Non-Equilibrium Thermodynamics in Porous Media: Porous Materials
advertisement

Non-Equilibrium Thermodynamics in Porous Media:
Battery Degradation, and Sorption and Transport in
Porous Materials
by
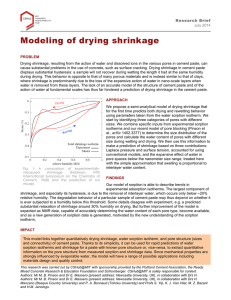
Matthew Bede Pinson
PhB, The Australian National University (2008)
Submitted to the Department of Physics
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
February 2015
c Massachusetts Institute of Technology 2015. All rights reserved.
○
Author . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Department of Physics
December 22, 2014
Certified by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Martin Z. Bazant
Professor of Chemical Engineering and Professor of Mathematics
Thesis Supervisor
Certified by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Mehran Kardar
Francis Friedman Professor of Physics
Thesis Supervisor
Accepted by . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Krishna Rajagopal
Associate Department Head for Education
2
Non-Equilibrium Thermodynamics in Porous Media: Battery
Degradation, and Sorption and Transport in Porous Materials
by Matthew Bede Pinson
Submitted to the Department of Physics on December 22, 2014, in partial
fulfillment of the requirements for the degree of Doctor of Philosophy
Abstract
Porous media offer many interesting problems in physics and engineering due to the
interaction of phase transitions, surface effects and transport. In this thesis I examine two such problems: the degradation of lithium-ion batteries, and sorption and
transport of fluids in porous materials.
The dominant capacity fade mechanism in many lithium-ion batteries is the loss
of cyclable lithium to a solid-electrolyte interphase layer on the surface of the negative
electrode. I develop a single-particle model of this fade mechanism, based on diffusion
of the reacting species through the growing layer and reaction at the surface of the
active material. This analytical model is justified by comparison with a computational
porous electrode model.
Temperature is identified as the most important variable
influencing the capacity fade rate, and the model is able to make predictions for
accelerated aging tests as well as the effect of mismatched internal resistances in
battery packs.
The quantity of a fluid taken up by a porous material as a function of the partial
pressure of the fluid relative to saturation can be used to measure the pore size distribution of the material. However, hysteresis between the wetting and drying paths
complicates the interpretation of experimental results. I present a unified model of
hysteresis that accounts for both single-pore and network effects, enabling the calculation of not only the pore size distribution but also a parameter measuring the
connectivity between large and small pores.
I then use the ideas of the model to
examine drying shrinkage in hardened cement paste, demonstrating that the hysteresis in this shrinkage is primarily due to water inserted between molecular layers
in calcium-silicate-hydrate. Finally, I outline a model of transport of a sorbing fluid
with hysteresis, and suggest possible extensions to allow quantitative comparison with
experimental results.
Thesis Supervisor: Martin Z. Bazant
Title: Professor of Chemical Engineering and Professor of Mathematics
Thesis Supervisor: Mehran Kardar
Title: Francis Friedman Professor of Physics
3
4
Acknowledgments
I am very grateful to many people who have supported me during my graduate studies,
including:
∙
Anna, Patrick and other friends who sometimes made me feel like I never left
Australia,
∙
My fellow music-lovers in the MIT Gilbert and Sullivan Players, who helped me
keep my horizons broad while at MIT,
∙
All my friends among the Tech Catholic Community, for the fun, food and faith
that we have shared,
∙
Prof.
David Williams, who first urged me to consider MIT and continues to
help me in my academic career,
∙
The Bazant research group, for their friendly faces each day and useful tips
whenever I needed them,
∙
Prof.
Hamlin Jennings, Enrico and the rest of the Dome collaboration and
Radu, for interesting collaborations leading to the results in this thesis,
∙
My friends in the MIT physics program, for many hours of productive study
and enjoyable socializing,
∙
Jordan and Patrick and the many visitors who made PPP Junction feel like a
true home,
∙
My thesis committee, Profs Mehran Kardar, Leonid Levitov and Robert Jaffe,
for insightful questions and suggestions that helped guide my work,
∙
My family, who encouraged me to make the most of the opportunities that came
my way and whose Skype calls have been the highlight of many weekends, and
∙
My supervisor, Prof. Martin Bazant, for finding the right projects for me and
never failing to offer suggestions and help.
5
6
Contents
1 Introduction
15
2 Degradation of lithium-ion batteries
17
2.1
Lithium-ion batteries . . . . . . . . . . . . . . . . . . . . . . . . . . .
17
2.2
Degradation mechanisms . . . . . . . . . . . . . . . . . . . . . . . . .
18
2.2.1
Solid-electrolyte interphase formation . . . . . . . . . . . . . .
18
2.2.2
Other degradation mechanisms
. . . . . . . . . . . . . . . . .
19
2.2.3
Degradation of silicon anodes
. . . . . . . . . . . . . . . . . .
20
. . . . . . . . . . . . . . . . . . . . . . . . . .
20
2.3
Degradation modeling
3 Modeling of solid-electrolyte interphase formation
3.1
Advantages of a physical model
3.2
A porous electrode model
3.3
3.4
23
. . . . . . . . . . . . . . . . . . . . .
23
. . . . . . . . . . . . . . . . . . . . . . . .
24
Single particle model . . . . . . . . . . . . . . . . . . . . . . . . . . .
30
3.3.1
Model formulation
. . . . . . . . . . . . . . . . . . . . . . . .
30
3.3.2
Comparison with experiment . . . . . . . . . . . . . . . . . . .
34
3.3.3
Multiple reacting species . . . . . . . . . . . . . . . . . . . . .
34
Temperature effects . . . . . . . . . . . . . . . . . . . . . . . . . . . .
35
3.4.1
Accelerated aging . . . . . . . . . . . . . . . . . . . . . . . . .
35
3.4.2
Resistance mismatch in battery packs . . . . . . . . . . . . . .
39
3.5
Lifetime prediction and statistics
. . . . . . . . . . . . . . . . . . . .
46
3.6
Extensions for rapid capacity fade . . . . . . . . . . . . . . . . . . . .
47
3.6.1
48
Fresh surface
. . . . . . . . . . . . . . . . . . . . . . . . . . .
7
3.6.2
Unstable SEI
. . . . . . . . . . . . . . . . . . . . . . . . . . .
4 Overview of models of sorption in porous materials
4.1
Modeling of sorption in a single pore
53
4.1.1
Surface adsorption
. . . . . . . . . . . . . . . . . . . . . . . .
53
4.1.2
Pore filling . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
54
Modeling of sorption hysteresis in porous materials
4.3
Modeling of transport in porous materials
. . . . . . . . . .
56
. . . . . . . . . . . . . . .
58
5 A model of sorption in multiscale porous materials
5.2
5.3
6.2
6.3
61
Single pore hysteresis model . . . . . . . . . . . . . . . . . . . . . . .
61
5.1.1
Model
61
5.1.2
Application to simple porous materials
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Network effects
. . . . . . . . . . . . .
63
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
63
5.2.1
Model
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
66
5.2.2
Insertion within the “solid” . . . . . . . . . . . . . . . . . . . .
68
5.2.3
Homogeneous nucleation . . . . . . . . . . . . . . . . . . . . .
70
5.2.4
Application of the model to experimental data . . . . . . . . .
72
5.2.5
Scanning isotherms . . . . . . . . . . . . . . . . . . . . . . . .
76
Potential for further developments . . . . . . . . . . . . . . . . . . . .
78
6 Drying shrinkage in hardened cement paste
6.1
53
. . . . . . . . . . . . . . . . . .
4.2
5.1
49
81
Overview of hardened cement paste . . . . . . . . . . . . . . . . . . .
81
6.1.1
Structure
. . . . . . . . . . . . . . . . . . . . . . . . . . . . .
81
6.1.2
Shrinkage hysteresis . . . . . . . . . . . . . . . . . . . . . . . .
82
Classification of water in cement paste
. . . . . . . . . . . . . . . . .
85
6.2.1
Interlayer water . . . . . . . . . . . . . . . . . . . . . . . . . .
85
6.2.2
Gel pore water
85
6.2.3
Capillary pore water
. . . . . . . . . . . . . . . . . . . . . . .
85
6.2.4
Surface adsorbed water . . . . . . . . . . . . . . . . . . . . . .
86
Water sorption isotherm of hardened cement paste . . . . . . . . . . .
86
. . . . . . . . . . . . . . . . . . . . . . . . . .
8
6.4
6.5
6.3.1
Interlayer water . . . . . . . . . . . . . . . . . . . . . . . . . .
86
6.3.2
Gel and capillary pore water . . . . . . . . . . . . . . . . . . .
87
Continuum modeling of reversible drying shrinkage
. . . . . . . . . .
6.4.1
Macroscopic length change due to Laplace pressure
6.4.2
Macroscopic length change due to surface energy
87
. . . . . .
89
. . . . . . .
89
6.4.3
Macroscopic length change due to loss of interlayer water . . .
90
6.4.4
Comparison with experiment . . . . . . . . . . . . . . . . . . .
91
Potential future extensions . . . . . . . . . . . . . . . . . . . . . . . .
93
7 Modeling of transport with hysteresis in porous materials
95
7.1
Model formulation
. . . . . . . . . . . . . . . . . . . . . . . . . . . .
95
7.2
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
98
7.2.1
Time-dependent hysteresis curves . . . . . . . . . . . . . . . .
98
7.2.2
Saturation-dependent apparent diffusivity
7.3
7.4
. . . . . . . . . . .
101
Humidity-dependent permeability . . . . . . . . . . . . . . . . . . . .
101
7.3.1
Pore size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
103
7.3.2
Pore connectedness . . . . . . . . . . . . . . . . . . . . . . . .
103
Inclusion of detailed modeling of hysteresis mechanisms . . . . . . . .
104
8 Conclusions and outlook
107
8.1
Battery degradation
. . . . . . . . . . . . . . . . . . . . . . . . . . .
107
8.2
Sorption and transport in porous materials . . . . . . . . . . . . . . .
108
9
10
List of Figures
2-1
Intercalation (desired) and SEI formation (unwanted) reactions . . . .
18
3-1
Experimental and modeled capacity fade . . . . . . . . . . . . . . . .
27
3-2
Simulated capacity fade depends on time, not on number of cycles.
.
28
3-3
Voltage-capacity curves with and without SEI resistance
. . . . . . .
29
3-4
Spatial variation in SEI formation . . . . . . . . . . . . . . . . . . . .
31
3-5
Experimental and modeled capacity fade at several temperatures . . .
36
3-6
Accelerated aging prediction for a Li-Li cell . . . . . . . . . . . . . . .
37
3-7
Accelerated aging prediction for a Li−LiCoO2 cell . . . . . . . . . . .
38
3-8
Current distribution between parallel-connected cells
40
3-9
𝐷
. . . . . . . . .
as a function of C rate, fit to measured capacity fade.
. . . . . . .
41
. . . . . . . . .
42
. . . . . . . . . . . . .
43
3-12 Modeled capacity fade according to Wang et al. [178] . . . . . . . . .
44
3-10 Experimental and model capacity fade for two cells.
3-11 Maximum charging current vs excess capacity
3-13 Experimental and predicted lifetime as a function of resistance mismatch 46
3-14 Lifetime distribution statistics . . . . . . . . . . . . . . . . . . . . . .
47
3-15 Capacity fade of a full cell with a Si anode . . . . . . . . . . . . . . .
50
3-16 Capacity fade with SEI dissolution or delamination
. . . . . . . . . .
52
4-1
Adsorbed layer thickness and example configurations
. . . . . . . . .
55
4-2
Illustration of radial filling and axial emptying . . . . . . . . . . . . .
57
5-1
Experimental and model sorption isotherms in carbon black plugs
. .
64
5-2
Calculated PSD of carbon black plugs . . . . . . . . . . . . . . . . . .
65
11
5-3
Hysteresis due to the absence of a liquid-vapor interface . . . . . . . .
66
5-4
A network of small and large pores is represented by a Bethe lattice .
68
5-5
Emptying probability vs fraction of pores below emptying pressure . .
69
5-6
Example sorption prediction showing single-pore and network hysteresis 73
5-7
Experimental and model isotherms in various porous materials . . . .
74
5-8
Calculated PSD of various porous materials
75
5-9
Experimental and model scanning isotherms in dental enamel
5-10 Cavitation pressure vs reduced temperature
. . . . . . . . . . . . . .
. . . .
77
. . . . . . . . . . . . . .
80
6-1
Water sorption and shrinkage of cement, clay and Vycor
. . . . . . .
83
6-2
Schematic illustration of water sorption in cement paste . . . . . . . .
84
6-3
Water sorption in cement divided by location of water . . . . . . . . .
88
6-4
Experimental and predicted drying shrinkage of cement paste . . . . .
92
7-1
Dynamic hysteresis of water content with no resting . . . . . . . . . .
99
7-2
Dynamic hysteresis of water content with resting . . . . . . . . . . . .
100
7-3
Effective diffusivity of water in cement vs saturation . . . . . . . . . .
102
12
List of Tables
5.1
Initial fraction of exposed pores,
𝑓,
calculated by applying the model
presented here to a variety of published experimental data.
13
. . . . .
76
14
Chapter 1
Introduction
This thesis examines the problems of lithium-ion battery degradation, and the sorption and transport of fluids in porous materials. Although these problems may seem
rather far removed, they are in fact linked by several important features.
First, in both systems phase transformations are crucial in determining behavior,
and these phase transitions are strongly influenced by surfaces and other features of
the microstructure of the materials. For example, the potential at which a lithium
iron phosphate particle will phase separate into lithium-rich and lithium-poor regions
is dependent on its size [41], while the relationship between sorbate partial pressure
and pore filling has long been used as a tool to measure the pore size distribution of
porous materials [14].
Second, measurements are usually made at the system level. In batteries, measurements generally relate voltage, current and state of charge, while sorption experiments
control the sorbate pressure and measure sorbed mass (or equivalent) as a function
of time. Information about the microscopic behavior of system constituents is thus
indirect, relying on models that relate microscopic and macroscopic properties.
Third, there is an interaction between transport and phase separation processes.
In lithium-ion batteries, transport effects can suppress phase separation during fast
discharge [7], or mitigate the degradation that results from the formation of unwanted
products, as discussed in this thesis. In porous materials, hysteresis in the sorption
process can complicate transport modeling, and knowledge of the location of con-
15
densed fluid may be crucial in accurately determining permeability.
Fourth, both problems are of enormous technological importance.
Lithium-ion
batteries are widespread in electronic devices, and their prevalence is only expected
to increase with further demands such as electric vehicles. Concrete is one example of
an extremely heavily used porous material, and its degradation is heavily influenced
by sorption and transport processes.
Chapter 2, provides an overview of the modeling of lithium-ion batteries, especially
of their degradation.
Chapter 3 then presents my model of degradation and its
application to accelerated aging testing as well as understanding the degradation
of battery packs. Switching to porous solids, chapter 4 outlines accepted models of
sorption and transport in these materials. Chapter 5 combines accepted hysteresis
mechanisms with a new analytic model of pore blocking in a network, to obtain a
method for determining pore size and connectivity from sorption isotherms. Chapter
6 uses such information in the specific case of water in hardened cement paste to
explain drying shrinkage and its hysteresis. Chapter 7 presents a model of transport
through porous media, presenting some results and discussing future extensions of
the model. Finally, chapter 8 discusses the application of the models of this work to
systems of technological importance, and how they may be extended and combined
with other ideas to provide further insights.
16
Chapter 2
Degradation of lithium-ion batteries
2.1 Lithium-ion batteries
Lithium-ion batteries comprise two active materials into which lithium can be intercalated, divided by a separator that allows the transmission of lithium ions but not
electrons [88, 151].
The materials are generally present in the form of a fine pow-
der, to minimise the impact of the slow transport of lithium through the solid.
A
conductive powder such as carbon is mixed with the active materials to allow the
transport of electrons between the active material and a current collector located in
each electrode. When these current collectors are connected by an external circuit,
lithium ions can move from one active material to the other, while the associated
electrons travel around the circuit. The existence of a free energy difference between
lithium atoms located in each active material means that the battery can provide
a voltage and sustain a current, while lithium moves from the high to the low free
energy material. Alternatively, applying an external voltage across the terminals can
drive electrons in the opposite direction, causing the transport of lithium ions from
the low to the high free energy material, recharging the battery.
Lithium-ion batteries are widespread in electronic appliances [164], and considerable research is focused on broadening their application, such as to electric vehicles [5]
and power grid load leveling [50]. To be competitive in such applications, battery life
of thousands of cycles and years to decades is required. The important degradation
17
Figure 2-1: The desired electrochemical reaction is the intercalation of lithium, but
lithium can also react with components of the electrolyte to form a solid-electrolyte
interphase.
mechanisms of lithium-ion batteries must therefore be understood and mitigated.
2.2 Degradation mechanisms
2.2.1
Solid-electrolyte interphase formation
One important degradation mechanism is the loss of lithium to a solid-electrolyte
interphase layer (SEI). This layer forms when, instead of intercalating into the negative electrode material, lithium ions react with substances present in the electrolyte,
as indicated schematically in figure 2-1. Such substances can include the electrolyte
solvent itself, as typical electrolytes are not stable at the charging potential of the
negative electrode. This reaction takes place on the surface of the active material,
and forms an SEI here [164]. The SEI can include an enormous variety of compounds
including lithium carbonate, lithium fluoride, lithium oxide, and many organic compounds [165, 172].
The initial formation of an SEI is beneficial to battery operation, as it protects
against solvent decomposition at large negative voltage. However, there are also two
ways in which SEI formation can degrade a battery: a loss of capacity as previously
active lithium is trapped in the SEI [127], and an increase in impedance due to the
18
resistance of the SEI to the diffusion of lithium ions [186, 173].
Additionally, the
compounds forming the SEI can react exothermically if the temperature becomes too
high, potentially leading to hazardous thermal runaway [189].
Many researchers have studied the role of the SEI in battery degradation, and the
factors that influence its formation. Ozawa found a gradual decline in capacity and
increase in impedance due to SEI formation, and identified lithium, carbon, oxygen
and fluorine as the main constituents of the SEI [120].
Peterson et al.
observed
slow degradation of batteries containing graphite electrodes under simulated driving
conditions (95% capacity remaining after 2000 days), with capacity loss proportional
to the number of intercalation and deintercalation events [131]. Zhang et al. studied
degradation as a function of temperature, finding that the increase in impedance due
to SEI formation was far more important at low temperature than at high temperature
[188].
2.2.2
Other degradation mechanisms
SEI formation is not the only mechanism by which lithium-ion batteries can degrade.
Degradation due to damage to thecrystal structure of the active material has been
found in cells using lithium manganese oxide [181] and lithium nickel cobalt oxide
[162] as the positive electrode active material. Komaba et al. studied the dissolution
of manganese from LiMn2 O4 , used as a positive electrode. They found that a small
quantity of dissolved manganese was sufficient to severely decrease the capacity of the
battery, as it formed a film on the surface of the negative electrode, preventing intercalation [82]. Amine et al. showed that this dissolution could be prevented by using
an electrolyte that does not contain fluorine [2]. Shim, Striebel and Cairns observed
capacity fade due to the dissolution of lithium polysulfides [154].
Mikhaylik and
Akridge studied this mechanism further, showing that increasing salt concentration
in the electrolyte lowers the rate of lithium dissolution [109]. Shim, Kostecki et al.
observed the build-up of an organic layer on the positive electrode, with much higher
resistance than the SEI generally found on the negative electrode [153]. Kostecki et
al. examined a similar system and found that the surface resistance of active material
19
particles in the positive electrode increased over time [84].
Despite this variety of degradation mechanisms, SEI formation is generally considered to dominate capacity fade in lithium-ion batteries suitable for commercial use
[173], and thus I focus on this mechanism in this thesis. The model presented here
will not be applicable when other sources of fade dominate.
2.2.3
Degradation of silicon anodes
Most studies of lithium-ion battery degradation, including those cited in subsection
2.2.1, have focused on batteries with a graphite negative electrode, as graphite is
found in almost all commercial lithium-ion batteries. A promising electrode material
for future application is silicon. Silicon has a much higher capacity for lithium intercalation than graphite does [27], but it experiences large expansion on intercalation, and
this large volume change can lead to degradation [115]. Cracking and pulverization
can lead to the loss of electrical contact and hence capacity loss [3], though this can be
minimised by using small silicon particles as the active material [21, 177, 81, 37, 45].
An SEI also forms on silicon: it is very similar to that on graphite but shows cracks
due to the volume change of the underlying silicon [38].
2.3 Degradation modeling
The most common approach to the modeling of lithium-ion battery degradation has
been based on fits of data to empirical models, not a theory of degradation mechanisms. For example, Rong and Pedram developed a single-cycle model of battery
behavior that was built on physical processes, but described degradation empirically,
using a very large number of parameters [146]. He et al. described capacity fade by
a sum of exponentials, and used measured capacity over early cycles to predict the
continuing capacity fade [76].
A model will be more useful, especially in the prediction of fade for a system
different from that used to determine model parameters, if it is based on the real
physical processes that lead to degradation. Various authors have modeled different
20
degradation processes, such as crack propagation, modeled using an equivalent circuit
model [110], but we focus here on models of fade due to SEI formation.
Two approaches to modelling capacity fade due to SEI formation have been put
forward.
The first uses a porous electrode model, developed to model the single-
cycle behavior of a cell or electrode, and adds a side reaction forming SEI. This
enables spatial differences in SEI formation to be examined, but previous examples
of such models developed by Ramadass et al. [139] and Sikha et al. [157] included
the dependence of the SEI formation rate on local potential, but not on lithium ion
concentration or the thickness of the existing SEI. Neglect of the latter in particular
means that the predicted capacity fade is linear in time, which contradicts experimental observations of a decreasing fade rate [159]. The second approach has been
to assume that the reaction forming SEI is indeed hindered by the existing layer as
it grows, but to ignore any possible spatial variation in SEI formation through the
electrode, and the reaction limitation of SEI formation when the existing layer is very
thin [135].
In chapter 3, I present a porous electrode model that combines features of both
approaches. The model accounts for gradients in lithium ion concentration as well as
potential, and also the effect of the growing layer. This model shows that gradients
in SEI formation are unimportant, so a single particle model may be used. The single
particle model I present is a reaction-diffusion model, an important step forward from
a previous diffusion model that assumed very fast reactions [135].
21
22
Chapter 3
Modeling of solid-electrolyte
interphase formation
The content of this chapter is based heavily on the published articles “Theory of SEI
Formation in Rechargeable Batteries: Capacity Fade, Accelerated Aging and Lifetime
Prediction” by the author and Bazant [132], and “Internal Resistance Matching for
Parallel-Connected Lithium-Ion Cells and Impacts on Battery Pack Cycle Life” by
Gogoana, the author et al. [67]. Material from these articles is copyright 2013, The
Electrochemical Society and copyright 2014, Elsevier, respectively and reproduced
with permission.
3.1 Advantages of a physical model
One way of assessing capacity fade is purely experimental: operate a battery in the
expected use conditions, and measure its capacity over time.
An important disad-
vantage of this method is that it is necessarily very time-consuming, especially for
a good, long-lived battery.
To overcome this difficulty, experiments are frequently
performed at high temperature, where the rate of capacity fade is increased [168]. To
interpret such experiments, and make predictions that are valid at use temperatures,
it is necessary to have a model of degradation that is based on the physical processes
that lead to capacity fade.
23
In this chapter, I present a theory of SEI growth and capacity fade. In section 3.2, I
use a porous electrode model to quantify capacity fade due to SEI formation, focusing
especially on any spatial variations through the electrode.
Since spatial variations
are insignificant, I present in section 3.3 a simple, single particle model. This model
improves upon previous models that simply assert a
√
𝑡
dependence of capacity fade
due to the increasing thickness of the SEI layer [135, 160], as it takes into account the
rate of the reaction forming SEI. In section 3.4, I examine the effect of temperature,
treating the two situations of accelerated aging tests and mismatched cell resistances
in battery packs. In section 3.5, I use the theory to predict battery lifetime statistics.
In section 3.6, I discuss extensions to account for additional degradation mechanisms
that accelerate capacity fade, such as area changes and SEI delamination resulting
from volume expansion of the active particles.
3.2 A porous electrode model
This work models capacity fade by considering only the loss of lithium to the SEI on
the negative electrode, assuming that other sources of capacity fade can be neglected.
The SEI is produced by the reaction between lithium ions and various other species
at the surface of the active material in one of the electrodes. If SEI formation were
sustained throughout battery operation, it would render Li-ion batteries unusable
due to the continual loss of lithium. The reason that Li-ion batteries can operate is
that the SEI does not conduct electrons, and is almost impenetrable to electrolyte
molecules [172].
Once an initial SEI layer has formed, the inability of electrolyte
molecules to travel through the SEI to the active material surface, where they could
react with lithium ions and electrons, suppresses further SEI growth [135]. Intercalation is suppressed far less, because lithium ions can easily pass through the SEI
through the exchange of ions between the solvent, SEI compounds and the lithium
intercalated in the active material [172]. Thus the battery is able to experience many
charge-discharge cycles with little additional SEI build-up.
The model presented here is a one-dimensional porous electrode model [179, 48,
24
62, 117, 60] that accounts for concentration gradients in the primary direction of
lithium propagation.
The porous electrode model is based on a diffusion-reaction equation,
𝜕𝑐𝑖
𝜕𝑁𝑖 ∑︁
=−
−
𝐽(𝑖),
𝜕𝑡
𝜕𝑥
(3.1)
𝑐𝑖 is the concentration
∑︀
𝐽(𝑖) is the sum of all
which expresses mass balance in a volume-averaged sense. Here
of species i in the electrolyte,
𝑁𝑖
is the flux of species i and
interfacial reactions consuming species i. We let i range over positive and negative
ions, assuming that the consumption of electrolyte molecules by reactions forming
SEI is slow enough that it does not significantly affect electrolyte concentration.
The fluxes
𝑁𝑖
+
are calculated using the Nernst-Planck equation.
reaction Li(dissolved) + e
–
The rate of the
−−→ Li(intercalated) is modeled using a symmetric Butler-Volmer
equation:
𝐽 = 2𝑖0 sinh(
Here
𝜂
−𝑒𝜂
).
2𝑘𝐵 𝑇
(3.2)
is the overpotential driving the reaction: the potential difference across the
surface of the active material, where the reactions take place, minus the equilibrium
potential difference, i.e.
𝜂 = 𝜑𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑑𝑒 − 𝜑𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑙𝑦𝑡𝑒 − ∆𝜑𝑒𝑞𝑢𝑖𝑙𝑖𝑏𝑟𝑖𝑢𝑚 + 𝐽𝑅.
(3.3)
The equilibrium potential for each electrode, relative to lithium, is drawn from figure
5 of Liu et al. [93]: these potentials are approximated by the functions
−0.425
∆𝜑𝑔𝑟𝑎𝑝ℎ𝑖𝑡𝑒
− 0.0903)
𝑒𝑞𝑢𝑖𝑙𝑖𝑏𝑟𝑖𝑢𝑚 = (0.132𝑐
V
𝑒𝑃 𝑂4
11
∆𝜑𝐿𝑖𝐹
𝑒𝑞𝑢𝑖𝑙𝑖𝑏𝑟𝑖𝑢𝑚 = (−0.0039𝑐 + 3.405 − 908.954(𝑐 − 0.470) )
where
𝑐
V,
is the concentration of intercalated lithium, divided by the concentration of
lithium in LiC6 . The slight dependence of
that the concentration
∆𝜑𝑒𝑞𝑢𝑖𝑙𝑖𝑏𝑟𝑖𝑢𝑚
on
𝑐+
is ignored. We assume
𝑐 is uniform throughout a given active material particle, that is,
25
we neglect transport and phase transformations [75] in the solid.
𝑅, in the case of the
graphite electrode, is the resistance of the SEI layer, proportional to SEI thickness.
The positive electrode is assumed to have no interfacial resistance.
current density
𝑖0
The exchange
depends on concentration: we assume that
√
𝑖0 = 𝑘 𝑐+
for the intercalation, where
𝑘
(3.4)
is a constant: this is the simplest implementation of
a symmetric Butler-Volmer equation. The concentration of lithium ions,
𝑐+ ,
varies
through the electrode but is assumed to be constant through the depth of the SEI
layer.
Capacity fade is modeled through a competing current that leads to SEI
formation, with rate
𝐽𝑆𝐸𝐼 = 2𝑖0𝑆𝐸𝐼 sinh(
+
this models the reaction Li(dissolved) + E + e
–
−𝑒𝜂𝑆𝐸𝐼
):
2𝑘𝐵 𝑇
(3.5)
−−→ LiE where E is some species from the
electrolyte that can react with lithium, and LiE is the product, which forms the SEI
layer. Overpotential is calculated in the same way as for the intercalation current,
𝜂𝑆𝐸𝐼 = 𝜑𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑑𝑒 − 𝜑𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑙𝑦𝑡𝑒 − ∆𝜑𝑆𝐸𝐼 + 𝐽𝑅.
Here
∆𝜑𝑆𝐸𝐼
[51, 33, 172].
(3.6)
is the equilibrium potential for SEI formation, approximately 0.8 V
In this case the reaction requires not only a lithium ion but also an
electrolyte molecule, so
√
𝑖0𝑆𝐸𝐼 = 𝑘𝑆𝐸𝐼 𝑐+ 𝑐𝑠 .
The electrolyte concentration
𝑐𝑠
(3.7)
should be measured at the bottom of the SEI layer.
Assuming that the concentration outside the SEI layer is constant, and also assuming
linear diffusion, we can write
𝑐𝑠 = (1 − 𝑍𝑐𝑆𝐸𝐼 𝐽𝑆𝐸𝐼 )𝑐0 ,
where
𝑍
(3.8)
is a constant inversely proportional to the diffusivity of electrolyte in SEI,
26
Figure 3-1: The single particle model (solid line) and porous electrode model (dotted
line) both closely fit experimental data (points).
Data from Liu et al.
[93] with a
lithium iron phosphate opposite electrode.
𝑐𝑆𝐸𝐼
measures the amount of SEI present at a location in space, given by
𝜕𝑐𝑆𝐸𝐼
= 𝐽𝑆𝐸𝐼 ,
𝜕𝑡
and
𝑐0
(3.9)
is the concentration of the relevant electrolyte molecule outside the SEI; its
value can be absorbed into
𝑘𝑆𝐸𝐼 .
Using these equations along with standard modeling of transport [48, 161, 47],
SEI formation was modeled over many cycles. Figure 3-1 compares experimental [93]
capacity fade with the predictions of the single particle and porous electrode models.
Using the values of
𝜌, 𝑘 , 𝑘𝑆𝐸𝐼
and
𝑍
needed to fit these experimental data, capacity
fade was simulated over a wide range of discharge rates. The results are plotted as a
function of cycle number and of time in figure 3-2. It is clear that time, rather than
cycling, is the dominant factor in SEI growth, under this model.
27
Figure 3-2: Simulated capacity fade depends on time, not on number of cycles.
28
Figure 3-3: The top graph shows simulated voltage-capacity curves for the 1st, 161st,
366th, 1054th, 1914th and 2628th cycles, assuming that the SEI has infinite lithium
ion conductivity.
The bottom graph shows the same curves for finite lithium ion
conductivity. Voltage is decreased in the latter case, while the capacity fade is not
substantially affected.
The best fit to the experimental results of Liu et al. [93] includes zero SEI resistivity. To ensure that this parameter choice, which simplifies computation substantially,
does not interfere with the accuracy of the results, simulations were performed with
a non-zero resistance. Results are shown in figure 3-3. The SEI resistance lowers the
output voltage, but has little effect on capacity fade.
An important feature of the porous electrode model is that it can assess the degree of spatial variation in SEI formation.
Results of the model demonstrate that
in realistic battery operating conditions, SEI formation is very uniform. Only in ex-
29
treme charging conditions, where the lithium concentration gradient in the electrolyte
is large enough to cause substantial underutilization of the battery, do significant
non-uniformities in SEI formation occur. Figure 3-4 compares SEI formation in an
electrode of width 50
𝜇m
𝜇m,
which is uniform even for fast charging, with that in a 250
electrode, where fast charging causes a moderate degree of non-uniformity. This
non-uniformity is dependent on strong depletion of the electrolyte during charging.
Thus although the variation of SEI through the electrode should be considered when
a battery is operated in extreme conditions such as at ultra-high rate or with very
thick electrodes, a single particle model ignoring this spatial non-uniformity will be
valid in normal circumstances.
3.3 Single particle model
3.3.1
Model formulation
Our single particle model relies on the following assumptions.
Uniformity
We assume that the amount of lithium in the SEI can be param-
eterized by a single variable
𝑠,
the thickness of the SEI. This thickness is assumed
to be independent of location within the negative electrode, meaning that the electrode is homogeneous and is thin enough in the direction of lithium propagation that
significant concentration gradients do not form.
Negligible solid deformation All intercalation compounds experience local expansion (and in some cases, also anisotropic contraction [40] due to lattice mismatch)
on lithium insertion.
If the lattice mismatch is not too large (<
5%
strain), as in
graphite [49, 15], then the solid expands reversibly with a concomitant, small increase
in the internal surface area. The area available for SEI growth is thus fluctuating, but
for practical battery electrodes it is reasonable to assume a constant surface area as
we do in our initial model. The SEI layer also experiences small enough strain that
it is likely to remain uniformly adhered to the surface.
For certain emerging electrode materials, it is well known that solid deformation
30
Figure 3-4: Computational results show that a thin electrode (top) displays no significant spatial variation in SEI formation, while a thicker electrode (bottom) does
display some variation when charged at a high rate.
chosen to achieve equal average SEI density.
31
The numbers of cycles were
plays a major role in limiting the performance and cycle life by accelerating the rate
of SEI formation. In particular, silicon is a very attractive high-energy-density anode
material for Li-ion batteries, but lithium insertion causes isotropic volume expansion
by a factor of four [27]. During expansion and contraction of the active particles, the
area for SEI growth changes, and the SEI layer is placed under large cyclic stresses
that can lead to delamination. We neglect these effects until section 3.6.
Constant base reaction rate We assume that, if electrolyte concentration were
constant at the reaction surface, the rate of SEI formation would remain constant. At
first glance, this assumption may seem shaky because it ignores any dependence of
SEI formation rate on the state of charge of the electrode and even on the magnitude
and direction of the intercalation current. However, the SEI formation rate should
depend on these factors only through its dependence on potential, and open circuit
potential is close to constant over a wide range of lithium concentration in graphite
[183].
The local potential will not differ too much from the open circuit potential
provided the battery is not cycled at an extremely high rate, so the potential at the
graphite surface remains rather constant.
First-order solvent decomposition kinetics
We make the further assump-
tion that the SEI formation rate is proportional to the concentration of the reacting
electrolyte species. The most important part of this assumption, particularly in the
limit of long times, is the observation that the formation rate must go to zero with
reactant concentration. Experimental data (see subsection 3.3.2 for a comparison of
this model with experimental results [93, 159]) suggest that the large time limit is
reached within a few days.
Linear solvent diffusion in the SEI
The very slow transport of electrolyte
molecules through the SEI is the rate-limiting part of the SEI formation process.
We assume that this transport can be modeled as being driven by a concentration
gradient between the outer and inner surfaces of the SEI, and that the concentration
difference is directly proportional to the thickness of the SEI and the reaction rate
consuming electrolyte at the inner surface.
Under these assumptions, the SEI thickness can be described by two equations.
32
The first,
𝐽𝑚
𝑑𝑠
=
,
𝑑𝑡
𝜌𝐴
(3.10)
relates the rate of increase of SEI thickness to the reaction rate
formed by a single reaction, the density of SEI
𝐽,
the mass
𝑚 of SEI
𝜌 and the graphite surface area 𝐴.
We
arbitrarily choose lithium fluoride [172] as a typical SEI component: its molar mass
is 26 g mol
−1
−3
and density is 2.6 g cm . The reaction rate
𝐽
can be described by
𝐽 = 𝑘𝐴(𝑐 − ∆𝑐),
(3.11)
where
𝐽𝑠
𝐴𝐷
∆𝑐 =
(3.12)
is the concentration difference between the outside and the inside of the SEI, needed
to transport electrolyte molecules through with flux
𝐽/𝐴.
reaction rate constant, a fitting parameter in our model.
𝐷
In equation 3.11,
is a
is the diffusivity through
the SEI of the species that reacts with lithium to form SEI, while
of this species in the bulk of the electrolyte.
𝑘
𝑐 is the concentration
Since we do not know exactly which
species is dominant in the continuing formation of SEI during cycling, we assume
that
𝑐
is 1 M, a typical concentration of anions.
assumptions in order to present
𝐷
and
𝑘
We have made several arbitrary
in useful units; their values should scale
appropriately (e.g. with temperature) but will only be a vague guess at the correct
values.
Equations 3.10 and 3.11 can be solved analytically to give the SEI thickness as a
function of time:
In the limit of large
√︀
2𝑐𝜌𝑚𝑘 2 𝐷𝑡 + 𝐷2 𝜌2 − 𝐷𝜌
𝑠=
.
𝜌𝑘
𝑡,
this becomes
√︃
𝑠=
which reproduces the
(3.13)
√
𝑡
2𝑐𝑚𝐷𝑡 𝐷
− ,
𝜌
𝑘
(3.14)
dependence of Ploehn et al. [135] and Smith et al. [160].
33
3.3.2
Comparison with experiment
To assess the ability of this simple model to explain SEI formation, we compare the
prediction of equation 3.14 to experimental results, treating
𝑘
and
𝐷
as adjustable
parameters. For example, we apply the model to the results of Liu et al. [93] Inferring
the SEI thickness from the percentage capacity loss (assuming an average particle
radius of approximately 5
𝜇m),
we find that the approximation of equation 3.14 is
valid even after the first charge-discharge cycle.
2 × 10−17
cm
2
−1
∘
s
at 15 C and
𝐷 = 3 × 10−16
cm
Using this equation we find
2
s
−1
at 60
∘
𝐷 =
C: see figure 3-1. In the
latter case, capacity loss between 707 and 757 cycles is much larger than is expected
under the model. This demonstrates that some other loss mechanism is important in
this instance. The most likely mechanism is the delamination of graphite from the
current collector: figure 10 of Liu et al. [93].
3.3.3
Multiple reacting species
The above analysis has considered only one species from the electrolyte reacting to
form SEI. Experimental work [165] has shown that many reactions can take place to
form SEI, involving a wide range of solvent species, anions or impurities.
A more
complete model would consider multiple reacting species, each with a characteristic
𝑘
and
𝐷.
However, since experimental results very quickly reach the large time
limit, we can model the process effectively simply by assuming that the
by experiment is given by
𝐷=
∑︀
𝑖
𝐷𝑖
𝐷
implied
where i references the different species. The
capacity fade will be dominated by the species with higher diffusivity through the
SEI. This suggests that capacity fade can be reduced by constructing the cell in a
way that avoids the presence of substances with high diffusivity in SEI, for instance
small molecules.
The presence of multiple reacting species also provides a mechanism by which
denser SEI may form at higher temperature. At higher temperature, all diffusivities
are higher, and the dominance of the most easily diffusing species will be lower. If the
less diffusive species form a denser SEI, as seems likely from experimental observation
34
of denser SEI towards the bottom of the layer [172], this will result in lower diffusivity
for the remainder of the aging process and hence slower continued SEI formation.
3.4 Temperature effects
Since the single particle model, whose important parameter is the diffusivity of electrolyte molecules through the SEI, appears to completely explain the loss of capacity
due to SEI formation, it is interesting to provide a theoretical model attempting to explain the temperature dependence of this diffusivity. The model of this work applied
to the experimental data of Liu et al. [93] shows a clear temperature dependence of
diffusivity. To quantify this dependence, we compared the single particle model with
the experimental data of Smith et al. [159], which span many days at temperatures
from 15
∘
C to 60
∘
C. The diffusivities calculated from these data were consistent with
an Arrhenius dependence,
𝐷 = 𝐴𝑒
with activation energy
𝐸 = 0.52
− 𝑘 𝐸𝑇
𝐵
,
(3.15)
eV. Figure 3-5 shows the prediction of the single
particle model, with diffusivities (and reaction rate constants) fit to the Arrhenius
description.
3.4.1
Accelerated aging
The observed temperature dependence enables the single particle model to be used to
draw quantitative conclusions from accelerated aging experiments, where an increased
temperature is used to hasten capacity fade.
A simple application is to estimate
battery life at room temperature from data taken at elevated temperatures.
The
most accurate way to do this is to measure the progress of capacity fade at different
temperatures, and use the results to calculate
data from only the first 105 days at 30-60
used to calculate
𝐷(𝑇 )
and
𝑘(𝑇 ),
∘
𝐷(𝑇 ).
As a test of this procedure,
C in the work of Smith et al. [159] were
using equation 3.14. Results for these times and
temperatures were fit to an Arrhenius equation. The resulting values of
35
𝐷
and
𝑘
at
Figure 3-5: The single particle model accurately describes capacity fade over a range
of temperatures, with diffusivity fit using an Arrhenius dependence (shown in the
inset). Data from Smith et al. [159] with a lithium opposite electrode.
36
Figure 3-6: The single particle model enables data from up to 105 days at 30-60
∘
C to
∘
C;
be used to provide a reasonable prediction of capacity fade up to 400 days at 15
assuming a simple
√
𝑡 dependence of capacity fade is less predictive.
Data from Smith
et al. [159] with a lithium opposite electrode.
15
∘
C were used to predict capacity fade for 400 days: results are shown in figure
3-6. Though the agreement is not perfect, it is reasonable given that no data at the
temperature or time of the predicted region were used in producing the prediction.
In particular, simply assuming
√
𝑠=𝛼 𝑡
[135, 160] (i.e. neglecting the second term
in equation 3.14) and using an Arrhenius equation to find
𝛼(𝑇 )
is not as predictive:
the best prediction using this method is shown by the dotted line in figure 3-6.
In addition, we applied the model to the data of Broussely et al. [30], predicting
capacity fade at 15
∘
C for 14.5 months using data at 30-60
∘
C for 6 months. In this
case, the second term in equation 3.14 was statistically indistinguishable from zero,
meaning that a simple
𝑡1/2
capacity fade model would give the same result.
It is
possible that the second term cannot be distinguished in this case because capacity
loss was not measured directly, as in Smith et al. [159], but inferred from measured
37
Figure 3-7: The single particle model, using accelerated aging data from 6 months
∘
∘
at 30-60 C, can be used to predict aging over 14.5 months at 15 C. Data from
Broussely et al. [30] with a lithium cobalt oxide opposite electrode.
capacity. Figure 3-7 compares the prediction with experimental data.
A more sophisticated application of the single particle model would be to predict
the total capacity fade of a battery subject to varying temperature. This information
could be used to construct a protocol to optimize lifetime given a particular set of
unavoidable temperature constraints. For example, is it possible to increase battery
life by cycling it a small number of times at particularly high or low temperature,
in order to produce an initial SEI with low electrolyte diffusivity?
Answering this
question will require experimental work to check whether the measured electrolyte
diffusivity depends on the temperature history of the battery as well as the current
temperature.
38
3.4.2
Resistance mismatch in battery packs
The discussion above considers temperature differences that are purely external, but
high rate use of a battery can lead to significant heating due to the battery’s internal
resistance.
Since this can alter the temperature of the battery, it must be taken
into account in order to accurately predict capacity fade. One instance where this is
important is the case of battery packs comprising two cells charged and discharged
in parallel. Experiments on this system were performed by my collaborator, Radu
Gogoana. I then analysed the experimental results with respect to my capacity fade
model.
When two cells with different internal resistance are charged in parallel, the current
experienced by each cell is not constant.
Early in the charging process, the less
resistive cell experiences a higher current.
This may cause it to approach a fully
charged state sooner than the more resistive cell, resulting in an increase in current
to the more resistive cell towards the end of charging. A large difference in internal
resistance thus results in high maximum charging current to both cells in the cell
group, which is shown in figure 3-8. The less resistive cell 2 has higher current early
in discharge and charge; the later increase in current to cell 1 is more apparent on
discharge than on charge.
It is important to note that the shape of the current distribution curve in figure
3-8 is linked to shape of the voltage vs capacity curve of LiFePO4 cells, which is
relatively flat between approximately 10% and 90% state of charge. For other Li-ion
cell chemistries that have more sloped voltage vs capacity curves (e.g.
LiMn2 O4 ,
LiCoO2 ), we expect a “balancing-out” effect to occur throughout the charge and
discharge cycles.
Because capacity and voltage are more closely related for those
chemistries, we expect that both cells in a parallel-connected configuration will reach
end-of-discharge more evenly without a large difference in current between the two
cells toward the end of the cycle.
Gogoana’s experimental results showed that cells with higher mismatch in internal
resistance fade more quickly. This implies that a higher maximum current increases
39
Current (A)
[positive indicates charging]
15
10
5
0
0
500
1000
-5
1500
Time (s)
Cell 1 Current
Cell 2 Current
-10
-15
Figure 3-8:
Current distribution within two parallel-connected cells upon cycling
(initial resistance difference of 18%).
the capacity fade rate, even when average current is fixed. This could arise because
resistive heating causes an increase in temperature, which exponentially increases the
rate of capacity fade [6]. The impact of temperature on the cycle life of lithium-ion
cells has been studied by several groups [6, 173, 26, 118]. Although the temperature
of all cells increased during cycling, there was no significant correlation between the
external temperature of an individual cell and either current or the rate of capacity
fade. Thus, if the dependence of capacity fading on current is due to a temperature
increase, this temperature increase must be highly localized inside the cell. Alternatively, the accelerating effect of very high current on capacity fade may be separate
from any temperature effect.
To apply the model of this work to explaining the effect of mismatched internal
resistances, it is necessary to hypothesize a particular dependence of
𝐷
𝐷
on C rate. If
has an Arrhenius dependence on temperature,
(︂
)︂
𝐸
𝐷 ∝ exp −
,
𝑘𝐵 𝑇
40
(3.16)
D (nm2/s)
8
4
0
3
4
5
6
C rate
Figure 3-9:
𝐷
as a function of C rate, fit to measured capacity fade.
the cell temperature is elevated above room temperature by an amount proportional to
the C rate, and this elevation is much smaller than room temperature, then equation
3.16 can be expressed as
𝐷 = 𝐷0 exp(𝛼𝐼),
where
𝐼
is the C rate and
dependence of
𝑘
𝛼 is a constant.
(3.17)
Combining this with an assumption that any
on C rate was unimportant, we used equation 3.10 to model capacity
fade. A least squares fit to Gogoana’s experimental data was used to calculate values
of the parameters
𝐷0 , 𝛼
and
𝑘.
The value of
𝐷
as a function of C rate is shown in
Figure 3-9. The model was sufficient to describe capacity fade until 75% capacity for
most of the cells studied by Gogoana.
One cell showed anomalously slow capacity
fade and was excluded from the analysis, as including it caused the calculated fade
rate of other cells to differ from the obvious cluster.
Figure 3-10 compares experimental capacity fade with the model prediction for
the two cells with smallest and largest error.
Using this model of capacity fade, lifetime to 75% of total capacity can be predicted
for a pair of cells with arbitrary difference in internal resistance. This prediction relies
41
2.4
Capacity (Ah)
Capacity (Ah)
2.4
Experiment
Model
2
2
Model
Experiment
1.6
1.6
1
1001
2001
Cycles
3001
4001
1
1001
Cycles
2001
Figure 3-10: Experimental and model capacity fade for two cells.
on the assignment of a maximum charging current to each cell at each cycle. For the
cell with lower resistance, this can be calculated assuming that the ratio of resistances
does not change with degradation.
The maximum charging current to the cell with higher resistance will come at the
end of charging, when the other cell nears a fully-charged state. It was observed that
the maximum charging current measured experimentally for the more resistive cell in
a pair was linearly related to the “excess capacity”
𝑄𝑥 = 𝑄1 −
where
𝑄1
and
𝑅1
𝑄𝑥 ,
defined by
𝑄2 𝑅2
𝑅1
(3.18)
are the capacity and resistance of the more resistive cell and
𝑅2 the capacity and resistance of the less resistive cell.
𝑄2
and
The excess capacity is thus the
charging capacity remaining in the more resistive cell when the less resistive cell has
finished charging, assuming that the charging rates remain constant. The observed
linear relationship, shown in Figure 3-11, was used as an input into the model-based
prediction.
This increase in maximum charging current from 9.5A to 12A (4.3C to 5.5C) can
be a significant factor in the capacity fading of the cell. Observed data by Wang et al.
42
Maximum current (A)
12
11
10
9
0
0.25
Excess capacity (Ah)
0.5
Figure 3-11: Maximum charging current to the more resistive cell as a function of
the excess capacity of this cell (defined by equation 3.18). More capacity remaining
in one cell at the end of a charge cycle leads to higher maximum charge current to
that cell.
43
100
% Capacity Loss
75
50
25
0
0
2
4
6
8
10
12
14
16
18
20
C‐Rate
Figure 3-12: Modeled capacity fade as a function of C rate according to Wang et al.
[178].
[178] on the same LiFePO4 cells that Gogoana tested showed an exponential increase
in capacity fade with C-rate. The curve-fitted model that they obtained from their
experimental data is shown in Figure 3-12. Although they only tested the impact of
higher discharge currents (their charge current in testing was a constant 2C), it is
possible that a similar relationship exists on charging C-rate, and that the jump from
4.3C to 5.5C on charging was responsible for this difference in capacity fade.
The model of this work includes a degradation rate that is independent of current. In fact, since the model identifies time rather than charge throughput as the
determinant of capacity fade, cells exposed to higher C rates are expected to have
a longer cycle life. The model does display a strong dependence of degradation rate
on temperature, and it is likely that the importance of fast charging is that it increases temperature, through resistive heating within the cell. In the experiments of
Gogoana, there was no apparent relationship between the measured temperature at
the outside of the cell and fade rate, so if heating is indeed the cause of increased
capacity fade, it must be somewhat localized within the spiral-wound electrode. This
localized temperature rise can be estimated by comparing the change in the rate of
44
capacity fade due to a C-rate increase, with that due to an increase in ambient air
temperature.
According to the SEI formation model, a 10
∘
Cincrease in tempera-
ture increases the capacity fade rate by approximately 40%, due to a doubling of the
diffusivity of electrolyte molecules through the SEI. The same increases were found
in Gogoana’s experiments when maximum charging current was increased by 0.7C.
In other words, a 10
∘
Cincrease in background temperature or a 0.7C increase in
maximum charging rate apply approximately equal stress to the battery in terms of
capacity fade rate.
Figure 3-13 shows the predicted lifetime as a function of relative resistance difference, calculated by combining equations 3.10, 3.17 and 3.18 with the empirical
relationship shown in figure 3-11. The top curve accounts only for gradual capacity
fade due to SEI formation. In addition, many cells experienced a sudden capacity loss
of up to 100 mAh towards the beginning of the cycling process. These capacity losses
could result from isolation of active material from the conductive matrix, due to SEI
growth or physical separation. The bottom curve represents a worst-case scenario,
where both cells experience a 100 mAh capacity loss at the beginning of cycling, followed by gradual SEI formation. The model explains the observed decrease of lifetime
with an increase in internal resistance mismatch, and that the observed scatter is due
to random sudden capacity losses. These random losses can drastically reduce cycle
life, and so are important to monitor (and ideally prevent), even though on average
most capacity fade is due to gradual SEI formation.
The modeling described in this subsection could be applied to any situation where
cells are charged at a C rate high enough to increase temperature. In order to obtain
a prediction of lifetime, it is vital to know the actual C rate to which a cell is exposed.
For instance, if more than two cells are connected in parallel, the extra current imposed when one cell reaches capacity will be shared between the others, and a detailed
measurement or model of this sharing is necessary to allow a quantitative prediction
of pack lifetime.
45
6000
Life to 75% (cycles)
Model without sudden losses
Experiment
3000
Model with sudden losses
0
0
0.1
0.2
Relative internal resistance mismatch
0.3
Figure 3-13: Experimental and predicted lifetime as a function of internal resistance
mismatch.
3.5 Lifetime prediction and statistics
Another application of the single particle model is to understanding the statistics of
battery lifetime. If the typical diffusivity of electrolyte through the SEI is
the battery is defined to fail when the SEI thickness reaches
life will be
𝑠0 ,
𝐷𝑚
and
the average battery
𝜏𝑚 = (𝜌𝑠20 )(2𝑐𝑚𝐷𝑚 ) (we have neglected the second term in equation 3.14).
Since a battery is made up of many, presumably identical, active particles i and the
total SEI formation is proportional to
√
∑︀ √
𝐷
𝑡
,
we
expect
the
measured
𝐷
𝑖
𝑖
a normal distribution, assuming the distribution of the true diffusivity
𝐷𝑖
to have
meets the
conditions of the central limit theorem. Thus the inverse of the lifetimes of a collection
of batteries should be normally distributed; SEI formation as modeled here will not
cause anomalously short battery lifetime.
This prediction can be compared with other predicted battery lifetime distributions, such as the Weibull distribution from the theory of extreme order statistics
[52, 122]. A Weibull distribution is expected when failure occurs in a “weakest link”
situation, where failure of a single element causes failure of the entire cell.
46
Math-
Figure 3-14: Lifetimes (calculated from data in Park et al. [122]) are consistent with
a normal distribution (solid line), but not with a Weibull distribution, which would
appear as a straight line on this Weibull plot. However, it is not possible to establish
the presence or absence of a Weibull tail at low lifetime (dashed line).
ematically, it is the limiting distribution for the smallest outcome of a large set of
independent identically distributed random variables which are bounded below, with
a power law tail [66]. Additionally, a Weibull tail at short lifetime occurs in quasibrittle fracture when the system can be modeled as a bundle of fibers arranged both
in series and in parallel [89].
In order to examine the experimental lifetime distribution, we converted the data
in table 2 of Park et al. [122] to lifetime data by defining the lifetime to be the number
of cycles taken to reach a relative capacity of 85.6
3.6 Extensions for rapid capacity fade
Until now, we have focused on gradual capacity fade due to the formation of a stable
SEI film, well adhered to an electrode internal surface of nearly constant area. This
47
is the situation in practical Li-ion cells, but various emerging technologies, which
offer certain benefits, such as high energy density, suffer from much faster capacity
fade related to accelerated SEI formation. The canonical and most intensely studied
example is silicon, which is a very attractive anode material for Li-ion batteries due
to its theoretical high energy density of 4200 mAh g
−1
[45], but, as a result of its
enormous volume expansion upon lithiation (> 300
3.6.1
Fresh surface
The single particle model assumes that the SEI is made up of a single layer growing
smoothly from the surface of the active material. In a material that experiences a large
volume change on lithiation, such as silicon [27], this assumption is no longer valid.
Instead, the expansion during lithiation produces fresh surface area, on which SEI
can form without hindrance from an already-existing layer. As the material shrinks
during delithiation, the SEI formed on the newly exposed surface will be forced into
the existing layer or possibly detached from the active material. In either case, it is
likely that a substantial fraction of the new area formed on the next cycle is once
again freshly exposed. This leads to capacity fade at a much faster rate than would
be expected from the model in which the SEI layer remains uniform and area changes
are ignored.
The effect of the fresh surface area can be modeled simply by applying the result of
the single particle model, equation 3.13 or 3.14, to a single charge-discharge cycle, and
finding the total capacity fade by adding that of each cycle. The total SEI thickness
is
𝑠=
where
𝜏
𝑡
(
𝜏
√︃
2𝑐𝑚𝐷𝜏
𝐷
− ),
𝜌
𝑘
(3.19)
is the time spent during one cycle at a voltage where SEI forms: we assume
that this is the total charge and discharge time.
This simple model predicts that
capacity fade is linear in time, which is precisely what is seen for full cells using silicon
electrodes [10, 45, 123, 96, 80]. Furthermore, since
rate of increase of
𝑠 per cycle should be linear in
48
√
𝜏
𝑡/𝜏
is the number of cycles, the
. Figure 3-15 shows experimental
−1
−1
data from figure 4c of Ji et al. [80], which exhibit capacity fade of 4 mAh g
cycle
at C/15 and 2 mAh g
−1
cycle
−1
at C/4, consistent with this prediction. Assuming
a characteristic particle size of 1 Âțm, this implies
𝐷 = 4 × 10−15
cm
2
s
−1
. This is
two orders of magnitude higher than that found in the case of graphite, for which we
offer two possible explanations. The first is that, even in the case of graphite, the
new SEI formed each cycle may form primarily on the newly exposed area, though in
this case the shape change is small enough that the SEI formed in the previous cycle
is not fully lost. In this case, the smaller formation area means that the actual SEI
thickness is higher than that calculated assuming the SEI forms evenly over the entire
active surface, since capacity loss, which is measured, is proportional to SEI volume.
This would mean that the true value of
assumptions of the simple model.
𝐷
is higher than that calculated under the
The second possibility is that the expansion of
silicon is large enough to disturb the structure of the SEI, even within a single cycle.
Such a disturbance might increase SEI porosity and is likely to increase
𝐷.
It is vital that full cell, not half cell, aging data are used to assess the impact of SEI
growth on capacity fade. SEI growth leads to capacity fade due to the consumption
of lithium that would otherwise be available for cycling.
If an electrode is cycled
against a lithium foil electrode containing excess lithium, this loss will not be observed,
potentially leading to an overestimate of battery life.
Indeed, experimental results
demonstrate a substantially shorter lifetime for a full cell than a half cell using a
silicon electrode: figure 4 of Baranchugov et al. [10] shows this particularly clearly.
Considering only half cell data could lead to a falsely optimistic assessment of the
lifespan of a silicon anode.
3.6.2
Unstable SEI
Another situation in which SEI formation could progress faster than suggested by the
single particle model is when SEI is lost from the layer, by delamination or dissolution
in a form from which Li remains unavailable.
Significant SEI removal is known to
occur in silicon anodes, as confirmed by direct observation [38]. Assuming that this
loss is not so great as to reduce the layer thickness to zero or almost zero, it can be
49
Figure 3-15: A full cell with a silicon anode shows linear capacity fade at two chargedischarge rates. The main graph plots capacity against cycle number. The inset plots
capacity against cycle number weighted by the square root of the charge-discharge
time, as the model of this work predicts that capacity fade is proportional to this
quantity.
The experimental data, from Ji et al.
[80], match the theory very well,
after accounting for the constant offset due to the change in charge-discharge rate,
which is well understood though not explicitly modeled in this work.
50
modeled by including a loss term in the differential equation for SEI formation:
𝐽𝑚
𝑑𝑠
=
− 𝑓 (𝑠),
𝑑𝑡
𝜌𝐴
where
𝑓
(3.20)
is a general function characterizing the dependence of the SEI loss rate on
SEI thickness. The rate of capacity loss remains
𝑑𝑄
= −𝐽,
𝑑𝑡
and the dependence of
𝐽
capacity. Assuming that
on
𝑓
𝑠
(3.21)
is unchanged. This alters the long time behavior of
is an increasing function of
𝑠 (strictly all that is necessary
is that the right hand side of equation 3.20 is ultimately negative; in particular,
a constant
𝑓
is sufficient),
𝑠
𝑠𝑚 .
Once this
𝐽(𝑠𝑚 ).
The overall
will eventually reach some maximum
maximum SEI thickness is reached, the rate of capacity loss will be
result is a capacity fade rate that is linear due to reaction limitation for a short time,
before moving through a
𝑡1/2
transition region to a second linear region with a lower
rate. This behavior, which could be expected for a system where the charge-discharge
process places a greater burden on the SEI layer than that in graphite but less than
that in silicon, is represented in figure 3-16. In such a situation, capacity fade could
be reduced not only by decreasing
Although for most choices of
for
𝑄(𝑡),
𝐽(𝑠),
𝑓 (𝑠)
but also by decreasing
𝑓.
it is not possible to find an analytic formula
we can make progress in the simple case
𝑓 (𝑠) = 𝑠/𝑡0 ,
in the limit
𝑘→∞
(which experiments [93] suggest is reasonable: see above). Under these conditions,
√︂
𝑄(𝑡) = 𝑄0 − 𝐴
where
𝑄0
1/2
(︂
)︂
√︀
𝑡
+ ln(1 + 1 − 𝑒−2𝑡/𝑡0 ) ,
𝑡0
(3.22)
is the initial capacity. We see that the long-term linear fade has the same
prefactor as the
1/(2𝑡0 ).
𝜌𝑐𝐷𝑡0
𝑚
𝑡1/2
fade of the stable case, except for the dimensionally-required
This long-term behavior is not influenced by the assumption
51
𝑘 → ∞.
Figure 3-16: When SEI dissolution or delamination occurs, capacity fade occurs in
three regimes.
Initially the SEI thickness
𝑠
is small, the SEI formation rate
very large, limited only by the reaction rate, and the SEI loss rate
𝑠
increases rapidly while capacity decreases rapidly.
𝑓
𝐽
is
is small, so
For intermediate times, SEI
formation is limited by diffusion through the existing SEI, but 𝐽 is still greater than
𝑓 , leading to an approximate 𝑡1/2 rate of capacity fade. Eventually the SEI formation
and loss rates will be equal, leading to constant
52
𝑠
and a steady rate of capacity loss.
Chapter 4
Overview of models of sorption in
porous materials
4.1 Modeling of sorption in a single pore
4.1.1
Surface adsorption
The quantity of a fluid taken up by a porous material, as a function of the partial
pressure of this fluid, is frequently used to analyse the pore structure of the porous
material. In order to carry out this analysis, models of the relationship between pore
properties, partial pressure and sorption are required.
If the sorption process begins at sorbate pressure zero, the conceptually simplest
approach though not necessarily the technique used in experiment, the first mechanism of sorption is adsorption of the sorbate molecules to the solid material.
Two
simple models of this adsorption process have been developed, based on different
assumptions.
Langmuir assumed that molecules from the vapor could adsorb onto the solid
surface in a single layer, with heat of adsorption
𝐸𝑠𝑙 .
Assuming that adsorption occurs
at independent sites, and that the adsorbed layer and vapor are in equilibrium, the
coverage of the solid is then
𝑡=
𝑐ℎ
1 + 𝑐ℎ
53
(4.1)
where
𝑐 = 𝑒(𝐸𝑠𝑙 −𝐸𝑙 )/𝑘𝐵 𝑇 , 𝐸𝑙
being the heat of liquefaction of the sorbate, and
ℎ
is the
partial pressure of the sorbate, relative to the saturation pressure [87]. This equation
is known as the Langmuir equation.
Brunauer et al.
assumed that many layers could adsorb on the surface.
also assumed adsorption at independent sites and heat of adsorption
𝐸𝑠𝑙
They
for the first
molecule at a site, but assumed that other molecules could adsorb in a tower above the
first, with heat of adsorption equal to the heat of liquefaction. The average thickness
of the adsorbed layer, in molecules, is then [32]
𝑡=
𝑐ℎ
.
((𝑐 − 1)ℎ + 1)(1 − ℎ)
(4.2)
This equation is known as the BET equation.
The Langmuir and BET equations predict the same behavior at low pressure,
but very different behavior as the pressure approaches the saturation pressure:
𝑡
approaches a constant below 1 and infinity respectively. The BET equation is widely
used in the calculation of surface areas of porous materials, though an area calculated
in this way is not necessarily equal to the actual surface area of the porous material
[63]. Figure 4-1 shows the predicted adsorbed layer thicknesses for the two models
with
𝑐 = 4,
along with sample configurations of adsorbed molecules at 25%, 50% and
75% relative pressure.
4.1.2
Pore filling
Even below its saturation pressure, a condensed phase of a sorbate can exist in a
porous material. The Kelvin equation describes the radius of curvature of an interface
between liquid and vapor at a particular partial pressure, in equilibrium [176]:
ln ℎ = −
where
𝛾
2𝛾𝑣
,
𝑟𝑘𝐵 𝑇
is the surface tension between liquid and vapor and
(4.3)
𝑣 is the average molecular
volume in the liquid state. The Kelvin equation allows the calculation of the partial
54
8
Adsorbed layer thickness
6
4
BET
2
Langmuir
0
0
0.2
0.4
0.6
0.8
1
Relative pressure
Figure 4-1: Adsorbed layer thickness in monolayers according to the BET and Langmuir equations, along with example configurations of adsorbed molecules at 25%,
50% and 75% relative pressure.
55
pressure at which the filled and empty (apart from adsorbed layers) state will be
in equilibrium for a particular regularly-shaped pore, provided the contact angle
𝜃
between liquid, vapor and adsorbed fluid is known or can be assumed. For instance,
a cylindrical pore of radius
𝑟
has this equilibrium transition at
ln ℎ = −
2𝛾𝑣 cos 𝜃
.
(𝑟 − 𝑡(ℎ)𝑎)𝑘𝐵 𝑇
(4.4)
Equivalent equations can be found for other geometries.
Experimentally, hysteresis is often found between sorbed quantities on increasing
and decreasing partial pressure. One source of this hysteresis is when the filling and
emptying processes follow different paths. For example, in the case of a cylindrical
pore, the equilibrium interface between liquid and vapor is a hemisphere, and emptying occurs by the propagation of such an interface along the axis of the cylinder. If the
pore has open ends, however, such an interface will not exist at low partial pressure,
and the pore must fill radially from the wall to the axis. Neglecting the effect of the
changing adsorbed layer thickness, the mean curvature of this cylindrical interface is
half that of the hemispherical interface, so the pore fills at a relative pressure higher
than that at which it will empty. Figure 4-2 illustrates a cycle of relative pressure.
This difference between filling and emptying paths has been noted as an important
source of sorption hysteresi [42, 29, 85].
4.2 Modeling of sorption hysteresis in porous materials
The existence of a network of pores can greatly increase hysteresis, and various models
of sorption in a porous network have been proposed, applicable to different materials.
The fundamental principle on which these models are based is that, since emptying
occurs by propagation of an interface between the vapor phase and the condensed
fluid, a pore cannot empty unless it is exposed to such an interface: that is, it is
adjacent to an empty pore or the boundary of the system. The emptying of pores
56
Figure 4-2: A schematic illustration of radial filling during wetting (bottom) and axial
emptying during drying (top).
can thus be described by a percolation model on some suitably designed lattice.
Mason [103] proposed a model of pores connected by windows, each with a characteristic radius drawn from a distribution [104]. Parlar and Yortsos [125] extended
this model to more completely describe the case of scanning isotherms. Seaton [152]
applied the model to the calculation of the average coordination number of pores in
various materials, while Tanev and Vlaev [163] considered the relationship between
assumptions about the pore shape and the corresponding hysteresis loop.
The models presented in these publications focus primarily on the percolation
threshold. They make certain assumptions about the pore network, and assume that
no emptying of a significant number of pores occurs until the vapor region is able to
percolate through this network.
This assumption seems to work well for materials
where the pore size distribution is not too wide, such as porous glass [104].
An
interesting and neglected case is that of a material with a broad PSD, such that an
extensive number of macropores is empty when the partial pressure is immediately
below the saturation pressure. This could be because these pores do not fill, or because
they are able to empty without hindrance due to a connection to a continuous network
of large pores.
57
Liu et al. modeled such a system, treating the pores as bonds of a simple cubic
lattice, which in the undisturbed state has connectivity
𝑍 = 10.
They controlled
the actual average connectivity by randomly deleting bonds from the lattice. They
also used a finite lattice to model the effect of finite-sized microparticles, between
which condensation does not occur.
The wetting process was assumed to occur at
equilibrium, and the empty fraction during drying was calculated computationally[95].
Liu and Seaton then extended this work to allow for the case that some pores are
never filled, and hence provide additional locations at which the vapor phase may be
nucleated [94]. A missing element from these models was single-pore hysteresis. Kruk
and Jaroniec studied sorption hysteresis in systems with single-pore hysteresis, but
in simpler systems comprising large pores with small necks, rather than a network
of similar pores [86]. Network modeling of sorption with single-pore hysteresis was
carried out by Rojas et al. [145]. Cordero et al. extended this modeling to account
for the nucleation and suppression of filling by adjacent pores [44].
These examples of network modeling of pore filling and emptying were carried out
computationally on lattices produced using pores with sizes drawn randomly from
a distribution.
This technique can be effective, but a simpler method, more easily
applicable to calculating parameters based on a particular experimental data set,
would be to find analytic expressions for the relevant quantities. I present such an
analytic model in this work.
4.3 Modeling of transport in porous materials
Models of transport in porous media are based on Darcy’s law, which can be written
q=
q
is flux density,
𝜅
𝜅
(−∇𝑝 + 𝜌g).
𝜇
is the intrinsic permeability of the porous medium, proportional
to the square of a characteristic pore size,
fluid,
𝜌
(4.5)
is fluid density and
g
𝜇
is fluid viscosity,
is acceleration due to gravity.
58
𝑝
is pressure in the
Gravity is dominant
in saturated flow but negligible in unsaturated flow because the scale of capillary
pressure is around 100 MPa, so the gravity term is dropped hereafter. Darcy’s law
can be derived by coarse-graining Stokes flow through the medium [180].
Two important sources of hysteresis have been included in various models of fluid
flow in porous media. The first is that the permeability can depend not only on the
current saturation, but also on the local saturation history. A reason for this is that
permeability depends on which pores are occupied by the flowing fluid.
If it takes
some time for fluid in large and small pores to reach equilibrium locally, there will be
a corresponding time-dependence of permeability. Barenblatt et al. model this effect
through the use of an effective saturation that relaxes to the true saturation [11].
The second source of hysteresis is in the relationship between saturation and pressure. Various authors have used empirical relationships to represent the dependence
of saturation on pressure history as well as the current pressure [170, 83, 124, 97]. A
particularly important example of such a model, accounting for the microscopic source
of the hysteresis, is due to Mualem. This work represents the fraction of pores that
have emptied during drying as the product of the fraction that could have emptied
based on an equilibrium consideration of the local pore structure, and the fraction of
such pores that are not blocked by water in the broader pore system [112]. However,
the model assumes a particular relationship between the equilibrium state of pores on
wetting and drying: namely, that the fraction of pores empty on drying is the square
of the fraction empty on wetting, assuming no blocking. A more general assumption
would be that the equilibrium state is the same, and that any difference in filling
must be due to some kind of blocking effect. In other words, blocking by local effects
such as pore necks is considered in the same way as blocking by larger-scale effects of
the pore network. It is this assumption that I use in my modeling of transport with
hysteresis.
59
60
Chapter 5
A model of sorption in multiscale
porous materials
The content of this chapter is based heavily on the article in preparation “Theory of
Sorption Hysteresis in Multiscale Porous Media” by the author, Jennings and Bazant
[133].
5.1 Single pore hysteresis model
5.1.1
Model
The starting point for modeling of sorption hysteresis in a pore network must be
a model of sorption in isolated pores.
I assume that the initial sorption mode is
adsorption on the pore walls, and that the thickness of the adsorbed layer can be
described by the BET equation, equation 4.2. I futher assume that the Kelvin equation, equation 4.3 can be used to relate the radius of mean curvature of an interface
between condensed fluid and vapor to the relative pressure of the sorbate. The use of
the Kelvin equation relies on the assumption that a continuum treatment of water is
valid down to small length scales, as suggested by the explanation of freezing point
suppression by an analogous continuum equation [155]. I ignore any dependence of
the surface tension on curvature, as this dependence is small and there is not even
61
consensus regarding its sign [91, 22].
Using the above equations describing sorption in mesopores, is is possible to analyse experimentally observed sorption isotherms to obtain the PSD of a porous material. Here I assume cylindrical pores since, in some cases below, assuming slit pores
predicts a greater than observed hysteresis loop even when neglecting network ef-
𝑟,
fects. If the distribution of pore radii
at relative pressure
∫︁
ℎ
by pore volume, is
𝑣(𝑟),
the sorbed volume
during drying is
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
∞
∫︁
𝑣(𝑟) 𝑑𝑟 +
𝑚𝑑 (ℎ) =
0
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
𝑟2 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟,
𝑟2
(5.1)
where
𝑟𝐾 (ℎ) =
2𝛾𝑎3
𝑘𝑇 ln ℎ
(5.2)
is the radius, measured to the inner surface of the adsorbed layers, of a pore where
the full and empty states are in equilibrium.
The two terms on the right are the
contribution of full pores and of the adsorbed layers on the walls of otherwise empty
pores, respectively. The sorbed volume during wetting has the same form, but half
the Kelvin radius instead of the Kelvin radius itself must be used in calculating the
radius that divides full and empty pores, due to the cylindrical shape of the meniscus.
This gives
∫︁
𝑚𝑤 (ℎ) =
𝑟𝐾 (ℎ)/2+𝑎𝑡(ℎ)
∫︁
∞
𝑣(𝑟) 𝑑𝑟 +
0
𝑟𝐾 (ℎ)/2+𝑎𝑡(ℎ)
𝑟2 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟.
𝑟2
In principle, equation 5.1 allows the calculation of
data.
(5.3)
𝑣(𝑟) directly from experimental
However, the discreteness of experimental data and the difficulty in distin-
guishing adsorbed and condensed water can lead to spurious bumps in the PSD thus
obtained. I therefore make the simple assumption of a log-normal distribution of pore
widths, which is sufficient for good agreement with experimental data.
62
5.1.2
Application to simple porous materials
I apply the model to experimental sorption isotherms for dichlorofluromethane in
plugs of carbon black [34]. Experimental and modeled isotherms are shown in Fig.
5-1, and the PSDs in Fig. 5-2. The parameters of the model are the total volume in
which fluid is sorbed, the mean and variance of the PSD, the offset of the experimentally measured “dry” mass from the truly dry mass, and the BET constant
𝑐,
which
mostly influences the low pressure sorption.
5.2 Network effects
This simple model, accounting for hysteresis due to differences in the shape of the
liquid-vapor interface in a single pore on wetting and drying, accurately describes
sorption in this particular porous material, which has a narrow distribution of small
pores. In a system with a wide range of pore sizes, network effects can act to broaden
hysteresis. The additional hysteresis is due to some pores remaining full below the
relative pressure at which the empty state is thermodynamically favored, because
they lack the connection with the vapor phase that is necessary to nucleate the liquid
to vapor transition. This is frequently known as “pore blocking” [163] [141], though
this term has the potential to mislead by suggesting a dynamic effect, whereas in
fact a metastable state is achieved.
Figure 5-3 illustrates a large pore kept full at
low partial pressure during drying due to a small pore providing the only path to the
atmosphere.
Various models of this hysteresis have been developed, and were discussed in
section 4.2. I present here a unified model combining several important features. It
allows for single-pore as well as network hysteresis, includes the effect of large pores
that provide nucleation points for the liquid-vapor interface, and is analytic rather
than computational. These all make the model, which is based on a simple, mean-field
treatment of percolation, ideal for studying complex, hierarchical pore structures.
63
8.5
Sorbed quantity (mmol/g)
Sorbed quantity (mmol/g)
6
5.5
5
4.5
4
3.5
0
0.2
0.4
0.6
0.8
8
7.5
7
6.5
6
5.5
5
4.5
4
1
0
0.2
Relative pressure
0.8
1
16
11
Sorbed quantity (mmol/g)
Sorbed quantity (mmol/g)
0.6
Relative pressure
12
10
9
8
7
6
5
4
0.4
0
0.2
0.4
0.6
0.8
14
12
10
8
6
4
1
Relative pressure
0
0.2
0.4
0.6
0.8
1
Relative pressure
Figure 5-1: Experimental (points) and modeled (lines) sorption isotherms on wetting
(blue) and drying (red) for dichlorofluromethane in carbon black formed into plugs
with porosity of (a) 0.50, (b) 0.58, (c) 0.66 and (d) 0.74 [34].
64
1.2
1
Increasing porosity
Volume fraction density
0.8
0.6
0.4
0.2
0
1
10
Pore radius (nm)
Figure 5-2: Pore size distributions of carbon black formed into plugs, obtained by
applying the model of this work to the experimental data of Carman and Raal [34],
as shown in Fig. 5-1.
65
Figure 5-3: Illustration of a system comprising a large pore whose only path to the
atmosphere passes through a small pore, exposed to a cycle of relative pressure. The
large pore remains full until the small pore empties and provides a meniscus where
the liquid-vapor transition can be nucleated.
5.2.1
Model
Since the pore network provides a hindrance only to drying, the sorbate content
during wetting is the same as that in the independent pore model given in equation
5.3.
To calculate the sorbate content during drying, we must derive a formula for
the probability that a pore is able to empty, as a function of relative pressure. The
first step is to turn the volume probability density
𝑣(𝑟)
into a number probability
density, which requires an assumption about pore shape. Since the model is already
built around cylindrical pores, the simplest assumption is that the volume of a pore
is proportional to the square of its radius, i.e., pores of different radii have the same
length.
At a particular relative pressure
ℎ,
it is then possible to define the fraction of
pores that are below their emptying transition pressure: this fraction
∫︀ ∞
𝑞=
The goal is to calculate the fraction
𝑣(𝑟)
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ) 𝑟2
∫︀ ∞ 𝑣(𝑟)
𝑑𝑟
𝑟2
0
𝑄
𝑞
is given by
𝑑𝑟
.
(5.4)
of such pores that has actually emptied. The
66
total sorbed volume during drying will then be
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
∫︁
𝑣(𝑟) 𝑑𝑟
𝑚𝑑 (ℎ) =
0
∫︁
∞
+ (1 − 𝑄)
∫︁
𝑣(𝑟) 𝑑𝑟
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
2
∞
𝑟 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟,
𝑟2
+𝑄
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
(5.5)
where the three terms on the right are the contributions of pores that are full due
to the pressure being above their emptying pressure, pores remaining full due to the
absence of a liquid-vapor interface, and adsorbed water in empty pores, respectively.
To perform this calculation, I model the pore network by a Bethe lattice of connectivity
𝑧 , where nodes correspond to pores.
Although in principle
describing the material, to avoid overfitting I fix
𝑧 = 4,
𝑧
is a free parameter
a reasonable value for a
random network in three dimensions and typical of values obtained if
𝑧
is allowed to
vary.
The Bethe lattice has been used previously to describe sorption in a porous material [103, 152]. I introduce an important extension by quantifying the effect of already
exposed pores on the drying behavior. Such pores are included through a parameter
𝑓,
defined to be the fraction of all pores exposed to a liquid-vapor interface as soon
as drying commences, so that the pores empty as soon as their equilibrium transition
pressure is reached. Figure 5-4 illustrates the model and the principles of the system
it represents: the exposed fraction
𝑓
accounts for the effect of large pores, which have
very different filling behavior and may remain empty throughout an experiment.
There are thus two mechanisms by which a particular pore may have access to
the vapor phase. The first is direct exposure, with probability
it could be connected to the vapor by a neighbor: I define
𝑋
𝑓.
The second is that
to be the probability
that an individual neighbor provides such a connection. This gives
1 − 𝑄 = (1 − 𝑓 )(1 − 𝑋)𝑧 .
67
(5.6)
Figure 5-4: A schematic diagram of a network of small (blue) and large (white) pores
formed by a solid skeleton (grey), and its representation by a Bethe lattice, here with
𝑧 = 3.
The holes in the boundary of some pores, as noted by green arrows, represent
an interface between the network of small pores and larger pores.
Taking advantage of the self-similarity of the Bethe lattice, we can further write
𝑋 = 𝑞[𝑓 + (1 − 𝑓 )(1 − (1 − 𝑋)𝑧−1 )].
I solve equation 5.7 self-consistently to find
𝑄.
Figure 5-5 shows
𝑄
as a function of
𝑞
(5.7)
𝑋 , and then use equation 5.6 to calculate
for
𝑓 = 0.2,
a typical value.
This percolation model is based on long-established ideas described by Mason and
others [105, 103, 152]. In fact, equations 5.6 and 5.7 are equivalent to equations 20
and 21 of the paper by Parlar and Yortsos [125] (there applied to scanning rather
than primary isotherms), where
𝑃𝑆 (𝑞; 𝑞𝑖 ) and 𝑞𝑖
in that paper are equal to
𝑄𝑞
and
𝑓𝑞
respectively in this paper.
5.2.2
Insertion within the “solid”
Equations 5.3 and 5.5 describe sorption in mesopores. Many porous materials possess
a hierarchical pore structure, and sorbate can be found not only in the mesopores, but
68
1
0.8
Q
0.6
0.4
0.2
0
0
0.2
0.4
0.6
0.8
1
q
Figure 5-5: Probability that a pore below its equilibrium emptying pressure is empty,
as a function of the fraction of pores below their equilibrium emptying pressures, for
an example case with an intially exposed fraction of 0.2.
69
also in even smaller spaces within what would be considered the “solid” part of the
structure [143] [144]. Simple models that treat the sorbate as a condensing fluid are
not applicable to these very small (approximately one nanometer or smaller) spaces,
because they neglect strong chemical interactions with the sorbent material, as well
as effects arising due to the discreteness of the sorbate molecules [16]. One approach
is to use various atomistic simulation techniques [23], but for the purposes of this
model a simpler approach is desired. In hardened cement paste, a material in which
inserted water is particularly important, the inserted water content as a function of
relative humidity is well approximated by assuming no loss of water on drying until
15% relative humidity is reached, linear loss of water below this, and linear reinsertion
on wetting [56, 79, 113]. The fluid content is then
𝑚𝑖𝑛𝑠 = 𝑚*
ℎ : 1 → 0.15
(5.8)
ℎ
𝑚*
0.15
ℎ : 0.15 → 0
(5.9)
𝑚𝑖𝑛𝑠 =
𝑚𝑖𝑛𝑠 = ℎ𝑚*
ℎ : 0 → 1.
This leaves only the total inserted water capacity
𝑚*
(5.10)
as an new unknown parameter.
If the direction of the pressure change is switched at a point other than 0 or 1, I
assume that
𝑚𝑖𝑛𝑠
retains its value at the turning point until this is reached by the
value predicted by equations 5.8 to 5.10.
5.2.3
Homogeneous nucleation
One further effect controls desorption at low relative pressure. This effect has been
called cavitation [169] or the tensile strength effect [70]. The cause of desorption is
that the condensed fluid becomes unstable at sufficiently low pressure. I assume that
the primary result of this desorption mechanism is the homogeneous nucleation of the
vapor phase in blocked large pores. In other words, I neglect the effect of the tensile
strength on the metastable sorption-desorption isotherm of an isolated single pore.
The vapor phase will nucleate in homogeneous bulk liquid when turning a small
70
quantity of liquid to vapor decreases the free energy: this will happen at some characteristic chemical potential. I denote the relative pressure in the vapor phase corresponding to this chemical potential by
ℎ𝑛 (∞).
Now when vapor is nucleated in a
pore of finite size, the same free energies are involved, but also the surface energy resulting from emptying the pore, which may be accounted for most simply by dividing
it equally among all the molecules, exactly as was done when considering pore filling
or emptying at equilibrium.
vapor in a pore of width
𝑟
The chemical potential difference between nucleating
and in the bulk is therefore (approximately) the same as
the chemical potential difference between condensation/evaporation in the pore and
in the bulk. In terms of pressures in the vapor phase, this can be written
2𝛾𝑣
ℎ𝑛 (𝑟) = ℎ𝑛 (∞)𝑒− [𝑟−𝑎𝑡(ℎ𝑛 )]𝑘𝑇 ,
The value of
ℎ𝑛 (∞)
(5.11)
should depend on the sorbate and temperature but not on the
sorbent.
Equation 5.11 gives a relationship between relative pressure and the width above
which all pores must be empty. Defining
∫︀ ∞
𝑞𝑛 =
𝑣(𝑟)
𝑟𝑛 𝑟2
∫︀ ∞ 𝑣(𝑟)
𝑟2
0
𝑑𝑟
𝑑𝑟
,
(5.12)
we can then write
𝑋 = 𝑞𝑛 + (𝑞 − 𝑞𝑛 )[𝑓 + (1 − 𝑓 )(1 − (1 − 𝑋)𝑧−1 )]
71
(5.13)
and
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
∫︁
𝑣(𝑟) 𝑑𝑟
𝑚𝑑 (ℎ) =
0
∫︁
𝑟𝑛
+ (1 − 𝑄)
∫︁
𝑟𝑛
𝑣(𝑟) 𝑑𝑙
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
2
+𝑄
∫︁
𝑟𝐾 (ℎ)+𝑎𝑡(ℎ)
∞ 2
+
𝑟𝑛
𝑟 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟
𝑟2
𝑟 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟.
𝑟2
(5.14)
The four terms on the right are the contributions of pores that are full due to the
pressure being above their equilibrium emptying pressure, pores remaining full due to
the absence of a liquid-vapor interface, adsorbed water in pores that could have been
kept full by the network but happen to have been emptied through propagation of a
meniscus, and adsorbed water in pores that must have emptied due to cavitation.
Figure 5-6 plots an illustrative prediction of this theory for a pore size distribution
with
𝜇 = 1.5, 𝜎 = 0.8
and
𝑓 = 0.3.
The figure divides the contributions to hysteresis
from single pore and network effects, and also shows the adsorbed quantity predicted
by the BET and Langmuir equations with no influence of the finite pore size.
5.2.4
Application of the model to experimental data
Figure 5-7 compares the model results with experimental observations for various
systems [92, 98, 61, 12]. The model parameters were determined in each case by a
least squares fit.
materials.
Figure 5-8 shows the calculated pore size distributions for these
The median radius of 13 nm found for the case of carbon nanotubes is
consistent with the transmission electron microscope image of these nanotubes (figure
12 of Frackowiak and Béguin [61]).
Values of
𝑓
and
𝑧
calculated for various materials are presented in table 5.1.
Uncertainty ranges are calculated by comparing isotherms of samples produced with
different specifications (hardened cement paste), or taken at different temperatures
(dental enamel) or using different sorbates (porous silica glass and Vycor). This is
72
1
BET
0.8
Network hysteresis
Saturation
0.6
Single-pore hysteresis
Tensile strength knee
0.4
Wetting
0.2
Langmuir
0
0
0.2
0.4
0.6
0.8
1
Relative pressure
Figure 5-6: An example sorption and hysteresis prediction of the model presented
here, showing the contributions to hysteresis of single pore and network effects, along
with the adsorbed quantity predicted by the BET and Langmuir equations.
73
1.2
1
15
Saturation
Water content (% of dry mass)
20
10
0.8
0.6
0.4
5
0.2
0
0
0.2
0.4
0.6
0.8
0
1
0
Relative humidity
0.4
0.6
0.8
1
Relative humidity
800
1.2
700
1
600
500
Saturation
Sorbed quantity (mL/g)
0.2
400
300
0.8
0.6
0.4
200
0.2
100
0
0
0.2
0.4
0.6
0.8
0
1
Relative pressure
0
0.2
0.4
0.6
0.8
1
Relative pressure
Figure 5-7: Experimental (points or dashed lines) and modeled (solid lines) sorption
isotherms on wetting (blue) and drying (red) for (a) water in hardened cement paste
[12], (b) water in Vycor [92], (c) nitrogen in carbon nanotubes [61] and (d) xenon in
silica glass [98].
74
1.4
1.2
Cement
1
Vycor
Volume probability density
Silica glass
0.8
0.6
Carbon nanotubes
0.4
0.2
0
1
10
Pore radius (nm)
Figure 5-8:
Pore size distributions, obtained by applying the model of this work
to published experimental data, for hardened cement paste [12], Vycor [92], carbon
nanotubes [61] and silica glass [98].
75
Table 5.1:
Initial fraction of exposed pores,
𝑓,
calculated by applying the model
presented here to a variety of published experimental data.
Material
Sorbate/s
Reference
f
CNT
Nitrogen
[61]
0.4
Cement
Water
[59, 12]
Enamel
Water
[187]
Silica glass
CCl2 F2 , C4 H10 , Xe, O2
[98]
Vycor
N2 , H2 O, C6 H14
[28, 92, 121]
0.33 ± 0.06
0.54 ± 0.05
0.13 ± 0.09
0.06 ± 0.01
statistical uncertainy only and does not account for model uncertainty.
5.2.5
Scanning isotherms
The principles of the model can be applied directly to the case of scanning isotherms,
where the partial pressure is cycled over some subsection of the possible range.
ℎ𝑚𝑖𝑛
If the decrease in partial pressure is halted at
increased to
ℎ,
and the partial pressure then
the total sorbed volume will be
∫︁
𝑟𝐾 (ℎ𝑚𝑖𝑛 )+𝑎𝑡(ℎ𝑚𝑖𝑛 )
𝑚↑𝑠𝑐𝑎𝑛 (ℎ) =
𝑣(𝑟) 𝑑𝑟
0
∫︁
𝑟𝑛 (ℎ𝑚𝑖𝑛 )
+ [1 − 𝑄(ℎ𝑚𝑖𝑛 )]
∫︁
𝑣(𝑟) 𝑑𝑟
𝑟𝐾 (ℎ𝑚𝑖𝑛 )+𝑎𝑡(ℎ𝑚𝑖𝑛 )
𝑟𝑛 (ℎ𝑚𝑖𝑛 )
2
+ 𝑄(ℎ𝑚𝑖𝑛 )
∫︁
𝑟𝐾 (ℎ𝑚𝑖𝑛 )+𝑎𝑡(ℎ𝑚𝑖𝑛 )
2
2
∞
+
𝑟𝑛 (ℎ𝑚𝑖𝑛 )
𝑟 − [𝑟 − 𝑎𝑡(ℎ)]2
𝑣(𝑟) 𝑑𝑟
𝑟2
𝑟 − [𝑟 − 𝑎𝑡(ℎ)]
𝑣(𝑟) 𝑑𝑟,
𝑟2
where any non-zero integral bound must be replaced by
𝑟𝐾 (ℎ)/2 + 𝑎𝑡(ℎ)
when the
latter surpasses the former.
If the increase in partial pressure is halted at
decreased to
ℎ𝑚𝑎𝑥
and the partial pressure then
ℎ, the total sorbed mass can be calculated using equations 5.12 and 5.14,
but using the smaller of
𝑟𝑛 (ℎ)
and
𝑟𝐾 (ℎ𝑚𝑎𝑥 )/2 + 𝑎𝑡(ℎ𝑚𝑎𝑥 )
in place of
𝑟𝑛 (ℎ),
i.e. the
radius of the largest pores that could be filled.
Figure 5-9 compares the model with experimental scanning isotherms for dental
enamel [187].
76
0.6
Water content (% of dry mass)
0.5
0.4
0.3
0.2
0.1
0
0
0.2
0.4
0.6
0.8
1
Relative humidity
Figure 5-9: Primary and scanning sorption isotherms calculated using the model of
this work (solid lines), along with experimental sorption isotherms for water in dental
enamel at 298 K [187] (points).
77
5.3 Potential for further developments
The model presented provides an overall description of hysteresis sorption in mesoporous media, especially in materials in which macropores are also present. Nevertheless, there remain some complications that increase the uncertainty in the level to
which the model can quantitatively describe pore size and network structure.
First, the model as formulated assumes cylindrical pores, and a particular amount
of single-pore hysteresis due to differences in the shape of the liquid-vapor interface.
Other assumptions, such as slit pores with larger hysteresis or equilibrium filling
and hence no hysteresis, could have been made instead and built into the model in
a straightforward way.
Agreement with TEM observations for the case of carbon
nanotubes gives some confidence that the assumption of cylindrical pores is reasonable.
However, the assumption still influences the obtained results.
Specifically, if
cylindrical pores are assumed when the system is really made up of slit pores, the
obtained pore sizes will be larger and obtained value of
𝑓
smaller than in reality (the
discrepancies are opposite if in fact the pores are able to reach local equilibrium on
filling).
Second, the model uses a very simple model of surface adsorption. The effect of
this can be seen in some discrepancy between model and experimental results at low
pressure. Where the data are available, this could be rectified by using experimental
t curves [156], at the loss of some generality.
Third, the assumption of a fixed value of
𝑧
precludes distinguishing between sys-
tems based on their connectivity. For example, we might expect carbon nanotubes to
have
𝑧 ≈ 2,
higher
𝑧.
while disordered porous systems such as glasses and cement paste have
Sorption data alone are not sufficient to allow such an identification to be
made. Also, it seems to be
𝑓
rather than
𝑧
that is most influential in determining
the sorption behavior.
One tantalising prospect is the use of experiments like those cited in this work to
measure
ℎ𝑛 (∞)
for various fluids as a function of temperature. In only two distinct
cases: water around 25
∘
Cand at 50
∘
C, was the characteristic knee observed.
78
ℎ𝑛 (∞)
was larger at higher temperature as expected. A systematic study could allow this
quantity to be measured as a function of temperature, providing insight into the
nature of forces between molecules in a liquid.
Values of
ℎ𝑛 (∞)
can be calculated for some simple models of interacting fluids.
For example, the regular solution model predicts
√
ℎ𝑛 (∞) =
where
𝑡𝑎𝑢 = 𝑇 /𝑇𝑐
𝑒−2
√
(1 + 1 − 𝜏 )
√
,
1− 1−𝜏
1−𝜏 /𝜏
(5.15)
is temperature divided by the critical temperature of the sorbate.
A solution can also be found for a van der Waals fluid, though not in closed form.
Writing
𝑝(𝑣) =
8𝜏 𝑣 2 − 27𝑣 + 27
,
27(𝑣 − 1)𝑣 2
(5.16)
which is the van der Waals equation of state in units where the parameters are both
1, we have
ℎ𝑛 (∞) =
where
𝑣𝑐𝑎𝑣
𝑝(𝑣𝑐𝑎𝑣 )
,
𝑝𝑒𝑞
(5.17)
is the root of
4𝜏 𝑣 3 − 27𝑣 2 + 54𝑣 − 27 = 0
𝑝𝑒𝑞
that lies between 1 and 3 and
∫︁
(5.18)
is found by applying the Maxwell equal area rule,
𝑣2
𝑝(𝑣) 𝑑𝑣 = 𝑝𝑒𝑞 (𝑣2 − 𝑣1 )
(5.19)
𝑣1
with
𝑝(𝑣1 ) = 𝑝(𝑣2 ) = 𝑝𝑒𝑞 .
(5.20)
Figure 5-10 compares these equations with the two existing data points. Although
these simple models are not necessarily good approximations of a sorbing fluid, the
independence of the curves on material specifics suggests that there might be a general
trend across sorbates, though there is no reason to expect a strictly universal curve.
79
1
Relative pressure of bulk cavitation
0.8
Water, 323 K
0.6
Water, 296-298 K
0.4
van der Waals
Regular solution
0.2
0
0
0.2
0.4
0.6
0.8
1
Reduced temperature
Figure 5-10: Relative pressure at which homogeneous nucleation occurs in the bulk
in the regular solution and van der Waals models, along with two experimental data
points.
80
Chapter 6
Drying shrinkage in hardened cement
paste
The content of this chapter is based heavily on the article in preparation “Hysteresis from multiscale porosity: water sorption and shrinkage in cement paste” by the
author, Masoero et al. [134].
In chapter 5, I showed that although sorption hysteresis can make it more difficult
to interpret sorption isotherms, it can also allow the calculation of an additional
parameter describing the pore system, namely the connectedness of the mesopores
to larger pores.
The situation is similar for drying shrinkage in hardened cement
paste: an understanding of the processes leading to hysteresis helps to understand
the overall shrinkage process.
6.1 Overview of hardened cement paste
6.1.1
Structure
The primary constituent of cement paste is calcium-silicate-hydrate (C–S–H), a semicrystalline and non-stoichiometric solid with a layered molecular structure [167, 166,
143, 130], which is formed by hydration reactions between cement clinkers (calcium
silicate minerals) and water during the setting of cement paste. The C–S–H precipi-
81
tation process produces a solid network containing many pores several nanometers in
size: the overall material comprising both solid and pore is referred to as C–S–H gel
[167, 171]. Water can be (and, in normal environmental conditions is) located within
these pores, both adsorbed on pore walls and condensed into a liquid state. Additionally, the solid network itself contains water within its layered molecular structure, in
spaces smaller than a nanometer [79].
The water found in cement paste is important because it influences important
mechanical properties such as drying shrinkage [18, 175, 147]. This influence arises
due to forces exerted on the solid structure by the water itself and ions dissolved in
it.
Shrinkage plays a fundamental role in cracking of cement and concrete, and so
a model of shrinkage could contribute to efforts to perform quality controle, assess
degradation, and design new cement-based materials that are more resilient against
damage due to shrinkage and cracking.
To understand and predict shrinkage and
its effect on cracking, it is necessary to know where water is located as a function
of humidity [12, 79, 140], what forces are present and most significant in pores of a
particular size, and how the pore structure of hardened cement paste influences bulk
transport of water [64].
6.1.2
Shrinkage hysteresis
In many mesoporous materials, such as Vycor, a porous borosilicate glass, shrinkage
during drying is greater than at the same relative humidity during wetting. Furthermore, hysteresis is not present at low humidity, when water is not condensed in the
pores but only adsorbed on pore walls. These observations are easily explained using
Kelvin-Laplace theory: there is a negative pressure in the water in the pores, and
since there is more water present at the same humidity on drying than wetting (as
explained in chapter 5), the shrinking effect is greater [19, 142].
Hardened cement paste has the opposite behavior:
shrinkage is greater during
wetting than during drying, and the hysteresis extends over the entire range of relative
humidity. This hysteresis behavior is also found in clays [128, 35]. In both cement
paste and clay, evaporable water can be found in molecular-scale spaces between layers
82
!"&
(a)
(b)
(c)
2
2
!"%
!"$
!"#
!
Cement paste
! !"# !"$ !"% !"& '
*0-,4.70289+.1.4:2;<=>
!
?!"#
Clay
222
!
!"# !"$ !"% !"&
<=
Vycor
'
222
222
!
!"# !"$ !"% !"&
<=
'
222
(d)
(e)
(f)
2
?!"$
2
()*+,-./0123,40*25)64064
()*+,-./012@4*,.62
2;+,A"2@8*.6B,C02@0424)2?'>
'
?!"%
?!"&
?'
Cement paste
! !"# !"$ !"% !"& '
*0-,4.70289+.1.4:2;<=>
Clay
!
!"# !"$ !"% !"&
<=
Vycor
'
!
!"# !"$ !"% !"&
<=
'
Figure 6-1: Water sorption and shrinkage isotherms for (a, d) Portland cement paste
[58], (b, e) raw clay with calcium ions in the molecular layers [36], and (c, f ) Vycor
glass [73]. Notice that the drying (red) and wetting (blue) strain paths for cement
paste and clay (d, e) are upside-down compared with those of Vycor (f ).
Figure
courtesy of Enrico Masoero.
of the solid skeleton, suggesting that it is the removal and reinsertion of this water
that causes the “upside-down” shrinkage hysteresis.
Figure 6-1 compares sorption
(in which hysteresis is in the same direction, though quantitatively different) and
shrinkage isotherms for cement paste, clay and Vycor.
Figure 6-2 illustrates the
water content of hardened cement paste over a drying and wetting cycle.
In this chapter, I present an overall picture of the pore spaces in which water may
be found, and a model of how water sorption in these spaces determines shrinkage.
Section 6.2 outlines the different types of water found within hardened cement paste.
Section 6.3 explains how ideas from chapter 5 can be used to deduce the quantity
of water present in each location as a function of relative humidity, while section 6.4
uses the division by type to develop a continuum model of shrinkage.
83
'
(a)
3"
()*+,-./0123,40*25)64064
(b)
8%91:/0;,:"
<,&,%;"+/0=%"
.>9=;,2"
"
'0?=--0/1"
?@/,"
.A-0<B2"
5 nm
'()(*"+,-".+/012"
"
6"7&"
!"
"
!"%
'"
"
!"$
C"
!"#
!
#$"%&"
"
!"&
4"
!
"
"
!"#
5 nm
(c) 3"
!"
Capillary
"
"
4"
'"
"
Capillary
Gel
Pore
'
C"
"
Gel
Pore
Capillary
Gel
Pore
!"$
!"%
!"&
*0-,4.70289+.1.4:2;<=>
5"
"
"
Capillary
Capillary
"
5"
Capillary
Gel
Pore
Gel
Pore
Gel
Pore
Capillary
Gel
Pore
Capillary
Capillary
Gel
Pore
Capillary
Capillary
Gel
Gel
Pore
Gel
Pore
Capillary
Gel
Pore
Capillary
Gel
Pore
Pore
Drying
Capillary
Gel
Pore
Water
IGP
Surface
Surface
Capillary
IGP
Surface
Figure 6-2:
Gel
Pore
Capillary
Gel
Pore
Wetting
Interlayer
space
Interlayer
(dry)
Calcium
oxide layer
SiO4
tetrahedra
Interlayer
(a) Backscattered scanning electron micrograph of a polished cement
paste surface. (b) Experimental water sorption isotherm [59]. (c) Schematic diagrams
illustrating the C–S–H water content at the six points marked on the isotherm. A:
saturated state with large capillary pores empty.
B, F: configurations at the same
relative humidity, but different water content, on drying and wetting. C, E: same, at
a lower humidity. D: an almost completely dried configuration. Figure courtesy of
Enrico Masoero and Hamlin Jennings.
84
This work was performed in collaboration with Enrico Masoero et al. I developed
the quantitative model of shrinkage, as well as contributing to the development of the
overall ideas regarding the importance of water present in different locations.
6.2 Classification of water in cement paste
Water can be divided into four categories based on its local environment within the
cement paste microstructure.
6.2.1
Interlayer water
Interlayer water is found in spaces with width less than about one nanometer. It is
bound strongly and removed only at low relative humidity. If C–S–H is considered
to have a particulate or granular structure [79], interlayer water includes both that
between layers within a single particle, and that between particles where they are in
close contact. It is well documented [126, 79] that there is substantial collapse and
swelling of the interlayer space as water is removed and re-inserted, respectively.
6.2.2
Gel pore water
The C–S–H gel formed during cement hydration is inevitably porous, as the product density is higher than the average density of the reactants. The gel porosity is
defined in the cement literature as the minimum porosity achievable: that resulting
when reaction products uniformly (on a large enough length scale) fill the originally
water-filled space, leaving no capillary pores [136].
Gel pores have width between
approximately 1 and 10 nanometers.
6.2.3
Capillary pore water
Capillary pores are the voids left in regions not filled by C–S–H during hydration.
They range in size from 10 nanometers up to hundreds of micrometers. The smaller
end of this range may include some pores intrinsic to the C–S–H gel [12, 114], but
85
since pore properties depend fundamentally on size, we include these pores in the
capillary pore category.
6.2.4
Surface adsorbed water
When gel and capillary pores are empty of condensed water, they retain an adsorbed
layer of water on their pore walls due to the hydrophilic nature of the C–S–H surface.
This adsorbed water is influential in determining shrinkage because it affects the
surface energy of the solid/pore interface.
6.3 Water sorption isotherm of hardened cement paste
6.3.1
Interlayer water
The Kelvin equation (equation 4.3) implies that water will not be removed from
the interlayer space until low relative humidity, around 14%. This is supported by
molecular simulations [24, 25] and nuclear magnetic resonance (NMR) experiments,
which are able to determine the size of the pore in which water is located [113]. The
removal of water at lower humidities is a complex process which depends on chemical
and electrostatic interactions between water, ions and the C–S–H surface. I make the
very simple approximation that water is removed from the interlayer space linearly
between 15% and 0% relative humidity.
Modeling the reinsertion of water requires more care.
Direct measurements of
interlayer water content on wetting are not yet available, so I use Jennings’ analysis
[79] of the experimental data of Feldman [56].
To experimentally determine the
interlayer water content on the adsorption branch at a particular relative humidity, the
sample is first fully dried, then equilibrated at the desired humidity. This procedure
ensures that the interlayer water content reaches (local) equilibrium at that humidity,
and for sufficiently high relative humidity will also result in some water being present
in the gel pores.
The humidity is then decreased from the level of interest down
to a level at which the gel pores are empty of condensed water: in this case, 11%
86
[56].
From the water that remains, the quantity adsorbed on pore walls must be
subtracted: this was estimated as 0.25 moles per mole of C–S–H [79].
is the interlayer water content at the turning point on adsorption.
The result
Possessing an
estimate of the interlayer water content on adsorption and desorption, the remainder
of the water must be adsorbed and condensed in the gel and capillary pores.
6.3.2
Gel and capillary pore water
The total water content of the gel and capillary pores is the experimentally observed
water content minus the interlayer water content. Equations 5.3, 5.6 and 5.14 can
then be used to rationalize this observed pore water content in terms of a pore size
distribution and connectivity to the largest pores. In particular, these equations allow
the calculation of the quantity of adsorbed and condensed water at any particular
humidity.
These quantities, along with the interlayer water content, are shown in
figure 6-3 and used in the model of drying shrinkage presented in section 6.4.
6.4 Continuum modeling of reversible drying shrinkage
Knowing the quantity of water in each category as a function of humidity on wetting
and drying, it is possible to model reversible drying shrinkage due to the changing
water content.
“Reversible” as a descriptor of drying shrinkage means that a bulk
sample returns to its original dimensions when fully resaturated: there is still extensive
hysteresis in the strain at intermediate humidity values. Irreversible drying shrinkage,
when the sample does not return to its original dimensions on resaturation, is observed
primarily on the first cycle of drying and rewetting, and the mechanisms for this are
not part of the models discussed here.
We assume throughout that shrinkage is
isotropic, so the linear strain measured in experiment is one third of the volumetric
strain.
Several continuum models of deformations induced by humidity changes have been
87
@+,)(/><:,):,/5A/<B/3(4/2+==6
%
Wetting
(a)
%!
Surface
(d)
Total sorption isotherm
$
$&
Drying
!
/
/
$% (b)
#
"
!
/
/
& (c)
/
/
/
/
Gel + Capil.
$%
#
/
/
/
789)(-2):,
;<3)*
789)('/=>+::-:?
;<3)*/=>+::-:?
"
"
%
!
ing
Scann
/
Interlayer
!
!'% !'" !'& !'#
()*+,-.)/012-3-,4/5(06
$
!
!
!'%
!'"
!'&
()*+,-.)/012-3-,4/5(06
!'#
$
Figure 6-3: Contributions to the water sorption isotherm from (a) water adsorbed on
pore walls, (b) water present in filled gel and capillary pores, and (c) water present
in the interlayer space, along with (d) the overall model result (red) obtained by
adding the three curves at left, along with the experimental isotherm (black) for
bottle-hydrated Portland cement [58]. Figure courtesy of Enrico Masoero, from data
calculated by the author.
88
proposed [18, 175, 147].
These models are based on poromechanics and relate the
volumetric strain to the Laplace pressure in the pore water (also known as capillary
stress). We extend such modeling to account also for the effects of surface energy,
and swelling caused by water in the interlayer space [129].
Since cement paste is isotropic at the bulk scale, and there is no external load,
drying shrinkage is isotropic and hence the linear strain is one third of the volumetric strain. The linear strain (which is less than 0.5%) is sufficiently small that the
constitutive equation for cement paste is linear, so the strain can be decomposed into
three contributions:
𝜀𝐿
to surface energy, and
from Laplace pressure in the gel and capillary pores,
𝜀𝐼
𝜀𝑆
due
from swelling caused by the interlayer water. These contri-
butions can be associated with three categories of water described in section 6.2 (gel
and capillary pore water considered together since water has bulk properties in both;
surface-adsorbed water; and interlayer water). The low strain magnitude also means
that there is negligble change in gel and capillary pore widths.
I also neglect the
effect of any gradient in saturation, as this will be small when the relative humidity
is changed in steps and the system allowed to equilibrate.
6.4.1
Macroscopic length change due to Laplace pressure
A simple and widely-used constitutive model describing the strain of a porous material
due to Laplace pressure in the gel pore water is [19, 175]
1 𝑘𝑇
𝜀𝐿 =
ln ℎ𝑆
3 𝑎3
Here
𝑘𝑇 ln ℎ/𝑎3
1
1
−
𝐾𝑏 𝐾𝑠
)︂
.
is the Laplace pressure in the pore water,
modulus of the macroscopic sample,
part of the C–S–H gel [99] and
6.4.2
(︂
𝑆
𝐾𝑠 = 50
(6.1)
𝐾𝑏 = 19
GPa is the bulk
GPa is the bulk modulus of the solid
is the filling fraction of the gel and capillary pores.
Macroscopic length change due to surface energy
To calculate the macroscopic length change associated with a change in surface energy,
the Bangham equation [8, 138, 74] must be modified to account for additional surface
89
area as pores empty. The linear strain due to surface energy is then
𝜀𝑆 = −
where
𝜎
∆(𝜎𝛾)
,
3𝐾(1 − 2𝜈)
(6.2)
is the surface area of empty pores per volume of porous material and
𝛾
is
the total surface tension of the pore wall with its adsorbed layer (note that “empty”
pores retain a layer of water adsorbed on their walls).
change in the total surface energy;
𝜈
Consequently
∆(𝜎𝛾)
is the
is Poisson’s ratio of the bulk macroscopic volume,
approximately 0.2 (see e.g. [150]). At 100% RH all pores are full, with the possible
exception of some very large pores with low surface area, so
∆(𝜎𝛾) = 𝜎𝛾 .
The surface tension is given by Gibbs’ equation [57, 138]:
𝑘𝑇
𝛾 = 𝛾0 − 2
𝑎
where
𝛾0
is the surface tension at
ℎ0 .
∫︁
ℎ
𝑡
ℎ0
𝑑ℎ
,
ℎ
(6.3)
It is convenient to use complete drying, 0%
relative humidity, as the reference point. The solid-air surface tension at this humidity
must be assumed: I estimate a value twice that of water-air.
6.4.3
Macroscopic length change due to loss of interlayer water
It is well established that many layered materials experience expansion when a substance is intercalated into them [49, 46, 185, 90], but the details of this expansion
can be complex. An accurate model of the relationship between the quantity of interlayer water in C–S–H and the associated length change of the macroscopic cement
paste sample will be rather difficult to develop. The presence of dissolved ions produces a repulsive osmotic pressure, but this is small: a mean field treatment with
walls separated by 0.5 nm gives a pressure around 1 MPa or less, regardless of ion
concentration [4]. More important are effects arising due to the discreteness of ions:
correlation effects can even result in a force that is joining instead of disjoining [129].
In the absence of a detailed model that accurately predicts length changes due
90
to changes in interlayer water content, useful progress can be made with the simple assumption that the strain of the bulk cement paste is isotropic and is linearly
proportional to the quantity of interlayer water, such that
𝜀𝐼 =
where
𝑣𝐼
𝜆
𝑣𝐼
3
(6.4)
is the volume of interlayer water (which can be calculated using the method
described in the previous section:
stant. Although
𝜆
see figure 6-3), and
𝜆
is a proportionality con-
is found empirically in this work and thus may be considered a
fitting parameter, it has an important physical meaning: it measures the proportion
of microscopic strain that is translated into bulk strain, rather than simply being
accommodated by the porous material. The value calculated here could potentially
be used to assess a microscopic model linking microscopic and macroscopic strains in
cement paste. The quantity may also be estimated from experimental measurements.
X-ray diffraction experiments have found 20% shrinkage of interlayer space while the
macroscopic shrinkage of the bulk was only 3$ [72].
Similarly, digital image-based
deformation mapping has found local deformations around 10% while overall strain
was less than 0.5% [116]. These observations suggest that the heterogeneous structure
of cement paste is capable of taking up most of the microscopic length change, and
only a small fraction is transmitted to overall strain.
𝜆
should therefore have a value
around 0.1.
6.4.4
Comparison with experiment
Figure 6-4 shows the overall predicted shrinkage calculated using this model, along
with the contributions from each of the three sources, and experimental data measured
by Feldman [58]. The unknown parameter
𝜆
is used as the only fitting parameter:
the best-fit value obtained is 0.07. The combined model sucessfully reproduces the
magnitude and general shape of the hysteresis in the reversible drying strain across
the entire range of humidity.
The model explains the sources of the observed shrinkage and its hysteresis. There
91
"
(a)
!#"&
Wetting
Surface
!#")
C/+.0?28D;
!#"B
"2
(d)
"
2
2
2
2
Drying
!"#(
2
!"#'
!#"&
!#")
Gel + Capil.
!#"B (b)
"2
2
2
2
2
!"#&
2
!#"*
Total shrinkage isotherm
!"#%
<=>,+05,?/
@A6,-
!#()
(c)
!#'%
"
Interlayer
"#' "#% "#) "#*
+,-./01,2345060/7289:;
(
!"#$
"
"#'
"#%
"#)
+,-./01,2345060/7289:;
"#*
(
Figure 6-4: Predicted shrinkage due to water (a) on the surface of C–S–H, (b) in
the gel pores, and (c) in the interlayer spaces, along with (d) total predicted and
experimentally measured [58] shrinkage.
Figure courtesy of Enrico Masoero, from
data calculated by the author.
is little hysteresis in the shrinkage caused by surface tension, and the hysteresis in
the shrinkage caused by Laplace pressure is in the opposite direction to the overall
shrinkage hysteresis. Only by accounting for the swelling effect of interlayer water can
the observed hysteresis in the shrinkage be understood. For this reason, models based
exclusively on Laplace pressure have been unable to explain experimental results [175].
The importance of interlayer water also means that if a sample is not dried below
approximately 15% relative humidity, much of the shrinkage hysteresis will disappear,
and the remaining hysteresis will be in the opposite direction. That is, shrinkage as
a function of relative humidity will be greater during desorption than adsorption,
as is observed in many other porous materials such as Vycor. This claim is partly
confirmed by the data of Feldman and Sereda, who observed that the upside-down
shrinkage hysteresis disappears when re-wetting is started before drying below 25%
RH [57].
92
6.5 Potential future extensions
The model presented here used indirect methods to determine the water content of
each type of space, and thence calculate shrinkage. Since NMR techniques are able to
more directly measure the water content of the various spaces [114], if NMR data were
available over a full wetting and drying cycle, a more confident prediction of shrinkage
could be made. Nevertheless, this work has shown that classifying the water in cement
paste according to its location and then modeling the physical behavior of each of
these water types separately allows the overall behavior of the paste to be better
understood.
The understanding of the fundamental importance of the difference between water
in different locations, and of the different humidity ranges over which these locations
empty and fill, opens the door to a new perspective for designing experiments that
extract information about the microstructure of cement based materials. For example,
the gel pores can be probed without interference from interlayer space by only drying
to about 25% RH. The size distribution of the gel pores can then be analyzed from
the adsorption isotherm, and their connectivity to the larger pores by analysis of the
hysteresis. This could be used, for example, to assess the gel pore ratio and initial
water to cement ratio of an existing sample of cement. The structure of gel pores can
change due to drying, load, or even deliberate alteration such as by filling these 10100 nanometer-scale spaces with polymer [108]. Since such a change in structure will
greatly influence properties, this technique for assessing the gel pore structure would
be a valuable research tool. In addition, it can complement and support the validation
of a growing body of mesoscale coarse-grained simulations aimed at elucidating the
mechanisms of formation, the nanoscale structure, and the mechanical properties of
the C–S–H gel [69, 107, 101, 68, 102, 77, 53]. There is also much scope for interesting
research on the kinetics of water leaving each kind of space.
Chapter 7 examines
water dynamics in the larger pores, but the problem of dynamics in the interlayer
space remains open.
93
94
Chapter 7
Modeling of transport with hysteresis
in porous materials
I now turn to the problem of transport of a sorbing fluid through a porous material.
One reason that such transport is important is that it plays a role in the degradation
of many porous materials [158, 106], such as creep, corrosion and contamination
in concrete [55, 149, 20, 65].
Other important areas of application include carbon
dioxide sequestration in sorbents such as zeolites or activated carbon [39], and shale
gas extraction [78].
7.1 Model formulation
For a sorbing fluid, Darcy’s law (equation 4.5) must be written as
q=−
where
𝑐
𝑐𝜅
∇𝑝,
𝜏𝜇
(7.1)
is the saturation of fluid (that is, its coarse-grained concentration relative to
the maximum possible), since flow only happens where there is fluid.
𝜏
is a correc-
tion for tortuosity: a typical model, known as the Bruggeman relation, is that it is
proportional to
𝑐−1/2
[31]. Since the pore network itself has a tortuous structure in
the porous material, there should be an additional constant factor: I use a factor of 3
95
as suggested by Gruener et al. [71]. The other fundamental principle describing flow
is conservation of mass, which can be written as
𝜕𝑐
= −∇.q
𝜕𝑡
(7.2)
To close this system of equations, it is necessary to know the relationship between
pressure and saturation, and obtaining a suitable relationship is the most important
step in the development of the model presented here.
First, I note that there is a well-defined relationship between relative pressure
in the vapor phase and pressure
𝑝
in the liquid phase, given by
𝑝=
where
𝑣
ℎ
𝑘𝑇
ln ℎ,
𝑣
(7.3)
is the average molecular volume in the liquid phase. Air pressure may be
ignored because it is so much smaller than liquid pressures at partial pressures below
saturation. Since the adsorption isotherm gives saturation as a function of relative
presure, it gives an upper bound for liquid pressure as a function of saturation.
A general expression relating liquid pressure and saturation relies on assumptions
about the processes that govern sorption. The assumption I use is that the adsorption isotherm, which marks the minimum quantity that can be sorbed at a particular
partial pressure, gives the stable quantity of fluid based on local considerations. Any
additional sorption must be trapped due to non-local considerations such as network
effects. Note that the term “trapped” simply refers to the absence of any monotonically decreasing path on a free energy landscape from the full to the empty state.
There is no kinetic blocking and the sorbate is free to move through the region occupied by trapped water. The measured saturation at some partial pressure can be
divided into stable and trapped parts
𝑐𝑠
and
𝑐𝑡 :
𝑐(ℎ, ...) = 𝑐𝑠 (ℎ) + 𝑐𝑡 (ℎ, ...),
where
𝑐𝑠 (ℎ)
(7.4)
is given by the adsorption isotherm and is an empirical input to the
96
model. The form of the functions
𝑐
𝑐𝑡
and
denotes that they depend on more than
just the current local partial pressure.
I propose as a reasonable assumption that the maximum fraction of the pore space
not filled with stable fluid that can be filled with trapped fluid is a function of the
total fluid content, and denote this function as
isotherm
𝑐↓ (ℎ)
then allows the calculation of
𝑔¯(𝑐).
An experimental desorption
𝑔¯:
𝑐↓ (ℎ) = 𝑐𝑠 (ℎ) + 𝑔¯[𝑐↓ (ℎ)][1 − 𝑐𝑠 (ℎ)].
Likewise, for any state
(ℎ, 𝑐),
(7.5)
not necessarily accessed by a desorption isotherm, it is
possible to define an analogous fraction
𝑔
by
𝑐 = 𝑐𝑠 (ℎ) + 𝑔[1 − 𝑐𝑠 (ℎ)].
Since we know the relationship between
𝑐𝑠
and
ℎ,
(7.6)
our description of the problem
will be complete if we determine a method for allocating changes in total saturation
to
𝑐𝑠
and
𝑐𝑡 .
I do this by making the approximation that the probability that a pore
is full of trapped water is independent of the radius of the pore and hence the partial
pressure at which it will fill (assuming of course that the partial pressure is above the
filling pressure). In this case, increasing
𝑐
will not affect
fluid turned into stable fluid to new fluid added will be
𝛿𝑐𝑠 = 𝛿𝑐 +
𝑔
𝛿𝑐.
1−𝑔
𝑔,
and the ratio of trapped
𝑔/(1 − 𝑔),
so
(7.7)
When sorbate pressure is decreased, stable fluid will be turned into trapped fluid
and lost from the local region in proportion
will cause
𝑔
𝑔¯ to 1 − 𝑔¯.
Additionally, the loss of fluid
to decrease. The assumption that trapping is independent of pore size
implies that
1 𝑑𝑔
1 𝑑¯
𝑔
=
,
𝑔 𝑑𝑐
𝑔¯ 𝑑𝑐
97
(7.8)
so
𝛿𝑐 = (1 − 𝑔¯)𝛿𝑐𝑠 +
𝑔
𝑐𝑡 𝑑¯
𝛿𝑐.
𝑔¯ 𝑑𝑐
(7.9)
Using equations 4.5-7.3, 7.7 and 7.9, it is now possible to simulate transport in a
porous medium with arbitrary initial and boundary conditions.
7.2 Results
As an interesting example of transport of a sorbing fluid through a porous medium,
I look at water sorption in hardened cement paste.
Experimental adsorption and
desorption curves are taken from the work of Baroghel-Bouny [12]. Simulations are
performed for a prismatic slab 50 cm in thickness, assuming an intrinsic permeability
2
of (2 nm) , since 2 nm is a typical pore radius.
7.2.1
Time-dependent hysteresis curves
One problem that can be tackled with this method is the identification of the contribution of transport effects to dynamic hysteresis curves, obtained when the atmospheric
humidity is changed too rapidly to be equilibrated across the sample. To simulate
these curves, the humidity at the surface of the sample was allowed to oscillate between 0 and 1 with period ranging from 1 hour to 1 month. The total mass of water
sorbed in the cement is plotted in figure 7-1 as a function of the instantaneous humidity (in the limit of long time, when the response is periodic). The mass change
can be seen to shrink as the humidity variation becomes too fast to be transmitted
through the sample.
Very different results are obtained if the sample is dried from complete saturation
to 2% relative humidity over a time equal to half of each of the periods tested and
allowed to equilibrate at this humidity for a month, then humidity is increased back
to 100% over the same timespan. In this case, faster humidity changes lead to wider
hysteresis curves: figure 7-2
These two tests are simply examples of the conditions that can be simulated with
98
1
Average saturation
0.8
0.6
0.4
0.2
0
0
0.2
0.4
0.6
0.8
1
Instantaneous relative humidity
Figure 7-1: Average saturation as a function of instantaneous relative humidity when
humidity is cycled with a period of 1 month (pink), 1 week (green), 1 day (blue) and
1 hour (red).
99
1
Average saturation
0.8
0.6
0.4
0.2
0
0
0.2
0.4
0.6
0.8
1
Instantaneous relative humidity
Figure 7-2: Average saturation as a function of instantaneous relative humidity when
humidity is cycled with a period of 1 month (pink), 1 week (green), 1 day (blue) and
1 hour (red), with a month’s equilibration at 2% relative humidity between drying
and wetting.
100
this model of transport, sorption and hysteresis.
7.2.2
Saturation-dependent apparent diffusivity
A different approach to the analysis of transport through porous materials has been
to assume that a diffusion equation,
q = −𝐷∇𝑐,
can be written, with an effective diffusivity
𝐷
(7.10)
that depends on
𝑐
[100, 13]. Although
this approach does not offer insight into the physical processes governing transport,
it can be a convenient way of expressing experimental observations. Using equation
7.1, we can write the effective diffusivity as
𝐷=
Using the above value for
𝜅,
the term
𝑐𝜅𝑘𝑇
.
𝑑𝑐
𝜏 𝜇𝑣ℎ 𝑑ℎ
𝜅𝑘𝑇 /(𝜇𝑣)
(7.11)
has value
6 × 10−7
m
2
−1
s .
The
calculated effective diffusivity as a function of saturation for a particular sample of
hardened cement paste [12] is shown in figure 7-3.
7.3 Humidity-dependent permeability
Although this method seems to be a useful approach to modeling transport in a
porous material with sorption hysteresis, there remains the fundamental problem
that it substantially overpredicts transport rates. The intrinsic permeability of hardened cement paste is not around
[9, 174, 184].
10−17
2
m as may be expected, but around
10−20
m
2
It may be that a more detailed treatment of where water is present
and absent in the porous material can correct this discrepancy. I propose here two
stages of development by which this could be incorporated into the transport model.
Although the current version of the model does not include this development, continuing work in the Bazant research group, especially by Tingtao Zhou, is focused on
101
0.5
Effective diffusivity (mm^2/s)
0.4
0.3
Drying
0.2
0.1
Wetting
0
0
0.2
0.4
0.6
0.8
1
Saturation
Figure 7-3:
Calculated effective diffusivity along the wetting (blue) and drying
(brown) isotherms for a sample of hardened cement paste [12].
102
its implementation.
7.3.1
Pore size
The current version of the transport model uses an intrinsic permeability that is, apart
from a simple model for tortuosity, independent of humidity. Since smaller pores fill
at lower humidity than larger pores, this independence is certainly an approximation
and perhaps an unreasonable one. A simple first step towards including the effect of
filling pores of different sizes would be to keep track of the size of pores filled, which
can be determined by applying the model of chapter 5 to the adsorption isotherm.
Taking a weighted average over the full pores, it will then be possible to calculate an
intrinsic permeability.
One immediately apparent consequence of such a technique is that we can expect
higher permeability during drying than wetting, as larger pores remain full due to
pore blocking. The technique is, however, unlikely to fully account for the discrepancy
between predicted and observed intrinsic permeabilities, as a permeability of
10−20
could naively be associated with pores of width smaller than a molecule, clearly an
unreasonable proposition.
7.3.2
Pore connectedness
A potential solution to this difficulty would come from a consideration of the connectedness of pores. Throughout this thesis, pores of various sizes have been considered
without any discussion of the configuration of the solid skeleton that forms these
pores. A proposed model of the structure of hardened cement paste suggests that it
may be thought of as a colloidal or granular material with grains of a few nanometers
in size [79]. In such a case, the first water to condense as humidity is increased will
be located in thin bridges near the contact points of grains.
Such bridges will not
be connected, and if the adsorbed water is held in place by strong forces from the
grain surfaces, there will be no flow. The permeability will thus be much lower than
expected, especially at low humidity.
103
A percolation model will be needed to allow the calculation of permeability of configurations approximating the actual distribution of water. A network with pore sizes
assigned randomly from an appropriate distribution would be insufficient, because
the true porous material most likely enhances the probability that small pores are
disconnected, due to the alternating arrangement of tiny bridges and larger interstices
surrounded by more than two particles.
A better model can be built by beginning with a random packing of particles:
identical spheres are probably sufficient. The Young-Laplace equation can be used
to find a stable configuration of condensed water in the material, as a function of
humidity. Some suitable measure of permeability can then be found on this system.
Below some saturation, which could be found with this model, there will be no
connected path of condensed water spanning the system and hence no possibility of
fluid flow. In this case, Knudsen diffusion in the vapor phase may be the dominant
transport mechanism.
7.4 Inclusion of detailed modeling of hysteresis mechanisms
The transport model of this chapter is largely independent of the sorption model of
chapter 5.
There are simplifications in the model that could be refined using the
sorption model. For example, the transport model assumes at every time that the
trapped water is distributed uniformly across all pore sizes above those filled with
stable water.
In some situations, this is manifestly false.
a system that is uniformly brought from humidity
where
ℎ1 < ℎ2 < ℎ3 ,
filled just below
ℎ2 ,
ℎ2
ℎ3
to
Consider, for example,
ℎ1 ,
and humidity then decreased again from
then returned to
ℎ2 .
Those pores that
will now contain trapped water with probability
while those that fill above
ℎ2
ℎ2 ,
𝑔¯
calculated at
were never refilled during rewetting, and contain
trapped water only with probability
𝑔¯
calculated at
ℎ1 .
This difference is ignored in
the model here. A model could be developed that keeps track of
104
𝑔
as a function of
pore size, and indeed the model of Mualem is set up in this way [112]. For simplicity,
however, I chose not to include this feature, assuming that for many typical applied
humidity protocols it would be unimportant.
105
106
Chapter 8
Conclusions and outlook
8.1 Battery degradation
We have developed a general theoretical framework to model capacity fade and lifetime statistics in rechargeable batteries, focusing on the mechanism of SEI formation
at the anode.We have shown that a very simple single particle reaction model is
sufficient to quantitatively understand SEI formation on graphite, since it provides
a good fit to observed capacity fade. Moreover, computational results with a more
complicated porous electrode model of capacity fade show that no significant spatial
variations form at the cell level.
The temperature dependence of the diffusivity of
the limiting reacting species through SEI can be modeled using an Arrhenius dependence. This model can be used to model the degradation of rechargeable batteries in
a variety of thermal conditions, to guide and interpret accelerated aging experiments,
and to understand the statistics of battery lifetime. The model can also be extended
to account for rapid SEI growth due to area changes and SEI loss, which occur, for
example, in nanostructured silicon anodes.
Although we have focused on SEI formation in Li-ion batteries, some of our models and conclusions may have broader relevance to other battery technologies, not
involving ion intercalation. It is important to emphasize that, although SEI growth
leads to capacity fade, it also plays a crucial role in rechargeable battery engineering.
The formation of a stable SEI layer protects the anode at large negative potentials
107
and allows the design of high-voltage batteries operating outside the âĂIJvoltage windowâĂİ for electrochemical stability of the electrolyte.
The same general principle
enables the stable operation of lead-acid rechargeable batteries (at a voltage of over
2 V), where the half-cell reactions at the lead anode and the lead oxide cathode both
form amorphous lead sulfate films, which allow active ions to pass, while suppressing
water electrolysis outside the voltage window of the sulfuric acid electrolyte (approximately 1 V) [137]. In that case, capacity fade can also occur by irreversible reactions
involving active species via lead sulfate crystallization (“sulfation”) [43, 182, 111, 148].
Although this capacity fade mechanism differs from SEI formation in Li-ion batteries in the microscopic details, the engineering consequences are the same (increased
interfacial resistance and loss of active material), and it may be possible to apply
and extend our models of capacity fade and lifetime prediction to this and other
rechargeable battery systems.
8.2 Sorption and transport in porous materials
The latter portion of this thesis has presented modeling covering various aspects of
sorption in porous media, with particular relevance to water in cement paste. Chapter
5 showed that a simple model can explain sorption and its hysteresis in a range of
porous materials.
This model accounts for some degree of hysteresis at the single-
pore level, as well as the effect of the porous network in suppressing nucleation of
the vapor phase by a liquid-vapor interface. The model allows calculation of the pore
size distribution as well as a measure of the connectedness of the mesopores to larger
pores in the material.
Some more detailed questions related to this model remain open. The assumed
single-pore sorption behavior, in particular the hysteresis between filling and emptying, was validated only for carbon nanotubes, through comparison of the peak in the
pore size distribution found by the model with the nanotube size measured by transmission electron microscopy. In other cases, especially materials with pores of a very
different shape, it may be that a different single-pore isotherm is more appropriate.
108
This would not harm the general principles of the model, but would change the relative contribution of single-pore and network effects to hysteresis, and the calculated
pore size.
Also, the assumed surface adsorption is based on a very simple model, lacking
both interactions between molecules within an adsorbed layer, and interactions of the
surface with molecules beyond the first adsorbed layer. A more accurate quantification
of surface adsorption, which may need to be specific to each sorbate-sorbent pair,
could improve the agreement of the model with experimental data at lower relative
pressure.
The model accounts, in an empirical way, for the existence of fluid in spaces at a
scale smaller than the mesopores in which a continuum description of condensation is
sufficient. This is especially important for water in hardened cement paste. Molecular
simulation techniques such as grand canonical Monte Carlo [25] may be able to explain
the observed quantity of water as a function of humidity, based on first principles.
Even without such an explanation, the recognition of the role of this water enables
substantial progress in the modeling of drying shrinkage, as shown in chapter 6.
Chapter 7 describes a simple model of transport through a porous material that
accounts for sorption hysteresis.
The model is able to capture dynamic hysteresis,
and can be used to simulate arbitrary applied humidity (or more generally, sorbate
partial pressure) conditions.
Although the principles of the model seem sound, it does not yet agree quantitatively with experimental results: specifically, it predicts faster transport of water
through cement paste than is actually observed.
This could be due to the porous
structure producing a disconnected or poorly-connected configuration of water at all
but rather high humidities. I have proposed some ways to extend the model to account
for this.
Other important extensions of the models presented here will be aimed at applying
the models to applications of particular relevance to engineering. An example of such
an application is moisture-buffering materials [119]. These materials take up water
from, and release water to, the atmosphere and moderate humidity changes, and are
109
used in construction for purposes such as to increase comfort or reduce damage to
fragile objects. If the design of such materials is to be optimized, it is important to
understand what factors influence water sorption. Also, a dynamical model that accounts for the link between sorption and transport is needed to assess the effectiveness
of materials in proposed operating conditions.
Another situation where transport modeling is very important is understanding
the degradation of porous materials such as concrete.
Important degradation pro-
cesses in concrete include corrosion of reinforcements [1], reactions between ions dissolved in the pore water and the concrete itself [65], and the freezing and thawing of
pore water [54]. All of these degradation mechanisms rely on the presence of water, so
an understanding of its transport through the material is an important ingredient in
the modeling of degradation. The transport model will need to be extended when it
is not directly the water that causes damages, but ions dissolved in the water. In this
case, convection of ions carried by flowing water as well as diffusion of ions must be
considered, as well as electrostatic effects that may influence the relative prevalence
of ions in pores of different sizes and reactions that consume or produce ions.
Another degradation process is cracking due to differential shrinkage on nonuniform drying [17]. Modeling of this process will require a combination of transport
and shrinkage models such as those presented here. Additionally, since cracking occurs
predominantly during first drying, when the hydration reactions that lead to cement
setting are still proceeding, a complete model must account for the consumption of
water and changes in the pore structure due to chemical reactions.
The complex
interaction of several physical processes is a reason why models that elucidate the
underlying mechanisms are needed, and why scientific and engineering research in
these areas will continue.
110
Bibliography
[1] Abdullah A. Almusallam, Ahmad S. Al-Gahtani, Abdur Rauf Aziz, Fahd H.
Dakhil, and Rasheeduzzafar. Effect of reinforcement corrosion on flexural behavior of concrete slabs.
127, 1996.
Journal of Materials in Civil Engineering
, 8(3):123–
[2] K Amine, J Liu, S Kang, I Belharouak, Y Hyung, D Vissers, and G Henriksen.
Improved lithium manganese oxide spinel/graphite Li-ion cells for high-power
applications.
Journal of Power Sources
, 129(1):14–19, April 2004.
[3] A. A. Anani, S. Crouch-Baker, and R. A. Huggins. Investigation of a Ternary
Lithium Alloy Mixed-Conducting Matrix Electrode at Ambient Temperature.
Journal of the Electrochemical Society: Solid-State Science and Technology
,
56(12):2103–2105, 1988.
[4] D Andelman. Electrostatic properties of membranes: the Poisson-Boltzmann
theory.
Handbook of biological physics
, 1:603–641, 1995.
[5] M. Armand and J.-M. Tarascon. Building better batteries.
2008.
Nature
, 451:652–657,
[6] Pankaj Arora, Ralph E White, and Marc Doyle. Capacity Fade Mechanisms
and Side Reactions in Lithium-Ion Batteries.
Society
, 145(10):3647–3667, 1998.
Journal of The Electrochemical
[7] Peng Bai, Daniel A. Cogswell, and Martin Z. Bazant.
Suppression of phase
separation in LiFePO4 nanoparticles during battery discharge.
11(11):4890–6, November 2011.
Nano Letters
,
[8] D. H. Bangham and R. I. Razouk. The wetting of charcoal and the nature of the
adsorbed phase formed from saturated vapours.
Society
, 33(D):1463, 1937.
Transactions of the Faraday
[9] Nemkumar Banthia and Sidney Mindess. Water permeability of cement paste.
Cement and Concrete Research
, 19:727–736, 1989.
[10] V Baranchugov, E Markevich, and E Pollak. Amorphous silicon thin films as
a high capacity anodes for Li-ion batteries in ionic liquid electrolytes.
chemistry Communications
, 9:796–800, 2007.
111
Electro-
[11] Grigory Isaakovich Barenblatt, Jesús Garcia Azorero, Arturo De Pablo, and
Juan Luis Vazquez. Mathematical model of the non equilibrium water oil displacement in porous strata.
[12] Véronique Baroghel-Bouny.
Applicable Analysis
, 65(1-2):19–45, June 1997.
Water vapour sorption experiments on hardened
cementitious materials part I: essential tool for analysis of hygral behaviour and
its relation to pore structure.
March 2007.
[13] Véronique Baroghel-Bouny.
Cement and Concrete Research
, 37(3):414–437,
Water vapour sorption experiments on hardened
cementitious materials. Part II: Essential tool for assessment of transport properties and for durability prediction.
454, March 2007.
Cement and Concrete Research
, 37(3):438–
[14] Elliott P. Barrett, Leslie G. Joyner, and Paul P. Halenda. The determination of
pore volume and area distributions in porous substances. I. Computations from
Journal of the American Chemical Society
nitrogen isotherms.
, 73:373–380,
1951.
[15] Neil Bartlett and B. W. McQuillan. Graphite Chemistry. In M Stanley Whittingham and Allan J. Jacobson, editors,
page 31. Academic Press, New York, 1982.
Intercalation Chemistry
, chapter 2,
[16] Zdeněk P. Bažant and Martin Z. Bazant.
Theory of sorption hysteresis in
Journal of the Mechanics
nanoporous solids: Part I Snap-through instabilities.
and Physics of Solids
, 60(9):1644–1659, May 2012.
[17] Zdenek P. Bazant and Warren J. Raftshol.
shrinkage specimens.
Effect of cracking in drying and
Cement and Concrete Research
, 12:209–226, 1982.
[18] F. Benboudjema, F. Meftah, and J.M. Torrenti.
Interaction between drying,
shrinkage, creep and cracking phenomena in concrete.
27(2):239–250, January 2005.
Engineering Structures
,
[19] Dale P. Bentz, Edward J. Garboczi, and Daniel A. Quenard. Modelling drying
shrinkage in reconstructed porous materials: Application to porous Vycor glass.
Modelling and Simulation in Materials Science and Engineering
, 6:211–236,
1998.
[20] Luca Bertolini, Bernhard Elsener, Pietro Pedeferri, and Rob Polder.
of Steel in Concrete Prevention, Diagnosis, Repair
.
Corrosion
Wiley-VCH, Weinheim,
2004.
[21] J. O. Besenhard, J. Yang, and M. Winter. Will advanced lithium-alloy anodes
have a chance in lithium-ion batteries?
September 1997.
Journal of Power Sources
, 68(1):87–90,
[22] Edgar M Blokhuis and Joris Kuipers. Thermodynamic expressions for the Tolman length.
The Journal of chemical physics
, 124(7):74701, February 2006.
112
[23] P. A. Bonnaud, B. Coasne, and R. J.-M. Pellenq.
Molecular simulation of
Journal of Physics: Condensed Matter
water confined in nanoporous silica.
22(28):284110, July 2010.
,
[24] P. A. Bonnaud, Q Ji, B Coasne, R. J.-M. Pellenq, and K. J. Van Vliet. Thermodynamics of water confined in porous calcium-silicate-hydrates.
28(31):11422–32, August 2012.
[25] Patrick A. Bonnaud, Qing Ji, and Krystyn J. Van Vliet.
Langmuir
,
Effects of elevated
temperature on the structure and properties of calcium-silicate-hydrate gels:
the role of confined water.
Soft Matter
, 9(28):6418, 2013.
[26] Gerardine G. Botte, Bradley A. Johnson, and Ralph E. White.
Influence of
Some Design Variables on the Thermal Behavior of a Lithium-Ion Cell.
of The Electrochemical Society
, 146(3):914–923, 1999.
[27] B. A. Boukamp, G. C. Lesh, and R. A. Huggins.
trodes with Mixed-Conductor Matrix.
128(4):725, 1981.
Journal
All-Solid Lithium Elec-
Journal of The Electrochemical Society
,
[28] D. F. Brewer and D. C. Champeney. Sorption of helium and nitrogen on Vycor
porous glass.
Proceedings of the Physical Society
, 79(855-868), 1962.
[29] J. C. P. Broekhoff and J. H. De Boer. Studies on Pore Systems in Catalysts IX.
Calculation of pore distributions from the adsorption branch of nitrogen sorption isotherms in the case of open cylindrical pores A. Fundamental equations.
Journal of Catalysis
, 9:8–14, 1967.
[30] M Broussely,
S Herreyre,
P Biensan,
P Kasztejna,
K Nechev,
and R.J
Staniewicz. Aging mechanism in Li ion cells and calendar life predictions.
nal of Power Sources
, 97-98:13–21, July 2001.
Jour-
[31] D. A. G. Bruggeman. Berechnung verschiedener physikalischer Konstanten von
heterogenen Substanzen. I. Dielektrizitätskonstanten und Leitfähigkeiten der
Mischkörper aus isotropen.
Annalen der Physik
, 416(7):636–664, 1935.
[32] Stephen Brunauer, P. H. Emmett, and Edward Teller. Adsorption of gases in
multimolecular layers.
1938.
Journal of the American Chemical Society
, 60:309–319,
[33] H. Bryngelsson, M. Stjerndahl, T. Gustafsson, and K. Edström. How dynamic
is the SEI?
Journal of Power Sources
, 174(2):970–975, December 2007.
[34] P. C. Carman and F. a. Raal. Physical Adsorption of Gases on Porous Solids.
Proceedings of the Royal
Society A: Mathematical, Physical and Engineering Sciences
I. Comparison of Loose Powders and Porous Plugs.
, 209(1096):59–69,
October 1951.
113
[35] Benoit Carrier, Linlin Wang, Matthieu Vandamme, Roland J-M Pellenq, Michel
Bornert, Alexandre Tanguy, and Henri Van Damme.
humidity-induced swelling of clay film.
2013.
ESEM study of the
Langmuir
, 29(41):12823–33, October
[36] J. M. Cases, I. Berend, M. Francois, J. P. Uriot, L. J. Michot, and F. Thomas.
Mechanism of adsorption and desorption of water vapor by homoionic montmorillonite; 3, The Mg2+, Ca2+, and Ba3+ exchanged forms.
Minerals
, 45(1):8–22, 1997.
Clays and Clay
[37] Candace K. Chan, Hailin Peng, Gao Liu, Kevin McIlwrath, Xiao Feng Zhang,
Robert A. Huggins, and Yi Cui. High-performance lithium battery anodes using
silicon nanowires.
Nature Nanotechnology
, 3(1):31–35, January 2008.
[38] Candace K. Chan, Riccardo Ruffo, Seung Sae Hong, and Yi Cui. Surface chemistry and morphology of the solid electrolyte interphase on silicon nanowire
lithium-ion battery anodes.
2009.
Journal of Power Sources
, 189(2):1132–1140, April
[39] Sunho Choi, Jeffrey H. Drese, and Christopher W. Jones. Adsorbent materials for carbon dioxide capture from large anthropogenic point sources.
SusChem
, 2(9):796–854, January 2009.
Chem-
[40] Daniel A Cogswell and Martin Z Bazant. Coherency strain and the kinetics of
ACS Nano
phase separation in LiFePO4 nanoparticles.
, 6(3):2215–25, March
2012.
[41] Daniel A. Cogswell and Martin Z. Bazant.
phase-separating nanoparticles.
Nano lLtters
Theory of coherent nucleation in
, 13(7):3036–3041, July 2013.
[42] Leonard H. Cohan. Sorption hysteresis and the vapor pressure of concave surfaces.
Journal of the American Chemical Society
, 60(2):433–435, 1938.
[43] A Cooper.
Development of a lead-acid battery for a hybrid electric vehicle.
Journal of Power Sources
, 133(1):116–125, May 2004.
[44] Salomon Cordero, Fernando Rojas, Isaac Kornhauser, Marcos Esparza, and
Giorgio Zgrablich. Menisci interactions during adsorption on mesoporous materials: Evaluation of delayed and advanced adsorption.
2005.
[45] Li-Feng Cui, Yuan Yang, Ching-Mei Hsu, and Yi Cui.
Adsorption
, 11:91–96,
Carbon-Silicon Core-
Shell Nanowires as High Capacity Electrode for Lithium Ion Batteries.
Letters
, 9(9):3370–3374, 2009.
[46] J. R. Dahn, D. C. Dahn, and R. R. Haering.
intercalation compounds.
1982.
Elastic energy and staging in
Solid State Communications
114
Nano
, 42(3):179–183, April
[47] S. Dargaville and T. W. Farrell.
Predicting Active Material Utilization in
LiFePO4 Electrodes Using a Multiscale Mathematical Model.
Electrochemical Society
, 157(7):A830–A840, 2010.
Journal of The
[48] Marc Doyle, TF Fuller, and John Newman. Modeling of galvanostatic charge
and discharge of the lithium/polymer/insertion cell.
chemical Society
, 140(6):1526–1533, 1993.
[49] M. S. Dresselhaus and G Dresselhaus.
Advances in Physics
Journal of the Electro-
Intercalation compounds of graphite.
, 30(2):139–326, 1981.
[50] Bruce Dunn, Haresh Kamath, and Jean-Marie Tarascon. Electrical energy storage for the grid: a battery of choices.
35, November 2011.
Science (New York, N.Y.)
, 334(6058):928–
[51] Kristina Edström, Marie Herstedt, and Daniel P. Abraham. A new look at the
solid electrolyte interphase on graphite anodes in Li-ion batteries.
Power Sources
, 153(2):380–384, February 2006.
Journal of
[52] Seung-Wook Eom, Min-Kyu Kim, Ick-Jun Kim, Seong-In Moon, Yang-Kook
Sun, and Hyun-Soo Kim. Life prediction and reliability assessment of lithium
secondary batteries.
Journal of Power Sources
, 174(2):954–958, December 2007.
[53] Merlin A. Etzold, Peter J. McDonald, and Alexander F. Routh.
Growth of
Cement and
sheets in 3D confinements - a model for the C-S-H meso structure.
Concrete Research
, 63:137–142, September 2014.
[54] G Fagerlund.
The critical degree of saturation method of assessing the
Materials and Structures
freeze/thaw resistance of concrete.
, 10(58):217–229,
1977.
[55] P. Faucon, F. Adenot, J. F. Jacquinot, J. C. Petit, R. Cabrillac, and M. Jorda.
Long-term behaviour of cement pastes used for nuclear waste disposal: review
of physico-chemical mechanisms of water degradation.
Research
, 28(6):847–857, 1998.
[56] R. F. Feldman.
Cement and Concrete
Helium flow characteristics of rewetted specimens of dried
hydrated Portland cement paste.
1973.
[57] R. F. Feldman and P. J. Sereda.
Cement and Concrete Research
, 3:777–790,
Sorption of Water on Compacts of Bottle-
Hydrated Cement. II. Thermodynamic Considerations and Theory of Volume
Change.
Journal of Applied Chemistry
, 14:93–104, 1964.
[58] R. F. Feldman and P. J. Sereda. A model for hydrated Portland cement paste
as deduced from sorption-length change and mechanical properties.
et Construction
, 1(6):509–520, 1968.
115
Matériaux
[59] Rolf F. Feldman. Sorption and length-change scanning isotherms of methanol
and water on hydrated Portland cement. In
Chemistry of Cement, Volume 3
V International Symposium on the
, pages 53–66, 1968.
[60] T. R. Ferguson and M. Z. Bazant. Nonequilibrium Thermodynamics of Porous
Electrodes.
tober 2012.
Journal of The Electrochemical Society
, 159(12):A1967–A1985, Oc-
[61] Elzbieta Frackowiak and François Béguin.
chemical storage of energy in capacitors.
Carbon materials for the electro-
Carbon
, 39(6):937–950, May 2001.
[62] Thomas F. Fuller, Marc Doyle, and John Newman. Simulation and Optimization of the Dual Lithium Ion Insertion Cell.
Society
, 141(1):1, 1994.
Journal of The Electrochemical
[63] Anne Galarneau, Helene Cambon, Francesco Di Renzo, and Francois Fajula.
True microporosity and surface area of mesoporous SBA-15 silicas as a function
of synthesis temperature.
Langmuir
, 17(26):8328–8335, 2001.
[64] E. J. Garboczi and D. P. Bentz.
cement-based materials.
Computer simulation of the diffusivity of
Journal of Materials Science
, 27(8):2083–2092, 1992.
[65] Fredrik P. Glasser, Jacques Marchand, and Eric Samson. Durability of concrete
- Degradation phenomena involving detrimental chemical reactions.
and Concrete Research
, 38(2):226–246, February 2008.
[66] B. V. Gnedenko, Yu. K. Belyayev, and A. D. Solovyev.
of Reliability Theory
. Academic Press, New York, 1969.
Cement
Mathematical Methods
[67] Radu Gogoana, Matthew B. Pinson, Martin Z. Bazant, and Sanjay E. Sarma.
Internal Resistance Matching for Parallel-Connected Lithium-Ion Cells and Impacts on Battery Pack Cycle Life.
Journal of Power Sources
, 252:8–13, 2014.
[68] R. Gonzalez-Teresa, J. S. Dolado, A. Ayuela, and Jean-Christophe Gimel.
Nanoscale texture development of C-S-H gel: A computational model for nucleation and growth.
Applied Physics Letters
, 103(23):234105, 2013.
[69] R. González-Teresa, Víctor Morales-Florez, Hegoi Manzano, and Jorge S.
Dolado.
Modelos estructurales del empaquetamiento aleatorio de partículas
esféricas de Tobermorita: una aproximación computacional sencilla.
de Construcción
, 60(298):7–15, May 2010.
Materiales
[70] Johan C Groen, Louk A. A. Peffer, and Javier Perez-Ramirez. Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in
gas adsorption data analysis.
June 2003.
Microporous and Mesoporous Materials
116
, 60:1–17,
[71] Simon Gruener, Tommy Hofmann, Dirk Wallacher, Andriy Kityk, and Patrick
Huber.
Capillary rise of water in hydrophilic nanopores.
79(6):067301, June 2009.
Physical Review E
,
[72] W. A. Gutteridge and L. J. Parrott. A study of the changes in weight, length
and interplanar spacing induced by drying and rewetting synthetic CSH (I).
Cement and Concrete Research
, 6:357–366, 1976.
[73] R. S. Haines and R. McIntosh.
Length Changes of Activated Carbon Rods
Caused by Adsorption of Vapors.
1947.
The Journal of Chemical Physics
, 15(1):28,
[74] Will Hansen. Drying shrinkage mechanisms in Portland cement paste.
of the American Ceramic Society
, 70(5):323–328, 1987.
Journal
[75] Stephen J. Harris, Adam Timmons, Daniel R. Baker, and Charles Monroe.
Direct in situ measurements of Li transport in Li-ion battery negative electrodes.
Chemical Physics Letters
, 485(4-6):265–274, January 2010.
[76] Wei He, Nicholas Williard, Michael Osterman, and Michael Pecht.
Prognos-
tics of lithium-ion batteries based on Dempster-Shafer theory and the Bayesian
Monte Carlo method.
ber 2011.
Journal of Power Sources
, 196(23):10314–10321, Decem-
[77] Katerina Ioannidou, Roland J-M Pellenq, and Emanuela Del Gado. Controlling local packing and growth in calcium-silicate-hydrate gels.
10(8):1121–1133, February 2014.
Soft Matter
,
[78] F. Javadpour, D. Fisher, and M. Unsworth. Nanoscale Gas Flow in Shale Gas
Sediments.
2007.
Journal of Canadian Petroleum Technology
, 46(10):55–61, October
[79] Hamlin M. Jennings. Refinements to colloid model of C-S-H in cement: CM-II.
Cement and Concrete Research
, 38(3):275–289, March 2008.
[80] Liwen Ji, Honghe Zheng, Ariel Ismach, Zhongkui Tan, Shidi Xun, Eric Lin,
Vincent Battaglia, Venkat Srinivasan, and Yuegang Zhang. Graphene/Si multilayer structure anodes for advanced half and full lithium-ion cells.
1(1):164–171, January 2012.
Nano Energy
,
[81] Uday Kasavajjula, Chunsheng Wang, and A. John Appleby. Nano- and bulksilicon-based insertion anodes for lithium-ion secondary cells.
Sources
, 163:1003–1039, 2007.
[82] Shinichi Komaba, Naoaki Kumagai, and Yoichi Kataoka.
Journal of Power
Influence of man-
ganese(II), cobalt(II), and nickel(II) additives in electrolyte on performance of
graphite anode for lithium-ion batteries.
February 2002.
117
Electrochimica Acta
, 47(8):1229–1239,
[83] J. B. Kool and J. C. Parker. Development and evaluation of closed-form expressions for hysteretic soil hydraulic properties.
23(1):105–114, 1987.
Water Resources Research
,
[84] Robert Kostecki, Jinglei Lei, Frank McLarnon, Joongpyo Shim, and Kathryn
Striebel.
Diagnostic Evaluation of Detrimental Phenomena in High-Power
Lithium-Ion Batteries.
2006.
Journal of The Electrochemical Society
, 153(4):A669,
[85] M Kruk, M Jaroniec, and A Sayari. Application of large pore MCM-41 molecular sieves to improve pore size analysis using nitrogen adsorption measurements.
Langmuir
, 7463(12):6267–6273, 1997.
[86] Michal Kruk and Mietek Jaroniec.
Argon Adsorption at 77 K as a Useful
Tool for the Elucidation of Pore Connectivity in Ordered Materials with Large
Chemistry of Materials
Cagelike Mesopores.
[87] Irving Langmuir.
, 15(15):2942–2949, July 2003.
The constitution and fundamental properties of solids and
liquids. Part I. Solids.
1917.
Journal of the Franklin Institute
, 38(11):2221–2295,
[88] M. Lazzari. A Cyclable Lithium Organic Electrolyte Cell Based on Two Intercalation Electrodes.
Journal of The Electrochemical Society
, 127(3):773, 1980.
[89] Jia-Liang Le and Zdeněk P. Bažant. Unified nano-mechanics based probabilistic
theory of quasibrittle and brittle structures: II. Fatigue crack growth, lifetime
and scaling.
July 2011.
Journal of the Mechanics and Physics of Solids
, 59(7):1322–1337,
[90] S Lee, H Miyazaki, Sd Mahanti, and Sa Solin. Composition-driven c-axis expansion of intercalated layered solids: 1D non-Vegard’s-law behavior in a 2D
solid solution.
Physical Review Letters
, 62(26):3066–3069, June 1989.
[91] Yi An Lei, Tikhon Bykov, Soohaeng Yoo, and Xiao Cheng Zeng. The Tolman
length: is it positive or negative?
127(44):15346–7, November 2005.
Journal of the American Chemical Society
,
[92] J.-C. Li, D. K. Ross, and M. J. Benham. Small-angle neutron scattering studies
of water and ice in porous Vycor glass.
pages 794–802, 1991.
Journal of Applied Crystallography
,
[93] Dongqiang Liu, Jiantao Han, Martin Dontigny, Patrick Charest, Abdelbast
Guerfi, Karim Zaghib, and John B. Goodenough. Redox Behaviors of Ni and
Cr with Different Counter Cations in Spinel Cathodes for Li-Ion Batteries.
Journal of The Electrochemical Society
, 157(7):A770, 2010.
[94] Hailing Liu and N. A. Seaton. Determination of the connectivity of porous solids
from nitrogen sorption measurements-III. Solids containing large mesopores.
Chemical Engineering Science
, 49(11):1869–1878, 1994.
118
[95] Hailing Liu, Lin Zhang, and Nigel A Seaton. Sorption Hysteresis as a Probe of
Pore Structure.
Langmuir
, 9:2576–2582, 1993.
[96] Nian Liu, Liangbing Hu, Matthew T McDowell, Ariel Jackson, and Yi Cui.
Prelithiated silicon nanowires as an anode for lithium ion batteries.
5(8):6487–93, August 2011.
[97] L. Luckner, M. Th. van Genuchten, and D. R. Nielsen.
ACS Nano
,
A consistent set of
parametric models for the two-phase flow of immiscible fluids in the subsurface.
Water Resources Research
, 25(10):2187–2193, 1989.
[98] William D. Machin. Temperature dependence of hysteresis and the pore size
distributions of two mesoporous adsorbents.
1994.
Langmuir
, (May 1993):1235–1240,
[99] Hegoi Manzano, Enrico Masoero, Iñigo Lopez-Arbeloa, and Hamlin M. Jennings. Shear deformations in calcium silicate hydrates.
2013.
[100] Nicos S. Martys and Chiara F. Ferraris.
concrete.
Cement and Concrete Research
Soft Matter
, 9(30):7333,
Capillary transport in mortars and
, 27(5):747–760, 1997.
[101] E. Masoero, E. Del Gado, R. J.-M. Pellenq, F.-J. Ulm, and S. Yip. Nanostructure and Nanomechanics of Cement: Polydisperse Colloidal Packing.
Review Letters
, 109(15):155503, October 2012.
Physical
[102] Enrico Masoero, Emanuela Del Gado, Roland J.-M. Pellenq, Sidney Yip, and
Franz-Josef Ulm. Nano-scale mechanics of colloidal C-S-H gels.
10(3):491–499, January 2014.
Soft Matter
,
[103] G Mason. A model of adsorption-desorption hysteresis in which hysteresis is
primarily developed by the interconnections in a network of pores.
of the Royal Society of London. A.
, 390:47–72, 1983.
[104] G. Mason.
Proceedings
Determination of the Pore-Size Distributions and Pore-Space In-
terconnectivity of Vycor Porous Glass from Adsorption-Desorption Hysteresis
Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences
Capillary Condensation Isotherms.
, 415(1849):453–486, February 1988.
[105] Geoffrey Mason.
The effect of pore space connectivity on the hysteresis of
capillary condensation in adsorption-desorption isotherms.
and Interface Science
, 88(1):36–46, July 1982.
Journal of Colloid
[106] K. Ulrich Mayer, Emil O. Frind, and David W. Blowes. Multicomponent reactive transport modeling in variably saturated porous media using a generalized formulation for kinetically controlled reactions.
38(9):13–1–13–21, September 2002.
119
Water Resources Research
,
[107] T. Mazumdar, S. Mazumder, and D. Sen.
Temporal evolution of mesoscopic
Physical
structure of some non-Euclidean systems using a Monte Carlo model.
Review B
, 83(10):104302, March 2011.
[108] Fabrice Merlin, Helene Lombois, Stephane Joly, Nicolas Lequeux, Jean-Louis
Halary, and Henri Van Damme. Cement-polymer and clay-polymer nano- and
meso-composites:
spotting the difference.
12(11):3308–3315, October 2002.
[109] Yuriy V. Mikhaylik and James R. Akridge.
Li/S Battery System.
2004.
[110] Alan Millner.
Journal of Materials Chemistry
,
Polysulfide Shuttle Study in the
Journal of The Electrochemical Society
, 151(11):A1969,
Modeling Lithium Ion battery degradation in electric vehicles.
2010 IEEE Conference on Innovative Technologies for an Efficient and Reliable
Electricity Supply
, pages 349–356, September 2010.
[111] Patrick T. Moseley. High rate partial-state-of-charge operation of VRLA batteries.
Journal of Power Sources
[112] Yechezkel Mualem.
Science
, 127(1-2):27–32, March 2004.
A modified dependent-domain theory of hysteresis.
, 137(5):283–291, 1984.
Soil
[113] A. C. A. Muller, K. L. Scrivener, A. M. Gajewicz, and P. J. McDonald. Use of
bench-top NMR to measure the density, composition and desorption isotherm
of C-S-H in cement paste.
September 2013.
Microporous and Mesoporous Materials
, 178:99–103,
[114] Arnaud C. A. Muller, Karen L. Scrivener, Agata M. Gajewicz, and Peter J.
McDonald. Densification of CâĂŞSâĂŞH Measured by 1 H NMR Relaxometry.
The Journal of Physical Chemistry C
, 117(1):403–412, January 2013.
[115] Reinhard Nesper. Structure and chemical bonding in Zintl-phases containing
lithium.
Progress in Solid State Chemistry
, 20:1–45, 1990.
[116] C. M. Neubauer, E. J. Garboczi, and H. M. Jennings. The use of digital images to determine deformation throughout a microstructure Part I Deformation
mapping technique.
Journal of Materials Science
Electrochemical Systems
, 35:5741–5749, 2000.
[117] John Newman and Karen E. Thomas-Alyea.
. Wiley-
Interscience, Berkeley, third ed edition, 2004.
[118] Gang Ning and Branko N. Popov. Cycle Life Modeling of Lithium-Ion Batteries.
Journal of The Electrochemical Society
, 151(10):A1584–A1591, 2004.
[119] Olalekan F. Osanyintola and Carey J. Simonson. Moisture buffering capacity
of hygroscopic building materials: Experimental facilities and energy impact.
Energy and Buildings
, 38(10):1270–1282, October 2006.
120
[120] K Ozawa.
trodes:
Lithium-ion rechargeable batteries with LiCoO2 and carbon elec-
the LiCoO2/C system.
1994.
Solid State Ionics
, 69(3-4):212–221, August
[121] J. H. Page, J. Liu, B. Abeles, E. Herbolzheimer, H. W. Deckman, and D. A.
Weitz. Adsorption and desorption of a wetting fluid in Vycor studied by acoustic
and optical techniques.
Physical Review E
, 52(3):2763–2777, 1995.
[122] Jong In Park, Jung Won Park, Minho Jung, Yang Hyun Huh, and Suk Joo
Bae. Cycle-life Test Time Reduction in Secondary Rechargeable Batteries by
Journal of the Society of Korea
Combining Different Types of Acceleration.
Industrial and Systems Engineering
, 31(4):153–161, 2008.
[123] Mi-Hee Park, Min Gyu Kim, Jaebum Joo, Kitae Kim, Jeyoung Kim, Soonho
Ahn, Yi Cui, and Jaephil Cho. Silicon nanotube battery anodes.
9(11):3844–7, November 2009.
Nano Letters
,
[124] J. C. Parker and R. J. Lenhard. A model for hysteretic constitutive relations
governing multiphase flow: 1. Saturation-pressure relations.
Research
, 23(12):2187–2196, 1987.
Water Resources
[125] M. Parlar and Y. C. Yortsos. Percolation theory of vapor adsorption-desorption
Journal of colloid and interface science
processes in porous materials.
124(1):162–176, 1988.
,
[126] L. J. Parrott. The effect of moisture content upon the elasticity of hardened
Magazine of Concrete Research
cement paste.
, 25(82):17–20, March 1973.
[127] E. Peled, C. Menachem, D. Bar-Tow, and A. Melman.
Anode for Lithium-Ion Batteries.
143(1):L4–L7, 1996.
Improved Graphite
Journal of The Electrochemical Society
,
[128] R. J.-M. Pellenq, J. M. Caillol, and A. Delville. Electrostatic attraction between
two charged surfaces: a (N, V, T) Monte Carlo simulation.
Physical Chemistry B
, 101:8584–8594, 1997.
The Journal of
[129] R. J.-M. Pellenq, N. Lequeux, and H. van Damme. Engineering the bonding
scheme in C-S-H: The iono-covalent framework.
38(2):159–174, February 2008.
Cement and Concrete Research
,
[130] Roland J.-M. Pellenq, Akihiro Kushima, Rouzbeh Shahsavari, Krystyn J. Van
Vliet, Markus J. Buehler, Sidney Yip, and Franz-Josef Ulm. A realistic molecular model of cement hydrates.
Proceedings of the National Academy of Sciences
,
106(38):16102–16107, September 2009.
[131] Scott B. Peterson, Jay Apt, and J.F. Whitacre. Lithium-ion battery cell degradation resulting from realistic vehicle and vehicle-to-grid utilization.
Power Sources
, 195(8):2385–2392, April 2010.
121
Journal of
[132] Matthew B. Pinson and Martin Z. Bazant.
Theory of SEI Formation in
Rechargeable Batteries: Capacity Fade, Accelerated Aging and Lifetime Prediction.
2013.
Journal of the Electrochemical Society
, 160(2):A243–A250, December
[133] Matthew B. Pinson, Hamlin M. Jennings, and Martin Z. Bazant.
Sorption Hysteresis in Multiscale Porous Media.
In preparation
Theory of
, 2014.
[134] Matthew B. Pinson, Enrico Masoero, Patrick A. Bonnaud, Hegoi Manzano,
Qing Ji, Sidney Yip, Jeffrey J. Thomas, Martin Z. Bazant, Krystyn J. Van
Vliet, and Hamlin M. Jennings.
Hysteresis from multiscale porosity:
sorption and shrinkage in cement paste.
In preparation
water
, 2014.
[135] Harry J. Ploehn, Premanand Ramadass, and Ralph E. White. Solvent Diffusion
Model for Aging of Lithium-Ion Battery Cells.
Society
, 151(3):A456, 2004.
Journal of The Electrochemical
[136] T. C. Powers. Structure and physical properties of hardened Portland cement
paste.
Journal of the American Ceramic Society
Electrochemical Engineering Principles
, 41(1):1–6, 1958.
[137] Geoffrey Prentice.
. Prentice Hall, En-
glewood Cliffs, 1991.
[138] V. S. Ramachandran, R. F. Feldman, and J. J. Beaudoin.
Heyden & Son Ltd, London, 1981.
Concrete Science
.
[139] P. Ramadass, Bala Haran, Parthasarathy M. Gomadam, Ralph White, and
Branko N. Popov.
Li-Ion Cells.
Development of First Principles Capacity Fade Model for
Journal of The Electrochemical Society
, 151(2):A196, 2004.
[140] H. Ranaivomanana, J. Verdier, A. Sellier, and X. Bourbon.
Toward a bet-
ter comprehension and modeling of hysteresis cycles in the water sorptiondesorption process for cement based materials.
41(8):817–827, August 2011.
Cement and Concrete Research
,
[141] Peter I. Ravikovitch and Alexander V. Neimark. Experimental confirmation of
different mechanisms of evaporation from ink-bottle type pores: equilibrium,
pore blocking, and cavitation.
Langmuir
, (10):9830–9837, 2002.
[142] Peter I. Ravikovitch and Alexander V. Neimark.
model of adsorption deformation.
2006.
[143] I. G. Richardson.
Langmuir
Density functional theory
, 22(26):10864–10868, December
Tobermorite/jennite- and tobermorite/calcium hydroxide-
based models for the structure of C-S-H: applicability to hardened pastes of
tricalcium silicate,
𝛽 -dicalcium
silicate, Portland cement, and blends of Port-
land cement with blast-furnace slag, metakaol.
34(9):1733–1777, September 2004.
122
Cement and Concrete Research
,
[144] I.G. Richardson. The calcium silicate hydrates.
38(2):137–158, February 2008.
Cement and Concrete Research
,
[145] F. Rojas, I. Kornhauser, C. Felipe, J. M. Esparza, S. Cordero, A. Domínguez,
and J. L. Riccardo. Capillary condensation in heterogeneous mesoporous networks consisting of variable connectivity and pore-size correlation.
Chemistry Chemical Physics
, 4(11):2346–2355, May 2002.
Physical
[146] Peng Rong and Massoud Pedram. An analytical model for predicting the remaining battery capacity of lithium-ion batteries.
Large Scale Integration (VLSI) Systems
IEEE Transactions on Very
, 14(5):441–451, 2006.
[147] Thomas Rougelot, Frédéric Skoczylas, and Nicolas Burlion. Water desorption
and shrinkage in mortars and cement pastes: Experimental study and poromechanical model.
Cement and Concrete Research
, 39(1):36–44, January 2009.
[148] Paul Ruetschi. Aging mechanisms and service life of lead-acid batteries.
of Power Sources
, 127:33–44, March 2004.
Journal
[149] E. Samson, J. Marchand, and J. J. Beaudoin. Modeling the influence of chemical
reactions on the mechanisms of ionic transport in porous materials: an overview.
Cement and Concrete Research
, 30(2000):1895–1902, 2000.
[150] Julien Sanahuja, Luc Dormieux, and Gilles Chanvillard. Modelling elasticity of
a hydrating cement paste.
October 2007.
Cement and Concrete Research
, 37(10):1427–1439,
[151] Bruno Scrosati. Lithium rocking chair batteries: An old concept?
The Electrochemical Society
, 139(10):2776–2781, 1992.
Journal of
[152] N. A. Seaton. Determination of the connectivity of porous solids from nitrogen sorption measurements.
January 1991.
Chemical Engineering Science
, 46(8):1895–1909,
[153] J. Shim, R. Kostecki, T. Richardson, X. Song, and K. A. Striebel. Electrochemical analysis for cycle performance and capacity fading of a lithium-ion battery
cycled at elevated temperature.
Journal of Power Sources
, 112:222–230, 2002.
[154] Joongpyo Shim, Kathryn A. Striebel, and Elton J. Cairns. The Lithium/Sulfur
Rechargeable Cell.
A1325, 2002.
[155] Steven Shimizu.
Journal of The Electrochemical Society
, 149(10):A1321–
Exploring Transport, Adsorption and Phase Behavior in
Nanoporous Materials Final Progress Report. Technical report, Massachusetts
Institute of Technology, Cambridge, 2014.
[156] C. G. Shull. The determination of pore size distribution from gas adsorption
data.
Journal of the American Chemical Society
, 70(4):1405–1410, 1948.
123
[157] Godfrey Sikha, Branko N. Popov, and Ralph E. White. Effect of Porosity on
the Capacity Fade of a Lithium-Ion Battery.
Society
, 151(7):A1104, 2004.
Journal of The Electrochemical
[158] Jiri Simunek and Donald L. Suarez. Two-dimensional transport model for variably saturated porous media with major ion chemistry.
search
, 30(4):1115–1133, 1994.
Water Resources Re-
[159] A. J. Smith, J. C. Burns, D. Xiong, and J. R. Dahn. Interpreting High Precision
Journal of The Electrochemical Society
Coulometry Results on Li-ion Cells.
,
158(10):A1136, 2011.
[160] A. J. Smith, J. C. Burns, Xuemei Zhao, Deijun Xiong, and J. R. Dahn. A High
Precision Coulometry Study of the SEI Growth in Li/Graphite Cells.
of The Electrochemical Society
, 158(5):A447, 2011.
Journal
[161] Venkat Srinivasan and John Newman. Discharge Model for the Lithium IronPhosphate Electrode.
2004.
Journal of The Electrochemical Society
, 151(10):A1517,
[162] K. A. Striebel, J. Shim, E. J. Cairns, R. Kostecki, Y.-J. Lee, J. Reimer, T. J.
Richardson, P. N. Ross, X. Song, and G. V. Zhuang.
Diagnostic Analysis of
Electrodes from High-Power Lithium-Ion Cells Cycled under Different Conditions.
Journal of The Electrochemical Society
, 151(6):A857–A866, 2004.
[163] Peter T. Tanev and Lyubomir T. Vlaev. An attempt at a more precise evaluation of the approach to mesopore size distribution calculations depending on the
degree of pore blocking.
1993.
Journal of Colloid and Interface Science
, 160:110–116,
[164] J.-M. Tarascon and M. Armand.
lithium batteries.
Nature
Issues and challenges facing rechargeable
, 414:359–367, 2001.
[165] Ken Tasaki, Alex Goldberg, Jian-Jie Lian, Merry Walker, Adam Timmons,
and Stephen J. Harris.
Solubility of Lithium Salts Formed on the Lithium-
Ion Battery Negative Electrode Surface in Organic Solvents.
Electrochemical Society
Cement Chemistry
, 156(12):A1019, 2009.
[166] H. F. W. Taylor.
Journal of The
. Thomas Telford, London, 2nd edition,
1997.
[167] Harry F. W. Taylor. Proposed structure for calcium silicate hydrate gel.
of the American Ceramic Society
, 69(6):464–467, 1986.
[168] E.V. Thomas, I. Bloom, J.P. Christophersen, and V.S. Battaglia.
Journal
Statistical
methodology for predicting the life of lithium-ion cells via accelerated degradation testing.
Journal of Power Sources
124
, 184(1):312–317, September 2008.
[169] Matthias Thommes, Bernd Smarsly, Matthijs Groenewolt, Peter I. Ravikovitch,
and Alexander V. Neimark. Adsorption hysteresis of nitrogen and argon in pore
networks and characterization of novel micro- and mesoporous silicas.
22(2):756–764, January 2006.
Langmuir
,
[170] M. Th. Van Genuchten and D. R. Nielsen. On describing and predicting the
hydraulic properties of unsaturated soils.
1985.
Annales Geophysicae
, 3(5):615–628,
[171] Krystyn Van Vliet, Roland Pellenq, Markus J. Buehler, Jeffrey C. Grossman,
Hamlin Jennings, Franz-Josef Ulm, and Sidney Yip. Set in stone? A perspective
on the concrete sustainability challenge.
2012.
MRS Bulletin
, 37(04):395–402, April
[172] Pallavi Verma, Pascal Maire, and Petr Novák.
A review of the features and
analyses of the solid electrolyte interphase in Li-ion batteries.
Acta
, 55(22):6332–6341, September 2010.
Electrochimica
[173] J. Vetter, P. Novák, M. R. Wagner, C. Veit, K.-C. Möller, J. O. Besenhard,
M. Winter, M. Wohlfahrt-Mehrens, C. Vogler, and A. Hammouche.
mechanisms in lithium-ion batteries.
September 2005.
Journal of Power Sources
[174] Wilasa Vichit-Vadakan and George W. Scherer.
, 147:269–281,
Measuring Permeability of
Journal of the
Rigid Materials by a Beam-Bending Method: III, Cement Paste.
American Ceramic Society
, 85(6):1537–1544, 2002.
[175] Ivan Vlahinić, Hamlin M. Jennings, and Jeffrey J. Thomas.
model for drying of a partially saturated porous material.
rials
, 41(3):319–328, March 2009.
[176] Robert von Helmholtz.
Ageing
A constitutive
Mechanics of Mate-
Untersuchungen über Dämpfe und Nebel, besonders
über solche von Lösungen.
Annalen der Physik
, 263(4):508–543, 1886.
[177] C. S. Wang. Lithium Insertion in Carbon-Silicon Composite Materials Produced
by Mechanical Milling.
1998.
Journal of The Electrochemical Society
, 145(8):2751,
[178] Feng Wang, Jason Graetz, M Sergio Moreno, Chao Ma, Lijun Wu, Vyacheslav
Volkov, and Yimei Zhu. Chemical distribution and bonding of lithium in intercalated graphite: identification with optimized electron energy loss spectroscopy.
ACS Nano
, 5(2):1190–7, February 2011.
[179] K. West, T. Jacobsen, and S. Atlung. Modeling of Porous Insertion Electrodes
with Liquid Electrolyte.
1982.
Journal of The Electrochemical Society
, 129(7):1480,
[180] Stephen Whitaker. Flow in porous media I: A theoretical derivation of Darcy’s
law.
Transport in Porous Media
, 1:3–25, 1986.
125
[181] Yongyao Xia and Masaki Yoshio. An investigation of lithium ion insertion into
spinel structure Li-Mn-O compounds.
143(3):825–833, 1996.
Journal of The Electrochemical Society
,
[182] J. H. Yan, W. S. Li, and Q. Y. Zhan. Failure mechanism of valve-regulated leadacid batteries under high-power cycling.
140, May 2004.
Journal of Power Sources
, 133(1):135–
[183] R. Yazami and Ph. Touzain. A reversible graphite-lithium negative electrode
for electrochemical generators.
Journal of Power Sources
, 9:365–371, 1983.
[184] Guang Ye. Percolation of capillary pores in hardening cement pastes.
and Concrete Research
, 35(1):167–176, January 2005.
Cement
[185] B. R. York and S. A. Solin. Effect of composition on charge exchange, lattice expansion, and staging in potassium-ammonia graphite intercalation compounds.
Physical Review B
, 31:8206, 1985.
[186] Arie Zaban and Doron Aurbach. Impedance spectroscopy of lithium and nickel
electrodes in propylene carbonate solutions of different lithium salts A comparative study.
Journal of Power Sources
[187] R. T. Zahradnik and E. C. Moreno.
, 54:289–295, 1995.
Structural Features of Human Dental
Enamel as Revealed by Isothermal Water Vapour Sorption.
Biology
, 20:317–325, 1975.
[188] Yancheng Zhang, Chao-Yang Wang, and Xidong Tang.
of an automotive LiFePO4 lithium-ion battery.
196(3):1513–1520, February 2011.
Archives of Oral
Cycling degradation
Journal of Power Sources
,
[189] Z. Zhang, D. Fouchard, and J. R. Rea. Differential scanning calorimetry material studies: implications for the safety of lithium-ion cells.
Sources
, 70:16–20, 1998.
126
Journal of Power