SPATIAL VARIATION OF CARDIAC RESTITUTION AND THE ONSET OF ALTERNANS Hana Dobrovolny
advertisement

SPATIAL VARIATION OF CARDIAC RESTITUTION
AND THE ONSET OF ALTERNANS
by
Hana Dobrovolny
Department of Physics
Duke University
Date:
Approved:
Daniel J. Gauthier, Supervisor
Joshua Socolar
Henry Greenside
Ronen Plesser
Patrick Wolf
Dissertation submitted in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
in the Department of Physics
in the Graduate School of
Duke University
2008
ABSTRACT
SPATIAL VARIATION OF CARDIAC RESTITUTION
AND THE ONSET OF ALTERNANS
by
Hana Dobrovolny
Department of Physics
Duke University
Date:
Approved:
Daniel J. Gauthier, Supervisor
Joshua Socolar
Henry Greenside
Ronen Plesser
Patrick Wolf
An abstract of a dissertation submitted in partial fulfillment of the
requirements for the degree of Doctor of Philosophy
in the Department of Physics
in the Graduate School of
Duke University
2008
c 2008 by Hana Dobrovolny
Copyright All rights reserved
Abstract
Instability in the propagation of nonlinear electro-chemical waves in the heart is
responsible for life-threatening disease. This thesis describes an investigation of the
effects of boundaries on cardiac wave propagation that arises from a site where an
electrical stimulus is applied or from boundaries beyond which current does not flow.
It is generally believed that the spatial scale for boundary effects is approximately
equal to the passive length constant λ of the tissue, the distance over which a a voltage
pulse decays when it is below the threshold for wave generation. From the results of
in vitro experiments with bullfrog cardiac tissue and through numerical simulations, I
find that boundaries affect wave propagation over a much larger spatial scale and that
the spatial variation in some cardiac restitution properties is correlated statistically
with the onset of alternans, a possible precursor to fibrillation in the human heart.
An optical imaging system using novel illumination based on LEDs is used to
determine the spatial dependence of action potential duration (APD) and the slope
of the dynamic restitution curve SDRC , which describes the relationship between
steady-state APD and diastolic interval. For tissue with nearly identical cells, I find
that APD is longest near the stimulus and shortest near the physical boundary with
significant changes (∼100 ms) over a distance of ∼10λ. SDRC decreases with distance
from the stimulus at a constant rate (∼0.1-1.5 /mm) over the surface of the tissue.
Simulations using a two-variable cardiac model confirm that spatial patterns of APD
and SDRC can be induced by boundaries.
Additional measurements with the simultaneous impalement of two microelectrodes are used to determine the spatial differences of other restitution properties.
iv
These studies indicate that APD and SDRC , as well as the slopes of the constant-BCL
and S1S2 restitution curves, vary in space and that the spatial differences and onset
of alternans at rapid pacing are correlated. If similar correlations are evident in humans, such measurements may identify patients who are susceptible to arrhythmias
and allow for early treatment.
v
Acknowledgements
The work described in this document would not have been possible without the assistance of many people. I would like to thank the many people who helped make these
experiments possible. Soma Kalb taught me basic biological experimental techniques
and provided invaluable advice and assistance for my own experiments. Ninita Brown
spent long nights in the lab performing tedious tasks and keeping the atmosphere
cheery. Carolyn Berger helped setup and run experiments. Salim Idriss provided
advice on physiology and clinical practices. Wanda Krassowska and Daniel Gauthier
provided guidance and assistance in improving experimental design and implementation.
In addition, many friends and family provided moral support that got me through
rough patches along the way. My parents were always available when I needed to
vent. Suzie Zeunges and Jamye Gaster on many occasions dragged me out of the lab
and made sure I took the time to relax and give my brain a rest. My officemates,
Heejeong Jeong, Andy Dawes, Michael Stenner and John Blakely kept the work
atmosphere pleasant and provided some of the most interesting conversations I’ve
ever had. Finally, Ashish Talwar shared in all my successes and failures and never
complained about having to put up with the craziness.
vi
Contents
Abstract
iv
Acknowledgements
vi
List of Figures
xv
List of Tables
xxxiii
1 Introduction
1.1
Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1
1.1.1
1
1.1.2
1.2
1.3
1
Historical Background . . . . . . . . . . . . . . . . . . . . . .
How Cardiac Cells Work
. . . . . . . . . . . . . . . . . . . .
3
Nonlinear Dynamics of Cardiac Tissue . . . . . . . . . . . . . . . . .
6
1.2.1
Single Cell Dynamics . . . . . . . . . . . . . . . . . . . . . . .
7
1.2.2
Spatial Dynamics . . . . . . . . . . . . . . . . . . . . . . . . .
9
Thesis Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
13
2 Background
16
2.1
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
16
2.2
Excitable Media . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
16
2.2.1
Introduction to Excitable Media . . . . . . . . . . . . . . . .
17
2.2.2
Cardiac Tissue as an Excitable Medium . . . . . . . . . . . .
22
Effect of Boundaries . . . . . . . . . . . . . . . . . . . . . . . . . . .
28
2.3
2.3.1
Passive Length Constant . . . . . . . . . . . . . . . . . . . . .
vii
28
2.3.2
Changes in Membrane Resistance . . . . . . . . . . . . . . . .
29
2.3.3
Blocked Current Flow . . . . . . . . . . . . . . . . . . . . . .
31
2.4
Stability of Plane Waves . . . . . . . . . . . . . . . . . . . . . . . . .
34
2.5
Experiment Overview . . . . . . . . . . . . . . . . . . . . . . . . . . .
35
2.5.1
Choice of Experimental Substrate . . . . . . . . . . . . . . .
35
2.5.2
Size of the Experimental Substrate . . . . . . . . . . . . . . .
38
2.5.3
Measurement Techniques . . . . . . . . . . . . . . . . . . . . .
39
3 Spatial Variation of Action Potential Duration
3.1
3.2
3.3
43
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
43
3.1.1
Background . . . . . . . . . . . . . . . . . . . . . . . . . . . .
43
3.1.2
Experiment Overview . . . . . . . . . . . . . . . . . . . . . . .
44
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
44
3.2.1
Tissue Preparation . . . . . . . . . . . . . . . . . . . . . . . .
44
3.2.2
Optical Recordings . . . . . . . . . . . . . . . . . . . . . . . .
45
3.2.3
Pacing Protocol . . . . . . . . . . . . . . . . . . . . . . . . . .
46
3.2.4
Data Analysis . . . . . . . . . . . . . . . . . . . . . . . . . .
48
3.2.5
Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . . .
53
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
54
3.3.1
Spatial Heterogeneity . . . . . . . . . . . . . . . . . . . . . . .
54
3.3.2
APD Maps . . . . . . . . . . . . . . . . . . . . . . . . . . . .
56
3.3.3
Width of the Boundary Layer . . . . . . . . . . . . . . . . . .
62
viii
3.3.4
3.4
3.5
APD Gradients . . . . . . . . . . . . . . . . . . . . . . . . . .
62
Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
69
3.4.1
Comparison of Experiment and Simulations . . . . . . . . . .
69
3.4.2
Spatial Heterogeneity of APD . . . . . . . . . . . . . . . . . .
70
3.4.3
Width of the Boundary Layer . . . . . . . . . . . . . . . . . .
71
3.4.4
Stability of Complex Rhythms . . . . . . . . . . . . . . . . . .
73
3.4.5
Study Limitations and Future Work . . . . . . . . . . . . . . .
73
Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
75
4 Spatial Variation of Dynamic Restitution
4.1
76
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
76
4.1.1
Background . . . . . . . . . . . . . . . . . . . . . . . . . . . .
76
4.1.2
Spatial DRC Slope Gradients . . . . . . . . . . . . . . . . . .
78
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
79
4.2.1
Pacing Protocol . . . . . . . . . . . . . . . . . . . . . . . . . .
79
4.2.2
DRC Slopes . . . . . . . . . . . . . . . . . . . . . . . . . . . .
80
4.2.3
Spatial Gradients . . . . . . . . . . . . . . . . . . . . . . . . .
81
4.3
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
82
4.4
Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
84
4.4.1
Spatial Variation of SDRC . . . . . . . . . . . . . . . . . . . .
84
4.4.2
∆SDRC and the Onset of Alternans . . . . . . . . . . . . . . .
87
Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
88
4.2
4.5
ix
5
Spatial Heterogeneity in a Two-Variable Cardiac Model
5.1
5.2
5.3
5.4
5.5
6
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
89
5.1.1
Two-Variable Model . . . . . . . . . . . . . . . . . . . . . . .
89
Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
91
5.2.1
Cardiac Model . . . . . . . . . . . . . . . . . . . . . . . . . .
91
5.2.2
Pacing Protocol . . . . . . . . . . . . . . . . . . . . . . . . . .
93
5.2.3
Data Analysis . . . . . . . . . . . . . . . . . . . . . . . . . .
93
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
96
5.3.1
Spatial Heterogeneity . . . . . . . . . . . . . . . . . . . . . . .
96
5.3.2
Predicting the Propensity to Exhibit Alternans . . . . . . . .
99
Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
5.4.1
Spatial Heterogeneity of Restitution Properties . . . . . . . . 105
5.4.2
Predicting Tissue’s Propensity to Exhibit Alternans . . . . . . 106
5.4.3
Study Limitations . . . . . . . . . . . . . . . . . . . . . . . . . 108
Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 109
Spatial Heterogeneity and the Onset of Alternans
6.1
89
110
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 110
6.1.1
Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . 110
6.1.2
Restitution Curves . . . . . . . . . . . . . . . . . . . . . . . . 110
6.1.3
Maps and Restitution Curves . . . . . . . . . . . . . . . . . . 112
6.1.4
Experiment Overview . . . . . . . . . . . . . . . . . . . . . . . 115
x
6.2
6.3
6.4
6.5
Methods
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
6.2.1
Tissue Preparation . . . . . . . . . . . . . . . . . . . . . . . . 116
6.2.2
Pacing Protocol
6.2.3
Electrical Recordings . . . . . . . . . . . . . . . . . . . . . . . 118
6.2.4
Restitution Portrait
6.2.5
Spatial Differences . . . . . . . . . . . . . . . . . . . . . . . . 120
6.2.6
Slope Criteria for the onset of Alternans . . . . . . . . . . . . 122
. . . . . . . . . . . . . . . . . . . . . . . . . 116
. . . . . . . . . . . . . . . . . . . . . . . 118
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
6.3.1
Restitution Portraits . . . . . . . . . . . . . . . . . . . . . . . 122
6.3.2
Steady State APD . . . . . . . . . . . . . . . . . . . . . . . . 126
6.3.3
S1S2 Restitution Curve . . . . . . . . . . . . . . . . . . . . . . 127
6.3.4
Constant-BCL Restitution Curve . . . . . . . . . . . . . . . . 128
6.3.5
Dynamic Restitution Curve . . . . . . . . . . . . . . . . . . . 128
6.3.6
Slope Criteria for the Onset of Alternans . . . . . . . . . . . . 129
Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
6.4.1
Spatial Differences in Restitution Properties . . . . . . . . . . 132
6.4.2
Predicting the Tissue’s Propensity to Alternans . . . . . . . . 133
6.4.3
Study Limitations . . . . . . . . . . . . . . . . . . . . . . . . . 135
6.4.4
Clinical Implications . . . . . . . . . . . . . . . . . . . . . . . 137
Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
7 Conclusions and Future Work
139
xi
7.1
7.2
Research Findings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
7.1.1
Spatial Variation . . . . . . . . . . . . . . . . . . . . . . . . . 139
7.1.2
Correlation to Alternans . . . . . . . . . . . . . . . . . . . . . 142
Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 144
7.2.1
Spatial Variation . . . . . . . . . . . . . . . . . . . . . . . . . 144
7.2.2
Onset of Alternans . . . . . . . . . . . . . . . . . . . . . . . . 146
7.3
Future Work . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 147
7.4
Final Thought . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149
A Ultra-high Power Light Emitting Diodes
150
A.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 150
A.2 Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153
A.2.1 LED characteristics . . . . . . . . . . . . . . . . . . . . . . . . 154
A.2.2 In vitro Experiments . . . . . . . . . . . . . . . . . . . . . . . 156
A.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
A.3.1 Intensity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
A.3.2 Thermal Effects . . . . . . . . . . . . . . . . . . . . . . . . . . 159
A.3.3 Noise . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
A.3.4 Signal Amplitude . . . . . . . . . . . . . . . . . . . . . . . . . 164
A.4 Discussion and Conclusion . . . . . . . . . . . . . . . . . . . . . . . . 167
B
Determination of Action Potential Duration
170
B.1 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
xii
B.2 Techniques for Finding APD . . . . . . . . . . . . . . . . . . . . . . . 171
B.2.1 Threshold Method . . . . . . . . . . . . . . . . . . . . . . . . 171
B.2.2 Slope Method . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
B.2.3 Phase Method . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
B.3 Effect of Noise . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177
B.3.1 Threshold Method . . . . . . . . . . . . . . . . . . . . . . . . 178
B.3.2 Slope Method . . . . . . . . . . . . . . . . . . . . . . . . . . . 181
B.3.3 Phase Method . . . . . . . . . . . . . . . . . . . . . . . . . . . 183
B.3.4 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
B.4 Filters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
B.4.1 Mean Filter . . . . . . . . . . . . . . . . . . . . . . . . . . . . 187
B.4.2 Median Filter . . . . . . . . . . . . . . . . . . . . . . . . . . . 190
B.4.3 Frequency Filter . . . . . . . . . . . . . . . . . . . . . . . . . 193
B.5 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
B.5.1 Application to an Optical Signal . . . . . . . . . . . . . . . . . 199
B.6 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
C
Considerations in Tissue Preparation
202
C.1 Experimental Limitations . . . . . . . . . . . . . . . . . . . . . . . . 202
C.1.1 Tissue Viability . . . . . . . . . . . . . . . . . . . . . . . . . . 202
C.1.2 Tissue Size
. . . . . . . . . . . . . . . . . . . . . . . . . . . . 205
C.1.3 Repeatability . . . . . . . . . . . . . . . . . . . . . . . . . . . 206
xiii
C.1.4 Tissue Damage . . . . . . . . . . . . . . . . . . . . . . . . . . 206
C.2 Suggested Tissue Preparations . . . . . . . . . . . . . . . . . . . . . . 207
C.2.1 Whole Heart Preparation . . . . . . . . . . . . . . . . . . . . . 207
C.2.2 Whole Ventricle . . . . . . . . . . . . . . . . . . . . . . . . . . 209
C.2.3 Anterior Ventricular Surface . . . . . . . . . . . . . . . . . . . 210
C.2.4 Ventricular Strip . . . . . . . . . . . . . . . . . . . . . . . . . 212
C.3 Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212
D MatLab Codes
213
D.1 Simulation Code . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
D.2 Data Analysis Code . . . . . . . . . . . . . . . . . . . . . . . . . . . . 218
E
Passive Length Constant of the Two-Variable Model
231
F
Core Conductor Model
234
G Glossary
237
H Guide to Symbols and Acronyms
241
Bibliography
246
Biography
265
xiv
List of Figures
1.1
1.2
1.3
Cardiac action potentials recorded from bullfrog ventricle. Recordings
are made using a microelectrode in a paced in vitro preparation. The
time between the beginning and end of one action potential is the
action potential duration (APD). The time between the end of one
action potential and the start of the next is the diastolic interval (DI).
The time between stimuli is the pacing period or basic cycle length
(BCL). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4
Cardiac action potential alternans. At rapid pacing, cardiac cells can
exhibit a long-short alternation in APD known as a 2:2 response or
alternans. This example was recorded from bullfrog ventricular myocardium. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
6
Cobweb diagram. The restitution curve (RC, solid line) represents
the relationship between APD and previous DI. The dotted line is the
BCL = AP Dn+1 + DIn . Starting at a particular DI, find the resulting
APD by drawing a line up to the RC. To determine the following DI,
draw a horizontal line across to the dotted line. This new DI then
leads to another APD and so on. Depending on the exact details of
the RC, several different results are possible, two of which are shown
here. (A) If the slope of the RC is less than one, the system will
eventually settle down to a single APD, indicated by the star (the 1:1
response). (B) When the slope of the RC is greater than 1, the system
oscillates between two APDs, indicated by the stars (the 2:2 response).
8
xv
1.4
2.1
Spatial variation of APD in a cardiac cable. A two-variable model (Eq.
2.4) is implemented for a 5-cm-long cable. The tissue is paced on the
left end at a BCL of 500 ms. The cable has two distinct boundaries,
the stimulus site and the physical boundary at the far end of the
cable. The APD is constant in the center of the cable, away from
the boundaries, but increases near the stimulus and decreases near the
opposite end. The passive length constant for this model is 1 mm, yet
the APD changes from ∼382 ms to ∼377 ms over a distance of ∼1
cm near the stimulus. Spatial variation of APD also occurs near the
insulated end of the cable over a similar distance. . . . . . . . . . . .
11
Excitable wave at a boundary. In one dimension, as the wave approaches the end of the medium, it cannot move forward and it cannot
move backward because of the refractory tissue behind the wave. . . .
19
2.2
Collision of two waves in excitable media. In the top panel, two waves
approach each other. There is a region of refractoriness behind each
wave. In the middle panel, each wave begins to run into the refractory region of the other wave, preventing them from propagating any
further. In the bottom panel, the two waves are completely annihilated. 21
2.3
Response of the 2-variable cardiac model to a subthreshold stimulus.
After a subthreshold current pulse is injected into the cell, (A) the
transmembrane voltage simply decays back to the rest state, B) the
gate remains open, C) the inward current decays back to the rest
state, and D) the outward current also decays back to the rest state.
Parameter values for this example are Vc = 0.13, τin = 0.2 ms, τout =
10 ms, τopen = 130 ms, τclose = 150 ms, and Iext /Cm = 0.05 /ms. . . .
xvi
24
2.4
2.5
2.6
2.7
2.8
Response of the 2-variable cardiac model to a suprathreshold stimulus. After a small suprathreshold current pulse is injected into the
cell, (A) the transmembrane voltage rapidly increases and then slowly
decreases, B) the gate variable decreases (the gate closes) and recovers once the transmembrane voltage returns below the threshold, C)
the inward current rapidly increases causing the upstroke of the action
potential before diminishing as the gate closes, and D) the outward
current is initially much smaller than the inward current, but eventually becomes larger causing the decrease in the voltage. Parameter
values for this example are Vc = 0.13, τin = 0.2 ms, τout = 10 ms,
τopen = 130 ms, τclose = 150 ms, and Iext /Cm = 0.2 /ms. . . . . . . . .
26
Subthreshold response in a cable. A subthreshold injection of current
into the cable causes a small increase in transmembrane voltage that
decays in space and time. Parameter values for this example are Vc =
0.13, τin = 0.2 ms, τout = 10 ms, τopen = 130 ms, τclose = 150 ms, and
D = 0.001 cm2 /ms. . . . . . . . . . . . . . . . . . . . . . . . . . . . .
27
Waves in excitable media. The cardiac model described by Eq. 2.4 is
implemented in a 5 cm cable. An external current is applied at the
left side of the cable. The action potential is initiated in the cell at the
left end and propagates through voltage diffusion to neighboring cells.
28
Definitions of length scales of APD variation. The figure shows the
insulated boundary of Fig. 1.4. The total spatial variation of APD,
that is the distance over which the APD varies from AP D0 to AP Dmid
is ∼0.7 cm or ∼ 7λ. The effective length constant, as defined by Eq.
2.6 is 1.57λ. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
31
Charge buildup at an insulated boundary. Since the current cannot
flow past the boundary, charge builds up in the cells near the boundary.
This causes the cells to repolarize more rapidly than cells in the middle
of the cable. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
32
xvii
2.9
Phases of the action potential. The action potential begins with the
depolarization phase (also called the upstroke), characterized by a
rapid increase in transmembrane voltage. This is followed by a plateau
where the voltage remains nearly constant. The voltage returns to the
rest state during the repolarization phase (also called the downstroke).
Start and end times of each of the phases of the action potential are
typically defined as a percentage of the amplitude (See App. B). Data
is from a microelectrode recording of an action potential in bullfrog
ventricular myocardium. . . . . . . . . . . . . . . . . . . . . . . . . .
33
2.10 Frog heart histology. (A) A longitudinal cross-section of a bullfrog
ventricle stained with hematoxylin and eosin. The ventricle consists
of clumps of tissue interspersed with empty space. (B) A magnified
view of the same piece of tissue. The clumps consist of cardiac cells
oriented in random directions. . . . . . . . . . . . . . . . . . . . . . .
37
2.11 Wave propagation in frog cardiac tissue. Contour lines denote the
wave front initiated from the electrode at 0.5 ms intervals. The wave
initially propagates slightly faster along the vertical direction (slightly
elliptical contour near the electrode), but then begins to propagate
slightly faster along the horizontal direction (compare width of the
contour indicated by the double arrows). This suggests that there is
no fixed anisotropy in frog cardiac tissue. . . . . . . . . . . . . . . . .
38
2.12 Microelectrode signals. (A) shows the signal form a properly impaled
microelectrode. (B) shows the signal from a microelectrode that is not
properly impaled for the first 3000 ms and exhibits motion artifact
once impalement is achieved. Both signals are recorded from a small
piece of bullfrog ventricle that is paced at BCL = 1000 ms. The signal
is passed through an amplifier with 10x gain. . . . . . . . . . . . . . .
40
2.13 Optical signal. The optical signal is recorded from a small piece of
bullfrog ventricle that was stained with di-4-ANEPPS, a potentiometric dye, and is paced at BCL = 1000 ms. Intensity is negative since the
signal has been inverted to assist in comparison to the microelectrode
signal. Raw optical are the inverse of traditional electrode recordings
since fluorescence decreases with increasing voltage. . . . . . . . . . .
42
xviii
3.1
Tissue Chamber. The tissue is pinned down in a custom-made tissue
chamber. Oxygenated solution is pumped into the chamber (lower
hole in back) and taken out through a hole on the other side of the
chamber to be re-oxygenated and recirculated. . . . . . . . . . . . . .
46
Experimental setup. Light from two cyan LEDs is focused onto a small
piece of cardiac tissue that has been stained with di-4-ANEPPS. The
fluoresced light emitted by the tissue is filtered through a high-pass
filter and collected by a high-speed CCD camera. . . . . . . . . . . .
47
Electrode Placement. Three unipolar silver electrodes are placed along
the three edges of the tissue. . . . . . . . . . . . . . . . . . . . . . . .
48
Calculation of boundary width. (A) Lines used to calculate the width
of the boundary layer from electrode 1 and (B) lines used to calculate
the width of the boundary layer from electrode 3. . . . . . . . . . . .
51
3.5
Examples of complex rhythm. Both examples are at BCL=300 ms. .
52
3.6
Range of BCLt . The transition BCL ranged from 200 ms to 400 ms.
See Table 3.3 for more details. . . . . . . . . . . . . . . . . . . . . . .
53
Frozen-in heterogeneity. APD varies from ≈500-650 ms (blue=500 ms
and red=650 ms) over the surface of the tissue. Note that even thought
the pacing location changes in each of the three panels, this does not
cause large changes in the spatial APD pattern in this experiment;
the longest APDs remain near the upper left side of the tissue. Data
shown is from experiment #1 of Table 3.1. . . . . . . . . . . . . . . .
55
Experimental spatial patterns of activation and deactivation. Maps of
steady state activation and deactivation when pacing at BCL = 1000
ms from (A,B) the upper right electrode, (C,D) the left electrode, and
(E,F) the lower right electrode. Contour lines are 5 ms apart. Data
taken from experiment #8 of Table 3.1. . . . . . . . . . . . . . . . . .
58
3.2
3.3
3.4
3.7
3.8
xix
3.9
Experimental spatial patterns of APD. Maps of steady-state APD produced when pacing at BCL = 1000 ms from (A) the upper right electrode, (C) the left electrode, and (E) the lower right electrode. Figures
B, D, and F show the APD along the lines indicated in A, C, and E,
respectively. When pacing from electrodes 1 and 3, the longest APDs
are near the stimulus electrode. When pacing from electrode 2, the
longest APDs are near electrode 3 in this experiment. Data shown is
from experiment #8 of Table 3.1. . . . . . . . . . . . . . . . . . . . .
59
3.10 Effect of BCL on spatial APD distribution. APD maps produced when
pacing at BCL=1000, 800, 600, 400 ms. To produce these images,
experimental data has been fit to a cubic function. Data shown is
from experiment #12 of Table 3.1. . . . . . . . . . . . . . . . . . . .
60
3.11 Spatial patterns of activation and deactivation in a two-variable model.
Maps of steady-state activation and deactivation when pacing at BCL
= 1000 ms from (A,B) the upper right electrode, (C,D) the left electrode, and (E,F) the lower right electrode. Contour lines are 2.5 ms
apart. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
61
3.12 Simulated spatial patterns of APD. Maps of steady state APD produced when pacing at BCL = 1000 ms from (A) the upper right electrode, (B) the left electrode, and (C) the lower right electrode. Figures
B, D, and F show the APD along the lines indicated in A, C, and E,
respectively. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
63
3.13 Sample experimental data used to calculate λef f . In the example, the
boundary width is calculated for the insulated end of the cable. I find
that λef f ∼ 2λ and the total distance over which APD varies is ∼ 8λ.
3.14 Experimental spatial patterns of APD gradient. Maps of steady state
APD gradient produced when pacing at BCL = 1000 ms from (A) the
upper right electrode, (B) the left electrode, and (C) the lower right
electrode. Figures B, D, and F show the APD gradient along the lines
indicated in A, C, and E, respectively. . . . . . . . . . . . . . . . . .
xx
64
66
3.15 Simulated spatial patterns of APD gradient. Maps of steady state
APD gradient produced when pacing at BCL = 1000 ms from (A) the
upper right electrode, (B) the left electrode, and (C) the lower right
electrode. Figures B, D, and F show the APD gradient along the lines
indicated in A, C, and E, respectively. . . . . . . . . . . . . . . . . .
67
3.16 Mean spatial APD gradient. The mean spatial APD gradient averaged
over all animals is shown as a function of BCL. The APD gradient is
independent of pacing electrode and shows a slight decrease as BCL
increases. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
68
3.17 ∆AP D and complex rhythms. (A) ∆AP D for trials that exhibit complex rhythms and those that go directly to 2:1. (B) P values below
0.05 (dashed line) indicate ∆AP D is significantly different in the two
groups. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
68
4.1
4.2
4.3
4.4
Dynamic Restitution Curve. The DRC is determined by the steadystate DI and APD at different BCLs. The tissue is paced for 2-3 times
the time constant of accommodation, (τ ), and the final (DI, AP D)
pair is one point on the DRC. The process is repeated at different
BCLs to determine the entire restitution curve. . . . . . . . . . . . .
78
Range of BCLt . The transition BCL ranged from 200 ms to 400 ms.
See Table 4.1 for more details. . . . . . . . . . . . . . . . . . . . . . .
82
Spatial variation of SDRC in a piece of bullfrog ventricle. The lower row
shows the results of pacing from an electrode placed along the upper
right side of the tissue. The lower row shows the results of pacing from
an electrode placed along the upper right side of the tissue. Results
are shown for BCLs of 900, 700 and 500 ms. Images are produced by
fitting experimental data to a cubic surface. . . . . . . . . . . . . . .
83
Spatial gradient of SDRC in a piece of bullfrog ventricle. The lower row
shows the results of pacing from an electrode placed along the upper
right side of the tissue. The lower row shows the results of pacing from
an electrode placed along the upper right side of the tissue. Results
are shown for BCLs of 900, 700 and 500 ms. ∆SDRC for each map is
given below the image. . . . . . . . . . . . . . . . . . . . . . . . . . .
85
xxi
4.5
5.1
5.2
5.3
5.4
Mean spatial gradients of SDRC as a function of BCLN . (A) ALT
and NoALT trials show differences in mean spatial gradient of SDRC .
ALT trials show a marked increase in the mean spatial gradient as
the transition to alternans is approached. (B) The t-test shows that
differences in ∆SDRC are significant at slow (BCLN > 400 ms) and
rapid (BCLN < 300 ms) pacing. . . . . . . . . . . . . . . . . . . . . .
86
Restitution and accommodation of the two-variable model. (A) The
restitution portrait for the two-variable model. The SRC and BRC
have not split from the DRC; there is a single restitution curve (B)
The two-variable model exhibits no accommodation. A single cell is
paced at a BCL of 1000 ms from initial conditions of V=0 and h=1.
The APD remains constant from the second beat on. After a change
in BCL from 1000 ms to 900 ms, the APD again remains constant
from the second beat on. . . . . . . . . . . . . . . . . . . . . . . . . .
90
Bifurcation diagrams of the two-variable model. (A) The bifurcation
diagram for the two variable model when the parameters in the second
column of Table 5.1 are used. These parameters result in 2:1 behavior
at BCL∼450 ms. (B) The bifurcation diagram for the two variable
model when the parameters in the third column of Table 5.1 are used.
These parameters result in 1:1 behavior changing to 2:1 behavior at
BCL∼200 ms. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
92
Spatial gradients in a two variable model. Three spatial gradients are
measured: the gradient between points A and B, the gradient between
points C and D, and the gradient between points E and F. . . . . . .
95
Spatial variation of steady state APD in the two-variable model. The
tissue is paced from the center of the left side; the resulting APD
maps at BCLs of (A) 1000 ms (B) 800 ms (C) 600 ms are shown. The
APD is longest near the stimulus and decreases as the wave propagates
away from the stimulus. Cross-sections taken along the horizontal line
indicated in (A) are shown in panels D,E, and F. The cross-sections
show that the APD drops sharply near the stimulus and near the far
end of the tissue, but does not change much in the middle. Parameters
used for this simulation are listed in the ALT column of Table 5.1. . .
97
xxii
5.5
Spatial gradient of steady state APD in the two-variable model. The
tissue is paced from the center of the left side; the resulting gradients
at BCLs of (A) 1000 ms (B) 800 ms (C) 600 ms are shown. The APD
gradient is largest near the boundaries and near zero in the center
of the tissue. Cross-sections taken along the horizontal line indicated
in (A) are shown in panels D,E, and F. The cross-sections show that
the APD gradient drops sharply near the stimulus and increases again
near the far end of the tissue. Parameters used for this simulation are
listed in the ALT column of Table 5.1. . . . . . . . . . . . . . . . . .
98
5.6
Spatial variation of slope of DRC in the two-variable model. The tissue
is paced from the center of the left side; the resulting DRC slope maps
at BCLs of (A) 1000 ms (B) 800 ms (C) 600 ms are shown. At slow
pacing, the slope of the DRC shows little spatial variation, but as
the BCL decreases, a gradient begins to appear. Cross-sections taken
along the horizontal line indicated in (A) are shown in panels D,E, and
F. The cross-sections show that the DRC slope decreases at a fairly
constant rate over the length of the tissue. Parameters used for this
simulation are listed in the first column of Table 5.1. . . . . . . . . . 100
5.7
Mean and maximum APD gradient. (A) The mean APD gradient is
slightly larger in ALT cases than in noALT cases. The mean APD
gradient can differentiate between ALT and noALT cases at almost all
BCLs. (B) There is no clear trend in ∇AP Dmax for either the ALT
or noALT case. At some BCLs, ALT and noALT cases have the same
∇AP Dmax , at others, ∇AP Dmax differs for ALT and noALT cases. . 101
5.8
APD spatial gradients. (A) ∇AP DAB is slightly larger in ALT cases
than in noALT cases, though the measurements agree within error.
(B) ∇AP DCD is essentially the same for both ALT and noALT cases.
(C) ∇AP DEF is essentially the same for both ALT and noALT cases. 102
xxiii
5.9
Mean and maximum SDRC gradient. (A) Both ALT and noALT cases
show a rapid increase in mean gradient of SDRC as BCL nears the transition point. At long BCLs, noALT cases exhibit an initial decrease in
mean gradient of SDRC while ALT cases exhibit a small increase. (B)
The maximum SDRC gradient is larger in ALT cases than in noALT
cases, although the measurements agree within error. Thus, the maximum SDRC gradient cannot differentiate between ALT and noALT
cases. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
AB
5.10 DRC spatial gradients. (A) ∇SDRC
is slightly larger in ALT cases than
in noALT cases at short BCLs with the difference becoming larger than
the measurement error about 100 ms from the transition point. (B)
CD
∇SDRC
is slightly larger in ALT cases than in noALT cases at short
EF
BCLs, though the measurements agree within error. (C) ∇SDRC
is
slightly larger in ALT cases than in noALT cases at long BCLs and
reverses at short BCLs, though the measurements agree within error
at all BCLs. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
6.1
S1S2 Restitution Curve. The SRC is determined by the responses to
perturbations in BCL. The tissue is paced at a constant BCL (the S1
rate) until steady-state is reached. A single pace at a different BCL
(the S2 rate) is applied and the resulting APD and previous DI are
used to create the SRC. Upon returning to the S1 rate, the tissue does
not need to be paced at a constant BCL for very long since it typically
recovers from a single perturbation very quickly. Further S2 paces at
different BCLs are applied to complete the entire RC. . . . . . . . . . 112
6.2
Restitution portraits of cardiac mapping models. (A) A one-variable
cardiac mapping model produces a single RC regardless of the pacing
protocol. (B) A two-variable model has different curves for the DRC
(steady-state responses), SRC (perturbations) and BRC (transients).
(C) A three-variable model produces a fourth RC, with the transient
response becoming split into two curves: transients associated with a
permanent change in BCL (BRC-D) and transients associated with a
perturbation (BRC-S). . . . . . . . . . . . . . . . . . . . . . . . . . . 113
xxiv
6.3
Restitution portrait from a frog ventricular myocyte. The RP from
frog cardiac cells shows four distinct restitution curves: the DRC,
SRC, BRC-D, and BRC-S. Steady state points are indicated by ‘*’
and form part of the DRC. Initial transients are indicated by ‘.’ and
form the BRC-D. Long and short perturbations are indicated by ‘+’
and ‘x’, respectively and along with the steady-state points form the
SRC. Finally, the transients after a perturbation are indicated by ‘o’
and along with the steady-state points form the BRC-S. This is qualitatively similar to the RP of a three-variable mapping model. . . . . 114
6.4
Perturbed downsweep pacing protocol. The tissue is paced at a constant BCL for 60 s (transient response, small dots). An additional 5
paces at steady state are applied (diamonds) followed by an S2 pace
at BCL+50 ms (’+’), 5 recovery paces at the original BCL (filled circles), an S2 pace at BCL-50 ms (’x’), and 5 more recovery paces (filled
circles). The entire sequence is repeated at progressively shorter BCLs
until the myocardium transitions to a 2:1 or 2:2 stimulus:response pattern. The downstep in BCL, denoted by ∆, is 50 or 100 ms. . . . . . 117
6.5
Sketch of a bullfrog ventricular preparation. Two microelectrodes are
placed 1-2 mm apart with the proximal one placed 1 mm from the
bipolar pacing electrode. . . . . . . . . . . . . . . . . . . . . . . . . . 119
6.6
Range of BCLt . The transition BCL was 200 ms for all 12 trials that
exhibited 2:1 behavior. The transition BCL ranged from 300 ms to
450 ms for trials that exhibited 2:2 behavior. See Table 6.1 for more
details. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
6.7
Segments of restitution curves for a single BCL. At each BCL, I collect the transient response (small dots), the steady state responses
(diamonds), the S2 pace at BCL+50 ms (‘+’), the S2 pace at BCL-50
ms (‘x’) and the recovery paces (circles). The DRC (dashed line) is
the curve that connects all steady state responses. Segments of SRCs
(grey lines) are determined by the S2 paces and the steady state response; segments of BRCs (black lines) are determined by the recovery
paces and the steady-state response. . . . . . . . . . . . . . . . . . . 123
xxv
6.8
Restitution portraits collected simultaneously from the electrode proximal (A) and distal (B) to the pacing site. The restitution portraits
contain all the responses of the perturbed downsweep protocol of Fig.
1B: the transient response (small dots), the steady state responses (diamonds), the S2 pace at BCL+50 ms (‘+’), the S2 pace at BCL-50 ms
(‘x’) and the recovery paces (circles). The DRC (dashed line) is the
curve that connects all steady state responses. At each BCL, segments
of SRCs (grey lines) are determined by the S2 paces and the steady
state response; segments of BRCs (black lines) are determined by the
recovery paces and the steady-state response. For clarity, panels (A)
and (B) show data for every second BCL collected in this trial. . . . . 124
6.9
Restitution properties as a function of BCL. The (A) APD, (B) SDRC ,
(C) SSRC , and (D) SBRC are determined at steady-state for the trial
shown in figure 6.8 for both the proximal (circles) and distal (diamonds) electrodes. SSRC and SBRC are almost the same at both electrodes. APD has a spatial difference that remains roughly constant as
BCL changes. The spatial difference in SDRC increases as BCL decreases.125
6.10 Steady state APD difference. (A) ∆AP D for ALT and NoALT trials
as a function of BCLN . (B) P values below 0.05 (dashed line) indicate
∆AP D is significantly different from zero. . . . . . . . . . . . . . . . 127
6.11 SRC slope difference. (A) ∆SSRC for ALT and NoALT trials as a
function of BCLN . (B) P values below 0.05 (dashed line) indicate
∆SSRC is significantly different from zero. . . . . . . . . . . . . . . . 128
6.12 BRC slope difference. (A) ∆SBRC for ALT and NoALT trials as a
function of BCLN . (B) P values below 0.05 (dashed line) indicate
∆SBRC is significantly different from zero. . . . . . . . . . . . . . . . 129
6.13 DRC slope difference. (A) ∆SDRC for ALT and NoALT trials as a
function of BCLN . (B) P values below 0.05 (dashed line) indicate
∆SDRC is significantly different from zero. . . . . . . . . . . . . . . . 130
6.14 Spatial variation and alternans. The p values returned from a t-test
comparing (A) ∆AP D, (B) ∆SSRC , (C) ∆SBRC and (D) ∆SDRC of
ALT and noALT trials. The dashed line indicates a p value of 0.05. . 130
xxvi
6.15 Slope criteria. The mean slopes of (A,B) SRC, (C,D) BRC (E,F) DRC
and (G,H) the mean memory criterion indicate that none of these are
predictive of alternans in spatially extended tissue since they do not
satisfy the requirements detailed in Section 6.2.6. The legend in panel
(H) applies to all panels. . . . . . . . . . . . . . . . . . . . . . . . . . 131
A.1 Spectra of the Luxeon Star/O LEDs. . . . . . . . . . . . . . . . . . . 154
A.2 Experimental setup for in vivo epifluorescence measurement of cardiac action potentials. (A) A Langendorff-perfused rabbit heart is
mounted in front of a CCD camera. Two LEDs with filters to block
long-wavelength emission illuminate the tissue. Images are collected
through a cut-off filter by a CCD camera. (B) A small piece of bullfrog ventricular tissue is placed in a tissue dish and superfused with
oxygenated Ringer’s solution. Two LEDs with filters to block longwavelength emission provide excitation illumination. Images are collected with a CCD camera equipped with a cut-off filter. . . . . . . . 157
A.3 Intensity of the green LED as a function of distance. . . . . . . . . . 159
A.4 Transverse intensity distributions of the green Star/O LED at (A) 1
cm from the source and (B) 5 cm from the source. The 1 cm distribution shows the 4x1 array of diodes that make up the LED (central
bright region), while the pattern is more uniform at 5 cm. (C) An
intensity profile of the 1 cm distribution. The high peak in intensity
corresponds to the central bright spot. (D) An intensity profile of the
5 cm distribution. The large peak in intensity has been replaced by a
fairly flat plateau. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
A.5 Time dependence of the output intensity of the green LED measured
every 20 seconds for ten minutes after applying power to the device. . 161
A.6 Noise of the green LED, cyan LED, and ND:YLF laser. If the source is
operating at the quantum limit, we would expect to see a square-root
relationship between intensity and noise, as is seen for the green and
cyan LEDs. The laser, however, has an additional source of noise since
it deviates from this dependence. . . . . . . . . . . . . . . . . . . . . 163
xxvii
A.7 Noise of the laser. We conjecture that the large scatter in standard
deviation at high intensities is caused by laser speckle and motion of
the card. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
A.8 Optically recorded action potentials from rabbit and frog hearts. Pacing interval was 300 ms for rabbit (A-F) and 800 ms for frog (G-J).
Data was filtered with a 3×3 spatial Gaussian filter and three-point
temporal averaging. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
A.9 Recorded action potential signal as a function of mean illumination
intensity for (A) rabbit and (B) frog hearts. The slope of the line
gives the percent change in intensity during the action potential. . . . 166
B.1 Threshold method for determining APD. The action potentials shown
here are bullfrog ventricular APs measured with a glass microelectrode.
The threshold method defines the start or end of an action potential
as the time at which the voltage crosses a specified threshold value.
Shown in this figure are 90% and 70% threshold crossings. The specific
APD value will vary depending on the chosen threshold. . . . . . . . 172
B.2 Slope method for determining APD. The action potentials shown here
are bullfrog ventricular APs measured with a glass microelectrode. (A)
The slope as approximated by Eq. B.1. (B) The slope method defines
the start (or end) of the action potential as the time at which there is
a maximum (or minimum) in the temporal derivative of the voltage. . 174
B.3 Phase space trajectory of an action potential. An action potential
forms a closed loop in phase space since the voltage returns to initial
rest state after the action potential. The clusters at the ends are the
rest state and the plateau which are joined by the upstroke (upper
curve) and downstroke (lower curve). . . . . . . . . . . . . . . . . . . 175
B.4 Phase during an action potential. The action potentials shown here are
bullfrog ventricular APs measured with a glass microelectrode. The
phase increases sharply during the upstroke, remains constant during
the plateau, falls sharply during the downstroke and remains constant
during the rest state. For (A) τ = 5 ms and c is the mean of the time
series. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175
xxviii
B.5 Effects of parameter changes on phase. (A) Changes in c move the
phase representation of the downstroke closer or further from the upstroke, thereby changing the measured APD. (B) Increases in τ cause
the upstroke and downstroke in the phase representation to be less
sharp. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
B.6 Effect of time delay on phase space trajectory. The value of τ controls
the width of the loop in phase space. . . . . . . . . . . . . . . . . . . 177
B.7 Microelectrode recording. A sample microelectrode recording that
shows five steady-state action potentials and has an SNR of 310. . . . 178
B.8 Multiple threshold crossings of noisy electrophysiological data. Noisy
electrophysiological signals will cross a threshold multiple times. Within
the box on the first downstroke, there are 18 downward crossings and
17 upward crossings. . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
B.9 Effect of noise on calculation of action potential amplitude. The measured APA increases as the noise in the signal increases. . . . . . . . 180
B.10 Effect of noise on APD found by the threshold method. As the SNR
decreases (increasing noise), the error in the measured APD becomes
larger. The correct value of APD is indicated by the dashed line. Using
the first threshold crossing to find APD produces the most accurate
APD measurements in noisy signals. . . . . . . . . . . . . . . . . . . 180
B.11 Derivatives of noisy electrophysiological signals. Noise washes out the
minimum of the derivative, which corresponds to the downstroke, but
the maximum remains unchanged. . . . . . . . . . . . . . . . . . . . . 182
B.12 Effect of noise on the measurement of APD using the slope method.
The correct value of APD is indicated by the dashed line. The slope
method is not accurate or precise below SNRs of 300. . . . . . . . . . 182
B.13 Phase of a noisy electrophysiological signal. Noise in an electrophysiological signal causes multiple crossings in the phase representation of
the downstroke. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 183
xxix
B.14 Effect of noise on the measurement of APD using the phase method.
The ‘correct’ value of APD is indicated by the dashed line. The phase
method using the mean crossing time returns the correct APD even at
low SNRs. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
B.15 A noisy electrophysiological signal. This signal was created by adding
Gaussian noise to the microelectrode signal of Fig. B.7. It has an SNR
of 22.4. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
B.16 Effect of the mean filter on a sample time series. (A) The mean filter
creates a smoother curve by averaging nearby points. A larger filter
size produces a smoother curve. (B) The SNR increases as the filter
size increases. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 189
B.17 Signal changes due to mean filtering. (A) The MSE shows an initial
drop due to the removal of noise from the signal, but then increases
as the signal becomes distorted by the filter. (B) The dashed line
is the original microelectrode signal while the solid line is the same
signal after Gaussian noise was added and then removed with a 40point mean filter. The filtered signal has a slower upstroke and a lower
amplitude than the original signal. . . . . . . . . . . . . . . . . . . . 190
B.18 Effect of the mean filter on calculation of APD. (A) APD calculated
using the threshold method on a signal filtered with a mean filter of
various sizes. (B) APD calculated using the phase method on a signal
filtered with a mean filter of various sizes. In both figures, the solid
line indicates the APD of the original signal. . . . . . . . . . . . . . . 191
B.19 Effect of the median filter on a sample time series. (A) The median
filter creates a smoother curve by determining the median of nearby
points. A larger filter size produces a smoother curve. (B) The SNR
increases as the filter size increases. . . . . . . . . . . . . . . . . . . . 192
xxx
B.20 Signal changes due to median filtering. (A) The MSE decreases with
increasing filter size. (B) The dashed line is the original signal while the
solid line is the same signal after Gaussian noise was added and then
removed with a 40-point median filter. The two signals show slight
deviations at the start of the upstroke, at the end of the downstroke,
and at the peak of the action potential. . . . . . . . . . . . . . . . . . 193
B.21 Effect of the median filter on calculation of APD. (A) APD calculated
using the threshold method on a signal filtered with a median filter of
various sizes. (B) APD calculated using the phase method on a signal
filtered with a median filter of various sizes. In both figures, the solid
line indicates the APD of the original signal. . . . . . . . . . . . . . . 194
B.22 Frequency spectrum of action potentials. (A) The frequency spectrum
of the microelectrode signal of Fig. B.7. (B) The frequency spectrum
of the same signal with Gaussian noise. . . . . . . . . . . . . . . . . . 195
B.23 Effect of the frequency filter on a sample time series. (A) The frequency
filter creates a smoother curve by removing noise in the frequency
spectrum. (B) The SNR increases as the filter size increases. . . . . . 196
B.24 Signal changes due to frequency filtering. (A) The MSE decreases
until a filter size of 3800 where it rises sharply because the frequency
filter removes parts of the action potential. (B) The dashed line is
the original microelectrode signal of Figure B.7. The solid line is the
same signal after Gaussian noise was added and then removed with
a 3800-point frequency filter. The filtered signal still has some noise,
but the overall shape of the action potential remains unchanged. . . . 197
B.25 Effect of the frequency filter on calculation of APD. (A) APD calculated using the threshold method on a signal filtered with a frequency
filter of various sizes. (B) APD calculated using the phase method on
a signal filtered with a frequency filter of various sizes. In both figures,
the solid line indicates the APD of the original signal. . . . . . . . . . 198
xxxi
B.26 Finding APD in an optical signal. (A) The original optical APs from
bullfrog ventricular myocardium collected with a CCD camera. The
upper trace is at a BCL of 1000 ms while the lower trace is at a
BCL of 300 ms. (B) The same signals after they have been filtered
with a 3-point median filter. The squares indicate the start of the
action potential and the circles indicate the end of the action potential
both found using the threshold method. The dashed line indicates the
threshold. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 200
C.1 Signal degradation due to cell death. (A) The optical signal from a
piece of bullfrog ventricular myocardium paced at BCL=1000 ms at
the beginning of an experiment. (B) The optical signal from the same
location about two and a half hours later. The recording taken at the
beginning of the experiment has an SNR of 22 and the recording taken
at the end of the experiment has an SNR of 4. . . . . . . . . . . . . . 203
C.2 Spatial variation of APD of a 2-variable cardiac model in a paced cable.
(A) When the cable is short (1 cm) there is no region of constant APD
in the center of the cable. (B) In a longer cable it is clear that APD
variation is a boundary effect. . . . . . . . . . . . . . . . . . . . . . . 205
C.3 Examples of whole heart tissue preparation. The two images show
the whole frog heart after application of the potentiometric dye. The
lack of dye in the auricles is intentional since they will not be used for
recording because the tissue there is not homogeneous. Note that in
panel B, it is difficult to visually discern the boundary between auricles and ventricle, making consistent placement of electrodes difficult.
Also, there is quite a bit of difference in the size and shape of the two
preparations, which may lead to differences in observed spatial patterns.208
C.4 Examples of the anterior surface ventricular preparation. Although
there is still some variation in size and shape of the tissue samples,
there is more consistency than in the whole heart preparation (Fig. C.3)211
F.1 Core conductor model. Cardiac fibers are modelled as electrical circuits. Individual cardiac cells are coupled through the intracellular
and extracellular space, modelled by resistors. . . . . . . . . . . . . . 235
xxxii
List of Tables
2.1
Physical meanings of the two-variable model parameters. . . . . . . .
3.1
Values of ∆AP D for all experiments. APD maps collected at BCL=1000
ms were used for the calculation of ∆AP D. The three experiments
marked with * show similar spatial APD variation from all three pacing sites. Error is determined by standard error. . . . . . . . . . . . . 56
3.2
Width of the boundary layer. APD maps collected at BCL=1000 ms
were used for the calculation of the width of the boundary layer. . . .
64
Summary of experimental trials indicating the occurrence of complex
rhythms, the BCL at which a change in response pattern was observed,
and the pacing electrode. . . . . . . . . . . . . . . . . . . . . . . . . .
65
Summary of experimental trials indicating the occurrence of alternans
and the BCL at which a change in response pattern was observed . .
86
Model parameters used to simulate tissue that exhibits alternans and
tissue that does not exhibit alternans. . . . . . . . . . . . . . . . . . .
91
3.3
4.1
5.1
6.1
23
Summary of experimental trials indicating the occurrence of alternans,
the BCL at which a change in response pattern was observed, and the
electrode at which alternans appeared. . . . . . . . . . . . . . . . . . 126
A.1 Values of the parameter used to fit the noise data of the three light
sources. R is determined by fitting the experimental data to Eq. A.2.
The reduced χ2 is a measure of goodness of fit. . . . . . . . . . . . . . 164
A.2 Results of the noise and action potential amplitude (APA) measurements.167
xxxiii
Chapter 1
Introduction
The human heart provides a simple, but critical, service for the body. The heart
is simply a pump whose contractions circulate blood through the body. In order
to achieve the pumping action, millions of cardiac cells have to work together in
what turns out to be a very complicated process. In a healthy functioning heart,
pacemaker cells in the sino-atrial node initiate an electro-chemical wave that is sent
along a specialized conduction system in the heart. The wave then moves through
the bulk of the cardiac tissue, causing a mechanical contraction that results in blood
being pumped through the body. Due to injury or disease, however, this process
can be disrupted causing a condition known as fibrillation. During fibrillation, the
electrochemical wave degenerates into disorganized electrical behaviour that hinders
the ability of the heart to pump blood, resulting in sudden cardiac death [1]. Sudden
cardiac death is one of the leading causes of death in the United States [2], and thus
there is great interest in determining the mechanisms that lead to arrhythmias.
1.1
1.1.1
Background
Historical Background
Just before the turn of the 20th century, it was determined that the heart functions
without the aid of a stimulus from the nervous system [3] and that conduction within
the heart was also unaided by nerve fibers [4]. This discovery lead to an interest
1
in studying the electrical dynamics of the heart [5–7] and their role in arrhythmias
[8, 9]. Such measurements were technically difficult at the time [5], so progress in
understanding the origin of the electrical response was slow.
A major advance occurred with the experiments and theory of Hodgkin and Huxley [10]. They developed an ionic model that reproduced experimentally measured
electrical responses. Although their model was developed for nerve cells, it was
quickly adapted to model the cardiac response [11–13]. As our understanding of the
movement of ions in and around the cell has increased, the original models have been
extended to include several more ionic currents as well as details of ionic movement
within the cell [14, 15]. These models form the basis of our current understanding of
the electrical response of cardiac cells and for many years were the primary method
of investigating propagation in cardiac tissue since spatially extended measurements
using electrode arrays in real tissue were technically difficult.
This problem was remedied with the advent of optical mapping. Early systems
used intrinsic optical properties of tissue to visualize electrical activity on the surface
of the tissue [16–20]. Although these systems could measure cardiac signals at many
spatial locations, the SNR was poor and it wasn’t until the development of fluorescent
voltage-sensitive dyes [21, 22] that the idea of optical measurement of cardiac signals
bore fruit. The recent development of high-resolution, high-speed cameras and advances in computer memory, permitting storage of the vast amount of data collected,
make direct observation of cardiac waves commonplace. Such studies have given
new insights into the behavior of cardiac fibrillation [23, 24], its termination using
electrical shocks [25] and the response of cardiac tissue to point stimulation [26].
2
1.1.2
How Cardiac Cells Work
To begin the study of cardiac dynamics, I introduce some of the basic concepts and
common terminology used in cardiac electrophysiology. Cardiac cells communicate
with each other by means of ionic currents. A change in the concentration of ions
within and around a cell causes the cell membrane to open or close channels allowing
ions to flow into or out of the cell [27]. These changes can cause the cell to produce
an excitable response known as an action potential. An action potential is a rapid
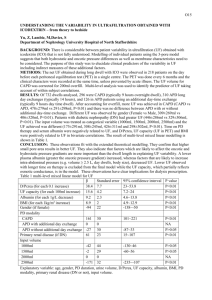
depolarization of the cell, followed by a slower repolarization (See Fig. 1.1). The
length of time that the cell remains depolarized is known as the action potential duration (APD). The time between successive action potentials is the diastolic interval
(DI).
Although the exact ionic currents that produce an action potential vary from
animal to animal [28, 29], the primary ions involved are Ca2+ , Na+ and K+ . When
the cell is at rest, the electrical and chemical gradients of the ions are in equilibrium
resulting in a transmembrane voltage of ∼-90 mV. In the rest state, the concentration
of K+ is larger inside the cell, while the concentrations of Ca2+ and Na+ are larger
outside the cell. When the cardiac cell receives a stimulus that causes the transmembrane voltage to increase beyond a threshold voltage (∼-70 mV), Na+ channels open
and Na+ ions rush along the chemical gradient into the cell. This creates the rapid
depolarization seen in the action potential. The Na+ channels close once the cell has
depolarized. The plateau phase voltage is maintained by an inward Ca2+ and an outward K+ . The Ca2+ channels close before the K+ channels allowing the cell to return
to its rest voltage through the net outward current. Although the transmembrane
3
Figure 1.1: Cardiac action potentials recorded from bullfrog ventricle. Recordings
are made using a microelectrode in a paced in vitro preparation. The time between
the beginning and end of one action potential is the action potential duration (APD).
The time between the end of one action potential and the start of the next is the
diastolic interval (DI). The time between stimuli is the pacing period or basic cycle
length (BCL).
4
voltage is now at the rest value, the ion concentrations take a little longer to recover,
creating a refractory period where further stimulus will not elicit an action potential.
Beyond the refractory period, there is a further period of time where an action
potential can be elicited, but because the ion concentrations are not completely recovered, the shape of the action potential will be different. In particular, the amplitude
is lower and the APD is shorter. Although the shape of the action potential is
essentially independent of the stimulus amplitude once it is above threshold, as is
characteristic of excitable systems, it is dependent on the period at which the tissue
is paced, known as the basic cycle length (BCL). This rate-dependence makes cardiac
cells a complex and interesting example of an excitable medium (excitable media are
discussed in detail in Chapter 2).
When cardiac tissue is paced at a constant BCL, it typically responds in what
is known as a 1:1 response pattern. That is, one action potential is produced for
every stimulus, with every action potential duration being the same, as in Fig. 1.1.
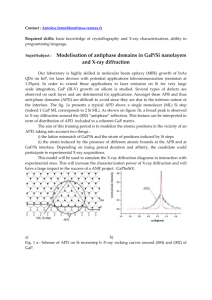
As the tissue is paced more rapidly, the APD shortens and the tissue may undergo
a bifurcation to a 2:2 response pattern. This pattern, also known as alternans, is a
long-short alternation of action potential duration (see Fig. 1.2). At even more rapid
pacing, the tissue will respond with a 2:1 response pattern; that is, one response for
every two stimuli. In this situation, the second stimulus arrives during the refractory
period of the previous action potential, so the cell is unable to respond to the stimulus.
5
Figure 1.2: Cardiac action potential alternans. At rapid pacing, cardiac cells can
exhibit a long-short alternation in APD known as a 2:2 response or alternans. This
example was recorded from bullfrog ventricular myocardium.
1.2
Nonlinear Dynamics of Cardiac Tissue
The transition from a 1:1 to a 2:2 response discussed in the previous section is of
particular interest to researchers. Although alternans is not a dangerous or lifethreatening response pattern in the human heart, it has been shown to be a precursor
to the deadly state of fibrillation. Briefly, alternans in the single cell manifest themselves clinically as T-wave alternans, a small beat-to-beat variation in the T wave of
the electrocardiogram [30]. T-wave alternans have been shown to be a predictor of
arrhythmias in clinical studies [31]. A mechanism linking cellular alternans to the
onset of conduction block and arrhythmia (discussed more fully in Sec. 2.4) was
proposed by Pastore et al. [30] and experimentally confirmed in both guinea pig [30]
and dog hearts [32].
6
Further, the transition from a 1:1 to a 2:2 response looks very much like a perioddoubling bifurcation [33]. A period-doubling bifurcation is a transition from constant
to oscillatory behavior seen in some nonlinear systems. Since this transition has been
studied and analyzed in other systems [34–36], it was hoped that this research could
be used to predict the onset of alternans in the heart. Further, researcher in the
nonlinear dynamics community have developed techniques that enable researchers to
control or suppress the 2:2 response pattern and return a system’s response to the
1:1 behavior [37]. The possibility of using nonlinear dynamics and control techniques
to predict and eventually suppress alternans, thus preventing the fatal cascade to
fibrillation, led researchers to apply the techniques of nonlinear dynamics to the
heart.
1.2.1
Single Cell Dynamics
The first attempt to understand the transition to alternans was made by Nolasco and
Dahlen [38]. They used the idea of a restitution curve (RC), which is the functional
relationship between APD and previous DI, and simple graphical methods (Fig. 1.3)
to show that alternans is the result of a period doubling bifurcation that occurs when
the slope of the RC becomes greater than 1.
Although this analysis has proven to be inadequate [39–43], the idea that nonlinear
analysis can be used to predict the onset of alternans has taken hold. This has lead
to studies of the different response patterns in single cells using bifurcation diagrams
(dependence of APD on BCL) [39, 44] and RCs [45, 46] and an effort to find simple
mapping models to predict this behavior [43, 45, 47].
7
Figure 1.3: Cobweb diagram. The restitution curve (RC, solid line) represents the relationship between APD and previous DI. The dotted line is the
BCL = AP Dn+1 + DIn . Starting at a particular DI, find the resulting APD by
drawing a line up to the RC. To determine the following DI, draw a horizontal line
across to the dotted line. This new DI then leads to another APD and so on. Depending on the exact details of the RC, several different results are possible, two of
which are shown here. (A) If the slope of the RC is less than one, the system will
eventually settle down to a single APD, indicated by the star (the 1:1 response). (B)
When the slope of the RC is greater than 1, the system oscillates between two APDs,
indicated by the stars (the 2:2 response).
8
1.2.2
Spatial Dynamics
With the advent of optical mapping systems and the ability to measure the electrical
waves propagating through cardiac tissue, there is now interest in applying the ideas
of nonlinear dynamics to describe and explain the observed spatiotemporal dynamics
[48–51]. Specifically, experimental, computational and theoretical studies have all
tried to determine whether the spatiotemporal dynamics of electrical activity can
lead to instability of the 1:1 response.
A long-standing assumption underlying many theories of instability of the 1:1
response in spatially extended cardiac tissue is that spatially homogeneous tissue will
result in spatially homogeneous dynamics except for possibly a small boundary effect
near the site where current is injected and near any physical boundary beyond which
current cannot flow. The boundary effect was assumed to be limited to spatial scales
on the order of the passive length constant of the tissue, λ, the length over which
subthreshold disturbances decay. In cardiac tissue, the passive length constant is
very small, ranging from ∼0.2-2 mm [52–56]. Any boundary effects on this scale were
thought to be negligible when performing studies on whole hearts, which are several
centimeters in size. This assumption was confirmed by early computational studies
that found that many action potential properties (action potential amplitude [57–59],
the sharpness of the upstroke as measured by dV /dtmax [58, 59], and the conduction
velocity [57, 59]) exhibited changes as they approached an insulated boundary, but
that all these changes occurred within ∼ λ of the boundary.
More recent simulations, however, have found that the APD exhibits a much
greater boundary effect [59–63]. Simulations show that, during the steady-state 1:1
9
response, APD shows a distinct spatial variation, with longer APDs occurring near
the stimulus site and shorter APDs occurring at insulated boundaries (Fig. 1.4).
Although a boundary effect is not surprising, the spatial scale over which the APD
varies is unexpected. Spatial variation of APD over distances of ∼10λ, much larger
than any previously observed boundary effect, have been observed in one [60, 61,
63] two [62] and three [60] dimensional simulations. In particular, Sampson and
Henriquez used the same model in three-dimensional and one-dimensional tissue and
found that APD was longest at the stimulus site, constant over most of the tissue
and shortest at physical boundaries far from the stimulus site no matter the number
of spatial dimensions used in the simulation [60].
Computational studies suggest that the large length scale of APD variation is
independent of the specific details of the model and is actually an intrinsic property
of cardiac tissue. Sampson and Henriquez determined that cell heterogeneity did not
significantly alter the length scale over which APD varied. In one simulation, different
cell types (with different intrinsic APDs and restitution properties) were assigned to
different regions of the heart, while in another simulation, the heart consisted of
completely identical cells. Similar spatial APD patterns were observed in both cases,
with a slightly larger APD variation being observed in the heterogeneous hearts. The
two-dimensional simulations of Lesh et al. support this finding [62]. They simulated
a sheet of heterogeneous cells, each cell having a slightly different, randomly assigned
inward current conductance and found that APD showed a larger than expected
boundary effect. Finally, the complexity of the computational model also seems to
have little effect on the observed boundary effect. The model used by Cain et al. [63]
is a very simple model consisting of a single inward current and a single outward
10
Figure 1.4: Spatial variation of APD in a cardiac cable. A two-variable model (Eq.
2.4) is implemented for a 5-cm-long cable. The tissue is paced on the left end at a
BCL of 500 ms. The cable has two distinct boundaries, the stimulus site and the
physical boundary at the far end of the cable. The APD is constant in the center of
the cable, away from the boundaries, but increases near the stimulus and decreases
near the opposite end. The passive length constant for this model is 1 mm, yet the
APD changes from ∼382 ms to ∼377 ms over a distance of ∼1 cm near the stimulus.
Spatial variation of APD also occurs near the insulated end of the cable over a similar
distance.
11
current, yet they still observe lengthening of the APD at the paced end of a cable of
cardiac tissue and shortening of APD at the insulated end of the cable over distances
of ∼10λ, similar to the boundary effect observed in more complicated models [60–62].
An advantage of computational models is that all possible variables can be tracked
and mathematical analysis can be performed on the models. This advantage has lead
to the development of two conjectures that may explain the observed spatial variation
of APD. The first conjecture is that changes in membrane resistance during the action
potential lead to increases in the passive length constant [60]. The second conjecture
is that, near the insulated end of the cable, the APD shortens because the current
cannot flow beyond the boundary [63]. Both theories are discussed in more detail in
Sec. 2.3. It is unclear if either of these theories play a role in spatial heterogeneity
of APD in real cardiac tissue since real cardiac tissue is more complicated than what
can be captured by the models.
Recent experiments indicate that APD is not homogeneous during the 1:1 response
[40, 64–72]. Unfortunately, these experiments do not agree on the details of the
observed spatial patterns. Some experiments showed an increased APD near the
stimulus site [40, 64, 72], suggesting a boundary effect similar to that observed in
computational studies. Other experiments did not see this effect [66–69,73], observing
instead that the spatial pattern of APD seemed to be determined by the underlying
structure of the heart.
Determining whether real cardiac tissue exhibits an exaggerated boundary effect
as predicted by computational studies is important because some experiments have
linked heterogeneity of APD to the onset of arrhythmias [30, 66, 67, 69, 73–75] and
some have even suggested that spatial heterogeneity of APD causes arrhythmias
12
[30–32, 76]. The mixed experimental results mentioned above suggest the need to
determine whether real cardiac tissue exhibits a large boundary effect, the extent to
which this boundary effect can be modified by underlying tissue heterogeneity, and
whether the APD boundary effect can be linked to instability of the 1:1 response.
1.3
Thesis Overview
This dissertation documents my research on the large boundary effect of APD and its
role in determining the stability of the 1:1 response. It is divided into seven Chapters,
the first of which has provided the context for my research.
Chapter 2 discusses concepts of excitable systems as they pertain to the electrodynamics of the heart. It reviews ideas and terminology necessary to understand the
later following chapters. In particular, I discuss why cardiac tissue is an especially
interesting example of an excitable system and why it provides a good substrate for
the study of nonlinear waves.
The spatial variability of APD is studied in detail in Chapter 3. It describes
experiments that use an optical imaging system to study the spatial variation of APD
in a small piece of bullfrog ventricle. The bullfrog ventricle consists of a single type of
cell and has almost no structures or anisotropy, so the only boundaries in the system
are the stimulus site and the physical boundaries of the tissue. This substrate is the
closest possible approximation to the homogeneous tissue with insulated boundaries
used in simulations. I find that the APD varies near the boundaries over a distance
much larger than the passive length constant of bullfrog ventricular tissue (0.3 mm
[52]). Specifically, APD varies over a distance of ∼10λ with an effective length
13
constant of ∼1.5-2λ.
One of the possible implications of the spatial variation of APD is that restitution
curves may also vary over the surface of the tissue. In particular, Chapter 4 takes
a closer look at the spatial variation of the slope of the DRC (SDRC ), which is
determined by steady-state APD and DI, and whether the spatial variation can be
correlated to the onset of alternans. Again, the optical mapping system is used to
study slope of the DRC at all points in a piece of bullfrog ventricle. I find that the
slope of the DRC is largest near the pacing electrode and diminishes at a constant
rate (∼0.1-1.5 /mm) over the surface of the tissue. I also find that the spatial gradient
of slope of the DRC can be an indicator of the tissue’s propensity to exhibit alternans
at rapid pacing. Tissue that exhibits alternans has a larger spatial gradient of SDRC
than tissue that does not exhibit alternans. Moreover, the increased spatial gradient
is evident at BCLs as much as 200 ms slower than the transition point.
In Chapter 5, I use a simplified cardiac model to study the spatial variation of
APD and SDRC in more detail. I use a homogeneous sheet of cardiac tissue with
insulated boundaries to determine the dynamically induced spatial patterns of APD
and SDRC . I also change the model parameters to simulate both tissue that exhibits
alternans and tissue that does not exhibit alternans to determine whether the spatial
gradients of restitution properties are different in the two cases. I find that, even in a
simplified cardiac model, APD and SDRC show dynamically-induced spatial gradients
and that the gradients can be correlated to the onset of alternans.
Finally, Chapter 6 extends the study of spatial variation in cardiac tissue to other
restitution properties. This study uses simultaneous measurement of transmembrane
voltage at two locations in small pieces of bullfrog ventricle to measure spatial vari14
ation in restitution properties. This chapter introduces the idea of the restitution
portrait (RP), a visualization of several restitution properties of cardiac tissue, and
uses it to determine spatial variability of steady state APD, and the slopes of the
dynamic restitution curve (DRC), S1S2 restitution curve (SRC), and constant-BCL
restitution curve (BRC). Statistical analysis is used to determine whether spatial
variation of any of these properties is correlated to the onset of alternans. I find that
all restitution properties show some spatial variability and that spatial variation in
steady-state APD, slope of the DRC and slope of the SRC can be correlated to a
tissue’s propensity to alternans.
Chapter 7 summarizes the results of these experiments and their implications
for our understanding of the stability of cardiac rhythms. It also discusses several
directions for future research.
15
Chapter 2
Background
2.1
Introduction
As mentioned in Sec. 1.1.2, cardiac tissue is an example of an excitable medium.
The heart is a particularly interesting excitable medium to study because the proper
operation of the heart is so crucial to human life. The heart is also a challenging system to study since there are several cardiac properties, such as the three-dimensional
nature of the heart and the specialized structures within the heart, that add to the
complexity of the system. This chapter provides an introduction to excitable media,
nonlinear dynamics and their application to cardiac electrodynamics and explains
the fundamental concepts needed to understand the following chapters.
2.2
Excitable Media
Linear waves, their behavior in various media and their interactions with boundaries have been studied for centuries. The equations describing their behavior are
well-known and have been used extensively both as exact descriptions and as first
approximations of a wide variety of phenomena [77–79]. More recent research has
focused on nonlinear waves, particularly in chemical and biological systems [80], and
the more interesting phenomena, such as solitons, that they display [81, 82]. Here I
study a particular kind of nonlinear medium, excitable media, and how waves propagate through it.
16
2.2.1
Introduction to Excitable Media
An excitable system is one in which small perturbations decay quickly to a global
rest state, but perturbations larger than some threshold value cause a large excursion
through phase space before returning to the rest state. After the excursion, the
excitable system cannot be re-excited for some period of time, known as the recovery
or refractory period.
Forest fires are the classic example of an excitable medium [83, 84]. Let us first
consider a fire started near a single isolated tree. This is the equivalent of an isolated
excitable oscillator. If the fire is small, it will die out on its own and the tree remains
essentially unchanged. This is the subthreshold response of the excitable element. A
fire larger than some threshold will consume the entire tree before dying away due
to lack of fuel. This is the suprathreshold response. Note that, although the fire is
gone, the tree has not quite returned to its original state. Another fire cannot be
started until a new tree has grown. This is the refractory period.
When several excitable oscillators are linked together, such as having many trees
next to each other in a forest, waves of activation can be formed. Due to the unique
properties of excitable media, particularly the threshold and the refractory period,
these waves behave very differently from linear waves.
Method of Propagation
When one oscillator is activated by some external stimulus, the coupling causes the
state of neighboring oscillators to be altered. Below threshold, the propagation is
similar to linear waves where elements are dragged along by their neighbors. If the
17
state of an element is pulled above threshold, the element is activated. It now acts
nearly independently of its neighbor and follows the usual path through phase space.
Thus, a wave in excitable media propagates by regenerating the pulse at each point
on the medium. In the forest fire example, a single burning tree generates heat that
can ignite neighboring trees, which then ignite their neighbors and so on. The fire
spreads element by element eventually consuming the entire forest.
This method of propagation leads to an interesting property of excitable media.
If the underlying medium is homogeneous, with all individual oscillators behaving
identically and starting in the same state, and in the absence of boundaries, the
shape of the wave will remain the same as it propagates through the medium. If the
coupling between elements dissipates too much energy, inactive elements will not be
pulled over threshold and the wave will not propagate at all. Thus, the all or nothing
characteristic of individual excitable oscillators is also true in the spatially extended
medium.
Behavior at a Boundary
Waves travelling in a homogeneous excitable medium, away from any boundary will
not change size or shape. When the wave approaches a boundary, however, an inhomogeneity is introduced that may alter the wave properties. In one-dimensional
excitable media, when a wave hits the end of the medium the wave does not get
reflected since the refractory region behind the wave prevents the wave from moving
back in the direction from which it came. Since the wave cannot move forward and
cannot move backward, it dies off. As an example, imagine a forest fire encountering
a highway. The fire cannot move forward since the highway provides no fuel, but
18
Figure 2.1: Excitable wave at a boundary. In one dimension, as the wave approaches
the end of the medium, it cannot move forward and it cannot move backward because
of the refractory tissue behind the wave.
it also cannot move back since those trees have already been consumed by fire and
cannot be reignited. In this scenario, the forest fire will be forced to die off. This
process is shown in Fig. 2.1.
The introduction of a boundary may also lead to more subtle changes in the size
and shape of the wave before it dies off. In cardiac tissue, changes in amplitude
[57–59], speed [57, 59], and shape [58, 59] have all been observed near the boundary.
Similar changes in speed and amplitude of electrical waves in frog muscle fibers occur
near the end of the fiber [85, 86]. These changes occur on a small spatial scale and
so have been typically considered negligible when studying propagation phenomena
in excitable media.
19
Wave Annihilation
Waves in excitable media can also be altered when two waves interact. When two
waves meet in an excitable medium, they annihilate each other. The reason for this
is the refractory period associated with an excitable medium. Behind each wave,
there is a region of the medium that cannot be excited. When a wave propagating
in the opposite direction encounters the region of refractoriness, the tissue cannot be
excited, so the wave dies. In the forest fire example, two forest fires that meet will
not be able to continue propagating since all the trees in the wake of each fire have
been burned and there is no fuel to feed the fires.
Figure 2.2 shows this process. In the top panel, we begin with wave A traveling
to the right and wave B traveling to the left. These can be thought of as forest
fires moving towards each other. Note that, behind each wave, there is a region of
non-excitable medium. In the case of forest fires, the non-excitable medium consists
of trees that are completely burned and cannot fuel another fire. In the middle panel,
the two waves meet and attempt to pass through each other. However, wave A hits
the refractory region associated with wave B, while wave B hits the refractory region
associated with wave A and so neither wave can continue. In the forest fire example,
fire A cannot propagate any further to the right since fire B has already consumed
all the fuel on the right and fire B cannot propagate any further to the left since fire
A has already consumed all the trees on the left. Finally, in the last panel, the two
waves have annihilated, leaving a region of refractoriness. In the case of forest fires,
we are left with a region of burned trees where a new fire cannot be started until the
trees have regrown.
20
Figure 2.2: Collision of two waves in excitable media. In the top panel, two waves
approach each other. There is a region of refractoriness behind each wave. In the
middle panel, each wave begins to run into the refractory region of the other wave,
preventing them from propagating any further. In the bottom panel, the two waves
are completely annihilated.
21
The collision of two waves in an excitable medium is physically similar to one wave
encountering a physical boundary. Not only does the excitation disappear in both
situations, but the changes in amplitude, speed and shape of the wave that occur near
a physical boundary will also occur when two waves approach each other [59]. Again
these more subtle changes occur over a small scale and so are typically neglected
when wave interactions in excitable media are studied.
2.2.2
Cardiac Tissue as an Excitable Medium
Single Cell
As mentioned before, cardiac cells are an example of an excitable medium. To see
this, consider the simplified cardiac model [87, 88] given by the equations
δt V =
V
Iext
h 2
V (1 − V ) −
−
.
τin
τout
Cm
δt h =
(
1−h
τopen
h
− τclose
V < Vc
V > Vc ,
(2.1)
(2.2)
where V is the transmembrane voltage, δt is the derivative with respect to time, h
is a gating variable, Vc is the threshold voltage, Iext is an externally applied current
(usually a small perturbation that initiates the pulse), Cm is the membrane capacitance, and τopen , τclose , τout , and τin are parameters that determine the size and shape
of the action potential (Table 2.1). The transmembrane voltage, V , has been scaled
to range between 0 and 1 using the following change of variables
V =
V − Vmin
,
Vmax − Vmin
22
(2.3)
Parameter
Physical Meaning
τopen
Time constant with which gate opens
τclose
Time constant with which gate closes
τout
Time constant with which voltage decays
τin
Time constant with which voltage rises during the upstroke
Table 2.1: Physical meanings of the two-variable model parameters.
where V is the physiological transmembrane voltage, Vmax is the maximum physiological transmembrane voltage, and Vmin is the minimum physiological transmembrane
voltage. Note that the voltage is dimensionless.
The inward current, given by Iin = hV 2 (1 − V )/τin , is a simplified version of
the sodium current of the Noble model [89]. The sodium current undergoes a rapid
initial increase when the membrane depolarizes, followed by a slower decrease even
if the depolarization remains. The outward current, given by Iout = −V /τout , is a
simplified version of the potassium current of the Noble model [89]. In real cardiac
cells, the process is much more complicated because there are many more currents
in a real cell. However, this model captures the essence of the excitable properties of
cardiac cells and thus serves as a good model to develop an intuitive understanding
of the underlying dynamics.
If the cell is at equilibrium and we apply an external current (Iext ) that keeps the
transmembrane voltage below the threshold (Vc ), the gate remains open resulting in
a simple voltage and current decay back to the rest state as shown in Fig. 2.3. This
is the subthreshold response.
If the cell is at equilibrium and we apply an external current (Iext ) that pushes the
transmembrane voltage beyond the threshold (Vc ), the gate begins to close. When
the gate closes, an inward current begins to flow. The inward current causes a rapid
23
Figure 2.3: Response of the 2-variable cardiac model to a subthreshold stimulus.
After a subthreshold current pulse is injected into the cell, (A) the transmembrane
voltage simply decays back to the rest state, B) the gate remains open, C) the inward
current decays back to the rest state, and D) the outward current also decays back
to the rest state. Parameter values for this example are Vc = 0.13, τin = 0.2 ms,
τout = 10 ms, τopen = 130 ms, τclose = 150 ms, and Iext /Cm = 0.05 /ms.
24
increase in voltage. As the gate closes, the inward current diminishes and is balanced
by and eventually overtaken by an outward current. This causes voltage to decrease,
eventually returning below the threshold. At this point, the inward current is turned
off and the cell returns to the initial rest state. This is the suprathreshold response
of the cardiac cell. Figure 2.4 shows this process in a single cell. The top panel
shows the transmembrane voltage; this is the action potential that we are studying.
The next panel is the gate variable that controls the strength of the inward current.
The lower two panels show the inward and outward currents. Note that, even after
the voltage returns to its initial state, the gating variable is still recovering from the
action potential. This delayed recovery of the gating variable creates a refractory
period during which another suprathreshold stimulus will not result in an action
potential.
Spatially Extended Tissue
When we put this model into a spatially extended medium, we assume that each
cell behaves according to Eq. 2.1 and that the cells are coupled through diffusion of
the voltage (App. F). So the only equation that is modified is the voltage equation,
which becomes
δt V = Dδx2 V +
V
Iext
h 2
V (1 − V ) −
−
,
τin
τout
Cm
(2.4)
where D is the diffusion constant, δx is the derivative with respect to space, and h
is the gating variable as described by Eq. 2.2. Similar to the behavior seen in the
single cell, propagation in a cable is only initiated if Iext exceeds a threshold value.
Note that, the threshold value for Iext in the single cell will differ from the threshold
25
Figure 2.4: Response of the 2-variable cardiac model to a suprathreshold stimulus.
After a small suprathreshold current pulse is injected into the cell, (A) the transmembrane voltage rapidly increases and then slowly decreases, B) the gate variable
decreases (the gate closes) and recovers once the transmembrane voltage returns below the threshold, C) the inward current rapidly increases causing the upstroke of
the action potential before diminishing as the gate closes, and D) the outward current is initially much smaller than the inward current, but eventually becomes larger
causing the decrease in the voltage. Parameter values for this example are Vc = 0.13,
τin = 0.2 ms, τout = 10 ms, τopen = 130 ms, τclose = 150 ms, and Iext /Cm = 0.2 /ms.
26
Figure 2.5: Subthreshold response in a cable. A subthreshold injection of current
into the cable causes a small increase in transmembrane voltage that decays in space
and time. Parameter values for this example are Vc = 0.13, τin = 0.2 ms, τout = 10
ms, τopen = 130 ms, τclose = 150 ms, and D = 0.001 cm2 /ms.
value of Iext in one-dimension because the injected current can flow from one cell to
its neighbor. If the externally applied current is below threshold, the injected current
will simply decay exponentially in both space and time (Fig. 2.5).
If, however, Iext exceeds the threshold, an action potential will be initiated in
that cell. The large voltage change during the action potential in that cell will cause
the voltage of neighboring cells to be pulled above threshold. In this way, the action
potential propagates along the cable. Figure 2.6 shows the action potential as it
propagates along the cable of tissue. Away from the stimulus site and the physical
end of the cable, the action potential is simply shifted along the cable, without
changing size or shape.
27
Figure 2.6: Waves in excitable media. The cardiac model described by Eq. 2.4 is
implemented in a 5 cm cable. An external current is applied at the left side of the
cable. The action potential is initiated in the cell at the left end and propagates
through voltage diffusion to neighboring cells.
2.3
Effect of Boundaries
Figure 2.6 shows that an electrical wave in cardiac tissue will propagate without
changing size and shape when not near any boundaries. When the wave hits a
boundary, we know that it must die off (see Sec. 2.2.1) because the excitation cannot
move forward or backward at that point. It turns out that this process of dissipation
can lead to changes in the size and shape of the wave as previously observed in models
of cardiac tissue [60–63] (Fig. 1.4). Over the years, several possible explanations for
this boundary effect have been put forward.
2.3.1
Passive Length Constant
A well-known property of cardiac tissue is the passive or subthreshold response of
cardiac tissue. As shown in Fig. 2.5, a subthreshold injection of current will decay
28
over time and space. The length over which one expects the passive response to
decay is known as the passive length constant, λ. Early experiments suggested that
any spatial variation of action potential shape only occurred over scales on the order
of λ. Some action potential properties such as action potential amplitude [57–59],
the sharpness of the upstroke as measured by dV /dtmax [58, 59], and the conduction
velocity [57, 59] only vary spatially within ∼ λ of boundaries.
Unfortunately, the passive electrical response does not explain the spatial variation of APD observed in most cardiac models; the spatial scale of APD variation is
typically much larger than the passive length constant. For example, the two-variable
√
model (Eqs. 2.1 and 2.2) has a passive length constant given by λ = Dτout (see
App. E for a derivation of this result). For the parameters used to generate Fig. 1.4,
λ = 1 mm. This is substantially shorter than the ∼1 cm over which the APD varies.
A similar discrepancy between the spatial scale of APD variation and λ was seen by
Sampson and Henriquez in simulations using more detailed cardiac models [60]. Passive length constants in real cardiac tissue are similarly short, ranging from ∼0.2-2
mm [52–56].
2.3.2
Changes in Membrane Resistance
A proposed explanation for the observed spatial variation in APD is that the passive length constant changes during the action potential. The reasoning behind this
conjecture is that the passive length constant is determined by cellular resistances,
λ=
s
Rm
,
(Ri + Re )β
29
(2.5)
where Rm is the cellular membrane resistance, Ri is the intracellular resistance, Re is
the extracellular resistance, and β is the ratio of membrane surface to tissue volume
[90]. Simulations of cardiac models show that the membrane resistance varies during
the action potential [60] and experiments reveal that both the membrane resistance
and the intracellular resistance vary during the action potential in canine cardiac
tissue [56].
In particular, a computer simulation study by Sampson and Henriquez suggests
that the membrane resistance increases during the action potential, with a particularly large increase around the downstroke and the beginning of the refractory period [60]. In this study, two different models were used: a mouse cardiac model
developed by Pandit et. al [91] and a modified version of the Luo-Rudy cardiac
model [92] that models rabbit cardiac dynamics. Sampson and Henriquez found that
changes in Rm resulted in an increased length constant, λef f . They found that, during the downstroke, λef f reached maximum values of 1.64λ for the Pandit model and
1.98λ for the Luo-Rudy model. Similar changes were seen experimentally in canine
Purkinje fibers by De Mello [56]. De Mello measured the passive length constant during the downstroke and beginning of the diastolic interval and found that λef f ∼ 1.5λ.
Unlike computer simulations, the change in length constant during experiments can
be attributed to changes in both Rm and Ri .
Note that λef f is not the same as the total distance over which APD varies (Fig.
2.7). In one dimension, λef f is the distance x for which
1
AP Dmid − AP D(x)
= ,
AP Dmid − AP D0
e
30
(2.6)
Figure 2.7: Definitions of length scales of APD variation. The figure shows the
insulated boundary of Fig. 1.4. The total spatial variation of APD, that is the
distance over which the APD varies from AP D0 to AP Dmid is ∼0.7 cm or ∼ 7λ. The
effective length constant, as defined by Eq. 2.6 is 1.57λ.
where AP Dmid is the APD away from the boundary and AP D0 is the APD at the
boundary. In Fig. 2.7, λef f = 1.57λ while the total distance over which APD varies
is ∼ 7λ.
2.3.3
Blocked Current Flow
The idea of blocked current flow was first proposed by Goldstein and Rall to explain
action potential shape changes that occur over spatial scales on the order of λ [57].
They suggested that the observed changes were due to charge buildup in cells near
the boundary. Figure 2.8 shows this process. Away from the boundaries, ions can
flow freely from a cell to its neighbor. Near the boundary, however, the current flow
is stopped since we must have dV /dx = 0 at the boundary and the ions build up in
the cells.
Although this idea was proposed to explain changes seen over length scales on the
31
Figure 2.8: Charge buildup at an insulated boundary. Since the current cannot flow
past the boundary, charge builds up in the cells near the boundary. This causes the
cells to repolarize more rapidly than cells in the middle of the cable.
order of λ, more recent mathematical analysis, motivated by the experimental results
presented in this thesis, has shown that charge buildup actually occurs over a length
scale longer than λ [63]. Cain and Schaeffer analyzed the APD of the two-variable
cardiac model (Eqs. 2.4 and 2.2) near an insulated boundary. They found that the
APD varies over a length scale of
λef f ∼
Note that
√
τout
τclose
−1/6 q
Dτout .
(2.7)
Dτout is the passive length constant of the two-variable model (See App.
E). For the example shown in Fig. 1.4, the width of the boundary layer is 1.57λ.
This result can be extended, at least approximately, to real cardiac tissue. Note
that for the two-variable model the duration of the plateau phase is ∼ τclose and the
1/3
1/3
duration of the repolarization phase is ∼ τclose τout [63]. Thus, a possible method for
32
Figure 2.9: Phases of the action potential. The action potential begins with the
depolarization phase (also called the upstroke), characterized by a rapid increase in
transmembrane voltage. This is followed by a plateau where the voltage remains
nearly constant. The voltage returns to the rest state during the repolarization
phase (also called the downstroke). Start and end times of each of the phases of the
action potential are typically defined as a percentage of the amplitude (See App. B).
Data is from a microelectrode recording of an action potential in bullfrog ventricular
myocardium.
calculating the boundary width in real cardiac tissue is to use the approximation
λef f ∼
trep
tplat
!−1/4
λ,
(2.8)
where trep is the duration of the repolarization phase of the action potential, tplat is the
duration of the plateau phase of the action potential (Fig. 2.9), and λ is the passive
length constant of the tissue. For typical bullfrog action potentials, tplat > trep ,
so λef f > λ. Unfortunately, this prediction is difficult to test since tplat and trep
are sensitive to the definition of the end time of the plateau phase. Changing the
definition of the end of the plateau phase from 20% of the action potential amplitude
to 30% of the action potential amplitude leads to changes in λef f of as much as λ.
33
2.4
Stability of Plane Waves
One of the reasons that the spatial variation of APD near the boundaries of cardiac
tissue is of interest is because spatial variation of APD has been implicated in the
development of alternans. The theory postulates that large gradients in APD lead
to conduction block, which can then degenerate into fibrillation [30, 31, 76]. To understand why this postulate is reasonable, consider an electrical wave propagating
through a piece of cardiac tissue that is accompanied by a steep APD gradient. The
next wave will propagate until it reaches the region of steep APD gradient, where it
will be forced to stop since that region of tissue has not yet recovered from the first
wave. In this way, steep APD gradients can lead to conduction block. The conduction block can itself lead to more complex arrhythmias. Suppose the region of steep
APD gradient is smaller than the length of an approaching wave front. Only the
part of the wave that encounters the steep gradient will be blocked. The remainder
of the wave will continue propagating. The next wave will still be affected by the
region of steep APD gradient because that region has had longer to recover than the
surrounding tissue. The APD in that region will be longer than the APD in regions
where there was no conduction block. This situation sets up different regions of steep
APD gradient, leading to conduction block in different parts of the tissue. Eventually, these moving regions of conduction block can disrupt subsequent plane waves to
such an extent that the rhythm degenerates into spatiotemporal chaos. Thus, spatial
heterogeneity of APD as an indicator of arrhythmogenecity is not only supported by
some experimental evidence [30, 66, 67, 69, 73–75], but can be plausibly linked to the
development of arrhythmias.
34
There is other experimental evidence that suggests that spatial heterogeneity of
APD does not necessarily lead to arrhythmias. The experiments of Qin et al. [68]
did not see the link between APD and arrhythmias, although they did not observe
gradients as large as those observed by other groups. Many experiments [40,42,64–72]
have observed spatial heterogeneity of APD in mammalian tissue, even during stable
1:1 response. These results suggest that, although steep APD gradient may be a route
to arrhythmia, it is either not the only route to arrhythmia or it requires some other
condition to act in conjunction with the steep APD gradient to lead to arrhythmias.
2.5
2.5.1
Experiment Overview
Choice of Experimental Substrate
I am interested in studying the spatiotemporal patterns induced near the boundaries
of cardiac tissue. The simulations that predict an increased boundary effect initially
assume homogeneous tissue with two effective boundaries: a stimulus site, where
current flows into the system, and one or more insulated boundaries, beyond which
current cannot flow. To attempt to reproduce these results, I need a test-bed that
ideally consists of a homogeneous piece of tissue without any specialized structures or
different cell types upon which I can then impose boundaries to study the boundary
effect.
Most studies of cardiac dynamics are done in mammalian cardiac tissue because
mammalian hearts are anatomically similar to human hearts. Mammalian hearts
consist of four chambers: two atria and two ventricles. Heartbeats are initiated by
pacemaker cells in the sino-atrial node and propagate first through the atria. The
35
electrical signal then passes along specialized conduction pathways, called Purkinje
fibers, that pass the electrical signals from the atria to the ventricles [27]. The
ventricles themselves consist of cylindrically-shaped cells aligned in a brick-wall type
pattern whose orientation changes as we move from the outer wall to the inner wall
of the ventricle [93]. The fiber structure of mammalian cardiac tissue leads to a
preferred direction of propagation along the long axis of the cylinder [94]. A further
complication in mammalian tissue is that there is evidence for different types of
cells as we move from the outer wall to the inner wall [95]. Finally, mammalian
hearts have blood vessels running throughout the tissue to supply the cardiac cells
with nutrients [27]. Mammalian cardiac tissue is clearly not suitable for this type of
research.
Most amphibians have hearts that consist of three chambers: two atria (also
called auricles) and one ventricle. The heartbeat is again initiated by pacemakers
in the atria, but amphibian hearts do not have Purkinje fibers to rapidly conduct
the electrical signal [96]. There is also no evidence for different types of cardiac
cells in the ventricle nor any evidence of fiber structure [97, 98]. Finally, there is no
vasculature in amphibian ventricles [97–99]. The tissue receives nutrients entirely by
diffusion [100]. Figure 2.10A shows a longitudinal slice of bullfrog ventricle. The heart
has been stained with hematoxylin and eosin, a common method used in histology and
anatomy. Nuclei are stained “blue” with hematoxylin while connective and all other
tissues are counterstained “red” with eosin. The figure shows clumps of cardiac tissue
separated by empty space. The empty space is filled with blood and then emptied
with each contraction. The clumps of cardiac tissue do not show any specialized
structures and are arranged in a seemingly random pattern. Figure 2.10B shows a
36
A
B
Figure 2.10: Frog heart histology. (A) A longitudinal cross-section of a bullfrog
ventricle stained with hematoxylin and eosin. The ventricle consists of clumps of
tissue interspersed with empty space. (B) A magnified view of the same piece of
tissue. The clumps consist of cardiac cells oriented in random directions.
magnified version of the same piece of tissue. Here we can see more clearly that
the cells within the clumps of tissue are oriented randomly meaning there is no fiber
structure within the tissue. Thus amphibian ventricular tissue is a more homogeneous
than mammalian cardiac tissue and so is a much better fit for the purposes of my
experiment.
Although frog hearts are not anatomically similar to human hearts, their response
patterns are similar to those observed in humans [101, 102], so there is reason to
assume that results of experiments performed in frog tissue will be relevant to human
hearts. Unlike mammalian hearts, which may be anatomically closer to human hearts,
frog hearts show little anisotropy (Fig. 2.11) and so frogs can provide a homogeneous
substrate on which to study spatial patterns and thus determine whether spatial
patterns can be dynamically induced in cardiac tissue.
37
Figure 2.11: Wave propagation in frog cardiac tissue. Contour lines denote the
wave front initiated from the electrode at 0.5 ms intervals. The wave initially propagates slightly faster along the vertical direction (slightly elliptical contour near the
electrode), but then begins to propagate slightly faster along the horizontal direction
(compare width of the contour indicated by the double arrows). This suggests that
there is no fixed anisotropy in frog cardiac tissue.
2.5.2
Size of the Experimental Substrate
The subthreshold response of frog cardiac tissue is characterized by the passive length
constant and the passive time constant. These constants are the temporal and spatial lengths over which a subthreshold stimulus decays to 1/e. The passive length
constant, λ, of frog tissue is 0.3 mm and the passive time constant is 4 ms [52].
Simulations suggest that spatial variation of APD occurs over a much larger scale
and thus is likely not driven by passive tissue processes alone. Simulations suggest
that APD variation has an effective length constant of ∼1.5-2λ, but can vary over a
total distance of ∼ 10λ (Fig. 2.7). Thus, I expect to see APD vary over distances of
∼3 mm in frog cardiac tissue. Since there are two boundaries (the stimulus site and
the cut edge), I require the tissue to be at least 6 mm to properly resolve the spatial
38
variation of APD. The tissue samples used in my experiments are approximately
10 mm by 10 mm, larger than the expected total spatial variation of APD so my
experiments should be able to resolve whether spatial variation of APD is a boundary
effect similar to those predicted by computer simulation.
2.5.3
Measurement Techniques
Two different techniques are used to measure the transmembrane voltage in the experiments described in the following chapters. This section provides a brief introduction
to these techniques and some of the rationale for using them in our experiments.
Microelectrode
The microelectrode represents the gold standard for measuring the transmembrane
voltage. The microelectrode is a small glass capillary one end of which has been
pulled to a very fine tip just a few microns wide. The capillary is filled with 3 M KCl
and a wire is inserted into the wide end. The fine tip is inserted directly into a cell
and provides a direct electrical connection to the intracellular solution [103].
Figure 2.12A shows the signal from a properly impaled microelectrode. We can
clearly see the rapid depolarization, the plateau and slow repolarization of the classic
action potential. Figure 2.12B, on the other hand, exhibits some of the problems
that can arise when using microelectrodes. The first 3000 ms show the signal from a
microelectrode that is not properly impaled. It may be sitting just outside the cell
or may have punctured the cell, but the cell membrane has not formed a seal around
the electrode, permitting current to leak out of the cell. At around 3000 ms, the
cell becomes properly impaled, but we do not yet see the classic action potential.
39
Figure 2.12: Microelectrode signals. (A) shows the signal form a properly impaled
microelectrode. (B) shows the signal from a microelectrode that is not properly impaled for the first 3000 ms and exhibits motion artifact once impalement is achieved.
Both signals are recorded from a small piece of bullfrog ventricle that is paced at
BCL = 1000 ms. The signal is passed through an amplifier with 10x gain.
The depolarization of the action potential is disrupted by a spurious signal that is
caused by the contractile motion of the tissue. The contractile motion can not only
cause distortions of the electrical signal, but can also break the electrode, or more
commonly, push the electrode out of the cell. For this reason it is difficult to collect
microelectrode signals over long periods of time. It is also extremely difficult to record
simultaneous microelectrode signals at multiple locations, since each microelectrode
must be constantly monitored and frequently manually adjusted. I use microelectrodes in studies where I do not require measurement at many spatial locations and
can take advantage of the high signal-to-noise ratio (SNR) of these signals.
40
Optical Mapping
Optical cardiac signals are produced with the aid of a fluorescent voltage-sensitive
dye. The most common voltage-sensitive dyes in use today are electrochromic dyes
which embed themselves inside the cell membranes. These dyes consist of molecules
that have a cloud of electrons on one end. When an electric field is applied parallel
to the length of the molecule, as is the case when the molecule is bound in the cell
membrane, the electron cloud is shifted along the molecule. This changes the energy
of the excited state, which in turn changes the absorption and emission spectra of
the dye. Electrochromic dyes are so popular today largely because they are very fast
(time constants on the order of 10−6 to 10−12 s) [104]. These dyes are also thought to
interfere very little with the cellular processes that cause the action potential [105].
It has also been shown that changes in fluorescence intensity accurately follow the
time course of the transmembrane voltage [106].
To take advantage of the voltage sensitive dyes, we need a light source to illuminate the tissue (white-light sources [25,107,108], lasers [109–111] and LEDs [112,113]
are commonly used) and a measurement device to capture the emitted light. In Appendix A, I discuss the development, testing and calibration of an optical imaging
system using new ultra-high-power LEDs. By using spatially extended measurement
devices such as cameras or photodiode arrays, we can easily make simultaneous measurement of the optical signal at many spatial locations. Unfortunately, the drawback
to using optical measurement lies in the nature of the fluorescence process. Since fluorescence is a random process, optical signals tend to be rather noisy (Fig. 2.13).
In comparison to the microelectrode signal, the baseline and plateau of the action
41
Figure 2.13: Optical signal. The optical signal is recorded from a small piece of
bullfrog ventricle that was stained with di-4-ANEPPS, a potentiometric dye, and is
paced at BCL = 1000 ms. Intensity is negative since the signal has been inverted
to assist in comparison to the microelectrode signal. Raw optical are the inverse of
traditional electrode recordings since fluorescence decreases with increasing voltage.
potential are not clearly defined. Thus, optical signals are used when some of the
SNR, and accuracy of APD measurements, can be sacrificed to the need for making
widespread spatial measurements.
42
Chapter 3
Spatial Variation of Action Potential
Duration
3.1
Introduction
Computer simulations of cardiac models suggest that APD exhibits spatial variation
in the form of a larger than expected boundary effect. Specifically, the models predict
that the APD is longest near the stimulus site, where current flows into the system,
and shortest at insulated boundaries, beyond which current cannot flow. In this
chapter, I present the results of experiments using optical mapping techniques that
permit simultaneous measurement of APD at all points on the surface of the tissue
and will determine whether a similar boundary effect occurs in real cardiac tissue.
3.1.1
Background
Many cardiac imaging studies have shown spatial variation in action potential duration (APD) during stable 1:1 response [40, 64–72]. None of these experiments, were
specifically looking for a boundary effect, however, so the observations were largely
made away from any boundaries. It is likely then that the observed spatial variation
of APD may have been due to underlying tissue heterogeneity [114]. Of the experiments that noted a boundary effect [40, 64, 72], in the form of increased APD near
the stimulus site, none measured the spatial extent of the variation in APD.
43
3.1.2
Experiment Overview
In this chapter, I present measurements of the spatial variation of APD in a small
piece of bullfrog ventricular myocardium. As discussed in section 2.5.1, there is no
evidence of fiber structure [97, 98], vasculature [97–99] or Purkinje fibers [115] in the
bullfrog ventricle.
In my experiments, I introduce boundaries by cutting a small piece of the ventricle
to use as an experimental substrate (See App. 3.2.1). Early experiments on the effect
of cutting cardiac tissue show that the boundary created by cutting the tissue can be
treated as having infinite resistance [116]. Thus, the tissue in my experiments can be
treated as a homogeneous sheet with insulated boundaries, similar to the substrate
assumed in computer simulations. This makes it a good substrate to determine
whether the spatial variation of APD is a boundary effect similar to the boundary
effect observed in computer simulations. I also present simulations of the simplified
two-variable cardiac model (Eqs. 2.1 and 2.2) in two dimensions to compare with
my experimental results. Finally, I use the mean spatial gradient as a measure of
the amount of spatial variation in APD and determine whether this measure can
differentiate between tissue that exhibits complex rhythms and tissue that transitions
directly to 2:1 behavior at rapid pacing.
3.2
3.2.1
Methods
Tissue Preparation
This study was performed in accordance with a protocol that conforms to the Research Animal Use Guidelines of the American Heart Association and was approved
44
by the Duke University Institutional Animal Care and Use Committee. Twelve American bullfrogs (Rana Catesbeiana) were anesthetized by immersion in a 1% solution
of aminobenzoic acid ethyl ester in distilled water and double-pithed. The heart was
excised and a cannula was inserted into the ventricle through a small incision in the
left auricle. The heart was perfused with standard Ringer’s solution (100 mM NaCl,
2.70 mM KCl, 5.6 mM glucose, 1 mM hepes, 2.8 mM Na2 HPO4 , 25 mM NaHCO3 ,
1.5 mM MgCl2 , 1.80 mM CaCl2 [117]) and 5 µM di-4-ANEPPs, a voltage-sensitive
dye. The staining solution was re-circulated for a minimum of 10 minutes (longer if
the tissue was not adequately stained, i.e., the emitted fluorescence did not saturate
the camera). Once the tissue was stained, the cannula was removed and the anterior
surface of the ventricle was cut from the heart and pinned in a dish (Fig. 3.1). The
tissue was superfused with Ringer’s solution bubbled with 95% O2 /5% CO2 . The tissue was paced with a silver unipolar electrode at a constant basic cycle length (BCL)
of 1,000 ms for 20 minutes, to allow it to recover, before any pacing protocols were
performed. While the tissue was recovering, between 10 mM and 20 mM diacetyl
monoxime (DAM) was added to the Ringers solution to eliminate tissue contraction.
3.2.2
Optical Recordings
A schematic of the experimental setup is shown in Fig. 3.2. The tissue is illuminated
with ultra-high power cyan LEDs (Lumileds Star/O, see App. A). The light is
absorbed by the voltage-sensitive dye and is emitted at higher wavelengths. As the
transmembrane voltage changes, the absorption and emission spectra of the dye shift
slightly, permitting us to track changes in the transmembrane voltage. It has been
45
Figure 3.1: Tissue Chamber. The tissue is pinned down in a custom-made tissue
chamber. Oxygenated solution is pumped into the chamber (lower hole in back) and
taken out through a hole on the other side of the chamber to be re-oxygenated and
recirculated.
shown that the change in intensity is linearly proportional to the change in voltage,
so the optical signal accurately tracks the action potential [118]. The emitted light
is passed through a high-pass cutoff filter (Edmunds Optics, OP590) to prevent any
emission light from entering the detection device. I collect the emitted light with
a high-speed 14-bit CCD camera (Ixon, Andor Technologies). The camera collects
128x128 pixels at 500 frames per second. The camera is controlled by a PCI camera
controller (Andor Technologies) installed on a Dell computer.
3.2.3
Pacing Protocol
Once the tissue has stabilized and the DAM has taken effect, three unipolar silver
electrodes are placed on the tissue (Fig. 3.3). Electrode 2 is placed along the edge
that was connected to the auricles while electrodes 1 and 3 were placed along the
remaining two edges. The following pacing protocol is used to determine spatial
46
Figure 3.2: Experimental setup. Light from two cyan LEDs is focused onto a small
piece of cardiac tissue that has been stained with di-4-ANEPPS. The fluoresced light
emitted by the tissue is filtered through a high-pass filter and collected by a high-speed
CCD camera.
heterogeneity of APD:
1. Tissue is paced with 2 ms current pulses from one of the three electrodes (chosen
at random) at a constant BCL for two minutes until steady state is achieved.
(In previous studies the time constant to reach steady state was measured at
∼30 s in bullfrog ventricular myocardium [43].)
2. Images of steady state pacing are collected for 10 s.
3. Pacing is switched to another electrode and steps 1 and 2 are repeated.
4. Pacing is switched to the third electrode and steps 1 and 2 are repeated.
5. The BCL is decreased by 50 or 100 ms and steps 1-4 are repeated at the new
BCL.
The use of three electrodes at three pacing sites is to determine whether any spatial
patterns that are observed are caused by spatial heterogeneity of the tissue or are
dynamically induced. Although bullfrog ventricular tissue is basically homogeneous
[99], individual frogs may have congenital defects or patches of dead tissue may
47
Figure 3.3: Electrode Placement. Three unipolar silver electrodes are placed along
the three edges of the tissue.
develop during tissue preparation. Previous work by Sampson and Henriquez [119]
has shown that wave fronts get stuck around regions of spatial heterogeneity. By
sending waves from three different directions, we will be able to identify any frozen
in heterogeneity if we see the same spatial pattern emerging from waves coming from
different directions.
3.2.4
Data Analysis
Only pixels with a mean intensity greater than 4000 digital numbers (DN, the measurement unit of the CCD camera) and an action potential amplitude greater than
150 DN are processed. 4000 DN is the dark noise of the camera, as measured by
collecting 10 s of images with the lens cap on. The data is filtered with a 3-point
temporal median filter before a custom-written Matlab code (App. D) determines
48
depolarization and repolarization times at 70% of the amplitude of the wave (App.
B). Action potential duration (APD) is determined by subtracting the depolarization times from the subsequent repolarization times. The wavefront is defined by the
depolarization time or start of the action potential and the waveback is defined by
the repolarization time or end of the action potential. APDs presented in the maps
are the mean steady-state APD.
Tissue Heterogeneity
As mentioned in the previous section, it is possible for tissue from some animals to
have defects that will cause frozen in spatial APD patterns. To determine whether
tissue in our experiments exhibits such frozen in APD patterns, I study APD difference maps defined by:
∆AP Di,j = |Ai − Aj | i, j = 1, 2, 3,
(3.1)
where Ai is the APD map produced by pacing from electrode i. If the spatial variation
in APD is due to underlying tissue heterogeneity, then changing pacing location will
not change the APD map; thus, we will have ∆AP Di,j = 0. Since this is experimental
data, the ideal value will not be achieved, so I will use a threshold of ∆AP Di,j < 10
ms. As a measure of the amount of frozen in heterogeneity, I determine the mean
value of ∆AP Di,j . A value of ∆AP Di,j less than the measurement error of 10 ms
(See App. B) suggest that changing the pacing location did not cause changes in the
spatial APD distribution.
49
Width of the Boundary Layer
Following the example of Cain and Schaeffer [63], the width of the boundary layer,
λef f , in one dimension is defined as the x value for which
1
AP Dmid − AP D(x)
= ,
AP Dmid − AP D(0)
e
(3.2)
where AP Dmid is the APD away from the boundary and AP D(0) is the APD at the
boundary. The definition of the boundary layer is applied along lines emanating from
electrodes 1 and 3 and going to the opposite edge of the tissue (Fig. 3.4). Electrode
2 is not used since the electrical activity initiated by electrode 2 seems to follow a
specialized conduction pathway (see Results and Discussion) and does not produce
a spatial APD pattern that agrees with simulations. Experimentally, AP Dmid is
determined by the mean of the 5 APDs at the center of the line and AP D(0) is the
APD at the insulated boundary. I calculate the mean boundary width along the
lines using only lines that cross more than 4 mm (twice the estimated width of the
boundary width) of tissue.
APD Gradient
For each pixel, I determine the local APD gradient:
∆AP Di,j =
s
AP Di+1,j − AP Di−1,j
2d
2
AP Di,j+1 − AP Di,j−1
+
2d
2
,
(3.3)
where ∆AP Di,j is the APD gradient at the pixel (i, j), AP Di,j is the APD measured
at the pixel (i, j), and d is the distance between pixels.
50
Figure 3.4: Calculation of boundary width. (A) Lines used to calculate the width
of the boundary layer from electrode 1 and (B) lines used to calculate the width of
the boundary layer from electrode 3.
I also attempt to use the spatial APD gradient to determine whether spatial
variation in APD can be correlated to the onset of arrhythmias. In six animals, at
least one trial lasted until either 2:1 or an arrhythmia, either an M:N response with
N 6= 1 or an irregular response, was observed (in other experiments the tissue stopped
responding to stimuli before either of these behaviors was seen). In these six animals,
4 trials exhibited a complex rhythm (Fig. 3.5) while 10 went from a 1:1 response to
a 2:1 response. The data is separated into two groups: those that exhibit a complex
rhythm and those that go directly to a 2:1 response. I calculate the mean APD
gradient (∆AP D is the average of the local APD gradients ∆AP Di,j (Eq. 3.3) over
the entire tissue) as a measure of the amount of spatial variation of APD. The mean
APD gradient has been used in previous studies as a measure of the amount of spatial
heterogeneity of APD [65,68,70,74]. Other measures of spatial heterogeneity are also
commonly used (spatial gradients measured between two specific sites [67,69,73,120]
and maximum gradient [66, 73]), but the measurement error of optical experiments
51
Figure 3.5: Examples of complex rhythm. Both examples are at BCL=300 ms.
causes these measures to be too imprecise to be useful (see Sec. 5.2.3 for a detailed
discussion). Note that the mean spatial APD gradient likely neglects too much
information about the spatial variation of APD to enhance our understanding of the
role of APD spatial heterogeneity in the onset of arrhythmias, but it may provide a
simple measure that can differentiate between tissue that exhibits complex rhythms
and tissue that does not exhibit complex rhythms, which may prove to be clinically
useful.
To allow proper comparison of ∆AP D between trials, BCL is normalized by
subtracting the BCL at which the transition occurs (BCLt ), BCLN = BCL − BCLt .
The range of BCLt is shown in Fig. 3.6. Differences in ∆AP D between the two
groups are statistically analyzed using SAS 9.1 (SAS, Cary, NC). Differences are
considered significant if p<0.05.
52
Figure 3.6: Range of BCLt . The transition BCL ranged from 200 ms to 400 ms.
See Table 3.3 for more details.
3.2.5
Simulations
To improve understanding of the spatial APD patterns that I observe in my experimental preparation, I ran simulations of steady-state pacing using the two-variable
cardiac model [87, 88] presented in Sec. 2.2.2. The tissue has a complicated shape,
so, for easier comparison between experiment and simulation, I use an image of the
tissue taken during one experiment to create a digital mask that defines the simulated tissue. The mask consists of a 2x2 matrix of 1s and 0s, where a 1 indicates
a location with intensity greater than 4000 DN and a 0 indicates a location with
intensity less than 4000 DN. The mask is modified slightly to correct for shadows
cast by the electrodes and by the pins that hold the tissue in the dish by changing
the 0s to 1s at these locations. Finally, the mask was subdivided into cells of size
0.1 mm on which the voltages were calculated using time steps of 0.01 ms. Three
53
stimulus locations were used, matching the stimulus sites of the experimental tissue.
The following parameter values are used: D = 0.001 cm2 /ms, τin = 0.2 ms, τout = 10
ms, τopen = 130, τclose = 150, and Vc = 0.13. I apply boundary conditions of δx V = 0
and δy V = 0 on the boundaries. The boundary width is calculated using same procedure as for experimental data. Note that the passive length constant for the model
is 1 mm, somewhat longer than the passive length constant of 0.3 mm for bullfrog
cardiac tissue. Simulation code is presented in App. D.
3.3
3.3.1
Results
Spatial Heterogeneity
Table 3.1 shows ∆AP D for all experiments. All experiments have one ∆AP D < 10
ms. A small value of ∆AP D indicates that, at most locations on the tissue, there
is little difference in the APD resulting from pacing at two different locations. In
all experiments, the APD spatial pattern produced when pacing from electrode 2 is
similar to the spatial APD pattern produced when pacing from either electrode 1 or
electrode 3. This will be discussed further in the next section. Three experiments
(denoted by *) show similar APD patterns when pacing from all three electrodes.
Upon visual inspection of these three experiments, it is noted that APD maps are
nearly identical no matter which pacing electrode is used (Fig. 3.7). I conclude that
these three experiments show evidence of frozen-in heterogeneity, potentially due to
structural heterogeneity of the tissue. These three experiments will not be included in
further analysis since I am interested in studying dynamically induced heterogeneity
of APD.
54
Figure 3.7: Frozen-in heterogeneity. APD varies from ≈500-650 ms (blue=500 ms
and red=650 ms) over the surface of the tissue. Note that even thought the pacing
location changes in each of the three panels, this does not cause large changes in the
spatial APD pattern in this experiment; the longest APDs remain near the upper
left side of the tissue. Data shown is from experiment #1 of Table 3.1.
55
Experiment Number ∆AP D1,2 (ms) ∆AP D1,3 (ms) ∆AP D2,3 (ms)
1*
5.2±0.1
5.4±0.1
5.5±0.1
2
9.6±0.1
19.5±0.3
16.1±0.2
3*
6.9±0.2
7.4±0.2
6.4±0.2
4
7.6±0.2
12.9±0.3
11.5±0.2
5
6.1±0.1
11.1±0.2
13.0±0.2
6
19.2±0.3
12.4±0.2
9.5±0.2
7
13.5±0.3
14.3±0.2
4.2±0.1
8
14.2±0.2
15.4±0.3
6.7±0.3
9*
5.9±0.2
6.4±0.2
5.2±0.2
10
20.7±0.4
13.4±0.4
9.1±0.3
11
5.0±0.1
19.6±0.3
18.9±0.3
12
15.2±0.3
17.4±0.2
7.4±0.2
Table 3.1: Values of ∆AP D for all experiments. APD maps collected at BCL=1000
ms were used for the calculation of ∆AP D. The three experiments marked with *
show similar spatial APD variation from all three pacing sites. Error is determined
by standard error.
3.3.2
APD Maps
Figure 3.8 shows the activation and deactivation patterns from a single BCL of an
experiment that did not show frozen-in heterogeneity. These maps are constructed
by determining the time of activation (or deactivation), as defined in section 3.2.4
at each point on the tissue. The activation (or deactivation) times are then used to
construct contours as seen in Fig. 3.8. The contours give an indication of how the
wavefront (or waveback) spread across the tissue since each contour represents the
location of the wavefront (or waveback) at a moment in time. The contours also give
an indication of how quickly the wave moves from one location to the next; contours
that are further apart indicate a higher velocity since the wavefront (or waveback)
has traveled a larger distance in a given time period. The activation pattern when
pacing from electrode 2 shows a very rapid spread of the wave through the center
56
of the tissue. The activation waves from electrodes 1 and 3 show a more constant
wavefront velocity as the wave propagates across the tissue. The waveback typically
propagates faster than the wavefront. However, when pacing from electrode 2 the
waveback does not show the same initial rapid spread as the wavefront.
Figure 3.9 shows the APD maps resulting from the activation and deactivation
maps of Fig. 3.8. The APD map is created by finding the APD at each location on
the surface of the tissue and applying a color map to the values. In Fig. 3.9, long
APDs are indicated by red and short APDs are blue; the exact range is indicated
by the color scale along the right side of each panel. When pacing from electrodes
1 and 3, the longest APDs are near the pacing electrode and decrease with distance
from the electrode. APD maps produced by pacing from electrode 2 show a pattern
similar to pacing from electrode 3. APD maps produced by pacing from electrode 2
also showed patterns similar to pacing from electrode 1 in some experiments. I never
saw APD maps where the APD was longest near the site of electrode 2.
The effect of changing BCL on APD maps is shown in Fig. 3.10. As the BCL
is decreased, the spatial APD maps also show slight changes, although the longest
APD remains near the stimulus electrode when pacing from electrodes 1 and 3. In
addition, the range of APD (AP Dmax − AP Dmin ) remains about 200 ms at all BCLs.
Figure 3.11 shows the activation and deactivation patterns from computer simulation using the model described in 3.2.5. The activation spreads from the stimulus
location, traveling at a fairly constant rate. The deactivation wave, on the other
hand, does not produce a target-like pattern and changes velocity as it propagates
over the tissue.
57
Figure 3.8: Experimental spatial patterns of activation and deactivation. Maps
of steady state activation and deactivation when pacing at BCL = 1000 ms from
(A,B) the upper right electrode, (C,D) the left electrode, and (E,F) the lower right
electrode. Contour lines are 5 ms apart. Data taken from experiment #8 of Table
3.1.
58
Figure 3.9: Experimental spatial patterns of APD. Maps of steady-state APD produced when pacing at BCL = 1000 ms from (A) the upper right electrode, (C) the
left electrode, and (E) the lower right electrode. Figures B, D, and F show the APD
along the lines indicated in A, C, and E, respectively. When pacing from electrodes
1 and 3, the longest APDs are near the stimulus electrode. When pacing from electrode 2, the longest APDs are near electrode 3 in this experiment. Data shown is
from experiment #8 of Table 3.1.
59
Figure 3.10: Effect of BCL on spatial APD distribution. APD maps produced when
pacing at BCL=1000, 800, 600, 400 ms. To produce these images, experimental data
has been fit to a cubic function. Data shown is from experiment #12 of Table 3.1.
60
Figure 3.11: Spatial patterns of activation and deactivation in a two-variable model.
Maps of steady-state activation and deactivation when pacing at BCL = 1000 ms from
(A,B) the upper right electrode, (C,D) the left electrode, and (E,F) the lower right
electrode. Contour lines are 2.5 ms apart.
61
Figure 3.12 shows the APD maps resulting from the activation and deactivation
maps of Fig. 3.11. In all three cases, the longest APDs are near the stimulus site and
decrease at the insulated boundary. As Figs. 3.12B, D, and F show, it is difficult
to distinguish whether there are two distinct boundary effects, one caused by the
injection of current by the stimulus and one caused by the inability of the current to
flow beyond the boundary, because there is no clear region of constant APD in the
center of the tissue.
3.3.3
Width of the Boundary Layer
The width of the boundary layer was calculated for the nine experiments that did
not exhibit frozen in heterogeneity. Sample experimental data used to calculate the
boundary width is shown in Fig. 3.13. In the example, λef f ∼ 2λ and the total distance over which APD varies is ∼ 8λ. Only spatial patterns generated from electrodes
1 and 3 are used to calculate the width of the boundary layer since spatial patterns
generated by electrode 2 appear to be influenced by tissue heterogeneity. Results for
all animals are presented in Table 3.2. λ1 refers to the width of the boundary layer
when pacing from electrode 1 and λ2 refers to the width of the boundary layer when
pacing from electrode 3. The boundary layer varies from 0.43-0.64 mm or ∼1.5-2.1λ
in experiment and 1.7-2.3λ in simulation.
3.3.4
APD Gradients
APD gradient is largest near the stimulus electrode, smallest in the center of the
tissue and increases again slightly near the insulated boundary (Figs. 3.14 and 3.15).
Note that the APD gradients observed in this experiment are much larger than 3
62
Figure 3.12: Simulated spatial patterns of APD. Maps of steady state APD produced when pacing at BCL = 1000 ms from (A) the upper right electrode, (B) the
left electrode, and (C) the lower right electrode. Figures B, D, and F show the APD
along the lines indicated in A, C, and E, respectively.
63
Experiment Number
2
4
5
6
7
8
10
11
12
Simulation
λ1 (mm)
0.62±0.06
0.43±0.04
0.53±0.02
0.53±0.07
0.56±0.04
0.64±0.08
0.44±0.02
0.55±0.03
0.60±0.05
2.3±0.1
λ3 (mm)
0.63±0.03
0.47±0.05
0.53±0.07
0.52±0.09
0.58±0.04
0.61±0.07
0.47±0.05
0.54±0.05
0.59±0.04
1.7±0.2
Table 3.2: Width of the boundary layer. APD maps collected at BCL=1000 ms
were used for the calculation of the width of the boundary layer.
Figure 3.13: Sample experimental data used to calculate λef f . In the example, the
boundary width is calculated for the insulated end of the cable. I find that λef f ∼ 2λ
and the total distance over which APD varies is ∼ 8λ.
64
Animal Pacing Electrode Complex Rhythm BCLt
1
1
no
300
1
2
yes
300
1
3
yes
400
2
2
no
200
2
3
yes
300
3
3
no
400
4
1
no
200
4
2
no
200
4
3
no
200
5
2
yes
300
5
3
no
200
6
1
no
300
6
2
no
300
6
3
no
300
Table 3.3: Summary of experimental trials indicating the occurrence of complex
rhythms, the BCL at which a change in response pattern was observed, and the
pacing electrode.
ms/mm, the value postulated to cause conduction block and arrhythmias.
The spatial APD gradient, ∆AP D, is shown as a function of BCL in Fig. 3.16.
The BCL dependence is shown separately for each pacing electrode. The pacing
location does not have a significant effect on ∆AP D, as the measured values agree
within error at all BCL.
Table 3.3 summarizes the trials used to study APD gradient differences in tissue
that exhibits complex rhythms, as defined in Sec. 3.2.4, and tissue that does not
exhibit complex rhythms. When trials are separated into those that exhibit complex
rhythms and those that do not exhibit complex rhythms, we see that trials that
exhibit complex rhythms tend to have larger ∆AP D (Fig. 3.17A). Statistical analysis
indicates that this difference is significant (Fig. 3.17B) at only some BCLs.
65
Figure 3.14: Experimental spatial patterns of APD gradient. Maps of steady state
APD gradient produced when pacing at BCL = 1000 ms from (A) the upper right
electrode, (B) the left electrode, and (C) the lower right electrode. Figures B, D, and
F show the APD gradient along the lines indicated in A, C, and E, respectively.
66
Figure 3.15: Simulated spatial patterns of APD gradient. Maps of steady state
APD gradient produced when pacing at BCL = 1000 ms from (A) the upper right
electrode, (B) the left electrode, and (C) the lower right electrode. Figures B, D, and
F show the APD gradient along the lines indicated in A, C, and E, respectively.
67
Figure 3.16: Mean spatial APD gradient. The mean spatial APD gradient averaged
over all animals is shown as a function of BCL. The APD gradient is independent of
pacing electrode and shows a slight decrease as BCL increases.
Figure 3.17: ∆AP D and complex rhythms. (A) ∆AP D for trials that exhibit
complex rhythms and those that go directly to 2:1. (B) P values below 0.05 (dashed
line) indicate ∆AP D is significantly different in the two groups.
68
3.4
3.4.1
Discussion
Comparison of Experiment and Simulations
I ran simulations using a two-variable model in two dimensions with the simulated
tissue having the same boundaries as the experimental preparation, permitting direct comparison of simulation and experiment. The simulation results were in good
agreement with experiment in the cases of pacing from electrodes 1 and 3. Both experiment and simulation showed longer APDs near the pacing electrode and shorter
APDs at the far boundaries. Both simulation and experiment wavefronts propagate
radially from the pacing electrode. Both simulation and experiment showed similar
boundary widths (∼ 1.5−2λ) when pacing from electrodes 1 and 3. Simulation APDs
were shorter than those seen in experiment and the range of APDs seen over simulated tissue was also smaller than in experiment. However, since this model is not
meant to accurately model frog action potentials, exact agreement is not expected.
Moreover, these discrepancies did not seem to affect the overall spatial pattern that
was observed.
The largest difference between experiment and simulation was seen when pacing
from electrode 2. In simulations, pacing from electrode 2 produced long APDs near
pacing electrode 2, whereas experiments showed long APDs either near electrode 1
or electrode 3, but never near pacing electrode 2. Experiments also showed a rapid
propagation of the wavefront along the center of the tissue (Fig. 3.8). This was
not seen in the simulations (Fig. 3.11). Finally, recovery propagation in experiment
showed two distinct pathways, either propagating towards electrode 3 or electrode
1, instead of radial propagation. These differences between experiment and simula69
tion could perhaps be explained by a conduction pathway in the tissue. Although
frog myocardium does not contain Purkinje fibers, the rapid conduction pathways
of mammalian myocardium, there is some recent evidence that frogs have trabecular
bands that extend from the auricles into the ventricle and may act as a conduction
pathway for electrical excitation [115]. Since electrode 2 is placed along the edge
where the auricles were connected and where these trabecular bands may be present,
an electrical impulse at that location may cause activation of the trabecular bands.
This would explain the rapid wavefront propagation through the center of the tissue
and the disagreement between simulation and experiment when pacing from electrode
2.
3.4.2
Spatial Heterogeneity of APD
Nine of the twelve experiments presented here showed spatial APD patterns similar
to the patterns produced by simulation of cardiac models [59–63], suggesting that
the boundary effect is present in bullfrog cardiac tissue. As the remaining three
experiments show, however, this boundary effect can be over-ridden by underlying
tissue heterogeneity since these three experiments exhibit a frozen-in spatial pattern
of APD. Also, when pacing from electrode 2, the possible activation of a specialized
conduction pathway leads to a spatial APD pattern that does not agree with that
predicted by simulation. Previous simulations confirm that tissue anisotropy can alter
the intrinsic spatial variation of APD, creating large APD gradients across rather than
along cardiac fibers [62]. This may explain the mixed results of other experiments,
with some tissue preparations exhibiting a boundary effect [40, 64, 72] and others
exhibiting different patterns of spatial APD variation [65, 66, 73, 74]. My studies
70
also suggest that some tissue heterogeneity can exist without completely over-riding
the APD boundary effect. All my experiments showed evidence of some specialized
conduction pathway that was activated when pacing from electrode 2. Yet when
pacing from either electrode 1 or 3, the APD boundary effect was observed despite
the existence of the trabecular bands.
The results of my experiments indicate that a state of spatially homogeneous APD
does not exist in paced cardiac tissue. Even if there are no insulated boundaries in
the tissue under study, the current injected at the stimulus site will cause changes in
the dynamics of the wave over scales larger than previously expected. Further, any
tissue inhomogeneities, such as physical damage to the tissue or dynamically induced
inhomogeneities, will also change the dynamics of wave propagation over distances
larger than previously expected.
3.4.3
Width of the Boundary Layer
I found that the width of the boundary layer is ∼1.5-2 times the passive length
constant of bullfrog cardiac tissue. Simulations of the Pandit model and the LuoRudy model found that the boundary layer increased to 1.64λ and 1.98λ [60] due to
changes in membrane resistance. More importantly, De Mello found that changes in
membrane and intracellular resistance increased the length constant 1.52λ [56] in dog
Purkinje fibers. Although membrane resistance changes during the action potential
are likely different in canine and bullfrog cardiac tissue, it is possible that changes
in membrane resistance can account for the increased boundary layer observed in
my experiment. The theory of charge buildup may also play a role in the observed
spatial pattern of APD. It too can account for the observed width of the boundary
71
layer. The two-variable model exhibits increased boundary layers of roughly the same
order of magnitude (1.7-2.3λ) as those observed in experiment. Without explicitly
tracking changes in membrane resistance or the movement of ions in the tissue, these
experiments cannot determine which of the two effects may be causing the increased
boundary width of APD.
The increased boundary width has some repercussions for the dynamics of electrical waves in cardiac tissue. Several simulation studies have indicated that obstacles
within the tissue, such as regions of dead cells or non-conducting structures within
the heart, can lead to breaks in the wavefront of propagating waves [121–123], if they
are large enough. My experiments suggest that the effects of an obstacle may be felt
over distances much greater than λ, so that even small obstacles may lead to changes
in wave propagation and possible arrhythmias.
The increased boundary width may also have an advantageous repercussion for
control of cardiac electrical activity. Several groups have attempted to control abnormal rhythms using small perturbations in the BCL to nudge the tissue back to
a stable 1:1 response [124–132]. Many of these experiments successfully controlled
cardiac dynamics locally [124–127], many researchers were unsure whether this technique could be used to control cardiac dynamics over a large piece of tissue or the
whole heart. The increased boundary width observed in this experiment indicates
that a stimulus injection site will alter APD as far as 10λ from the stimulus. This
distance greatly reduces the number of control sites that may be needed to properly
control an abnormal rhythm in the whole heart.
72
3.4.4
Stability of Complex Rhythms
Previous research has linked large spatial gradients (> 3 ms/mm) of APD to the
onset of cardiac arrhythmias [120], with subsequent research suggesting a possible
mechanism [30,133]. In this study, I observe even larger APD gradients (∇AP D ∼ 4−
10 ms/mm) during stable 1:1 response. There are also regions of very large local APD
gradients (>30 ms/mm) near the stimulus site and near the insulated boundaries,
yet these regions do not inhibit the propagation of the wave in my experiments. This
differs from the results of experiments by Laurite et al. [120], who observed that
regions of large spatial APD gradient led to conduction block, and Aiba et al., who
observed that ventricular fibrillation originated in regions of large APD gradient.
Other experiments seem to confirm my findings. Large APD gradients during stable
1:1 response have also been observed in other experiments [40, 134], suggesting that
APD gradients may not be a sufficient condition for the onset of arrhythmias.
The mean APD gradient was used as a measure of spatial heterogeneity to determine whether tissue that exhibits complex rhythms shows a different spatial pattern
of APD than tissue that does not exhibit complex rhythms. Although tissue that
exhibits complex rhythms tends to have a larger mean APD gradient, the difference
was only significant at some BCLs.
3.4.5
Study Limitations and Future Work
This study determined that an increased boundary effect, similar to what is observed
in computer simulation, is present in bullfrog ventricular tissue. The study could not,
however, explain the cause of the boundary effect. To properly determine the cause of
73
the large boundary width observed in my experiments, further experiments that track
the change in membrane resistance during an action potential or optical experiments
that track the movement of ions in the tissue may be needed. Such experiments will
help elucidate whether changes in membrane resistance or charge buildup near the
boundary are responsible for the large length constant of APD spatial variation in
bullfrog ventricle.
The study was also limited by the use of mean APD gradient as a measure of the
amount spatial heterogeneity in APD. Tthe mean APD gradient is not ideal since it
neglects directional information and ignores the spatial variation in the gradient, information that could be useful in distinguishing tissue that exhibits complex rhythms
from tissue that does not. It may be the neglect of the directional information that
leads to the discrepancy between the results presented in this chapter (Fig. 3.17) and
the results of the microelectrode study (Ch. 6). The use of a photodiode array may
permit more accurate measurement of a quantity that better characterizes the spatial
variation of APD and may provide a stronger correlation to the onset of arrhythmias.
Natural animal-to-animal variation also limited this study by affecting the size,
shape and physiological properties of the tissue sample. These differences make
direct comparison of spatial variation of APD from one animal to the next difficult
because each sample has different boundaries and has stimulus electrodes at slightly
different locations. Since the tissue is also slowly dying as the experiment progresses,
regions of dead cells will increase in size or will appear at new locations in the tissue
which will also cause differences in the observed spatial variation of APD. The ideal
tissue sample for this experiment is a one-dimensional strip of tissue. This would
allow propagation in only one direction and provide more consistent sample size and
74
placement of the stimulus electrode. Due to cell death near the boundaries, this type
of tissue sample does not work experimentally (See App. C). A possible solution is
to use cultured monolayers [135], which are a single layer of cardiac cells grown on
a slide. This substrate reduces the experiment to truly two-dimensions and has the
added advantage of being better able to control the size and shape of the sample and
better control the placement of electrodes.
3.5
Conclusions
The experiments presented in this chapter show that large APD gradients occur in
homogeneous cardiac tissue during stable 1:1 responses. The spatial variation appears
to be a boundary effect similar to that observed in computer simulations, although
in some cases the boundary effect appears to be over-ridden by tissue heterogeneity.
Although the APD gradient only correlates to the onset of alternans at some BCLs,
it may cause spatial variation of other cardiac restitution properties that may play a
role in the stability of the 1:1 response.
75
Chapter 4
Spatial Variation of Dynamic Restitution
4.1
Introduction
In the previous chapter, I found that APD exhibits spatial variation primarily in the
form of a boundary effect. Although the spatial variation of APD, as measured by
the mean APD gradient, and the development of complex rhythms at rapid pacing
were correlated at only some BCLs, the spatial variation of APD may affect the
stability of the 1:1 response through other mechanisms. The dynamic restitution
curve (DRC), determined by steady-state APD and DI, is thought to play a role
in the stability of the 1:1 response. Since the steady-state APD exhibits spatial
variation, it is likely that the DRC will also vary in space. In this chapter, I present
the results of experiments using optical mapping techniques that permit simultaneous
measurement of the DRC and the slope of the DRC, SDRC , at all points on the surface
of the tissue.
4.1.1
Background
Modern researchers believe that the diastolic interval (DI) determines the APD [136].
In other words, its the amount of time that the tissue has had to recover that determines the duration of the next action potential. In mathematical terms, this is
written as
AP Dn+1 = F (DIn ).
76
(4.1)
The function F that relates APD to the previous DI is known as a restitution curve
(RC). Many specific forms for the RC have been proposed [39, 137], but a specific
function is not needed for further analysis. Using non-linear analysis techniques [138],
we find that there is a single steady-state value of APD and DI (AP D∗ , DI ∗ ) when
|F (DI ∗ )′ | < 1, but that this fixed point becomes unstable and leads to a long-short
alternation in APD and DI once |F (DI ∗ )′ | > 1. This prediction is known as the
restitution hypothesis.
Several different pacing protocols have been proposed to experimentally determine
the RC (see Sec. 6.1.2 for full details). One of the most commonly used protocols
used to test the restitution hypothesis produces the dynamic restitution curve. The
DRC is constructed using steady-state APD and DI pairs. The experimental pacing
protocol that is used to determine the DRC is as follows:
1. Pace at a slow constant BCL until steady-state is achieved. In the case of
cardiac tissue, steady-state pacing is reached after pacing for 2-3 times the
time constant of accommodation, τ (typically, τ = 20 − 40 s [43, 139]).
2. Record the final APD and previous DI as a single point on the DRC.
3. Step down to a new faster BCL and repeat the process.
The process is depicted in Figure 4.1.
It is tempting to believe that this analysis can predict the BCL at which bifurcations occur and that the problem of cardiac stability is solved. Unfortunately, the
complex nature of cardiac tissue means that the solution is not quite so simple. The
restitution hypothesis was confirmed by experimental [46] and modeling [140, 141]
studies, but other experimental studies, by the Duke Cardiac Dynamics group [39]
and in other studies [40], have observed stable 1:1 responses when the slope of the
77
Figure 4.1: Dynamic Restitution Curve. The DRC is determined by the steady-state
DI and APD at different BCLs. The tissue is paced for 2-3 times the time constant
of accommodation, (τ ), and the final (DI, AP D) pair is one point on the DRC. The
process is repeated at different BCLs to determine the entire restitution curve.
RC was greater than 1 or stable 2:2 responses when the slope of the RC was less than
1 [41, 42].
4.1.2
Spatial DRC Slope Gradients
One possible reason for the failure of the restitution hypothesis is the multicellular
nature of the experimental preparations used to test the hypothesis [39–42]. The
results of the previous chapter suggest that there will be spatial variation in the
DRC, caused by the spatial variation of steady-state APD.
Several studies have measured the DRC at two or more spatial locations [64, 65,
67, 68]. Although these studies all noted that there were spatial differences in DRC,
the only study that performed a detailed study of the slope of the DRC and its spatial
variation was that of Qin et al. [68]. In their study, a 504 electrode plaque was used
to measure activation-recovery intervals (ARI) as a porcine heart was paced using a
78
dynamic restitution protocol. They found that the slope of the DRC did not exhibit
any consistent spatial gradient. Since this experiment was performed in tissue that
was not homogeneous, one cannot determine whether the observed spatial pattern
on SDRC was due to underlying tissue heterogeneity or whether it was dynamically
induced.
In this chapter, I present results of an experiment that determines whether there
is a dynamically induced spatial gradient of SDRC in real cardiac tissue and whether
spatial differences in SDRC can be linked to the tissues propensity to exhibit alternans
at rapid pacing.
4.2
Methods
The tissue was prepared as described in section 3.2.1. The optical mapping system
described in section 3.2.2 was used to measure transmembrane voltage changes during
the following pacing protocol.
4.2.1
Pacing Protocol
Once the tissue has stabilized and the DAM has taken effect, the following pacing
protocol is implemented.
1. Pace at a constant BCL for 1 minute.
2. Images of steady state pacing are collected for 10 s.
3. The BCL is decremented by 100 ms and steps 1 and 2 are repeated.
4. Repeat steps 1-3 until 2:2 or 2:1 behavior is observed.
5. Move the electrode to a new location and all the tissue to recover by pacing at
1000 ms for 10 minutes before repeating steps 1-4.
79
This chapter presents results from 18 trials from 10 animals (the tissue did not survive
to the end of a second trial in two of the animals).
There are several differences between this pacing protocol and the one used to
measure steady state APD maps. First, in this experiment we use a bipolar electrode
instead of a unipolar electrode. In the course of the experiments presented in the
previous chapter, it was found that the threshold current for initiation of the wave
was much higher when using the unipolar electrode. Due to equipment limitations,
it was often difficult to supply enough current to consistently initiate a wave at rapid
pacing (the threshold increases with decreasing BCL [142]) when using a unipolar
electrode. Since it was crucial that I be able to initiate waves at fast pacing in this
experiment, I decided to use the bipolar electrode. Also in this experiment, the entire
downsweep is performed with one electrode at a time instead of switching to different
electrodes at each BCL. This is done so that the entire downsweep can be collected
before tissue characteristics change due to the slow death of the tissue. It is for this
reason also that the pacing is performed for only 1 minute instead of two.
4.2.2
DRC Slopes
Only pixels with a mean intensity greater than 4000 DN and a signal greater than 150
DN are processed. The data is filtered with a 3-point temporal median filter before a
custom-written Matlab code (App. D) determines depolarization and repolarization
times at 70% of the amplitude of the wave (App. B). Action potential duration
(APD) is determined by subtracting the depolarization times from the subsequent
repolarization times.
80
For each pixel, the (APD,DI) pairs at each BCL are fit to an exponential
DI
,
AP D = A − B exp −
τ
(4.2)
where A, B, and τ are parameters determined by the fitting process. The slope is
then given by the derivative of the exponential
SDRC =
4.2.3
B
DI
.
exp −
τ
τ
(4.3)
Spatial Gradients
The local spatial gradient is calculated as
i,j
∆SDRC
=
v
!
u
u S i+1,j − S i−1,j 2
t
DRC
DRC
2d
i,j+1
i,j−1
SDRC
− SDRC
+
2d
!2
,
(4.4)
i,j
i,j
where ∆SDRC
is the SDRC gradient at the pixel (i, j), SDRC
is the slope measured
at the pixel (i, j), and d is the distance between pixels. The mean SDRC gradient,
i,j
∆SDRC is the average of ∆SDRC
over the entire tissue.
Trials are divided into two groups: trials that exhibit alternans at rapid pacing
(ALT) and trials that do not exhibit alternans (noALT). There were 3 ALT trials
and 15 noALT trials. To properly compare measurements from different trials, the
BCL is normalized by subtracting the BCL at which either a 2:1 or 2:2 behaviour was
first observed (BCLt ), BCLN = BCL − BCLt . The range of BCLt is shown in Fig.
4.2. Statistical analysis using SAS 9.1 (SAS, Cary, NC) is performed to determine if
ALT and noALT trials show significant differences in ∆SDRC . A value of p < 0.05 is
81
Figure 4.2: Range of BCLt . The transition BCL ranged from 200 ms to 400 ms.
See Table 4.1 for more details.
considered significant.
4.3
Results
Figure 4.3 shows the spatial variation of the slope of the DRC when pacing from
two different locations on the same piece of bullfrog ventricle. The largest slopes are
near the stimulus and the slope decreases as we move away from the stimulus. The
slope increases as the BCL decreases, though the effect seems to be larger near the
stimulus site.
Figure 4.4 shows the SDRC gradient for the two trials shown in Fig. 4.3. While
both stimulus locations show some increase of the spatial gradient as BCL decreases,
pacing from the lower right side shows a more dramatic increase in the gradient.
This is significant because, when pacing at stimulus site 1, the tissue exhibited 2:2
82
Figure 4.3: Spatial variation of SDRC in a piece of bullfrog ventricle. The lower row
shows the results of pacing from an electrode placed along the upper right side of the
tissue. The lower row shows the results of pacing from an electrode placed along the
upper right side of the tissue. Results are shown for BCLs of 900, 700 and 500 ms.
Images are produced by fitting experimental data to a cubic surface.
83
response at BCL=300 ms, while pacing from stimulus site 2 resulted in a 2:1 response
at BCL=200 ms. This observation suggests that SDRC gradient is larger just before
a transition from 1:1 to 2:2 response than just before a transition from 1:1 to 2:1
response.
Table 4.1 summarizes the trials used to study ∆SDRC differences in ALT and
noALT trials. Figure 4.5 shows that there is a dfference in ∆SDRC between ALT
and noALT trials. The mean spatial gradients in trials that do not exhibit alternans
remain below 0.5 /mm for all BCLs. In trials that exhibit alternans, however, the
mean spatial gradient rises above 0.5 /mm when pacing at a BCL about 200 ms
faster than the transition to alternans. Statistical analysis indicates that differences
in ∆SDRC are significant at slow (BCLN > 400 ms) and rapid (BCLN < 300 ms)
pacing.
4.4
4.4.1
Discussion
Spatial Variation of SDRC
Unlike the experiments of Qin et al., who did not see a consistent gradient in SDRC
[68], my experiments show that SDRC decreases as I move away from the stimulus
site. This difference could be because of the different experimental substrates used in
the two experiments. The underlying tissue structure of the guinea pig hearts used by
Qin et al. may have overridden any dynamically-induced spatial variation. Although
there is no direct evidence that this is the case for SDRC , the results of the previous
chapter have shown that tissue heterogeneity can affect the observed spatial patterns
of APD, although there is no evidence from the experiments presented here that SDRC
84
Figure 4.4: Spatial gradient of SDRC in a piece of bullfrog ventricle. The lower row
shows the results of pacing from an electrode placed along the upper right side of the
tissue. The lower row shows the results of pacing from an electrode placed along the
upper right side of the tissue. Results are shown for BCLs of 900, 700 and 500 ms.
∆SDRC for each map is given below the image.
85
Animal Trial Alternans BCLt
1
1
ALT
300
1
2
noALT
200
2
1
noALT
300
2
2
noALT
200
3
1
noALT
200
3
2
noALT
300
4
1
ALT
200
5
1
ALT
300
5
2
noALT
300
6
1
noALT
300
6
2
noALT
300
6
3
noALT
300
7
1
noALT
300
8
1
noALT
200
9
1
noALT
300
9
2
noALT
300
10
1
noALT
400
10
2
noALT
300
Table 4.1: Summary of experimental trials indicating the occurrence of alternans
and the BCL at which a change in response pattern was observed
Figure 4.5: Mean spatial gradients of SDRC as a function of BCLN . (A) ALT
and NoALT trials show differences in mean spatial gradient of SDRC . ALT trials
show a marked increase in the mean spatial gradient as the transition to alternans is
approached. (B) The t-test shows that differences in ∆SDRC are significant at slow
(BCLN > 400 ms) and rapid (BCLN < 300 ms) pacing.
86
spatial variation is affected by tissue heterogeneity. No animals showed evidence of
frozen-in spatial SDRC patterns. It is possible, however, that Qin et al. observed
spatial variation of SDRC that was influenced by frozen-in heterogeneity, while my
experiment observed SDRC gradients that were primarily dynamically induced.
As was the case for APD gradients, SDRC gradients varied over a distance much
larger than the 0.3 mm passive length constant of bullfrog cardiac tissue [52]. This
is not very surprising since I saw spatial APD variation over distances greater than
the passive length constant and the DRC is determined by steady state APDs. Unfortunately, it is not clear from this experiment whether the variation of SDRC is
a boundary effect. It could be that the tissue was not large enough to to discern
whether SDRC becomes constant away from any boundaries.
4.4.2
∆SDRC and the Onset of Alternans
This study shows a large increase of the spatial gradient of SDRC as the transition
to alternans is approached. Trials that do not exhibit alternans at rapid pacing
show only a small increase in spatial gradient of SDRC as BCL decreases. Statistical
analysis confirms that the difference in gradient of SDRC is significant at rapid pacing.
The observed difference in SDRC gradient between ALT and noALT trials may
have a clinical application, although there are questions that need to be resolved
before this could be implemented. One complication is that my experiments included
ALT and noALT trials in the same tissue. It is possible that the restitution properties
change as the tissue is dying, leading to changes in the tissue’s propensity to exhibit
alternans. This conjecture will need to be tested with further experiments. Another
87
complication is that similar correlations may not be seen in human hearts. The
experiment of Qin et al. [68] indicates that tissue heterogeneity may over-ride any
dynamically-induced spatial variation of SDRC and it is not clear that a correlation
between mean spatial gradient of SDRC and the propensity to exhibit alternans would
still hold in such a case.
If these questions can be addressed, measuring mean gradient of SDRC to predict
a patient’s susceptibility to alternans provides some benefits over current diagnostic
procedures. Current or proposed diagnostic procedures analyze spatially-averaged
temporal response patterns (microvolt T-wave alternans in the ECG [143]) or temporal response patterns at a single location (steepness of the restitution curve [102] or
increase in gain during alternate pacing [144]). My findings suggest that measurement
of spatial gradient of SDRC may provide another alternative method for diagnosis of
arrhythmias in patients. The benefit of this method is that since increased ∆SDRC is
apparent in tissue that exhibits alternans at BCLs 200 ms longer than BCLt , there
is no need to put the patient at risk by pacing at BCLs near the transition.
4.5
Conclusions
This optical mapping study has shown SDRC varies over the surface of the tissue, with
larger slopes near the stimulus site and smaller slopes near the insulated boundaries.
Unlike APD, the spatial gradient of SDRC is essentially constant over the surface
of the tissue. Finally, the spatial gradients of SDRC observed in trials that exhibits
alternans increase dramatically before the onset of alternans.
88
Chapter 5
Spatial Heterogeneity in a Two-Variable
Cardiac Model
5.1
Introduction
In the previous two chapters, I observed that APD and SDRC exhibit spatial heterogeneity and that, in the case of SDRC this heterogeneity may be able to predict
cardiac tissue’s propensity to exhibit alternans. In this chapter, I use simulations
to explore the spatial variation of APD and slope of the DRC and it’s role in the
stability of the 1:1 response in a truly homogeneous medium.
5.1.1
Two-Variable Model
I use simulation of a cardiac model to study the spatial variation of steady-state APD
and the slope of the DRC in a homogeneous two-dimensional sheet of cardiac tissue
and its effect on the stability of the 1:1 response. The model I use is the two-variable
model presented in section 2.2.2. This model was chosen because its simplicity permits mathematical analysis [63, 88], yet it is complex enough to reproduce much of
the complex behavior seen in real cardiac tissue. One drawback of this model is that
it has a single unique restitution curve and that it does not show accommodation
(Fig. 5.1). Since I want to focus only on the steady-state APD and the slope of the
DRC, these shortcomings should not be an obstacle in this experiment.
89
Figure 5.1: Restitution and accommodation of the two-variable model. (A) The
restitution portrait for the two-variable model. The SRC and BRC have not split
from the DRC; there is a single restitution curve (B) The two-variable model exhibits
no accommodation. A single cell is paced at a BCL of 1000 ms from initial conditions
of V=0 and h=1. The APD remains constant from the second beat on. After a change
in BCL from 1000 ms to 900 ms, the APD again remains constant from the second
beat on.
Simulations
The simulations are used to study two questions: (a) Is there a boundary effect in
steady-state APD and the slope of the DRC? (b) Can spatial differences in APD
and slope of the DRC be correlated to the onset of alternans? The first question
is answered by implementing the pacing protocol in a homogeneous sheet of tissue
with insulated boundaries and studying observing the resulting spatial patterns of
steady-state APD and slope of the DRC.
To determine whether spatial gradients in APD or SDRC can be correlated to the
onset of alternans, I use two different sets of parameters, one of which causes the
model to exhibit alternans (ALT) and one of which causes the model to go directly
from a 1:1 to a 2:1 response (noALT). The parameters were chosen based on previous
90
Parameter
ALT
noALT
Vc
0.13
0.13
D
0.001 cm2 /ms 0.001 cm2 /ms
τopen
130 ms
350 ms
τclose
150 ms
150 ms
τout
4 ms
3 ms
τin
0.2 ms
0.2 ms
Table 5.1: Model parameters used to simulate tissue that exhibits alternans and
tissue that does not exhibit alternans.
analysis by Mitchell and Schaeffer [88] and are listed in Table 5.1. Changes in the
parameters will alter not just the behavior of the model at rapid pacing. In particular,
changing τopen changes the refractory period of the model and changing τout will alter
the length of the repolarization phase of the action potential. The corresponding
single-cell bifurcation diagrams are shown in Fig. 5.2. I then characterize the amount
of spatial heterogeneity of APD or SDRC and compare the results of ALT and noALT
trials to determine if they show differing amounts of spatial heterogeneity.
5.2
5.2.1
Methods
Cardiac Model
I use a simplified cardiac model to study the spatial variation of APD and SDRC .
The model was described in some detail in section 2.2.2, so I will simply restate the
equations in two-dimensions here:
δt V = D(δx2 V + δy2 V ) +
V
Iext
h 2
V (1 − V ) −
−
.
τin
τout
Cm
91
(5.1)
Figure 5.2: Bifurcation diagrams of the two-variable model. (A) The bifurcation
diagram for the two variable model when the parameters in the second column of
Table 5.1 are used. These parameters result in 2:1 behavior at BCL∼450 ms. (B)
The bifurcation diagram for the two variable model when the parameters in the third
column of Table 5.1 are used. These parameters result in 1:1 behavior changing to
2:1 behavior at BCL∼200 ms.
δt h =
(
1−h
τopen
h
− τclose
V < Vc
V > Vc ,
(5.2)
This model is used because it reproduces much of the complex behavior seen in real
cardiac tissue, but is still simple enough to be analyzed mathematically [63, 88].
The model is implemented in Matlab 6.5.1 (The MathWorks, Natick, MA) on a
Dell PC with a 3.2 GHz Intel processor and 2 GB RAM. The custom-written code
(App. D) uses the forward Euler method of advancing the model in space and time.
Simulations are run on a 2x2 cm2 sheet divided into a 64x64 grid with a time step of
0.05 ms. A full downsweep takes approximately 4 days to run.
92
5.2.2
Pacing Protocol
Since this model has a single unique restitution curve and does not exhibit accommodation, I use a simple pacing protocol to measure both the steady-state APD and
the DRC. The tissue is paced for ten paces at a constant BCL. The final APD is the
steady-state APD and the final (DI,APD) pair is a point on the DRC. The BCL is
then decreased and the process is repeated. Every downsweep begins at BCL=1000
ms and continues in steps of 10 ms until either 2:1 or 2:2 behavior is observed.
5.2.3
Data Analysis
The slope of the DRC is calculated as described in section 6.2.4. The gradients are
defined as
~ = ∂P x̂ + ∂P ŷ,
∇P
∂x
∂y
(5.3)
here P is the particular restitution property (APD or SDRC ) we are measuring. Since
I am using a discrete grid, the gradient is approximated by differences,
~ i,j = Pi+1,j − Pi−1,j x̂ + Pi,j+1 − Pi,j−1 ŷ,
∇P
2dx
2dy
(5.4)
~ i,j is the spatial gradient of P at the point (i, j), and Pi,j is the value of the
where ∇P
restitution property at the point (i, j).
To compare ALT and noALT cases, I use several different measures of the amount
of spatial heterogeneity: the mean gradient, the maximum gradient and the gradient
as measured from two locations on the tissue. All of these measures have been
used previously by other research groups to compare tissue that exhibits arrhythmias
93
and tissue that does not exhibit arrhythmias [42, 67, 70] and each has benefits and
limitations as a characteristic measure.
The mean gradient is defined as
∇P =
P
i,j
~ ∇Pi,j # of spatial points
,
(5.5)
where all spatial points on the tissue are used to calculate the mean. The mean
gradient includes information from all points on the surface of the tissue and is not
strongly influenced by noisy measurements. In modeling studies, noise is not a big
concern, but in experiments, particularly optical experiments, noise can impact the
accuracy of measurements. By minimizing the effects of noise, the mean spatial
gradient is an attractive measure of spatial heterogeneity for experiments. Unfortunately, by averaging the gradient at all points on the tissue and using only the
magnitude, information about the spatial pattern of the gradient and information
about the direction of the gradient is lost.
The maximum gradient (denoted by ∇P max ) is defined as the maximum magnitude of the spatial gradient on the surface of the tissue. The maximum gradient also
does not retain information about the spatial pattern of the gradient or information
about the direction of the gradient. Further, the maximum gradient uses the information from only a single pixel which, particularly in experiments, may have a large
amount of measurement error.
Finally, I measure the spatial gradient between two locations on the surface of
the tissue. I use three pairs of locations to try to capture a cross-section of the
spatial patterns (Fig. 5.3). The gradients are measured between points A and B
94
Figure 5.3: Spatial gradients in a two variable model. Three spatial gradients are
measured: the gradient between points A and B, the gradient between points C and
D, and the gradient between points E and F.
(denoted by ∇P AB ), which are both 0.5 cm from the vertical edges of the tissue
and centered horizontally, between points C and D (denoted by ∇P CD ), which are
both 0.5 cm from the horizontal edges and centered vertically, and between points E
and F (denoted by ∇P EF ), which are 0.5 cm from both the horizontal and vertical
edges. These spatial gradient measurements are similar to the spatial differences
measured in Ch. 6 since they use the information from only two locations. This
measurement neglects information from most of the locations on the tissue, but it
does retain information about the direction of the gradient. Also, the results may
vary depending on the locations that are chosen to make the measurements. Finally,
gradients measured in this way are susceptible to inaccuracies due to measurement
error and noise.
To properly compare results from different trials, BCL was shifted by subtracting
the transition BCL at which either 2:2 or 2:1 behavior was observed (BCLt ); that
is, BCLN = BCL − BCLt . The mean gradients are determined as a function of
95
BCL and are compared for ALT and noALT cases to determine whether they can
differentiate between tissue that exhibits alternans and tissue that does not exhibit
alternans.
5.3
5.3.1
Results
Spatial Heterogeneity
Steady-State APD
Figure 5.4 shows the steady-state APD at several BCLs when pacing from the center
on the left side. In both cases, the APD is largest near the stimulus site and decreases as the wave propagates away from the stimulus. Cross-sections of the maps
(Figs. 5.4D,E,F) show that there is a sharp drop in APD near the stimulus site, a
slight plateau in the middle of the tissue followed by another sharp drop as the wave
approaches the far end of the tissue. It is fairly clear from these simulations, that
the two-variable model exhibits boundary effects, one near the stimulus and one near
the insulated boundary, in two dimensions.
Figure 5.4 indicates that the gradient in APD varies over the surface of the tissue.
Figure 5.5 shows the spatial variation of gradient on the tissue. The gradient is
large near the edges and near zero in the center of the tissue. Note that, near the
boundaries, the gradients are much larger than 3 ms/mm even during stable 1:1
response. APD gradients larger than 3 ms/mm have been linked to the onset of
arrhythmias in other experiments [31].
96
Figure 5.4: Spatial variation of steady state APD in the two-variable model. The
tissue is paced from the center of the left side; the resulting APD maps at BCLs of
(A) 1000 ms (B) 800 ms (C) 600 ms are shown. The APD is longest near the stimulus
and decreases as the wave propagates away from the stimulus. Cross-sections taken
along the horizontal line indicated in (A) are shown in panels D,E, and F. The
cross-sections show that the APD drops sharply near the stimulus and near the far
end of the tissue, but does not change much in the middle. Parameters used for this
simulation are listed in the ALT column of Table 5.1.
97
Figure 5.5: Spatial gradient of steady state APD in the two-variable model. The
tissue is paced from the center of the left side; the resulting gradients at BCLs of (A)
1000 ms (B) 800 ms (C) 600 ms are shown. The APD gradient is largest near the
boundaries and near zero in the center of the tissue. Cross-sections taken along the
horizontal line indicated in (A) are shown in panels D,E, and F. The cross-sections
show that the APD gradient drops sharply near the stimulus and increases again near
the far end of the tissue. Parameters used for this simulation are listed in the ALT
column of Table 5.1.
98
Dynamic Restitution Curve
Figure 5.6 shows the slope of the DRC at several BCLs when pacing from the center
on the left side. At long BCLs, there is little change in the slope of the DRC over
the tissue, but as BCL increases, a gradient begins to appear with the largest slopes
near the stimulus site and smaller slopes at the far end of the tissue. Cross-sections
of the maps (Figs. 5.6D,E,F) show that the DRC slope decreases at a fairly constant
rate as the wave propagates from the stimulus site. If spatial variation of SDRC is a
boundary effect similar to that observed for APD, it has boundary width even larger
than the boundary width for APD since there is no region of constant SDRC in the
center of the tissue.
5.3.2
Predicting the Propensity to Exhibit Alternans
Steady-State APD
The mean APD gradient as a function of BCL is shown in Fig. 5.7A for both ALT
(alternans appears at BCL=510 ms) and noALT (2:1 response appears at BCL=640
ms). In both cases, the mean APD gradient decreases slightly as the BCL decreases.
At most BCLs, the mean APD gradient can differentiate between ALT and noALT
cases. The maximum spatial gradient ∇AP Dmax (Fig. 5.7B) shows no clear trend as
a function of BCL. Although ∇AP Dmax differs for ALT and noALT cases at many
BCLs, it is not a good measure for discriminating between the two cases because, at
some BCLs, the noALT case has a larger ∇AP Dmax and sometimes the maximum
gradient is the same for both cases. The remaining spatial gradients, ∇AP DAB
(Fig. 5.8A), ∇AP DCD (Fig. 5.8B), ∇AP DEF (Fig. 5.8C) show a slight difference
99
Figure 5.6: Spatial variation of slope of DRC in the two-variable model. The tissue
is paced from the center of the left side; the resulting DRC slope maps at BCLs of
(A) 1000 ms (B) 800 ms (C) 600 ms are shown. At slow pacing, the slope of the
DRC shows little spatial variation, but as the BCL decreases, a gradient begins to
appear. Cross-sections taken along the horizontal line indicated in (A) are shown in
panels D,E, and F. The cross-sections show that the DRC slope decreases at a fairly
constant rate over the length of the tissue. Parameters used for this simulation are
listed in the first column of Table 5.1.
100
Figure 5.7: Mean and maximum APD gradient. (A) The mean APD gradient is
slightly larger in ALT cases than in noALT cases. The mean APD gradient can
differentiate between ALT and noALT cases at almost all BCLs. (B) There is no
clear trend in ∇AP Dmax for either the ALT or noALT case. At some BCLs, ALT
and noALT cases have the same ∇AP Dmax , at others, ∇AP Dmax differs for ALT
and noALT cases.
between ALT and noALT cases, but the measurements all agree within error. Thus,
these measures of spatial heterogeneity cannot differentiate between ALT and no
ALT cases.
Dynamic Restitution Curve
The mean DRC slope gradient as a function of BCL is shown in Fig. 5.9A for
both ALT and noALT. In the ALT case, the mean DRC slope gradient increases as
BCL decreases. In the noALT case, the mean DRC slope gradient shows an initial
decrease followed by a rapid increase as BCL decreases. The mean SDRC gradient can
differentiate between ALT and noALT cases at nearly all BCLS, with the difference
being particularly dramatic at both slow and rapid pacing. The maximum SDRC
gradient shows a trend similar to the mean spatial gradient (Fig. 5.9B), although
101
Figure 5.8: APD spatial gradients. (A) ∇AP DAB is slightly larger in ALT cases
than in noALT cases, though the measurements agree within error. (B) ∇AP DCD
is essentially the same for both ALT and noALT cases. (C) ∇AP DEF is essentially
the same for both ALT and noALT cases.
102
Figure 5.9: Mean and maximum SDRC gradient. (A) Both ALT and noALT cases
show a rapid increase in mean gradient of SDRC as BCL nears the transition point.
At long BCLs, noALT cases exhibit an initial decrease in mean gradient of SDRC
while ALT cases exhibit a small increase. (B) The maximum SDRC gradient is larger
in ALT cases than in noALT cases, although the measurements agree within error.
Thus, the maximum SDRC gradient cannot differentiate between ALT and noALT
cases.
max
the larger error in the measurement of ∇SDRC
means that this measure of spatial
heterogeneity cannot differentiate between ALT and noALT cases. The remaining
AB
CD
spatial gradients give mixed results. Measurements of ∇SDRC
(Fig. 5.10A), ∇SDRC
EF
(Fig. 5.10B), and ∇SDRC
(Fig. 5.10B) exhibit slight differences in ALT and noALT
cases, though at most BCLs these differences are within measurement error. Only
AB
∇SDRC
can differentiate between ALT and noALT cases within ∼100 ms of the
transition point.
103
AB
Figure 5.10: DRC spatial gradients. (A) ∇SDRC
is slightly larger in ALT cases
than in noALT cases at short BCLs with the difference becoming larger than the
CD
measurement error about 100 ms from the transition point. (B) ∇SDRC
is slightly
larger in ALT cases than in noALT cases at short BCLs, though the measurements
EF
agree within error. (C) ∇SDRC
is slightly larger in ALT cases than in noALT cases
at long BCLs and reverses at short BCLs, though the measurements agree within
error at all BCLs.
104
5.4
5.4.1
Discussion
Spatial Heterogeneity of Restitution Properties
Since the sheet of cardiac tissue used in these simulations is homogeneous, any spatial variation of restitution properties is dynamically induced. Further, in the case of
APD, the spatial variation is in the form of a boundary effect with little variation of
APD observed far from the boundaries. This finding is important for several reasons.
In real cardiac tissue, observed spatial heterogeneity may be due to a combination
of structural heterogeneity of the underlying tissue and dynamically induced heterogeneity. It is important to understand the role each plays the stability of the 1:1
response of cardiac tissue. Also, if the onset of alternans is mediated by spatial heterogeneity of restitution properties, as is suggested by some of the experimental and
simulation results presented here, then determining the cause of the heterogeneity
may lead to methods to reduce or eliminate it.
Steady-State APD
The steady-state APD shows a great deal of spatial heterogeneity. In particular, there
is a large drop in APD near the stimulus site and again at the far end of the tissue. A
similar pattern of APD spatial variation has previously been seen in one [59–61, 63],
two [62], and three [60] dimensional simulations of cardiac models. Mathematical
analysis of the one-dimensional version of the model used here has shown that, at the
insulated end of the tissue, APD shortening is due to the inability of the current to
flow beyond the boundary [63]. It is not immediately obvious that this same effect
should be evident in two dimensions. In two dimensions, when the current reaches a
105
boundary, the ions can still flow parallel to the boundary, potentially lessening or even
eliminating the charge buildup that occurs in a one-dimensional system. The spatial
variation of APD seen in these two-dimensional simulations (Fig. 5.4) show a striking
resemblance to the spatial APD variation seen in simulations in one-dimension (Fig.
1.4). Thus, it is likely that the same effect plays some role in the spatial variation of
APD in two dimensions.
Dynamic Restitution Curve
The slope of the DRC showed little spatial heterogeneity at long BCLs. As the
BCL approaches the transition point, the slope of the DRC begins to show some
spatial heterogeneity. In particular, the slope of the DRC is largest near the stimulus
and smallest at the far end of the tissue with a constant gradient over the surface
of the tissue. This finding is consistent with the results of Ch. 4, and is in stark
contradiction to the experimental results of Qin et al. who found that SDRC showed
no consistent gradient in porcine hearts.
5.4.2
Predicting Tissue’s Propensity to Exhibit Alternans
The results presented here indicate that mean APD gradient can differentiate between ALT and noALT cases. The results do not support the theory that large APD
gradients cause alternans. Although I find that mean APD gradient is larger in the
ALT case than in the noALT case, the mean APD gradient is ≈0.8 ms/mm, much
lower than the 3 ms/mm experimentally predicted threshold for alternans [30,31,76].
Not only does the mean gradient not reach the appropriate threshold for alternans,
but because of the wide range of gradients over the surface of the tissue, some regions
106
of the tissue have gradients larger than the threshold while still exhibiting stable 1:1
behavior. Thus, it is unlikely that steep APD gradients cause alternans. Nonetheless, there is a correlation between the mean spatial gradient of APD and a tissue’s
propensity to exhibit alternans that can potentially be exploited for clinical use (See
section 6.4.4), even if it doesn’t lead to insight into the origin of alternans.
The other measures of spatial heterogeneity of APD presented here are not useful
as characteristics that can differentiate between ALT and noALT cases. Spatial
gradients measured from two selected locations agree within measurement error for
ALT and noALT cases. Selecting other locations may yield different results, but this
leads to difficulty in the clinical application of this measure. If spatial differences from
only a few specific locations on the tissue can differentiate between ALT and noALT
cases, then we must first identify those specific locations before this measurement can
be used. The maximum APD gradient is also not a clinically useful measurement for
differentiating between ALT and noALT cases. Although ∇AP Dmax for ALT and
noALT cases differs significantly at some BCLs, there is no consistent trend over all
BCLs. Thus it would be difficult to classify the tissue type based on measurement of
∇AP Dmax at a single (or even a few) BCLs.
The mean gradient in SDRC can also be used to differentiate between ALT and
noALT cases. As in the previous chapter, the ALT case shows a larger increase in
gradient of SDRC than the noALT case. Although the maximum spatial gradient of
SDRC shows a trend similar to the mean spatial gradient of SDRC , the measurements
max
of ∇SDRC
for ALT and noALT cases agree within error an so cannot differentiate
between the two cases. Spatial gradients of SDRC measured from two specific locations
may be useful for differentiating between ALT and noALT cases, although only certain
107
specific locations yield significantly different measurements for ALT and noALT cases,
making this measure impractical for clinical applications.
5.4.3
Study Limitations
Although the use of computer simulation to study cardiac tissue can provide insights
into the dynamics of cardiac tissue, this type of study also has limitations. In this
simple study, I have not captured the full complexity of cardiac tissue. The simulations were performed in two-dimensional sheets of cardiac tissue while real tissue
is three-dimensional. It is not immediately clear how the extra dimension will affect
the observed spatial patterns, although previous studies suggest that at least steadystate APD exhibits a similar spatial pattern in three dimensions [60]. Further, this
is a highly simplified model and does not contain detailed information about the
actual currents that create the action potential. Other studies using more complex
models have also exhibited similar spatial patterns in steady state APD [60–62], but
little is known about the effect of individual currents on the spatial patterns of other
restitution properties.
The result linking spatial variation of restitution properties to the onset of alternans may also be of limited use in real cardiac tissue. Real cardiac tissue contains
specialized structures and tissue fibers that may override any dynamically induced
heterogeneity, so the effects seen here may not hold.
108
5.5
Conclusion
Through the use of computer simulation, we have seen that APD and SDRC exhibit
spatial heterogeneity that is dynamically induced. Further, I have shown that the
mean spatial gradients of SDRC and APD can differentiate between tissue that exhibits alternans and tissue that does not exhibit alternans. The experimental results
of the previous chapters suggest that the results of the computer simulation may also
hold in frog cardiac tissue. In the following chapter, I extend the study of spatial
variation of restitution properties by determining whether there is spatial variation
in other restitution curves and whether spatial variation of restitution properties can
be linked to tissue’s propensity to exhibit alternans at rapid pacing.
109
Chapter 6
Spatial Heterogeneity and the Onset of
Alternans
6.1
Introduction
This chapter extends the results of the previous chapters by studying the spatial
variation of all restitution properties. The study is a spatially limited study meant to
determine which cardiac restitution properties exhibit spatial variation and whether
the spatial variation of restitution properties and the onset of alternans are correlated.
6.1.1
Background
As described in Sec. 4.1.1, many researchers believe that the stability of the 1:1
response is determined by the restitution curve (RC). Unfortunately, the restitution
hypothesis, in its original form, has been shown to fail in experiments [39–42].
6.1.2
Restitution Curves
The restitution hypothesis is known to be inadequate in two ways: cardiac tissue is
not accurately described by a one-variable map [43], and the RC varies with the pacing
protocol used to obtain it [43,145]. Modified stability criteria have been developed for
two and three-variable cardiac mapping models [43, 47], but, to properly understand
them, we need to understand the different RCs commonly used to characterize cardiac
tissue.
110
Dynamic Restitution Curve
The DRC is described in detail in Sec. 4.1.1, but recall that it consists of steady-state
(APD,DI) pairs determined at different BCLs. There is a single unique DRC for each
tissue sample.
S1S2 Restitution Curve
The S1S2 restitution curve (SRC) measures the tissue’s response to perturbations.
The experimental pacing protocol that is used to determine the SRC is as follows:
1. Pace at a slow constant BCL until steady-state is achieved. This initial BCL is
known as the S1 rate.
2. Apply a single perturbation at a different BCL (the S2 BCL). The APD produced by the S2 pace and the preceding DI are used to determine one point on
the restitution curve.
3. Return to the S1 rate and repeat the process with a new S2 BCL.
The process is depicted in Fig. 6.1. Unlike the DRC, there are many different SRCs
for a given piece of tissue. Different S1 BCLs produce different SRCs.
Constant-BCL Restitution Curve
The constant-BCL restitution curve (BRC) consists of all (DI, AP D) pairs collected
while pacing at a constant BCL. This curve includes all points during the transient
after a change in BCL as well as the steady state response. Like the SRC, there are
many different BRCs for a given piece of tissue since pacing at different constant
BCLs will produce different curves.
111
Figure 6.1: S1S2 Restitution Curve. The SRC is determined by the responses to
perturbations in BCL. The tissue is paced at a constant BCL (the S1 rate) until
steady-state is reached. A single pace at a different BCL (the S2 rate) is applied and
the resulting APD and previous DI are used to create the SRC. Upon returning to
the S1 rate, the tissue does not need to be paced at a constant BCL for very long
since it typically recovers from a single perturbation very quickly. Further S2 paces
at different BCLs are applied to complete the entire RC.
6.1.3
Maps and Restitution Curves
The multitude of RCs can be captured by turning to mathematical models slightly
more complex than the one-variable map. In a one-variable mapping model, the different pacing protocols produce a single unique RC (Fig. 6.2A). In a two-variable model,
where AP Dn+1 = f (DIn , AP Dn ), the three restitution curves are distinct (Fig.
6.2B). Finally, a three-variable model, where AP Dn+1 = F(DIn , AP Dn , DIn−1 ), actually has four distinct restitution curves (Fig. 6.2C). The BRC has two different
components: the transients associated with a change in BCL fall along a different
line (BRC-D) than the transients associated with a one-beat perturbation in BCL
(BRC-S). A full treatment of the RCs produced by models with arbitrary numbers
of variables was done by Kalb et al. [146], but they will not be discussed here since
112
Figure 6.2: Restitution portraits of cardiac mapping models. (A) A one-variable
cardiac mapping model produces a single RC regardless of the pacing protocol. (B)
A two-variable model has different curves for the DRC (steady-state responses), SRC
(perturbations) and BRC (transients). (C) A three-variable model produces a fourth
RC, with the transient response becoming split into two curves: transients associated with a permanent change in BCL (BRC-D) and transients associated with a
perturbation (BRC-S).
experiments in frog tissue produce RPs qualitatively similar to RPs produced by a
three-variable model [43] (Fig. 6.3).
Since the three-variable map seems to capture the dynamics of frog cardiac cells,
the stability criterion for a three-variable map should also give the condition for
stability of the 1:1 response in frog cardiac cells. The stability criterion is determined
by the total derivative of F and is given by
δF
dAP Dn
δF dDIn
δF dDIn−1 +
+
< 1,
δAP Dn dAP Dn
δDIn dAP Dn δDIn−1 dAP Dn (6.1)
with all derivatives evaluated at the fixed point. The RCs can be written in terms of
the derivatives of F [47]
SDRC =
δF
δDIn
1−
113
δF
δDIn−1
δF
δAP Dn
−
(6.2)
Figure 6.3: Restitution portrait from a frog ventricular myocyte. The RP from
frog cardiac cells shows four distinct restitution curves: the DRC, SRC, BRC-D, and
BRC-S. Steady state points are indicated by ‘*’ and form part of the DRC. Initial
transients are indicated by ‘.’ and form the BRC-D. Long and short perturbations
are indicated by ‘+’ and ‘x’, respectively and along with the steady-state points form
the SRC. Finally, the transients after a perturbation are indicated by ‘o’ and along
with the steady-state points form the BRC-S. This is qualitatively similar to the RP
of a three-variable mapping model.
SSRC =
δF
δDIn
(6.3)
with all derivatives evaluated at the fixed point. Substituting these expressions into
6.1 leads to the stability criterion for a 3-variable model:
Smem
SSRC
SSRC
SSRC (1 − SDRC )
1 − SSRC −
+
+ SBRC−S ≤ 1,
= 1 − SSRC −
SDRC
SDRC − SSRC
SDRC
(6.4)
where SSRC , SDRC , SBRC−S are the slopes of the SRC, DRC, and BRC-S measured
at the fixed point [43]. However, when the quantity on the left-hand side of Eq.
6.4 is measured experimentally, we find that it is less than 0.5 just before alternans
appears [43]. One of the possible reasons for the failure of the stability criterion is
because the measurements were made in a single cell of a multi-cellular preparation.
114
The single cell from which the measurements were made is subjected to the effects
of coupling to its neighbors, an effect that was not considered when the stability
criterion was derived.
6.1.4
Experiment Overview
In the following sections, I describe the results of experiments that study the role of
spatial heterogeneity of different characteristics of cardiac tissue in the propensity of
tissue to exhibit alternans [147]. The experiments are performed in small pieces of
bullfrog ventricular myocardium that have little inherent spatial heterogeneity, so any
heterogeneity that is observed is dynamically-induced. I use the restitution portrait
to measure the steady-state APD, and slopes of the DRC, SRC, and BRC simultaneously at different spatial locations. I then determine whether the spatial heterogeneity of any of these characteristics is indicative of a particular tissue’s propensity
to alternans.
The measurements are performed using two microelectrodes at different spatial
locations. Optical imaging is not used in these experiments because it takes 40-60
minutes to collect data for a restitution portrait over which time the tissue would
significantly degrade due to the phototoxic nature of di-4-ANEPPS [148]. The degradation of the tissue not only changes the tissue properties we are trying to measure,
but also decreases the SNR of the measured signals, making it difficult to measure
small changes in APD, thereby preventing accurate measurement of the slopes.
115
6.2
6.2.1
Methods
Tissue Preparation
This study was performed in accordance with a protocol that conforms to the Research Animal Use Guidelines of the American Heart Association and was approved
by the Duke University Institutional Animal Care and Use Committee. Seven bullfrogs were anesthetized and double-pithed. The heart was excised and the anterior
surface of the ventricle was removed and pinned in a dish. The tissue was superfused
with a standard Ringer’s solution (100 mM NaCl, 2.70 mM KCl, 5.6 mM glucose, 1
mM hepes, 2.8 mM Na2 HPO4 , 25 mM NaHCO3 , 1.5 mM MgCl2 , 1.80 mM CaCl2 [117],
buffered by CO2 ) at room temperature. The tissue was paced with a silver bipolar
electrode at a constant basic cycle length (BCL) of 1,000 ms for 20 minutes before
any pacing protocols were performed.
6.2.2
Pacing Protocol
The tissue was paced using a perturbed downsweep protocol [43] that allowed me
to collect all the data needed to construct the RP. A pacing sequence for one BCL
is shown in Fig. 6.4. Beginning at a long BCL (typically 1000 ms), the tissue is
paced for 60 s (small dots) until steady state is achieved. Five steady-state paces
(diamonds) are applied at the initial BCL before an S2 pace at BCL+50 ms (’+’)
is applied. This is followed by five recovery paces (filled circles) at the initial BCL
and another S2 pace at BCL-50 ms (’x’). The sequence ends with five recovery paces
(filled circles) at the initial BCL. The BCL is then decremented by 50 or 100 ms
and the sequence of Fig. 1 is repeated. Pacing at decreasing BCLs continues until
116
Figure 6.4: Perturbed downsweep pacing protocol. The tissue is paced at a constant
BCL for 60 s (transient response, small dots). An additional 5 paces at steady state
are applied (diamonds) followed by an S2 pace at BCL+50 ms (’+’), 5 recovery
paces at the original BCL (filled circles), an S2 pace at BCL-50 ms (’x’), and 5 more
recovery paces (filled circles). The entire sequence is repeated at progressively shorter
BCLs until the myocardium transitions to a 2:1 or 2:2 stimulus:response pattern. The
downstep in BCL, denoted by ∆, is 50 or 100 ms.
either a 2:1 or 2:2 response is seen. A typical trial consisted of 15 BCLs and lasted
20 minutes. Several trials were performed on each animal. Only trials where both
electrodes remained impaled for the majority of the BCLs were analyzed. Results
from 19 trials in 7 animals are presented here.
Seven trials in 4 animals showed steady-state alternans in at least one electrode,
which I denote as ’ALT’ trials; the remaining trials that did not show alternans are
denoted as ‘noALT’ trials. Specifically, the final 4 steady-state responses (diamonds
117
in Fig. 1B) were used to determine whether a trial was classified as ALT or noALT.
For each BCL, I determined δA = AP Dn+1 − AP Dn where n = 1...4 are the steadystate beats. A response pattern was classified as displaying steady-state alternans
if δA alternated in sign from beat to beat and |δA| > 2 ms (the error in APD
measurement).
6.2.3
Electrical Recordings
Electrical signals were recorded simultaneously from two locations in the tissue using
glass microelectrodes filled with 3 M KCl. Microelectrodes were placed 1-2 mm apart
perpendicular to the line connecting the two terminals of the pacing electrode with
the proximal microelectrode placed ∼1 mm from the pacing electrode (Fig. 6.5).
This spacing is 3-6 times longer than the 0.3 mm passive length constant of bullfrog
ventricular tissue [52]. Voltage signals acquired at 1 kHz were low-pass filtered (1 kHz
3-dB bandwidth) with analog circuitry and stored on a computer for later processing.
6.2.4
Restitution Portrait
Action potential durations and diastolic intervals were found using a threshold of
70% of the action potential amplitude. Restitution portraits were generated for each
electrode by plotting APD versus previous DI as shown in Fig. 6.8. RPs contain
the transient response (small dots), which ends with the steady state responses (diamonds); the responses to S2 paces at BCL+50 ms (‘+’) and at BCL-50 ms (‘x’);
and the response to recovery paces (filled circles). These responses form the following RCs: the DRC (dashed line) that runs through all the steady state responses;
118
Figure 6.5: Sketch of a bullfrog ventricular preparation. Two microelectrodes are
placed 1-2 mm apart with the proximal one placed 1 mm from the bipolar pacing
electrode.
segments of SRCs (grey lines) that are determined by the S2 paces and the steady
state response at each BCL; and segments of BRCs (black lines) that are determined
by the two sets of five recovery paces and the steady-state response at each BCL.
Each RP provides four measures that are used to characterize the tissue at each
BCL:
1. The steady-state APD, computed as an average of the five steady-state responses (diamonds in Fig. 6.8).
2. Slope of the SRC (SSRC ). To compute SSRC , the S2 (‘+’ and ‘x’) and steady
state (diamonds) paces forming a segment of SRC are fit to a straight line using
least-square regression (grey lines in Fig. 6.8). The slope of this line determines
SSRC at the steady-state of the BCL under consideration.
3. Slope of the BRC (SBRC ) is computed like SBRC but using the recovery (circles)
and steady state (diamonds) paces forming a segment of BRC (black lines in
Fig. 6.8).
4. Slope of the DRC (SDRC ). Steady-state APDs for all BCL are fit to an exponential function,
DI ∗
∗
AP D = A − B exp −
,
(6.5)
τ
119
where A, B, and τ are parameters. SDRC is computed by evaluating the derivative of Eq. 6.5 at the BCL under consideration.
These four characteristics were determined for each electrode at every BCL in
the downsweep. In six frogs, I also determined the time constant of the transient
response following a downstep in BCL. Spatial differences in the time constant were
not found to be statistically significant, and this characteristic is not included in
further analysis.
6.2.5
Spatial Differences
The four characteristics of the RP were used to investigate spatial differences in
restitution. Differences in APD were computed by subtracting steady-state APD at
the distal electrode from the one at the proximal electrode: ∆AP D = AP Dprox −
AP Ddist . Differences in slopes, ∆SSRC , ∆SBRC , ∆SDRC were computed likewise. All
computations were performed in Matlab 6.5.1 (The MathWorks, Natick, MA).
Spatial differences were analyzed as a function of the BCL. Since I am trying to
determine whether spatial differences in restitution properties vary as the transition
point is approached, I must eliminate the trial-to-trial variability of the transition
point by normalizing the BCL. To properly compare results from different trials, BCL
was shifted by subtracting the transition BCL at which either 2:2 or 2:1 behavior was
observed (BCLt ); that is, BCLN = BCL − BCLt . The range of BCLt is shown in
Fig. 6.6. ALT and noALT trials were analyzed separately.
To assess whether the measurements made at the two different spatial locations
differ, my study uses the statistical method proposed by Altman and Bland [149].
This method determines whether there is a spatial difference in, say, APD by de120
Figure 6.6: Range of BCLt . The transition BCL was 200 ms for all 12 trials that
exhibited 2:1 behavior. The transition BCL ranged from 300 ms to 450 ms for trials
that exhibited 2:2 behavior. See Table 6.1 for more details.
termining ∆AP D for all trials and testing whether the mean of ∆AP D (denoted
by ∆AP D) is zero using a t-test [150]. A ∆AP D significantly different from zero
suggests that APDs differ at the two spatial locations. We then compare ∆AP D
for ALT and noALT trials. In this case, the t-test compares the means of ∆AP D
of ALT and noALT trials to determine if there is a significant difference. The same
method is used to investigate spatial differences in SRC, BRC, and DRC slopes. The
analysis is performed separately for each BCLN . Statistical tests were performed in
Excel (Microsoft, Redmond, WA). In all t-tests, p value less than 0.05 was considered
significant.
121
6.2.6
Slope Criteria for the onset of Alternans
For completeness, I analyzed our data to determine whether the traditional slope
criteria can distinguish between ALT and noALT trials at either proximal or distal
electrode. I extracted from the data the mean values of SSRC , SBRC , and SDRC at all
BCLN > 0. In addition to examining individual slopes, I also evaluated the memory
criterion of Eq. 6.4 that involves a combination of SRC, BRC, and DRC slopes and
accounts for one possible way that short-term memory affects rhythm stability [43].
These four quantities were evaluated separately for data from the proximal and distal
electrode.
For any quantity S, the criterion for alternans is considered satisfied when: 1) S
is less than one for all BCLN > 0 for the ALT and noALT trials; 2) S increases as
BCLN decreases for the ALT and noALT trials; and 3) S approaches one as BCLN
approaches zero for the ALT trials but not for the noALT trials.
6.3
6.3.1
Results
Restitution Portraits
For each BCL at each spatial location, I find the APD, DRC, SRC, and BRC as
shown in figure 6.7. Combining the RC segments at different BCLs produces a restitution portrait. The restitution portrait provides a visual method for qualitatively
comparing important restitution properties at different spatial locations. Figure 6.8
shows the restitution portraits generated from simultaneous measurements with two
microelectrodes proximal and distal to the stimulus site for one of the ALT trials.
These restitution portraits have the same qualitative features as those measured pre122
Figure 6.7: Segments of restitution curves for a single BCL. At each BCL, I collect
the transient response (small dots), the steady state responses (diamonds), the S2
pace at BCL+50 ms (‘+’), the S2 pace at BCL-50 ms (‘x’) and the recovery paces
(circles). The DRC (dashed line) is the curve that connects all steady state responses.
Segments of SRCs (grey lines) are determined by the S2 paces and the steady state
response; segments of BRCs (black lines) are determined by the recovery paces and
the steady-state response.
viously in bullfrog [43], rabbit, and guinea pig [139]. RPs from both locations are
qualitatively similar, although there is evidence of quantitative differences between
them. In particular, at all BCLs, there is a large difference in APDs between the two
locations, about 30-50 ms, and the DRC for the proximal electrode is visibly steeper
at short BCLs (Fig. 6.9).
Overall, 19 pairs of simultaneously-measured RPs are constructed and the differ-
123
Figure 6.8: Restitution portraits collected simultaneously from the electrode proximal (A) and distal (B) to the pacing site. The restitution portraits contain all the
responses of the perturbed downsweep protocol of Fig. 1B: the transient response
(small dots), the steady state responses (diamonds), the S2 pace at BCL+50 ms
(‘+’), the S2 pace at BCL-50 ms (‘x’) and the recovery paces (circles). The DRC
(dashed line) is the curve that connects all steady state responses. At each BCL,
segments of SRCs (grey lines) are determined by the S2 paces and the steady state
response; segments of BRCs (black lines) are determined by the recovery paces and
the steady-state response. For clarity, panels (A) and (B) show data for every second
BCL collected in this trial.
124
Figure 6.9: Restitution properties as a function of BCL. The (A) APD, (B) SDRC ,
(C) SSRC , and (D) SBRC are determined at steady-state for the trial shown in figure
6.8 for both the proximal (circles) and distal (diamonds) electrodes. SSRC and SBRC
are almost the same at both electrodes. APD has a spatial difference that remains
roughly constant as BCL changes. The spatial difference in SDRC increases as BCL
decreases.
125
Animal Trial Alternans BCLt
1
1
ALT
300
1
2
noALT
200
1
3
noALT
200
2
1
ALT
300
3
1
noALT
200
4
1
ALT
400
4
2
ALT
450
4
4
4
5
5
6
6
6
7
7
7
7
3
4
5
1
2
1
2
3
1
2
3
4
ALT
ALT
noALT
ALT
noALT
noALT
noALT
noALT
noALT
noALT
noALT
noALT
400
350
200
300
200
200
200
200
200
200
200
200
Electrode
both
both
both
distal only at BCL=450 ms,
proximal only at BCL=350 ms
both
distal only
both
Table 6.1: Summary of experimental trials indicating the occurrence of alternans,
the BCL at which a change in response pattern was observed, and the electrode at
which alternans appeared.
ences between them are analyzed below. Table 6.1 summarizes my finding of ALT
and noALT trials, the BCL at which a change in response pattern was observed
(BCLt ), and the electrode in which alternans appeared.
6.3.2
Steady State APD
At all BCLs, the spatial difference of APD is positive (Fig. 6.10A), indicating that
the APD decreases as the wave propagates away from the stimulus. ∆AP D is sig126
Figure 6.10: Steady state APD difference. (A) ∆AP D for ALT and NoALT trials
as a function of BCLN . (B) P values below 0.05 (dashed line) indicate ∆AP D is
significantly different from zero.
nificantly different from zero at all BCLs below BCLN = 500 ms and for both ALT
and noALT trials (p values given in Fig. 6.10B). Furthermore, ∆AP Ds in ALT trials are larger than in noALT trials with the difference becoming significant below
BCLN = 200 ms (p values shown in Fig. 6.14A).
6.3.3
S1S2 Restitution Curve
The mean SRC slope difference (∆SSRC ) is positive in ALT trials and negative in
noALT trials (Fig. 6.11A). ∆SSRC moves toward zero as the BCL increases, still
remaining positive in ALT trials and negative in noALT trials (p values given in Fig.
6.11B). ∆SSRC in ALT and noALT trials differs significantly at most BCLN s, the
exceptions being BCLN = 600, 350, 50 ms (Fig. 6.14C).
127
Figure 6.11: SRC slope difference. (A) ∆SSRC for ALT and NoALT trials as a function of BCLN . (B) P values below 0.05 (dashed line) indicate ∆SSRC is significantly
different from zero.
6.3.4
Constant-BCL Restitution Curve
The mean BRC slope difference (∆SBRC ) is generally positive in ALT trials and
negative in noALT trials (Fig. 6.12A). ∆SBRC moves toward zero as BCL decreases,
still remaining positive in ALT trials and negative in noALT trials. This is similar to
what is seen with ∆SSRC , but the effect is smaller in ∆SBRC (p values given in Fig.
6.12B). Note that ∆SBRC values in ALT and noALT trials differ significantly only at
BCLN = 550, 250 ms (Fig. 6.14D).
6.3.5
Dynamic Restitution Curve
The mean DRC slope difference (∆SDRC ) is mostly positive in both ALT trials and
noALT trials (Fig. 6.13A). ∆SDRC increases as BCL decreases, particularly in ALT
trials. P values given in Fig. 6.13B show that ∆SDRC is significantly different from
zero when BCLN ≤ 250 ms for ALT trials; for noALT trials, ∆SDRC is never signif128
Figure 6.12: BRC slope difference. (A) ∆SBRC for ALT and NoALT trials as
a function of BCLN . (B) P values below 0.05 (dashed line) indicate ∆SBRC is
significantly different from zero.
icantly different from zero. Note that ∆SDRC values in ALT and noALT trials differ
significantly at when BCLN ≤ 200 ms (Fig. 6.14B).
6.3.6
Slope Criteria for the Onset of Alternans
I have found that none of the slopes of individual restitution curves (SSRC , SBRC ,
SDRC ) or the memory criterion, evaluated from Smem , correlate with alternans (Fig.
6.15). Specifically, the SRC and BRC are generally very shallow and their slopes
remain well below one (less than or of the order of 0.3) for all experiments. In
contrast, DRC becomes quite steep at small BCL with slopes well above one seen in
59% of all measurements that show stable 1:1 responses for both ALT and noALT
trials. The memory criterion fails because Smem decreases as BCLN decreases and it
is much larger than one for slow pacing (large BCLN ).
129
Figure 6.13: DRC slope difference. (A) ∆SDRC for ALT and NoALT trials as
a function of BCLN . (B) P values below 0.05 (dashed line) indicate ∆SDRC is
significantly different from zero.
Figure 6.14: Spatial variation and alternans. The p values returned from a t-test
comparing (A) ∆AP D, (B) ∆SSRC , (C) ∆SBRC and (D) ∆SDRC of ALT and noALT
trials. The dashed line indicates a p value of 0.05.
130
Figure 6.15: Slope criteria. The mean slopes of (A,B) SRC, (C,D) BRC (E,F) DRC
and (G,H) the mean memory criterion indicate that none of these are predictive
of alternans in spatially extended tissue since they do not satisfy the requirements
detailed in Section 6.2.6. The legend in panel (H) applies to all panels.
131
6.4
6.4.1
Discussion
Spatial Differences in Restitution Properties
My study found spatial differences in several characteristics of restitution. In particular, steady-state APD and the slope of the SRC showed significant spatial differences
at many BCLs in both ALT and noALT trials. APD showed a consistent positive
difference, suggesting that the APD was larger close to the stimulus, as was observed
in optical experiments (Ch. 3). SRC slope showed a mean positive difference in
ALT trials and a mean negative difference in noALT trials, although this trend did
not hold for every trial. Results for ∆SDRC and ∆SBRC were mixed with significant
spatial differences detected only at some BCLs. Specifically, the slope of the DRC
showed significant spatial differences only at fast pacing in ALT trials.
I believe that the spatial differences I observed are dynamically induced because
of our choice of experimental substrate. The nearly homogeneous nature of bullfrog
ventricular tissue suggests that any observed spatial differences in restitution properties are not simply reflecting underlying tissue inhomogeneity, but are instead created
dynamically during pacing of the tissue. The optical studies presented in Chs. 3 and
4 confirm that spatial variation of SDRC is entirely dynamically induced and that
spatial variation of APD can also be dynamically induced.
Some spatial differences reported here have been seen in experiments using mammalian cardiac tissue. Large APD gradients seem to be a common characteristic of all
cardiac tissue, having been observed in guinea pig [30, 65, 67, 70, 134] and pig [64, 68]
hearts. The APD gradients observed in those experiments were smaller than those observed in our experiments. In simulations of homogeneous myocardium [60,61], APD
132
gradients were also larger than in experiments, suggesting that tissue heterogeneity may decrease spatial variation of APD. Choi and Salama [134] observed shorter
APDs at the apex and longer APDs at the base, further suggesting that spatial tissue heterogeneity may alter APD gradients. Gradients in the slope of the SRC have
also been observed in previous studies [69–71,151], however these measurements were
made in humans [69] and guinea pigs [70,71,151], where the gradients may have been
caused by fiber structure or other heterogeneities. In fact, Akar et al. note that the
SRC slope gradient is parallel to the cardiac fibers [70]. Finally, our observation that
the DRC slope differences were primarily positive differs from the findings of Qin et
al. [68], who observed no consistent gradient in DRC. These findings suggest that
tissue heterogeneity in mammals may mask dynamically-induced differences in slope
of the DRC.
6.4.2
Predicting the Tissue’s Propensity to Alternans
The restitution hypothesis [38], in which the stability is determined by the slope of the
restitution curve, failed in all trials, regardless of the slope tested or the measurement
location. The slopes of the SRC and BRC remained well below one in ALT trials,
even near the onset of alternans. In fact, the slopes of the SRC and BRC were slightly
larger in noALT trials just before the transition to a 2:2 response. The slopes of the
DRC were quite steep even during stable 1:1 response. This result has also been
observed in other experiments [39, 41, 43] and in simulations [39, 42, 43, 152].
The failure of the restitution hypothesis is not surprising since my experiments
show rate-dependence and short-term memory (Fig. 6.8). However, even a stability
criterion that takes into account these effects in one particular form (Eq. 6.4) does
133
not accurately predict the tissue’s propensity to alternans. In our experiments, Smem
was well above one during stable 1:1 response in ALT and noALT trials. The failure
of all slope-based criteria suggests the models of local APD dynamics, from which
these criteria are derived, do not completely capture the factors responsible for the
loss of stability in spatially-extended cardiac tissue.
Some of the factors not accounted for by the slope criteria include border-collision
bifurcations, intracellular calcium, and spatial interactions. Border-collision bifurcation occurs in systems whose dynamics is described by a piece-wise smooth function [153]; a bifurcation occurs when the parameter crosses the border between two
smooth pieces of the function. Recent research has suggested that the transition to
alternans in paced cardiac tissue may be mediated by a bifurcation that has bordercollision characteristics [154]. If this is the case, it may not be possible to predict the
onset of alternans by examining the 1:1 response. It would explain why the slopebased criteria that are derived from models with smooth dynamics fail to predict the
onset of alternans.
Another possibility is that APD alternans is driven by calcium alternans [155–
160]. In mammals, the stability of calcium transients is thought to be determined
by the feedback gain of calcium release from the sarcoplasmic reticulum [159]. This
possibility shifts the problem of predicting the onset of APD alternans to one of
predicting the onset of calcium alternans. A stability criterion for calcium alternans
[159] involves measures of calcium changes in the cytosol and sarcoplasmic reticulum,
but the measurement of subcellular calcium has only recently been achieved during
repeatable fast pacing [160]. However, calcium cycling in frog myocardium relies
much less on the sarcoplasmic reticulum [161], so calcium alternans may not drive
134
APD alternans or they may do so by a different mechanism in my experiment.
Finally, all stability criteria used to date were derived from models of single
cardiac cell dynamics. My experiments, and most other cardiac experiments related to rhythm stability, are performed in spatially extended pieces of tissue where
neighboring cells are coupled through gap junctions. Experiments and simulations
show that cell-to-cell coupling strongly affects cardiac rhythm dynamics: it decreases spatial heterogeneity [61, 62] and changes the BCL at which alternans is
observed [49, 61, 162, 163].
The results presented here indicate that spatial interactions may also play a critical role in the stability of the 1:1 response. I found that spatial differences in APD
and SRC slope are predictive of alternans at many BCLs. ALT trials had greater
∆AP D than noALT trials and ∆SSRC was positive in ALT trials but negative in
noALT trials. The slope of the DRC is predictive of alternans at fast pacing, with
∆SDRC becoming positive and growing in magnitude at fast BCLs in ALT trials.
Finally, the slope of the BRC is predictive of alternans at BCLN = 250, 550 ms.
Most importantly, all four characteristics are predictive of alternans at some BCLs
much slower than BCLt , which may have a clinical advantage, as discussed below.
6.4.3
Study Limitations
My findings are purely empirical. In contrast to the traditional slope criterion, I do
not have an underlying model and cannot offer a theoretical explanation of why the
spatial differences should be predictive of alternans. Likewise, without an underlying
model, I cannot propose a specific quantitative threshold for alternans. Nevertheless, the results are compelling enough to stimulate future work on developing the
135
theory linking spatial differences in restitution properties and tissue’s propensity to
alternans.
The first step in building such a theory is to obtain a better picture of the observed spatial differences. Use of only two microelectrodes can tell us whether spatial
differences exist, but it does not give us any details of the spatial distribution. The
repeatability of my results over seven animals suggests that these differences result
from consistent spatial gradients of restitution properties, rather than from random
patterns. Nevertheless, there is a need for a follow-up study that would use potentiometric dyes and an optical imaging system to provide information about the spatial
distribution of multiple restitution properties throughout the tissue as a function of
BCL.
The results presented in this study may be specific to bullfrog tissue, which is
relatively uniform, both structurally and electrophysiologically [97–99, 115]. To be
clinically useful, my results will need to be confirmed in mammalian tissue. The
main question is whether the more complex tissue structure in mammals may alter dynamically-induced spatial gradients of restitution. As discussed above, spatial gradients of APD, SSRC , and SDRC have also been observed in mammalian
tissue [30, 64, 66–71, 134, 151], suggesting that some of my observations may hold
in larger, heterogeneous hearts. A simulation study has shown that dynamicallyinduced spatial gradients in APD appear on the surface only in small and relatively
uniform hearts [60]; in larger and more heterogeneous hearts, these gradients tend to
appear transmurally. It is not known whether these transmural gradients show the
same correlation to the onset of alternans as observed in our experiments on bullfrogs. Studies of rabbit hearts are currently underway in our laboratory to confirm
136
the findings presented here.
6.4.4
Clinical Implications
My results, if they are confirmed in mammalian tissue, may have important implications for the diagnosis and treatment of patients with arrhythmias. Diagnostic
procedures, either currently in use or proposed based on theory and experiments,
analyze spatially-averaged temporal response patterns (microvolt T-wave alternans
in the ECG [143]) or temporal response patterns at a single location (steepness of
the restitution curve [102] or increase in gain during alternate pacing [144]). My
study suggests an entirely new approach, based on spatial differences in restitution
properties. Likewise, while some current pharmacological treatments aim to prevent
arrhythmias by decreasing steepness of the restitution curve [164], my results may
open the possibility of alternative pharmacological treatments that target spatial gradients of restitution properties. Thus, my findings may lead to entirely new methods
of determining a patient’s vulnerability to arrhythmias and to the design of novel
antiarrhythmic medications.
The proposed spatial approach may have significant advantages. Existing interventional diagnostic methods include pacing at fast rates or delivering multiple
shortly-coupled extrastimuli in order to induce an episode of an arrhythmia. My
results show that spatial differences are predictive of alternans at cycle lengths much
longer than the cycle length of the transition to alternans. This suggests that the
new procedure may be safer and better tolerated by patients. Additionally, it can
be implemented using existing technology because diagnostic catheters already have
multiple recording sites.
137
6.5
Conclusion
This study examined the spatial variation of the slopes of the DRC, SRC, and BRC
as well as the spatial variation of the steady-state APD simultaneously by using the
restitution portrait. The spatial differences in APD and in the slope of the DRC were
primarily positive, indicating that these variables decreased as the wave propagates
away from the tissue, as was observed in the optical experiments. The differences of
the slopes of the BRC and the SRC were primarily positive in tissue that exhibits
alternans and primarily negative in tissue that does not exhibit alternans. Finally,
I determined that spatial heterogeneity in the slope of the DRC at rapid pacing
was significantly larger in tissue that exhibits alternans than in tissue that does not
exhibit alternans.
138
Chapter 7
Conclusions and Future Work
7.1
Research Findings
I built an optical imaging system using the novel light source of ultra-high power
LEDs that was used to record electrical activity in small pieces of bullfrog ventricular
tissue. The optical imaging system was used to determine the spatial variation of
steady-state APD and SDRC over the surface of the tissue. Experimental results were
compared to simulations using a simplified cardiac model. Finally, spatial differences
in SBRC and SSRC , as well as APD and SDRC , were studied using microelectrode
measurements made simultaneously at two spatial locations in the tissue.
7.1.1
Spatial Variation
Steady-State APD
The spatial variation of steady-state APD was found to be a boundary effect in
simulations (Fig. 5.4) with a decrease in APD near the site of the stimulus and
another decrease in APD near the insulated boundaries. There is no variation of
APD in the middle of the tissue, making it clear that the changes in APD near the
stimulus site and the insulated boundaries are due to the presence of the imposed
tissue heterogeneity at these locations.
The evidence for a similar boundary effect in bullfrog ventricular tissue is not as
clear-cut. Although I observe a decrease in APD as the wave propagates away from
139
the stimulus site and near the insulated boundaries (Fig. 3.9) in some trials, I also
observe other spatial patterns of APD. In particular, when pacing from a site along
the top of the ventricle (where the auricles had been), the electrical wave propagated
rapidly through the center of the tissue and resulted in a spatial pattern of APD
in which the longest APDs were not at the stimulus site, but were instead located
along one of the two insulated boundaries. Further, three of twelve animals exhibited
evidence of “frozen-in” tissue heterogeneity. In these three animals, the spatial APD
pattern remained the same despite changes in stimulus location (Fig. 3.7, Table 3.1).
These experimental observations suggest that even if spatial patterns of APD can be
dynamically induced, underlying tissue heterogeneity, such as specialized conduction
pathways (see Sec. 3.4.1), will affect the observed APD spatial pattern.
Even in trials where the APD spatial pattern was similar to that observed in
simulations, with longest APDs near the stimulus site, this study cannot conclusively
state that the experimentally observed spatial pattern of APD is a result of similar
boundary effects. Some data show a region of little APD variation in the center of
the tissue (Fig. 3.13), while other data are not so clear (Fig. 3.8). Simulations in
a sheet with boundaries identical to the experimental substrate also did not show
a large region of constant APD in the center of the sheet (Fig. 3.12). Thus, it is
difficult to determine the influence of each type of boundary, one where current is
injected into the system and one where current cannot flow out of the system, on
the resulting observed spatial pattern of APD. Nonetheless, the fact that spatial
patterns of APD can be altered by changing pacing location suggests that spatial
patterns of APD are at least partially determined by the location of the stimulus and
any insulated boundaries. Simulations with a two-variable cardiac model confirm
140
that spatial patterns of APD are tied to the locations of the stimulus and insulated
boundaries (Fig. 3.12). I found that the width of the boundary layer in experiments
was 1.6-2λ (λ = 0.3 mm) and that the width of the boundary layer in simulations
was 1.6-2.5λ (λ = 1 mm). The total spatial variation of APD, however, extended
over distances significantly longer than the width of the boundary layer, often varying
over the entire surface of the tissue (∼10 mm).
Slope of the Dynamic Restitution Curve
SDRC also varies spatially over the surface of the tissue, both in experiment (Fig. 4.3)
and in simulation (Fig. 5.6). SDRC is largest near the stimulus site and decreases
near the insulated boundaries. Unlike APD, which shows increased gradients near
boundaries, SDRC exhibits a nearly constant gradient over the surface of the tissue.
Simulations confirm that SDRC has a constant gradient, at least over a 2 cm sheet
of tissue (Fig. 5.6). Also unlike APD, none of my experiments showed evidence that
spatial variation of SDRC was influenced by underlying tissue heterogeneity. In every
tissue sample, when the pacing location was changed, the resulting spatial pattern
of SDRC also changed with the largest slopes always located near the stimulus site.
Finally, I found that the spatial variation of SDRC , as measured by mean SDRC
gradient in Ch. 4 or spatial difference in Ch. 6, increases with decreasing BCL.
Slope of the S1S2 Restitution Curve
Although spatial variation of SSRC was not studied over the entire surface of the
tissue, the two-microelectrode study determined that SSRC also shows spatial differences in bullfrog ventricular tissue (Fig. 6.11) primarily at large BCL. The spatial
141
difference of SSRC decreases as BCL decreases even though the slope of the S1S2 RC
increases with decreasing BCL.
Slope of the Constant-BCL Restitution Curve
The evidence for spatial variation of SBRC is limited. The two-microelectrode studies
indicate that SBRC exhibits statistically significant spatial differences at some BCLs
(Fig. 6.12), with the general trend that the spatial difference decreases as the BCL
decreases.
7.1.2
Correlation to Alternans
Steady-State APD
Experimentally, two measures of the amount of spatial variation of APD were used to
determine whether there is a statistical correlation between spatial variation of APD
and the propensity to exhibit alternans at rapid pacing. In the optical experiments
presented in Ch. 3, I found that the mean spatial APD gradient could differentiate
between trials that exhibited complex rhythms at rapid pacing and trials that went
directly to 2:1 at rapid pacing, at least at some BCLs (Fig. 3.17). The spatial
difference in APD, as measured by simultaneous microelectrode recordings, could
also be used to differentiate between ALT and noALT trials (Figs. 6.10A and 6.14A)
with statistically significant differences evident as far as 200 ms from the transition
point. Steady-state APD exhibited larger mean spatial gradient and spatial difference
in ALT trials than in noALT trials.
142
Slope of the Dynamic Restitution Curve
Measures of spatial variation of SDRC could also differentiate between ALT and
noALT trials. I found that mean spatial gradient of SDRC could differentiate between ALT and noALT trials at BCLs 200 ms larger than BCLt (Fig. 4.5). Similarly,
the spatial difference in SDRC could also be used to differentiate between ALT and
noALT trials at BCLs 200 ms larger than BCLt (Figs. 6.13A and 6.14B). ALT trials
exhibited an increase in mean spatial gradient and in spatial difference of SDRC as
BCL decreased, whereas the mean spatial gradient and spatial difference of SDRC
in noALT trials showed little increase as BCL decreased. Simulations confirm that
mean spatial gradient of SDRC can be used to differentiate between ALT and noALT
trials, with ALT trials exhibiting a large increase in mean spatial gradient as BCL
decreases (Fig. 5.9A).
Slope of the S1S2 Restitution Curve
Spatial differences in SSRC were significantly different for ALT and noALT trials
at most BCLs (Figs. 6.11A and 6.14C). ALT trials exhibited primarily positive
spatial differences particularly at large BCLs, while noALT trials exhibited primarily
negative spatial differences particularly at large BCLs.
Slope of the Constant-BCL Restitution Curve
Although spatial differences in SBRC were primarily positive for ALT trials and primarily negative for noALT trials (Figs. 6.12A), the trend was less conclusive than for
SSRC . In particular, the observed spatial differences of SBRC could not differentiate
143
between ALT and noALT at most BCLs.
7.2
7.2.1
Discussion
Spatial Variation
Several other studies have also seen spatial differences in steady-state APD [30, 40,
64–70,73,74,134]. These studies gave conflicting results on the possible causes of the
observed heterogeneity. Some studies have reported long APDs near the stimulus
site [40, 64, 72], a boundary effect predicted by simulations [59–63]. Other studies
report that the observed spatial heterogeneity of APD is determined by underlying
tissue heterogeneity [66–69,73]. My study suggests that both of these effects occur in
cardiac tissue. I observed a boundary effect similar to that predicted by simulations,
where APD increased near the stimulus site and decreased near insulating boundaries.
I also observed trials where the spatial pattern of APD was driven by structure within
the tissue, In particular, activating a specialized conduction pathway within the tissue
produced a spatial pattern of APD where the longest APDs were not near the stimulus
electrode. In addition, three animals produced spatial patterns of APD that did not
change with changes in stimulus location.
My study could not determine whether the APD boundary effect was caused
by increased membrane resistance or blocked current flow (Sec. 2.3). Both conjectures predict similar increases in boundary width (∼1.5-2λ), so they cannot be
differentiated by measurement of the boundary width alone. Increased membrane
resistance that leads to an increased length constant, as proposed by Sampson and
Henriquez [60], requires measurement of the membrane resistance during the action
144
potential and diastolic interval and inability of current to flow beyond insulating
boundaries, as proposed by Cain and Schaeffer [63], requires that the movement of
ions be tracked during the action potential, neither of which was monitored during
my experiments.
Regardless of the cause of the increased boundary effect, increased boundary
width has several repercussions for cardiac dynamics. Regions of non-propagating
tissue, possibly caused by cell death or even by previous electrical activity, will be
felt over distances larger than previously expected. It is known that regions of nonpropagating tissue can lead to breaks in the wavefront and conduction block [121–
123] if they are large enough. The increased boundary effect suggests that even
small obstacles to propagation may cause wavebreaks and lead to arrhythmias. The
increased boundary width may also have consequences for the application of control
techniques to the heart. Most control techniques involve injecting small amounts
of current into the tissue in an attempt to alter the local dynamics [128–132]. My
studies indicate that the injected current may affect the dynamics over a greater
distance than previously expected. This means that control of large pieces of tissue
or even whole hearts may be possible with the application of only a few controllers.
Spatial variation of the slope of the DRC was previously measured by Qin et
al. [68]. They observed that SDRC did not have a consistent spatial gradient in
porcine hearts. My studies indicate that SDRC can, in fact, show a constant gradient
over the surface of cardiac tissue. Although my studies did not show any evidence that
spatial variation of SDRC was affected by underlying tissue structure (i.e. no animal
exhibited “frozen-in” patterns of SDRC ), it is possible that tissue heterogeneity plays a
role in the conflicting observations of spatial variation of SDRC . For example, porcine
145
hearts consist of different types of cardiac cells [95] whereas bullfrog ventricular tissue
consists of a single type of cardiac cell [99], a difference that may affect observed
spatial patterns of SDRC .
7.2.2
Onset of Alternans
These experiments have shown that the prevalent theories of when the onset of alternans occurs in single cells do not hold in spatially extended tissue. The most
prevalent theory suggests that alternans occur when the slope of the restitution curve
becomes greater than 1. Although several studies have already called into question
this theory [39–41, 41, 43, 68], my study further emphasizes this result by measuring
the slopes of all the restitution curves at two different spatial locations simultaneously and finding that none of the slopes agree with the hypothesis (Fig. 6.15). I
also tested a stability criterion derived from a mapping model with memory (Eq.
6.4) [43] and found that it also did not accurately predict the onset of alternans in
our experimental preparation.
The prevalent theory of arrhythmogenesis in spatially extended tissue suggests
that arrhythmias are caused by large spatial gradients in APD [30,31,76]. Specifically,
experiments found that spatial gradients larger than 3 ms/mm lead to arrhythmias.
My experiments consistently showed much larger spatial gradients (4-8 ms/mm) during stable 1:1 responses. Also, I found that the mean spatial APD gradient decreased
as BCLt was approached (Figs. 6.10A, 3.16, and 3.17A). If large APD gradients
caused arrhythmias, I would expect the onset of the arrhythmia to occur at the BCL
with the largest APD gradient. I do not observe this behavior.
My experiments suggest additional correlations between spatial variation of resti146
tution properties and the propensity to exhibit alternans. Spatial variation of SDRC
exhibits a larger increase in ALT trials than in noALT trials as the transition point is
approached. Spatial differences in SSRC and to some extent SBRC are primarily positive in ALT trials and primarily negative in noALT trials. Although my experiments
cannot determine if the observed spatial variation of restitution properties causes
alternans, the observed statistical correlations are worthy of further investigation to
determine whether there is a link between spatial variation of RCs and alternans.
Even without a firm link, the statistical correlations may be clinically useful. If
similar correlations are found in human cardiac tissue, measurement of spatial variation of restitution properties may offer a safer method of predicting which patients
are at risk for alternans. Current methods of assessing vulnerability to arrhythmias
often involve rapid pacing which puts the patient at risk. The advantage of measuring spatial differences in RCs is that, particularly for SSRC , the spatial difference at
slow pacing may be able to predict the onset of alternans at rapid pacing.
7.3
Future Work
The spatial variation of SSRC and SBRC should be studied using the optical mapping
system. The spatial differences of these two restitution properties were smaller than
the differences seen in APD and SDRC so the evidence for a consistent gradient is
less conclusive. It is possible that the spatial differences we observe are simply due
to cell-to-cell differences in the tissue. Maps of SSRC and SBRC over the entire tissue
would provide conclusive evidence of gradients or lack thereof.
The spatial variation of restitution properties also needs to be studied in mam147
malian tissue. Studies in mammalian tissue have seen spatial gradients of APD
similar to those seen in our experiments [40,66], but spatial gradients of SSRC [70,71]
and SDRC [68] different than those seen in my experiments. These differences need
to be explored, perhaps using a systematic simulation study such as the one used
by Lesh et al. [62] to show that tissue heterogeneity tends to decrease and alter the
direction of spatial APD gradients. Such studies will lead to a better understanding
of the roles of dynamics and underlying tissue heterogeneity in the behavior of the
heart.
My experiments cannot determine whether the theories proposed to explain spatial variation of APD in simulation can explain experimentally observed spatial variation of APD. Experiments that track the change in membrane resistance during the
course of an action potential and experiments that track the movement of ions near
the boundaries of cardiac tissue will help determine what role each of these plays in
the observed boundary effect.
Although my experiments discounted the prevalent theories of the onset of alternans, they also suggest new correlations which can be exploited to predict the onset
of alternans. Spatial differences in slopes of the DRC, SRC, and BRC show significant differences between trials that exhibit alternans and trials that do not exhibit
alternans. Several tissue samples exhibited alternans in one trial and transitioned
directly to 2:1 in another. It is possible that the restitution properties of the tissue
changed over time, causing the change in observed behavior, but detailed studies of
how restitution properties change in dying tissue and how that affects the behavior
of the tissue are needed.
Finally, in order to use correlations between spatial variation of restitution prop148
erties and the propensity to exhibit alternans in a clinical setting, further experiments
are needed in mammalian tissue to determine whether these correlations hold in more
heterogeneous tissue. As mentioned above, tissue heterogeneity can affect spatial gradients of restitution properties. However, even if tissue heterogeneity alters dynamically induced gradients, it remains unclear whether this will affect the correlations
between spatial differences in restitution properties and the onset of alternans. A
study by Pak et al., which found that patients with inducible ventricular tachycardia
had larger spatial differences in SSRC [69] provides hope that correlations between
spatial gradients of restitution properties and arrhythmias holds in more complex
hearts.
7.4
Final Thought
The work presented here has shown that the current understanding of spatial variation of restitution properties and its role in the stability of the 1:1 response in
spatially extended cardiac tissue is incomplete. It is hoped that the studies of spatial
variation of restitution properties will lead to a better understanding of the role of
restitution in the onset of alternans and other arrhythmias.
149
Appendix A
Ultra-high Power Light Emitting Diodes
A.1
Introduction
The development of voltage-sensitive (potentiometric) dyes has revolutionized the
study of electrical activity in spatially extended biological systems such as the heart
[165] and brain [166]. In a typical optical mapping study, the electrical activity
at different spatial locations can be visualized directly using a fluorescent dye in
combination with an illumination source to excite the dye and a detector array to
record dye fluoresence. Detectors range from a single photomultiplier tube (used with
a laser scanner) [25], to photodiode arrays and CCD cameras. Such studies have given
new insights into the behavior of cardiac fibrillation [23, 24], its termination using
electrical shocks [25] and the response of cardiac tissue to point stimulation [26].
The absorbance and fluorescence emission spectra of the voltage-sensitive dyes
exhibit voltage-dependent shifts that can be used to determine the transmembrane
action potential [167], where in a typical experiment the fluorescent power changes by
∼8 to 10% during an action potential for myocardium stained with the best available
dyes. Therefore, the transmembrane voltage can be measured using a narrow-band
excitation source in combination with a long-pass filter placed before the detector
array.
Over the years, a variety of excitation sources have been used successfully in
optical mapping systems. A typical excitation source for use with di-4-ANEPPS
150
(specifications for other dyes are available from Molecular Probes) needs to provide
an intensity of ∼10-100 mW/cm2 at the tissue surface, a spectral bandwidth less
than ∼35 nm so that it is less than the ∼100 nm dye absorption bandwidth, and the
variation in the power of the source over time must be much less than the anticipated
change in fluorescent power when the cell depolarizes. Early systems used high-power
white-light sources (such as tungsten-halogen filament lamps and mercury/xenon arc
lamps [25,107,108]) in combination with a narrow band-pass filter to select the desired
excitation wavelength and spectral bandwidth [168]. Since the filter bandwidth needs
to be much smaller than the emission spectrum of the white light source, only a
small fraction of the power makes it through the filter and thus the system is very
inefficient. However, these sources are still in widespread use [169]. In addition, the
spatial pattern generated by white light sources tends to vary over time, thereby
degrading the quality of the voltage-sensitive map. For this reason, many researchers
have used lasers (e.g., argon-ion [109, 110] and frequency-doubled neodymium-doped
yttrium aluminum garnet [111]), although lasers with sufficient power and stability
tend to be very expensive (>$10,000).
As an alternative, some groups have investigated the use of light emitting diodes
(LEDs) as illumination sources [112, 113]. LEDs consist of contacted p-type and ntype semiconductors; when current is passed through the junction, excess electrons
from the n-type material combine with holes in the p-type material, resulting in an
emitted photon. LEDs are very efficient light sources, ideally emitting one photon
for every electron injected, with all the optical energy emitted in the desired narrow
(∼30-50nm) spectral band. Due to this efficiency, the amount of noise in the emitted
light is largely determined by the noise of the current source [170]. The narrow
151
bandwidth, high efficiency, and the potential for low-noise operation of LEDs satisfy
the illumination source requirements for succesful optical imaging. LEDs are also
significantly less expensive than either lasers or white light sources. Thus, they offer
an attractive new option for use in optical imaging experiments if they can achieve a
signal to noise ratio comparable to current sources.
Low-power LEDs have been used succesfully in cardiac optical imaging experiments where small areas of tissue were imaged. Kodama et al. used LEDs to study
high voltage DC stimulation in rabbit hearts [112]. They used seven LEDs with a
total illumination power of 0.25 mW in conjunction with an optical fiber bundle to
illuminate and collect emission from an area of 0.2 cm2 . The small imaging area
in this experiment did not require a powerful light source, so the low-power LEDs
sufficed. They also noted that the light output of the LEDs varied significantly over
time, but did not offer an explanation of this time-dependent behavior. More recently, Entcheva et al. used 10 mW LEDs to study the dynamics of a monolayer of
cardiac cells [113]. In their experiment, the LEDs were used to illuminate the tissue
directly and an optical fiber bundle with a total area of 1.1 cm2 was used to collect
the emitted light. Here again, the area requiring illumination was quite small, so
the excitation source did not need to be very powerful. Although the LEDs used in
these experiments are too low power to image large sections of cardiac tissue, new
generations of LEDs are produced every year, so we expect rapid progress that will
make the LED useful for imaging large sections of the heart.
In this paper, we report on the use of recently available ultra-high power LEDs
(Lumileds, model Luxeon Star/O and Star/V) as an illumination source in cardiac
optical mapping systems. They are available in two different models: Star/0 (∼$15),
152
lower power (35-85 mW) with collimating optics, and Star/V (∼$40), higher power
(200-400 mW) without collimating optics. The Star/V is mounted on a hexagonal
base about 2 cm in diameter and operates at a maximum current of 700 mA. The LED
chip consisting of 4 diodes is mounted inside a plastic lens to increase light extraction
efficiency. The Star/O LED is mounted on a 2 cm×2 cm square base and operates
at a maximum current of 350 mA. The LED chip, again consisting of 4 diodes, is
mounted inside a plastic lens that has collimating optics mounted on it. The optics
create a beam about 1.5 cm in diameter with an angular divergence of 10◦ . The
new LEDs are significantly more powerful than those used in previous experiments
and are available in a variety of emission wavelengths (Fig. A.1), so the LEDs can
be used with a variety of different dyes and for ratiometry experiments [171, 172].
Thus, we expect that the LEDs will be useful in a broad variety of optical imaging
experiments. The purpose of our study is to demonstrate that the Luxeon Star LEDs
perform as well as currently used sources and so are an appealing alternative in many
imaging experiments that require illuminating a large tissue area.
A.2
Methods
We performed experiments to measure properties of the LEDs that are relevant to
their use in imaging experiments. We determined the intensity and spatial uniformity
of the LEDs, as well as the noise and long-term light output stability. Finally, we
conducted in vitro experiments in both rabbit and frog cardiac tissue to measure
the performance of the LEDs in optical mapping experiments and to compare its
performance to that of a frequency-doubled Nd:YLF laser. The Nd:YLF laser is a
153
Figure A.1: Spectra of the Luxeon Star/O LEDs.
light source that is commonly used in optical imaging experiments [111,173–175] and
was used in our experiments as a standard that the LEDs had to meet or exceed.
The cardiac experiments were performed using the potentiometric fluorescent dye di4-ANEPPS, which requires an excitation source with a central wavelength between
470 and 570 nm. The green and cyan LEDs both have central wavelengths within
this range (Fig. A.1), thus we performed experiments with both color LEDs to assess
the fluorescence signal quality.
A.2.1
LED characteristics
We measured the intensity of the LEDs with a New Focus photodetector (model
#2031, calibrated to a Newport optical power meter, model 1830-C) at various distances for both the Star/O and Star/V models. Both models were run at their maximum current (700 mA for the Star/V and 350 mA for the Star/O) by a low-noise,
154
constant current power supply (Agilent model E3615A). We made measurements using both colors. Since the results were similar for both, only the results for the green
LED will be presented here.
To measure the uniformity of the intensity, we captured images of the transverse
intensity distribution of the Star/O model at 1 cm and 5 cm from the LED. We measured the distributions by illuminating a piece of paper with the LED and capturing
100 images with a CCD camera (DALSA model CA-D1-0128T). We then averaged
the images to determine the resulting distributions.
Since these LEDs are so much more powerful than previous models, heating of
the junction may affect their performance. We mounted the LEDs on CPU heat
sinks and fans to help dissipate heat. We then turned on the LEDs and recorded the
intensity with the New Focus detector for 20 seconds every 2 minutes over a span of
10 minutes and every 10 minutes after that for 1 hour. Measurements were made on
both the Star/O and Star/V models.
Finally, we measured the noise of the LEDs by illuminating a card stained with
fluorescent paint that mimics the fluorescence of the dye in an experiment without
the constant disruption of action potentials. This type of measurement ensures that
any change in intensity from frame to frame is solely due to noise in the experimental
setup and not due to changes in electrical activity in the tissue. We illuminated the
card with one of three light sources: the green LED, the cyan LED and a frequencydoubled Nd:YLF laser for comparison. We used the DALSA camera to collect 500
images at a frame rate of 490 Hz. We calculated the mean and standard deviation of
the intensity for each pixel to determine the relationship between intensity and noise.
155
A.2.2
In vitro Experiments
We used the LEDs to perform optical mapping experiments in rabbit tissue. This
study was performed in accordance with the Research Animal Use Guidelines of the
American Heart Association and the Public Health Service Policy on Humane Care
and Use of Laboratory Animals. The experimental protocol was approved by the
Vanderbilt Institutional Animal Care and Use Committee. Two New Zealand white
rabbits were anesthetized and their hearts were excised and moved to a Langendorff
perfusion system. The hearts were perfused with oxygenated Tyrode’s solution and
the temperature and pH were maintained at 37◦ C and 7.4, respectively. We stained
the hearts with 200 µL of di-4-ANEPPS, which was delivered through a bubble trap
above the aorta. We also added diacetyl monoxime (DAM) at a concentration of
15 mM to the Tyrode’s solution to block muscle contraction and prevent motion
artifacts. The heart was paced at a constant basic cycle length of 300 ms.
We illuminated the tissue with one of three sources: the cyan LED, the green
LED or the Nd:YLF laser. Since both LEDs emit some light at wavelengths greater
than the cut-off for our high-pass filter, additional dichroic filters (Edmund Industrial
Optics H52-538 and H52-535) were placed in front of the LEDs to block any long
wavelength emission that could be mistaken for fluorescence. We used an OG 590
filter (Edmund Industrial Optics H46-064) to filter the fluorescent emission and we
used the DALSA camera to capture images at 490 Hz (Fig. A.2A). We made several
3,000-frame recordings to determine the signal amplitude and signal-to-noise ratio
(SNR) of all three sources.
We performed similar experiments using frog cardiac tissue to determine if there
156
A
B
Stimulus
Stimulus
CCD
Oxygenated
LED
Camera
Solution
OG 590
Filter
CCD
Camera
Oxygenated
OG 590
Filter
Solution
Dichroic
LED
Dichroic
Filter
Filter
Figure A.2: Experimental setup for in vivo epifluorescence measurement of cardiac
action potentials. (A) A Langendorff-perfused rabbit heart is mounted in front of a
CCD camera. Two LEDs with filters to block long-wavelength emission illuminate
the tissue. Images are collected through a cut-off filter by a CCD camera. (B) A
small piece of bullfrog ventricular tissue is placed in a tissue dish and superfused
with oxygenated Ringer’s solution. Two LEDs with filters to block long-wavelength
emission provide excitation illumination. Images are collected with a CCD camera
equipped with a cut-off filter.
157
were any tissue-specific effects. This study was performed in accordance with the
Research Animal Use Guidelines of the American Heart Association. The protocol
was approved by the Duke University Institutional Animal Care and Use Committee.
We stained small pieces (about 5×5×3 mm) of bullfrog ventricular myocardium with
50 µM di-4-ANEPPS and placed them in a tissue chamber. We maintained the
viability of the tissue by superfusion with a standard Ringer’s solution. The tissue
was paced at a constant basic cycle length of 1,000 ms and illuminated with either
the cyan or green LED with the appropriate filters. We captured images at 490 Hz
for 2 s with the DALSA camera (Fig. A.2B) to determine the signal size and SNR.
A.3
A.3.1
Results
Intensity
Most imaging experiments require a minimum excitation source intensity of ∼10
mW/cm2 . Since intensity decreases with distance from the source, we need to determine how rapidly it decreases and at what distance it no longer meets the requirements for optical imaging.
Figure A.3 shows the effectiveness of the collimating optics. The Star/O manages
to produce intensities larger than those of the Star/V (more powerful) LED once
we are more than ∼1 cm from the source. Thus, for imaging experiments where
the source needs to be some distance from the tissue, the Star/O LED is the better
choice.
Though the LEDs provide enough intensity for imaging experiments, we also
require that that intensity be uniform over the imaging surface. The intensity distri158
Figure A.3: Intensity of the green LED as a function of distance.
bution of the green Star/O LED at 1 cm shows the 4 diodes that make up the source
(Fig. A.4A). By the time we have moved about 5 cm away (∼1 cm for the Star/V),
the intensity is essentially uniform (Fig. A.4B).
In summary, we have found that both the Star/O and Star/V LEDs meet the
minimum intensity requirement of 10 mW/cm2 . The Star/O meets the requirement
up to a distance of ∼4 cm while the Star/V meets the requirement up to a distance
of ∼2.5 cm. The LEDs provide somewhat non-uniform (bright central spot) intensity
at close distances, but are uniform light sources beyond 5 cm for the Star/O and 1
cm for the Star/V.
A.3.2
Thermal Effects
When an LED is turned on, the junction temperature increases. As the junction
heats up, the light intensity of the LED decreases, primarily due to a decrease in
159
Figure A.4: Transverse intensity distributions of the green Star/O LED at (A) 1
cm from the source and (B) 5 cm from the source. The 1 cm distribution shows the
4x1 array of diodes that make up the LED (central bright region), while the pattern
is more uniform at 5 cm. (C) An intensity profile of the 1 cm distribution. The high
peak in intensity corresponds to the central bright spot. (D) An intensity profile of
the 5 cm distribution. The large peak in intensity has been replaced by a fairly flat
plateau.
160
Figure A.5: Time dependence of the output intensity of the green LED measured
every 20 seconds for ten minutes after applying power to the device.
internal efficiency of the LED. Although we mounted the LEDs to a heat sink and
fan to minimize heating, we expect that once the LEDs are turned on, the light
intensity will drop as the junction heats up. Results of this experiment using the
green LED are shown in Fig. A.5.
We see a drop in light output of 6.3% for the Star/V and 5.0% for the Star/O.
Although this is a fairly large drop in intensity (about the same magnitude as an
action potential), it only happens over a short period of time; the time constant for
the Star/V is 11.2 s and for the Star/O it is 8.32 s. The LEDs reach steady state
within 30 s and the light output remains very stable (within the digitization error of
our data acquisition card: ±0.001 mW/cm2 ) beyond the initial decrease in intensity.
161
A.3.3
Noise
In an optical experiment, there are two primary sources of noise: noise due to the
emitted light (shot noise) and noise from the detector (dark noise). The resulting
total noise is given by [176]
∆2total = ∆2shot + ∆2dark ,
(A.1)
where ∆ is a measure of the amount of noise. In our experiments, we use the standard
deviation of our signal as the measure of the amount of noise.
Dark noise arises from thermally generated electrons creating current fluctuations
on the camera sensing elements in the absence of light and is a constant for a particular device (∆2dark = 2.15 for our camera). Shot noise is the noise due to the
randomness of photon emission. Since photon emission events are uncorrelated, they
are described by a Poisson distribution. For a Poisson distribution [176], the standard
√
deviation is equal to the square root of the mean number of photons, ∆shot = n.
Since our camera converts the number of photons to a digital number, this can be
√
re-written as ∆shot = R · N , where R is the camera conversion factor. Therefore,
an ideal light source would have the following relationship between noise and mean
intensity:
∆N =
q
R · N + ∆2dark ,
(A.2)
Any deviation from the form of this equation indicates the presence of technical noise,
which is any other source of noise, such as variation in the light output of the emission
source or noise from the power supply [177].
To determine how well the data fit the above relationship, we fit the mean and
162
Figure A.6: Noise of the green LED, cyan LED, and ND:YLF laser. If the source
is operating at the quantum limit, we would expect to see a square-root relationship
between intensity and noise, as is seen for the green and cyan LEDs. The laser,
however, has an additional source of noise since it deviates from this dependence.
standard deviation values for each source to Eq. A.2. Experimentally determined
values of R are given in Table A.1. As a measure of the goodness of fit, reduced χ2
values were calculated [178]. Values of χ2 much greater than or much lower than 1
indicate a poor fit. Both the green and cyan LEDs fit the theoretical curve well, but
the laser does not fit this curve well. This implies that the laser has an additional
source of noise that was not considered in our simple model.
The raw data for the laser (before binning) clearly shows an additional source of
noise, as seen in Fig. A.7. The large scatter in standard deviation at high intensities
is likely due to a combination of laser speckle and minute motion of the card. Laser
speckle is caused by the interference of coherent waves when they are reflected off
a rough surface. If the surface happens to be moving, the phase of the reflected
163
Source
R
(N)
Green LED 0.0568±0.006
Cyan LED 0.0569±0.006
Laser
0.07±0.01
Reduced χ2
1.9
2.5
4.7
Table A.1: Values of the parameter used to fit the noise data of the three light
sources. R is determined by fitting the experimental data to Eq. A.2. The reduced
χ2 is a measure of goodness of fit.
waves will shift making some spots darker and others brighter. This is picked up by
the camera as a large change in intensity leading to a misleadingly high standard
deviation. Speckle can interfere with measurements of action potential duration and
amplitude, as noted by Lin and Wikswo [111], but there are techniques for reducing
its effect (spinning ground-glass filters [179]). Speckle occurs when the coherence
length of the source is larger than the height of the surface features [180]. Lasers
have a coherence length on the order of kilometers, while the LEDs have a coherence
length on the order of 10 µm. With this short coherence length, the LEDs are not as
sensitive to motion.
A.3.4
Signal Amplitude
We analyzed results of the in vitro experiments by examining the time series from
single pixels. Each pixel gives a series of action potentials, as shown in Fig. A.8.
Panels A through F show a time course of optical signals in rabbit tissue, paced at
a cycle length of 300 ms. Both raw data (A.8A,A.8C, and A.8E) and data filtered
with a 3×3-spatial Gaussian filter and a 3-point temporal averaging filter (A.8B,
A.8D, and A.8F) are shown. The laser and green LED recordings from the rabbit are
similar in shape and amplitude. The cyan LED, however, produces a much weaker
164
Figure A.7: Noise of the laser. We conjecture that the large scatter in standard
deviation at high intensities is caused by laser speckle and motion of the card.
signal. Panels G through J show a time course of optical signals in frog tissue, paced
at a cycle length of 800 ms. Again, both raw data (A.8G and A.8I) and filtered data
(A.8H and A.8J) are shown. In the frog tissue, both LEDs produce action potentials
with roughly the same amplitude.
We determined the average action potential amplitude (APA) for each pixel and
results were binned and plotted as a function of mean intensity. We then fitted
the data to a straight line, the slope of which gives the percent change in intensity
during the action potential. Figure A.9 shows the results for both frog and rabbit
experiments.
The biggest difference in the results for the two different types of tissue is the
performance of the cyan LED. In the rabbit tissue, the signal size of the cyan LED
is significantly smaller than that of either the laser or the green LED, while in the
165
RABBIT
FROG
Figure A.8: Optically recorded action potentials from rabbit and frog hearts. Pacing
interval was 300 ms for rabbit (A-F) and 800 ms for frog (G-J). Data was filtered
with a 3×3 spatial Gaussian filter and three-point temporal averaging.
Figure A.9: Recorded action potential signal as a function of mean illumination
intensity for (A) rabbit and (B) frog hearts. The slope of the line gives the percent
change in intensity during the action potential.
166
Source
Rabbit
APA
SNR
Laser
4.4±0.4%
11±2
Green LED 4.6±0.4%
12±2
Cyan LED 2.6±0.2% 7.0±0.9
Frog
APA
N/A
4.7±0.8%
5.3±0.8%
SNR
N/A
13±3
14±3
Table A.2: Results of the noise and action potential amplitude (APA) measurements.
frog both LEDs have almost equal signal size. Although this type of tissue-specific
difference in amplitude has been seen before [181], the underlying cause is unknown.
Finally, we computed the signal-to-noise ratio (SNR) by dividing the APA by
the standard deviation of the intensity. The results are summarized in Table A.2.
In rabbit tissue, the green LED provides the largest SNR, while in the frog tissue,
the cyan LED provides the best SNR. The laser was out-performed by the LEDs in
all cases except for the cyan LED in rabbit tissue. Thus, the LEDs are a suitable
alternative to currently used light sources in optical imaging experiments.
A.4
Discussion and Conclusion
We have shown that the characteristics of the Luxeon Star LEDs satisfy the requirements for an illumination source in an epifluorescence measurement of transmembrane potential. The Star/O provides sufficient intensity (≥10 mW/cm2 ) up to 4
cm from the source, while the Star/V provides sufficient intensity up to 2.5 cm from
the source. If greater intensity is needed, several LEDs can be used simultaneously
to illuminate the same region of tissue. Alternatively, a collimating lens could be
mounted to the Star/V LED to achieve higher intensities over a narrower field. We
have also shown that the intensity is spatially uniform beyond 5 cm from the Star/O
167
and 1 cm from the Star/V. Finally, there are no large temporal fluctuations in light
output after an initial rapid drop in intensity due to warming of the junction. More
than just meeting some minimum standards, the characteristics of the LED make it
an attractive alternative to the currently used white-light sources and lasers.
Although the LED is not as powerful as an unfiltered white-light source, it has
several advantages over this traditional excitation source. White-light sources are
very inefficient since most of the light is not used. The LEDs are much more efficient
since all the light they produce can be used for excitation of the dye. Moreover, the
process by which the light is produced is much more efficient in the LED. Most of the
power put into the white-light source is lost to heat. LEDs produce much less heat
for a given light intensity. White-light sources also tend to produce a time-varying
spatial pattern, which reduces the quality of the voltage signal. Although LEDs show
some spatial pattern close to the source, this pattern may be sufficiently attenuated
at normal working distances to not pose a problem.
Lasers provide the same advantages over white-light sources as the LEDs. They
are also energy efficient and provide spatially uniform intensity. However, they have
drawbacks of their own. The light output of many lasers fluctuates over time. We
have shown here that the light output of the LED is stable after an initial transient.
Furthermore, we have shown here that the LED operates closer to quantum noise
limits than the laser. In fact, the laser has the added disadvantage of being sensitive
to motion due to laser speckle [111]. Although, there are techniques for minimizing
this effect, they only add to the cost of imaging with a laser. Finally, lasers that
are sufficiently powerful and stable to satisfy experimental requirements are very
expensive. The same power and stability can be achieved with the LED for a fraction
168
of the cost.
This study has limitations. There was no direct comparison between a whitelight source and the LEDs. However, since the LEDs operate at the shot-noise limit
(a fundamental noise limit), we expect the LEDs to be no worse than the whitelight source. Also, in vitro experiments were performed in only two types of animal
tissue, using the same dye. Although the range of available LED spectra permits
experiments in other tissue using dyes with other emission and absorption spectra,
specific results, such as SNR, will differ.
Given the short timescale for new generations of LEDs, we expect that the LEDs
will only become an even more attractive option in the future as more powerful
LEDs become available. There will likely also be additional colors, producing better
matches for a larger variety of dyes.
169
Appendix B
Determination of Action Potential
Duration
B.1
Introduction
Electrical signals recorded from cardiac tissue are continuous. We analyze these
signals by defining a discrete object, the action potential, for which we must define
start and end times. Several different methods are used to define the start and
end of the action potential: threshold [182–185], maximum slope [186], and phase
[24, 187]. In low-noise electrophysiological signals, such as those obtained from glass
microelectrodes, all three can determine consistent APD values, so the choice of a
particular method for data analysis is mostly a matter of preference. When signals
are noisy, such as optical signals, these methods may not produce consistent results,
if they work at all.
In this section, I describe the effect of noise on the APD measured by each of
these three methods and I determine the best method for calculating APD of an
optical signal. There are three criteria that are used to determine the best method
for calculating APD.
The first is whether the method is accurate. That is, if we use this method on a
signal without noise and on the same signal with noise added, do we, within error,
get the same APD? With real optical signals, we don’t know what the clean signal
looks like, so we must be assured that the method we use to calculate APD returns
170
the correct value.
The second criterion is the precision or the amount of error in the measurement.
A method may accurately find APD, but, if the error is large, we cannot be confident
of the measurement. We also would like to be able to detect beat-to-beat differences
in APD to study transient behavior and complex rhythms like alternans. Beat-tobeat differences during transients can be as small as 2 ms to 4 ms and alternans
of 4 ms can be detected with microelectrodes. We are unlikely to achieve the same
resolution with optical signals, but we would like to get as close as possible.
Finally, we need a method that does not require much computation time. Although the calculation of one APD does not take much time, a typical image file
will have ∼ 105 action potentials, so any extra computations will add a significant
amount of time to the calculation.
B.2
Techniques for Finding APD
We begin this section with outlines of the most commonly used methods for determining APD.
B.2.1
Threshold Method
The threshold method is the easiest and fastest of the three methods to implement. It
does not require calculation of a new quantity, reducing the computation time of the
method. The threshold method simply defines the start (or end) time of the action
potential as the time at which the voltage crosses a specified threshold value (See
Fig. B.1). The actual value of APD will vary depending on the chosen threshold,
171
Figure B.1: Threshold method for determining APD. The action potentials shown
here are bullfrog ventricular APs measured with a glass microelectrode. The threshold
method defines the start or end of an action potential as the time at which the voltage
crosses a specified threshold value. Shown in this figure are 90% and 70% threshold
crossings. The specific APD value will vary depending on the chosen threshold.
which may affect restitution relationships [188, 189].
Typical threshold values are between 50% and 90% [182, 184] of the action potential amplitude. A fixed voltage is not used as a threshold for experimental data
because the baseline voltage will change from one recording to the next [182,185,190].
In microelectrode recordings the baseline voltage is partially determined by the quality of the impalement and will change if the microelectrode pulls out or is moved
to another cell. In optical recordings, the baseline voltage differences are due to
differences in intensity from pixel to pixel. Thus, using a fixed voltage rather than
a percentage of the amplitude requires adjustment of the processing parameters for
every recording (or every pixel in an optical signal).
172
B.2.2
Slope Method
The slope method defines the start (or end) of the action potential as the time at
which there is a maximum (or minimum) in the time derivative of the voltage. Since
experimental data is discrete, the derivative is approximated by differences,
Vt+1 − Vt
dV
≈
,
dt
δt
(B.1)
where Vt+1 and Vt are successive transmembrane voltage measurements. Figure B.2
shows the derivative of transmembrane voltage of a microelectrode signal over the
course of an action potential. The slope has a large positive spike at the upstroke and
a smaller negative dip at the downstroke. The slope method is more commonly used
on neural action potentials [186], which have a much sharper downstroke. In the case
of cardiac action potentials, the slope method is sometimes used to find the start of
the action potential with the threshold method used for the downstroke [191].
B.2.3
Phase Method
The phase method is based on the observation that a cell returns to its original rest
state after an action potential. Thus a plot in phase space, that is, a plot of Vt versus
Vt+τ , where τ is a constant, should be a closed loop (See Fig. B.3). The loop is
D-shaped with clusters of points at both ends of the D. These clusters correspond to
the rest and plateau phases of the action potential. The depolarization (upstroke)
and repolarization (downstroke) are the curves connecting the two clusters of points.
We can define a new variable, the phase θ calculated from the voltage time series by
173
Figure B.2: Slope method for determining APD. The action potentials shown here
are bullfrog ventricular APs measured with a glass microelectrode. (A) The slope
as approximated by Eq. B.1. (B) The slope method defines the start (or end) of
the action potential as the time at which there is a maximum (or minimum) in the
temporal derivative of the voltage.
the equation [187]
!
Vt − c
,
θ(t) = arctan
Vt+τ − c
(B.2)
where c is a constant that defines the center about which we measure the angle θ. The
phase is the angle that the current position in phase space makes with the horizontal
that emanates from the center c. In practice, both τ and c are chosen to produce the
best results, as described below.
The time course of θ during an action potential is shown in Fig. B.4. The phase
increases sharply during the upstroke, remains constant during the plateau and then
drops sharply again during the downstroke. We define the start (or end) of the action
potential as the time at which the phase crosses a threshold value.
The effect of different choices of c and τ are shown in Fig. B.5. Changes in c
174
Figure B.3: Phase space trajectory of an action potential. An action potential forms
a closed loop in phase space since the voltage returns to initial rest state after the
action potential. The clusters at the ends are the rest state and the plateau which
are joined by the upstroke (upper curve) and downstroke (lower curve).
Figure B.4: Phase during an action potential. The action potentials shown here are
bullfrog ventricular APs measured with a glass microelectrode. The phase increases
sharply during the upstroke, remains constant during the plateau, falls sharply during
the downstroke and remains constant during the rest state. For (A) τ = 5 ms and c
is the mean of the time series.
175
Figure B.5: Effects of parameter changes on phase. (A) Changes in c move the
phase representation of the downstroke closer or further from the upstroke, thereby
changing the measured APD. (B) Increases in τ cause the upstroke and downstroke
in the phase representation to be less sharp.
change the value of APD while changes in τ make the upstroke and downstroke less
sharp. For this reason, τ should remain small. Unfortunately, τ also determines the
width of the closed loop (See Fig. B.6), so A value of τ that is too small will not
separate the upstroke and downstroke enough to use this method.
Although this method is very similar to the threshold method, using the phase
instead of the original time signal has several advantages. The upstroke and particularly the downstroke are much sharper and therefore much more clearly defined
than in the original time signal. Also, the phase will always remain between −π and
π, so a fixed threshold point can be defined instead of a percentage as is used in
the threshold method. The phase method, however, requires computation of a new
variable before computing thresholds and so it requires more computation time than
the threshold method.
176
Figure B.6: Effect of time delay on phase space trajectory. The value of τ controls
the width of the loop in phase space.
B.3
Effect of Noise
In this section, we determine the effect of noise on the measured value of APD using
the three methods described above. We use a sample microelectrode recording taken
from bullfrog ventricular myocardium (See Fig. B.7) that contains five steady-state
action potentials. Increasing amounts of Gaussian noise are added to the signal and
APD for each action potential is determined using the three methods described in
the previous section. We plot the mean and standard deviation of the five APDs as
a function of the signal-to-noise ratio (SNR), a measure of the amount of noise in a
signal. The SNR is calculated by dividing the action potential amplitude by the noise
of the signal [192]. The noise is determined by calculating the standard deviation
of the signal during a time period when there is no action potential. The plots of
APD versus SNR will help determine the effect of noise on the measured APD as
calculated by each of the methods discussed above.
177
Figure B.7: Microelectrode recording. A sample microelectrode recording that
shows five steady-state action potentials and has an SNR of 310.
The goal of this study is to determine which method of calculating APD will give
the most accurate results when used on optical signals, which typically have an SNR
of 20-30.
B.3.1
Threshold Method
Although the threshold method is fast and easy to implement, it is extremely sensitive
to noise. A noisy signal may have multiple threshold crossings at both the start and
the end of the action potential (See Fig. B.8). If this is the case, do we define the start
(or end) time as the first crossing, the last crossing or something in the middle? The
processing code also needs to be modified to be able to determine whether a particular
downward (upward) threshold crossing point is part of the upstroke (downstroke) in
which case it should be ignored or whether it is a true downward (upward) crossing.
This adds to the computational effort and time required to implement the method.
Noise adds a further complication when used with a percentage threshold. The
178
Figure B.8: Multiple threshold crossings of noisy electrophysiological data. Noisy
electrophysiological signals will cross a threshold multiple times. Within the box on
the first downstroke, there are 18 downward crossings and 17 upward crossings.
amplitude of the action potential (APA) is typically determined by subtracting the
minimum voltage from the maximum voltage. Noise will add error to the measurement of these two values and thus to the measurement of the amplitude. Figure B.9
shows the mean and standard deviation of the APA measured from the test signal
with varying amounts of noise. As the noise increases, the measured APA also increases, effectively changing the threshold voltage. Different thresholds will lead to
different APD values.
When implementing the method described above to determine the effect of noise
on APD calculated using the threshold method, I looked at three possible variations:
using the first crossing time, last crossing time and mean crossing time (taking the
mean time of all crossings). In all cases I used a threshold of 70%. The mean and
standard deviation of the APDs are presented in Fig. B.10.
At signal-to-noise ratios (SNRs) above 100, there are no multiple crossings, so the
179
Figure B.9: Effect of noise on calculation of action potential amplitude. The measured APA increases as the noise in the signal increases.
Figure B.10: Effect of noise on APD found by the threshold method. As the
SNR decreases (increasing noise), the error in the measured APD becomes larger.
The correct value of APD is indicated by the dashed line. Using the first threshold
crossing to find APD produces the most accurate APD measurements in noisy signals.
180
first, last and mean crossing are all the same. Below an SNR of 100, the three options
begin to give different results: using the first crossing gives the shortest APD, the
last crossing gives the longest APD, and the mean crossing gives an APD between
the two extremes. Below an SNR of about 40, there is a large divergence in the three
options with the first crossing remaining the most accurate method. The error in
APD, as measured by the standard deviation, is ∼2 ms at SNRs above 50. Below an
SNR of 50, the error in APD ranges from 3-5%.
If we are to use the threshold method with optical signals, using the first crossing
point will give the most accurate result, although the error is too large to differentiate
small beat-to-beat variations in APD.
B.3.2
Slope Method
The derivative of the voltage signal has a sharp, distinct maximum at the upstroke
that is not affected much by noise, but the minimum of the derivative is fairly small
and is easily washed out by noise. Figure B.11 shows the derivative of a sample signal
with increasing amounts of noise added to it. The maximum shows little change as
noise is added, but the minimum, barely visible at an SNR of 310, cannot be seen at
an SNR of 170.
The disappearance of the minimum makes calculation of APD using this method
difficult at low SNRs. Figure B.12 shows that the slope method is only accurate for
SNRs of ∼300 or higher. This SNR is difficult to achieve with microelectrodes and
nearly impossible with optical techniques. Even at these high SNRs, the error in the
measurement is about ±10 ms, much higher than the threshold method.
181
Figure B.11: Derivatives of noisy electrophysiological signals. Noise washes out the
minimum of the derivative, which corresponds to the downstroke, but the maximum
remains unchanged.
Figure B.12: Effect of noise on the measurement of APD using the slope method.
The correct value of APD is indicated by the dashed line. The slope method is not
accurate or precise below SNRs of 300.
182
Figure B.13: Phase of a noisy electrophysiological signal. Noise in an electrophysiological signal causes multiple crossings in the phase representation of the downstroke.
B.3.3
Phase Method
The phase method runs into some of the same problems as the threshold method
when used on a noisy signal. In particular, there may be multiple crossing points
(See Fig. B.13), primarily in the downstroke. Thus, in studying the effect of noise
on the phase method, I will again use three cases: first crossing point, mean crossing
point, and last crossing point.
The plot of APD calculated using the phase method versus SNR is shown in Fig.
B.14. The phase begins to exhibit multiple crossings at and SNR of just over 100,
roughly the same SNR at which there are multiple crossings of voltage threshold.
Below an SNR of 100, using the first crossing point leads to shorter APDs while
using the last crossing point leads to longer APDs. Using the mean crossing time
leads to the most accurate APDs with the least amount of error in the measurement
(∼10 ms at low SNRs).
183
Figure B.14: Effect of noise on the measurement of APD using the phase method.
The ‘correct’ value of APD is indicated by the dashed line. The phase method using
the mean crossing time returns the correct APD even at low SNRs.
B.3.4
Discussion
Typical optical signals have an SNR of 20-30. At this low SNR, the slope method
cannot be used since the minimum in the derivative is completely hidden by noise.
The threshold method using the first crossing point and the phase method using the
mean crossing point provide comparable results at low SNRs.
The error in APD measurement is fairly large for both of these methods at low
SNRs. In order to achieve smaller errors, the signals can be filtered to reduce the
amount of noise before we attempt to determine APD. The following section discusses
some common filtering techniques and their effect on calculation of APD using the
phase method and the threshold method.
184
B.4
Filters
In the previous section, we found two methods for finding APD in noisy signals that
produce accurate measurements of APD. For signals with SNRs of 20-30, the error in
the measured APD is ±10 ms, too large to discern small beat-to-beat changes. From
Figs. B.14 and B.10, we see that even a modest improvement in the SNR, to ∼50, will
greatly reduce the error. The SNR of a signal can be improved with the use of filtering
techniques that can remove some of the noise. Unfortunately, filtering techniques will
also distort the signal, so we need to determine which filtering techniques will remove
the maximum amount of noise with the minimum amount of distortion.
This section describes several temporal filtering techniques: mean filter, median
filter and frequency filter. Other filters, such as the wavelet filter [23], are also
sometimes used, but will not be discussed here as they are more computationally
intensive. We test each filter on a sample signal (See Fig. B.15), which is just the
microelectrode signal of Fig. B.7 (which we will refer to as the original signal) with
Gaussian noise of standard deviation 0.05 added to it (the noisy signal). This signal
has an SNR of 22.4, in the range of a typical optical signal. The Gaussian noise is
a good model of the noise in an optical signal since most of the noise in the optical
signal is shot noise (See Appendix A). The noisy signal is filtered (the filtered signal)
using one of the three filters. We test the effectiveness of the filter by measuring the
SNR. We also determine if the filter is distorting the original signal by determining
the mean square error (MSE) [193], defined as
M SE =
N
1 X
(y(t) − x(t))2 ,
N t=1
185
(B.3)
Figure B.15: A noisy electrophysiological signal. This signal was created by adding
Gaussian noise to the microelectrode signal of Fig. B.7. It has an SNR of 22.4.
where x(t) is the original signal, y(t) is the filtered signal, and N is the length of
the signal. Finally, the APD of the filtered signal is calculated using either the
threshold method or the phase method. The calculated APD is compared to the
APD calculated from the original signal.
A Note on Spatial Filters
This section discusses temporal filters, that is the application of the filter to a time
series. However, the optical data collected in our experiments is spatially extended,
so we could alternatively apply a spatial filter. The temporal filter treats each pixel
individually by using information from the temporal evolution of the intensity at that
pixel to modify the signal. The spatial filter does not use any information from the
temporal evolution of the pixel, but instead assumes pixels that are spatially close
will have similar intensities. For this reason, information from neighboring pixels can
be used to modify the value of the pixel of interest. The mean and median filters
186
discussed in the next section are quite often extended to two dimensions and applied
spatially [173]. Many groups use both spatial and temporal filters when processing
optical signals from cardiac tissue [111, 194].
We will not discuss spatial filters in any great detail since they were not used to
process our data. Spatial filters are more likely to degrade the temporal signal and
it is the temporal signal that we use to determine the start and end of the action
potential. The degradation of the temporal signal by spatial filtering is because we
are combining information from signals that are slightly shifted in time. For example,
if we have an electrical wave moving from pixel A to pixel B, the cells in pixel A will
depolarize before the cells in pixel B. If we take a picture at this moment and apply a
spatial filter, the depolarizing signal in pixel A will be modified by the signal in pixel
B where the cells are still at rest. A filter that corrects for this time shift has been
developed by Sung et al. [195], but it is computationally intensive. For this reason
spatial filters will only be used if a suitable temporal filter is not found.
B.4.1
Mean Filter
The mean filter uses the mean of neighboring points to adjust the value of the data
at time t [196]. Specifically, we define a new data set y(t) as
y(t) =
t+k
X
1
x(i),
2k + 1 i=t−k
(B.4)
where k is the size of the filter [197]. The mean filter assumes noise in neighboring
points is random and so should have a mean of zero. For the mean filter to work
correctly, the mean of neighboring points must equal the value of the data at the
187
center. This is not always the case if data varies rapidly. For this reason, the mean
filter has been extended by use of a weighting function that allows closer points to
have more influence than points further away. In this case, the new series is given by
y(t) =
t+k
X
wi x(i).
(B.5)
i=t−k
where wi is the weight function and we require that
PN
i=1
wi = 1. These extensions
of the mean filter will not be considered here because they are more computationally
intensive and typically they are not as effective in removing noise. They are usually
used with rapidly varying signals because they do a better job of preserving the
original signal.
The mean filter moves outliers closer to their neighbors, creating a smoother curve
(See Fig. B.16A). As the size of the filter increases, more of the noise is removed.
This can be seen by improvement in the SNR, as shown in Fig. B.16B. Even a 2-point
mean filter greatly improves the SNR.
Unfortunately, a mean filter can alter the original curve. Figure B.17A shows that
the MSE initially drops, as is expected because the signal is now cleaner. Beyond
a filter size of ∼10, the MSE begins to increase, indicating that the filtered curve
deviates from the original curve. Figure B.17B shows that the mean filter has two
effects on action potentials: the upstroke is slower, and the amplitude is slightly
lower. Both of these effects could alter the APD calculated by the threshold method.
The filtered signal crosses the threshold at a different time than the original signal.
Because of the change in amplitude, the threshold itself will also be at a different
voltage in the filtered signal than in the original signal. The phase method will only
188
A
B
Figure B.16: Effect of the mean filter on a sample time series. (A) The mean filter
creates a smoother curve by averaging nearby points. A larger filter size produces a
smoother curve. (B) The SNR increases as the filter size increases.
be affected by the washout of the upstroke. The phase representation of the upstroke
will also be slower in the filtered signal than in the original signal and so will cross the
threshold in phase space at a different time. The effect of the washout is more severe
at lower BCLs where the DI can be as low as 50 ms. As the upstroke is distorted, it
can run into the downstroke of the previous action potential, completely eliminating
the diastolic interval and making it difficult to distinguish where one action potential
ends and the next one begins.
The effect of different size filters on the measurement of APD is summarized in
Fig. B.18. In Fig. B.18A the APD was found using the threshold method, while in
Fig. B.18B the APD was found using the phase method. In both figures, the bold
line indicates the APD of the original microelectrode signal. The threshold method
returns APD values larger than the original APD when the signal is filtered, while
the phase method returns APD values lower than the original APD for filters of 3
189
A
B
Figure B.17: Signal changes due to mean filtering. (A) The MSE shows an initial
drop due to the removal of noise from the signal, but then increases as the signal
becomes distorted by the filter. (B) The dashed line is the original microelectrode
signal while the solid line is the same signal after Gaussian noise was added and then
removed with a 40-point mean filter. The filtered signal has a slower upstroke and a
lower amplitude than the original signal.
points or larger. The error in the APD measurement by threshold method falls to
∼3 ms using a mean filter larger than 4 points. The error in APD measurement by
phase method falls to ∼2 ms using mean filters between 10 and 25 points in size;
filters larger than 25 points have errors of ∼3 ms.
B.4.2
Median Filter
The median filter is similar to the mean filter in that it replaces a point at time t
with some function of its neighbors. In this case, instead of using the mean, the new
series is found by using the median of neighboring points:
y(t) = median{x(i)|t − k ≤ i ≤ t + k},
190
(B.6)
A
B
Figure B.18: Effect of the mean filter on calculation of APD. (A) APD calculated
using the threshold method on a signal filtered with a mean filter of various sizes.
(B) APD calculated using the phase method on a signal filtered with a mean filter of
various sizes. In both figures, the solid line indicates the APD of the original signal.
where k is the size of the filter [197]. Again, generalizations of the median filter
have been proposed [198], but we will not consider them here because they are more
computationally intensive and do not provide much benefit over the basic median
filter.
The median filter, like the mean filter, moves outliers closer to their neighbors,
creating a smoother curve (See Fig. B.19A). Again, the SNR ratio improves (See
Fig. B.19B) with increasing filter size. Unlike the mean filter, the median filter does
not have a rapid increase in SNR at small filter sizes. Larger median filters have a
greater SNR than mean filters of the same size.
The median filter also preserves rapid changes much better than the mean filter.
Figure B.20B shows the original signal and the noisy signal after it was filtered with
a 40-point median filter. The upstroke of the filtered signal aligns well with that of
the original signal. In fact, it has been shown that signals that are locally monotone
191
A
B
Figure B.19: Effect of the median filter on a sample time series. (A) The median filter creates a smoother curve by determining the median of nearby points. A
larger filter size produces a smoother curve. (B) The SNR increases as the filter size
increases.
will pass through a median filter unchanged [199, 200], so it’s not surprising that
these features are preserved. Anywhere there is a change of direction, at the peak,
at the start of the upstroke, and at the end of the downstroke, we see a deviation of
the filtered signal from the original signal. Overall though, the signal filtered with a
median filter follows the original signal more closely than the signal filtered with a
mean filter. This is confirmed by the decrease in MSE with increasing filter size (See
Fig. B.20A). Small median filters lead to a dramatic decrease in the MSE and not
much is gained by using larger filters.
The effect of different size filters on the measurement of APD is summarized in
Fig. B.21. In Fig. B.21A the APD was found using the threshold method, while in
Fig. B.21B the APD was found using the phase method. In both figures, the bold
line indicates the APD of the original microelectrode signal. The threshold method
returns APD values slightly larger than the original APD when the signal is filtered,
192
A
B
Figure B.20: Signal changes due to median filtering. (A) The MSE decreases with
increasing filter size. (B) The dashed line is the original signal while the solid line is
the same signal after Gaussian noise was added and then removed with a 40-point
median filter. The two signals show slight deviations at the start of the upstroke, at
the end of the downstroke, and at the peak of the action potential.
although they agree within error. The phase method returns APD values lower than
the original APD for filters of 3 points or larger. The error in the APD measurement
by threshold method falls to ∼3 ms using a median filter larger than 6 points, falling
to ∼2 ms with filters greater than 12 points. The error in APD measurement by
phase method falls slightly with increasing filter size.
B.4.3
Frequency Filter
The frequency filter is based on the idea that periodic signals have a finite number of
frequency components. We find the frequencies of the signal, the frequency spectrum,
by taking the Fourier transform of the data. For discrete data, the Fourier transform
is [197]
X(k) =
−1
k
1 NX
x(j)e−2πi N j ,
2N j=0
193
(B.7)
A
B
Figure B.21: Effect of the median filter on calculation of APD. (A) APD calculated
using the threshold method on a signal filtered with a median filter of various sizes.
(B) APD calculated using the phase method on a signal filtered with a median filter
of various sizes. In both figures, the solid line indicates the APD of the original
signal.
where N is the number of points in the data set. The original data can be recovered
by taking the inverse transform,
x(j) =
−1
k
1 NX
X(k)e−2πi N j .
2N k=0
(B.8)
Since a series of action potentials is periodic, it should have a distinct frequency
spectrum. Figure B.22A shows the frequency spectrum of the original microelectrode
signal. The spectrum has large peaks at both ends, but is essentially zero in the
middle. Contrast this to the frequency spectrum of the noisy signal (See Fig. B.22B),
where the central region is not zero. The conclusion is that this non-zero center is
the frequency representation of the noise. If we remove the noise from the frequency
representation and take the inverse transform (Eq. B.8), we should have a cleaner
signal.
194
A
B
Figure B.22: Frequency spectrum of action potentials. (A) The frequency spectrum
of the microelectrode signal of Fig. B.7. (B) The frequency spectrum of the same
signal with Gaussian noise.
We implement this filter by setting all points in the frequency representation
between two points yet to be chosen to zero. The size of the filter is the number of
points that are set to zero. We would like to keep the symmetry of the spectrum,
so setting the size of the filter will determine the endpoints. For example a 3000
point filter will set all points between 500 and 3500 to zero. One drawback to the
frequency filter is that the frequency spectrum differs for signals at shorter BCLs. At
faster BCLs, there is a smaller central region were the spectrum is zero. Thus in the
practical implementation of this filter, we should ideally use a different filter size for
each BCL.
Examples of filtered signals are shown in Fig. B.23A. Even large frequency filters
do not remove all the noise from the signal. The noise is primarily in the plateau and
the rest stages of the action potential. The upstroke and the downstroke, which for
our purposes are the most important parts of the action potential, are fairly clean.
195
A
B
Figure B.23: Effect of the frequency filter on a sample time series. (A) The frequency filter creates a smoother curve by removing noise in the frequency spectrum.
(B) The SNR increases as the filter size increases.
The frequency filter does not improve the SNR as much as either the mean or median
filters (See Fig. B.23B), but because it cleans the upstroke and downstroke, it may
do a better job of reducing the error in APD measurement. The sudden drop in
SNR after a filter size of 3800 is because those filters are removing parts of the action
potential and not just the noise.
Even though the frequency filter is not the best filter for removing noise, it preserves the shape of the action potential better than either the median or mean filter.
Figure B.24A shows that the MSE decreases with increasing filter size until a filter
size of 3800 points. The large increase in MSE beyond this is because the filter is
removing parts of the action potential. Except for these large filters, the MSE for a
frequency filter is lower than that of a mean or median filter even though the SNR
is larger. Figure B.24B shows the original signal and the noisy signal after it was
filtered with a 3800-point frequency filter. There is very little difference between the
196
A
B
Figure B.24: Signal changes due to frequency filtering. (A) The MSE decreases
until a filter size of 3800 where it rises sharply because the frequency filter removes
parts of the action potential. (B) The dashed line is the original microelectrode signal
of Figure B.7. The solid line is the same signal after Gaussian noise was added and
then removed with a 3800-point frequency filter. The filtered signal still has some
noise, but the overall shape of the action potential remains unchanged.
two curves. In particular, the upstroke and the downstroke line up exactly.
The effect of different size filters on the measurement of APD is summarized in
Fig. B.25. In Fig. B.25A the APD was found using the threshold method, while in
Fig. B.25B the APD was found using the phase method. In both figures, the bold
line indicates the APD of the original microelectrode signal. The threshold method
returns APD values slightly larger than the original APD when the signal is filtered.
This difference is because there is a slight difference in the amplitudes of the original
signal and the filtered signal (See. Fig. B.24). The threshold for the two signals
is thus at a slightly different voltage, causing the different APD measurement. The
phase method returns APD values that agree with the original APD for all filter
sizes. The error in the APD measurement by threshold method is not much better
than the noisy signal for many of the filter sizes, typically between 6 ms and 9 ms.
197
A
B
Figure B.25: Effect of the frequency filter on calculation of APD. (A) APD calculated using the threshold method on a signal filtered with a frequency filter of
various sizes. (B) APD calculated using the phase method on a signal filtered with a
frequency filter of various sizes. In both figures, the solid line indicates the APD of
the original signal.
The error in APD measurement by phase method remains between 2 ms and 4 ms.
B.5
Discussion
Although the frequency filter preserves the shape of the action potential better than
the median or mean filters and returns APD measurements more consistent with the
APD of the original signal, it is computationally expensive and does not reduce the
error in the APD measurement any better than the mean or median filters. Since
the cost in computation time does not outweigh the benefit, the frequency filter will
not be used for cleaning noisy signals.
The mean filter reduces the SNR well, but distorts the signal. The result of this
is that, when the filter is large enough to reduce the error in the APD measurement,
neither the phase nor threshold methods finds the true APD.
198
The median filter effectively reduces the error in APD measurement while preserving the shape of the signal. Computationally, the median filter takes longer to
compute than the mean filter, particularly for larger filters. Even a small filter will
reduce the error in the measurement to a tolerable level. Of the two methods used to
calculate APD, the threshold method is more accurate than the phase method when
used on a signal filtered with a median filter. The threshold method also requires less
computational effort than the phase method, so we will use the threshold method in
conjunction with a median filter to calculate APD of the optical signals. The median
filter will be a 3-point filter, since this size reduces the error in the measurement to
below 4 ms and does not take too long to compute.
B.5.1
Application to an Optical Signal
As a check that this method of calculating APD will work as expected in a real optical
signal, we apply the method to the two signals shown in Fig. B.26A. Both signals
were collected by a CCD camera from bullfrog ventricular myocardium stained with
di-4-ANEPPs and illuminated with a cyan LED. The signals were collected after 2
minutes of pacing at the same BCL and are taken from the same pixel in the center
of the tissue. Note that fluorescent intensity decreases when the transmembrane
voltage increases, so the action potentials are upside down. The upper signal is at
a BCL of 1000 ms and has a SNR of 22.6. This signal is similar to the test signal
used in the previous sections. The lower signal is at a BCL of 300 ms and has
a SNR of 15.2. Optical signals at shorter BCLs typically have a lower SNR than
signals at longer BCLs. As the pacing rate increases, the action potential amplitude
decreases [201], resulting in a lower SNR. Note that this signal also shows an irregular
199
A
B
Figure B.26: Finding APD in an optical signal. (A) The original optical APs from
bullfrog ventricular myocardium collected with a CCD camera. The upper trace is at
a BCL of 1000 ms while the lower trace is at a BCL of 300 ms. (B) The same signals
after they have been filtered with a 3-point median filter. The squares indicate the
start of the action potential and the circles indicate the end of the action potential
both found using the threshold method. The dashed line indicates the threshold.
response; sometimes activating for every stimulus and sometimes skipping every other
stimulus. This signal is a test of the robustness of the chosen method.
Figure B.26B shows the signals after they have been filtered with a 3-point median
filter. After the signals are filtered, the threshold method is applied to determine the
APD. The 70% threshold is indicated by the dashed line. The start time of the
action potential is indicated by the squares while the end time is indicated by the
circles. The method correctly identified every action potential and did not mislabel
multiple threshold crossings as separate action potentials. Both signals are steadystate responses, but only the signal at a BCL of 1000 ms consists of identical action
potentials. For this signal, we can average the measured APDs to find a single steadystate APD with the standard deviation of the measured APDs giving an estimate of
the error in the measurement. The steady-state APD at a BCL of 1000 ms is 428±4
200
ms. We see that the error is similar to the error found in the previous section.
B.6
Conclusion
The threshold method for finding APD works well in determining APD in optical
signals filtered with a 3-point median filter. The method determines APD with an
error of ±4 ms and can handle irregular cardiac responses.
201
Appendix C
Considerations in Tissue Preparation
The purpose of the experiments is to determine whether cardiac tissue exhibits spatial variation in APD and other restitution properties and whether spatial variation
in restitution properties can be correlated to the onset of alternans. The ideal experimental tissue preparation for these experiments is a one-dimensional homogeneous
piece of cardiac tissue. The tissue should be homogeneous, except for boundaries,
since I wish to eliminate any underlying tissue heterogeneity which could create spatial patterns in restitution properties. The ideal preparation is one-dimensional for
ease of comparison to the analysis and simulations of Cain and Schaeffer [63]. Unfortunately, both of these ideals are rather difficult to achieve with real cardiac tissue, so
compromises were made when designing the experiments. This appendix addresses
some of the considerations that went into the experimental design as well as the
advantages and limitations of the tissue preparation used in my experiments.
C.1
Experimental Limitations
Before deciding on the method for tissue preparation, several factors were considered.
C.1.1
Tissue Viability
Once the heart is excised from the body, it will begin to die due to lack of nutrients.
The slow death of cells in the tissue changes the restitution properties and thus
202
Figure C.1: Signal degradation due to cell death. (A) The optical signal from a
piece of bullfrog ventricular myocardium paced at BCL=1000 ms at the beginning of
an experiment. (B) The optical signal from the same location about two and a half
hours later. The recording taken at the beginning of the experiment has an SNR of
22 and the recording taken at the end of the experiment has an SNR of 4.
the dynamics of the tissue. Additionally, dead cells introduce heterogeneity into the
tissue, potentially affecting the observed spatial patterns. Finally, dead cells decrease
the SNR of optically recorded signals since dead cells contribute to the baseline
fluorescence but do not produce action potentials which decreases the amplitude of
the recorded optical action potentials. Figure C.1 shows the degradation of an optical
signal due to cell death over the course of an experiment. At the beginning of the
experiment the signal has an SNR of 22 (Fig. C.1A), but by the end of the experiment
the SNR is only 4 (Fig. C.1B).
Tissue death can be slowed by providing the tissue with the appropriate nutrients
through perfusion or superfusion of Ringer’s solution [117]. Perfusion of Ringer’s
solution involves delivery of the solution into the whole heart through a cannula and
allowing the natural pumping action of the heart to deliver the nutrients to the tissue.
203
Superfusion can be done using either the whole heart or pieces of cardiac tissue.
The tissue is placed in a circulating bath of Ringer’s solution and the nutrients are
delivered to the tissue primarily through diffusion. Perfusion generally does a better
job of delivering nutrients to all the tissue, but as noted above, this method can only
be used if the whole heart is used in the experiment.
There are additional complications that add to cell death in optical experiments.
Optical experiments require the addition of a voltage-sensitive dye. The most commonly used dyes are phototoxic [202], accelerating cell death when the stained tissue
is exposed to light. Additionally, to properly record electrical activity with a CCD
camera, the contractile motion of the tissue must be stopped. If the tissue is contracting, cells will move from one pixel to a neighboring pixel during an action potential,
causing inaccurate measurements of electrical activity. Contraction is commonly
stopped through the addition of an electro-mechanical decoupler. Unfortunately, the
decoupler is also known to alter restitution properties of cardiac tissue [203,204]. An
alternate method is to stabilize the imaging surface by pressing it against a piece of
glass. This method is difficult to use when superfusing the tissue since the imaging
surface will not be exposed to the Ringer’s solution and will thus experience more
rapid cell death.
Each of the three pacing protocols presented in this thesis take approximately
20-40 minutes to complete, so I would like to keep changes due to cell death to a
minimum over this time frame.
204
Figure C.2: Spatial variation of APD of a 2-variable cardiac model in a paced cable.
(A) When the cable is short (1 cm) there is no region of constant APD in the center
of the cable. (B) In a longer cable it is clear that APD variation is a boundary effect.
C.1.2
Tissue Size
The observed spatial variation of APD in computer simulations is known to be a
boundary effect. To determine whether experimentally observed spatial variation is
also a boundary effect, the tissue needs to be large enough to be sure that there is
little spatial away from the boundaries. Fig. C.2 shows the results of a simulation
in a 1-dimensional cable. In Fig. C.2A, the cable is 1 cm and the APD appears to
decrease linearly along the cable. However, Fig. C.2B shows that variation in APD
is actually a boundary effect since the 5 cm cable is larger than the length constant
of the variation.
In computer simulations, the APD varies over a length of ∼2λ. The passive length
constant for bullfrog ventricular tissue is 0.3 mm [52], so I estimate that APD will
vary over a length of 0.6 mm, if a similar boundary effect occurs in real cardiac tissue.
Thus I require the tissue preparation to be longer than 4 mm (∼3λ at each end) in
205
at least one direction.
C.1.3
Repeatability
Since cardiac tissue is a biological system, there will be natural animal-to-animal
variation in cardiac tissue properties. Although this natural variation cannot be
eliminated, other factors that can cause variation in experimental results should be
minimized. In particular, the following design issues may affect repeatability of the
experiments:
• the size and shape of the tissue sample.
• the placement of electrodes, both stimulus electrodes and recording electrodes
in the case of the microelectrode experiments.
• placement of the pins that hold the tissue in place.
• unequal application of any chemicals such as voltage-sensitive dyes or electromechanical decouplers. Due to variation in cardiac tissue properties, some
samples require more decoupler or dye to achieve the same desired effect.
C.1.4
Tissue Damage
Any cuts in the tissue cause cells to die along the cut boundary [116]. Although this
is an advantage since the cut boundary is an insulated boundary of the type assumed
in the analysis of Cain and Schaeffer [63], it can also cause problems if there are too
many cuts too close to each other. When the cuts are close together, boundaries of
dead cells will meet and there will not be enough viable tissue for proper propagation
of the electrical signal.
Additionally, tissue damage can occur during the tissue preparation process.
When the tissue is stained, a cannula is inserted from the auricle into the ventri206
cle which may cause damage to the inner walls of the ventricle. As the tissue is cut
and moved from one location to the next, the tweezers used to hold the tissue may
crush and damage some of the cells. Finally, pins used to hold the tissue in place during preparation and during the experiment can cause further damage to the tissue.
These localized regions of damaged tissue may alter the propagation and therefore
the observed spatial patterns of restitution properties. Aside from minimizing the
amount of handling during tissue preparation, the effect of localized tissue damage
can also be minimized by handling the tissue only along cut edges where there is
already a layer of dead cells.
C.2
Suggested Tissue Preparations
This section describes possible tissue preparations and the advantages and limitations
of each.
C.2.1
Whole Heart Preparation
The advantage of the whole heart preparation is that it minimizes cell death and
damage. The whole heart preparation can be perfused rather than superfused, resulting in better delivery of nutrients to the tissue and slower tissue degradation.
Additionally, cutting and handling of the tissue is kept to a minimum so there are
fewer regions of localized cell damage or death. The whole heart preparation also
allows for the largest possible surface area on which to observe spatial patterns.
Unfortunately, the whole heart preparation has several disadvantages. Since I
am interested in spatial patterns in homogeneous tissue, the auricles of the bullfrog
207
Figure C.3: Examples of whole heart tissue preparation. The two images show the
whole frog heart after application of the potentiometric dye. The lack of dye in the
auricles is intentional since they will not be used for recording because the tissue
there is not homogeneous. Note that in panel B, it is difficult to visually discern the
boundary between auricles and ventricle, making consistent placement of electrodes
difficult. Also, there is quite a bit of difference in the size and shape of the two
preparations, which may lead to differences in observed spatial patterns.
heart cannot be used as part of the imaging surface since they contain specialized
structures and different cell types. Even if recordings are constrained to the ventricular surface, the pacemaker cells that remain in the whole heart may initiate waves
that interact with the waves initiated by pacing electrodes. Additionally, there is no
control over the size and shape of the tissue preparation (Fig. C.3) which may lead
to differences in observed spatial patterns and also makes it difficult to consistently
place electrodes from one sample to the next. Finally, since there are no cut edges,
there are no insulated boundaries so spatial patterns near an insulated boundary
cannot be studied. However, spatial patterns induced near the stimulus electrode
can be determined and compared to patterns observed in computer models.
208
C.2.2
Whole Ventricle
Another possible tissue preparation is to remove the auricles and use the entire ventricle. As Fig. C.3 shows, it is sometimes difficult to visually determine the boundary
between the auricles and ventricle. To ensure that only ventricular tissue remains
after the dissection, the dissected tissue is observed for spontaneous contractions.
Spontaneous contraction is initiated by pacemaker cells in the auricles, so if the dissected tissue contracts it means that some tissue from the auricles remains. If this is
the case, more tissue is cut off until the spontaneous contraction stops, so that I am
sure that I am left with only ventricular tissue.
Removal of the auricles eliminates one of the limitations of the whole heart preparation since there are no longer pacemaker cells that can initiate unwanted electrical
activity. However, removal of the auricles also means that this preparation cannot be
perfused. Instead, the whole ventricle must be superfused and due to the thickness of
the tissue, cells on the inner surface of the ventricle may not receive enough nutrients.
The deterioration of these cells can alter the propagation of waves in the tissue.
The removal of the auricles also introduces a cut edge to the preparation. This
edge can be used to study spatial patterns near an insulated boundary and since there
is plenty of undamaged tissue remaining in the ventricle, this boundary should not
inhibit normal propagation through the remainder of the ventricle. Additionally, since
there is only one cut edge, this preparation offers a large surface area for observing
spatial patterns although there is still little consistency in the size and shape of the
preparation. Finally, the single cut edge leads to an asymmetry when pacing from
different edges. If stimulus electrodes are placed along the three edges of the tissue,
209
two electrodes will be initiating waves from undamaged edges of the tissue while the
third electrode will initiate waves near a boundary that contains dead cells.
C.2.3
Anterior Ventricular Surface
To remove the asymmetry introduced by a single cut edge, the anterior and posterior surfaces of the ventricle can be cut apart, thus introducing two more insulated
boundaries. The anterior surface of the ventricle is used for experiments since the
posterior surface contains a dead spot, a region of non-cardiac cells, where the pericardium attaches to the heart. In addition to providing three symmetrical edges, this
preparation improves nutrient delivery to the inner surface of the ventricle since the
inner surface is now directly exposed to the circulating Ringer’s solution. Finally, by
cutting all three edges, there is slightly more control over the size and shape of the
samples (Fig. C.4).
The biggest drawback of this preparation is that it decreases the undamaged
surface area. However, as long as care is taken not to remove more tissue than is
necessary, the samples will still be longer than 6 mm, the estimated length needed
to discern the boundary effect, in at least one direction.
A variation of this method of tissue preparation is to cut the anterior surface of
the ventricle into a shape (such as a square) that could be consistently reproduced
from one experiment to the next. Although, this would aid in the repeatability of
the experiments, it also makes the surface area smaller, potentially making the tissue
shorter than 6 mm in all directions. This would make it difficult to determine whether
spatial variation of restitution properties is a boundary effect.
210
Figure C.4: Examples of the anterior surface ventricular preparation. Although
there is still some variation in size and shape of the tissue samples, there is more
consistency than in the whole heart preparation (Fig. C.3)
211
C.2.4
Ventricular Strip
The tissue preparation that would be closest to the one-dimensional homogeneous
ideal is a ventricular strip produced by unfolding the ventricle and cutting a strip
from the center and placing electrodes at either end of the strip. The size of the strip
preparation can be controlled better than the ventricle preparation actually used in
the experiments since length and width could standardized. Electrode placement
would still vary somewhat but not to the extent that it does in the anterior surface
preparation since there is not as much surface area to potentially place the electrode
in the strip. However, when the strip was attempted in experiments, waves could
not be initiated in the preparation. This was probably due to cell death at the
boundaries. Since the sides of the strip are cut edges, there is a layer of dead cells
along each side leaving a narrow band or possibly no cells alive in the center.
C.3
Conclusion
Essentially, the experimental design comes down to trying to balance the need to
have consistent size, shape and electrode placement between experiments and the
need to retain as much undamaged surface area as possible in order to effectively
determine the spatial variation of restitution properties. I decided that using the
anterior surface of the ventricle was the best way to achieve this balance.
212
Appendix D
MatLab Codes
D.1
Simulation Code
This is the Matlab code used to run simulations of the 2-variable model in a twodimensional sheet. Results are presented in chapters 4 and 5. The variable mask is an
mxn matrix of ones and zeros where ones denote cells that are part of the tissue and
zeros denote empty space. This variable is used to define tissue of arbitrary shape
and size to match experimental tissue samples.
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
dynamic_2d.m
%
%
by Hana Dobrovolny
%
%
%
%
This program runs a dynamic pacing protocol.
%
%
subroutines:
%
%
schaeffer.m
%
%
%
%
Last modified 03/24/06
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%%%%%%%%%%%%%%%%%%%%%%%%% parameters %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
213
dt = .05;
dx = 2/64;
dy = 2/64;
xmax=size(mask,1)
ymax=size(mask,2)
stimx = 33;
stimy = 2;
bcl_max = 100;
bcl_min = 440;
bcl_step = 10;
paces = 10;
num_steps = bcl*paces/dt;
num_steps2 = bcl*10/dt;
%%%%%%%%%%%%%%%%%%%%%%%% initialize %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
for i=1:xmax
for j=1:ymax
V(i,j) = 0;
h(i,j) = 1;
end;
214
end;
count = 1;
%%%%%%%%%%%%%%%%%%%%%%% main loop %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
for i=bcl_max:-bcl_step:bcl_min
fname = strcat(’g:\sim\schaeffer_’,num2str(bcl),’_a.asc’)
fid=fopen(fname,’w’);
%%%%%%%%%%%%%%%%%%%%%%%%% Calculate time series %%%%%%%%%%%%%%%%%%
% Pre-pace to reach steady state
Iext=zeros(xmax,ymax);
Vout=V;
hout=h;
for j=1:num_steps
% Set up stimulus
if (rem(j,bcl/dt)<2/dt)
Iext(stimx,stimy) = 3;
else
Iext = zeros(xmax,ymax);
end;
215
% Calculate voltage
schaeffer
V = Vout;
h = hout;
end;
% Collect time series
Iext=zeros(xmax,ymax);
for j=1:num_steps2
% Set up stimulus
if (rem(j,bcl/dt)<2/dt)
Iext(stimx,stimy) = 3;
else
Iext = zeros(xmax,ymax);
end;
% Calculate voltage
schaeffer
if rem(j,10)==0;
fprintf(fid,’%7.3f’,Vout);
fprintf(fid,’\n’);
216
end;
V = Vout;
h = hout;
end;
fclose(fid)
end;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
schaeffer.m
%
%
by Hana Dobrovolny
%
%
%
%
This program does one time step of the 2-variable cardiac
%
%
model in a 2D sheet.
%
%
%
%
Last modified 03/26/06
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%%%%%%%%%%%%%%%%%%%%%%%%% parameters %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
D = 0.001;
Vc = 0.13*ones(size(V));
TauOut = 10*ones(size(V));
TauIn = 0.2*ones(size(V));
217
TauOpen = 130*ones(size(V));
TauClose = 150*ones(size(V));
Epsilon = 0.0001*ones(size(V));
%%%%%%%%%%%%%%%%%%%%%%%% main loop %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% Step h and V forward
for k=2:xmax-1
for m=2:ymax-1
if V(k,m)<=Vc
hout(k,m) = ((1-h(k,m))/TauOpen(k,m))*dt + h(k,m);
else
hout(k,m) = (h(k,m)/TauClose(k,m))*dt + h(k,m);
end;
Vout(k,m) = ((D/dx^2*(mask(k+1,m)*V(k+1,m)-2*V(k,m)+mask(k-1,m)*V(k-1,
end;
end;
D.2
Data Analysis Code
This is the Matlab code used to find start and end times of optical action potentials. It
is used to determine activation and deactivation times and APD for the experiments
presented in chapters 5 and 6.
218
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
apd_map.m
%
%
by Hana Dobrovolny
%
%
%
%
This program finds activation and deactivation times of
%
%
optical action potentials.
%
%
Subroutines:
%
%
find_mean_maxmin.m
%
%
make_mask.m
%
%
find_crossings.m
%
%
extra_cleaning.m
%
%
get_apds.m
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%%%%%%%%%%%%%%%%%%%%%%%%% parameters %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
exp_no=[1:10];
dates = [30306 31606 42605];
for n=1:length(dates)
for k=1:length(exp_no)
for bcl=1000:-50:200
clear m maximum minimum mask thresh starts ends times even odd apds
filename = strcat(’g:\’,dates,’\’,dates,’_exp’,num2str(exp_no(k)),’_’,
num_images = 2500;
219
thresh_coeff = .7;
window = 10;
apd_thresh = 150;
filter_size = 3;
cutoff=4000;
%%%%%%%%%%%%%%%%%%%%%%%%% main loop %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
fid=fopen(filename);
if fid~=-1
% Find mean intensity, maximum intensity and minimum intensity for each pixel
[m, maximum, minimum, flag] = find_mean_maxmin(fid,num_images,filt
if flag==0
% Decide which pixels will be processed -- must have an intensity over
% background noise (cutoff) and large enough APA (apd_thresh)
mask = make_mask(m,cutoff,maximum-minimum,apd_thresh);
% Define the threshold for activation and deactivation
thresh = (maximum-minimum)*thresh_coeff+minimum;
% Find activation times
starts = find_crossings(fid,’s’,thresh,num_images,mask,filter_
% Find deactivation times
ends = find_crossings(fid,’e’,thresh,num_images,mask,filter_si
% Remove multiple crossings
times = extra_cleaning(mask,starts,ends,window,bcl);
220
% Calculate APD
[even,odd,apds] = get_apds(times);
% Save all to file
fname = strcat(’c:\maps\’,dates,’\’,dates,’_’,num2str(bcl),’_e
save(fname,’starts’,’ends’,’times’,’even’,’odd’,’apds’)
end;
fclose(fid);
end;
end;
end;
end;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
find_mean_maxmin.m
%
%
by Hana Dobrovolny
%
%
%
%
This maximum, minimum and mean intensity of an optical signal. %
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
function [m, maximum, minimum, flag]=find_mean_maxmin(fid,num_images,filter_size)
%%%%%%%%%%%%%%%%%%%%%%%%% initialize %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
frewind(fid)
221
sum=zeros(128,128);
maximum=zeros(128,128);
minimum=ones(128,128)*2^14;
flag=0;
%%%%%%%%%%%%%%%%%%%%%%%%% main loop %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% Initialize a sliding window to apply the temporal filter
count = 1;
while count<=filter_size*2+1
a=(fscanf(fid,’%d’,[129,128]));
v=find(a(:,:)==0);
if isempty(v)~=0
if size(a)==[129,128]
A(:,:,count)=a(2:129,:);
sum=sum+a(2:129,:);
count=count+1;
else
flag=1;
end;
end;
end;
% Find maximum, minimum and mean intensity of temporally filtered signal
222
A1=median(A,3);
s=size(A,3);
for i=filter_size+1:num_images-filter_size-1
if flag==0
minimum=(A1<minimum).*A1+(A1>=minimum).*minimum;
maximum=(A1>maximum).*A1+(A1<=maximum).*maximum;
A=circshift(A,[0 0 -1]);
a=(fscanf(fid,’%d’,[129,128]));
if size(a)==[129,128]
sum=sum+a(2:129,:);
count=count+1;
A(:,:,s)=a(2:129,:);
A1=median(A,3);
else
if i<num_images-100
flag=1;
end;
end;
end;
end;
m=sum/count;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
223
%
make_mask.m
%
%
by Hana Dobrovolny
%
%
%
%
This program determines which pixels will undergo further
%
%
processing by creating a binary mask.
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
function y=make_mask(m,cutoff,amp,amp_thresh)
y=zeros(size(m,1),size(m,2));
for i=1:size(m,1)
for j=1:size(m,2)
if m(i,j)>cutoff & amp(i,j)>amp_thresh
y(i,j)=1;
end;
end;
end;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
find_crossings.m
%
%
by Hana Dobrovolny
%
%
%
%
This program determines activation or deactivation times of an %
%
optical signal.
%
224
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
function y=find_crossings(fid,type,thresh,num_images,mask,filter_size,bcl)
% Initialize
frewind(fid)
crossings=zeros(128,128,num_images);
for i=1:filter_size*2+1
a=(fscanf(fid,’%d’,[129,128]));
A(:,:,i)=a(2:129,:).*mask;
end;
A1=median(A,3);
s=size(A,3);
% Find threshold crossings
for i=filter_size+1:num_images-filter_size-1
A=circshift(A,[0 0 -1]);
a=(fscanf(fid,’%d’,[129,128]));
A(:,:,s)=a(2:129,:).*mask;
A2=median(A,3);
if type==’s’
crossings(:,:,i)= (A2<=thresh & A1>thresh);
end;
225
if type==’e’
crossings(:,:,i)=(A2>=thresh & A1<thresh);
end;
A1=A2;
end;
% Remove multiple crossings
count=ones(128,128);
y=zeros(128,128);
for i=1:num_images
for j=1:128
for k=1:128
if count(j,k)<20*bcl/1000
if mask(j,k)==1
if crossings(j,k,i)==1
y(j,k,count(j,k))=i;
count(j,k)=count(j,k)+1;
end;
end;
end;
end;
end;
end;
226
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
extra_cleaning.m
%
%
by Hana Dobrovolny
%
%
%
%
This program removes multiple crossing points.
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
function times=extra_cleaning(mask,starts,ends,window,bcl)
times=zeros(128,128);
for i=1:128
for j=1:128
if mask(i,j)==1;
count = 1;
clear all_times
all_times=0;
v=find(starts(i,j,:)~=0);
if length(v)<20*bcl/1000
for k=1:size(starts,3)
if starts(i,j,k)~=0
all_times(count)=starts(i,j,k);
count=count+1;
end;
end;
227
for k=1:size(ends,3)
if ends(i,j,k)~=0
all_times(count)=ends(i,j,k);
count=count+1;
end;
end;
end;
if all_times~=0
all_times=sort(all_times);
diffs=diff(all_times);
count = 1;
for k=1:length(diffs)
if diffs(k)>window
times(i,j,count)=all_times(k);
count=count+1;
end;
end;
end;
end;
end;
end;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
%
get_apds.m
%
228
%
by Hana Dobrovolny
%
%
%
%
This program calculates APD and DI.
%
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
function [a,d,apd]=get_apds(times)
apd=zeros(size(times,1),size(times,2));
count = ones(size(times,1),size(times,2));
a=zeros(128,128);
d=zeros(128,128);
for i=1:size(times,1)
for j=1:size(times,2)
for k=1:size(times,3)-1
if times(i,j,k+1)~=0
apd(i,j,count(i,j))=times(i,j,k+1)-times(i,j,k);
count(i,j) = count(i,j) + 1;
end;
end;
clear apds dis
if count(i,j)>1
apds=0;
dis=0;
for m=1:count(i,j)-1
229
if rem(m,2)==0
apds(m/2)=apd(i,j,m);
else
dis((m+1)/2)=apd(i,j,m);
end;
end;
if apds~=0
a(i,j)=mean(apds);
end;
if dis~=0
d(i,j)=mean(dis);
end;
end;
end
end
230
Appendix E
Passive Length Constant of the
Two-Variable Model
In this appendix, I present the derivation of the passive length constant for the
two variable cardiac model. Recall that the one-dimensional equations for the twovariable model are
δt V = Dδx2 V +
δt h =
h 2
V
Iext
V (1 − V ) −
−
,
τin
τout
Cm
(
1−h
τopen
h
− τclose
V < Vc
V > Vc ,
(E.1)
(E.2)
We begin by applying a subthreshold stimulus at x = 0 in an infinite cable,
Iext = I0 δ(x),
(E.3)
where δ(x) is the delta function. We are interested in the steady-state response to
the stimulus so δt V = δt h = 0. We also note that we are in the regime where V < Vc ,
so the gate remains open and h = 1. With these assumptions, equation E.1 becomes
Dδx2 V −
V
I0
V 2 (1 − V )
−
−
δ(x) = 0.
τin
τout Cm
231
(E.4)
Integrating equation E.4 from x = 0− to x = 0+ gives the boundary condition
δx V |x=0+ − δx V |x=0− =
I0
.
Cm D
(E.5)
All the terms involving V have disappeared since V must be continuous at x = 0.
Away from x = 0, there is no stimulus, so equation E.1 simplifies further:
Dδx2 V −
V 2 (1 − V )
V
−
= 0.
τin
τout
(E.6)
The solution of Eq. E.6 has the form
x
V = Ae λ + Be
−x
λ
+k
(E.7)
where A and B are constants determined by the boundary conditions, λ is the length
constant and k is given by
1 τin
k= +
2
2
s
1
τin
1
4
.
−
τin τout
(E.8)
We must have V = 0 when x → ±∞. This means that A = 0 when x < 0 and B = 0
when x > 0. We also insist that V is continuous at x = 0, so A = B. Thus we have
the following equation for the voltage
V =
(
x
Ae λ + k x ≤ 0
−x
Ae λ + k x ≥ 0,
(E.9)
Applying the boundary condition given by equation E.5, we find that spatial
232
variation in voltage is
I0
V =−
2Cm
|x|
τout − √Dτ
out .
e
D
(E.10)
Dτout ,
(E.11)
r
Thus the passive length constant,
λ=
q
depends on the diffusion coefficient as well as the voltage decay time constant τout .
233
Appendix F
Core Conductor Model
Models of cardiac cells are based on the idea that cardiac cells can be treated as
electrical circuits. This is commonly known as the core conductor model. In this
appendix, I use the core conductor model to derive the relationship between transmembrane voltage and transmembrane current in a cardiac fiber and find that cardiac
cells in a fiber are coupled through the second derivative of transmembrane voltage.
We assume that a single fiber of cardiac tissue has axial symmetry and so can be
treated as essentially one-dimensional (Fig. F.1). Cardiac cells are coupled through
the intracellular and extracellular space, modelled as resistors in the core conductor
model. Ie is the current through the extracellular space; Ii is the current through the
intracellular space. Φe is the extracellular potential; Φi is the intracellular potential.
re is the resistance per unit length in the extracellular space; ri is the resistance per
unit length in the intracellular space. The transmembrane current, im , is specified
by the choice of cellular model (for example, Eqs. 2.1 and 2.2 for the two-variable
model).
The transmembrane voltage is given by Vm = Φi − Φe . By Ohm’s law, we know
δΦe
= −Ie re
δx
(F.1)
δΦi
= −Ii ri .
δx
(F.2)
and
234
Figure F.1: Core conductor model. Cardiac fibers are modelled as electrical circuits.
Individual cardiac cells are coupled through the intracellular and extracellular space,
modelled by resistors.
The intracellular current can only be increased or decreased by changes in the transmembrane current,
δIi
= −im .
δx
(F.3)
The extracellular current can be changed in one of two ways: through the transmembrane current, or by leaving (or entering) the preparation through external electrodes,
δIe
= im + ip ,
δx
(F.4)
where ip is the current per unit length applied from outside the preparation.
The spatial derivative of transmembrane voltage is given by
δVm
δΦi δΦe
=
−
= −ri Ii + re Ie
δx
δx
δx
235
(F.5)
Differentiating with respect to space, we find
δ 2 Vm
δIi
δIe
= −ri
+ re
= (ri + re )im + re ip .
2
δx
δx
δx
(F.6)
This equation tells us that the transmembrane currents of coupled cardiac cells are
related through the second spatial derivative of transmembrane voltage, or as is
commonly said, cells are coupled through diffusion of voltage.
236
Appendix G
Glossary
Action Potential – The response of an excitable cell to an external stimulus, where
the transmembrane potential rapidly increases and more slowly decreases to it’s initial
state.
Action Potential Amplitude – The difference between the maximum transmembrane voltage achieved during an action potential and the transmembrane voltage of
a cell in the rest state.
Action Potential Duration (APD) – The time between the start and end of the
action potential.
Alternans (2:2) – A response pattern in which tissue exhibits a long-short alternation of APD.
Auricles – A region of cardiac tissue consisting of two chambers and veins located
in the upper portion of amphibian hearts.
Arrhythmia – A heart rhythm that cannot be characterized as an M:N response.
Basic Cycle Length (BCL) – The length of time between externally applied stimuli.
Charge-coupled device (CCD) camera – A camera that collects images by converting photons incident on the device to electrons.
237
Concordant Alternans – Spatially extended manifestation of alternans where the
entire tissue oscillates in phase.
Conduction Block – Failure of a wave to propagate beyond a certain spatial location, typically caused by the wave encountering a region of tissue still in the refractory
period.
Constant-BCL Restitution Curve (BRC) – A restitution curve produced by
any APD and previous DI resulting from pacing at a constant BCL.
Diacetyl monoxime (DAM) – An electro-mechanical decoupler that when applied
to cardiac tissue causes the tissue to stop contracting without inhibiting electrical
propagation.
Diastolic Interval (DI) – The period of time between the end of one action potential
and the beginning of the following action potential.
Dichroic Filter – A color filter that selectively passes a small range of colors while
reflecting all others.
Discordant Alternans – Spatially extended manifestation of alternans where parts
of the tissue oscillate out of phase.
Digital Number (DN) – Measurement unit of digital cameras.
Dynamic Restitution Curve (DRC) – A restitution curve produced by the steady
state APD and DI.
Excitable Medium – A medium in which an external stimulus below a certain
threshold causes a rapid decay to a global rest state and an external stimulus above
the threshold causes a large excursion through phase space before a return to the
238
global rest state. Following the excursion, there is a refractory period during which
further suprathreshold stimuli will not elicit the excitable response.
Fibrillation – A disorganized contraction of cardiac muscle.
Intensity – The number of photons per pixel collected during a single frame of the
camera.
Light-Emitting Diode – A semiconductor device that emits light in a narrow spectral band.
Microelectrode – A small glass capillary which has one end pulled to a fine tip
(∼10 µm diameter). The tip is inserted into a cell to measure the transmembrane
voltage.
M:N response – A response pattern in which tissue produces N distinct APDs for
every M stimuli.
Perturbed Downsweep Protocol (PDP) – The pacing protocol used to produce
the restitution portrait.
Plane Wave – A constant-frequency wave whose wavefronts are infinite parallel
planes of constant amplitude.
Potentiometric Dye – A fluorescent dye whose emission and absorption spectra
change in the presence of an applied voltage.
Refractory Period – The period of time after an action potential during which
another action potential cannot be elicited.
Restitution Curve (RC) – A curve representing the relationship between APD
239
and previous DI.
Restitution Portrait (RP) – A visual representation of the relationships between
the different restitution curves.
Ringer’s Solution – A solution meant to mimic blood that provides nutrients to
an in vitro tissue preparation. Standard Ringer’s solution consists of 100 mM NaCl,
2.70 mM KCl, 5.6 mM glucose, 1 mM hepes, 2.8 mM Na2 HPO4 , 25 mM NaHCO3 ,
1.5 mM MgCl2 , 1.80 mM CaCl2 .
S1S2 Restitution Curve (SRC) – A restitution curve produced by the APD and
DI following a perturbation in BCL.
Spatial Gradient – The rate of change of a property in space.
Threshold Voltage – The transmembrane voltage at which the cell will be induced
to elicit an action potential.
Transmembrane Voltage – The voltage difference measured across the cell membrane.
Ventricle – The main pumping chamber of the heart.
240
Appendix H
Guide to Symbols and Acronyms
AP - Action potential
APA - Action potential amplitude
APD - Action potential duration
∆AP D - Spatial difference in APD
∆AP D - Mean of all trials of spatial difference in APD
∇AP D - Spatial gradient of APD
∇AP D - Mean of all points on the surface of the tissue of spatial gradient of APD
ARI - Activation-recovery interval
ALT - Trials which exhibit alternans at rapid pacing
BCL - Basic cycle length
BCLt - Basic cycle length at which a transition to either a 2:1 or 1:1 response occurs
BCLn - Normalized basic cycle length, defined as BCLN = BCL − BCLt
BRC - Constant-BCL restitution curve
BRC-D - Constant-BCL restitution curve produced after a permanent change in
241
basic cycle length
BRC-S - Constant-BCL restitution curve produced after a perturbation in basic
cycle length
Cm - Cellular membrane capacitance
CCD - Charge-coupled device
DAM – Diacetyl monoxime
DI - Diastolic interval
DN - Digital number
DRC - Dynamic restitution curve
h - Gate variable of the two-current model
Iext - Externally applied stimulus current
Iin - Inward current
Iout - Outward current
LED - Light-emitting diode
MSE - Mean square error (See Eq. B.3)
noALT - Trials which went directly from 1:1 to 2:1 response at rapid pacing
PDP - Perturbed downsweep protocol
RC - Restitution curve
242
SBRC - Slope of the constant-BCL restitution curve
∆SBRC - Spatial difference in slope of the BRC
∆SBRC - Mean of all trials of spatial difference in slope of the BRC
SDRC - Slope of the dynamic restitution curve
∆SDRC - Spatial difference in slope of the DRC
∆SDRC - Mean of all trials of spatial difference in slope of the DRC
∇SDRC - Spatial gradient of slope of the DRC
∇SDRC - Mean of all points on the surface of the tissue of spatial gradient of slope
of the DRC
Smem - Slope criterion derived using a 3 variable cardiac mapping model with memory
(See Eq. 6.4)
SSRC - Slope of the S1S2 restitution curve
∆SSRC - Spatial difference in slope of the SRC
∆SSRC - Mean of all trials of spatial difference in slope of the SRC
SNR - Signal to noise ratio
SRC - S1S2 restitution curve
τclose - Time constant of gate closing in the two-variable model
τin - Time constant of the inward current in the two-current model
243
τopen - Time constant of the gate opening in the two-variable model
τout - Time constant of the outward current in the two-variable model
V - Transmembrane voltage
Vc - Threshold voltage
244
Bibliography
[1] AM Katz. Physiology of the Heart. Raven Press, New York, 1992.
[2] American Heart Association. Heart disease and stroke statistics 2007 update.
Dallas, TX, 2007.
[3] GW Gaskell. On the tonicity of the heart and blood vessels. Journal of Physiology - London, 3(1):48–89, 1880.
[4] EG Martin. The rise of the present conceptions as to the cause of the heartbeat. ii. the myogenic theory, and modern studies of rhythmicity. Bulletin of
the Johns Hopkins Hospital, 16:377–84, 1905.
[5] GR Mines. On dynamic equilibrium in the heart. Journal of Physiology London, 46(4/5):349–83, 1913.
[6] GR Mines. On pulsus alternans. Proceedings Of The Cambridge Philosophical
Society, 17:34–41, 1914.
[7] E Bordet, E Donzelot, and C Pezzi. A case of mechanical and electric cardiac
alternation observed in man. Comptes Rendus Des Seances De La Societe De
Biologie Et De Ses Filiales, 75:468–70, 1913.
[8] JH Musser. The myogenic doctrine of the cardiac activity and its relation to
arrhythmia. American Journal of the Medical Sciences, 141:360–74, 1911.
[9] GW Norris. Cardiac arrhythmias. American Journal of the Medical Sciences,
136:46–66, 1908.
[10] AL Hodgkin and AF Huxley. A quantitative description of membrane current
and its application to conduction and excitation in nerve. Journal of Physiology,
117:500–44, 1952.
[11] D Noble. On cardiac action and pacemaker potentials based on the hogkinhuxley equations. Nature, 188(4749):495–97, 1960.
[12] D Noble. A description of cardiac pacemaker potentials based on the hogkinhuxley equations. Journal of Physiology - London, 154(3):P64–65, 1960.
245
[13] D Noble. Computation of cardiac action and pacemaker potentials. Journal of
Physiology - London, 157(1):P14, 1961.
[14] GW Beeler and H Reuter. Reconstruction of action potential of ventricular
myocardial fibers. Journal of Physiology - London, 268(1):177–210, 1977.
[15] CH Luo and Y Rudy. A model of the ventricular cardiac action potential
depolarization, repolarization, and their interaction. Circulation Research,
68(6):1501–26, 1991.
[16] LB Cohen, RD Keynes, and B Hille. Light scattering and birefringence changes
during nerve activity. Nature, 218:438–441, 1968.
[17] LB Cohen, B Hille, and RD Keynes. Light scattering and birefringence changes
during activity in the electrical organ of Electrophorus electricus. Journal of
Physiology, 203:489–509, 1969.
[18] I Tasaki, R Watanabe, R Sandlin, and L Carnay. Changes in fluorescence,
turbidity, and birefringence associated with nerve excitation. Proceedings of
the National Academy of Sciences USA, 61:883–888, 1968.
[19] LB Cohen and RD Keynes. Changes in light scattering associated with the
action potential in crab nerves. Journal of Physiology, 212:239, 1971.
[20] LB Cohen, B Hille, and RD Keynes. Changes in axon birefringence during an
action potential. Journal of Physiology, 211:495–515, 1971.
[21] LB Cohen, BM Salzberg, HV Davila, WN Ross, D Landowne, AS Waggoner,
and CH Wang. Changes in axon fluorescence during activity: molecular probes
of membrane potential. Journal of Membrane Biology, 19:1, 1974.
[22] G Salama and M Morad. Merocyanine-540 as an optical probe of transmembrane electrical activity in the heart. Science, 191(4226):485, 1976.
[23] FX Witkowski, LJ Leon, PA Penkoske, RB Clark, ML Spano, WL Ditto, and
WR Giles. A method for visualization of ventricular fibrillation: Design of
a cooled fiberoptically coupled image intensified ccd data acquisition system
incorporationg wavelet shrinkage based adaptive filtering. Chaos, 8(1):94–102,
1998.
[24] RA Gray, AM Pertsov, and J Jalife. Spatial and temporal organization during
cardiac fibrillation. Nature, 392:76–78, 1998.
246
[25] KF Kwaku and SM Dillon. Shock-induced depolarization of refractory myocardium prevents wave-front propagation in defibrillation. Circulation Research, 79:957–973, 1996.
[26] JP Wikswo Jr., S-F Lin, and RA Abbas. Virtual electrodes in cardiac tissue: A
common mechanism for anodal and cathodal stimulation. Biophysical Journal,
69:2195–2210, 1995.
[27] D Dubin. Ion Adventure in the Heartland. Cover Publishing, Hong Kong, 2003.
[28] KW Linz and R Meyer. Profile and kinetics of l-type calcium current during the
cardiac ventricular action potential compared in guinea-pigs, rats and rabbits.
European Journal of Physiology, 49(5):588–99, 2000.
[29] A Varro, DA Lathrop, SB Hester, PP Nanasi, and JGY Papp. Ionic currents
and action-potentials in rabbit, rat, and guinea-pig ventricular myocytes. Basic
Research in Cardiology, 88(2):93–102, 1993.
[30] JM Pastore, SD Girouard, KR Laurita, FG Akar, and DS Rosenbaum. Mechanism linking t-wave alternans to the genesis of cardiac fibrillation. Circulation,
99:1385–94, 1999.
[31] DS Rosenbaum, LE Jackson, JM Smith, H Garan, JN Ruskin, and RJ Cohen.
Electrical alternans and vulnerability to ventricular arrhythmias. New England
Journal of Medicine, 330:235–41, 1994.
[32] JM Cao, Z Qu, YH Kim, TJ Wu, A Garfinkel, J Weiss, HS Karagueuzian, and
PS Chen. Spatiotemporal heterogeneity in the induction of ventricular fibrillation by rapid pacing: Importance of cardiac restitution properties. Circulation
Research, 84:1318–31, 1999.
[33] TR Chay. Bifurcations in heart rhythms. International Journal of Bifurcation
and Chaos, 5(6):1439–86, 1995.
[34] L Stone and S Ezrati. Chaos, cycles and spatiotemporal dynamics in plant
ecology. Journal of Ecology, 84(2):279–91, 1996.
[35] GR Chen. An overview of bifurcation, chaos and nonlinear dynamics in controlsystems. Journal of the Franklin Institute-Engineering And Applied Mathematics, 331B(6):819–58, 1994.
247
[36] DM Wolf, M Varghese, and SR Sanders. Bifurcation of power electroniccircuits. Journal of the Franklin Institute-Engineering And Applied Mathematics, 331B(6):957–99, 1994.
[37] GR Chen, ML Moiola, and HO Wang. Bifurcation control: Theories, methods,
and applications. International Journal of Bifurcation and Chaos, 10(3):511–
48, 2000.
[38] JW Nolasco and RW Dahlen. A graphic method for the study of alternation
in cardiac action potentials. Journal of Applied Physiology, 25:191–6, 1968.
[39] GM Hall, S Bahar, and DJ Gauthier. Prevalence of rate-dependent behaviors
in cardiac muscle. Physical Review Letters, 82(14):2995–8, 1999.
[40] I Banville and RA Gray. Effect on action potential duration and conduction
velocity restitution and their spatial dispersion on alternans and the stability
of arrhythmias. Journal of Cardiovascular Electrophysiology, 13:1141–9, 2002.
[41] RF Gilmour, NF Otani, and MA Watanabe. Memory and complex dynamics in
cardiac purkinje fibers. American Journal of Physiology, 272:H1826–32, 1997.
[42] EJ Vigmond and LJ Leon. Restitution curves and the stability of reentry in
three-dimensional simulations of cardiac tissue. Computing and Visualization
in Science, 4:237–47, 2002.
[43] SS Kalb, HM Dobrovolny, EG Tolkacheva, SF Idriss, W Krassowska, and
DJ Gauthier. The restitution portrait: A new method of investigating ratedependent restitution. J Cardiovasc. Electrophys., 15:698–709, 2004.
[44] MR Boyett and BR Jewell. Study of the factors responsible for rate-dependent
shortening of action potential in mammalian ventricular muscle. Journal of
Physiology, 285:359–80, 1978.
[45] M Watanabe, NF Otani, and RF Gilmour. Biphasic restitution of action potential duration and complex dynamics in ventricular myocardium. Circulation
Research, 76(5):915–21, 1995.
[46] ML Koller, ML Riccio, and RF Gilmour. Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. American
Journal of Physiology, 275:H1635–42, 1998.
248
[47] EG Tolkacheva, DG Schaeffer, DJ Gauthier, and W Krassowska. Condition
for alternans and stability of the 1:1 response pattern in a ‘memory’ model of
paced cardiac dynamics. Physical Review E, 67:031904, 2003.
[48] AV Holden and AV Panfilov. Graphical interpretation of spatiotemporal chaos.
Computers and Graphics, 15(2):301–2, 1991.
[49] B Echebarria and A Karma. Instability and spatiotemporal dynamics of alternans in paced cardiac tissue. Physical Review Letters, 88(20):208101, 2002.
[50] H Hayashi, Y Shiferaw, D Sato, M Nihei, SF Lin, PS Chen, A Garfinkel,
JN Weiss, and ZL Qu. Dynamic origin of spatially discordant alternans in
cardiac tissue. Biophysical Journal, 92(2):448–60, 2007.
[51] JGC Ponard, AA Kondratyev, and JP Kucera. Mechanisms of intrinsic beating
variability in cardiac cell cultures and model pacemaker networks. Biophysical
Journal, 92(10):3734–52, 2007.
[52] RA Chapman and CH Fry. An analysis of the cable properties of frog ventricular myocardium. Journal of Physiology, 283:263–82, 1978.
[53] ML Pressler. Membrane properties of the cardiac conduction system: Comparative properties. Proceedings of the Koninklikje Nederlandse Akademie van
Wetenschappen, 93:477–478, 1990.
[54] FG Akar, BJ Roth, and DS Rosenbaum. Optical measurement of cell-to-cell
coupling in intact heart using subthreshold electrical stimulation. American
Journal of Physiology, 281(2):H533–42, 2001.
[55] S Poelzing, BJ Roth, and DS Rosenbaum. Optical measurements reveal nature
of intercellular coupling across ventricular wall. American Journal of Physiology, 289:H1428–35, 2004.
[56] WC De Mello. Increased spread of electrotonic potentials during diastolic depolarization in cardiac muscle. Journal of Molecular and Cellular Cardiology,
18(1):23–9, 1986.
[57] SS Goldstein and W Rall. Changes of action potential shape and velocity for
changing core conductor geometry. Biophysical Journal, 14:731–57, 1974.
249
[58] MS Spach and JM Kootsey. Relating the sodium current and conductance to
the shape of transmembrane and extracellular potentials by simulation: Effects
of propagation boundaries. IEEE Transactions on Biomedical Engineering,
32(10):743–55, 1985.
[59] BM Steinhaus, KW Spitzer, and S Isomura. Action potential collision in cardiac
tissue – computer simulations and tissue experiment. IEEE Transactions on
Biomedical Engineering, 32:731–42, 1985.
[60] KJ Sampson and CS Henriquez. Electrotonic influences on action potential
duration dispersion in small hearts: A simulation study. American Journal of
Physiology, 289:H350–60, 2005.
[61] RW Joyner.
Modulation of repolarization by electrotonic interactions.
Japanese Heart Journal, 27:167–83, 1986.
[62] MD Lesh, M Pring, and JF Spear. Cellular uncoupling can unmask dispersion
of action potential duration in ventricular myocardium. a computer modeling
study. Circulation Research, 65(5):1426–40, 1989.
[63] J Cain and DG Schaeffer. Shortening of cardiac action potential near an insulating boundary. submitted to, 2007.
[64] I Banville, N Chattipakorn, and RA Gray. Restitution dynamics during pacing
and arrhythmias in isolated pig hearts. Journal of Cardiovascular Electrophysiology, 15(4):455–63, 2004.
[65] DS Rosenbaum, DT Kaplan, A Kanai, L Jackson, H Garan, RJ Cohen, and
G Salama. Repolarization inhomogeneities in ventricular myocardium change
dynamically with abrupt cycle length shortening. Circulation, 84(3):1333–45,
1991.
[66] T Aiba, W Shimizu, I Hidaka, K Uemura, T Noda, C Zheng, A Kamiya,
M Inagaki, M Sugimachi, and K Sunagawa. Cellular basis for trigger and
maintenance of ventricular fibrillation in the brugada syndrome model. Journal
of the American College of Cardiology, 47(10):2074–85, 2006.
[67] M Restivo, EB Caref, DO Kozhevnikov, and N El-Sherif. Spatial dispersion
of repolarization is a key factor in the arrhythmogenicity of long qt syndrome.
Journal of Cardiovascular Electrophysiology, 15(3):323–31, 2004.
250
[68] H Qin, J Huang, JM Rogers, GP Walcott, DL Rollins, WM Smith, and
RE Ideker. Mechanisms for the maintenance of ventricular fibrillation: The
nonuniform dispersion of refractoriness, restitution properties, or anatomic heterogeneities? Journal of Cardiovascular Electrophysiology, 16(8):888–97, 2005.
[69] H-N Pak, SJ Hong, GS Hwang, HS Lee, S-W Park, JC Ahn, YM Ro, and
Y-H Kim. Spatial dispersion of action potential duration restitution kinetics
is associated with induction of ventricular tachycardia/fibrillation in humans.
Journal of Cardiovascular Electrophysiology, 15(12):1357–63, 2004.
[70] FG Akar, KR Laurita, and DS Rosenbaum. Cellular basis for dispersion of
repolarization underlying reentrant arrhythmias. Journal of Electrocardiology,
33:23–31, 2000.
[71] EJ Pruvot, RP Katra, DS Rosenbaum, and KR Laurita. Role of calcium cycling
versus restitution in the mechanism of repolarization alternans. Circulation
Research, 94:1083–90, 2004.
[72] A Nygren, AE Lomax, and WR Giles. Heterogeneity of action potential durations in isolated mouse left and right atria recorded using voltage-sensitive dye
mapping. American Journal of Physiology, 267(6):H2634–43, 2005.
[73] C-S Kuo, K Munakata, CP Reddy, and B Surawicz. Characteristics and possible mechanism of ventricular arrhythmia dependent on the dispersion of action
potential durations. Circulation, 67(6):1356–67, 1983.
[74] KR Laurita, SD Girouard, FG Akar, and DS Rosenbaum. Modulated dispersion
explains changes in arrhythmia vulnerability during premature stimulation of
the heart. Circulation, 98:2774–80, 1998.
[75] LC Baker, B London, BR Choi, G Koren, and G Salama. Enhanced dispersion of repolarization and refractoriness in transgenic mouse hearts promotes
reentrant ventricular tachycardia. Circulation Research, 86(4):396–407, 2000.
[76] JM Smith, SM Clancy, R Valeri, JN Ruskin, and RJ Cohen. Electrical alternans
and cardiac electrical instability. Circulation, 77:110–21, 1988.
[77] H Wang and M Thoss. Quantum dynamical simulation of electron-transfer
reactions in an anharmonic environment. Journal of Physical Chemistry A,
111(41):10369–75, 2007.
251
[78] A Mokhtari. First-principles investigation of electronic and structural properties and bowing parameters in srfclxbr1-x alloy. Journal of Physics - Condensed
Matter, 19(43):436213, 2007.
[79] M Wagner and F Schmitz. P-modes of a polytropic convection zone with an
overlying hot envelope. Astronomy and Astrophysics, 472(3):897–903, 2007.
[80] MC Cross and PC Hohenberg. Pattern formation outside of equilibrium. Reviews of Modern Physics, 65(3):851–1112, 1993.
[81] DS Ricketts, XF Li, N Sun, K Woo, and D Woo. On the self-generation of
electrical soliton pulses. IEEE Journal of Solid-State Circuits, 42(8):1657–68,
2007.
[82] S Rudiger, EM Nicola, J Casademunt, and L Kramer. Theory of pattern forming systems under traveling-wave forcing. Physics Reports, 447(3-6):73–111,
2007.
[83] P Bak, K Chen, and C Tang. A forest fire model and some thoughts on
turbulence. Physical Letters A, 147(5-6):297–300, 1990.
[84] I Karafyllidis and A Thanailakis. A model for predicting forest fire spreading
using cellular automata. Ecological Modelling, 99(1):87–97, 1997.
[85] A Gydikov, L Gerlikovsky, and N Trayanova. Influence of the muscle fibre
end geometry on the extracellular potentials. Biological Cybernetics, 54(1):1–
8, 1986.
[86] GH Hakansson. Conduction velocity and amplitude of the action potential as
related to circumference in the isolated fibre of frog muscle. Acta Physiologica
Scandinavica, 37:14–34, 1956.
[87] A Karma. Spiral breakup in model equations of action potential propagation
in cardiac tissue. Physical Review Letters, 71:1103–7, 1993.
[88] CC Mitchell and DG Schaeffer. A two-current model for the dynamics of cardiac
membrane. Bulletin of Mathematical Biology, 65:767–93, 2003.
[89] D Noble. A modification of the hodgkinhuxley equations applicable to purkinje
fibre action and pacemaker potentials. Journal of Physiology, 160:317–52, 1962.
252
[90] R Plonsey and RC Barr. The four-electrode resistivity technique as applied
to cardiac muscle. IEEE Transactions on Biomedical Engineering, 29:541–546,
1982.
[91] SV Pandit, RB Clark, WR Giles, and SS Demir. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophysical
Journal, 81:3029–51, 2001.
[92] GM Faber and Y Rudy. Action potential and contractility changes in [na]i
overloaded cardiac myocytes: a simulation study. Biophysical Journal, 78:2392–
2404, 2001.
[93] RH Anderson, SY Ho, D Sanchez-Quintana, K Redmann, and PP Lunkenheimer. Heuristic problems in defining the three-dimensional arrangement of
the ventricular myocytes. Anatomical Record A, 288(6):579–86, 2006.
[94] N Bursac, KK Parker, S Iravanian, and L Tung. Cardiomyocyte cultures with
controlled macroscopic anisotropy - a model for functional electrophysiological
studies of cardiac muscle. Circulation Research, 91(12):E45–54, 2002.
[95] B Taccardi, BB Punske, F Sachse, X Tricoche, P Colli-Franzone, LF Pavarino,
and C Zabawa. Intramural activation and repolarization sequences in canine
ventricles. experimental and simulation studies. Journal of Electrocardiology,
38(4):131–7, 2005.
[96] D Sedmera, M Reckova, A deAlmeida, M Sedmerova, M Biermann, J Volejnik, A Sarre, E Raddatz, RA McCarthy, RG Gourdie, and RP Thompson.
Functional and morphological evidence for a ventricular conduction system in
zebrafish and xenopus hearts. American Journal of Physiology, 284(5):1152–60,
2002.
[97] JR Sommer and EA Johnson. Cardiac muscle: A comparative ultrastructural
study with special reference to frog and chicken hearts. Zeitschrift fur Zellforschung und Mikroskopische Anatomie, 98(3):437–68, 1969.
[98] B Kisch. Electronmicroscopy of the frog’s heart. Experimental Medicine and
Surgery, 19:104–42, 1961.
[99] NS Staley and ES Benson. The ultrastructure of frog ventricular muscle and
its relationship to mechanisms of excitation-contraction coupling. Journal of
Cell Biology, 38:99–114, 1968.
253
[100] HL Sharma. The circulatory mechanism and anatomy of the heart of the frog,
Rana pipiens. Journal of Morphology, 109:323–49, 1961.
[101] F Bode, M Kilborn, P Karasik, and MR Franz. The repolarzation-excitability
relationship in the human right atrium is unaffected by cycle length, recording
site and prior arrhythmias. Journal of the American College of Cardiology,
37(3):920–5, 2001.
[102] MP Nash, CP Bradley, PM Sutton, RH Clayton, P Kallis, MP Hayward, DJ Paterson, and P Taggert. Whole heart action potential duration restitution in
cardiac patients: A combined clinical and modeling study. Experimental Physiology, 91(2):339–54, 2006.
[103] LA Geddes. Electrodes and the Measurement of Bioelectric Events. John Wiley
and Sons, New York, 1972.
[104] LM Loew, LB Cohen, BM Salzberg, AL Obaid, and F Bezanilla. Charge-shift
probes of membrane potential - characterization of aminostyrylpyridinium dyes
on the squid giant axon. Biophysical Journal, 47(1):71–77, 1985.
[105] A Grinvald, R Hildesheim, IC Farber, and L Anglister. Improved fluorescentprobes for the measurement of rapid changes in membrane-potential. Biophysical Journal, 39(3):301–308, 1982.
[106] SD Girouard, JM Pastore, KR Laurita, KW Gregory, and DS Rosenbaum.
Optical mapping in a new guinea pig model of ventricular tachycardia reveals
mechanisms for multiple wavelengths in a single reentrant circuit. Circulation,
93(3):603–613, 1996.
[107] JM Davidenko, PF Kent, DR Chialvo, DC Michaels, and J Jalife. Sustained
vortex-like waves in normal isolated ventricular muscle. Proceedings of the
National Academy of Sciences USA, 87:8785–9, 1990.
[108] I Banville, RA Gray, RE Ideker, and WM Smith. Shock-induced figure-of-eight
reentry in the isolated rabbit heart. Circulation Research, 85:742–52, 1999.
[109] SF Lin, RA Abbas, and JP Wikswo. High-resolution high-speed synchronous
epifluorescence imaging of cardiac activation. Reviews of Scientific Instruments, 68(1):213–217, 1997.
[110] RT Bove and SM Dillon. Optically imaging cardiac activation with a laser
system. IEEE Engineering in Medicine and Biology, 17(1):84–94, 1998.
254
[111] S-F Lin and JP Wikswo. Panoramic optical imaging of electrical propagation
in isolated heart. Journal of Biomedical Optics, 4(2):200–207, 1999.
[112] I Kodama, I Sakuma, N Shibata, SB Knisley, R Niwa, and H Honjo. Regional
differences in arrhythmogenic aftereffects of high intensity dc stimulation in the
ventriclees. PACE, 23:807–817, 2000.
[113] E Entcheva, Y Kostov, E Tchernev, and L Tung. Fluorescence imaging of
electrical activity in cardiac cells using an all-solid-state-system. IEEE Transactions on Biomedical Engineering, 51(2):333–41, 2004.
[114] C Antzelevic and J Fish. Electrical heterogeneity within the ventricular wall.
Basic Research in Cardiology, 96(6):517–27, 2001.
[115] D Sedmara, M Reckova, A deAlmeida, M Sedmerova, M Biermann, J Volejnik, A Sarre, E Raddatz, RA McCarthy, RG Gourdie, and RP Thompson.
Functional and morphological evidence for a ventricular conduction system in
zebrafish and xenopus hearts. American Journal of Physiology, 284:1152–60,
2003.
[116] S Weidmann. The electrical constants of purkinje fibers. Journal of Physiology,
118:348–60, 1952.
[117] RF Burton. Ringer Solutions and Physiological Salines. Wright Scientechnica,
1975.
[118] E Fluhler, VG Burnham, and LM Loew. Spectra, membrane-binding, and potentiometric responses of new charge shift probes. Biochemistry, 24(21):5749–
55, 1985.
[119] KJ Sampson and CS Henriquez. Simulation and prediction of functional block
in the presence of structural and ionic heterogeneity. American Journal of
Physiology, 281:H2597–H2603, 2001.
[120] KR Laurita and DS Rosenbaum. Interdependence of modulated dispersion and
tissue structure in the mechanism of unidirectional block. Circulation Research,
87:922–8, 2000.
[121] G Bub and A Shrier. Propagation through heterogeneous substrates in simple
excitable media models. Chaos, 12(1):747–53, 2002.
255
[122] N Maglaveras, F offner, FJL van Capelle, MA Allessie, and AV Sahakian. Effects of barriers on propagation of action potentials in two dimensional cardiac
tissue. Journal of Cardiovascular Electrophysiology, 28(1):17–31, 1995.
[123] KHWJ Ten Tusscher and AV Panfilov. Wave propagation in excitable media with randomly distributed obstacles. Multiscale Modeling and Simulation,
3(2):265–82, 2005.
[124] A Garfinkel, ML Spano, WL Ditto, and JN Weiss. Controlling cardiac chaos.
Science, 257(5074):1230–35, 1992.
[125] GM Hall and DJ Gauthier. Experimental control of cardiac muscle alternans.
Physical Review Letters, 88(19):198102, 2002.
[126] K Hall, DJ Christini, M Tremblay, JJ Collins, L Glass, and J Billette. Dynamic
control of cardiac alternans. Physical Review Letters, 78(23):4518–21, 1997.
[127] WL Ditto, ML Spano, V In, J Neff, B Meadows, JJ Langberg, A Bolmann,
and K McTeague. Control of human atrial fibrillation. International Journal
of Bifurcation and Chaos, 10(3):593–601, 2000.
[128] H Zhang, Z Cao, NJ Wu, HP Ying, and G Hu. Suppress winfree turbulence by
local forcing excitable systems. Physical Review Letters, 94:188301, 2005.
[129] DJ Christini, KM Stein, SM Markowitz, S Mittal, DJ Slotwiner, MA Scheiner,
S Iwai, and BB Lerman. Nonlinear-dynamical arrhythmia control in humans.
Proceedings of the National Academy of Sciences USA, 98(10):5827–32, 2001.
[130] Z Cao, H Zhang, F Xie, and G Hu. Controlling turbulence in excitable media
by applying boundary periodic pacing and gradient force. Europhysics Letters,
75(6):875–81, 2006.
[131] U Brandt-Pollmann, D Lebiedz, M Diehl, S Sager, and J Schloder. Realtime nonlinear feedback control of pattern formation in (bio)chemical reactiondiffusion processes: A model study. Chaos, 15:033901, 2005.
[132] MA Allessie, C Kirchof, GJ Scheffer, F Chorra, and J Brugada. Regional control of atrial fibrillation by rapid pacing in concious dogs. Circulation, 84:1689–
97, 1991.
[133] S Narayan. T-wave alternans and the susceptibility to ventricular arrhythmias.
Journal of the American College of Cardiology, 47:269–81, 2006.
256
[134] BR Choi and G Salama. Simultaneous maps of optical action potentials and
calcium transients in guinea-pig hearts: mechanisms underlying concordant
alternans simultaneous maps of optical action potentials and calcium transients
in guinea-pig hearts: mechanisms underlying concordant alternans. Journal of
Physiology, 529(1):171–88, 2000.
[135] SM Hwang, KH Yea, and KJ Lee. Regular and alternant spiral waves of
contractile motion on rat ventricle cell cultures. Physical Review Letters,
92(19):198103, 2004.
[136] BR Choi, T Liu, and G Salama. Adaptation of cardiac action potential durations to stimulation history with random diastolic intervals. Journal of Cardiovascular Electrophysiology, 15:1188–97, 2004.
[137] A Vinet, DR Chialvo, DC Michaels, and J Jalife. Nonlinear dynamics of ratedependent activation in models of single cardiac-cells. Circulation, 67(6):1510–
24, 1990.
[138] SH Strogatz. Nonlinear Dynamics and Chaos: with Applications in Physics,
Biology, Chemistry, and Engineering. Addison-Wesley, Reading, MA, 1994.
[139] EG Tolkacheva, JMB Anumonwo, and J Jalife. Action potential duration restitution portraits of mammalian ventricular myocytes: Role of calcium current.
Biophysical Journal, 91(7):2735–45, 2006.
[140] A Karma. Electrical alternans and spiral wave breakup in cardiac tissue.
Chaos, 4:461–72, 1994.
[141] Z Qu, JN Weiss, and A Garfinkel. Spatiotemporal chaos in a stimulated ring
of cardiac cells. Physical Review Letters, 78:1387–90, 1997.
[142] O Orias, CM Brooks, EE Suckling, JL Gilbert, and AA Siebens. Excitability
of the mammalian ventricle throughout the cardiac cycle. American Journal of
Physiology, 163(2):272–82, 1950.
[143] T Chow, DJ Kereiakes, C Bartone, T Booth, EJ Schloss, T Waller, ES Chung,
S Menon, BK Nellamothu, and PS Chan. Prognostic utility of microvolt twave alternans in risk stratification of patients with ischemic cardiomyopathy.
Journal of the American College of Cardiology, 47(9):1820–7, 2006.
257
[144] A Karma and Y Shiferaw. New pacing protocol for the induction of t-wave
alternans at slower heart rate and improving the prediction of sudden cardiac
death. Heart Rhythm, 1:S190, 2004.
[145] V Elharrar and B Surawicz. Cycle length effect on restitution of actionpotential duration in dog cardiac fibers. American Journal of Physiology,
244(6):H782–92, 1983.
[146] SS Kalb, JJ Fox, EG Tolkacheva, DJ Gauthier, and W Krassowska. Parameter
estimation in mapping models with memory. Anatomical Record A, 288(6):579–
86, 2006.
[147] HM Dobrovolny, CM Berger, W Krassowska, and DJ Gauthier. Predicting
the propensity to alternans from spatial differences in the restitution portrait.
American Journal of Physiology, submitted, 2007.
[148] P Schaffer, H Ahammer, W Muller, B Koidl, and H Windisch. Di-4-anepps
causes photodynamic damage to isolated cardiomyocytes. Pfluger’s Archive European Journal of Physiology, 426(6):548–51, 1994.
[149] DG Altman and JM Bland. Measurement of medicine: The analysis of method
comparison studies. The Statistician, 32(3):307–17, 1983.
[150] R Khazanie. Elementary Statistics in a World of Applications. Harper Collins,
USA, 1990.
[151] JM Pastore, KR Laurita, and DS Rosenbaum. Importance of spatiotemporal
heterogeneity of cellular restitution in mechanism of arrhythmogenic discordant
alternans. Heart Rhythm, 3(6):711–9, 2006.
[152] DG Schaeffer, JW Cain, DJ Gauthier, SS Kalb, RA Oliver, and EG Tolkacheva. An ionically based mapping model with memory for cardiac restitution.
Bulletin of Mathematical Biology, in press.
[153] HE Nusse, E Ott, and JA Yorke. Border-collision bifurcations: An explanation
for observed bifurcation phenomena. Physical Review E, 49(2):1073–7, 1994.
[154] X Zhao and DG Schaeffer. Alternate pacing of border-collision period-doubling
bifurcations. Nonlinear Dynamics, accepted.
[155] Y Shiferaw, MA Watanabe, A Garfinkel, JN Weiss, and A Karma. Model
of intracellular calcium cycling in ventricular myocytes. Biophysical Journal,
85(6):3666–3686, 2003.
258
[156] Y Shiferaw and A Karma. Turing instability mediated by voltage and calcium diffusion in paced cardiac cells. Proceedings of the National Academy of
Sciences USA, 103(15):5670–5, 2006.
[157] LD Wilson, X Wan, and DS Rosenbaum. Cellular alternans: A mechanism
linking calcium cycling proteins to cardiac arrhythmogenesis. Annals of the
New York Academy of Sciences, 1080:216–34, 2006.
[158] JL Goldhaber, LH Xie, T Duong, C Motter, K Khuu, and JN Weiss. Action
potential duration restitution and alternans in rabbit ventricular myocytes:
The key role of intracellular calcium cycling. Circulation Research, 96:459–66,
2005.
[159] DA Eisner, ME Diaz, Y Li, SC O’Neill, and AW Trafford. Stability and instability of regulation of intracellular calcium. Experimental Physiology, 90(1):3–12,
2004.
[160] GL Aistrup, JE Kelley, S Kapur, M Kowalczyk, I Sysman-Wolpin, AH Kadish,
and JA Wasserstrom. Pacing-induced heterogeneities in intracellular ca2+ signaling, cardiac alternans, and ventricular arrhythmias in intact raw heart. Circulation Research, 99:E65–73, 2006.
[161] P Tijskens, G Meissner, and C Franzini-Armstrong. Location of ryanodine
and dihydropyridine receptors in frog myocardium. Biophysical Journal,
84(2):1079–92, 2003.
[162] Z Qu. Dynamical effects of diffusive cell coupling on cardiac excitation and
propagation: A simulation study. American Journal of Physiology, 287:2803–
12, 2004.
[163] D Sato, Y Shiferaw, A Garfinkel, JN Weiss, Z Qu, and A Karma. Spatially
discordant alternans in cardiac tissue: Role of calcium cycling. Circulation
Research, 99:520–7, 2004.
[164] A Garfinkel, YH Kim, O Voroshilovsky, Z Qu, JR Kil, MH Lee, S Karagueuzian,
JN Weiss, and PS Chen. Preventing ventricular fibrillation by flattening
cardiac restitution. Proceedings of the National Academy of Sciences USA,
97(11):6061–6, 2000.
[165] M Morad and G Salama. Optical probes of membrane potential in heart muscle.
Journal of Physiology, 292, 1979.
259
[166] A Grinvald, L Anglister, JA Freeman, R Hildesheim, and A Manker. Realtime optical imaging of naturally evoked electrical activity in intact frog brain.
Nature, 308(5962):848–850, 1984.
[167] G Salama. Optical measurements of transmembrane potential in the heart.
In L. M. Loew, editor, Spectroscopic Membrane Probes, pages 137–199. CRC
Press, 1988.
[168] JY Wu, YW Lam, CX Falk, LB Cohen, J Fang, L Loew, JC Prechtl, D Kleinfeld, and Y Tsau. Voltage-sensitive dyes for monitoring multineuronal activity
in the intact central nervous system. Journal of Histochemistry, 30(3):169–187,
1998.
[169] JL Byars, WM Smith, RE Ideker, and VG Fast. Development of an optrode
for intramural multisite optical recordings of v-m in the heart. Journal Cardiovascular Electrophysiology, 14(11):1196–1202, 2003.
[170] EF Schubert. Light-Emitting Diodes. Cambridge University Press, Cambridge,
UK, 2003.
[171] RY Tsien and AT Harootunian. Practical design criteria for a dynamic ratio
imaging system. Cell Calcium, 11(2-3):93–109, 1990.
[172] W Kong, GP Walcott, WM Smith, PL Johnson, and SB Knisley. Emission
radiation for simultaneous calcium and action potential measurements with
coloaded dyes in rabbit hearts: Reduction of motion and drift. Journal of
Cardiac Electrophysiology, 14(1):76–82, 2003.
[173] VY Sidorov, RR Aliev, MC Woods, F Baudenbacher, P Baudenbacher, and
JP Wikswo Jr. Spatiotemporal dynamics of damped propagation in excitable
cardiac tissue. Physical Review Letters, 91(20):208104–1–208104–4, 2003.
[174] VY Sidorov, MC Woods, and JP Wikswo Jr. Effects of elevated extracellular
potassium on the stimulation mechanism of diastolic cardiac tissue. Biophysical
Journal, 84(5):3470–9, 2003.
[175] S-F Lin, BJ Roth, and JP Wikswo Jr. Quatrefoil reentry in myocardium: An
optical imaging study of the induction mechanism. Journal of Cardiovascular
Electrophysiology, 10(4):574–86, 1999.
[176] RW Boyd. Radiometry and the Detection of Optical Radiation. John Wiley &
Sons, New York, 1983.
260
[177] M Maolinbay, Y El-Mohri, LE Antonuk, K-W Jee, S Nassif, X Rong, and
Q Zhao. Additive noise properties of active matrix flat-panel imagers. Medical
Physics, 27(8):1841–1854, 2000.
[178] PR Bevington and DK Robinson. Data Reduction and Error Analysis for the
Physical Sciences. McGraw-Hill, Boston, 1992.
[179] E Schroeber. Elimination of granulation in laser beam projections by means of
moving diffusers. Optics Communications, 3:68–72, 1971.
[180] E Hecht. Optics. Addison-Wesley, San Francisco, 4 edition, 2002.
[181] WN Ross and LF Reichardt. Species-specific effects on the optical signals of
voltage-sensitive dyes. Journal of Membrane Biology, 48(4):343–356, 1979.
[182] RJ Myerburg, JW Stewart, SM Ross, and BF Hoffman. On-line measurement
of duration of cardiac action potentials and refractory periods. Journal of
Applied Physiology, 28(1):92–3, 1970.
[183] MB Waxman, ND Berman, E Downer, and P Mendler. A method for the online automatic beat-to-beat digital display of cardiac action potential duration.
Medical and Biological Engineering, 14(2):136–40, 1976.
[184] DJ Sheridan, AJ Bell, WJ Penny, and W Culling. A microcomputer system for
real-time analysis of cardiac action potentials. Journal of Medical Engineering
and Technology, 7(5):238–42, 1983.
[185] PA Guse and JS Burrow. Measurement of transmembrane cardiac potentials
using a real-time microcomputer analysis system. Biomedical Sciences Instrumentation, 23:107–110, 1987.
[186] E Soto, E Saleda, R Cruz, A Ortega, and R Vega. Microcomputer program for
automated action potential waveform analysis. Computer Methods and Programs in Biomedicine, 62(2):141–144, 2000.
[187] AN Iyer and RA Gray. An experimentalist’s approach to accurate localization of phase singularities during reentry. Annals of Biomedical Engineering,
29(1):47–59, 2001.
[188] TK Knilans, A Varro, PP Nanasi, RJ Murray, FC Kaiser, and DA Lathrop. Electrophysiological effects of fpl-13210 on canine purkinje-fiber actionpotential duration and vmax conparison to disopyramide. Cardiovascular
Drugs and Therapy, 5(1):139–146, 1991.
261
[189] MR Franz, PF Kirchhof, CL Fabritz, and M Zabel. Computer-analysis of
monophasic action-potentials - manual validation and clinically pertinent applications. PACE, 18(9):1666–78, 1995.
[190] DR Dickenson, DR Davis, and GN Beatch. Development and evaluation of a
fully automated monophasic action potential analysis program. Medical and
Biological Engineering and Computing, 35(6):653–60, 1997.
[191] DS Coulshed, A Rudenski, JC Cowan, SJ Coulshed, and R Hainswon. The
use of a microcomputer to automate measurement of action potential duration for both transmembrane and monophasic action potentials. Physiological
Measurement, 14(3):347–358, 1993.
[192] CTJ Alkemade, W Snelleman, GD Boutilier, BD Pollard, JD Winefordner,
TL Chester, and N Omenetto. Review and tutorial discussion of noise and
signal-to-noise ratios in analytical spectrometry: Fundamental principles of
signal-to-noise ratios. Spectrochimica Acta B, 33:383–99, 1978.
[193] EB Dagum and A Luati. Relationship between local and global nonparametric
estimators measures of fitting and smoothing. Studies In Nonlinear Dynamics
And Econometrics, 8(2):17, 2004.
[194] FG Evans, RE Ideker, and RA Gray. Effect of shock-induced changes in transmembrane potential and reentrant waves on outcome during cardioversion of
isolated rabbit hearts. Journal of Cardiovascular Engineering, 13(11):1118–27,
2002.
[195] D Sung, J Somayajula-Jagai, P Cosman, R Mills, and AD McCulloch. Phase
shifting prior to spatial filtering enhances optical recordings of cardiac action
potential propagation. Annals of Biomedical Engineering, 29(10):854–61, 2001.
[196] SM Dillon. Optical recordings in the rabbit heart show that defibrillaiton
strength shocks prolong the duration of depolarization and the refractory period. Circulation Research, 69(3):842–856, 1991.
[197] RL Allen and DW Mills. Signal Analysis: Time, Frequency, Scale and Structure. John Wiley & Sons, New York, 2004.
[198] YH Lee and SA Kassam. Some generalizations of median filters. In Proceedings
of the International Conference on Acoustics, Speech, and Signal Processing,
pages 411–414, 1981.
262
[199] NC Gallagher and GL Wise. A theoretical analysis of the properties of median filters. IEEE Transactions on Acoustics, Speech, and Signal Processing,
29(6):1136–41, 1981.
[200] E Ataman, VK Aatre, and KM Wong. Statistical properties of median filters.
IEEE Transactions on Acoustics, Speech, and Signal Processing, 29(5):1073–75,
1981.
[201] HS Karagueuzian, SS Khan, WJ Mandel, K Hong, and GA Diamond. Nonlinear
dynamic analysis of temporally heterogeneous action-potential characteristics.
PACE, 13(12):2113–18, 1990.
[202] M Novakova, J Bardonova, I Provaznik, E Taborska, H Bochorakova, H Pavlova,
and D Horsky. Effects of voltage sensitive dye di-4-anepps on guinea pig and
rabbit myocardium. General Physiology and Biophysics, 27(1):45–54, 2008.
[203] M Biermann, M Rubart, A Moreno, JS Wu, A Josiah-Durant, and DP Zipes.
Differential effects of cytochalasin d and 2,3 butanedione monoxime on isometric
twitch force and transmembrane action potential in isolated ventricular muscle:
Implications for optical measurements of cardiac repolarization. Journal of
Cardiovascular Electrophysiology, 9(12):1348–57, 1998.
[204] B Kettlewell, NL Walker, SM Cobbe, FL Burton, and GL Smith. The
electrophysiological and mechanical effects of 2,3-butane-dione monoxime and
cytochalasin-d in the langendorff perfused rabbit heart. Experimental Physiology, 89(2):163–72, 2004.
263
Biography
Hana Maria Dobrovolny was born in Winnipeg, Manitoba, Canada on September 12,
1976. Her family lived on a small farm outside the city before settling in Winnipeg
where Hana attended Balmoral Hall School. Upon graduation in 1993, Hana began
her study of physics at the University of Winnipeg. She received a B.Sc. in physics
and mathematics in 1997. She continued her studies at Bryn Mawr College in Bryn
Mawr, Pennsylvania, receiving an M.A. in physics in 2000, after which she began her
Ph.D studies at Duke University.
Honors and Awards
Riverbend Scholarship, Balmoral Hall School
Canada Scholarship, Natural Sciences and Engineering Research Council, held at
University of Winnipeg
Special Entrance Scholarship, Henry Doidge Memorial Scholarship in Physics, Academic Proficiency Scholarship, S.K. Sen Scholarship in Physics, B.G. Hogg Memorial
Scholarship in Physics, Lawson Scholarship in Mathematics, Duckworth Scholarship
University of Winnipeg
University Gold Medal in Physics, University of Winnipeg
NSERC Postgraduate Scholarship, Natural Sciences and Engineering Research Council, held at Bryn Mawr College
Charles H. Townes Fellowship, Jenkins Family Graduate Fellowship, Duke University
264
Publications
H. M. Dobrovolny, N. H. Brown, C. M. Berger, S. F. Idriss, D. G. Schaeffer, W.
Krassowska, D. J. Gauthier, Spatial heterogeneity of restitution properties in homogeneous cardiac tissue, in preparation
C.M. Berger, X. Zhao, D.G. Schaeffer, W. Krassowska, H.M. Dobrovolny, and D.J.
Gauthier, ’Evidence for an unfolded border-collision bifurcation in paced cardiac
tissue,’ Phys Rev Lett 99, 058101 (2007)
N. H. Brown, H. M. Dobrovolny, P. D. Wolf, D. J. Gauthier,A Fiber-Based Ratiometric Optical Cardiac Mapping Channel using a Diffraction Grating and Split Detector,
Biophys J. 93, 254 (2007)
H. M. Dobrovolny, H. Elmariah, S. S. Kalb, J. P. Wikswo, Jr., D. J. Gauthier,
Imaging cardiac dynamics using low-cost ultra-high-power light emitting diodes and
voltage-sensitive dyes, arXiv:physics/0702241
S.S. Kalb, H. Dobrovolny, E. Tolkacheva, S.F. Idriss, W. Krassowska, and D.J. Gauthier, ‘The resitution portrait: A new method for investigating rate-dependent restitution,’ J Cardiovasc Electrophys 15, 698 (2004)
C.J. Cellucci, P.D. Brodfuehrer, R. Acera-Pozzi, H. Dobrovolny, E. Engler, J. Los, R.
Thompson, A.M. Albano, ’Linear and nonlinear measures predict swimming in the
leech’, Phys Rev E, 62, 4826 (2000)
H. Dobrovolny, On nonlinear time series analysis, Master’s thesis, Bryn Mawr College,
Bryn Mawr, PA
Presentations
C.M. Berger, X. Zhao, D.G. Schaeffer, H. Dobrovolny, W. Krassowska, D.J. Gauthier, ‘Evidence for an unfolded border-collision bifurcation in paced cardiac tissue,’
Dynamics Days 2007, Boston, MA, Jan. 2-6, 2007
D.J. Gauthier, C.M. Berger, X. Zhao, D.G. Schaeffer, H. Dobrovolny, and W. Krassowska, ‘Discovery of a new type of bifurcation in paced cardiac muscle,’ Third
Wrokshop Promotionskolleg, Helmholtz Center for Brain and Mind Dynamics, Lieben265
walde, Germany, July 14, 2006.
C.M. Berger, H.M. Dobrovolny, X. Zhao, D.G. Schaeffer, W. Krassowska and D.J.
Gauthier, ‘Investigating a Period-Doubling Bifurcation in Cardiac Tissue Using Alternate Pacing,’ Dynamics Days 2006, Bethesda, MD, Jan. 4-7, 2006.
C.M. Berger, H. Dobrovolny, D.G. Schaeffer, W. Krassowska, D.J. Gauthier, ’Evidence for a border-collision bifurcation in paced cardiac tissue,’ Southeastern Section
of the APS, Gainesville, FL, November 10-12, 2005.
H. Dobrovolny, C. Berger, S. Kalb, S. Idriss, D. Schaeffer, W. Krassowska, D. Gauthier,‘Spatial heterogeneity of the restitution portrait correlates with alternans in
paced cardiac tissue,’ Heart Rhythm, New Orleans, LA, May 4-7, 2005. [Heart
Rhythm, 2: S297 (2005)]
C.M. Berger, H.M. Dobrovolny, S.S. Kalb, S.F. Idriss, D.G. Schaeffer, D.J. Gauthier,
W. Krassowska,‘Investigating a Period-Doubling Bifurcation in Cardiac Tissue using
Alternate Pacing,’ American Physical Society, Los Angeles, CA, March 21-25, 2005.
H.M. Dobrovolny, E.G. Tolkacheva, D.J. Gauthier, ‘Spatiotemporal dynamics and
control of alternans in cardiac tissue with short-term memory,’ APS March meeting,
Los Angeles, CA, Mar. 21-25, 2005.
H. Dobrovolny, R. Oliver, S. Kalb, E. Tolkacheva, W. Krassowska, and D. Gauthier,‘Conduction velocity dispersion in cardiac tissue,’ 2004 CAP Congress, Winnipeg, Canada, June 13-16, 2004
Hana Dobrovolny, Robert Oliver, Soma Kalb, Elena Tolkacheva, David Schaeffer,
Wanda Krassowska, Daniel Gauthier, ’Action Potential and Conduction Velocity
Restitution in Cardiac Tissue’, APS March Meeting, Montreal, Canada, March 2126, 2004
H. Dobrovolny, ’Recent Developments in Cardiac Dynamics,’ University of Cambridge, Cambridge, UK, November 26, 2003
H. Dobrovolny, S. Sau, H. Elmariah, D. Gauthier, J. Gilligan, J. Wikswo, ’Use of
LEDs for imaging cardiac tissue,’ Gordon Research Conference on Cardiac Arrhythmia Mechanisms, New London, NH, August 10-15, 2003.
S. Sau, H. Dobrovolny, E. Tolkacheva, D. Schaeffer, W. Krassowska, D. Gauthier,
266
’New Experimental Protocol for Simultaneous Measurement of the S1S2, ConstantBCL and Dynamic Restitution Curves,’ Gordon Research Conference on Cardiac
Arrhythmia Mechanisms, New London, NH, August 10-15, 2003 (Winner of Best
Experimental Poster Award).
H. Dobrovolny, S. Sau, H. Elmariah, D. Gauthier, J. Gilligan, J. Wikswo, ’Use of
LEDs for imaging cardiac tissue,’ 2nd annual Fitzpatrick Center Conference, Durham,
NC, May 27-28, 2003
H. Dobrovolny, S. Sau, E. Tolkacheva, D. Schaeffer, W. Krassowska, D. Gauthier,
’New Experimental Protocol for Simultaneous Measurement of the S1S2, ConstantBCL and Dynamic Restitution Curves,’ NASPE, Washington, DC, May 14-17, 2003.
H. Dobrovolny, S. Sau, E. Tolkacheva, D. Schaeffer, W. Krassowska, D. Gauthier,
’Stability of cardiac response patterns’, Dynamics Days, January, 2003
267