondrial DNA control region (Black and grey represent different clades in the area 97 Figure 4-4 Principal Component Analysis (PCA) of the ABC simulations for the mitochondrial data for a model depicting multisource nature of recolonization of the Banks Peninsula.The yellow circle shows the position of the observed data. The X and Y axes respectively illustrate the first and second principal components, which together explain over 50% of the variation in posterior distributions. 98 The logistic regression of the posterior probabilities of ABC analysis strongly rejected both the model with expansion from a small founder population without external recruitment, and the model with a single massive recolonization event, in favour of multi-source re-colonization dynamics (P value < 0.00001; Figure4.5). In fact all estimated summary statistics for the two other models were significantly less than the observed value. The mean value for g (exponential growth rate) as explained in (Kuhner 2006) for the area was estimated at 30.544 consistent with an expanding population. 99 Figure 4-5 Posterior probability of multisource pattern of recolonization of Banks Peninsula (scenario1) and a single massive recolonization (scenario2) and gradual increase from a small population of size 100 (scenario3) calculated from a subset of 2000 closest simulated data set in a logistic regression estimation method. X axis is subset of closest data set and Y axis is the posterior probability. Mantel test results did not show significant correlation between genetic and geographical distances (r = 0.014, P value = 0.35; Figure 4.6). The lack of significant spatial correlation is also obvious in the spatial autocorrelation test (Figure 4.7). However, the allelic aggregation analysis showed that the most abundant allele in the area show a barely significant (P value = 0.06) suggesting slightly aggregated allelic structure. The landscape of the genetic variation show two picks in the southern edge of the area corresponding to the Horseshoe Bay specific allelic structure (Figure 4.8). 100 Mantel Test Results 0.72 0.7 0.68 0.66 0.64 0.62 0.6 0.58 0.56 0.54 0.52 0.5 0.48 0.46 0.44 Genetic Distance 0.42 0.4 0.38 0.36 0.34 0.32 0.3 0.28 0.26 0.24 0.22 0.2 0.18 0.16 0.14 0.12 0.1 0.08 0.06 0.04 0.02 0 0 2,000 4,000 6,000 8,000 10,000 12,000 14,000 16,000 Geographical Distance 18,000 20,000 22,000 24,000 26,000 28,000 30,000 Figure 4-6 the mantel test on mitochondrial DNA control region from Banks Peninsula. X axis is geographical distance and Y Axis is genetic distance. 101 Spatial Autocorrelation Analysis Results 0.078 0.076 0.074 0.072 0.07 0.068 0.066 0.064 0.062 0.06 0.058 0.056 0.054 0.052 0.05 0.048 0.046 0.044 Ay 0.042 0.04 0.038 0.036 0.034 0.032 0.03 0.028 0.026 0.024 0.022 0.02 0.018 0.016 0.014 0.012 0.01 0.008 0.006 0.004 0.002 0 0 2,000 4,000 6,000 8,000 10,000 12,000 14,000 16,000 Geographical Distance 18,000 20,000 22,000 24,000 26,000 28,000 30,000 Figure 4-7 Spatial autocorrelation of the mitochondrial DNA control region from Banks Peninsula. X axis is distance classes and Y axis (Ay) is genetic distance. . 102 5 10 15 20 25 30 35 X (Southern edge) 40 45 50 55 60 5 10 15 20 25 45 40 35 30 Y (Eastern edge) 50 55 60 Figure 4-8 Genetic landscape of mitochondrial DNA control region from Banks Peninsula. X and Y axis are geographic coordinates of the sampled colonies and Z axis is genetic distance among haplotypes. 4.4 Discussion Recolonization of Banks Peninsula by New Zealand fur seals has happened in the past 50 years. Wilson (1981) reported the presence of only 50-100 individuals around Banks Peninsula in 1973, mainly nonbreeding males and immature juveniles. The lack of significant local population structure in the New Zealand fur seal population at Banks Peninsula is consistent with observed patterns reported for other fur seals at other locations ((Matthee, Fourie et al. 2006, Dickerson, Ream et al. 2010, Lancaster, Arnould et al. 103 2010, Berry, Spiller et al. 2012). This finding indicates that the amplitude of matrilineal gene flow among neighbouring colonies is substantial, causing a homogenous dispersion of genetic variation at a local scale. This finding supports the evidence for high natal site fidelity reported for fur seal species in various tagging studies (Baker, ANTONELIS et al. 1995) as uniform distribution of maternal mitochondrial DNA will occur when females are extremely philopatric and their offspring form new colonies in the vicinity of their parent colonies. The observed pattern in the mitochondrial data is consistent with the spill-over theory of colony expansion suggested first by Bradshaw, Lalas et al. (2000). The first founder colonies on Banks Peninsula probably profited from an abundance of vacant spaces and food resources available in the area and grew rapidly in size. At some stage, an absolute shortage of space in a colony forced some breeding females, probably first-time breeders (Roux 1987, Bradshaw, Lalas et al. 2000), to emigrate to suitable habitat in the vicinity of the founder colonies. These newly-established colonies may be further re-colonized, to a lesser extent, by immigrants from other colonies around New Zealand (Taylor, Barton et al. 1995). Accepting this hypothesis requires the assumption of some degree of site philopatry (repeated return to natal sites) and site fidelity (repeated return to non-natal sites), which is not unusual for otariids (Kenyon and Wilke 1953, Trites and Antonelis 1994, Gentry 1998). The spill-over dynamics of colony expansion is obvious in most of the colonies as the highest numbers of shared haplotypes are usually found in colonies situated in close proximity to one another. This pattern is particularly obvious in the Island, Damon and Otanerito Bays colonies. The Island Bay colony is one of the oldest colonies in the region, reported by (Wilson 1981) as a potential haul out site. However, two other colonies in the vicinity of Island Bay (Damons Bay and Otanerito Bay) were not reported as breeding colonies until 2011 (Baird 2011). Interestingly, both of these colonies show the highest number of shared haplotypes with the older Island Bay colony, and an Fst 104 value of zero, which strengthens the idea of spilling over of older colonies in to new colonies in their vicinity. In addition, the allelic aggregation analysis show that the most abundant haplotype in the area which in other term could have corresponded to the oldest lineage in the area still show a slightly aggregated allelic structure. The presence of two well separated haplotype groups around Banks Peninsula strongly supports the hypothesis of the existence of two separate population refuges for New Zealand fur seals that escaped the large-scale human harvest as was initially suggested by Lento, Mattlin et al. (1994). The fact that both of these hypothetical haplotype groups have been identified at a local scale may suggest the random initial re-colonisation of Banks Peninsula by individuals originating from these different clades. The Approximate Bayesian Computation results also strongly support the presence of at least two putative source populations recolonizing the area in the past 50 years. The Horseshoe Bay colony was genetically the most differentiated from other colonies in the area. Most of the haplotypes in this colony are private haplotypes that are found in no other colony. The Horseshoe Bay colony was the first colonies reported from Banks Peninsula after recent re-colonization of the area (Wilson 1981). It is possible that the origin of the first recolonization event in the area was different from subsequent recolonization events. Alternatively, this colony might represent a relict lineage that survived human harvest in hard to access population refuges around Banks Peninsula. The number of haplotypes at Banks Peninsula was also substantial for a population that has passed through a bottleneck and mirrors the pattern observed in other seal species (Lento, 105 Haddon et al. 1997, Weber, Stewart et al. 2004, Baker, Loughlin et al. 2005, Matthee, Fourie et al. 2006, Coltman, Stenson et al. 2007). To what extent the observed pattern reflects presealing demographic dynamics in this species is unknown. A high level of genetic diversity has already been reported for this region. Banks, Mitchell et al. (2002) found that little penguins (Eudyptula minor) around Banks Peninsula originated from populations all around New Zealand and suggested that the particular location of Banks Peninsula in New Zealand makes it an ecological hotspot for marine species travelling through New Zealand waters. The populations of New Zealand fur seals on Banks Peninsula are probably in the early stages of the maturity phase of re-colonization dynamics. There is growing evidence that population genetics parameters may vary through time (Viard, Justy et al. 1997, PIERTNEY, MacCOLL et al. 1999, Garant, Dodson et al. 2000, Nussey, Coltman et al. 2005), for example fixation index estimations are considerably influenced by factors such as dispersal, mating system, and effective population size (Nussey, Coltman et al. 2005). FST values for matrilines in the current study are mostly not significantly different from zero, but this does not definitively rule out the presence of some kind of population structure in earlier stages of the re-colonization of the area or later on when new mutations start accumulating in the recently-established colonies. The level of diversity observed on Banks Peninsula is more or less representative of the overall genetic diversity of the species observed throughout New Zealand. The data also support the idea that there is enough diversity in mitochondrial DNA for this marker to be useful when used in conjunction with high through put computation for more detailed population genetics studies. 106 Chapter 5 A mitogenomic announcement: complete mitochondrial genome of the stoat (Mustela erminae) and New Zealand fur seal (Arctocephalus forsteri) and their significance for mammalian phylogeny Abstract The complete mitochondrial genome of three mustelid species, stoats (Mustela erminae), weasels (Mustela nivalis) and ferrets (Mustela putorius), and the New Zealand fur seal (Arctocephalus forsteri) were sequenced using direct mitochondrial DNA extraction and overlapping long PCRs. The usual 37 mammalian mitochondrial genes (13 protein coding genes, 22 t-RNA and 2 r-RNA) were identified in all four mitogenomes. The divergence of stoats from other members of the subfamily Mustelinae was dated at 4.5 million years ago. The mitogenomic data were consistent with a bear-like origin of seals. Mustelidae are important for understanding the evolutionary history of the pinnipeds (seals and sea lions). Pinnipeds as a monophyletic clade whose closest relatives are ursidae (bears) (Hunt Jr and Barnes 1994, Lento, Hickson et al. 1995, Higdon, Bininda-Emonds et al. 2007)or a diphyletic clades with mustelidae as the closest relative of otaridae. (Flynn, Finarelli et al. 2005, Arnason, Gullberg et al. 2006, Sato, Wolsan et al. 2006). Mustelidae are the most diverse group of the mammalian order Carnivora (Yu, Peng et al. 2011). Mustelidae comprises eight subfamilies that live in all five continents, although their occurrence in Australasia is the result of recent human introduction (Nowak 1999). The evolutionary history of this group is characterized by a rapid early radiation and several recent speciation events (Koepfli and Wayne 1998, Marmi, López‐Giráldez et al. 2004), which make phylogenetic inferences concerning the evolutionary history of this family challenging. 107 Three species of Mustelidae, stoats (Mustela erminae), weasels (Mustela nivalis) and ferrets (Mustela putorius) were introduced into New Zealand in the 1870s-1880s to control expanding populations of rabbits that were damaging farming industries at that time (King 1989). Unsurprisingly, the mustelids soon began impacting the native fauna, especially bird species (King 1990). The devastating impacts of introduced mustelid species continues, and large scale ecosystem management operations, in the form of active control of these species, are now indispensable for the survival of many vulnerable native species (King 1989). I de novo sequenced and assembled whole mtDNA from three introduced mustelid species in New Zealand and the New Zealand fur seal. These data were used in a Bayesian phylogenetic framework to highlight the phylogenetic position of stoats in the Mustelidae family. The divergence time of these three species from other members of Mustelid family was estimated. I also investigated pinniped evolutionary history in the light of the new mitochondrial DNA. This study is the first mitogenome assembly for stoats. Ferret and weasel assemblies from their native distribution range have already been submitted to the NCBI (Yu, Peng et al. 2011), but these are the first whole mitochondrial sequences for these species from outside their native range. New Zealand fur seal mitochondrial DNA (mtDNA) has been submitted to the NCBI (Lin, McLenachan et al. 2002), but despite being labelled as a complete mitochondrial genome, it was missing approximately 1kb of sequences. This assembly is, therefore, the first complete mtDNA sequence for this species. Mitochondrial DNA was extracted from liver tissue from three dead mustelid specimens and followed the protocol of Williams, Foster et al. (2014) with minor modifications : 1- Cut a piece (25-100 mg) of liver Tissue from Ferret, Stoat and Weasel from a big piece of liver Tissue preserved in Ethanol 95%. 2- Following Williams et al, first press the liver tissue onto an absorbent tissue to remove the excess of ethanol. 108 3- Immerse the tissue in 1 mL of PBS for 10 min, keeping it on ice. 4- Remove the tissue from the buffer, pressed against absorbent tissue, placed over a cover slip on ice, cut it into small pieces and transfer to a dounce homogeneizer (1 mL Wheaton), with 1 mL of chilled PBS. 5- Homogenized the tissue with 30 strokes (or until no more pieces of tissue were seen), keeping the homogenizer on ice. 6- Transfer the homogenate to a 1.5 ml microcentrifuge tube. 7- Centrifuged at 1,000 x g for 15 minutes at 4 °C (nuclei and cellular debris will precipitate). 8- Transfer the supernatant to a 2ml tube and keep it on ice. 9- Re-suspend the nuclear pellet in 600 μl of PBS. 10- Centrifuged at 1,000 x g for 10 minutes at 4 °C. 11- Discard the nuclei and cellular debris pellet. Combine the supernatants. 12- If cellular debris are contaminating the combined supernatant, centrifuge this last one at 1,000 x g for 10 minutes at 4 °C. 13- Transfer the clear pellet to a new 2 ml tube and centrifuged at 12,000 x g, 30 minutes at 4 °C. 14- Re-suspend the mitochondrial pellet in 180 µl of 1X Reaction Buffer (Epicentre) of the Plasmid-Safe ATP-Dependent DNase enzyme (Epicentre). (Prepare the 1X Reaction Buffer with PBS, to keep the isotonic solution). 15- Add 8 µl of ATP (25 mM, Epicentre), and 3 µl of the Plasmid Safe DNase (Epicentre). 16- Mix gentle and incubate for 1 h at 37°C. 17- Centrifuged at 12,000 x g, 30 minutes at 4 °C. 18- Re-suspend the mitochondrial pellet with 180 μl of ATL buffer (Qiagen). 19- Incubate 30 min at 70°C to denature the Plasmid Safe DNase 20- Add 20 µl of proteinase K (Qiagen) and incubated O.N. at 56°C. 21- Add 200 µl of AL buffer (Qiagen) and incubate 10 min at 70°C. 22- Add 200 µl of Ethanol. Mix gentle. 23- Transfer everything through mini columns, 600 µl at a time 109 24- Centrifuge at 6,000 x g, 1 min. 25- Wash the mini columns with 500 µl of AW1 buffer (prepared fresh. Qiagen). Wait 2 minutes. 26- Centrifuge at 6,000 x g, 1 min. 27- Wash the mini Wash the mini columns with 500 µl of AW2 buffer (prepared fresh. Qiagen). Wait 2 minutes. 28- Centrifuge at 6,000 x g, 1 min. 29- To dry the mini columns, centrifuge at 10,000 x g, 1 min twice, rotating the columns 180°. 30- Elute the DNA with 10 µL of Elution Buffer (Qiagen) pre warmed at 65°C. Wait 5 min. 31- Centrifuge at 6,000 x g, 1 min. 32- Repeat steps 30 to 31. 33- Measure by absorbance at 260 and run a 0.8% agarose gel. This was a two day protocol, starting at noon on the first day, and finishing at noon on the following day. Primers were designed to amplify New Zealand fur seal mtDNA in 3-6 kb fragments from genomic DNA with approximately 500 bp overlap. DNA libraries were constructed from each specimens using an Illumina Nextera kit and sequenced at 120 X coverage on an Illumina Miseq sequencer following the manufacturer’s instructions. Whole mtDNA sequences were assembled using CLC Genomics software package v7.5 (CLC Bio), using default parameter settings. The stoat and New Zealand fur seal assembled mitochondrial genomes were annotated using the MITOS metazoans annotation pipeline (Bernt, Donath et al. 2013). The MITOS pipeline identified the usual 13 mammalian protein coding genes, 22 tRNA genes and 2 rRNA genes in both mitochondrial genomes (Table 6.1 – 6.2 and 6.3). 110 Complete mitochondrial genomes from two seal species (Arctocephalus forsteri sequenced in current research and Leptonychotes weddellii), eleven mustelid species (Martes martes, Martes foina, Mustela atlantica, Mustela erminae, Mustela putorius, Mustela nivalis, Mustela nigripes, Mustela sibrinica, Mustela kathiah, Mustela frenata, and Lutra lutra), three New Zealand mustelid species sequenced in this research, and two bear species (Ursus maritimus and Ursus arctos) were used to construct Bayesian phylogenies using BEAST v2 (Bouckaert, Heled et al. 2014). To calculate the divergence dates for the three mustelid species, nodes were calibrated using time estimates for the most recent common ancestor of all the Mustelinae and Martinae, and the common origin of Mustelinae and Lutrinae, based on the fossil record (Yu, Peng et al. 2011). To calibrate the age of the Mustelinae and Lutridea node, the oldest Lutridae genera were considered: Kenyalutra, Potamotherium, Mionictis, and Paralutra. Paralutra has been suggested as the oldest Lutrinae lineage Sato, Hosoda et al. (2003). Fossil records dated this genus to the middle Miocene in the European Neogene Land Mammal Zone MN 7+8, approximately 11.1 to 13.5 million years ago (Steininger 1999). The origin of the Martinae was estimated based on the fossil records of Martes wenzensis (Anderson 1970) which is dated to the Pliocene deposits of Weze 1, Poland (Steininger 1999) around 3.3– 4.0 million years ago (Glazek, Sulimski et al. 1976). The resulting log file was visualized after 5 x 108 Markov chain Monte Carlo simulations in the software package Tracer v 1.5 (Rambaut and Drummond 2003). The tree file was summarized using the Tree annotator software included in the BEAST package and visualized in the software package Fig Tree v1.4.2 (http://tree.bio.ed.ac.uk). The mustelids were found to be monophyletic, with M. frenata and M. kathiah diverging early. The mitogenomic sequences dated the split of the stoat (M. erminae) from other mustelid 111 species at approximately 4.7 (95% HPD 3.2-5.8) million years ago. Around 2.5 (95%HPD 1.2 – 3.7) million years ago M. atlantica and the weasel (M. nivalis) diverged from other members of the mustelid family. The ferret (M. putorius) diverged from M. nigripes 1.06 (95% HPD 0.35 – 2.10) million years ago. The New Zealand fur seal and Weddell seal were also monophyletic and most closely related to the bear species rather than mustelids Data strongly supports a bear–like ancestry for the modern pinnipeds. The data dated the common ancestor of the Otarioidea and Phocidae at almost 30 million years ago ( 95% HPD of the node height 25 34.5) is consistent with Hammond, Hauton et al. (2012). The mitogenomic data reported here consolidate our knowledge of the evolutionary history of the Mustelidae and the origin of modern pinnipeds. 112 Figure 5-1 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent posterior distribution and scale bar is time in millions of years. 113 Figure 5-2 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent estimated divergence date (Millions years ) and scale bar is time in millions years.Figure 5-3 A Bayesian Phylogenetic tree constructed for eleven members of Mustelidae, two member of Ursidea and two pinnipeds (New Zealand fur seal (Arctocephalus forsteri) and Weddell seals (Leptonychotes weddelii)). Numbers of each node represent posterior distribution and scale bar is time in millions of years. 114 Table 5-1 Annotation of the protein coding genes in New Zealand fur seal complete mitogenome. Name Start Stop Strand Length nad1 nad2 2754 3921 3704 4949 + + 951 1029 cox1 cox2 5354 7039 6886 7722 + + 1533 684 atp8 atp6 7794 7955 7991 8629 + + 198 675 cox3 nad3 8635 9489 9417 9833 + + 783 345 nad4l nad4 nad5 nad6 cob 9905 10195 11795 13587 14182 10198 11562 13573 14105 15315 + + + + 294 1368 1779 519 1134 115 Table 5-2 Annotation of the tRNA genes in New Zealand fur seal complete mitogenome. Name Start Stop Strand Length trnF(gaa) 1 71 + 71 trnV(tac) 1032 1098 + 67 trnL2(taa) 2676 2751 + 76 trnI(gat) 3710 3779 + 70 trnQ(ttg) 3777 3850 - 74 trnM(cat) 3852 3920 + 69 trnW(tca) 4963 5030 + 68 trnA(tgc) 5041 5109 - 69 trnN(gtt) 5111 5183 - 73 trnC(gca) 5218 5284 - 67 trnY(gta) 5285 5352 - 68 trnS2(tga) 6896 6964 - 69 trnD(gtc) 6972 7038 + 67 trnK(ttt) 7724 7792 + 69 trnG(tcc) 9419 9488 + 70 trnR(tcg) 9836 9904 + 69 trnH(gtg) 11573 11641 + 69 trnS1(gct) 11642 11700 + 59 trnL1(tag) 11701 11770 + 70 trnE(ttc) 14109 14177 - 69 trnT(tgt) 15322 15393 + 72 trnP(tgg) 15393 15458 - 66 116 Table 5-3 Annotation of the rRNA genes in New Zealand fur seal complete mitogenome. Name Start Stop Strand Length rrnS 72 1031 + 960 rrnL 1097 2675 + 1579 117 Chapter 6 Identifying prey and parasite items from New Zealand fur seal faeces using massive parallel sequencing Abstract The New Zealand fur seal (Arctocephalus forsteri) in one of many pinniped species that has shown a remarkable recovery from the brink of extinction after cessation of commercial sealing during the 19th century. It is commonly believed that this species competes with recreational and commercial fisheries. I identified prey and parasites items from New Zealand fur seal faecal samples using massive parallel sequencing across the species distribution. The data support generalist feeding behaviour for this species. The diet composition showed significant geographical and inter-seasonal variation. As many as 46 species of fish and 18 species of cephalopod and 5 parasitic worms were identified from a single colony. The data suggest cartilaginous species (sharks, rays and skates) constitute an important part of the New Zealand fur seal diet. Approximately 10% of the species identified in the seal diet are of significant commercial value and indicates limited qualitative food competition between New Zealand fur seals and commercial fisheries in exploiting marine species. 6.1 Introduction A thorough knowledge of a species’ feeding behaviours is the first step to studying the ecological roles it plays in an ecosystem (Deagle, Jarman et al. 2005). Understanding the position of an animal in a food web requires detailed knowledge of its food and feeding habits (Pauly, Trites et al. 1998). Oceans are a vast source of food diversity, from planktonic plants and animals, to fish and large marine mammals (Riedman 1990). Predators, including humans, 118 depend on the ocean’s food resources to fulfil their energetic requirements. Among marine animals, mammals consume a wide array of different species from all trophic levels (Bowen 1997). While sirenians (dugong and manatees, collectively known as “sea cows”) are mainly grazers of marine plants at the base of food webs, other predatory mammals, like leopard seals (Hydrurga leptonyx), killer whales (Orcinus orca), and polar bears (Ursus maritimus), prey on marine birds and mammals. Pinnipeds (true seals, fur seals, sea lions and walruses) are the top predators in many marine ecosystems. Studies of pinniped ecology provide ecosystem managers with information for monitoring the health and dynamics of marine ecosystems (Boyd and Murray 2001, Reid and Croxall 2001, Arim and Naya 2003, Lundström, Hjerne et al. 2010). The number of fish species consumed by marine mammals is unknown. Trites, Christensen et al. (1997) used population size, total biomass, and daily consumption rate to estimate that the 84 species of marine mammals inhabiting the Pacific Ocean would have consumed about three times as many fish as humans harvested from this region between 1940 and 1990. Catch statistics from the Food and Agriculture Organization (FAO) show that the total catch by humans in Oceania has increased from 77,030 tons in 1950 to around 1,267,031 tons in 2012, a 16-fold increase in just over 60 years. Total catch made by humans in this area has remained around 1.0-1.3 million tons since 1997 (except for two record years of 2004 and 2005). A similar trend can also be seen in the global catch statistics, which have remained around 90 million tons since 2005. This plateau may be due to severe depletion of global fish stocks, or the reaching of limitations of the current techniques used in fishing. After two centuries of over-exploitation, the populations of many species of marine mammals, including pinnipeds, are recovering from near extinction (Macdonald and Norris 2001). Although a large proportion of most marine mammal diets are known to be species, such as deep sea squid or small fish species that are of limited commercial value (Trites, Christensen 119 et al. 1997), there is little doubt that marine mammals do consume species of commercial value, and compete with commercial species in the consumption of mutual prey (food web competition). Trites, Christensen et al. (1997) suggested that pinnipeds, with 60% overlap of species in common with those taken by human fishing industries, rank ahead of dolphins and porpoises in competing with fishing industries. Expansion of human fishing areas, and inclusion of new species in the long list of commercial species, will definitely increase the extent of current competition between humans and marine mammals (Trites, Christensen et al. 2006). Following severe population decline in the 18th and early 19th, the recent increase in the population of New Zealand fur seals (Arectocephalus fosteri) has raised concerns in the fishing industry, and among coastal inhabitants, concerning the potential conflict with commercial interests and human health (Street 1964, Boren 2010). It is commonly believed that fur seals that inhabit inshore waters compete with recreational and industrial fisheries (Carey 1992). The extent of such conflict in New Zealand waters is yet to be comprehensively studied (Lalas and Bradshaw 2001). Having a precise and comprehensive knowledge of fur seal diets is essential before conservation and management issues can be considered. New Zealand fur seals appear to feed mainly on fish and squid (Crawley 1990, Fea, Harcourt et al. 1999, Harcourt, Bradshaw et al. 2002). Some populations in the southern part of the species’ distribution are known to prey on penguins (Bailey and Sorensen 1962) and this may be common in some areas (Riedman 1990). The New Zealand fur seal diet has traditionally been studied by analysing diagnostic hard parts (e.g., flesh, skeleton, sagittal otoliths, cephalopod beaks) directly from stomach contents, faeces, and regurgitates (Boren 2010 for a complete review). Scats and regurgitates are relatively abundant and the non-invasive nature of sample collection minimises disturbance in the colony compared to other invasive or lethal methods. Teleost sagittal otoliths and cephalopod beaks are typically recovered from 120 these sources (Dellinger and Trillmich 1988). However, there are several potential difficulties with reconstructing seal diets from stomach contents, faeces and regurgitates. First, Gales and Cheal (1992) showed that in the Australian sea lion (Neophoca cinerea), cephalopod beaks tend to cluster near the pyloric sphincter for a few days before being regurgitated out of the body, which will bias the sample. Second, even though New Zealand fur seals are known to swallow small prey whole, larger prey are often broken into smaller, more manageable pieces. Hard part analysis methods rely mainly on fish sagittal otoliths; for a fish to be included in a diet reconstruction therefore, the seal has to have eaten the fish’s head, which will bias diet reconstruction against larger fish that could be an important part of the diet. Species that do not have sagittal otoliths, or those that hardly ever leave any hard parts behind (e.g. cartilaginous fish such as sharks, rays, and skates) may be missed using these methods. Differential recovery and digestion of diagnostic hard parts hinders precise diet reconstruction (Murie and Lavigne 1985, Gales and Cheal 1992, Fea and Harcourt 1997). In captive breeding trials, between 0% and 89% of fish otoliths were recovered from a controlled feeding trial. Otolith digestion resulted in a 16-51% underestimation of fish length (Tollit, Steward et al. 1997) and a 35% underestimation of fish mass (Dellinger and Trillmich 1988). An alternative to the traditional method of diet analysis is the application of DNA-based methods to DNA extracted from stomach contents, faeces and regurgitates. Since their advent in 1992 (Höss, Kohn et al. 1992), a variety of DNA-based methods have been used to study feeding behaviour and trophic dynamics of a range of organisms in different terrestrial and marine ecosystems. Early applications of these methods used electrophoresis or cloningsequencing to identify prey species. Deagle, Jarman et al. (2005) studied the diet of giant squid (Architeuthis dux) from gut contents of captured and stranded individuals. Purcell, Mackey et al. (2004) and (Parsons, Piertney et al. 2005) used a DNA-based method to identify salmonid species in seal faeces. 121 Development of massively parallel sequencing in 2005 revolutionized the application of DNAbased methods in ecological studies. In one of the first studies of its kind Deagle, Kirkwood et al. (2009) used this newly available technology to successfully reconstruct the diet of Australian fur seals (Arctocephalus pusillus) and little penguins (Eudyptula minor) (Deagle, Chiaradia et al. 2010). Later, Bohmann, Monadjem et al. (2011) used a DNA-based method to study the diet of African free-tail bats (Molossidae). (For a full history of the method see (Pompanon, Deagle et al. 2012).) Any DNA fragments that survive the digestion process can be used for diet reconstruction (Symondson 2002). Captive breeding trials and field studies show that DNA-based methods reconstruct animal diets more consistently than traditional hard parts methods, especially for species with cartilaginous skeletons (Casper, Jarman et al. 2007, Deagle and Tollit 2007). Prey with larger body sizes, or those that are more frequently eaten, are likely to contribute proportionately more DNA to the total faecal DNA (Deagle, Kirkwood et al. 2009), which may provide semi-quantitative dietary data (Deagle and Tollit 2007). DNA-based methods are more sensitive for identifying rare prey items (Purcell, Mackey et al. 2004, Deagle, Tollit et al. 2005, Deagle, Kirkwood et al. 2009). Barros and Wells (1998) showed that the diversity of prey items recovered from five faecal samples is equivalent to 16 stomach contents in bottlenose dolphins (Tursiops truncatus) in Sarasota Bay, Florida. Deagle, Kirkwood et al. (2009) identified 35 prey species in 90 seal faecal samples, which was 2.5 times more than the total number of species found by hard part analysis of 490 faecal samples over three years in the same area. However, the application of faecal DNA methods to field studies depends on several key issues including, but not restricted to, (1) the length and copy number of DNA that can be extracted and amplified from faeces, (2) elimination of contamination, (3) prevention of DNA degradation in the field and laboratory, and (4) removal of dietary inhibitors (Wasser, Houston et al. 1997). Designing appropriate primers to amplify informative markers from prey DNA is 122 a challenging task, particularly for generalist predators for which little or no a priori knowledge is available. Another major problem that must be considered is PCR errors and their effect on species assignment (Pompanon, Deagle et al. 2012). This study is the first application of a DNAbased methodology to identify prey species of New Zealand fur seals. Here the emphasis is on colony-level diet reconstruction, which will provide marine ecosystem managers with a level of resolution necessary for most conservation plans. 6.2 Material and Methods Fresh, moist faecal samples were collected between 2012-2014 from sites in the North Island: Bay of Plenty (37°44'28.69"S, 176°32'30.40"E ), Cape Palliser ( 41°35'21.31"S, 175°17'12.72"E); South Island: Kaikoura (42°13'57.73"S, 173°50'30.91"E), Sandymount, Otago Peninsula (45°52'48.79"S, 170°44'41.58"E), Breaksea Island, Fiordland (45°34'37.58"S, 166°38'18.26"E ); and Codfish Island, near Stewart Island (46°46'25.27"S, 167°37'55.68"E). The collection locations and timing (winter- summer) were chosen to cover the full geographical range of New Zealand fur seals throughout the year Whole scats were transferred immediately to 99% ethanol and kept cold (-4°C) until DNA could be extracted in the Molecular Ecology Laboratory at Lincoln University, usually within one month. DNA from prey species is known to be heterogeneously distributed within faecal samples (Matejusová, Doig et al. 2008), therefore, samples were mixed thoroughly using a disposable paint stirrer mounted on an electric drill. Whole genomic DNA was extracted from 5-10 g of faecal slurry using MO BIO Power Max Soil (Catalogue number 12988-10). DNA was preserved at -20°C until downstream analysis could be performed. Samples from each colony were pooled prior to library construction. Libraries were prepared from pooled DNA samples as follow: four PCRs were performed on each pool of extracts amplifying different parts of the 123 mitochondrial and nuclear genomes using primers already developed (Deagle, Kirkwood et al. 2009). Primer set A (F_ CGAGAAGACCCTRTGGAGCT, F_GACGAGAAGACCCTAWTGAGCT,R_GGATTGCGCTGTTATCCCT, R_AAATTACGCTGTTATCCCT) amplifies 16S mitochondrial DNA from both Chordata and Cephalopoda. Primer set B (F_CGAGAAGACCCTRTGGAGCT, R_CCTNGGTCGCCCCAAC) amplifies a shorter region of Chordata 16S mitochondrial DNA. Primer set C (F_AAAAGAAACCAACCGGGATT, R_CAAGCAACCCGACTCTCG) amplifies a region of nuclear 28S rDNA from Cephalopoda. Primer set D (F_AGAGGTGAAATTSTTGGAYCG, R_CCTTTAAGTTTCAGCTTTGCA) amplifies a region of nuclear 18S rDNAfrom all Bilateria. Each PCR reaction consisted of 0.25 µM of each primer, 4µL of template DNA, and PCR master mix. Thermal cycling conditions were: 95°C for 15 min followed by 15 cycles of: 94°C for 20s, a primer specific annealing temperature (52°C for primer set A, 57°C for primer sets B and D, and 55°C for primer set C) for 90s, and 72°C for 45s. This was followed by a final extension at 72 °C for 2 min. The PCR products were cleaned up using QIAquick columns (Qiagen) prior to the second PCR step. In the second step, primers with additional sequences corresponding to Illumina Nextera XT adaptor sequences were used in five more rounds/cycles. Only a portion of the first step was used as a template in the second step. The second step PCR was column-purified using a QIAquick kit. The third step involved using Nextera XT indexing primers and 12 more cycles/rounds. Only 1 ng of second step product was used in the indexing reaction. After the indexing PCR, samples were cleaned up using AMPure XP beads (Beckman Coulter Genomics). The samples were quantified and normalized using Life technologies Qubit® Fluorometric Quantitation method prior to sequencing on an Illumina MiSeq usin SE X 250 chemistry , following the manufacturer’s instructions. The resulting sequences were adaptor trimmed and low quality sequences filtered. Filtered sequences were searched against the NCBI database using the program KLAST, which is known to be faster than BLAST for comparing large data 124 sets (Lavenier 2009). The three best hits for each sequence were chosen. An in-house Python script was used to search the KLAST output files for the three best matches against a list of New Zealand fish and cephalopod species (Gordon 2009). In most cases, the first three hits allowed conclusive assignment of sequences to their species of origin. In the cases where the first three hits shared the same parameter values, the hit was considered inconclusive and discarded from further analysis. The exceptions were instances where the first three hits had the same values, but only one or two of the assigned hit species were known from New Zealand waters. In those cases, the hit was considered to have originated from New Zealand species, even though the sequence was not completely conclusive. Uncertainty exists regarding sequences where only the third hit matched species known from New Zealand waters. There is a remote possibility that the fourth hit and beyond might have had the same values as the third hit, but were ignored due to the design of the experiment and selection algorithm. For example, a group of sequences were found to match the giant squid (Architeuthis dux) as the third best hit (the first and second hits were species not known from New Zealand waters). There is a possibility that one of the hits beyond the third match is the real origin of the sequence. Given the exploratory nature of this study I assigned such sequences to their origin less conservatively to include a larger set of potential prey species for future studies. 6.3 Results The faecal samples resulted in a total of 3383340 unique reads that passed all quality filters. 125 6.3.1 Stewart Island At Stewart Island, a total of 21 species of fish and 14 species of cephalopods were identified (Table 6-1). With a few exceptions, the recovered DNA could usually be assigned to a particular species or genus. One group of sequences matched Salmo salar; I believe that these sequences are likely the results of predation on King salmon (Onkorhyncus nerka), which is a known aquaculture species in the area, with a recent escape of around 80,000 individuals in to the ocean (Brent Beaven, New Zealand Department of Conservation , pers. comm.). Two groups of sequences matched the freshwater fish species Cyprinus carpio and Perca fluviatilis. The origin of these species is unknown but they might be the results of direct, or more likely secondary, predation on these species. They are known to be favourite prey items of salmonid species. Alternatively, these sequences might come from unknown (or at least un-sequenced) fish species whose closest matches were these species. The DNA fragments were also unable to differentiate between two closely related shark species, blunt nose six-gill shark (Hexanchus griseus), and broad nose seven-gill shark (Notorynchus cepedianus). One cestode and one nematode species were identified from this colony. 6.3.2 Fiordland At Breaksea Island, 46 species of fish and 18 species of cephalopods were identified from faecal samples and regurgitates. Except for inconclusive assignment of Hexanchus griseus and Notorynchus cepedianus, the remaining species were assigned with acceptable levels of certainty. Three species of freshwater fish were also identified, whose origin remains unknown. Fiordland was the most diverse colony regarding seal parasites. Four species of parasitic nematode and one cestode species were identified from faecal samples in this colony. 126 6.3.3 Kaikoura Samples collected from Kaikoura in winter suggest the presence of 24 species of fish and 12 species of cephalopods. 24 species of fish and 15 species of cephalopods were identified in the same site in summer. The presence of rainbow trout (Oncorhynchus mykiss) is not surprising given the widespread distribution of this non-native species in New Zealand, even though the exact circumstances of predation remain unresolved. The presence of the brook char (Salvelinus fontinalis) in Kaikoura in summer is more of surprise and I cannot definitively rule out the possibility that this species might be an artefact of the assignment algorithm. Four species of parasitic nematode and one cestode species were identified from faecal samples in this colony. 6.3.4 Cape Palliser There were 20 species of fish and 12 species of cephalopods identified from samples collected from Cape Palliser. Even though the samples were far fewer and more degraded than the rest of the study sites, the success rate was substantial. Three species of parasitic nematode and one cestode species were identified from faecal samples in this colony. 6.3.5 Bay of Plenty At the Bay of Plenty colony, 29 species of fish and 8 species of cephalopods were recovered. Cyprinus carpio and/or Tinca tinca occurred at this colony and were also identified from Kaikoura in winter and Cape Palliser. The lack of these species in Kaikoura summer diet, despite the presence of salmonid species, adds to the uncertainty surrounding identification 127 of the origin of these freshwater species. Three species of parasitic nematode and one cestode species were identified from faecal samples in this colony. Cape Palliser and Kaikoura in winter show the highest species overlap for all of the colonies sampled. Given the relative geographical proximity of these two sites, and the fact that they were both sampled in the winter, this similarity is not surprising. The Fiordland colony showed the highest diversity of prey items of all the colonies. 6.4 Discussion Deagle, Eveson et al. (2006) reported that 19% of prey DNA fragments extracted from sea lion scat samples were ≤ 226 bp in length and that, in most instances, <2 % of prey DNA was > 500 bp in length. The reconstruction of animal diet using faecal samples typically relies on short fragments of DNA that still show enough interspecific variation to identify species (Dunshea 2009). Even though the DNA fragments chosen for this study were initially developed for a closely related species (Deagle, Kirkwood et al. 2009) they still showed a high degree of interspecific variation in the current study. The specific design of primers allowed us to assign DNA sequences to species in most cases. In cases where a sequence could not be confidently assigned to a single species, the second assigned species was usually a closely related species. 6.4.1 New Zealand fur seal parasites Most of the parasite species found in our samples were nematodes (round worms). Round worms are commonly found in marine mammals. Most of these species spend their larval stages inside fish and infect seals when infected fish are consumed. The Parafilaroides nematodes identified in this research are associated with a hypersensivity reaction in 128 California sea lions (Zalaphus californianus) (Sweeney and Gilmartin 1974), which results in acute bronchitis and bronchopneumonia (Nicholson and Fanning 1981) and stimulates mucous secretion, which can lead to asphyxiation (Geraci and Aubin 1987). Anisakine nematodes were identified in most of the colonies. These are the most cosmopolitan of all marine mammals parasites (Geraci and Aubin 1987). They usually live freely in the stomach cavity or are attached to the gastric mucosa. Penetration by larva and adults into host tissue can lead to stomach ulcers (Schroeder and Wegeforth 1935). Two heartworm species, Dirofilaria immitis and Dipetalonema sp., were also identified. Pulmonary and cardiovascular complications associated with heartworms can have negative impacts on seal diving and feeding behaviour (Geraci and Aubin 1987). 6.4.2 New Zealand fur seal prey Items The considerable diversity of prey items identified in the current research is consistent with earlier studies indicating a generalist diet for fur seals (Harcourt, Bradshaw et al. 2002). Hume, Hindell et al. (2004) found a range of 1-3 cephalopod species and up to 7 different species of fish from a single Australian fur seal faecal sample. A few species of cephalopods and fish, including giant squid (Architeuthis dux), Sepia officinalis, Nototodarus gouldi, Martialia hyadesi, Spirula spirula, Nototodarus sloanii, and elephant fish (Callorhinchus milii) were common among all the colonies studied, while the rest of the diet differed among colonies. These results support the findings by Hume, Hindell et al. (2004) who identified geographical location as the major cause of variation in Australian fur seal diet. One colony was sampled twice in a year. Winter and summer diet in Kaikoura showed little species overlap and could indicate the dynamic state of prey communities 129 around New Zealand. Elephant fish (Callorhinchus milii), hoki (Macruronus novaezelandiae), giant squid (Architeuthis dux), red baitfish (Emmelichthys nitidus), Hexanchus griseus, Notorynchus cepedianus, Sepia officinalis, Abralia sp., Nototodarus gouldi, Lampanyctodes hectoris, Martialia hyadesi, Spirula spirula, arrow squid (Nototodarus sloanii), Octopoteuthis sp., and Moroteuthis sp. were identified in both seasons. I suggest that the remaining differences reflect seasonal changes in prey abundance, which is consistent with earlier research (e.g., Carey (1992)). At the first glance some identified species, such as goblin shark (Mitsukurina owstoni), giant squid (Architeutix dux) and tiger shark (Galeocerdo cuvier), might seem very unlikely to have been eaten by New Zealand fur seals. These species live far below the known dive depth of fur seals. Knowledge concerning the biology of these elusive species is minimal. In early stages of their life cycles these species are found at shallower depths where they may come into contact with fur seals. It is known that shark nurseries are usually in shallower areas of the sea compared to the distribution of adults (Heupel, Carlson et al. 2007). The fact that tiger sharks do not have a defined nursery area (Heupel, Carlson et al. 2007) also does not preclude the possibility that they might have been eaten in earlier stages of their life cycle. In addition, fur seals have been observed preying on shark species far larger than their usual prey items. For example, Cape fur seals (Arctocephalus pusillus) have been observed successfully hunting up to five 1.4 meter blue sharks(Prionace glauca) in Cape Point, South Africa (Fallows, Benoît et al. 2015) where the seals only consumed the shark’s fish-filled stomachs and energy-rich livers. I suggest that predation on juveniles of large body-sized species, especially sharks, might be a more common practice among fur seals than initially thought. However, the possibilities of KLAST software misidentification with a closely related species, or the presence of prey items that do not have reference sequences available in public databases, need further investigation. 130 In 2009, around 20 marine species constituted more than 91% of the New Zealand fishery (New Zealand Statistics February 2010). From these commercially important species, hoki (Macruronus novaezelandiae), ling (Genypterus blacodes), warehou (Seriolella sp.), tarakihi (Nemadactylus macropterus), bluenose (Hyperoglyphe antarctica), barracouda (Thyrsites atun), jack mackerel (Trachurus declivis), and arrow squid (Nototodarus sloanii) were identified from seal diets. These species constitute a small proportion of the total number of species identified from New Zealand fur seals. Three out of 34 species identified from Stewart Island (8%) are of major commercial value. In Fiordland, seven species from 63 species of marine prey (11%) are classified as major commercial fish species. In Kaikoura in winter, four out of 35 species (11%) and in Kaikoura in summer three out of 38 species (8%) are commercial species. These numbers are three out of 31 species (9.6%) for Cape Palliser and six out of 37 species (16%) for Bay of Plenty respectively; which gives a mean of approximately 10.6 % of the species consumed by fur seals overlapping with commercial species across the whole New Zealand in the current study. This study shows that New Zealand fur seals consume a large variety of prey species, of which only a small proportion are of commercial interest. However, our study design does not allow any quantitative estimate regarding prey species consumption by volume. The absolute contribution of each of these species in term of biomass to the seal diet is to be studied. Rapid depletion of global fish stock diversity, coinciding with global warming, could potentially exacerbate the extent of current competition between seals and fishing industries. In the absence of alternative food sources, seals are forced to feed on whatever species are available, many of which have significant commercial or recreational value for people . Sequencing of pooled DNA samples from different colonies can provide marine ecosystem managers with information that is needed in decision making. Even though individual diet variation cannot be assessed using this approach, the financial gains from pooling colony-level 131 DNA samples make intra-annual sampling a more realistic and affordable option. Appropriate pooling strategies and standardisation of the pooling method can also provide acceptable semi-quantitative data for addressing management issues. 132 Table 6-1 Fish and squid species identified from New Zealand fur seal faecal samples across 5 different colonies across New Zealand 133 134 Table 6-2 Parasite species identified from New Zealand fur seal faecal samples across 5 different colonies across New Zealand 135 136 Chapter 7 Conclusion The final stages of the preparation of this thesis coincided with reports of a mysterious death of dozens of grey seals (Halichoerus grypus) at Cornwall Bay England (BBC 2014). It was only mere chance that a similar incident did not happened in the peak of commercial sealing of the 18th and -19th centuries, where the number of New Zealand fur seals was at its lowest ever, around several hundred breeding females, otherwise the New Zealand fur seal may have gone extinct for ever. The rapid increases in the population of New Zealand fur seal in the current socio-economics conditions has given rise to concerns among both experts and the public. The current research addressed some of the controversial issues regarding increasing population of New Zealand fur seals. I answer two main questions in my research: how much fur seal diet overlaps with recreational- commercial fisheries? And what was the size of population before human arrival and how the population was affected after New Zealand settlement. The second question is important because it will help conservation authorities to have a better picture of this species demographic past for future management plans. These finding have a major role to play in years to come in conservation and management of this species. The data show that re-colonization of New Zealand from two potential refuges are likely to have followed a spill-over dynamics. The founder colonies, at least at a local scale, spill-over and form new colonies in their vicinities. The new colonies will further recruit immigrant seals to change in to a new mature larger colony. This pattern is of pivotal management importance since it implies that in any future local management plans the whole population of the species as a dynamic entity needs to be taken into consideration. 137 The size of the New Zealand fur seal bottleneck, its historical and pristine population size show a population that has witnessed a severe population decline to approximately 1% of its original size and a subsequent recovery to 20% of it this original size before human arrival in the area. The dramatic decline in population size means loss of substantial genetic diversity from the population genetic pool. Whatever genetic diversity is left in the species needs to be preserved if the prospect of long-term evolutionary survival of the species is to be considered. The question of whether Maori dramatically impacted the fur seal population is addressed in this study. It is likely that the fur seal population reduced by about half after Maori settlement. This reduction probably had little impact in the population as a whole but may have removed some colonies from throughout the range as suggested by Lalas and Bradshaw (2001) The long dispersal capability of New Zealand fur seals and spill-over pattern of colony expansion means reduced population density in a local scale will result in re-invasion of the area by new individuals seeking suitable habitat. The presence of an ectopic gene conversion in ZFY-ZFX region in a fraction of individuals may support that there are two major patrilineal haplotype group in the species. The origin of the gene conversion and why it is observable only in a fraction of males yet to be studied in more details. The population of New Zealand fur seals is still an order of magnitude less than what it was on human arrival. The current research used two genetic markers with completely different inheritance pattern to study population genetics in New Zealand fur seals. However, the lack of obvious recombination, at least in mitochondrial DNA, makes it a very large single locus 138 which only represents a single realization of evolutionary history involved in the species population structure. Coalescent simulation involves uncertainty particularly when a single marker without recombination is considered. The coalescence genealogy of complete mitochondrial DNA is no more than a single realization of the evolutionary history involving fur seal evolutionary history. Population size estimated in each coalescent interval is subjected to certain amount of uncertainty (Ho and Shapiro 2011). The process is similar to an estimation of the mean of an exponential distribution using only a single value from distribution (Minin, Bloomquist et al. 2008) but still highly informative to have a general picture of what has happened in evolutionary history of the species. It is suggested that in any future study a reduced representation of the whole genome (preferably the whole genome even if in fewer individuals) be considered to answer questions more precisely. However the computational pipeline, especially in Bayesian frameworks, lags behind the wet laboratory technologies, making the interpretation of large amounts of generated data sets a major challenge. It is also recommended that a sequence capture study of the whole Y and X chromosome could unwind the complex evolutionary history of the sex chromosomes in New Zealand fur seals. This study was the first attempt to reconstruct the diet of New Zealand fur seals from faecal DNA (diversity capsules (Boyer et al 2015)). In the absence of a purposed-made genetic data base of prey species of New Zealand fur seals, the NCBI data based-were used for analysing data. Using such a big data base as NCBI resulted in many false negative and false positive hits which were unavoidable. It is highly recommended that in the future a data base of fish, 139 cephalopods and crustaceans from New Zealand waters become available to increase sensitivity of the DNA-based method in all New Zealand marine mammal species. One of the major area of conflict among New Zealand fur seals and humans is in competing for shared marine resources. This research used the latest technology in the field of molecular ecology to investigate the degree of competition. It was found that only 10% of the prey species identified from faecal samples are in fact species of major economic value. 90% of prey elements in the diet were deep sea fish and cephalopods with no major commercial value. Competition between seals and humans will be probably more severe in areas already depleted by intense harvests. Seals in such areas do not have any other option but to compete with humans for whatever food item is left. The breadth of the generalist diet of the New Zealand fur seal does suggest that they will be able to persist in areas where certain fish species are heavily targeted by humans. The specific design of the current research does not allow us to reliably estimate quantitative contribution of each prey item in the fur seal diet. Quantitative estimation of prey items from faecal samples is an area of growing interest (Deagle and Tollit 2007). Our pilot study on inter-individual variation in diet composition shows a considerable variation among individual faeces in DNA recovery (Christine Bezic Alpeñes, personal communication). It is highly recommended that a quantitative study be initiated to complement our results. Another issue that needs to be dealt with is that as more in-depth sequencing technology become available and more affordable, contaminant DNA may be detected and sequenced. A major concern in studies like this is DNA contamination from sea water. Sea water could contain DNA from marine animals and when DNA come to contact with faecal samples could it could result in false positive species matches. This source of contamination is of major concern when faecal samples are collected directly from sea water columns rather than 140 shorelines. Therefore, a thorough examination of possible contamination of faecal samples collected from sea waters is strongly recommended. It is suggested that even a trace amount of fish or squid DNA is detectable from faecal samples. This study coincides with rapid developments in methodologies using reduced representation of genomes to study the past evolutionary history. The reduced representation methods has the advantage of studying a genome in few thousands to hundreds of thousands of Independent loci each representing a single realisation of the evolutionary history involving in the focal species past history. The advantage of studying many independent loci is reconstructing more precisely the evolutionary history of the focal species which will not be possible when a single locus such as mitochondrial DNA or Y chromosome are considered. The new methods require high quality DNA. The biological samples collected for this research were collected over the span of four years, mainly from remote areas of New Zealand and many specimens were stranded individuals whose exact (and sometimes even approximate) geographical origin was unknown. Suboptimal storage and transport to the laboratory resulted in degradation of some samples. Based on heterogeneous nature of DNA quality, the best option was to target mitochondrial DNA and /or short nuclear genes which are more likely to survive degradation at the expense of losing some degree of precision. It would have been useful to have invested more time and financial resources into using more advanced biological sample storage and transport systems to better profit from unique opportunities like the one that I had in the current research. In conclusion I believe that the current thesis will serve as a good step towards using new advances in genetic technology to study the biology and evolutionary past of the New Zealand fur seal (and other pinnipeds) with unprecedented precision. The data in the current thesis 141 could also provide marine ecosystem managers with necessary information desperately required to address current challenges faced by the New Zealand fur seals. 142 Appendix A Brief history of Zinc finger marker Most of the proteins are very long chains consisting of many amino acid residues while only few of them are intensively involved in the biochemical functions of proteins. The main reason for this special design is to provide the protein with stable, functional structure. The longer amino acid chain means more chemical bonds among residues which will result in a stronger protein structure. Some proteins use a shortcut to consolidate their structure: profiting from a Zinc (Zn) atom in their middle to allow the protein to fold around it. These proteins do not need very long chains to hold their chemical structure, indeed they are far shorter than other proteins in length. These proteins were first discovered in a frog eggs transcription factor TFIIIA and are generally called zinc fingers. Sticky protruded arms of their external amino acids gave them a special structure very similar to a hand’s finger. The main function of these diverse family of protein is modular recognition of the DNA strand which in turn will interfere with transcription of DNA strands and post transcriptional RNA interactions. 143 Zinc finger structure adapted from (Lee, Gippert et al. 1989) (Pavletich and Pabo 1991) Zinc finger structure adapted from (Lee, Gippert et al. 1989) (Pavletich and Pabo 1991) 144 145 References Acevedo-Whitehouse, K., F. Gulland, D. Greig and W. Amos (2003). "Inbreeding: disease susceptibility in California sea lions." Nature 422(6927): 35-35. Alacs, E., A. Georges, N. FitzSimmons and J. Robertson (2010). "DNA detective: a review of molecular approaches to wildlife forensics." Forensic science, medicine, and pathology 6(3): 180-194. Amos, W., J. W. Wilmer, K. Fullard, T. Burg, J. Croxall, D. Bloch and T. Coulson (2001). "The influence of parental relatedness on reproductive success." Proceedings of the Royal Society of London. Series B: Biological Sciences 268(1480): 2021-2027. Anderson, E. (1970). "Quaternary evolution of the genus Martes (Carnivora, Mustelidae)." Acta Zool Fenn 130: 1-132. Arim, M. and D. E. Naya (2003). "Pinniped diets inferred from scats: analysis of biases in prey occurrence." Canadian Journal of Zoology 81(1): 67-73. Armstrong, E. (1982). "A look at relative brain size in mammals* 1." Neuroscience Letters 34(2): 101104. Arnason, U., A. Gullberg, A. Janke, M. Kullberg, N. Lehman, E. A. Petrov and R. Väinölä (2006). "Pinniped phylogeny and a new hypothesis for their origin and dispersal." Molecular phylogenetics and evolution 41(2): 345-354. Avise, J. C. (1994). Molecular Markers, Natural History and Evolution: Natural History and Evolution, Springer. Bailey, A. M. and J. H. Sorensen (1962). Subantarctic Campbell Island, Denver Museum of Natural History. Baird, S. (2011). New Zealand Fur Seals: Summary of Current Knowledge, Ministry of Fisheries. Baker, A. R., T. R. Loughlin, V. Burkanov, C. W. Matson, R. G. Trujillo, D. G. Calkins, J. K. Wickliffe and J. W. Bickham (2005). "Variation of mitochondrial control region sequences of Steller sea lions: the three-stock hypothesis." Journal of Mammalogy 86(6): 1075-1084. Baker, J. D., G. A. ANTONELIS, C. W. FOWLER and A. E. YORK (1995). "Natal site fidelity in northern fur seals, Callorhinus ursinus." Animal Behaviour 50(1): 237-247. Baker, J. R., M. A. Fedak, S. S. Anderson, T. Arnbom and R. Baker (1990). "Use of a tiletaminezolazepam mixture to immobilise wild grey seals and southern elephant seals." Veterinary Record 126(4): 75. Bandelt, H.-J., P. Forster and A. Röhl (1999). "Median-joining networks for inferring intraspecific phylogenies." Molecular biology and evolution 16(1): 37-48. Banks, J. C., A. D. Mitchell, J. R. Waas and A. M. Paterson (2002). "An unexpected pattern of molecular divergence within the blue penguin (Eudyptula minor) complex." Notornis 49(1): 29-38. Barrett Lennard, L. G., T. G. Smith and G. M. Ellis (1996). "A cetacean biopsy system using lightweight pneumatic darts, and its effect on the behavior of killer whales." Marine mammal science 12(1): 1427. Barros, N. B. and R. S. Wells (1998). "Prey and feeding patterns of resident bottlenose dolphins (Tursiops truncatus) in Sarasota Bay, Florida." Journal of Mammalogy 79(3): 1045-1059. Barton, k. (1996). "Fur seals flourish." New Zealand Science Monthly 7(5)(3). BBC. (2014). "Mass seal strandings in Cornwall: Experts 'baffled'." Retrieved 10/12/2014, from http://www.bbc.com/news/uk-england-cornwall-30402678. Bean, K., W. Amos, P. Pomeroy, S. Twiss, T. Coulson and I. Boyd (2004). "Patterns of parental relatedness and pup survival in the grey seal (Halichoerus grypus)." Molecular Ecology 13(8): 23652370. BearzI, G. (2000). "First report of a common dolphin (Delphinus delphis) death following penetration of a biopsy dart." Journal of Cetacean Research and Management 2(3): 217-222. Beaumont, M. (2004). "Recent developments in genetic data analysis: what can they tell us about human demographic history?" Heredity 92(5): 365-379. 146 Beaumont, M. A. (2008). "Joint determination of topology, divergence time, and immigration in population trees." Renfrew C Matsumura S, Forster P, editor, Simulation, Genetics and Human Prehistory, McDonald Institute Monographs: 134-1541. Beebee, T. J. C. and G. Rowe (2004). An introduction to molecular ecology, Oxford University Press Oxford. Bernt, M., A. Donath, F. Jühling, F. Externbrink, C. Florentz, G. Fritzsch, J. Pütz, M. Middendorf and P. F. Stadler (2013). "MITOS: Improved de novo metazoan mitochondrial genome annotation." Molecular phylogenetics and evolution 69(2): 313-319. Berry, O., L. C. Spiller, R. Campbell, Y. Hitchen and W. J. Kennington (2012). "Population recovery of the New Zealand fur seal in southern Australia: a molecular DNA analysis." Journal of Mammalogy 93(2): 482-490. Berta, A. (2002). Pinniped evolution. Encyclopedia of marine mammals. W. F. Perrin, B. G. Würsig and J. G. M. Thewissen. San Diego, Academy Press. Berta, A., J. L. Sumich and K. M. Kovacs (2006). Marine mammals: evolutionary biology, Academic Press. Bertilsson‐Friedman, P. (2006). "Distribution and frequencies of shark‐inflicted injuries to the endangered Hawaiian monk seal (Monachus schauinslandi)." Journal of Zoology 268(4): 361-368. Bohmann, K., A. Monadjem, C. L. Noer, M. Rasmussen, M. R. Zeale, E. Clare, G. Jones, E. Willerslev and M. T. P. Gilbert (2011). "Molecular diet analysis of two African free-tailed bats (Molossidae) using high throughput sequencing." PLoS One 6(6): e21441. Bonferroni, C. E. (1936). Teoria statistica delle classi e calcolo delle probabilita, Libreria internazionale Seeber. Bonin, C. A., M. E. Goebel, J. Forcada, R. S. Burton and J. I. Hoffman (2013). "Unexpected genetic differentiation between recently recolonized populations of a long‐lived and highly vagile marine mammal." Ecology and evolution 3(11): 3701-3712. Boren, L. (2010). Diet of New Zealand fur seal. DOC Research and development series. Welington, Departement of Conservation. 319. Bouckaert, R., J. Heled, D. Kühnert, T. Vaughan, C.-H. Wu, D. Xie, M. A. Suchard, A. Rambaut and A. J. Drummond (2014). "BEAST 2: a software platform for Bayesian evolutionary analysis." PLoS computational biology 10(4): e1003537. Bowen, W. (1997). "Role of marine mammals in aquatic ecosystems." Marine Ecology Progress Series 158(267): 74. Boyd, I. and A. Murray (2001). "Monitoring a marine ecosystem using responses of upper trophic level predators." Journal of Animal Ecology 70(5): 747-760. Bradshaw, C. J., C. Lalas and C. M. Thompson (2000). "Clustering of colonies in an expanding population of New Zealand fur seals (Arctocephalus forsteri)." Journal of Zoology 250(1): 105-112. Bradshaw, C. J. A., C. Lalas and C. M. Thompson (2000). "Clustering of colonies in an expanding population of New Zealand fur seals (Arctocephalus forsteri)." Journal of Zoology 250(1): 105-112. Bright, J.-A. and S. F. Petricevic (2004). "Recovery of trace DNA and its application to DNA profiling of shoe insoles." Forensic science international 145(1): 7-12. Brinkman, T. J., D. K. Person, F. S. Chapin, W. Smith and K. J. Hundertmark (2011). "Estimating abundance of Sitka black‐tailed deer using DNA from fecal pellets." The Journal of Wildlife Management 75(1): 232-242. Brooks, R., J. Williamson, A. Hensley, E. Butler, G. Touchton and E. Smith (2003). "Buccal cells as a source of DNA for comparative animal genomic analysis." Biotechnology letters 25(6): 451-454. Brown, M. R., P. J. Corkeron, P. T. Hale, K. W. Schultz and M. M. Bryden (1994). "Behavioral responses of east Australian humpback whales Megaptera novaeangliae to biopsy sampling." Marine mammal science 10(4): 391-400. Brown, M. W., S. D. Kraus and D. E. Gaskin (1991). "Reaction of North Atlantic right whales(Eubalaena glacialis) to skin biopsy sampling for genetic and pollutant analysis." Special Issue Report. International Whaling Commission[REP. INT. WHALING COMM.(SPEC. ISSUE).]. 1991. Burger, R. and M. Lynch (1995). "Evolution and extinction in a changing environment: a quantitativegenetic analysis." Evolution: 151-163. Carey, P. W. (1989). "Behavioural thermoregulation and polygyny in the New Zealand fur seal." 147 Carey, P. W. (1992). "Fish prey species of the New Zealand fur seal (Arctocephalus forsteri, Lesson)." New Zealand Journal of Ecology 16(1). Caro, T. and M. K. Laurenson (1994). "Ecological and genetic factors in conservation: a cautionary tale." Science-AAAS-Weekly Paper Edition-including Guide to Scientific Information 263(5146): 485486. Carr, D., J. Bowman, C. J. Kyle, S. M. Tully, E. L. Koen, J. F. ROBITAILLE and P. J. Wilson (2007). "Rapid homogenization of multiple sources: genetic structure of a recolonizing population of fishers." The Journal of Wildlife Management 71(6): 1853-1861. Carreon-Martinez, L. and D. Heath (2010). "Revolution in food web analysis and trophic ecology: diet analysis by DNA and stable isotope analysis." Molecular ecology 19(1): 25-27. Casper, R. M., S. N. Jarman, B. E. Deagle, N. J. Gales and M. A. Hindell (2007). "Detecting prey from DNA in predator scats: A comparison with morphological analysis, using< i> Arctocephalus</i> seals fed a known diet." Journal of Experimental Marine Biology and Ecology 347(1): 144-154. Castella, V. and P. Mangin (2008). "DNA profiling success and relevance of 1739 contact stains from caseworks." Forensic Science International: Genetics Supplement Series 1(1): 405-407. Caudron, A., S. N. Sandra, G. M. Christopher, J. B. Laura and J. G. Neil (2006). "Hair sampling and genotyping from hair folliccles : A minimally-invasive alternative for genetics studies in small,mobile pinnipeds and other mammals " Marine mammal Science Chesser, R. K. (1983). "Genetic variability within and among populations of the black-tailed prairie dog." Evolution: 320-331. Chesser, R. K. (1991). "Gene diversity and female philopatry." Genetics 127(2): 437-447. Chikhi, L., V. C. Sousa, P. Luisi, B. Goossens and M. A. Beaumont (2010). "The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes." Genetics 186(3): 983-995. Choisy, M., P. Franck and J. M. Cornuet (2004). "Estimating admixture proportions with microsatellites: comparison of methods based on simulated data." Molecular Ecology 13(4): 955-968. Coltman, D., J. Pilkington and J. Pemberton (2003). "Fine‐scale genetic structure in a free‐living ungulate population." Molecular Ecology 12(3): 733-742. Coltman, D., G. Stenson, M. Hammill, T. Haug, C. Davis and T. Fulton (2007). "Panmictic population structure in the hooded seal (Cystophora cristata)." Molecular Ecology 16(8): 1639-1648. Coltman, D. W., W. D. Bowen and J. M. Wright (1998). "Birth weight and neonatal survival of harbour seal pups are positively correlated with genetic variation measured by microsatellites." Proceedings of the Royal Society of London. Series B: Biological Sciences 265(1398): 803-809. Corkeron, P. J., R. J. Morris and M. M. Bryden (1987). "A note on healing of large wounds in bottlenose dolphins, Twsiops truncates." Cornuet, J.-M., P. Pudlo, J. Veyssier, A. Dehne-Garcia, M. Gautier, R. Leblois, J.-M. Marin and A. Estoup (2014). "DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data." Bioinformatics 30(8): 1187-1189. Crawley, M. C. (1990). The Hand Book Of New Zealand Mammals. Auckland, Oxford University Press. Crawley, M. C. and R. Warneke (1979). " New Zealand fur seal in Mammals in the seas." FAO fisheries series 2(5). Crawley, M. C. and G. J. Wilson (1976). "The natural history and behaviour of the New Zealand fur seal (Arctocephalus forsteri)." Tuatara 22: 1-29. Curtis, C., B. S. Stewart and S. A. Karl (2007). "Sexing pinnipeds with ZFX and ZFY loci." Journal of Heredity 98(3): 280. Curtis, C., B. S. Stewart and S. A. Karl (2007). "Sexing pinnipeds with ZFX and ZFY loci." Journal of heredity 98(3): 280-285. 148 de Oliveira, L. R., J. I. Hoffman, E. Hingst-Zaher, P. Majluf, M. M. Muelbert, J. S. Morgante and W. Amos (2008). "Morphological and genetic evidence for two evolutionarily significant units (ESUs) in the South American fur seal, Arctocephalus gazella." Conservation Genetics 9(6): 1451-1466. Deagle, B., S. Jarman, D. Pemberton and N. Gales (2005). "Genetic screening for prey in the gut contents from a giant squid (Architeuthis sp.)." Journal of Heredity 96(4): 417-423. Deagle, B. E., A. Chiaradia, J. McInnes and S. N. Jarman (2010). "Pyrosequencing faecal DNA to determine diet of little penguins: is what goes in what comes out?" Conservation Genetics 11(5): 2039-2048. Deagle, B. E., J. P. Eveson and S. N. Jarman (2006). "Quantification of damage in DNA recovered from highly degraded samples–a case study on DNA in faeces." Frontiers in Zoology 3(1): 11. Deagle, B. E., R. Kirkwood and S. N. Jarman (2009). "Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces." Molecular Ecology 18(9): 2022-2038. Deagle, B. E. and D. J. Tollit (2007). "Quantitative analysis of prey DNA in pinniped faeces: potential to estimate diet composition?" Conservation Genetics 8(3): 743-747. Deagle, B. E., D. J. Tollit, S. N. Jarman, M. A. Hindell, A. W. Trites and N. J. Gales (2005). "Molecular scatology as a tool to study diet: analysis of prey DNA in scats from captive Steller sea lions." Molecular Ecology 14(6): 1831-1842. Dellinger, T. and F. Trillmich (1988). "Estimating diet composition from scat analysis in otariid seals (Otariidae): is it reliable?" Canadian Journal of Zoology 66(8): 1865-1870. Dickerson, B. R., R. R. Ream, S. N. Vignieri and P. Bentzen (2010). "Population structure as revealed by mtDNA and microsatellites in northern fur seals, Callorhinus ursinus, throughout their range." PLoS One 5(5): e10671. Dickie, G. S. and S. M. Dawson (2006). "Age, growth, and reproduction in New Zealand fur seals." Marine mammal science 19(1): 173-185. Dix, B. (1993). Population changes and diet preferences of the New Zealand fur seal Arctocephalus forsteri in eastern Cook Strait, Victoria University of Wellington. Dix, B. (1993). "Population changes and diet preferences of the New Zealand fur seal Arctocephalus forsteri in eastern Cook Strait." Unpublished MSc thesis, Victoria University of Wellington, Wellington. Doming, D. P. (2002). Sirenian Evolution. Encyclopedia of marine mammals. W. F. Perrin, B. G. Würsig and J. G. M. Thewissen. San Diego Academic Press. Dracopoli, N., F. Brett, T. Turner and C. Jolly (1983). "Patterns of genetic variability in the serum proteins of the Kenyan vervet monkey (Cercopithecus aethiops)." American journal of physical anthropology 61(1): 39-49. Drummond, A. J. and A. Rambaut (2007). "BEAST: Bayesian evolutionary analysis by sampling trees." BMC evolutionary biology 7(1): 214. Drummond, A. J., A. Rambaut, B. Shapiro and O. G. Pybus (2005). "Bayesian coalescent inference of past population dynamics from molecular sequences." Molecular biology and evolution 22(5): 11851192. Dunshea, G. (2009). "DNA-based diet analysis for any predator." Plos One 4(4): e5252. Dupanloup, I., S. Schneider and L. Excoffier (2002). "A simulated annealing approach to define the genetic structure of populations." Molecular Ecology 11(12): 2571-2581. Ellison, J., X. Li, U. Francke and L. Shapiro (1996). "Rapid evolution of human pseudoautosomal genes and their mouse homologs." Mammalian Genome 7(1): 25-30. Ernest, H., M. Penedo, B. May, M. Syvanen and W. Boyce (2000). "Molecular tracking of mountain lions in the Yosemite Valley region in California: genetic analysis using microsatellites and faecal DNA." Molecular Ecology 9(4): 433-441. Excoffier, L., A. Estoup and J.-M. Cornuet (2005). "Bayesian analysis of an admixture model with mutations and arbitrarily linked markers." Genetics 169(3): 1727-1738. Excoffier, L., G. Laval and S. Schneider (2005). "Arlequin (version 3.0): an integrated software package for population genetics data analysis." Evolutionary bioinformatics online 1: 47. Excoffier, L., P. E. Smouse and J. M. Quattro (1992). "Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data." Genetics 131(2): 479-491. 149 Ezaki, N., M. Koyama, Y. Kodama, T. Shomura, K. Tashiro, T. Tsuruoka, S. Inouye and S. Sakai (1983). "Pyrrolomycins F1, F2a, F2b and F3, new metabolites produced by the addition of bromide to the fermentation." The Journal of antibiotics 36(11): 1431. Fallows, C., H. Benoît and N. Hammerschlag (2015). "Intraguild predation and partial consumption of blue sharks Prionace glauca by Cape fur seals Arctocephalus pusillus pusillus." African Journal of Marine Science(ahead-of-print): 1-4. Fea, N. and R. Harcourt (1997). "Assessing the use of fecal and regurgitate analysis as a means of determining the diet of New Zealand fur seals." Marine mammal research in the southern hemisphere 1: 143–150. Fea, N. I., R. G. Harcourt and C. Lalas (1999). "Seasonal variation in the diet of New Zealand fur seals (Arctocephalus forsteri) at Otago Peninsula, New Zealand." Wildlife Research 26(2): 147-160. Flynn, J. J., J. A. Finarelli, S. Zehr, J. Hsu and M. A. Nedbal (2005). "Molecular phylogeny of the Carnivora (Mammalia): assessing the impact of increased sampling on resolving enigmatic relationships." Systematic Biology 54(2): 317-337. Fordyce, R. E. (2002 ). Cetacean evolution. Encyclopedia of marine mammals. W. F. Perrin, B. G. Würsig and J. G. M. Thewissen. New York, Academic Press. Frère, C. H., M. Krützen, A. M. Kopps, P. Ward, J. Mann and W. B. Sherwin (2010). "Inbreeding tolerance and fitness costs in wild bottlenose dolphins." Proceedings of the Royal Society B: Biological Sciences: rspb20100039. Gales, N. J. and A. J. Cheal (1992). "Estimating diet composition of the Australian sea-lion (Neophoca cinerea) from scat analysis: an unreliable technique." Wildlife Research 19(4): 447-456. Garant, D., J. J. Dodson and L. Bernatchez (2000). "Ecological determinants and temporal stability of the within‐river population structure in Atlantic salmon (Salmo salar L.)*." Molecular Ecology 9(5): 615-628. Gardner, B., J. A. Royle, M. T. Wegan, R. E. Rainbolt and P. D. Curtis (2010). "Estimating black bear density using DNA data from hair snares." The Journal of Wildlife Management 74(2): 318-325. Gauthier, J. and R. Sears (1999). "Behavioral response of four species of balaenopterid whales to biopsy sampling." Marine mammal science 15(1): 85-101. Gemmell, N. J. and P. Majluf (1997). "Projectile biopsy sampling of fur seals." Marine mammal science 13(3): 512-516. Gentry, R. L. (1998). Behavior and ecology of the northern fur seal, Princeton University Press. Geraci, J. R. and D. J. S. Aubin (1987). "Effects of parasites on marine mammals." International Journal for Parasitology 17(2): 407-414. Glazek, J., A. Sulimski and T. Wysoczanski-Minkowicz (1976). On the stratigraphic position of Weze 1 locality (Middle Poland). Proceedings of the 6th International Congress of Speleology, Olomouc-CSSR. Volume. Goldsworthy, S., J. Francis, D. Boness and R. Fleischer (2000). "Variation in the mitochondrial control region in the Juan Fernandez fur seal (Arctocephalus philippii)." Journal of Heredity 91(5): 371-377. Gordon, D. P. (2009). New Zealand Inventory of Biodiversity: Radiata, Lophotrochozoa, Deuterostomia. Kingdom Animalia, Canterbury University Press. Gorgone, A. M., P. A. Haase, E. S. Griffith and A. A. Hohn (2008). "Modeling response of target and nontarget dolphins to biopsy darting." The Journal of Wildlife Management 72(4): 926-932. Greenwood, P. J. (1980). "Mating systems, philopatry and dispersal in birds and mammals." Animal behaviour 28(4): 1140-1162. Gribble, G. W. (2003). "The diversity of naturally produced organohalogens." Chemosphere 52(2): 289-297. Guerrant, E. O., K. Havens and M. Maunder (2004). Ex situ plant conservation: supporting species survival in the wild, Island Press. Hamilton, M. (2011). Population genetics, John Wiley & Sons. Hammond, J. A., C. Hauton, K. A. Bennett and A. J. Hall (2012). "Phocid seal leptin: tertiary structure and hydrophobic receptor binding site preservation during distinct leptin gene evolution." PloS one 7(4): e35395. Harcourt, R. G., C. Bradshaw, K. Dickson and L. S. Davis (2002). "Foraging ecology of a generalist predator the female New Zealand fur seal." Marine Ecology-Progress Series 227: 11-24. 150 Harcourt, R. G., C. J. A. Bradshaw, K. Dickson and L. S. Davis (2002). "Foraging ecology of a generalist predator, the female New Zealand fur seal." Marine Ecology Progress Series 227: 11-24. Harris, R. B., J. Winnie, S. J. Amish, A. BEJA‐PEREIRA, R. Godinho, V. COSTA and G. Luikart (2010). "Argali abundance in the Afghan Pamir using capture–recapture modeling from fecal DNA." The Journal of Wildlife Management 74(4): 668-677. Hartl, D. L. and A. G. Clark (1997). Principles of population genetics, Sinauer associates Sunderland. Heath, R. B., D. Calkins, D. McAllister, W. Taylor and T. Spraker (1996). "Telazol and isoflurane field anesthesia in free-ranging Steller's sea lions (Eumetopias jubatus)." Journal of Zoo and Wildlife Medicine 27(1): 35-43. Heller, R., L. Chikhi and H. R. Siegismund (2013). "The confounding effect of population structure on Bayesian skyline plot inferences of demographic history." PLoS One 8(5): e62992. Heupel, M. R., J. K. Carlson and C. A. Simpfendorfer (2007). "Shark nursery areas: concepts, definition, characterization and assumptions." Marine Ecology Progress Series 337: 287-297. Heyning, J. E. and G. M. Lento (2002). The evolution of marine mammals. Marine Mammal Biology : An Evolutionary Approach A. R. Hoelzel. Oxford, Blackwell Sience. Higdon, J. W., O. R. Bininda-Emonds, R. M. Beck and S. H. Ferguson (2007). "Phylogeny and divergence of the pinnipeds (Carnivora: Mammalia) assessed using a multigene dataset." BMC Evolutionary Biology 7(1): 216. Ho, S. Y. and B. Shapiro (2011). "Skyline‐plot methods for estimating demographic history from nucleotide sequences." Molecular Ecology Resources 11(3): 423-434. Hoberecht, L. K., D. J. Vos and G. R. VanBlaricom (2006). "A remote biopsy system used to sample Steller sea lion (Eumetopias jubatus) blubber." Marine mammal science 22(3): 683-689. Hoelzel, A. (1998). "Genetic structure of cetacean populations in sympatry, parapatry, and mixed assemblages: implications for conservation policy." Journal of Heredity 89(5): 451-458. Hoelzel, A., J. Halley, S. O'brien, C. Campagna, T. Arnborm, B. Le Boeuf, K. Ralls and G. Dover (1993). "Elephant seal genetic variation and the use of simulation models to investigate historical population bottlenecks." Journal of Heredity 84(6): 443-449. Hoelzel, A. R., R. Fleischer, C. Campagna, B. Le Boeuf and G. Alvord (2002). "Impact of a population bottleneck on symmetry and genetic diversity in the northern elephant seal." Journal of Evolutionary Biology 15(4): 567-575. Hoffman, J. and W. Amos (2005). "Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion." Molecular Ecology 14(2): 599-612. Hoffman, J., J. Forcada and W. Amos (2006). "No relationship between microsatellite variation and neonatal fitness in Antarctic fur seals, Arctocephalus gazella." Molecular Ecology 15(7): 1995-2005. Hoffman, J., C. Matson, W. Amos, T. Loughlin and J. Bickham (2006). "Deep genetic subdivision within a continuously distributed and highly vagile marine mammal, the Steller's sea lion (Eumetopias jubatus)." Molecular Ecology 15(10): 2821-2832. Hoffman, J. I. and J. Forcada (2009). "Genetic analysis of twinning in Antarctic fur seals (Arctocephalus gazella)." Journal of Mammalogy 90(3): 621-628. Hoffman, J. I., J. Forcada and W. Amos (2006). "No relationship between microsatellite variation and neonatal fitness in Antarctic fur seals, Arctocephalus gazella." Molecular Ecology 15(7): 1995-2005. Hoffman, J. I., J. Forcada, P. N. Trathan and W. Amos (2007). "Female fur seals show active choice for males that are heterozygous and unrelated." Nature 445(7130): 912-914. Hoffman, J. I. and H. J. Nichols (2011). "A novel approach for mining polymorphic microsatellite markers in silico." PloS one 6(8): e23283. Hohenlohe, P. A., S. Bassham, P. D. Etter, N. Stiffler, E. A. Johnson and W. A. Cresko (2010). "Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags." PLoS genetics 6(2): e1000862. Holdaway, R. N. and C. Jacomb (2000). "Rapid extinction of the moas (Aves: Dinornithiformes): model, test, and implications." Science 287(5461): 2250-2254. Höss, M., M. Kohn and S. Pääbo (1992). "Excrement analysis by PCR." Nature 359: 199. Hume, F., M. Hindell, D. Pemberton and R. Gales (2004). "Spatial and temporal variation in the diet of a high trophic level predator, the Australian fur seal (Arctocephalus pusillus doriferus)." Marine Biology 144(3): 407-415. 151 Hunt Jr, R. and L. Barnes (1994). Basicranial evidence for ursid affinity of the oldest pinnipeds. Proc. San Diego Soc. Nat. Hist. Hutton, F. W. and J. Drummond (1923). The Animals Of New Zealand. Auckland, Whitcombe And Tombs Limited. Inoue, E., M. Inoue-Murayama, O. Takenaka and T. Nishida (2007). "Wild chimpanzee infant urine and saliva sampled noninvasively usable for DNA analyses." Primates 48(2): 156-159. Janssens, J. C. A., H. Steenackers, S. Robijns, E. Gellens, J. Levin, H. Zhao, K. Hermans, D. De Coster, T. L. Verhoeven and K. Marchal (2008). "Brominated furanones inhibit biofilm formation by Salmonella enterica serovar Typhimurium." Applied and environmental microbiology 74(21): 6639. Jefferson, T. A. and S. K. Hung (2008). "Effects of biopsy sampling on Indo-Pacific humpback dolphins (Sousa chinensis) in a polluted coastal environment." Aquatic Mammals 34(3): 310-316. Jinks-Robertson, S., S. Sayeed and T. Murphy (1997). "Meiotic crossing over between nonhomologous chromosomes affects chromosome segregation in yeast." Genetics 146(1): 69-78. Jones, R. S. (2001). "Editorial II." British Journal of Anaesthesia 87(6): 813. Karesh, W. B., F. Smith and H. Frazier‐Taylor (1987). "A remote method for obtaining skin biopsy samples." Conservation Biology 1(3): 261-262. Kawamoto, Y. (1996). "Population genetic study of Sulawesi macaques." Variations in the Asian Macaques: 37-65. Keller, L. F., P. Arcese, J. N. Smith, W. M. Hochachka and S. C. Stearns (1994). "Selection against inbred song sparrows during a natural population bottleneck." Nature 372(6504): 356-357. Kenyon, K. W. and F. Wilke (1953). "Migration of the northern fur seal, Callorhinus ursinus." Journal of Mammalogy: 86-98. King, C. (1989). "natural history of weasels & stoats." King, C. M. (1990). "The handbook of New Zealand mammals." King, J. E. (1983). Seals of the world. London, British Museum of Natural History. Kingman, J. F. C. (1982). "The coalescent." Stochastic processes and their applications 13(3): 235-248. Kiszka, J., B. Simon-Bouhet, F. Charlier, C. Pusineri and V. Ridoux (2010). "Individual and group behavioural reactions of small delphinids to remote biopsy sampling." Animal Welfare 411: 417. Koepfli, K.-P. and R. Wayne (1998). "Phylogenetic relationships of otters (Carnivora: Mustelidae) based on mitochondrial cytochrome b sequences." Journal of Zoology 246(04): 401-416. Kovacs, K. M., A. Aguilar, D. Aurioles, V. Burkanov, C. Campagna, N. Gales, T. Gelatt, S. D. Goldsworthy, S. J. Goodman and G. J. Hofmeyr (2012). "Global threats to pinnipeds." Marine Mammal Science 28(2): 414-436. Kretzmann, M., L. Mentzer, R. DiGiovanni, M. S. Leslie and G. Amato (2006). "Microsatellite diversity and fitness in stranded juvenile harp seals (Phoca groenlandica)." Journal of Heredity 97(6): 555-560. Krützen, M., L. M. Barré, L. M. Möller, M. R. Heithaus, C. Simms and W. B. Sherwin (2002). "A biopsy system for small cetaceans: darting success and wound healing in Tursiops spp." Marine mammal science 18(4): 863-878. Kuhner, M. K. (2006). "LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters." Bioinformatics 22(6): 768-770. Lacy, R. C. (1997). "Importance of genetic variation to the viability of mammalian populations." Journal of Mammalogy: 320-335. Lahn, B. T. and D. C. Page (1997). "Functional coherence of the human Y chromosome." Science 278(5338): 675-680. Lalas, C. and C. J. Bradshaw (2001). "Folklore and chimerical numbers: review of a millennium of interaction between fur seals and humans in the New Zealand region." New Zealand Journal of Marine and Freshwater Research 35(3): 477-497. Lalas, C. and C. J. A. Bradshaw (2001). "Folklore and chimerical numbers: review of a millennium of interaction between fur seals and humans in the New Zealand region." New Zealand journal of marine and freshwater research 35(3): 477-497. Lalas, C. and R. Harcourt (1995). "Pup production of the New Zealand fur seal on Otago Peninsula, New Zealand." Journal of the Royal Society of New Zealand 25(1): 81-88. Lancaster, M., J. Arnould and R. Kirkwood (2010). "Genetic status of an endemic marine mammal, the Australian fur seal, following historical harvesting." Animal conservation 13(3): 247-255. 152 Larkin, M. A., G. Blackshields, N. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm and R. Lopez (2007). "Clustal W and Clustal X version 2.0." Bioinformatics 23(21): 2947-2948. Lavenier, D. (2009). "PLAST: parallel local alignment search tool for database comparison." BMC bioinformatics 10(1): 329. Lee, M. S., G. P. Gippert, K. V. Soman, D. A. Case and P. E. Wright (1989). "Three-dimensional solution structure of a single zinc finger DNA-binding domain." Science 245(4918): 635-637. Lefort, M.-C., S. Boyer, A. Barun, A. E. Khoyi, J. Ridden, V. R. Smith, R. Sprague, B. R. Waterhouse and R. H. Cruickshank (2014). Blood, sweat and tears: non-invasive vs. non-disruptive DNA sampling for experimental biology, PeerJ PrePrints. Lehman, N. (1998). "Conservation biology: genes are not enough." Current biology 8(20): R722-R724. Lento, G., M. Haddon, G. Chambers and C. Baker (1997). "Genetic variation of southern hemisphere fur seals (Arctocephalus spp.): investigation of population structure and species identity." Journal of heredity 88(3): 202-208. Lento, G. M., R. E. Hickson, G. K. Chambers and D. Penny (1995). "Use of spectral analysis to test hypotheses on the origin of pinnipeds." Molecular Biology and Evolution 12(1): 28-52. Lento, G. M., R. H. Mattlin, G. K. Chambers and C. S. Baker (1994). "Geographic distribution of mitochondrial cytochrome b DNA haplotypes in New Zealand fur seals (Arctocephalus forsteri)." Canadian journal of zoology 72(2): 293-299. Lin, Y.-H., P. A. McLenachan, A. R. Gore, M. J. Phillips, R. Ota, M. D. Hendy and D. Penny (2002). "Four new mitochondrial genomes and the increased stability of evolutionary trees of mammals from improved taxon sampling." Molecular Biology and Evolution 19(12): 2060-2070. Losos, J. B., D. A. Baum, D. J. Futuyma, H. E. Hoekstra, R. E. Lenski, A. J. Moore, C. L. Peichel, D. Schluter and M. C. Whitlock (2013). The Princeton Guide to Evolution, Princeton University Press. Loughlin, T. R. and T. Spraker (1989). "Use of Telazol to immobilize female northern sea lions (Eumetopias jubatus) in Alaska." Journal of Wildlife Diseases 25(3): 353. Lundström, K., O. Hjerne, S.-G. Lunneryd and O. Karlsson (2010). "Understanding the diet composition of marine mammals: grey seals (Halichoerus grypus) in the Baltic Sea." ICES Journal of Marine Science: Journal du Conseil: fsq022. Macdonald, D. W. (2001). "The Encyclopedia of Mammals (New Ed.)." Macdonald, D. W. and S. Norris (2001). The encyclopedia of mammals, Facts On File. Majluf, P. and M. E. Goebel (1992). "The capture and handling of female South American fur seals and their pups." Marine Mammal Science 8(2): 187-190. Marjoram, P. and S. Tavaré (2006). "Modern computational approaches for analysing molecular genetic variation data." Nature Reviews Genetics 7(10): 759-770. Marmi, J., J. F. López‐Giráldez and X. Domingo‐Roura (2004). "Phylogeny, evolutionary history and taxonomy of the Mustelidae based on sequences of the cytochrome b gene and a complex repetitive flanking region." Zoologica Scripta 33(6): 481-499. Martin, A. R. and R. R. Reeves (2009). Marine Mammal Biology An Evolutionary Approach. Oxford, Blackwell. Maruyama, T. and P. A. Fuerst (1985). "Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck." Genetics 111(3): 675-689. Matejusová, I., F. Doig, S. Middlemas, S. Mackay, A. Douglas, J. Armstrong, C. Cunningham and M. Snow (2008). "Using quantitative real‐time PCR to detect salmonid prey in scats of grey Halichoerus grypus and harbour Phoca vitulina seals in Scotland–an experimental and field study." Journal of Applied Ecology 45(2): 632-640. Matthee, C., F. Fourie, W. Oosthuizen, M. Meyer and K. Tolley (2006). "Mitochondrial DNA sequence data of the Cape fur seal (Arctocephalus pusillus pusillus) suggest that population numbers may be affected by climatic shifts." Marine Biology 148(4): 899-905. Mattlin, R. H., N. J. Gales and D. P. Costa (1998). "Seasonal dive behaviour of lactating New Zealand fur seals (Arctocephalus forsteri)." Canadian Journal of Zoology 76(2): 350-360. McGlone, M. (1989). "The Polynesian settlement of New Zealand in relation to environmental and biotic changes." New Zealand journal of ecology: 115-129. 153 McKim, K. S., B. L. Green-Marroquin, J. J. Sekelsky, G. Chin, C. Steinberg, R. Khodosh and R. S. Hawley (1998). "Meiotic synapsis in the absence of recombination." Science 279(5352): 876-878. McLintock, A. H. (1966). "encyclopaedia of New Zealand." McWethy, D. B., C. Whitlock, J. M. Wilmshurst, M. S. McGlone, M. Fromont, X. Li, A. DieffenbacherKrall, W. O. Hobbs, S. C. Fritz and E. R. Cook (2010). "Rapid landscape transformation in South Island, New Zealand, following initial Polynesian settlement." Proceedings of the National Academy of Sciences 107(50): 21343-21348. McWilliam, H., W. Li, M. Uludag, S. Squizzato, Y. M. Park, N. Buso, A. P. Cowley and R. Lopez (2013). "Analysis tool web services from the EMBL-EBI." Nucleic acids research 41(W1): W597-W600. Meldgaard, M., P. Bollen and B. Finsen (2004). "Non-invasive method for sampling and extraction of mouse DNA for PCR." Laboratory Animals 38(4): 413-417. Melnick, D. (1987). "The genetic consequences of primate social organization: a review of macaques, baboons and vervet monkeys." Genetica 73(1-2): 117-135. Melnick, D. J. (1987). "Cercopithecines in multimale groups: genetic diversity and population structure." Primate societies. Miller, E. H. (1975). "Annual cycle of fur seals, Arctocephalus forsteri (Lesson), on the Open Bay Islands, New Zealand." Miller, M. (2005). "Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information." Journal of Heredity 96(6): 722-724. Miller, P. and P. Hedrick (2001). "Purging of inbreeding depression and fitness decline in bottlenecked populations of Drosophila melanogaster." Journal of Evolutionary Biology 14(4): 595601. Minin, V. N., E. W. Bloomquist and M. A. Suchard (2008). "Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics." Molecular biology and evolution 25(7): 1459-1471. Mirimin, L., R. Miller, E. Dillane, S. Berrow, S. Ingram, T. Cross and E. Rogan (2011). "Fine‐scale population genetic structuring of bottlenose dolphins in Irish coastal waters." Animal Conservation 14(4): 342-353. Murie, D. J. and D. M. Lavigne (1985). "Digestion and retention of Atlantic herring otoliths in the stomachs of grey seals." Marine mammals and fisheries. Edited by JR Beddington, RJH Beverton, and DM Lavigne. George Allen and Unwin, London: 292–299. Nei, M., T. Maruyama and R. Chakraborty (1975). "The bottleneck effect and genetic variability in populations." Evolution: 1-10. Nicholson, A. and J. Fanning (1981). Parasites and associated pathology of the respiratory tract of the Australian sea lion: Neophoca cinerea. Wildlife disease of the Pacific Basin and other countries, ME Fowler (ed.). Proceedings of the Fourth International Conference of the Wildlife Disease Association, Sydney, Australia. Nielsen, R. and M. A. Beaumont (2009). "Statistical inferences in phylogeography." Molecular ecology 18(6): 1034-1047. Nielsen, R. and M. Slatkin (2013). An introduction to population genetics: theory and applications, Sinauer Associates Sunderland, MA. Nino-Torres, C. A., S. C. Gardner, T. Zenteno-Savín and G. M. Ylitalo (2009). "Organochlorine pesticides and polychlorinated biphenyls in California sea lions (Zalophus californianus californianus) from the Gulf of California, México." Archives of environmental Contamination and Toxicology 56(2): 350-359. Novak, R. M. (2003). Walker's marine mammals of the world. Baltimore and London, The Johns Hopkins University Press Nowak, R. M. (1999). Walker's mammals of the world, JHU Press. Nowak, R. M. (2003). Walker's marine mammals of the world, JHU Press. 154 Nussey, D., D. Coltman, T. Coulson, L. Kruuk, A. Donald, S. Morris, T. CLUTTON‐BROCK and J. Pemberton (2005). "Rapidly declining fine‐scale spatial genetic structure in female red deer." Molecular Ecology 14(11): 3395-3405. Nybom, H. (2004). "Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants." Molecular ecology 13(5): 1143-1155. O'brien, S., M. Roelke, L. Marker, A. Newman, C. Winkler, D. Meltzer, L. Colly, J. Evermann, M. Bush and D. Wildt (1985). "Genetic basis for species vulnerability in the cheetah." Science 227(4693): 1428-1434. O'brien, S. J., D. E. Wildt, D. Goldman, C. R. Merril and M. Bush (1983). "The cheetah is depauperate in genetic variation." Science 221(4609): 459-462. Oberbauer, A., D. Grossman, M. Eggleston, D. Irion, A. Schaffer, N. Pedersen and J. Belanger (2003). "Alternatives to blood as a source of DNA for large-scale scanning studies of canine genome linkages." Veterinary research communications 27(1): 27-38. Obrien, S. J. (1994). "The cheetah's conservation controversy." Conservation Biology: 1153-1155. Obrien, S. J., J. S. Martenson, C. Packer, L. Herbst, V. DEVOS, P. Joslin, J. OTTJOSLIN, D. E. Wildt and M. Bush (1987). "Biochemical Genetic-Variation in Geographic Isolates of African and Asiatic Lions." National Geographic Research 3(1): 114-124. Packer, C., A. Pusey, H. Rowley, D. Gilbert, J. Martenson and S. O'brien (1991). "Case study of a population bottleneck: lions of the Ngorongoro Crater." Conservation Biology 5(2): 219-230. Page, B., J. McKenzie and S. D. Goldsworthy (2005). "Inter-sexual differences in New Zealand fur seal diving behaviour." Marine Ecology Progress Series 304: 249-264. Pamilo, P. and N. Bianchi (1993). "Evolution of the Zfx and Zfy genes: rates and interdependence between the genes." Molecular biology and evolution 10(2): 271-281. Parsons, K. M., S. B. Piertney, S. J. Middlemas, P. S. Hammond and J. D. Armstrong (2005). "DNA‐ based identification of salmonid prey species in seal faeces." Journal of Zoology 266(3): 275-281. Pastor, T., J. Garza, P. Allen, W. Amos and A. Aguilar (2004). "Low genetic variability in the highly endangered Mediterranean monk seal." Journal of Heredity 95(4): 291-300. Pauly, D., A. Trites, E. Capuli and V. Christensen (1998). "Diet composition and trophic levels of marine mammals." ICES Journal of Marine Science: Journal du Conseil 55(3): 467-481. Pavletich, N. P. and C. O. Pabo (1991). "Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A." Science 252(5007): 809-817. Peter, B. M., D. Wegmann and L. Excoffier (2010). "Distinguishing between population bottleneck and population subdivision by a Bayesian model choice procedure." Molecular Ecology 19(21): 46484660. Peters, R. H. (1986). The ecological implications of body size, Cambridge Univ Pr. Pidancier, N., C. Miquel and C. Miaud (2003). "Buccal swabs as a non-destructive tissue sampling method for DNA analysis in amphibians." Herpetological Journal 13(4): 175-178. PIERTNEY, S. B., A. D. MacCOLL, X. Lambin, R. Moss and J. F. DALLAS (1999). "Spatial distribution of genetic relatedness in a moorland population of red grouse (Lagopus lagopus scoticus)." Biological Journal of the Linnean Society 68(1‐2): 317-331. Pompanon, F., B. E. Deagle, W. O. Symondson, D. S. Brown, S. N. Jarman and P. Taberlet (2012). "Who is eating what: diet assessment using next generation sequencing." Molecular Ecology 21(8): 1931-1950. Pope, T. R. (1992). "The influence of dispersal patterns and mating systems on genetic differentiation within and between populations of the red howler monkey (Alouatta seniculus)." Evolution: 11121128. Purcell, M., G. Mackey, E. LaHood, H. Huber and L. Park (2004). "Molecular methods for the genetic identification of salmonid prey from Pacific harbor seal(Phoca vitulina richardsi) scat." Fishery Bulletin 102(1): 213-220. Rambaut, A. and A. Drummond (2003). "Tracer, version 1.5, MCMC trace analysis package." Website http://tree. bio. ed. ac. uk/software. Rambaut, A. and A. Drummond (2003). Tracer, version 1.5, MCMC trace analysis package. 155 Raymond, J. J., R. A. van Oorschot, S. J. Walsh, C. Roux and P. R. Gunn (2009). "Trace DNA and street robbery: a criminalistic approach to DNA evidence." Forensic Science International: Genetics Supplement Series 2(1): 544-546. Reeves, R. R. and B. S. Stewart (2003). Walker's Marine Mammals of the World. R. M.Nowak. Baltomore and London, The Johns Hopkins University Press. Reeves, R. R., B. S. Stewart, S. Leatherwood and P. A. Folkens (1992). The Sierra Club handbook of seals and sirenians, Sierra Club Books San Francisco. Reid, K. and J. P. Croxall (2001). "Environmental response of upper trophic-level predators reveals a system change in an Antarctic marine ecosystem." Proceedings of the Royal Society of London. Series B: Biological Sciences 268(1465): 377. Repenning, C. A. R. H. T. (1977). "Otariod seal of the Neogene." U.S.Geol.Surv.Prof.Pap(992). Riedman, M. (1990). The pinnipeds: seals, sea lions, and walruses, Univ of California Press. Robertson, B. C. and N. J. Gemmell (2005). Microsatellite DNA markers for the study of population structure in the New Zealand fur seal Arctocephalus forsteri, Department of Conservation. Rosser, Z. H., P. Balaresque and M. A. Jobling (2009). "Gene conversion between the X chromosome and the male-specific region of the Y chromosome at a translocation hotspot." The American Journal of Human Genetics 85(1): 130-134. Roux, J. P. (1987). Recolonisation processes in the subantarctic fur seal,Arctocephalus tropicalis, on Amstedam Island. Status, biology, and ecology of fur seals. J. P. G. Croxall, R.L. Cambridge, MA., NOAA Technical Report: 189-194. Ryan, C. J., G. Hickling and K.-J. Wilson (1997). "Breeding habitat preferences of the New Zealand fur seal (Arctocephalus forsteri) on Banks Peninsula." Wildlife research 24(2): 225-235. Sato, J. J., T. Hosoda, M. Wolsan, K. Tsuchiya, M. Yamamoto and H. Suzuki (2003). "Phylogenetic relationships and divergence times among mustelids (Mammalia: Carnivora) based on nucleotide sequences of the nuclear interphotoreceptor retinoid binding protein and mitochondrial cytochrome b genes." Zoological science 20(2): 243-264. Sato, J. J., M. Wolsan, H. Suzuki, T. Hosoda, Y. Yamaguchi, K. Hiyama, M. Kobayashi and S. Minami (2006). "Evidence from nuclear DNA sequences sheds light on the phylogenetic relationships of Pinnipedia: single origin with affinity to Musteloidea." Zoological science 23(2): 125-146. Saundry, P. (2009). "New Zealand Fur Seal ", from www.eol.org. Sawyer, S. (1999). "GENECONV: A computer package for the statistical detection of gene conversion." Distributed by the author, Department of Mathematics, Washington University in St. Louis. Schneider, S. and L. Excoffier (1999). "Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA." Genetics 152(3): 1079-1089. Schroeder, C. R. and H. M. Wegeforth (1935). "The occurrence of gastric ulcers in sea mammals of the California coast, their etiology and pathology." J. Am. Vet. Med. Assoc 87: 333-347. Schultz, J. K., J. D. Baker, R. J. Toonen and B. W. Bowen (2009). "Extremely low genetic diversity in the endangered Hawaiian monk seal (Monachus schauinslandi)." Journal of Heredity 100(1): 25-33. Schwartz, O. A. and K. B. Armitage (1980). "Genetic variation in social mammals: the marmot model." Science 207(4431): 665-667. Sellas, A. B., R. S. Wells and P. E. Rosel (2005). "Mitochondrial and nuclear DNA analyses reveal fine scale geographic structure in bottlenose dolphins (Tursiops truncatus) in the Gulf of Mexico." Conservation Genetics 6(5): 715-728. Sewell, J., I. Quinones, C. Ames, B. Multaney, S. Curtis, H. Seeboruth, S. Moore and B. Daniel (2008). "Recovery of DNA and fingerprints from touched documents." Forensic Science International: Genetics 2(4): 281-285. Simberloff, D. (1988). "The contribution of population and community biology to conservation science." Annual review of ecology and systematics: 473-511. Simonsen, K. L., G. A. Churchill and C. F. Aquadro (1995). "Properties of statistical tests of neutrality for DNA polymorphism data." Genetics 141(1): 413-429. Skulason, S. and T. B. Smith (1995). "Resource polymorphisms in vertebrates." Trends in ecology & evolution 10(9): 366-370. 156 Slattery, J. P. and S. J. O'Brien (1998). "Patterns of Y and X chromosome DNA sequence divergence during the Felidae radiation." Genetics 148(3): 1245-1255. Slattery, J. P., L. Sanner-Wachter and S. J. O'Brien (2000). "Novel gene conversion between XY homologues located in the nonrecombining region of the Y chromosome in Felidae (Mammalia)." Proceedings of the National Academy of Sciences 97(10): 5307-5312. Soule, M. E. (2005). "Conservation biology: the science of scarcity and diversity." Spielman, D., B. W. Brook and R. Frankham (2004). "Most species are not driven to extinction before genetic factors impact them." Proceedings of the National Academy of Sciences of the United States of America 101(42): 15261-15264. Spong, G., J. Stone, S. Creel and M. Björklund (2002). "Genetic structure of lions (Panthera leo L.) in the Selous Game Reserve: implications for the evolution of sociality." Journal of Evolutionary Biology 15(6): 945-953. Städler, T., B. Haubold, C. Merino, W. Stephan and P. Pfaffelhuber (2009). "The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations." Genetics 182(1): 205-216. Statistics, N. Z. (February 2010). Fish Monetary Stock Account: 1996–2009, New Zealand Statistics. Steele, D., M. E. Morris and S. Jinks-Robertson (1991). "Allelic and ectopic interactions in recombination-defective yeast strains." Genetics 127(1): 53-60. Steininger, F. F. (1999). "Chronostratigraphy, geochronology and biochronology of the Miocene “European land mammal mega-zones”(ELMMZ) and the Miocene “Mammal-zones (MN-zones)”." The Miocene land mammals of Europe: 9-24. Stewart, B. S., P. K. Yochem, R. L. DeLong and G. A. Antonelis (1993). Trends in abundance and status of pinnipeds on the southern California Channel Islands. Third California Islands symposium: recent advances in research on the California Islands. Santa Barbara Museum of Natural History, Santa Barbara, California, USA. Stirling, I. (1970). "Observations on the behavior of the New Zealand fur seal (Arctocephalus forsteri)." Journal of Mammalogy 51(4): 766-778. Storz, J. F. (2005). "Nonrandom dispersal and local adaptation." Jay F. Storz Publications: 11. Street, R. J. (1964). "Feeding habits of the New Zealand fur seal." New Zealand Marine Department of Fisheries Technical Report 9: 20. Sugg, D. W., R. K. Chesser, F. Stephen Dobson and J. L. Hoogland (1996). "Population genetics meets behavioral ecology." Trends in Ecology & Evolution 11(8): 338-342. Surridge, A., K. Ibrahim, D. Bell, N. Webb, C. Rico and G. Hewitt (1999). "Fine‐scale genetic structuring in a natural population of European wild rabbits (Oryctolagus cuniculus)." Molecular Ecology 8(2): 299-307. Sweeney, J. and W. Gilmartin (1974). "Survey of diseases in free-living California sea lions." Journal of Wildlife Diseases 10(4): 370-376. Symondson, W. O. C. (2002). "Molecular identification of prey in predator diets." Molecular Ecology 11(4): 627-641. Taberlet, P., L. P. Waits and G. Luikart (1999). "Noninvasive genetic sampling: look before you leap." Trends in Ecology & Evolution 14(8): 323-327. Tajima, F. (1983). "Evolutionary relationship of DNA sequences in finite populations." Genetics 105(2): 437-460. Tajima, F. (1989). "Statistical method for testing the neutral mutation hypothesis by DNA polymorphism." Genetics 123(3): 585-595. Tavaré, S., D. J. Balding, R. C. Griffiths and P. Donnelly (1997). "Inferring coalescence times from DNA sequence data." Genetics 145(2): 505-518. Taylor, R. (1982). "New Zealand fur seals at the Bounty Islands." New Zealand journal of marine and freshwater research 16(1): 1-9. Taylor, R., K. Barton, P. Wilson, B. Thomas and B. Karl (1995). "Population status and breeding of New Zealand fur seals (Arctocephalus forsteri) in the Nelson‐northern Marlborough region, 1991–94." New Zealand journal of marine and freshwater research 29(2): 223-234. Taylor, R. H. (1982). "New Zealand fur seals at the Bounty Islands." New Zealand journal of marine and freshwater research 16(1): 1-10. 157 Taylor, R. H., K. J. Barton, P. R. Wilson, B. W. Thomas and B. J. Karl (1995). "Population status and breeding of New Zealand fur seals (Arctocephalus forsteri) in the Nelson-northern Marlborough region, 1991–94." New Zealand journal of marine and freshwater research 29(2): 223-234. Todd, B. (2002). Seals and Sea lions. Auckland, Reed Publishing Ltd. Tollit, D. J., M. J. Steward, P. M. Thompson, G. J. Pierce, M. B. Santos and S. Hughes (1997). "Species and size differences in the digestion of otoliths and beaks: implications for estimates of pinniped diet composition." Canadian Journal of Fisheries and Aquatic Sciences 54(1): 105-119. Trites, A., V. Christensen and D. Pauly (2006). "Effects of fisheries on ecosystems: just another top predator?". Trites, A. W. and G. A. Antonelis (1994). "The influence of climatic seasonality on the life cycle of the Pribilof northern fur seal." Marine Mammal Science 10(3): 311-324. Trites, A. W., V. Christensen and D. Pauly (1997). "Competition between fisheries and marine mammals for prey and primary production in the Pacific Ocean." Journal of Northwest Atlantic Fishery Science 22: 173-187. Trotter, M. M. (1972). "A Moa-hunter site near the mouth of the Rakaia River, South Island." Records of the Canterbury Museum 9(2): 129-150. Turner, T. (1981). "Blood protein variation in a population of Ethiopian vervet monkeys (Cercopithecus aethiops aethiops)." American journal of physical anthropology 55(2): 225-232. Urian, K. W., S. Hofmann, R. S. Wells and A. J. Read (2009). "Fine‐scale population structure of bottlenose dolphins (Tursiops truncatus) in Tampa Bay, Florida." Marine Mammal Science 25(3): 619638. Valqui, J., G. B. Hartl and F. E. Zachos (2010). "Non-invasive genetic analysis reveals high levels of mtDNA variability in the endangered South-American marine otter (lontra felina)." Conservation genetics 11(5): 2067-2072. Van Den Hoff, J. and M. G. Morrice (2008). "Sleeper shark (Somniosus antarcticus) and other bite wounds observed on southern elephant seals (Mirounga leonina) at Macquarie Island." Marine mammal science 24(1): 239-247. Van Oorschot, R., R. Weston and M. Jones (1998). Retrieval of DNA from touched objects. Proceedings of the 14th International Symposium on the Forensic Sciences of Australian and New Zealand Forensic Science Society, Adelaide. van Staaden, M. J., R. K. Chesser and G. R. Michener (1994). "Genetic correlations and matrilineal structure in a population of Spermophilus richardsonii." Journal of Mammalogy: 573-582. Vetter, W. and W. Jun (2003). "Non-polar halogenated natural products bioaccumulated in marine samples. II. Brominated and mixed halogenated compounds." Chemosphere 52(2): 423-431. Vetter, W., W. Jun and G. Althoff (2003). "Non-polar halogenated natural products bioaccumulated in marine samples. I. 2, 3, 3', 4, 4', 5, 5'-Heptachloro-1'-methyl-1, 2'-bipyrrole (Q1)." Chemosphere 52(2): 415-422. Vetter, W., E. Scholz, C. Gaus, J. F. Müller and D. Haynes (2001). "Anthropogenic and natural organohalogen compounds in blubber of dolphins and dugongs (Dugong dugon) from northeastern Australia." Archives of environmental Contamination and Toxicology 41(2): 221-231. Viard, F., F. Justy and P. Jarne (1997). "Population dynamics inferred from temporal variation at microsatellite loci in the selfing snail Bulinus truncatus." Genetics 146(3): 973-982. Wakeley, J. (1999). "Nonequilibrium migration in human history." Genetics 153(4): 1863-1871. Walker, A. H., D. Najarian, D. L. White, J. Jaffe, P. A. Kanetsky and T. R. Rebbeck (1999). "Collection of genomic DNA by buccal swabs for polymerase chain reaction-based biomarker assays." Environmental health perspectives 107(7): 517. Warnock, W. G., J. B. Rasmussen and E. B. Taylor (2010). "Genetic clustering methods reveal bull trout (Salvelinus confluentus) fine-scale population structure as a spatially nested hierarchy." Conservation Genetics 11(4): 1421-1433. Wasser, S. K., C. S. Houston, G. M. Koehler, G. G. Cadd and S. R. Fain (1997). "Techniques for application of faecal DNA methods to field studies of Ursids." Molecular Ecology 6(11): 1091-1097. Watterson, G. (1975). "On the number of segregating sites in genetical models without recombination." Theoretical population biology 7(2): 256-276. 158 Wayne, R. K., A. Meyer, N. Lehman, B. Van Valkenburgh, P. W. Kat, T. K. Fuller, D. Girman and S. J. O'Brien (1990). "Large sequence divergence among mitochondrial DNA genotypes within populations of eastern African black-backed jackals." Proceedings of the National Academy of Sciences 87(5): 1772-1776. Weber, D., B. Stewart and N. Lehman (2004). "Genetic consequences of a severe population bottleneck in the Guadalupe fur seal (Arctocephalus townsendi)." Journal of Heredity 95(2): 144-153. Weber, D., B. S. Stewart, J. C. Garza and N. Lehman (2000). "An empirical genetic assessment of the severity of the northern elephant seal population bottleneck." Current Biology 10(20): 1287-1290. Wegmann, D., C. Leuenberger, S. Neuenschwander and L. Excoffier (2010). "ABCtoolbox: a versatile toolkit for approximate Bayesian computations." BMC bioinformatics 11(1): 116. Wickenheiser, R. A. (2002). "Trace DNA: a review, discussion of theory, and application of the transfer of trace quantities of DNA through skin contact." Journal of forensic sciences 47(3): 442-450. Wildt, D., M. Bush, K. Goodrowe, C. Packer, A. Pusey, J. Brown, P. Joslin and S. O'Brien (1987). "Reproductive and genetic consequences of founding isolated lion populations." Nature 329(6137): 328-331. Wilkinson, G. S. (1985). "The social organization of the common vampire bat." Behavioral Ecology and Sociobiology 17(2): 123-134. Williams, C. L., K. Blejwas, J. J. Johnston and M. M. Jaeger (2003). "A coyote in sheep's clothing: predator identification from saliva." Wildlife Society Bulletin: 926-932. Williams, S., P. Foster and D. Littlewood (2014). "The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny." Gene 533(1): 38-47. Wilmshurst, J. M., A. J. Anderson, T. F. Higham and T. H. Worthy (2008). "Dating the late prehistoric dispersal of Polynesians to New Zealand using the commensal Pacific rat." Proceedings of the National Academy of Sciences 105(22): 7676-7680. Wilson, G. J. (1981). "Distribution and Abundance of the New Zealand fur seal Arctocephalus forsteri." Fisheries Research Division Occasional Publication(20). Work, T. M., R. L. DeLong, T. R. Spraker and S. R. Melin (1993). "Halothane anesthesia as a method of immobilizing free-ranging California sea lions (Zalophus californianus)." Journal of Zoo and Wildlife Medicine 24(4): 482-487. Worthington Wilmer, J., P. Allen, P. Pomeroy, S. Twiss and W. Amos (1999). "Where have all the fathers gone? An extensive microsatellite analysis of paternity in the grey seal (Halichoerus grypus)." Molecular Ecology 8(9): 1417-1429. Wright, S. (1938). "Size of population and breeding structure in relation to evolution." Science 87(2263): 430-431. WÜRsig, B. and M. WÜRsig (1977). "The photographic determination of group size, composition, and stability of coastal porpoises (Tursiops truncatus)." Science 198(4318): 755. Wynen, L. P., S. D. Goldsworthy, C. Guinet, M. N. Bester, I. L. Boyd, I. Gjertz, G. J. Hofmeyr, R. W. White and R. Slade (2000). "Postsealing genetic variation and population structure of two species of fur seal (Arctocephalus gazella and A. tropicalis)." Molecular Ecology 9(3): 299-314. Wynen, L. P., S. D. Goldsworthy, S. J. Insley, M. Adams, J. W. Bickham, J. Francis, J. P. Gallo, A. R. Hoelzel, P. Majluf and R. W. White (2001). "Phylogenetic relationships within the eared seals (Otariidae: Carnivora): implications for the historical biogeography of the family." Molecular phylogenetics and evolution 21(2): 270-284. Xue, Y., Q. Wang, Q. Long, B. L. Ng, H. Swerdlow, J. Burton, C. Skuce, R. Taylor, Z. Abdellah and Y. Zhao (2009). "Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree." Current Biology 19(17): 1453-1457. Yu, L., D. Peng, J. Liu, P. Luan, L. Liang, H. Lee, M. Lee, O. A. Ryder and Y. Zhang (2011). "On the phylogeny of Mustelidae subfamilies: analysis of seventeen nuclear non-coding loci and mitochondrial complete genomes." BMC evolutionary biology 11(1): 92. Zasloff, M. (2011). "Observations on the Remarkable (and Mysterious) Wound-Healing Process of the Bottlenose Dolphin." Journal of Investigative Dermatology. 159 160