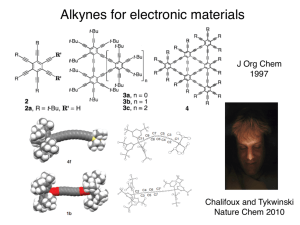
PATHWAYS IN COAL THERMOLYSIS: A THEORETICAL AND EXPERIMENTAL by
advertisement

PATHWAYS IN COAL THERMOLYSIS: A THEORETICAL AND EXPERIMENTAL STUDY WITH MODEL COMPOUNDS by Ini A. Ekpenyong and Preetinder S. Virk Energy Laboratory Report No. MIT-EL 82-007 January 1982 -- kilill Doctoral Thesis Digest PATHWAYS IN COAL THERMOLYSIS: A THEORETICAL AND EXPERIMENTAL STUDY WITH MODEL COMPOUNDS by II A. EKPENYONG Submitted to the Department of Chemical Engineering on January 15, 1982 in partial fulfillment of the requirements for the Degree of Doctor of Science at the Massachusetts Institute of Technology Thesis Supervisor: Prof. Preetinder S. Virk 4I i - - ~1611 1 -1CHAPTER 1: SUMMARY Paae Contents 1.1 ............................. Thesis Abstract........... Background and Thesis Objectives............................ 1.1.1 Introduction and Previous Work Review................... 1.1.2 Thesis Objectives and Strategy....................... PART A: 2 3 3 6 THEORETICAL STUDIES 1.2 i Theoretical ethods......................................... 10 1.2.1 LCAO-MO Calculations................................... 1.2.2 Perturbation and FO Treatment of Reactivity........... 14 1.3 Theoretical Results and Discussion: (I) Reactivities of H-donors and Acceptors...................................... 1.3.1 Special Nomenclature................................. 1.3.2 Factors Affecting Reactivity of H-donors ................ .............. 1.3.3 Reactivity of H-Acceptors ......... 1.4 32 Retroene Reactivity ............................................. Reactivities of Aromatic Cope/Claisen Rearrangements........... 34 1.5 i.6 Some Previous Coal Model Experiments Explained by the Theoretical Results......................................... PART 8: 1.7 1.8 1.9 1.10 16 16 17 26 35 EXPERIMENTAL STUDIES Experimental Methods.......................................... Experimental Results.............. ...... ............... 1.8.1 Product Spectra........... 1.8.2 Product Distribution and Kinetics ...................... 1.8.2a The BPE/1,4-DHN System...................... 1.8.2b The BPE/1,2-DHN System ....................... 1.8.2c The BPE/Tetralin System..................... 1.8.2d The BPE/trans-Decalin System.................... 1.8.3 Tabular Summary of Experimental Results ................. 37 38 39 40 40 47 52 58 61 Discussion of Experimental Results.......................... .......................... 1.9.1 Reaction Pathways. 1.9.2 Reaction Mechanisms.................................. 1.9.3 Comparison with Previous Work......................... Conclusion and Significance of the Thesis................... 61 61 62 72 72 ............... ...... 76 -- List of References ................... -- List of Recurrent Abbreviations.......................... ......... 87 -2PATHWAYS IN COAL THERMOLYSIS: A THEORETICAL AND EXPERIMENTAL STUDY WITH MODEL COMPOUNDS by Ini Akpan Ekpenyong and Preetinder S. Virk Department of Chemical Engineering Massachusetts Institute of Technology, Cambridge, MA 02139 ABSTRACT Fundamental aspects of coal thermolysis were investigated, including how the chemical structures of aromatics, hydroaromatics, and alcohols affect their reactivities as hydrogen donors and acceptors in coal processing. The susceptibilities of substructural entities in coals to fragmentation via a number of thermal pericyclic and free radical mechanisms were probed, as were the factors governing relative reactivities within series of such coal model compounds. The theoretical part of the work applied perturbation molecular orbital (P.10) and frontier orbital theories, in conjunction with -- and pseudo-7 MG's, to the study of model compound reactivity. This enabled prediction of reactivity patterns of H-donors, H-acceptors and coal-like structures as functions of their ,- and a-bond configurations, including heteroatomic effects. Experimentally, the liquid phase reactions of the coal model compound PhOCH2Ph (Benzyl phenyl ether, BPE) were detailed for the first time in eacR of the four hydronaphthalene H-donor solvents, I(Gi (1,4dihydronaphthalene, or 1,4-OHN), § (1,2-DHN), 9 (tetralin), and (trans-decalin), in the temperature range 220*-300 0C. The thermolysis of BPE exhibited a pronounced dependence on solvent structure, both with respect to product selectivities and reaction kinetics. The most preferential formation of light products, the most efficient h)drogen In contransfer, and the fastest kinetics were obtained with 1,4-DHN. trast, the overall BPE transformation rates in 1,2-DHN, tetralin and t-decalin were roughly comparable, despite qualitative differences in kinetic detail and heavy product (>C1 30) formation. BPE thermolysis pathways were delineated as involving (a) rearrangement, leading to isomerization, (b) hydrogenations, leading ultimately to PhOH and PhCH 3 products,and (c) addition reactions, engendering heavy products. Pathways (b) and (c) are competitive and, in each, self-reactions of 8PE-derivatives vie against reactions between these and the donor solvent. Of the detailed free radical and pericyclic reaction mechanisms postulated, the latter rationalized many more facets of the BPE results than the former. The theoretical and experimental results were appraised against previous coal thermolysis literature. Thesis Supervisor: Title: Dr. Preetinder S. Virk Associate Professor of Chemical Engineering " -- 'IIIIlinEl III -- hi ,,Lilill -3- Chapter 1 SUMMARY 1.1 1.1.1 Background and Thesis Objectives Introduction and Previous Work Review A prime objective of coal chemistry is to predict and rationalize coal reactions based on an understanding of the reactivity patterns of substructural groups ("moieties") in coal, the macromolecular structure of the latter being uncertain and dependent on the coal's rank and geologic source. It is known 1 '2 '4 ,6-17 that all coals have con- siderable unsaturation in the form of aromatic ring clusters (typically one to four rings per cluster), interconnected by aliphatic and alicyclic segments. And, as the basic goal in coal liquefaction and pyrolysis is to fragment the coal macromolecule into lower-molecularweight products, the partial reduction (hydrogenation.) of the unsaturation is desirable, as it not only increases the extent to which the coal can fragment but also promotes the solubility properties of highmolecular-weight fragmentation products. Thus, there is a continuing quest for fuller understanding of the fissionabilities of various structural linkages in coal, as well as of the effectiveness of various compounds ("H-donors") in transferring hydrogen to coal, and the abilities of various ,-bond types in coals to accept hydrogen. Studying coal with model compounds rather than with coal itself offers the advantage of scientific control of variables and unambiquous attribution of effects to particular functional groups. Deduc- tions about reactivities of particular moieties can then be cautiously generalized to all coals in which they occur, (with the understanding that the exact structural environment immediately flanking a moiety in -4an actual coal may have some secondary effect on its reactivity). Model compounds are chosen to fit the particular chemical aspect of coal being investigated; e.g., simple polynuclear aromatics like naphthalene, phenanthrene and anthracene are candidates in modelling coal as an H-acceptor. Compounds of the type ArX(CH 2 )m+1 Ar' and ArX(CH 2 )m+ R -- where X is CH2 or 0 and m is an integer >0 -- by combining fissionable aliphatic linkages with aromatic nuclei, are choice, multipurpose models for coal: They can model the effects of the specific identity of Ar on the decomposition rates of the molecule, and/or act as H-acceptors or donors, and/or undergo thermal rearrangements compatible with segments of their structure. phanyl ethar (BPE), PhOCH 2 Ph, and bibenzy', Benzyl Ph(CH 2 )2Ph are the proto- typical compounds of that kind studied in the thesis. The thermolysis of bibenzyl has been widely studied, particularly 19-24 or with an H-donor for the compound reacting neat in the gas phase agent, usually tetralin, in the liquid or gas phases. 20 '29 -35 works of Sato, et al., 34,35 Poutsma, 27 and Cronauer, et al. special mention.) 32 (The deserve Earlier studies of BPE, on the other hand, have been fewer, both for the neat thermolysis 3 7 93 9 presence of H-donors like tetrahydroquinoline and for reaction in the 3 6 ' 39 and tetralin. 38 '39 Most were conducted under limited-range and severe conditions, resulting in incomplete characterizations of product distributions, kinetics and/or mechanism. This applies too to previously reported 38 ,39 exam- inations of the effects of varying the H-donor solvent, for a given PhXCH2Ph substrate. More specific reviews of related- previous work on coal model compounds are given in Sections 2.3 and 8.4 of the thesis. The work of Curran, et al.,26,45 dealing with the effectiveness ^ S--ipiM I, ,hhn -5Table 2.T of Hydrogen Donor Activity Towards Coal, A Comoarison I ,i roll III I of Various Solvents* Temp Exptl Set H-Donor (aC) 09 Contact Solvent Activity Ratio' Time (Tetralin = 1) (Min) Perhydrophenanthrene 380 60 0.18 Decalin: 390 / 0.25 Indane: 380 / 0.29 r / / 1 (c-Cyclohexylphenol) / Tetralin: 0.88 1.08 (Dihydrophenat rene) 327 60 1 / / 1.91 50% Tetralin 50% Decalin 427 20 1 50% Isopropanol 50% Decalin / O Tetralin: B Cyclohexanol OH : C 0.84 *Ireland mine coal used in all cases. ,Activity of each H-donor was originally measured as % wt gain of the MAF coal due to H uptake. Adapted from Curran, et al. (Ref. 26). -6of various hydroaromatic and seconday alcohols, and those of Ross and Blessing,46,59 comparing classes of alcohols as H-donors to coals, also pertain directly to the objectives of this thesis. The results of Curran, recast as Table 2.1, clearly suggest the desirability of aromatic content in H-donors. Yet if, as is commonly believed, tet- ralin owes its effectiveness as an H-donor solely to the ease of abstraction of its benzylic H's by radicals of the type ArX, then tetralin and indan (see Table 2.1) should reasonably be of comparable practical effectiveness, which they are not. Likewise, secondary alcohols, despite lacking -bonds for delo- calization of unpaired electrons, are good H-donors 4 5 4659 to coals, while priniary and tertiary alcohols are not. 46 4 6 ' 59 'o 46 Ross and Blessing 4 6 explain that in terms of the operation of an ionic H-transfer mechanism, whereby a strong base, M-, internal to coal or added during processing, catalyzes the H-transfer. But (as analyzed in Section 2.4.1 of the thesis) that argument fails to explain the superiority of secondary, over primary alcohols in the absence 46 of externally added strong bases. [For if there is an internal base, M-, in coal that promotes ionic H-transfer from secondary alcohols to coal, it ought to do the same with primary alcohols, since the associated conjugate acid/base systems R'CHROH/R'CHRO- and RCH 2 OH/RCH20- are comparable in strength. ] 1.1.2 Thesis Objectives and Strategy In brief, the thesis set out to address the following shortfalls which emerged from a review of the previous literature on coal thermolysis fundamentals: * The popular free radical and ionic rationalizations for rela- -_1 ii -7tive H-donor activities fail to explain a number of important exceptions. * No systematic theoretical study of the basic factors that govern H-donor and H-acceptor reactivity had hitherto been undertaken. Neither had a rigorous, comparative experimental study of several H-donors been made using a particular H-acceptor substrate of the type PhXCH 2 Ph. * The quantitative data base on the reactions of PhOCH 2 Ph (BPE) in a good H-donor solvent was spotty, at best, notwithstanding the important implications to coal processing which differences between the characteristics of PhOCH 2 Ph and Ph(CH 2 ) 2 Ph, in such donors, would signify. * The susceptibility of the aromatic, hydroaromatic and aliphatic structure of coals to pericyclic reactions61-64 had, with few recent exceptions,18,60 been ignored when explaining the observed chemistry of coals and their model compounds. Conse- quently, the specific factors governing such reactions in the coal context had not been studied beyond what is known for such reactions in general. Strategy: There are only a few known elementary reactions through which it is hypothesized that the aromatic-hydroaromatic-aliphatic moieties in coal can dismember spontaneously when heated neat or in nonpolar Hdonor or H-shuttler media. As illustrated in principle by the model compound reactions Rl.l-R1.8, they are (1) homolysis (RT.1), (ii) Diels-Alder reversions (Rl,2; requires a tetrahydrobenzene moiety -- -8naturally occurring or arising via partial hydrogenation of an aromatic ring); (iii) endothermic retroene reactions (RI.3); and (iv) ex- othermic retroenes (Rl.6 and R1.8), [which would be preceded by the Claisen/Cope rearrangement (R1.4) of an ArXCH 2 Ar' moiety, and hydrogenation (i.e., concerted 2H-transfer, R1.5 or R1.7)]. R1.1 RI 2H "2* R R1.2 R2 R2 0 o1 0 OH O1 Rl . R.8 Of these basic fragmentation pathways, only homolysis is not pericyclic. Of the pericyclic ones, the Diels-Alder prototype has 6 1 ,63,94,96,97,122-124 been the most exhaustively studied theoretically IIIIIIIYY 111i 1111 ---- 1,nllia/llmlIili ill/4 WiI I -9and experimentally, 6 8 , 7 1 though not in the particular context of coal thermolysis, and is the only one not formally explored in the thesis. The thesis comprises two parts. Part A (Chapters 3-5) is purely theoretical and, presupposing the applicability of pericyclic reactions to coal chemistry, uses methods of physical chemistry (LCAO,* perturbation molecular orbital and frontier orbital theories) to investigate factors which determine reactivities of'H-donors in pericyclic 2H-exchange. Retroene reactions and Claisen/Cope rearrangements are likewise analyzed. The likelihoods of actual occurrence of these reactions for coal/model-compounds are assessed, and the present theoretical results are compared with literature results, where possible. Part B (Chapters 6-8) is experimental, and consists mainly of detailed studies of the (liquid phase) reactions of PhOCH 2 Ph in hydronaphthalenes in the combinations shown in Table 2.2. Table 2.2. Experimental Work Grid MOBIUS DONOR HUCKEL DONOR REACTIVE CONTROL 'INERT' CONTROL (1,2-DHN) (1,4-DHN) TETRALIN trans-DECALIN PhOCH 2 Ph (BPE) PhCH2 CH2 Ph (DPE) The discussion refs. 34,35,32 / Executed In Detail; t Executed In Part; x Not Executed. Neat Studies: See Section 2.3 of text. Key: *LCAO = linear combination of atomic orbitals. Wlililil m -10(Chapter 8) of the experimental results considers both free radical and pericyclic rationalizations, and cross-compares with features of Ph(CH2 ) 2 Ph reactions. The conclusions and significance of the thesis are then outlined (Sec. 1.10, and Chapter 9). PART A: 1.2 THEORETICAL STUDIES Theoretical Methods LCAO-MO Calculations 1.2.1 Fundamental studies of reactivity of the type attempted in the thesis require the availability of molecular orbitals (MO's) of the reactants (or reactant segments) and the first task was to select and adapt an LCAO-MO calculation method that was both simple and adequate for the afore-stated theoretical objectives. The Huckel molecular or- bital (HMO) approximation 67 a,84 -87 and a number of its basic variants, 88 ,91,108a which usually work well for most applications to all- carbon -*-bond system, and some strictly hydrocarbon ,r/pseudo-r applications, proved insufficient here because many coal-model-compounds and H-donors are heteroatomic (with significant roles for C-O and C-H bonds of various kinds). The weakness of HMO in predicting atomic or- bital (AO) coefficient distributions, and relative spacings of MO energy levels, is largely a consequence of (i) complete neglect of overlap (in the off-diagonal elements of the secular matrix, see Sections 3.1-3.4, or any of the cited textbooks), and (ii) the inherent neglect of electron repulsion effects in most "naive" LCAO-MO methods. Short of using considerably more intricate self-consistent-field 83 the (e.g., semi-empirical CNDO/2, MINDO, or ab initio) methods, adopted naive LCAO-MO method that best attempts to address some of the /II -11shortcomings of Huckel-like methods was the Mulliken-Wheland-Mann (,MWM) method. 98 -100 It is iterative, retains overlap (adjusted after each iteration) and takes some cognizance of electron-repulsion by adjusting.the Coulomb integrals (main-diagonal elements ai = (Hii-a)/W of the secular matrix, see Secs. 3.3 and 3.4), based on updated charge distributions after each iteration. Unfortunately, the original MWM version was developed, and works best for charged, strictly hydrocarbon ,-and-pseudo- systems. For purposes of this thesis, it had to be modified to handle charge-neutral, as well as oxygen-containing, molecules. The main modifications (see Sec. 3.4 for details) were the following: (i) Initialization values were chosen for Coulomb integral parameters for oxygen atoms: bonyl groups, and nitial nta0. 0 .815 2 71 for 0 in car- for 0 in ethers, alcohols 8initial was set from considerations of total 0 w-energy and HOMO-_nergy differences between butadiene, initial was then set on aO . S, and acrolein, (y and phenols. the basis of the differential electronegativity constraint a -60 = SC+-"C potential of = 0 s'oC .. Me ' and the fact that the ionization is about the same124 as butadiene's (meaning that their HOMO energies are near-equal). bonds, Streitwieser's 07 values for ij - For C-0 Sij/0.25 were re- tained. (ii) The formula for updating Si between iterations was generalized to handle oxygen sp3-lone-pair orbitals as well as C and H AO's. 08 -12(iii) To differentiate between assorted types of pseudo- bonds, like C=H 2 , C=(H,C), and C=(O,H), encountered in coal chemistry, new group overlap values Sij, calculated with strict inclusion of internal overlaps using Mulliken's methods 10 1 were used. For investigating the concerted 2H-transfer characteristics of alcohols, a pseudo-7 representation of alcohol/phenolic OH groups was devised and parametrized, (Sec. (iv) 3.4.3). The number of iterations was restricted to two (including the initial) for optimum results with this set of parametrizations and modifications of the MWM method. Detailed for- rrulation and soltition procedures are provided in Sections 3.3 and 3.4. Table 3.1 compares the -,-MO's of acrolein, calculated with the basic Huckel, the modified MWM, and the more advanced CND00/2 methods. The critical considerations in a set of MO's are not so much the specific numerical values of energy (eigenvalues) Ej = +,\ Bo , or AO-co- efficients (eigenvectors) cij, as the relative spacings of Ej and the polarization, J(c i - ci+ 1 ) 2 , of AO-coefficients at each energy level. On both these counts, the modified MWM method is significantly better than the Huckel, and the margin of improvement increases as the number of AO's in the i/pseudo-r system increases, a desirable feature in coal model compound chemistry. MWM cMWM - DO cCNO0/2 [Note that at each energy level in Table 3.1, MOc HMO i is significantly smaller than CNiO/2 CND0/2 1 1 The modified MWM method does, however, exaggerate the AO coefficients of carbonyl oxygen atoms. But because during reactive interac- tions, such oxygen coefficients are always multiplied by interaction C Table 3.1 w-MO's of CI2 (CII) 2 CO, as calculated with CNDO/2)d methods. the modified MWM, a luckelb and 1 2 3 4 C -. C 0 - C Orbital No. (j) 1 2 (IIHOMO) 3 (LUMO) (Ej-a)/B o C1 c2 c3 c4 1.22 a (1.88)b 0.43 (0.66) 0.61 (0.58) 0.55 (0.43) 0.37 (0.23) [-14.5eV]d 0.71 [0.58] 0.63 [0.30] 0.32 [-0.48] -0.40 [-0.581 -0.59 (1.00) (0.58) (0.00) (-0.58) (-0.58) [+2.5eV]d -0.47 -0.35 [0.51] 0.62 (0.43) [-0.48] -0.50 (-0.58) [-0.39) -0.27 (-0.23) [0.591 0.54 (0.66) -2.32 0.28 -0.53 0.66 -0.45 -1.53 (0.23) (-0.58) (0.66) (-0.43) (a) Modified Mulliken-Wheland-Mann method, see Section 3.4 of text, parameters used are contained in Table 3.4. (b) luckel MO's are in ( ) parentheses; obtained with 6C=0.0, pO=P12=23= p3 4 =1.0. go's are different for Iluckel and MWM methods (of Section 3.3). (d) Values in square [ ] brackets are from an advanced semi-empirical SCF (CNDO/2) method, as reported by lHouk and Strozier, 9 2 (also, see Alston and Shillady 93 ); corresponding Ej are in eV, not dimensionless. -14resonance integral ratios,140 (C--O/C--C) 0.86, or (aO--H /C--H) 0.88, that exaggeration is safely offset in all the applications considered in the thesis. 1.2.2 [For elaboration, see Secs. 5.1.2, 5.1.3.] Perturbation and FO Treatment of Reactivity Perturbation MO techniques, specifically 1st and 2nd order intra/ intermolecular perturbations ill and FO methodology,119-124 were the main tools used for relative reactivity analyses, based on MO's calculated with the modified MWM method. With MO energies measured in units of 8o as EHO MO ELUMO = a+ LUMOR, energy, IEF, HOMO o and (cf., Sec. 1.2.1) the HOMO-LUMO (or FO) interaction between reactants A and A' has the form (c LUMO HOMO L(caCa -2:EFO= LUMO HOMO( bb/Aaa )) b' Cb , HOMO LUMO A "A HOMO LUMO HOMO LUMO b' (bb'/ +Cb Cal (Ca HOMO +LUMO 2 aa' • 2 aa' - the AO-coefficient at site a, in the LUMO of molecule A. where cLUMO a cLUMO b and the AO-coefficient at site b, in the LUMO of molecule A. aa, and ebb' are the interaction resonance integrals, see Fig. 1.1. Subject to the usual assumptions (cf., Sec. 3.5.2) of frontier orbital theory, Eq. 1.1 (or the fuller form involving all occupied MO - unoccupied MO interactions) allows qualitative or semi-quantitative prediction of relative rates or reactivities, as one or both of ~-~~'~~ --- IIYIIIIIIY IIYIYII I IIo I I IIlIII -15- 8aa' a' a , (LUMO)A (LUMO)A A' A (HOMO)A - - It (HOM)A' b' b Bbb' Fig. 1.1: Interacting AO sites and frontier orbitals, at the inception of reaction between molecules A and A'. the reactants A and A' are varied.* In Eq. 1.1, the two terms represent both FO pairs of interactions, i.e., (LUMO)A - (HOMO)A , and (HOMO)A - (LUMO)A. Often, one of these terms is decisive, and further simplification can be achieved by dropping the other. That is the case in 2H-exchange reactions, in which the controlling interaction is between the HOMO of the H-donor and the LUMO of the H-acceptor; (see Sec. 4.1). By arbitrarily selecting and holding one reactant constant (e.g., an "ideal" H-acceptor), a relative reactivity index -- proportional to the larger term in Eq. 1.1 -- may be defined for a hierachial comparison of the coreactants (the H-donors). The numerical values of the index will then *For the benefit of the general reader, it may be briefly noted that (i) AEFO relates to the activation energy (the larger -AEFo, the smaller the intrinsic activation energy); (ii) the sole use of occupied MO - unoccupied MO interaction energies for reactivity comparisons assumes kinetic, rather than thermodynamic, control of reaction rates. ll0- . .II. . i depend only on the FO energies and AO coefficients of members of the class of reactants being compared. In practice (as developed in detail in Chapters 4 and 5), it was necessary to selectively (i.e., guided by perturbational following of MO's) incorporate non-FO's into the reactivity index, depending on the specific type of pericyclic reaction under consideration. 1.3 1.3.1 Theoretical Results and Discussion: and H-acceptors (1) Reactivities of H-donors Soecial Nomenclature: Because of selection rules 6 1 - 6 4 , 6 6 , 67 (i.e., reactants' symmetry compatibility requirements)* of pericyclic reactions, and geometrical constraints, thermally induced concerted 2H-exchange between an Hdonor and an H-acceptor will normally take place via an untwisted aromatic transition state (TS) involving 4n+2 electrons, where n = 1,2,3,---. Accordingly in the simplest prototype of such an exchange, the H-acceptor moiety is a single -bond (= 2e-), and the H-donor moiety would consst of twc vicinal, mutually-cis (i the parent structure is cyclic) a-bonds, contributing 4e- to the 6e- aromatic TS, as in the (hypothetical) example R1.9. For terminological conven- ience, we designate such as an 2H-exchange between a Huckel H-acceptor (HA) and a Mobius H-donor (MD).* % R1 .9 *Further discussed in Sections 2.5 and 4.1 **In this new usage "Huckel" and "Mobius" refer only to the electron count, 4p+2 and 4q, respectively, and to the associated AO-coefficient symmetries of the reactant segment. Not to be confused with Huckel and Mobius transition state topologies. luilill -17- PIa La SR1i.10 SHb tH In the other prototype, illustrated in principle by R1.10, the Hacceptor sites are the termini of a cis butadiene (or cis acrolein, S) moiety, and the donor's H's are mutually-cis termini of a ara segment (i.e., a "2-butene" moiety). change would then involve the donor. The aromatic TS for the 2H-ex- O1e- : 4e- from the acceptor and 6e- from Such a 2H-exchange is here said to be between a Mobius H- acceptor (MA) and a Huckel H-donor (HO). All thermally induced pericyclic H-transfers are thus classifiable as either of the HA-MD or MA-HD types. Factors Affecting Reactivity of H-donors 1.3.2 Table 4.1 ranks a selection of Mobius H-donors, in descending order (alcohols excepted) of predicted intrinsic reactivity; likewise Table 4.2 for Huckel H-donors. The hierachy of MO reactivity in Table 4.1 indicates that the most potent Mobius H-donors are those in which both C-H bonds of the moiety C (inwhich the hydrogens Ha and Hb are cis) are flanked by unsaturation (r-bonds or an aromatic ring). For this pur- pose, non-aromatic r--bonds or "linear" polyenic. environments are more effective than aromatic ones. Thus, 1,3-cyclohexadiene is intrinsi- caliy a better H-donor than 1,2-dihydronaphthalene, which in turn is better than 9,10-dihydrophenanthrene; i.e., DM2 > DM3 > DM4, in Table I .. Table 4,1: Predicted Ranking of the Iintrinsic Reactivities of Selected Mbius ll-Donors, for 21i-Tratis er t.o an Ideal Ilickel Accegtor.d Donor Code/Rank DMI Donor Identi ty 0 y Reactivity Index e Donor S 110MO 0.41 S0. 4 7 f 0.217 ,-0. 260 Donor NilOMO bIIa DM2 0.32 0.526 0. 215,-0.215 note g 0.25 0.553 0.138,-0.206 0.759 0.179,-0.012 DM4 0.22h 0.577 0.28,-0.128 0.757 0.143,-0.143 DM5 0.22 0.486 0. 013,+0.129 0.625 0.1 95 ,-0.15 DM6 0.19 h 0.744 0.182,-0.182 0,754 0.31,+0.131 a DM3 (Carried Forward) llb b Table 4.1 (Colt'd) Donor Code/Rank Donor Identity Ila DM7 DM8 lla Reactivity Sindex e Donor 111MO Donor NIIOMO 0.19 0.619 0.150,-0.149 0.760 0.169,-0.014 0.13 IIb h 0.726 0.254,-0.039 DM9 0.13 0.520 0.181,-0.019 i 0.794 0.160,-0.037 DMI0 0.12 0.721 0.108,-0.065 0.106,-0.000 0.10 0.722 0.193,-0.029 0.757 0.109,-o.021 0.10 0.765 0,192,-0.037 0.802 0.117,-0,024 11 IIj a DM11 DM12 (Carried Forward) 0.760 b, Table 4.1 (Cont'd) Donor Donor Code/Rank Reactivity Index Identity Donor O1MO Donor NIIOMO II 0.00 DMI13 Oa ll1b 0.722 0.29,+0.029 0.757 0.021,-0.021 071k ~1~_~1_~ Oil 01.1 SAI SA2 / 0-11 -11b 0.39 0.71k 0.27,-0.05 0.71 -I.12,0.27 (Table Notes Carried Forward) 1.31 0.47,-0.51 1.31 +0. 06 ,+0. 47 - I -- I ~' -- IIIIidIY WEIIIIIIY W6Y IIMM1N1III ItYYN -21Table 4.1 Notes: (d) The symmetrical HUckel acceptor, PhCH 2 =CH2Ph (cis-stilbene) with LUMO energy X = -0.641 (and coefficients c1 = c2 = 0.430) has been assumed in calculating the reactivity index. However, the relative ordering of donor activity is independent of the H-acceptor used, provided the latter gives a heat of reaction,.,H < 0 with each of the donors. For comparing the donors, an elementary pericyclic transfer of the vicinal, cis hydrogens Ha and Hb is assumed. But in oractice some of the donors shown may not hydrogenate cis-stilbene (or other chosen model acceptor) pericyclically. (e) (f) LUMO Reactivity index defined by Eq. 4.3, with 'acceptor = -0.641. When a donor's NHOMO is not explicitly listed in the last column of the table, the index is based on the orbital parameters given in the "Donor HOMO" column only. See note g below. Abbreviated designation of pertinent orbital parameters , where X a(E-a)/S o = orbital energy in units of s., and caaand c b are the appropriate AO coefficients, derived with the modified MM MO method. (g) and >1.0O, the When the energy of the donor's NHOMO is E = 3+x\ exclusion from their emphasize to listed, not are parameters NHOMO 4.2.2. Section text, see index; reactivity The of the evaluation (h) Ties in the rounded reactivity indices of two or more donors are broken by considering their HOMO contributions only: the one with larger HOMO contribution is accorded a higher reactivity (rank). (i) In this special case, the third highest occupied MO (THOMO) is listed instead of the NHOMO because it gives a much larger contribution than the latter and still meets the condition that the orbital energy is such that X<l. However, the overall index shown is the sum of the contributions of the HOMO, NHOMO and THOMO. (j) Not directly comparable with the reactivity indices of the donors DM1 to DM12, because of the absence of a i-bond environment in 2propanol (and cyclohexanol). (k) MO's for secondary alcohols were based on the model discussed in Section 3.4.3. The full MO's from which these particular partial MO's were abstracted are included in Appendix A. immediately following Chapter 5. (1) This highly reactive intermediate can arise in the Claisen rearrangement of the benzyl phenyl ether and like moieties in coal, (cf., reaction R2.17). -22(Note, however, that polyenic unsaturation also promotes the 4.1. tendency of the "H-donor" to polymerize, or add to the H-acceptor, rather than donate H.) If only one side of the moiety is flanked by unsaturation (as in tetralin, DM11, and indan, DM10), the H-donation potential falls accordingly. The intrinsic effectiveness of substituents R and R in enhancing the H-donor activity of the moiety (a "2 b are in the order: linear, conjugated polyene > single r-bond > phenyl > naphthyl > phenoxy > alkyl. Secondary alcohol superiority over primary and tertiary ones, in the absence of strong bases, appears, from this investigation, to be Ho for the X due to the preference in secondary alcohols donation of the pair (H ,H ) instead of (H ,H): The contributions of both the HOMO and NHOMO to the reactivity must be considered, whereupon the (H,H a ) pair wins, because this pair of hydrogens has correct MD orbital symmetry in both these MO's, :ontributing to a larger overall reactivity index. The combination of factors which leads to this effect is lacking in primary and tertiary alcohols, -- namely that H is a tertiary hydrogen, one position removed from an oxygen atom. (See also Secs. 2.4 and 4.2.2.) The Huckel H-donor reactivity hierachy in Table 4.2 reveals two prominent patterns: * The best Huckel H-donors (HD's) are those in which concerted transfer of (Ha,Hb) would result in an aromatic ring in the dehydrogenation product. ODH Within the subgroup of all such HD's, e.g., entries to DH4 in Table 4.2, the order of increase of reactivity coincides Table 4.2: Predicted Ranking of the Intrinsic Reactivities of Selected liiuckel H-Donors, for 211-Transfer to an Ideal Mobius Accepor.mu Reaclivity Index"n Donor Identity Donor Code/Rank Donor 110140 0.80 0.668 0.322,0.322 D112 0.66 0.679 DH3 0.62 0.687 t 0.276,0.276 0114 0.50 D11l D115 D116 (Carried Forward) Ia- - Ila -- a tib lib 0.689 06.256,0.256 0.47 r 0.726 0.254,0.254 0.47r,s 0.520 0.181,0.120 Donor TIIOM10 P note q 0.799 0.059,0.059 0.787 0.079,0.079 0.780 o.00. U.o- note q 0.974 0.160,0.237 Table 4.2 (Coint'd) Donor Code/Rank Donor Identity Reactivi tv Index" 0117 0.46 Donor JlOMO Donor TIIOMOp 0.730 note q 0.251,0.251 DI)18 01110 0.29 0.721 0.198,0.198 note q 0.27 0.722 . 193,0.193 note q 0.14 0,559 0.083,0.119 0.-711 u 0.008,-0o.82 II1 0.01 Dl111i I1b (Table Notes Carried Forward) 0.722 +0.029,+0.029 note q ol -25a' Table 4.2 Notes: (m) The symmetrical M6bius acceptor, 0 to be hydrogenated at a', b' positions, and having LUMO energy X = -0.387, has been assumed in calculating the reactivity index (see note n below), because it would give a Hreaction < 0 with each of the HUickel Hdonors in this table. However, it is not implied that each and every one of these donors has a favorable pericyclic path for transferring Ha , Hb to said acceptor in an elementary reaction. (See also note d, Table 4.1). (n) Reactivity index defined by Eq. 4.5, with LUptor = -0.387. The index is evaluated with the donor orbital parameters given in the last two columns of the table. Where only the "Donor HOMO" parameters are explicitly given, the reactivity index was based on just those. See note (q) below. (p) THOMO: Third highest molecular orbital. The symmetry properties of HUckel H-donors are such that the THOMO (rather than NHOMO) complements the HOMO contribution; see text. and >11.0, the (q) When the energy of the donor's THOMO is E = a+xh, THOMO parameters are not listed, to emphasize their exclusion from the evaluation of the reactivity index. (r) Ties in the rounded reactivity indices of two or more donors are broken by considering their HOMO contributions, only, to the index: The one with the larger HOMO contribution is accorded a higher reactivity (rank). (s) The overall index listed for this compound contains a small but significant NHOMO contribution, in addition to the main ones from the HOMO and THOMO. (t) Special case: the listed orbital is the NHOMO rather than the HOMO, because the later, with parameters X/(ca,cb) = 0.529/ (-0.085,+0.085), has wrong symmetry. (u) NHOMO listed, rather than THOMO which is excluded because its x>1.0. However, in contrast with NHOMO's of most other Huckel donors, this particular NHOMO contributes significantly to the reactivity index. -26with the order of incremental gain in resonance energy (aromatic stability) of the dehydrogenation product. Thus, DHI > DH2 > 0H3 > DH4. * If functioning as a Huckel H-donor would result in the disruption of a benzene ring, the intrinsic HD reactivity of such a compound is severely reduced. [Note the low ranks of indan (DH8), tetralin (DH9) and o-cyclohexylphenol (DH10), concertedly donating those particular hydrogens indicated in Table 4.2.] It is noteworthy that these results arose from FO considerations only, but coincided, as special cases, with thermochemical intuition. (Cf., ,.3.3 Sec. 4.3.2). Reactivity of H-acceotors Relative reactivity patterns of H-acceptors are important because they relate to the relative ease at which various kinds of unsaturation in coal (and model compounds) can be hydrogenated, in continuous coal liquefaction processes. The optimum H-donors (and H-shutt- lers 2 5 , 1 3 8 ) must therefore not only give up their H's to coal readily, but must also be fairly easy to rehydrogenate. Separate reactivity scales for Huckel and Mobius H-acceptors are necessary, for pericyclic 2H-exchange, as already noted in Secs. 1.3.1 and 1.2.2. The HA and MA reactivity hierachies shown in Tables 4.3 and 4.4 were obtained with the same methods used for analyzing H-donor reactivities. The LUMO and NLUMO* energy levels and AO-coefficient distribution of an unsaturated compound are what primarily determine its intrinsic H-acceptor reactivity, and are, in turn, functions of the compound's -bond configuration and heteroatomic content. *NLUMO = next lowest unoccupied molecular orbital. detailed The struc- * Table 4.3: Acceptor Code/Rank 4 Predicted Relative Reducibilities.of i-Donds ("lluckel u II- Acceptors!!) in 211-Transfer Reaction witlh an Ideal fiobius II-Donor, Acceptor Identity/Site Reactivity Il(eox v K.bW 1.7 y A113 Acceptor LUMO NLUMO -0.332 1.7 y A[12 Acceptor -1.53 0.518,-0.417 0.322-0.515 -0.322 0.428,-0.152 0.471,--0.510 -0.620 0.502,-0.22 -1.87 0.445,-0.57f -1.53 A114 1.7 -0.620 0.475,-0.262 -1.87 0.435,-0.510 AHI1 1.3y -0.322 0.288,-0.442 -1.53 +0. 381 ,+0. 063 -0.641 -1.33 b A116 1.3 0.430,0.430 (Carried Forward) .n .o Table 4.3 (Cont'd) Acceptor Code/Rank Acceptor Identity/Si te Acceptor LUMO Acceptor NLU4MO -0.620 0.428,-0.405 +0.216,9+0.191 1.2 -0.862 0.542,-0. 380 -1.32 0.035,-0.035 1.1 y -0.713 0.D415,-0.415 +0.101,+0. 101 1.1 -0.462 0.311 ,-0.220 -1.33 0.379,-0.321 0.93 -0.731 0.425,-0.263 -1.33 0.0,0.408 0.83 -0.713 0.340,-0.042 -0.952 0.273,-0.389 Reactivity Index v 1 .2 All7 ab a L, Al 18 b7 AI 19 Al AIIl 0 -1.87 =b -0.952 a All 12 AM2I ~OO ~wa (Carried Forward) b Table 4.3 (Cont'd) Acceptor Code/Rank Acceptor Identity/Site a b AHI14 Cob Acceptor LUMO Reactivity IndexV -1.02 0.81 0.72 Acceptor NLUMO 0.223,-0.468 -1.44 0.269,-0.162 -0.731 +0.263,+0.263 -1.33 0.408,-0.408 Notes: (u) An elementary pericyclic reaction with the syinetrical Mobius Hl-donor 1,3-cyclohexadiene is assumed. This donor has AIOM0 = 0.526 and would give a Allrxn<O with each of the acceptors in the table. The comparative purpose of this table (as everywhere else in Part A of this thesis) is stressed, since some of the reactions (e.g., with the acceptors A1113 and A1114) would not occur pericyclically, if at all. (v) Reactivity index, RAH as defined by Eq. 4.6b, with XdoIor = 0.526; see note (u). (w) The heptatrienic and hexadienonic moieties shown in square brackets in this table (and Table 4.4 also) would arise transiently in Claisen and Cope rearrangement products when coal or some of its Imodel compounds (e.g., benzyl phenyl ether) are heated; (cf., reaction R2.17). (x) For II transfer, the resonance integral a for CII--O=C is 10.88 times the value of 1 for CII--C=C in the transition state. (See Section 5.1.3). This is effected by reducing the actual (listed) AO coefficients of oxygen by a factor of 0.88 when calculating the reactivity inldex. (y) Ties in the rounded reactivity indices of two or more acceptors are broken in favor of the acceptor whose LUMO contribution alone to the index is larger. Table 4.4: Acceptor Code/Rank Predicted Relative Reducibilities of cis-13utadiene-floieties ("Mobus ll-Acceptors") in 211-Transfer Reaction with an Ideal llUckel ll-Donor.d Acceptor Terminfni Indexe 2.2 AMI AM2 Reactivity ba a1 AM3 Acceptor LUMO Acceptor NLUMO -1.70 -0. 38 7 g 0.511,0.511 0.272,0.272 1.7 -0.332 0.288,0.428 -1.53 0.381,0.471 1.6 -0.620 0.4280.475 -1.87 0.216,0.435 1.6 -0.834 0.560,0.560 -2.63 +0.361 ,-0.361 1.4 -0.462 0.440,0.440 0.0,0u0- a AM4 b a AM5 b b (Carried Forward) -0.462 1.2 AM6 0-. ,311W -1.33 -1.33 oThU2~7T Table 4.4 (Cop!!t'l). Acceptor Code/Rank Acceptor Teriniii Acceptor Reactivity Index e Acceptor LUMO 1.0 o.A4 -0.731 -0- 45 -- 1.33 0.98 -0.713 0.340,0.233 -0.952 0.273,0.369 NLUMO a AM7 a A/48 b9 Notes: (d) For this comparison, the syunetrical Huckel II-donor 1,4-dihydronaphthalene (1)112 in Table 4.2) <rxn<0 for all the acceptors considered. is assumed so that A (e) Reactivity index, RAM, as defined by Eq. 4.6a, with 1acc hydronaphthalene. (f) = 0.679, the acceptor being 1,4-di- This entity arises fairly readily from the [1,5] hydrogen shift of 1,2-dihydronaphthalene. -32ture-versus-reactivity patterns apparent in Tables 4.3 and 4.4 are further discussed in Section 4.5. 1.4 Retroene Reactivity Retroene reactions for substrates of the type ArX(CHR) 2R' [of which RI.3 (Section 1.1.2) is an example] are generally substantially endothermic (though generally not more so than homolytic fission). In theoretically studying their relative reactivities (Secs. 5.1 and 5.2), cognizance was taken of Hammond's postulatel 25 by estimating relative TS stabilizations from the product side; i.e., relative TS stabilizations -- as substrate structure was varied -- were based on pro- duct side MO's. All occupied MO - unoccupied MO interactions were summed, since no sufficiently general simplifications based on FO's only were feasible. In addition, as perturbing the product-side MO's allows only intrinsic activation energies of a series of such endothermic reactions to be compared (whereas the total activation energy, Ea , is the sum of the energy of reaction, AErxn, and the intrinsic activationr energy, ,.intr ), it was necessary to analyze the endcthermic Ea retroene reactions within subgroups in which the enthalpy of reaction, H298K, were comparable. Let the endothermic retroene reaction be represented by R1.11, with (51) as the (generalized) substrate: R R (51) X2 R1 .! - IYMM111111MINI -33Then: * (RI ,R2 ) being H or alkyl makes for higher reactivity than their being part of an aryl system; of the latter, 2-naphthyl > 1naphthyl > phenyl. * If either X1 or X2 (but not both) is oxygen, decomposition will be faster when X2 = 0 than when X= 0; either of those promotes faster retroene than if X = X2 = CH2. * X3 = 0 accords greater reactivity than X3 = CH2 , other things being equal. * The reactivity promoting abilities of R3 are in the order: aryl > ArCH 2 > alkyl > H. Thus, for a subseries of substrates such as Ph(CH 2 )nPh, retroene reactions are imoossible for n<2, and fastest for n=3, followed by n=4. Retroene reactivities for n>5 would be smaller still and mutually equal. al., 20 This trend fits the experimental observations of Benjamin, at for the thermolysis of such a series in tetralin; (the pre- sence of a suitable H-donor results in the hydrogenation of the unsaturated retroene products, such as styrene to ethylbenzene). Reactivity comparisons for highly exothermic reactions (typified by R1.6) were also examined (Sections 5.1.3 and 5.2.2) by partitioning the reactant into two segments and studying the second order interactions of the MO's of these segments. While some differences in pre- dicted reactivity were thus revealed, the pre-eminent conclusion is intr that their exothermicity (hence, low Ea ) makes such retroenes so facile as to be most likely non-rate-determining in pericyclic fragmentation schemes for coal and its aromatic-cum-aliphatic model compounds. -341.5 Reactivities of Aromatic Claisen/Cope Rearrangements The methodology used in studying aromatic Claisen and Cope re- arrangements of ArXCH 2 Ar' substrates (cf., reaction RI.4) was similar to that used for endothermic retroene reactions -- namely a combination of enthalpy considerations and perturbation of product-side MO's. LEach rearrangement product was partitioned into two segments whose 2nd order (occupied MO - unoccupied MO) interactions gave a measure of relative TS stabilizations. Details are given in Section 5.3.] The results may be summarized as follows. arranging ether. Let ROCH 2 R' be the re- Then for a series of substrates in which R' (or R) is constant, relative reactivities depend on R (or R' if R is fixed) in the order: vinyl > 2-naphthyl* > l-naphthyl > phenyl; 9-phenanthryl > 1-anthryl z l-naphthyl; (Si) 2-anthryl* > 2-naphthyl. Of the two isomers, ROCH2R' and R'OCH 2 R, when RfR', the one predicted to undergo the Claisen rearrangement faster is that in which the oxygen of the -OCH 2 - bridge is adjacent to the aryl group occupying the higher position in the sequence (S2): 9-phenanthryl > l-naphthyl > 2-naphthyl* > 1-anthryl > 2-anthryl* > phenyl *There are two possible rearrangement products when R = 2-naphthyl or 2-anthryl: One involves the [1,2] canonical w-bond position of the naphthyl or anthryl group, and is the one implied here. The other involves the [2,3- canonical ,r-bond positions, and is highly disfavored because it causes disruption of all the aromatic rings of the naphthyl or anthryl groups. ---------------- -------- illY -35E.g., when (R,R') = (2-naphthyl, phenyl), the 2-naphthoxy ether's Claisen rearrangement is predicted to be faster than that of the isomeric phenoxy ether. Claisen rearrangements (involving ArOCH 2 Ar') were concluded to be much likelier to occur in practice than the analogous Cope rearrangements of Ar(CH 2 )2Ar', primarily because of enthalpic effects; :'ow- ever, the relative reactivities of aromatic Cope rearrangements depends in theory on Ar (when Ar' is fixed) according to the ordering Sl. : The substantially positive enthalpy of reaction for Claisen re- arrangement of ArOCH2Ar' is nontheless significantly lower than the homolytic bond dissociation energy (BDE) of the latter, (thus making the rearrangement -- where structurally possible -- at least competitive with homolytic fission*; at least one closely related such rearrangement has been observed 148). The opposite seems probable for Cope rearrangements of ArCH2 CH2 Ar', versus homolysis of the latter in- to ArCH 2 and Ar'CH2 radicals. These results have important implications for the thermolysis mechanisms of ArXCH 2Ar' moieties/compounds, as discussed in Sections 1.1.2, 1.8 and 1.9. 1.6 Some Previous Coal Model Experiments Explained by the Theoretical Results While reaction mechanisms have not generally been unequivocally established for coal model compounds, much less for coals, examples are cited below where the sparse experimental data available in the literature fit the theoretically-derived reactivity patterns summar*For example, for PhOCH 2 Ph, AH298K = 168 kJ/mol for the Claisen rearrangement, and BDE= 214 kJ/mol for dissociation into PhO and PhCH2 radicals. For Ph(CH 2 )2 Ph, on the other hand, the corresponding estimates are 241 and 234 RJ/mol for Cope rearrangement and homolysis. -36ized in the preceding sections. (Additional examples of qualitative agreement were noted in passing in the text.) It will be recalled that the theoretical results were founded on assumptions of pericyclic reaction mechanisms. But it must be noted that some of the referenced experimental results can also be explained by free radical hypotheses. 1.6.1 Hydrogen Transfer Reactions Corresponding Theoretical Experimental Result Acceptor > 165 OM3 > DM11 (Table 4.1) 165 DM3 > DM11 > Coal >0 0 45 1,> W > Co > M _n_t_ H 2 1 1.6.2 1.3.2) Ref. Coal* Coal* Result (Sec. Donor Activity 6 > (Cc > t DM4 > DM11 165 DH2 > DH9 (Table 4.2) 166 D > DH2 > DH9 Ref Corresoondina Theoretical Result (Sec. 1.3.3) 53,167 AM5 > AM7 (Table 4.4) 165 AH8 > AH9 (Table 4.3) Retroene Reactions As discussed in Secs. 1.4 and 5.2.1, fragmentation via the re- troene mechanism is open to coal-like substrates of the type ArX(CH 2 )m+2R, where m>O particularly when X is oxygen. If a good H- donor is also present, the immediate retroene products may be hydrogenated. *Under confirmed kinetic control conditions. -~~E 111,1 IIII -37Predicted (This Work): 0 > >1> 0> Actual % conversions 1 66 56 10 5 <1 at 400cC, I hr, in C) (ref. 18) ( ------- ref. 20 -------- ) 1.6.3 Claisen/Cope Rearrangements Molecules of the structure ArXCH 2Ar', where X = 0 or CH2, were earlier noted to be capable of Claisen (X = 0) or Cope (X = CH2 ) rearrangements, a possible initial and rate determining step in their thermolysis; (however, when X = CH2, the rearrangement is not necessarily preferable to homolytic fission). Theoretical reactivity pat- terns for the rearrangement are discussed in Sections 1.5 and 5.3. Theoretical hierachy when X = CH2: Actual 5 conversions at 400 C, I hr, in Of - 19 5 (ref. 35) (ref. 20) Predicted for X = 0: - 0.06 (refs. 35,32) > 130:1 at 350 0C (refs. 36,35) Experimental: PART B: :: 160:1 at 400 0C (ref. 168) EXPERIMENTAL STUDIES Section 1.1.2, and the accompanying Table 2.2 summarized the objectives of this part of the thesis. 1.7 Exoerimental Methods All .experiments were conducted in the batch mode, with reactants charged in sufficient amounts to assure that reaction was in the -38liquid phase (>0.98 liquid fraction). Reactors consisted of small, precisely machined tubes (0.794 cm OD x 0.464 cm ID x 4.44 cm, which, with end-caos, had internal volume of 1.165 ml at room temperature), immersed in a fused-salt (NaNO 2 -NaNO 3 -KNO 3 eutectic) bath. Heat-up times were about 20 seconds for the reactions of benzyl phenyl ether (BPE) in hydronaphthalenes, conducted in the 2200-300 C range. bibenzyl reactions were conducted in the range 330 -415"C. were by 1H-NMR Neat Analyses and GC (column and conditions described in Section 6.1). Biphenyl was used as inert internal standard in the reactions of BPE in hydronaphthalenes. The basic reactant proportions used (at 3 or 4 equally spaced temperatures in tne aforesaid interval) were hydronaphthalene : BPE : biphenyl = -2.0:1:-0.30 (molar); this was complemented (at one or two of those temperatures) with scans for the effects of dilution on reaction rate and product selectivity, using initial hydronaphthalene : BPE : biphenyl mole ratios = -5.0:1:-0.30 and -10:1:-0.30. 1.8 Exoerimental Results Experimental results are summarized below for the reactions of BPE in 1,4-dihydronaphthalene (1,4-OHN), decalin, in turn. 1,2-DHN, tetralin and trans- This is followed in Sec. 1.8.3 by a tabular summary for easy cross-comparison of their salient features, and in Sec. 1.9 by a discussion of the results.* *Fuller details of the sented in Chapter 7. lysis of bibenzyl are digest as they are in experimental results summarized here are preAlso, results on the neat liquid phase, thermogiven in Sec. 6.3, but are omitted from this accord with recently published accounts. 2 ,3 ,35 vlffi M11" -391.8.1 Product Soectra In all the BPE reactions in the hydronaphthalenes, the following BPE-derived, low-molecular-weight products and isomers were confirmed. Present: (HMBPh; 56) Absent: Inferred but unconfirmed absence: rTraces of products resemblina these, in GC retention times, appeared at high extents of reaction, but the probable absence of these compounds was inferred from the confirmed absences of benzylphenols, bibenzyl, and (Ph) 2 CHOH, as co-products.] Further, the following C10 products derived from the hydronaphthalene "H-donors" were identified: From 1,4-dihydronaphthalene (1,4-OHN), ' : From 1,2-DHN, From tetralin, ~, , From trans-decalin, (3 : ; and traces of 9 : (probable structure); and 0 (subsequent co-product). In all the BPE/hydronaphthalene systems, products of unconfirmed structure heavier than CT30 were also apparent, including those suspected of being C17's and C16 0's; suspected C20 's were also in evi- -40- dence in the BPE/1,2-OHN and BPE/l ,4-HN systems, especially the for(GC-elutable heavy products were generally more pronounced in mer. the BPE/1,2-OHN system than in the others; see Sec. 1.8.2b below.) Compounds heavier than C20 1s were not eluted under the GC conditions used. Product Distribution, and Kinetics 1.8.2 The SPE/I,4-DHN reaction system -- which in many ways exhibited the simplest characteristics -- will be described first, in some detail, to provide a basis for the briefer, comparative description of the quantitative results obtained with the other BPE/hydronaphthalene systems. 1.8.2a The BPE/1,4-DHN system PhOH and PhCH 3, the principal 3PE-derived products, were formed in near-exactly T:1 mole ratios (see Fia. 7.1), being independent of reaction temperature and initial 1,4-DHN : BPE ratio, but mildly dependent on extent of reaction. As apparent from the selectivity diagram, Fig. 1.2a, the moles of PhOH formed per mole of BPE reacted was generally in the vicinity of 0.80, the exact value depending weakly on both temperature and BPE conversion, (<0.80 at low BPE conversions, and rising to a steady limit >0.80 at higher conversions). The yield of toluene followed the same trends as that of PhOH, although, at 0.72+0.05 moles PhCH3 per mole BPE reacted, it was usually slightly lower than that of phenol. Other than PhOH and PhCH 3, the BPE isomer 2-hydroxy-2'-methylbiphenyl (HMBPh) was the most significant BPE product, as indicated in Fig. 1.2a. First detectable at overall BPE conversions of -10%, it I -% (A~~ Ps .Arrfl (P -1 I r -42- rC 6 0 0.8hJ- P LCIIT ~t 6 %% u .. -N :- - ± -L .- , S.c - ....- 1.0T"< PhCH 3 0.6-1 Ph0H i o BSPE 5- 0.4- rC60 - PhOH + HIMBPh PhCH3 + HMBPh C7 Cr- 0.2- .1~ 0.0 HlDr II A 8 Reaction Time (mrin) 0 _ 2 ,,.. 9 at 30o 0C ----6 4 r 9 8 . .. . ..... ZCvfq A- Naph d -J 5- 0.80.6- (N/No C. 0.4- 0 1, 4-DHN (EC10 = Naph + Hydronaph's) 0.2- ( Tetralin 0 2 4 6 8 2-DHN 9 Fig. 1.2a,b : Selectivity diagrams for light products of the BPE/1,4-DHN system at 300±0.5 0 C, liquid phase. Initial mo!e ratios of 1,4-OHN : BPE : inert = 2.04:1:0.446. [(N/N ) i unconverted fraction of reactant i; HMBPh = 2-hydroxy-2'methylbiphenyl (BPE isomer); Naph = naphthalene.] 0l0ll MIllIIIllIlM i -43grew to a maximum at which it represented 6-9% of the BPE converted, at the temperatures studied. It should be noted that the summation of C,O units from phenol and HMBPh (shown as ZC 0 in Fig. 1.2a) typically amounted to > 0.95, implying excellent closure of the oxygen balance, since, as BPE is a C1 30 compound, each mole of it reacted is equivalent to one mole each of C6 0 and C7 segments in products. Thus the departures of )C60 and SC 7 from unity, in Fig. 1.2a, measure the extents of incorporation of C6 0 and C7 fragments from reacted BPE into unidentified C13 0 and heavier products (other than those ruled out in Sec. 1.8.1 above). In this light, the fact that TC7 = 0.85, while representing good closure, suggests the inclusion of more C7 than C6 0 segments in heavy products of the BPE/1,4-OHN system. The accompanying selectivity diagram, Fig. 1.25, for 1,4-DHNderived products is indicative of the dominance of naphthalene among the Co 10 products: typically -0.80 mole of naphthalene was formed per mole of 1,4-DHN reacted. Note that ZC10 , the sum of all C10 products, ranges between 0.80 and 0.90 in Fig. 1.2b, suggesting that the extent of incorporation of 1,4-DHN into heavy products was minor. For the BPE/dihydronaphthalene systems, a partial H-balance based on light products was monitored as the (molar) quotient, [2(CM)-~))/(PhOH + PhCH 3)], which measures how much of the H required for formation of PhOH and PhCH 3 from BPE is stoichiometricaliy accounted for by the net dehydrogenation of the OHN to naphthalene. (By using the difference between naphthalene and tetralin, any distortion by background disproportionation rea.ctions of the DHN, cf., 6.4, are eliminated). Sec. For the BPE/1,4-DHN reaction system, this H- iiiilililitiiITSiiil!Wi"llj ] l1 I11, e Ill: _________________________________________________________________________________ lIII.hIIII -45- FIG. 7,6: ORDER TEST WITH RESPECT TO BPE, REACTING IN 1,4-DHN ; 7 .".. -u i ~E is,. -- -I i A A rI 11 EI11 (1 1RL 1L T\Eil E isIi 9i I toD, 1 RI D IN CF ,E I I ' I . . ... - I ,2 ert c . . . 1 . l - Ii' I " -Il#latv -( ----- j() -- . .. . ... . .I ", llIil -47balance index stood at 100+10%, over the entire range of conditions (220*-300*C, initial 1,4-OHN : BPE ratios = 2:1 to -12:1) studied. All material balances thus pointed to the simplicity of the product spectrum of the BPE/1,4-OHN system, as already described, and to the relative insignificance of heavy (>C13 0) products. Regarding the kinetics, the intial hydronaphthalene : SPE mole ratios (>2:1) scanned allowed meaningful estimates of overall reaction order with respect to BPE concentration, only. Accordingly, the over- all BPE reaction rate was modelled simply'as d(CBPE)/dt = -kC n which, with the pseudo-first-order treatment,* gives (n-l) as the slope of the linear plot of log(Co,BPE) versus log(keff), as Co,BP E is For the BPE/I,4-OHN system, a true first order, n=l, was varied. found, see Figs. 7.5 and 7.6. The Arrhenius plot for that system's k is shown corresponds [iog 0 A(s 1.8.2b in Fig. 7.9 and to the parameters ),Ea (kJ/mol)] = (11.1+2.3,150+22]. The 8PE/1,2-DHN System The behavior of BPE in 1,2-DHN medium was strikingly different from that of the BPE/1,4-OHN system. In the first place, the PhOH : PhCH 3 product ratio was always substantially greater than unity, ranging between 3.5 and 9.5, and decreasing markedly with increasing reaction temperature and with increasing extent of reaction, see Fig. 7.10. Selectivity diagrams for this system are shown as Figs. 1.3a,b. In Fig. 1.3a, it is seen that the BPE conversion to phenol, 0.7±0.1, *Detailed description of the methodology is given in Sec. 7.1.2. Co,BPE and keff are the initial concentration (mol/liter) and pseudofirst-order BPE rate constant (s-l), respectively, where keff = kCn-1. 1. l I I I II 11 I 1[. 04 V, 64L a II t -m ill ~~ _~ S. iM..UM liIlI S -49- 1.0 0.8 0.6 0.4, 0.2, 0.0 0 5 10 15 20 Reaction Time (min) at 300*C 1.0 S. 0.8 .w 0.6 0 CC E 0.4 Fig. 1.3a,b : Selectivity diagrams for light products of the BPE/1,2-DHN system at 300-0.5°C, liquid phase. Initial mole ratios of 1,2-OHN : BPE : biphenyl = 2.01:1:0.299. [(N/N )i = unconverted fraction of reactant i; HMBPh .= 2-hydroxy-2'methylbiphenyl (BPE isomer); Naph = naphthalene.] hiIl,0 10lAO -50far exceeded that to toluene, 0.15+0.05. ably, the indices Further, and quite remark- ZC6 0 and C7 are of the orders 0.80+0.15 and 0.26+ 0.16, respectively, indicating that only -20-40% of the C7 fragments of reacted SPE are accounted for in light products, the remaining 8060% being presumably attributable to heavy products. (Contrast this with the situation in the BPE/1,4-OHN system, Sec. 1.8.2a.) In Fig. 1.3b, it is apparent that at 300 0 C, 0.15+0.03 mole of tetralin was formed, per mole of 1,2-OHN reacted, as against 0.35+0.03 mole of naphthalene. The high yields of tetralin imply considerable disproportionation, while the difference between naphthalene and tetralin measures the dehydrogenation of 1,2-DHN via other reactions. [The partial H-balance index, defined as the molar quotier.t 2(naphthalene-tetralin)/(PhOH + PhCH 3 ), as explained in Sec. .S.2a, was generally significantly less than -100%, in contrast with the BPE/ 1,4-OHN system, hinting that a significant part of the net hydrogen transferred from the 1,2-DHN was embodied in heavy (>C13 0) products.] The fact that C1 = 0.55+0.05, rather than -1, in Fig. 1.3b, points to the incorporation of about half of all reacted 1,2-DHN into heavy products. Regarding the kinetics of BPE thermolysis in 1,2-OHN, the initial reaction rates were appreciably slower than in 1,4-OHN. The convexity of the (N/N o)pE (= fraction of BPE unconverted) versus time curves, evident in Fig. 1.3a, persisted at all 4 temperatures studied. The overall BPE reaction rate at low conversions was second order in BPE concentration. Because of such convexity of the BPE concentration-ver- sus-time profiles, two sets of rate constants were abstracted: one for low BPE conversions, xBPE < 0.15, and another for moderate conver- I* 10 TO I It h1 .IItI V,, INf II , X1i 1. ,1 ,) 700 460700 IL1 1 i i '' 1 t i* ., :1 . , . '4 -52sions 0.1 5 <XBPE<0. 5 . Arrhenius plots for both are given in Fig. 7.16. The associated initial rate parameters, [logl 0 A(s Ea(kJ/mol) *liter*mol'), = (7.8+1.8,124+19), are the appropriate ones for cross- comparison with those of other BPE/hydronaphthalene systems. 1.8.2c The BPE/tetralin System The selectivity diagrams, Figs. 1.4a,b, for the BPE/tetralin system at 300 0 C, was typical of the light product distribution of that system, for the initial proportion of tetralin : BPE : biphenyl = 2:1:0.3. The diagrams show that, under the stated conditions, PhOH was formed in appreciable excess over PhCH 3 (but much less so than in the BFE/1,2-DHN system). A'so, 1,2-DHN was initia'ly the sole tetra- lin-dehydrogenation product, whose yield curve ultimately fell as it became involved in reaction, including formation of naphthalene. (Note, however, that tetralin always remained in large excess, ensuring relatively low concentrations of 1,2-DHN.) A role for 1,2-DHN formed in situ in the system is further suggested by Fig. 7.17 which shows the dependence of the PhOH : PhCH 3 product ratio on initial tetralin : BPE ratio, extent of reaction, and temperature (B /Ao , xA, and T). At constant T, PhOH/PhCH 3 increases to an asymptotic maximum as xA increases, which is the outcome expected (cf., Sec. 1.8.2b) as 1,2-OHN increases in concentration. At constant T, progressively higher B /A o values diminish the effective concentration of nascent 1,2-0DHN, creases until at 8 /A o0 and PhOH/PhCH 3 commensurately de- 10, PhOH : PhCH 3 is effectively 1:1 at all extents of reaction. Returning to the selectivity diagrams Figs. 1.4a,b, it is seen that IC6 0 = 0.90+0.05 is of order unity as was the case in the -53- --- -- 1.0 I--- -1 V' EC60 - - - -t '+.0.81 PhOH ZC7 0= a o_ c. 0. 6- PhCH3 _o 0.4 0.2 (ZC6 0 (C 0.2 7 " (N/No)BPE PhOH + C130's) E PhCH 3 + C1 3 O 's) C,30's c ............ ~ U.U 10 5 0 15 Reaction Time (min) at 300 0 C 0 1.0 5 r 10 t i .. 15 (/N)o tetralin 0.8-S .C n- C10 0.6i Naph + Hydronaph's I- 0.4 1,2-DHN o E C. 0.21 Naph 0r Sr .., 0.0 r- -, , - - HHN Fig. 1.4a,b : Selectivity diagrams for light products of the BPE/tetralin system at 300_0.5C, liquid phase. Initial mole ratios, tetralin : BPE : biphenyl = 2.134:1:0.300. [(N/No) i = unconverted fraction of reactant i; C13 0's * all 0130derivatives of BPE, mainly 2-hydroxy-2'-methylbiphenyl; Naph = naphthalene; HHN = hexahydronaphthalene.] ri -65BPE/DHN systems, whereas ZC7 = 0.65 is appreciably less than unity, implying -- as in the BPE/1,2-OHN reaction system, see Fig. 1.3a -that more C7 than C6 0 fragments of reacted BPE went into heavy products. Also in Fig. 1.4b, the fact that C10 = 0.55+0.05 indicates that, as with the BPE/1,2-DHN system, (cf., Sec. 1.8.2b), about half of the converted H-donor, tetralin, ends up in heavy products. Related to this, the partial H-balance based on light products [as measured by the quotient [2(~4 +20~ )/(Ph0H + PhCH3 )] stood at only 0.50+0.10, signifying that, on a net basis, only about half of the hydrogen originating from tetralin was incorporated into light products. With regard to reaction rates, it was found that the onset of perceptible consumption of reactants in the BPE/tetralin system was strongly temperature-, and weakly concentration-, dependent. fied by Fig. 7.23, when the unconverted fraction (N/i'o) i As typi- was plotted as ln(N/No) i against time, the zero conversion intercept z ("induction time") on the time axis was consistently and significantly positive. Thus, with the initial tetralin : BPE : biphenyl mole ratios fixed at 2.1:1:0.30, the induction times, r, were 53.5, 10.9, and 2.55 min. at 223.5*, 260.20, and 300 0 C, respectively -- too large and con- sistent to be attributable to reactor heat-up times of -20 seconds. (Besides, the phenomenon was not found in the other BPE/hydronaphthalene systems.) The mathematical description of the kinetics of the system was therefore broken into separate induction and post-induction reaction regimes. As T increased markedly with increasing T, and to some lesser extent with increasing B /Ao at constant T, the weaker dependence on rl I I LO I I ~ II N IN I'~OtAl S JA 7,12F: l i BEIr e I t II0 ,LCGIO J) 1 ' It .(21)2---- rII OOLO 9P 44 es' X II ) 1 IaI I -58concentration was ignored and the induction rate was modelled as a temperature-activated function: (7.7) (l/T) = (/o)exp(-Ea,J/RT) with Cl lo 010with 1 o (s 1 ),Ea,r (kJ/mol)] = (5.83+1.7,88.8+18). The post-in- duction reaction rate of BPE was estimated to be approximately second order with respect to SPE concentration, with Arrhenius parameters, Fig. 7.24, v'ery similar to those of the initial BPE reaction rate in 1,2-DHN. 1.7.2d The BPE/trans-decalin System Exploration of this "control" case was limited to 30000. Io in- duction phenomenon was apparent. The product selectivity patterns of this system were appreciably. different from those in the other BPE/hydronaphthalene systems, especially in the low fraction (<30%) of reacted BPE accountable as light products (PhOH, PhCH 3 and BPE isomer). This implied extensive forma- tion of heavy products (molecular weights exceeding C17 's and C1 6 0's) not eluted by GC. PhOH and PhCH 3 yields were comparable. The overall consumption of t-decalin was always very small (<2%). The overall BPE reaction rate appeared to fit n=1.5 order with respect to SPE concentration better than n=2, with associated rate constant k = 3.92+0.07E-4 (mol/litre)-1/ 2 s-1 . However, the effective initial BPE transformation rate at 300 0 C was comparable to the initial rate in the BPE/1,2-OHN system as well as to the post-induction rate in the BPE/tetralin system; Call of which were significantly slower than that of the BPE/1,4-OHN system). L Table 1.1: r ComparaLive Suniiary of Reaction 8il1 in1 Ilydrona !hthalenes. eCharacteristics of- --~-~----- Solvent: PhOH/PhCI13 mole ratio Mol PhOll/mol BPE reacted Mol 1 /mol BPE reacted (1,4-DIIN) (1,2-DIIN) (tetralin) 1.0-1.2 3.5-9.5 1.0-2.0 f(xA)a f(T,xA) f(T, o/Ao,xA) 0.7-0.9 f(xA) 0.5-0.9 f(xA,T) 0.5-0.8 f(xA,T) 0.05-0.5 f(Bo/Ao,xA,T) 0-0.1 0-0.1 0-0.1 0-0 f(xA) f(xA) f(xA) 0 No No 1 2 2 1.5 90s 400s 360s 1800s 300s(150)b 1700s(630) .360s (t-decalin) Reaction Kinetics: Temperature activated (E 1 (Ea,r_88.8kJ/IIl) induction process? Order w*r*t BPE Time for 15% BPE conversion at mol/1 CoBPE=1.5+0. T=300 0C T=260 C 0 I i - --- -I-c~---~-l-- --------- - 7.8 .8 c 1241 19 3 CC~------ - - --- I- Yes 888 kJ/ol)No -- 8.11+1.0 123.5-20 _post-inductio)__ Table 1.1 (Cont'd): 1,4-1DIN 1,2-DIIN tetralin t-decalin 0.5-0.91 f(xA,T) 0.05-0,5 f(Bo/A ,xA,T) Material balances on Sight (C 1 3 0) products: YC6 0 (per mnol BPE reacted) 0.7-1.0 f(xA) 0.5-0.91- j.C7 (per mol BPE reacted) 0.6-0.91- 0.1-0.4 C10 (per mol solvent reacted) Partial I1-balance Notes: (a) f(xA) 0.4-0.6 f(T,B /A ,xA ) 0.05-0.4 f(Bo/A o ,x A , T) f(xA) f(xA'T) 0.8-0.9 0.3-0.55 f(xA, Bo/Ao,T) 0.4-0.7 f(T,B 0/Ao ) 100110% 20-200% 50±10% 0-30% f(xA,B13/AoT) f(xA ,B /Ao , T) f(B /Ao IxA ) f( ) denotes functional dependence on x , Bo/A conversion of BPE, Bo/A 0 = and/or T, where xA initial mole ratio of solvent to BPE, = fractional and T = temp- erature. (b) for tetralin, induction time, in seconds, is shown in parentheses. (c) These Arrhenius parameters for BPE in 1,2-DIIN are valid only for O<xBPE<O.15. (d) Definition depends on the solvent identity, see text Sec. 1.8.2. -611.8.3 Tabular Summary of the Experimental Results The main features of the experimental results discussed above for all four BPE/hydronaphthalene reaction systems are tabulated in Table 1.1. 1.9 Discussion of Exoerimental Results Discussion of the present experimental results is undertaken in three steps, namely (i) qualitative identification of the principal pathways apparently operating during BPE thermolysis in H-donor solvents; (ii) postulation and assessment of detailed mechanisms (both free radical and pericyclic) consistent with these pathways; and (iii) comparison with previous work. 1.9.1 Reaction Pathways An examination of the results outlined in Sec. 1.8 suggests the existence of three main reaction paths in BPE thermolysis: (a) re- arrangement of BPE to form, ultimately, the observed isomer, HIMBPh; (b) hydrogen transfer to (some derivative form of) BPE leading to the observed PhOH and PhCH 3 fragmentation products; and (c) addition reactions involving BPE-derived moieties, leading to heavy products. Path (a) appears to operate to the same (small) degree, regardless of the solvent identity, but paths (b) and (c) are competitive. over, in both paths (b) and (c), BPE (or one tives) may react either with itself of with Whether the "self-reaction" of BPE wins, More- of its C13 0 derivathe H-donor solvent. or its cross-reaction with the H-donor solvent, depends on the H-transfer and addition abilities of the solvent, relative to BPE (or its active form). For example, in trans-decalin, an essentially inert solvent, the BPE thermolysis would be dominated by its self-reactions, whereas in 1,4-DHN, which is an excellent H-donor but a poor adder, pathway (b) would dominate. In 1,2-OHN, which is both a good H-donor and a powerful adder, pathways (b) and (c) are both in evidence, as is lesser the case in tetralin. Within the framework of these pathways, the observed reaction kinetics shed light on the sequence of events. Since the BPE thermo- lysis kinetics are substantially similar in 1,2-DHN, tetralin and t- decalin as solvents, the rate-determining step (RDS) in these cases is likely dominated by the self-reaction of BPE (or derivative) moieties, with the product-selectivity-determining solvent intervention occurring only subsequently. The fact that the reaction rate is enhanced, and the kinetic order with respect to BP. reduced, in 1,4-OHN solvent, suggest that both BPE and 1,4-OHN are involved in the RDS, and that the RDS is probably a hydrogen transfer. As the reaction of 1,4-OHN with SPE (or derivative) is thus more facile than the self-reactions of the latter, pathway (b) would dominate over (c), and lead to the observed clean product spectrum when 1,4-OHN is the H-donor solvent. Reaction Mechanisms 1.9.2 The basic mechanistic interest lies in the observed, similarities and differences between the BPE/1,4-DHN and BPE/l,2-OHN systems, since the experimental results pointed convincingly to a role for 1,2-OHN, generated in situ, in the BPE/tetralin reaction system, 1.9.2a Free Radical Mechanisms The postulated free radical mechanisms for these systems are shown in Fig. 8.1. At low extents of reaction, the pivotal steps, following the dissociation of BPE into PhO and PhCH 2 radicals, are the F 1lllllrluM 11 wamam w -63- Fig. 8.1: Generalized Free Radical Mechanism for BPE Reactions in Dihydronaphthalene (A) Chain-initiating Eauilibrium: (R8.1) -----Ph + PhCH 2 PhOCH 2 Ph (8PE) Radical Additions to R8.2a R8.2b a XPhX XPh (C) H-Abstractions: (R8.4a) (RBS.4b) PhR - + " PhXH + (RBS.c) PhOCHPh (SPE -) PhOCH 2 Ph (3PE) JI (R8.5a) + 00" (58) (R8. 5b) (D)~1,2] Phenyl Shifts: PhOCHPh (R8.5c) BPE* BPE (Ph) 2 CHOH . 8.6 (Ph)2CHO H-abst./disprop (E) Radical Disoroportionations: PhX or (Ph) 2 C==O '8 .7) PhXH (R8 .8) BPE 8-PE* -(R8.9) BPE .%. (F) Termination Products: 00Ph "H 3Ph In=,e QaI*I-bP , etc. -64competitive addition (R8.2) versus abstraction (R8.4) reactions of those radicals with a DHN molecule: addition leads to a C17, C16 0 or heavier product, H-abstraction results in PhOH or PhCH 3. Consider first the case of 1,2-DHN as the H-donor/addend with which PhCH 2 or PhO would react. The only way in which a free radical mechanism can explain the fact that the PhOH : PhCH 3 product is substantially higher than unity (actually 3-10) in the BPE/1,2-HN reaction system is by assuming that the differential tendency of PhO to Habstract rather than add to 1,2-DHN is higher than that of PhCH . 2 Now, because PhCH 2 has the higher spin electron density at the X position of the PhX radical than does PhO, it would add to the non-aromatic sr-bond of 1,2-OHN faster than PhO would; but, by tne same token, PhCH 2 would also abstract H from 1,2-DHN faster than PhO would. This runs contrary to the essential assumption needed for the free radical rationalization of high PhOH : PhCH 3 product ratio in 1,2-DHN. In fact, for a given PhX radical, the quotient of rate constants (kab/kadd)PhX -- which measures the relative rates of H-abstraction versus addition to the nonaromatic w-bond of the OHN -- is determined by the difference in energetics between the transition states [l,2-DHN---XPh]ab, for H-abstraction and [1,2-DHN---XPh]ad d , for addi- The effect of whether X is actually CH2 or 0 is thus minimized tion. or eliminated by the implicit substraction of the two transition states. Therefore, (kab/kadd)Ph 0 = (kab/kadd)PhCH2) , 8.2 and this would assure that the PhOH : PhCH 3 ratio would be just as -65nearly 1:1 in 1,2-OHN as in 1,4-OHN, even though the absolute yields (mole per mole of BPE reacted) of PhOH and PhCH 3 might change in going from 1,2-OHN to 1,4-OHN. This counterargument is one of the primary reasons for doubting free radical explanations for experimental results presented in this thesis; (see Table 1.2 of Sec. 1.9.2c). In the free radical framework, the induction phenomenon observed in the BPE/tetraTin system might be attributable to the time needed to generate a critical minimum concentration of 1,2-DHN in situ from tetralin, after which 1,2-OHN supplants tetralin as the preferred H-donor (as well as addend for PhX radicals). (Initially the benzylic H's of tetralin would be the only H source besides BPE itself.) How the post-induction activity of nascent 1,2-OHN explains the kinetics and overall PhOH : PhCH 3 selectivity of the BPE/tetralin system (irrespective of whether the mechanism is free radical or pericyclic) has already been noted in Section 1.8.2c above. In the BPE/trans-decalin system, the PhX radicals would mostly have to abstract H from SPE or its derivatives, t-decalin being a poor H-donor. This would lead to heavy PhOCHPh and PhX recombination and condensation products, as further discussed in Sec. 8.3.4. 1.9.2b Pericyclic Mechanisms Whereas the free radical mechanism for PhOCH 2 Ph reactions is initiated by homolytic dissociation of the 0-C bond, the pericyclic pathway hinges on the Claisen rearrangement (reaction R8.10 of Figs. 8.2 and 8.3), which, as noted in Sec. 1.5, is less endothermic than BPE homolysis by -46 kJ/mol. Once formed, the Claisen-rearranged BPE, (60), has three options (besides reverting to BPE): -66Fia. 8.2: Postulated Pericyclic Mechanism for BPE in 1,2-OHN Claisen rearrancement (and subsequent tautomerism): R8.10 o S- " .I R8. 11 u (60) (59) s6) 2H-transfer to rl,2 or S ,61 positions of (50), folowec ov retro-ene framentation to ?PhH and PhCHr. (60) (6o) (Go) R8.12 Q8.la> (Go) + (61) SR8.13 (63) (60) + R8.15 > (61) Diels-Alder addition of 1,2-OHN to r9,12] positions of 50) (and sequel reactions: (60) + o R8.17 R8.16 (64) (55) (R8.1 8) (60) OR R8.19 (66) OH (67) IMillIM -67Fig. 8.3: Postulated Pericyclic Mechanism for BPE in 1,4-HN Claisen rearrangement (and subsequent tautomerism): OH R8.11 R8.10 • ., (59) 2H-transfer to [3,5 or ,9,121 cositions of (60), : follo,,ed by retro-ene fragentation to PhOH -- and PhCH3 --- .2R8.21 R8.20 (60) + 0 H H68) r68) R8.23 (60) + (60) "--- - - (62) R8.22 (69 (69) (70, -t-2 . (61) Diels-Alder addition of 1,4-DHN to [9,l]2 positions of (60) (and sequel reaactions): (60) (60) ++o (gslow) (7)..Co (71) (72) (R8.27) R8,28 (73) + )WOK L -68(i) It may tautomerize to 2-hydroxy-2'-methylbiphenyl (56), via known 14 3 , 14 8 but non-pericyclic thermal [1,3] H-shifts. Or, (ii) It can be hydrocenated by concerted 2H-transfer, in accordance with selection rules of pericyclic reactions, [meaning that 1,2-DHN and 1,4-OHN can only hydrogenate different ,rsegments of (60); see reactions R8.12 in Fig. 8.2 and R8.20 in Fig. 8.3, and cf., Sec. 1.3.1]*. Such hydrogenation sets the stage for an exothermic and highly facile retroene fragmentation to PhOH and PhCH 3 , in 1:1 ratio (R8.13, Fig. 8.2, or R8.21, Fig. 8.3). (iii) Or, The Claisen-rearranged BPE may undergo Diels-Alder addition to a DHN (or to another Claisen-rearranged BPE molecule, if there is not a good H-donor source in abundance; e.g., in the neat thermolysis of BPE, or in the SPE/trans-decalin system). It is shown in Section 8.3 that, on theoretical grounds, and by analogy with documented experimental cases, tha CHN's would rather add to the C7 side than to the C60 of the bicyclic structure (60), as depicted in R8.16, Fig. 8.2, and R8.25, Fig. 8.3, and that the DielsAlder addition would be 102 +1 times faster in the BPE/1 ,2-DHN system *The situation is slightly complicated, in the BPE/1,2-OHN systems, by the fact that the H's attached to the bridgehead carbons -- positions (7,8) -- of (60) are of the same symmetry type as, and superior to, the dimethylenic hydrogens of 1,2-DHN. Consequently, the regenerative H-transfer path R8.12 + R8.14 in Fig. 8.2 may be preferred to the direct hydrogenation R8.15. This may be partly why the overall kinetic order with respect to BPE is higher than unity in the BPE/ 1,2-OHN and BPE/tetralin systems. By contrast, the indirect path R8.22 + R8.23 + R8.24 in Fig.8.3 is disfavored, in the BPE/1,4-DHN system, as it is both non-regenerative and energetically disadvantageous to R8.20; that assures unit order with respect to SPE. -69than in the BPE/1,4-DHN one. These considerations -- plus the fact that hydrogenation of the Diels-Alder adduct is still feasible, ultimately yielding PhOH in excess over PhCH 3 , via path R8.16 + R8.18 + 8.19 in Fig. 8.2, or path R8.25 + R8.27 + R8.28 in Fig. 8.3 -- would explain why the overall PhOH : PhCH 3 selectivity is high in 1,2-DHN, but very close to 1:1 in 1,4-OHN, medium. By the same token, there would be more C17 and other heavy products [presumably, structures (67) and (65)] in the BPE/1,2-DHN system than in the BPE/1,4-OHN one. Because tetralin is a poorer H-donor than the DHN's, and because its preferred pericyclic dehydrogenation product is 1,2-OHN (cf., Secs. 1.3.2 and 4.2.2), matters in the BPE/tetralin system would be strongly influenced by 1,2-DHN generated in situ, both with respect to product selectivities and kinetics, as observed in fact (cf., Sec. 1.8.2c above). A partial answer to the induction phenomenon in the BPE/tetralin system would thus be the time needed to produce the ,.threshold concentration of 1,2-DHN, below which the latter is too dilute to be effective. In the BPE/trans-decalin system, some PhOH and PhCH 3 would be formed, in comparable amounts, but heavy products would prevail, due to the likely predominance of self-additions of Claisen-rearranged BPE under such H starving conditions. 1.9.2c Comparative Summary of Mechanisms The preceding arguments are summarized in Table 1.2. It is evi- dent therefrom that, in the end, the pericyclic pathway was more successful than the free radical in rationalizing most facets of the results. -70Table 1.2: Scoresheet for adequacy of free radical and pericyclic rationalizations for the experimental results Result Free Radical Pericyclic 1. Disjoint temperature intervals, -2200300'C versus -360 0 -410 0 C, for roughly equal reactivities of PhOCH 2 Ph and PhCH7CH 2 Ph, vis-a-vis their small (-20+22 kJ/mol) difference in bond dissEciation energy into PhX radicals. x 2. Main BPE isomer in all BPE/hydronaphthalene systems is 2-hydroxy-2'-methylbiphenyl, even at low extents of reaction. x v 3. o-Benzylphenol and (Ph) 2 CHOH absent; (absence of p-benzylphenol and (Ph) 2 C=O also suspected). x / 4. PhOH : PhCH 3 mole ratios in BPE reactions in 1,2-OHN differ strikingly from those in 1,4-OHN. x 5. Formation of heavies (>C1 7 ) much more significant in SPE/1,2-OHN than in BPE/1 ,4-OHN. / 6, PhOH : PhCH 3 ratios for BPE/tetralin is a multivariate function, f(BO/A 0 ,xAT). y 7. Temp.-activated induction process in BPE/tetralin system. 8. Initial BPE reaction rate in 1,4-OHN significantly higher than in 1,2-DHN. 9. Kinetic order w.r.t. BPE: (a) n=1 in 1,4-DHN, (b) n=2 in 1,2-OHN, tetralin and transdecalin 10. Very low yields of light products (<C13 0) in the BPE/trans-decalin system. * x V x / / Table 8.3 Sumiinary of Results of Previous Workers on BPE Reactions in Ii-donor Media Schl osber9 39 Brucker and Kolling36 lt-donor solvent TIIQ* neat Tetralin Whitehurst28 1(Q Tetralin "Large Excess" 0 2.73 2.73 0.5 Reaction Temp. (°C) 300-390 375 375 375 399 Reaction Time (min.) 2.10 Donor solvent : BPE- Sitchin 37 lieat mole ratio BPE Conversion PhOll : PICH3 mole ratio 1.03 at 3000C 1.00 at >375'C Variable 1 17% at 300 C 70.5% at 3500C 00% at 375SC --- 30-40% ---- 2.8 1.2 100% Variable 7- 1.3 *TIIQ = tetrahydroquinoline, -200-310 -"c---- - -72Comoarison with Previous Work 1.9.3 While no other extensive study exists of BPE thermolysis in Hdonor solvents, previous experimental findings by Schlosberg, 39 Whitehurst,45 Brucker and Kolling,36 and Sitchin,37 are summarized in Table 8.3. Comparisons between this and our Table 1.1 reveal the pre- vious results to be either special cases or analogs of the present It should also be noted that the free radical explanations sug- ones. gested by some 39 '45 of those previous authors for BPE are beset by much the same fundamental problems as noted above for that pathway, vis-a-vis the pericyclic. For bibenzyl, the findings of the thesis agreed well with those of Sato, 34 ' 35 Poutsma22 and ot?,ers, anc the operation of a radical mechanism was corroborated as the more likely. The theoretical studies of H-donor, H-acceptor reactivities, etc., undertaken in Part A in the context of coal chemistry is, to the author's knowledge, unprecedented. 1.10 1. Conclusions and Significance of the Ihesis Product distributions and kinetics of benzyl phenyl ether (BPE) reactions in 1,4-, and 1,2-dihydronaphthalene (DHN) and tetralin were studied for the first time in the temperature range 220 0 C-300 0 C, as were the reactions of BPE in trans-decalin (control) at 300 0 C. Neat, liquid-phase reactions of bibenzyl were also examined in the range 360 0 -410 0C. These represent a sianificant broadening of the quantitative data-base on coal-model-comoound reactions in H-donor solvents. 2. The controlled, comparative study of the four hydronaphthalenes -73used as H-donor media for BPE reactions revealed that ostensibly minor perturbations in the chemical structure of H-donors can affect or alter the prevailing reaction rates and/or mechanism of a given coal/model-compound. In particular, the significant differences in characteristics of the BPE/1,4-DHN, BPE/1,2-DHN and BPE/tetralin systems demonstrated that, even amona "qood H-donors," the identity of the H-donor can influence the selectivities of light and heavy products of the coal-(model-compound) substrate as well as the reaction rates. 3. Three major pathways were delineated for BPE thermolysis: (a) re- arrangement leading to isomerization; (b) hydrogenations, leading ultimately to PhOH and PhCH 3 products and (c) addition reactions, engendering heavy (>C13 0) products. Paths (b) and (c) are competitive and may each include self-reactions of BPE-derivatives and reactions of the latter with the solvent, to relative extents determined by the solvent's H-donor versus rr-bond-addition propensities. The best sol- vents, e,g., 1,4-OHN, suppress path Cc) and lead to predominantly (and desirable) light products. 4. Detailed pericyclic and free radical reaction mechanisms based on the above (item 3) pathways were proposed (Chapter 8) to explain the experimental results for'the four BPE/hydronaphthalene systems considered, and the relative merits of the two schemes were assessed. The postulated pericyclic mechanisms were judged to be more successful, in these cases, than the free radical ones, for the temperature interval studied. For bibenzyl, on the other hand, the free radical pathways (laid out in Section 6.3.3) were strongly supported. ably competitive nature of these two sets of mechanisms in coal The vi- -74chemistry was thereby brought across. This is a departure from the near-exclusive bias towards the free radical pathways in most previous coal-related literature. 5. The broad predictive powers of perturbation molecular orbital (PMO) theory -- including frontier orbital (FO) theory -- was introduced and applied extensively to the study of coal chemistry, apparently for the first time. In Part A of the thesis (Chapters 3-5), the methods were adapted and applied to studying the factors that govern the reactivities of potential H-donors to coal, H-acceptor structures in coal model compounds, and certain Cope/Claisen and retroene reactions to which coals are susceptible. In part B, some re- sults and methods of Part A were invaluably applied to rationalizing experimental results on coal-model compounds. This illustrates the ultimate applicability of theoretical PMO techniques and results to process-oriented coal research. 6. To cite some theoretical results: it was argued that the most im- portant determinants of reactiv-ty for H-dcnors and H-acceptors are their r- and pseudo-w-bond configurations, which define their FO characteristics -- particularly the HOMO (for H-donors) and LUMO (for Hacceptors) and the occupied/vacant MO's closely adjoining those. It was thereby deduced that,among strictly hydrocarbon compounds, having C-H bonds flanked by non-aromatic r-bond(s) is more contributory to Hdonor activity than having them flanked by aryl groups, although nonaromatic unsaturation sometimes fosters undesirable addition reactions. For these reasons, and n, %are predicted to be among the best practical, structurally simple H-donors. Tetralin owes its acclaimed H-donor activity in part to in situ formation of 4 -75, a better H-donor, Cwhose undesirable tendency to polymerize and ( disproportionate can be prevented with the use of high tetralin : Hacceptor-substrate ratios). The same theoretical tools were also used in tackling a number of known "anomalies" in H-donor activity. 7. It was also concluded, on the joint basis of the theoretical and experimental parts of this work, that as pericyclic mechanisms appear to apply to PhOCH 2 Ph, they most likely are also feasible for most ArOCH2 Ar' and ArOCH2R moieties in coals. Entities like Ar(CH2 )m+2 Ar' (where m>O) are, on the other hand, likelier to prefer fragmentation through free radical mechanisms. -76LIST OF REFERENCES I. Lowry, H.H., Ed., "Chemistry of Coal Utilization, Suppl. Vol.; John Wiley, New York (1963). 2. Van Krevelin, D.W., "Coal"; Elsevier Publ. Amsterdam/New York (1961); chapter XI. Co./Van Nostrand, 3. See, for example, (a) Hottel, H.C., and Howard, J.B., "New Energy Technology -- Some Facts and Assessments"; M.I.T. Press, Cambridge, Mass. (1971). (b) Corey, R.C., in "Riegel's Handbook of Industrial Chemistry," 7th Ed.; J .A. Kent, Ed.; Van Nostrand Reinhold, New York (1974); chapter 3: "Coal Technology." 4. Gould, R.F Ed., "Coal Science," Advances in Chemistry Series, 55, Americ;an Chemical Society, Washington, D.C. (1966). 5. Tingey, G.L., and Morrey, J.R., "Coal Structure and Reactivity," Battelle Energy Report; Battelle Pacific Northwest Lab., Richland, Washington (1973). 6. For more recent work on structure of coals and coal-derived substances, see for example, (a) Whitehurst, D.D., Farcasiu, M., Mitchell, T.O., and Dickert Jr., J.J.: "The Nature and Origin of Asphaltenes in Processed Coals," Annual Report No. EPRI AF-480; Mobil R&D Corp., Princeton, N.G. (1977); pp. 7.92-7.93. fb) Far,:asiu, M., Fuel, 56(1), 9 (1977). (c) Deno, N.C., Greiger, B.A., and Stroud I S.G., Fuel, 57(8), 455 (1978); Deno, et al., Fue l, 59(10), 701 (1980); ibid., 59(10), 694, 699 (1980). (d) Ruberto, R.G., Cronauer, D.C., Jewell , D.M., and Seshadri, K.S., Fuel, 56(1), 17, 25 (1977). (e) Bartle, K.D., Martin, T.G., and Willi ams, D.F., Fuel, 54(4), 226 (1975); Bartle, K.D., Calimli, A., et al., Fuel, 58(6), 423 (1979). (f) Bodzek, D. and Marzec, A., Fuel, 60(1), 47 (1981). 7. Ref. 2, chapter VI. 8. Oka, M., Chang, H.C., and Gavalas, G.R., Fuel, 56(1), 3 (1977). 9. Fuchs, W., and Sandhoff, A.G., Ind. Ena. Chem., 34, 567 (1942). -7710. Given, P.H.,'Fuel, 39, 147 (1960). 11. Hill, G.R., and Lyon, L.B., 12. Wiser, W.H., Preprints, Div. Fuel Chem., Am. Chem. Soc., 20(2), Ind. Eng. Chem., 54(6), 36 (1962). 116 (1975). 13. Suuberg, E.M., Sc. D. Thesis, Massachusetts Institute of Technology, August 1977; chapter 3. 14. Whitehurst, D.D., Farcasiu, M., and Mitchell, T.O., "The Nature and Origin of Asphaltenes in Processed Coals," Annual Report No. EPRI AF-252; Mobil R&D Corp., Princeton, N.J. (1976); page 8.32. See also, ref. 6a, pages 7.99 and 7.117. 15. Dryden, I.G.C., in "Chemistry of Coal Utilization," Suppl. Vol.; Lowry, H.H., Editor; John Wiley, New York (1963); chapter 6, pp. 256, 260. 16. Heredy, L.A., and Wender, I., Preprints, Div. Fuel Chem., Am. Chem. Soc., 25(4), 38 (1980); Fig. 1. 17. Wender, I., Preprints, Div. Fuel Chem., Am. Chem. Soc., 20(4), 16 (1975). 18. Klein, M.T., Sc.D. Thesis, Massachusetts Institute of Technology, February 1981. 19. Sweeting, J.W., and Wilshire, J.F.K., Austral. J. Chem., 15, 89 (1962). 20. Benjamin, 8.M., Raaen, V.F., Maupin, P.H., Brown, L.L., and Collins, C.J., Fuel, 57, 269 (1978). 21. Horrex, C., and Miles, S.E., Disc. Faraday Soc., 10, 187 (1951). 22. Poutsma, M.L., Preprints, Div. Pet. Chem., Am. Chem. Soc., 25(1), (1980). 23. Vernon, L.W., Fuel, 59, 103 (1980). 24. Livingstone, R., Zeldes, H., and Conradi, M.S., J. Am. Chem. Soc., 101, 4312 (1979). 25. Neavel, R.C., 26. Curran, G.P., Struck, R.T., and Gorin, E., Ind. Eng. Chem., Process Des. Dev., 6(2), 166 (1967). 27. Hill, G.R., Hairiri, H., Reed, R.I., and Anderson, L.L., in ref. 4, chapter 27, p. 427. Fuel, 55, 237 (1976). -7828. Whitehurst, D.D., et al., ref. 6a, chapter 8. 29. Coll i ns, C.J., Raaen, V.F., Benjamin, B.M., and Kabalka, G.W., Fuel, 56, 107 (1977). 30. Benjamin, B.M., 31. Ref. 6a, pp. 9.40 - 9.42. 32. Cronauer, D.C., Jewell, D.M., Shah, Y.T ., and Kueser, K.A., Eng. Chem. Fundam., 17(4), 291 (1978). 33. Vernon, L.W., 143 (1979). 34. Sato, Y., Yamakawa, T., Onishi, R., Kameyama, H., and Amano, A., J. Japan Petrol. Inst., 21(2), 110-115 (1978). 35. Sato, Y., 36. Bricker, V.R., and Kolling, G., Brennst. Chem., 37. Sitchin, Ricardo: nology, May 1980. 38. Ref. 6a, p. 9.45. 39. Schlosberg, R.H., Davis Jr., W.H., and Ashe, T.R., 201 (1981). 40. Schlosberg, R.H., Ashe, T.R., Fuel, 60(3), 155 (1981). 41. Reifarth, K., Ph.D. Thesis, University of Berlin, 1977. 42. "InCronauer, D.C., Jewell, D.M., Shah, Y.T., and Modi, R.J.: vestigatio n of Mechanism of Hydrogen Transfer in Coal Hyd rogenation," Pha se 1 Final Report. Under Dept. of Energy Contr act No. E(49-18)-2 305, February (1978). 43. Hellyar, M.J., Sc.D. Thesis in preparation, M.I.T. (1981). 44. Ruberto, R.G., Cronauer, D.C., Fuel, 56( 1), 25 (1977). 45. Ref. 26, p. 168, Table IV. 46. Ross, D.S., and Blessing, J.E., Fuel, 47. Hooper, R.J., Battaerd, H.A.J., and Evans, D.G., Fuel, 58(2), 132 (1979). Fuel, 57(6), Preprints, 378 (1978). Div. Fuel Chem., Ind. Am. Chem. Soc., 2 -(2), Fuel, 58, 318 (1979). 46, 41 (1965). B.S. Thesis, Massachusetts Institute of Tech- Pancirov, R.J. Fuel, 60(3), and Donaldson, M., Jewell, D.M. and Seshadri, K.S., 58(6), 433 (1981); Table 1. -79- 48. Benjamin, B.M., Hagaman, E.W., Raaen, V.F., and Collins, C.J., Fuel, 58(5), 386 (1979). 49. Bredael, P., 50. Gill, G.B., and Hawkins, S., J. Chem. Soc.,- Chem. Commun. (1974), p. 742. 51. Debiccari, A.: B.S. Thesis, Massachusetts Institute of Technology, May 1981. 52. Heesing, A., and Millers, W., Chem. Ser., 113, 9-18 (1980). 53. Bass, D.H.: May 1981. 54. Geiser, D.J.: June 1979. 55. Bredael, P., and Rietvelde, D., Fuel, 58(3), 215 (1979). 56. De Mare, G.R., Huybrechts, G., and Toth, M., Perkin Trans. 2, (1972), p. 1256. 57. Benson, S.W., and Shaw, R., 58. Ref. 6a, p. 8.57. 59. Ross, D.S., 60. Virk, P.S., Fuel, 58(2), 151 (1979), 61. Gilchrist, T.L., and Storr, R.C., "Organic Reactions and Orbital and Vinh, T.H., Fuel, 58(3), 211 (1979). M.S. Thesis, Massachusetts Institute of Technology, B.S. Thesis, Massachusetts Institute of Technology, J. Chem. Soc., J. Am. Chem. Soc., 89, 5351 (1967). and Blessing, J.E., Fuel, 58(6), 438 (1979), Table 3. Symmetry," 2nd Ed., Cambridge Univ. Press, London (1979). "Pericyclic Reactions," Chapman and 62. Gill, G.B., and Willis, M.R., Hall, London (1974). 63. Fleming, I., "Frontier Orbitals and Organic Chemical Reactions," John Wiley, London and New York (1976); chapters 4, and 5 in particular. 64. Woodward, R.B., and Hoffmann, R., "The Conservation of Orbital Symmetry," Verlag Chemie/Academic Press (1970). pp. 115, 179-182. 65. Ref. 61, 66. Zimmerman, H.E., J. Am. Chem. Soc., 88, 1564, 1566 (1966); Angew. Chem. Int. Ed., 8, 1, (1969); Acc. Chem. Res., 4, 272 (1971). 67. (a) Dewar, M.J.S., "The Molecular Orbital Theory of Organic Chemistry," McGraw Hill, New York (1969); -80- (b) Angew. Chem. Int. Ed., 10, 761 (1971); (c) Dewar, M.J.S. and Kirschner, S., 4290-2 (1971); J. Am. Chem. --- Soc., 93, (d) Dewar, J.M.S. and Dougherty, R.C., "The PMO Theory of Organic Chemi stry," Plenum, New York (1975), p. 345. 68. Wassermann, A., "0iels Alder Reactions"; El sevi er, London and New York (1965); (a) p. 50, Table 13; (b) p. 63, Table 19. 69. Rowley, D., and Steinner, H., Discuss. Faraday Soc., (1951). 70. K'ichler, L., Nachr. Akad. Wissensch G6ttingen, Math. Physik. K1. III, 1, 231 (1939). 71. Robinson, P.J., and Holbrook, K.A., "Unimolecular Reactions"; John Wiley, London and New York (1972), chapter 7; (a) p. 212, Table 7.16; (b) p. 234, Table 7.30; (c) p. 210, Table 7.15; (d) p. 232, Table 7.28; (e) p. 238, Table 7.24; (f) p. 209, Table 7.14. 72. 2, 311 (1970). Tsang, W., Internat. J. Chem. -- Kinet., -- 73. Schuler, F.W., and Murphy, G.W., J. Am. Chem. Soc., 72, 3155 (1950). 74. Frey, 75. Amano, A., and Uchiyama, M., J. Phys. Chem., 69, 1278 (1965). 76. Blades, A.T., and Murphy, G.W., (1952). 77. Smith, G.G., and Yates, B.L., J. Chem. Soc., 7242 (1965). 78. Blades, A.T., Canad. J. Chem., 32, 366 (1954). 79. Blades, A.T., and Gilderson, P.W., Canad. J. Chem., 38, 1407 (1960). 80. Frey, H.M., and Pope, B.M., J. Chem. Soc. (A), 81. O'Neal, H.E., (1970). 82. Ref. 64, p. 143. 83. See, for example, H.M., Solby, R.K., Trans. 10, 370 Faraday Soc., 64, 1858 (1968). J. Am. Chem. Soc., 74, 1039 1701 (1966). and Frey, H.M., Internat. J. Chem. Kinet., 2, 243 -81- (a) Pople, J., and Beveridge, D.L., "Approximate Molecular Orbital Theory," McGraw-Hill, New York (1970; also Pople, J.A., and Segal, G.A., J. Chem. Physics, 44(9), 1966. (b) Murrell, J.N., and Harget, A.J., "Semi-empirical Sel f-consistent-field Molecular Orbital Theory of Molecules," WileyInterscience, London (1972). (c) Zimmerman, H.E., ref . 84, chapter 5. (d) Dewar, M.J.S., ref. 67a, chapters 2, 3, 10. 84. Zimmerman, H.E., "Quantum Mechanics for Organic Chemists," Academic Press, New York (1975). 85. Streitwieser, A., "Molecular Orbital Theory for Organic Chemists," John Wiley, New York (1961). 86. Roberts, J.D., "Notes on Molecular Orbital Calculations," W.A. Benjamin, Inc., Publishers, Reading, Mass. (1961). 87. Smith, W.B., "Molecular Orbital Methods in Organic Chemistry, HMO and PMO," Marcel Dekker, Inc., New York (1974). 88. Ref. 85, chapter 5. 99. Ref. 63, p. 141. 90. Zimmerman, H.E., Tetrahedron, 16, 169 (1961). 91. Streitwieser Jr., A., and Nair, P.M. Tetrahedron, 5, 149 (1959). 92. 95(12), 4094Soc., Houk, K.N., and Strozier, R.W., J. Am. Chem.__ _ 7 (1973). 93. Alston, P.V., and Shillady, D.D., J. Org. Chem., 39(23), 3402 (1974). 94. (a) Eisenstein, 0., Lefour, J.-M., and Anh, N.T., Chem. Comm., 969 (1971); (b) Eisenstein, 0., and Anh, N.T., Tetrahedron Letters, 17, 1191 (1971); (c) Eisenstein, 0., and Anh, N.T., Bull. Soc. chim. France, 2721 and 2723 (1973).. 95. Ref. 63, chapter 5; ref. 85, chapters 11-13; and original references cited therein. 96. Salem, L., J. Am. Chem. Soc., 90, 558 (1968); Salem, L., ibid., 91, 3793, note 3c (1969). Devaquet, A., and -8297. Woodward, R.S., and Katz, T.J., Tetrahedron, 5, 70 (1959). 98. Zimmerman, H.E., 99. (a) Wheland, ref. 84, pp. 153, 154. G.W., J. Am. Chem. Soc., 64, 900 (1942); (b) Wheland, G.W., and Mann, D.E., J. (1949). 100. Chem. Physics, 17(3) (a) Muller, N., Pic kett, L.W., and Mulliken, R.S., J. Am. Chem. Soc., 76, 4770 (1954); (b) Muller, N., and Mulliken, R.S., ibid., 80, 389 (1958). and Orloff, H., J. Chem. 101. Mulliken, R.S., Rieke, C.A., Orloff, D., Physics, 17(12), 1248 (1949). 102. Hoijtink, G.J., and van Schooten, J., Rec. Tray. Chim. 71, 1089 (1952) ; ibid., 72, 691, 903 (1953). 103. Ref. 84, pp. 130-131. 104. Ref. 34, pp. 190-191. 105. Streitwieser, A., ref. 85, p. 115; and original therein. 106. Murrell, N.N., Kettle, S.F.A., and Tedder, J.M., "The Chemical Bond," John Wiley, Chichester (1978); p. 255. 107. Streitwifeser Jr., A., 108. (a) Pays-Bas, references cited ref. 85, p. 120. Streitwieser Jr., A., J. Am. Ch.m. Soc. 82, 4123 (1960); (b) Ref. 85, pp. 118, 119. 109. Benson, S.W., Cruickshank, F.R., Golden, D.M. Haugen, G.R., O'Neal, H.E., Rodgers, A.S., Shaw, R., and Walsh, R., Chem. Rev., 69, 279-324 (1969). 110. Bender, C.M., and Orszag, S.A., "Advanced Mathematical Methods for Scientists and Engineers," McGraw-Hill, New York (1978); pp 369-375. 111. For an introductory discussion of the relationship between orbital symmetry, energy and quantum mechanical mixing, see Dewar, M.J.S. and Dougherty, R.C., ref. 67d, chapter 1. 112. For further discussion, see, for example, Streitwieser Jr., A., ref. 85, pp. 97-99. 113. Mulliken, R.S. Rieke, C.A. and Brown, W.G., J. Am. Chem. Soc., 63, 41 (1941); Mulliken, R.S., and Parr, R.G., J. Chem. Phys., -83114. Streitwieser Jr., A., ref. 85, pp. 131-135. 115. Ref. 84, pp. 116. Jorgensen, W.L., and Salem, L., "The Organic Chemist's Book of Orbitals," Academic Press, New York (1973). 117. Coulson, C.A., and Longnet-Higgins, H.C., Proc. Roy. Soc.-A, 192, 16 (1947). 118. Dewar, M.J.S., J. Am. Chem. Soc., 74, 3341, 3345, 3350, 3353, and 3357; (1952). 119. Fukui, K., Yonezawa, T., and Nagata, C., J. Chem. Phys., 26, 831 (1954); for a review of the contributions of the Fukui group see also, refs. 123 and 124. 120. Klopman, G., J. Am. Chem. Soc., 90, 223 (1968). 121. Salem, L., 122. Fujimoto, H., and Fukui, K., in "Chemical Reactivity and Reaction Paths," Klopman, G., Ed.; Wiley-Interscience, New York (1974); chapter 3. 124. Houk, K.N., in "Pericyclic Reactions, Vol. II"; Marchand, A.P. and Lehr, R.E., (Ed.); Academic Press, N.Y. (1977); chapter 4. 125. See, for example, ref. 67d, p. 220. 126. See, for example, ref. 122, p. 10. 127. See, for example, ref. 67d, pp. 212-220. 128. Streitwieser Jr., A., ref. 85, p. 196. 129. Ref. 116, pp. 101, 102, eclipsed ethane orbitals 2E', 1E", iE'; pp. 225-227, cyclopentadiene orbitals 2A2 , 381, 1A2 , and 2 1 . 130. [1,5] H-shifts are common in thermal pericyclic reaction systems. In the case of iso-indene isomerization to indene, it has been proven with deuterium labelling: See (a) Roth, W.R., Tetrahedron Letters, (1964), 1009; Woodward and Hoffmann, ref. 64, p. 124; Gilchrist and Storr; ref. 61, p. 248. 131. See, for example, ref. 85, p. 119, Table 5.1; ref. 101, Table I, and ref. 106, p. 41, Table 4.6. 132. Fleming, I., ref. 63, pp. 115-120. 133. Nishiguchi, T., Ohki, A., Sakakibara, H., and Fukuzumi, K., J. Org. Chem., 43(14), 2803 (1978). 131-145. J. Am. Chem. Soc., 90, 543 and 553 (1968). -84134. Streitwieser Jr., A., ref. 85, p. 241. 135. Lewis, K.E., and Steiner, H., J. Chem. Soc., 3080 (1964). 136. Goldfarb, T.D., and Lindquist, L., J. Am. Chem. Soc., 89, 4588 (1967). 137. Robinson, P.J., and Holbrook, K.A., 138. Whitehurst, D.D., 139. Jorgensen, W.L., and Salem, L., 140. Houk, K.N. , Sims, J., Watts, C.R., and Luskus, L.J., J. Am. Chem. Soc., 95, 7301 (1973); see also ref. 61,p. 150. 141. Kwart, H., Slutsky, J., and Sarner, S.F., J. Am. Chem. 5254 (1973). 142. Pauling, L., "The Nature of the Chemical Bond," Cornell Univ. Press (1960); p. 255. 143. Bailey, J., and Baylouny, R.A., 'J.Org. Chem., 27, 3740 (1962). 144. Oppolzer, W., and Snieckus, V., Angew. _Chem., int. mEd. Encl., 17, 476 (1978). 145. Hoffmann, H.M.R., Angew. 146. Kwart, H., Sarner, S.F. , and Slutsky, J., 5234 (1973); also ref. 141. 147. Reviews: ref. 11, p. 208, Table 7.3. et al., ref. 6a, p. 8.20. ref. 116, p. 140, orbital 2A". Soc., 95, Chem., Int. Ed. Engl., 8, 556 (1969). Am. Chem. Soc. , 95, (a) Rhoads, S.J., and Raulins, N.R., Organic Reactions, 22, (1975). (b) Hansen, H.J., and Schmid, H., Chem. in Britain, 5, 111 (1960). (c) Jefferson, A., and Scheinmann, F., Quart. Rev., 22, 391 (1968). 148. Thyagarajan, B.S., Balasubramanian, K.K., and Bhima Rao, R., Tetrahedron, 21, 2289 (1965). 149. (a) Shaw, R., Golden, D.M., and Benson, S.W., J. Phys. Chem., 81, 1776 (1977). & Eng. Data, 15, (b) O'Neal, H.E., and Benson, S.W., J. Chem. --266 (1970). -85- (c) Stei n, S.E., Golden, D.M., and Benson, S.W., J. Phys. Chem., 81, 314 ( 1977). (d) Murrel, J.N., et al., ref. 106, p. 292. 150. Reid, R.C., Prausnitz, J.M., and Sherwood, T.K., "The Properties of Gases and Liuqids," 3rd Ed.; McGraw-Hill, New York (1977). 151. Weast, R.C., (Ed.): "Handbook of Chemistry and Physics," 56th Ed., CRC Press, Cleveland, Ohio (1975). 152. National Research Council: "International Critical Tables," Washburn, E.W., (Ed.-in-Chief); McGraw-Hill, New York (1925); Vol. 1, C-Tables, pp. 176-275. 153. "Chemical Engineers' Handbook," Perry, R.H., and Chilton, C.H.York 5th Ed.; McGraw-Hill, New York (1973); Section 3. 154. Yamada, T., and Gunn, R.D., J. Chem. Eng. Data, 18, 234 (1973). , 155. "Introduction See, for example, Smith, J.M., and Van Ness, H.C. McGraw-Hi ll, to Chemical Engineering Thermodynamics," 3rd Ed. ; New York (1975); chapter 8, pp. 314 ff. 156. Soave, G., Chem. Eng. Sci., 27, 1197-1203 (1972); also in Reid, et al., ref. 150, pp. 174 and 37 157. Peng, D.-Y., and Robinson, D.3., 59 (1976). 158. Robinson Jr., R.L., and Chao, K.-C., Ind. Eng. Chem. Process Des. Dev., 10(2), 221 (1971). 159. Hoy, K.L.: "Tables of Solubility Parameters," Union Carbide Corp., Chemi cals and Plastics R&D Dept., South Charleston, W. Va. (1969); p. ( iii). 160. See, for example, Nonhebel, D.C., and Walton, J.C., "Free Radical Chemistry," Cambridge Univ. Press, London (1974); chapter 8, pp. 195-259. 161. (a) Nonhebel, D.C., and Walton, J.C., ref. 160, pp. 500-510. Ind. Eng. Chem. Fundam., 15(1), i na "i i , ,r c s, w., (b) Ingold, K.U., in "Free Radicals, Vol. 1"; Kochi, J.K., Edi - to r; John Wiley, New York (1973); chapter 2, pp. 95-96. 162. (a) Walling, C., "Free Radicals in Solution," John Wiley, New York (1957); p. 152, Table 4.12. (b) Gregg, R.A., and Mayo, F.R., Discussions Faraday Soc., 2, 328 (1947). i i, -86163. Kochi, J.K., in "Free Radicals, Vol. 2" Kochi, J.K. (Ed.); John Wiley, New York (1973); chapter 23, pp. 665-710. 164. (a) Wassermann, A., "Diels-Alder Reactions"; Elsevier Publishing Co., London and New York (1965); p. 9. (b) ibid., p. 52. 165. Virk, P.S., Bass, D.H., Eppig, C.P., and Ekpenyong, D.J., in "Coal Liquefaction Fundamentals"; Whitehurst, D.D. (Editor); ACS Symposium Series 139, American Chemical Society, Washington, D.c. (1980). 166. Eppig, C.P., M.S. Thesis, Massachusetts institute of Technoiogy, January 1980. 167. Hellyar, M.J., and Virk, P.S., Fuel, in press, 1982. 168. Frey, H.M., and Walsh, R., Chem. Rev., 69, 103 (1969). - I-- l~l----;uL =l---~ LIST OF RECURRENT ABBREVIATIONS AO atomic orbital Bo/A o BPE benzyl phenyl ether, PhOCH 2 Ph cyclohexadiene CHD Co initial mole ratio of hydrogen donor to hydrogen acceptor; initial hydronaphthalene to benzyl phenyl ether ratio i initial concentration (mol/liter) of component i in reaction mixture DHN dihydronaphthalene DPE FO 1,2-diphenylethane (alias bibenzyl), PhCH 2 CH2 Ph frontier orbital GC gas chromatograph(y) HMO HUckel molecular orbital HA H 'ckel H-donor HMBPh 2-hydroxy-2'-methylbiphenyl HOMO highest occupied molecular orbital ID inside diameter LCAO linear combination of atomic orbitals LUMO lowest unoccupied molecular orbital MA Mobius H-acceptor MD Mobius H-donor MO molecular orbital MWM Mull i ken-Wheland-Mann NHOMO next highest occupied molecular orbitdl NLUMO next lowest unoccupied molecular orbital NMR (N/No) i nuclear magnetic resonance spectroscopy unconverted fraction of component i, = (1 - fractional conversion of i) 00 outside diameter PM0 perturbation molecular orbital RDS rate determining step THOMO third highest occupied molecular orbital TS transition state w.r.t. with respect to xA extent of reaction; fractional converstion of H-acceptor substrate X; fractional conversion of component i.