Adil Nasir for the degree of Master of Science in
advertisement

AN ABSTRACT OF THE THESIS OF
Adil Nasir for the degree of Master of Science in
Chemical Engineering presented on July 5th ,1995.
Title: Sequential and Competitive Adsorption of Bovine Serum
Albumin and 0-lactoglobulin, and Their Resistance to
Exchange with a-lacta bumin and (3- casein.
Abstract approved:
Redacted for Privacy
J
The sequential and
EPH MCGUIRE
petitive adsorption of bovine
serum albumin (BSA) and 0-lactoglobulin at silanized silica
surfaces were investigated using protein radiolabeling and
in-situ ellipsometry.
Simultaneous adsorption of
It-labeled BSA and 0-lactoglobulin revealed an initial
dominance of the surface by 0-lactoglobulin, with subsequent
replacement by BSA.
In sequential adsorption experiments,
where the surface was contacted with 14C- labeled BSA,
followed by contact with protein-free buffer and addition of
P-lactoglobulin, most of the adsorbed BSA was not removed.
Listeria monocytogenes adhesion to protein films prepared
with BSA and 0-lactoglobulin adsorbed in sequence and
simultaneously was explainable with reference to the
passivating character of adsorbed BSA, and its resistance
to elution as measured here.
The effect of adding a-lactalbumin, 0-casein and
0-lactoglobulin to adsorbed BSA as well as the effect of
adding a-lactalbumin, 0-casein and BSA to adsorbed
P-lactoglobulin was investigated using in-situ ellipsometry.
0-casein was the most effective eluting agent, while
a-lactalbumin was the least effective.
a-lactalbumin and
The abilities of
0-casein to exchange with adsorbed BSA
and 0-lactoglobulin were evaluated with reference to a
simple kinetic model for protein adsorption and exchange.
The model is based on a mechanism allowing for reversible
adsorption followed by either a surface-induced
conformational change yielding an irreversibly adsorbed form
or exchange with the dissimilar protein. Differences in
exchange behavior were generally found to be consistent with
the rate constant for surface-induced conversion of the
adsorbed protein as opposed to describing exchange of
adsorbed protein with a dissimilar protein.
This suggests
that under these experimental conditions exchange was
mediated by a protein-surface interaction, i.e., a
displacement mechanism, where protein-protein associations
were of minor significance.
Sequential and Competitive Adsorption of BSA and
0-lactoglobulin, and their Resistance to Exchange
with a-lactalbumin and 0-casein.
by
Adil Nasir
A THESIS
submitted to
Oregon State University
in partial fulfillment of
the requirements for the
degree of
Master of Science
Completed July 5th, 1995
Commencement June 1996
Master of science thesis of Adil Nasir presented on
July 5th, 1995
APPROVED
Redacted for Privacy
s4,r, representing Chemical Engineering
Major
Redacted for Privacy
Head o fheparment of Chemical Engineering
Redacted for Privacy
Dean of G
uate S
I understand that my thesis will be part of the permanent
collection of Oregon State University libraries. My
signature below authorizes release of my thesis to any
reader upon request.
Redacted for Privacy
Adil Nasir, Author
1
ACKNOWLEDGEMENTS
I would like to express my sincere gratitude to Dr
Joseph McGuire for his invaluable guidance. I would also
like to thank Dr Viwat Krisdhasima for help with the
ellipsometer and Dr Sridhar Babu for help with
radiolabeling.
I would like to thank, Dr Michael Penner, Dr Goran
Jovanovic, and Dr Bruce D'Ambrosio for kindly serving as
committee members.
I would like to acknowledge the support and
encouragement of my family.
This work was supported by the USDA National Research
Initiative Competitive Grants Program and Western Center for
Dairy Proteins.
ii
TABLE OF CONTENTS
1. INTRODUCTION
1
2. LITERATURE REVIEW
3
2.1
Properties of the milk proteins
3
2.2
Adsorption at solid-liquid interfaces
5
2.3
Factors affecting the surface activity of
the protein
6
2.4
Sequential and competitive adsorption
11
2.5
Studies related to sequential and competitive
adsorption
12
Protein modification
15
2.6
3.
MATERIALS AND METHODS
18
3.1 Preparation of Protein Solutions
18
3.2 Surface preparation
18
3.3 Adsorption kinetics
19
3.4 Protein radiolabelling
22
3.4.1 Protein purification
22
3.4.2 Radiolabelled product analysis
23
3.4.3 Sequential and competitive adsorption with
14C-BSA
4.
RESULTS AND DISCUSSION
23
25
iii
4.1 Competitive adsorption
25
4.2 Sequential adsorption
29
4.2.1 Sequential adsorption of BSA and
0-lactoglobulin
31
4.2.2 Sequential adsorption of BSA and
a-lactalbumin
37
4.2.3 Sequential adsorption of BSA and 0-casein
43
4.2.4 Sequential adsorption of 0-lactoglobulin
and BSA
49
4.2.5 Sequential adsorption of 0-lactoglobulin and
a-lactalbumin
54
4.2.6 Sequential adsorption of 0-lactoglobulin and
0-casein
60
4.3 Comparison of the abilities of the proteins to
exchange with adsorbed BSA and 0-lactoglobulin
66
5.
CONCLUSIONS
71
6.
RECOMMENDATIONS
73
BIBLIOGRAPHY
74
APPENDIX
81
iv
LIST OF FIGURES
Figure
2.3.1
2.3.2
4.1a
4.1b
Page
Schematic representation of how a protein
molecule may bind to a surface
7
Schematic showing (a) low probability of desorption
for a protein adsorbed via several binding sites and
(b) concerted cooperative mechanism of exchange
between dissolved and adsorbed protein
9
Competitive adsorption of BSA and 0-1g on
hydrophobic silica with t-adsorption = 480 min
26
Competitive adsorption of BSA and 0-1g on
hydrophilic silica with I--adsorption = 480 min
27
4.2.1a Sequential adsorption of BSA and 0-1g on
hydrophobic silica with tmA = 240 min,
trinse = 30 min, tp_ig = 240 min
4.2.1b Sequential adsorption of BSA and 0-1g on
hydrophobic silica with tmA= 60 min,
trinse = 30 min, t13-1g = 60 min
4.2.1c Sequential adsorption of BSA and 0-1g on
hydrophilic silica with tBsA = 240 min,
trinse = 30 min, tv1g = 240 min
32
33
34
4.2.1d Sequential adsorption of BSA and 0-1g on
hydrophilic silica with tElsA = 60 min,
trinse = 30 min, t13-1g = 60 min
35
4.2.2a Sequential adsorption of BSA and a-lac on
hydrophobic silica with tmA = 240 min,
trinse = 30 min, ta-lac = 240 min
38
4.2.2b Sequential adsorption of BSA and a-lac on
hydrophobic silica with tmA= 60 min,
trinse = 30 min, ta-lac = 60 min
39
LIST OF FIGURES (Continued)
Figure
Page
4.2.2c Sequential adsorption of BSA and a-lac on
hydrophilic silica with tBSA = 240 min,
trinse= 30 min, to -lac = 240 min
4.2.2d Sequential adsorption of BSA and a-lac on
hydrophilic silica with tBSA = 60 min,
trinse = 30 min, to -lac = 60 min
40
41
4.2.3a Sequential adsorption of BSA and 0-casein on
hydrophobic silica with tBsp, = 240 min,
trinse = 30 min, tp-casein = 240 min
44
4.2.3b Sequential adsorption of BSA and 0-casein on
hydrophobic silica with tBsA= 60 min,
trinse = 30 min, tp-casein = 60 min
45
4.2.3c Sequential adsorption of BSA and 0-casein on
hydrophilic silica with tBsp, = 240 min,
trinse = 30 min, tp-casein = 240 min
46
4.2.3d Sequential adsorption of BSA and 0-casein on
hydrophilic silica with tBSA = 60 min,
trinse =
30 min, tp-casein = 60 min
47
4.2.4a Sequential adsorption of 0-1g and BSA on
hydrophobic silica with tBSA = 240 min,
trinse = 30 min, tvig = 240 min
50
4.2.4b Sequential adsorption of 0-1g and BSA on
hydrophobic silica with tBSA
60 min,
trinse = 30 min,
ti3-1g = 60 min
4.2.4c Sequential adsorption of 0-1g and BSA on
hydrophilic silica with tBSA = 240 min,
trinse = 30 min, tp-lg = 240 min
51
52
vi
LIST OF FIGURES (Continued)
Figure
Page
4.2.4d Sequential adsorption of 0-1g and BSA on
hydrophilic silica with t-BSA = 60 min,
trinse = 30 min, tf3-1g = 60 min
53
4.2.5a Sequential adsorption of 0-1g and a-lac on
hydrophobic silica with twm = 240 min,
trinse = 30 min, ta-lac = 240 min
55
4.2.5b Sequential adsorption of 0-1g and a-lac on
hydrophobic silica with tBsA = 60 min,
trinse =
30 min,
ta-lac =
60 min
4.2.5c Sequential adsorption of 0-1g and a-lac on
hydrophilic silica with tBSA = 240 min,
trinse = 30 min, ta-lac = 240 min
56
57
4.2.5d Sequential adsorption of 0-1g and a-lac on
hydrophilic silica with t-BSA = 60 min,
trinse =
30 min,
ta-lac =
60 min
58
4.2.6a Sequential adsorption of 0-1g and 0-casein on
hydrophobic silica with tev, = 240 min,
trinse =
30 min,
to-casein = 240 min
61
4.2.6b Sequential adsorption of 0-1g and 0-casein on
hydrophobic silica with t-BSA = 60 min,
trinse = 30 min, tilt-casein = 60 min
62
4.2.6c Sequential adsorption of 0-1g and 0-casein on
hydrophilic silica with timm = 240 min,
trinse = 30 min, tp-casein = 240 min
63
4.2.6d Sequential adsorption of 0-1g and 3- casein on
hydrophilic silica with twm = 60 min,
trinse = 30 min, tvcasein = 60 min
64
vii
LIST OF TABLES
Table
2.1.1
4.1a
4.1b
4.2a
Page
Some Physical Properties of the Four
Proteins
Adsorbed mass determined by ellipsometry after
competitive adsorption of BSA and 0-1g, on
hydrophobic and hydrophilic silica for lh, 4h,
8h
25
Average radioactivity detected in the
protein layers (Competitive adsorption)
28
Adsorbed mass of each protein on hydrophobic and
hydrophilic silica following contact for 4h
and lh
31
4.2.1a Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and 0-1g on
hydrophobic and hydrophilic silica for lh
and 4h
4.2.2
4.2.3
4.2.4
4
31
Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and a-lac on
hydrophobic and hydrophilic silica for lh
and 4h
37
Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and 0-casein on
hydrophobic and hydrophilic silica for lh
and 4h
43
Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-1g and BSA on
hydrophobic and hydrophilic silica for lh
and 4h
49
viii
LIST OF TABLES (Continued)
Table
4.2.5
4.2.6
4.3.1
4.3.2
Page
Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-1g and a-lac on
hydrophobic and hydrophilic silica for lh
and 4h
54
Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-1g and (3- casein on
hydrophobic and hydrophilic silica for lh
and 4h
59
Summary of the removability of the adsorbed
protein in the lh tests as compared to the 4h
tests
69
Summary of whether differences in exchange of
BSA and P-lactoglobulin can be explained by sl
or ks
70
ix
NOMENCLATURE
DDES
NV
A
A
v
s1
ks
t
ti
is
81
82
81,ti
Dichlorodiethylsilane
Adsorbed mass, gg/cm2
The arctangent of the factor by which the
amplitude ratio changes
The change in phase of the light, degrees
Molar refractivity, cm3/gmole
Partial specific volume, cm3 /gm
Surface-induced conversion rate constant, min-1
Exchange rate constant, min-1
Time, min
Time at which at which contact of the adsorbed
protein with protein-free buffer is initiated,
min
Time at which the second protein is introduced,
min
Fractional surface coverage of reversibly adsorbed
protein
Fractional surface coverage of irreversibly
adsorbed protein
Fractional surface coverage of reversibly
adsorbed protein at time ti
SEQUENTIAL AND COMPETITIVE ADSORPTION OF BSA AND
0-LACTOGLOBULIN, AND THEIR RESISTANCE TO EXCHANGE WITH
a-LACTALBUMIN AND 0-CASEIN.
1. INTRODUCTION
Protein adsorption studies are highly relevant to
various technical applications, ranging from protein
deposition on food processing equipment to biocompatibility
of blood-contacting implants.
Blood coagulation, complement
activation, and the reactions of tissue to artificial
implants are all affected by
protein adsorption.
Purification of proteins (from
plasma and other complex
media) often involves solid-liquid chromatography,
where separation depends on interaction with the
chromatographic support by hydrophobic, ion exchange, or
charge-transfer mechanisms.
Adsorption of protein molecules onto surfaces alters
interfacial properties and affects adhesion of bacteria,
cells, and blood platelets (18, 34, 68).
The presence of
adsorbed milk proteins is known to affect microbial adhesion
(63,
64, 65).
Listeria monocytogenes is a pathogenic
bacterium that can exist in a variety of food processing
environments (66, 67).
Contamination of contact surfaces
used in food processing by Listeria monocytogenes and other
microorganisms has resulted in product recall and
destruction, food-borne illnesses in humans and death
(76,77).
It is difficult to control adhesion due in part to
the complicated nature of biofilm development, which is
affected by competitive adsorption events among different
proteins.
2
Protein molecules in a solution may desorb from a
surface while other protein molecules adsorb in their place.
These events are referred to as exchange reactions.
Protein
exchange reactions occur when proteins are introduced to a
surface in sequence or simultaneously (9, 14, 35). When
proteins are introduced in mixtures,
initially accomodates
the interface
protein having the highest rate of
arrival, possibly consistent with the highest concentration
in solution.
However, adsorbed molecules may be gradually
eluted by others that have a higher affinity.
The major objective of this research was to study the
kinetics of adsorption of selected milk proteins contacted
with silica surfaces in sequence and simultaneously.
The
experiments were motivated by the need for this information
in an ongoing study to evaluate bacterial adhesion to
surfaces following sequential and competitive
adsorption (APPENDIX).
protein
The milk proteins a-lactalbumin,
0-lactoglobulin, bovine serum albumin and 0-casein were
chosen because they are highly surface active and among
other things differ in size, charge, hydrophobicity, amino
acid composition and sequence in known ways.
In this work,
in-situ ellipsometry and protein radiolabeling were used to
study adsorption and exchange of proteins on silanized
silica surfaces of high and low hydrophobicity.
3
2. LITERATURE REVIEW
2.1 Properties of the milk proteins.
The proteins a-lactalbumin (a-lac), 0-lactoglobulin,(0-1g)
and Bovine serum albumin (BSA) are all major components in milk
whey.
Of these, 0-lactoglobulin and a-lactalbumin are present
in the highest concentration, 54% and 21% of the total whey
protein, respectively (51) and are of primary importance in the
overall properties of whey (52). On the average, common bovine
milk contains 3 mg/ml 0-lactoglobulin, 1 mg/ml a-lactalbumin,
0.4 mg/ml BSA and 10 mg/ml 0-casein (53). Some essential
properties of these proteins are tabulated in Table 2.1.1.
0-lactoglobulin (162 amino acid residues, MW 18,320
for the monomer) exists as a dimer in solution at normal pH
because of electrostatic interactions between Aspl" and
Glu134 of one monomer with a corresponding Lys residue of
another monomer (54).
Each monomer contains two disulphide
bridges and one free thiol group (-SH).
The thiol group is
important since it appears to facilitate SHIS -S interchange
reactions which allow the formation of new structures or
intermolecular disulphide-bonded dimers and polymers upon
heating (55).
The 0-lactoglobulin conformation is pH and
temperature senstive, even at pH 6.5, it undergoes some
internal reorganization (51).
a-lactalbumin (123 amino acid residues, MW 14,161)
on the other hand, is a very compact, nearly spherical
single chain globulin with four disulphide bonds but no
thiol group.
a-lactalbumin has a biological function as
well, that being to modulate the substrate specifity of
galactosyltransferase in the lactos synthetase complex,
Table 2.1.1
Some physical properties of the four proteins (78).
Oaa
Protein
Residue
Mol. Wt
Shape and
Charge
Dimension(nm)
in neutral
solution
%a-helix:
Hydrophobicity
P-sheet :
(kcal/residue)
0 S-S
pI
(per molec)
unorder
oblate
a-lac
123
14,161111
ellipsoid
-2.0("'"I
45:45:10
1,019"
4.2-4.5
-15.0"(2)
26:14:60
1,077111
5.13
2 + 1 SH
ellipsoid with
_ le.om
-55:16:29
4.7-4.9
17 + 1
3 domains
(-9,-8,-1)
4
(2.3x3.7x3.2)
2-spheresP-lg
162
18,320111
conjugated121
(3.58x6.93)
prolate
BSA
582
66,267
995
SH
(4.2x14.1)
random/unord.
0-casein
209
24,000
coil
(unknown)
-12
10:-13:70
1330
N 5.2
0
Notes: [1]. averaged between genetic variants A and B;[2].dimer;[3].based on Ref. (79); [4]. values in
parentheses represent charge of each domain. All properties of
P-casein based on Ref. (80).
5
which is responsible for the synthesis of lactose in
lactating mammary tissue (56).
Calcium binding, which may
stabilize thermal denaturation, is another characteristic of
a-lactalbumin (57).
BSA (MW 66,267) is a large globular protein in whey
with a single polypeptide chain containing 582 amino acid
residues with 17 intra chain disulphide bonds and one thiol
group.
Synthesized in the liver tissue, BSA gains entrance
to milk through the secretory cells (58).
The BSA structure
consists of three domains and nine subdomains (59).
The
multidomain structure of BSA in responsible for the
anomalous behavior of the protein observed under
denaturation conditions (59).
The 0-casein molecule consists of 209 amino residues
and has molecular weight of 23,980 Da (62).
The molecule is
a single chain with five phosphoserine residues.
The
N-terminal portion (residues 1-43) contains all five
phosphoserine residues and carries essentially all of the
protein's net charge.
The remainder of the molecule is very
hydrophobic, particularly the region of residues 136-209.
0-casein is thus a linear amphiphile, and its structure is
largely unordered. 0-casein undergoes endothermic
aggregation at temperatures above 4°C.
2.2 Adsorption at solid-liquid interfaces.
The surface activity at the solid -liquid interface is
usually described by isotherms showing the adsorbed amount
as a function of protein concentration in bulk, at
equilibrium.
For most proteins at low bulk concentration
(< lmg/m1), a plateau in the adsorption isotherm is
observed.
The adsorbed amounts in the plateau
usually in the range of several mg/m2.
region are
This coverage is
6
close to the expected amounts in a close-packed monolayer of
molecules in the native configuration.
However, in some
cases, as for human plasma albumin adsorbed on polystyrene
latex, it was observed that the concentration at the
surface, at saturation, is considerably lower than the
expected value.
This was attributed to the spreading and
unfolding of the molecule at the interface (12,13).
Thermodynamic analysis of protein adsorption reveals
several parameters that contribute to the adsorption energy;
hydrophobic dehydration, ion incorporation, electrostatic
interactions, and structural changes in the protein
molecule.
It was concluded that in most cases adsorption in
entropically driven and that the adsorption is usually
irreversible(9,35).
The irreversibility results from
high
adsorption due to the large number of adsorbing segments and
from the unlikely event of simultaneous desorption
of all the segments.
2.3 Factors affecting the surface activity of a protein.
The surface activity is derived from the amino-acid of
each protein and from the possible attachment of various
segments to the interface.
The main parameters that result
in variance in surface activity, as reviewed by Horbett and
Brash(9)are:
Molecule size: It is assumed that larger molecules may
have more contact points, thus increasing the total
adsorption energy (which leads to irreversible adsorption).
However, it appears that this parameter is less significant
than the protein structure, for example, hemoglobin is far
more surface active than fibrinogen, although it has a much
lower molecular weight (65,000 and 330,000 respectively).
7
Fig 2.3.1 Schematic representation of how a protein molecule
may bind to a surface. From left to right: a few strong bonds,
many (weak) bonds,and a combination of both (35).
8
Amphipathicity and hydrophobicity: It is expected that
increased hydrophobicity of the protein would lead to
increased surface activity, as indeed was observed.
A native ovalbumin was denatured for various degrees of
hydrophobicities(10).
It was concluded that increasing the
hydrophobicity leads to greater reduction in interfacial
tension and better emulsifying properties.
Flexibility: It is very likely that proteins which can
unfold to a greater degree, or that unfold more rapidly,
would be more surface active due to increased adsorbed sites
at the interface, and due to entropic gain caused by
unfolding of the molecule.
Thus the flexible 0-casein
molecules are more surface active than the globular BSA and
lysozyme molecules, which contain many disulfide bonds(11)
Charge: It is frequently observed that the highest
adsorption and surface pressure are obtained near the
iso-electric point of the protein.
This observation results
from decreased electrical barriers to adsorption, decreased
repulsion among adsorbed molecules, and minimal solubility
of the protein in the bulk phase
Time dependency: It takes time for a protein molecule
to form all its possibe bonds with a surface, which maybe
connected with a conformational change of the molecule.
This makes spontaneous desorption and also exchange
reactions more difficult with time, especially if not only
more but also stronger bonds are formed with the surface
with increasing time.
A protein molecule has in general multiple contact or
"bonds" with a solid surface.
These bonds may or may not
develop gradually after the protein molecule has reached the
surface (35).
The longer a given molecule resides on a
surface, the larger is the probability for the formation of
multiple bonds.
The molecule maybe adsorbed through a few
9
a.
PURE DESORPTION
rA
b.
EXCHANGE
Figure 2.3.2 Schematic showing (a) low probability of
desorption for a protein adsorbed via several binding sites
and (b) concerted cooperative mechanism of exchange between
dissolved and adsorbed protein (9).
10
(strong) bonds or through a number of weak bonds or a
combination thereof as seen in Fig 2.3.1.
Exchange vs Desorption:
The difference between
desorption and exchange is that the former can be very slow
and exchange can be rapid.
Lundstrom and Elwing(35) have
described exchange reactions as the study of proteins which
stick to a given surface as a function of time and the rate
of escape from the surface of different types (kind and
conformation) of protein molecules.
Exchange and desorption can be differentiated by the
depiction in
Fig 2.3.2.
Desorption can be depicted by
assuming protein with multiple binding sites (9).
The
protein binds to the site using an imaginary `foot'.
foot may lift up or down randomly.
Each
For desorption to occur
all feet would be required to lift off altogether which is
statistically improbable proving thereby that the protein is
irreversibly bound.
To explain explain exchange we consider protein
molecules present in the local environment.
At random a
protein molecule may be desorbed, while another protein
molecule may implant a foot on the site vacated by the foot
of the desorbing protein. Exchange occurs when many
adsorbing protein molecules may encourage the desorption of
already adsorbed protein molecules.
In essence, a new
protein molecule implants its feet and induced the removal
of a pre-adsorbed molecule.
This is the basis of the
explanation of the exchange processes by Vroman and workers
(14).
The more "feet" and energy per foot (total adsorption
energy), the lower the probability that exchange can occur.
Hence in a competitive adsorption process, one finds that
those molecules present in the highest amounts and of
smaller size bind first. (due to diffusion and collision
11
arguments) and are then
exchanged by more strongly
adsorbing components.
2.4 Sequential and competitive adsorption.
Sequential adsorption is a process in which a protein
is adsorbed and a second protein is added to the adsorbed
protein.
The second protein may exchange some or most of
In an exchange reaction, a second
the adsorbed protein.
molecule gradually replaces the originally adsorbed one, and
the original bonds are broken one by one which may be a much
probable process.
A lower total free energy of the second
molecule, when it binds to the
surface may be a
thermodynamic driving force for the exchange reaction.
To
describe sequential adsorption, many variable are relevant
including the amount adsorbed and time of adsorption of the
first protein (55-57) as well as the conditions supplied,
e.g., pH, adsorption time of the second protein, and
concentration (55-58 ).
Sequential adsorption is important
in the development of diagonastic test systems in which
immunologically active antibodies (IgG's) are preadsorbed on
a carrier.
To suppress non-specific interactions of the
complement antigen, non-occupied parts of the adsorbent
surface have to be covered with a second protein (59-61).
Besides this additional adsorption, partial displacement of
preadsorbed protein could be desirable to obtain a
(homogeneous) population of immunoglobulins that are
strongly attached to the surface.
During adsorption, different amino acid residues
(segments) of one and the same protein attach themselves to
the surface, increasing the molar Gibbs energy of adsorption
to large values (6).
As a consequence, the trend is that
proteins are extremely difficult to remove by diluting the
12
On the other hand, if the solution contains a
solution.
displacer or other protein whose molecules have an affinity
for the adsorbent, any desorbing segment can be replaced by
another.
Desorption of the molecule is now virtually an
exchange process and, as AexchangeG
is much more likely.
AdesorptionGt
this process
Competitive adsorption from a
mixture of proteins is a dynamic scene of protein
adsorption, desorption, and displacement.
After the protein
contacts the surface, the interface will initially
accomodate the protein molecules that (i) have the highest
rate of arrival, i.e.,the largest diffusion coefficient, and
(ii) are most abundantly present in the solution.
However,
the adsorbed molecules may be gradually displaced by others
that have a higher affinity.
The final composition of the
adsorbed layer at a.given interface is determined by the
concentrations of the various kinds of the proteins in the
solution, the intrinsic adsorption affinities, and the
possibilities of the proteins to desorb (6).
2.5 Studies related to sequential and competitive
adsorption.
Sequential adsorption of proteins was done by Pitt,
Park and Cooper (18). They used techniques that included
125
I-labelled protein, Fourier transform infra-red spectroscopy
and immunogold labelling techniques to clarify how the
sequence of protein adsorption affected blood response.
It
was observed that platelet deposition and thrombus formation
were strongly influenced by the sequence of protein
adsorption.
The first protein adsorbed in sequential
adsorption seemed to dominate the response
observed in a
nonanticoagulated canine ex vivo shunt experiment.
They
attributed this behavior to the first adsorbed protein
13
binding tightly to high energy surface sites and thereby
affecting the blood response.
In the case of competitive
adsorption, the high energy sites were occupied by a
distribution of proteins depending upon the diffusivities
and concentrations of the proteins.
Arnebrant and Nylander (16), studied sequential and
competitive adsorption of 0-lactoglobulin and x-casein on
Metal surfaces.
The effect of a monolayer of one protein on
the subsequent adsorption of another protein, and
competitive adsorption of both the proteins, was studied
using ellipsometry and 14C-labelled 0-lactoglobulin.
It was
observed that x-casein sequentiallly adsorbs onto a layer of
f3- lactoglobulin whereas 0-lactoglobulin cannot adsorb onto a
layer of x-casein.
In competition, the surface energy of
the substrate influenced the amount adsorbed and the
composition of the adsorbed layer.
Ruzgas et al (7) studied sequential adsorption of
y-Interferon and BSA on hydrophobic silicon surfaces using
ellipsometry and radiolabelling.
They found that the
displacement degree of y-Interferon from the hydrophobic
silicon surface by BSA molecules is increased with decreased
electrostatic interactions between the proteins.
They
observed that the displacement of y-Interferon by BSA is
followed by formation of loose protein layers exhibiting low
refractive indices.
Competitive and Sequential adsorption of 0-casein and
0-lactoglobulin on hydrophobic surfaces was done by Nylander
and Wahlgren (8) in order to elucidate the interfacial
structure of 0-casein.
It was observed in earlier studies
(16), that 3- lactoglobulin could not adsorb on a layer of
14
By cleaving the molecule at residues Asp 43 and
x-casein.
Asp 47, using an enzyme namely endoproteinase Asp-N, allowed
the adsorption of 0-lactoglobulin onto 0-casein.
Shirahama,Lyklema and Norde (5), studied the adsorption
of lysozyme,ribonuclease and a-lactalbumin.
They selected
proteins with similar size and shape but with different
isoelectric points and structural stabilities. The
adsorption was done on negatively charged surfaces such as
hydrophilic silica and hydrophobic polystyrene coated
silica.
The adsorption process was monitored by
reflectometry which indicates the total adsorbed amount with
a combination of streaming potential measurements, which
provided information on the composition of the adsorbed
layer.
They found in general sequential and competitive
adsorption on hydrophilic silica is ruled by electrostatics.
In some cases electrostatics were accompanied by the
hydrophobicity of the protein exterior and structural
re-arrangements.
On hydrophobic surfaces electrostatics
played a minor role.
Wahlgren et al (17), studied the adsorption of pure
protein solutions and binary mixtures of proteins with
opposite net charges at pH 7 by in situ ellipsometry.
They
found that electrostatic attraction between proteins and a
change in ionic strength affects the adsorption behavior.
The mixtures studied were lysozyme/P-lactoglobulin and
0-lactoglobulin/lactoferrin.
Elgersma et al (36) studied the adsorption competition
between BSA and monoclonal immuno-gammaglobulins(IgG's), by
using sequential and simultaneous addition of the proteins
to differently charged polystyrene(PS) latices as the
adsorbates.
They found that preadsorbed IgG was more easily
displaced by BSA than the converse, and also that
15
electrostatics did not play a major role, under certain
conditions their effect was absent. They also found that the
displacement of the first protein was not always a
prerequisite for the adsorption of the second protein.
Both
sequential and competitive adsorption additions of the
proteins indicated faster conformational rearrangements in
adsorbates of BSA than in those of the IgG's
2.6 Protein modification.
The following section is a brief discussion of
the advantages of using reductive methylation to label
proteins.
Each type of protein can be modified by reductive
alkylation as described by Means and Feeney.
For a wide
range of proteins, the covalent modification proceeds as
follows, between either aliphatic aldehydes or ketones, and
protein amino groups:
Protein-NH2+RHCOPProtein-NH=CHR-* Protein-NH-CH2R
(2.5.1)
The first step is very rapid and highly reversible, and the
adduct can be reduced to a stable alkylamino group.
Under
mild, slightly alkaline conditions,extensive alkylation of
protein amino groups takes place while other side chains do
not form stable derivatives.
Means and Feeney (23)
indicated that extensive modification was easily obtained
using a low concentration of sodium borohydride as the
reductant; disulphide bonds are not cleaved in this
environment.
The reaction is strongly dependent, and best
results are obtained near pH 9 (and 0°C), although similar
modification can be obtained at pH 7. Means and Feeney
(23) used formaldehyde as the carbonyl component of choice
16
for the reductive methylation and they observed that
monomethyllysine was formed initially, but was converted to
dimethyllysine very rapidly; the modified proteins exhibited
c-N,N-dimethyllysine residues:
Protein-NH2+2H2C0+ 1/2 NaBH4
Protein-N(CH3)2 +
1/2 NaH2B03-F 1 / 2H20
(2.5.2
)
However, it was discovered that the use of NaBH4 as the
reducing agent has several disadvantages, including its
ability to reduce aldehydes directly, thus decreasing the
efficiency of the reaction, its instability at neutral pH,
its ability to reduce disulphide linkages,and its potential
for breaking peptide bonds (21).
It was found that these
drawbacks can be overcome by using NaCNBH3, a much weaker
reducing agent which, as described by Borch et al(22),
reducing Schiff bases but not aldehydes at neutral pH.
Dottavio-Martin et al (30) observed that the
advantages of using sodium cyanoborohydride as opposed to
sodium borohydride, was that the reaction could be carried
out at neutral pH for extended periods of time and greater
incorporation of [19C] methyl groups could be attained.
NaCNBH3can selectively and quantitatively convert free
amino groups to their dimethyl derivatives at neutral pH,
under very mild conditions(24).
Although most aldehydes
give the mono-substituted alkylamine shown in reaction
(2.5.1) as the final product, reaction with formaldehyde
preferentially gives a dimethyl product as shown in reaction
(2.5.3) with the second methylation step proceeding more
rapidly than the first.
17
6HCHO + 3 Protein-NH2 + 2 NaCNBH3
-+ 3 Prot-N(CH3)2 + 2 HCN + 2 NaH2BO3
(2.5.3)
Jentoft and Dearborn (31) indicate that the reaction
(2.5.3) is extremely specific supported by the fact that
model compound studies (24 ), as well as amino acid analysis
and nuclear magnetic resonance (NMR) studies of several
proteins (26,27,28
)
have shown that only the NH2 terminus
and lysyl residues are labeled.
It was noted by Jentoft and
Dearborn (31) that unless the protein contained an essential
lysyl residue, the changes in its physical and chemical
properties were minimal because of the small size of the
added methyl groups and because the charge of the amino
group was retained with only a small alteration in pica
value.
A comparison of radioiodination of proteins with
reductive methylation by
'4C
indicates the latter to be the
method of choice for this study.
radioactive source than "C
,
Although 1251 is a stronger
several reports have pointed
out that iodination of proteins may affect their adsorption
at solid surfaces and their chromatographic behavior
(69-73)
.
18
3.
MATERIALS AND METHODS
3.1 Preparation of Protein Solutions.
The proteins from bovine milk were purchased from Sigma
Chemical Co.
(St. Louis, MO).
a-lactalbumin (type III, L-
6010, Lot 128F8140), P-lactoglobulin (L-0130, Lot 51H7210),
bovine serum
albumin (A-7906, Lot 15F0112), and 0-casein
(C-6905, Lot 12H9550) were of the highest native pure grade
prepared from bovine milk.
Prior to an experiment the
protein was carefully weighed (Mettler Model AE 240, Mettler
Instrument Corp., Hightstown, NJ ) and dissolved in a
phosphate buffer solution.
Each protein solution was
stirred for approximately 20 minutes then used after
dilution to the molar equivalent of 1.000mg/m1 0-1g
(27.22gM).
Buffer solutions (pH 7.00) were prepared by
mixing a solution of 0.01M sodium phosphate monobasic
monohydrate (NaH2PO4.H20) and 0.01M sodium phosphate dibasic
(NaHPO4).
Sodium Azide (NaN3), used as an antimicrobial
agent, was also added to the solutions at a concentration of
0.02%(mass per volume) prior to mixing.
Both buffer and
protein solutions were filtered (0.22 or 0.45gm type
GV,Millipore Corp., Bedford, MA) prior to use.
3.2 Surface preparation.
All surfaces were prepared from a single type of
silicon (Si) wafer (hyperpure, type N, phosphorus doped,
plane 1-0-0, Wacker Siltronic Corp., Portland, OR).
First,
the Si Wafers were cut into small plates of approximately
19
They were subsequently
1 X 2 cm using a tungsten pen.
treated to exhibit hydrophilic of hydrophobic surfaces.
The silanizations were modified slighty from the method
described by Jonsson et al (37).
Each small Si plate was
placed into a test tube and 5m1 of the mixture NH4OH:H20: H2O
(1:1:5) was added to the tube which was then heated to 80 °C
The Si plates were then
in a water bath for 15 minutes.
rinsed
with 20m1 distilled-deionized water(Corning Megapure
System, Corning, NY) and immersed in 5m1 HC1: H202:
H20(1:1:5) for 15 min at 80 °C.
Each plate was then rinsed
with 30m1 distilled-deionized water and stored in 20m1 of a
50% ethanol/water solution.
Each hydrophilic Si plate was
rinsed with 40m1 distilled-deionized water, dried with N2,
and stored in a desiccator for 24 h.
Dried, hydrophilic Si
plates were then immersed in a stirred solution of
dichlorodiethylsilane (DDES, Aldrich Chemical Co., Inc.,
Milwaukee, WI) in xylene for lh.
The degree of silanization
was controlled by the concentration of DDES.
The
concentrations used in this work were 0.01% DDES (for
hydrophilic surfaces) and 0.1% DDES (for hydrophobic
surfaces) in xylene.
Finally, the silanized silica
surfaces were sequentially rinsed in 100 ml xylene,
acetone and ethanol.
The plates were dried with N2 and then
kept in a desiccator, and their relative hydrophobicities
were measured using contact angle analysis.
3.3 Adsorption kinetics.
The kinetic data was monitored is in situ, with
ellipsometry (Model L116C, Gaertner Scientific Corp.,
Chicago. IL) according to procedures developed in our
laboratory(38).
A silanized silica surface was placed into
a fused quartz trapezoid cuvette(Hellma Cells, Germany).
20
The cuvette had a volume of about 8 ml; its fuzed quartz
windows were placed perpendicular to the incident and
reflected beams(angle of incidence=70°C).
The ellipsometer
sample stage was then adjusted to obtain a maximum in
reflected light intensity.
Filtered buffer solution (sodium
phosphate, 0.01M, pH 7) was then injected into the cuvette.
The surface was left to equilibrate with the buffer for 30
min.
Fine adjustments of the stage were conducted in
parallel with ellipsometric measurements of bare surface
optical constants ws and Az until steady values were
obtained.
Final measurements of bare surface properties
were then recorded.
The buffer solution was carefully
removed from the cuvette and replaced with the protein
solution.
The sequential experiments were performed in
three parts.
a.Introduction of the first protein.
The buffer was carefully replaced by a solution of the first
protein using a syringe by continual withdrawal of buffer
and subsequent introduction of the protein.
The values of
w and A were ellipsometrically measured and recorded every
15 seconds for 4h for long term experiments and every 6
seconds for lh for short term experiments under static
conditions, i.e., no stirring and no flow.
b. Rinse.
The first protein solution was carefully replaced by buffer
using a syringe by continual withdrawal of the first protein
and subsequent introduction of buffer.
This was done after
4h for long term experiments and lh for short term
experiments.
The pre-adsorbed protein layer was allowed to.
equilibrate in buffer for 30 min during which values of w
and A were ellipsometrically measured and recorded every 15
seconds for long term experiments and 6 seconds for short
term experiments under static conditions.
21
c.Introduction of the second protein.
The buffer was carefully replaced by.the second protein
solution using a syringe by continual withdrawal of the
buffer and subsequent introduction of the second protein.
Similar to the first step the values of w and A were
ellipsometrically measured for the same duration and same
time interval.
In competitive adsorption an equimolar solution of the
two proteins wer used, ie., such that the total molar
concentration of protein was equal to that of the single
protein solutions.
The procedure for competitive adsorption
was similar to the first step in sequential adsorption
except that the optical properties were recorded every 30
seconds for 8h.
Recorded values of
were stored on a floppy disk.
, and A were stored
A computer program based on
McCrackin's calculation procedure (39) was used to import
the data from the disk and determine the refractive index
and thickness corresponding to each pair of w and A, which
were then used to calculate the adsorbed mass of protein
according to the Lorentz-Lorentz relationship (40).
The
required molecular weight: molar refractivity ratios(M /A)
were calculated to be 3.816g/ml for a-lac, 3.8140g/ml for
0-casein, 3.796g/ml for 0-1g, and 3.837 giml for BSA (41).
The partial specific volumes (v) for each protein are 0.733
ml/g for a-lac, 0.748 ml/g for 0-casein, 0.751 ml/g for
0-1g, and 0.729 ml/g for BSA.
In sequential tests, the MIA
and v used at any time was that pair associated with the
protein in solution at that time; in competitive tests an
averaged M/A and v were used.
At least two replicate tests
were performed in each case, with the maximum error in
adsorbed mass generally being 5 %.
22
3.4 Protein radiolabelling.
Bovine Serum Albumin was labeled by reductive
methylation of lysine residues using "C- formaldehyde
according to methods slightly modified from Jentoft and
Dearborn (17). The above technique has been used to study
protein adsorption from mixed solutions as well as protein
exchange reactions at solid-water and air water
interfaces(16, 42-43).
Alkylation of lysine residues in
this was does not affect their basicity (23).
The protein
was methylated by dissolving 0.32mg Bovine Serum Albumin in
15ml 0.05M pottassium phosphate(pH 7), and mixing with 2 ml
0.10M
sodium cyanoborohydride(Sigma), and 250gCi
"C-formaldehyde (30-50 mCi/mmol, ICN Biomedicals) at 4°C for
48h. The reaction mixture was gently shaken at intervals.
3.4.1
Protein purification
Unreacted species, side products and buffer salts from
the unreacted protein were ultrafiltered in a Centriprep
ultrafiltration cell (10,000 molecular weight cut-off;
Amicon, Beverly, MA).
The protein was further purified by
adding distilled, deionized water and concentrating the
solution; this was repeated five times to remove 99% of the
species smaller than 10,000 Da.
The protein solution
(concentration > 20 mg/ml) was diluted with sodium phosphate
buffer(0.01 M, pH 7) to a final concentration of 1.55 mg/ml,
as quantified by the Bradford assay (45).
Sodium phosphate
buffer was used to allow more direct comparison with results
concerning unlabeled BSA reported here.
23
3.4.2 Radiolabeled product analysis.
100g1 of the labeled protein was taken and 10p1 of
trichloracetic acid was added.
on ice for 15 minutes.
The solution was left to sit
Thereafter it was spun in a
microfuge to allow the precipitate to separate from the
supernatant.
The supernatant was decanted and added to 15
ml of Optifluor (Packard Instrument Co, Meridien, CT) and
counted for 10 minutes.
The precipitate was left to dry and
then was redissolved in 0.01M sodium phosphate buffer and
counted in 15 ml of Optifluor scintillation fluid.
Most of
the radioactivity was in the protein and the specific
activity of the protein was found to be 2.86 pCi/mg.
3.4.3 Sequential and competitive adsorption with 14C -BSA.
The hydrophobic surfaces used were the same as that
used in ellipsometry and were silanized with
0.100%
dichlorodiethylsilane dissolved in xylene.
All adsorption experiments were carried out in Nunclon
multi-well dishes (Nunc Inc, Naperville, IL 60563). In a
typical experiment 250g1 of the labeled protein solution was
pipetted into a compartment of the multi-well dish.
A carefully weighed silicon plate was placed onto the
surface of the solution.
Care was taken to ensure no
bubbles were present between the solid and water interface.
The surface area of the plate was calculated by the weight.
Adsorption was allowed to proceed for 1 or 4 h, and the
surface was rinsed by introducing 250g1 buffer, then
withdrawing 250g1 of the solution, this sequence was
repeated ten times.
After 30 minutes, 250g1 P-lg (1 mg/ml)
was introduced followed by withdrawal of 250g1 of the
24
solution, this sequence being repeated ten times.
Contact
with 0-1g was maintained for the same period of time allowed
for 'AC-BSA.
The surface was then rinsed as before, and
left in buffer for an additional 30 min.
The adsorbed
protein layer was removed from the surface by immersion of
the silica plate in 1 mL 48% hydrofluoric acid for 5
minutes(17).
To this was added 15mI, Gold AB scintillation
fluid(Packard Instrument Co.) and 1mL Triton-X.
The sample
was well-mixed, then analyzed within one day with a Beckman
scintillation counter.
Competitive adsorption tests were run similarly, with
adsorption occuring for 1 or 4 h from a solution of 250g1
'AC-BSA combined with 250g1 0-1g solution.
The surface was
rinsed, and the adsorbed layer removed and analyzed, as
described before.
In each case experiments were compared to
controls in which the tests proceeded exactly as described
above, with the exception that 0-1g solutions were replaced
with equal volumes of protein-free phosphate buffer.
25
4.
RESULTS AND DISCUSSION
4.1 Competitive adsorption.
Competitive adsorption occurs when proteins are
introduced in mixtures.
In this study BSA & 0-lactoglobulin
were simultaneously introduced and their adsorption kinetics
on hydrophobic and hydrophilic surfaces were monitored over
a period of 8h.
Figures 4.1a and 4.1b depict the adsorbed amounts
versus time from a mixture of BSA and 0-lactoglobulin on a
hydrophobic surface and a hydrophilic surface respectively.
The adsorbed mass values determined from these figures are
given in Table 4.1a.
Table 4.1a. Adsorbed mass determined by ellipsometry after
competitive adsorption of BSA and 0-lactoglobulin, on
hydrophobic and hydrophilic silica for lh, 4h, 8h.
Surface
adsorbed mass
adsorbed mass
adsorbed mass
after lh
after 4h
after 8h
[11g/cm2]
hydrophobic
0.170
[4g/cm2]
0.175
[4g/cm2]
0.175
(0.164,0.140)
hydrophilic
0.130
0.150
0.155
(0.150,0.140)
The adsorbed mass in parentheses indicate single-component kinetic
adsorbed mass values of p-lactoglobulin and BSA respectively.
The experimentally determined average radioactivity
(disintegrations.min-l.cm-2) detected in the protein layers
removed from hydrophobic silica after adsorption of NC-BSA
and 0-lactoglobulin for lh and 4h and incubation in buffer
for 30min are given in Table 4.1b.
26
02
DO
0.18
0.16
0.14
0.12
0.1
0.08
0.06
0.04
0.02
0
1
0
50
1
100
i
150
I
200
r
F
250
300
i
350
i
400
1
450
1
500
Time (min)
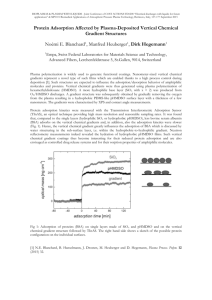
Figure 4.1a. Competitive adsorption of BSA and 0-1g on
hydrophobic silica with tadsorption = 480 min.
27
0
50
100
150
250
Ti m. lain)
900
350
400
450
5a)
Figure 4.1b. Competitive adsorption of BSA and 13-1g on
hydrophilic silica with I--adsorption = 480 min.
28
Table 4.1b. Average radioactivity (DPM/cm2) detected in the
protein layers.
The figures in the parentheses indicate the
deviation from the mean of two replicates.
Experiment
DPM/cm2
Control, lh adsorption
5861 (14%)
lh competitive adsorption
1480
(1%)
Control, 4h adsorption
5717
(14%)
4h competitive adsorption
2295 (27%)
Fig 4.1a and Figs 4.1b show a higher mass adsorbed from
a mixture of BSA and P-lactoglobulin on hydrophobic
hydrophilic surfaces.
than on
The adsorbed mass from a mixture of
BSA and 0-lactoglobulin on hydrophobic silica was higher
than that attained by either BSA or 0-lactoglobulin
when adsorbed from single-protein solutions as indicated by
Table 4.1a.
This indicates the presence of a mixed film,
possibly involving more than one layer.
Figs 4.1a and 4.1b
show faster adsorption kinetics on hydrophobic surfaces than
on hydrophilic surfaces.
This is consistent with the
general observation that these negatively charged proteins
exhibit slower kinetics and attain a lower adsorbed mass on
hydrophilic, negatively-charged surfaces (3).
Table 4.1b
shows that 25% of the "C-BSA originally adsorbed remained
on the hydrophobic surface after adsorption of
'4C
-BSA and
0-lactoglobulin for lh followed by incubation in buffer for
30min, and this increased to 40% after 4h.
Applying the
radiolabelling results directly to the ellipsometric data
(Fig 4.1a) would indicate that the mixed layer was comprised
of about 0.043gg/cm2BSA and 0.127gg/cm20-lactoglobulin
29
after lh.
However, after 4h the composition of the mixed
layer was 0.07gg/cm2 BSA and 0.105gg/cm2 0-lactoglobulin,
i.e. an increase in the amount of BSA and a decrease in the
amount of f3- lactoglobulin with time.
Krisdhasima et al (4) proposed a mechanism for
irreversible protein adsorption consisting of two steps.
In
step 1, corresponding to short contact time, the protein
reversibly adsorbs to the surface (governed by rate
constants kl and k-1), with its adopted surface conformation
closely approximating its native form. In step 2, a surfaceinduced conformational change occurs (governed by rate
constant si)
changing the reversibly adsorbed molecule to
an irreversibly bound form. In a later study (3), the
adsorption kinetics and surfactant mediated elutability of
a-lactalbumin, 0-casein, 0-lactoglobulin and BSA was studied
at hydrophobic silica.
0-lactoglobulin was characterized by
the highest k1 and the lowest s1, whereas BSA had the lowest
k1 and highest s1.
This suggests that 0-lactoglobulin would
dominate adsorption initially from a mixture of BSA and
0-lactoglobulin, but once adsorbed, would be more likely to
exchange with BSA than adsorbed BSA would be to exchange
with 3- lactoglobulin.
This is consistent with the
competitive adsorption behavior recorded here.
4.2 Sequential adsorption.
Sequential adsorption is a two-step process in which a
protein is adsorbed onto a surface, left there for a certain
time after which a second protein is added to the
protein-covered adsorbent.
30
In sequential adsorption, it is of interest to
compare the final adsorbed mass value attained after
introducing the second protein to the single component
plateau values of the second protein in order to gain
information concerning the final adsorbed layer (6,8).
Table 4.2a shows the single-component adsorbed mass values
used in this study which were obtained from the data of
Krisdhasima et al (3).
The lh and 4h single-component
plateau values obtained for BSA and 0-lactglobulin in this
study were not used for this comparison due to their
subsequent contact with protein-free buffer.
The plateau
values recorded for BSA and 0-lactoglobulin after contact
with protein-free buffer varied somewhat among experiments
as is evident from Figs 4.2.1a through 4.2.6d.
However the
.change in adsorbed mass recorded upon introduction of the
second protein was reproducible.
Table 4.2a. Adsorbed mass (11g/cm2) of each protein on
hydrophobic and hydrophilic silica surfaces following
contact for 4h and lh (3).
4h
lh
Protein
Hydrophobic
Hydrophilic
Hydrophobic
Hydrophilic
BSA
0.136
0.136
0.124
0.120
13-1g
0.156
0.140
0.128
0.112
a-1ac
0.132
0.128
0.132
0.112
13-casein
0.254
0.255
0.233
0.236
31
4.2.1
Sequential adsorption of BSA and 0-lactoglobulin.
Figures 4.2.1a through 4.2.1d show the sequential adsorption
of BSA and 0-lactoglobulin on hydrophobic and hydrophilic
surfaces for 4h and 1h. The adsorbed mass values from these
figures are given in Table 4.2.1a
Table 4.2.1a. Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and f3- lactoglobulin on
hydrophobic and hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
protein'
mass
protein
mass
[p.g/cm2]
[gg/cm2]
hydrophobic
4h
BSA
0.155
0-1g
0.180
hydrophobic
lh
BSA
0.135
P-1g
0.151
hydrophilic
4h
BSA
0.133
P-lg
0.175
hydrophilic
lh
BSA
0.134
P-lg
0.159
'adsorbed mass values are after contact with protein-free buffer
The experimentally determined average radioactivity
(disintegrations.min-1.cm-2) detected in the protein layers
removed from hydrophobic silica, after sequential adsorption
of '4C-BSA and
0-lactoglobulin, for lh and 4h, and
incubation in buffer for 30min are given in Table 4.2.1b.
32
02
Q1B
as
1Q14
"- Q12
to
0
a
Q1
QCB
QC6
O m`Pr y
01 `A
Q02
0
0
ID
2D
3D
4D
5:D
Irme (min)
Figure 4.2.1a. Sequential adsorption of BSA and p-lg on
hydrophobic silica with tBsA = 240 min, triame
30 min, tp_ig = 240 'min.
33
Q2
az
5216
14
'1112
to al
OS
0
AWB
0
O4
W2
0
0
40
83
1C0
123
140
103
Tine (rain)
Figure 4.2.1b. Sequential adsorption of BSA and 0-1g on
hydrophobic silica with twm = 60 min,
trine = 30 min, t 0-1g = 60 min.
34
Rinse
02
13-ig
Q18
ela
as
0)014
-Q12
m Q1
od
6
QCB
0
0
100
ZO
33)
403
633
Tine (min)
Figure 4.2.1c. Sequential adsorption of BSA and 13-1g on
hydrophilic silica with tESA = 240 min,
t,
30 min, and tvig = 240 min.
35
02
Frag
Q18
15016
0.12
to
Co
al
.0 OMB
C)
LI acs
0
Ts Q0'
002
0
100
Tine
133
140
103
(min)
Figure 4.2.1d. Sequential adsorption of BSA and 0-1g on
hydrophilic silica with tssA= 60 min,
trinm,= 30 min, to_ig = 60 min.
36
Table 4.2.1b. Average radioactivity (DPM/cm2) detected in
the protein layers. The figures in the parentheses indicate
the deviation from the mean of two replicates.
Experiment
DPM/cm2
Control, lh adsorption
2695
(21%)
lh sequential adsorption
1935
(7%)
Control,
adsorption
2217
(11%)
4h sequential adsorption
1599
(4%)
4h
Figs 4.2.1a through 4.2.1d show an increase in adsorbed mass
following addition of 0-lactoglobulin to adsorbed BSA in all
experiments.
The final adsorbed mass exceeded that expected
of either protein contacted alone on hydrophobic and
hydrophilic surfaces (Table 4.2.1a, Table 4.2), indicating
the presence of a mixed film.
Figures 4.2.1a through 4.2.1d
show that the adsorbed layer of BSA was largely irreversible
to dilution in both long and short term experiments.
Experiments with 0-lactoglobulin sequentially added to
adsorbed 140-BSA on hydrophobic surfaces (Table 4.2.1b),
indicate 72% 14C-BSA remained on the surface after the lh
and 4h experiments. For the lh experiment (Fig 4.2.1b,
Table 4.2.1a), direct application of the radiolabeling
results to ellipsometric data indicates that 0.038gg/cm2
BSA was removed and substituted by 0.054gg/cm2
J3- lactoglobulin adsorbed.
For the 4h experiment
(Fig 4.2.1a, Table 4.2.1a), direct application of the
radiolabeling results to ellipsometric data indicates that
37
0.043 gg/cm2ESA was removed and 0.068gg/cm20-lactoglobulin
adsorbed.
This suggests that although the composition of
0-lactoglobulin increased in the mixed layer, the extent of
exchange of 0-lactoglobulin with adsorbed BSA at the
hydrophobic surface did not similarly increase.
The higher
adsorbed mass at longer time maybe attributed to the partial
development of an outer layer, or a closer packing among
these proteins than that adopted for either protein alone,
in a monolayer.
4.2.2 Sequential adsorption of BSA and a-lactalbumin.
Figures 4.2.2a through 4.2.2d show the sequential adsorption
of BSA and a-lactalbumin on hydrophobic and hydrophilic
surfaces for 4h and lh. The adsorbed mass values from these
figures are given in Table 4.2.2
Table 4.2.2. Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and a-lactalbumin on
hydrophobic and hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
proteins
mass
protein
mass
[gg/cm2]
[11g/cm2]
hydrophobic
4h
BSA
0.139
a-lac
0.132
hydrophobic
lh
BSA
0.142
a-lac
0.142
hydrophilic
4h
BSA
0.127
a-lac
0.123
hydrophilic
lh
BSA
0.140
a-lac
0.131
'adsorbed mass values are after contact with protein-free buffer
Figs 4.2.2a through 4.2.2d and Table 4.2.2 show that there
was not much variation in the total adsorbed mass on the
38
42
a18
"6 Eris
tnaist
0.12
to a1
0.C6
0to 401
4 042
0
0
103
Zn
333 -
400
SD
eco
Tine (min)
Figure 4.2.2a. Sequential adsorption of BSA and a-lac on
hydrophobic silica with taim = 240 min,
trinae = 30 min,
= 240 min.
39
02
Q18
T.)
Q16
tn
ta
0
0.1
0.03
0.C6
o 0.04
0.02
0
0
23
40
eo
80
to
ix
140
VD
Mrs onin)
Figure 4.2.2b. Sequential adsorption of BSA and a-lac on
hydrophobic silica with tium = 60 min,
trine = 30 min, to -lac = 60 min.
40
02
018
el0
als
1a14
CO al
os
0
ics
o)
N
0ta
WE
acs
Q04
002
0
0
ZO
3M
4:0
5:0
Time Win)
Figure 4.2.2c. Sequential adsorption of BSA and a-lac on
hydrophilic silica with tBSA = 240 min,
triase = 30 min, and ta- lac = 240 min.
41
40
CO
E0
103
123
140
1E0
Time (min)
Figure 4.2.2d. Sequential adsorption of BSA and a-lac on
hydrophilic silica with twm = 60 min,
trinse = 30 min, to -lac = 60 min.
42
addition of a-lactalbumin to adsorbed BSA.
In particular,
the adsorbed mass was constant or decreased a little.
Adsorbed BSA has shown to inhibit cell adhesion
(34) and adsorption of other proteins.
BSA is a multi-
domain protein where one domain is neutral and the entire
negative out-of-balance charge resides on the other twothirds of the molecule (33).
Presumably BSA would adsorb
with its neutral domain adjacent to either a hydrophobic or
a hydrophilic surface with its negatively charged domains
oriented toward solution.
This relatively high negative
charge density would be expected to repel a negatively
charged a-lactalbumin molecule.
Spreading pressure data of
Suttiprasit et al (32) at the air-water interface suggests
that the number of non-covalent contacts made by one
a-lactalbumin molecule roughly equals that of one BSA
molecule, even though BSA is larger in size than
a-lactalbumin.
This would suggest thermodynamically that a
BSA molecule would be replaced by one molecule of
a-lactalbumin with no change in interfacial energy.
It
must be noted that the adsorption isotherms obtained by
Suttiprasit et al (32) at the air-water interface indicated
that, in the region of the plateau, the number of moles of
adsorbed a-lactalbumin was about twice that of either BSA
or J3- lactoglobulin.
In this study at the solid-water
interface the number of moles of adsorbed a-lactalbumin was
about four times that of BSA and twice that of
P-lactoglobulin.
Using the spreading pressure data obtained
by Suttiprasit et al
(32) and the adsorption isotherms used
in this study, a-lactalbumin would make twice as many
non-covalent contacts as BSA.
This would suggest that one
molecule of a-lactalbumin could be replaced by two
molecules of BSA without any change in interfacial energy.
43
Such a replacement would result in a noticeable decrease in
adsorbed mass.
However, on addition of a-lactalbumin to
adsorbed BSA the adsorbed mass remained constant or decrease
a little. (Figs 4.2.2a through 4.2.2d, Table 4.2.2),
indicating very less or no exchange.
This is consistent
with the fact that BSA has a very high sl and would be
resistant to exchange by an adsorbing protein.
4.2.3 Sequential adsorption of BSA and 0-casein
Figures 4.2.3a through 4.2.3d show the sequential adsorption
of BSA and 0-casein on hydrophobic and hydrophilic surfaces
for 4h and lh. The adsorbed mass values from these figures
are given in Table 4.2.3
Table 4.2.3. Adsorbed mass determined by ellipsometry after
sequential adsorption of BSA and 0-casein on hydrophobic and
hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
protein'
mass
protein
mass
[4g/cm2]
[pg/cm2]
hydrophobic
4h
BSA
0.159
0-casein
0.290
hydrophobic
lh
BSA
0.145
0-casein
0.239
hydrophilic
4h
BSA
0.135
0-casein
0.222
hydrophilic
lh
BSA
0.150
0-casein
0.245
'adsorbed mass values are after contact with protein-free buffer
Due to the large difference in the final adsorbed
mass expected of single-component solutions of BSA and
0-casein, it would be interesting to compare the adsorbed
44
0
100
2:0
3:0
403
500
ECO
Time (min)
Figure 4.2.3a. Sequential adsorption of BSA and 0-casein on
hydrophobic silica with tBSA = 240 min, trinee =
= 240 min.
30 min, and tv
45
0
23
40
eo
83
100
123
140
183
Time tnin)
Figure 4.2.3b. Sequential adsorption of BSA and 0-casein on
hydrophobic silica with tssA= 60 min,
tree
30 min, to. wm = 60 min.
46
0
ZO
103
XO
403
E03
Time (min)
Figure 4.2.3c. Sequential adsorption of BSA and 0-casein on
hydrophilic silica with twm = 240 min,
= 30 min, to_i, = 240 min.
tree
4-7
.... Q3
0
tn
Q25
Q2
010.15
cd
o Q1
w
$4 0 .05
o
4
0
0
20
40
83
8D
100
120
140
1E0
Time Wit)
Figure 4.2.3d. Sequential adsorption of BSA and 0-casein on
hydrophilic silica with to = 60 min,
truce = 30 min, tp-casein = 60 min.
48
mass at the end of each experiment, to that expected of
0-casein alone (6,8).
The final plateau value attained in the lh
experiment on both hydrophobic and hydrophilic surfaces
(Fig 4.2.3b, Fig 4.2.3d, Table 4.2.3), were quite similar
to that of single-component adsorbed mass values of 0-casein
alone on hydrophobic and hydrophilic surfaces (Table 4.2a).
This would indicate complete removal of BSA.
The final
plateau values for the 4h experiment with hydrophilic
surface (Fig 4.2.3c, Table 4.2.3) failed to reach the
single- component adsorbed mass value of 0-casein alone on a
hydrophilic surface (Table 4.2a), indicating the presence of
a mixed layer of BSA and 0-casein, and incomplete removal.
For the 4h experiment on the hydrophobic surface
(Fig 4.2.3a, Table 4.2.3), the final adsorbed mass exceeded
that of 0-casein alone (Table 4.2a).
This is consistent
with incomplete removal as well, but could suggest a more
tighter packing of the mixed layer at this interface or
possibly an outer layer.
0-casein has an unordered structure, with regions
of marginal stability that have a high degree of segmental
motion (74).
The N-terminal 21-amino acid sequence of
0-casein contains one-third of its charged residues at pH 7,
and this portion of the protein is highly solvated and
flexible.
The remainder of the molecule is nonpolar and
very hydrophobic, make 0-casein distinctly amphiphilic.
Due to its amphiphilic nature, 0-casein may behave like a
large surfactant molecule.
Graham and Phillips (11)
observed that 0-casein molecules were much more surface
active than the globular molecules that they studied, and
this would be consistent with its ability to remove BSA in
the lh experiments on hydrophobic and hydrophilic surfaces
49
(Fig 4.2.3b, Fig 4.2.3d, Table 4.2.3).
The inability of
0-casein to completely remove BSA in the 4h experiments on
hydrophobic and hydrophilic surfaces (Fig 4.2.3a,
Fig
4.2.3c, Table 4.2.3) can be attributed to more extensive
conformational adaptation undergone by BSA.
4.2.4 Sequential adsorption of f3- lactoglobulin and BSA.
Figures 4.2.4a through 4.2.4d show the sequential adsorption
of 0-lactoglobulin and BSA on hydrophobic and hydrophilic
surfaces for 4h and lh. The adsorbed mass values from these
figures are given in Table 4.2.4
Table 4.2.4. Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-lactoglobulin and BSA and on
hydrophobic and hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
protein'
mass
protein
mass
[fIgicm2]
[4g/an2]
hydrophobic
4h
0-1g
0.150
BSA
0.177
hydrophobic
lh
P-lg
0.145
BSA
0.171
hydrophilic
4h
p-lg
0.143
BSA
0.175
hydrophilic
lh
P-1g
0.130
BSA
0.155
'adsorbed mass values are after contact with protein-free buffer
Table 4.2.4 (Figures 4.2.4a through 4.2.4d)
indicates an increase in the total adsorbed mass on addition
of 0-lactoglobulin to adsorbed BSA in all experiments.
The
final adsorbed mass exceeded single-component adsorbed mass
values of either protein in the 4h experiments (Fig 4.2.4a,
Fig 4.2.4c, Table 4.2.4a, Table 4.2a), indicating the
presence of a mixed film possibly involving more than one
50
02
418
"041s
to
to 01
IS
X406
444
a404
g0
40.02
0
0
103
ZO
ZO
407
91)
so
Tine (ilia)
Figure 4.2.4a. Sequential adsorption of 0-1g and BSA on
hydrophobic silica with tp_ig = 240 min,
= 30 min, and t. = 240 min.
51
02
Q18
Q16
13/1314
'"" Q12
to
Q1
403
aas
O am
04 402
0
120
0
143
1E0
This (min)
Figure 4.2.4b. Sequential adsorption of 0-1g and BSA on
hydrophobic silica with tvia = 60 min,
30 min, tssA= 60 min.
trinae
52
02
0.18
016
a 0.14
- 0.12
0.1
ms ac
6
k acs
ve Q04
0.02
0
0
100
200
300
400
9:0
9:0
Ting boku)
Fig 4.2.4c. Sequential adsorption of 13-1g and BSA on
hydrophilic silica with tog = 240 min, trine
30 min, tmm = 240 min.
53
Q2
Q18
iM8
-4114
Da
od
Q1
0
,6003
a)
0
id OM
Q02
0
0
100
1D
140
Time (min)
Figure 4.2.4d. Sequential adsorption of 13-1g and BSA on
hydrophilic silica with tvig = 60 min,
= 30 min, t
= 60 min.
54
layer.
The ellipsometric data was not sufficient to
qualitatively determine the extent of removal of
0-lactoglobulin.
However the results of the competitive
adsorption experiments with "C-BSA (Table 4.1.4b) indicated
that BSA proceeded to displace 0-lactoglobulin with time,
implying that BSA would displace 0-lactoglobulin to some
extent in these tests.
4.2.5 Sequential adsorption of 0-lactoglobulin and
a-lactalbumin
Figures 4.2.5a through 4.2.5d show the sequential adsorption
of 0-lactoglobulin and a-lactalbumin on hydrophobic and
hydrophilic surfaces for 4h and lh. The adsorbed mass values
from these figures are given in Table 4.2.5.
Table 4.2.5. Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-lactoglobulin and a-lactalbumin
on hydrophobic and hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
protein'
mass
protein
mass
[1.1.g/cm2)
[11g/cm2)
hydrophobic
4h
P-1g
0.165
a-lac
0.150
hydrophobic
lh
P-lg
0.129
a-lac
0.129
hydrophilic
4h
P-lg
0.144
a-lac
0.135
hydrophilic
lh
f3 -lg
0.130
a-lac
0.130
'adsorbed mass values are after contact with protein-free buffer
The sequential addition of a-lactalbumin to
adsorbed 0-lactoglobulin on a hydrophobic and hydrophilic
surface for lh (Fig 4.2.5b, Fig 4.2.5d, Table 4.2.5), did
not result in a change in adsorbed mass.
However, for the
55
Q2
,..
Q18
El Q16
In
=. Q14
crt2
ca
Kidd
Q1
03Q
444 QC6
co
0co
V
4
Q04
ac2
0
0
ZO
103
33) -'
4C0
533
E03
Tine (min)
Figure 4.2.5a. Sequential adsorption of 0-1g and a-lac on
hydrophobic silica with tp_ig = 240 min,
tr-
e = 30 min, tti_i = 240 min.
56
Q2
Q18
016
Mau
to
OA
aa3
acs
0 am
01
4 0.02
0
0
23
40
0
83
ED
123
143
'ED
Figure 4.2.5b. Sequential adsorption of 13-1g and a-lac on
hydrophobic silica with to_ig = 60 min,
tr.
e = 30 min, ta_la, = 60 min.
5'7
02
0.18
6016
10.14
0.12
co
0.1
is
ei
ns0.06
ACM
s4
o20.04
0/2
0
0
1C0
203
3:0
me
5C0
03
(min)
Figure 4.2.5c. Sequential adsorption of 13-1g and a-lac on
hydrophilic silica with to_ig = 240 min,
= 30 min, ta_l = 240 min.
58
02
el; 0.18
0.16
tri
=014
at2
ca
to
es
0.1
00.03
.0
0.C6
O 0.04
ta
' 0 0 .02
0
0
2)
40
E0
83
103
120
140
1E0
Time (lain)
Figure 4.2.5d. Sequential adsorption of 13-1g and a-lac on
hydrophilic silica with to_ig = 60 min,
= 30 min, ta_i = 60 min.
tra
59
4h experments, addition of a-lactalbumin to adsorbed
f3- lactoglobulin (Fig 4.2.5a, Fig 4.2.5b, Table 4.2.5),
resulted in a decrease in adsorbed mass on each surface.
The surface pressure data of Suttiprasit et al
(32), suggests that the number of contacts made by one
a-lactalbumin molecule roughly equals the number of
contacts made by one 0-lactoglobulin dimer at the air-water
interface. As mentioned in section 4.2.2 this argument would
hold only if the adsorption isotherms at the air-water
interface obtained by Suttiprasit et al
(32) were similar to
those at the solid-water interface used in this study.
A
comparison of the adsorption isotherms at the air-water
interface obtained by Suttiprasit et al
(32) indicated that
the number of moles of adsorbed a-lactalbumin was twice
that of 0-lactoglobulin, and was similar to that obtained at
the solid-water interface, used in this study.
This would
imply that adsorbed P-lactoglobulin dimers could be replaced
by the same number of a-lactalbumin molecules with no
change in interfacial energy.
Such a replacement would
result in a decrease in adsorbed mass. The lh experiments
indicate no change in adsorbed mass upon addition of
a-lactalbumin (Fig 4.2.5b, Fig 4.2.5d, Table 4.2.5),
suggesting no exchange.
However, the 4h experiments
indicate a decrease in adsorbed mass in each case
(Fig 4.2.5a, Fig 4.2.5c, Table 4.2.5), suggesting some
removal of adsorbed 0-lactoglobulin.
60
4.2.6 Sequential adsorption of 0-lactoglobulin and 0-casein
Figures 4.2.6a through 4.2.6d show the sequential adsorption
of f3- lactoglobulin and 0-casein on hydrophobic and
hydrophilic surfaces for 4h and lh. The adsorbed mass values
from these figures are given in Table 4.2.6
Table 4.2.6. Adsorbed mass determined by ellipsometry after
sequential adsorption of 0-lactoglobulin and 0-casein on
hydrophobic and hydrophilic surfaces for lh and 4h.
Surface
Time of
first
adsorbed
second
adsorbed
Adsorption
protein'
mass
protein
mass
[Dg/cre]
[ggicre]
hydrophobic
4h
P-1g
0.135
f3- casein
0.235
hydrophobic
lh
0-1g
0.135
P-casein
0.227
hydrophilic
4h
P-1g
0.169
P-casein
0.265
hydrophilic
lh
0-1g
0.130
0-casein
0.230
'adsorbed mass values are after contact with protein-free buffer
The adsorbed mass following 0-casein addition to
adsorbed 0-lactoglobulin on a hydrophobic surface in the 4h
tests (Fig 4.2.6a, Table 4.2.6) was less than that expected
for 0-casein alone (Table 4.2a),
indicating that the final
layer was a mixture of 0-lactoglobulin and 0-casein.
However, the adsorbed mass attained in the lh tests for each
surface (Fig 4.2.6b, Fig 4.2.6d, Table 4.2.6) was nearly
equal to that expected for 0-casein alone (Table 4.2a),
indicating a large removal of 0-lactoglobulin.
Similarly
the 4h tests with the hydrophilic surface (Fig 4.2.6c, Table
4.2.6) indicated a large removal of
0-lactoglobulin.
The large flexibility and amphiphilicity of the
0-casein
molecule contributes to its surface active nature (11), as
61
0.3
0casein
. 0.25
Rinse
0.2
w 0.15
0.1
O
0.05
til
'CS
100
200
300
400
500
600
Time (min)
Figure 4.2.6a. Sequential adsorption of 0-1g and 0-casein on
hydrophilic silica with to_lg = 240 min,
240 min.
trine 30 min,
tv,,,, =
62
0
23
40
a)
EO
11:0
123
14D
1E0
The fazin)
Figure 4.2.6b. Sequential adsorption of 13-1g and 0-casein on
hydrophobic silica with tp_1g = 60 min,
= 30 min,
= 60 min.
tree
to_in
63
0
1C0
2:0
3:0
401)
5:0
Tine (min)
Figure 4.2.6c. Sequential adsorption of 13-1g and 13-casein on
hydrophilic silica with tp_ig = 240 min,
ti. = 30 min, tp-casein = 240 min.
64
0
23
40
E0
83
100
120
140
1E0
Time (min)
0-1g and 0-casein on
Figure 4.2.6d. Sequential adsorption of
hydrophilic silica with to_ig = 60 min,
tr=0,= 30 min, tp_cein = 60 min.
65
mentioned with reference to the BSA and
0-casein tests.
Complete removal of 0-lactoglobulin is consistent with this
property of 0-casein.
Incomplete removal of 0-lactoglobulin
following contact with the hydrophobic surface for 4h can be
attributed to adsorbed 0-lactoglobulin having attained a
more tightly bound state.
66
4.3
Comparison of the abilities of the proteins to exchange
with adsorbed BSA and P-lactoglobulin.
Lundstrom and Elwing (35) considered an experimental
situation in which adsorption to a solid surface was allowed
to occur from a single-component solution of protein A,
followed by incubation in buffer, after which time a second,
dissimilar protein B was added.
They showed under certain
experimental conditions their model would lead to an
expression that relates the fraction of protein A that is
non-exchangeable to rate constants governing conversion of
protein A to an irreversibly adsorbed form, and exchange of
adsorbed protein A by protein B.
Krisdhasima et al (3) adapted that model to study the
surface-mediated elution of a-lactalbumin, (3- casein,
P-lactoglobulin and BSA at hydrophobic surfaces.
They
referred to e1 as the fractional surface coverage of
removable protein and 02 as the fractional surface coverage
that is not removable.
During incubation of protein A in
protein-free buffer, and in the absence of spontaneous
desorption,
dOi
dt
= s101
(4.3.1)
This leads to
=
and consequently
-s,t
(4.3.2)
67
ei,t, [1
12
(4.3.3)
esit
where 01J is the value of 01 at t=ti (the initiation of
adsorbed protein contact with protein-free buffer).
At
t=ts, the second protein is introduced and
dOi
dt
= siel +
ksei
(4.3.4)
where ks is a rate constant describing exchange of
A by protein B.
protein
Lundstrom and Elwing(35) defined the
exchange rates as dependent on protein B concentration as
well.
For t>ts
01 =01,te
x e
-(si +10(t-ts)
(4.3.5)
Since only that fraction of 01 molecules depleted by
conversion according to rate constant s1 contributed to
formation of irreversibly adsorbed protein, the consequence
of eqn (4.3.4) and eqn (4.3.5) was that
02 =
-sit, .{
1
1- e-
Si
(si+Ics)(t-ts)
si
+ks +el
t
[1esits]
(4.3.6)
68
After a sufficiently long time, the fraction of
non-removable protein molecules would be, according to eqn
(4.3.6)
92
s
esit,
(4.3.7)
91,t.
The left-hand side of equation 4.3.7
can be approximated as
the fraction of originally adsorbed protein A that is
non-removable.
protein A.
This fraction would
increase with the sl of
BSA has a relatively high sl whereas
P-lactoglobulin has a relatively low sl, indicating that
proteins would be expected to more readily exchange
P-lactoglobulin than BSA, assuming ks was independent of the
protein being exchanged.
The model shown by equation 4.3.7 assumes that at the
end of an experiment the originally adsorbed protein is
present only in the the non-removable state.
In these
experiments, the effect of varying the adsorption times of
both proteins from lh to 4h can be used to verify the
applicability of the model.
An increase in the removal of
the adsorbed protein on the introduction of the second
protein would imply that at the end of the lh experiments
some molecules were present in the removable state.
A
decrease in the removed amount or no change in the degree of
removal would indicate that most of the molecules had
converted to the non-removable state.
An analysis of each experiment is shown in Table 4.3.1
69
indicating whether an increase, decrease or no measurable
difference in the degree of removal was observed upon
addition of the second protein to the adsorbed layer.
Table 4.3.1 The removability of the adsorbed protein
in the lh experiment compared to that of the 4h experiment.
Experiment
Hydrophobic
(increase in time from
BSA and a-lactalbumin
no measurable
difference
BSA and P-casein
decrease
P-lactoglobulin and
a-lactalbumin
increase
P-lactoglobulin and
0-casein
decrease
1-4h)
Hydrophilic
(increase in time from
1-4h)
no measurable
difference
decrease
increase
no measurable
difference
Table 4.3.1 shows that an increase in time of adsorption
of 0-lactoglobulin and the contact time of a-lactalbumin
resulted in an increase in removal of P-lactoglobulin. This
indicates that some P-lactoglobulin remained in the
removable state in the lhr experiments.
Therefore, while
comparing the ability of a-lactalbumin to exchange with BSA
and P-lactoglobulin, only the 4h experiments were considered
applicable to the model.
However, while comparing the
ability of P-casein to exchange with adsorbed BSA and
P-lactoglobulin, both lh and 4h experiments were considered
applicable.
The ability of a-lactalbumin to exchange with adsorbed
BSA was compared to its ability to exchange P-lactoglobulin
70
using equation 4.3.7.
Similarly the ability of (3- casein to
exchange with adsorbed BSA and P-lactoglobulin was compared.
The results of the analysis are summarized in Table 4.3.2.
Table 4.3.2 Summary of whether differences in exchange of
BSA and P-lactoglobulin can be explained by slor ks based on
equation 4.3.7.
Experiments
BSA and a-lactalbumin vs
P-lactoglobulin and
a-lactalbumin
4h
lh
4h
lh
(0.1%)
(0.1%)
(0.01%)
(0.01%)
s,
NA
s
NA
a
b
s
b
BSA and 13- casein vs
P-lactoglobulin and
(3- casein
a
:
in both cases an increase or decrease in removal was
undistinguishable.
b : in both cases there was a large removal and an increase
or decrease in removal was undistinguishable.
With reference to Table 4.3.2, in all the allowable 4h
tests differences in exchange was governed by sl as opposed
to k,
The finding that ks was of lesser importance may
suggest a protein-surface interaction, i.e, displacement
mechanism where protein-protein associations were of minor
significance.
71
CONCLUSIONS
1. Ellipsometric measurements showed a higher adsorbed
mass of BSA and 0-lactoglobulin on hydrophobic than on
hydrophilic surfaces.
Faster adsorption kinetics were
observed from a mixture of BSA and 0-lactoglobulin
on hydrophobic as opposed to hydrophilic surfaces.
Radiolabeling experiments indicated 25% "C-BSA
remained on a hydrophobic surface after lh, which
increased to 40% for the 4h test.
This suggests an
initial dominance of adsorption by 0-lactoglobulin,
with subsequent replacement by BSA.
2. Sequential adsorption experiments with 0-lactoglobulin
added to "C-BSA on hydrophobic surfaces indicated that
72% "C-BSA remained on the surface after the lh and 4h
experiments.
This indicates most of adsorbed BSA was
resistant to removal by j3- lactoglobulin.
3. The kinetic data for sequential adsorption of BSA and
a-lactalbumin on hydrophobic and hydrophilic surfaces
showed that the adsorbed mass decreased a little or
remained constant, indicating little or no exchange.
a-lactalbumin was unable to remove adsorbed
0-lactoglobulin on hydrophobic and hydrophilic surfaces
in the lh tests.
The 4h tests showed some removal of
0-lacto globulin by a-lactalbumin.
4. 0-casein was able to completely elute adsorbed BSA in
the lh tests as compared to the 4h tests on both
hydrophobic and hydrophilic surfaces.
However,
72
0-casein was able to completely remove 0-lactoglobulin
in the lh tests and on hydrophilic surfaces for the 4h
tests.
Incomplete removal was observed for 0-casein
addition to adsorbed f3- lactoglobulin on hydrophobic
surfaces for the 4h tests.
Based on these results
0-casein was the most effective eluting agent
and a-lactalbumin was the least effective eluting
agent.
5. Differences in exchange behavior of a-lactalbumin and
0-casein with adsorbed BSA and 0-lactoglobulin were
found to be consistent with sl as opposed to ks.
This suggests that exchange was mediated by a
protein-surface interaction, i.e, a displacement
mechanism, where protein-protein associations were of
minor significance.
73
RECOMMENDATIONS
1.
In order to quantify k5 of the exchanging protein
with reference to equation 4.3.7 in section 4, it would
be necessary to know s1 of the adsorbed protein. It
would be impossible to quantify sl of the proteins
used in this study due to the vast differences in their
properties.
However, in our laboratory si of
lysozyme mutants with similar properties but differing
in one aspect such as charge and stability, have been
quantified.
Performing sequential adsorption
experiments with labeled lysozyme mutants with a known
sl and subsequent contact with a dissimilar protein will
enable us to determine k5 of the exchanging protein.
2.
Radiolabeling of one or more proteins in a mixture and
it subsequent detection is one of the most reliable
methods of directly determining the composition of the
adsorbed layer. However this technique is accompanied
with the disadvantages of being time-consuming and
hazardous due to contamination.
Development of a method
of labeling a protein with a fluorescent-probe and
direct quantification using colorimetric detection
techniques would eliminate the drawbacks offered by
the radiolabeling method.
74
BIBLIOGRAPHY
1. Vroman,L,Adams,A.L.,J.Colloid Interface Sci.111,
391(1986).
2. Lok,B.K.,Cheng,Y.L.,and Robertson,C.R, J. Colloid
Interface Sci,91,104(1983).
3. Krisdhasima V.,Vinarophong P,and McGuire J.,J.Colloid and
Interface Sci 161,325-334(1993).
4. Krisdhasima V.,McGuire J.,J.Colloid and Interface Sci
(1992)
.
5. Arai T., and Norde W.,Colloids and Surfaces,51(1990)1-15.
6. Shirahama H.,Lylema J., and Norde Willem.,J.Colloid and
Interface Sci 139,177(1990).
7. Ruzgas T.A.,Razumas V.J.,Kulys J.J.,J.Colloid and
Interface Sci 151,136(1992).
8. Nylander T., and Wahlgren N.M.,J. Colloid and Interface
Sci 162,151-162(1994).
9. Brash J.L., and T.A.Horbett,eds.,Proteins at Interfaces
ACS Symposium Series 343,American Chemical Society,
Washington DC,1987.
10.Kato A., and S.Nakai,Biochem.Biophysics.Acta
624:13(1980).
11.Graham D.E.and M. C. Phillips, J. Colloid Interface
Sci.70:427(1979).
12.Norde W., and J.Lyklema,J.Colloid Interface Sci.66:266
(1978) .
13.Norde W.,and J.Lyklema,J.Colloid.Interface Sci.71:350
(1979)
.
75
14. Vroman L., and A.L.Adams,Multiple plasma/surface
interactions,B/ood,(1984).
15. Kondo A.,S.Oku and K.Higashitani, J.Colloid Interface
Sci.143:214(1991).
16. Arnebrant T., and T.Nylander, J.Colloid Interface
Sci.111:529(1986).
17. Wahlgren M.C.,T.Arnebrant and M.A.Paulsson, J.Colloid
Interface Sci.158:46(1993).
18. Pitt W.G.,K.Park and S.L.Cooper, J.Colloid Interface
Sci.111:343(1985).
19. Green, D.W. and Aschaffenburg,R. , J.Mol.Biol. 1, 54
(1959)
20. Witz,J.,Timasheff,S.N. and Luzzati,V.,J.Amer.Soc.86,168
(1964) .
21. Crestfield A.M., and S. Moore ,J.Biol.Chem.238,622(1963).
22. Borch R.F.,M.D.Bernstein,and H.D.Durst,J.Am.Chem.Soc.93,
2897.
23. Means G.E., and R.E.Feeney,Biochemistry 7,2191(1968).
24. Jentoft N., and Dearborn,J.Biol.Chem.254,4359(1979).
25. Geoghegan K.F., J.C.Cabacungan, H.B.F.Dixon,and
R.E.Feeney,Int.J.Pept.Protein Res.17,345(1981).
26. Jentoft J.E., N.Jentoft,T.A.Gerken,and D.G.Dearborn,
J.Bio/.Chem.,254,4366(1979).
27. Jentoft J.E., N.Jentoft,T.A.Gerken,and D.G.Dearborn,
J.Bio/.Chem.,256,231(1981).
76
28. Jentoft J.E., N.Jentoft,T.A.Gerken,and D.G.Dearborn,
J.Biol.Chem.,257,2894 (1982) .
29. Udenfriend S., S.Stein,P.Bohlen,W.Dairman,W.Leimgruber,
and H. Weigle,Science 178,871(1972).
30. Dottavio-Martin D., and J.M.Ravel,Analytical
Biochemistry 87,562-565(1978).
31. Jentoft N., and Dearborn,Methods in Enzymology,Vo1,91,
570(1983).
32. Suttiprasit P., Viwat Krisdhasima,and J McGuire,J.
Colloid Interface Sci. 316,154(1992).
33. Andrade, J.D., Hlady,V.,Wei,A.-P., and Gilander,
C.-G.,Croat. Chem. Acta 63,527(1990).
34. A1-Makhlafi,H.,J.McGuire,and M. Daeschel.J.App/.
Environ.Microbiol. 60,3560(1994).
35. Lundstrom I., and H.Elwing ,
136,68(1990).
J.Colloid Interface Sci.
36. Elgersma A.V., R.L.J.Zsom, J.Lyklema,and W.Norde.
J. Colloid Interface Sci. 152,410,(1992).
37. Jonsson U., B.Ivarsson,I.Lunstrom,and L.Berghem,
J.Colloid Interface Sci. 148,90,(1982).
38. Krisdhasima V., J.McGuire,and R.Sproull., J. Colloid
Interface Sci. 154,337(1992).
39. Krisdhasima V.,J.McGuire,and R.Sproull.,Surf.
Interface Anal. 18,453(1992).
40. Cuypers P., J.Corsel,M.Janssen,J.Kop,W.Hermens,H.
Hemker.,J. Biol.Chem. 258,2426(1983).
77
41. Pethig R.,"Dielectric and Electronic Properties of
Biological Materials ",p.64.Wiley,New York,1979.
42. Hunter J.R., P.K.Kilpatrick and R.G.Carbonell.,J.
Colloid Interface Sci. 137,462(1990).
43. Hunter J.R., P.K.Kilpatrick and R.G.Carbonell.,J.
Colloid Interface Sci. 142,429,(1991).
44. Hunter J.R., P.K.Kilpatrick and R.G.Carbonell.,J.
Colloid Interface Sci. 143,37,(1991).
45. Bradford M.M., Analytic.Biochem. 72,248(1976).
46. Duncan J.F., and G.B.Cook,/sotopes in Chemistry,
Clarendon,Oxford,1968,p.97.
47. Robertson R.F., C.A.Winkler, and S.G.Mason,Can.J.Chem.,
34, 716(1956).
48. Rabinovitch W., R.F.Robertson, and S.G.Mason,J.Colloid
Sci.,13,600(1958).
49. Doyle W.P., and A.H.Ellison,Advan. Chem.,43,268(1964).
50. Archer R.J., "Ellipsometry," Gaertner Scientific
Corporation, Chicago, IL(1968).
51. Kinsella J.E., 1987. Milk proteins: Physicochemical
and Functional Properties. CRC Crit. Rev. Food Sci.
Nutr. 21,197.
52. Barraquio, V.L. and F.R. Van de Voort. 1988. Milk
and Soy proteins: Their Status in Review. Can Inst.
Food Sci. Technol.J. 21, 477.
53. Eigel W.N., J.E. Butler, C.A. Ernstrom, H.M.
Farrell, Jr., V.R. Harwalkar, R. Jenness, R. McL.
Whitney. 1984. Nomenclature of Proteins of Cow's
Fifth Revision. J. Dairy Sci. 67, 1599.
Milk.
78
54. Creamer, L.K., D.A. Parry, and G.N. Malcolm. 1983.
Secondary Structure of Bovine 0-lactoglobulin B.
Arch. Biochem. Biophys. 227,98.
55. Watanabe, K. and H. Klostermeyer. 1976. Heat Induced
Changes in Suiphydryl and Disulphide Levels of
a-Lactalbumin A and the Formation of Polymers.
J. Dairy Res. 43,411.
56. Hill, R.L. and K. Brew. 1975. Lactose Synthetase.
Adv. Enzymol. Relat. Areas Mol. Biol. 72,105.
57. Hiraoka, Y. and S. Sugai. 1984. Thermodynamics of
Thermal Unfolding of Bovine Apo-a-lactalbumin.
Int. J. Pept. Protein Res. 23,535.
58. Bessinger, R. L., and Leonard, E. F., J. Colloid
Interface Sci. 85, 521 (1982).
59. Klein, F., Bronsveld, W., Norde, W., van Rommunde,
L. K. J., and Singer, J. M., J. Clin. Path. 32,
90 (1979)
.
60. Chow, S.-N., Ho-Yuen, B., and Lee, C.-Y. G.,
Appl. Biochem. 7, 114 (1985).
J.
61. Lahav, J., J. Colloid Intei:face Sci. 119,262
(1987) .
62. Farrell.,
Chemistry
and E. H.
Reinhold,
K. M., in " Fundamentals of Dairy
" (N. P. Wong, R. Jenness, M. Keeney,
Marth, Eds.), p.461. Van NostrandNew York, 1988.
63. M. Fletcher, J. General Microbiol. 94, 400 (1976).
64. Meadows P. S., Arch. Mikrobiol. 75, 374(1971).
65. Tamada Y., and Y. Ikada, J. Colloid Interface Sci.
155, 334 (1993)
.
79
66. Cox L. J., T. Kleiss, J. L. Cordier, C. Cordelana,
P. Konkel, C. Pedrazzini, R. Beuener, and A.
Siebenga, Food Microbiol. 6, 49, (1989)
.
67. Gellin B. G., and C. V. Broome, J. Am. Med. Assoc.
261, 1313 (1989)
.
68. Baier R. E., and R. C. Dutton, J. Biomed. Mater.
Res. 3,
191 (1969)
.
69. Koshland M. E., F. M. Engleberger, M. J. Erwin,
S. M. Gaddone, J. Biol. Chem. 238, 1343 (1963).
70. Greenwood F. C. in
Principle,of Competitive
Protein Binding Assay, 288, (1971).
:
72. Van der Scheer A., J. Feijen, J. K. Elhorst,
P. G. L. C. Krugers-Dagneaux, J. Colloid Interface
Sci. 66, 136, (1978)
.
73. Crandall R. E., J. Janatova, J. D. Andrade, Prep.
Biochem. 11, 111, (1981)
.
74. Swaigood H. E., in " Developments in Dairy
Chemistry-1"
P. F. Fox, Ed. ), p. 1. Applied
Science Publishers, London,, 1982.
(
75. Wahlgren M. C., M. A. Paulsson and T. Arnebrant,
Colloids and Surfaces A: Physicochemical and
Engineering Aspects, 70, 139 (1993).
76. Cox L.J., T. Kleiss, J.L. Cordier, C. Cordelana,
P. Konkel, C. Pedrazzini, R. Beuner, and A.
Siebenga. 1989. Listeria spp. in food processing,
non-food, and domestic environments. Food Microbiol.
6:49-61.
77. Gellin B.G, and C.V.Broome. 1989. Listeriosis.
JAMA 261:1313-1320.
78. Suttiprasit, P., Phd. Thesis. Oregon State
University. (1992).
80
79. Walstra, P. and R. Jenness. 1984. Dairy Chemistry and
physics. John Wiley and Sons, New York: 117-118.
80. Swaisgood, H.E., in "Development of Dairy Chemistry.1" (P.F. Fox Ed.), Applied Science Publishers,
London, 1982.
81
APPENDIX
APPENDIX
82
Vol. 61. No. 5
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 1995, p. 2013-2015
0099-2240/95/$04.00+0
Copyright © 1995, American Society for Microbiology
Adhesion of Listeria monocytogenes to Silica Surfaces after
Sequential and Competitive Adsorption of Bovine
Serum Albumin and 13-Lactoglobulin
HAMOOD AL- MAKHLAFI,' ADIL NASIR,23 JOSEPH McGUIRE,1.3* AND MARK DAESCHEL'
Departments of Bioresource Engineering,2 Chemical Engineering,3 and Food Science & Technology.'
Oregon State University, Corvallis, Oregon 97331
Received 14 November 1994/Accepted 16 February 1995
Adsorbed bovine serum albumin was resistant to exchange with f3- lactoglobulin, and when albumin was
adsorbed from a mixture, its surface concentration increased with time. The passivating character of adsorbed
albumin and its resistance to desorption were consistent with the level of Listeria monocytogenes adhesion
evoked by albumin-containing protein films.
Adsorption of soluble macromolecules such as proteins to
at the interface, adsorption experiments for which BSA was
cells (15). Adsorbed proteins can inhibit or facilitate attachment of subsequently arriving microorganisms or be displaced
[14C]formaldehyde according to the methods of Jentoft and
Dearborn (10) were conducted. These techniques have been
used to study protein adsorption from mixed solutions as well
as protein exchange reactions at solid-water and air-water interfaces (2, 7-9, 19). Alkylation of lysine residues in this way
minimally affects their basicity (18) and does not alter protein
hydrophobicity insofar as it relates to surface activity (7-9). In
labeled by reductive methylation of lysine residues with
solid surfaces may affect adhesion of microorganisms and other
by other surface-active species (17). Listeria monocytogenes is a
pathogenic bacterium that can exist in a variety of food processing environments (3, 5) and is capable of adhering to food
contact surfaces (6, 16). Problems for food processors arise
because adhered microorganisms may exhibit an increased resistance to sanitizers and other antimicrobial agents (13, 14).
Consequently, the microbial-macromolecular matrix (i.e., the
biofilm) can serve as a continuing source of contamination in
food processing systems. Recently, Al-Makhlafi et al. (1) reported the effect of four milk proteins (a-lactalbumin, (3-lac-
these tests, all surfaces were made hydrophobic by silanization
with 0.100% dichlorodiethylsilane, as opposed to dichlorodimethylsilane (1), dissolved in xylene.
In a typical sequential adsorption experiment, 250 p.I of
14C-BSA solution was placed in one well of a Nunclon multiwell dish (Nunc Inc., Naperville, Ill.). A silanized silica plate of
known surface area was placed onto the surface of the solution.
toglobulin [(3-Lg], (3-casein, and bovine serum albumin [BSA])
on the adhesion of L. monocytogenes to silica surfaces exhib-
iting either high or low hydrophobicity. The protein-specific
Adsorption was allowed to proceed for 1 or 4 h, and the
effects on adhesion were different, with P-Lg and BSA evoking
the most dissimilar responses. The purpose of this study was to
evaluate bacterial adhesion following adsorption of p-Lg and
BSA in sequence and from mixtures to surfaces exhibiting high
and low hydrophobicities. Such a study has practical relevance,
as processes involving contact between surfaces and biological
fluids can involve many different surface-active species.
Protein adsorption and cell adhesion. All materials used in
the experiments were described previously, as were the procedures for silica surface modification, cell culture and adhesion,
surface was rinsed by introducing 250 p.I of buffer and then
withdrawing 250 p.1 of the solution, with this sequence being
repeated 10 times. After 30 min, 250 p.1 of p-Lg (1 mg/mi) was
introduced and then 250 ill of the solution was withdrawn, and
this sequence was repeated 10 times. Contact with P-Lg was
maintained for the same period of time as that allowed for
contact with 14C-BSA. The surface was then rinsed as before
and left in buffer for an additional 30 min. The adsorbed
protein layer was removed from the surface by immersion of
the silica plate in 1 ml of 48% hydrofluoric acid for 5 min (19).
To this was added 15 ml of Gold AB scintillation fluid (Packard Instrument Co.) and 1 ml of Triton X. The sample was well
mixed and then analyzed within 1 day with a Beckman scintillation counter.
Competitive adsorption tests were run similarly, with ad-
rinsing, image analysis, and statistical analysis (1). In the
present experiments, however, surfaces were allowed to contact BSA and (3-Lg either sequentially or simultaneously before
cell culture contact. Individual proteins were always used at the
molar equivalent of 1.000 mg/ml of (3-Lg. In sequential adsorp-
tion, surfaces were allowed to contact the second protein for
8 h if the second contact had been preceded by an 8-h contact
with the first protein and for 1 h if preceded by a 1-h contact.
For the cell adhesion experiments, cultures of L. monocytogenes were diluted to a cell density of 109 CFU/ml after reaching the stationary phase.
Sequential and competitive adsorption with 14C -BSA. In
sorption from a solution of 250 p.1 of 14C-BSA combined with
250 p.1 of (3 -Lg solution occurring for 1 or 4 h. The surface was
order to provide an independent, more direct indication of
adsorption and exchange events undergone by BSA and P-Lg
rinsed, and the adsorbed layer was removed and analyzed, as
described above. The results of the experiments were compared with those of the controls, which proceeded exactly as
described above, with the exception that the P-Lg solutions
were replaced with equal volumes of protein-free phosphate
buffer. Interpretation of adsorption kinetics for BSA and 3 -Lg
with reference to first-order kinetic models shows data recorded for 4 and 8 h to be indistinguishable (11, 12).
Adhesion following sequential protein adsorption. The
* Corresponding author. Mailing address: Department of Bioresource Engineering, Oregon State University, Gilmore Hall 116, Corvallis, OR 97331-3906. Phone: (503) 737-6306. Fax: (503) 737-2082.
numbers of cells adhered to hydrophobic and hydrophilic silica
surfaces following sequential contact with proteins for 8 h are
2013
83
2014
NOTES
APPL. ENVIRON. M1CROBIOL.
TABLE 1. Population of L. monocytogenes adhered to each type of
surface immediately following sequential contact with p-Lg and BSA
for 1 and 8 h
Mean number of adhered cells/cm:
0-LgBSA
Surface
Hydrophobic°
Hydrophilic'
BSA-0-Lg
1h
8h
1h
8h
1.7 x 106
1.4 x 106
1.3 x 107
1.4 x 107
1.5 x 106
1.3 x 106
2.2 x 106
9.0 x 105
° Mean number of adhered cells on protein-free surface = 1.6 x 107 cells per
cm2.
° Mean number of adhered cells on protein-free surface = 2.9 x 10° cells per
cm2.
shown in Table 1, along with those measured on protein-free
surfaces. These data indicate that the film formed by adsorption of 3 -Lg followed by BSA (3 -Lg BSA) encouraged adhe-
sion more than did the film formed by adsorption of BSA
followed by p-Lg (BSAP-Lg) (P < 0.05), although the extent
of adhesion to P-LgBSA remained lower than that measured
on bare hydrophobic surfaces. Moreover, BSA -3 -Lg reduced
the number of adhered cells to values well below that evoked
by the bare hydrophilic surfaces, with BSAp-Lg inhibition of
cell adhesion on hydrophilic surfaces being more extensive
than that observed on hydrophobic surfaces (P < 0.05).
The mechanisms by which these two proteins may affect
adhesion to both hydrophobic and hydrophilic silica surfaces
when each adsorbs from a single-component solution for 1 and
8 h were discussed previously (1). The surfaces treated with
p-LgBSA evoked an adhesion response similar to that measured following 3 -Lg adsorption alone, while those treated
with BSAp-Lg evoked a response closer to that measured
following adsorption of BSA alone, although the extents of cell
adhesion are higher in the present case. Al-Makhlafi et al. (1)
measured about 4 x 105 cells per cm2 following adsorption of
BSA for 8 h to each type of surface. If only on the basis of the
relative rates at which each of these proteins, once adsorbed,
achieves a nonexchangeable (or more tightly bound) state, we
would expect 13-Lg to experience more resistance in exchang-
ing with adsorbed BSA than BSA would experience in ex-
changing with adsorbed P-Lg (1, 12). Table 1 suggests that 8 h
is sufficient for rendering the first protein effectively nonre-
movable, i.e., the kinetics associated with the adoption of a
more tightly bound form are not relevant after 8 h of proteinsurface contact.
Results of sequential adsorption tests performed with hydrophobic silica and "C-BSA are shown in Table 2, which
shows that about 72% of adsorbed "C-BSA resisted exchange
with P-Lg when protein contact was allowed for 4 h.
The fact that greater inhibition of Listeria adhesion is seen
with hydrophilic surfaces in the BSAp-Lg tests is consistent
with the facts that (i) less adhesion would be expected on
exposed hydrophilic silica than on exposed hydrophobic silica
and (ii) while BSA shows roughly equal affinities for the two
surfaces, 3 -Lg shows greater affinity for hydrophobic surfaces
and may exchange more readily with BSA adsorbed to hydrophobic surfaces than to hydrophilic surfaces (12). These two
points are also consistent with the increase in adhesion measured on both surfaces following BSAP-Lg contact relative to
that measured following adsorption of BSA alone (1).
Table 1 also shows results from the 1-h sequential adsorption tests. For both surfaces, cell adhesion to p-LgBSA as well
as to BSAp-Lg was lower than that observed with bare hydrophilic surfaces. These data would suggest that adsorbed
TABLE 2. Average radioactivity detected in the protein layers
removed from the silica surfaces at the end of each experiment
Experiment
Average radioactivity
(dpm /cm2)"
Control, 1-h sequential adsorption'
1-h sequential adsorption
Control, 4-h sequential adsorption
4-h sequential adsorption
2,695 (21)
1.935 (7)
2.217 (11)
1.599 (4)
Control, 1-h competitive adsorption'
1-h competitive adsorption
Control, 4-h competitive adsorption
4-h competitive adsorption
5.861
1.480
5,717
2.295
(14)
(1)
(14)
(27)
° Values shown are the means of duplicate experiments: the percent deviation
of each replicate from the mean is in parentheses.
° Adsorption of "C-BSA. followed by adsorption of I3-Lg.
' Adsorption of a mixture of '4C-BSA and (1-Lg.
p-Lg was readily exchanged with BSA while adsorbed BSA
more effectively resisted exchange with P-Lg, at least to the
extent that the surfaces evoked an adhesion response similar to
that seen for the 8-h tests. Table 2 shows that about 72% of
adsorbed 14C -BSA resisted exchange with P-Lg when protein
contact was allowed for 1 h. The similarity of the numbers of
adhered cells with hydrophobic silica following the 8-h and 1-h
BSA -3 -Lg adsorptions is consistent with the similar BSA contents of these layers.
Earlier, we reported adhesion following a 1-h contact with
BSA to be greater than that measured in the present tests (1).
It was argued that because of its low initial binding constant
relative to that of p-Lg (1, 12), the population of BSA molecules tightly adsorbed in the passivating orientation would be
relatively low. Apparently, the presence of P-Lg in this case
facilitated BSA adsorption in the most passivating orientation.
In the cases in which BSA was adsorbed first, the results of
Table 1 would suggest that after 2 h of contact, the BSA film is
more similar to one formed during 8 h of contact than to one
formed during 1 h of contact for films formed from a singlecomponent solution on either surface (1).
Adhesion following competitive adsorption. Following adsorption from a mixture of 3 -Lg and BSA for 8 h, adhesion to
protein-coated hydrophobic silica was greater than that observed on protein-coated hydrophilic silica (1.0 x 106 versus
3.4 x 105 cells per cm2; P < 0.05), but it remained below that
recorded on bare hydrophilic silica. The adhesion behavior
measured on the hydrophilic surface is quantitatively similar to
that observed in the case of BSA adsorbed alone on each
surface (1).
The reduced number of cells in the case of the hydrophilic
surfaces could be related to the faster conformational rear-
rangement undergone by BSA at that surface relative to that of
P-Lg (4, 12) as well as to the expectation of less adhesion on
regions of exposed hydrophilic surfaces compared with that on
hydrophobic surfaces. The concentrations of these two pro-
teins in solution are equal, and these concentrations are
greater than those corresponding to diffusion-limited adsorption. It is expected that P-Lg would initially bind to the surface
faster than BSA, but it might readily exchange with BSA as its
rate of conversion to a nonexchangeable form is relatively low
(1, 12). Adsorbed BSA, on the other hand, may be much more
difficult to displace.
In competitive adsorption experiments with "C-BSA, the
amount of "C-BSA present on the surface after 4 h was about
40% of what it would have been in the absence of 3 -Lg (Table
2). This value is lower than expected, since the extents of
84
VOL. 61, 1995
adhesion recorded following adsorption from a mixture of
P-Lg and BSA for 8 h are not only similar to those recorded
following BSA-p-Lg sequential contact (Table 1) but also are
close to that measured following 8 h of contact with BSA
alone. This may be due in part to the concentration of "C-BSA
being of a molarity about 17% lower than that of 3 -Lg (which
was used at 1 mg/ml), while the concentrations in the cell
adhesion experiments were equimolar. While the fact of the
lower concentration would have had little effect on adsorption
from single-component solutions, it may have compromised
"C-BSA's ability to compete with P-Lg for surface sites when
they were mixed, especially considering that the diffusion coefficient of BSA is nearly half that estimated for p-Lg (11).
Following 1 h of protein contact with either surface, the
mean numbers of adhered cells recorded for hydrophobic and
hydrophilic silica were 1.6 x 106 and 7.3 x 105 cells per cm',
respectively. These levels are greater than those recorded for
each surface after contact with the protein mixture for 8 h but
are lower than that recorded on the bare hydrophilic surface.
The amount of 14C -BSA present on the surface after 1 h was
about 25% of what it would have been in the absence of P-Lg,
according to Table 2. Again, this is lower than expected. But
both proteins show high affinities for hydrophobic silica. Resolving the adsorption of each protein into two steps (arrival in
an exchangeable form followed by conversion to a nonexchangeable form), Krisdhasima et al. (12) observed that among
the four milk proteins tested, p-Lg was fastest in completing
the first step and slowest in completing the second while the
opposite was true for BSA. Thus, domination of the interface
by P-Lg at a short contact time with the presence of BSA
increasing with time should be expected, and that was observed. The higher number of cells adhered after 1 h as op-
posed to 8 h of contact with these protein solutions could also
be related to both the lower surface coverage and the higher
rinsability of the protein following a 1-h contact, which would
leave bare hydrophobic and hydrophilic surface regions.
The adhesion response following 1 h of contact with the
protein mixture was lower than that recorded following BSA
contact from a single-component solution for 1 h (1). The
reason for this is not obvious, but the finding is analogous to
that associated with the P-Lg-BSA tests of Table 1. These
results suggest that adsorption of BSA with concomitant exchange of adsorbed 3 -Lg may facilitate BSA adsorption in a
favorable, passivating orientation.
This work was supported in part by the Western Center for Dairy
Protein Research and Technology, the USDA National Research Ini-
NOTES
2015
tiative Competitive Grants Program (award number 92-37201-8176),
and the Sana'a University Faculty of Agriculture Project funded by
USAID.
REFERENCES
I. Al-Makhlafi, H., J. McGuire, and M. Daeschel. 1994. The influence of
preadsorbed milk proteins on adhesion of Listeria monocytogenes to hydrophobic and hydrophilic silica surfaces. Appl. Environ. Microbiol. 60:35603565.
2. Arnebrant, T., and T. Nylander. 1986. Sequential and competitive adsorption
of 13-lactoglobulin and K-casein on metal surfaces. J. Colloid Interface Sci.
111:529-533.
3. Cox, L J., T. Kleiss, J. L Cordier, C. Cordelana, P. Konkel, C. Pedrazzini,
R. Benner, and A. Siebenga. 1989. Listeria spp. in food processing, non-food,
and domestic environments. Food Microbiol. 6:49-61.
4. Elgersma, A. V., R. L J. Zsom, J. Lyldema, and W. Norde. 1992. Adsorption
competition between albumin and monoclonal immuno-gamma-globulins on
polystyrene lattices. J. Colloid Interface Sci. 152:410-428.
5. Gellin, B. G., and C. V. Broome. 1989. Listeriosis. JAMA 261:1313-1320.
6. Herald, P. J., and E. A. Zottola. 1988. Attachment of Listeria monocytogenes
to stainless steel surfaces at various temperatures and pH values. J. Food Sci.
53:1549-1552.
7. Hunter, J. R., R. G. Carbonell. and P. K. Kilpatrick. 1991. Lysozyme and
13-casein adsorption at the air/water interface. J. Colloid Interface Sci. 143:
37-53.
8. Hunter. J. R.. P. K. Kilpatrick. and R. G. Carbonell. 1990. Lysozyme adsorption at the air/water interface. J. Colloid Interface Sci. 137:462-482.
9. Hunter, J. R., P. K. Kilpatrick, and R. G. Carbonell. 1991.13-Casein adsorption at the air/water interface. J. Colloid Interface Sci. 142:429-447.
10. Jentoft, N., and D. G. Dearborn. 1979. Protein labeling by reductive alkylation. J. Biol. Chem. 254:4359-4365.
11. Krisdhasima, V., J. McGuire, and R. Sproull. 1992. Surface hydrophobic
influences on (3-lactoglobulin adsorption kinetics. J. Colloid Interface Sci.
154:337-350.
12. Krisdhasima, V., P. Vinaraphong, and J. McGuire. 1993. Adsorption kinetics and elutability of a-lactalbumin. (3-casein, I3-lactoglobulin and bovine
serum albumin at hydrophobic and hydrophilic interfaces. J. Colloid Interface Sci. 161:325-334.
13. Krysinski, E. P., L J. Brown, and T. J. Marchisello. 1992. Effect of cleaners
and sanitizers on Listeria monocytogenes attached to product contact surfaces. J. Food Prot. 55:246-251.
14. Le Chevallier, M. W., C. C. Cawthon. and R. G. Lee. 1988. Inactivation of
biofilm bacteria. Appl. Environ. Microbiol. 54:2492-2499.
15. Loeb, G. I. 1985. The properties of nonbiological surfaces and their charac-
terization, p. 111-129. In D. C. Savage and M. Fletcher (ed.). Bacterial
adhesion: mechanisms and physiological significance. Plenum Press. New
York.
16. Mafu, A. A.. D. Roy, J. Goulet, and P. Magny. 1990. Attachment of Listeria
tnonocytogenes to stainless steel. glass. polypropylene. and rubber surfaces
after short contact times. J. Food Prot. 53:742-746.
17. Marshall, K. C. 1985. Mechanisms of bacterial adhesion at solid-water interfaces, p. 133-161. In D. C. Savage and M. Fletcher (ed.). Bacterial adhesion: mechanisms and physiological significance. Plenum Press. New York.
18. Means. G. E., and R. E. Feeney. 1971. Chemical modification of proteins.
Holden-Day, San Francisco.
19. Wahlgren. M. C.. T. Arnebrant. and M. A. Paulsson. 1993. The adsorption
from solutions of 13-lactoglobulin mixed with lactoterrin or lysozyme onto
silica and methylated silica surfaces. J. Colloid Interface Sci. 158:46-53.