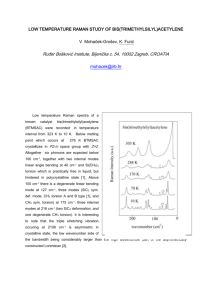
Accepted Manuscript
advertisement

Accepted Manuscript Excited-State Proton Transfer of Photoexcited Pyranine in Water Observed by Femtosecond Stimulated Raman Spectroscopy Fangyuan Han, Weimin Liu, Chong Fang PII: DOI: Reference: S0301-0104(13)00148-1 http://dx.doi.org/10.1016/j.chemphys.2013.03.009 CHEMPH 8832 To appear in: Chemical Physics Please cite this article as: F. Han, W. Liu, C. Fang, Excited-State Proton Transfer of Photoexcited Pyranine in Water Observed by Femtosecond Stimulated Raman Spectroscopy, Chemical Physics (2013), doi: http://dx.doi.org/ 10.1016/j.chemphys.2013.03.009 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. Excited-State Proton Transfer of Photoexcited Pyranine in Water Observed by Femtosecond Stimulated Raman Spectroscopy Fangyuan Han, Weimin Liu, and Chong Fang* Department of Chemistry, Oregon State University, Corvallis, OR 97331 U.S.A. *Corresponding author. Tel.: +1 541 737 6704. E-mail address: Chong.Fang@oregonstate.edu (C. Fang). ABSTRACT We use femtosecond stimulated Raman spectroscopy (FSRS) to illuminate the choreography of intermolecular excited-state proton transfer (ESPT) of photoacid pyranine (8-hydroxypyrene-1,3,6-trisulfonic acid, HPTS) in water. The multidimensional reaction coordinate responsible for photoacidity is revealed to involve sequential activation of characteristic skeletal motions during the ca. 1 ps preparation stage preceding ESPT. The initial ring-coplanarity breaking follows in-plane ring breathing (191 cm-1), and is facilitated by HPTS ring wagging (108 cm-1) and ring-H out-of-plane motions (321, 362, 952 cm-1), which largely decay within ~1 ps. ESPT then occurs with intrinsic inhomogeneity via various number of intervening water molecules over relatively larger distances than those in acetate-water system. The intricate relationship between the time-resolved excited-state vibrational modes of HPTS reveals the essential role of coherent low-frequency skeletal motions gating ESPT, and the multi-staged proton-transfer process having the kinetic isotope effect (KIE) value of 3–4 in aqueous solution on the 5–200 ps timescale. Keywords: Femtosecond stimulated Raman spectroscopy; excited-state proton transfer; molecular conformational dynamics; low-frequency skeletal motions; hydrogen bond dynamics; photoacid 1. Introduction Proton transfer (PT) in water plays a ubiquitous and important role in many chemical and biological processes [1-4], particularly concerning the omnipresent hydrogen bonds (H-bonds). In living systems, protons catalyze a myriad of aqueous reactions and serve as an important means for transient energy transport and storage, so the significance of investigating proton functionality cannot be overstated. A widely used experimental approach to study PT is to precisely activate it via photoexcitation of a photoacid. For these molecules, absorption of a photon leads to a significant increase of the molecular acidity, and therefore triggers a series of events contributing to the overall processes of intermolecular excited state proton transfer (ESPT) [3]. These molecular processes can be distinguished as electronic redistribution and hydrogen bond rearrangement in sub-femtosecond (sub-fs) to fs time range, proton dissociation and 1 solvation processes in sub-picosecond (sub-ps) to ps time scale, and proton diffusion as well as proton recombination and quenching reactions in ps to nanosecond (ns) time regime. The multidimensionality of the potential energy surface (PES) of photoacids needs to be studied in considerable detail to reveal the anharmonic coupling matrix within the molecule, and the structural origin of chemical reactivity [5-10]. Pyranine (8-hydroxypyrene-1,3,6-trisulfonic acid, or HPTS), a commonly used photoacid, has a pKa value of ~7 in the electronic ground state (S0) that drops to ~0 upon photoexcitation [11-27] and serves as a pragmatic system to scrutinize the reaction mechanism for a transducing photosensitive molecule undergoing ESPT. HPTS has strong photoabsorption near 400 nm and can thus be easily triggered through optical excitation, which induces a vertical transition between S0 and the electronic excited states (commonly the singlet excited state S1), with the subsequent ESPT followed by fluorescence and other electronic absorption changes. The vibronic wavepacket generated by the optical excitation samples the excited-state PES to relax energy, and eventually finds ways to go back to S0. The rate and mechanism of intermolecular ESPT of HPTS in water (e.g., initial step ROH*+H2O→RO *+H3O+) have been monitored with different electronic and vibrational spectroscopies, including the time-resolved fluorescence spectroscopy [11-13, 15], transient visible [18, 22] or mid-IR spectroscopy [17, 19-21, 23-26]. In particular, Tran-Thi, et al. and Leiderman with co-workers used both timeresolved fluorescence and transient absorption spectroscopy to investigate ESPT from HPTS to water [15, 18]. They found that the ESPT processes involve ultrafast solvation dynamics (0.3–0.8 ps), two steps of proton dissociation (the formation of a contact ion pair RO *∙∙∙H3O+ in ~3 ps and separation of ion pair RO *----H3O+ in 87–100 ps), and a subsequent long-lifetime RO * emission/fluorescence step (~5.3 ns). In D2O, the dynamics of the two proton-dissociation steps are delayed to ~5 ps and 210–300 ps, respectively [17-19, 22, 24]. This represents a kinetic isotope effect (KIE) of ca. 3 for the long time constant observed in the ESPT dynamics [3]. Recently, ESPT of HPTS and different carboxylate base complexes dissolved in aqueous solution was extensively studied using fs UV-pump mid-IR-probe spectroscopy [17, 20, 21, 23-25], wherein the PT reactions from HPTS to the base exhibit multiexponential decay processes with sub-ps to hundreds of ps time constants. Upon addition of stronger bases such as acetate to aqueous solution, the ESPT dynamics speed up significantly due to the intermolecular PT from HPTS to the base molecule, which forms a greater amount of more tightly bound H-bonding complexes over relatively short distances, and dominates the reaction through the water medium. It was reported that the optimal PT distance is ~0.75 nm, corresponding to approximately two water molecules in between the proton donor (acid) and acceptor (base) in the Eigen-Weller model [1, 21, 25]. A recent theoretical study on the ESPT dynamics of 6-hydroxyquinolinium (6HQc), a photoacid smaller than HPTS, found that the threshold of the water cluster size is 3 for its ESPT in aqueous solvent [28]. Water participates in the PT reaction by receiving a proton from the proton-donating group and then releasing a proton to the protonaccepting group in the sequential or concerted manner depending upon the base concentration [23, 25]. In protic solvents such as water, a single H-bond or a H-bonded water matrix connects the proton donor and accepter, in which protons are highly mobile and can be transferred via intricate energy relaxation pathways. In addition, water will 2 stabilize and/or destabilize various charge-transferred species as PT occurs. However, after years of active research on ESPT from HPTS to water, the mechanism still remains unclear particularly concerning the involved conformational dynamics of the photoacid. To examine the ESPT process in solvated HPTS is to understand its photochemistry using a direct bottom-up approach. We need to go beyond measuring the electronic response of the molecular system or collecting the time-resolved fluorescence data, because those data can only infer the electronic PES with no detailed vibrational or structural information. The visible-pump mid-IR probe studies have their shortcomings including the limited time resolution and spectral range, and some uncertainty correlating relatively broad spectral features with specific molecular motions. In order to capture the molecular snapshots of the photoexcited HPTS immediately following photoexcitation and to track the choreography of the transferring proton in conjunction with all the coupled atomistic motions, the time-resolved vibrational spectra of HPTS on S1 need to be analyzed in detail. The ability to concomitantly track the acceptor response is an added bonus [27]. The emerging femtosecond stimulated Raman spectroscopy (FSRS) [29-31] provides that level of information with simultaneously high spectral and temporal resolution, perfectly suitable to probe the non-equilibrium atomic motions of photoacids following photoexcitation. Since fluorescence is typically on the ns timescale, the fs resolution of the FSRS setup ensures the unravelment of earlier (i.e., pre-fluorescence) processes, mainly skeletal motions and ESPT, following the vibronic wavepacket motion out of the Franck-Condon (FC) region of ROH* (PA*) toward the deprotonation barrier crossing, and before the nascent RO * (PB*) state relaxes back to PB via fluorescence. One of the most appealing advantages of FSRS is the resolution achievable in both the spectral and time domain, which merits some discussion. This is accomplished by the unique sequence of laser pulses with either the fs or ps time duration, and the dispersive signal detection scheme without temporally resolving the Raman scattering photon carrying the impulsively excited vibrational coherence free induction decay (FID) information. The spectral resolution is thus determined by the FID time as well as the Raman pump pulse duration, because the Fourier transform of the coherence decay convoluted with the Raman pump pulse duration will dictate the frequency linewidth of the observed vibrational mode. The typical FID time of a vibrational coherence is on the timescale of 1–3 ps, and since we can generate the Raman pump with ~4 ps full-width-athalf-maximum (fwhm) [27], the spectral resolution can be well below 10 cm-1. The temporal resolution, on the other hand, is decoupled from the pulse duration of the Raman pump and can be smaller than 30 fs. This is due to the fact that the vibrational coherence is precisely generated upon the simultaneous arrival of the Raman pump and probe photons on the sample spot, and because the Raman probe is a broadband pulse with ~30 fs pulse duration, its cross correlation with the preceding fs actinic photoexcitation pulse is well defined and can be very small. It is therefore key to have that fs-ps-fs pulse sequence to enable us precisely trigger vibrational coherences and probe structural evolution in real time [31, 32]. The ability to track non-equilibrium conformational dynamics thus makes FSRS a unique and powerful technique to study photosensitive molecules in condensed phase. Furthermore, the advantages of FSRS include the broad spectral window spanning more than 1200 cm-1 for one spectrograph grating position, selective excitation of the chromophore (e.g., it could be embedded inside a big protein such as wild-type green fluorescent protein, a.k.a. wtGFP) [9], 3 convenience of using water as the solvent, fluorescence rejection, moderate sample concentration requirement, fast data acquisition (e.g., the CCD detector is synchronized with the main laser repetition rate of 1 KHz) with high signal-to-noise ratio (SNR), and versatile control of each individual pulse across the UV to NIR regime [27, 33]. In our previous report [27], we used the newly developed FSRS setup [29-31, 34] to capture the molecular structural snapshots of HPTS in S1 as it pushes the hydroxyl proton toward the acetate ion in aqueous solution. The temporal evolution of excited-state Raman modes, especially in the low-frequency regime, reveals the remarkable ultrafast structural dynamics events along the ESPT multidimensional reaction coordinate of photoexcited HPTS starting from time zero. The corresponding vibrational frequency range of 100−500 cm−1 cannot be directly observed in the mid-IR region in water. The molecular skeletal motions therein reduce the distance between the proton donor and acceptor, which may adopt a largely different electronic character between S0 and S1 states, and result in lowering the effective barrier to proton hopping [27, 35-39]. Our experimental results show that the vibrational marker bands attributed to the deprotonated form of HPTS appear earlier and faster than the nascent monomeric acetic acid peak, indicating that various number of water molecules actively participate in the ESPT chain. The goal of this contribution is to delineate the choreography of intermolecular ESPT reaction of photoexcited HPTS in pure water. The absence of strong bases such as acetate excludes the complexity of various degrees of driving forces to attract the hydroxyl proton of HPTS, and provides a clean background to investigate the bilateral interaction between HPTS and the H-bonded water matrix. Upon photoexcitation of HPTS, water will act as the proton acceptor because its pKa is higher than zero (the pKa for the S1 state of HPTS). But since the proton-accepting strength of water is weaker than that of acetate, slower ESPT dynamics may be observed. Also, the accompanying skeletal motions of HPTS that become Raman active and show pronounced activities should be exposed by the time-resolved FSRS spectra of the S1 state of HPTS surrounded by water molecules only. The comparison with previous results on HPTS in acetate water solution will provide deep insights on the dynamic variation of the H-bonding network and the driving force for intermolecular ESPT, as well as the conserved skeletal motions gating ESPT between photoexcited HPTS and proton acceptor molecules in aqueous solution. 2. Experimental methods The photoacid pyranine (HPTS) was purchased from TCI America (≥85%). It was previously found to have no detectable spectral difference from the ≥97% purity sample from Aldrich, so we used the ≥85% purity sample (11 mM) in millipore water solution (pH≈6) after 0.22-m filtration but without further purification [27]. The Raman spectra of HPTS through the ESPT reaction are collected in both H2O and D2O (99.9% D, Cambridge Isotope Laboratories, Inc.) solutions. Though we did not intentionally use DPTS to start with, the concentration difference between HPTS (11 mM) and D2O (~55.3 M) leads to the almost complete deuteration at the phenolic hydroxyl end of HPTS after 1-2 days of storage and H/D exchange in solution at room temperature. The UV/Vis spectrum shows OD≈26/mm at the protonated HPTS (PA) absorption peak (PA≈24,000 M-1·cm-1) and this is to ensure that we have enough SNR for both ground-state and 4 excited-state FSRS data. The S0 and S1 PES of PA dictates the electronic resonance conditions here and the 800 nm Raman pump pulse we use does not induce strong Raman vibrational features, in contrast to the previously studied wtGFP chromophore case [9]. The experimental procedure has been reported in detail elsewhere [27, 34]. Briefly, our FSRS setup (Fig. 1) uses a fs Ti:Sapphire laser amplifier system (Legend Elite USP-1K-HE, Coherent Inc.), which provides ~35 fs, 4 W laser pulse centered at 800 nm with 1 kHz repetition rate. Half of the output laser beam is split into three beams required for FSRS, which are depicted in different colors in Fig. 1. Around 200 μJ/pulse of the laser output is frequency-doubled using a β-barium borate crystal (BBO, type-I, phase matching angle θ=27.8°, 0.3 mm thickness) to generate the actinic pump pulse at 400 nm with the pulse energy of 50 μJ/pulse, then compressed by a prism pair (Suprasil1, CVI Melles Griot) to ~40 fs. The average power of the 400 nm photoexcitation pulse is then attenuated to ca. 1 mW for excited-state FSRS measurement, to ensure a stable spectral baseline as well as enough ground-state (S0) depletion to observe excited-state vibrational features of HPTS in water in the linear regime [34]. Persistent and unchanged S0 depletion is observed as spectral dips at S0 vibrational frequencies when higher photoexcitation power (e.g., 1.5 mW) is used, whereas lower power at 0.5 mW yields smaller depletion than the 1 mW photoexcitation case. About 15 μJ/pulse of the laser output is focused on a Z-cut single crystal sapphire plate with 2 mm thickness to generate the supercontinuum white light. The wavelength range from 805–915 nm of the white light that corresponds to ca. 100–1600 cm−1 Stokes Raman shift to the 800 nm fundamental is selected using a long-wavelength pass filter, and then compressed by a fused silica prism pair (Thorlabs, Inc.) to produce ~35 fs broadband Raman probe pulse. The Raman pump pulse with ~10 cm−1 bandwidth and pulse duration of 3.5 ps is produced by a homemade grating-based spectral filter (1200 grooves/mm, wavelength first order at 750 nm, blaze angle θ=26.7°). The collimated Raman pump, probe, and the photoexcitation beams are all focused onto the sample cell using an off-axis parabolic reflective mirror (to avoid chirp), with the focus size of ~150 m for the Raman pump and actinic pump beams, and ~100 m for the much weaker Raman probe beam. The dispersed FSRS spectra are first calibrated using carbon tetrachloride (CCl4) and ethanol (CH3CH2OH) mixed solution as a standard that spans the Raman frequency range from ca. 200–1600 cm−1 [27]. The ground-state FSRS signal of the standard solution is maximized through spatial and temporal overlap adjustment of the Raman pump and probe beams, which are at a crossing angle of ~3.5º to ensure a relatively long interaction length. The average power of the 800 nm Raman pump pulse is ~6 mW and we set it to balance the signal strength, the peak width and the stability of the baseline. A higher Raman pump power will increase the signal strength but at the expense of broader peaks and fluctuating baselines particularly in the low-frequency region. The probe beam after the sample is sent into a spectrograph and dispersed by a 600 grooves/mm grating (wavelength first order at 1000 nm, blaze angle θ=17.5°), and then imaged onto a frontilluminated charge-coupled device (CCD) camera (Princeton Instruments, PIXIS 100F) consisting of a 1340 × 100 pixel array, synchronized with the main laser repetition. The Raman pump beam is chopped at half of the laser repetition rate (i.e., 500 Hz) to measure the single-shot Raman probe spectrum, with the sequential Raman pump on and off conditions repeatedly. The FSRS spectrum is collected, calculated, averaged and displayed on-screen using an updated LabVIEW program incorporating STIK scientific 5 imaging toolkit (R-Cubed software, NJ) for the CCD camera. The spatial and temporal overlap between the two fs pulses, the 400 nm photoexcitation and the Raman probe continuum at a crossing angle of ~4º, is finely adjusted by optimizing the Optical Kerr Effect (OKE) signal at the front portion in the liquid standard sample, with the typical cross-correlation time measured to be ~140 fs full width at half maximum (fwhm) for our time-resolved FSRS experiments in the aqueous solution. The sample volume of ~800 µL is used in a 1-mm pathlength flow cell (48-Q-1, Starna Cells) to avoid sample degradation under intense pulsed laser excitation. Higher excitation power at ~1.5 mW causes irreversible photodegradation despite rapid flow of the sample solution, showing strong ground-state vibrational peak depletion even when the 400 nm photoexcitation pulse arrives later (e.g., 3 ps) than the Raman pump-probe pair. Less than 5% population change (mainly from the protonated to the deprotonated form of HPTS) is observed from the UV-Vis spectra before and after ~2 hours of excited-state FSRS measurement with 1 mW photoexcitation power used, so the population variation effect on our FSRS spectral analysis is negligible. The kinetics of the stimulated Raman intensities in the excited-state FSRS spectra (see below) are multi-exponentially fitted and convoluted with the aforementioned instrument response time of ~140 fs. 3. Experimental results The ground-state FSRS spectrum of HPTS has been measured at two grating positions of the spectrograph (Fig. 1), in order to cover the wide spectral range of 50– 2000 cm-1. The slit width in Raman pump generation is set below 0.1 mm still with enough output power to induce the Raman transition, enabling the observation of the narrow linewidth of the Raman peak close to the natural linewidth seen in the continuous wave (cw) excitation case. Fig. 2 shows the FSRS spectrum of the S0 state of HPTS, with the vibrational modes above 1000 cm-1 dominating the spectrum. It is notable that the maximum electronic absorption peak of HPTS in water is at ~404 nm for the protonated form (PA) of the photoacid [27], therefore we rely on the two-photon absorption cross section to obtain the ground-state FSRS spectrum of HPTS with the ~800 nm Raman pump pulse. The weaker spectral features observed in the excited-state FSRS spectra of HPTS in water in comparison to the wtGFP chromophore data [9] suggest that the resonance enhancement factor for S1 vibrational features is insignificant for HPTS because the 800 nm Raman pump does not closely match the energy gap between S1 and other higher-lying electronic states of the PA or PB form [40, 41]. Density functional theory (DFT, RB3LYP and UB3LYP) calculations using Gaussian 09 [42] and the 6-311G++(2d,2p) basis sets on the protonated HPTS molecule (–3 charge, singlet state), with the water solvation effect included by a polarizable continuum model (PCM) using the integral equation formalism (IEF-PCM-H2O), yield Raman peak frequencies to be compared with experimental results using a typical scaling factor of 0.96. The calculated ground-state frequencies are considered to be more accurate in the low-frequency region due to the collective nature of those participating atomic motions, which should remain largely unshifted in the first singlet electronic excited state S1 [9, 39]. The detailed assignment of key vibrational modes that show significant activities in H2O is listed in Table 1, and can also be found in our earlier 6 report [27]. In brief, the skeletal modes below 1000 cm-1 assume weak Raman activities in the ground state due to the coplanarity of the aromatic four-ring system of HPTS, which agrees with DFT calculation results. The ring-H rocking, C–O stretching and C=C stretching motions above 1000 cm-1 exhibit strong peak intensities due to their relatively high tendency for electron redistribution and large Raman polarizabilities upon electronic excitation [40, 41]. Reckoning the two-photon absorption of HPTS at the 800 nm Raman pump wavelength, it suggests that these high-frequency modes are along the PES slope in the FC region of PA* with the bottom well of S1 displaced from the equilibrium position of S0. It is notable that the deprotonated (PB) HPTS has relatively higher intensities of the low-frequency modes in comparison with the protonated (PA) HPTS, consistent with a more twisted structure of the chromophore after the departure of the phenolic proton [27] even in S0. A pictorial representation of the ESPT process from HPTS to water through the H-bonding chain can be found in the Fig. 2 insert. Fig. 3 shows the time-resolved FSRS spectra of HPTS in water following ~1 mW of 400 nm photoexcitation pulse, from –1 ps to 150 ps. The relatively high peak power is used for excitation due to the high concentration of HPTS being used (OD≈26/mm at the PA absorption peak), making it essentially nontransparent to the 400 nm laser pulse. The local heating effect is minimized by rapidly flowing the sample through a sealed reservoir away from light at room temperature. The ground-state (S0) Raman spectrum is collected periodically throughout the excited-state FSRS measurement and averaged, followed by subtraction from each time-delayed spectrum to discern the difference spectrum with various dips and peaks, then a percentage of the fitted S0 spectrum is added back to consistently fill those dips and reveal the positive excited-state features across the wide spectral detection window. The negative-time spectra corroborate the robustness of the current experimental approach in that no chemistry is happening before photoexcitation, and the sample also holds well without photodegradation throughout the FSRS scan. The maximum ground-state depletion achieved in this experiment is ~12%. It is possible that individual vibrational modes might assume slightly different bleaching dynamics, but to a generally good approximation, we envision the ground-state vibrational peaks to be depleted simultaneously upon reaching a different electronic state. Though we commonly attribute the remaining positive peaks to S1 vibrational transitions, it is conceivable that these transient features might be associated with S0 vibrational transitions [9, 43]. The latter case requires the photoexcited wavepacket to be directly generated on S0, or for the initially generated S1 wavepacket to quickly relax back to S0 on the timescale of our FSRS measurement, which is fs to ps, thus excluding the fluorescence pathway that is typically on the ns timescale. However, the fact that the excited-state spectrum at T=0 fs differs from the ground-state spectrum and also shows wider linewidth indicates that the vibronic wavepacket and vibrational coherence is generated on S1, with significantly different electronic distribution over the HPTS ring system. Furthermore, the pronounced activities of the low-frequency modes following photoexcitation correlate well with the spectral activities of the high-frequency modes, with the peak kinetic analysis intimately reflecting the ESPT dynamics responsible for its photoacidity in solution phase. There are a number of prominent low-frequency modes in Fig. 3 that show PA* characteristics, which grow in following photoexcitation, and gradually decay away. These include 108, 125, 143, 191, 321, 362, 630, and 952 cm-1 (Table 1), which exhibit different dynamics that will be analyzed and discussed in detail below. The correlated 7 high-frequency modes appear at 1048, 1180, and 1285 cm-1. In particular, the vibrational modes at 143, 191, 952, and 1180 cm-1 are predominantly active before ~1 ps, and are labeled in red in Fig. 3 and also highlighted by red boxes. Evidence of quantum beats shows up in the kinetic plot of the 143 cm-1 mode intensity. The 870 cm-1 mode shows complex dynamics within the experimental time window of 150 ps, and involves ringhydrogen out-of-plane (HOOP) motions from both the PA* and PB* modes as shown by calculations. The mode at 1138 cm-1 is an interesting case in that the ephemeral feature before 1 ps might be closely associated with PA* adopting different electronic character from the ground state in association with photoacid skeletal motions, while the gradual increase of the peak intensity after 1 ps can be attributed to the accumulation of PB* as ESPT occurs. The modes at 276 and 460 cm-1 have a delayed onset in comparison to the promptly emerged PA* modes, and display the gradual increase within the temporal window of the experiment, so we assign them to PB* modes (labeled in blue in Fig. 3) that will eventually decay away on the ns timescale due to fluorescence. Fig. 4 displays the temporal evolution of the 1048 cm-1 mode that is characteristic of the PA* dynamics up to 150 ps. The integrated peak area is used to best capture the essence of the stimulated Raman peak intensity. After deconvolution from the crosscorrelation time of 140 fs measured from the OKE signal of the actinic pump and Raman probe pulses, the 1048 cm-1 mode rises within ~210 fs, and decays bi-exponentially with time constants of 4.5 ps (32%) and 150 ps (68%). Fitting results of various vibrational modes are listed in Table 1, and the relative weight of the fitted exponentials is shown in parentheses. The 150 ps time constant is less accurately determined than the 4.5 ps time constant due to the detection time window, limited by the 1-inch motorized translation stage that we used. The insert in Fig. 4 shows the detailed time-dependent peak intensity plot up to 10 ps, with the signal strength indicated by the double-arrowed vertical line. The time correlation between the nascent PA* and PB* modes can be found in Fig. 5. The 1285 cm-1 PA* mode rises exponentially with a time constant of ~190 fs, and decays with two time constants of 4.3 ps (40%) and 215 ps (60%). Note that the long decay time constant is larger than that of the 1048 cm-1 mode, which may hint an overlapping PB* mode contribution around 1285 cm-1. The 460 cm-1 PB* mode rises biexponentially with time constants of 680 fs (26%) and 130 ps (74%); while the 1138 cm-1 PB* mode after 1 ps can be fitted by two rising exponentials with time constants of 5 ps (12%) and 78 ps (88%). The difference between the two PB* modes can be explained by the atomistic motions responsible for them: the 460 cm-1 mode is primarily an HPTS ring asymmetric wagging motion and may start gaining Raman intensity in an earlier stage of ESPT, whereas the 1138 cm-1 mode is the HPTS phenolic CO···(H) inplane rocking motion that takes longer to appear but rises faster on the tens of ps timescale overall (78 ps vs. 130 ps). The sub-ps onset of the 460 cm-1 mode is indicative of the activation of some coherent low-frequency modes preceding ESPT, in which the ring wagging motion plays an important role in modulating the intermolecular distance between the hydroxyl group of HPTS and the neighboring proton acceptors, e.g., water molecules in this work. The transient coherent PA* ring wags can potentially generate a small proportion of (~26% if estimated from the fast rise component of the 460 cm-1 mode) PB* state, with its ring wagging mode on the rise with a 680 fs time constant that is delayed to some of the fastest PA* in-plane modes (e.g., the 191 cm-1 mode). 8 A collage of low-frequency modes of photoexcited HPTS is plotted in Fig. 6, and their time-resolved peak intensity analysis is very informative. The most transient PA* peak, the 191 cm-1 mode, shows a rising exponential time constant of 320 fs and a decay constant of 540 fs. The 321 cm-1 PA* mode rises with a time constant of 650 fs, and has a biphasic decay with time constants of 1.1 ps (80%) and 75 ps (20%). The 276 cm-1 PB* mode intensity kinetic trace shows a biphasic rise with time constants of 550 fs (85%) and 110 ps (15%), which has some interesting differences from the 460 cm-1 PB* mode dynamics and will be discussed later. The peak kinetics and the frequency shift from ground-state peaks, as well as the intricate relationship between various time-resolved vibrational modes in FSRS, exclude the possibility of a simple scheme of Raman pump attenuation-induced ground state depletion. The matching rise and decay time constants such as ca. 550 and 650 fs (see Table 1) from multiple vibrational peak kinetic analysis infer the ESPT reaction mechanism of HPTS in water, showing vivid atomistic details as the chemical reaction proceeds on the anharmonic multidimensional PES in real time. To further investigate the structural origin of the observed kinetic processes of photoexcited HPTS, we have also conducted FSRS measurements on HPTS in D2O. The S1 vibrational modes at 1048 and 1138 cm-1 in H2O redshift to 1042 and 1136 cm-1 in D2O, respectively, which agrees with the retardation of mode vibrations upon deuteration. It is evident from the blue (H2O) and red (D2O) traces in Fig. 7 that the PA* decay and PB* rise all slacken in D2O, wherein the 1042 cm-1 mode rises with the time constant of 610 fs, and decays bi-exponentially with time constants of 24 ps (40%) and 650 ps (60%) (see Table 2). This represents a KIE of ~5.3 for the short time component, and ~4.3 for the long component of the peak intensity decay. The 1136 cm-1 mode in D2O has a biphasic rise with time constants of 21 ps (12%) and 200 ps (88%). A detailed account on the corresponding KIE appears below in the Discussion section. The enlarged PB* peak intensity kinetic plot up to 10 ps is shown in the insert, manifesting the complex peak dynamics of the HPTS phenolic CO···(H/D) in-plane rocking mode before 1 ps, which might be affected by the aforementioned transient PA* feature in that spectral region. It is notable that the short component of the PB* peak rise matches the short component of the PA* peak decay (5 vs. 4.5 ps in H2O, and 21 vs. 24 ps in D2O), but the long component is significantly shorter for the PB* rise than the PA* decay (78 vs. 150 ps in H 2O, and 200 vs. 650 ps in D2O). This can be explained by the fact that although ESPT is the dominant energy relaxation pathway for PA* to cross the barrier and produce PB*, there might be other relaxation pathways present to dissipate the photoexcitation energy being absorbed by HPTS. For instance, the direct fluorescence from the PA* state to PA leads to a decay time constant of ~4.5 ns, which can contribute to the elongation of the overall decay dynamics of PA*. On the contrary, the initial PB* peak intensity rise is a direct consequence of ESPT and is a more reliable parameter to report on the PT process and to compare the rate difference upon deuteration. The two lowest-frequency modes at 108 and 125 cm-1 observed in the timeresolved excited-state FSRS spectra in Fig. 3 are fitted with two overlapping gaussian profiles, and plotted against the time delay in Fig. 8. The 108 cm-1 mode rises with the 630 fs time constant, and has a biphasic exponential decay of 1.1 ps (75%) and 1.7 ns (25%). The 125 cm-1 mode has a delayed onset of ~1 ps, which can then be fitted with the biphasic rise time constants of 650 fs (55%) and 5.3 ps (45%), along with an exponential decay time constant of ~270 ps. Since we have observed a similar 106 cm-1 mode 9 previously in 11 mM HPTS in 2 M acetate water solution [27], and the conservative nature of such skeletal collective motions leads to similar vibrational frequencies in various aqueous solutions, we attribute the 108 cm-1 mode to the HPTS 4-ring out-ofplane (OOP) wagging motion of the photoexcited protonated chromophore (PA*). The more pronounced 125 cm-1 mode that gains intensity after a dwell of 1 ps but decays away after reaching the maximum at ~20 ps may be associated with a related ring translational motion of PA*, which might better facilitate the later stage of ESPT (e.g., nascent ion-pair separation) after the initial stage (e.g., heavy atom optimization of the Hbonding chain) is largely over on the ca. 5 ps timescale. Evidence of the coherent quantum beats can be clearly seen in the time-dependent intensity plot of the 952 cm-1 mode (Fig. 9). The convoluted multi-exponential fitting with the 140 fs cross-correlation time yields the rise time constant of ~140 fs, and the biphasic decay time constants of ~600 fs (78%) and 90 ps (22%). The insert of Fig. 9 manifests the mode intensity quantum beats with a period of ~360 fs, which is prominent up to 1 ps. Considering that this mode can be attributed to HPTS ring-HOOP motions mixed with some in-plane ring deformation from DFT calculations (Table 1), it is justifiable to correlate this particular PA* motion with one of the early-time reaction coordinates to set up the stage for subsequent ESPT from HPTS to water. 4. Discussion The ability to simultaneously monitor a multitude of vibrational modes on the electronically excited state (e.g., S1) in real time makes FSRS a powerful structural dynamics tool to study photosensitive molecules in solution phase [30, 31]. Here we investigate the commonly used photoacid HPTS in pure water, without the presence of any other proton acceptor, to ensure that ESPT only occurs between HPTS and water molecules within the H-bonded network upon photoexcitation. The simplicity of this system construct leads to the direct correlation between experimental data and theoretical calculations regarding the chromophore in complex with water molecules in condensed phase. The transient disappearance of PA* and appearance of PB* modes should in principle be correlated with ESPT, in competition with other radiative/nonradiative relaxation pathways from PA* or radiative pathways from PB*, on the fs to ns timescale. Furthermore, the excited-state vibrational peak temporal evolution (Figs. 3–9) from the well-defined time zero reveals the detailed elementary steps of molecular conformational dynamics responsible for ESPT and the photoacidity of HPTS in aqueous solution with unprecedented precision. It is important to point out that we are not temporally resolving the vibrational coherence FID once it is generated by the Raman pump-probe pair because the Raman pump pulse is ps in duration and the CCD camera keeps recording the FSRS signal within every 1 ms at the laser repetition rate. Therefore, the vibrational mode intensity evolution revealed in our FSRS data probes the instantaneous molecular conformation (not the FID or vibrational population relaxation typically on the few ps timescale) at a certain time delay after photoexcitation, and reports on (1) the coherent nuclear motion of the molecule and/or molecular complex navigating the S1 PES from the FC region [9, 31, 44-47] to (2) further structural evolution going beyond the vibrational coherence decay time window. These exemplary vibrational excitations (besides 10 characteristic modes that are strongly coupled to the optical transition hence the generation of transient vibronic wavepackets) and their temporal evolution throughout the ESPT reaction on the fs to ps timescale thus provide vivid molecular conformational snapshots of HPTS as it navigates the dynamic excited-state PES (varying with the solvent rearrangement) to transfer the phenolic proton to water before fluorescence. Excited-state FSRS spectrum of HPTS in H2O and D2O. A number of prominent vibrational modes from 50—1450 cm-1 appear in the excited-state FSRS spectrum of HPTS in water (Fig. 3) upon adding back a certain percentage of the multiple-gaussianfitted ground-state spectrum. There are several transient low-frequency modes below 1000 cm-1 that are either associated with PA* or PB*, and the most effective way to assign them is by the individual kinetic analysis and Gaussian calculations (main results shown in Table 1). Considering the typical ps timescale of ESPT in solvated systems, excited-state vibrational features that appear promptly following photoexcitation but decay on the ps timescale can normally be assigned to PA*, while the modes that appear later but keep rising are possibly associated with PB*. This is due to the temporal resolution provided by the fs photoexcitation and Raman probe pulse, and the temporal precision of the vibrational coherence being generated and detected in excited-state FSRS spectrum. The main reasons for the PA* vibrational modes to diminish can include the loss of resonance enhancement as the vibronic wavepacket moves out of the FC region (e.g., akin to the case of the wtGFP neutral chromophore [9]), or the wavepacket movement toward another electronic state (e.g., barrier crossing from PA* to PB*), and further structural evolution to convert more PA* to PB* population. As a result, unless there is a mode with similar vibrational frequency to PA* and similar resonance condition with the Raman pump pulse, the peak intensity dynamics of PA* will report on the decay of PA* via ESPT and other energy relaxation pathways. On the other hand, the PB* mode emerges with shifted frequency from the ground state PA modes, and will probe the rise of PB* state via ESPT only with high specificity, because other relaxation processes from PB* generally occur on much longer timescales. The ~4.5 ps short decay time constant of the 1048 cm-1 mode for photoexcited HPTS in water is longer than the ~1.6 ps decay observed in water with 2 M acetate [27], and also with a reduced amplitude (32% in water vs. 60% in acetate water). This result indicates that the absence of the stronger proton acceptor largely shuts off the direct HPTS-base ESPT channel through pre-existing highly polarized H-bonds, wherein the solvent dynamics within the first solvation shell of the photoacid dominate the ESPT pathway on the 1–2 ps timescale. ESPT that solely relies on the HPTS-H2O H-bonding network is more subject to the labile water molecular dynamics, and likely takes longer time as well as distance to establish the effective proton transfer chain to accommodate the significantly increased acidity of HPTS. Previous experiments using time-resolved fluorescence/absorption and visible pump-probe or mid-IR probe spectroscopies [3, 15, 19, 22] have revealed comparable short decay time constant at ~3 ps and the long decay time constant of ~90 ps for the deprotonation of HPTS in water. The latter temporal component is close to our result here, ~150 ps for the long decay time constant of the 1048 cm-1 mode, and ~78 ps for the long rise time constant of the 1138 cm -1 mode (see Table 1). We consider that the 1138 cm-1 mode, attributed to PB* that represents the deprotonated state of HPTS, reports on the ESPT process more faithfully. In contrast, the 11 decay dynamics of the 1048 cm-1 mode of PA* is more susceptible to other relaxation pathways such as the direct emission from PA* and the geminate recombination of the separated ion pairs [13, 14], which is diffusion controlled and occurs on a typical timescale of 300–400 ps at room temperature for the PA* population that has dissociated. Ground state FSRS results on HPTS in D2O (data not shown) corroborate the aforementioned peak assignment. Almost all the vibrational peaks of HPTS remain unshifted upon solvation in pure D2O, except that the 1050 cm-1 mode redshifts to 1048 cm-1, and the 1292 cm-1 mode blueshifts to 1298 cm-1 that is associated with the deprotonated chromophore. The kinetic analysis of the characteristic PB* mode in D2O at 1136 cm-1 shows two rise time constants of 21 ps (12%) and 200 ps (88%) (Table 2). The relative amplitudes of the exponential fitting are the same with the two rising components in H2O, but with significantly larger time constants. This result is reminiscent of the ESPT process from HPTS to the solvent, pure H2O or D2O here, which reports on the proton motion on the ps timescale and the complimentary rise of an aromatic ring vibrational response that is characteristic of PB*. The slowing down of ESPT upon deuteration will be quantitatively addressed below with reference to KIE. The most significant difference of this work in comparison to our previously reported FSRS data for HPTS in 2 M acetate water solution [27] can be appreciated from the kinetic analysis of the observed excited-state vibrational modes. It is expected that at thermal equilibrium and the electronic ground state (S0), water solvates the HPTS molecules by providing a solvation shell around them, while still maintaining various Hbonding geometries with several neighboring water molecules. There is intrinsically an inhomogeneity regarding H2O molecules in the H-bonding chain involving HPTS, i.e., different number of H2O molecules might be participating in the chain starting or ending with the phenolic hydroxyl group of HPTS. It has been proposed that the Grotthuss hopping model of the proton [3, 48] plays a dominant role in ESPT within these Hbonded reactive complexes, via different numbers of intervening H-bonded H2O molecules, and by interchanging of the covalent and H-bonds. As a result, the proton does not need to diffuse over large distances; rather its charge is transferred between the donor and acceptor through the highly directional H-bonding chain. This mechanism is supported by the previously reported KIE of 1.4–1.5 of the proton/deuteron continuum peak dynamics at early time (<10 ps) in 10 mM HPTS, 2 M acetate solutions in H2O/D2O [21, 24]. It is worth mentioning that the acetate ion represents a stronger proton acceptor than water, thereby providing a significant amount of driving force to facilitate ESPT following photoexcitation and the increased acidity of HPTS. In other words, ESPT between HPTS and acetate can be attributed to an adiabatic PT process as the proton moves along a preformed highly polarizable H-bond, without significant barrier tunneling or H-bonding reorganization, and mainly on the sub-ps to ps timescale [24]. However, in pure water the observed KIE is a factor of ~5.3 for the short component, and ~4.3 for the long component of the 1048 cm-1 PA* peak intensity decay. We consider this KIE to lack in accuracy when compared to that from the PB* increase (e.g., 1138/1136 cm-1 modes in H2O/D2O, with a KIE of ~4.2 for the short component, and ~2.6 for the short component), which has to be a direct consequence of ESPT. This is still larger than the KIE of ~1.8/2.4 derived from previous visible pump-probe measurements on HPTS in H2O/D2O [22], but similar to ~2.8 for the long time component deduced from visible-pump mid-IR-probe experiments [19] and ~3 from 12 time-resolved ps fluorescence studies [12]. The driving force for ESPT is significantly weaker in pure water than in the acetate-water system, hence requiring more proton tunneling through a higher reaction barrier. Therefore we observe the retardation of both PA* decay and PB* rise on the 4.5–5 ps timescale in water, in comparison to the initial 1.6 ps PA* decay time constant in 2 M acetate water solution [27]. Caution is needed though when comparing these results because the spectral source from which the time constants are derived is fundamentally different, and various electronic and vibrational contributions might be involved. Our kinetic analysis is based on the S1 vibrational peak intensity over a broad spectral range (instead of a broad electronic response, or broad vibrational peaks over limited spectral range), which should in principle be more accurate in capturing the structural snapshots of photoexcited molecules in action. The observed KIE for the short time component of PA* decay (e.g., the 1048/1042 cm-1 mode in H2O/D2O) and PB* rise (e.g., the 1138/1136 cm-1 mode in H2O/D2O) of HPTS is similar in magnitude to the ESPT process in solvated wtGFP, wherein the observed C=N stretching mode redshift has a time constant of 5.1/22.0 ps for the chromophore embedded in the protein pocket in H2O/D2O, respectively, so the KIE is 4.3 [9]. This similarity suggests that both the short and long time components of ESPT from HPTS to water involve some significant and active proton transfer motions, accompanied by the optimization of nearby heavy atoms likely within the ESPT chain on the ps timescale, not merely the formation of contact pairs through highly specific proton wires [18, 22]. The long time constant (>80 ps) sees diffusion playing a dominant role, as PB* and the proton acceptor molecule are likely separated by 4 or more water molecules [21], so ESPT and the charge transfer take much longer to complete. It is also notable that these high-frequency vibrational modes mainly probe structural evolution after ESPT efficiently proceeds following the initial preparation stage, wherein the impulsively excited low-frequency modes emerge with various time constants (discussed below) on the dynamic multidimensional PES and are sensitive to the instantaneous molecular conformations of asymmetric HPTS on the fs to ps timescale. Sequential emergence of various low-frequency modes gating ESPT. Following the 400 nm photoexcitation, we observe the multi-staged onset of PA* and PB* vibrational modes. The optical excitation accesses the singly excited 1* state [10, 15, 19, 49], and generally donates the photoexcitation energy to the totally symmetric Raman-active vibrational modes that exhibit strong FC activities [19]. The sub-ps time constants observed in our FSRS experiments on photoexcited HPTS in water range from 140–680 fs, a pre-ESPT timescale. The 952, 1048, and 1285 cm-1 modes rise with time constants of 140, 210, and 190 fs, respectively, which strongly argues that these are readily accessible PA* modes such as ring-HOOP motions, in-plane ring deformation, and ring C–O(H) stretching motions. The prompt electronic redistribution over the aromatic ring system upon photoexcitation induces these atomic motions, which have high Raman polarizabilities along the FC slope and are strongly coupled to the electronic redistribution process and exhibit excited-state vibrational modes at shifted positions from the ground-state peaks. It is noteworthy that the emergence of various excited-state vibrational modes particularly in the low-frequency region is intimately related to FC dynamics of photoexcited HPTS as it undergoes conformational motions starting from time zero. This ultrafast initial phase of excited-state structural evolution is likely 13 accompanied by intramolecular vibrational energy redistribution (IVR) processes [5, 43, 50-54], however, it is the relative equilibrium geometry displacement between S1 and higher lying electronic excited states (or virtual state that is off-resonance) dictating the FSRS peak intensity detected here by the Raman pump-probe pair. FSRS thus provides the unique and laudable capability to monitor a multitude of vibrational modes simultaneously and pinpoint the structural evolution pathway as ESPT initiates and proceeds on the anharmonic S1 PES that is changing in real time. Certain vibrational modes are promptly Raman active upon initial vibronic wavepacket generation on S1, whereas some other vibrational modes become Raman active later in time because they need a rather distorted molecular conformation to acquire significant Raman intensity. Furthermore, the vibrational peak kinetics plot in FSRS is not limited by the vibrational coherence decay typically on the few ps timescale because the time-delayed Raman pspump-fs-probe pair can generate multiple vibrational coherences due to the broad bandwidth of the probe pulse to monitor the evolving molecular conformation along multidimensional reaction coordinates. Therefore, the microscopic molecular snapshot (i.e., HPTS conformational dynamics in S1 before fluorescence) is vividly available in time-resolved excited-state FSRS. In particular, the observed transient vibrational modes promptly following photoexcitation might be the dominant atomic motions contributing to the proposed Stokes shift of photoexcited HPTS in H2O with the time constant of ~1 ps [22], which is a separate and distinct phenomenon (i.e., preparation stage) from the subsequent deprotonation process for PA*PB* population transfer (i.e., ESPT stage). Regarding the low-frequency region, the most transient 191 cm-1 mode is impulsively and coherently excited by the broadband photoexcitation pulse. It rises with a ~320 fs time constant and decays with a ~540 fs time constant. This in-plane ring skeletal symmetric breathing motion has a large projection onto the initial reaction coordinate for ESPT, coherently modulates the dihedral angle between the HPTS hydroxyl group and the H-bonded water molecule (with a less optimal geometry in S0), and promotes some immediate solvation process that accommodates the increased acidity of photoexcited HPTS. The 540 fs decay component indicates that HPTS starts to break the original ring coplanarity on that timescale, and should match the onset of various ring wagging, ringHOOPs, and asymmetric ring deformation motions of PA*. This is indeed the case: the 108, 321, and 362 cm-1 modes show the rise time constants of ~630, 650, and 650 fs, respectively, and match well with the calculated frequencies at ~109, 311, and 355 cm-1 (Table 1). The 630 cm-1 mode in Fig. 3 likely involves the PA* ring in-plane and OOP deformations, e.g., a combination of ring breathing and wagging motions, as confirmed by DFT calculations at ~631 cm-1 (frequency scaling factor 0.96). The peak area plot shows strong oscillations before 10 ps with a period of ~3 ps (data not shown, see below the frequency quantum beat discussion for some modes below 250 cm-1). This mode rises after 300 fs and mainly decays on the 90 ps timescale, and appears to be more prominent in water than the acetate water solution previously studied [27]. The interwoven transient vibrational peak kinetics suggests that the ESPT reaction coordinate on the excited-state PES is multidimensional and anharmonic in nature to efficiently facilitate PT, and is finely tuned by the molecular environment surrounding the photoacid in solution. On a related note, the 191 cm-1 mode in Fig. 3 is weaker and decays faster in water than that in 2 M acetate water [27], suggesting that this intrinsically conserved coherent skeletal motion of photoexcited HPTS takes on different dynamics upon photoexcitation in 14 response to an altered microenvironment for ESPT, and is thus an integral part of the initial chemical reaction coordinate in the PES for the photoacidity of HPTS. A small population of PB* is generated on the sub-ps timescale as well, evinced by the fact that the characteristic PB* modes of 276 and 460 cm-1 exhibit the fast rise time constants of 550 and 680 fs, respectively, although with distinct relative amplitudes (85% vs. 26%, Table 1). This result indicates that the ring-HOOP motions at the phenolic end of the nascent PB* state rise earlier and faster than the pure wags of the HPTS 4-ring system. Some HOOP motions may facilitate the immediate solvation of PB*. In contrast, the other PB* mode at 1138 cm-1 is fitted after 1 ps to show the biphasic rise time constants of 5 and 78 ps. We cannot precisely comment on the sub-ps dynamics of this mode due to complication from the overlapping ephemeral PA* feature (see Figs. 5 and 7), however, the two time constants directly report on two sequential stages for ESPT, with the latter one dominating the accumulation of PB* due to the diffusion-assisted distance-dependent ESPT. The electronic charge flow from the HPTS hydroxyl group back to the aromatic ring system occurs strongly in the anion [3, 27, 55], likely after the initial PT within the HPTS–O···H–OH2 complex to generate HPTS–O( )···H(+)–OH2, and prepares for further PT between the nascent ion pair and beyond. The aforementioned ~1 ps delay or preparation stage (Figs. 5 and 7) at 1138 cm-1 shows complex vibrational dynamics, likely associated with the Stokes shift of HPTS in conjunction with solvation but not PT [22]. The ephemeral signal is ca. 2 cm-1 to the blue side of the PB* mode (see Fig. 3, within the rightmost red box), and appears within our instrument response time dictated by the cross correlation between the actinic pump and Raman probe pulses (~140 fs). It then quickly decays away with a ~600 fs time constant, similar to the transient 1180 cm-1 mode dynamics nearby. We speculate this transient spectral feature at very early time being the PA* mode in the FC region way before the PB* formation at barrier crossing, correlated with the disruption of the equilibrium solvation shell possibly involving some immediate solvent motions around HPTS. Therefore the sub-ps decay is indicative of the vibronic wavepacket moving out of the initial FC region, toward the ESPT region on the PES. The PB* formation then becomes the dominant process, and the peak kinetics tracks the ESPT progress as HPTS gradually transfers its phenolic proton to nearby water molecules through H-bonding chains. It is useful to accentuate that we observe prompt appearance of the ~191 cm-1 mode following photoexcitation, analogous to the 195 cm-1 mode previously observed in the HPTS-acetate-water system [27], suggesting that this particular low-frequency mode is characteristic of the HPTS in-plane ring symmetric breathing motion that modulates the intermolecular O···O bending within the immediate solvation shell. In other words, this conserved PA* mode samples the phase space that includes better H-bonding geometry between the proton donor and neighboring acceptor, and might be a direct consequence of the electronic redistribution that weakens the original HPTS–OH···OH2 Hbond in S0 [3]. This vibrational motion is likely ubiquitous for HPTS in water-based solution and has a large projection onto the ESPT reaction coordinate. As a result, this mode effectively receives the photoexcitation energy and helps the impulsively generated vibronic wavepacket slide down the PES slope in the FC region. Once this process occurs, the other coherent low-frequency modes associated with PA* (e.g., 108, 321, 362 cm-1, primarily ring wags and ring HOOPs) start to facilitate the proton motion through the immediate H-bond to one acceptor water molecule on the typical hopping time of 15 ~1.2 ps [21, 48, 56]. That is why we see a prominent decay component of all these ringwagging motions on the 1 ps timescale (Table 1), since the solvent rearrangement also affects PA* vibrational motions, particularly those concerning H-bonding at the phenolic hydroxyl end. An enlightening comparison can also be made to our previous results for HPTS in 2 M acetate water solution: the 106 and 321 cm-1 modes observed therein both rise on the ~630 fs timescale and exhibit the dominant decay time constant of ~1 ps [27], with a relative exponential fitting amplitude of 80–85% that agrees very well with the component percentage derived here in pure water. As more nearby water molecules start to align along the ESPT chain connecting to the HPTS hydroxyl group, the ring wags associated with the nascent charge-separated HPTS(–)–O···H(+)–OH2 at ~460 cm-1 report on the formation of extended H-bonds with increased number of better-oriented water molecules, presumably with a reduced coordination number in comparison to that in bulk water [14, 24]. It is crucial to point out that photoexcited HPTS in pure water represents a different case from the 2 M acetate water environment studied before [27]. In pure water, there is no longer the strong donor-acceptor potential gradient between HPTS and acetate to drive ESPT, and there are less effectively H-bonded or closer HPTS-acceptor pairs already formed in S0. As a result, particularly after the vibrant molecular events (i.e., coherent skeletal motions) within the first 1 ps and some proton hopping to the immediate H-bonded water molecule, a significantly greater amount of work is needed to bring more nearby H2O molecules, which are over larger distances than those in the acetate water case, into optimized H-bonding geometry with the nascent charge-separated HPTS(–)– O···H(+)–[OH2]n=1,2... complex. That is why we observe a ~4.5 (24) ps PA* decay component and a concomitant ~5 (21) ps PB* rise component in H2O (D2O), representing further structural evolution of HPTS on S1 beyond the FC region. The sub-ps rise time constant of 220 fs for the 1139 cm-1 PB* mode in the 2 M acetate water system is absent in pure water, partially obscured by the overlapping PA* mode that shows strong activity in the same spectral region up to 1 ps (Figs. 5 and 7). This finding indicates that the absence of an effective proton acceptor such as the acetate ion largely turns off direct ESPT over short distances through highly polarizable H-bonds and much reduced proton transfer barrier on the <300 fs timescale. We suspect that a small portion of the HPTS hydroxyl proton still manages to hop over to the immediately adjacent H2O molecule that is originally H-bonded to the hydroxyl group of HPTS, but only after some small-scale molecular reorientation of H2O occurs in ~1 ps to strengthen that existing H-bond following photoexcitation. This agrees with the aforementioned subps emergence of various PA* low-frequency modes, and the accompanying solvent motions within the first solvation shell. Except for the 276 cm-1 mode, a small percentage of the PB* peak intensity rises on the timescale of ca. 1–20 ps, while the majority of the PB* peak intensity rises on the timescale of ca. 80–200 ps in H2O and D2O (Tables 1 and 2). The ensemble-average approach of the FSRS measurement dictates that we observe the overall time constants, with the clear distinction of two processes attributed to establishing effective H-bonds with a few water molecules nearby, and to separating the deprotonated HPTS (PB*) from the rest of the H-bonding chain as well as solvating the nascent charge-separated molecules. The latter process is very likely to be diffusioncontrolled, but since the H-bonding chain is longer and more asymmetric [48] by then, the deduced KIE of ~2.6 is smaller than the value (4.2) from the former PT process. The 16 majority of the PA* decay and PB* rise occur within that latter diffusion-controlled process, meaning that ESPT can only effectively occur after the ~5 (21) ps heavy-atom geometric optimization of the H (D)-bonding chain connecting the photoexcited HPTS and the acceptor H2O (D2O) molecule via a few bridging H2O (D2O) molecules. The time-resolved frequencies of the relatively sharp 172 and 215 cm-1 modes (marked by vertical dashed lines but without labels in Fig. 3) after ~1 ps are compared with the frequencies of the 108 and 125 cm-1 modes. The 125 cm-1 mode clearly shows a delayed onset of ~1 ps (Fig. 8), whereas the 108 cm-1 mode rises promptly after photoexcitation. It is interesting to find that the frequency quantum beats (data not shown) displayed by these low-frequency modes (below 250 cm-1) are rather pronounced on the 1–10 ps timescale, with a period of ~3 ps that corresponds to a modulation vibrational mode at ~11 cm-1. The frequency oscillation of the 172 cm-1 mode is almost 180° out-of-phase to that of the 215 cm-1 mode. It is thus plausible to attribute the two modes to PA* and PB*, respectively, assuming that the actively modulating 11 cm-1 vibrational mode survives the ESPT barrier crossing, and plays integral roles during both the reactant (PA*) consumption and the product (PB*) generation. The frequency oscillations of the 108 and 125 cm-1 modes are approximately in phase with each other, commensurate with the mechanistic picture of sequentially observed PA* modes in gating multi-staged ESPT. The peak intensities are expected to exhibit the quantum beating effect as well, given that the nonlinear Raman polarizabilities of HPTS vary during the low-frequency modulation period. This can be seen clearly in Fig. 9 wherein the 952 cm-1 peak intensity is strongly modulated by a vibrational mode at ~93 cm-1 within 1 ps, suggesting that both modes are anharmonically coupled (likely mechanical coupling) during the ESPT preparation stage [9, 39, 57]. Further experiments are planned to unravel the low-frequency modes that remain active up to ~5 ps and anharmonically couple to other low-frequency modes as seen here. More stable white-light probe pulse and finer time delay steps are needed with improved SNR to reveal the quantum beats of various vibrational modes particularly in the low-frequency region at early time, and to provide deep insight on the multi-faceted PES of HPTS following photoexcitation. Scheme 1. Multidimensional reaction coordinate and multi-staged ESPT from HPTS to water: fs activation of skeletal modes gates the ps heterogeneous PT through optimizing intervening water molecules Multi-staged ESPT from HPTS to water with vivid conformational details. The fs fluorescence up-conversion and pump-probe spectroscopies on HPTS in water have revealed two ultrafast steps (300 fs and 2.5 ps) preceding the relatively slow diffusionrelated proton transfer step (87 ps) [3, 15]. The proposed mechanism involves solvation dynamics, the formation of an ion contact pair, and the dissociation into free ions. Solvent reorganizations play an important role to reduce the reaction barrier and to accommodate the solvent-bridged ion contact pairs, and we have found previously that the heterogeneity of this H-bonding network for ESPT affects the overall dynamics observed for individual vibrational modes of the photoexcited HPTS in the presence of acetate base [27]. The reported time dependence of the photoexcited PA* in water is 17 biphasic with decay time constants of ~3 ps (30%) and 100 ps (70%) from the pumpprobe measurements [18]. In another fs UV-pump IR-probe study on HPTS in water, the transient absorbance for frequencies below 2850 cm-1 shows the multiexponential decay with 300±200 fs, 3.0±1.5 ps, 90±30 ps, and 200±50 ps time components [19]. Our kinetic analysis of the 1048 cm-1 PA* mode of HPTS in water reveals the bi-exponential decay time constants of ca. 4.5 and 150 ps, which can be considered similar to previous results. However, we believe that the values derived from the current time-resolved FSRS peak kinetic analysis are more accurate since a number of vibrational marker bands are spectrally separated from each other (Fig. 3), and can be analyzed simultaneously and independently (Figs. 4–9). We also find that within the preparation stage for ESPT, multiple low-frequency PA* modes coupled with some motions of the immediate Hbonding partner (Table 1) show pronounced activities on the 300 fs–1.2 ps timescale. In addition, there is the subtle difference between the vibrational mode intensity observed here and the concentration of the molecular species, and it is the interplay between electronic and vibrational motions that leads to the polarizability change dictating Raman peak intensity, not simply due to the concentration of the species involved. Therefore it is more appropriate to discuss the non-equilibrium vibrational dynamics and KIE deduced from FSRS data in the context of specific atomic motions of transient conformational states, rather than a simple physical kinetic model that builds on equilibrium constants and thermodynamics of various molecular species involved. In addition, the timeresolved FSRS peak intensity reported here effectively tracks the detailed structural evolution starting from the FC region toward the product state on the excited-state PES, because the Raman polarizability continues to change for vibrational normal modes as the photoexcited molecule evolves on the multidimensional reaction surface with a complex landscape and various equilibrium positions for participating vibrational modes. The 1138 cm-1 PB* mode displays the biphasic rise with time constants of ~5 and 78 ps, which corroborates the short time component of PA* decay as the initial phase of ESPT involving a few water molecules, although the predominant ESPT process for PB* production occurs on the 78 ps timescale. This observation confirms the existence of some initial stage that modifies the population of PA* but does not significantly generate PB*. This could be due to some intermediate conformations of photoexcited HPTS that have already broken the initial coplanarity of the aromatic ring system, have formed contact pairs with neighboring proton acceptors reorienting on the ca. 5 ps timescale in water, but have yet to conduct the long-range diffusion-controlled proton/charge transfer and eventually separate the ion pairs. The relative large KIE observed in our FSRS data confirms that after the preparation stage wherein the sequential observation of various low-frequency modes is staple, heavy atom optimization is needed during the first phase of ESPT on the few ps timescale, as the proton hops incoherently through more or less randomly coiled water wires [25] that are asymmetric. The ~5 ps timescale is consistent with the previously reported optimal ESPT distance of going across two water shells [1, 21, 25], where each water molecule donates two H-bonds over which the proton charge gets transferred. The ~1.6 ps time constant previously observed for HPTS in 2 M acetate water solution [27] can be understood in that the acetate ion effectively establishes a large number of H-bonded ―reactive complexes‖, and reduces the initial acid-base distances to sufficiently small values. This also explains the small KIE value of ~1.4 observed in 18 those tightly H-bonded systems involving the acetate ion as the adiabatic PT process resembles the case for largely asymmetric, strongly downhill reactions. The wealth of structural dynamics information provided by our time-resolved excited-state FSRS spectra reveals that upon photoexcitation, the asymmetric HPTS undergoes ESPT in water through multiple reaction stages: (1) the almost instantaneous electronic redistribution leads to some swift in-plane ring deformation and ring-HOOP motions on the 200 fs timescale (e.g., PA* S1 modes at 1048, 952 cm-1); (2) the facile inplane ring-breathing motion (e.g., 191 cm-1, PA*) then emerges with the 320 fs rise time constant, modulating the relative geometry between the directly H-bonded HPTS hydroxyl group and H2O molecule; (3) the ring wagging and phenolic HOOP motions (e.g., 108, 321, 362 cm-1, PA*) subsequently gain Raman intensity with the 630–650 fs rise time constant, in correlation with the diminishment of the in-plane ring breathing motion with a 540 fs decay time constant, as well as the waning of the ring-HOOP motion adjacent to the phenolic ring with a 600 fs decay time constant; (4) a small amount of proton transfer starts to occur with the appearance of these PA* ring wagging motions in S1, and characteristic ring wags associated with the deprotonated HPTS (PB*, 276 and 460 cm-1 modes) begin to accumulate on the 550–680 fs timescale; (5) The PA* wagging motions start to diminish on the ca. 1 ps timescale, indicative of a solvation process that mainly reorients the directly H-bonded H2O molecule to be in a more favorable H-bonding geometry with HPTS [21, 27, 48], and the initial ground-state 4-ring coplanarity is broken as the molecule reaches the charge-transfer state [15, 19, 49]; (6) ESPT occurs through various-length H-bonding chains, accompanied by some tardy PA* motions such as the 125 cm-1 mode, generating PB* with two distinct time constants of ~5 ps and 80 ps. The few ps component suggests that ESPT is a heterogeneous process for HPTS in the labile H-bonded water matrix, since more H2O molecules nearby need to be optimized to establish efficient H-bonds to transfer that phenolic proton. The observed KIE of 3–4 is indicative of the weak H-bonding nature and possibly also longer Hbonding chains between HPTS and water, characteristic of the nonadiabatic PT where the solvent fluctuations modulate both the PA* and PB* PES of the chromophore. Besides the fundamental importance in elucidating the chemical reaction mechanism of photoacidity, HPTS is also a fluorescent dye and pH sensor [58], and small organic fluorophores have been powerful research tools to enable bioimaging with novel insights into both cellular and molecular processes [59, 60]. This puts our work into perspective for bioengineering and bioimaging. When the chromophore is part of a protein sequence and strategically embedded in the protein pocket, wtGFP being the perfect example for these genetically encodable biomarkers, the biofunctions can in principle be illuminated step by step with unprecedented precision [9]. Whether or not this precision extends into both the spatial and temporal regimes for practical bioimaging is another question, and extensive work has been done in both areas to develop ultrahighresolution microscopy to overcome the diffraction limit [61], as well as to collect timeresolved cellular-level images in situ and in real time [62]. All these exciting applications depend upon a photostable, bright, and controllable photosensitive reagent, which we can understand deeply using the powerful toolset such as FSRS described here and then perform targeted design at the molecular level to efficiently improve their biofunctionality. 19 5. Conclusion We have used the newly developed femtosecond stimulated Raman spectroscopy (FSRS) to study the excited-state structural dynamics of HPTS in pure H2O and D2O following 400 nm photoexcitation. The simultaneously high spectral and temporal resolution of the apparatus enable the collection of time-resolved excited-state FSRS spectra of the photoexcited chromophore as it transfers its phenolic proton to the labile molecular water H-bonded network in real time. The non-equilibrium spectroscopic approach reveals the multidimensional reaction coordinate on the excited-state PES for intermolecular ESPT, wherein the sequentially emerged low-frequency skeletal motions gate and/or facilitate ESPT in H2O on multiple timescales of 620±50 fs, ~4.5 ps, and ~100 ps. The observed KIE upon deuteration is in the vicinity of 3–4 for the latter two time constants. We attribute the first sub-ps component to the preparation stage for ESPT, which involves the decay of the transient 191 and 952 cm-1 PA* modes, and the rise of the 108, 125, 276, 321, 362, and 460 cm-1 modes. The 276 and 460 cm-1 mode intensities show the biphasic exponential rise, and are most likely associated with the ring wagging and some HOOP motions of PB*. The 108, 321 and 362 cm-1 modes also show a predominant (relative exponential fitting amplitude at 75–80%) decay time constant of 1.1 ps, matching the reorientation dynamics of a single water molecule possibly in direct contact with the phenolic hydroxyl group of HPTS. The solvent rearrangement along the H-bonding chain connecting HPTS and water molecules following photoexcitation plays a dominant role in the ps dynamics of ESPT, and can be attributed to the nonadiabatic proton transfer in comparison to the faster adiabatic ESPT when strong bases are present in water solution. The observed ESPT on the 5–200 ps timescale in H2O/D2O involves several water molecules being brought into better H-bonding geometry with the photoexcited HPTS molecule, which later undergo diffusion-assisted ion-pair separation. It is noteworthy to summarize the convincing evidences for a number of coherent low-frequency vibrational modes to play the important functional gating role [4, 9, 27, 43, 63-65] in ESPT of HPTS in aqueous solution, besides the kinetic analysis of their FSRS intensities. (1) They get impulsively excited pre-ESPT and exhibit different dynamics. The deprotonated form PB* has the characteristic 1138 cm -1 mode that redshifts from the 1154 cm-1 for PA at S0, and Fig. 7 shows that the 1138 cm-1 mode starts to rise after 1 ps. The dwell between the observed low-frequency modes and ESPT is small, indicative of causality. (2) These low-frequency modes significantly modulate the intermolecular distance and/or geometry between HPTS and the neighboring H-bonding acceptors, providing the appropriate atomic displacements to start optimizing H-bonding chains and gate ESPT. (3) Quantum beats exist for a number of vibrational modes, indicative of anharmonic coupling between various conformational motions of HPTS capable of ESPT in water. (4) Certain conserved low-frequency modes exhibit different dynamic behavior in response to different acceptor molecules. The key to retrieve the underlying system Hamiltonian [9, 66, 67] is to observe mode-dependent vibrational dynamics starting from time zero for HPTS in various external microenvironments, and to simultaneously monitor a wide array of vibrational modes including the reactant, intermediate and product with enough (i.e., fs) time resolution. We can then firmly establish the causative connection between the observed coherent low-frequency modes and ESPT via the temporal and structural correlation of the associated transient 20 molecular motions. FSRS thus renders an emerging powerful structural dynamics tool to elucidate the choreography of ESPT from HPTS to water throughout the reaction, and paves the way to study other photosensitive molecules with biological relevance on their intrinsic reaction timescales in aqueous solution. Acknowledgments This paper is dedicated to Robin M. Hochstrasser who for over 50 years pioneered in many fields of modern molecular spectroscopy and contributed deeply to our understanding of the interplay between conformational dynamics and chemical reactions. We thank the financial support from the Oregon State University Faculty Research Startup Fund, and the College of Science Venture Fund Award to C. Fang. We are also grateful to Yanli Wang and Longteng Tang for sample preparation, and to Breland Oscar and reviewers for helpful discussion. 21 TABLE 1: Representative Vibrational Peaks of HPTS in H2O Observed in FSRS FSRS peak freq.a (cm-1) cal. peak freq.b (cm-1) kinetics of the Raman peak area major species symbol vibrational mode assignment PA* HPTS 4-ring OOP wags PA* In-plane ring translation with huge nearby water translational motionc PA* Intermolecular O···O stretch between the hydroxyl and the acceptor PA* In-plane ring skeletal breathing with intermolecular O···O bending at the phenolic ende PB* HPTS ring wags with the phenolic COH HOOP motions, and H-bonded water HOOP motions PA* In-plane ring deformation with some ring HOOPs PA* In-plane ring deformation with significant COH rocking motion with some phenolic COH HOOPs PB* HPTS ring asymmetric wagging motion PA* HPTS ring-H HOOPs and in-plane ring deformation PA, PB / PA* In-plane asymmetric ring deformation with some phenolic CO stretching PB* Phenolic CO···(H) rocking and nearby ring-H rocking (+) 630 fs 108 109 (–) 1.1 ps (75%); 1.7 ns (25%) (+) 650 fs (55%); 125 132 5.3 ps (45%) (–) 270 ps 143 150 Quantum beats with ~350 fs periodd (+) 320 fs 191 208 (–) 540 fs (+) 550 fs (85%) 276 271f 110 ps (15%) (+) 650 fs 321 311 (–) 1.1 ps (80%); 75 ps (20%) (+) 650 fs 362 355 (–) 1.2 ps (80%); 80 ps (20%) (+) 680 fs (26%); 460 453f 130 ps (74%) (+) 140 fs; 952 950 (–) 600 fs (78%) 90 ps (22%) (+) 210 fs 1050 / 1048 1004g (–) 4.5 ps (32%); 150 ps (68%) (+) 5 ps (12%); 1138 1137f 78 ps (88%) h 22 1154 1149i N/A PA Phenolic COH rocking and nearby ring-H rocking PA* Phenolic CO stretch and strong ring-H & COH rocking motions (+) 190 fs 1285 1287 (–) 4.3 ps (40%); 215 ps (60%) a Observed Raman frequencies of the excited state as well as ground state FSRS peaks of 11 mM HPTS in pure water solution (pH≈6). b RB3LYP-DFT calculations are performed using 6-311G++(2d,2p) basis set for PAHPTS in aqueous solution in complex with a H-bonding water molecule at the phenolic hydroxyl end. Calculation results with three H-bonding water molecules nearby show slightly non-coplanar ring structure of HPTS in the ground state, and some OOP motions mixed with the above-mentioned in-plane motions. Solvent effects are included by the IEF-PCM-H2O model. The calculated vibrational frequencies are all scaled by a factor of 0.96. c This mode mainly involves the translational motion of the nearby H-bonded water molecule at the phenolic hydroxyl end of HPTS. If the ring coplanarity is disrupted due to the presence of more water molecules within the H-bonding distance, some slight OOP motions are then mixed in. The main effect of this vibration is to significantly modulate the intermolecular O···O distance between HPTS and the water molecule nearby. d The detailed kinetic plot is not attempted due to the strong oscillatory pattern and ephemeral nature of the time-resolved peak integrated intensity (i.e., area) data. The mode disappears within 1 ps. This mode involves some in-plane ring translational motions and significant modulations between the ring hydroxyl and the neighboring water molecule. e This ring skeletal breathing motion modulates the intermolecular (HPTS–)O–H···O(– H2) angle and distance between the donor and H-bonded acceptor molecule in the same ring plane. Slight OOP ring motions might be present if more water molecules are within the H-bonding distance with the phenolic hydroxyl group, and modify the ground-state geometry of HPTS in aqueous solution to some extent. f These PB* modes are approximated using the RB3LYP 6-31G+(d,p) calculation for PBHPTS in aqueous solution with a H-bonding water molecule at the phenolic hydroxyl end (now with a C=O bond). In reality, the excited-state mode of PB* is in a partially deprotonated configuration, which could significantly deviate from the simple calculation of the corresponding ground-state mode in completely deprotonated PB. g This rather large discrepancy between the calculated and observed PA, PB mode is interesting, as the DFT calculation correctly captures the trend of this mode being unshifted from PA to PA···H2O, and from PB to PB···H2O. This mode seems to be relatively insensitive to the electronic distribution over the ring system but deviates from 23 the realistic solution configuration. Given that it consists of large-scale in-plane ring deformation and CO stretch at the phenolic hydroxyl end, it probably has cancellation effect when electrons redistribute. This mode broadens in S1 compared with S0, and shows characteristic rise-decay kinetics attributed to PA*, which has a ~2 cm-1 redshift to the ground-state vibrational frequency of PA. h The double-exponential fit of the peak intensity kinetics plot shows a time-zero offset of ca. 1 ps (Figs. 5 & 7). This is consistent with the delayed onset of this PB* vibrational mode after the overlapping PA* mode disappears in the spectral region within ~1 ps (Fig. 3, within the rightmost red box), the preparation stage for ESPT. i Upon adding three water molecules within H-bonding distances to the phenolic hydroxyl end of HPTS in the geometrically optimized Raman frequency calculation, the DFT results show a slightly blueshifted mode at 1161 cm-1. Since the observed groundstate frequency is at ~1154 cm-1, it probably suggests that the first solvation shell of HPTS at the phenolic end contains 1–2 H-bonded water molecules. TABLE 2: Representative Vibrational Peaks of HPTS in D2O Observed in FSRS FSRS peak freq.a in D2O (cm-1) kinetics of the Raman peak area major species symbol vibrational mode assignment PA* In-plane asymmetric ring deformation with some phenolic CO stretching PB* Phenolic CO···(D) rocking and nearby ring-H rocking (+) 610 fs 1042 (–) 24 ps (40%); 650 ps (60%) (+) 21 ps (12%) 1136 200 ps (88%) b a Observed Raman frequencies of two excited-state FSRS peaks of 11 mM HPTS in D2O solution (pD≈6). Both peaks show some small frequency redshift to the corresponding vibrational modes in H2O, probably due to the collective atomistic motions involved. b The double-exponential fit of the peak intensity kinetics plot shows a time-zero offset of ca. 1 ps (Fig. 7). This is consistent with the aforementioned delayed onset of the PB* vibrational mode at 1138 cm-1 in H2O. 24 Figure captions Fig. 1. Schematic of the newly developed femtosecond stimulated Raman spectroscopy (FSRS) in our laboratory [27]. The output laser beam from a Coherent femtosecond regenerative amplifier is split to generate three beams: the actinic pump beam at 400 nm (40 fs, 1 mW), Raman pump beam at 800 nm (3.5 ps, 6 mW), and Raman probe beam with the wavelength range of 805–940 nm (30 fs, 100 nW). BS: beamsplitter, G: reflective ruled diffraction grating (1200 grooves/mm, wavelength first order at 750 nm, blaze angle θ=26.7°), CL: cylindrical lens, UM: pick-up mirror, ND: neutral density filter, L: bi-convex lens (f=10 or 5 cm), SA: sapphire plate, LPF: long-wavelength pass filter, PR1: fused silica prism pair, PR2: Suprasil-1 prism pair, P: polarizer, λ/2 WP: halfwavelength waveplate, and DL: delay line stage. Fig. 2. Ground-state FSRS spectrum of HPTS in pure water (pH≈6). The two spectra collected at two different spectrograph grating positions to expose the low-frequency and high-frequency regions (10–2000 cm-1) are shown in red and black, respectively. The chemical structures of HPTS in different H-bonding geometries with nearby H2O molecules are depicted in equilibrium upon photoexcitation, transferring the proton from its phenolic hydroxyl end to a proton acceptor H2O molecule, via intervening H2O molecules. The two forms of HPTS, protonated (PA) and deprotonated (PB), are shown in red and blue, respectively. The temporal evolution of PA* converting to PB* on the excited state S1 can be found in the excited-state FSRS spectra in Fig. 3. Fig. 3. Time-resolved excited-state FSRS spectra of 11 mM HPTS in water following ~1 mW 400 nm photoexcitation. The Raman pump is at 802 nm. The water-subtracted ground-state FSRS spectrum of HPTS is scaled by 0.3 and plotted at the bottom for comparison. The time delay up to 150 ps between the photoexcitation and Raman probe pulses is noted beside each individual ground-state-subtracted excited-state spectrum, with the vertical dashed lines marking the vibrational modes of interest from HPTS. Peak frequencies in cm-1 are noted in the top portion of the figure: red for transient PA* modes, black for other PA* modes, and blue for PB* modes. The red boxes enclose the transient PA* modes that are predominantly active up to 1 ps. The blue box emphasizes the low-frequency modes that rise after 1 ps. The magnitude of the stimulated Raman peak strength is indicated by the double-arrowed vertical line in the middle of the figure. Fig. 4. Time evolution of the 1048 cm-1 excited-state mode of 11 mM HPTS in water following 400 nm electronic excitation up to 150 ps. The time-dependent stimulated Raman peak intensity is fitted with a rising exponential (210 fs) and two decaying exponential functions (~4.5 and 150 ps), convoluted with the ~140 fs instrument response function determined from the cross correlation between the photoexcitation and Raman probe pulses. The insert shows the very early time dynamics of the PA* mode up to 10 ps. The fits are shown in solid lines. The error bar represents one standard deviation from the average value of multiple fitting procedures of the experimental data sets. Fig. 5. Time evolution plots of three excited-state vibrational modes of HPTS in water following 400 nm photoexcitation. The 1285 cm-1 (green), 1138 cm-1 (red), and 460 cm-1 25 (blue) modes can be attributed to PA*, PB*, and PB* modes, respectively (see text). The fitting results are listed in Table 1. It is notable that due to the overlap of Franck-Condon PA* modes and the nascent PB* mode around the ~1138 cm-1 spectral region, the doubleexponential fit of that vibrational mode is performed after ~1 ps (indicated by the vertical dashed line). Fig. 6. Time evolution of three low-frequency vibrational modes of photoexcited HPTS in water following 400 nm electronic excitation. The stimulated Raman peak intensity is taken as the integrated gain of the Raman peak with the solid lines showing the multiexponential fits convoluted with instrument response time of 140 fs. The detailed fitting results are listed in Table 1 for the 191, 276 and 321 cm-1 modes. It is evident (see text) that the 191 and 321 cm-1 modes are two sequentially emerged PA* modes, while the 276 cm-1 mode can be attributed to the nascent PB* state. Fig. 7. Comparison between the peak intensity kinetic plots of the 1048/1042 cm-1 modes in H2O/D2O (blue/red solid), and the 1138/1136 cm-1 modes in H2O/D2O (blue/red dashed), respectively. The rise and decay time constants of HPTS in D2O are all prolonged compared with those in H2O, shown as the disappearance of PA* and appearance of PB* modes. Detailed fitting results regarding this isotope effect can be found in Tables 1 and 2. The insert (expanded kinetics plot up to 10 ps) displays the PA* preparation stage for ESPT in the HPTS-water system, before PB* starts to grow in gradually after 1 ps. It is notable that following the preparation stage, the PB* mode intensity rises faster in H2O than that in D2O. Fig. 8. Temporal evolution of the peak intensities of the 108 and 125 cm-1 modes of HPTS in water following 400 nm photoexcitation up to 150 ps. The 108 cm-1 mode rises slower than the 1048 cm-1 mode in Fig. 4, and decays with time constants of ca. 1.1 ps and 1.7 ns. The delayed onset of the 125 cm-1 mode is conspicuous from the figure, which is also highlighted by the leftmost blue box in Fig. 3. The decaying behavior on the ps timescale suggests that both modes are associated with PA*, but with different mechanism to facilitate multi-staged ESPT from HPTS to water. Fig. 9. Time-resolved peak intensity plot of the 952 cm-1 mode of 400 nm-photoexcited HPTS in water. Pronounced intensity oscillations show up before 1 ps with a period of ~360 fs (similar to the kinetic plot of the 143 cm-1 mode intensity), which corresponds to a modulating low-frequency mode at ~93 cm-1. The observed 108 cm-1 mode primarily involves 4-ring OOP wags (Table 1) and has an closely matching frequency, hence could be the modulating source via mechanical coupling. The structural origin for the proposed anharmonic coupling between these vibrational modes in S1 along the ESPT reaction coordinate is described in the text. 26 References [1] M. Eigen, Angew. Chem. Int. Ed. 3 (1964) 1. [2] L.M. Tolbert, K.M. Solntsev, Acc. Chem. Res. 35 (2002) 19. [3] N. Agmon, J. Phys. Chem. A 109 (2005) 13. [4] A. Douhal, S.K. Kim, A.H. Zewail, Nature 378 (1995) 260. [5] M.H. Vos, F. Rappaport, J.-C. Lambry, J. Breton, J.-L. Martin, Nature 363 (1993) 320. [6] A.H. Zewail, Femtochemistry: Ultrafast Dynamics of the Chemical Bond, World Scientific, Singapore, 1994. [7] R.M. Hochstrasser, Proc. Natl. Acad. Sci. USA 104 (2007) 14190. [8] C. Fang, J.D. Bauman, K. Das, A. Remorino, E. Arnold, R.M. Hochstrasser, Proc. Natl. Acad. Sci. USA 105 (2008) 1472. [9] C. Fang, R.R. Frontiera, R. Tran, R.A. Mathies, Nature 462 (2009) 200. [10] W. Domcke, A.L. Sobolewski, Science 302 (2003) 1693. [11] E. Pines, D. Huppert, J. Phys. Chem. 87 (1983) 4471. [12] D.H. Huppert, A. Jayaraman, R.G. Maines, Sr., D.W. Steyert, P.M. Rentzepis, J. Chem. Phys. 81 (1984) 5596. [13] E. Pines, D. Huppert, N. Agmon, J. Chem. Phys. 88 (1988) 5620. [14] L. Genosar, B. Cohen, D. Huppert, J. Phys. Chem. A 104 (2000) 6689. [15] T.-H. Tran-Thi, T. Gustavsson, C. Prayer, S. Pommeret, J.T. Hynes, Chem. Phys. Lett. 329 (2000) 421. [16] J.T. Hynes, T.-H. Tran-Thi, G. Granucci, J. Photochem. Photobiol. A Chem. 154 (2002) 3. [17] M. Rini, B.-Z. Magnes, E. Pines, E.T.J. Nibbering, Science 301 (2003) 349. [18] P. Leiderman, L. Genosar, D. Huppert, J. Phys. Chem. A 109 (2005) 5965. [19] O.F. Mohammed, J. Dreyer, B.-Z. magnes, E. Pines, E.T.J. Nibbering, ChemPhysChem 6 (2005) 625. [20] O.F. Mohammed, D. Pines, J. Dreyer, E. Pines, E.T.J. Nibbering, Science 310 (2005) 83. [21] B.J. Siwick, H.J. Bakker, J. Am. Chem. Soc. 129 (2007) 13412. [22] D.B. Spry, A. Goun, M.D. Fayer, J. Phys. Chem. A 111 (2007) 230. [23] M.J. Cox, H.J. Bakker, J. Chem. Phys. 128 (2008) 174501. [24] B.J. Siwick, M.J. Cox, H.J. Bakker, J. Phys. Chem. B 112 (2008) 378. [25] M.J. Cox, R.L.A. Timmer, H.J. Bakker, S. Park, N. Agmon, J. Phys. Chem. A 113 (2009) 6599. [26] K.J. Tielrooij, M.J. Cox, H.J. Bakker, ChemPhysChem 10 (2009) 245. [27] W. Liu, F. Han, C. Smith, C. Fang, J. Phys. Chem. B 116 (2012) 10535. [28] Y.-H. Liu, T.-S. Chu, Chem. Phys. Lett. 505 (2011) 117. [29] M. Yoshizawa, M. Kurosawa, Phys. Rev. A 61 (1999) 013808. [30] P. Kukura, D.W. McCamant, R.A. Mathies, Annu. Rev. Phys. Chem. 58 (2007) 461. [31] R.R. Frontiera, C. Fang, J. Dasgupta, R.A. Mathies, Phys. Chem. Chem. Phys. 14 (2012) 405. [32] S. Mukamel, J.D. Biggs, J. Chem. Phys. 134 (2011) 161101. [33] W. Liu, L. Zhu, C. Fang, Opt. Lett. 37 (2012) 3783. 27 [34] D.W. McCamant, P. Kukura, S. Yoon, R.A. Mathies, Rev. Sci. Instrum. 75 (2004) 4971. [35] C. Chudoba, E. Riedle, M. Pfeiffer, T. Elsaesser, Chem. Phys. Lett. 263 (1996) 622. [36] M. Pfeiffer, A. Lau, K. Lenz, T. Elsaesser, Chem. Phys. Lett. 268 (1997) 258. [37] S. Lochbrunner, A.J. Wurzer, E. Riedle, J. Chem. Phys. 112 (2000) 10699. [38] S. Lochbrunner, K. Stock, E. Riedle, J. Mol. Struct. 700 (2004) 13. [39] S. Lochbrunner, A. Szeghalmi, K. Stock, M. Schmitt, J. Chem. Phys. 122 (2005) 244315. [40] L.A. Peteanu, R.A. Mathies, J. Phys. Chem. 96 (1992) 6910. [41] J.L. McHale, Molecular Spectroscopy, Prentice-Hall, Upper Saddle River, NJ, 1999. [42] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Revision B.1, Gaussian, Inc., Wallingford, CT, 2009. [43] P. Kukura, D.W. McCamant, S. Yoon, D.B. Wandschneider, R.A. Mathies, Science 310 (2005) 1006. [44] S.Y. Lee, E.J. Heller, J. Chem. Phys. 71 (1979) 4777. [45] R.M. Hochstrasser, Nature 336 (1988) 621. [46] Z. Sun, J. Lu, D.H. Zhang, S.-Y. Lee, J. Chem. Phys. 128 (2008) 144114. [47] A.B. Myers, R.A. Mathies, Resonance Raman intensities: A probe of excited-state structure and dynamics, in: T.G. Spiro (Ed.) Biological Applications of Raman Spectroscopy, John Wiley & Sons, Inc., New York, 1987, pp. 1. [48] N. Agmon, Chem. Phys. Lett. 244 (1995) 456. [49] D.B. Spry, A. Goun, C.B. Bell, III, M.D. Fayer, J. Chem. Phys. 125 (2006) 144514. [50] Q. Wang, R.W. Schoenlein, L.A. Peteanu, R.A. Mathies, C.V. Shank, Science 266 (1994) 422. [51] L. Zhu, J.T. Sage, P.M. Champion, Science 266 (1994) 629. [52] D.V. Kurochkin, S.R.G. Naraharisetty, I.V. Rubtsov, Proc. Natl. Acad. Sci. U.S.A. 104 (2007) 14209. [53] M. Schade, P. Hamm, J. Chem. Phys. 131 (2009) 044511. [54] J.R. Ambroseo, R.M. Hochstrasser, J. Chem. Phys. 89 (1988) 5956. [55] G. Granucci, J.T. Hynes, P. Millie, T.-H. Tran-Thi, J. Am. Chem. Soc. 122 (2000) 12243. [56] S. Meiboom, J. Chem. Phys. 34 (1961) 375. [57] D. Madsen, J. Stenger, J. Dreyer, E.T.J. Nibbering, P. Hamm, T. Elsaesser, Chem. Phys. Lett. 341 (2001) 56. [58] N. Barrash-Shiftan, B. Brauer, E. Pines, J. Phys. Org. Chem. 11 (1998) 743. [59] M.S.T. Goncalves, Chem. Rev. 109 (2009) 190. 28 [60] S. van de Linde, M. Heilemann, M. Sauer, Annu. Rev. Phys. Chem. 63 (2012) 519. [61] B. Huang, H. Babcock, X. Zhuang, Cell 143 (2010) 1047. [62] C.W. Freudiger, W. Min, B.G. Saar, S. Lu, G.R. Holtom, C. He, J.C. Tsai, J.X. Kang, X.S. Xie, Science 322 (2008) 1857. [63] A. Sanchez-Galvez, P. Hunt, M.A. Robb, M. Olivucci, T. Vreven, H.B. Schlegel, J. Am. Chem. Soc. 122 (2000) 2911. [64] J.D. Coe, B.G. Levine, T.J. Martínez, J. Phys. Chem. A 111 (2007) 11302. [65] K. Heyne, N. Huse, E.T.J. Nibbering, T. Elsaesser, Chem. Phys. Lett. 369 (2003) 591. [66] G.S. Engel, T.R. Calhoun, E.L. Read, T.-K. Ahn, T. Mancal, Y.-C. Cheng, R.E. Blankenship, G.R. Fleming, Nature 446 (2007) 782. [67] E. Collini, C.Y. Wong, K.E. Wilk, P.M.G. Curmi, P. Brumer, G.D. Scholes, Nature 463 (2010) 644. 29 Figure 1 Figure 2 Figure 3 Figure 4 Figure 5 Figure 6 Figure 7 Figure 8 Figure 9 Scheme in Discussion Text Graphical Abstract TOC Figure A number of low – frequency modes are sequentially observed in photoexcited HPTS. Evidence of coherent quantum beat in several low – frequency modes with anharmonic coupling. The most transient low – frequency mode is the symmetric ring breathing of HPTS. Excited – state proton transfer occurs nonadiabatically on hte 5-200ps timescale. Kinetic isotope effect is 3-4 fot the two – stage ESPT components in water.