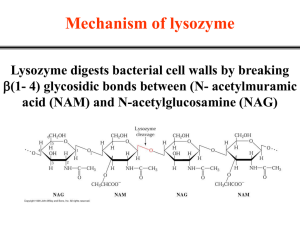
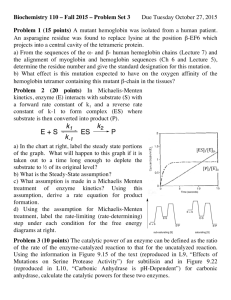
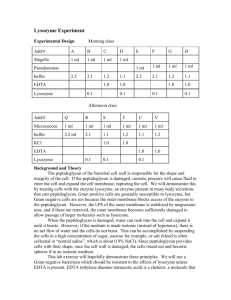
AN ABSTRACT OF THE DISSERTATION OF
advertisement

AN ABSTRACT OF THE DISSERTATION OF Yawen Wu for the degree of Doctor of Philosophy in Food Science and Technology presented on March 19. 2004. Title: Immobilization of Hen Egg White Lvsozyme by the Sole Histidine Residue to Polystyrene Beads through Peptide Spacers Abstract approved: Mark A. Daeschel Lysozyme is a natural antimicrobial agent that is effective against many food spoilage and pathogenic microorganisms by disintegrating their cell walls. Immobilization of lysozyme has attractive applications for use in the food industry: (1) The enzyme could be readily separated from treated foods and beverages and re-used while the foods could still be claimed additive-free; (2) It could impart stable antimicrobial capability to the surface of food packaging polymers. In this study, a novel method is described for the preparation of a highly active immobilized lysozyme system. The method addressed three key issues in the covalent attachment of a biological active protein to an insoluble support: 1.) The protein should be attached to the matrix by the fewest possible bonds to minimize conformational change; 2.) The binding site(s) on the enzyme to the supports should be located as far as practical from its active center and be nonessential for its tertiary structure; 3.) The binding method should minimize the steric interference between the support and the immobilized enzyme. Using polystyrene resin beads as support matrix, peptide spacers of various lengths composed of 6-aminocaproic acid were synthesized with the solid phase peptide synthesis method. Then the amino terminals of the spacers were derivatized with bromoacetyl bromide and coupled to the protein's only histidine residue (His-15) that is nonessential for its lytic activity. Immobilized lysozyme with a spacer composed of three 6-aminocaproic acid units displayed the best lytic result against lyophilized M. lysodeikticus cells: 2736 U/g resin with a protein load of 2.21 mg/ resin. Retained activity was 14.2% of that of the free enzyme. Preparations with longer spacers yielded higher protein load yet the retained activity remained at about 14 % level. A control consisted of random coupling of lysozyme to polystyrene beads without spacer gave an activity of 158U/G with a protein load of 1.24 mg/g resin and 1.4% of retained activity, Properties of the immobilized lysozyme system were studied, including stability, effect of pH, surface characteristics of the support. A kinetics study of the system using Eadie-Hofstee plot demonstrated strong external diffusion effects, which resulted in deviation from classic Michaelis-Menton kinetic behavior. Immobilization of Hen Egg White Lysozyme by the Sole Histidine Residue to Polystyrene Beads through Peptide Spacers by Yawen Wu A DISSERTATION submitted to Oregon State University in partial fulfillment of the requirement for the degree of Doctor of Philosophy Completed March 19, 2004 Commencement June, 2004 Doctor of Philosophy dissertation of Yawen Wu Presented on March 19. 2004. APPROVED: t Major Professor, representing Food Science and Technology c? Head of the Department of Food Science and Technology T3T- U. Dean of the Graduate School I understand that my dissertation will become part of the permanent collection of Oregon State University libraries. My signature below authorizes release of my dissertation to any reader upon request. en Wu, Author ACKNOWLEDGEMENTS I would like to express my sincere appreciation to my major adviser, Dr. Mark Daeschel, for his constructive guidance and continuous support during this research and in the preparation of this dissertation. Special thanks and appreciation are also extended to: Professor Floyd Bodyfelt, Dr. Joseph McGuire, Dr. Ronald Wrolstad, Dr. Mina McDaniel, and Dr. Charles Boyer for kindly serving as committee members when this project started eight years ago, and especially, for their continual support and encouragement during the writing of this dissertation five years after I left the program! (Please feel free to use me as an example for any of your graduate students who dares to think about taking a job before finishing his writing. Don't even think about it!) I would like to thank the Eckelman Foundation for granting me the fellowship to pursue this project. And I am also grateful to Kerry Group, for supporting me financially to complete this project through its employee continual education program. I wish to thank Mr. Dustan Doud, my dear colleague at Kerry, for proof reading this dissertation and his insightful comments. I wish to thank my wife, Tianying Jiang, for her love, help, and encouragement during these long years; my six year-old son, Tommy; four year-old daughter, Tina, for the enormous joy, love and meaning they have added to my life (giving them a piggy-back ride was my favorite break after hours of writing, or, hair-pulling, on a weekend day; the problem part, though, was who got to ride first). Finally, I want to thank my parents for kindling my passion for science and their never-ending support of my dreams and quests. Without their unconditional love and help, I would never have been able to come this far. TABLE OF CONTENTS Page 1. INTRODUCTION 1 I. Lysozyme 1 Structure of Lysozyme Lytic Mechanism of Lysozyme Properties of Lysozyme Assays of Lysozyme Application of Lysozyme in Foods II. Enzyme Immobilization - An Overview Physical Adsorption Entrapment Covalent Bonding Properties of Covalently Immobilized Enzymes 1 7 10 17 19 23 24 27 28 34 III. Immobilization of Lysozyme 40 IV. Approach of This Study-The His-15 and Spacer Strategy 43 2. MATERIALS AND METHODS I. Solid Phase Spacer Synthesis Preparation of t-(Boc)-6-Aminocaproic acid Purity of t-(Boc)-6-Aminocaproic acid TLC Attachment Boc-6-Aminocaproic Acid to the Resin Stepwise Synthesis of 6-Aminocaproic Acid peptide on the Resin Quantification of 6-Aminocaproic acid by HPLC II. Attachment of Lysozyme to the Spacer thorough His-15 47 47 49 49 50 51 53 55 Bromoacetylation of 6-Aminocaproic Acid 55 Coupling of Lysozyme onto the Support via Bromoacetylated Spacers- 57 Amino Acid Compositions of the Immobilized Lysozyme 58 III. Controls Covalent Binding via EDC Coupling As Controls 58 58 TABLE OF CONTENTS (Continued) IV. Determination of Lysozyme Activity 63 Activity against M. lysodeikticus cells Free Enzyme Immobilized Enzyme Activity against Glycol Chitin Free Enzyme Immobilized Enzyme 63 63 64 64 65 65 V. Others 66 Effects of pH on the Activity of Lysozyme - Free and Immobilized 66 Effect of Inhibitor Pentane on the Activity of Immobilized Lysozyme —66 Kinetics Study of Immobilized Lysozyme 66 3. RESULTS AND DISCUSSION 67 I. Solid Phase Spacer Synthesis 67 II. Attachment of Lysozyme 69 Covalent Coupling through Carboxyl Groups of Lysozyme Covalent Coupling through His-15 Efficacy of the His-15 with Spacer Strategy Activity against Soluble and Insoluble Substrates Kinetics Study of the Immobilized System Effect of pH on Activity of the Immobilized Lysozyme Hydrophobicity of the Polystyrene Support Stability of the Immobilized Lysozyme 69 72 74 78 78 86 88 89 4. CONCLUSION 93 BIBLIOGRAPHY 95 APPENDICES 107 Appendix A. On the Initial OD Rise in the First Few Seconds in the Turbidity Assay of Lysozyme - the Puff Theory 107 Appendix B. HPLC Assay of Lysozyme in Wine and Beer 111 LIST OF FIGURES Figure Page 1. Lysis of the NAG-NAM polysaccharide composing cell walls 2 2. Amino acid sequence of hen egg-white lysozyme 4 3. Two representative cartoon maps of the three dimensional structure of hen-white lysozyme 5 4. Lytic mechanism of lysozyme (part one) 8 5. Lytic mechanism of lysozyme (part two) 8 6. Possible modes of enzyme immobilization 25 7. Schematic illustration of conformational changes and steric hindrances 37 8. Proposed model for the lysozyme immobilization scheme 46 9. The basic plan and chemical reactions of solid phase peptide spacer synthesis 10. The modified seperatory funnel used as the reaction vessel for solid phase spacer synthesis 11. Chemical reactions involved in attaching 6-aminocaproic acid to the £2-nitrogen of His-15 of lysozyme 12. Coupling of lysozyme to the reactive amine group on the support via 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC) activation of carboxyl groups of the enzyme 13. Eadie-Hofstee plot of immobilized lysozyme (100 mg SAA.His-IS) against M. lysodeikticus (0.05-0.175mg/ml) 14. Lineweaver-Burk plot from the same data set of Figure 13. 15. Effect of pH on the lytic activity of free and bound lysozyme (3AA,His-15) 48 52 56 60 81 83 87 LIST OF FIGURES (Continued) 16. Stability of bound and free lysozyme over a period of 14 days when stored at 40C 91 A 1. The OD rise observed during the first few seconds of turbidity assay lysozyme activity of samples with three different concentrations: top line - 2.5 ppm, middle line - 1 ppm , bottom line - 0.1 ppm 108 A 2. The initial OD rise of a lysozyme sample at extremely low concentration - 0.001 ppm 110 B 1. Detected lysozyme concentration of lysozyme samples without heat treatment from the HPLC and Turbidity methods 114 B 2. Detected lysozyme concentration of lysozyme samples treated at 750C for 1,2,3, and 4 hours 115 B 3. Detected lysozyme concentration of lysozyme samples treated at 95C for 1,2,3, and 4 hours 116 LIST OF TABLES Table Page 1. Properties of hen egg-white lysozyme 11 2. Reactive residues of proteins 30 3. Average amino acid composition of proteins reactive residues 4. Number of reactions in which amino acid participates 32 5. Reactivity of low-molecular-weight reagents used to chemical Modification of amino acid residues of enzymes 33 6. Desired characteristics for a protein support 35 7. Summery of reported studies on lysozyme immobilization 42 8. Deprotection and coupling cycles for solid-phase peptide synthesis using Na - Boc-6-aminocaproic acid and ester linkage to polystyrene-resins 54 Amino acid analysis of the peptide spacers synthesized on the polystyrene beads 68 10. Activity of lysozyme covalently attached polystyrene beads via 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), with and without NAG protection 71 11. Amino acid compositions of lysozyme immobilized through His-15 73 12. Characterization of polystyrene-lysozyme catalysts 75 13. Specific activity of lysozyme against two different substrates 79 14. The inhibition effect of 4%(v/v) pentane on the activity of free and immobilized lysozyme against M. lysodeikticus 90 9. 32 B 1. Comparison of Lysozyme activity in red wine and beer samples detected with HPLC and turbidity methods, respectively 117 B 2. Releasing lysozyme from lysozyme-tannin complex in red wine with addition of NaCI at 2% and 8% levels 118 Immobilization of Hen Egg White Lysozyme by the Sole Histidine Residue to Polystyrene Beads through Peptide Spacers CHAPTER 1 INTRODUCTION I, Lysozyme Structure of Lysozyme Lysozyme [EC 3.2.1.17] is an enzyme that catalyzes the hydrolysis of a polysaccharide that is the major constituent of the cell wall of certain bacteria. The polymer is formed from p (1 —> 4) linked alternating units of Nacetylmuramic acid (NAM) and /V-acetylglucosamine (NAG). The enzyme cleaves the glycosidic bond between the C-1 atom of NAM and 0-4 of NAG of in the bacterial cell wall polysaccharide (Figure 1.) This action weakens the covalent structure of the wall and reduces its resistance to osmotic swelling, as a result of which the cells burst. First discovered in 1921 by Fleming in the mucous of humans (Fleming, 1922), lysozyme has since been found widely distributed in animals, plants and microorganisms - hen egg albumin and other avian egg-whites (Fukamizo et al. 1983), animal tissues and serum (Jolles and Jolles, 1961; Wardlaw, 1962; Pavlovskii et al., 1976), fruits and vegetables (Chandan and Ereifej, 1981; GlcNAc MurNAc GlcNAc MurNAc Glc NAc MurNAc CHj 0,=— CH,CH R, = —NH— C — CH, I ^= — 01— CO^H Figure 1. Lysis of the NAG-NAM polysaccharide composing cell walls. The hexasaccharide composed of alternating A/-acetylmuramic acid (NAM) and Nacetylglucosamine (NAG) residues as found in the natural cell wall peptidoglycan of gram(+)bacteria. The saccharine units are thought to bind to successive sites on lysozyme labeled A through F. Law and Goodenough, 1995), and bacteria (Richmond, 1959). It is considered a natural defense mechanism of its host system against bacterial invasion (Boiler etal, 1983) Hen egg white lysozyme is the best-characterized member of the lysozyme family. Some unique properties of the enzyme such as abundant, easily crystallized, and highly stable have made it an excellent model for extensive protein and enzyme studies. The solution of the structure of lysozyme is one of the triumphs of X-ray crystallography. In 1963, an investigation using X-ray crystallography at 6 A (1 A = 10nm) resolution revealed that hen egg white lysozyme consists of relatively small single polypeptide chain of 129 amino acids cross-linked by four disulfide bridges, which contribute to its high stability (Canfield, 1963). The enzyme has a mass of 14.6 kdal and its amino acid sequence is shown In Figure 2. Two years later, three-dimensional structure of hen egg white lysozyme was determined through 2 A resolution X-ray crystallography (Blake et al., 1965). Results showed that lysozyme is a compact molecule, roughly ellipsoidal in shape, with dimensions 450 X 300 X 300 nm. The high resolution X-ray crystallography also revealed that the oval fold of the protein can be divided into two lobes or domains with the active site of enzyme being situated in a well-defined cleft between them for binding the substrate (Figure 3). One of the domains is almost entirely (3-sheet structure (encompassing residues 40 to 85), whereas the other is comprised of the N and C-terminal segments (residues from 1 to 39 and 101 to 129) and Figure 2. Amino acid sequence of hen egg-white lysozyme. (Adapted from Canfieldand Liu, 1965) ACTIVE SITE Figure 3. Two representative cartoon maps of the three dimensional structure of hen-white lysozyme (Adapted from Acharya et al., 1989) is more helical in nature. The two domains are linked by an a-Helix (residues 89 to 99). The cleft in between is partly lined with nonpolar side chains of amino acids for binding the nonpolar region of the substrate, and it also has hydrogenbonding sites for the acylamino and hydroxyl groups (Fersht.igSS). Subsequently, the structure of hen egg-white lysozyme has also been determined from tetragonal crystals under a variety of conditions of temperature and pressure (Kundrot and Richards, 1987) and from triclinic, monochlinic and orthorhomic cystal forms (Joynson et al., 1970; Moult et al., 1976; Artymiuk et al., 1982; Hodsdon et al., 1990). All of these studies indicate that the conformation of lysozyme is essentially identical under the different conditions and crystalline environments. In the meantime, studies have also been done to compare the structures of the enzyme in crystalline state and in solution. NMR studies of hen eggwhite lysozyme in solution gave data consistent with its crystal structure (Dobson, 1977; Blake et al., 1981). A more recent NMR study (Smith et al., 1992) indicated that the main-chain fold of hen egg-white lysozyme is very similar between the two states. Further studies using Raman spectroscopy, NMR and inelastic neutron scattering have shown that the incorporation of water produces only small changes in protein structure and flexibility necessary for its enzymatic activity (Poole, 1994). These research results suggest that the structural data obtained from crystallized lysozyme can be applied to lysozyme molecules in solution with good confidence. Lytic Mechanism of Lysozyme The currently accepted mechanism of lysozyme action was elucidated mainly from examining the 2 A resolution X-ray crystallography data of the native enzyme and the complexes with inhibitors through model building (Blake et al., 1967). The catalysis begins when the enzyme's active site or the cleft selectively meshes with a six sugar segment (termed A through F in Figure 1.) of the alternating NAG-NAM polysaccharide substrate. When the segment binds, the D residue is distorted into a half-chair form from its normal chair form to fit into the active site, thereby weakening the glycosidic bond. This is an important aspect of the mechanism because the half-chair form is assumed to be the conformation of the transition state. In other words, in the process of binding, the enzyme forces the substrate to assume the geometry of the transition state. The remaining catalytic mechanism is comprised of the following five steps: 1 The -COOH group of Glu-35 donates an H+to the bond between C-1 of the D residue and the glycosidic oxygen atom, which thereby cleaves the bond (Figure. 4); 2.This creates a positive charge on C-1 of the D residue or a carbonium ion; Glu 35 V V V ;c- Asp 52 Asp 52 Figure 4. Lytic mechanism of lysozyme (part one). A key step in the catalytic mechanism for lysozyme is the transfer of an hTfrom Glu 35 to the oxygen atom of the glycosidic bond. The glycosidic bond is thereby cleaved, and a carbonium ion intermediate is formed. Glu 35 1/H oA D V NAG, Glu 35 <\ Asp >- 52 -o > A~-H k r-\ H0N H /s D V 0 >-\3\ |N;*GI| Figure 5. Lytic mechanism of lysozyme (part two). The hydrolysis reaction is completed by the addition of OH" to the carbonium ion and of H+ to the side chain of Glu 35. 3. Negatively charged Asp 52 stabilizes the carbonium ion by interacting electronically with the positive charge on C-1 of the D residue; 4. The carbonium ion intermediate then reacts with OH" from the solvent. The hydrolyzed product diffuses away from the enzyme (Figure 5) 5. Glu 35 becomes protonated, and the enzyme is ready for another round of catalysis. The crucial roles of Glu 35 and Asp 52 in the catalytic mechanism have been proved by selective modification experiments. Lysozyme derivatives modified at Glu 35 and Asp 52 were totally inactive, whereas it remained active when all of its carboxyl groups except those of Glu 35 and Asp 52 were esterfied (Parsons et al., 1969; Eshdat et al., 1973,1974; Yamada et al., 1982). When lysozyme was pretreated with NAG oligosaccharide - a substrate analog, however, Glu 35 and Asp 52 could not be modified by the esterfying reagents and the enzyme remained active (Lin and Koshland,1969). In the meantime, genetic modification of the two amino acids has been reported to examine their roles in the proposed mechanism. Site-directed mutagensis in which Glu-35 and Asp-52 were converted to the corresponding Asn and Gin demonstrated the critical requirement for Glu-35 - 0% activity from the Gin mutant, and slightly lesser essentially of Asp 52 - 0.5% activity from the mutant (Malcolm, et al., 1989). Studies also showed that the electrostatic force of attraction between lysozyme molecule and cell wall of M. lysodeikticus plays an important role in the initial binding process. The cell wall of the bacteria is negatively charged 10 when the cells are suspended in buffer solutions with normal pH range (Yamasaki et al. 1968; Maurel and Douzo, 1976; Price and Pethig, 1986). Electrophoresis measurements on the bacterial cells have shown that the net surface charge density on the cell wall is constant at around -1 .SpC/cm for the pH range 4-8. Since lysozyme has an isoelectric point around pH 11.2 (Imoto et al., 1972), it carries a net positive charge in the same pH range. Therefore from a macroperspective, the lysis of bacterial cells by lysozyme consists of the following three steps: The first step is an interaction between the positive charges of lysozyme and the negative charges on the surface of the bacterial cells. The second step of lysis is hydrolysis of p-1,4glucosaminide linkage in the polysaccharide of the cell wall. The last step is dissolution of the damaged cell wall. Properties of Lysozyme Just like other enzymes the stability and activity of lysozyme are affected by temperature, pH, ionic strength, and specific inhibitors and enhancers. Some of the general properties of lysozyme are summarized in Table 1. Factors Affecting Lytic Activities of Lysozyme Lysoyzme is highly heat stable. In phosphate buffer at pH 6.2 highly purified lysozyme showed only 5% loss of lytic activity after being exposed to 11 Table 1. Properties of hen egg-white lysozyme. Notice the two pH optima depending on the ionic strength (/). MW (kDa) 14.6 Shape/Size (nm) Oval/450x300x300 pi 11.2 Opt.pH Opt./* Opt.Temp (0C) 6.2 0.06-0.07 25-28 But heat stable up to 100 at Opt.pH & Opt./ 9.2 0.04-0.05 ' / = Vz Z C/Z,2, where c = ion concentration in moles; z = valence. 12 80 0C for 80 min. When exposed to100 0C for 20 min it lost only 25% (Smolelis and Hartsellf1952). Increasing the proton concentration has shown to increase the heat stability of the lysozyme. Beychock and Warner (1959) reported that stability in the temperature range of 85 to 950C was maximum at pH 5.5, 5% more active than that at pH 6.5. The extraordinary heat stability of lysozyme is mainly attributed to the four disulfide bridges formed between Cys64 - Cys80, Cys76 - Cys94, Cys30 Cys115, and Cys6 -Cys127. It has been shown that the Cys6-Cys127 bridge, which basically ties the head and tail of the polypeptide together (remember lysozyme has 129 amino acid residues), is not required for the complete folding of the protein since the protein already folds efficiently to its native state with other three disulfide bridges (Eyles et al., 1994). The role of the disulfide bridge appears to be solely strengthening the structure of the already fully functional enzyme rather than inducing structure into the polypeptide chain. In addition, the hydrophobic core of the enzyme is also thought to contribute to its stability, which is formed with hydrophilic amino acid side chains protruding toward the surface. The lytic activity of lysozyme varies markedly with the ionic strength of the solution in which is dissolved. When ionic strength is at 0.06-0.07, it has an optimum pH of 6.2; when at 0.04 - 0.05, it has a second optimum pH of 9.2 (Davies et al., 1968). In the complete absence of salts, or at an ionic strength of 0.005 or less, lysozyme has little or no activity on cell walls. This observation 13 was confirmed by Chang and Carr (1971) who found lysozyme to be inactive in distilled water. While many other investigators only observed the first pH optimum of 6.2-6.7 using strong buffer solutions, the appearance of the second pH optimum at lower ionic strength was also reported by Rupley (1967) and Neuberger and Wilson (1967). It was suggested that at pH 9.2 the zammonium group of Lys-97 acts as a proton donor to form the glycoside carbonium ion and that the negative charge on the carboxyl group of Asp-101 stabilizes the intermediate carbonium ion in accordance with the Glu-35 and Asp 52 mechanism at lower pH of 6.2 (Neuberger and Wilson, 1967, Davies et al. 1968) Inhibitors Knowledge on the inhibitors and activators of lysozyme is very important for studying its properties as well as its practical application. Lysozyme has been shown to be inhibited by a serious of agents which can be summarized into three groups: (1) surfactants, (2) nonpolar hydrocarbons, and (3) acidic polymers. When lysozyme is used as an antimicrobial agent, efforts should be made to minimize the presence of these agents in the system. Surface-active reagents inhibit lysozyme by binding to the enzyme at or near the active site. Lysozyme has been shown to be inhibited by sodium dodecyl sulfate (Meyer et al., 1947) and in general by alkyl sulfates, fatty acids, and aliphatic long-chain alcohols of 12 carbons or more (Smith and Stocker, 14 1949). It has been reported that 2.8% sodium dodecyl sulfate abolishes interaction between the substrate tri-A/-acetylchitotriose and lysozyme (Rupley, 1967). In addition, this concentration has been shown to produce no change in the helix content of the protein, but to alter the character of its tryptophan chromophores through specific interaction with them (Glazer and Simmons, 1965). A Tryptophan residue has been shown to be in involved in the formation of the enzyme-substrate complex between lysozyme and poly-A/acetylglucosamine (Hayashi et al., 1968). The second group inhibitors are nonpolar hydrocarbons, such as isobutane and pentane. They interact with the hydrophobic regions of lysozyme by hydrophobic bonding, thus render the enzyme inactive due to conformational change (Watanabe and Takesue, 1972, 1974). The higher the concentration of pentane and the lower the ionic strength, the stronger the inhibition. The last group of inhibitors are acidic organic polymers, including submaxillary mucoid (Simmons, 1952), hyaluronic acid, pneumococcus polysaccharide, glutamyl polypeptide, DNA, RNA (Skarnes and Watson, 1955; Wang et al., 1990), E. coli lipopolysaccharide (Ohno and Morrison, 1989), and tannin (Green, 1995). It is established that this group of agents inhibits lysozyme by forming lysozyme-polymer complex through electrostatic interaction between the basic enzyme and the acidic polymers. The inhibition tends to be very strong and yet usually pH sensitive and can be reversed by concentrated urea or sodium chloride solutions. It is worth pointing out that 15 majority of the above polymers are produced by bacteria which serve as their defense against lysozyme. Lytic activity of lysozyme on Gram-negative bacteria While lysozyme lyses many, though not all, Gram-positive bacteria in their vegetative forms, it is generally inactive against gram-negative bacteria because their outer membranes prevent access to the underlying peptidoglycan that is the enzyme's substrate. Many efforts, however, have been made to broaden the antimicrobial spectrum of lysozyme, by either changing the structure of the enzyme or pretreating the target microbial cells. All these efforts have potential value in food preservation and safety. Conjugation of lysozyme with dextran through the Maillard reaction increased its activity against both gram-positive and gram-negative bacteria (Nakamura et al., 1990). Further study from the same group indicated that the lysozyme-dextran conjugate also has greater heat stability and emulsifying properties than native lysozyme. In addition, its activity against gram-negative bacteria significantly increased if the temperature was raised during the conjugation treatment (Nakamura et al., 1992). Lysozyme modified with four perilaldehyde residues exerted a markedly enhanced antimicrobial activity against both gram-negative bacteria (Escherichia coli K12) and gram-positive bacteria (Staphylococcus aureus) (Ibrahim et al., 1994) More recently, Ibrahim et al. (1996) reported an interesting finding that partial unfolding of lysozyme with heat treatment at certain pH could switch its 16 antimicrobial activity to include Gram-negative bacteria without a significant detrimental effect on the inherent bactericidal effect against Gram-positive ones. It was found that heat denaturation of lysozyme at increasing temperatures for 20 min at pH 6.0 resulted in progressive loss of enzyme activity which greatly promoted its antimicrobial action to Gram-negative bacteria. The most potent antimicrobial lysozyme to either Gram-negative or positive bacteria was that heated at 80°C and pH 6.0, retaining 50% of the native enzymatic activity. The partially unfolded enzyme exposed two thiol groups and exhibited a 14-fold increase in surface hydrophobicity. Addition of the heat-treated enzyme to E. coli phospholipid vesicles resulted in a blue shift in the intrinsic tryptophan fluorescence accompanied by an increase in the size of the vesicle, indicating enhanced protein-membrane binding and subsequent fusion of liposomes. The authors claimed that the unique antimicrobial action of unfolded lysozyme attributed to membrane binding and subsequent perturbation of its functions. According to Samuelson et al.(1985), pretreatment with ethylenediaminetertraacetate (EDTA) significantly sensitized Salmonella to the action of lysozyme. Gram-negative bacteria, including salmonellae, became lysozyme-sensitive following a quick osmotic downshift - achieved by sudden dilution of a salt solution. This process has been proposed as the basis for a dip- or spray-decontamination treatment for poultry and other animal carcasses (Chatzolopou etal., 1993). After being treated with 0.5 to 5mM trisodium phosphate for 10 min, Campylobacterjejuni, Escherichia coli, Pseudomonas 17 fluorescens, and Salmonella enteritidis showed greatly increased susceptibility to lysozyme (Carneiro et al., 1998) Assays of Lysozyme A survey of literature reveals that most methods for determining lysozyme activity involve measuring turbidity change of its substrate spectrophotometrically. The lysis of bacterial cells suspended in a buffer system results in decreasing solution turbidity. Under proper conditions, there is linearity between the concentration of cell suspension and the turbidity, or absorbance at certain wavelength. Therefore the absorbance-changing rate of a cell suspension can be used as an indirect measure for the activity lysozyme in the system. Most of the assays were developed in the 1950s. Some workers have used Sarcina lutea cells as the substrate and a wavelength of 440 nm to follow the cell lysis (Dickman and Proctor, 1952; Smith et al. 1955), while others have used Micrococcus lysodeikticus cells as substrate and wavelengths of 450nm (Shugar, 1952) and 540nm (Smolelis and Hartsell, 1952; Kerby and Eadie, 1953). The Shugar (1952) method is a simple assay that measures initial velocity and has good reproducibility (± 3%). It has become the most common method used or adapted by most of the lysozyme research groups nowadays. One unit of enzyme decreases the absorbance at 450 nm by 0.001/min at 250C 18 in 66mM phosphate buffer, pH 7.1, using a suspension of live Micrococcus lysodeikticus as substrate in 3 ml reaction mixture (0.2mg/ml; 1 cm light path). Two modifications are often made to improve the original method: (1) replacing the vegetative Micrococcus lysodeikticus cells with standardized lyophilized cells that are commercially available; (2) changing the buffer pH from 7.1 to 6.24, which is closer to the optimum pH for the enzyme under the assay condition. It has also been reported that preparing the cell suspension at least 6 hours prior to use can improve reproducibility of the method after the cells had been allowed to equilibrate with the buffer for this period (Parry et al., 1965). A limitation for the spectrophotometric method, however, is that it requires that the enzyme sample must be in a clear solution, which often makes it impractical for measuring lysozyme in many food systems, such as milk and cheese. An extracting and decolorizing process might destroy the lysozyme being measured. When this is the case, an agar plate method developed by Gosnell et al. (1975) is especially useful to measure lysozyme in non-clear background. Agar plates are prepared consisting of 1.0 agarose with 0.1g NaCI in 100 ml of a pH 7.0 phosphate buffer seeded with 0.020 g lyophilized Micrococcus lysodeikticus. Several equidistant holes are punched into the agar and filled with lysozyme at various concentrations. After being incubated at 47°C for 3 hours, clear zones of lysed bacterial cells will appear around those wells containing lysozyme. The base 10 logarithms of the clear zone diameters are directly proportional to lysozyme concentration. As low as 5pg egg white 19 lysozyme per 100 ml of samples could be determined by this method. The agar plate method can be modified by seeding the agar with living bacterial cultures and using filter disks instead of punching holes (Vakil et al. 1969). Another more fundamental limitation for the turbidity assay is that the polymer substrate -cell walls is insoluble in buffer, albeit a homogeneous suspension. Theoretically, we want to measure the glycosidic cleavage, but what is actually measured is the reduction in turbidity of a suspension of dried cells of Micrococcus lysodeikticus in aqueous buffer solutions. The change in turbidity has not been correlated directly with the number of glycosidic bonds the enzyme splits and therefore use of an insoluble substrate is especially unsatisfactory for kinetic studies. Efforts have been made to address the ill-defined insoluble substrate issue. Assays have been developed using soluble oligo or poly NAG derivatives as the substrate, such as carboxymethylchitin (Marzotto and Galzigna, 1969), and glycol chitin (Imoto and Yagishita, 1971). These two assays were monitored by viscosimetry and reducing group analysis. Application of Lysozyme in Foods Hen egg white lysozyme has been the most commercially used animalderived antimicrobial to improve the safety and shelf stability of foods. Lysozyme constitutes 3-4% of the total egg white protein, so that it is readily available and relatively inexpensive (Powrie and Nakai, 1986). In addition, it's 20 natural origin is very appealing to the food industry due to growing public concerns about the safety of chemical preservatives and additives. Thus numerous studies and applications have been developed to use lysozyme as natural food additive to a wide range of foods with promising results. While most of the pioneer work on using lysozyme as a preservative in foods was done in Japan in late 1960s and early 1970s, concentrating on sea food, meat, beverage, and vegetables, European researchers have mainly focused on its application in dairy products in the past two decades. Lysozyme is a legal and popular preservative in cheese and dairy products in Germany, Italy and France (Anon, 1987). Lysozyme is recently granted GRAS status in USA. When lysozyme is used as a preservative in meat products, synergies are often found between the enzyme and other conventional preservatives. Akashi (1969) found that cured ground beef was preserved more effectively with the combination of 3% of NaCI, 12.5 ppm of NaN02, and 50 or 200 ppm of lysozyme than either by lysozyme alone or by the salts alone. In later work with Vienna sausage Akashi (1971) found that dipping them in 500 ppm lysozyme in pH 6.5 phosphate buffer gave the best results than using lysozyme alone, or nitro-furylacrylamide and sorbic acid. In a Japanese patent by the Eisai Company (1971), fresh seafoods were claimed to be preserved in aqueous solutions containing a lysozyme salt, amino acids, and NaCI. Later the same company patented another process to preserve oysters and shrimps by soaking them in aqueous solutions of 21 lysozyme and NaCI (Eisai, 1972). The Eisai Company (1971) also patented a process in which wine and sake were stabilized by incorporation of lysozyme together with p-hydroxybenzoic esters. Lysozyme was added to soy milk to preserve Tofu bean curd (Taiyo Food Co., 1972) and dried milk compositions for pediatric use were preserved by addition of lysozyme derived from egg whites along with ovalbumin and ovomucin (Morinaga Milk Industry Co., 1970). Kanebo Ltd (1973) patented a process to extend the shelf life of fresh vegetables, fish, meat and fruits by coating the surface with lysozyme. The major successful use of lysozyme commercially has been eradicating cells of Clostridium tyrobutyricum as they outgrow from germinated spores (Carminati et al, 1985; Bottazzi et al, 1993), thus control the so-called "late blowing" in certain semi-hard and hard cheeses caused by fermentation of lactate by the bacterium (Wasserfall and Prokopek, 1976; Carini and Lodi, 1982). The International Dairy Federation (1987) recommended the use of lysozyme as good practice in controlling late blowing when the number of C. tyrobutyricum spores in milk is in the order of hundreds. It has been estimated that more than 100 tons of lysozyme are used annually for this purpose (Scott etal, 1987). Lysozyme has been reported to kill resting vegetative cells of C. tyrobutyricum at room temperature and severely inhibit actively growing ones at concentration of 500U/ml (Wasserfall and Teuber, 1979; Bester and Lombard 1990). Its effect on the spores involves a two-step process: It first stimulates its 22 germination; then significantly inhibits the outgrowth of spore cells into vegetative cells (Wasserfall and Teuber, 1979; Bester and Lombard 1990). Wasserfall and Teuber (1979) suggested that the inhibition of the outgrowth of spore cells into vegetative cells was the basis for using lysozyme to control late blowing. Lysozyme has potential in the wine industry to be used as a control measure for malolactic fermentation and preservative against bacterial spoilage during wine conservation (Amati et al., 1992). In the course of winemaking, malolactic fermentation (MLF) - the conversion of malic acid to lactic acid and carbon dioxide by mainly Leuconotoc oenos, reduces acidity, thus has significant effects on the sensory profile and microbial stability of wines (Henick-Kling 1993). While MLF can be beneficial for high acid wines, MLF may reduce acidity too much in high pH wines and cause proliferation of spoilage lactic acid bacteria, such as Pediococcus sp. and Lactobacillus sp.(Rankine and Bridson, 1971; Radler, 1975) According to Green et al. (1994), lysozyme concentrations of 500 and 1000 ppm are effective at preventing MLF in red wine. The ability of lysozyme to stop MLF midway through fermentation was demonstrated in the 300 ppm lysozyme treatment. Similar results were reported later by Gerbaux et al. (1997) that an addition of 500 ppm lysozyme to grape must inhibited MLF, while addition of 250ppm to red wines, after MLF, promoted microbiological stability. In the wines to which lysozyme was added, there was no increase in the content of acetic acid and biogenic amines during a period of six months at 18 23 degree C. Whereas the levels of volatile acidity and biogenic amines in the controls were 20% and 400% higher, respectively. II. Enzyme Immobilization - An Overview Enzyme immobilization is a physical or chemical process that fixes enzymes onto a solid support, or traps them into an insoluble matrix. By so doing, a soluble enzyme is converted into bound or insoluble form but remains biologically active with regards to its substrate. The technique has two major advantages: the enzymes can be recovered and used again for research or application purposes; the enzyme can be used in a variety of configurations of bioreactors that allow continuous operation. Immobilized enzymes also often have improved storage and operational stability. Enzyme immobilization methods may be divided into three categories: The enzyme can be (1) adsorbed onto, (2) physically entrapped or encapsulated within, and (3) covalently bound to a distinctive phase that allows exchange with, but is separated from, the bulk phase in which substrate is dispersed. Figure 6 lists possible modes of enzyme immobilization. The covalent binding approach has been by far the most widely investigated and it has been the top choice thus far for most of the lysozyme immobilization studies. Therefore, this overview will rather be strongly oriented towards methods based on the covalent immobilization of enzymes. 24 Immobilized enzymes are a qualitatively new type of biocatalyst different from the free enzymes in solution, not only in their physical properties, but also in many other biochemical characteristics. After being fixed to an insoluble support the enzyme is in a new state in which its properties are influenced by different factors related to the chemical and structural nature of the support, as well as to the way that the enzyme is attached to the matrix structure. A discussion of the factors affecting the characteristics of immobilized enzymes is also included at the end of this overview. Physical Adsorption The oldest and simplest method of enzyme immobilization is that of physically adsorbing the enzyme on to a solid matrix. Eighty eight years ago Nelson and Griffin (1916), reported that invertase retained its catalytic ability after adsorbed on to activated charcoal. Most of the later developments in this area took place between 1965 and 1975. Wide ranges of materials have 25 (1) Covolent bonding (2) Eleclrostotic bonding (3) Copolymen'zcjfion (A) Polymei entropment ':;[$ Enzyme molecule I (5) Hydrophobic interoction /A, Phospholipid Polymer matrix Pf©© (6) Liposmal entrapment (7) Encapsulation Figure 6. Possible modes of enzyme immobilization (from Trevan, 1980). 26 been used as absorbents for various proteins. Synthetic ion exchange resins, though originally designed for chromatographic purposes, such as Dowex 50 (Barnett and Bull, 1956), DEAE- and CM-cellulose (Mitz, 1956), have been the most extensively used. Other materials often used, but usually with lower adsorption capacities, include polystyrene resins, kaolinite, collagen, aluminum, silica gel, and glass. Immobilization by physical adsorption is a very easy to perform: the adsorbent and enzyme are stirred together for sometime. Yields (enzyme bound per unit of adsorbent) for physical adsorption can be high. However, the process is non-specific and a real or apparent decrease in activity will ensure if adsorption involves groups at the active site. Protein-protein interaction resulted from overloading can also lead to apparent loss in catalytic activity (Bernath and Veith, 1974). During operational use adsorbed enzymes may leak from the carrier because of the binding force(s) - ionic, hydrophobic, hydrogen bonds, or Van der Waals' interactions, between the protein and the carrier is weak. Another factor which often causes desorption is the addition of substrate to the enzyme preparation (Trevan, 1980). This is particularly problematic, for although other factors which might cause desorption (such as pH, temperature, or ionic strength) can be controlled, no enzyme can work without its substrate. One interesting development of the technique of adsorption is in the use of an activator of an enzyme as a coupling agent between a support and that enzyme. Fukui et al. (1975) demonstrated the immobilization of tyrosinase and tryptophanse by adsorption on to an insoluble derivative of pyridiosal-5' 27 phosphate, an activator of these enzymes. Thus the enzyme is not only specifically adsorbed on to the polymer, but is also activated by the same process. Entrapment The entrapment technique is a growing development in enzyme immobilization technology (Kumaraswamy et al., 1981; Stevenson and Sefton, 1987). The method is based on enclosing enzymes in the lattice of a polymeric matrix or semi -permeable membranes, such that the substrate and products are able to diffuse through the enclosure while the enzyme proteins are too large to escape. A minor loss of biological activity is one of the main positive features of the entrapping technique, simply because the enzyme is not actually attached to anything. There are none of the steric problems associated with covalently binding the enzyme in such a way that its active site is obstructed by a portion of the polymer matrix. On the other hand, the diffusion limitations are the most negative feature of the method. This type of enzyme immobilization is obviously not suitable for enzymes with high molecular weight substrate, such as lysozyme. 28 Covalent Bonding Compared to physical adsorption and entrapment methods, the immobilization of enzymes on solid supports by covalent coupling usually leads to very stable preparations with extended active life. Large scale experimentation in this field was first carried out by Grubhofer and Schleith in 1953, who used a diazo derivative of poly-p-aminostyrene to immobilize pepsin, amylase, and carboxypeptidase (Grubhofer and Schleith,1954). Since then covalent binding has become the most frequently used and investigated approaches for enzyme immobilization, mainly due to the growing discovery of various bonding reactions and of matrices with functional groups capable of covalent coupling, or susceptible to activation to provide such groups. The compositional and structural complexity of proteins has not allowed, however, the application of general rules to predict the best method suited for a specific immobilization task. Accumulated experience in the field has emphasized the importance of two factors that have to be evaluated when choosing a method for the covalent immobilization of an enzyme: (1) the type of functional group on the protein through which the covalent bonds with the support matrix are formed and hence the chemical reaction to be used; (2) the physical and chemical characteristic of the support matrix onto which the reactive groups are to be coupled. 29 Functional Groups and Coupling Reactions The rule of thumb for choosing the functional groups on the enzyme through which the covalent bond with the support is to be formed is that they should be naturally nonessential for the catalytic activity of the enzyme. In the meantime the binding reactions that can be carried out under relatively mild condition should be preferred. Such reactions should exhibit, under ideal conditions, relatively high specificity toward one type of functional group on the protein and minimal side reactions with other functional groups. In practice, however, such a situation is seldom if ever realized. For the immobilization of a given enzyme, the information is often limited on its amino acid composition, the amino acids involved in the active site, the effects of specific chemical modifications on activity, the protection of the active site region by inhibitors, as well as the three-dimensional structure of the enzyme. Indeed, so far the reaction and support selecting process is still very much empirical, heavily relying on the methods used in the chemical modification of proteins. It is not uncommon that several different reactions and supports have to be tested for a particular immobilization task before a satisfactory preparation can be achieved (Cabral and Kennedy, 1991). The selection of methods applied to the chemical modification of enzymes has been based on knowledge of the reactivity, frequency of occurrence (relative concentration) and the accessibility of the amino acid residues of the enzyme. Table 2 summarizes the residue of amino acids that have functional groups in the side chain suitable for linking to a support. 30 Table 2. Reactive residues of proteins (from Taylor 1991) f-Amino of r.-lysinc (i.-Lys) and N-tcrminus amino group Thiol of i.-cysteine (i.-Cys) Carboxyi of i.-aspartate (i.-Asp) and i.-glutamate (1..-GI11) and C-terminus carboxyi group -NH2 -SH -COOH <y OH Phenolic of L-tyrosine (r-Tyr) NH N-C \ Guanidino of i.-arginine (i.-Arg) NH, Imidazole of i.-hislidinc (i.-His) Disulfide of i.-cystinc Indolc of i-tryptopluin (i.-Trp) CH3-S-CH2OH Thioether of i..-nietl)ionine (i-Met) Hydroxyl of i.-serine (i.-Scr) and i.-threoninc (i.-Thr) 31 From this list, notice that the amino acids with amide groups (glutamine and asparagine), those with hydrocarbon side chains (alanine, leucine, isoleucine, valine, phnenylalanine, and proline), and glycine are absent due to their relatively low concentration on the exposed protein surfaces and, more importantly, due to their non reactive hydrophobic nature. For enzymes with limited information of their structure and amino acid composition, data in Tables 3, 4 are often used to access the most convenient residues in the enzyme that is to be covalently immobilized. Comparing the data in the two tables, it appears that the most convenient residues for immobilization are the lysyl residues, followed by cystein, tyrosine, histidine, aspartic acid, glutamic acid, arginine, tryptophan, serine, threonine, and emthionene. In practice, most of the common covalent coupling reactions involve amino groups, carboxyls, or the aromatic rings of tyrosine and histidine. After the residues are chosen, suitable reactions may be selected according to the rules governing chemical modification of protein, which is summarized in Table 5 by Means and Feeney (1971). Polymeric Supports After the coupling reaction is selected that would preserve the most activity, proper support(s) can be chosen to fit the final application 32 Table 3. Average amino acid composition of proteins reactive residues (from Means and Feeney, 1971). Residue Percent Ser Lys Thr Asp Glu Arg Tyr Cys His Met Trp 7.8 7.0 6.5 4.8 4.8 3.8 3.4 3.4 2.2 1.6 1.2 Table 4. Number of reactions in which amino acid participates (from Means and Feeney, 1971). Residue Number of Reactions Cys Lys Tyr His Met Trp Arg Glu Asp Ser Thr 31 27 16 13 7 7 6 4 4 0 0 Table 5. Reactivity of low-molecular-weight reagents used to chemical modification of amino acid residues of enzymes (from Means and Feeney, 1971). Reactivc amino acid residue of enrvmc' Reaecm -NH, Acetic anhydride Aldchyde/NaBHj N-Carboxyanhydrides Cyanogen bromide Diiizonium salts 5,5'-Dithiot!!S(2-ni'roben7.0!c acid) Formaldehyde Clyoxal Haloacctatcs Maieic anhydride p- Mercu riben/oa EC Sodium borohydride Succitnc anhydride Thiols Wator-solublt carbodiimide and iuicicophik- -SH vL --COOH K> .S--S- SCH, z I -r -f + 4- -•-- +++ +++ ' - . • . • -. dnd • - • (ndrc.Hc rciativf rcaciivitirf -^. ± ±. ami •*: ± ~ Ufctfu-tv indicate ttm-.^r f^Aciivittci which toa? nr tn;(;-- n'.« nt :u;;uricd depcndirj; upon the E-i/uthuor^ t?iRpi>tu:J. ' Spunliitieuy.tiy n'vrr*:h:c im-Jirr ihi: reaciion ^vniiuiv-^ or wpon diiuhon. rcgcncrnling origimi! aroup. Fasiiv rcscrsibir. ri-r.cncrstin* orijiina! jiroiir-. 34 accordingly. A carrier judiciously chosen can enhance the immobilized protein preparation. The primary base supports for enzyme covalent bonding are mainly polysaccharides and other polymers, including: agarose, cellulose, dextrans, chitosan, alginate, collagen polyacrylamide, nylons, and polystyrenes, silica gel and glass beads, etc. The properties of these supports are thoroughly reviewed by Cabral and Kennedy (1991). Although there is no universal carrier, a number of desirable characteristics should be common to any material considered for immobilizing enzymes (Table 6). Important support properties, such as morphology (shape, size, surface, porous vs. nonporous, surface area, etc.), mechanical and microbial stability, and modification, should be assessed with respect to the specific application prior to the selection for evaluation. Properties of Covalently Immobilized Enzymes While in many cases the stability of enzyme is increased by immobilization (Zaborskey, 1973) mainly because of the prevention of autolysis by restricting intermolecular contact, according to most studies the activity of immobilized of immobilized enzymes has been lower than that of the same amount of soluble enzyme at a given concentration of substrate and effectors (Engasser and Horvath, 1976). The changes in the enzymatic behavior 35 Table 6. Desired characteristics for a protein support. Large surface area Good permeability Hydrophilic character Chemical, mechanical, thermal, microbial stability High rigidity Suitable shape and particle size Regenerability 36 due to immobilization in a heterogeneous medium can be attributed to two main factors. The first involves changes in the conformation of the enzyme molecule and steric hindrance dependent on the mode of enzyme binding to the carrier. The second factor concerns the non-uniform distribution of substrate, products or protons between the immediate vicinity of bond enzyme, often called the microenvironment, and the bulk solution, or macroenvironment, where effects related to mass transfer often arise, such as diffusion restrictions of substrate and products to the active site of the enzyme and back into the bulk solution. Conformational Change and Steric Hindrance The decrease in enzymatic activity upon immobilization is often caused by conformational changes in the enzyme structure or by steric hindrance s in the immediate vicinity of the enzyme molecules. These two effects are illustrated in Figure 7. Multiple covalent bonds between the enzyme and the matrix can stretch the whole molecule and thus alter the three-dimensional structure of the active site. Datta et al. (1973) have suggested that such conformational alterations may account for the significant loss in the lysozyme activity upon immobilization. Thus an important consideration in the covalent attachment of a biologically active protein to an insoluble support is that the protein should be attached to the matrix by the fewest possible bonds (Cuatrecasas, 1969). This will increase the probability that the attached macromolecules will retain 37 Enxymo \ Substrato a fl Enzyme in free solution Immobilized enzyme Conformational chango Steric hindrance Figure 7. Schematic illustration of conformational changes and steric hindrances that may affect the kinetic behavior of an enzyme upon immobilization (from Engasserand Horvath, 1976). 38 its native tertiary structure, and its properties may more nearly resemble those of the native protein in solution. Steric hindrances are caused by the shielding effect of the matrix, which makes part of the enzyme molecules less accessible to the substrate. When an enzyme molecule is randomly bound to a matrix there is no guarantee that it will bind the correct way round and, often as not, it is likely to be bound with its active site completely blocked by the support matrix (Figure 7). This steric effect is particularly troublesome for enzymes with high molecular weight substrates, such as lysozyme. In order to reduce the shielding of the active sites, which may accompany the binding of an enzyme to a carrier, a "spacer" can be used to keep the enzyme at a certain distance from the matrix. This approach has found wide application in affinity chromatography, where steric hindrances would otherwise impair the binding of high molecular weight substances to the matrix-bound moiety (Steers et al., 1971; Mosbach et al., 1974). Diffusional Restrictions and Partition Effects When an enzyme is bound to a support, its substrate diffuses from the bulk solution to the catalytic sites, and the product of reaction diffuse back to the bulk solution. Thus concentration gradients are established in the surroundings of the bound enzyme so that concentrations of substrate and product both differ between the immediate vicinity of the bound enzyme and the 39 macroenvironment. These processes can cause diffusional resistance -either external and/or internal diffusional barriers. The external diffusion barrier is a result of the thin, unstirred layer of solvent that surrounds the polymer particle (Trevan, 1980). Solutes diffuse in this layer by a combination of passive molecular diffusion and convection. The thickness of this layer is affected, to a certain extent, by the speed at which the solvent around the immobilized enzyme particle is stirred. Increasing the stirring rate will reduce this external diffusion barrier. Thus an important factor in any experiment involving an immobilized enzyme preparation is the stirring speed. Internal diffusion resistances are limitations to free diffusion within the polymer matrix imposed by the polymer matrix. Within the polymer matrix diffusion within usually takes place by passive molecular diffusion only, and is not affected by stirring speed. Obviously internal diffusion effects will be more marked if enzyme is immobilized by entrapment within the polymer matrix rather than attachment to the surface of the polymer matrix. As expected, the decrease in enzymatic activity due to immobilization has been found to be greater with high than with low molecular weight substrates (Silman et al., 1966). It is well established that electrostatic, hydrophobic, and hydrophilic interactions among the carrier the substrate and solutes often produce an unequal distribution of these species between the micro- and macroenvironment. A good example is partitioning of protons that causes 40 shifting of the pH optimum of the enzyme that is immobilized on polyonic matrices. Polyanions will tend to concentrate protons (thus lowering pH) around the enzyme while polycations will tend to expel protons (raising pH). Goldstein (1972) observed a shift of 1 pH unit towards alkaline values of the pH optimum of chymotrypsin acting on acetyl-L-tyrosine ethyl ester, when immobilized on the polyanion ethylene-maleic anhydride copolymer. Conversely a downward shift of the pH optimum by 1 unit was observed when chymotrypsin was immobilized on the polycation polyornithine. III. Immobilization of Lysozyme A highly active immobilized lysozyme system can be exploited for food industry application. Some promising advantages of the technology are: (1) the enzyme can be readily separated from treated foods and beverages and be repeatedly used while the foods can still be claimed additive-free, which is something now preferred by more and more consumers;(2) It can provide stable microbial inhibition capability to surface of polymers to develop antimicrobial food packaging system to improve shelf life and food safety. Despite the appealing potential benefits of immobilized lysozyme systems, however, studies on lysozyme immobilization have been relatively sporadic since early 1970s. Covalent binding appeared to be the major coupling method employed by various research groups, and retained specific 41 activities against M. lysodeikticus of the systems were very low - only 0.015.5% of the activity of the free enzyme, hardly of any practical potential (Table 7). It is concluded that diffusion limitation, steric hindrance, excessive coupling, and coupling through essential amino acid residues are the main reasons responsible for the significantly low retained activities (Datta et al., 1973; Moser et al., 1988; Crapisi et al., 1992; Chen and Chen, 1996). The significant diffusion limitation of immobilized lysozyme likely results from the insoluble particulate substrate. It is a system of a doubly heterogeneous nature: a situation where the enzyme attached to one solid reacting with a substrate located in a separate solid material, the cell wall. Since the diffusivties of micron sized particles are several orders of magnitude less than those of proteins (the diameter of the globule protein is about 1/200 of that of the cell of M. lysodeikticus), the substantial rate loss is at least partially attributable to collision limitations (Datta et al.1973). Additionally, steric factors may be more significant than when the substrate is totally soluble. An reciprocal relation between binding yield and specific activity of the immobilized protein was, first observed by Datta et al., in 1973, and repeatedly confirmed by subsequent reports (Moser et al., 1988; Crapisi et al., 1992). In general, the higher the immobilization yield, the lower the specific activity. Table 7. Summery of reported studies on lysozyme immobilization. Turbidity reduction rate measurement of M. lysodeikticus cell suspension is the principle of the assays used by all the studies. Support Method Polyacrylamide Covalent/Diazonium Nonporous iron particles Covalent/Glutaraldehyde Silicate Covalent/Shift base Glass beads Enzyme Load (mg/g support) 2.85 Retained Activity (%) "appreciable" Reference Dattaetal. 1973 <0.01 Hailing etal. 1979 2.2 1.5 Moseretal. 1988 Covalent/BrCN 2.0 2.9 Chitosan Adsorption 2.5 0.24 Silica gel Adsorption 0.8 0.8 Polystyrene Ionic Binding 0.2 10.7 Nonporus glass beads Covalent/Glutaraldehyde 0.4 5.5 AS-La Covalent/EDC 31.4 14.0b 3 b Not Reported Crapisietal. 1992 Chen etal. 1996 An enteric coating polymer which shows reversibly soluble-insoluble characteristics with pH change. Activity measured when AS-L - lysozyme complex was in the soluble state. to 43 It is generally observed that with covalent coupling the binding yield is proportional to the density of protein coupling groups on the support surface and the number of activated amino acid residues on the surface of the protein. Therefore, while an highly-derivatized surface and/or an highly-derivatized protein can yield higher protein load (Datta, 1973; Crapisi et al, 1992), it can result in excessive bonding between the two as to significantly change or even denature the enzyme. And lastly, any binding through or close to essential amino residues would render the protein inactive. A review of the reported studies suggested that most of the coupling methods chosen in the studies were directly adopted from the methods used for other enzymes. The "trial -and -error" approach was often used to choose a coupling reaction. It is a common observation that several different reactions and supports have to be tested before a suitable immobilization protocol forms the basis of experimentation. (Datta et al., 1973; Crapisi et al., 1992). Moreover, it appears that little advantage was taken of the comprehensive and detailed information available on lysozyme structure, catalytic mechanism, and other enzymatic behaviors under different conditions to formulate immobilization methods . IV. Approach of This Study - The His-15 and Spacer Strategy Based on above and earlier discussion, rules for maximizing the activity of a covalently immobilized enzyme system can be summarized as the following 1.) The protein should be attached to the matrix by the fewest 44 possible bonds to minimize conformational change; 2.)The binding site(s) on the enzyme to the supports should be located as far as practical from its active center and nonessential for its tertiary structure; 3.) The binding method should minimize the steric interference between the support and the immobilized enzyme. The extensive knowledge and understanding of the structure and properties of lysozyme make it possible to apply these parameters to its immobilization approaches.. The basic strategy of this study is to establish a single bond between a single nonessential amino acid residue in lysozyme and its support to minimize any conformational change of the bound enzyme, and to introduce a suitable spacer between the enzyme and its support to reduce steric hindrance. The IS"1 amino acid residue in lysozyme molecule is the only histidine (His-15) in the enzyme and has been shown nonessential to its lytic activity (Kravchenko et al., 1964; Goux and Allerhand 1979). Moreover His-15 is located on the opposite side of the activity center (Blake et al., 1967) and the imidazole group is well exposed on the surface and readily reactive to alkylating agents, such as bromoacetamide derivatives (Yamada et al, 1984). These two fortuitous facts make His-15 the ideal reaction group on the enzyme for this study. The role of the so-called spacer molecules involved in the binding of biospecifically active groups at the surface of matrices was the subject of extensive discussion in 1970s (Cuatrecasas, 1970; Cuatrecasas and Anfinsin, 45 1971; O'Carra et al., 1974; Costerton, et al., 1978. Turkova, 1978). The introduction of flexible spacers lengthens the distance between the protein and the matrix surface, thus reduce the steric hindrance. Solid phase peptide synthesis was developed by Merrifield (1963) in order to simplify and accelerate peptide synthesis. The fundamental premise of the technique is that amino acids can be assembled into a peptide of any desired sequence of significant length while one end of the chain is anchored to an insoluble support. The most commonly used solid support in solid peptide synthesis is fine polystyrene beads prepared by copolymerization of styrene with 2 per cent divinylbenzene, which happened to be a common support used for many enzyme immobilization studies (Grubhofer and Schleith, 1953; Filippusson and Hornby, 1970; Taylor and Swaisgood, 1972). Thus solid phase peptide synthesis process is readily adaptable to be used in this study for spacer synthesis, using polystyrene beads as the support for lysozyme. With the amino group on the to carbon, 6-amino-caproic acid is a good amino acid to be used as the building block for the spacer. Each coupling reaction will increase the length by 6 carbon units, instead of 2 as it will get from using glycine. The proposed model of the lysozyme immobilization scheme is given in Figure 8. 46 A 9 ? /^\ ^—(O-C-CH,-CH,-CI K-CH^CI-U-NH^C-CM, —Hisf-15 < ' x / Figure 8. Proposed model for the lysozyme immobilization scheme: The His-15 of the enzyme, which is on the opposite side of its active center, is covalently linked to a spacer that is anchored to a solid support. 47 CHAPTER 2 MATERIALS AND METHODS I. Solid Phase Spacer Synthesis The method of spacer synthesis was adopted from the process of Solid Phase Peptide Synthesis (Stewart and Young, 1969). The procedure and chemical reactions used in the spacer synthesis are summarized in Figure 9. The solid support is a synthetic resin, 200-400 mesh, a copolymer of styrene and divinylbenezene, which bears reactive chloromethyl groups (also known as Merrifield Polymer, Fluka). Four basic steps involved in the synthesis of the peptide spacer: (1 )Protect the amino group of 6-Aminocaproic acid with a f-butyloxycarbonyl(f-Boc) group, (2)The salt of the f-Boc-6-Aminocaproic reacts with the chloro group on the resin to form an ester bond between the amino acid and the resin,(3)Activate the amino group of the attached amino acid by removing the f-Boc protecting group, (4) Attach the carboxyl group of the next tBoc-6-aminocaproic acid to the activated amino group of the first amino acid along with a coupling agent. Steps (3) to (4) were repeated till the desired length of the spacer was achieved. Throughout all these steps, reagents and reaction conditions were chosen so that all reactions went to 100 per cent completion. 48 0 II HzN—(CH2)5-C-OH Step*!. PROTECT O Ph II I (O^C-O-C-O-N^C-CN O O II II (CH3)3C-0-C-NH-(CH2)5-C-OH Step 2. ATTACH Cl-CH2-Pofyrrer O II O II (CH3)3C-0-C-NH-(CH2)5-C-0-CH2-Polymsr Step 3. DEPROTEC O II NH2-(CH2)5-C-0-CH2-Po]yrrer Step 4 COUPLE o II o II (CH3)3C-0-C-NH-(CH2)5-C-OH OOO II II II (CH3)3C-0-C-NH-(CH2)5-C-NH-(CH2)5-C-0-CH2-Polyn£r Figure 9. The basic plan and chemical reactions of solid phase peptide spacer synthesis. 49 Preparation of t-butyloxycarbonyl (Boc)-6-Aminocaproic acid The procedure was modified from the Sampson and Barttlett method (1990). The amount of 5.5 mmol Boc-On (Fisher Chemical) and 5 mmol 6aminocaproic acid (Aldrich Chemical) were suspended in a 300 ml bottomed round bottle with 200 ml 50% aqueous dioxane and 10 ml triethyl amine. The mixture was magnetically stirred at room temperature for 22 hours. Then high vacuum (755 mmHg) was applied to the bottle to evaporate excessive dioxane and triethyl amine. The remaining content in the bottle was transferred to a 300 ml seperatory funnel and extracted 3 times with 100 ml ethyl acetate to remove unreacted reagents. The aqueous phase was acidified to pH 1 with 12 N HCI and then extracted 4 times with ethyl acetate. The total of 400 ml ethyl acetate extract was combined into a 1000 ml Erlenmeyer flask and dried over Na2S04. Finally, the ethyl acetate phase was carefully decanted into a 1000 ml beaker and let evaporate overnight. White crystals of Boc-6aminocaproic acid precipitated out on the bottom of the beaker. Purity of the Boc amino acid determination by TLC (Thin Layer Chromatography) TLC analysis was performed to check the purity of the Boc-amino acid, using the Stewart and Young (1969) method. Approximately 1 pi of the amino acid derivative solution (50 mg per ml in ethyl acetate) was spotted onto a TLC plate (Brinkman Silica Gel H, 4X5 inch). Same amount of 6-aminocaproic acid was also spotted as control. Then the plate was run in a solvent composed of 50 chloroform (85), methanol (10), and acetic acid (5) in a sealed glass chamber in which the atmosphere was saturated with the solvent. After solvent development the TLC plate was dried in the air and placed in a closed chamber containing a beaker of fresh, concentrated HCI. After 15 min of exposure to the HCI fumes, the plate was heated in a 105 0C oven for 10 min. Then the plate was sprayed with ninhydrin solution (0.2% in acetone) to develop color. Finally the purity of Boc-6-aminocaproic acid was assessed based on the shape and Rf s of the color spots Attachment Boc-6-Aminocaproic Acid to Chloromethylstyrene Resin The procedure was adopted from the book "Solid Phase Peptide Synthesis" by Stewart and Young (1969). Chloromethyl polystyrene resin (5 grams) (Merrifield Polymer, Fluka), 5 grams of Boc-6-Aminocaproic acid and 3 mis of triethyl amine were combined in a 250 ml round-bottom flask with 25 ml absolute alcohol added to cover the resin. The mixture was refluxed gently for 24 hours at 81 0C in a Clas-Col electric heater with a 500-mm H2O condenser with a CaCb drying tube attached. The molar ratio for the reactants was 1.0 mole of Boc-6-aminocaproic acid (230.18 g per mole): 1.0 mole of Cl of the chloromethyl resin (232.6 g per mole ): 0.9 mole triethyl amine ( 0.14 ml per millimole; sp. gr. 0.723). The reacted resin was transferred to coarse-fritted Buchner funnel and washed successively with EtOH, H2O, MeOH, and CH2CI2, three times with 51 each solvent, allowing one minute contact time for the solvent to penetrate the resin beads and for solutes to diffuse out of the beads. The washed resin, still suspended in CH2CI2, was then transferred to a 250-ml seperatory funnel, stirred with CH2CI2 (approximately 20 ml per g), and let stand until the bulk of the resin floated to the top and a fairly sharp demarcation line appeared at the bottom of the floating resin. The solvent, carrying the suspended finest particle of resin, was run out of the funnel and discarded. This separation process was repeated three times to prevent troublesome clogging of the glass wool filter in the synthesis vessel. After removing all volatile solvents with a water aspirator, the resin was dried overnight in a desiccator with silicon gel. The amount of 50 mg resin was weighed and hydrolyzed to determine the degree of substitution of the amino acid on the resin. Stepwise Synthesis of 6-Aminocaproic Acid peptide on the Resin Figure 10 shows the apparatus used for the synthesis of the peptide. The reaction vessel was modified from a 250-ml seperatory funnel with the upper-half of the hole in the stopcock enlarged into a "pit" - 1/4 inch in diameter and 1/4 inch deep by an electric drill, The lower half of the hole was left intact. Glass wool was pressed into the pit to form a filter such that liquid could pass through but not the resin beads when the stopper turned to the drain position. 52 N ^ Drilled pit stuffed with glass wool Figure 10. The modified seperatory funnel used as the reaction vessel for solid phase spacer synthesis. A pit was drilled in the stopcock and filled with glass wool to serve as filter to keep the resin beads inside the seperatory funnel when the stopcock was turned to drain position to drain the solvents. 53 Boc-6-Aminocaproic acid-resin (1 gram) was loaded into the reaction vessel. The sequence of steps used to add each amino acid to the resin is outlined and explained in Table 8. Temporary co-amino protection is provided by the Boc group, removed at each step by the moderately strong acid trifluroacetic acid (TFA) Quantification of 6-Aminocaproic acid by Reverse-Phase High-Performance Liquid Chromatography (HPLC) The amino acid was quantitatively determined based on the HPLC amino acid analysis originally developed by Heinrikson and Meredith (1984), using precolumn derivatization with phenylisothiocyanate (PITC). The analysis was performed at room temperature with a Shimadzu LC10AS HPLC system using an Econosil C18 column (250 mm X 4.6 mm, 10 (jm) and Shimadzu SPD-6A UV spectrophotometric detector at 254 nm. Isocratic mobile phase was composed of 0.1 M ammonium acetate, pH6.8 in methanol: water (80:20)v/v. Pump speed was set at 1.0 ml/min. PITC reagent and 6aminocaprioc acid were purchased from Aldrich Chemical Co. All other chemicals were analytical grade. Standard Preparation The amount of 10[jl of 2.5 mM standard 6-aminocaproic acid was vacuum dried in a small test tube and dissolved in 100 pi of coupling buffer (acetonitrile: pyridine: triethylamine:H20, 10:5:2:3). To this solution was added 5 pi of PITC. After a 10-min reaction at room temperature, the solution 54 Table 8. Deprotection and coupling cycles for solid-phase peptide synthesis using Na - Boc-6-aminocaproic acid and ester linkage to polystyrene-resins. Step No. 1. Reagent and Solvent Tinne(nnin) X Treatment No. Dichloromethane(DCM) wash 3X 1 2 TFA-DCM 1 X3 3 Dichloromethane wash 4X1 4 Diisopropylethylamine-DCM (1:19) 2X2 5 DCM wash 6 X1 6a Boc-amino acid (3 equiv.) in DCM 5 (no filtering) 6b DCC (3equiv.) in DCM 60 7 DCM wash 6X2 55 was evaporated to dryness by rotary evaporation under high vacuum. The PITC-derived amino acid was then dissolved in 250 pi of 5 mM ammonium acetate water solution. Quantity of 20 pi of the solution was injected as standard to the HPLC system described above. Sample Preparation Dioxane (5 ml) and 5 ml concentrated hydrochloric acid were added to 50 mg resin sample in a 50-ml round glass flask. The mixture was re-fluxed over night in a Clas-Col electric heater with a 0.5 m air condenser. A converter such controlled the temperature dialed that mild simmering was maintained. The resulting hydrolysate was filtered through a 0.45 pm filter and 10 pi of the filtration was derivatized with PITC by the procedure described above. II. Attachment of Lysozyme to the Spacer thorough His-15 Bromoacetylation of 6-Ammocaproic Acid. The Yamada method (Yamada et al. 1984) was used to bromoacetylate the amine group of 6-aminocaproic acid at the end of spacer. The bromoacetylated 6-aminocaproic acid was then coupled to the imidazole group of His-15 of lysozyme. Figure 11 summarizes the chemical reactions involved. After step 5 in Table 8, the resins were washed with distilled water 4 times, each for 1 minute. The resins were transferred to a 100 ml beaker containing 20 ml distilledwater. The beaker was cooled in an ice bath and 500 pi bromoacetyl 56 0 II H2N—(CH2)5-C—O—CH2-Potymer O II BrCH2C—Br 0 II O II BrCH2C—NH—(CH2)5-C—0—CH2-Po^mer His-i5 NH Nc-y 0 II His-i5 0 N-CH2C-N—(CH2)5-C—0—CH2-Po^mer NW Figure 11. Chemical reactions involved in attaching 6-aminocaproic acid to the £2-nitrogen of His-15 of lysozyme. 57 bromide (5.7 mmol; MW 201.86, d 2.317) was added dropwise over 15 min with vigorous stirring. During the reaction the pH of the suspension was maintained at 7 with 4 N NaOH. After addition of all bromoacetyl bromide, the suspension was stirred for another 15 min. Finally the resins were transferred to Buchner funnel with coarse fritted disc, washed thoroughly with distilled water, and stored at 4°C. Coupling of Lysozyme onto the Support via Bromoacetylated Spacers Coupling reactions were performed between lysozyme and resins with spacer lengths of one 6-aminocaproic acid (1AA), 2AA, 3AA and 4AA respectively, using the following procedure. A mixture of 20 mg lysozyme (Fordras Sa, Switzerland) and 400 mg resin with bromoacetylated 6-aminocaproic acid in 10 ml of 0.5 M sodium acetate-acetate acid buffer (pH 5.5) containing 20% methanol was incubated at 40oC for 12 hr. The resin beads were then submerged in 6 M urea solution for 30 min with gentle stirring to dissociate any adsorbed protein; followed by washing with 1.0 liter 66mM phosphate buffer, pH 6.24. From monitoring absorbance at 280nm of the wash and its volume, the amount of enzyme removed by washing was calculated. Accordingly, the amount of immobilized lysozyme was determined. Beads (100 mg) with immobilized lysozyme were reserved for amino acid composition analysis of the immobilized protein by HPLC to verify the bonding through His-15 of lysozyme. 58 Amino Acid Compositions of the Immobilized Lysozyme Amino acid analysis of the immobilized lysozyme was performed by Protein Structure Core Facility of University of Nebraska Medical Center, Omaha, Nebraska, using the following procedure: 2000 pmoles of noreleucine was added to the samples as internal standard. The samples were then evaporated to dryness in a speedvac. The samples were then hydrolyzed in HCL vapor under argon atmosphere. (6N HCI with 1% phenol and 0.5% sodium sulfite were used in the hydrolysis mix). After 20 hours of hydrolysis at 11CC, the samples were dissolved in 200 (jl of Beckman Na-S sample buffer. The samples were then brought up in 200 pi Na-S. Sample of 50 pi was injected automatically onto the Beckman 6300 Amino Acid Analyzer. In data analysis, correction was made to the amount of the internal standard to minimizing dilutional errors. III. Controls Lysozyme was also immobilized to the polystyrene beads as controls with three different methods: (1) Covalent binding via 1-ethyl-3dimethylaminopropyl carbodiimide (EDC) coupling without spacer; (2) Covalent binding via EDC coupling with one 6-amino-caproic acid as spacer, and (3) Covalent binding through His-15 without spacer. Covalent Binding via EDC Coupling As Controls The carboxyl groups in the enzyme were activated by 1-ethyl-3(3- 59 dimethylaminopropyl)carbodiimide (EDC) in 0.1 M phosphate buffer, pH 4.75 at 25 0C to form O-acyl isourea derivatives. Then the pH was raised to 7 and the temperature lowered to 4 0C to allow the highly reactive intermediates to condense with the amine groups on the surface of the beads to yield the corresponding amides, or peptide bonds. The chemical reactions involved in the coupling process are given in Figure 12. 1. Preparation of the Support - Attachment of Boc-Glycine Boc-Glycine (Sigma Chemical) was attached to the chloromethyl polystyrene beads to introduce amine groups. Five grams of the resin, 3.8 grams of Boc-Glycine, and 2.7 ml of triethyl amine were mixed in a 250 ml round-bottom flask with 20 ml absolute alcohol added to cover the reactants. The mixture was refluxed gently for 24 hours at 81 "C in a Clas-Col electric heater with a 500-mm H2O condenser with a CaCb drying tube attached. The molar ratio for the reactants was 1.0 mole of Boc-Glycine (175.2 g per mole): 1.0 mole of Cl of the chloromethyl resin (232.6 g per mole ): 0.9 mole triethyl amine (0.14 ml per mMole; sp. gr. 0.723). The reacted resin was transferred to coarse-fritted Buchner funnel and washed successively with 60 o II Protein—C—OH CH3CH2-N=C=N—(CH2)3—N; EDC ,CH3 *CH3 O Protein—C—O I /CH3 CH3CH2—HN-C=N—(CH2)3—N^ H2N Polymer O Protein—C—NH—Polymer and O CH3CH2-HN-C-NH-(CH2)3—N: ,CH3 CH, Figure 12. Coupling of lysozyme to the reactive amine group on the support via 1-ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC) activation of carboxyl groups of the enzyme. 61 EtOH, H2O, MeOH, and CH2CI2, three times with each solvent, allowing one minute contact time for each solvent to penetrate the resin beads and for solutes to diffuse out of the beads. The washed resin, still suspended in CH2CI2, was then transferred to a 250-ml seperatory funnel, stirred with CH2CI2 (approximately 20 ml per g), and let stand until the bulk of the resin floated to the top and a fairly sharp demarcation line appeared at the bottom of the floating resin. The solvent, carrying the suspended finest particle of resin, was run out of the funnel and discarded. After removing all volatile solvents with a water aspirator, the resin was dried overnight in a desiccator with silicon gel. The amount of 50 mg resin was weighed and refluxed in 20 ml HCI - Dioxane (50:50, v/v) for 24 hr. The amount of glycine in the hydrolysate was determined HPLC (Heinrikson and Meredith (1984) to determine the degree of substitution of the amino acid on the resin. 2. Deprotect the Amine Groups on the Resin Beads Five hundred mg of the treated beads were soaked and stirred in a trifluroacetic acid - CH2CI2 mixture (1:4) for 30 min to remove the Boc protection on the amine groups. The beads were then transferred to a coarse-fritted Buchner funnel and washed with C^C^and later with distilled water. Finally the washed beads were suspended in 20ml of 0.1 M phosphate buffer, pH 7.0, for 1 hr at 4 0C. 62 3. Protection of the active site of lysozyme with N-acetylglucosamine NAG NAG oligosaccharide (50 mg) (trimer, tetramer and pentamer. Sigma) was added to 50 ml 2 mg/ml solution of lysozyme in 0.1 M phosphate buffer, pH 4.75. The mixture was stirred gently at room temperature for 30 min to protect the carboxyl groups of Glu-35 and Asp-52 from being reacted to EDC (Lin and Koshland, 1969). 4. Activation of lysozyme with EDC To activate the carboxyl groups in lysozyme, 500 mg EDC were mixed into the 50 ml lysozyme solution treated with NAG. After stirring gently at 25 0C for 2 hr, the pH of the mixture was raised to 7 and the temperature dropped to 40C. As control, 10 ml of 2 mg/ml solution of lysozyme not treated with NAG was also activated with EDC under the same condition. 5. Coupling of Lysozyme to the Resin Beads. Same coupling reactions were carried out for both the NAG-protected and non-NAG-protected lysozymes. Four hundred milligrams (400 mg) glycine derivatized beads were transferred to 10 ml buffer containing 20 mg activated lysozyme. The mixture was stirred at 4 0C for 2 hr. The resin beads were then soaked in 6 M urea solution to remove any adsorpted protein and dialyzed against water for 4 hours (to remove the protecting substrate in the NAG treated experiment and for control purpose in the non-treated experiment) 63 followed by washing with 1.0 liter 66mM phosphate buffer pH 6.24. From the absorbance at 280nm of the wash and its volume, the amount of immobilized lysozyme was determined. The same coupling procedure was performed for the resin beads with one 6-amino-caproic acid as spacer. The third control - covalent binding through His-15 without spacer, was prepared with the same protocol as with spacers except for using the glycine attached resin beads as support. IV. Determination of Lysozyme Activity Activity against M. lysodeikticus cells The activity of both free lysozyme and immobilized lysozyme was determined by measuring the absorbance decrease rate of lyophilized M. lysodeikticus cells in a 66 mM pH 6.24 phosphate buffer at 25 0C. One activity unit is defined as the decrease of one milli-absorbance per minute. Free Enzyme Lyophylized M. lysodeikticus (Sigma, No. M-3770) cell suspension of 0.02% (w/v) was prepared in the 66 mM phosphate buffer, which gave an A450nm between 0.7 and 0.8. The lytic reaction was carried out in a 3 ml quartz cuvette with 1 cm light path at 25 0C using a Shimatzu UV-Vis 2100 spectrophotometer. Assay was started by adding 0.1 ml of sample to the 64 bottom of the cuvette and then 2.5 ml of the substrate suspension was added and mixed well. The difference of /Wjnm between 20 and 40 seconds (linear part of the curve) of reaction was recorded with the spectrophotometer and the activity was calculated using kinetic software (Shimadzu Corp.). A blank assay using 0.1 ml of the phosphate buffer instead of lysozyme was also performed to account for any autolytic degradation of the substrate. Immobilized Enzyme The activity of immobilized lysozyme was assayed by addition of 100mg of lysozyme beads into a 5 ml suspension of 0.02% w/v lyophilized M. lysodeikticus cells in 66 mM phosphate buffer at 25 0C, and agitated at 240 rpm in a 25-ml centrifuge tube. After one minute, the cell suspension was quickly aspirated into a 25-ml syringe to separate it from the enzyme beads through a small coarse-fritted glass Buchner funnel. The A45onm of the enzyme-free cell suspension was then determined with the same method for free enzyme assay and subtracted from the initial A45onm before the enzyme was added. The difference of A45onm was recorded to calculate the activity - units/g resin. A blank assay with 100mg polystyrene beads without lysozyme was done to account for any A45onm loss due to non-enzymatic reasons. Activity against Glycol Chitin Glycol chitin, a water-soluble polymer of substituted Nacetylglucosamine with P-1,4-glucosminide linkages, was also used as 65 substrate to measure the activity of lysozyme of both free and immobilized, based on the method of Imoto and Yagishita (1971) Free Enzyme For free enzyme, 0.5 ml of lysozyme solution was added to 1 ml of 0.05% glycol chitin (Sigma) solution and incubated for 30 min at 37 0C. Both solutions were buffered in 0.1 M acetate at pH 4.5 and preincubated at 37 0C. To this reaction mixture was added 2.0 ml of 0.025% potassium ferricyanide in 0.5 M sodium carbonate, and the mixture was immediately placed in boiling water for 15 min to develop color in a test tube stopped with aluminum foil. After cooling to room temperature, the A42onm was measured against water in the same Shimadzu spectrophotometer used for the cell lysis method. The difference (A A42onm) between a given sample and the blank was referred to as the amount of reducing power generated by the tested lysozyme. Immobilized Enzyme For immobilized enzyme, in a continuously stirred 15-ml beaker, 100 mg beads mixed with 5 ml of 0.05% glycol chitin (Sigma) solution buffered in 0.1 M acetate at pH 4.5 and incubated for 30 min at 37 0C. The mixture was then filtered and 1.5 ml of the clear solution was mixed with the color reagent and completed the remaining steps the same as for the free enzyme. 66 V. Others Effects ofpH on the Activity ofLysozyme - Free and Immobilized The effect of pH on both free and immobilized lysozymes was conducted by measuring activities against lyophilized M. lysodeikticus substrate (0.02% w/v) suspended in 66mM phosphate buffer of pH values from 4.0-9.2. Buffer pH was adjusted with 0.1 N HCL and 0.1 N NaOH accordingly. Effect of Inhibitor Pentane on the Activity of Immobilized Lysozyme Lyophilized M. lysodeikticus substrate solution (0.25 mg/ml in 66mM phosphate buffer, pH 6.24), containing 4% (v/v) pentane (Sigma Chemical) (%,v/v) was used to study its inhibition effects on the lytic activity lysozyme free (2 ppm) and immobilized (3 AA, His-15). Kinetics Study of Immobilized Lysozyme The turbidity assay described earlier in this chapter was used to measure the initial activity of immobilized lysozyme (3 AA, His-15) for six substrate concentrations: 0.05, 0.075, 0.1, 0.125, 0.15, and 0.175, mg/ml. The data were evaluated with the Eadie-Hofstee plot and the Lineweaver-Burk plot, respectively. 67 CHAPTER 3 RESULTS AND DISCUSSION I. Solid Phase Spacer Synthesis The amount of 6-aminocaproic acid attached to the polystyrene resin beads and the length of the spacers determined by HPLC are summarized in Table 9. The data indicate that the adopted solid phase peptide synthesis protocol worked successfully for synthesizing the spacers with different predetermined lengths on the polystyrene bead support. The achieved spacer lengths were very close to the desired integer numbers, indicating all the reactions throughout the synthesis process went to completion. Several properties of polystyrene beads make it a good candidate as solid support to immobilize enzyme. It is both mechanically and microbially stable. It is relatively inexpensive and its fine particle size offers large surface area to volume ratio to bind large quantities of enzyme as necessary. Polystyrene has been reported as a preferred support for immobilizing Pfructofuranosidase (Filippusson and Hornby, 1970) and trypsin (Taylor and Swaisgood, 1972). Besides, the "coincidence" that polystyrene has been used as the solid support for solid phase peptide synthesis (Stewart and Young, 1969) made it an ideal support for the purpose of this search. The solid support is composed of fine beads (70-140 pm in diameter) of a synthetic resin prepared by copolymerization of styrene with 2 per cent 68 Table 9. Amino acid analysis of the peptide spacers synthesized on the polystyrene beads. Spacer length designed (No. of amino acid) Amino acid attached (mM/g resin) Spacer length synthesized (No. of amino acid) 1 0.86 1.00 2 1.69 1.97 3 2.67 3.10 4 3.37 3.92 69 divinylbenzene. The long alkyl chains bear a phenyl ring on every second carbon. These chains are cross-linked at approximately every fiftieth carbon by p-diethylphenyl residues derived from the divinylbenzene. The crosslinking causes the polymer to be completely insoluble in all ordinary solvents, yet swell extensively in the organic solvents used in the peptide synthesis. Because of the swelling, many of the peptide chains on the resin are attached within the pores of the beads. Therefore the spacers synthesized in this study (0.86 mM/g resin) should be viewed as evenly distributed throughout whole matrix, not just on the surface of the beads. However, since the polystyrene matrix should not be swollen under the lysozyme attachment condition, the enzyme is presumed only bound to the spacers on the surface. II. Attachment of Lysozyme Covalent Coupling through Carboxyl Groups ofLysoyzme EDC coupling is a fairly common covalent bonding method that has been used for immobilizing a variety of enzymes: rabbit muscle lactate dehydrogenase (Cho and Swaisgood, 1971), trypsin (Taylor and Swaisgood, 1972), and lysozyme (Chen & Chen, 1996) In order to get a consistent support environment between the test and control experiments, the same polystyrene resin beads were used as the 70 support for controls in this study to match that of the spacer and His-15 specific binding system. Since amino groups are the reactive groups at the tip of the spacers, carbodiimide or EDC coupling was chosen as the covalent binding method for the controls in this study, which allowed the carboxyl groups on the surface of lysozyme molecules attach to the amino groups on the support surface or at the tip of spacers. When lysozyme was directly attached to the polystyrene support through its carboxyl groups without the pretreatment of NAG oligosaccharide, the bound enzyme suffered great activity loss: only 0.7% of the activity was retained compared to the activity of the same amount of free enzyme (Table 10). When pretreated with NAG oligosaccharide, the retained activity increased from 0.7% to 1.8%, while the protein load remained at about the same level. This result suggests the very low activity observed for the bound enzyme was at least in part due to some bonding through the key carboxyl groups - i.e. Glu- 35 and/or Asp-52 involved in the catalysis. However, even the improved 1.8% retained activity is still a very disappointing result. When free lysozyme was derivatized with a carbodiimide-nucleophile procedure very similar to the EDC reaction employed in this study, the presence of NAG oligosaccharide protected Asp-52 and Glu-35 and over 50% activity was retained (Lin and Koshland, 1969). Therefore there should be other adverse changes happened during the immobilization process that caused the major activity loss. 71 Table 10. Activity of lysozyme covalently attached polystyrene beads via 1ethyl-3(3-dimethylaminopropyl)carbodiimide (EDC), with and without NAG protection . Method of Attachment Lysozyme bound (mg/g resin) Activity (U/g resin) Retained activity compared Jto free enzyme* (%) EDC w/o NAG protection 1.24 158 0.7 EDC with NAG protection 1.30 213 1.8 Based on the specific activity of free lysozyme at 9.09 U/pg protein. 72 Since the 11 free carboxyl groups (2 glutamic acid residues, 8 aspartic acid residues, and 1 COOH-terminal leucine residue) are all on the surface of the molecule, it is very likely that lysozyme will have multiple attachment sites to its support through EDC coupling reaction. Multiple attachment could strain or even disrupt the tertiary structure of the enzyme and make steric hindrance more evident. In addition, it has been reported that chemical modification of the carboxyl group of Asp-101 of lysozyme could significantly weaken substrate binding (Yamada et al. 1981) Covalent Coupling through His-15 In contrast to the "shot gun" strategy that couples lysozyme to the solid support randomly through the carboxyl groups on its surface, coupling through the single histidine residue of the protein that is nonessential for its activity seems much more appealing. Amino acid composition analysis by HPLC of polystyrene-lysozyme beads obtained with the His-15 coupling protocol is given in Table 11. No natural histidine residue was detected in the hydrolysate of the lysozyme-coated polystyrene beads, whereas a newly formed 3carboxylmethylhistidine was found. The numbers of the other amino acid residues matched very well to those of the control and the calculated values. These results clearly indicated that binding did occur and only through the alkylation of His-15 residues by the bromoacetylated 6-aminocaproic acid groups at the end of the spacers. 73 Table 11. Amino acid compositions of lysozyme immobilized through His-15. Native lysozyme Immobilized lysozyme Amino acid Theory Control Asp 21 21.6 20.1 21.9 Thr 7 6.1 6.2 6.7 Ser 10 9.3 10.6 10.7 Glu 5 4.8 5.7 4.9 Pro 2 1.8 1.8 2.3 Gly 12 11.7 12.0 11.6 Ala 12 12.9 11.7 12.0 3-CM-Hisa m - 0.8 0.7 Val 6 6.0 6.1 6.1 Met 2 1.7 1.8 1.7 lie 6 5.7 5.4 5.5 Leu 8 8.0 8.0 7.9 Tyr 3 2.8 3.1 2.8 His 1 1.1 0.0 0.0 Lys 6 5.7 6.3 6.1 Arg 11 10.4 11.1 10.7 ' 3-Carboxylmethylhistidine 1 AA His-15 2 AA His-15 74 The observation that no native histidine residues were detected also indicated that the extensive post-coupling washing steps, especially exposure to 6M urea for 30 min, worked well at removing the lysozyme molecules that had been noncovalently adsorbed to the beads. Although urea is perhaps the most commonly used denaturing agent for proteins, exposure to 6 M urea has been used successfully for removing adsorbed enzyme without inactivating coupled enzyme in several studies (Weetall, 1976). Lysozyme has been reported to survive up to 7 M urea without any unfolding occurred (Leonis, 1956). This was confirmed in this study by observing that free lysozyme (20 mg/L) displayed no activity loss after immersion in 6 M urea for 30 min at room temperature (data not shown). Efficacy of the His-15 with Spacer Strategy Compared to the random EDC binding no spacer control, coupling through His-15 binding without spacer already produced significant improvement in terms of total activity of the bound enzyme (Table 12). The test displayed higher activity (188 vs. 158 U/g resin), although its protein load was only half of that of the control's (0.59 vs. 1.24 mg/ resin). This further suggested that there was a significant amount of non-productive coupling occurred in the random EDC binding control. 75 Table 12. Characterization of polystyrene-lysozyme catalysts Method of attachment Activity Retained activity compared to free enzyme* Lysozyme bound (mg/g resin) (U/g resin) (%) No spacer, EDC 1.24 158 1.4 No spacer, His-15 0.59 188 3.5 1 AA spacer, EDC 1.44 314 2.4 1 AA spacer, His-15 0.84 801 10.5 2 AA spacer, His-15 1.31 1631 13.7 3 AA spacer, His-15 2.21 2736 14.2 4 AA spacer, His-15 2.13 2536 13.1 Based on the specific activity of free lysozyme at 9.09 U/pg protein. 76 Conceivably, the His-15 binding caused much less change in the tertiary structure of the enzyme. In the meantime, since the His-15 residue is on the back of side of the activity center, it could be pictured that lysozyme molecules bound to the matrix through this group would have their activity centers all facing outwards, rendering much higher accessibility to substrate. A similar yet more dramatic benefit of His-15 binding is illustrated in Table 12, when the total and retained activity of 1 AA spacer, His-15 is compared to that of the 1 AA spacer, EDC configuration. The nonessential role of the single histidine residue for the lytic function of lysozyme has been previously confirmed by a series of studies. According to Parsons et al. (1967), alkylation of the residue by three different reagents (iodoacetic acid, iodoaceamide, and 4-bromoacetamide-2-nitrophenol) did not have any effect of either the binding properties of the enzyme or its catalytic efficiency. These results are consistent with the similar alkylation studies by Kravchenko et al. (1964) and Goux and Allerhand (1979) employing iodoacetic acid. Furthermore, X-ray structural analysis have demonstrated that the single histidine residue of the enzyme occurs on the surface of the molecule remote from the binding cleft (Blake et al., 1965,1967; Imoto et al., 1972) 13 C NMR spectroscopy was used by Goux and Allerhand (1979) to study the reaction of iodoacetate with His-15 of hen egg white lysozyme. With the use of various reaction conditions, His-15 was carboxymethylated in detectable quantities only at NE2of the imidazole group. A comparison of the spectrum of chromatographically pure [NE2-cqarboxymethylhistidine-15] lysozyme with that 77 of the intact protein indicated that the chemical modification did not affect the conformation of the enzyme. The introduction of spacer also significantly benefited the total and retained activity of the bound enzyme, specifically comparing the result of 1 AA spacer/His-15 to that of no spacer/His-15; and 1 AA spacer/ EDC to no spacer/ EDC, respectively. Presumably the benefit is a result of reduced steric hindrances. A not fully expected side benefit, however, is that the increasing length of the spacer significantly increased the protein load - bound lysozyme which went from 0.59 mg/g resin with no spacer to 0.84 mg/g resin with 1 AA spacer; and kept increasing with the length of the spacer to 2.21 mg/g resin with 3 AA spacer. However, further spacer length increase from 3 AA to 4 AA did not result in further improvement in binding capacity. More importantly, with the increasing protein load, the specific activity of the bound enzyme also increased and eventually stabilized at a level of about 14%. This is the opposite of the reciprocal relation between the protein load and specific activity of the bound enzyme as observed by many previous studies on lysozyme immobilization (Datta et al., 1973; Moser et al., 1988; Crapisi et al., 1992) It is suggested that under the given coupling conditions, as length of the spacer increased the bromoacetylated amino groups at the ends of the spacers became more accessible to the His-15 side chains of lysozyme molecules, thus resulting in the higher and productive immobilization yields. 78 Activity against Soluble and Insoluble Substrates The retained activity of free and bound lysozyme against the glycol chitin - a soluble substrate and lyophilized M. lysodeikticus cells were compared in Table 13. When soluble chitin was the substrate, lysozyme immobilized with the His-15/spacer strategy showed very high retained activity - around 80%, regardless the different spacer lengths. This again suggests the approach did preserve the natural conformation and catalytic function for most of the bound protein molecules. Nonetheless, when insoluble cells was the substrate, the retained activity only ranged from 10.5 -14.2%, albeit 10 times higher than that of the no spacer/EDC control. This drastic difference of retained activity between the two substrates suggests that the diffusional limitation due to the bulky insoluble substrate remains to be the major hurdle for further improving the lytic efficiency of the immobilized Lysoyzme system. Kinetics Study of the Immobilized System The Eadie-Hofstee plot (v against v/[S]) is a linear transformer of the Michaelis-Menton equation and is an appropriate graph to use when one is interested in detecting deviations from Michaelis-Menton behavior. It is especially valuable at diagnosing the presence of diffusion effects in immobilized enzyme system. This method yields straight lines only when the enzyme obeys Michaelis-Menton kinetics and diffusion effects are negligible 79 Table 13. Specific activity of lysozyme against two different substrates. Retained activity compared to free lysozyme (%) Lysozyme type Free enzyme Soluble Chitin M. lysodeikticus cells 100 100 No Spacer EDC 33 1.4 No Spacer His-15 72 3.5 1AAHis-15 80 10.5 2AAHis-15 83 13.7 3AAHis-15 79 14.2 4AAHis-15 81 13.1 - 80 or fully excluded. With increasing diffusion limitations, however, the curves depart from straight lines. Internal diffusion resistance yields a sigmoidal curve, whereas external diffusion limitations are manifested in arched curves (Engasserand Horvath, 1976; Gemeiner, 1992). The Eadie-Hofstee plot method was used in this study to further explore the diffusion limitation effects suggested by the huge difference between the retained activity of the immobilized lysozyme against whole cell and soluble chitin substrates. When the initial velocity data of 3AA/His-15 for six substrate concentrations (0.05-0.175 mg/ml, see Chapter two) were converted to generate the Eadie-Hofstee plot, a highly convex curve was obtained (Figure 13), suggesting a strong external diffusion effects. That is, when insoluble cells are used as substrate, the rate of the substrate breakdown by the bound enzyme is faster than that of the substrate transfer. The rate of the enzyme reaction depends on the diffusion of the cells from the bulk solution to the enzyme. This was not unexpected given the double-heterogeneous nature of the system and the size of the cells. So far there have been no reported studies on kinetic behavior of immobilized lysozyme against M. lysodeikticus cells, presumably because the retained activities were too low to conduct Michaelis-Menton kinetics studies. There have been reports, however, that free lysozyme obeyed MichaelisMenton kinetics when lyophilized M. lysodeikticus was used as substrate (Neville and Eyring, 1972; Maurel and Douzou 1976; Ohno and Morrison 1989). All three studies reached the conclusion based on the linear fit of their 81 Figure 13. Eadie-Hofstee plot of immobilized lysozyme (100 mg 3 AA,His-15) against M. lysodeikticus (0.05-0.175 mg/ml) 82 data to the double-reciprocal or Lineweaver-Burk plot. It is worth pointing out that while the Lineweaver-Burk plot does have the advantage that the values of v for given values of [S] are easy to read from it, it has the big disadvantage of compressing the data points at high substrate concentration into a small region and over emphasizing the points at lower concentration. According to Dowd and Riggs (1965), it tends to give a deceptively "good" fit, even with unreliable data. Figure 14. is the Lineweaver-Burk plot from the same set of raw data that yielded the significantly arched curve in the Eadie-Hofstee plot in Figure 13. Clearly the fit is fairly linear (at least as good as the fit in the Ohno and Morrison paper (1989), if not better). So if based on the data fit from Figure13, it would have been concluded that the immobilized lysozyme system obeys Michaelis-Menton kinetics when it hydrolyzes M. lysodeikticus, which is clearly not the case as the data suggested in this study. A comparison between the two plots generated with the same set of raw data demonstrate that the data points in the Eadie-Hofstee plot are equally weighed, whereas in the Lineweaver-Burk plot the points at high substrate concentrations are highly compressed and the points at lower concentrations are stretched out. In other words the data at lower concentrations have much bigger impact on the overall shape of the curve in the Lineweaver-Burk plot than the ones at higher concentrations. This disadvantage of the LineweaverBurk plot could be particularly problematic for lysozyme study using the turbidity assay method. Because the assay tends to give less reliable data when 83 0.009 0.008 0.007 0.006 0.005 0.004 0.003 0.002 0.001 0 10 15 20 25 1/[S] Figure 14. Lineweaver-Burk plot from the same data set of Figure 13. 84 the substrate concentration is low. Therefore the studies on kinetic behavior of lysozyme using the Lineweaver-Burk plot should be viewed with caution. Using Eadie-Hofstee plot Green (1995) studied the kinetics of lysozyme inhibition caused by wine acids, ethanol, and wine phenolics. It was concluded that lysozyme did not follow Michaelis-Menton kinetics when M. lysodeikticus is used as its substrate based on the arc shaped curve obtained in the EadieHofstee plot. The proposed explanations are: 1) Changes in absorption (Mabs/min) may not linearly correspond to individual P(1-4) NAG-NAM bonds broken; the degree of cell wall degradation corresponding to change in optical density is unknown (Yamasaki et al., 1968), 2) The substrate is presented in an immobilized status; polysaccharide sections suitable for lysing are not in solution but bound in the cell wall matrix, 3) Non-productive binding is a consideration; NAG-NAM polymer 3 units in length will reversibly bind to the catalytic cleft but are not hydrolyzed. Among the three explanations, the lack of direct correlation between the turbidity decreasing rate and the number of glycosidic bonds split is the most fundamental one. In fact the initial decreasing rate of turbidity may very likely underestimate the actual activity of lysozyme in the system and thus cause the nonconformity of the Michaelis-Menton if the following scenario is true: In the lysis of cells by lysozyme the first step must be assumed to be the absorption of the lysozyme molecules onto the very large surface of the cell wall (Diameter ceii/Diameter lysozyme = 200 /1, (Datta et al., 1973), which electrostatic forces play a large part in the binding process (Price and Pethig, 85 1986). Assume all cells are identical and the (3-1,4 bond are evenly distributed over the surface of the cells; and after a certain amount of the P-1,4 linkage on the surface of a cell is the broken, say 10,000, the whole cell bursts, or collapses, into pieces of debris or fragments. All enzyme molecules start hydrolyzing cell walls simultaneously, because substrate is in great excess and the system is being thoroughly agitated. The generated fragments, which still have many of the intact glycosidic bonds, would become more accessible substrate for the enzyme than the intact cells, because they would diffuse faster to the activity site of the enzyme. Then, only a portion of the original enzyme molecules will move on to the next intact cells, while a significant amount of the enzyme will keep on hydrolyzing the cell wall fragments from the first round attack. Therefore the latter portion of the enzyme would contribute significantly less to the further decrease of turbidity, since according to Prasad and Litwack (1963) the whole cells would produce a greater turbimetric change when they collapse than that produced by fragmented cell walls being continuously lysed. When lysozyme is immobilized the underestimation of activity or nonconformity of Michaelis-Menton becomes only worse because of the diffusion limitation caused by the immobilization. Shortly after the turbidity starts decreasing, two different groups of substrates are generated for the bound enzyme: the small cell wall pieces still with the intact glycosidic bonds and the big whole cells yet to be broken. Naturally the former group would diffuse much faster to the bound enzyme than the latter and become the favored substrate in 86 the system. Consequently, as the hydrolysis proceeds, more and more of the enzyme reactions become less contributing to the reduction in turbidity of the cell suspension. Thus a severe deviation from Michaelis-Mention was observed. Effect ofpH on Activity of the Immobilized Lysozyme There was no significant shift of the pH optimum between the free enzyme and the bound enzyme (3AA/His-15) (Figure 15). Both showed maximum activity at about pH 6.4. But the curve for bound enzyme is slightly constricted compared to the control. The second pH optimum around 9 was not observed in this experiment presumably due to the high ionic strength of the buffer used. The bell-shaped pH-optimum curve can be explained on the basis of the charge status of the critical carboxyl groups of Glu-35 and Asp-52 residues. It can be assumed that at pH 6.2 in the given buffer system, the -COOH groups in Glu-35 is un-ionized and Asp-52 is ionized, which is the requirement for the enzyme to be fully active (Lipscomb, 1971; Imoto et al., 1972). Then the decrease on the alkaline side is the result of the ionization of Glu-35, whereas the decrease on the acid side reflects the gradual protonation of Asp-52. 87 Figure15. Effect of pH on the lytic activity of free and bound lysozyme (3AA,His-15) 88 The same pH optimum observed for both the free and bound enzyme would indicate that there is little or no charge-charge interaction involved in the immobilized system that would affect the charge status of Glu-35 and Asp-52 at the optimum pH of free enzyme. Hydrophobicity of the Polystyrene Support- Study on Inhibition Effect of Pentane on Lysozyme Immobilized on Polystyrene Beads It is observed that, in general, the level of activity of an immobilized hydrolase, which lysozyme is, depends on the degree of hydration of the polymer matrix (Manecke, 1962). Therefore the inherent hydrophobicity of the polystyrene matrix used in this study could actually be a disadvantage. It could also adversely affect the coupling yields of the protein. Nevertheless, since the surface of the matrix is heavily derivatized with chloro groups, about 4.3 mmol/g resin, which would form hydrogen bonds with the water molecules around them, it is possible that the highly hydrophilic chloro groups would offset the hydrophobicity of the polystyrene. It has been reported that pentane inhibited lysozyme activity against M. lysodeikticus by 56% when added at 5% (v/v) to the reaction (Wantanabe and Takesue, 1974). Pentane was used in this study to probe the level of hydrophobicity of surface of the polystyrene matrix based on the hypothesis that if the surface is highly hydrophobic the concentration of pentane near the surface of the matrix would be higher than that in the bulk due to hydrophobic interaction between the two, therefore pentane would exhibit stronger inhibition 89 to the immobilized lysozyme than to the free enzyme even though the concentration of pentane in the bulk for the two systems was the same. This proved to be the case (Table 14). When 4% (v/v) pentane was added to M. lysodeikticus substrate solution (0.2mg/ml in 66mM phosphate buffer, pH 6.24), the activity of the free enzyme (2ppm) reduced by 32%, while that of the bound enzyme (3AA, His-15) reduced by 51%. This suggests that the concentration of pentane in the microenvironment of the immobilized lysozyme was significantly higher than that in the bulk due to the hydrophobicity of the polystyrene matrix. The surface of the support remains hydrophobic despite the heavy chloro-derivatization. Stability of the Immobilized Lysozyme The operational stability of an immobilized lysozyme system is important for its practical use. Figure 16 shows the result of the stability study of the immobilized Lysoyzme (3AA/His15). Activities of free and two bound enzymes against lyophilized M lysodeikticus were monitored daily with the methods describe earlier. After each assay, the bound enzymes were rinsed with 500 ml of 66mM phosphate buffer and stored at 40C along with the free enzyme stock (2ppm). After 14 days the 3AA/His-15 sample retained 90% of its original activity, demonstrating a good stability that rivals that of the free enzyme. The control of no-spacer/EDC sample, however, retained only 45 percent of its activity after 14 days. For both bound enzymes, no protein loss 90 Table 14. The inhibition effect of 4%(v/v) pentane on the activity of free and immobilized lysozyme against M. lysodeikticus. Lysozyme Activity Lost (%) Free (2ppm) 32 Immobilized (0.5 g resin; 3AA,His-15) 51 91 -♦— EDC no spacer -■—3AA,l-lis15 Free lysozyme 20 10 0 * 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Time (day) Figure 16. Stability of bound and free lysozymes over a period of 14 days when stored at 4°C. 92 was detected in the wash done each time after the activity assay, indicating a stable support and bond linkage. The result again suggests that the His-15/spacer strategy minimized the conformational change to the lysozyme molecule to an extent that the bound enzyme had practically the same stability of the free enzyme. However, with the control the stress caused by multiple bonding between the protein and support eventually took a toll on its conformation and consequently the stability over time. 93 CHAPTER 4 CONCLUSION The three key issues in the covalent attachment of a biological active protein to an insoluble support are: 1.) The protein should be attached to the matrix by the fewest possible bonds to minimize conformational change; 2.) The binding site(s) on the enzyme to the supports should be located as far as practical from its active center and be nonessential for its tertiary structure; 3.) The binding method should minimize the steric interference between the support and the immobilized enzyme. The fortuitous facts that the 15th amino acid residue in lysozyme molecule is the only histidine (His-15) that is located on the opposite side of its lytic activity center and nonessential for its tertiary structure allowed us to apply the rules above rather successfully in this study. By coupling through the sole histidine residue on the enzyme to minimize conformational change and introducing spacers to reduce steric hindrance, the total and retained activity of the preparation were 15 and 9 folds higher than those of the control prepared with random covalent coupling protocol using the same polystyrene support. In addition to reducing steric hindrance, the spacer also significantly increased the immobilization yield. 94 The minimal conformation change of the immobilized lysozyme was also supported by the observations from this study that its optimum pH and operational stability were virtually identical to those of the free enzyme. However, the drastic difference of retained activity between the soluble substrate glycol chitin and insoluble substrate lyophilized M. lysodeikticus cells (80% vs. 14%) suggested that the collision and diffusion limitations remained the major hurdle for further improving the lytic efficiency of the immobilized lysozyme system. Kinetics study of the system using the Eadie-Hofstee plot confirmed that there was strong external diffusion effect occurring when lyophilized M. lysodeikticus cells was used as substrate. This was not unexpected given the double-heterogeneous nature of the system and the bulky size of the insoluble substrate. For future research, it would be interesting to test different hydrophilic supports, which may improve the yield and lytic activity of the system. In the meantime, other reagents that react to the imidazole group of His-15 are worth exploring, such as, diazonium salts and formaldehyde. 95 BIBLIOGRAPHY Artymiuk, P.J., Blake, C.C.F., Rice, D.W. and Wilson, K.S. 1982. The structures of the monoclinic and orthorhombic forms of hen egg-white lysozyme at 6 .ANG. resolution. Acta Crystallogr. Sect. B. 38:778 Acharya, K.R., Sturat.D.I., Walker, N.P.C., Lewis, M. and Philips, D.C. 1989. Refined structure of baboon alpha-lactalbumin at 1.7 A resolution. Comparison with C-type lysozyme. J. Mol. Biol. 208:99. Akashi, A., 1969. Preservative effect of egg white lysozyme added to cooked sausage. Jap. J. Zootechnical Sci. 40:243 (Cited in Food Sci. Technolo. Abst. 1:12S868. Akashi, A. 1971. Preservative effect of egg white lysoyzme on Vienna sausage. Jap. J. Zootechnical. Sci. 42:289. (Cited in Food Sci. Technolo. Abst. 4:1S66) Amati, A., Arfelli, G.,Simoni, M.,Gandini, A., Gerbi, V., Tortia, C, and Zironi, R. 1992. Inibizione della fermentazione maloattica mediante il lisozima: aspetti micobioloici e tecnologici. Biologia Oggi. 1-2 Anon. 1987. The use of lysozyme to control butyric acid fermentation. Bull. Int. Dairy. 216:5. Barany, G., Kneib-Cordonier, N. and Mullen, D.G. 1987. Solid-phase peptide synthesis: a silver anniversary report. Int. J. Peptide Protein Res. 30:705 Barnett, L.B. and Bull, H.B. 1956. The optimum pH of adsorbed ribonuclease. Biochim. Biophys. Acta. 36:244. Bernath, F.R. and Vieth, W.R. 1974. The optimum pH of adsorbed ribonuclease. In: Immobilized Enzymes in Food and Microbial Processes. Olson, A.C. and Cooney, C.L. (eds). Pp 157-185. Plenum Press, New York. Bester, B.H. and Lombard, S.H. 1990. Influence of lysozyme on selected bacteria associated with Gouda cheese. J. Food Prot. 54:306 Beychok, S. and Warner, R.C. 1959. Denaturation and electrophoretic behavior of lysoyzme. J. Am. Chem. Soc. 81:1892. Blake, C.C.F., Koenig.D.F., Mair, G.A., North, A.C.T., Phillips, D.C. and Sarma, V.R. 1965. Structure of hen egg-White lysozyme - a three-dimensional Fourier synthesis at 2 A resolusion. Nature. 206:757 96 Blake, C.C.F., Johnson, L.N., Mair, G.A., North, A.C.T., Philips, D.C. and Sarma, V.R. 1967. Crystallographic studies of the activity of hen eggwhite lysozyme. Proc. Roy. Soc. London, Ser. B. 167:378 Blake, C.C.F., Cassels, R., Dobson, CM., Poulsen, F.M., Williams, R.J.P. and Wilson, K.S. 1981. Structure and binding properties of hen lysozyme modified at tryptophan 62. J. Molec. Biol. 147:73 Boiler, T., Gehri, F., Mauch, F., Vogeli, U. 1983. Chitinase in bean leaves: induction by ethylene, purification, properties, and possible function. Planta, 157:22 Bottazzi, V., Battistotti, B., Cappa, F.,Rebecchi, A., Bertuzzi, S., and Brambilia, E. 1993. Clostridium spore germination and lysozyme action in Grana cheese. Seienza-e-Technica-Lattiero-Caseria. 44:79 Cabral, J.M.S. and Kennedy, J.F. 1991. Covalnetand coordination immobilization of proteins. In: Protein Immobilizaiton - Funddamentals and Applications. Taylor, R.F. (ed). pp. 73-138. Dekker, New York. Canfield, R.E.1963. The amino acid sequence of egg white lysozyme. J. Biol.Chem. 238:2689 Canfield, R.E. and Liu, A.K. 1965. The disulfide bonds of egg white lysozyme (Muramidase). J. Biol. Chem. 240:2000. Carini, S., and Lodi, R. 1982. Inhibition of germination of clostridial spores by lysozyme. Ind. Latte. 18:35 Carminati, D., Nevianti, E. and Muchetti, G. 1985 Activity of lysozyme on vegetative cells of Clostridium trybutyricum. Latte. 10:194 Carneiro, D., Melo, A.M.S., and Miles, R.J. 1998.Trisodium phosphate increases sensitivity of gram-negative bacteria to lysozyme and nisin. J. Food Protection 61:839. Chandan, R.C. and Ereifej. 1981. Determination of lysozyme in raw fruits and vegetables. J. Food. Sci. 46:1278. Chang, K.Y. and Carr, CW. 1971. Studies on the structure and function of lysoyzme. I. The effect of pH and cation concentration on lysozyme activity. Biochim. Biophys. Acta. 229:496 Chatzolopou, A., Miles, R.J., and Anagnostopoulos, G. 1993. Destruction of gram-negative bacteria. Int. Pat. Appl. WO 93/00822. 97 Chen, J. P. and Chen Y.C. 1996. Improvement of cell lysis activity of immobilized lysozyme with reversibly soluble-insoluble polymer as carrier. Biotech. Techniq. 10:749 Cho, I. And Swaisgood, H. 1972. The reactivation of an unfolded subunit enzyme covalently linked to a solid surface. Biochim. Biophys. Acta. 258:675 Crapisi, A., Lante, A., Pasini, G., and Spettoli, P. 1992. Enhanced microbial cell lysis by the use of lysozyme immobilized on different carriers. Process Biochem. 32:17. Cuatrecasas.P. 1969. Interaction of insulin with the cell membrane: primary action of insulin. Proc. Natl. Acad. Sci. USA 63:450 Cuatrecasas.P. 1970. Protein purification by affinity chromatography. J. Biol. Chem. 245:3059 Cuatrecasas.P.,, Anfinsen, C.B., 1971. Affinity chromatography. Methods Enzymol., 22:345 Costerton, J.W., Gillsex, G.G. and Cheng, K.J., 1978. How bacteria stick. Sci. Amer., 238:86 Datta, R., Arminger, W., and OIlis, D.F. 1973 Lysis of Mlcrococcus lysodeikticus by lysozyme covalently immoblized on cellulose and polyacrylamide. Biotech. Bioeng. 15:993. Davies, R.C., Neugerger, A. and Wilson, B.M. 1968. The dependence of lysozyme activity on pH and ionic strength. Biochim. Biophys. Acta. 178:294. Dickman, S.R. and Proctor, CM. 1952. Factors affecting the activity of egg white lysozyme. Arch Biochem Biophys. 40:364. Dobson, CM. 1977. In: NMR in Biology(Dwek, R.A.,Campbell, I.D., Richards, R.E., and Williams, R.J.P., Eds.), Academic Press, London, p.63. Dowd, J.E. and Riggs, D.S. 1965. A comparison of Estimates of MichaelisMenten Kinetic constants from various linear transformations. J. Biol. Chem. 240:863 Eisai Company. 1971. Seafood Preservation. Japanese Patent 19576/71. Eisai Company. 1972. Fish Product Preservation. Japanese Patent 5710/72. 98 Engasser, J-M., and Horvath, C. 1976. Diffusion and kinetics with immobilized enzymes. In: Applied Biochemistry and Bioengineering. Vol 1. Pp 128178. Academic Press. New York. Eshdat, Y., McKelvy, J. F., and Sharon, N. 1973. Identification of aspartic acid 52 as the point of attachment of an affinity label in hen egg white lysozyme. J. Biol. Chem. 248:5982. Eshdat, Y., Dunn, A., and Sharon, N. 1974. Chemical conversion of aspartic acid 52, a catalytic residue in hen egg-white lysozyme, to homoserine. Proc. Natl. Acad. Sci. U.S.A. 71:1658. Eyles, S.J., Radford, S.E., Robinson, C. V., and Dobson, CM. 1994. Kinetic concequences of the removal of a disulfied bridge on the folding of hen lysozyme. Biochemistry 33:13038 Fersht, A. 1985. Enzyme Structure and Mechanism. W.H. Freeman and Company, New York, p. 30. Filippusson, H. and Hornby, W.E. 1970. The preparation and properties of yeast p-fructofuranosidase chemically attached to polystyrene. Biochem. J. 120:215 Fleming, A. 1922. On a remarkable bateriolytic element found in tissues and secretions. Proc. Roy. Soc. London Ser. B 93:306. Fukamizo.T., Torikata, T., Nagayama, T., Minematsu, and Hayashi, K. 1983. Enzymatic activity of avian egg-white lysozyme. J. Biochem. 94:115 Fukui, S., Ikeda, S-l., Fujimura, M., Yamada, H., and Kumagai, H. 1975. Comparative studies on the properties of tryptophanase and tyrosine phenol-lyase immobilized directly on Sepharose or by use of Sepharosebound pyridoxal 5'-phosphate. Eur. J. Biochem. 51:155. Gemeiner, P. 1992. Enzyme Engineering - Immobilized Biosystems. Ellis Horwood, New York. Gerbaux, V., Villa, A., Monay, C, and Betrand, A. 1997. Use of lysozyme to inhibit malolactic fermentation and to stablilize wine after malolactic fermentation. Am. J. Enol. Vitic. 48:49. Glazer, A.N. and Simmons, N.S. 1965. The cotton effect associated with certain tyrosine residues in ribonuclease. J. Am. Chem. Soc. 87:2287. Goldstein L. 1972. Microenvironmental effects on enzyme catalysis. A kinetic study of polyanionic and polycationic derivatives of chymotrypsin. Biochemistry 11:4072 99 Goux, W.J. and Allerhand, A. 1979. Studies of chemically modified histidine residues of proteins by carbon 13 nuclear magnetic resonance spectroscopy - reaction of hen egg white lysozyme with iodoacetate. J. Biolog. Chem. 254:2210 Green, J.L. 1995. The use of lysozyme in winemaking: the interaction of lysozyme with wine and efficacy in preventing maloactic fermentation in Oregon Pinot Noir and Chardonnay. M.S. Thesis. Oregon State University, Corvallis, Oregon, USA. Green, J.L., Watson, B.T., and Daeschel, M.A. 1994. Efficacy of lysozyme in preventing malolactic fermentation in Pinot noir wines. Am. J. Enol. Vitic. 45:355. Grubhofer, N. and Schleith, L. 1954. Protein coupling with diazotized polyaminostyrene. Hoppe Seylers Z Physiol Chem. 297:108 Hailing P.J., AsenjoJ.A. and Dunnill P. 1979 Nonporous magnetic supports , for proteases, cell-lytic enzymes, and ribonuclease: limits of reactant size. Biotech. Bioeng.21:2359 Hayashi, K., Kugimiya, M., Imoto, T., Funatsu, M., and Bigelow, C.C. 1968. The inhibitory interaction of cationic detergents with the active center of lysozyme. I. Site of interaction. Biochemistry 7:1461 Heinrikson, R.L. and Meridith, S.C. 1984. Amino acid analysis by ReversePhase High-Performance liquid chromatography: precolumn derivatization with phenylisothiocyanate. Analyt. Biochem. 136:65 Henick-Kling, T. 1993. The Malolactic fermentation. In: Wine Microbiology and Biotechnology. Fleet G.H. Hardwood Academic Publishers, pp 289-326. Hodsdon, J.M., Brown, CM., Sieker, L.C. and Jensen, L.H. 1990. Refinement of triclinic lysozyme. I. Fourier and least-squares methods Acta Crystallogr. Sect. B. 46:54. Hogle, J., Rao, ST., Mallikarjunan, M., Beddell, C, McMullan, R.K., and Sundralingam, M. 1981. Studies of monoclinic hen egg white lysozyme. I. Structure solution at 4 .ANG. resolution and molecular-packing comparisons with tetragonal and triclinic lysozymes. Acta Crystallogr. Sect. B. 37:591 100 Husain, Q. Iqdal, J., and Saleemuddin, M. 1985. Entrapment of concanavalin A-glycoenzyme complexes in calcium alginate gels. Biotech. Bioeng. 27:1102. Ibrahim, H.R.,Hatta, H., Fujiki, M., Kim M., and Yamamoto, T. 1996. Enhanced antimicrobial action of lysozyme against gram-negative and grampositive bacteria due to modification with perilaldehyde. J. Agri. Food Chem. 42:1813. Imoto, T. and Yagishita, K. 1971. A simple activity measurement of lysozyme. Agr. Biol. Chem. 35:1154 Imoto, T., Johnson, L.N., North, A.C.T.,Philips, D.C. and Rupley, J.A. 1972. Vertebrate lysoyzmes. In Ii The Enzymes, 3rd Ed. Boyer, P.D., (ed), Academic Press. 7:665 International Dairy Federation. 1987. The use of lysozyme in the preservation of late blowing in cheese. Bulletin of IDF No. 216 (Brussels, Belgium: IDF) Jolles, P., and Jolles., J. 1961. Lysozymes from human milk. Nature. 192:1187. Joynson, M.A., North, A.C.T., Sarma, V., Dickerson, R.E., and Steinrouf, L.K. 1970. Low-resolution studies on the relation between the triclinic and tetragonal forms of lysozyme. J. Mol. Biol. 50:137. Kanebo Ltd. 1973. Preservative, Japanese Patent. 4831-905 Kerby, G.P. and Eadie, G.S. 1953. Inhibition of lysozyme by heparin. Soc.Exptl. Biol. Med. 83:111. Proc. Kravchenko, N., Kleopina, G., and Kaverzneva, E. 1964. Active sites of lysozyme. Carboxymethylation of the imidazole group hitstidine and of the e - amino group of lysine. Biochim. Biophys. Acta. 92:412 Kumaraswamy, M.D.K., Rao, K.P., Joseph, K.T.Santappa, M 1981. Immobilization of enzymes on alginic acid-polyacrylamide copolymers. Biotechnol. Bioeng. 23:1889 Kundrot, C.E. and Richards, F.M. 1987. Crystal structure of hen egg-white lysozyme at a hydrostatic pressure of 1000 atmospheres. J. Mol. Biol. 193:157. Law, B.A. and Goodenough, P.W.1995. Enzymes in milk and cheese production, In: Enzymes in Food Processing.2nd. Tucker, G.A. and Woods, L.F.J. (eds) Blackie Academic & Professional. Glasgow, UK. 101 Leonis, J. 1956. Behavior of lysoyzme in urea solutions. Arch. Biochem. Biophys. 65:182. Lin, T-Y. and Koshland, D.E., Jr. 1969. Carboxyl group modification and the activity of lysoyzme. J. Biol. Chem. 244:505 Lipscomb, W.N. 1971. Structures and mechanisms of enzymes. Proc. Robert A. Welsh Conf. Chem. Res. 15:131. Malcolm, B.A., Rosenberg, S., Corey, M.J., Allen, J.S., de Baetselier, A. and Kirsch.J.F. 1989. Site-directed mutagenesis of the catalytic residues Asp-52 and Glu-35 of chicken egg white lysozyme. Proc Natl Acad Sci USA. 86:133. Manecke, G. 1962. Reactive polymers and their use for the preparation of antibody and enzyme resins. Pure Appl. Chem. 4:507. Marzotto, A. and Galzigna, L. 1969. On the enzymatic hydrolysis of carboxymethylchitin by lysozyme. Hoppe-Seyler's Physiol. Chem. 35:1154. Maurel, P. and Douzou, P. 1976. Catalytic implications of electrostatic potentials: the lytic activity of lysozyme as a model. J. Mol. Biol. 102:253 Means, G.E. and Feeney, R.E. 1971. Chemical Modification of Proteins. Holden-Day, San Francisco. Merrifield, R.B. 1963. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85:2153 Meyer, K., Prudden, J.F., Lehman, W.L. and Steinberg, A. 1947. Lysozyme content of the stomach and its possible relationship to peptic ulcer. Proc. Soc. Exptl. Biol. Med. 65:220. Mitz, M.A. 1956. New insoluble active derivative of an enzyme as a model for study of cellular metabolism. Science 123:1076. Moser I., Dworsky P. and Pittner, F. 1988. Degradation of bacterial cell walls by immobilized lysozyme. Appl Biochem Biotechnol. 19:243. Moult, J^Yonath, A., Traub, W., Smilansky, A. Podjarny, A., Rabinovich, D. and Saya, A. The structure of triclinic lysozyme at 2-5 A resolution. 1976. J. Mol. Biol. 100:179. 102 Mosbach, K., Larsson, P.O., Brodlius, P., Guilford, H., and Lindberg, M. 1974. Synthesis and application of matrix bound AMP, NAD, and other adenine nucleotides. In Enzyme Engineering, Vol.2, pp.237-242. Plenum, New York. Nakamura, R., Kato, A. and Kobayashi, K. 1990. Novel bifunctional lysozymedextran conjugate that acts on both gram-negative and gram-positive bacteria. J. Agric. Food Chem. 54:3057. Nakamura, R., Kato, A. and Kobayashi, K. 1992. Bifunctional lysozyme galactomannan conjugate having excellent emulsifying properties and bactericidal effect. J. Agric. Food Chem. 40:735. Nelson, J.M. and Griffin, E.G. 1916. Adsorption of invertase. J. Am. Chem. Soc. 38:1109. Neuberger, A. and Wilson, B.M.1967. Inhibition of lysozyme by N-acyl-Dglucosamine derivatives. Nature. 215:524. Neville, W.M. and Eyring, H. 1972. Hydrostatic pressure and ionic strength effects on the kinetics of lysozyme. Proc. Natl. Acad. Sci. USA. 69:2417. O'Carra, P., Barry, S., and Corcoran, E. 1974. Affinity chromatographic differentiation of lactate dehydrogenase isoenzymes on the basis of differential abortive complex formation. FEBS Letters 43:163 Ohno, N. and Morrison, D.C. 1989. Lypopolysaccharide interaction with lysozyme - binding of lipopolychaccharide to lysozyme and inhibition of lysozyme enzymatic activity. J. Biol. Chem. 264:4434 Parry, R.M., Chandan.R.C. Jr., and Shahani, K.M. 1965. A rapid and sensitive assay of muramidase. Proc. Soc. Experim. Biol. Med. 119:384 Parsons, S.M., Jao, L., Dahlquist, F.W., Borders.Jr, C.L., Groff, T., Racs, J., and Raftery, M.A. 1969. The nature of amino acid side chains which are critical for the acivity of lysozyme. Biochemistry. 8:700. Pavlovskii, P.E. Cherkasov, I.A., and Chikima, N.S. 1976. Content and some properties of lysozyme in organs of slaughter cattle. Prikl. Biokhim. Mikrobiol. 12:134. Phillips, D.C. 1973. The three-dimentional structure of an enzyme molecule, In: Organic chemistry of life: Readings from Scientific American. Calvin, M. and W.A. Proyor (eds). W.H.Freeman and Co. San Francisco. 103 Poole, P.L. 1994. The role of hydration in lysozyme structure and activity: relevance in protein engineering and design. J. Food. Eng. 22:349 Powrie, W.D. and Nakai, S. 1986. Chemistry of eggs and egg proteins. In: Egg Science and Technology, 3rd Edn., W. J. Stadelman and D.J. Cotterill (eds), AVI Publishing, Westport, CN pp.97-128. Prasad, A.L.N. and Litwack, G. 1963. Measurement of the lytic activity of lysozymes (muramidases) Anal. Biochem. 6:328. Price, A.R. and Pethig, R. 1986. Surface charge measurements on Micrococcus lysodeikticus and the catalytic implications for lysozyme. Biochim. Biophys. Acta 889:128 Radler, F. 1975 The metabolism of organic acids by lactic acid bacteria. In Lactic Acid Bacteria in Beverages and Food. Carr, J.G., Cutting, C.V., and Whiting, G.C. (eds). Academic Press, London, pp 17-27. Rankine.B.C, and Bridson, D.A. 1971. Bacterial spoilage in dry red wine and its relationship to malo-lactic fermentation. Autral. Wine Brew. Spirit. Rev. 90:44 Richmond, M. H. 1959. Lytic enzymes of Staphylococcus aureus 524. Biochim. Biophys. Acta. 31:564. Rupley, J.A. 1967. Enzymic activity of lysozyme. II. The hydrolysis and transfer reactions of N-acetylglucosamine oligosaccharides. Proc. Natl. Acad. Sci. USA. 57:49 Sampson N.S. and Barttlett, P.A. 1990. Peptide phosphonylating agents as irreversible inhibitors of serine proteases and models of the tetrahedral intermediates. Biochemistry. 30:2256 Samuelson, K.J., Rupnow, J.H., and Froning, G.W. 1985. The effect of lysozyme and ethylenediaminetetraacetic acid on Salmonella on broiler parts. Poultry Sci. 64:1488. Scott, D., Hammer, F.E., and Szalkucki, T.J. 1987. Bioconversions: enzyme technology. In Food Biotechnology, Knorr, D (ed), Marcel Dekker, New York, pp.78-93. Schinitzky, M., Katchoilski, E., Grisaro, V. and Sharon, N. 1966. Inhibition of lysozyme by imidazole and indole derivatives. Arch. Biochem. Biophys. 116:332 104 Sela, M., and Steiner, L.A. 1962. Inhibition of lysozyme by some copolymers of amino acids. Biochemistry 2:416. Shugar, D. 1952. The measurement of lysozyme activity and the ultra-violet inactivation of lysozyme. Biochim. Biophys. Acta. 8:309 Silman, H.I., Albu-Weisenbur, M., and Katchalski, E. 1966. Some waterinsoluble papain derivatives. Biopolymers. 4:441 Simmons, N.S. 1952. Studies on the defense mechanisms of the mucous membranes with particular reference to the oral cavity. Oral Surg. Oral Med. Oral Pathol. 5:513 Skarnes, R.C. and Watson, D.W. 1955. The inhibition of lysozyme by acidic polymers from pathogenic bacteria. J. Bacteriol.70:110. Smith, E.L., Kimmei, J.R., Brown, D.M. and Thompson, E.O.P. 1955. Isolation of properties of a crystalline mercury derivative of a lysozyme from papaya latex. J. Biol. Chem. 215:67. Smith, G.N. and Stocker, C. 1949. Inhibition of crystalline lysozyme. Biochem. 21:383. Arch. Smith, L.J., Stutcliffe, M.J., Redfield, C. and Dobson.C.M. 1992. Structure of hen lysozyme in solution. J. Mol. Biol. 229:930 Smolelis, A.N. and Hartsell, S.E. 1952. Factors affecting the lytic activity of lysozyme. J. Bacteriol. 63:665. Steers, E., Cuatrecasas, P., and Pollard, H. 1971. The purification of pgalactosidase from Escherichia coli by affinity chromatography. J. Biol. Chem. 246:196 Stevenson, W.T.K. and Sefton, M.V. 1987. Graft copolymer emulsions of sodium alginate with hydroxyalkyl methacrylates for microencapsulation. Biomaterials. 8:449 Stewart, J.M. and Young, J.D. 1969. Solid Phase Peptide Synthesis. W.H. Freeman and Co., San Fancisco, Calif., 1969. Stone, I. 1955. U.S. Patent 2,717,852. Taiyo Food Co. 1972. Bean Curd Preservation, Japanese Patent. 46-336/72 Taylor, J.B. and Swaisgood, H.E. 1972. A kinetic study of the effect of coupling distance between insoluble trysin and its carrier matrix. Biochim. Biophys. Acta. 284:268. 105 Taylor, R.F. 1991. Commercially available supports for protein immobilization. In: Protein Immobilization - Fundamentals and Application. Pp.139-160. Dekker, New York Trevan, M. 1980. Immobilized Enzymes: An Introduction and Applications in Biotechnology. Wiley, New York. Turkova, J. 1978. Affinity Chromatography, vol. 12. Elsevier, Amsterdam. Vakil, J.R., Chandan, R.C., Parry, R.M. and Shahani, K.M. 1969. Susceptibility of several microorganisms to milk lysozyme. J. Dairy Sci. 52:1192. Wang S., Murao S. and Arai, M. 1990. Lysozyme inhibitory activity of DNA from Bacillus subtilis 1-139. Agric. Biol. Chem. 54:1289. Wardlaw, A.C. 1962. The complement-dependent bacteriolytic activity of normal human serum. I. The effect of pH and ionic strength and the role of lysozymes. J. Exp. Med. 115:1231. Wasserfall, F., Voss, E., and Prokopek. 1976. Experiments on cheese ripening: the use of lyozyme instead of nitrite to inhibit late blowing of cheese. Kiel. Milchwirsch. Forschungsber. 28:3. Wasserfall, F. and Teuber, M. 1979. Action of egg white lysozyme on Clostridium tyrobutyricum. Appl. Environ. Mcirobial. 38:197 Watanabe, K., and Takesue, S. 1972. Inhibitory effect of some gaseous hydrocarbons on the cell-lysis of Micrococcus lysodeikticus by egg-white lysozyme. Arg. Biol. Chem. 36:825. Watanabe, K., and Takesue, S. 1974. Pentane inhibition of the egg-white lysozyme-induced cell lysis of Micrococcus lysodeikticus. Arg. Biol. Chem. 38:1477. Weetall, H. 1976. Covalent coupling methods for inorganic support materials. Methods Enzym. 44:134 Yamada, H., Uozumi, F., Ishikawa, A. and Imoto, T. 1984. Nature of the binding site around and reactivity of histidine-15 in lysozyme. J. Biochem. 95:503 Yamada, H., Imoto, T., Fujita, K., Okazaki, K., and Motomura, M. 1981. Selective modification of aspartic acid-101 in lysozyme by carbodiimide reaction. Biochemistry. 20:4836. Yamada, H., Imoto, T., and Noshita, S. 1982. Modification of catalytic groups in lysozyme with ethylenimine. Biochemistry. 21:2187. 106 Yamasaki, N., Hayashi, K. and Funatsu, M. 1968. Acetylation of lysozyme. II. Mechanism of lysis by lysozyme. Agr. Biol. Chem. 32:64 Zaborskey, O. 1973. Immobilized Enzymes. CRC Press, Cleveland, Ohio. 107 APPENDICES APPENDIX A On the Initial OD Rise in the First Few Seconds in the Turbidity Assay of Lysozyme - the Puff Theory The rule of thumb of enzyme kinetics study is always to measure the initial rate of a reaction. When initial rates are used, complicating factors such as the reverse reaction, product inhibition and inactivation of the enzyme can be avoided and a much simpler rate equation can be used. However, an interesting observation from this study when meaning lysozyme activity with the turbidity method is that, instead of decreasing immediately, the OD of M. lysodeikticus suspension often rises during the first 5 seconds or so right after the enzyme is injected into the cuvette (Figure A.1). To avoid this interference, the activity of lysozyme samples was actually determined from the slope of the curve between 20 and 40 seconds, NOT 0 to 40 seconds. Otherwise the computer calculated activities would be significantly less or even negative! A plausible explanation is that in that the initial attack of lysozyme on the cell walls caused the cells to expand, or puff up, before they burst into pieces due to the further hydrolysis from lysozyme. It is the initial puffing of the cells made the OD rise. 108 Data supported the hypotheses: When extremely small amount of lysozyme was added - 0.001 ppm, the OD did not decline after the initial rise, because the enzyme was only enough to make limited damages on the 0.560, l-t ft* / 0.55$ '* fr^-.S * * ^ ,''^* .'- ^..*t in 0>\f? /A ■ ■ i-',-----ri !> •'•'.«■ ;,-i.:,V /■iA4,s:. :.y^ ; it* 0.00 40.00 Ti.!ns! (sec? Figure A 1. The OD rise observed during the first few seconds of turbidity assay of lysozyme activity of samples with three different concentrations: top line 2.5 ppm, middle line - 1 ppm , bottom line - 0.1 ppm. 109 cells that caused them to expand, i.e. OD increase, but not enough to make the cells disintegrate (Figure A.2). On the other hand, when the assay was done for 10 ppm lysozyme sample, the initial OD rise was almost not noticeable (data not shown here). The cells started to be broken apart immediately after exposed to the overwhelming amount of lysozyme. This hypothesis could be easily tested by the following two experiments: 1. Simply look for visual evidence of the puffing under microscope during the first a few seconds of the hydrolysis reaction. A real-time imaging device would be helpful; 2. Use cell walls instead of whole cells as substrate for the assay. If the hypothesis is correct, the OD rising should be not be observed. If the cell walls is not commercially available, it could be prepared by mechanically grinding the whole cells in a mortar with sand. 110 O.SiO. 8^^v'^sn/,"-- ■ ^ ' *<* <"' ,.„^,-. ^>' V ". f^-: O.505- O.SOOi 0.00 20.00 40.00 sa.os Figure A 2. The initial OD rise of a lysozyme sample at extremely low concentration - 0.001 ppm. Ill APPENDIX B HPLC Assay of Lysozyme in Wine and Beer Objectives Fordras HPLC method was optimized and compared to the classical turbidity method to: I. Test the heat stability of lysozyme samples treated at different temperatures: room temperature, 750C, and 950C; II. Detect lysozyme in red wine and beer samples treated with the enzyme; I. Monitor lysozyme released from lysozyme-tannin participation in red wine due to addition of NaCI. Observations and Discussions I. Heat Stability Lysozyme samples were treated at different temperatures as described below and analyzed at room temperature with the HPLC and turbidity methods. A. 500ppm, 250ppm and 125ppm prepared in distilled water (Milli-Q). At room temperature highly consistent and linear results were obtained for the two methods (Figure B.1) B. 500 ppm in distilled water and heated to 75 'C for 1, 2, 3, and 4 hours. 112 As showed in the figure B.2, significantly different results were given by the two methods. While the HPLC method showed a moderate decline of detected concentration as the treatment time increased, the turbidity method indicated significant increase of activity, especially for samples treated for 2 and 3 hours. C. 500ppm in distilled water and heated to 95 V for 1, 2, 3, and 4 hours. At this higher temperature, both methods showed decline of detected concentration. However, the turbidity method revealed the remarkable heat stability of the enzyme (Figure B.3). The HPLC method separates lysozyme from background and detects it as protein with UV281. An important aspect that need to be explored is that how sensitive this method is to the tertiary and secondary structure change of the enzyme. It is noticed that there is a small peak that always eludes about 20 seconds before the main lysozyme peak, and the two peaks are very poorly separated (chromatograms not shown). The small peak is suspected to represent a lysozyme protein with a minor different structure. Interestingly, as the heating time increased, the main peak shrunk while the small peak grew significantly. At room temperature, the small peak's area is about 5% of that of the main peak. After being kept at 950C for 4 hours, the two peaks had about the same area. Obviously, more protein was transformed into the different structure. It would be interesting to see whether the transformation will 113 eventually complete if the heating process keeps going longer, and to examine the activity with the turbidity method. II. Lysozyme in Red Wine and Beer Samples Red wine (Pinor Noir) and Pilsner style beer were spiked with 500 ppm lysozyme and assayed for lysozyme concentration 48 hours later. The separation of the lysozyme peaks were not very satisfactory. Attempts to improve the separation by changing gradient have not been successful. Data indicated that lysozyme content decreased quickly in the red wine, while fairly stable in beer. Again different results were obtained from the two different assay methods for red wine. This time more protein was detected with less activity (Table B.1). It has been proposed that the significant loss of lysozyme in red wine was due its conjugation with the phenolic compounds through electrostatic interaction and raising ionic strength of system could reverse the combining process. A. Releasing Lysozyme from Lysozyme-Tannin Complex in Red Wine with Addition of NaCI The same red wine sample was spiked with 500ppm lysozyme. Two days later 2% and 8% NaCI were added to the samples and analyzed with the HPLC 114 method 24 hours later. Results showed that with 8% NaCI the absorbed lysozyme was completely released (Table B.2) o HHPLC rl' ■ Turbidity 125 Prepared Lysozyme Concentration (ppm) Figure B.1 Detected lysozyme concentration of lysozyme samples without heat treatment from the HPLC and Turbidity methods. hV'' 115 HHPLC ■ Turbidity Time (hr) Figure B.2. Detected lysozyme concentration of lysozyme samples treated at 750C for 1,2,3, and 4 hours. 116 I • Figure B.3. Detected lysozyme concentration of lysozyme samples treated at 95C for 1,2,3, and 4 hours. 117 Table B 1. Comparison of Lysozyme activity in red wine and beer samples detected with HPLC and turbidity methods, respectively. Sample HPLC Cone. (ppm) Turbidity Cone. (ppm) SOOppmStd 500 516 R. Wine w/500 ppm 212 176 Widmer Beer w/500 ppm 489 118 Table B. 2 Releasing lysozyme from lysozyme-tannin complex in red wine with addition of NaCI at 2% and 8% levels. Sample HPLC Cone, (ppm) Percentage of original 500 ppm Std 500 100 R. Wine w/500 ppm 234 47 R. Wine w/500 ppm + 2% NaCI 396 80 R. Wine w/500 ppm + 8% NaCI 500 100