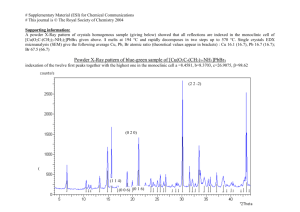
MAY 2014 LIBRARIES 14
advertisement

Design of Novel Lithium Storage Materials with a Polyanionic Framework MASACUS-- E M by j ASOF TECHNOLOGY JAE CHUL KIM MAY 14 2014 B.S., M.S. Materials Science and Engineering Korea University (2005, 2007) LIBRARIES Submitted to the Department of Materials Science and Engineering in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2014 0 2013 Massachusetts Institute of Technology. All rights reserved. Signature of A uthor ............................... ..................................... Department of Materials Science and Engineering December 18, 2013 C ertified by ............................................. Gerbrand Cedei? R. P. Simmons Professor of Materials Science and Engineering Thesis Supervisor Accepted by .......................................... R. P. Simmons Professor of Materials Science and Engineering Chair, Departmental Committee on Graduate Students 1 , Design of Novel Lithium Storage Materials with a Polyanionic Framework by JAE CHUL KIM Submitted to the Department of Materials Science and Engineering on December 18, 2013 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Emerging, Fundamental, and Computational Studies in Materials Abstract Lithium ion batteries for large-scale applications demand a strict safety standard from a cathode material during operating cycles. Lithium manganese borate (LiMnBO3) that crystallizes into a hexagonal or monoclinic framework is one prominent polyanionic compound to cope with such requirement since it can possess high safety and high energy density simultaneously, without trading one for the other, theoretically. However, the hexagonal phase was nothing but a disregarded composition due to its negligible Li intercalation capacity. In contrast, the monoclinic LiMnBO3 compound exhibited much more electrochemical activity than the hexagonal polymorph. In this thesis work, the discharge capacity of 100 mAh g 1 with acceptable capacity retention was achieved by simple optimization. The different electrochemical behaviors between them were understood in relation to their structural difference as it affects the Li migration barrier, structural stability of Li-deficient states, and even particle morphology. However, although promising, monoclinic LiMnBO3 needed further improvement in terms of the achievable capacity and cyclability. Electrochemical analysis showed that the limited capacity of LiMnBO3 mostly originated from transport limitation, a hindered Li migration through the one-dimensional diffusion channel. It also struggled from the phase decomposition and Mn dissolution due to the instability of the delithiated state, which led to gradual capacity fading in prolonged cycles. As an effective materials design strategy to overcome such limitations, systematic substitution of transition metal elements was proposed. To increase achievable capacity, Mn was partially substituted by Fe. Also, to fortify the structural integrity, Mg replaced Mn. In order to obtain both improved capacity and cyclability, Fe and Mg are co-doping led to an optimized composition. Prepared by cold-isostatic pressing, LiMgo.1Mno.5Feo.4BO3 showed near theoretical capacity of 200 mAh g-1 with much improved capacity retention. These newly established materials outperformed most of the polyanionic cathode compounds. Therefore, it can be concluded a new promising candidate as a Li storage material has been developed through this thesis research. Thesis Supervisor: Gerbrand Ceder Title: R.P. Simmons Professor of Materials Science and Engineering 3 Acknowledgement First and foremost, I would like to thank my thesis advisor, Professor Gerbrand Ceder, for his thoughtful guidance and ample support to complete this work. Through his profound scientific knowledge and insights, Prof. Ceder has enlightened and broadened my perspective for materials science and engineering and the electrochemical system. His passion and vision for the emerging energy storage materials made my research more fruitful and mature. I am also grateful to my thesis committee members, Professors Donald R. Sadoway and Jeffrey C. Grossman for sharing their invaluable time and expertise. Their extraordinary comprehensions in materials chemistry and quantum physics were of great value to my research. I must thank Professor Sahn Nahm, my M.S. advisor, for his constant encouragement and mentorship, which have formed the firm basis of my Ph.D. study. I express my very special thanks to ShinYoung Kang who has shared all the joys and sorrows in my MIT life. In my long and rough journey, I was able to tough it out because of her warm encouraging words and gestures. She has been my best friend, colleague, and mentor to whom I can really open up myself and ask for advices. I sincerely thank Prof. Jae Kyung Kim, Jaehoon Lee, Prof. Hyowon Gweon, and Yale Song for their mental communion with me in my graduate school years. Good memories we have shared shall never be forgotten. It was a pleasure to interact with Jinhyuk Lee and Dr. Dong-Hwa Seo in the same group. Their enthusiasm and creativity were an invaluable inspiration to me. I am indebted to Charles J. Moore and Dr. Xin Li, who have always been great devotees to my research. It was more than an exciting experience to collaborate closely with them. I am also lucky to work with the experimental team in Ceder group. Dr. Plousia Vassilaras, Nancy Twu, Dr. Lei Liu, Ian Matts, Di Wu, and Yuechuan Lei have been helpful and a great asset to me at all time. I thank all other Ceder group members and alumni, as well. Prof. Byoungwoo Kang, Prof. Byungchan Han, Prof. Kisuk Kang, Dr. Xiaohua Ma, Prof. Yifei Mo, Dr. Yabi Wu, Dr. Rahul Malik, Dr. Ruoshi Sun, Prof. Hailong Chen, Prof. Shyue Ping Ong, Dr. Geoffroy Hautier, Dr. Anubhav Jain, Dr. Kristin Persson, Dr. Pieremanuele Canepa, Dr. Sai Jayaraman, Dr. Alexander Urban, Dr. Eric Wang, Dr. Bo Xu, Dr. Hong Zhu, Aziz Abdellahi, Stephen Dacek, Wenxuan Huang, Daniil Kitchaev, William Richards, Ziqin Rong, Wenhao Sun, Alexandra Toumar, Lusann Wren Yang, and Kathryn E. Simons have provided an intellectually stimulating research environment and administrative support to me. I must express my gratitude to my Korean community in DMSE, especially for Heechul Park, Dr. Hyeongho Shin, Dr. Hyunjung Yi, Jaebyum Chang, Dr. Eunseon Cho, Dahyun Oh, Jae Jin Kim, Jeongyun Kim, Mansoo Park, Alan Gyehyun Kim, Donghun Kim, Jiyoun Christina Chang, Sangtae Kim, Sangjin Lee, Kyoung-Won Park, Intak Jeon, Hyoungwon Park, Dr. Sehoon Chang, Dr. Dong Hun Kim, Prof. Hojong Kim, and Dr. Jae Hyung Yi, for making my life at MIT more involved and livable. 5 I must praise my beloved vehicle, the ultimate driving machine, for giving me a shear pleasure that sets me free from the stress and struggle. It has been more than just transportation to me as I have found my peace of mind while driving it. I cannot thank my parents more than enough for their endless and unconditional love and unlimited support, which I shall never forget as long as I live. Without their encouragement, this work would never be accomplished. I also thank my sister, Jay Young for her devotion and dedication to our family. My gratitude goes to parents-inlaw and sister-in-law for their bottomless care, as well. Most of all, I give my sincerest thanks to my wife, Kiyoung Yoo. I was able to depend on her encouragement all the time when I was in doubt. She always raised me up when I was down and let me walk at a steady pace when I was too hasty. Her patience, understanding, and devotion were beyond all description and essential to make this work possible. 6 Table of Contents List of Figure Captions 11 List of Table Captions 20 PART ONE 23 CHAPTER 1. INTRODUCTION AND MOTIVATION, OVERVIEW OF THE THESIS 25 1.1. 27 Introduction and motivation 27 1.1.1. Li-ion battery as a mobile energy source 1.1.2. An increasing demand of high performance Li-ion batteries and motivation to find a new cathode material 1.2. 30 Overview of the thesis CHAPTER 2. LITHIUM ION BATTERY GENERALS 2.1. 28 33 Li-ion battery generals: fundamental thermodynamics of an electrochemical system 36 2.1.1. Voltages 36 2.1.2. Capacities 39 2.1.3. Cyclability 40 2.1.4. Rate capability 40 2.1.5. Safety 41 2.2. Commercially available Li intercalation compounds 42 2.2.1. Layered oxides 42 2.2.2. Spinel oxides 43 2.2.3. Phospho-olivines 45 2.3. Characteristic features of polyanionic cathode materials 2.3.1. 47 47 Stability and safety 7 2.3.2. Inductive effect 49 2.3.3. Electronic conductivity 51 2.3.4. Gravimetric / volumetric energy density 51 2.3.5. Packing density / processing cost 52 2.3.6. Summary 53 PART TWO 55 CHAPTER 3. DESIGNING A NEW CATHODE MATERIAL 3.1. Lithium manganese borate compounds (LiMnBO 3) 57 60 3.1.1. Motivation 60 3.1.2. Literature review 63 3.1.2.1. Structural framework of lithium (transition) metal borates 63 3.1.2.2. The monoclinic and hexagonal polymorphs of LiMnBO 3 64 3.1.2.3. The electrochemical properties of lithium transition metal borates 3.1.2.4. 68 High-throughput computation driven material screening and filtering to discover LiMnBO3 as a new Li intercalation cathode 71 3.2. Results 3.2.1. 75 Experimental procedure 75 3.2.1.1. Solid-state synthesis 75 3.2.1.2. Structure and morphology analysis 77 3.2.1.3. Electrochemical performance 78 3.2.2. Temperature-dependent phase formation of LiMnBO3 79 3.2.2.1. In-/ex-situ X-ray diffraction patterns of Li-Mn-B-O mixture 79 3.2.2.2. Monoclinic LiMnBO 3 82 3.2.2.3. Hexagonal LiMnBO3 84 3.2.3. Preliminary electrochemical properties of LiMnBO3 polymorphs 8 86 3.2.3.1. Monoclinic LiMnBO 3 86 3.2.3.2. Hexagonal LiMnBO3 87 3.2.3.3. Optimizing the electrochemical properties of LiMnBO 3 poly88 morphs by planetary ball-milling and carbon coating 3.3. 96 Discussion 96 3.3.1. Diffusion behaviors of monoclinic and hexagonal LiMnBO3 3.3.2. Delithiated state stabilities of monoclinic and hexagonal LiMnBO3 99 3.4. 100 Particle size and polarization 3.3.3. 101 Conclusion CHAPTER 4. UNDERSTANDING PERFORMANCE-LIMITING FACTORS OF MONOCLINIC LITHIUM MANGANESE BORATE 103 4.1. Factors influencing the electrochemical performance of LiMnBO3 105 4.2. The limited specific capacity achievable 106 4.2.1. 4.2.1.1. Temperature and rate dependence of Li activity 106 4.2.1.2. Galvanostatic intermittent titration test (GITT) 109 4.2.1.3. Potentiostatic intermittent titration test (PITT) 116 4.2.1.4. Diffusivity of LiMnBO 3 118 4.2.1.5. Channel blocking antisite defects 122 4.2.2. 4.3. 106 Transport limitation 125 Discussion 129 Capacity fading upon cycling Limitation from structural instability 129 4.3.1.1. Computed formation enthalpy 130 4.3.1.2. Electrochemical charging 132 4.3.1.3. Chemical delithiation 135 4.3.1. 4.3.2. 138 Discussion 4.3.2.1. Decomposition of delithiated states 9 138 4.3.2.2. 4.4. Mn dissolution 140 Conclusion 142 PARTTHREE 143 CHAPTER 5. DESIGNING HIGH CAPACITY POLYANIONIC CATHODES 145 5.1. 147 Substitution strategy 5.1.1. Motivation 147 5.1.2. The substitution criteria 149 5.1.3. Inactive element substitution 151 5.1.3.1. LiMgyMnl-yB03 (0 y 0.2) 153 5.1.3.2. LiZnyMn1yBO3 (0 y ! 0.2) 157 5.1.4. 5.2. Active element substitution 159 z ! 0.35) 5.1.4.1. LiMni-zFezBO 3 (0 5.1.4.2. LiMnj-zMzBO 3 (M =Co, Ni, and Cu, 0 ! z 160 0.35) Inactive and active element co-doping: LiMgo.lMnl-zFezBO3 (0.3 5 z 165 0.4) 170 5.2.1. 5.3. Optimizing composition 170 Cycling performance improvement by processing control 175 5.3.1. Motivation 175 5.3.2. Cold-isostatic pressing (CIP) 177 5.3.3. Improved cyclability of LiMnBO 3 by CIP 179 5.3.4. Improved cyclability of LiMgo.lMno.5Feo.4BO3 by CIP 183 5.4. 189 Conclusion CHAPTER 6. CONCLUDING THESIS STATEMENTS 191 195 References 10 List of Figure Captions Figure 1-1. Energy densities of various kinds of commercial rechargea- 28 ble batteries Figure 1-2. Examples of the industrial applications that adopt Li-ion 29 batteries as a power source. Figure 2.1. Schematic (a) configuration of conventional Li-ion battery 34 cell and (b) its voltage/energy diagram during operation. The electrical work deliverable is defined by Equation 2-1. In the ideal case, V 2 oc-Vioc = V is the maximum voltage dif- ference available. Figure 2-2. Schematic illustration of a layered oxide with 03 stacking. Figure 2-3. Schematic 42 oxide, 44 Schematic diagram of a representative phosphor-olivine, 45 diagram of a representative spinel LiMn204. Figure 2-4. LiFePO4. Figure 2-5. Schematic diagrams of the electron energy versus density of states of 3d-band in transition metal (Mn+/M(n+l)+) 48 and 2p- band in oxygen (02-) (a) with overlapping and (b) without overlapping. Figure 2-6. (a) The energy diagram of M-0 bonds, (b) various Fe-O pol- 50 yhedra with different coordination numbers, and (c) energy level of Fe 2+/Fe3+ redox elements with respect to metallic lithium in the NASICON structure with different M-0-X bonds Figure 3-1. Schematic diagrams of LiMnBO 3 polymorphic structures (Yellow; Li, Green; Mn, White; B, Shade; 0 polyhedra). The space group for each phase is (a) C2/c(monoclinic, #15) and (b) P-6(hexagonal, #174), respectively. 11 65 Figure 3-2. Schematic diagrams of (a) Idealized 2D layer of LiFeBO3, (b) 66 LiFeBO3 structure in the literatures including anisotropic displacement for 0 atoms, and (c) LiFeBO 3 fragment from the modulated superstructure. Figure 3-3. Representative electrochemical properties of lithium transi- 69 tion metal borates in the literature: (a) Hexagonal LiMnBO3 displays large irreversibility at the initial cycle and only intercalates 0.02 Li per formula unit (4 mAh g-1). 4 2 (b) Hexag- onal LiMnBO3 shows 75.5 mAh g-1, but it is most likely to originate from a conversion reaction below 1.5 V.58 (c) LiFeB0 3 outperforms other borates in terms of a capacity. 43 (d) LiCoBO3 shows a negligible capacity. Figure 3-4. Calculated formation enthalpies of (a) monoclinic and (b) 73 hexagonal LiMnBO 3, and (c) their corresponding voltage profiles with respect to Li contents. Figure 3-5. The flowchart of experimental procedure for LiMnBO 3 syn- 76 thesis. Figure 3-6. In-situ XRD patterns of pelletized Li-Mn-B-0 with respect 80 to temperature change. Figure 3-7. XRD patterns of Li-Mn-B-0 mixture calcined at 623 K and 81 followed by sintering at (a) 773 K and (b) 1073 K. Figure 3-8. Rietveld refinement and profile matching of monoclinic 82 LiMnBO 3 sintered at 773 K, whose space group is C2/c (ICSD# 200535). The calculated pattern matches well with the observed one. Figure 3-9. SEM image of monoclinic LiMnBO3 sintered at 773 K. 83 Figure 3-10. Rietveld refinement and profile matching of hexagonal 84 LiMnBO 3 sintered at 1073 K, whose space group is P-6 (ICSD# 94318). 12 Figure 3-3. SEM image of hexagonal LiMnBO3 sintered at 1023 K. 85 Figure 3-4. Voltage versus capacity profiles of monoclinic LiMnBO3 cy- 86 cled at a 0.05 C rate. Figure 3-5. Voltage versus capacity profiles of hexagonal LiMnBO 3 cy- 88 cled at a 0.05 C rate. Figure 3-6. SEM image of monoclinic LiMnBO3 and carbon black mixed 89 by planetary ball-milling. Figure 3-7. Voltage versus capacity profiles of monoclinic LiMnB03 90 cathode prepared by planetary ball-milling (solid) and its comparison to the manually mixed one (dashed). Both were cycled at a 0.05 C rate. Figure 3-8. SEM image of carbon coated monoclinic LiMnBO3 prepared 91 by planetary ballmilling after annealing. Figure 3-9. HR-TEM image of carbon-coated monoclinic LiMnBO3 par- 92 ticles. Figure 3-10. Voltage versus capacity profiles of carbon-coated (a) mono- 93 clinic and (b) hexagonal LiMnBO 3 (solid lines) cycled at a 0.05 C rate. Figure 3-19. Ex-situ XRD patterns of the cycled monoclinic LiMnBO3 94 cathode films which are collected (a) before cycling (discharged), (b) after the first charge, (c) after the first discharge, and (d) after the 10th charge. To obtain charged and discharged state, the electrodes were charged and discharged in CCCV mode within 4.5-2.0 V with a 0.05 C rate. Figure 3-11. Schematic diagrams of Li diffusion path and activation bar- 97 riers of Li diffusion in delithiated states of (a) monoclinic and (b) hexagonal LiMnBO 3. Figure 4-1. Voltage vs. capacity profiles of LiMnBO3 in various conditions: (a) pristine and (b) carbon-coated LiMnBO3 at a 0.05 13 107 C rate with respect to temperature, and (c) carbon-coated LiMnBO 3 at room temperature (RT) with respect to a rate. Figure 4-2. Schematic diagrams of Li concentration profile in LiMnBO3 111 cathode with respect to Li extraction and corresponding voltage profiles: (a-e) ideal and (f-j) real cases. Figure 4-3. (a) Schematic diagram of cell configuration used in GITT 112 and (b) applied galvanostatic current pulse and its response in voltage with respect to time. Figure 4-4. Voltage profile with respect to x in Lil-xMnBO 3/C and 114 elapsed time according to SOC: (a) 15%, (b) 25%, and (c) 45% obtained by GITT. Figure 4-5. Estimated sequential non-equilibrating Li concentration 115 profile of LiMnBO cathode during charging by GITT. Figure 4-6. Schematic plots of one step of a potentiostatic input and its 116 corresponding current response in PITT. Figure 4-7. Linear fittings of current with respect to time to derive dif- 119 fusivity. Figure 4-8. Incremental charge with respect to voltage obtained by 120 PITT at RT with interval of 10 mV. Data points represent the value at every 50 mV. Figure 4-9. Li chemical diffusivities obtained by PITT: the values were 121 collected with a 10 mV voltage step from 2-4 V charging and 4.5-2.5 V discharging but plotted with 50 mV intervals for clarity. The diffusivity displays three distinctive regions in charge (I, II, and III) and discharge (IV, V, and VI). Figure 4-10. Profile matching of the XRD pattern of monoclinic LiMnBO 3 (top) with and (bottom) without antisite defects (Mn in Li sites) whose agreement indexes are Rp=7.66, RWP=9.84 and Rp=5.86, Rwp=8.24, respectively. 14 123 Figure 4-11. Particle size distribution and its correlation with the antis- 124 ites. (a) SEM image of LiMnBO 3/C particles (scale bar: 500 nm) and (b) Representative particle size distribution and volume fraction. Figure 4-12. Schematic illustration of one-dimensional diffusion path 126 parallel to [001] direction in monoclinic LiMnBO3 assuming 4.47% antisites in the channel. Figure 4-13. Integrated incremental charge (dQ dV- 1) with respect to 127 voltage (V). The voltage profile as a function of specific capacity acquired by integrating the incremental charges over voltage from PITT. Figure 4-14. Li-Mn-B-O quaternary phase diagram. 131 Figure 4-15. (a) Voltage vs. capacity curves of LiMnBO3/C in the second 132 cycle at 0.01 C and 0.05 C, RT, (b) their capacity retention for 10 cycles with photographs of the anode after the designated discharge cycles, and (c) ex-situ XRD patterns of charged Lil-xMnBO3/C electrodes with different obtained capacity. Figure 4-16. (a) XRD patterns, (b) HR-TEM images (scale bar: 5 nm), and 135 (c) 7Li MAS NMR spectra of pristine and chemically delithiated Lil-xMnBO3 respectively. Figure 4-17. Schematic diagram of c-lattice parameter change upon deli- 136 thiation. Figure 4-18. XRD pattern and HR-TEM image (scale bar: 5 nm) of chem- 136 ically delithiated Lil-xMnBO3 for 10 days. The inset is electron diffraction pattern of the specimen showing a typical diffused amorphous ring pattern. Figure 4-19. Profile matching of the XRD pattern of chemically delithiated MnBO3. 15 138 Figure 4-20. Graphical representation of Li grand-potential phase dia- 139 gram with competing phases of LiixMnBO 3 at pLi=4.64 eV. Red dots stand for stable phases. LiixMnBO 3 is marked with the blue cross, which is unstable with respect to Li 4B 2 0 7 , Mn 2BO 4 , and Mn 30 4 . Figure 4-21. EDS of the designated spot on the Li anode in Figure 4- 140 11(b) verifies that the identity of the stain is Mn deposited from the cathode during cycling, excluding P and F from the soaked electrolyte salt and C and 0 from the equipment. Figure 5-1. Computed average voltage in phosphates versus maximum 147 gravimetric capacity achievable. Specific energy curves at 600 and 800 Wh/kg are drawn on the figure (blue dashed lines). The red dashed line indicates the upper voltage which we consider safe against decomposition of the electrolyte. Figure 5-2. Schematic diagram of edge-sharing trigonal bipyramidal 150 Mn coordinated by five 0. Mn sits off-centered from the trigonal bipyramidal sites and tends to occupy either the upper or lower tetragonal-like site. Figure 5-3. Graphical representation of computed Mn-B-0 2 ternary 152 phase diagram. Red dots stand for stable phases. MnBO 3 is marked with the blue cross, which is unstable with respect to MnO 2 , Mn2O 3, and MnB4 0 7 . Figure 5-4. (a) XRD patterns of LiMgyMnl-yBO 3 (0 5 y 5 0.2) fired at 154 773-823 K and (b) their refined lattice parameters. Figure 5-5. Five consecutive charge and discharge curves for LiMgyMni. yBO 3 (0 5 y < 0.2): (a) y=0.05, (b) y=0.1, and (c) y=0.2 cycled at a 0.05 C rate. For comparison, the representative second cycles of each y are plotted in (d). The dotted line represents 16 155 the undoped LiMnBO3. Figure 5-6. (a) STEM image and (b) the EELS line scan of designated 156 location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMgo. 2Mno.8BO3/C particle. Figure 5-7. (a) STEM image and (b) the EELS line scan of designated 156 location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMnBO 3/C particle. Figure 5-8. XRD patterns of LiZnyMnlyBO3 (0 < y < 0.2) fired at 157 773-823 K Figure 5-9. Five consecutive charge/discharge curves of LiZnyMnl-yBO3 158 (0 s y < 0.2): (a) y=0.05 and (b) y=0.1 cycled at a 0.05 C rate. The dotted line represents the undoped LiMnBO3. Figure 5-10. (a) XRD patterns of LiMni-zFezBO3 (0 < z 5 0.35) fired at 160 773-823 K and (b) their refined lattice parameters. Figure 5-11. Five consecutive charge/discharge curves for LiMni-zFezBO3 161 (0 < z < 0.35): (a) z=0.05, (b) z=0.1, (c) z=0.2, (d) and z=0.35 cycled at a 0.05 C rate. For comparison, the representative second cycles of each z are plotted in (e). The dotted line represents the undoped LiMnBO3. (f) The first three voltage-capacity profiles of LiMno.65 Feo.35BO3 initiated by discharging (red) and their comparison to those of (d). Figure 5-12. (a) STEM image and (b) the EELS line scan of designated 164 location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMno.8 Feo. 2 BO3/C particle. Figure 5-13. XRD patterns of LiMni-zMzBO3 (0 < z 5 0.35) fired at 166 773-823 K: (a) M = Co, (b) M = Ni, and (c) M = Cu. Figure 5-14. Refined lattice parameters of LiMnl-zMzBO3 (0 5 z < 0.35) 167 fired at 773-823 K: (a) M = Co, (b) M = Ni, and (c) M = Cu. Figure 5-15. The representative second cycles of LiMni-zMzBO3 (0 5 17 Z < 168 0.35) for (a) M = Co, (b) M = Ni, and (c) M = Cu. Note that LiMno.6 5 Cuo. 3 5 BO 3 was not able to be tested due to repeated cell failure after the first charging. Figure 5-16. (a) XRD patterns of LiMgo.1Mno. 9 -zFezBO3 (0.3:5 z 5 0.4) fired 171 at 823 K and (b) their refined lattice parameters. Figure 5-17. Five consecutive charge/discharge curves for LiMgo.1Mno.9. 172 zFezBO 3 (0.3 < z _ 0.4): (a) z = 0.3, (b) z = 0.35, and (c) z = 0.4 cycled at a 0.05 C rate. For comparison, the representative second cycles of each z are plotted in (e). Figure 5-18. Simplified schematic diagrams of pressure application dur- 177 ing (a) uniaxial pressing and (b) CIP. Figure 5-19. Optimal change in particle size distribution to minimize 177 side reaction by removing very small particles Figure 5-20. Measurement of pellet diameter after firing at 773 K for 10 179 h prepared by (a) uniaxial pressing and (b) CIP. Figure 5-21. Rietveld refinement and profile matching of LiMnBO 3 pre- 180 pared by (a) uniaxial pressing and (b) CIP, which are fired at 773 K and annealed at 773 K for carbon coating. Figure 5-22. SEM images of carbon-coated LiMnBO 3 prepared by (a) 181 uniaxial pressing and (b) CIP in different magnifications, and (c) particle size distribution comparison between them. Figure 5-23. (a) Five consecutive charge and discharge curves for carbon- 182 coated LiMnBO 3 at a 0.05 C rate prepared by CIP and (b) capacity retention during 10 cycles. For reference, LiMnBO 3 without CIP is also plotted (open triangle). Figure 5-24. Rietveld refinement and profile matching of LiMgo.1Mno.5 Feo. 4BO 3 prepared by (a) uniaxial pressing and (b) CIP, which are fired at 823 K and annealed at 798 K for carbon coating. 18 184 Figure 5-25. SEM images of carbon-coated LiMgo.iMno.5Feo. 4 BO 3 pre- 185 pared by (a) uniaxial pressing and (b) CIP in different magnifications, and (c) particle size distribution comparison between them. Figure 5-26. (a) Five consecutive charge and discharge curves for carbon- 186 coated LiMgo.lMno. 5 Feo.4BO3 at a 0.05 C rate prepared by CIP and (b) capacity retention during 10 cycles. For reference, LiMnBO 3 without CIP is also plotted (open circle). Figure 5-27. Rate capability comparison at discharge between (a) 188 LiMnBO 3 and (b) LiMgo.1Mno. 5 Feo.4BO3 prepared by CIP. Each curve was charged at a respective 0.05 C rate prior to discharging. Figure 5-28. Schematic diagram of one-dimensional Li diffusion and lattice planes surrounding the path. 19 188 List of Table Captions Table 3-1. Structural parameters of lithium metal borates 63 Table 3-2. Computed (theoretical) properties of the LiMnBO3 poly- 71 morphs Table 3-3. Rietveld refinement result of monoclinic LiMnBO 3 83 Table 3-4. Site occupancy of monoclinic LiMnBO 3 83 Table 3-5. Rietveld refinement result of hexagonal LiMnBO 3 85 Table 3-6. Site occupancy of hexagonal LiMnBO 3 85 Table 4-1. Computed stability of LiixMnBO 3 (0 _ x < 1) and its ground 130 states with respect to Li concentration at zero K. AE (meV/atom) stands for difference in energy from the computed ground states. Table 4-2. Computed stability of LiixFeBO 3 (0 < x 5 1) and its ground states with respect to Li concentration 134 at zero K. AE (meV/atom) stands for difference in energy from the computed ground states. Table 4-3. Atomic ratio of m-LiMnBO 3 specimen before and after 141 chemical delithiation by inductive coupled plasma (ICP, ASTM E 1097-12) and inert gas fusion (ASTM E 1019-11). The excess amount of Li and B may be due to the LiBF 4 residue Table 5-1. Ionic radius (A) of possible substituents for Mn in LiMnBO 3 151 Table 5-2. Achieved electrochemical performance in LiMni.zFezBO 3 163 cathode Table 5-3. Computed properties of lithium metal borates 169 Table 5-4. Electrochemical performance of LiMgo.iMno.9 -zFezBO 3 (0.3 5 173 z < 0.4) cathodes 20 Table 5-5. Rietveld refined lattice constants and derived parameters 180 of LiMnBO3 prepared by cold-isostatic pressing and fired at 773 K. Table 5-6. Rietveld refined lattice constants and derived parameters of LiMgo.lMno.5Feo.4BO3 prepared by cold-isostatic pressing and fired at 823 K. 21 180 [This page is intended to be blank.] 22 PA RT ON E Introduction, motivation, and overview of the thesis Lithium-ion battery generals 23 [This page is intended to be blank.] 24 CHAPTER 1 INTRODUCTION AND MOTIVATION OVERVIEW OF THE THESIS The primary subject of this thesis is to discover and develop a new novel lithium intercalation material whose electrochemical performance can possibly surpass the contemporary cathode compounds for lithium-ion (Li-ion) batteries. During the thesis study, thorough investigation with scientific insights is devoted to analyze and understand the electrochemical behavior of such a new candidate material. Furthermore, on top of the scientific comprehension, engineering efforts are dedicated systematically to maximize the immanent performance of the material to assess its potential as a promising candidate for Li-ion battery cathode. The principal tools used in studying the material are the conventional solidstate mechano-chemical method for synthesis, X-ray diffraction and Rietveld re25 finement for structural analysis, electron microscopy for morphological and microstructural investigation. Elemental analysis and detection are completed by several spectroscopy techniques. Other supportive practices, for example and 7 Li magic angle spinning nuclear magnetic resonance, Brunauer-Emmett-Teller method and thermogravimetric analysis, are also adopted when necessary. Various electrochemical testing sequences such as galvanostatic cycling, cyclic voltammetry, impedance spectroscopy, and galvanostatic/potentiostatic intermittent titration test are applied to characterize and evaluate the energy storing performance in an electrochemical cell with two-electrode configuration. Also, as an efficient and effective way to discover materials properties and understand physical phenomena, the results predicted by ab initio computation are often referred and compared with the experimental outcomes. Moreover, active collaboration with other research groups has been conducted in order to facilitate the progress of this study. 26 Introduction and motivation 1.1. 1.1.1. Li-ion battery as a mobile energy source It is now the advancing era of ubiquitous network, mobile workstation, and hybrid electric vehicles. Such a high-tech lifestyle could have not been managed without mobile power sources represented by lithium ion (Li-ion) batteries. Therefore, its importance as the most promising portable energy storage device cannot be too emphasized. Moreover, there are even more to come in our future life due to the necessity of emission-free technology, such as electrification of transportation and smart grid system, to deal with numerous regulations caring for the environment and global warming problem and to use the world's limited resources more wisely in a controlled manner, worldwide.' 3 That being said, the Li-ion battery technology seems to hold a key to unlock the future development of the life, which will be so complex to be operated that it requires significantly larger capacity of portable energy. In this chapter, the requirements and prospects of research in Li-ion battery are discussed in brief. Also, to serve as an introductory chapter, the overview of this thesis will be laid out. 27 An increasing demand of high performance 1.1.2. Li-ion batteries and motivation to find a new cathode material Li-ion batteries can carry a substantially larger amount of energy than other rechargeable batteries. As illustrated in Figure 1.1,4 it dominates the energy density space over the lead-acid and nickel based materials. In this reason, almost all of the current portable electronics and some of the (hybrid) electric vehicles exemplified in Figure 1.2 utilize Li-ion batteries, which are composed of a graphite anode and LiCoO2 cathode, as their power sources. . 900 18650 4.0 Ah 18660 3.6 Ah 3 18650 2.6 Ah 600 Metal-air 500 5 400 Li-Ion Li-polymer Ni-MH L0-metal 300 E 553450 prismatic Li-titanate 200 Ni-C Developing 100 Developed Lead-acid 100 200 300 400 500 600 Gravimetric energy density (Wh kg 1 ) Figure 1-12. Energy densities of various kinds of commercial rechargeable batteries.4 28 Portable Electronics 5-50 Wh Plug-in Hybrid Vehicles Chevrolet Volt 16 kWh Electric Vehicles Tesla Model S 85 kWh Figure 1-13. Examples of the industrial applications that adopt Li-ion batteries as a power source (images from http:images.google.com). However, it is rather debatable to fulfill the further large-scale necessity like a long-range electric vehicle and smart grid system with the current Li-ion battery system due to the high material cost of Co and the explosive risk of LiCoO2 compound during operation. 5 Thus, one of the main focuses in the Li-ion battery community has been onto finding entirely new chemistry of materials to substitute LiCoO2. 6 Unfortunately, nevertheless of the hard work devoted to bring down the cost and improve the safety of Li-ion batteries, LiCoO2 cannot be replaced yet by any other candidate materials in terms of electrochemical performance, so that it is still the most frequently used cathode compound since Sony Corporation firstly introduced it into the market in 1990s. 29 1.2. Overview of the thesis The objectives of the thesis work are to develop a new Li intercalation cathode with the guaranteed safety and to understand its experimental electrochemical behaviors. Moreover, in the process of understanding, the high capacity cathode material is to be designed and optimized for maximal electrochemical performance by remedying identified performance-limiting factors. To be specific, the material of my interest in this thesis work is monoclinic lithium manganese borate, LiMnBO 3 . The primary focus was to synthesize the phase-pure LiMnBO 3 specimen and recognize its potential and limitation as a Li storage cathode since it had been completely unknown as a Li-ion battery cathode (until a part of this work testified its first electrochemical results and analysis).7 Afterwards, the thermodynamic and kinetic characteristics of material were analyzed in depth to define performance-limiting factors and understand its electrochemical behavior. The identified problems were sluggish Li transport originated by diffusion channel blockage and phase instability upon the high level of Li extraction. With much endeavor, such limiting obstacles were finally addressed by applying appropriate strategies for the well-defined problems. In the end, the LiMnBO 3 compound was suggested as a promising candidate material with high capacity and safety for Li-ion battery applications. 30 The thesis is mainly consisted of three parts; introduction and motivation (PART ONE), finding of a new material and understanding its electrochemical behaviors (PART TWO), and improving the performance (PART THREE). PART ONE Chapters 1 and 2 contain the motivation of the research and brief introduction to Li-ion batteries and its intercalation compounds. PART TWO Chapter 3 introduces and justifies LiMnBO3 as a new chemistry of cathode materials and reports its electrochemical performances. Chapter 4 is devoted to explain the electrochemical behavior of the newly developed LiMnBO3 cathode by analyzing its transport phenomena and charged state stability. PART THREE Chapter 5 highlights efforts to enhance the electrochemical performance of the electrode by forming solid solution compounds and controlling an experimental process. Finally, all of the work is summarized and concluded in Chapter 6. 31 [End of Chapter 1] 32 CHAPTER 2 LITHIUM ION BATTERY GENERALS A lithium ion (Li-ion) battery is an electrochemical energy storage device that can convert electrical energy into chemical energy and vice versa. The nomenclature, Li-ion battery, is globally accepted as Li+ ions are essentially involved in such energy conversion reaction. Since it can store and release charges reversibly, it is also known as a rechargeable Li battery or Li secondary battery. The battery (pack) is consisted of multiple electrochemical cells interconnected in series and/or parallel to provide a designated operation voltage and capacity. Each cell contains a cathode and an anode, which is separated by polymer films (separator) and merged into the electrolyte. The cathode and anode serve as a positive and negative electrode, respectively. During operation, the Li+ ions will spontaneously diffuse out of the negative electrode upon discharging and be delivered to the positive electrode by 33 (a) Charge Cathode Electrolyte Anode Conductive matrix Charge Discharge Active particle 0Discharge (b) Potential Cathode2 (delithiated) Cathode (11thated) V2 *C * voc Anode LI Figure 2.1. Schematic (a) configuration of conventional Li-ion battery cell and (b) its voltage/energy diagram during operation. The electrical work deliverable is defined by Equation 2-1. In the ideal case, V2**-V 1* = V is the maximum voltage difference available. trespassing through the electrolyte in between. In the meantime electrons shall come out of the anode and flow into the cathode along an external circuit to per- 34 form work. The reaction can be reversed by applying external potential to store the energy. This cyclic process is illustrated in Figure 2.1(a). Since the amount of energy to be performed and stored is determined by how much Li+ ion can be inserted and extracted into and from the electrodes, the battery performance strongly depends upon the solid state chemistry of them. In this chapter, it is demonstrated how such an electrochemical cell operates and discussed the role of the electrodes in the system. Also, a few representative cathode materials are listed, and general characteristics of polyanionic compounds are summarized. 35 2.1. Li-ion battery generals: fundamental thermodynamics of an electrochemical system The performance of Li-ion battery is generally discussed in terms of (a) voltage, (b) capacity, (c) cyclability, (d) rate capability, and (e) safety. Moreover, the manufacturing cost and the toxicity of the raw materials are also important factors to be considered in Li-ion battery industries. In this section, such key parameters are briefly stated. 2.1.1. Voltages Equation 2.1 defines the equilibrium potential (V), also known as the opencircuit voltage (OCV), where z and F stand for number of charge per formula unit and Faraday constant (96,485 C per mole), respectively. It is a thermodynamic quantity, which is set by the difference in Li chemical potential (UL) be- tween the anode and cathode. Thus, it can be adjustable by selecting various combinations of the electrodes. Cathode V = -- i xF 36 Anode [2.1] An anode is the material with high Ai such as Li metal, Li alloys, and Ligraphite. In commercial Li-ion batteries, the graphite is the anode of choice due to the safety issue of the others and reversibility during cycling. 8 A cathode must have smaller PU than the anode and intercalate Li feasibly with chemical stability with carbonate-based organic electrolyte. These materials are mostly lithium transition metal oxides and polyanionic (oxo-anionic) materials. 8 In battery research, charging means such a process to extract Li+ ions and thus electrons from the cathode and insert into the anode by convention. Equivalently, since Ai of a Li-full (lithiated) cathode is higher than that of the Liexhausted (delithiated) one, charging also stands for lowering 'Li of the cathode. It can be done by applying a potential to the cell with an external power supply. In order for the cathode to be charged (that is, to lower PL), the potential applied must be larger than OCV of the lithiated cathode since YU is negatively proportional to the electric potential by Nernst equation (G = -xFc). Whenever Li in the lithiated cathode is pumped to the anode, the cathode becomes capable of performing electrochemical work. Charging is therefore an energy storing process. When charging is completed, the cathode is fully excited from its lithiated state. Thus, it is now willing to accept both Li+ ions and electrons spontaneously once it forms the closed circuit. Ideally, the voltage in Eqn. 2.1 will drive Li+ ions and electrons into the cathode from the anode. In the process, the cell performs work to the environment. This is defined as discharging of the cathode. The 37 charging and discharging process as a scheme of PLi and electric potential is illustrated in Figure 2.1(b). In summary, the following reaction happens during charging and discharging in the cathode. LixMyZ Charging ) XLi+ + Xe- + MY xZ Discharging LixMyZ D xLi+ + xe- + MY-XZ [2.2] [2.3] The reaction in the anode during charging and discharging is therefore, xLi+ + xe- xLi+ + xe Charging [2.4] ischarging [2.5] The operating voltage has an upper bound due the limited stability of the electrolyte during charging. Also, the applying voltage cannot be too low because there is always a chance of metallic Li precipitation on the surface of a graphite anode, which likely leads to dendrite formation. Thus, it must be maintained above a certain voltage level to avoid a cell failure. In addition, since electrons must not trespass through the electrolyte, the lowest-unoccupied-molecularorbital (LUMO) of the electrolyte should be aligned higher than the Fermi level of a cathode. In similar, the highest-occupied-molecular-orbital (HOMO) must be located lower than the Fermi level of an anode. 6 38 2.1.2. Capacities A capacity is the conventional nomenclature in Li-ion battery industries to define the amount of charge that can be stored in the electrode. In general, the normalized capacity with respect to molecular weight (gravimetric capacity) or volume (volumetric capacity) of the electrode is used to define its performance. The gravimetric and volumetric capacities can be calculated by following equations. x 1000 -- x Mw 3600 x 1000 Volumetric capacity (mAh L-1) = - x VM 3600 Gravimetric capacity (mAh g-') = [2.6] [2.7] The product of the normalized capacity and voltage is defined as an energy density (Wh kg-1 or Wh L-1) In the equations, x is the number of moles of the charge carrier per formula unit participating in the reversible electrochemical reaction, and M, and VM stand for molecular weight and volume of the electrode material, respectively. In other words, x specifically indicates how many Li+ ions (or electrons) can be extracted from the electrode composition without provoking structural instability within the given voltage window. Actually, the maximum of x is preset by the chemical formula of electrode materials simply because it is determined by how many Li+ ions can be present per formula unit. 39 2.1.3. Cyclability Cyclability can be denoted by the percentage of a capacity change during operation per total cycle numbers, and therefore it is the indicator of the lifespan of batteries. It is affected by various factors such as parasitic electrolyte oxidation, surface-electrolyte interface layer formation, and the integrity of electrode materials. It often serves as a qualitative measure of stability of the electrode since it tends to influence primarily on the cyclability. 2.1.4. Rate capability Rate capability is another convention terminology in Li-ion battery research to quantify how fast a battery can be fully charged and discharged. Therefore, the rate capability of an electrochemical cell can qualitatively illustrate a power density, which is energy density per unit time. It is strongly related to the cell impedance generated by Li diffusion in the electrode, charge transfer at the electrode surface, electrolyte resistance through the electrolyte. A material with a good rate capability can maintain its slow rate capacity at high rate cycling. The (gravimetric) rate of cycling is defined by Equation 2.8 as Rate (mA g-) 40 = C n - [2.8] where C stands for a theoretical value of the gravimetric capacity and n is the time taking to obtain C. 2.1.5. Safety The safety of Li-ion batteries depends on oxidation of the electrolyte and gas evolution from the cathode during charging. Given that electrolyte is made of hydrocarbons, it is a highly flammable fuel when it gets seriously oxidized. Assuming the sealing of the battery is perfect, the oxidation occurs due to oxygen evolution from the charged cathode. Also, the cathode may release gases such as 02 and/or C0 2 , which potentially damage the sealing of the battery and lower quality of its packaging. In the worst case, it can cause an explosive reaction between battery components and air. 5 41 2.2. Commercially available Li intercalation compounds 2.2.1. Layered oxides A typical layered structure is in Figure 2-2.9 It consists of the hexagonal framework of close-packed oxygen, which arrays in A-B-C-A-B-C (03) stacking sequence. Due to this structure, Li diffusion in layered oxides can occur in twodimension parallel to the layered slabs. 10 Many of lithium transition metal oxides can have such a layered structure, and LiCoO2 is the first commercial Li-ion battery cathode, which is still prevailing in portable electronics.11 Layered oxide LIMO 2 Figure 2-2. Schematic illustration of a layered oxide with 03 stacking.9 Reprinted (adapted) with permission from Masquelier, C.; Croguennec, L., Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chemical Reviews, 2013, 113, (8), 6552-6591. Copyright 2013 American Chemical Society. 42 However, due to the high cost of Co and the structural instability issue at the charged state (LiiXCoO2, x > 0.5), a vigorous research activity is going on to develop a new chemistry within the layered framework. 6 The layered oxides with multiple cations such as LiNio. 3 3Mno. 33 Coo.3302 and LiNio.8 Coo.,5 Alo.o5 0 2 are being studied vastly in that it can possess high energy density and high power density with reduced materials cost.12 -16 However, the safety is yet the unsolved issue for the compounds.15-17 Also, as a derivative of the layered oxides, Li-excess layered compounds with typical composition of xLi 2 MnO 3 -(1-x)LiNio. 33 Mno. 33 Coo.3302 exhibits sub- stantial energy density since it involves more than one Li per formula unit.18, 19 However, it suffers from poor rate capability, irreversible loss of the capacity, and redox voltage fading and thus energy density loss upon cycling due to its phase transformation into spinel structure. 20 , 21 2.2.2. Spinel oxides Another type of cathode materials has a spinel structure in Figure 2-3.9 It possesses a three-dimensional diffusion path, so that it displays fast ionic and electronic conduction suitable for high power cathode applications. 9 , 22 The representative compound with the spinel framework is LiMn204. 2 2 Since it does not include the Co element, its fabrication cost is economical and the safety can be guaranteed compared to LiCoO2. However, LiMn204 is vulnerable to the perfor43 MnO6 LiO4 Spinel oxide LIMn 2O4 9 Figure 2-3. Schematic diagram of a representative spinel oxide, LiMn 2O4. Reprinted (adapted) with permission from Masquelier, C.; Croguennec, L., Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chemical Reviews, 2013, 113, (8), 6552-6591. Copyright 2013 American Chemical Society. 23 mance degradation at high temperature resulted from Mn dissolution. Merged 4 2 3 in the carbonate electrolytes, disproportionation of Mn + into Mn + and Mn + of- 24 ten occur, and Mn 2+ tends to end up with its dissolution. , 25 To deal with such a problem, the composition of LiMn 2 0 4 was modified by 26 doping foreign elements such as Al or Ni. , 27 Also the surface of the particles was coated to minimize the parasitic reaction with the electrolytes. Yet, the low energy density of LiMn204 is the inherent limitation unless the high voltage electrolyte is developed to account for the up to 5 V redox range of Ni substituted LiNio.5 Mn1 .5 0 4 .2 7 44 2.2.3. Phospho-olivines Concerning the safety issue, lithium iron phosphate, LiFePO 4 , with olivine structure in Figure 2-4 has aroused lots of interests as a new cathode material due to its thermal and structural stability, which lead to superb safety, as well as low cost and low toxicity of the elements. 9 , 28 The stability is mainly resulted from the characteristics of polyanionic structure providing strong covalency between oxygen and additional anions of a different kind, phosphorous in this case.6 , 28 Such an anion incorporation into the framework lead to a few characteristic features in all polyanionic category of materials. This will be laid out more in detail in the following Subsection 2.3.1. Triphylite LiFePO 4 FeO, P0 4 Ui -- 4 Figure 2-4. Schematic diagram of a representative phosphor-olivine, LiFePO 4 .9 Reprinted (adapted) with permission from Masquelier, C.; Croguennec, L., Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chemical Reviews, 2013, 113, (8), 6552-6591. Copyright 2013 American Chemical Society. 45 LiFePO 4 has a one-dimensional diffusion channel with low electric conductivity. 2 9 , 30 However, nanosized LiFePO 4 displays remarkably fast Li diffusion along the channel within the lattice that enables a 200 C rate (7.2 minutes) charging maintaining more than 70% of the theoretical capacity with good retention if its surface is properly treated. 31 If mass-produced, however, its low material density and packaging density become one big huddle to be a large-scale battery for automobiles since in many case the volumetric energy density is considered as a critical factor. 32 46 Characteristic features of polyanionic 2.3. cathode materials 2.3.1. Stability and safety Although utilized in the commercial portable electronics, LiCoO2 and its offspring layered oxide cathodes are not so practical for large-scale Li rechargeable batteries due to the safety concern. 8 If overcharged, such materials tend to lose oxygen,6 , 32 and the evolved oxygen atom and/or 02 gas will either oxidize electrolyte or damage cell packaging by increasing internal pressure. Moreover, since the evolution reaction is exothermic, it can eventually burn the electrolyte in run-away fashion, which may lead to a vigorous explosive reaction in the largescale battery. 8 On the other hand, polyanionic cathodes are likely to be more stable than the layered oxides against the oxygen loss during charging provided by the strong covalent bonding between anions., 28, 33-35 The difference in the stability can be rationalized by their contrast in the electronic structure. Figure 2-5 illustrates electron energy versus density of state (DOS) of the 3d-transition metal (Mn+/M(n+1)+) in the octahedral coordination of oxygen ions (02-).32 If the energy band of 3d-electrons and 2p-electrons overlaps as shown in the figure, the top of the 2p-orbital of 02- can possibly limit the amount of 3d-electrons extractable from Mn+/M(n+l)+. Overcharging beyond this limit can potentially lead to oxygen 47 and/or 02 gas evolution by removing 02- 2p-electrons instead of 3d-electrons. This energy scheme is found in LixCoO2, whose oxygen can be lost if it is charged more than 55%.32 In contrast, polyanionic compounds possess the transition metal-oxygenanion bonds (M-0-X), so that O-X bonds modify the whole energy band scheme of the M-0 bond.6 , 32 In Figure 2-5, 3d-electrons of Mn+/M(n+l)+ is free from overlapping with the 2p-electrons of 02-, and consequently full charging will not cause any oxygen evolution. For example, the energy diagram of LiCoPO4 does not 6 have any overlap between Co and 0 bands, in opposition to LiCOO2. Many of E E E E Co: 4s" ~ Ti: 4s" Co: 4s0 ~ -------- -------------------------------------------------------------- 0.2 eV 2.3 eV. i4+r1 3 + 4,0 eV4 2 S :3p C0 4 +C3+ Co 02 :2p6 N(s) LiC 6 N(s) N(s) LiCoO2 LixTiS2 & LI1 jTi2 IS 4 g 2 3 '*/Co o:2p 4 6 N(s) LixCoPO 4 Figure 2-5. Schematic diagrams of the electron energy versus density of states of 3dband in transition metal (M"*/M("*,) and 2p-band in oxygen (02-) (a) with overlapping and (b) without overlapping.3 2 Reprinted (adapted) with permission from Goodenough, J. B.; Kim, Y., Challenges for Rechargeable Li Batteries. Chemistry of Materials, 2010, 22, (3), 587-603. Copyright 2013 American Chemical Society. 48 other polyanionic materials also have the similar energy scheme. Therefore, provided by covalent nature of the O-X bond, polyanionic cathodes tend to be superior in stability against oxygen loss to plain oxides. 2.3.2. Inductive effect The electronic structure of M-O bonds can be affected by polarization present in proximity. 6 Hence, in polyanionic structures possessing M-O-X bonds, adjacently polarized O-X bonds can modify the character of the M-O bond. The degree of polarization largely depends on the electronegativity difference between elements in bonding. 6 As a result, the electronegativity of the anion can play an important role to determine the overall electronic structure of the M-O-X bond in polyanionic cathodes. 6 This consequence caused by incorporation of X into the M-O bond is entitled as the inductive effect. 6 The redox potential of transition metal (Mn+/M(n+l'+) versus lithium (Li/Li+) is often determined by the electronic structure of the M-O bond, as shown in Figure 2-5. Therefore, the voltage of cathode compounds can be tailored via the inductive effect by selecting anions with various electronegativities. If the electronegativity of X is high, it will pull nearby electrons, so that the covalency of M-O bond will be diminished, which leads to OCV elevation as shown in Figure 2-6(a). 36 49 For instance, the OCVs of the Fe 2+/Fe3+ redox couple with various Fe-O-X bonds are displayed in Figure 2-6(c). 32 Since sulfur has a greater electronegativity than phosphorous or boron, it is predictable that the OCV of Fe2 +/Fe3+ versus Li/Li+ in sulfate compounds has the highest OCV than the phosphate, borate, or silicates. This tunability of redox potential via the inductive effect is a powerful design strategy to develop new cathode materials, especially for polyanionic compounds. UIu- (a) E vOC (C) Li3 Fe2(XO4)3 JvOC J a - . I Li +/Lio IOW O W_ CA (b) U^IsW4 a /~O Fe3*/Fe2+ in X = As N"rCOVsg F" kon UFO, A, . . UOP04 A .. ......... Fe 3+/Fe2 + in X = P Fe3+/Fe2 +in X = Mo > Fe3 +/Fe2 +in X = S LVOPIOI _!J -------------------------------- Density of States N(E) LMW F*10* M&M COMM WWW PdOW W*W) Figure 2-6. (a) The energy diagram of M-O bonds, (b) various Fe-O polyhedra with different coordination numbers,3 and (c) energy level of Fe2+/Fe3 redox elements with respect to metallic lithium in the NASICON structure with different M-O-X bonds. 32 Reprinted (adapted) with permission from Gutierrez, A.; Benedek, N. A.; Manthiram, A., CrystalChemical Guide for Understanding Redox Energy Variations of M2 +13 + Couples in Polyanion Cathodes for Lithium-Ion Batteries. Chemistry of Materials 2013, Article ASAP. Copyright 2010 American Chemical Society, and Goodenough, J. B.; Kim, Y., Challenges for Rechargeable Li Batteries. Chemistry of Materials, 2010, 22, (3), 587-603. Copyright 2013, American Chemical Society. 50 2.3.3. Electronic conductivity Many of polyanionic cathodes are not considered as a good electronic conductor. This is because the O-X bond in polyanionic structure can confine the transition metal redox centers. 37, 38 In other words, the delocalization of electrons is disturbed by the presence of electronegative element X nearby the transition metals. However, this low electronic conductivity does not necessarily deteriorate the electrochemical performance in the cathode level. Many studies have demonstrated to circumvent such an issue by coating a conductive phase on the 31 particle surface or minimizing the particle size. , 37, 2.3.4. 39-41 Gravimetric / volumetric energy density Compared to simple oxides, one major drawback in polyanionic cathodes is the low gravimetric capacity (charge per unit mass, mAh g-1) due to additional weight of the polyanion group. If only one lithium per formula unit is active, its theoretical capacity hardly exceeds 200 mAh g-1, which can be often readily obtained from the layered oxides.' 0 , 20, 31 The exceptional case is found in lithium transition metal borate chemistries (LiMBO 3 , M: Mn, Fe, and Co), whose theoretical capacity is up to 222 mAh g- 1, the largest among polyanionic cathodes due to the simplest borate group. 7, 42-44 If more than one lithium can be activated, it 51 may surpass the 200 mAh g- 1 milestone, as demonstrated in Li2(Fe,Mn)SiO 4, but it involves serious engineering and optimization. 4 5-47 In addition, unlike the layered oxides, polyanionic compounds are not a close-packed structure but a relatively loose framework. Thus, the material density of polyanionic compounds must be inherently lower than that of oxides. Inevitably, the low density affects the volumetric energy density in negative manner. Since the theoretical volumetric energy density is obtained from multiplying gravimetric energy density by material density, the difference between energy densities of layered oxides and polyanionic compounds becomes even larger due to weight and density disadvantage of the polyanions. 2.3.5. Packing density / processing cost As stated in Subsection 2.3.4, due to lack of tightly stacked polyhedral configuration, the density of polyanionic materials is intrinsically smaller than that of layered oxides. Moreover, in many cases, the conductivity problem requires nanosized active particles with conductive phase coating. 31, 39, 40, 44 Thus, once packaged, on top of the low volumetric energy density, the overall tap density and battery pack energy density becomes even significantly compromised. In addition, in order to produce the same amount of energy, the overall manufacturing expense for polyanionic cathodes including material preparation 52 and handling may cost more than the layered oxides due to the nanosizing and surface coating processes. 2.3.6. Summary Polyanionic materials display a clear edge on the safety criterion over the contemporary layered oxides as a cathode. At the same time, additional anion involved causes some disadvantages with regard to the energy densities, electronic conductivity, and processing cost per energy output. Therefore, to maximize its strength and minimize its weakness, we simply have to pursue the polyanionic compounds with the highest energy density possible. With various possible selections of the polyanion groups, since the inductive effect readily offers an opportunity to design the redox voltage of the material, there is still a huge room to develop a new polyanionic compound that can outperform the competitors. In following PART TWO and THREE, such efforts devoted during this thesis work are enumerated. 53 [End of Chapter 2] 54 PART TWO The new polyanionic cathode material: Lithium manganese borate 55 [This page is intended to be blank.] 56 CHAPTER 3 DESIGNING A NEW CATHODE MATERIAL The development of new cathode materials that can store and release the significant amount of charges reversibly for an extended period of time is crucial for the future improvement of Li ion battery technology. It is, however, a nontrivial task to realize such materials possessing all of the necessities, so that on32 ly a limited number of oxides has been commercialized since 1970S. , 48 Most importantly, an incremental demand of large-scale batteries imposes on strict safety requirement during the cycling operation. 5 , 49 Unfortunately, the most successful and popular cathode material, LiCoO2, even failed to meet such a cri6 terion due to the oxygen evolution problem at the high state of charge. 32 Materials containing simple polyanion (oxo-anion) groups are advantageous to cope with the growing safety concern: the covalent nature between oxygen and 57 additional anion readily provides distinctive stability against oxygen loss for the polyanionic cathodes. 9 Thus, numerous researchers have tried to adopt the polyanion group to cathode materials when to design a safe electrochemical system. 7, 28, 31, 43, 47, 50-55 In chemistry, the combination of multiple anions to form the oxo- anion group can be diverse and even limitless. Yet, a great part of interest goes to simple oxo-anionic combination such as borate (B0 3), phosphate (P0 4), and silicate (Si0 4 ).28, 43, 47 Moreover, the attention is extended to more complex poly- anion groups like fluoro-phosphate (PO 4F), fluoro-sulphate (SO 4 F), and carbonophosphate (C0 3 PO4 ).50-53 Among them, materials with phosphate group have been studied intensively owing to the feasibility of synthesis. 56 Currently, LiFePO 4 is considered as the most successful polyanionic intercalation cathode due to its low cost, high stability, low toxicity and high rate capability as well as long cycle life. 28 , 31, 38 In pursuit of a new cathode material that can have the high capacity and safety at the same time, it is a proper materials design strategy to adopt the borate group so as to take advantage of its stable and simple chemistry. Therefore, in this chapter, as a promising candidate for Li intercalation compounds, the pioneering experimental findings of lithium manganese borate (LiMnBO 3) system in my thesis work are discussed. In Section 3.1, the LiMnBO 3 system is introduced as a promising candidate for the Li intercalation materials. Section 3.2 documents the utility of computation as a design and discovery tool of the new material. Moreover, experimental results on LiMnBO 3 in terms of synthesis, 58 electrochemical properties and corresponding optimizations are also discussed. Section 3.3 provides hypothetical explanation for electrochemical behaviors of the LiMnBO 3 system to acknowledge the impact of its polymorphic structure, and the chapter is concluded in Section 3.4. 59 3.1. Lithium manganese borate compounds (LiMnBO3) 3.1.1. Motivation Although successfully demonstrated high rate capability, excellent safety and long cycle-life, the theoretical gravimetric capacity of LiFePO 4 (170 mAh g- 1) is less attractive than that of state-of-the-art layered oxides (-200 mAh g-1). This is an obviously anticipated consequence in most polyanionic compounds utilizing one-electron redox reaction owing to the additional weight of the multiple anions. Therefore, it is legitimate to pursue a material that consists of the lightest elements in order to minimize the disadvantage in the specific capacity of such oxoanionic compounds. More specifically, the low weight-to-charge ratio of the anion group is a vital criterion for the material to compete with the state-of-the-art layered oxides as a Li intercalation cathode. Lithium transition metal borate system can be a strong contender in polyanionic compounds because the borate (B0 3) group is the lightest among simple oxo-anions. For instance, by simply converting the P0 4 group into B0 3 in LiMnPO 4 , the theoretical capacity increases from 171 to 222 mAh g-1. As a result, several results dealing with some of lithium transition metal borates (LiMBO3 : M=Mn, Fe, Co) have been reported in the same context. 4 2 , 57-59 However, the electrochemical outcomes were premature, especially for lithium manganese and co60 balt borate (LiMnBO 3 and LiCoBO 3) compounds, and general understanding on such a borate system was absent due to the lack of detailed and vigorous analy42 sis on the materials characteristics. , 57-59 Moreover, in the lithium transition metal borate system, most studies have focused on lithium iron borate (LiFeBO3).4 3, 59-63 Yet, it was very recent that ei- ther its meaningful structural and electrochemical properties were discussed in detail with the perspective as a Li intercalation cathode material. 4 3, 62-64 On the other hand, LiMnBO 3 and LiCoBO 3 have been barely studied since their preliminary electrochemical properties were so negligible that they were concluded as unsuccessful cathode materials. 42 , 58 Nevertheless, unbiased large-scale materials search via high-throughput computation still identifies the borates as potential high energy density Li-ion battery cathode materials. 7 Judging from the fact that essential battery properties such as voltage, stability, and safety can be estimated with a reasonable accuracy by computation, the electrochemical performance of LiMnBO 3 or LiCoBO3 may not be properly discovered by experiment yet. 33 , 46, 65, 66 They might have been so hastily abandoned at a preliminary stage that they were unable to be investigated thoroughly as much as LiFeBO 3 . Therefore, it may be possible to enhance their electrochemical properties by systematic and elaborate experimental efforts. However, in terms of practicality, such as the cost and environmentallyfriendliness of raw materials, LiCoBO 3 instantly loses its attraction as a new cathode material. 61 There is an interesting and also important feature in the LiMnBO 3 chemistry which had been overlooked until this thesis work identified it: unlike LiFeB0 3 or LiCoBO 3 , LiMnBO 3 can have polymorphs. 42 67 While both LiFeBO 3 and LiCoBO 3 crystallize only in monoclinic framework, LiMnBO 3 can have a hexagonal structure as well as the monoclinic one, depending on their synthesis conditions. 7 , 67, 68 Previously investigated LiMnBO3 was all about the hexagonal polymorph, and pure monoclinic LiMnB0 3 had never been investigated properly prior to this thesis work, except for the initial structural report since 1970s. 6 7 Thus, monoclinic LiMnBO was regarded as the only material in the uncharted territory of Li intercalation materials among the borate chemistry. In this chapter, the results of systematic investigation on the electrochemical properties of monoclinic LiMnBO 3 are discussed in order to assess its potential as a new cathode material. 7 Furthermore, differences in the electrochemical characteristics between monoclinic and hexagonal LiMnBO 3 polymorphs are distinguished and understood in relation to their crystal structures. 7 The scope of this chapter spans a literature review, experimental effort for synthesis and electrochemical characterization, and optimization of the properties in monoclinic LiMnBO 3 as well as the hexagonal phase. The results are also discussed to raise the importance of polymorphic structure on the materials properties of LiMnBO 3. 62 3.1.2. Literature review 3.1.2.1. Structural framework of lithium (transition) metal borates Lithium metal borates are reported to have either monoclinic or hexagonal structure. LiMgBO3, LiFeBO 3 , LiCoBO3, and LiZnBO3 are known to have the monoclinic framework whose space group is C2/c. 4 2, 67, 69 In contrast, LiCdBO 3 crystallizes in the hexagonal structure with P-6 space group. 70 In the case of LiMnBO 3, both structures are possible depending on the synthesis temperature and/or recipe. 7,68 The reported structural parameters of the lithium metal borate system are summarized in Table 3-1. The detailed review is following in the next section. 42 , 69, 70 Table 3-1. Structural parameters of lithium metal borates Structure Composition a (A) b (A) c (A) P (0) V (A 3) ICSD Ref No. Monoclinic LiMgBO 3 5.161 8.88 9.911 91.29 454.10 67226 Monoclinic LiMnBO 3 5.188 8.952 10.367 91.75 481.25 200535 Monoclinic LiFeBO 3 5.169 8.924 10.138 91.39 467.51 94317 Monoclinic LiCoBO 3 5.129 8.84 10.1 91.36 457.81 59346 Monoclinic LiZnBO 3 5.094 8.806 10.374 91.09 465.27 200534 Hexagonal LiMnBO 3 8.172 8.172 3.147 120 182.01 94318 Hexagonal LiCdBO 3 8.324 8.324 3.264 120 195.86 20835 63 3.1.2.2. The monoclinic and hexagonal polymorphs of LiMnBO 3 The crystal structure of monoclinic LiMnBO 3 is shown in Figure 3-1(a). It is isostructural to LiMgBO 3 and LiZnBO3 , which consists of columns of edgesharing trigonal-bipyramidal Mn sites (MnO5 ) joined by planar borate (B0 3) and trigonal-bipyramidal Li sites (LiO5 ).7,67,69 The structure is reported with a small amount of disorder, whereby each trigonal-bipyramidal Li and Mn site is split into both an upper site and a lower site. 7 ,64,67 Due to the close proximity of these sites, this splitting has been interpreted as follows: while any given lithium or manganese ion may sit in either the upper or lower portion of the bipyramid, they may not both be occupied simultaneously. 7 In turn, this implies that each trigonal-bipyramid will be occupied by a single ion at all times. The similarity in crystal structure is also found in monoclinic LiFeBO 3 and LiCoBO3 with a slight variation of lattice parameters and occupancy of the Li and transition metal sites. 6 7 Recently, Janssen et al. reported modulated LiFeBO 3 structure whose Li and Fe are not split. 62 According to single crystal X-ray diffraction, unlike the conventional disordered structural unit in Fig. 3-2(a) and (b), Li atoms may sit in the tetrahedral site, and Mn atoms can locate in the center of the trigonal bipyramidal site, as shown in Figure 3-2(c). 62 Unlike LiFeBO 3 or LiCoBO 3 , LiMnBO 3 can have a hexagonal polymorph, which possesses the same crystallographic features as LiCdBO 3 (P-6 space 64 group). 42 Figure 3-1(b) illustrates the crystal structure of the hexagonal LiMnBO 3 composed of columns of edge sharing square pyramidal Mn sites (MnO5) connected by planar borate (B0 3) and tetrahedral Li sites (LiO4), 42 and it (a) (b) o C b aW a b , o b b 0 a \-~a C 0 a ... a Monoclinic (C2/c) Hexagonal (P-6) Figure 3-1. Schematic diagrams of LiMnBO3 polymorphic structures (Yellow; Li, Green; Mn, White; B, Shade; 0 polyhedra). The space group for each phase is (a) C2/c(monoclinic, #15) and (b) P-6(hexagonal, #174), respectively. 65 68 is reported to appear at elevated temperatures in general. While the LiMnBO3 polymorphs may appear drastically different, they (a) Vina (b) 03 02 02 03 Fe Joe L C 'if 01 (c) 03 02 03 02 Fe Ue 011 2 Figure 3-2. Schematic diagrams of (a) Idealized 2D layer of LiFeBO3 ,6 (b) 2 LiFeBO 3 structure in the literatures including anisotropic displacement for 0 atoms, and (c) 2 Reprinted (adapted) with perLiFeBO 3 fragment from the modulated superstructure. mission from Janssen, Y.; Middlemiss, D. S.; Bo, S. H.; Grey, C. P.; Khalifah, P. G., Structural Modulation in the High Capacity Battery Cathode Material LiFeBO 3 . J. Amer. Chem. Soc. 2012, 134, (30), 12516-12527. Copyright 2012 American Chemical Society 66 share many commonalities. The nominal Mn coordination is five (MnO5), and all Li is approximately tetrahedrally coordinated (LiO 4 ) if considering only the top or bottom half of the trigonal-bipyramids in the monoclinic structure. 7 Both structures consist of columns of edge-sharing Mn polyhedra linked by planar borate groups and Li polyhedra. At the same time, the polymorphs differ in a few substantial ways. 7 First of all, the number of atoms in a conventional unit cell is obviously different: 24 atoms are present in the monoclinic unit cell (4 formula units), but 18 atoms in the hexagonal one (3 formula units). More importantly, Mn is trigonal-bipyramidal coordinated in the monoclinic structure and square pyramidal in the hexagonal form. 4 2 The network connectivity between the polyhedral sites is also different in the two polymorphs. In the monoclinic structure in Figure 3-1(a), three adjacent Mn columns are always joined by a repeating pattern of two borate groups followed by two Li groups. In the hexagonal form, three neighboring Mn columns are joined by either a stacked series of borate groups or a stacked series of Li groups, as shown in Figure 3-1(b). 7 In consequence, the LiO 4 chains along <001> direction are cross-linked to weave the MnO5 chains along <101> direction in monoclinic LiMnBO 3 lattice. However, for the hexagonal case, both LiO 4 and MnO5 chains are completed aligned with each other along <001> direction, so that one LiO 4 chain is completely isolated with another by neighboring MnO5 channels 67 3.1.2.3. The electrochemical properties of lithium transition metal borates Determined by its chemical composition, the theoretical capacity of lithium transition metal borates (LiMnBO 3 , LiFeB0 3 , and LiCoBO 3) is approximately 220 mAh g- 1. However, in the reported case of hexagonal LiMnBO3 in Figure 33(a), it has thus far displayed very low electrochemical activities with a negligible capacity less than 10 mAh g-1 even at very slow charging and discharging rate. 42 In consequence, it was concluded as an unsuccessful cathode material, and research attractions quickly receded away. Several recent reports on hexagonal LiMnBO 3 claim improved capacities, but almost all of them are obtained with a very wide voltage window range making it likely that the capacity reflects a conversion reaction rather than intercalation, as exemplified in Figure 3-3(b).58 The latest study on the hexagonal compounds claims 150 mAh g-1 synthesized by sol-gel method with reduced graphene oxide as a conducting media, but it still seems its large portion comes from conversion according to the cycling curve profile and the low discharge voltage cutoff.7 1 On the contrary, LiFeBO3 (monoclinic) seems to show more significant Li intercalation activity than the others, and it is the most studied compound in the borate system. 43, 44, 57, 59, 62, 63 Although it showed undesirable electrochemical properties at the early stage of the study, Abouimrane et al. was able to enhance its capacity up to 145 mAh/g by coating a conductive carbon layer on the particle surface. 42, 44, 57 The capacity enhancement was explained by reducing electronic 68 (a) (b) d c 5.0 4.5- 4.0 4i7 >3 0 0 3.5 3.5 -j 3.0 0 3 2.5 2.5 'S 0 2F 1.5p a b 4.5 2.0 1.5 1.0 0 -0.1 -0.2 Capacity as: A x Li / formula unit 91 ft a d 0 20 40 60 8 Capacity (mAhlg) (d) (9) A 4 3.5 298 K, C120rate 3 3 (D 2 1st --- 2nd 1 10th -6 2.5 > 2- 20th 0 50 100 Capacity C I mAh g 150 200 1.5 0 -0.01 -0.02 -0.03 Capacity as : Ax Li / formula unit Figure 3-3. Representative electrochemical properties of lithium transition metal borates in the literature: (a) Hexagonal LiMnBO displays large irreversibility at the initial 42 cycle and only intercalates 0.02 Li per formula unit (4 mAh g'). (b) Hexagonal LiMnBO shows 75.5 mAh g", but it is most likely to originate from a conversion reac43 tion below 1.5 V." (c) LiFeBO outperforms other borates in terms of a capacity. (d) LiCoBO 3 shows a negligible capacity.42 Reprinted from Solid State lonics, 139, (1-2), Legagneur, V.; An, Y.; Mosbah, A.; Portal, R.; La Salle, A. L.; Verbaere, A.; Guyomard, D.; Piffard, Y., LiMBO 3 (M = Mn, Fe, Co): synthesis, crystal structure and lithium deinsertion/insertion properties, 37-46., Copyright 2001, with permission from Elsevier, and from J. Alloy. Compd., 494, (1-2), Chen, L.; Zhao, Y.; An, X.; Liu, J.; Dong, Y.; Chen, Y.; Kuang, C., Structure and electrochemical properties of LiMnBO3 as a new cathode material for lithium-ion batteries. 415-419., Copyright 2010, with permission from Elsevier. Reprinted from Adv. Mater., Yamada, A.; Iwane, N.; Harada, Y.; Nishimura, S.; Koyama, Y.; Tanaka, I., Lithium Iron Borate as High-Capacity Battery Electrodes 3583-3587; Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim resistance of LiFeBO3 in the electrode. 44 Recently, Yamada et al. reported that 190 mAh g-1 of reversible capacity was achieved by not only carbon-coated but 69 also nano-sized LiFeBO3 along with 3.0 V of average voltage, as shown in Figure 3-3(c). 43 Note that such a discharge capacity was only realizable by the constant current and constant voltage (CCCV) charging mode. It is suggesting asymmetrical charge and discharge kinetics, so if charging were done galvanostatically, the discharge capacity may not as high as the reported. In the case of LiCoBO 3 in Figure 3-3(d), Li extraction and insertion were so much insignificant that it is considered as electrochemically inactive like the early stage of the hexagonal LiMnBO 3 compound even though it possesses the analogous monoclinic structural framework to LiFeBO 3 .42 One thing to point out is that the synthesis route of LiCoBO3 deviates from the processes for LiFeBO 3 or LiMnBO 3 .72 While LiFeBO3 or LiMnBO 3 can be feasibly obtained by a solidstate method reacting Li, Fe (or Mn), and B0 3 at the same time, LiCoBO3 was formed by either molten-salt technique or a stepwise solid-state reaction, which required C02B20 5 as an intermediate phase. 7, 43, 72 The firing temperature of 7 43 LiCoBO 3 was also considerably higher than those of LiFeBO 3 and LiMnBO3. , , 72 According to the up-to-date literatures dealing with lithium transition metal borates, majority of the efforts is very recent and/or focused mainly on LiFeB0 3 .4 3 , 59-63 This reflects not only a refreshing attention on the borate chemistries but also a biased interest to one of this new class of materials. Above all, the most surprising conclusion through the literature survey is that no electrochemical study had been devoted to monoclinic LiMnBO 70 3 as a phase-pure form until this thesis work started. 7 Therefore, in the following sections, the experimental efforts to synthesize phase-pure LiMnBO3 in both monoclinic and hexagonal form are explained. The first electrochemical performance on the monoclinic LiMnBO3 is also presented as well as the hexagonal counterparts with detailed and comprehensive discussion. 3.1.2.4. High-throughput computation driven material screening and filtering to discover LiMnBO3 as a new Li intercalation cathode First-principles or ab initio calculation can nowadays predict essential battery properties such as voltage, stability, and safety with a reasonable accuracy. 46 , 73-75 In principle, the scalability of computations can make such predictions be applied to thousands of compounds and potentially all known inorganic materials. 76 With this high-throughput computational tool, the ongoing Table 3-2. Computed (theoretical) properties of the LiMnBO polymorphs.7 Phase Vol. Avg. Grav. E. Vol. E. Voltage Density Density Capacity change (V) (Wh kg~) (Wh L~) (mAh g~) (%) Monoclinic 3.70 822 2635 222 2.5 Hexagonal 4.11 912 2922 222 0.6 71 Lithiated Delith. distance distance to hull to hull (meV per (meV per atom) atom) 140 4 0 216 research project at MIT, named as Materials Project, aims to accelerate materials discovery by developing a database of calculated properties and structural information on all known inorganic compounds. 76, 77 To search for a promising cathode material, the database was screened with certain criteria: voltage versus Li, theoretical capacity, stability as measured by the driving force for phase transformation of the charged and discharged state, and safety as measured by the oxidation strength of the charged cathode. 33 , 46, 65, 73 This large-scale and unbiased screening also identified both monoclinic and hexagonal LiMnBO 3 as potential high energy density lithium ion battery cathode materials. 7 The calculated battery properties such as the Li redox voltage and energy density with respect to a Li anode, volume change, and thermodynamic stability in the charged and discharged state at zero K for both the monoclinic and hexagonal LiMnBO 3 are summarized in Table 3-2.7 Distance to hull stands for difference in energy from the computed ground state, and volume change is taken as a percentage of the Lithiated state volume. The computationally predicted ground state for LiMnBO 3 is the hexagonal phase, although its energy difference with the monoclinic phase of 4 meV per atom is close to the limit of numerical accuracy for our methods, and is small enough to be easily overcome by entropic effects. 7 The computationally identified ground state for MnBO 3 is a stoichiometric mixture of Mn2O3, MnO2, and MnB 4 0 7 .7 72 Figures 3-4(a) and (b) show the calculated formation energies of monoclinic and hexagonal LiMnBO3 as a function of Li content, respectively, and the corre- L6 0 0 > 0 0o -50 . 4.15 W j -100 3.46 0 (a) 0 00 8 0 3.95 Is _150 0 38 1/4 1/2 3/4 x in monoclinic Li,,MnBO3 1 LA. 0 -50- 4.5 so0 3.96 4.30 S150(b) ) 1/4 1/2 3/4 x in hexagonal Li,MnBO3 5 I- 4.5 V q 1 # Hexagonal 4 3.5 Q 0 Monoclinic (C) 1/2 1/4 3/4 1 x in LiMnBO3 Figure 3-4. Calculated formation enthalpies of (a) monoclinic and (b) hexagonal LiMnBO 3, and (c) their corresponding voltage profiles with respect to Li contents.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society 73 sponding voltage profiles for both are plotted in Figure 3-4(c). 7 The average voltages over the full capacity range are approximately 3.70 V for the monoclinic phase and 4.11 V for the hexagonal phase. 7 74 3.2. 3.2.1. 3.2.1.1. Results Experimental procedure Solid-state synthesis The schematic chart of experimental workflow in Figure 3-5 illustrates how to obtain the LiMnBO 3 phase by the conventional (high-temperature) solid-state synthesis. The flow is to be repetitively looped until the desired phase is obtained by optimizing experimental variables such as selection of precursors, ballmilling time, and firing conditions. Here, before delving into the specifics of LiMnBO 3 , it is worthy to summarize and clarify some of the conventional terminologies used in the solid-state synthesis and in Figure 3-5. (1) Ball-milling is process to disperse agglomerates and aggregates of feed materials and to control the average particle size and its distribution by mechanical impact between the material and wear-resistant media. It can be done with or without a solvent depending on reactivity of the precursors or desirable viscosity. In this study, ball-milling with the acetone (C2 H 5 0) solvent is used to mix precursors homogeneously and break agglomerated particles so as to obtain uniformly blended and finely ground slurries of the feed materials. (2) Calcination is one of the firing processes applicable to inorganic/ceramic materials to induce phase decomposition and transition or evaporation of a volatile segment. It normally takes place below the melting temperatures of each precursor. Here, it 75 Stoichiometric mixture of precursors Li: Mn: B0 3 = 1 : 1 : 1 Ball-milling (mixing) C2 H5 0 solvent, 24h-72 h, 300 rpm IDrying & Grinding Pre-firing (calcination) 3500C, 10 h, Ar atmosphere Grinding & Pelletizing (forming) Manual & disc-shaped, 20 MPa Firing (sintering) 450-8000C, 10 h, Ar atmosphere XRD Is the phase obtained? No Yes Product Figure 3-5. The flowchart of experimental procedure for LiMnBO3 synthesis stands for the burnout process for organics evaporation and, at the same time, forming the desirable crystalline nuclei of LiMnBO 3 phase. (3) Sintering is also a thermal treatment process to remove pores and densify a green body from a forming process preceded. It involves a mass-transport, so the firing temperature is the most controlling variable to bring about an anticipated result. In addition to densification, nuclei crystallization and/or grain/crystallite growth of the 76 product can often occur during sintering. 78 Sintering in this thesis is intended to complete the crystallization reaction of LiMnBO 3 to increase the overall crystallinity of the specimen. Overall, as a successfully optimized synthesis procedure for LiMnBO3, a stoichiometric amount of Li 2 CO 3 (Alfa Aesar, 99.99%), MnC 2 0 4 -2H 2 O (Alfa Aesar, 98%), and H 3 BO 3 (Alfa Aesar, 99.8%) were mixed by ball-milling with C2 H 5 0 (Sigma Aldrich, 99%) solvent in a polypropylene bottle for 72 h and dried overnight in air. The mixed precursor batch was then calcined to evaporate organic residues at 350oC for 10 h under argon atmosphere. With intermediate grinding, the powder specimen was pressed into disc-shaped pellet and sintered to form the LiMnBO 3 phase at 450oC-800oC for 10 h under argon atmosphere. Some specimens were coated with a conductive carbon phase. 7 For such carbon coating, 10 wt% sucrose (EMD, 99%) was added into the synthesized LiMnBO 3 powder, and they were blended and ground thoroughly by planetary ballmilling (Retzsch, PM200) at a mild speed (350 rpm) for 5~10 h. The mixture was then annealed at 5000C for 5 h under flowing argon. 7 3.2.1.2. Structure and morphology analysis Once the synthesis was completed, X-ray diffraction (XRD), transmission electron microscopy (TEM), and scanning electron microscopy (SEM) were performed in order to analyze the crystal structure, particle morphology, and particle size distribution, respectively. The X-ray patterns were obtained on Rigaku 77 Diffractometer using Cu-Ka or Cr-Ka radiation by step scanning in the 2-theta position range of 10-80* (Cu) or 25-65o (Cr). Rietveld refinement and profile matching of the powder diffraction data were performed with X'pert HighScorePlus software. A high-resolution transmission electron microscopy (HR-TEM) images and electron diffraction patterns were obtained under an accelerating voltage of 200 keV on a JEOL 2010 FEG analytical electron microscope. The specimen was suspended on a copper grid with lacey carbon. SEM images were collected on Helios Nanolab 600, and the samples were coated with gold-palladium to prevent charging. Energy-dispersive X-ray spectroscopy (EDS) was also taken with the same equipment for an elemental analysis on a Li anode. 3.2.1.3. Electrochemical performance For battery performance testing, the cathode was composed of 80 wt% active material, 15 wt% carbon black (Timcal, Super P), and 5 wt% polytetrafluoroethylene (PTFE) (Dupont, Teflon 8C). The active material and carbon black were firstly mixed with agate mortar and pestle or planetary ballmilling for 30 min, and then PTFE was added and mixed manually in an argon-filled glovebox. 1 M of LiPF 6 in ethylene carbonate (EC)-dimethyl carbonate (DMC) solution (Techno Semichem), microporous.polymer film (Ce4gard, C480), and Li metal foil (FMC) was used as an electrolyte, separator, and counter electrode, respectively. A customized Swagelok-type cell was assembled inside the glovebox and tested on Maccor 2200 at various temperatures. The loading density of the cathode was 78 kept to be approximately 3 mg/cm 2 . A 1 C rate was estimated based on 222 mA g1, and all cell tests had a constant voltage step for 1 h followed by 1 min opencircuit rest at the end of each charge and discharge. 7 3.2.2. Temperature-dependent phase formation of LiMnBO3 3.2.2.1. In-/ex-situ X-ray diffraction patterns of Li-Mn-B-O mixture Figure 3-6 shows in-situ XRD patterns of the pelletized Li-Mn-B-O specimen, which were fired at various temperatures. Each pattern was obtained after 2 h of holding duration at the designated firing temperatures. Peaks for monoclinic LiMnBO3 start to appear at 623 K, and they intensify at 723 K and 823 K. At the same time, peaks for Mn3(BO3)2 are also detected at 823 K. Above 823 K, the monoclinic phase disappears, and peaks for other phases such as Mn 3 (BO3)2 and hexagonal LiMnBO 3 are indexed until the heating stopped at 1023 K. Therefore, the implication from diffraction patterns is that the phase formation and transformation of monoclinic LiMnBO 3 is quite abrupt so that the temperature window for the synthesis targeted is very narrow ranging from 623 K to 823 K in the case of the monoclinic polymorph. 79 A m-LiMnB03 . . 1023 K 923 K 823 K 723 K 623 K 298 K .. 4I-J -- 20 e .. .__ il 25 30 35 2 Theta (Degree, Cu) Figure 3-6. In-situ XRD patterns of pelletized Li-Mn-B-C with respect to temperature change Figure 3-7 shows the XRD patterns of LiMnBO3 synthesized by a conventional solid-state method. A homogeneous monoclinic LiMnBO3 phase was obtained by calcining at 623 K for 10 h under argon atmosphere and sintering at 773 K with the identical condition. In contrast, the phase pure hexagonal LiMnBO 3 was formed when it was calcined at 623 K for 10 h under argon atmosphere and fired at 1073 K with the same condition. Both the monoclinic and hexagonal phases co-existed if sintered at 873 K or 973 K. The finding here is that the monoclinic phase is the low temperature phase and the hexagonal phase is the high temperature phase in the LiMnBO3 system, and it agrees with the previous work. 7 , 42, 67, 68 Other than the temperature, phase purity is also significantly dependent on the stoichiometry of precursors and ballmilling time. The monoclinic phase was only precisely achievable in the 80 (a) Monoclinic 0 0 0 C', I- NN 0 0 N NN 4-, N CZ '- Ci N~ 20 24 28 32 36 N C 40 2 Theta (Degree, Cu) (b) Hexagonal - .0M0 JLC 4-' i 20 I 28 24 28 2 32 6 36 40 40 2 Theta (Degree, Cu) Figure 3-7. XRD patterns of Li-Mn-B-O mixture calcined at 623 K and followed by sintering at (a) 773 K and (b) 1073 K. stoichiometric mixture of precursors with the given condition. Slight nonstoichiometry with shorter or longer ballmilling time resulted in hexagonal LiMnBO 3 and some secondary phases below 1073 K. 81 3.2.2.2. Monoclinic LiMnBO 3 Figure 3-8 shows the Rietveld refinement and profile matching of the monoclinic LiMnBO 3 phase sintered at 773 K. The profile is fitted to the structure referenced in ICSD (#200535) with the space group of C2/c. 67 The phase is a composite of monoclinic and hexagonal LiMnBO 3 , but since the amount of hexagonal one is negligible (0.3%), it can be regarded as pure monoclinic LiMnBO 3. The fitting parameters and site occupancies are summarized in Table 3-3 and Table 3-4, respectively. The structural parameters are in good agreement with values previously reported. 67 Figure 3-9 shows the SEM image of monoclinic LiMnBO 3 sintered at 773 K. The average particle size is about 100 nm, but a o - Observed Calculated m-LiMnBO 3 (ICSD# 200535) -Obs. - Cal. I li 10 20 30 lIE II 40 lii I 11111 50 DuE| 60 I lNlllIll 70 80 2 Theta (Degree, Cu) Figure 3-8. Rietveld refinement and profile matching of monoclinic LiMnBO3 sintered at 773 K, whose space group is C2/c (ICSD# 200535). The calculated pattern matches well with the observed one. 82 rather wide size distribution ranging from approximately 80 to 250 nm is observed. Table 3-3. Rietveld refinement result of monoclinic LiMnBO3 Space group C2/c Ratio (%/) 99.7 P-6 0.3 a (A) b (A) c (A) V (A3) 5.1928 8.9641 10.3741 482.6791 8.2463 8.2463 3.1363 GOF Rp Rwp 3.70 7.81 10.9 184.7003 Table 3-4. Site occupancy of monoclinic LiMnBO 3 Li Mn B 0 Site Li1 Li2 Mn1 Mn2 B 01 02 03 Occupancy 0.6567 0.5 0.4784 0.4986 0.9934 0.8990 0.9824 1 ICSD 0.5 0.5 0.5 0.5 1 1 1 1 Figure 3-9. SEM image of monoclinic LiMnBO 3 sintered at 773 K.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society 83 3.2.2.3. Hexagonal LiMnBO3 Figure 3-10 plots the Rietveld refinement and profile matching of hexagonal LiMnBO3 sintered at 1023 K for 10 h under argon atmosphere. P-6 space group symmetry in hexagonal unit cell (ICSD #94318) was used to refine the obtained XRD pattern. 42 The specimen included the insignificant amount of (0.7%) monoclinic LiMnBO3, and therefore it can be assumed that the phase is homogenous hexagonal one. The fitting parameters summarized in Table 3-5 and site occupancy in Table 3-6 agree with the previously reported values in the literature. 42 Figure 3-9 shows an SEM image of the ground hexagonal LiMnBO3 particles after sintering at 1073 K. In this case, some of the particles reach up to 800 nm, which could be - Observed Calculated h-LiMnBO3 (ICSD# 94318) - Obs. - Cal. 0 0 4-J 10 20 30 40 50 60 70 80 2 Theta (Degree, Cu) Figure 3-10. Rietveld refinement and profile matching of hexagonal LiMnBO sintered at 1073 K, whose space group is P-6 (ICSD# 94318). 84 a result of the high temperature firing. The average particle size is approximately 500 nm, but particles smaller than 200 nm is also observable. Table 3-5. Rietveld refinement result of hexagonal LiMnBO 3 Space Ratio group (%/) P-6 99.3 C2/c a (A) b (A) c (A) 8.1710 8.1710 3.1508 6.0368 0.7 7.7000 10.7814 V (A3) GOF Rp Rwp 9.97 14.5 18.4 182.1806 496.6691 Table 3-6. Site occupancy of hexagonal LiMnBO 3 0 B Li Mn Li Mn B1 B2 B3 01 02 03 Occupancy 1 0.984 1 0.99 1 0.9319 1 0.9822 ICSD 1 1 1 1 1 1 1 1 Site Figure 3-14. SEM image of hexagonal LiMnBO 3 sintered at 1023 K.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society 85 3.2.3. Preliminary electrochemical properties of LiMnBO3 polymorphs 3.2.3.1. Monoclinic LiMnBO 3 For cathode performance testing, the cathode film is prepared by a dry mixing method, which is thorough manual blending of LiMnBO 3 powder, carbon black, and granular PTFE binder with an agate mortar and pestle in the glove box. The voltage output during the galvanostatic charge and discharge input with a 0.05 C rate was obtained within 4.5-2.0 V window. The first and yet preliminary electrochemical performance of monoclinic LiMnBO 3 is plotted in Figure 3-12. The open-circuit-voltage (OCV) of the fresh 4.5 4.0 3.5 4. MOnoclinic LiMnBO 3 3.0 cv Cr, 2.5 0 2.0 :10 51 0 10 20 30 40 50 Specific Capacity (mAh g 1 ) Figure 3-12. Voltage versus capacity profiles of monoclinic LiMnBO 3 cycled at a 0.05 C rate. 86 monoclinic LiMnBO3 cathode versus a Li anode is 3.17 V. The first charge and discharge capacities are 44 and 36.2 mAh g-1, respectively. Upon cycling, the average voltage is approximately 3.3 V, but a rather large hysteresis due to significant polarization is observed. The average charging and discharging voltages, if considered separately, are 3.8 V and 2.9 V respectively. After irreversible capacity loss at the first cycle, the charge and discharge capacity is stabilized at 35 mAh g-1 after 10 cycles. Such a loss may be due to the SEI layer formation, which is typical in many Li intercalation cathodes. 79 As a representative number, the second discharging capacity was 35.6 mAh g-1, which is 16% of the theoretical capacity. 3.2.3.2. Hexagonal LiMnBO 3 Figure 3-13 shows charge and discharge cycle curves for the hexagonal LiMnBO 3 cathode with respect to a Li anode. At the identical galvanostatic testing condition to the previous Section 3.2.3.1, the hexagonal phase displayed much less Li activity compared to the monoclinic counterpart. The hysteresis is much larger and the irreversible capacity loss is more prominent. The achievable capacity is merely 10 mAh g-1 at the first discharge, and it even fades quickly away to 6 mAh g-1 after the 10th cycle. This inactivity confirms the reported result, which concluded that the hexagonal phase is inappropriate for a Li intercalation material.4 2 87 4.0 ~ 3.5 3 .0 ...... Hexagonal LiMnBO 3 ------ > 10 51 0 20 5 10 15 Specific Capacity (mAh g-1) 25 Figure 3-15. Voltage versus capacity profiles of hexagonal LiMnBO 3 cycled at a 0.05 C rate. 3.2.3.3. Optimizing the electrochemical properties of LiMnBO 3 polymorphs by planetary ball-milling and carbon coating It is generally known that percolation between an active material and carbon in an electrode affects the degree of polarization. 39 Therefore, in order to ensure better physical coagulation between active particles and carbon and to reduce polarization, LiMnB03 and carbon black were mixed by planetary ballmilling instead of manual mixing. Planetary ball-milling (also known as high-energy ball-milling) was expected to affect LiMnBO3 particle size distribution as well since the process involves highly efficient mixing and grinding of the contents. 88 Figure 3-14. SEM image of monoclinic LiMnBQ3 and carbon black mixed by planetary ballmilling.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society Figure 3-14 shows the SEM image monoclinic LiMnBO 3 after planetary ballmilling for 10 h. Compared to Figure 3-9, there is no obvious particle size reduction on average, but its size distribution becomes less deviated from the average 100 nm. Figure 3-15 shows the representative voltage curves of the planetary ballmilled and manually mixed monoclinic LiMnBO 3 cathodes in their second cycles. The capacity is clearly improved by the ball-milling process, and a second cycle discharge capacity of 72 mAh g' is achieved at a 0.05 C rate. Although improved, the hysteresis between the charge and discharge cycle remains, which implies polarization is still prohibiting a facile charge transport throughout the cathode. Since both lithium ions and electrons must freely come into and go out of cathodes, such a material must possess good electronic conductivity as well as 89 fast Li+ diffusion characteristics. However, monoclinic LiMnBO3 is likely to be an intrinsic insulator due to the stable borate group that can contribute to isolation of its Mn 2+'Mn3+ redox center within the lattice, which is often found in other polyanionic cathode materials. 37 As a quick fixing settlement, it is widely accepted that the overall electric conductivity of polyanionic cathodes can be greatly enhanced by a conductive phase coating, especially as shown in LiFeBO3 studies. 4 3 , 44 Therefore, to circumvent the highly-probable electronic conductivity problem in LiMnBO3, carbon coating on the particle was attempted in order to form percolating electron conduction paths among particles in the cathode. Carbon-coated samples were obtained by adding 10 wt% sucrose into syn- 4.5 4.0 3.5 - Monodihic LUWE 03 ,T >2. vi3.0 S2.5 > 2.0 Manual mixed 0 Planetary ball-m illed 60 40 20 Specific Capacity (mAh g1 ) 80 Figure 3-15. Voltage versus capacity profiles of monoclinic LiMnBO cathode prepared by planetary ballmilling (solid) and its comparison to the manually mixed one (dashed). Both were cycled at a 0.05 C rate. 90 Figure 3-16. SEM image of carbon coated monoclinic LiMnBO prepared by 3 planetary ballmilling after annealing.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society thesized monoclinic LiMnBO 3, mixing by planetary ball-milling for 12 hours, and annealing at 773 K for 5 h in an argon atmosphere. Combustion infrared detection analysis revealed that 3.84 wt% of carbon remained after annealing. As shown in Figure 3-16, the particle size of carbon-coated monoclinic LiMnBO 3 is approximately 100 nm with a narrow size distribution. Figure 3-17 is a high resolution transmission electron microscopy (HR-TEM) image of the carbon-coated monoclinic LiMnBO . It can be clearly seen 3 that an amorphous carbon layer uniformly covers the region of lattice fringes with thickness of 2.5 nm, approximately. In other words, the conductive carbon phase has been successfully coated on the surface of the crystalline particle. Planetary ballmilling was again performed to prepare the cathode mix. 91 7 Figure 3-17. HR-TEM image of carbon-coated monoclinic LiMnBO 3 particles. Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society Figure 3-18(a) shows the voltage curve of the carbon-coated monoclinic LiMnBO 3 cathode as a function of a specific capacity. As expected, the polarization has become noticeably decreased, which suggests the carbon coating improves the charge transport. The representative discharge capacity at second cycle is much enhanced compared to the cases in the previous sections: 100 mAh g-1 at a 0.05 C rate within 4.5-2.0 V is achieved. The capacity decreases by approximately 3.4% per cycle during the first 10 cycles. This cycling performance is rather respectable considering this work is only the early stage of development. In comparison, Figure 3-18(b) illustrates capacity improvement of hexagonal LiMnBO3 by the identical carbon-coating 92 4.5 4.0 (a) Qarbon-coated 3.5 -- 4-J monoclinic LiMnB03 3.0 2.5 0 Uncoated 0 20 40 60 10 5 2 80 100 1 120 4.5 4.0 -+ (b) Carbon coated 3.5 hexagonal - 3.0 0) CU WiMnBQ 3 0.05 C, RT 2.5 2.0 U rcoated 10 1 0 20 40 60 80 100 120 Specific Capacity (mAh g-) Figure 3-18. Voltage versus capacity profiles of carbon-coated (a) monoclinic and (b) hexagonal LiMnBO 3 (solid lines) cycled at a 0.05 C rate. approach. Although it is enhanced by the factor of two, compared to uncoated one, the increase is almost insignificant compared to that of the monocinic phase. 93 * Mn3(BO 3)2 Discharged Shifted by charging 4-.. C,, (a) Before cycling (b) Charged: 1st C ~L) 4-.. C I I* * I * (c) Discharged: 1st (d) Charged: 10th 20 24 28 32 36 40 2 Theta (Degree, Cu) Figure 3-19. Ex-situ XRD patterns of the cycled monoclinic LiMnBO3 cathode films which are collected (a) before cycling (discharged), (b) after the first charge, (c) after the first discharge, and (d) after the 10t charge. To obtain charged and discharged state, the electrodes were charged and discharged in CCCV mode within 4.5-2.0 V with a 0.05 C rate. Figure 3-19 shows ex-situ XRD patterns of the carbon-coated monoclinic LiMnBO 3 before and after cycling. In order to obtain the charged state XRD patterns, the cathode was charged galvanostatically with a 0.05 C rate, after which constant voltage charging at 4.5 V was imposed for 10 hours (CCCV mode). For the XRD pattern of the discharged state, the same CCCV mode was applied at 2.0 V. The charge and discharge capacities were about 100 mAh g- 1, which indicated 45% Li per formula unit were involved in the electrochemical reaction. Comparison of the charged state XRD patterns in Figure 3-19(b) and (d) to the discharged state patterns in Figure 3-19(a) and (c), indicates no new phases 94 formed during cycling, providing some evidence that monoclinic LiMnBO3 functions by topotactic lithiation/delithiation. In other words, the patterns indicate a reversible solid-solution type intercalation/deintercalation reaction in the case where approximately half Li per formula unit is accessed since the major peaks shift slightly upon charging, as shown in Figure 3-19(b) and (d), and recover back to the original position during discharging, as shown in Figure 3-19(a) and (c). In summary, the electrochemical performance of monoclinic LiMnBO3 was noticeably improved by planetary ballmilling, and by carbon coating, as shown in Figures 3-15 and 3-18. Since LiMnBO3 is likely a poor electronic conductor, the homogenous network between the LiMnBO3 and carbon black in the electrode is likely to facilitate electron transport. Besides, it appears that the carbon coating on the particle surface is effective in reducing polarization and increasing capacity. Moreover, even though the capacity achieved for LiMnBO3 is still below what is needed for practical lithium ion battery cathodes, it is worth studying and optimizing it more in depth since the achieved capacity value of 100 mAh g-1 by simple carbon coating has already shown a very substantial improvement compared to the previous reports on the hexagonal LiMnBO3, as compared in Figures 3-18(a) and (b) 95 3.3. 3.3.1. Discussion Diffusion behaviors of monoclinic and hexagonal LiMnBO 3 The dissimilar electrochemical properties between monoclinic and hexagonal LiMnBO 3 can be understood by looking at the difference in Li diffusion behavior originated from the structural variation. As experiment-computation collaboration, activation barriers for Li diffusion of each polymorph are calculated for the fully lithiated and fully delithiated limits using the nudged elastic band method. 80 The diffusion pathways calculated correspond to Li diffusion via hops between nearest neighbor Li sites in both monoclinic and hexagonal LiMnBO 3 cases. For technical information, in order to avoid problems with charge ordering, plain GGA (as opposed to GGA+U) was used for the elastic band calculations. Calculations were performed on supercells containing either a single Li vacancy or a single Li atom for the lithiated or delithiated states respectively. The lattice parameters were fixed at the optimized GGA+U lattice parameters of the nondefected structure. The supercell dimension was 1 x 1 x 3 for the hexagonal cells, and 2 x 2 x 1 for the monoclinic cells. 7 96 Figures 3-20(a) and (b) show the schematic diagrams of the calculated Li diffusion pathways in the monoclinic and hexagonal LiMnBO 3 unit cell, respectively. To highlight the diffusion pathway, an iso-energy surface for Li position is plotted. This iso-energy surface was obtained from an empirical energy model consisting of screened electrostatics and a repulsive Li-O pair potential. Even though we used an empirical energy model to detect and highlight the possible (a) (al) 400 300 A2 200 A2 100A 0 1 2 3 4 5 Reaction Coordinate (b) (b1) 800- L. 0 0.5 1 1.5 2 2.5 3 3.5 Reaction Coordinate Figure 3-20. Schematic diagrams of Li diffusion path and activation barriers of Li diffusion in delithiated states of (a) monoclinic and (b) hexagonal LiMnBO 3.7 Reproduced with permission from J. Electrochem. Soc., 158, A309 (2011). Copyright 2011, The Electrochemical Society 97 Li diffusion pathway, the actual migration barriers for Li are calculated with ab initio methods. For both pathways Li needs to migrate through the faces of adjacent oxygen tetrahedra. 7 In the monoclinic structure in Figure 3-20(a), each stable Li tetragonal bipyramidal site consists of two tetragonal sites (Si and S2), and there are two symmetrically distinct activated sites between Li bipyramids (Al and A2). In the hexagonal structure, nearest neighbor stable sites (S) are separated by a single activated Li site (labeled A), as shown in Figure 3-20(b). 7 In both structures the low-energy migration path is one-dimensional. The ab initio calculated diffusion barriers are significantly lower for the monoclinic structure than for the hexagonal structure, as shown in Figures 3-20(al) and (bi). In the monoclinic structure the barriers are significantly lower with migration energies of 395 meV in the delithiated state and 509 meV in the lithiated state. In the hexagonal form the migration barrier is 529 meV for the delithiated structure, and 723 meV for the lithiated structure. Therefore, the calculated activation barriers suggest that differences in Li diffusion may explain the difference in electrochemical performance between the monoclinic and hexagonal phases. While slightly higher than the calculated relevant diffusion barriers in current cathode materials such as LiCoO 2 , LiFePO 4, and LiMn 2 04, 2 9, 8 1 , 82 the monoclinic barrier energies (395 meV and 509 meV) are sufficiently low to produce reasonable diffusion constants as shown in the analysis done by Kang et al. 83 The barriers are comparable to those in other potential 98 electrode materials such as I-Li2 NiO 2 , and Li 2 Ti 2O 4 spinel. 83, 84 In contrast, the hexagonal phase's lithiated barrier energy of 723 meV is large enough, to exclude the bulk of the material from significant electrochemical performance. 3.3.2. Delithiated state stabilities of monoclinic and hexagonal LiMnBO3 As summarized in Table 3-2, there is a substantial difference in the energy of the delithiated MnBO 3 state of each polymorph. The monoclinic MnBO3 is preferred over the hexagonal phase by 76 meV per atom, which equals to 456 meV per formula unit. Thus, the difference in average voltage, 3.70 V and 4.11 V for the monoclinic and hexagonal cathodes respectively is almost entirely due to the difference in stability of the delithiated phases. While the monoclinic MnBO3 phase is thermodynamically unstable by 140 meV per atom in Table 3-2, the delithiated hexagonal phase has a considerably higher driving force to decompose with respect to the computed ground states, which are MnO2, MnB40 7 , and Mn203. This difference in the stability of the delithiated states may have influenced the battery performance of them. Since the monoclinic MnBO3 phase is energetically preferred, monoclinic LiMnBO 3 may be less susceptible to decomposition upon Li extraction. This is also one possible explanation for the different electrochemical behaviors between LiMnBO polymorphs. 99 3.3.3. Particle size and polarization Considering that both the monoclinic and hexagonal phases suffer from large polarization, it is possible that their charging is incomplete at the 4.5 V cutoff. More critically, the hexagonal LiMnBO 3, whose calculated average voltage is to be 4.11 V (compared to 3.7 V for monoclinic LiMnBO 3), would be more limited in charging by such polarization. A significant factor in the capacity difference between the two polymorphs is surely the larger particle size of the hexagonal LiMnBO 3 , as shown in Figures 3-11(a) and 3-16(b). The larger particle size is likely due to the higher temperature required to obtain the hexagonal LiMnBO 3 phase over the monoclinic phase. While the synthesis condition for hexagonal LiMnBO 3 in this work is similar to that in previous reports, 42 , 57 it should not be excluded that hexagonal LiMnBO 3 with smaller particle size, prepared through a different route would have much better performance. Indeed, Afyon et al. demonstrated how small particles can improve the overall energy storing activity although a portion of the achieved capacity seemed from the conversion reaction. 7 1 Such a prevailing behavior of phase conversion is highly likely due to the instability of the delithiated hexagonal phase, as discussed in Subsection 3.3.2, aggravated by a significant surface activity due to serious particle size reduction. 100 3.4. Conclusion In this chapter of my thesis work, LiMnBO 3 compounds were synthesized by the solid-state method and evaluated their potential as new Li intercalation cathodes. For the first time, monoclinic LiMnBO3 was electrochemically tested, optimized, and showed 100 mAh g-1 discharging capacity with possibility of further development. It is a quite promising result considering this is the inchoate work. In the case of hexagonal LiMnBO 3 , it showed no Li-intercalating behavior, as previously reported. Also, with the calculated activation barriers for Li diffusion and structural stabilities of both lithiated and delithiated states in LiMnBO3 compounds, the different electrochemical activities between monoclinic and hexagonal polymorphs are understood. As a concluding statement, monoclinic LiMnBO3 deserves further investigation on understanding its electrochemical behavior to achieve the maximal performance. Considered the indications and implications obtained through this study, identifying limiting factors and its corresponding problem-solving strategies are to follow in next chapters. 101 [End of Chapter 3] 102 CHAPTER 4 UNDERSTANDING PERFORMANCELIMITING FACTORS OF MONOCLINIC LITHIUM MANGANESE BORATE In Chapter 3, the preliminary and fairly optimized electrochemical properties of LiMnBO 3 are demonstrated. However, they fail to meet the necessary criteria as the state-of-the-art cathode materials, especially for the achievable capacity and cycle life. To resolve these issues, several attempts to optimize LiMnBO 3 have been continued. In turn, enhanced performances by particle morphology modification, surface-coating optimization, and Fe substitution have been reported for monoclinic and even hexagonal LiMnBO 3 in recent studies. 7 1, 85 Yet, a comprehensive investigation and general understanding on the electrochemical behavior of LiMnBO 3 have not been fully posted. Most importantly, it is still not clear to explain where its limitation comes from. Therefore, in this chapter, focusing on monoclinic LiMnBO 3 , the materials characteristics by iden103 tifying performance-limiting factors are investigated in order to understand its electrochemical behavior in depth and promote a maximal performance. Since monoclinic LiMnBO 3 is about to be discussed only, it will be simply described as LiMnBO 3 throughout this chapter for simplicity, otherwise noted. 104 4.1. Factors influencing the electrochemical performance of LiMnBO 3 In general, the electrochemical performance of cathodes depends on not only transport parameters such as bulk Li diffusion, bulk electronic conduction, and surface charge transfer but also conditions imposed by electrode configuration like electrical "wiring" between particles and a current collector. 39 , 40 The former is to be said as an intrinsic property of materials, and the latter is to be said the external influence from the environment. Such external factors can be excluded in this study since the applied current level is low enough to neglect the resistive effect from the electrolyte and the electrode geometry. Moreover, the performance is also determined by the structural stability at the charged and discharged states of the material. This is also materials property governing the structural integrity during cycling. Therefore, to understand the electrochemical behavior, the major two materials characteristics, the transport phenomenon and structural stability, are to be investigated in following sections. In Section 4.2, transport limitation resulted from Li diffusion and electronic conduction of LiMnBO 3 is discussed. The analysis on structural stability and its influence on the electrochemical performance are also followed in Section 4.2. 105 The limited specific capacity achievable 4.2. 4.2.1. Transport limitation In Figure 3-18(a), although mostly optimized by planetary ballmilling and carbon coating, monoclinic LiMnB03 shows the first discharge capacity of 110 mAh g-1, which is approximately equivalent to 50% of its theoretical capacity. So far, this is the highest capacity that the monoclinic LiMnBO3 phase can get by intercalation since various attempts to optimize LiMnBO3 in the literature failed to achieve more than even 30% (60 mAh g-1) of the theoretical intercalation capacity. 7 ,71,85-87 In other words, nominal 50% Li is, somehow, only extractable and insertable. It is considered that such a limited capacity is originated from sluggish transport phenomena, for example, slow diffusion and poor electronic conduction. 4.2.1.1. Temperature and rate dependence of Li activity Figures 4-1(a) and (b) show the charge and discharge (second) cycle curves for LiMnBO3 compounds at a 0.05 C rate and different temperatures within 4.52.0 V. The discharge capacity of pristine LiMnB03 is 81 mAh g- 1 at high temperature (HT, 333 K) and 35 mAh g- 1 at room temperature (RT, 299 K), as shown in Fig. 4-1(a). 7 For carbon coated LiMnBO 3 (LiMnBO3/C) in Fig. 4-1(b), the discharge capacity is 135 mAh g-1 at HT and 100 mAh g- 1 at RT. In both cases, more 106 lithium can be extracted from and inserted into the cathode at HT accompanied with less overpotential and/or polarization than at RT. It is also noticeable that the carbon coating enhances the capacity, and its effect is more substantial at RT (from 35 mAh g-1 to 100 mAh g- 1) than HT (from 81 mAh g-1 to 135 mAh g- 1). (a) 4.5 4.0 3.5 -- 3.0 2.5 2.0 * I . 4.0 4) 0) 0 I I * I I . (b) - 4.5 _j No-coating, 0.05 C, RT No-coating, 0.05 C, 328 K - .- 3.5 3.0 - 2.5 -C-coated, 0.05 C, RT C-coated, 0.05 C, 328 K 2.0 4.5 (c) 4.0 ----C-coated, 3.5 0.05 C, RT C-coated, 0.02 C, RT 3.0 2.5 2.0 * 0 I 40 . I 80 . * 120 I 160 . I 200 Specific Capacity (mAh/g) 7 Figure 4-1. Voltage vs. capacity profiles of LiMnBO3 in various conditions: (a) pristine and (b) carbon-coated LiMnBO 3 at a 0.05 C rate with respect to temperature, and (c) carbon-coated LiMnBO 3 at room temperature (RT) with respect to a rate. 107 Figure 4-1(c) shows the influence of the rate on the Li activity of LiMnBO3/C at RT: A 0.01 C rate cycling delivers significantly larger capacity of 155 mAh g- 1 at the second discharge than the 0.05 C test. In Figures 4-1(a) and (b), the improved performance of pristine and carbon coated LiMnBO3 at HT directly indicates the transport, a typical thermally activated process, limitation. By observing decrease in overpotential (or polarization), both Li diffusion and electron transport throughout the particles must be enhanced by the thermal activation. Note that the capacity promotion due to carbon coating stands out more prominently at RT (from 36 to 100 mAh g- 1) than HT (from 80 to 135 mAh g- 1). It can be explained by effectiveness of the coating. At HT, since the charge transfer reaction is already accelerated even in pristine LiMnBO 3 , it may be set aside from the rate-limiting steps. Hence, the capacity increase at HT from Figure 4-1(a) to Figure 4-1(b) originates mostly from finer percolation between particles driven by carbon coating. This differs from the RT case where both charge transfer and electrical wiring are still significantly influenced by carbon coating. Although improved, however, the overall capacity is somewhat below the expectation because the wiring resistance is likely to be high due to lack of the conductive matrix. If carbon coated, as shown in Figure 4-1(b), the wiring resistance can be significantly reduced as the coating layer can facilitate the electron transport effectively throughout the electrode and at the particle surface. Therefore, when LiMnBO 3 is carbon coated, the resistive effect from the wiring 108 becomes relatively small, and it can be presumably ruled out from the performance limiting factor supplemented by low electrode loading (-3 mg) and thickness (< 50 jim) with the low applied current and small particle size in this study.7, 39 Voltage profiles of LiMnBO 3/C with respect to the cycling rate in Figure 4- 1(c) also represent the transport (Li+ and e-) related limitation, which shows more capacity at a 0.01 C rate than at 0.05 C. All in all, considering the dependence of Li activities on temperature and rate, we can conclude that LiMnBO 3 is inherently transport-limited. 4.2.1.2. Galvanostatic intermittent titration test (GITT) Galvanostatic intermittent titration test (GITT) is an experimental technique to determine many useful electrochemical quantities. 88 For Li intercala- tion materials research, it is often employed to obtain Li diffusivity as well as equilibrium voltage profile of an electrode. 89 The unit of test is simply consisted of transient- and steady-state measurement, and it is repeated until the necessary information is achieved. 89 Starting from the equilibrated state, the transient-state measurement stands for galvanostatic charging or discharging pulse on the electrode for the short period of time. By doing so, due to a kinetic effect, Li concentration gradient is developed from the bulk to the surface of an electrode, which is measured as overpotential or underpotential. The following steady-state measurement monitors relaxation of such non-equilibrium potential as a func109 tion of time, which visualizes redistribution of Li to remove the concentration gradient. After the redistribution, since the amount of Li in the electrode has been changed by the previous galvanostatic input, the relaxed voltage represents a new equilibrium value at that Li concentration. This set of experiment can be repeated to obtain equilibrium voltage profile for all Li concentration. Figure 4-2 illustrates how the Li concentration profile corresponds to the shape of a voltage curve during galvanostatic charging. In ideal or steady-state case, which is not kinetically limited, Li concentration is always uniform throughout the electrode along with Li extraction. The more Li is extracted, the lower Li concentration becomes, but no gradient develops in the concentration profile, as shown in Figures 4-2(a-e). Therefore, the Li chemical potential determined by Li concentration at the electrode surface is identical to the bulk, and it leads to the equilibrated voltage profile depicted in the bottom-left in Figure 4-2. In real Li extraction, however, it requires a certain amount of activation for Li to diffuse out of the electrode. Moreover, due to the inevitable kinetic effect, more Li is extracted at the electrode/electrolyte surface than the electrode bulk. Consequently, Li concentration becomes inhomogeneous, and the gradient develops. If Li diffusion is extremely sluggish, Li at surface is only extractable until it gets depleted while Li in bulk remains intact (Figures 4-2(f-j)). Since an electrochemical tester reads surface potential, the voltage profile will deviate from the equilibrium one. Such a real or transient-state case is shown in bottom-right of Figure 4-2. 110 (a) CLI (b) (c) Cm (f) c (g) CA (h) Ca 0 (d) CmI (e) CmI (0) C/ rx -x X j Real Li extraction Ideal Li extraction 0) o) 4-I 4- (g)(h) 0 fa) 90b) .. * d(e (f) x in Li 1-MnB03 1 0 x in Li 1-MnB03 I Figure 4-2. Schematic diagrams of Li concentration profile in LiMnBO3 cathode with respect to Li extraction and corresponding voltage profiles: (a-e) ideal and (f-j) real cases III Since GITT features both the transient- and state-state measurement, the expected concentration change during charging can be illustrated in the order of Figure 4-2(a) (or (f)), (g), (b), (h), (c), (i), (d), (j), and (e). The schematic diagram of GITT measurement setup is illustrated in Figure 4-3. Weppner and Huggins derived a useful relation between the GITT profile and Li diffusivity based on Fick's second law (Equation 4-1).89 With appropriate initial and boundary conditions, Equations 4.2, 4.3, and 4.4, imposed by GITT, the law could be modified as following Equation 4-5.89 (b) (a) 2 0wrSources U to 0 r L 0 X to+T Tim }I/Rdrop to t+T Tim Figure 4-3. (a) Schematic diagram of cell configuration used in GITT and (b) applied galvanostatic current pulse and its response in voltage with respect to time 19Cu(x,t) -a at D CLi(x,o) = co (0 112 2 CLi(x,t) ax 2 x L) [4.2] -D aX lx=O aCL aX D - 4 = SZLiq (t > 0) = 0 (t > 0) [4.3] [4.4] x=L rr S MB MB kAEM ) [4.5] t In this derivation of diffusivity (D), it assumes the infinite one-dimension diffusion source satisfying r<< L 2 D-1 condition, where L stands for diffusing distance, which can be presumed as a particle size. 90 S and mn are surface area and weight of the active material. VM and MB are molecular volume and mass of the active compound. AEt was taken as the difference between activated and equilibrated potentials during current pulse time (r), and zEs was taken as the difference between equilibrated potentials with respect to Li composition (x). The voltage profile of LiixMnBO 3/C with respect to x obtained by galvanostatic intermittent titration test (GITT) is shown in Figure 4-4. Each step was consisted of a constant current pulse applied for 15 min at 0.01 C rate and consecutive 5 h relaxation and repeated within 4.5-2.0 V. Prior to the test, the cathode was cycled within the same voltage window and held at 2.0 V for 20 h in order to ensure a fully discharged state in the beginning of GITT. In the figure, overall 70% nominal Li was (de)intercalated, which is estimated by normalizing the cumulated capacity (155 mAh g-1) over the theoretical one. 113 Figures 4-4(a)-(c) show the voltage profiles with respect to elapsed time at the designated state of charge (SOC). As SOC increases, the relaxation time gets longer, and even the overpotential is hardly relaxed within the given time frame in Figs. 4-4(c). Since the Weppner and Huggins modeling assumes the fully equilibration of voltage, the diffusivity cannot be calculated. 89 However, Figure 4-4 still qualitatively indicates the kinetic limitation, especially for Li diffusion, inferred from large overpotential. The development of overpotential (underpotential) upon charging (discharging) suggests sluggish Li transport from the electrode bulk (diffusion-front at surface) to diffusion-front at surface (bulk) since it is the consequence of inhomogeneous Li depletion (accu- x in Li 1 MnBO 3 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 ' ' ' 4.5 ' Charge 4.0 -soc-15% 3. 0 2.4r Discharge -+- 3.6 50 S 3.5- (a) 3.6 3.0- 305 115 2.5 - 4.5 2.0 3.3Ea, 0 40 70 4.2-0- SOC-25% (C) (b) 0 60 80 120 Specific Capacity (mAh g1) 160 125 soc-45% 155 ti 165 (C) 175 Elapsed time (h) Figure 4-4. Voltage profile with respect to x in Li 1.xMnBO 3/C and elapsed time according to SOC: (a) 15%, (b) 25%, and (c) 45% obtained by GITT. 114 mulation) at the surface, as shown in Figures 4-2 and 4-5. As a result, large hysteresis is observed, so that the average equilibrium voltage during charging and discharging is 3.75 V and 3.05 V, respectively. t (a) CLiI L- t (b) t (c) Cu CUI L L t (d) t (e) <LCu CLI L L I (f) t (g) Cu Cu L L Transient-state Steady-state Figure 4-5. Estimated sequential non-equilibrating Li concentration profile of LiMnBO cathode 3 during charging by GITT. 115 4.2.1.3. Potentiostatic intermittent titration test (PITT) Similar to GITT, potentiostatic intermittent titration test (PITT) also serves as one of the useful technique to understand electrochemical behaviors in Li intercalation materials. 91, 92 Its concept is basically identical to GITT in that it forces for a diffusing species, Li+ ion, to get excited from its equilibrated state and records a responding current. However, it differs from GITT that the input is a small potentiostatic voltage step of tens of millivolt, instead of a galvanostatic current, as illustrated in Figure 4-6. The experiment drives oxidation or reduction by applying an external voltage. Then, electrochemically active Li intercalation materials must react to such an input by varying its Li concentration because the Li chemical potential of a specimen must corresponds to the voltage. Therefore, the Li diffusion must occur. 0 0 to Time to+T C 05 Area = accumulated charge Background current 0 to to+T + Time Figure 4-6. Schematic plots of one step of a potentiostatic input and its corresponding current response in PITT. 116 As the potentiostatic driving force is huge in the beginning of the step, large amount of current will flow whose value is infinite in ideal case where there is no charge transfer resistance. As time elapses, the current will decay while the overall Li chemical potential of the specimen gets equilibrated toward the external voltage. It gives a current profile as a function of time shown in Figure 4-6, and this current response is directly related to the Li chemical diffusion coefficient of the material. Thus, by analyzing the profile, the diffusivity can be extracted. Also, if the voltage step proceeds to cover all redox voltage ranges, the Li diffusivities as a function of potential can be achieved. Under the initial and boundary conditions of PITT in Equations 4.6-4.8, Fick's second law can be solved as Equations 4.9 and 4.10. The details about calculating diffusivity are followed in the next section. 91 9CL = 0 (t > 0) [4.6] aX x=L C(X= cc(x,t)-- CS -c CO [4.7] CU (0, t) = cs (t > 0) [4.8] (--C1)n erf c (n(n +1)L + 1 L -x-x+ erfc nL+x) nL + X[4.9] CS - CO c(x, t) - CO CLi(x,o) = co (0 : x 5 L) -,D-t n=O 4 = - -7 1 = 2n + 1 . (2n + 1)nw 2L 117 exp [49 rDi ((2n + 1) 2w 2 Dt 4L2 [4.10] 4.2.1.4. Diffusivity of LiMnBO3 To examine the transport limitation quantitatively, the Li chemical diffusion coefficient (D) of LiMnBO3 was determined by the potentiostatic intermittent titration technique (PITT).91, 93 By solving Fick's second law, Wen et al obtained Equation 1 for time-dependent current for short time diffusion in accordance with t « L' D- condition. 91 (t) 1 _QVQiI [4.11] 1 In the long-time approximation where t >> L2 D- is satisfied, the current as a function of time can be also expressed by Equation 2.91 I(t) = 2QD exP ( 2 - D t [4.12] Therefore, by plotting I(t) versus t-0- 5 in Eqn. 1 or ln I(t) versus t in Eqn. 2, the diffusivities can be extracted from the slopes of each equation. Both equations are valid, but in this study, Equation 1 was used to obtain Li diffusivities from 2 to 2.8 V in charging and from 4.5 to 3.8 V in discharging since the data points in these ranges were collected for the short time only. Otherwise, Equations 2 was used. Here, L defines the diffusion length, which is taken as the half of the average particle size, 50 nm, and Q stands for an accumulated charge in the voltage step. 118 Representative examples of the current response with respect to time at 2.271 V, 2.994 V and 3.782 V with a 10 mV incremental step interval are shown D = 6.38 x 10-14 cm 2 s- 1 0.0090.006 Q 4.7894x 10-4 C L = 50 x 107 cm 0.003 V =2.271 V 0.000 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 te 5 (s-0) -12.0 -12.0 -L V = 2.994 V -12.5C = 50 x 10-7 CM -13.0 -13.5 D 0 = 3.49 x 10-15 cm 2 s-I 100 200 300 400 t (s) -12.0 - L=50x10-7 cm V =3.782 V <-12.5C -13.0 -13.5 _ D = 4.67 x 10-16 cm 2 S-1 0 300 600 t (s) 900 1200 Figure 4-7. Linear fittings of current with respect to time to derive diffusivity. 119 in Figure 4-7. The linear fittings concurs well with the observed profile, and the derived Li diffusivities from either Eqn. 4-6 or 4-7 for the each step are 6.38 x 1014, 3.49 x 10-15, and 4.67 x 10-16 cm 2 s- 1 respectively. The same process was done for all voltages in the testing window. Note that since there are observable anodic and cathodic side reactions above 4.3 V and below 2.2 V, as seen in the incremental charge (dQ dV-1, C V-1) versus voltage plot in Figure 4-8, the Li chemical diffusivities are acquired from 2 to 4 V in charging and 4.5 V to 2.5 V for discharging for every 10 mV step interval. o-Charge -- 1.0 < Discharge 0.5 ) 0.0 -0.5 - E -1.0 2.0 2.5 3.0 4.0 3.5 4.5 Voltage vs. Li/Li (V) Figure 4-8. Incremental charge with respect to voltage obtained by PITT at RT with interval of 10 mV. Data points represent the value at every 50 mV. Figure 4-9 shows Li chemical diffusion coefficients obtained by PITT. The diffusivity gradually decreases from 9.8 x 10-12 to 1 x 10-14 cm 2 s-1 as charging proceeds from 2 to 2.7 V (Region I). The value in Region II also declines from 1 x 120 10-14 to 8.3 x 10-16 cm 2 s- 1, and upon further delithiation, it goes through a sud- den drop at 3.4 V and becomes nearly constant at about 4.5 x 10-16 cm 2 s-1 above 3.5 V (Region III). When discharged, the Li diffusivity begins at 1.1 1 and x 10-11 cm 2 s- gets down to 7.8 x 10-15 cm 2 s- 1 in the voltage range from 4.5 to 3.8 V (Re- gion IV). Subsequently, the descending trend continues in Region V: the diffusivity gets lowered from 7.8 x 10-15 to 5.5 x 10-16 cm 2 s-1 . The drop is less prominent while discharging than charging, but it clearly levels off at 3 x 10-16 cm 2 s-1 below 3 V (Region VI). 011 I 0: EI 0,12 E 0-13 0 0> 0-15 _0) Charge 0,16 I VI I * 0,11 0-12 0 0_13 0_1 (o 3 0-15 K -+Discharge 016 2.0 2.5 3.0 3.5 4.0 4.5 Voltage vs. Li+/Li (V) Figure 4-9. Li chemical diffusivities obtained by PITT: the values were collected with a 10 mV voltage step from 2-4 V charging and 4.5-2.5 V discharging but plotted with 50 mV intervals for clarity. The diffusivity displays three distinctive regions in charge (1,11, and 111) and discharge (IV, V, and VI). 121 The settled values with the order of 10-16 cm 2 s-1 in both charging and dis- charging are significantly lower than those of the other cathode materials whose diffusivities range from 10-13 to 10-8 cm 2 s-' in general.11 , 29,82 Therefore, based on the scale of the Li chemical diffusivity, we can also conclude the electrochemical performance of LiMnBO3 is indeed diffusion-limited. The non-uniform diffusivity values with respect to voltage (and thus the Li content in LiMnBO 3), whose trend is skew-symmetrical upon charging and discharging voltages, could be related to inaccessible Li caused by channel-blocking defects and will be discussed in the following section. 4.2.1.5. Channel blocking antisite defects Some of the obtained Li chemical diffusivities in Figure 4-9 deep down to the order of 10-16 cm 2 s- 1. With this scale range, according to x = -fD/t estimation, Li in the center of the 100 nm particles is not likely to keep up with 0.05 C-rate Li extraction and insertion, so that only Li near surface may respond to the galvanostatic input. However, such low diffusivity values are somewhat unexpected since the Li migration barrier in the LiMnBO phase derived by ab initio computation (395-509 meV) implies feasible diffusion. 7 , 64 Based on the atomistic diffusion model, D = vc 2 fexp (-) k 122 exp kT [4.13] where c is atomistic hopping distance (5.2 A), v is natural jump frequency of solids (10-13 s-1), f is correlation factor (1), Sm is entropy of migration (~1 k), such migration barrier should give the order of 10-11-10-12 cm 2 s-1 for diffusivities. Giv- en that the computed activation barriers only assume the situation where the diffusion path is free from the defect, it can be inferred that additional disturbance such as channel-blocking antisite defects (Mn in Li site) may exist. If so, because Li diffusion in the LiMnBO 3 lattice occurs along the one-dimensional channel parallel to c-axis with less possibility of inter-channel diffusion, such antisite defects will considerably influence on its Li mobility.7, 64, 94 Indeed, it was possible to estimate the amount of channel blocking defects o - I I C ) ant isites a b C d . IN TI Observed Calculated Residue I LiMnBO IA 20 WI( a Residue LiMnBO 0 - b Observed Calculated Ili d WI ant isites IIl 25 Il 30 I 40 35 2 Theta (Degree, Cu) 25.9 26.6 34.3 35.0 2 0 (Degree) 2 0 (Degree) Figure 4-10. Profile matching of the XRD pattern of monoclinic LiMnBO 3 (top) with and (bottom) without antisite defects (Mn in Li sites) whose agreement indexes are Rp=7.66, Rep=9.84 and Rp=5.86, Rwp=8.24, respectively. 123 by simulating diffraction pattern with Mn in Li sites. Figure 4-10 shows profile matching and Rietveld refinement of monoclinic LiMnBO3. If the pattern is refined without considering the antisites, the discrepancies between the observed and calculated patterns are apparent especially for peaks enclosed by dashed boxes (a) and (b): (11-2) at 25.910, (112) at 26.630, (004) at 34.640, and (130) at 34.650. On the other hand, patterns refined with antisite defects (Mn in Li sites) show very good fitting agreement, as shown in Figure 4-10(c) and (d). Given that b n -Volume Particle count fraction Gaussian fit - a i.W C 0.5 C M 0 0 < 0 E, 3 0.0 0 50 100 150 ~ 200 Particle size (nm) Figure 4-11. Particle size distribution and its correlation with the antisites. (a) SEM image of LiMnBO/C particles (scale bar: 500 nm) and (b) Representative particle size distribution and volume fraction. 124 Mn is such an electron-rich element whose position can be readily identifiable by the diffraction pattern, we consider there are a fair amount of antisites in the channel. The refinement suggests 4.47% of them may exist in the structure. Most importantly, given that the LiMnBO3 electrode is consisted of particles with the size distribution ranging from 20 to 220 nm, as shown in Figure 411(a) and (b), with median and mean size of 71 and 101 nm respectively, our system can be under the detrimental influence of channel-blockage based on the analysis by Rahul et al. 94 4.2.2. Discussion Assuming uniform distribution of the antisites defects throughout the crystal, larger size particles will suffer from more severe channel blockage than smaller ones since there will be more chance for such defects to lie in the same channel, as illustrated in Figure 4-12. In turn, it is highly likely a large portion of Li in the large particles will be inaccessible. 94 At the same time, small particles with very low probability to have blocked Li diffusion paths can respond to external voltage or current input more instantaneously than larger ones no matter it drives oxidation or reduction. In consequence, the size dependent Li kinetics may occur: in the beginning of the redox reaction, because small particles can display nominally faster Li diffusion, they can be (de)lithiated prior to the larger ones. On the contrary, since the apparent Li diffusivity in the large particles will 125 Discharged Antisite I43tIv III1tGI H3tv 100nm 20nm 60nm Li vacancy 1-D Channel Charged Figure 4-12. Schematic illustration of one-dimensional diffusion path parallel to [001] direction in monoclinic LiMnBO assuming 4.47% antisites in the channel. be low to account for the retarded Li transport by the blocked channels, they have to defer the reaction. In the context, we can interpret the GITT profile in Figure 4-4 that it reflects the aspect of size-dependent kinetics in Li (de)intercalation. The beginning oxidation/reduction reaction with less over/under-potential represents feasible Li kinetics in smaller particles, and advancing large degree of over-/under-potential is attributed to sluggish diffusion in larger particles. Also, the size-dependence can be quantitatively analyzed and related to the skew-symmetrical Li diffusivities in Figure 4-9. The number of Li sites will be in direct proportion to the particle volume, so the total number of antisites in a sin126 gle particle can be considered proportional to the cube of the particle diameter. If the particle sizes are distributed to follow the Gaussian function as fitted in Figure 4-11(b), the number of particle whose diameter is smaller than 50 nm is about 12%. This translates to 3% of the total volume of the active mass, and therefore the achievable capacity from particles with 50 nm or less size is only about 3% of the theoretical capacity, which is about 7 mAh g-1. In other words, relatively facile diffusion can happen, but its impact on overall capacity is minor. In Figure 4-9, the region I whose diffusivity is relatively large (10-14-10-12 cm 2 s-1) lies below 2.78 V and above 3.8 V for charging and discharging, respec- Charge > 4.5 27 ++117+ VI 0 > Discharge -+- 89 ++ V IV 31 +4+ 1.5 0 40 80 120 160 Specific Capacity (mAh g~) Figure 4-13. Integrated incremental charge (dQ dV 1) with respect to voltage (V). The voltage profile as a function of specific capacity acquired by integrating the incremental charges over voltage from PITT. 127 tively. This corresponds approximately to the voltage where 7 mAh g- 1 can be achieved in integrated dQ dV-1 plot in Figure 4-13 (Region I). Region II seems to be comprised of particles with 75 nm size. The volume fraction of the regime is about 12% in Figure 4-11(b), which parallels to the capacity of 27 mAh g-1 (12% of the theoretical capacity) in Figure 4-13. This matches to the slow diffusion with order of 10-15 cm 2 s-1 . The rest of the capacity, which is the dominant por- tion, can be achieved in 75-150 nm particles but only in slow rates such as 0.05 C or 0.01 C to keep up with Li sluggish diffusion with the order of 10-16 cm 2 s-1 . When the diffusivities set the minimum value at about 10-16 cm 2 s-1, it infers the system at that point is dealing with larger particles with many blocked channels. Diffusivities reaches the lowest value since it embraces the hardly accessible Li securely capped by antisites in larger particles. Thus Li diffusion in this stage seems to occur by detouring the defects around to other channels. On the other hand, 30% of theoretical capacity will be hardly achievable at any rates because it is the portion of the largest particles, as black-shaded in Figure 4-11(b), whose diameter is more than 150 nm. It is expected almost every channel is likely to be blocked by antisites in particles with those sizes, so the capacity from them will be negligible. All in all, it can explain the diffusionlimited capacity of LiMnBO3, and therefore we conclude that the antisite defect plays a major role in the electrochemical performance of monoclinic LiMnBO 3 . 128 4.3. 4.3.1. Capacity fading upon cycles Limitation from structural instability The capacity retention of LiMnBO 3 in Figure 3-18 can be considered reasonable since it is only an initial stage of material development. However, to compete with the state-of-the-art cathode materials, it is also true that the cyclability must be improved to the large extent. To do so, the capacity fading mechanism must be understood and addressed. In order to understand the cycling behavior of a Li storage material, an important focus to investigate would be its structural stability. Especially, analyzing the stability of delithiated states is indispensable and can be more than critical since it is frequently undermined by an oxidizing environment during charging, particularly at high SOC. Such analysis, however, cannot have been made in the previous LiMnBO 3 studies because the specimens prepared by electrochemical charging and chemical delithiation were inappropriate to represent the charged state due to insufficient Li extraction or amorphization during oxidation. 85 In this section, the phase stability of monoclinic Lij xMnBO3 (0 x 1) with respect to Li contents, x, is investigated by experimental demonstration of electrochemically charged and chemical delithiated LiMnBO 3 for the first time. The results are also compared with computational results obtained from the first- 129 principles calculation. In the process, the possible capacity fading mechanism of LiMnBO 3 is accounted for. Besides, how to further improve its cycling performance is suggested. 4.3.1.1. Computed formation enthalpy Table 4-1 summarizes calculated relative formation enthalpies of the monoclinic LiixMnBO3 (0 < x < 1) phase at zero K with respect to the ground states based on convex-hull construction. 33 , 95, 96 The listed (competing) stable phases are the lowest energy states obtained from the quaternary phase diagram of the Li-Mn-B-O system, which is shown in Figure 4-14, projected onto certain Li chemical potentials (ptLi).33, 97 The AE (also known as the energy-above-hull) value, Table 4-1. Computed stability of Li 1.MnBO 3 (0 s x s 1) and its ground states with respect to Li concentration at zero K. AE (meV/atom) stands for difference in energy from the computed ground states. x in Li1.,MnBO3 Formula AE Ground states 0 LiMnBO 3 4 h-LiMnBO 3 0.12 LiO. 88 MnBO 3 23 h-LiMnBO 3 Li 2 B4 0 7 Mn 2BO 4 Mn 3 04 0.5 LiO. 5OMnBO 3 73 h-LiMnBO 3 L 2 B4 0 7 Mn 2 BO 4 Mn 304 0.62 LiO.3 8MnBO 3 100 Li 3B70 12 Mn 2BO 4 Mn 3 04 0.75 LiO.2 5MnBO 3 119 Li 3B70 12 Mn 4 B4 0 7 Mn 2 03 1 MnBO 3 140 MnB 407MnO 2 Mn 2O 3 130 0.8 0 LIBO? 0.7 L 2 B4 0 7 0.6 * LB 3LiB 701 0r..-- L*nBJ .nO -- L3803. 0.5 Mn 2 BO 4 Mn 3 (B0 3 )2 2 0.4 0.3 n203 -....--- Ui202 LiMMnO2 - i . 20 L'2MnO2 - Bl~ Mn Mn4 207 . - 0.2 30 4 .......--- M nO 00 -MnB4 MnB - L 0.0 -0.B .. 0.6 0.6 .5 0.2 3 0-4 0.2 06n YiMnO, 0.1 1.0 0,0 Figure 4-14. Li-Mn-B-O quaternary phase diagram. which scales with instability, gradually increases from 4 meV per atom at x = 0 to 140 meV per atom at x = 1. The result indicates the energetic status for all x is unstable against the ground states. However, we may be able to regard the status of Lil.xMnBO3 as 'relatively stable' where x 5 0.5 and 'relatively unstable' where x > 0.62 due to the statistical fact that 90% of the computed energies of ex96 isting compounds in ICSD range from 0 to 100 meV per atom. 131 4.3.1.2. Electrochemical charging a 4.5 4.0 (1 3.5 3. - C 0.01 C <>0.05 - 0 > 2.52.040 0 80 120 160 200 Specific capacity (mAh g-) b -2-00- Charge ~---Charge Discharge -0- Discharge o>200 .-< E 160 o ~120 M A". 0. 01 C -- .- - 80 o.5 c 0 1 2 3 4 5 6 7 8 9 10 ik Cycle number C - - Before cycling -- Charged, 105 mAh gCharged, 155 mAh g" --- * Mn 3 (B0 3 )2 Mn 3 C 30 34 38 42 46 50 54 58 62 2 Theta (Degree, Cr) Figure 4-15. (a) Voltage vs. capacity curves of LiMnBO 3/C in the second cycle at 0.01 C and 0.05 C, RT, (b) their capacity retention for 10 cycles with photographs of the anode after the designated discharge cycles, and (c) ex-situ XRD patterns of charged Li 1.xMnBO 3/C electrodes with different obtained capacity. 132 Figures 4-15(a) and (b) show the representative (second) charge and discharge profiles for carbon coated LiMnBO 3 (LiMnBO3/C) at room temperature with respect to a cycling rate and their corresponding capacity retentions for 10 cycles, respectively. It is clearly seen that although a 0.01 C rate cycling delivers significantly larger capacity of 155 mAh g-1 at the second discharge than that of the 0.05 C discharging (100 mAh g-1), its capacity fading is more severe (5.1% per cycle) than in 0.05 C cycling (3.4% per cycle). Figure 4-13(c) shows ex-situ XRD patterns of charged Lii-xMnBO 3/C electrodes with respect to the obtained charge capacity. The additional peak for Mn304, which should have been decomposed from the part of the delithiated state of LiMnBO 3/C, prominently appears along with significant amount of Li extraction. The trend of capacity fading in Figure 4-15(b) can be explained by looking at the predicted phase stability of LiixMnBO 3 (0 < x < 1) in Table 4-1. When the LiMnBO 3/C cathode was cycled at a 0.01 C rate, the charged state became un- stable due to the large amount of Li extraction (x ~ 0.69), and such instability could trigger partial phase decomposition into the lower energy phases like Mn304, as shown in ex-situ XRD pattern in Figure 4-15(c). Since the actual amount of the active mass had been lost at charging, less capacity was deliverable at following discharging, which led to severe capacity fading in consecutive cycles. On the contrary, the charged state might not be so unstable enough to drive phase decomposition at 0.05 C rate charging, whose states (x ~ 0.45) locat- 133 ed in the "relatively stable" regime. Thus, in this case, almost the same amount of Li could be extracted and inserted over multiple cycles at 0.05 C cycling. This computed stability discussion can be also extended to account for generally superior cyclability of LiFeBO 3 to LiMnBO 3 .43 The AE, the energy-abovehull, values of Lil-xFeBO 3 (0 _ x < 1) are summarized in Table 4-3. They range from 0 to 60 meV per atom for all x, which are estimated fairly stable against decomposition into the ground states. In other words, unlike LiMnBO 3 , there is more chance for LiFeBO 3 to remain intact upon charging and, therefore, cycling. Hence, its greater cycling performance to LiMnBO 3 can be attributed to the stability of its delithiated states. Table 4-2. Computed stability of Li-,FeBO 3 (0 s x < 1) and its ground states with respect to Li concentration at zero K. AE (meV/atom) stands for difference in energy from the computed ground states. x in Li 1 ,FeBO Formula AE Ground states o LiFeBO 3 0 LiFeBO 3 0.12 LiO. 88FeBO 3 13 LiFeBO 3 Fe 3BO 5 LiBO 2 0.5 LiO. 5OFeBO 3 42 LiFeBO 3 Li 2 B4 0 7 Fe 3 BO 5 LiBO 2 , Fe 2O 3 0.62 LiO. 38FeBO 3 58 0.75 LiO.2 5FeBO 3 60 1 FeBO 3 54 3 Li 2 B40 7 Fe 3BO 5 Li 3 B 7 0 Li 3 B 70 12 Fe 2O 3 Fe 2 B 2 0 Fe 2O 3 B2 0 3 134 12 Fe 2 O 3 B2 0 3 4.3.1.3. Chemical delithiation a --- Pristine * Mn (B0 3 )2 Chem. Delith. 52 53 58 62 -' C -Ilk 30 34 38 46 42 50 54 2 Theta (Degree, Cr) b C L' in LiMnB03 Pristine 2000 1000 0-201 -2000 No Li in LiMnBO 3 :*-j Chem. Delith. 2000 -1000 1000 0 7Li -1000 -2000 Shift (ppm) Figure 4-16. (a) XRD patterns, (b) HR-TEM images (scale bar: 5 nm), and (C) 7Li MAS NMR spectra of pristine and chemically delithiated Li1.xMnBO 3 respectively. 135 To evaluate the fully delithiated stability of the charged states, chemical delithiation was attempted. Figure 4-16(a) shows XRD patterns of LiMnBO 3 before and after chemical delithiation (for 1.5 days), and Figure 4-16(b) is their corresponding high resolution transmission electron microscopy (HR-TEM) images. When delithiated, the overall XRD peak intensity decreased while maintaining LiMnBO 3 Chem. Delith.> MnBO 3 a b Figure 4-17. Schematic diagram of c-lattice parameter change upon delithiation 4-J 10 15 20 30 35 40 45 50 55 2 Theta (Degree, Cu) 25 60 Figure 4-18. XRD pattern and HR-TEM image (scale bar: 5 nm) of chemically delithiated Li 1.xMnBO 3 for 10 days. The inset is electron diffraction pattern of the specimen showing a typical diffused amorphous ring pattern. 136 the monoclinic unit cell. Moreover, (004) peak-shift toward a higher angle indicates shrinkage of c-lattice parameter implying Li extraction from the lattice. 43 Such lattice parameter change is illustrated in Figure 4-17. HR-TEM for the identical specimen also shows definite lattice fringes representing a crystalline particle. Note that Mn 2 (BO 3)2 was present as an impurity phase in the pristine specimen, whose intensity did not change upon chemical delithiation. Also, if the delithiation continued for more than 1.5 days, the crystalline LijxMnBO 3 specimen suddenly turned into an amorphous phase, as shown in Figure 4-18. This amorphization, which agrees with the reported result, was accompanied with significant loss in collected mass of the specimen. 85 Figure 4-16(c) shows 7Li magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra of pristine and chemically delithiated LiMnBO 3. Isotropic shift is observed in the pristine specimen at -201 ppm in response to Li in the monoclinic lithium-metal-borate unit cell.6 3 On the other hand, there is no such a resonance occurred at the same frequency in the chemically delithiated specimen, and it is a convincing evidence that no Li exists in the structure. Therefore, combining the results in Fig. 4-16, it is considered the fully delithiated MnBO 3 is finally obtained via chemical delithiation, which has never been reported. It is worth noting that the resonance at 0 ppm is simply due to diamagnetic Liphases such as unwashed LiBF 4 from chemical delithiation or Li 2B 4 0 posed from Lii-xMnBO3 6. 3 ,98 137 7 decom- 4.3.2. 4.3.2.1. Discussion Decomposition of delithiated states Figure 4-19 shows profile matching of the XRD pattern obtained from the chemically delithiated MnBO 3 specimen. Using ICSD structure of LiMnBO 3 (#200535), the pattern was refined with no Li present in the unit cell. Fully delithiated MnBO 3 (51 wt%) is identified as a major phase, and Mn (BO ) (29 wt%) 3 3 2 as an impurity and a few secondary phases such as Mn30 4 (11 wt%) and Li2 B 4 07 (9 wt%) coexist. - I Li 0MnBO 3 I Mn 2(B0 3 )2 IMn3O4 I L 2 B40 7 o Observed - Calculated - Residue 30 34 38 42 46 50 54 58 62 2 Theta (Degree, Cr) Figure 4-19. Profile matching of the XRD pattern of chemically delithiated MnBO3 . The secondary phases produced by chemical delithiation exemplify the products by Li extraction at a constant pLi condition. 3 3 , 95 Indeed, they mostly 138 B L'4B3207 B6 Li .,MnBO3 MnB 40 7 Li B7 0 2 nB4 Mn2BO4 Mn 3 B4 B2Q3 L14B207 . :: 2 LiBOi** 1-x 3 Li3BO*. MnOV Mn 3 O4 MnO Mn4 L12 0 LM222 Li 2 Mn'nB2O1 3(803)2 B04 4 M Figure 4-20. Graphical representation of Li grand-potential phase diagram with competing phases of Li 1.xMnB0 3 at pLi=-4. 64 eV. Red dots stand for stable phases. Li1. ,MnBO 3 is marked with the blue cross, which is unstable with respect to Li 4B2O7 , Mn 2BO 4 , and Mn 3O4 . agree with the predicted phases at gLi=-4.64 eV in Figure 4-20. The phase diagram in the figure is a projection of Li-Mn-B-O quaternary phase diagram on the constant pLi=-4.64 eV, which is shaded as green in Figure 4-14. Although Li2 B4 07 and Mn3O4 do not appear at pLi=-5.1 eV, the equivalent potential of the NO 2+/NO2 oxidizer, 99 taking into account the kinetic effect of the delithiation reaction, it is considered the thermodynamic aspect of computed phase decomposition is well represented by the experiment. Therefore, taken together with phase decomposition in the charged electrode, it can be concluded the delithiated state of monoclinic LiMnBO3 becomes unstable and loses its structural integrity due to substantial Li extraction and/or the highly oxidizing environment. This instability driven phase decomposition can be one of the major issues causing irreversible cycling behavior in LiMnBO3. 139 4.3.2.2. Mn dissolution Mn dissolution from LiMnBO 3 and its dissolving tendency upon cycles is also a main obstacle for capacity retention. According to photographs in Figure 415(b) that represents Li anodes taken from the disassembled LiMnBO 3/C cell at the designated 0.05 C discharge cycle, the blemish on the anodes intensifies as cycling number increases. Energy-dispersive X-ray spectroscopy (EDS) in Figure 4-21 verifies that the identity of the stained spot in Figure 4-15(b) is deposited Mn from the cathode during cycling. As reported in Li-Mn-spinel oxides,100 - 02 it can be concluded the chemical instability of LiMnBO 3 in an acidic environment is involved with the problem. Moreover, in Figure 4-16 (a) and 4-19, the phase-identified (relative) amount of Mn3(BO 3) 2 in the chemically delithiated specimen is 29 wt%, which used to be 5 wt% in the pristine sample. Since the absolute peak intensity of 4-A 0 1 2 3 4 5 6 7 8 Energy (keV) Figure 4-21. EDS of the designated spot on the Li anode in Figure 4-11(b) verifies that the identity of the stain is Mn deposited from the cathode during cycling, excluding P and F from the soaked electrolyte salt and C and 0 from the equipment. 140 Mn2(BO3)2 is unchanged before and after chemical delithiation, it implies the some amounts of Lii-xMnBO 3 has been dissolved away during the process. Elemental analysis by inductively coupled plasma (ICP) in Table 4-3 confirms the amount of Mn is actually deficient from the stoichiometric ratio. Table 4-3. Atomic ratio of m-LiMnBO 3 specimen before and after chemical delithiation by inductive coupled plasma (ICP, ASTM E 1097-12) and inert gas fusion (ASTM E 1019-11). The excess amount of Li and B may be due to the LiBF 4 residue. Atomic ratio Chemical Delithiation Li Mn B 0 1.07 1.01 0.97 3 Li Mn B 0 0.38 0.63 1.55 3 Before After 141 4.4. Conclusion In summary, the stability of LiMnBO decreases significantly in the process of Li extraction, as demonstrated by chemical delithiation and ex-situ XRD on the charged electrode. Such results agree well with the predicted phase stability by first-principles modeling. The charged state instability leads to actual loss in the active mass by both phase decomposition and Mn dissolution, which are critically responsible for capacity degradation upon cycling. Therefore, the phase instability can be one of the major factors that limit the electrochemical performance of LiMnBO3 . It is unfortunate that stability is an inherent material property, which cannot be simply manipulated. One doable approach to enhance the stability of LiMnBO 3 can be elaborating Fe substitution into Mn sites since LiFeBO 3 has more strength to resist phase decomposition during charging. [ref] Also, preventing from the aggressive reaction between LiMnBO 3 and the electrolyte at the particle surface by introducing a protective coating can help to reduce the Mn dissolution. Since fully delithiated MnB0 3 has been obtainable, this infers that if the loss in the active mass can be avoided, full capacity cycling will be expected. [End of Chapter 4] 142 PART THREE Designing high capacity polyanionic cathode Concluding thesis statement 143 [This page is intended to be blank.] 144 CHAPTER 5 DESIGNING HIGH CAPACITY POLYANIONIC CATHODES In the previous chapter, it is learned that the performance of monoclinic LiMnBO 3 is largely limited by (1) sluggish diffusion originated from antisite defects and (2) structural instability of delithiated states during charging. When it comes to the transport limitation, since the presence of antisite defects seriously undermines overall Li activity in large particles of the LiMnBO 3 cathode, minimizing its particle size as small as possible will significantly boost its achievable intercalation capacity. However, it is almost unmanageable to further reduce the particle size by the current synthesis method because it seems to be already at its lowest limit.7, 43 Also, alternative conventional methods, such as sol-gel and carbothermal synthesis are not considered suitable to fabricate monoclinic LiMnBO 3 since the products tend to involve a great deal of impurities as well as 145 larger particle size, which make its Li-intercalation properties even worse. 7 103 Thus, to find a way promote a maximal performance, nanoparticle synthesis can be one doable solution in the LiMnBO 3 system. Yet, such nanosizing may not be an ultimate answer to deal with the immanent phase decomposition and Mn dissolution issue due to the increased surface activity. Therefore, in this chapter, the effort to enhance the structural stability is laid out. To do so, a portion of Mn is substituted by other elements. The first strategy stated in Section 5.1 is to replace Mn to inactive elements such as Mg and Zn partially so as to stabilize the intercalating host during cycling. The second strategy in Section 5.2 is to utilize multiple redox elements such as Fe, Co, Ni, and Cu along with Mn to achieve the maximal capacity. Section 5.3 covers the co-doping of Mg and Fe for synergetic effects on both cyclability and capacity. Also, the property improvement by controlling an experimental process is discussed in Section 5.4, and the chapter is concluded in Section 5.4. 146 Substitution strategy 5.1. 5.1.1. Motivation Geoffroy et al. computed a variety of the redox potential of transition metals in polyanionic cathodes, whose value and/or trend agree with the experimental observations with the reasonable accuracy. 75 In Figure 5-1, for instance, the reaction voltage of Fe2 +/Fe3+ redox couple typically sits lower than that of Mn 2+/Mn 3+ in phosphates. 75 For Co2 +/Co3 + and Ni 2 +/Ni3 + redox couples, their volt- 3+/4+ 3+/4+ A 213 3rN .i~,4+/4Ni23+/+ Bib Mn2+4C24 4 445 :PC1 +3 4 0 4- 0 5 .800 Wh/kg~ Sn.^-,+% M+ -44 / Fe2 e *o+s M0+6*Sn** V34 3 +/4+ 4-n tl- 2+'-- 41 Cr2 -.. 600 Wh./k M4+/5+ 0+ 100 Capacity (mAh/g) 200 Figure 5-1. Computed average voltage in phosphates versus maximum gravimetric capacity achievable. Specific energy curves at 600 and 800 Wh/kg are drawn on the figure (blue dashed lines). The red dashed line indicates the upper voltage which we consider safe against decomposition of the electrolyte. 75 Reprinted (adapted) with permission from Hautier, G.; Jain, A.; Mueller, T.; Moore, C.; Ong, S. P.; Ceder, G., Designing Multielectron Lithium-Ion Phosphate Cathodes by Mixing Transition Metals. Chemistry of Materials 2013, 25, (10), 2064-2074. Copyright 2013 American Chemical Society 147 ages tend to locate higher than that of Mn 2 +/Mn 3+, and particularly the Ni2 +/Ni3 + couple often exceeds the tolerance of currently available electrolytes, as heighted in the figure. 7 5 In general, Mn 2+/Mn 3+ redox reaction in many other polyanionic cathodes occurs at the optimal voltage permitted by the electrolyte. 7 5 , 104 However, cathode materials possessing Mn redox element often suffer from Jahn-Teller distortion, Mn dissolution, structural instability, and so on. 10 1, 105,106 In turn, most of Mn-containing polyanionic compounds are outperformed by their Fe siblings in terms of the achievable capacity, rate capability, and cyclability. 7 , 31, 43, 107 As a result, in many studies, optimization of the electrochemical performance via substituting a portion of Mn into Fe or other transition metals is attempted. 4 5 , 85, 108 Considering LiMnBO 3 is also confronted by the analogous obstacles, as discussed in PART TWO, the similar substitution strategy can be applied to improve the electrochemical properties of the monoclinic LiMnBO 3 cathode. Not only Fe but also other transition metals are expected to substitute Mn partially. For lithium metal borate chemistry, however, the similar study on redox voltage is not reported in the literature except for LiFeBO 3 , whose average voltage is about 2.8 V.43 Given that the redox reaction of Mn 2 +/Mn 3+ in the LiMnBO 3 occurs at 3.3 V on average (3.8 V at charging and 2.8 V at discharging),7 there may be still a margin in the voltage window for substituted Co or Ni to be activated along with Mn at the same time. Moreover, if substituted by nontransition metal elements, the substituent can possibly function as a structural 148 stabilizer. Thus, in following sections of this thesis, it is discussed how to approach to a systematic substitution. 5.1.2. The substitution strategy As a general and yet targeted strategy of improving performance in polyanionic cathode materials, a portion of Mn in LiMnBO 3 can be substituted by other elements. The expectations of substitution in LiMnBO 3 system can be (1) to prevent Mn dissolution and phase decomposition at the charged state by stabilizing the framework, (2) to promote more than 50% capacity by activating multiple redox elements, and (3) to perturb ordering between Li and its vacancy or Mn by introducing foreign element. For a systematic search for the Mn substituents, it is worth looking at coordination numbers and ionic radius to construct the trigonal bipyramid, which is the main framework of Mn 2 + coordination in monoclinic LiMnBO 3 . The trigonal bipyramidal Mn 2+ ion has a five-fold nominal coordination surrounded by 0 (MnO5), and the MnO5 polyhedra are edge-shared with each other, as shown in Figure 5-2. The polyhedron is neither close-packed nor equi-bond length structure. 67 In the literature, however, it is reported that Mn 2 + favors the upper or lower tetrahedral-like site more than the trigonal bipyramidal one. 67 Note that both Mnl and Mn2 sites in Figure 5-2 cannot be occupied at the same time but disor149 Figure 5-2. Schematic diagram of edge-sharing trigonal bipyramidal Mn coordinated by five 0. Mn sits off-centered from the trigonal bipyramidal sites and tends to occupy either the upper or lower tetragonal-like site. dered into either one of them due to the proximity between sites.7, 67 With this polyhedral arrangement, the size of Mnl site is approximated to be 63 pm, which agrees with the Shannon radii of Mn2+ (66 pm) whose coordination number is four.109 Therefore, it can be presumed that the coordination of Mn2+ in monoclinic LiMnB03 is likely to be more or less tetragonal other than trigonal bipyramidal, namely pseudo-tetragonal. There are only a few options for substituting transition and non-transition metal ions that satisfy the required criteria of valence, ionic radius, and coordi- nation number. They are Mg2+, Mn2+, Fe2+, C02+, Ni2+, CU2+, and Zn2+, as summarized in Table 5-1. Among them, Mg, Fe, Co, and Zn are already reported to form the identical monoclinic lithium (transition) metal borate structure. 42, 67, 69 Also, they can be divided into two categories: inactive and active elements. Inactive 150 Table 5-1. Ionic radius (A) of possible substituents for Mn in LiMnBO 3 Coordination number Mg Mn Fe Co Ni Cu Zn 5 0.66 0.75 n/a 0.67 0.63 0.65 0.68 4 0.57 0.66 0.63 0.58 0.55 0.57 0.6 5 n/a 0.58 0.58 n/a n/a 0.65 n/a 4 n/a n/a 0.49 0.61 n/a 0.65 n/a 2+ 3+ elements are to serve as a structural stabilizer upon substitution. Active elements are to act as a redox center. Based on computational studies on a redox potential, C02+/Co3+ and Ni 2 +/Ni3+ generally tend to show higher reaction voltages than Mn 2 +/3 + within the same chemical system,7 5 so it is expected to elevate the voltage if substituted. 5.1.3. Inactive element substitution As discussed in Chapter 3 and 4, although the structural stability of monoclinic LiMnBO 3 against decomposition is estimated to be stable, it is different for the delithiated Lii-xMnBO 3 states when x is large. 7 A computed Li-Mn-B-O quaternary phase diagram projected onto constant Li chemical potential plane in Figure 4-18 shows that Lil-xMnBO 3 is unstable with respect to Li 4 B 2 0 7, Mn 2 BO 4 and Mn304. Inferred from that, the partially delithiated LiixMnBO 3 state may 151 02 MnB0 3 MnO MnB 407 MnB3 B203 . Li3 701M2 MnO Mno2 Mn2BO4 - -Mn(B3)2 M 07 B6 0 Mn Mn 2B MnB MnB 4 B Figure 5-3. Graphical representation of computed Mn-B-0 2 ternary phase diagram. Red dots stand for stable phases. MnBO 3 is marked with the blue cross, which is unstable with respect to MnO 2 , Mn 20 3, and MnB 4 0 7. not be as stable as the fully lithiated LiMnBO 3. Moreover, fully delithiated MnBO3 is quite unstable, and it may undergo phase decomposition into MnO2, Mn2O3, and MnB 407, as shown in Figure 5-3.7 Therefore, to maintain the structural stability upon delithiation can possibly play a critical role to achieve the maximal capacity in a reversible manner. This structural stabilization can be achieved by incorporation of inactive elements, as demonstrated in LiCoO2 and LiMn204 studies.2 6 , 110 In the LiMnBO3 2 case, Mg and Zn may be able to serve as structural stabilizers since Mg + and Zn 2+ can have either four- or five-fold coordination (that is, pseudo-tetragonal co109 ordination) with proper ionic radius to reside in the trigonal bipyramids. In- deed suggested LiMgBO3 and LiZnBO 3 are known to form in the same monoclinic framework as LiMnB0 3.67,69 Therefore, both are expected to incorpo- 152 rate into the existing sites to form a solid solution with LiMnBO 3 . The substitution amount varies but it is set to be less than 20% for Mg and 10% for Zn so as not to compromise the theoretical capacity of LiMnBO 3 significantly. 5.1.3.1. LiMgyMn-yBO An accurate 3 (0 y 0.2) measure of stoichiometric Li 2 CO 3 (Alfa Aesar, 99.99%), MnC20 4 -2H 2 0 (Alfa Aesar, 98%), MgC20 4 -2H 2 0 (Alfa Aesar, 99.9%) and H 3B0 3 (Alfa Aesar, 99.8%) were mixed by rotational ballmilling with C2 H 5 0 (Sigma Aldrich, 99%) solvent in a polypropylene jar for 72 h. After dried overnight in air, the mixed batch was calcined at 623 K for 10 h under argon atmosphere. With intermediate grinding, the specimen was pressed into disc-shaped pellet and fired at 773-823 K for 10 h under argon atmosphere. For carbon-coating, 10 wt% sucrose (EMD, 99%) was added into the fired specimen, and they were blended thoroughly by planetary ballmilling (Retzsch, PM200) at a 350 rpm for 5 h. Annealing was conducted at 773-798 K for 5 h under flowing argon. Figure 5-4 shows the XRD patterns of LiMgyMniyBO 3 (0 < y < 0.2) specimens fired at 773-823 K and their lattice parameters changes obtained by Rietveld refinement. No outstanding peaks for impurities are detected other than Mn 3 (BO3)2, which also exists in the undoped LiMnBO 3 cases, at around 490 of 2-theta position (Cr-target). Its composition is less than 1%. Also, as y increases, the all of lattice parameters decrease reflecting the smaller unit cell volume 153 (a) LiMgYMn 1 -yBO 3 y=0.20 4-0 C s....y=0l 25 30 35 40 45 60 55 50 y=0.05 55 50 65 2 Theta (Degree, Cr) (b) 0? 5.200" a 5.195 -ECu CE 8.98 C 10.3 10.3 8.97 10.34. 5.190 1.. 5.185" 10.3 2 8.96 0 5 10 20 Mg substitution (%) 0 5 10 20 Mg substitution (%) 0 5 10 20 Mg substitution (%) Figure 5-4. (a) XRD patterns of LiMgyMnl-yBO 3 (0 s y 5 0.2) fired at 773~823 K and (b) their refined lattice parameters. of LiMgBO 3 . Thus, in all y, Mg successfully substituted Mn to form solid solution with satisfying Vegard's law. Figure 5-5 shows the voltage versus capacity curves for LiMgyMniyBO3 (0 y < 0.2) cycled at a 0.05 C rate. The 1 C rate for each composition is based on their individual theoretical capacities: 213, 205, and 187 mAh g-1 for respective y = 0.05, 0.1, and 0.2. In all y, Mg substitution noticeably improves the cycle retention over LiMnBO3. During five cycles, which is where the most severe fading usually occurs, the capacity fading rate per cycle obviously decreased from 5.7% in LiMnBO 3 to 1.5% in LiMgo.o5Mno.95BO3, 1.2% in LiMgo.lMno.9BO3, and 2.3% in 154 4.0 S2.5 LMnBO S2.0 - -3.5 >1.5 4.5 4.0 - M3 . ...... C .. LiMgo 2M I _ I..., ... I I ...... J .............A () BO 3 ....... .... LiM4g, MnB 3 0- 1.5 y=.0 0 20 40 60 80 100 120 0 20 40 60 80 01 . 100 120 Specific Capacity (mAh g') Specific Capacity (mAh g') Figure 5-5. Five consecutive charge and discharge curves for LiMgyMn1.yBO 3 (0sysO. 2): (a) y=0.05, (b) y=0.1, and (c) y=0.2 cycled at a 0.05 C rate. For comparison, the representative second cycles of each y are plotted in (d). The dotted line represents the undoped LiMnBO 3. LiMgo.2Mno.8B03. Other than cyclability, the substitution also enhances the discharge capacities in y = 0.1 and 0.2 cases. Note that since Mg substitution eventually leads to decrease in the theoretical capacity of the modified formula, 10% substitution is chosen to be an optimized composition. It was expected for the Mg substituted Li(Mg,Mn)B03 to provide the enhanced structural stability onto the intercalation host by diminishing the amount of fully oxidized Mn 3+ ions via Mg implantation at the particle surface. In addition, according to scanning tunneling electron microscopy (STEM) obser155 (b) C ed -1- a-ede Energy loss (EV) Figure 5-6. (a) STEM image and (b) the EELS line scan of designated location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMg 0.2Mno. 8BO/C particle. (b)3 (_) Energy loss (EV) Figure 5-7. (a) STEM image and (b) the EELS line scan of designated location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMnBO 3/C particle. vation and electron energy loss spectroscopy (EELS) line-profile in Figures 5-6(ab) and Figures 5-7(a-b), it turns out a uniform boron layer was formed in between the particle surface and carbon coating layer of LiMgo.2Mno.sBO3. However, there is no such a layer observed in LiMnBO 3 . Thus, it is considered that the layer effectively protected particle surface from dissolution. Moreover, super- 156 structure is not found in Figure 5-6(c) suggesting that introducing the foreign element could perturb Li-vacancy ordering. 5.1.3.2. LiZnyMn1.BO3 (0 y 0.2) LiZnyMn.yBO 3 (0 5 y 5 0.2) is prepared in similar with the Li(Mg,Mn)B03 case. In Figure 5-8, it seems Zn incorporated into the structure. However, the overall peak shape is not well defined at the same firing conditions to the Mg doped ones. This may be due to the fact that LiZnBO3 needs much higher temperature (-1273 K) than that of LiMnBO 3 (773 K). Since both the peak intensity and crystallinity of the Zn substituted LiMnBO3 are not suitable for refinement, the lattice parameter fitting is not conducted. Figure 5-9 shows five consecutive charge and discharge curves for LiZnyMnl-yBO3 (0 : y 5 0.1) cycled at a 0.05 C rate. 1 C is individually based on the theoretical capacities for LiZno.o5 Mno. 95 BO3 and LiZno.1Mno. 9 B03, which are .. ...... .. ... . LiZnMn,_,BO3 4-J y=0 .2 0 y=0.10 0.05 1= C I I.,.s.I.I.I.,. 25 30 35 40 ......... .................................. 45 50 55 60 65 y=0- 50 2 Theta (Degree, Cr) Figu re 5-8. XRD patterns of LiZnyMn 1.yBO 3 (0 < y s 0.2) fired at 773-823 K. 157 5E 4.5 54.0 -- 3.5 (b) (a)LZnMnBO 3.0 1*55+1 1.5* 0 40 20 BO ()MnB ,M2.0 0 LZnM 5 1 60 80 100 120 Specific Capacity (mAh g-) 0 20 40 60 80 100 120 Specific Capacity (mAh g-1) Figure 5-9. Five consecutive charge/discharge curves of LiZnyMn 1.yBO 3 (0 s y s 0.2): (a) y=0.05 and (b) y=0.1 cycled at a 0.05 C rate. The dotted line represents the undoped LiMnBO 3. 210 and 198 mAh g- 1, respectively. Similar to Mg substituted cases, Zn substitution brings about the improved cyclability for all substituted compositions. However, the capacities delivered, which are less than 80 mAh g-1 , are somewhat unsatisfactory for both compositions. It can be attributed to the absence of the long-range order from poor crystallization of the solid solution phases, so that the diffusion channel may not be defined to feasibly extract Li from the bulk, especially for large particles. Note that since the compromised theoretical capacity of LiZno.2Mno. 8BO 3 (175 mAh g- 1) seemed already unappealing, and crystallinity and phase formation is less prominent than others, its electrochemical test for that composition was not made at this time. Although the cyclability and capacity retention rate were apparently improved, as shown in Figure 5-9, it turned out Zn substitution was not so promis- 158 ing due to its weight disadvantage compared to Mg substituted phases. Therefore, it can be concluded that Zn is less optimistic than Mg as a substituent. 5.1.4. Active element substitution In LiMnPO 4 studies, one of the suggested routes to enhance its performance is to form a solid solution with LiFePO 4 .28 , 111 By doing so, in various compositions of LiMnl.xFexPO4, the solid solution compound has succeeded to achieve both the high rate property as well as high energy density by activating both Mn 2 +/Mn 3 + and Fe2 +'Fe3+ redox couples. Combining Mn and Fe redox couple is indeed quite general and effective in terms of optimizing properties, as also demonstrated in silicates and carbono-phosphates. 45 , 112 Thus, the similar ap- proach can be adopted to this LiMnBO 3 work. In this section, the effect of partial substitution in LiMnBO3 is discussed. Since LiFeBO 3 performs reasonably well, partial Fe substitution to form LiMni. zFezBO3 solid solution is expected to enhance the electrochemical activity of LiMnBO 3 . Note that Yamada et al. attempted the similar approach but due to the poor phase purity, the substitution did not make any synergy. 85 It is worth pointing out that since the average voltage of Mn 2+/Mn 3+ reaction is higher than Fe 2+/Fe3+, Mn 2+ is more stable than Fe 2 + from oxidation in air by moisture. Therefore, it is better to have the reduced amount of Fe as much as possible to minimize energy density compromise and surface contamination by the atmos159 phere. Thus, the amount of substitution will range from 5% to 35%, so that the 3 majority of redox reaction occurs in Mn 2 +Mn +. The substitution is also extended to other transition metals to promote multiple redox reactions in the premise that all of them can form a solid solution with Mn. LiMnl,.FeBO3 (0 5 z 5.1.4.1. 0.35) The preparation of LiMnl-zFezBO3 (0 5 z 0.35) was similar to the previous- ly stated. Additionally, with being extra careful, the specimens were handled to 2 minimize the air exposure due to reactivity of Fe + with oxygen and moisture. (a) -------------- LiMn1.,FeBO 3 z=0.35 z=. 2 0 Z0.10 z- 0.05 Z= 4-, 25 30 45 40 35 50 55 60 65 55 50 2 Theta (Degr ee, Cr) (b) . 0Em 5.20 - -.- --. 5.19- - 5.18 --.- -..-. 5.17.-...... 0510 20 b. 8.98 a - 8.96 - - 10.38 - C 10.34 8.94 8.92 35 10.36 . 0510 20 35 LO.32 -. .... LO.30........ 35 0510 20 Fe substitution (%) Fe substitution (%) Fe substitution (%) Figure 5-10. (a) XRD patterns of LiMn 1zFeBO3 (0 s z s 0.35) fired at 773~823 K and (b) their refined lattice parameters. 160 Figure 5-10(a) shows the XRD patterns of LiMn1.zFezBO3 (0 < z : 0.35) fired at 773~823 K. The peaks detected were almost pure monoclinic phases with no more than about 5% of Mn3(BO3)2 secondary phases. In the figure, peaks for the 4.5 4.0 3.5 (a 3.0 - LiMn Fe 0 BO3 b LiMn4.Fe 1 B0 3 2.5 0 2.0 1.5 5 4-1 4.5 tno 4.0 3.5 (C) 3.0 LiMn 0 Fe0 2 B0 3 Lim%.4 FeO gBO -J 4 .3 2.5 0e 00 --+.. ........ . 2.0 1.5 + i 1 4.5 0 4.0 -- 3.5 ... . .. ..... . ... e 3.0 LFeBO .fl 2.5 '1) 2.0 >0 1.5 y=.O5 0 40 80 0.1 0.2 0.35 120 160 0 40 80 120 160 Specific Capacity (mAh g1 ) Specific Capacity (mAh g-1 ) Figure 5-11. Five consecutive charge/discharge curves for LiMnj.zFezBO 3 (0 s z s 0.35): (a) z=0.05, (b) z=0.1, (c) z=0.2, (d) and z=0.35 cycled at a 0.05 C rate. For comparison, the representative second cycles of each z are plotted in (e). The dotted line represents the undoped LiMnBO3 . (f) The first three voltage-capacity profiles of LiMnO.6sFeO. 35BO 3 initiated by discharging (red) and their comparison to those of (d). 161 (004) plane show gradual shifting toward the higher angle as z increases, which indicates shrinking c-lattice parameter. 42,67 According to refined lattice parameters in Figure 5-10(b), the monotonic decrease in such numbers indicates Fe successfully substituted Mn and formed the solid solution with in a monoclinic framework, as intended. The voltage versus capacity curves for LiMnjpzFezBO3 (0 _ z 5 0.35) cycled at a 0.05 C rate are plotted in Figure 5-11. Since both Mn and Fe are assumed to be active, the 1 C rate, 222 mA g-1, was based on the theoretical capacity of LiMnBO 3 . As designed, it turns out the overall capacity is improved with a positively proportion to the amount of Fe substitution. In Figure 5-11(e), the achieved capacities from each composition of LiMni-zFezBO 3 at the first discharge are 123, 135, 147, and 167 mAh g- 1 for z = 0.05, 0.1, 0.2 and 0.35, respectively. Regarding to the profiles, distinctive features at 3.1 V at charging and 2.4 V at discharging show the signatures of Fe 2+/Fe3+ redox reaction. 85 Hence, the improved capacity can be attributed to activation of the Fe redox in addition to Mn. Note that although the Fe redox activity occurs at 2.7 V on average, the capacity gain can counterbalance more than the average voltage decrease from 3.3 V (LiMnBO 3) to 2.9 V (LiMno.6 sFeo. 35 B0 3), so that the energy density increases. If considering the electrochemical energy released by discharging, the energy densities obtained are 327, 360, 383, and 422 Wh kg-1 for z = 0.05, 0.1, 0.2 and 0.35, 162 Table 5-2. Achieved electrochemical performance in LiMn 1 zFezBO 3 cathode z in LiMn,_-FezBO 3 0 0.05 0.1 0.2 0.35 Discharge capacity (mAh g-') 98 122 135 147 167 Average voltage at discharge (V) 2.81 2.68 2.67 2.60 2.53 Energy density at discharge (Wh kg-1) 275 327 360 383 422 respectively. The energy density of LiMnBO 3 at discharge is 275 Wh kg-1. Table 5-2 summarizes the obtained electrochemical performances. According to the voltage profiles of LiMnl-zFezBO 3 (0 < z < 0.35) in Figures 5-11(a)-(d), the first discharging always delivers more capacity than the first charging. In other words, more Li can be inserted than extracted. Since this trend gets more noticeable as more Fe is incorporated, it can reflect spontaneous Li extraction or equivalent surface oxidation of Fe 2 + into Fe 3+ in the fresh cathode before cycling. The number of this active redox couple decrease consequently has led to the capacity loss in first charge. Such a phenomenon is more clearly seen from the voltage profile comparison of differently cycled LiMno.65 Feo.35BO 3 in Figure 5-11(f). When a LiMno.65 Feo.3 5BO 3 cathode at its fresh condition initiates the first cycle by discharging, it displays a significant amount of capacity, -100 mAh g-1. Given its magnitude, although the specimen was prepared with careful handling, it seems that almost all Fe 2 + at the surface had been already oxidized to its 3+ state during preparation. Also, it is possible for extracted Li to build a surface contami- 163 nant. The speculated forms of such contamination can be Li 2 CO 3 , Li-B-O, and/or Mn-B-O glassy phases, as often found in other cathode materials such as Li2FeSiO4 and LiFePO 4 .3 1 , 113 After the oxidized surface elements are reverted by completing first cycle, Li intercalates and deintercalates reversibly in the consecutive cycles. The similar behavior was also reported in the previous LiFeBO3 studies. 43 , 63 The irreversible loss of the discharge capacity from the first to second cycle is noticeable in all specimens, which may be related to the formation of an SEI layer. 101 However, at this point, it is not clear whether the surface contamination by air exposure affects the SEI layer formation or not. The capacity fading rates of LiMn1.zFezBO3 (0 < z 0.35) per cycle are not yet satisfactory. If compared to LiMnBO 3 (5.7%), Fe substitution only slightly enhances the cyclability for the first five cycles: 4.2%, 3.9%, 3.7%, and 3.2% for z = 0.05, 0.1, 0.2, and 0.35. This can be explained by observing STEM images and (b) Energy loss (EV) Figure 5-12. (a) STEM image and (b) the EELS line scan of designated location from bulk to surface, and HR-TEM image and electron diffraction pattern of LiMnO. 8FeO.2BO/C particle. 164 EELS line scan profile in Figures 5-12(a-b). Unlike the Mg substituted case in Figure 5-7, there is no surficial boron detected between the active particle and carbon coating layer, which can extenuate continuous Mn dissolution. It is believed that the absence of such a layer may responsible for the more prominent fading than the Mg substituted Li(Mg,Mn)B0 3 situation. Although the cyclability still needs further enhancement, the positive indication is that capacity retention demonstrates an improving trend as the amount of Fe substitution increases. It suggests that as the portion of Fe increases in the particle surface, the side reaction provoked by Mn dissolution at charged state can be decelerated due to its better stability, as summarized in Table 4-2 and 4-3. 5.1.4.2. LiMnlzM2BO 3 (M = Co, Ni, and Cu, 0 z 0.35) The substitution was extended to proper first-raw 3d-transition metals in order to confirm the additional redox activities, as proposed in Subsection 5.1.2. LiMn-zMzBO 3 (M = Co, Ni, and Cu, 0 < z < 0.35) samples were also prepared in the similar way as Li(Mn,Fe)BO 3 was done. Figures 5-13 and 5-14 show the XRD patterns and Rietveld-refined lattice parameters of the synthesized phases with respect to different substitution levels. As shown in Figure 5-13, no outstanding secondary peaks are detected in all the specimens but all the peaks are indexed as a typical monoclinic lithium transition metal borate phase, except for z = 0.35 substitution cases, which includes unidentifiable peaks. Note that in the figure, the (004) peaks do not seem shift165 ing much. This is because the contraction of c-lattice parameters in Figure 5-14 is not as much as that of the Fe substituted case. Therefore, it is considered each of substituent still formed a solid solution in the hosting structure. 3 2 3 2 According to computation results in Table 5-3, Co +'Co +, Ni +'Ni +, and 3 75 2 Cu2 +ICu3 + are expected to have higher redox potential than Mn +'Mn+. Howev- (a) - LiMn CoBO, z=0.35 - 4-J z0.20 C Z0.10 Z0.5 Z0 4-J C: 30 25 (b) 35 50 55 45 40 LiM n NiB03 60 ............ ........... 55 50 65 ................. C z0.35 z0.20 =0.1O z0.0 5 =0 !5 30 (c 35 i-z CBO 50 45 40 55 60 0 .......... .... ....... 55 50 65 . . . .. . . . .. 3 z0.35 z.20 -O.10 z-0.05 z0 - 25 30 35 40 45 50 55 60 65 ..... 50 .......... 55 2 Theta (Degree, Cr) Figure 5-13. XRD patterns of LiMn1 -zMzBO 3 (0 (b) M = Ni, and (c) M = Cu. 166 z S 0.35) fired at 773-823 K: (a) M = Co, (a) .. 10.38A 8 .98 . 5.197 .... 8.97 0- - - 5.196 5.195 . 0 510 20 35 0 510 35 20 35 Co substitution (%) Co substitution (%) Co substitution (%) U 5.196 1<.. - E 0.E 8.98 ---- -... 5.194 8.97 - 5.192 8.96 0 510 20 m (C) 10.36 8.95 0 510 20 (b) 10.37 8.96 --- C Ni substitution - 10.38 - LO.37 - 0 510 20 35 0 510 20 35 35 (%) Ni substitution (%) Ni substitution (%) b 10.38 5.196 * 8.98 5.192 8.96 10.37 5.188 8.94 10.36 5.184 8.92 10.35 0510 20 .. 35 0 510 20 35 Cu substitution (%) Cu substitution (%) 0 510 20 35 Cu substitution (%) Figure 5-14. Refined lattice parameters of LiMn 1-zMzBO 3 (0 s z : 0.35) fired at 773~823 K: (a) M = Co, (b) M = Ni, and (c) M = Cu. er it turned out they are hardly active in the borate system. In Figure 5-15, All the substituted Li(Mn,M)B03 (M = Co, Ni, Cu) cathodes perform even inferior to LiMnBO3 . The conversion-reaction-like tale at the end of each discharge strayed from the intercalation track is also observed and becomes longer as z amount increases. Other than that, there is no feature in the voltage profile showing the substituted transition metals have been activated. Rather, the polarization be167 4.5 4.0 _j 3 .5 13.0 . - ............. a) LiMn Co BO e 2.5 0 .5 0.220.0 ..... ......... O Lpk35, O. 4.5 4.0 ~3.5 (b) 2.5 tw 2.0 4- LiMnNiBO3 =0 zB3 1.5 t0.35 0. 4.5 4.0 -3.5 - 3 .0. w ...... 3C) LiMn,. CuzBO 3 .......... . ... .. .. 2.5 . .. . 2.0 o15 1.5 0 .. 0.2 0.05 0.1 120 80 40 Specific Capacity (mAh g-) 160 Figure 5-15.The representative second cycles of LiMn 1-MBO 3 (0 S z S 0.35) for (a) M = 3 was not able to be tested due Co, (b) M = Ni, and (c) M = Cu. Note that LiMno.65Cu. 3 BO0 to repeated cell failure after the first charging. comes severe as the substitutional amount increases, so that their capacities could not be realized within the given voltage window. The capacity aggravation upon substitution and noticeable conversion reaction can be related to the instability of the compound. Although the solid solu168 Table 5-3. Computed properties of lithium metal borates Compounds Avg. Voltage Capacity Lith Delith AE>hull AE>hull AV LiCoBO 3 3.9 V 215 mAh g 1 0.013 eV/at 0.131 eV/at 2.57% LiNiBO 3 4.58 V 214 mAh g-1 0.031 eV/at 0.214 eV/at 3.68% LiCuBO 3 4.53 V 211 mAh g-1 0.044 eV/at 0.223 eV/at 0.62% tion seems to be formed, a computational study predicts poor stabilities of LiCoB0 3 , LiNiBO 3 , and LiCuBO 3 compared to LiMnBO 3 no matter they are lithiated or delithiated. Given that the stability of LiMnBO 3 is already at the unstable side when delithiated, the substitution may not sustain its integrity upon charging and following discharging. The calculated stabilities for each phase are summarized in Table 5-3. In conclusion, the substitution strategy is very successful for Mg and Fe cases among the several attempted elements. Mg substitution improves cycling capacity significantly, and Fe substitution realizes the substantial increase of the energy density. Therefore, in the next section, the attempts to accomplish both the cyclability and capacity by substituting multiple elements at the same time are followed. 169 5.2. Inactive and active element co-doping: LiMgo.1Mno.9..FezBO 5.2.1. 3 (0.3:5 z 5 0.4) Optimizing composition In Subsection 5.1.3.1, the most optimized amount of Mg substitution is chosen to be 10% due to its consequence on both improving cyclability and compromising theoretical capacity. Besides, in Subsection 5.1.4.1, the Fe-substituted Li(Mn,Fe)B0 3 cathodes always deliver more capacities than pre-substituted LiMnBO 3 even though the average voltage decreases. However, due to surface contamination by unexpected oxidation of Fe2 + and possible energy density deficit by further lowering an average redox potential, there may be the optimal compositional limit of Fe substitution. Therefore, in this section, Mn is substituted by 30, 35, and 40% Fe in order to discover an optimized composition for the maximal performance. The starting composition is LiMgo.1Mno. 9 .2FezBO 3 with various substitutional ratios, z, where 0.3 < z < 0.4. A synthesis method is identical to the previous case for Li(Mn,Fe)B0 3 in Section 5.1. Also, specimens were mostly and strictly handled in the argon filled glovebox in order to minimize air exposure. After synthesis, the powder was carbon-coated and annealed at 798 K for 5 h so as to ensure firm physisorption as well as chemisorption of carbon layer onto the active particle surface. 170 Figure 5-16(a) shows XRD patterns of LiMgo.,Mno.9-zFezBO3 (0.3 < z < 0.4) fired at 823 K for 10 h. According to the patterns, both Mg and Fe are well incorporated into the monoclinic framework at once showing no additional peaks other than the targeted phases. The small peak for Mn3(BO3)2 is again detected but its amount is less than 3% according to phase identification. Rietveld-refined lattice parameters in Figure 5-16(b) can be linearly fitted satisfying Vegard's law. Consequently, the phases obtained are concluded as solid solution between LiMgo.lMno.9BO3 and LiFeBO3. Figure 5-17 shows the first five consecutive charge and discharge curves for (a) --------------------...------.-. LiMgOejMnO,..,Fe,,B03 z=0.40 -a Li z- z-035 =0 -----25 30 35 45 60 65 - - 55 50 50 55 2 Theta (Degree, Cr) (b) 5.2 0 _-1.-J 40 4 -0 SE 0.. - ... 8.97 8.94 .. 5.1 8 ... -.....-...-... 5.1 6 .....-. 0 - 8.91 . 35. 3040 Fe substitution (%) 8.88 ._ . 10.3 6 . - 10.3 2 . -. .- 10.2 8 -- _. _.35. 10.2 4 3C 40 0 - Fe substitutio n (%) 0 . 35. 30 40 Fe substitution (%) Figure 5-16. (a) XRD patterns of LiMgO.1Mno.9..zFezBO 3 (0.3 s z s 0.4) fired at 823 K and (b) their refined lattice parameters. 171 4.5 Z3.5 03.0 .... BIO - i -:a ..... 2.5 - 2.0 -.... >1.5 . . ... . ..... ... ... ... 4.0 4.5 3.0 2.5 2.0 .... .. .-.. -.. . -.. .. -.. .. >1.5 0.35i 0 40 80 120 160 200 0 40 80 120 160 200 Specific Capacity (mAh g-) Specific Capacity (mAh g-) Figure 5-17. Five consecutive charge/discharge curves for LiMgo.1Mno.g-zFezBO 3 (0.3 s z 5 0.4): (a) z = 0.3, (b) z = 0.35, and (c) z = 0.4 cycled at a 0.05 C rate. For comparison, the representative second cycles of each z are plotted in (e). LiMgo.Mno.-zFezBO3 (0.3 < z 5 0.4) at a 0.05 C rate. The 1 C rate, 205 mA g-1, was based on the theoretical capacity of LiMgo.1Mno.9BO3. According to the figure, an increasing trend of the overall capacity and Fe redox features in voltage profiles are almost identical to that of Fe substituted Li(Mn,Fe)B03: the surface oxidation is observed in all specimens displaying less capacity at charging than discharging, and this phenomenon disappeared in consequent cycles. The achieved discharge capacities from each composition of z at the first cycle are 160, 173, and 181 mAh g-1 for z = 0.3, 0.35, and 0.4, respectively. If con- 172 Table 5-4. Electrochemical performance of LiMgO1MnO.9-zFezBO 3 (0.3 z < 0.4) cathodes z in LiMgo.1Mno.9..zFezBO 3 0 0.3 0.35 0.4 Discharge capacity (mAh g-1 ) 101 160 173 181 Average voltage at discharge (V) 2.74 2.53 2.50 2.51 Energy density at discharge (Wh kg-1 ) 276 404 432 454 sidering the energy density output by discharging, calculated energy densities are 404, 432, and 454 Wh kg-1 where z equals to 0.3, 0.35, and 0.4, respectively. The obtained values of the electrochemical performance are summarized in Table 5-4. Although the capacity is greatly improved by Fe 2+/Fe3+ activation along with Mn 2+/n 3+ redox reaction in the LiMnBO host, the expected cyclability im- provement from Mg implantation has not appeared in Figure 5-18. Rather, the capacity fades quite severely, about 4-5% per cycle in all cases, similar to the case of LiMni-zFezBO 3 series. The possible explanation is that although the Mn dissolution problem could have been suppressed during cycling by reduced ratio of Mn and Mg substitution at the same time, the surface of the particle is inevitably reacted with the electrolyte because the cathode compound has to spend an extended period of absolute time experiencing highly delithiated states, which is not as stable as lithiated one for LiMnBO 3 and even LiFeBO repeatedly. The degree of such an electrode/electrolyte reaction mainly depends on the active surface area of the particles. Therefore, if the area can be adjusted with173 out increasing the average particle size, the overall performance will be improved. This may be accomplished by altering the particle size to have narrow distribution around the same mean size. The modification is expected to subsidize the overall cathode performance of LiMgo.iMno.5Feo. 4 BO 3 because (1) reducing the number of small particles, and thus the associated side reaction, will improve the cyclability and (2) removing large particles will enhance the capacity achievable by encountering less channel blockage in Li diffusion. The following section is to deal with such tuning of particle size distribution. 174 5.3. Cycling performance improvement by processing control 5.3.1. Motivation Controlling the particle morphology of electrode materials is one important factor influencing its overall performance since the electrochemical reaction mainly occurs at the surface of the active particle. Especially for LiMnBO 3 , it must be reminded that there is a prominent Mn dissolution problem caused by unwanted parasitic reaction at the particle surface. Therefore, its surface morphology is even more crucial and can dominantly impact the global cycling performance. In this sense, although the huge benefit to remedy transport-limitation, the nanosizing may not be the ultimate solution to resolve the capacity fading issue owing to its increasing surface area to volume ratio: the more surface is involved, the more side reactions there will be. Therefore, the approach must be toward realizing larger, but not too large to extract Li, particles. However, it is not a simple task in LiMnBO 3 at all due to following reasons. Assuming the identically prepared LiMnBO 3 specimens are in the same condition before firing, the phase morphology obtained the conventional solid-state reaction depends primarily on temperature-related variables. 78 , 114 Above all, since it relies on the solid state diffusion, the firing temperature is the most crit175 ical factor compared to the other experimental variables such as firing duration and ramping rate. 78, 114 Normally, a phase obtained at higher temperature is better crystallized and free from defects, and is consisted of well-defined uniform particles compared to the phase formed at lower temperature. 7 8 , 114 In the case of synthesizing monoclinic LiMnBO 3, the firing temperature is also considered as the major factor to determine the quality and morphology of the phase and particles. However, as shown in Figures 3-6 and 3-7, the hexagonal polymorph starts taking over when the temperature is raised to 873 K and dominates at above 1073 K. Thus, the synthesis temperature for monoclinic phase must be kept below 873 K, which makes a dead-end situation since firing at low temperature in the solid-state method does not always guarantee a quality of the product. In these reasons, soft-chemistry synthesis like the sol-gel method was adopted to achieve unconventional results. 7 1 If the temperature cannot be much adjustable, the next influential variable for the particle morphology can be the condition of a sample prior to firing (or sintering). 78, 114 In this thesis work, all of the calcined powders are pressed into disc shape pellets, and such a shaped specimen to be fired is conventionally called as a green body. Since a densely packed green body can provide uniform environment for solid-state diffukion, it is expected to promote phase formation at the same firing temperature, which in turn is expected to drive a bettercrystallized phase. In this section, the electrochemical properties from specimens 176 prepared by cold-isostatic pressing are compared to those from the specimen by uniaxial pressing. 5.3.2. Cold-isostatic pressing (CIP) Green bodies prepared by uniaxial pressing often get warped and even 78 cracked during the sintering process. , 114 This is mainly due to the unevenly applied pressure by enormous friction from die walls to the green body as well as (b) Oil bath (a) Steel mold I+ Figure 5-18. Simplified schematic diagrams of pressure application during (a) uniaxial pressing and (b) CIP. A Particle size , Particle size Figure 5-19. Optimal change in particle size distribution to minimize side reaction by removing very small particles. 177 the spring-back effect of the ceramic powder during uniaxial pressing. 78, 114 It leads to poor quality of the sintered body and, sometimes, meager crystallinity of the synthesizing phase. 7 8 , 114 Cold-isostatic pressing (CIP) is a well-known forming technique often used in ceramics synthesis to obtain a densely packed green body with uniform shrinkage. 7 8 , 114 It usually utilizes an oil bath at room temperature as a media to apply isostatic pressure onto a specimen through a flexible mold such as latex. 78 Since it is free from wall and friction, it can generally apply much higher pressure on the specimen than the uniaxial press, as illustrated in Figure 5-18. In turn, the green body obtained from CIP is more densely packed and it leads to better sintering process. 78 Thus, in my thesis work, CIP is utilized in order to prepare a green body and compared it with the previous results that were prepared by uniaxial pressing. The attempt aims to promote particle coarsening by providing uniform environment during firing, as illustrated in Figure 5-19, so that LiMnBO 3 electrodes can escape from its dissolution problem. Note that since CIP requires preforming, pellets for CIP were pre-formed by uniaxial pressing with mild applied pressure. The pre-forming pressure was 15 MPa and CIP pressure was 55 MPa. Therefore, in following section, 'with CIP' defines a sample prepared by both uniaxial and CIP. 'Without CIP' stands for a sample by uniaxial pressing only. Figure 5-20 captures the effect of CIP on sintering. In the figure, both pellets were fired at 773 K for 10 h under flowing argon. However, the size differ- 178 Figure 5-20. Measurement of pellet diameter after firing at 773 K for 10 h prepared by (a) uniaxial pressing and (b) CIP. ence is distinctive between the sintered pellets formed by uniaxial pressing and CIP: CIP clearly led to larger shrinkage of the pellet. Given that they are fired at the same temperature, it infers that the more it shrinks, the more its particle get coarsen. When particles grow, it consumes the smaller sized particle, so the anticipated particle size distribution is like the one in Figure 5-19. 5.3.3. Improved cyclability of LiMnBO3 by CIP Figures 5-21(a) and (b) shows the Rietveld refinement and XRD profile matching of LiMnBO3 fired at 773 K for 10 h in argon atmosphere, which had been prepared without CIP and with CIP, respectively. According to the patterns in the figure and fitting parameters listed in Table 5-5, there is no dramatic change in the overall peak ratio or intensity except for a diminished amount of 179 (a) - LiMnBO 3 o Observed Calculated m-LiMnBO3 (ICSD# 200535) . Mn 3(BO 3)2 Obs. - Cal. ;1 4-, C: 30 25 (b) 50 45 40 35 6! 60 55 LiMnBO 3 CIP o Observed - Calculated . Mn 3(BO 3)2 m-LiMnB0 3 (ICSD# 200535) :3 - Obs.-Cal. 41-J 0 4-, C li Il....Il I~... 25 30 45 40 35 50 55 60 65 2 Theta (Degree, Cr) Figure 5-21. Rietveld refinement and profile matching of LiMnBO prepared by (a) uniaxial pressing and (b) CIP, which are fired at 773 K and annealed at 773 K for carbon coating. Table 5-5. Rietveld refined lattice constants and derived parameters LiMgo.1Mno. 5Fe 0.4B0 3 prepared by cold-isostatic pressing and fired at 823 K. LiMnBO3 a (A) b (A) (004) FWHM Crystallite size Microstrain c (A) Uniaxial pressing 5.197 8.965 10.368 0.3500 438 A 0.208% 5.197 8.965 10.376 0.3330 462 A 0.105% CIP of Mn3(BO3)2 impurity. However, the change in full-width-half-maximum (FWHM) of detected peaks suggests that the crystallite size increases and microstrain de180 creases, as estimated by Scherer's equation. The morphology difference can be seen more apparently in Figure 5-22. By CIP, the particle becomes smoother at surface and the average size slightly increased. As intended, the particle size distribution also shifted, and the number of smaller particles considerably decreased. Figure 5-23(a) shows five consecutive charge and discharge curves at a 0.05 C rate for carbon-coated LiMnB03 fired and annealed at 773 K prepared by CIP. (c) (*)LiMnBO 3 CIP 0 (o) LiMnBO 3 4-J -0 .. 0 U i , 0 50 e e: 100 i 150 e i 1 200 Particle size Figure 5-22. SEM images of carbon-coated LiMnBO 3 prepared by (a) uniaxial pressing and (b) CIP in different magnifications, and (c) particle size distribution comparison between them. 181 The capacity fading rate per cycle during the five cycles is 1%. This is much improved cyclability over the previous case without CIP whose rate was 5.7% per cycle in Figure 5-5(a). The direct comparison of cyclability between LiMnBO3 without and with CIP is shown in Figure 5-23. By preventing the initial capacity 4.5 4.0 3.5 W CIP,-LiMgPO -- 3.0 2.5 0 4- 2.0 I 1.5 5*1 E .i 200 . 160 A LiMnBO3 CIP .A .LiMnBO- 120 AA C. 00 200 160 120 80 40 Specific Capacity (mAh g-) 0 -o 3 * A 80 C> -c 40 a------------ --------- 0 1 2 3 4 5 6 7 Cycle number 8 9 10 Figure 5-23. (a) Five consecutive charge and discharge curves for carbon-coated LiMnBO 3 at a 0.05 C rate prepared by CIP and (b) capacity retention during 10 cycles. For reference, LiMnBO 3 without CIP is also plotted (open triangle). 182 drop, the cathode prepared by CIP brings about better cyclability than the other. This suggests that minimized side reaction and Mn dissolution by modifying the particle morphology. 5.3.4. Improved cyclability of LiMgo.1Mno.5Feo. 4 BO3 by CIP Since the active powder preparation with CIP was successful to control particle size distribution of LiMnBO 3 , the identical strategy is also applied to LiMgo.iMno.5Feo. 4 BO 3 , which shows the most optimized capacity at its beginning but suffers from its fading in following cycles. Figures 5-24(a) and (b) shows the Rietveld refinement and XRD profile matching of carbon-coated LiMgo.iMno.5Feo.4BO 3 , which had been respectively prepared without CIP and with CIP. Both are fired at 823 K for 10 h in argon atmosphere and annealed at 798 K for 5 h in the identical argon environment. The patterns in the figure and corresponding refined parameters in Table 5-6 indicate that slight change in FWHM without alteration in the overall peak ratio or intensity. Just as in LiMnBO 3 , it infers that the crystallite size gets slightly increased and microstrain becomes relieved to the some extents by Scherrer's equation.115 Since CIP can allow more homogeneous environment 183 around LiMnBO3 nuclei in the process of firing the pelletized specimen, it may be able to form microstructurally less defected crystals at the same firing temperature. (a) LiMg0 .jMn0 .5Fe0 .4B03 o Observed Calculated m-LiMnBO 3(ICSD# 200535) -- Obs. -Cal. - . Mn 3(BO 3)2 C S? 30 !5 40 35 ?0 45 50 (b) LiMg01 Mn05 Fe0 4 B0 3 CIP .) o Observed - Calculated m-LiMnB0 3(ICSD# 200535) - Obs. - Cal. 65 60 55 . Mn ,3(B0 3)2 Lo 4-j .... ..... I 25 30 35 40 45 50 55 60 65 2 Theta (Degree, Cr) Figure 5-24. Rietveld refinement and profile matching of LiMgo. 1Mn 0.5Fe 0.4B0 3 prepared by (a) uniaxial pressing and (b) CIP, which are fired at 823 K and annealed at 798 K for carbon coating. Table 5-6. Rietveld refined lattice constants and derived parameters LiMg 0.1 Mn 0.5Fe 0.4B0 3 prepared by cold-isostatic pressing and fired at 823 K. (004) FWHM Crystallitesize Microstrain 0.186% 268 A 0.5430 5.159 8.872 10.223 0.112% 280 A 0.5550 5.156 8.879 10.173 LiMg0 .jMn0 .5Fe 0 .4 B0 3 a (A) b (A) Uniaxial pressing CIP of c (A) 184 (*) LiMgO. 1MnO. 5FeO.4BO 3 CIP (C) (A) LiMgO. 1MnO.:FeO.4 BO3 A 4-J C 0 0 0 0 50 A 0 5 0 150 200 A 100 Particle size Figure 5-25. SEM images of carbon-coated LiMgO.jMnO. 5FeO.4BO prepared by (a) uniaxial pressing and (b) CIP in different magnifications, and (c) particle size distribution comparison between them. Also, SEM images in Figure 5-25 can show the effect of CIP on the particle morphology: the particle size distribution becomes narrower around the mean value of 109 nm. In turn, the number of particles smaller than 50 nm significantly decreased suggesting that CIP indeed spurred highly reactive smaller-sized particle to grow and become narrowly-dispersed size distribution. 185 Figure 5-26 shows the how much CIP procedure can improve the electrochemical properties of LiMgo.lMno. 5Feo.4BO3. As shown in Figure 5-26(a) the first five charge/discharge curves showing enhanced capacity retention. A slight shift of the particle size distribution toward a large particle domain in Figure 5-25(c) (a) -... 4.5 --- 4.0 > 3.0 LMgMn .Fe..BO 2.50 1.5 40 0 80 120 200 160 1 Specific Capacity (mAh g ) (b) S200- < 160 E 4%120 S80 t 40 LiMgMnFe* 4B0 3 CIP Fe0 .4BO 3 0LiMg 1 Mn -... ------ 0 1 2 3 4 5 6 7 Cycle number 8 9 10 Figure 5-26. (a) Five consecutive charge and discharge curves for carbon-coated LiMgo.,Mno. 5Feo.4BO at a 0.05 C rate prepared by CIP and (b) capacity retention during 10 cvcles. For reference. LiMnBOA without CIP is also Dlotted (oDen circle). 186 does not affect the achievable capacity, so that the initial discharge capacity reaches 180 mAh g-1 at a 0.05 C rate. Most importantly, the degree of the irreversible loss decreases, and after five cycles, it still maintains discharge capacity of 160 mAh g- 1. The cyclability difference between samples without and with CIP gets larger in subsequent cycles in Figure 5-26(b). After the 10th discharge, the cell was disassembled to see whether Mn had been deposited on the anode side or not. For specimen prepared by CIP, there is no major deposition observable in opposition to the case without CIP, as compared in the photographic inset of Figure 5-26(b). Therefore, as intended, CIP indeed induces reduction of Mn dissolution by modifying the surface morphology. The alleviated capacity fading rate per cycle is estimated 2%, approximately. Figure 5-27 plots the rate capability of CIP processed LiMgo.iMno.5Feo.4BO3 cathode. First of all, at a 0.02 C rate cycling almost all of Li is activated, so that 200 mAh g-1 is achieved. This is equivalent to 98% of the theoretical capacity. It is more than a huge improvement in lithium transition metal borate systems to achieve such a progress. Moreover, it shows respectable rate capability in terms of discharge capacity that at 1 C discharge, 120 mAh g- 1 is achievable. The application of CIP, therefore, can address not only the dissolution issue but also the kinetic limitation. This can be understood by the effect of substitution as well. As shown in Figure 5-28, the substitution drives the shrinkage of the lattice, especially along the c-axis. Since Li now can hop along that direction with reduced 187 length by substitution and also experience less barrier due to the relieved microstrain. 4.5 (b) LiMg Mn1 J5Fe QBOJ CIP 4.0 3.5 3.0 2.5 4-.J 2.0 1.5 2d ic 0.5C O16C a 0 40 i 80 120 .02C-aOflc 160 0.02 i 200 Specific Capacity (mAh g-) Figure 5-27. Rate capability comparison at discharge between (a) LiMnBO 3 and (b) LiMgo. 1MnO. 5Feo.4BO 3 prepared by CIP. Each curve was charged at a respective 0.05 C rate prior to discharging. [010] [001] Figure 5-28. Schematic diagram of one-dimensional Li diffusion and lattice planes surrounding the path. 188 5.4. Conclusions Approached from the microstructural point of view, possible substitutional elements were proposed to design a high energy density polyanionic cathode within the monoclinic LiMnBO 3 framework. Among Mg, Fe, Co, Ni, Cu, and Zn, elements that satisfy the criteria for substitution for trigonal-bipyramidal Mn, Mg and Fe shine out of the batch in terms of their contribution to cyclability and achievable capacity, respectively. Mg substitution improves cyclability to the large extent mainly because stabilizing the structure to minimize the Mn dissolution. It was done by formation of a protective boron layer on the particle surface. Also, Fe substitution adds significant amount of capacity to the LiMnBO 3 thanks to its redox activity. Thus, to claim the both benefits, Mg and Fe are substitute Mn at the same time and the optimized composition was set to be LiMgo.lMno.5Feo.4BO3 considering the compromise of achievable energy density. However, if co-doped, the retention of its capacity was still inadequate and unsettled. Therefore, as a part of design strategy to seek for cyclability improvement, particle morphology of LiMnBO3 and LiMgo.1Mno.5 Feo. 4BO 3 were reengineered by applying CIP to the experimental procedure. A densely packed green body prepared by CIP induced a microstrain relieved quality crystallite for the both borates after firing. More importantly, as a consequence of additional CIP process, removal of small particles to reform its size distribution from widely dispersed to 189 narrowly concentrated spread enabled LiMnBO3 and LiMgo.lMno. 5 Feo. 4BO3 to have much improved cyclability. Especially, a (near) theoretical capacity of 200 mAh g-1 at 0.02 C is achieved in LiMgo.1Mno. 5 Feo.4BO 3 compound. If cycled at a 0.05 C rate, the capacity retention rate for first five cycles is approximately 90%, which is considered satisfactory as this beginning level of academic research. Moreover, the rate capability of the mostly optimized cathode stands in a good shape. At 1 C discharge, the achieved capacity is 120 mAh g- 1. All in all, it can be concluded that substitution and CIP strategies properly work for LiMnBO 3 to improve its electrochemical properties. The optimized LiMgo.lMno.5 Feo.4BO3 accomplished 200 mAh g- 1 milestone, which is truly rare in the polyanionic cathodes. It is believed, with its inherent safety against oxygen evolution, such a high capacity achieved makes the substituted LiMnBO3 compound a strong contender as a new cathode material for Li-ion batteries. [End of Chapter 5] 190 CHAPTER 6 CONCLUDING THESIS STATEMENTS The electrochemical results of the monoclinic LiMnBO 3 as a Li intercalation cathode are firstly reported as a result of this work. The initial and yet preliminary capacity (35 mAh g-1) achieved was suddenly optimized by particle size redistribution and conductive phase coating, so that 100 mAh g- 1 of a specific capacity in the second discharge was obtained. Also, this study has acknowledged the importance of a polymorphic structure of LiMnBO 3 to the electrochemical performance, for the first time. Compared to the hexagonal counterpart, monoclinic LiMnB0 3 displays substantially more electrochemical activity. This phenomenon has originated from the critical difference in the Li migration barrier and delithiated state stability as well as particle size, which is the consequence of the structural variation between them. 191 The characteristics of superior performing monoclinic LiMnBO3 were also understood. Although better than the hexagonal one, it still showed somewhat limited properties compared to the contemporary cathode materials. My research on the system has revealed that the LiMnBO3 system suffers from sluggish diffusion, but that is not because it is inherently slow but because it is impeded by the presence of channel blocking defects. Also, its instability during charging has been also rationalized by unprecedented analysis of the Li deficient LiixMnBO 3 (0.5 < x < 1) phases obtained from chemical delithiation as well as electrochemical charging. After two major obstacles were defined, the problem-solving strategies were followed. To promote more redox activity and reinforce the structural stability, a systematic substitution was designed. Endeavor to find an optimal substituted composition finally excavated LiMgo.lMno.5 Feo.4BO 3 as the most optimum composition in terms of the achieved energy density and cyclability. The composition indeed outperforms most of the polyanionic compounds with 200 mAh g-1 capacity at 0.02 C discharging with respectable cyclability. I, therefore, conclude that the objectives of this research have been successfully accomplished. In my thesis work, monoclinic LiMnBO 3 is discovered, examined, understood, modified, developed, and finally proposed as a new candidate cathode material for Li-ion batteries. This research has encompassed the broad aspects of the material from its synthesis of the phase to optimization of the performance based on firm understanding. It is also a successful example of synergetic collab- 192 oration between experimental and computational work in that physically observed quantities and phenomenon could be effectively accounted for by using computational analysis on top of experimental evidences. This work holds its value in a sense that a new favorable cathode material is proposed. Through my thesis work, I believe the chemical space that can be utilized as Li intercalation materials has been expanded to borate compounds. Moreover, not only the new chemistry suggestions but also the promising electrochemical property demonstrations were made in the process of thesis research. I consider my contribution to the Li-ion battery community can enrich the perspective of future research on the search for new materials for energy storage. [End of Chapter 6] 193 [This page is intended to be blank.] 194 REFERENCES 1. California exhaust emission standards and test procedures for 2005 and subsequent model zero-emission vehicles, and 2001 and subsequent model hybrid electric vehicles, in the passenger car, light-duty truck and medium-duty vehicle classes. In California Air Resources Board.: 2003. 2. United Nations framework convention on climate change: handbook; 2006. United Nations Climate Change Secretariat: 2006. 3. Bradley, T. H.; Frank, A. A., Design, demonstrations and sustainability impact assessments for plug-in hybrid electric vehicles. Renewable & SustainableEnergy Reviews 2009, 13, (1), 115-128. 4. http://www.iccnexergy.com 5. Guideline for the Safety Evaluation of Secondary Lithium Cells. Japan Battery Association: 1997. 6. Goodenough, J. B.; Park, K. S., The Li-Ion Rechargeable Battery: A Perspective. Journalof the American Chemical Society, 2013, 135, (4), 1167-1176. 7. Kim, J. C.; Moore, C. J.; Kang, B.; Hautier, G.; Jain, A.; Ceder, G., Synthesis and Electrochemical Properties of Monoclinic LiMnBO3 as a Li Intercalation Material. Journalof the Electrochemical Society, 2011, 158, (3), A309-A315. 8. G. -A. Nazri, G. P., Lithium Batteries: Science and Technology. Kluwer Academic/Plenum: Boston, 2004. 195 9. Masquelier, C.; Frameworks Croguennec, L., Polyanionic (Phosphates, Silicates, as Electrode Materials for Rechargeable Sulfates) Li (or Na) Batteries. Chemical Reviews, 2013, 113, (8), 6552-6591. 10. Kang, K. S.; Meng, Y. S.; Breger, J.; Grey, C. P.; Ceder, G., Electrodes with high power and high capacity for rechargeable lithium batteries. Science, 2006, 311, (5763), 977-980. 11. Mizushima, K.; Jones, P. C.; Wiseman, P. J.; Goodenough, J. B., LixCoO2 - a New Cathode Material for Batteries of High-Energy Density. Materials Research Bulletin, 1980, 15, (6), 783-789. 12. Ohzuku, T.; Makimura, Y., Layered lithium insertion material of LiCo,3Nil/Mnl302 for lithium-ion batteries. Chemistry Letters, 2001, (7), 642-643. 13. Park, B. C.; Kim, H. B.; Myung, S. T.; Amine, K.; Belharouak, I.; Lee, S. M.; Sun, Y. K., Improvement of structural and electrochemical properties of A1F3-coated Li[Nis3Co1/3Mn1/3 ]0 2 cathode materials on high voltage region. Journal of Power Sources, 2008, 178, (2), 826-831. 14. Park, S. H.; Kang, S. H.; Belharouak, I.; Sun, Y. K.; Amine, K., Physical and electrochemical properties of spherical Lii+x(NilI3Col/3Mni/3)(.x)02 cathode materials. Journalof Power Sources, 2008, 177, (1), 177-183. 15. Sun, Y. K.; Myung, S. T.; Park, B. C.; Prakash, J.; Belharouak, I.; Amine, K., Highenergy cathode material for long-life and safe lithium batteries. Nature Materials, 2009, 8, (4), 320-324. 16. Belharouak, I.; Lu, W. Li(Nio.sCoo.i5Alo.o 5)0 2 Q.; Vissers, D.; Amine, K., Safety characteristics of and Li(Ni/3CO13Mn/ 3 )0 2 . Electrochemistry Communications, 2006, 8, (2), 329-335. 17. Kim, H. B.; Park, B. C.; Myung, S. T.; Amine, K.; Prakash, J.; Sun, Y. K., Electrochemical and thermal characterization of AlF3-coated Li[Nio.8Coo.5Alo.o5]O2 cathode in lithium-ion cells. Journalof Power Sources, 2008, 179, (1), 347-350. 18. Thackeray, M. M.; Kang, S. H.; Johnson, C. S.; Vaughey, J. T.; Benedek, R.; Hackney, S. A., Li2MnO3-stabilized LiMO 2 (M = Mn, Ni, Co) electrodes for lithium- ion batteries. Journalof Materials Chemistry, 2007, 17, (30), 3112-3125. 19. Kang, S. H.; Kempgens, P.; Greenbaum, S.; Kropf, A. J.; Amine, K.; Thackeray, M. M., Interpreting the structural and electrochemical complexity of 0.5Li2MnO3* 196 0.5LiMO2 electrodes for lithium batteries (M = Mno.5_xNio.5-xCo2x, 0 _ x 0.5). Journalof Materials Chemistry, 2007, 17, (20), 2069-2077. 20. Kang, S. H.; Thackeray, M. M., Enhancing the rate capability of high capacity xLi2MnO3 - (1-x)LiMO2 (M = Mn, Ni, Co) electrodes by Li-Ni-P04 treatment. Electrochemistry Communications, 2009, 11, (4), 748-751. 21. Cabana, J.; Kang, S. H.; Johnson, C. S.; Thackeray, M. M.; Grey, C. P., Structural and Electrochemical Characterization of Composite Layered-Spinel Electrodes Containing Ni and Mn for Li-Ion Batteries. Journalof the Electrochemical Society, 2009, 156, (9), A730-A736. 22. Thackeray, M. M.; David, W. I. F.; Bruce, P. G.; Goodenough, J. B., Lithium Insertion into Manganese Spinels. MaterialsResearch Bulletin, 1983, 18, (4), 461- 472. 23. Gummow, R. J.; Dekock, A.; Thackeray, M. M., Improved Capacity Retention in Rechargeable 4v Lithium Lithium Manganese Oxide (Spinel) Cells. Solid State Ionics, 1994, 69, (1), 59-67. 24. Jang, D. H.; Shin, Y. J.; Oh, S. M., Dissolution of spinel oxides and capacity losses in 4V Li/LiMn204 coils. Journal of the Electrochemical Society, 1996, 143, (7), 2204-2211. 25. Cho, J. P.; Kim, T. J.; Kim, Y. J.; Park, B., Complete blocking of Mn 3+ ion dissolution from a LiMn204 spinel intercalation compound by Co3O4 coating. Chemical Communications, 2001, (12), 1074-1075. 26. Lu, W.; Belharouak, I.; Park, S. H.; Sun, Y. K.; Amine, K., Isothermal calorimetry investigation of Li+xMn2-yAl2O4 spinel. ElectrochimicaActa, 2007, 52, (19), 5837- 5842. 27. Ma, X. H.; Kang, B.; Ceder, G., High Rate Micron-Sized Ordered LiNio.5Mnl.50 4. Journalof the Electrochemical Society, 2010, 157, (8), A925-A931. 28. Padhi, A. K.; Nanjundaswamy, K. S.; Goodenough, J. B., Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. Journal of the ElectrochemicalSociety, 1997, 144, (4), 1188-1194. 29. Morgan, D.; Van der Ven, A.; Ceder, G., Li conductivity in LixMPO4 (M = Mn, Fe, Co, Ni) olivine materials. Electrochemical and Solid State Letters, 2004, 7, (2), A30-A32. 197 30. Sun, L.; Islam, R.; Ho, J. C.; Mujumdar, A. S., A diffusion model for drying of a heat sensitive solid under multiple heat input modes. Bioresource Technology, 2005, 96, (14), 1551-1560. 31. Kang, B.; Ceder, G., Battery materials for ultrafast charging and discharging. Nature, 2009, 458, (7235), 190-193. 32. Goodenough, J. B.; Kim, Y., Challenges for Rechargeable Li Batteries. Chemistry of Materials,2010, 22, (3), 587-603. 33. Ong, S. P.; Wang, L.; Kang, B.; Ceder, G., Li-Fe-P-02 phase diagram from first principles calculations. Chemistry of Materials,2008, 20, (5), 1798-1807. 34. Masquelier, C.; Padhi, A. K.; Nanjundaswamy, K. S.; Goodenough, J. B., New cathode materials for rechargeable lithium batteries: The 3-D framework structures Li3Fe2(XO 4 )3 (X = P, As). Journal of Solid State Chemistry, 1998, 135, (2), 228-234. 35. Koltypin, M.; Aurbach, D.; Nazar, L.; Ellis, B., On the stability of LiFePO 4 olivine cathodes under various conditions (electrolyte solutions, temperatures). Electrochemicaland Solid State Letters, 2007, 10, (2), A40-A44. 36. Gutierrez, A.; Benedek, N. A.; Manthiram, A., Crystal-Chemical Guide for Understanding Redox Energy Variations of M2+13+ Couples in Polyanion Cathodes for Lithium-Ion Batteries. Chemistry of Materials, 2013, Article ASAP. 37. Ellis, B.; Perry, L. K.; Ryan, D. H.; Nazar, L. F., Small polaron hopping in LixFePO4 solid solutions: Coupled lithium-ion and electron mobility. Journalof the American Chemical Society, 2006, 128, (35), 11416-11422. 38. Chung, S. Y.; Blocking, J. T.; Andersson, A. S.; Chiang, Y. M., Electronically conductive phospho-olivines as lithium storage electrodes. Solid State Ionics: Trends in the New Millennium, Proceedings,2002, 85-85. 39. Gaberscek, M.; Kuzma, M.; Jamnik, J., Electrochemical kinetics of porous, carbondecorated LiFePO 4 cathodes: separation of wiring effects from solid state diffusion. Physical Chemistry Chemical Physics, 2007, 9, (15), 1815-1820. 40. Gaberscek, M.; Dominko, R.; Bele, M.; Remskar, M.; Jamnik, J., Mass and charge transport in hierarchically organized storage materials. Example: Porous active materials with nanocoated walls of pores. Solid State Ionics, 2006, 177, (35-36), 3015-3022. 198 41. Ellis, B.; Herle, P. S.; Rho, Y. H.; Nazar, L. F.; Dunlap, R.; Perry, L. K.; Ryan, D. H., Nanostructured materials for lithium-ion batteries: Surface conductivity vs. bulk ion/electron transport. FaradayDiscussions, 2007, 134, 119-141. 42. Legagneur, V.; An, Y.; Mosbah, A.; Portal, R.; La Salle, A. L.; Verbaere, A.; Guyomard, D.; Piffard, Y., LiMBO3 (M = Mn, Fe, Co): synthesis, crystal structure and lithium deinsertion/insertion properties. Solid State Ionics, 2001, 139, (1-2), 37-46. 43. Yamada, A.; Iwane, N.; Harada, Y.; Nishimura, S.; Koyama, Y.; Tanaka, I., Lithium Iron Borates as High-Capacity Battery Electrodes. Advanced Materials, 2010, 22, (32), 3583-+. 44. Abouimrane, A.; Armand, M.; Ravet, N., Carbon Nano-painting: Application to Non-Phosphate Oxyanions e.g. Borate. Electrochemical Society Proceedings, 2003, 2003-20, 15-22. 45. Kokalj, A.; Dominko, R.; Mali, G.; Meden, A.; Gaberscek, M.; Jamnik, J., Beyond one-electron reaction in Li cathode materials: Designing Li2MnxFe-xSiO4. Chemistry of Materials,2007, 19, (15), 3633-3640. 46. Zhou, F.; Cococcioni, M.; Kang, K.; Ceder, G., The Li intercalation potential of LiMPO4 and LiMSiO4 olivines with M = Fe, Mn, Co, Ni. Electrochemistry Communications, 2004, 6, (11), 1144-1148. 47. Nyten, A.; Abouimrane, A.; Armand, M.; Gustafsson, T.; Thomas, J. 0., Electrochemical performance of Li2FeSiO4 as a new Li-battery cathode material. Electrochemistry Communications, 2005, 7, (2), 156-160. 48. Tarascon, J. M.; Armand, M., Issues and challenges facing rechargeable lithium batteries. Nature, 2001, 414, (6861), 359-367. 49. Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D., Challenges in the development of advanced Li-ion batteries: a review. Energy & Environmental Science, 2011, 4, (9), 3243-3262. 50. Ramesh, T. N.; Lee, K. T.; Ellis, B. L.; Nazar, L. F., Tavorite Lithium Iron Fluorophosphate Cathode Materials: Phase Transition and Electrochemistry of LiFePO 4F-Li2FePO4F. Electrochemical and Solid State Letters, 2010, 13, (4), A43A47. 199 51. Ellis, B. L.; Ramesh, T. N.; Davis, L. J. M.; Goward, G. R.; Nazar, L. F., Structure and Electrochemistry of Two-Electron Redox Couples in Lithium Metal Fluorophosphates Based on the Tavorite Structure. Chemistry of Materials, 2011, 23, (23), 5138-5148. 52. Barpanda, P.; Recham, N.; Chotard, J. N.; Djellab, K.; Walker, W.; Armand, M.; Tarascon, J. M., Structure and electrochemical properties of novel mixed Li(Fei. xMx)SO 4F (M = Co, Ni, Mn) phases fabricated by low temperature ionothermal synthesis. Journal of Materials Chemistry, 2010, 20, (9), 1659-1668. 53. Ati, M.; Sougrati, M. T.; Recham, N.; Barpanda, P.; Leriche, J. B.; Courty, M.; Armand, M.; Jumas, J. C.; Tarascon, J. M., Fluorosulfate positive electrodes for Liion batteries made via a solid-state dry process. Journal of the Electrochemical Society, 2010, 157, (9), A1007-A1015. 54. Chen, H. L.; Hautier, G.; Jain, A.; Moore, C.; Kang, B.; Doe, R.; Wu, L. J.; Zhu, Y. M.; Tang, Y. Z.; Ceder, G., Carbonophosphates: A New Family of Cathode Materials for Li-Ion Batteries Identified Computationally. Chemistry of Materials, 2012, 24, (11), 2009-2016. 55. Jain, A.; Hautier, G.; Moore, C.; Kang, B.; Lee, J.; Chen, H. L.; Twu, N.; Ceder, G., A Computational Investigation of Li9 M 3(P 20 7)3 (PO4 ) 2 (M = V, Mo) as Cathodes for Li Ion Batteries. Journalof the Electrochemical Society, 2012, 159, (5), A622-A633. 56. Malik, R.; Abdellahi, A.; Ceder, G., A Critical Review of the Li Insertion Mechanisms in LiFePO 4 Electrodes. Journal of the Electrochemical Society, 2013, 160, (5), A3179-A3197. 57. Allen, J. L.; Xu, K.; Zhang, S. S.; Jow, T. R., LiMBO 3 (M=Fe, Mn): Potential cathode for lithium ion batteries. Materials Research Society Symposium Proceedings,2002, 730, 9-14. 58. Chen, L.; Zhao, Y. M.; An, X. N.; Liu, J. M.; Dong, Y. Z.; Chen, Y. H.; Kuang, Q., Structure and electrochemical properties of LiMnBO3 as a new cathode material for lithium-ion batteries. Journal of Alloys and Compounds, 2010, 494, (1-2), 415419. 59. Dong, Y. Z.; Zhao, Y. M.; Shi, Z. D.; An, X. N.; Fu, P.; Chen, L., The structure and electrochemical performance of LiFeBO3 as a novel Li-battery cathode material. ElectrochimicaActa, 2008, 53, (5), 2339-2345. 200 60. Barpanda, P.; Yamashita, Y.; Yamada, Y.; Yamada, A., High-Throughput Solution Combustion Synthesis of High-Capacity LiFeBO3 Cathode. Journal of the ElectrochemicalSociety, 2013, 160, (5), A3095-A3099. 61. Aravindan, V.; Umadevi, M., Synthesis and characterization of novel LiFeBO3/C cathodes for lithium batteries. Ionics, 2012, 18, (1-2), 27-30. 62. Janssen, Y.; Middlemiss, D. S.; Bo, S. H.; Grey, C. P.; Khalifah, P. G., Structural Modulation in the High Capacity Battery Cathode Material LiFeBO3. Journal of the American Chemical Society, 2012, 134, (30), 12516-12527. 63. Bo, S. H.; Wang, F.; Janssen, Y.; Zeng, D. L.; Nam, K. W.; Xu, W. Graetz, J.; Yang, X. Q.; Du, L. S.; Q.; Zhu, Y. M.; Parise, J. B.; Grey, C. P.; Khalifah, P. G., Degradation and (de)lithiation processes in the high capacity battery material LiFeBO3 . Journalof Materials Chemistry, 2012, 22, (18), 8799-8809. 64. Seo, D. H.; Park, Y. U.; Kim, S. W.; Park, I.; Shakoor, R. A.; Kang, K., Firstprinciples study on lithium metal borate cathodes for lithium rechargeable batteries. PhysicalReview B, 2011, 83, (20). 65. Wang, L.; Maxisch, T.; Ceder, G., A first-principles approach to studying the thermal stability of oxide cathode materials. Chemistry of Materials,2007, 19, (3), 543-552. 66. Ong, S. P.; Jain, A.; Hautier, G.; Kang, B.; Ceder, G., Thermal stabilities of delithiated olivine MPO 4 (M = Fe, Mn) cathodes investigated using first principles calculations. Electrochemistry Communications, 2010, 12, (3), 427-430. 67. Bondareva, 0. S.; Simonov, M. A.; Egorovtismenko, Y. K.; Belov, N. V., CrystalStructure of LiZnBO3 and LiMnBO3. Soviet Physics Crystallography,1978, 23, (3), 269-271. 68. Li, R. K.; Chen, C. T.; Greaves, C., Magnetic order of LiMnBO3: A new type of chiral magnetic ground state. PhysicalReview B, 2002, 66, (5), -. 69. Norrestam, R., The Crystal-Structure of Monoclinic LiMgBO3. Zeitschrift Fur Kristallographie,1989, 187, (1-2), 103-110. 70. Kazanskaia, E. V.; Sandomirskii, P. A.; Simonov, M. A.; Belov, N. V., Crystal- 71. Structure of LiCdBO3 . Doklady Akademii Nauk Sssr, 1978, 238, (6), 1340-1343. Afyon, S.; Kundu, D.; Krumeich, F.; Nesper, R., Nano LiMnBO3, a high-capacity cathode material for Li-ion batteries. Journalof Power Sources, 2013, 224, 145-151. 201 72. Piffard, Y.; Rangan, K. K.; An, Y. L.; Guyomard, D.; Tournoux, M., Cobalt lithium orthoborate, LiCoBO3. Acta CrystallographicaC, 1998, 54, 1561-1563. 73. Aydinol, M. K.; Kohan, A. F.; Ceder, G.; Cho, K.; Joannopoulos, J., Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Physical Review B, 1997, 56, (3), 1354-1365. 74. Meng, Y. S.; Arroyo-de Dompablo, M. E., First principles computational materials design for energy storage materials in lithium ion batteries. Energy & Environmental Science, 2009, 2, (6), 589-609. 75. Hautier, G.; Jain, A.; Mueller, T.; Moore, C.; Ong, S. P.; Ceder, G., Designing Multielectron Lithium-Ion Phosphate Cathodes by Mixing Transition Metals. Chemistry of Materials,2013, 25, (10), 2064-2074. 76. Ong, S. P.; Richards, W. D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V. L.; Persson, K. A.; Ceder, G., Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. ComputationalMaterials Science 2013, 68, 314-319. 77. Hautier, G.; Jain, A.; Ong, S. P., From the computer to the laboratory: materials discovery and design using first-principles calculations. Journal of Materials Science, 2012, 47, (21), 7317-7340. 78. Reed, J. S., Principlesof Ceramics Processing.Wiley Int. Sci.: 1995. 79. Verma, P.; Maire, P.; Novak, P., A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. ElectrochimicaActa, 2010, 55, (22), 63326341. 80. Mills, G.; Jonsson, H.; Schenter, G. K., Reversible Work Transition-State Theory Application to Dissociative Adsorption of Hydrogen. Surface Science, 1995, 324, (23), 305-337. 81. Van der Ven, A.; Ceder, G., Lithium diffusion mechanisms in layered intercalation compounds. Journalof Power Sources, 2001, 97-8, 529-531. 82. Xu, B.; Meng, S., Factors affecting Li mobility in spinel LiMn204 A first-principles study by GGA and GGA plus U methods. Journalof Power Sources, 2010, 195, (15), 4971-4976. 83. Kang, K.; Morgan, D.; Ceder, G., First principles study of Li diffusion in I-Li 2NiO 2 structure. PhysicalReview B, 2009, 79, (1). 202 84. Bhattacharya, J.; Van der Ven, A., Phase stability and nondilute Li diffusion in spinel Lil+.Ti20 4. Physical Review B, 2010, 81, (10). 85. Yamada, A.; Iwane, N.; Nishimura, S.; Koyama, Y.; Tanaka, I., Synthesis and electrochemistry of monoclinic Li(MnxFei-x)BO3: a combined experimental and computational study. Journal of Materials Chemistry, 2011, 21, (29), 10690-10696. 86. Lee, K. J.; Kang, L. S.; Uhm, S.; Yoon, J. S.; Kim, D. W.; Hong, H. S., Synthesis and characterization of LiMnBO3 cathode material for lithium ion batteries. CurrentApplied Physics, 2013, 13, (7), 1440-1443. 87. Aravindan, V.; Karthikeyan, K.; Amaresh, S.; Lee, Y. S., LiMnBO3/C: A Potential Cathode Material for Lithium Batteries. Bulletin of the Korean Chemical Society, 2010, 31, (6), 1506-1508. 88. Weppner, W.; Huggins, R. A., Electrochemical Methods for Determining KineticProperties of Solids. Annual Review of MaterialsScience 1978, 8, 269-311. 89. Weppner, W.; Huggins, R. A., Thermodynamic Properties of Intermetallic Systems Lithium-Antimony and Lithium-Bismuth. Journal of the Electrochemical Society, 1978, 125, (1), 7-14. 90. Delacourt, C.; Ati, M.; Tarascon, J. M., Measurement of Lithium Diffusion Coefficient in LiyFeSO 4F. Journal of the Electrochemical Society, 2011, 158, (6), A741-A749. 91. Wen, C. J.; Boukamp, B. A.; Huggins, R. A.; Weppner, W., Thermodynamic and Mass-Transport Properties of Lial. Journal of the Electrochemical Society, 1979, 126, (12), 2258-2266. 92. Levi, M. D.; Levi, E. A.; Aurbach, D., The mechanism of lithium intercalation in graphite film electrodes in aprotic media .2. Potentiostatic intermittent titration and in situ XRD studies of the solid-state ionic diffusion. Journal of ElectroanalyticalChemistry, 1997, 421, (1-2), 89-97. 93. Montella, C., Apparent diffusion coefficient of intercalated species measured with PITT - A simple formulation. ElectrochimicaActa, 2006, 51, (15), 3102-3111. 94. Malik, R.; Burch, D.; Bazant, M.; Ceder, G., Particle Size Dependence of the Ionic Diffusivity. Nano Letters, 2010, 10, (10), 4123-4127. 95. Mo, Y. F.; Ong, S. P.; Ceder, G., First Principles Study of the Li1oGeP2S12 Lithium Super Ionic Conductor Material. Chemistry of Materials,2012, 24, (1), 15-17. 203 96. Wu, Y. B.; Lazic, P.; Hautier, G.; Persson, K.; Ceder, G., First principles high throughput screening of oxynitrides for water-splitting photocatalysts. Energy & Environmental Science, 2013, 6, (1), 157-168. 97. Jain, A.; Hautier, G.; Ong, S. P.; Moore, C. J.; Fischer, C. C.; Persson, K. A.; Ceder, G., Formation enthalpies by mixing GGA and GGA plus U calculations. Physical Review B, 2011, 84, (4). 98. Matsuo, T.; Shibasaki, M.; Katsumata, T.; Onoda, Y., Nuclear-Magnetic-Resonance of 7Li in Li2B407 Single-Crystal and Glass. Japanese Journal of Applied Physics, 1994, 33, (7A), 3913-3917. 99. Wizansky, A. R.; Rauch, P. E.; Disalvo, F. J., Powerful Oxidizing-Agents for the Oxidative Deintercalation of Lithium from Transition-Metal Oxides. Journal of Solid State Chemistry, 1989, 81, (2), 203-207. 100. Darling, R.; Newman, J., Modeling side reactions in composite LiyMn2O4 electrodes. Journalof the Electrochemical Society, 1998, 145, (3), 990-998. 101. Amatucci, G.; Du Pasquier, A.; Blyr, A.; Zheng, T.; Tarascon, J. M., The elevated temperature performance of the LiMn204/C system: failure and solutions. ElectrochimicaActa, 1999, 45, (1-2), 255-271. 102. Amatucci, G. G.; Schmutz, C. N.; Blyr, A.; Sigala, C.; Gozdz, A. S.; Larcher, D.; Tarascon, J. M., Materials' effects on the elevated and room temperature performance of C/LiMn204 Li-ion batteries. Journal of Power Sources, 1997, 69, (12), 11-25. 103. Li, S. L.; Xu, L. Q.; and performance Li, G. D.; Wang, M.; Zhai, Y. J., In-situ controllable synthesis investigation of carbon-coated monoclinic and hexagonal LiMnBO3 composites as cathode materials in lithium-ion batteries. Journal of Power Sources, 2013, 236, 54-60. 104. Delacourt, C.; Poizot, P.; Morcrette, M.; Tarascon, J. M.; Masquelier, C., One-step low-temperature route for the preparation of electrochemically active LiMnPO4 powders. Chemistry of Materials,2004, 16, (1), 93-99. 105. Yamada, A.; Tanaka, M.; Tanaka, K.; Sekai, K., Jahn-Teller instability in spinel Li-Mn-O. Journalof Power Sources, 1999, 81, 73-78. 106. Yamaguchi, H.; Yamada, A.; Uwe, H., Jahn-Teller transition of LiMn204 studied by X-ray-absorption spectroscopy. Physical Review B, 1998, 58, (1), 8-11. 204 107. Kang, B.; Ceder, G., Electrochemical Performance of LiMnPO 4 Synthesized with Off-Stoichiometry. Journal of the Electrochemical Society, 2010, 157, (7), A808A811. 108. Martha, S. K.; Grinblat, J.; Haik, 0.; Zinigrad, E.; Drezen, T.; Miners, J. H.; Exnar, I.; Kay, A.; Markovsky, B.; Aurbach, D., LiMno.8Feo.2PO4: An Advanced Cathode Material for Rechargeable Lithium Batteries. Angewandte Chemie-International Edition, 2009, 48, (45), 8559-8563. 109. Shannon, R. D., Revised Effective Ionic-Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta CrystallographicaA, 1976, 32, (Sepi), 751-767. 110. Prasad, R.; Benedek, R.; Thackeray, M. M., Dopant-induced stabilization of rhombohedral LiMnO2 against Jahn-Teller distortion. Physical Review B, 2005, 71, (13). 111. Yamada, A.; Chung, S. C., Crystal chemistry of the olivine-type Li(MnyFel-y)P04 and (MnyFel-y)P04 as possible 4 V cathode materials for lithium batteries. Journal of the Electrochemical Society, 2001, 148, (8), A960-A967. 112. Matts, I.; Chen, H.; Ceder, G., Electrochemical Properties of Li3 Feo.2Mno.8CO3PO4 as a Li-Ion Battery Cathode. ECS ElectrochemistryLetters, 2013, 2, (8), A81-A83. 113. Nyten, A.; Stjerndahl, M.; Rensmo, H.; Siegbahn, H.; Armand, M.; Gustafsson, T.; Edstrom, K.; Thomas, J. 0., Surface characterization and stability phenomena in Li2FeSiO4 studied by PES/XPS. Journal of Materials Chemistry, 2006, 16, (34), 3483-3488. 114. W. David Kingery, H. K. B., Donald R. Uhlmann, Introduction to ceramics. 2nd ed.; Wiley: 1976. 115. B. D. Cullity, S. R. S., Elements of X-ray diffraction. 3rd ed.; Addison-Wesley Publishing Company: 2001. 205 [End of thesis] 206