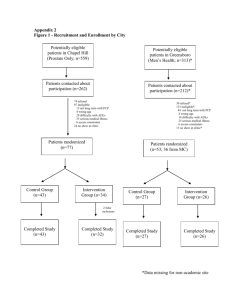
ARCHNES MAR 0 2010 LIBRARIES
advertisement

Nanomaterials for the Detection of Cancer-Associated Biomarkers by ARCHNES Chunyao Jenny Mu MASSACHUSETTS INS OF TECHNOLOGY B.S., Biomedical Engineering The Johns Hopkins University, 2000 MAR 0 5 2010 S.M., Mechanical Engineering Massachusetts Institute of Technology, 2005 LIBRARIES Submitted to the Harvard-MIT Division of Health Sciences and Technology in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mechanical and Medical Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2010 @ Massachusetts Institute of Technology 2010. All rights reserved. Author .......... Ha4rd-MfT Division of Health Sciences and Technology oe January 29, 2010 Certified by............. Bruce R. Zetter, Ph.D. Charles Nowiszewski Professor of Cancer Biology, HMS Thesis Supervisor Certified by.. Accepted by ...................... W\s' ....................... Robert S. Langer, Sc.D. David H. Koch Institute Professor Thesis Supervisor ............... . .............. Ram Sasisekharan, Ph.D. Director, Harvard-MIT Division of Health Sciences and Technology Edward Hood Taplin Professor of Health Sciences & Technology and Biological Engineering E Nanomaterials for the Detection of Cancer-Associated Biomarkers by Chunyao Jenny Mu Submitted to the Harvard-MIT Division of Health Sciences and Technology on January 29, 2010, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mechanical and Medical Engineering Abstract Prostate cancer persists as a major public health issue in the United States and remains the second leading cause of cancer death in men. Early detection and disease monitoring in prostate cancer can significantly improve a patient's prognosis. The advent of prostate-specific antigen (PSA) screening has allowed physicians to monitor the levels of a specific protein, or biomarker, as a correlate of disease progression. This thesis focuses on optical detection of prostate tumors through the development of biomarker-targeted molecular imaging probes. In the first part of this work, engineered human prostate cancer cell lines were developed and characterized to determine the dynamics of post-translational processing for PSA proteolytic activity and to establish potential small animal models for validating protease-activatable imaging probes. Target-activatable gold nanoparticle imaging probes that can be self-assembled in a one-step reaction were then developed to detect biomarker proteases in vivo. The activated probes demonstrated a 5 to 8-fold fluorescence signal amplification, extended circulation time, and high image contrast in a mouse tumor model. Lastly, differential phage display selection was performed on human prostate cancer cells with low and high metastatic potentials to (1) identify cell-surface biomarkers specific to highly aggressive tumors, and (2) develop molecular imaging probes for detecting prostate cancer metastases. One peptide, LN4P-1, demonstrated preferential binding to highly metastatic PC3M-LN4 cells and identified a highly expressed protein on their cell surface. Fluorescently labeled LN4P-1 was able to detect PC3MLN4 tumors in vivo. In summary, this thesis outlines the development of molecular imaging probes for targeting tumors both at the primary site, through evaluation of biomarker protease activity, and at the metastatic site, through affinity-based analysis of biomarker expression. Thesis Supervisor: Bruce R. Zetter, Ph.D. Title: Charles Nowiszewski Professor of Cancer Biology, HMS Thesis Supervisor: Robert S. Langer, Sc.D. Title: David H. Koch Institute Professor Acknowledgments I would like to express my deepest gratitude to my thesis advisors, Bob Langer and Bruce Zetter, for their guidance and support throughout my graduate career. They have afforded me a wealth of experiences at the intersection of medicine and engineering that few ever have the privilege to undergo. I will carry with me always the lessons I learned as a member of their research groups. My colleagues from both laboratories have been a constant source of knowledge, mentorship, encouragement, and friendship throughout my tenure for which I am deeply grateful. I am also greatly indebted to the members of my thesis committee, Sangeeta Bhatia, Fred Bowman, and Kim Hamad-Schifferli, for their invaluable insights and enthusiasm. My often tortuous path through graduate school and in my research endeavors has demanded a great deal of love and patience on the part of my friends and family. I continue to be in awe of the amazing and generous people who have shared this journey with me. I am eternally grateful for their dedication and unwavering support. This thesis is dedicated to my family who has always been the source of my strength. In particular, I would like to express my love and gratitude to David for being my biggest fan and courageous partner in all of life's wonderfully unpredictable adventures. This work was supported financially through a graduate fellowship from the Whitaker Foundation, HST MEMP program as well as research grants from the NIH. Contents 13 1 Introduction 1.1 M otivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13 1.2 Problem Identification . . . . . . . . . . . . . . . . . . . . . . . . . . 16 1.3 Specific A im s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17 19 2 Molecular Imaging 3 Characterization of Prostate Cancer Associated Proteases 25 3.1 A bstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25 3.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26 3.2.1 Prostate-specific antigen . . . . . . . . . . . . . . . . . . . . . 26 3.2.2 H epsin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 3.2.3 Urokinase-type plasminogen activator . . . . . . . . . . . . . . 33 Experimental Methods . . . . . . . . . . . . . . . . . . . . . . . . . . 34 3.3.1 Cell culture . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34 3.3.2 Conditioned media collection . . . . . . . . . . . . . . . . . . 35 3.3.3 Gel electrophoresis and immunoblotting . . . . . . . . . . . . 35 3.3.4 PSA enzyme activity assay . . . . . . . . . . . . . . . . . . . . 36 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 3.3 3.4 3.4.1 Engineering secretion of enzymatically active PSA: full-length KLK3 ........ 3.4.2 ............................... 36 Engineering secretion of enzymatically active PSA: pro-sequence deletion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 3.4.3 3.5 4 Engineering secretion of enzymatically active PSA: KLK2 + KLK3 co-transfection . . . . . . . . . . . . . . . . . . . . . . . 50 3.4.4 Hepsin expression and proteolytic activity . . . . . . . . . . . 53 3.4.5 Endogenous uPA secretion . . . . . . . . . . . . . . . . . . . . 57 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59 Self-assembled Gold Nanoparticle Molecular Probes 60 4.1 A bstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 4.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60 4.3 Experimental Methods . . . . . . . . . . . . . . . . . . . . . . . . . . 64 4.3.1 Fluorescence quenched AuNP probe preparation . . . . . . . . 64 4.3.2 Trypsin activation assay . . . . . . . . . . . . . . . . . . . . . 64 4.3.3 uPA activation assay . . . . . . . . . . . . . . . . . . . . . . . 65 4.3.4 kcat : Km determination . . . . . . . . . . . . . . . . . . . . . . 65 4.3.5 In vivo characterization . . . . . . . . . . . . . . . . . . . . . 66 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 4.4.1 Photophysical characterization of AuNP probes . . . . . . . . 67 4.4.2 Functional screens of AuNP probe libraries . . . . . . . . . . . 69 4.4.3 Effects of AuNP probe design on substrate kinetics and in vivo 4.4 4.5 biocompatibility . . . . . . . . . . . . . . . . . . . . . . . . . . 75 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78 5 Phage-derived Peptides for Targeting Highly Metastatic Cells 80 5.1 Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80 5.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80 5.3 Experimental Methods . . . . . . . . . . . . . . . . . . . . . . . . . . 85 5.3.1 Cell culture . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85 5.3.2 Phage display selection . . . . . . . . . . . . . . . . . . . . . . 85 5.3.3 Modified Jacobson's pellicle method for plasma membrane isolation 5.3.4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86 . . . . . . . . . . . . 88 Gel electrophoresis and immunoblotting 5.4 5.5 . . . . . . . . . . . . . 89 . . . . . . . . . . . . . . . . . . . . . . . . 90 5.3.7 FACS analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . 90 5.3.8 Confocal microscopy . . . . . . . . . . . . . . . . . . . . . . . 91 5.3.9 Cy5.5-labeled phage FACS analysis . . . . . . . . . . . . . . . 91 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92 5.3.5 Phage-facilitated immunoprecipitation 5.3.6 Mass spectrometry 92 5.4.1 Phage peptide selection converges on a common sequence 5.4.2 LN4P-1 phage preferentially binds PC3M-LN4 cell surface 97 5.4.3 Development of imaging probe from LN4P-1 phage peptide 101 5.4.4 Validation of Jacobson's pellicle method . . . . . . . . . . . . 109 5.4.5 LN4P-1 phage binds to ~ 130 kDa plasma membrane-associated . protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125 127 6 Conclusion 6.1 Sum m ary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127 6.2 Future Directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129 List of Figures 3-1 Schematic diagram of prostate-specific antigen (PSA) posttranslational processing. 3-2 . . . . . . . . . . . . . . . . . . . . . . . Total endogenous PSA protein secretion into conditioned media for LNCaP and LNCaP-LN3 cells. 3-3 38 . . . . . . . . . . . . . . 39 PSA protein overexpression in conditioned media from 293 HEK and PC3M-LN4 cells using pSecTag2/Hygro/PSA plasm id. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3-4 40 Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and stable PC3M-LN4 clones transfected 41 with PSA plasmids. ................................. 3-5 PSA protein overexpression in conditioned media from 293 HEK and PC3M-LN4 cells using pSecTag2/Hygro/PSA plasmid with additional control samples . ... 3-6 ............... 42 Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and stable PC3M-LN4 clones transfected with PSA plasmids. ................................. 3-7 43 Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK clones transfected with full-length Origene PSA plasm ids. 3-8 ............................ . 45 Enzyme kinetics of PSA secreted into conditioned media by transient PC3M-LN4 clones transfected with full-length Origene PSA plasmids. ................................. 46 3-9 Enzyme kinetics of PSA secreted into conditioned media by transient LNCaP and LNCaP-LN3 clones transfected with 47 full-length Origene PSA and pSecTag2-PSA plasmids. .... 3-10 Schematic diagram of prostate-specific antigen (PSA) post- translational processing and engineered pro-sequence deletion. 48 3-11 Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK, PC3M-LN4, LNCaP, and LNCaP-LN3 clones transfected with full-length Origene PSA and KLK349 deletion plasm ids. ............................ 3-12 Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and PC3M-LN4 clones co-transfected with 52 full-length Origene KLK2 and KLK3 plasmids. ............ 3-13 Reaction rate of PSA secreted into conditioned media by transient 293 HEK and PC3M-LN4 clones co-transfected with 53 full-length Origene KLK2 and KLK3 plasmids. ............ 3-14 Enzyme kinetics of PSA secreted into conditioned media by co-culture of stable PC3M clones transfected with full-length 54 Origene KLK2 and KLK3 plasmids. ..................... 3-15 Hepsin immunoprecipitation from 293 HEK, PC3M-LN4, LNCaP, HepG2, and LNCaP-LN3 cell lysates (Cayman polyclonal an. 55 tibody).. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3-16 Enzyme kinetics of cell surface hepsin in cell cultures of HepG2, 56 293 HEK, PC3M-LN4, LNCaP, and LNCaP-LN3 ........ 3-17 Hepsin immunoblot (Cayman polyclonal antibody) of HPN 57 overexpression in 293 HEK and PC-3 cells. ............ 3-18 Endogenous active uPA secretion by PC-3, PC3M, PC3MLN4, and HT1080 cells........................ 4-1 . 58 Schematic diagrams of gold nanoparticle (AuNP) probe synthesis and activation. ................................ 63 4-2 Photophysical characterization of AuNP probes. . . . . . . . . 68 4-3 Trypsin activation assay. . . . . . . . . . . . . . . . . . . . . . . . 70 4-4 Protease-induced fluorescence enhancement over unactivated 73 A uN P probe. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4-5 In vivo circulation retention of AuNP probes as compared to the equivalent mole quantity of uncomplexed Quasar 670labeled and BHQ2-labeled peptide substrates. 4-6 75 . . . . . . . . . In vivo near-infrared fluorescence imaging of subcutaneous tu. . . . . . . . . . . . . . . . . . . . . . . . . 77 5-1 Schematic diagram of subtractive phage display procedure. . 84 5-2 Background fluorescence as a result of nonspecific antibody mor phantom model. 98 binding during FACS. . . . . . . . . . . . . . . . . . . . . . . . . . 5-3 FACS analysis of LN4P-1 phage binding to PC3M and PC3M99 LN4 cells as quantified by fluorescence distribution. . . . . . . 5-4 Confocal microscopy images of PC3M and PC3M-LN4 cells labeled with LN4P-1 phage. LN4P-1 (green) and actin (red) are shown....... 100 .................................... 5-5 LN4P-1 peptide binding to PC3M and PC3M-LN4 cells ... 5-6 LN4P-1 peptide binding to PC3M and PC3M-LN4 cells in 104 105 serum-free media .................................. 5-7 FACS analysis of PC3M and PC3M-LN4 cells using Cy5.5. 106 labeled LN4P-1 phage ........................ 5-8 In vivo fluorescence imaging of intravenously administered Cy5.5labeled phage . .. ... .... .. .. ... .. ... ..... ... 5-9 Western blot analysis of membrane content in lysates ..... .. 108 111 5-10 Western blot analysis of membrane content in lysates ..... .112 5-11 Indirect phage Western blot of pellicle and WCL ......... 116 5-12 Indirect phage Western blot of fd-tet Ab immunoprecipitation 117 5-13 Indirect phage Western blot of fd-tet Ab immunoprecipitation 118 5-14 CDCP1 immunoprecipitation and evaluation of LN4P-1 bindingl2l 5-15 Indirect phage Western blot of SBED cross-linking immunoprecipitation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122 List of Tables 3.1 Concentration of Enzymatically Active PSA in PC3M-KLK2 51 and PC3M-KLK3 Co-Culture ......................... 4.1 AuNP Probe Surface Composition as Defined by Reaction Concentrations of Quasar 670-labeled Peptide Substrate . 5.1 . . 74 Peptide Sequences from Subtractive Phage Display Selection for Ligands with Preferential Affinity for Highly Metastatic Prostate Cancer Cells ............................... 5.2 LN4P-1 Peptide Sequence Matches to Expressed Human Proteins using BLASTP Engine .......................... 5.3 120 Mass spectrometry identified proteins from 130 kDa protein bands from phage immunoprecipitation .............. 5.7 119 Mass spectrometry identified proteins from 100-150 kDa protein bands from pellicle isolation .................. 5.6 107 Mass spectrometry identified proteins from 100-150 kDa protein bands from pellicle isolation .................. 5.5 96 Optimization of Cy5.5 loading on phage for maximal distinction in phage labeling of cells ......................... 5.4 95 123 Mass spectrometry identified proteins from 75 kDa protein bands from phage immunoprecipitation .............. 124 Chapter 1 Introduction 1.1 Motivation In the United States, prostate cancer is the second leading cause of cancer death in men. Since the discovery of prostate-specific antigen (PSA) as a potentially useful serum marker for prostate cancer, this form of neoplastic disease has become the most commonly diagnosed cancer in the male population. The main risk factors associated with prostate cancer are age, ethnicity, and family history. Men over the age of 65 account for more than 70% of all prostate cancer cases. Moreover, African-American men have the highest incidence rates in the world. In 5-10% of prostate cancer cases, there may be a strong genetic predisposition to the disease [1]. As with most degenerative disorders, early detection in prostate cancer can significantly improve a patient's prognosis. The advent of PSA screening in the 1980s has revolutionized prostate cancer diagnosis by allowing physicians to monitor the levels of a specific protein as an early indicator of prostate dysfunction. Elevated serum PSA levels can be detected even before neoplastic growth has reached a palpable mass. Tumors that are indistinguishable from normal prostate tissue using ultrasound can be detected through PSA screening. Consequently, annual PSA tests are recommended for men beginning at the age of 50 as a simple yet effective method of early detection. The utility of any biomolecular marker for disease, such as PSA, depends strongly on the sensitivity with which it can be detected and the level of correlation to disease progression. PSA has gained wide clinical acceptance in the diagnosis of prostate cancer due to high-sensitivity assays that can detect serum PSA at the level of 0.02 ng/ml and clinical studies that have established its correlation with cancer incidence. Moreover, the complexity of prostate cancer management has required the use of serum PSA levels as a clinical measure of treatment efficacy. As a result, in vitro assays for PSA have proven indispensable in the early detection and management of neoplasms. However, these assays only measure the total level of PSA secreted into the bloodstream and cannot provide tissue-specific characterization of biochemical activity. This thesis outlines several approaches towards establishing small animal models of proteolytically active PSA as an important tool in understanding its role in disease progression as well as for validating molecular imaging probes. Molecular imaging is an emergent technique that integrates clinical imaging modalities with our ever expansive understanding of molecular and cellular pathogenesis. Massoud and Gambhir defined molecular imaging as the visual representation, characterization, and quantification of biological processes at the cellular and subcellular levels within intact living organisms [2]. Clinical imaging techniques are currently used to detect nonspecific physiological and metabolic changes at the macroscopic level to distinguish pathological from normal tissue. The aim of molecular imaging is to provide a visual context for cellular and molecular pathways as an extension of the anatomic and physiologic information yielded by traditional imaging techniques, such as magnetic resonance imaging (MRI). Rapid growth in this field of biomedical research has been fueled by advances in molecular and cell biology techniques and the use of transgenic animal models to study the molecular basis of disease. The validation of conventional molecular assays in vitro has also driven translational research to make them applicable to medical imaging techniques. Molecular imaging has the potential to advance integrative biology, promote early detection and characterization of disease progression, and allow for greater ease in the evaluation of medical treatments. This new paradigm in medical imaging has the ability to visualize specific molecular events in intact living organisms and to track dynamic processes in real time. Applications that may benefit from molecular imaging include pheno- typic screening with respect to physiological abnormalities that arise from genetic mutations and biodistribution or pharmacokinetic studies. In both cases, biopsy is unnecessary for quantitative evaluation of tissue specimens, and temporal changes in expression can be monitored in the same living subject. Pathologic states that arise from a complex array of physiological interactions can thus be evaluated in vivo with greater authenticity. The additional insight that molecular imaging brings to traditional imaging techniques is derived from the use of contrast agents, or molecular indicators, targeted to specific biochemical functions. Disease-associated proteases, such as the family of matrix metalloproteinases (MMPs) involved in the progression of breast cancer, have become attractive targets for molecular imaging studies. Proteases are often upregulated in disease states and represent a natural means of signal amplification, as enzymatic processing of an optimized substrate can occur at a highly accelerated rate. With regards to prostate cancer, PSA has been shown to have enzymatic activity against semenogelin, a major component of human semen, and several researchers have attempted to find the optimal peptide substrate for PSA. Research has shown that there is upregulation of enzymatically-active PSA in patients with prostate cancer. All of this active PSA is confined within the extracellular space of the prostate; active PSA has not been detected in the bloodstream. Several factors make PSA an attractive biomarker and target for molecular imaging studies of human prostate cancer: (1) active PSA is found exclusively in prostatic fluids, (2) the literature suggests that the large majority of PSA can only be found in the prostate (i.e., PSA is tissue-specific), (3) proteolytic activity of PSA has been well-documented with several potential substrates that exhibit high specificity, (4) PSA is used in the clinic in association with prostate cancer screening, and (5) in vitro and in vivo models exist to simulate active and inactive PSA secretion. This thesis focuses primarily on the development of minimally-invasive yet highly correlative methods of detecting prostate cancer lesions to improve upon the stateof-the-art in current detection schemes that involve highly uncomfortable and often inaccurate techniques, such as digital rectal examination and biopsy. In the female correlate of prostate cancer, breast cancer, the use of mammography has significantly improved a woman's chances of finding lesions in their early stages. Unfortunately, no comparable modality exists for men at risk for prostate cancer. Transrectal ultrasound is currently the most widely used method of prostate cancer diagnosis along with ultrasound-guided biopsy, in which 6-12 suspicious tissue regions are removed for histological analysis. Despite the advantages of an image-guided approach to sampling potential neoplastic lesions, biopsy cores represent a very small fraction of the total prostate volume. As a consequence, many tumorigenic foci are never detected. The work described in this thesis attempts to address the shortcomings of current diagnostic methods by introducing targeted molecular probes that identify tissue regions with high levels of cancer-associated functional activity and/or biomarker expression. The additional molecular information provided by probe-enhanced imaging may improve diagnostic accuracy and serve as another clinical indicator for determining disease stage and prognosis. 1.2 Problem Identification Prostate cancer has continued to be a public health issue in the United States. Emerging technologies and innovations in genomics and proteomics have allowed researchers and clinicians to understand disease with respect to specific genes and proteins. These biomarkers, when used in conjunction with clinical observations, may lead to improved diagnostic accuracy as well as specific prognosis. Our current understanding of prostate cancer remains constrained within two disparate camps: molecular and cellular mechanisms and clinical pathology. Since prostate cancer remains a disease of aging, clinical monitoring of cancer progression is essential to patient care. PSA is a good candidate for clinical monitoring, as it is currently a means of detecting metastatic disease in patients post-prostatectomy. In addition, it is a protease with well-documented substrate specificity for human semenogelin. The research described herein seeks to understand the proteolytic activity of several prostate cancer-associated biomarkers and develop biomarker-activated and affinity- based optical contrast agent for identifying primary as well as metastatic prostate cancer lesions in vivo. 1.3 Specific Aims The primary objective of this work was to design, develop, and characterize molecular imaging probes for detecting primary and metastatic lesions based on biomarker function and expression level. Towards this end, the research described herein were focused in the following directions: 1. Characterization of protease biomarkers in human prostate cancer cell lines to determine the dynamics of post-translational processing and the appropriate cells for use in small animal models of disease 2. Design and develop nanoparticle-based molecular imaging probes for detecting cancer-associated proteases in vivo 3. Investigate differential cell surface protein expression between human prostate cancer cell lines with varying metastatic potential to determine a potential target protein for imaging metastases 4. Develop a molecular imaging probe to target highly metastatic human prostate cancer cells in vivo Efforts to determine the clinical relevance of cancer biomarkers have been predominantly focused on disease correlation with total protein levels. In Chapter 3, I present an often overlooked approach to assessing disease progression by examining the enzymatic activity levels of several prostate cancer associated proteases as they relate to cancer aggressiveness. Urokinase-type plasminogen activator (uPA) activity in vitro varied inversely with the human prostate cancer cell line's metastatic potential, as opposed to in vivo experimental data from the current literature that show the reverse correlation. This contradiction in observation suggests that the in vivo environment and its interaction with cancer cells influence uPA activity in a complex manner that cannot be recapitulated in vitro. Furthermore, I show that the protease activity of exogenously introduced PSA in previously non-PSA producing cell lines can be enhanced by the addition of an upstream convertase, hK2. These data represent one of the first demonstrations of an engineered cell line that secretes enzymatically active PSA. Prior attempts by other research groups have yielded cell lines that secreted incorrectly processed, and consequently, inactive, PSA. Chapter 4 outlines a self-assembly approach towards generating protease-activatable molecular probes for in vivo imaging applications. Through optimization studies that investigate the contributions of individual probe components, I demonstrate that the incorporation of a dark quencher and a polymer stabilizer is essential to overall probe performance. Since the majority of reported research in molecular imaging focuses on proof-of-principle demonstrations, little information exists on optimization parameters for molecular probe design. The data presented herein thus fill this gap in knowledge by highlighting the critical design considerations for synthesizing high performance molecular probes. Despite the high mortality rate attributed to metastatic disease, research efforts have largely ignored the distinction between primary and disseminated cancers. In Chapter 5, subtractive phage display screening is performed to identify peptide sequences that preferentially bind and identify highly metastatic cells. The selected phage was then fluorescently labeled to function as a metastasis homing contrast agent in vivo. I further demonstrate the discovery of a differentially expressed cell surface protein that is highly abundant in the more aggressive cell line. These results represent one of the first efforts to specifically target metastatic lesions in vivo and could potentially reveal novel biomarkers for disseminating cancer cells. Chapter 2 Molecular Imaging Current implementations of molecular imaging involve traditional imaging modalities combined with unique contrast agents that are designed to target specific molecular and cellular processes. Standard clinical imaging techniques include positron emission tomography (PET), single photon emission computed tomography (SPECT), optical bioluminescence and fluorescence imaging, magnetic resonance imaging (MRI), computed tomography (CT), and ultrasound. Contrast agents, or molecular probes, are designed for each technique to maximize the signal-to-background ratio for a particular tissue or biochemical pathway. A comparison of these imaging modalities can be made based on spatial and temporal resolution, depth of penetration, energy required in image generation, availability of molecular probes, and the probe detection threshold. PET is a highly sensitive imaging technique that utilizes positron-emitting isotopes to label species of interest. The positron collides with an electron to produce two y-rays whose paths are nearly 180' apart from each other. These high-energy 7-rays are detected by scintigraphic instrumentation to form a composite image of tracer distribution. PET exhibits spatial resolution at 1-2 mm with no limits in penetration depth. Its principal uses are in monitoring metabolic changes, receptor-ligand interactions, and enzyme targeting with sensitivity in the picomolar range. Distinct morphological features can be distinguished with tracer uptake in several million cells. However, isotope production for PET imaging relies on the use of a cyclotron or generator and results in high operational costs. Furthermore, these tracer molecules have short half-lives at -110 minutes. Molecular imaging strategies using PET must target biochemical processes that occur rapidly within the limit of the isotope half-life. SPECT is another form of radionuclide imaging that detects 7-emitting isotopes using a gamma camera that rotates around the subject. SPECT has spatial and temporal resolutions comparable to those of PET, since the signal source and detection schemes are nearly the same in both imaging applications. Due to the isotropic emission of 7-rays in this application, SPECT detection requires the use of a lead collimator to facilitate image reconstruction. However, the use of a collimator results in low detection efficiency and severely limits sensitivity to 10-10 - 10-" mole/L. SPECT does allow for simultaneous imaging of multiple tracers, since different isotopes emit y-rays of different energies. Using PET, tracer detection can only be performed in series. The two a-rays produced under positron emission have nearly identical energies and thus, cannot be modulated to distinguish multiple molecular events occurring simultaneously. Applications of PET and SPECT in molecular imaging involve receptor-ligand interactions and reporter-gene expression in which radiolabeled tracers are selectively internalized by certain cell populations. In order to achieve the highest spatial resolution for molecular imaging applications, researchers have explored the use of magnetic resonance imaging (MRI) in conjunction with tailored molecular probes. The underlying principle of MRI is that unpaired nuclear spins, or magnetic dipoles, align themselves when exposed to a magnetic field. A temporary radiofrequency (RF) pulse then perturbs the nuclear spin alignment. The time required for the magnetic dipole to return to the baseline orientation is then measured using the same RF coil that generated the disruption pulse. This relaxation time is the quantifiable parameter represented in the MRI image. MRI scanners incorporate both a strong magnet to generate the magnetic field as well as a RF coil to detect changes in magnetic dipole alignment. MRI is sensitive to soft-tissue differences and provides a spatial resolution of 25-100 pm and no limit to the penetration depth. However, temporal resolution is on the order of minutes to hours for one complete scan. MRI on the molecular level can be accommodated by the use of paramagnetic metal cations or superparamagnetic nanoparticles that are modified for targeting specificity. With the use of contrast agents such as chelated gadolinium or dysprosium, physiological/molecular characterization and anatomical information can be extracted simultaneously. The distinct disadvantage to MRI is that it is several orders of magnitude less sensitive than radionuclide and optical techniques. Signal-to-noise ratio can be improved with the use of high-powered magnets that generate fields as high as 14 T. However, the cost of such a scanner becomes prohibitive. One recent application in molecular imaging involves receptor-based MRI imaging, where transferrin-monocrystalline iron oxide nanoparticle probes were used to identify cell populations that overexpressed engineered transferrin receptors (TfR) in vivo [3]. Computed tomography is another widely-used clinical imaging system. CT images are constructed from the differential absorption of X-rays as they pass through component tissues in the body. Small animal CT scanners rely on high-resolution phosphor screen/CCD detectors to improve image quality. Typical spatial resolution is on the order of 50-200 pm with no limit in penetration depth. However, CT has been used principally for morphological characterization. It is not as sensitive as MRI in detecting soft-tissue contrast, necessitating the administration of iodinated contrast media to delineate anatomical features. Limited molecular applications have been explored for CT due to the inherent obstacles in adapting the technology. Recent research has focused on developing CT-based molecular probes that are tagged with X-ray absorbing species to provide contrast enhancement. Aside from issues with cell uptake and tissue accumulation of these probes, CT imaging exposes the subject to a significant radiation dose that precludes repeated scans of the same subject. Adverse health risks and biological interactions have made CT a less attractive target for developing molecular imaging techniques. However, in cases where tissue uptake is non-limiting, such as in bone and tumor, CT imaging with contrast media can provide a highly detailed anatomical view. Ultrasonography has emerged as the most prevalent clinical imaging modality used today. Advantages in using ultrasound include its low cost, availability, and safety. High-frequency sound waves are emitted from a transducer placed against the skin. Reflected sound waves from internal tissue structures are processed based on their backscatter and attenuation properties using a number of algorithms to produce a contrast image. Ultrasound applications have spatial resolution in the range of 50-500 pm and a penetration depth on the order of millimeters to centimeters. Molecularlevel imaging with ultrasound has not progressed as rapidly as applications in MRI and optical techniques, because of its traditional role as a means for morphological characterization. However, echo contrast agents have been produced that enable molecular imaging of cell-surface receptors. Acoustic nanoparticles are modified with ligand molecules specific to the target receptor. These molecular probes increase the regional echogenicity of the cell surface once receptor-ligand binding is achieved. Optical imaging is another class of imaging modalities that is being explored for use with fluorescent and bioluminescent probes to detect biochemical processes in vivo. For many years, optical imaging techniques have been the mainstay for applications in molecular and cell biology. Light photons serve as the signal source and are converted into an electrical charge pattern through a charge coupled device (CCD) detector. CCD chips have high sensitivity to light and can be cooled to reduce thermal noise for an improved signal-to-noise ratio. Bioluminescence imaging relies on the emission of photons by either endogenous or exogenous molecular species. It differs from fluorescence imaging in that no excitation source is required to trigger light emission. One common application of bioluminescence is in the use of Firefly luciferase, an enzyme that oxidizes D-luciferin into a luminescent molecule, as a reporter for gene expression. The Firefly luciferase gene (Fluc) can be expressed in selected cell populations by transfection. Bioluminescence activation is then achieved through systemic administration of the reporter probe, D-luciferin, and its oxidation by intracellular luciferase. In this manner, only cells expressing Fluc will exhibit luminescence upon substrate administration. This reporter gene/probe strategy may be particularly useful in cell trafficking studies for tracking metastases and monitoring cell proliferation and gene expression as a function of local environmental stresses or protein-protein interactions. A similar approach can also be used in fluorescence imaging. Fluorescent molecules are conjugated to proteins or ligands of interest. Selective cellular uptake via receptor-ligand interactions or activation via the target biochemical reaction results in a concentrated localization of fluorescent markers. Spatial and temporal resolutions for both bioluminescence and fluorescence imaging are comparable. However, bioluminescence detection exhibits higher sensitivity due to minimal background luminescence. Fluorescence imaging in intact living subjects suffers from tissue autofluorescence, which produces a much higher background signal. Optical imaging coupled with novel molecular probes has emerged as one of the more developed forms of molecular imaging. This phenomenon is due in large part to the ease with which fluorescence and luminescence-based in vitro assays have been adapted to the needs of in vivo imaging. However, the major drawback in all forms of optical imaging is the limited penetration depth of light. Bioluminescence imaging has sufficient detection capabilities to a depth of 1-2 cm, whereas fluorescence imaging can only detect molecular probes at a depth of less than 1 cm. Light is easily absorbed and scattered through tissue sections, but judicious selection of the operating wavelengths can increase optical transmission. The near-infrared wavelength range exhibits optimal transmission properties due to low interaction with the main absorbing species in tissue, hemoglobin and water. The promise of molecular imaging is in its ability to bridge the divide between clinical imaging techniques and molecular and cell biology tools. Both research areas have experienced tremendous growth and innovation within recent years and are poised to forge a new paradigm in medicine. Clinical medicine has embraced the concept of in vitro diagnostics, in which biological discoveries in the laboratory are being translated into clinically relevant assays. The next logical step is to bring the same strategy and approach to the development of in vivo diagnostics and monitoring. For many disease processes, pathogenesis, progression, and effects of treatment are poorly understood at the systemic level, the point at which most clinicians interact with their patients. Molecular imaging also bears tremendous scientific implications. Biochemical pathways that were elucidated in isolated cell culture systems could be visualized at the ultimate scale with complete physiological input. It is anticipated that this new technique will raise new and interesting questions in biomedicine as well as increase the number of targets available for medical treatments. All of the imaging modalities described in this section have come into the standard repertoire of medical care. However, only a few of these lend themselves readily to adaptation for molecular imaging applications. Because the ultimate goal is microscale imaging of biomolecular events in live human subjects, many obstacles and health concerns must be addressed to achieve successful implementation. The work described herein tackles the design and optimization of molecular probes for use with a novel optical imaging platform that overcomes some of the traditional barriers to in vivo monitoring with light. Chapter 3 Characterization of Prostate Cancer Associated Proteases 3.1 Abstract Cancer development and progression occurs under a confluence of growth factors, enzymes, and other proteins. Secreted biomarker proteins are of particular importance to clinical diagnosis and prognosis as their expression levels and changes in response to disease state or therapy can be assayed with minimal invasiveness. Within this group of biomarkers, proteases and their activity are optimal targets for contrast imaging due to the inherent amplification process involved in hydrolytic reactions. However, protease activity as a clinical indicator for cancer progression has only been investigated for a select few enzymes. This report studies the proteolytic activity of several prostate cancer biomarkers that were originally established in the literature based on positive correlations between their expression levels and clinical disease. Prostate-specific antigen (PSA), urokinase-type plasminogen activator (uPA), and hepsin (HPN) expression levels and proteolytic activity were assessed in several tumor models. 3.2 Introduction Cancer-associated upregulation of protease expression levels facilitates multiple disease progression mechanisms, such as metastasis, angiogenesis, and tumor growth. This study focused on three important proteases in prostate cancer development and dissemination and the relationships between the level of secreted enzymatically active protein and cell line aggressiveness. The resulting data is important in defining the role of various protease activities during cancer progression as well as in determining the appropriate cells for use in small animal modeling of specific protease activities. 3.2.1 Prostate-specific antigen PSA is a biomolecular marker that has gained wide clinical acceptance in prostate cancer screening and monitoring [4, 5]. Clinical studies have determined that a cutoff value of 4.0 ng/ml for PSA levels aids in the detection of suspicious lesions [6, 7]. Use of a lower cutoff value of 2.6 ng/ml has recently been reported to increase the detection rate for small, organ-confined tumors without becoming oversensitive to 'clinically insignificant' disease [8]. Produced primarily by prostate epithelium, PSA is an androgen-regulated serine protease that acts to cleave semenogelins in the seminal coagulum. One study has hypothesized that elevated serum PSA in pathologic states, primarily metastatic prostate cancer, is mediated by the biochemical breakdown of glandular and capillary basement membrane and stroma [9]. The disruption of extracellular matrix components as well as the endothelial cell layer allows PSA to effectively 'leak' into the microvasculature, primarily as a result of prostate cancer. The most recent studies suggest that the physiological functions of PSA have both beneficial and deleterious effects on cancer progression. PSA is an insulin-like growth factor binding protein-3 (IGFBP-3) protease [10]. IGFBP-3 acts as a carrier protein for insulin-like growth factors (IGFs) in human seminal plasma. Once IGFBP-3 is cleaved by PSA, IGF-I loses significant affinity for IGFBP-3 and is able to stimulate increased mitogenic activity and growth in prostate cancer cells [11]. PSA also acts to cleave extracellular matrix glycoproteins, such as fibronectin and laminin, to promote cancer cell invasion in metastatic disease [12]. Despite indications that PSA enables prostate cancer cells to proliferate and metastasize, PSA expression in xenograft prostate tumors derived from the PC-3 cell line failed to increase tumor size as compared to wild type tumors that do not express PSA [13]. The PSA protein secreted by transfected PC-3 cells was not enzymatically active, further supporting the idea that proteolytic action by PSA is an important function in cancer pathogenesis. Prostate cancer metastases occur predominantly in bone with primary osteoblastic lesions, although there is increasing evidence that suggests an interplay between osteoblastic and osteoclastic activity [14]. PSA appears to play a role in prostate cancer cell adhesion to bone marrow endothelial cells, which is presumably the first step towards invasion and metastasis. Addition of active, exogenous PSA increased cell-cell adhesion, whereas antibodies to PSA and downregulation of PSA mRNA expression using small interfering RNA (siRNA) attenuated cell-cell interactions [15]. Osteoblastic lesions can also promote prostate cancer cell proliferation and PSA expression through an androgen-independent pathway, further feeding the metastatic potential [16]. There is also evidence that PSA promotes antiangiogenic activity by converting Lys-plasminogen into angiostatin-like fragments through proteolysis. These fragments, when purified, inhibit proliferation and tubular formation in human umbilical vein endothelial cells (HUVECs) [17]. Interestingly, recombinant PSA that has been enzymatically inactivated through deletion of the first amino acid, inhibits angiogenesis in vivo to a similar degree as active recombinant and native PSA [18]. PSA is expressed in mammalian cells as an inactive pro-form, containing 244 amino acids [19, 20]. Once the pro-PSA is secreted into the lumen of the prostate, proteases in the seminal fluid cleave seven amino acids from the N-terminus to yield the enzymatically-active form of PSA. Activation by trypsin, human glandular kallikrein (hK2), and prostin has been demonstrated and releases a pro-sequence of Ala-ProLeu-Ile-Leu-Ser-Arg [21, 22]. PSA was first discovered and characterized as having a molecular weight of 33-34 kDa and an isoelectric point of 6.9 [23]. It can become complexed with several proteins in human serum, primarily oa-antichymotrypsin (ACT), a 2-macroglobulin (a 2M), and protein C inhibitor (PCI). Although PSA and ACT are found in human serum, prostatic fluid, and seminal plasma, PSA-ACT complex was found only in human serum by Western blot [24]. Qian and coworkers further discovered that the addition of exogenous ACT to prostatic fluid yielded quantities of PSA-ACT complex that varied with the amount of ACT added. However, the addition of exogenous PSA failed to produce the complex. This indicated that PSA is biologically active in prostatic fluid where ACT exists in an inactive form. Espafia and coworkers found varying levels of all complexes (PSA-ACT, PSA-a 2 M, and PSAPCI) in prostatic fluid, seminal plasma, and seminal vesicle fluid using a much more sensitive sandwich ELISA [25]. The reason for great interest in elucidating the different molecular forms of PSA is the potential application towards improving clinical diagnostics for prostate cancer. Prostate pathologies fall under several classifications, which include prostate cancer (PCa), prostate intraepithelial neoplasia (PIN), and benign prostatic hyperplasia (BPH). Significant efforts have been made towards identifying diagnostic tools that can distinguish between benign and malignant lesions with greater specificity than total PSA. Stenman and coworkers reported that PSA-ACT is the predominant form of serum PSA in patients with prostate cancer; patients with BPH had a significantly lower level of PSA-ACT [26]. The ratio between PSA-ACT complex and total PSA has been used to further discriminate between these two pathologic states. In men with high serum levels of total PSA (> 10 ng/ml), this ratio, at a cutoff of 0.62, becomes a highly sensitive and specific test for prostate cancer [27]. In the intermediate tPSA range of 4.1-20 ng/ml, a ratio of PSA-ACT to prostate volume, known as ACT density, is the most reliable predictor of cancer as compared to tPSA alone [28]. Other molecular forms of PSA, primarily free PSA (fPSA), when expressed as a ratio against total PSA, have also proven valuable in positively identifying PCa and eliminating unnecessary prostate biopsies in patients with BPH [29, 30, 31, 32], even in men with tPSA levels less than 4 ng/ml [33]. Lilja first characterized the enzymatic activity of endogenous PSA as a serine proteinase with direct action against semenogelin and semenogelin-related proteins [34, 35]. When purified from semen, approximately one-third of the PSA was found to be enzymatically inactive [36]. The remaining PSA formed stable complexes with both ACT and a 2 M, as documented in other reports. Complexation between PSA and ACT was initiated by chymotrypsinlike cleavage of ACT, although at a much slower rate than that observed between chymotrypsin and ACT. PSA and ACT complex formation can be reversed through prolonged incubation at 37'C in vitro to yield free active PSA [37]. Because PSA immunoreactivity is lost when it binds to a 2 M, presumably due to epitope shielding by the bound a 2 M, Leinonen and coworkers identified PSA-a 2 M complexes by determining the undetected fraction of PSA in a total mixture. The undetected fraction, presumed to be PSA complexed with a 2 M, is 66% of the total PSA content. This result led to the conclusion that a 2 M is the major inhibitor to PSA in serum. The enzymatic activity of PSA is highly dependent on its molecular form and complex state. Uncomplexed PSA, also known as free PSA, can act freely to cleave specific substrates. Subsequently, fPSA has become the standard measure of enzymatically active PSA. Clinical measurements of serum PSA level are typically performed using the Tandem-R assay (Hybritech/Beckman-Coulter, Brea, CA) and reflect the amount of total PSA, which includes fPSA and PSA-ACT complex (PSA-a 2 M is undetectable using these assays). To evaluate the levels of fPSA in the various fluid compartments accessible to the prostate, Denmeade and coworkers examined extracellular fluid and serum from human patients (normal and prostate cancer) as well as from tumor xenografts. Within the patient group, the researchers found that total PSA in the extracellular fluid, as measured by the Tandem-R and Tandem-MP assays, did not vary significantly between normal and diseased individuals. However, 89% of the total PSA in patients with prostate cancer was enzymatically active versus 78% for the normal individuals. In addition, total serum PSA levels were extremely low in both groups with no enzymatically active fractions [38]. Tumor xenografts in nude mice demonstrated that only 18% of PSA secreted by LNCaP cells are enzymatically active, as compared to 66% from PC-82 cells. In all cases, whether human primary tumors or xenograft, no active PSA was found in the serum. The absence of active PSA in human serum presents an unique opportunity to deliver proteolytically-activated fluorescent contrast agents, such as the ones described in this work, intravenously without fear of extra-glandular activation. Moreover, PSA enzymatic activity appears to be a better metric for differentiating between normal and disease states in patients than total protein level. PSA substrate specificity has been characterized as chymotrypsin-like towards its main physiological substrate, semenogelin I. Cleavage sites were shown to be predominantly after tyrosine or leucine residues [39]. Serine, glutamine, and aspartic acid have also been found at the P_ 1 position [40]. Malm and coworkers confirmed reports that divalent cations, particularly zinc, act to inhibit PSA proteolytic activity towards semenogelin-I as well as PSA complexation with ACT. However, Robert and coworkers demonstrated evidence that the enzyme inhibitory effect of zinc may be attributed to zinc binding to semenogelin-I and not PSA. In recent research, PSA, SgI, and SgII have all demonstrated binding affinity for zinc. Zinc-inhibited PSA can recover its enzymatic activity with the addition of SgI and SgII, to effect an indirect regulatory mechanism for PSA proteolytic activity [41]. Historically, many of the synthetic substrates used to evaluate PSA enzymatic activity were chymotrypsin substrates. More specific substrates were necessary for targeted drug delivery applications, so Denmeade and coworkers evaluated several peptide sequences corresponding to the cleavage map for semenogelin-I and semenogelinII to identify HSSKLQ as the best substrate with high specificity for PSA and low degradation in serum [42]. Yang and coworkers used single-position minilibraries to determine an optimal hexapeptide substrate for PSA. Several studies had demonstrated increased substrate specificity for tyrosine at position P_ 1 . Based on this data, PSA was then found to have preference for serine at position P1 and phenylalanine at position P- 2 , for a proposed optimal substrate of QFYSSN [43]. These synthetic substrates were optimized from base peptide sequences chosen arbitrarily from the semenogelin-I cleavage map, and thus, reflect the best substrates from a limited pool of candidates. To truly pan the SgI cleavage map for PSA substrate preferences and rules which govern amino acid arrangements, Coombs and coworkers employed an iterative optimization technique to systematically evaluate substrate ef- ficiency as a function of single-position changes in amino acid residue. As a means of corroborating peptide sequence results from iterative optimization, substrate phage display was implemented and found to converge on SS(Y/F)-S(G/S) [44]. The most labile substrate found through phage display, GAGLRLSSYY-SGAG, was cleaved by PSA at a kcat : Km value of 3100 M 1 s 1 , nearly 100-times greater than the kcat : Km value reported for QFYSSN by Yang and coworkers. However, chymotrypsin was able to cleave this same substrate at an efficiency that was 24-times greater than PSA, as measured by kcat : Km value. Rehault and coworkers managed to identify SSIYSQTEEQ as the best PSA substrate to date with a kcat : Km value of 60,000 M-s-1 [45]. These reports, taken together, illustrate the major barrier to developing efficient and specific substrates, an incomplete knowledge of PSA enzymatic mechanisms and interaction with its native substrate. Moreover, even the most optimal substrates presented in the literature demonstrate weak catalytic efficiency, at a rate that is nearly an order of magnitude less than chymotrypsin. Several studies have conjectured that there may be extended site interactions between PSA and its protein substrates which enhance its enzyme activity and which cannot be recapitulated by short peptide substrates. Interestingly, novel PSA-binding peptides have been found, through cyclic phage display, which increase PSA enzyme activity towards a chymotrypsin substrate [46]. Many of these substrates were originally developed for use in clinical assays for PSA. There has also been significant interest in using these substrates as cleavable linkers for prodrug activation in prostate tumor-targeted delivery [47, 48, 49, 50, 51]. Prodrugs have the advantages of reduced systemic toxicity and selective activation in the tumor for site-specific drug action. 3.2.2 Hepsin Hepsin is a type II transmembrane serine protease that was first identified from cDNA clones isolated from human liver cDNA libraries [52]. This 417 amino acid protein was expressed at the RNA level in multiple tissues assayed in baboon, with the highest level found in the liver. Immunoblot analysis of a human hepatocellular carcinoma cell line, HepG2, and baby hamster kidney (BHK) cells identified one major band at 51 kDa observed in both cell lines and a minor bind, presumably a degradation product derived from the catalytic subunit portion of the 51-kDa protein, at 28 kDa found only in HepG2 cells. The catalytic subunit is located on the carboxyl half of the protein, extending from the cell surface into the extracellular space [53]. Anti-hepsin antibodies directed to the extracellular portion of the protein on the cell surface greatly suppressed cell growth when assayed in human hepatoma cells. Furthermore, antisense oligonucleotides that inhibited hepsin biosynthesis not only reduced cell growth but also altered the cell morphology to result in a more enlarged and flattened appearance [54], suggesting that hepsin is required for mammalian cell growth. Its proteolytic activity is involved in activation of human factor-VII in the blood coagulation cascade [55], pro-urokinase-type plasminogen activator [56], hepatocyte growth factor [57], and cleavage of extracellular matrix component, laminin-332 [58]. Hepsin-deficient mice experienced normal embryonic development and hemostasis [59]. Hepsin gene expression is found in endothelial cells [60] and is associated with angiogenesis [61]. Initial evidence of hepsin involvement in prostate cancer was revealed by gene expression profiling of benign and malignant human prostate samples [62] with further validation at the protein level using tissue microarrays comprised of clinically stratified prostate-cancer specimens [63]. Hepsin overexpression was observed in prostate cancer tissue samples with predominant staining confined to the plasma membrane in immunohistochemical analysis and correlated with measures of clinical outcome. Klezovitch and coworkers elucidated the physiologic role of hepsin overexpression in vivo in a transgenic mouse model with probasin (PB) promoter-driven hepsin expression [64]. PB-hepsin transgenic mice exhibited disorganized basement membrane as evidenced by weak or absent laminin 5 staining and diffuse collagen IV localization. Double transgenic mice bred from crossing PB-hepsin mice with LPB-Tag mice to create a hepsin overexpression model against a model of prostate cancer demonstrated that hepsin promotes prostate cancer progression as well as metastasis. Hepsin may operate physiologically by proteolytically activating specific substrate proteins that lead to downstream morphologic and functional changes. As a serine protease, hepsin may rely heavily on its functional activity in contributing towards cancer progression. We sought to characterize this functional activity as most reports in the literature have only analyzed gene and protein expression levels. Herter and coworkers [57] identified an optimized protease substrate, KQLR4VNG, from the hepatocyte growth factor precursor. 3.2.3 Urokinase-type plasminogen activator Another member of the membrane-associated serine protease family, urokinase-type plasminogen activator (uPA), is involved in tumor invasion and metastasis in many different cancers. uPA is expressed ubiquitously and secreted by many different cell types during their life cycles in a single chain zymogen form. Pro-uPA is proteolytically converted into an active Mr 50, 000 enzyme comprised of two polypeptide chains attached via one disulfide bond [65]. This mature form of uPA contains an A chain that imparts uPAR binding affinity and the catalytic B chain [66]. The zymogen form of uPA has no activity against its natural substrate, plasminogen; its activated form can be recognized by binding to plasminogen activator inhibitor type-1 (PAI-1). uPA also catalyzes the activation of hepatocyte growth factor/scatter factor (HGF/SF) and macrophage-stimulating protein (MSP) [67]. Additional inhibition of cell surface-associated uPA is modulated by a novel serine protease inhibitor (serpin), maspin, on DU145 cells [68]. Cell surface association through uPA binding sites, such as the uPA receptor (uPA-R), appears to enhance uPA proteolytic activity. Overexpression of uPA in a rat prostate cancer cell line led to increased metastasis to skeletal as well as non-skeletal sites; skeletal lesions exhibited osteoblastic characteristics [69]. Moreover, uPA was differentially expressed at the protein level between human prostate cancer cell lines derived from primary (1013L) versus metastatic lesions (DU145). Homogenates derived from nonaggressive 1013L tumor xenografts exhibited nearly 300-fold lower levels of uPA as compared to DU145 tumor homogenates. uPA could not be detected in plasma from 1013L inoculated mice but was present in the plasma of DU145-bearing mice [70]. Metastatic spinal column tumors were also found to exhibit higher levels of uPA activity as compared to primary spinal tumors [71], suggesting that uPA proteolytic activity may be as important as protein expression levels in the establishment and progression of prostate cancer metastases. Miyake and coworkers extended these inquiries addressing the role of uPA in prostate cancer progression into human clinical samples and concluded that the elevation of uPA or uPAR serum levels may positively associate with patient prognosis [72]. The clinical association of uPA levels with metastasis suggested that uPA may contribute mechanistically to prostate cancer invasiveness and migration. Yerba and coworkers investigated these potential mechanisms by transfecting a nonuPA expressing prostate cancer cell line, LNCaP, with a uPAR encoding cDNA to induce a high level of surface expression. uPAR-uPA ligation resulted in augmented tumor cell migration on fibronectin and potentiation of integrin-mediated signaling [73]. Besides cell surface associated uPA activity, uPA associated with membrane vesicles shed by prostate tumor cells can exert its effects at a distance from the local lesion. Vesicle-associated uPA can degrade basement membrane components in the absence of cells as well as enhance the invasive potential of nonaggressive LNCaP cells in in vitro assays [74]. Once at the skeletal metastatic site, uPA derived from prostate cancer cells influences the bone tumor burden; uPA-silenced PC3 cells in bone xenografts induced a reduction in tumor burden and bone destruction [75]. Recent evidence has also suggested that human stem cells exhibit increased tropism to tumor cells with high uPA and uPAR expression [76]. 3.3 3.3.1 Experimental Methods Cell culture The human prostate cancer cell lines, LNCaP-LN3, PC3M and PC3M-LN4, were a gift from Dr. Isaiah J. Fidler (M. D. Anderson Cancer Center, The University of Texas, Houston, TX). PC-3, LNCaP, and HT1080 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). HEK-293 cells were obtained from Jacqueline Banyard (Children's Hospital Boston, Boston, MA). PC-3, PC3M, and PC3M-LN4 cells were maintained in RPMI media (Invitrogen, Carlsbad, CA) supplemented to a final composition containing 10% fetal bovine serum (FBS) and 1%L-glutamine. LNCaP and LNCaP-LN3 cells were maintained in RPMI media modified to contain 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/L D-glucose, and 1.5 g/L sodium bicarbonate. HT1080 cells were maintained in Eagle's Minimum Essential Medium (EMEM; ATCC) supplemented with 10% FBS. HEK-293 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 2 mM L-glutamine and 10% FBS. All cells were grown under 370C and 5% CO 2 atmosphere. 3.3.2 Conditioned media collection Cells were seeded in 10-cm dishes at densities that would yield near confluence after 72 h. At 48 h after cell seeding, the culture medium was removed and replaced with 2 ml of serum-free culture medium per dish. After 24 h of incubation in serumfree conditions, the conditioned medium was collected and centrifuged at ~ 900 x g for 5 min at room temperature to pellet residual cells and cellular material. The supernatant was then added to an Amicon@Ultra-4 centrifugal filter unit (Millipore, Billerica, MA) and centrifuged per the manufacturer's instructions to concentrate the medium. The resulting conditioned media concentrates were analyzed for total protein content by BCA analysis (Thermo Fisher Scientific, Waltham, MA). 3.3.3 Gel electrophoresis and immunoblotting Protein samples were quantified for total protein content by the bicinchoninic acid method using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL) and reduced in Laemmli sample buffer containing #-mercaptoethanol by boiling the mixture for 5 min. Proteins were resolved on Tris-HCl precast polyacrylamide gels (Bio-Rad, Hercules, CA) by one-dimensional sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE). Molecular weights were determined by comparison to the Precision Plus Protein Kaleidoscope standards (Bio-Rad, Hercules, CA). Proteins resolved electrophoretically were transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (0.45 pm pore size; Millipore, Billerica, MA) using a Bio-Rad electroblotting transfer apparatus. Membranes were blocked with 5% (w/v) nonfat dry milk in PBS for 1 h followed by five washes with PBS supplemented to contain 0.1% Tween 20 (PBS-T). Primary antibodies were prepared to the specified dilutions in PBS containing 2% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and 0.1% sodium azide. Membranes were incubated in primary antibody solution for 1 h at room temperature followed by three washes of PBS-T for 5 min each. Horseradish peroxidase-conjugated secondary antibodies were diluted in 5% milk in PBS and added to the membrane for 1 h incubation at room temperature. Membranes were then washed six times with PBS-T for 5 min each and developed with chemiluminescent substrates as indicated by manufacturer. 3.3.4 PSA enzyme activity assay Concentrated conditioned media (50 pl) was added to an opaque 96-well plate in duplicate or triplicate samples while on ice. Substrate solution (HSSKLQ-AFC, Calbiochem/EMD Biosciences) was prepared as a 10 mM stock in DMSO. The remainder of the reaction volume was comprised of 400 pM substrate in 50 mM Tris-HCl and 10 mM NaCl to a final total volume of 150 pl. PSA standards were prepared from human enzymatically-active PSA (Calbiochem/EMD Biosciences) diluted in PBS. Substrate fluorophore release was monitored for AFC fluorescence on a Spectramax Gemini M5 spectrofluorometer (Nexc/em 3.4 3.4.1 400/505 nm). Results Engineering secretion of enzymatically active PSA: fulllength KLK3 Enzymatically active PSA results from correct post-translational processing of its zymogen form. As shown in Figure 3-1, the full-length native PSA sequence contains a 17-amino acid intracellular signaling peptide followed by a 7-amino acid pro-sequence that is cleaved extracellularly. Under- and over-processed PSA protein sequences result in enzymatic inactivation. Consequently, correct translation and processing by intracellular and extracellular proteases are critical to the proteolytic activity of PSA. Several human prostate cancer cell lines have been reported to synthesize and secrete PSA. However, the proteolytic activity levels of the secreted PSA in these cells have not been characterized in detail. LNCaP cells represent androgen-responsive human prostate cancer and were originally derived from a needle aspiration biopsy of the left supraclavicular lymph node of a 50-year-old Caucasian male with confirmed metastatic prostate cancer. We analyzed the total secreted PSA levels from LNCaP cells and its associated metastatic variant, LNCaP-LN3, as well as androgenindependent PC-3 and its metastatic variants, PC3M and PC3M-LN4. Figure 3-2 demonstrates the higher secreted levels of PSA from LNCaP cells over LNCaP-LN3 cells; PC-3, PC3M, and PC3M-LN4 cells did not produce detectable levels of PSA. LNCaP cells are slow growing and do not produce PSA levels that compare to values observed in clinical patient samples. Consequently, we sought to engineer a fast- growing high PSA-secreting cell line to serve as a potential in vivo tumor model, as such cells have not been found or established in the literature and would serve as an important model for investigating the role of PSA in human prostate cancer. Using a plasmid containing the full length PSA gene preceded by a secretory tag and driven by the T7 promoter (Figure 3-3a), we demonstrated the overexpression of this form of PSA transiently in HEK-293 and PC3M-LN4 cells, as they are extremely fast growing cells (Figure 3-3b), and established stably transfected clones from PC3M-LN4 cells. As shown in Figure 3-3c, clones #1, 4, pools A and B express significant amounts of PSA as compared to vector control clones. However, in proteolytic activity assays of conditioned media from transiently transfected HEK-293 cells and stable PSA clones from PC3M-LN4 cells, there is no detectable levels of PSA activity against a fluorogenic peptide substrate (Figure 3-4). The inactivity of the secreted PSA may be attributed to the appended secretory tag at the N-terminus of the PSA protein that may interfere with its proteolytic function. However, to eliminate the possi- bility of neutralizing proteins in the cell culture environment that may hinder PSA proteolysis, we collected conditioned media under additional controls that included non-transfected cells whose culture media had been spiked with exogenous purified active PSA. PSA secretion from the transient and stable clones as well as controls were confirmed (Figure 3-5), but proteolytic activity was once again only observed for purified active PSA (Figure 3-6). Interestingly, exogenous PSA added to tissue culture dishes of culture medium but no cells was not found on immunoblots after media collection. We presumed that the PSA protein may have irreversibly adhered to the treated tissue culture dish surface and was not collected in the conditioned medium. Furthermore, the preserved proteolytic activity levels of exogenously introduced purified PSA led us to conclude that no neutralizing proteins or deactivating processes occur in the cell culture and that the lack of PSA activity from the overexpression clones was a result of incorrect PSA processing due to the added secretory tag. Intracellular Signal Peptidase -24 to -8 Extraceflular Protes (hK2?) +1 to +237 -7 to -1 APULSR PSA Protein IVGG..... Pr-Pro PSA (nctive) Pro PSA (Inactive) Mature PSA (Active) Figure 3-1: Schematic diagram of prostate-specific antigen (PSA) posttranslational processing. A plasmid encoding the full length KLK3-tvl (human gene for PSA protein, variant 1) without additional modifications was used to investigated the natural posttranslational processing capabilities in fast growing HEK-293 and PC3M-LN4 cells (Figure 3-7a). PSA expression and subsequent secretion into conditioned media was demonstrated for HEK-293 cells (Figure 3-7b) but no proteolytic activity was observed 12 10 - 8 6 42 F 01 LNCaP LNCaPLN3 Figure 3-2: Total endogenous PSA protein secretion into conditioned media for LNCaP and LNCaP-LN3 cells. in the conditioned media of full length KLK3-expressing transient clones (Figure 37c). Although transient overexpression in PC3M-LN4 cells resulted in high secreted levels of total PSA, as compared to LNCaP whole cell lysates, which do endogenously express PSA (Figure 3-8a), no proteolytic activity was observed in these clones (Figure 3-8b). As previously discussed, the post-translational processing of full-length PSA protein is not well understood. Extracellular proteases, such as human kallikrein 2 (hK2), are known to cleave the pro-sequence from PSA and potentiate its enzymatic activity. However, hK2 is only one of many such proteases and no definitive upstream activator protein has been identified to date. The conversion of PSA from the inactive to the active form requires precise control, as under- and over-processing results in inactive protein. PSA expression in LNCaP cells has been well documented in the literature, and we presumed that the proper post-translational processing machinery and proteins were present in this cell line. Consequently, we overexpressed the full-length PSA protein in the LNCaP and LNCaP-LN3 cells to dissect the origins of this post-translational control. Using two different transfection reagents, Lipofec- - ~--~--~ -v I pSecTag2 TY prmote*rming site: bases 863-82 Murine Igkappa-Chain V-J2-C signal popUde: bases 905-967 PSA gene: bases 1042-1732 Hygro/PSA 6.4 kb 0-myc epitope: bases 1745-1777 Polyhistidine tag: bases 1790-1807 (Adapted from initrogen) b Transient Transfection EJE myc PSA C PC3M-LN4 Stable Clones e PSA Figure 3-3: PSA protein overexpression in conditioned media from 293 HEK and PC3M-LN4 cells using pSecTag2/Hygro/PSA plasmid. 293 Transient Transfections with PSA Plasmid - 200 ng PSA MinIprep - Control - Invitrogen Ib 0 20 40 60 80 100 120 140 160 Time (min) PC3M-LN4 Stable PSA Clones 300 - 250 - 200 - """PSA #1 -UPSA #4 -- PSA Pd A -x-PSA Pool B 150- -9i-Control Al 10050- -+-Hurman PSA (200 ng) An -501 20 40 60 80 100 120 140 160 Time (min) Figure 3-4: Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and stable PC3M-LN4 clones transfected with PSA plasmids. IC 293 HEK Transient Transfection IC +a. PSA PC3M-LN4 Stable Transfection CC 0 4 0 0 PSA Figure 3-5: PSA protein overexpression in conditioned media from 293 HEK and PC3M-LN4 cells using pSecTag2/Hygro/PSA plasmid with additional control samples. a 293 HEK Transient Transfection 1000 900 800 700, 600, 500. 400. 300. 200. 100' -+-DNA 2 --- No ThrAse +No ThnseclanPSA -x-sP-DUEM.PSA -u-200 ng PSA 0 0 20 40 OD 80 100 120 140 Time (min) PC3M-LN4 Stable Transfection 1000- -- Clone #1 -U-Clone 4 CIOne Pool A 900__- -X- U. Clone POol B -cnrl 700-600 -- Cim Al et TIUbSSS -+-Na Trns90con+PSA 500400- 20ri SA 3002001000 -100 20 40 60 80100 120 140 Time (min) Figure 3-6: Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and stable PC3M-LN4 clones transfected with PSA plasmids. tamine 2000 and FUGENE, we confirmed overexpression of pSecTag2-PSA and full length PSA proteins in LNCaP and LNCaP-LN3 cells. Transfection with pSecTag2PSA plasmids resulted in doublet protein bands, presumably corresponding to the increased protein molecular weight as a result of the appended secretory tag (Figure 3-9a). Non-transfected LNCaP cell conditioned media contained higher levels of active PSA as compared to LNCaP-LN3 cell-derived conditioned media (Figure 3-9b). Moreover, overexpressing PSA using the full length KLK3 plasmid in LNCaP cells produced higher levels of active PSA than the wild-type LNCaP cells. Although the increase in PSA activity levels was modest from the LNCaP FL #3 clone, the data provided conclusive evidence that LNCaP cells possessed the proper post-translational machinery to modify full-length PSA to its active form, as compared to HEK-293 and PC3M-LN4 cells. Since post-translational processing can be incredibly complex, it was not feasible to recapitulate the proteins and machinery involved from LNCaP cells to our desired model cell line, PC3M-LN4. 3.4.2 Engineering secretion of enzymatically active PSA: prosequence deletion PSA is expressed as a prepro-protein, where the pre-sequence facilitates intracellular localization and processing and the pro-sequence is cleaved extracellularly to produce the active form. Since the pre-sequence is associated with proper PSA secretion from the cell, this region was not manipulated. However, we deleted the pro-sequence using site-directed mutagenesis to facilitate the production of active PSA in the absence of appropriate extracellular conversion proteins (Figure 3-10). The pro-sequence deleted plasmid (KLK3-Del) was expressed in and secreted by all cell lines tested (LNCaP, HEK-293, LNCaP-LN3 and PC3M-LN4), as verified by PSA immunoblots of conditioned media (Figure 3-11a). Using a fluorogenic PSA substrate to quantify proteolytic activity, we calculated the reaction rate, which is associated with the amount of active protease, as normalized by 200 ng of active purified PSA. As shown in Figure 3-11b, LNCaP and LNCaP-LN3 wild-type, or control, cell derived conditioned media ..................................... Poybkmr Seque~m of pCUVS.XL4, XLS and XIA A) PCMV6-XL4 and XL5 (EcoR1-Xhol/Sal) Ane pCMV6-XLA A&A=OCQC=ZCAGM 7 zt?'ZGCACC,.MA? 13re - sma I (4483bp) cem1 svA s owittau no ~--- WS1 * -1*0-o aria GMTCCGAGTG&2CCCG=U C---9MOCIeOa """"* ---- --- C el rCeACeCCCCThAQ&& GCCWCCCTCTGACCCCTCCC20TGCTC2CTCGC 293 HEK Transient Transfection + + PSA 900 800 700 600 LL -+-293+KLK3-FL1 500 -U-293+KKUC3L3 -293(-) + PSA 400 300 -23(-) 200- - PSA -4P-PSA(1@ ng) 100 0 -100 20 40 60 80 100 120 140 Time (min) Figure 3-7: Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK clones transfected with full-length Origene PSA plasmids. a PC3M-LN4 Transient Transfection with KLK3-tv1 a. o + V_ j j W~~ L Z zL_ I PSA 3000 2500 2000 E -+s-Centre FL #1 FL #3 1500 -*-No Trmnsfe6ein+ PSA -purined PSA 1000 500 0 -500 20 40 60 80 100 120 140 Time (min) Figure 3-8: Enzyme kinetics of PSA secreted into conditioned media by transient PC3M-LN4 clones transfected with full-length Origene PSA plasmids. ........................... LNCaP WCL * 0 'LIZ ~ -- PSA FUGENE Upofectanine a0. LNCaP-LN3 WCL W * *0 L a. c1 '2 -n wa Lpofectami. u PSA FUGENE b 800 700 -- LNCP-uN Cn -u-LN+P-LN i+pn.02 +LNCaP-LN8+ FL #1 -*-LNCP-LM -Xfect +PSA 60 500U. M S400- -4-LNCaP Contro 300 -+-LNCaP+ pSe #1 LNC*P + FL 3 -- Purtlid PSA 200100 0 -100 Time (min) Figure 3-9: Enzyme kinetics of PSA secreted into conditioned media by transient LNCaP and LNCaP-LN3 clones transfected with full-length Origene PSA and pSecTag2-PSA plasmids. contained the highest levels of active PSA. Exogenous modulation of PSA overexpression may interfere with proper cell function and viability and thus, lower PSA production. Interestingly, conditioned media from PC3M-LN4 cells that had been transfected with full-length KLK3 plasmid demonstrated the highest PSA activity levels, rivaling those of wild-type LNCaP conditioned media. HEK-293 conditioned media exhibited no detectable levels of PSA activity. Despite previous evidence of non-detectable PSA activity in full-length KLK3 transfected PC3M-LN4 cell conditioned media, the current data indicate the possibility that PC3M-LN4 can secrete proteolytically active PSA but at very low levels. a Intracellular Signal Peptidase -24 to -8 Extracellular Protease (hK2?) +1 to +237 -7 to -1 PSA Protein IVGG..... Pre-Pro PSA (Inactive) Pro PSA (inactive) Mature PSA (Active) b (M3 150 1a1 AMo of HF Trs~qbcnd Od Trad Trrdago'ID1 2-7.060c Aim of 1O1CQ6*063O&ATG^t Trsdsba, of KO82Y-406 lila of <D52-3T06200rATGoriy Gywm 19.3 ABU 8 2W 240 i 2i57 cc06200 (14 DTra2dion3I (1>_4362006 M<U(3tv1Gae6 (144) Tr-lIF063 (37) 37 (1)_______ (1) WL8DGVI (37) (1) (37) (1) (37) 1%VWFII0RL3vMIG PO 70 89 AM 018 ________ VARPWG A 'MnIggN Jt p Figure 3-10: Schematic diagram of prostate-specific antigen (PSA) posttranslational processing and engineered pro-sequence deletion. Conditioned Media from Transient Transfections *e PSA 23 HEK LNCP C A..3 U cc U. PSA PC3M4LN4 LNCSP-LN3 * * KLK3-Del KLK3-FL U Contro N wnend t Pn.Md PSA PC3M4N4 LNCaP LNCaP-LN3 Figure 3-11: Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK, PC3M-LN4, LNCaP, and LNCaP-LN3 clones transfected with full-length Origene PSA and KLK3-deletion plasmids. 3.4.3 Engineering secretion of enzymatically active PSA: KLK2 + KLK3 co-transfection Another strategy to increase extracellular levels of active PSA was to overexpress an upstream activator protein to correctly convert pro-PSA to PSA. Human kallikrein 2 (hK2) has been well documented as such an upstream protease. Consequently, we co-transfected HEK-293 and PC3M-LN4 cells with both full-length KLK2 and KLK3 plasmids and analyzed the secreted protein activities against a fluorogenic PSA substrate. Figure 3-12a demonstrates the secreted protein levels of cells transfected with KLK2 alone, KLK3 alone, and both KLK2 and KLK3. KLK2 has high sequence homology to KLK3/PSA, resulting in a faint band close to the molecular weight of KLK3 on PSA immunoblot. The secreted conditioned media from both HEK-293 and PC3M-LN4 cells co-transfected with KLK2 and KLK3 demonstrate a much higher level of PSA activity (Figure 3-12b). HEK-293 conditioned media contained higher levels of active PSA as compared to PC3M-LN4 conditioned media, but these same cells, when transfected with each plasmid alone, exhibited no appreciable PSA activity. This data suggest that the high PSA activity observed in KLK2 and KLK3 co-transfection in HEK-293 cells is not due to an additive effect of PSA and hK2induced non-specific substrate cleavage, but instead, the result of synergistic activity between the two proteins. The evidence for this synergism between PSA and hK2 is not as definitive in co-transfected PC3M-LN4 cells. However, the observed increase in substrate cleavage activity is significant compared to the individual contributions of each plasmid transfected alone. The combined overexpression of KLK3 and its upstream activator, KLK2, demonstrated the highest level of active PSA in conditioned media achieved using multiple methods. Based on the promising results of transiently co-transfecting PC3M-LN4 cells with KLK2 and KLK3 plasmids, we investigated the use of PC3M-KLK2 and PC3M-KLK3 stable clones in co-culture for generating high levels of active PSA. Multiple selection rounds had failed to select pure clonal populations of KLK2 and KLK3 overexpressing cells. However, we were successful in establishing PC3M stable clones and performed a co-culture assay as outlined in Figure 3-14a. One stable clone was seeded in a 6well tissue culture plate, while the other stable clone was grown on fibronectin-covered glass coverslips in a 12-well plate. After independent growth culture for 48 h, the cellseeded coverslips were aseptically inserted into the corresponding well of the 6-well plate to commence co-culture. Conditioned media was collected 24 h after the start of co-culture. Figure 3-14b shows the reaction kinetics of conditioned media with a PSA fluorogenic substrate. In this assay, PC3M-KLK3 cells seeded in the 6-well plate co-cultured with coverslip-confined PC3M-KLK2 vector control cells resulted in the highest levels of active PSA in the conditioned media. The next highest level of active PSA was produced by the combination of PC3M-KLK3 cells seeded in the 6well plate co-cultured with PC3M-KLK2 cells on the coverslip. The concentration of active PSA in the conditioned media after cell co-culture was quantified by comparison with a standard curve generated within the same assay. Table 3.1 summarizes these values and demonstrates that the highest concentration of active PSA is slightly less than three times that of the vector control background level. Despite the promising levels of active PSA demonstrated under transient co-transfection with KLK2 and KLK3 plasmids, the PC3M stable clones under co-culture did not produce a dramatic increase in active PSA levels. Table 3.1: Concentration of Enzymatically Active PSA in PC3M-KLK2 and PC3M-KLK3 Co-Culture Active PSA Co-Culture Conditions (ng/ml) 94 hK3 + GFP Vector Control (Coverslip) 76 hK3 + hK2 (Coverslip) 44 GFP Vector Control + hK3 (Coverslip) 35 DsRX Vector Control + GFP Vector Control (Coverslip) _j Conditioned Media from Transient Transfections + HE + PSA PC3M-LN4 293 HEK 300 250 -4--293 KLK2 200 M IL -- 293 Kuc3 -r-293 KLK2 + KLK3 -M-293 Control -- PcU3M KLK2 150 -- PC3M-L4 KLK3 -+-PC3M-LN4 KLK2 + KLK3 100 -PossuAs 50 cmontro -- Pwefd PSA 0 -50 20 40 60 80 100 120 140 Time (min) Figure 3-12: Enzyme kinetics of PSA secreted into conditioned media by transient 293 HEK and PC3M-LN4 clones co-transfected with full-length Origene KLK2 and KLK3 plasmids. ................. w ft 15 - EKLK2 12 - EKLK2+KLK3 I-KLK3 Control S 3 0 -3 L j M HIM Nm"Wflwd 18 PWVW MA PCX-LN Figure 3-13: Reaction rate of PSA secreted into conditioned media by transient 293 HEK and PC3M-LN4 clones co-transfected with full-length Origene KLK2 and KLK3 plasmids. 3.4.4 Hepsin expression and proteolytic activity Hepsin is a transmembrane serine protease that has been positively associated with human prostate cancer metastasis. Its expression at the mRNA and protein levels has been well characterized with respect to disease progression in clinical human samples and experimental models. We developed a hepsin proteolytic activity assay by incubating cell monolayer culture with a fluorogenic hepsin substrate (KQLRVVNGAFC). Due to the dearth of validated hepsin-specific antibodies, endogenous hepsin expression was evaluated with a hepsin immunoblot of immunoprecipitated samples from HepG2, LNCaP, LNCaP-LN3, HEK-293, and PC3M-LN4 whole cell lysates (Figure 3-15). Hepsin was found in HepG2, LNCaP, LNCaP-LN3, and HEK-293 cells but not in PC3M-LN4 cells. These results concur with data reported in the research literature [77, 53]. Using the pro-HGF derived hepsin substrate, we assayed the fluorescence release generated when incubated with each cell line in culture. Fluorescence signals were normalized to that generated by 1 x 104 HepG2 cells. As shown in Figure 3-16a, fluorescence generation did not associate with hepsin expression levels. Despite moderate hepsin expression, HEK-293 cells did not cleave the pro-HGF derived sub- Coverslips In 12-well plate seeded with stable clone cells Stable clone cells seeded in 6-well plate OOOO0 OOOO 48 h Place coversap Into appropriate wefl of 6-well plate Collect conditioned media after 24 h -- 2+3C 200 +3+2C +2+RC -+-3+GC *G+RC -- R+GC 150 -+-R+2C -G+3C 200 300 400 500 600 700 800 900 Time (min) Figure 3-14: Enzyme kinetics of PSA secreted into conditioned media by coculture of stable PC3M clones transfected with full-length Origene KLK2 and KLK3 plasmids. strate to any appreciable degree. Furthermore, PC3M-LN4 cells were able to cleave the substrates, presumably through other expressed proteases, despite not showing any hepsin expression. This non-specific substrate cleavage was also observed during the time progression of fluorophore release, where PC3M-LN4 cells generated a significant and relatively linear signal. The conflicting data led us to overexpress human hepsin in HEK-293 and PC-3 cells to determine the immunoblot antibody specificity as well as to determine the relevant protein bands and associated molecular weights. The major protein band was observed ~ 50 kDa with additional bands observed for hepsin overexpressed in HEK-293 cells (Figure 3-17). The presented data demonstrate that the reported pro-HGF derived substrate is not specific to hepsin cleavage. A 150, 100 75 Hepsin 50 37 Figure 3-15: Hepsin immunoprecipitation from 293 HEK, PC3M-LN4, LNCaP, HepG2, and LNCaP-LN3 cell lysates (Cayman polyclonal antibody). ........................................ 1.0 - 0.8 - 0.6 - Normanned to HIp02-10K Cens * 1E4 Cells/Well * 2.5E4 Cells/WelI 0.011 _rMMM HepG2 293 HEK PC3MLN4 LNCaP LNCaPLN3 20001600 - 1200 - 800 - +HepG2 -E-293 HEK - -PC34.N4 --*LNCaP -*-LNCaP-LNS 4000 10 20 30 40 50 60 70 80 90 100 Time (min) Figure 3-16: Enzyme kinetics of cell surface hepsin in cell cultures of HepG2, 293 HEK, PC3M-LN4, LNCaP, and LNCaP-LN3. b2 a0. z o + 'U 'U C I- C Z 0 U 50 37 Hepsin 25 Figure 3-17: Hepsin immunoblot (Cayman polyclonal antibody) of HPN overexpression in 293 HEK and PC-3 cells. 3.4.5 Endogenous uPA secretion uPA expression and protease activity play an important role in cancer progression and metastasis. We characterized the secreted levels of uPA and its enzymatic activity by ELISA. Active uPA is known to bind PAI-1 and can be quantified using indirect immunobinding in a 96-well plate assay. As shown in Figure 3-18a, total secreted active uPA levels as measured in conditioned media vary inversely with metastatic potential in PC-3-derived cell lines. HT1080 cells were included in the panel, because several reports in the literature have commented on its high secretion of uPA. In our assay, the total uPA level secreted by HT1080 cells was ~-95.9 ng per 1 x 106 cells as compared to ~,, 134.5 ng per 1 x 106 cells. Active uPA constituted at most ~ 5% of the total amount of secreted uPA in PC-3 cells. The percentage of active uPA also varied inversely with metastatic potential (Figure 3-18b). . ........... . ..... a 876 - 4- S3 - 01- PC-3 PC3M PC3MLN4 HT1080 PC-3 PC3M PC3MLN4 HT1080 b 7654. 3210 Figure 3-18: Endogenous active uPA secretion by PC-3, PC3M, PC3M-LN4, and HT1080 cells. 3.5 Conclusions Proteases play important regulatory roles in cancer progression and dissemination. We sought to evaluate the suitability of various cell lines to establish small animal cancer models that recapitulated the enzyme activity levels observed in human clinical samples. Efforts to engineer high levels of enzymatically active PSA secretion in a fast growing human prostate cancer cell line revealed the highest active PSA concentration in conditioned media from PC3M-LN4 cells co-transfected with KLK2 and KLK3 genes. Overexpression of the upstream protease activator for the secreted pro-form of PSA generated increased levels of active PSA not only in PC3M-LN4 cells but also in HEK-293 cells that had not shown any significant PSA activity using fulllength KLK3 transfection. However, it was not possible to establish double-stable clones for KLK2 and KLK3 expression in PC3M-LN4 cells for in vivo experiments. Consequently, singly-transfected stable PC3M clones were generated for use in coculture induction of PSA activation by hK2. The highest active PSA levels were generated by PC3M-KLK3 stable clones co-cultured with the PC3M-KLK2 vector control clone (~ 2.8 nM) followed by PC3M-KLK3 clone co-culture with PC3MKLK2 clone (- 2.3 nM) and represented less than 2% of the amount of active PSA found in extracellular fluid from human prostate cancer samples from patients [38]. Human prostate cancer cell lines, PC3M and PC3M-LN4, were shown to possess at least core post-translational processing machinery both intracellularly as well as extracellularly to convert a portion of overexpressed PSA to active PSA when neither cell lines constitutively express this protein. In relating levels of active uPA to cancer progression, we found that in cell culture, the amount of secreted active uPA varied inversely with the cell line's metastatic potential. The highest total and active uPA levels were secreted by PC-3 cells. This series of protease characterization in cancer cell lines will allow appropriate selection of in vitro and in vivo models for applying and relating protease function to progression-associated mechanisms. Chapter 4 Self-assembled Gold Nanoparticle Molecular Probes 4.1 Abstract Target-activatable fluorogenic probes based on gold nanoparticles (AuNPs) functionalized with self-assembled heterogeneous monolayers of dye-labeled peptides and poly(ethylene glycol) have been developed to visualize proteolytic activity in vivo. A one-step synthesis strategy that allows simple generation of surface defined AuNP probe libraries is presented as a means of tailoring and evaluating probe characteristics for maximal fluorescence enhancement after protease activation. Optimal AuNP probes targeted to trypsin and urokinase-type plasminogen activator required the incorporation of a dark quencher to achieve 5 to 8-fold signal amplification. These probes exhibited extended circulation time in vivo and high image contrast in a mouse tumor model. 4.2 Introduction Molecular imaging is an emergent discipline that has the potential to advance integrative biology, promote early detection and characterization of disease, and allow for greater ease in the evaluation of medical interventions. Current implementa- tions of molecular imaging couple the use of existing imaging modalities with unique contrast agents, or molecular probes, designed to target select biomarkers or molecular processes. Specifically, the development of near-infrared fluorescence (NIRF) imaging probes, as essential tools in optical imaging, has enabled researchers to study the real-time dynamics of cellular and biochemical processes in vivo. NIRF probe-enhanced optical imaging demonstrates particular promise in cancer diagnosis and treatment and has been applied towards two main areas of interrogation: (1) semiquantitative assessment of biomarker expression levels, and (2) evaluation of pathway-dependent biomolecular activity. Imaging studies to determine pathologic biomarker expression levels typically employ NIRF probes consisting of near-infrared fluorescent molecules or nanoparticles conjugated to affinity ligands, such as antibodies or peptides, to achieve target-specific image contrast and visualization of tumors [78, 79, 80, 81, 82, 83, 84] These same lesions could also be imaged and characterized by their biochemical activity signatures, such as increased proteolysis, using proteaseresponsive NIRF probes that generate an increased fluorescence signal upon cleavage [85, 86, 87]. One significant advantage of targeting protease activity to convert fluorescently quenched probes to a fluorescent state via proteolysis is the signal amplification capability inherent to the process. Many proteases are known to play essential roles in disease progression, and the ability to directly visualize their physiological activity provides an additional modality for understanding pathogenesis, progression, and treatment effects. However, the process of optimizing NIRF probes, specifically, their quenching efficiency, biocompatibility, targeting ability in vivo, and enzyme reaction kinetics, requires a combinatorial approach that proves cumbersome and impractical when applied to existing probe synthesis strategies. Protease-activatable NIRF contrast agents can take the form of single dye-labeled peptide substrates on a synthetic graft copolymer [88, 89] or other multivalent support or dual-labeled substrates with dye and quencher/acceptor molecules covalently linked to either terminus [90, 91]. The first design represents a homogeneous concentration of fluorescent substrates on a vehicle backbone and relies on self-quenching, while the second con- figuration implements fluorescence resonance energy transfer (FRET) between dye and quencher/acceptor molecules to suppress the pre-activation fluorescence signal. These strategies require multiple covalent labeling reactions and purification steps and do not allow the ease or modularity of "swappable" components, such as changes in dye molecules. To address the need for a simple combinatorial method of synthesizing defined NIRF probes, we report the development of fluorescence quenched peptide-based gold nanoparticle (AuNP) probes for the detection of trypsin and urokinase-type plasminogen activator (uPA) proteolytic activities. uPA has been implicated in the progression of multiple cancers, including breast and prostate, by promoting tumor cell invasion, survival, and metastasis. Trypsin was chosen as a simple model protease against which to test the fluorescence quenched AuNPs. Gold nanoparticles (20 nm diameter) were modified with a heterogeneous monolayer of fluorophore and dark quencher-labeled peptide substrates and thiol poly(ethylene glycol) (SH-PEG; M. 1000 Da). The fluorophore and dark quencher dyes were chosen to be Quasar 670 (Q670) and BHQ-2, respectively. Peptide substrates were synthesized with a cysteine residue at the C-terminus so that the peptides could passively adsorb onto the gold nanoparticle surface via the sulfhydryl group. Once all labeled substrates and SHPEG self assemble on the AuNP, effective fluorescence quenching is facilitated by the proximity between fluorophore and dark quencher on the nanoparticle surface as well as the AuNP itself. Figure 4-1 illustrates the one-step reaction method employed in synthesizing different mixed monolayer surfaces on the AuNP probes and the mode of fluorescence signal generation by target proteolysis of probe surface substrates. Stoichiometric ratios of each surface component at a molar excess over the estimated maximal mole quantity of peptide loading on the 20 nm AuNP surface were varied to generate probe surfaces of differing composition. Dye-labeled peptides and SHPEG were combined with AuNPs in a single 500 pl reaction mixture and rotated for 16 h at room temperature. The self-assembled AuNP probes were purified and separated from uncomplexed peptide substrate and SH-PEG by centrifugation and multiple washes with water. AuNP probe activation by the target protease is proposed + /0 16 hat RT + G / 20-nm AuNP Peptide-Cys W 0 Near4nfrared fluorophore (Quasar 670) Dark quencher (BHO-2) SH-PEG (MW -1000) ~Target protease Figure 4-1: Schematic diagrams of gold nanoparticle (AuNP) probe synthesis and activation. to effect a de-quenching state by cleavage-specific release of fluorescent and nonfluorescent substrates from the probe surface and subsequent separation of fluorophore and quencher. 4.3 4.3.1 Experimental Methods Fluorescence quenched AuNP probe preparation Gold nanoparticles (Ted Pella, Redding, CA) were pelleted from the stock solution by centrifugation at 10,000 x g for 15 min. The solvent was then replaced with distilled water. N-HSSKLQC-Wang resin was commercially synthesized (Anaspec, San Jose, CA) and modified with Quasar 670 carboxylic acid and BHQ-2 carboxylic acid (Biosearch Technologies, Novato, CA) at the MIT Biopolymers Laboratory (Cambridge, MA) using standard Fmoc protocol. Dye-labeled uPA substrates with core peptide sequence, GGSGRSANAKC, were also synthesized and modified in the same manner at the MIT Biopolymers Laboratory. Purified Quasar 670 and BHQ-2 labeled peptides were dissolved in distilled water. Stock solution concentrations were determined using absorbance at 579 nm (6 = 38, 000 M-1 cm- 1 for BHQ-2) and 644 nm (E = 187,000 M- 1 cm-1 for Quasar 670). Varying stoichiometric ratios of SH-PEG (Nanocs, New York, NY), B-HSSKLQC, and Q-HSSKLQC were added to AuNPs in water to a final volume of 500 pl. The conjugation mixture was rotated end-overend at room temperature overnight for 16 h. Functionalized AuNPs were pelleted at 10,000 x g for 15 min and washed three times with distilled water. AuNP probes were then stored in water at 4'C. 4.3.2 Trypsin activation assay Functionalized AuNPs were pelleted at 10,000 x g for 15 min and solubilized in 600 pl of 2% dimethyl sulfoxide (DMSO) in water (v/v). The solution was then aliquoted (200 pl per well) into opaque 96-well plates (Whatman UNIPLATE). Freshly prepared trypsin (100 U/pl; Sigma- Aldrich, St. Louis, MO) was added at 1,000 U per well concurrent with dithiothreitol (DTT; Sigma-Aldrich, St. Louis, MO) at 10 Pi per well and incubated at room temperature for 30 min. Fluorescence measurements for Q670 (Aexc/em = 644/670 nm) were made on a Spectramax Gemini M5 spectrofluorometer (Molecular Devices, Sunnyvale, CA). 4.3.3 uPA activation assay Functionalized AuNPs were reconstituted in phosphate-buffered saline (PBS) pH 7.4 to a final nanoparticle concentration of 1.61 nM and aliquoted as 180 pl per well (0.29 pmol AuNP per well) of an opaque 96-well plate (Whatman UNIPLATE). Human urokinase purified from urine (Calbiochem/EMD Biosciences, San Diego, CA) was added to each well in triplicate at 10 U (20 pl of 0.5 U/pl in PBS pH 7.4); 20 pl of DTT was added to separate wells in triplicate for a final concentration of 100 mM per well. Fluorescence kinetic measurements were recorded on a Spectramax Gemini M5 spectrofluorometer immediately after addition of reagents. Endpoint fluorescence measurements for Q670 (Aexc/em = 644/670 nm) were made after the microplate reactions had incubated at room temperature for 16 h overnight. 4.3.4 kcat : Km determination AuNP probes (50-2-5) at different concentrations were aliquoted into replicate wells of an opaque 96-well Whatman plate (final concentrations in each well ranging from 773 pM to 11.6 nM). Freshly prepared trypsin was then added at 1000 U per well; fluorescence (Aexc/em = 644/670 nm) was then monitored in a spectrofluorometer. Initial release rates of surface substrate were calculated based on a standard curve generated by measuring mixtures of AuNP, BHQ2-labeled and Quasar 670-labeled substrates corresponding to the assayed AuNP probe concentration range. Enzyme kinetics curve fitting was performed using Prism 5.0 software (GraphPad Software, La Jolla, CA) based on the Michaelis-Menten model. 4.3.5 In vivo characterization All animal studies were approved by and adhered to guidelines set forth by the Children's Hospital Boston Institutional Animal Care and Use Committee. Circulation studies Total surface dye-labeled substrates on each batch of 50-2-5 AuNP probes were determined by isolating 1 mg of AuNP probe by centrifugation (10,000 x g for 15 min at room temperature). AuNP probe surface components were isolated by addition of 60 pul DTT (1 M stock concentration) followed by 240 pl double-distilled water to the AuNP probe pellet. The reaction was allowed to incubate at room temperature for 16 h. The released surface molecules were collected by centrifuging the reaction at 10,000 x g for 15 min at room temperature. Aliquots (100 pl) of the supernatant were made into a UV-transparent 96-well plate. Absorbance readings were then made as previously described to determine dye composition. Male Nu/nu mice (Massachusetts General Hospital, Boston, MA; 6-8 weeks old) were anesthetized using Avertin (240 mg/kg) injected intraperitoneally. 50-2-5 AuNP probe or the equivalent amount of uncomplexed dye-labeled peptide in sterile water was injected intravenously via lateral tail vein (1 mg AuNP probe in 100 pl water per mouse). Mice were euthanized at the specified time points followed by immediate blood collection into BD Microtainer serum separator tubes (BD Biosciences, Franklin Lakes, NJ). Blood samples were allowed to clot at room temperature for 20-30 min prior to centrifugation at 10,000 rpm for 2 min at room temperature. Aliquots of 100 pl mouse serum were made into each well of an opaque 96-well plate (Whatman UNIPLATE). Dye labeled substrates from AuNP probes in the serum were released from the surface by addition of 40 pl DTT to each well (-, 286 mM final DTT concentration in each well) and allowed to incubate at room temperature for 16 h. Fluorescence measurements for Q670 (Aexc/em = 644/670 nm at 20 reads on medium PMT settings) were made on a Spectramax Gemini M5 spectrofluorometer. Imaging studies Male Nu/nu mice (Massachusetts General Hospital, Boston, MA; 6-8 weeks old) were anesthetized under isoflurane. Functionalized AuNPs in sterile water were mixed with PuraMatrix (BD Biosciences, Franklin Lakes, NJ) and stored on ice. Immediately prior to injection, 250 U trypsin was mixed with the AuNP/PuraMatrix solution. 100 pl of this solution was then injected subcutaneously on each flank. Mice were then imaged using a Xenogen IVIS 200 system (Caliper Life Sciences, Hopkinton, MA) employing the Cy5.5 filter set (Aexc/em = 615 - 655/695 - 770 nm). 4.4 4.4.1 Results Photophysical characterization of AuNP probes The self-assembled AuNP probe demonstrated an absorption spectrum bearing strong similarity to that of the bare AuNP, with peak absorbance at approximately 530 nm (Figure 4-2a). Although no localized surface plasmon resonance (LSPR) band shift was observed for the surface-modified AuNP probe as compared to the bare AuNP, broadening of the absorption spectrum in the longer visible wavelengths reflected the incorporation of BHQ2-labeled and Q670-labeled peptide substrates. The absence of a LSPR band shift indicated that the adsorption of surface components had no appreciable effect on the resulting nanoparticle size, shape, or dielectric constant of the surrounding medium and did not induce AuNP aggregation. BHQ2-labeled peptides exhibited a broad extinction profile that extended into the near-infrared wavelengths to overlap with the predicted emission wavelength range of the Quasar 670 fluorophore. Fluorescence emission spectra (Aexc = 400 nm) were collected for unlabeled AuNPs, Q670-labeled and BHQ2-labeled peptide substrates at molar concentrations matched to those of the assembled AuNP probe surface components in water (Figure 4-2b). As predicted, the BHQ-2 molecule did not fluoresce and acted only as a strong absorber, thus termed a dark quencher. The AuNP probe demonstrated negligible fluorescence and an emission spectrum mirroring that of the unlabeled AuNP. This 1.0 0.8 0.6 0.4 0.2 0.0 '- 400 57570 500 600 Wavelength (nm) 700 - 0670-Iabeled peptide - Bare AuNP AuNP probe -- BHQ2-abeled peptide 720 670 620 Wavelength (nm) 800 770 Figure 4-2: Photophysical characterization of AuNP probes. observation suggests that the combination of BHQ-2 and attachment to the gold surface provides sufficient fluorescence quenching for Quasar 670 emission. Fluorescence suppression is so complete that the only signal detected is attributable to the native emission properties of the unlabeled gold nanoparticle core. Several groups have demonstrated the application of gold nanoparticle-induced fluorescence quenching in biomolecular and cellular detection [92, 93, 94, 95, 96]. The primary mode of surfaceconfined quenching of fluorophores on AuNPs has been identified as a reduction in the radiative rate rather than energy transfer [97]. Studies have shown that strong fluorescence quenching occurs when fluorophores are confined within ~ 5 nm distance from the nanoparticle surface [98, 99]. The surface moieties used in assembling the AuNP probe were expected to fall well within this distance range and benefit from gold-induced quenching. However, since AuNPs are also capable of fluorescence enhancement [100, 1011, we hypothesized that the inclusion of a dark quencher on the molecular probe would further reduce the fluorescence background signal, beyond the signal suppression offered by the gold surface. 4.4.2 Functional screens of AuNP probe libraries We assessed the contributions of individual surface components to the overall AuNP probe performance by generating a library of trypsin-targeted probes. Trypsin preferentially hydrolyzes peptide bonds at the C-terminal side of arginine and lysine residues. The prevalence of these two amino acids in the proteome enables nonspecific trypsin cleavage of many optimized protease substrates. Consequently, initial proof-of-concept and characterization of the AuNP probe library were performed using trypsin as a low-stringency protease target. A prostate-specific antigen (PSA) selective protease substrate, His-Ser-Ser-Lys-Leu-Gln [42], served as the core peptide sequence for the activatable dye-labeled components on the AuNP probe surface. Q670 (or BHQ2)-(His-Ser-Ser-Lys-Leu-Gln)-Cys substrates were synthesized with the dye conjugated to the N-terminus and a cysteine residue appended to the C-terminus to impart thiol functionalization to the peptide for spontaneous adsorption to AuNPs. Maximum surface peptide density on a 20 nm AuNP was estimated 0.4 - *$S OB+6PEG 0 0B+15PEG 6B+15PEG LI 6B+6PEG 0.3- 0 0 0 o2 .2 0.1 0.0 6 15 30 150 Q670-Peptide reaction concentration (pM) b ** 0.4- 0 oh- 0.30.20.1 - a- 0.0 -- Figure 4-3: Trypsin activation assay. to be 1 peptide/nm 2 , resulting in - 1.29 nmol of peptide per pmol of AuNP. The nomenclature used herein to classify different AuNP probe formulations follows the scheme X- Y-Z, where X, Y, and Z refer to the molar fold excess over the estimated maximum surface peptide density of Q670-labeled substrate, BHQ2-labeled substrate, and SH-PEG, respectively, in the reaction mixture. An activation ratio, or fraction of total possible fluorescence, was determined by measuring the Quasar 670 fluorescence (Aee = 644 nm; Aem = 670 nm) generated after reacting 1000 U of trypsin with 0.39 pmol of assembled AuNP probe for 30 min and comparing that value to the fluorescence of all surface moieties after being displaced from the nanoparticle surface with dithiothreitol (DTT). The highest activation ratio (36.7 ± 1.4%) was observed for the 50-2-5 AuNP probe, which corresponds to a reaction mixture containing 150 pM Q670- substrate, 6 pM BHQ2-substrate, and 15 pM SH-PEG (Figure 4-3a). AuNP probes formulated with higher concentrations of Q670-substrate demonstrated a trend towards higher activation ratios, suggesting that the increased availability of Q670-substrate in the formulation mixture leads to greater availability of these same peptides on the nanoparticle surface for trypsin cleavage. Furthermore, the absence of BHQ2-substrates in the reaction mixture results in activation ratios that are much lower than the same AuNP probe formulations with BHQ2-substrates. AuNP probes assembled without BHQ2- substrate exhibited activations ratios that do not extend beyond 15%. To further explore these observations, we examined the activation ratios for the family of AuNP probes formulated with the optimal Q670-substrate concentration (150 pM) more closely for significant trends (Figure 4-3b). An increase in reaction concentrations of SH-PEG, from 6 pM to 15 pM, improved the activation ratios, independently of the amount of BHQ2-substrates. This observed effect may be attributed to increased hydrophilicity of the AuNP probe surface monolayer and greater probe stability in solution. AuNP probes formulated without SH-PEG precipitated from the aqueous solution, most likely due to the increased hydrophobicity of the probe surface created by the adsorption of multiple dye molecules. In addition, the incorporation of BHQ2-substrates further enhanced the AuNP probe activation ratio, irrespectively of the level of SH-PEG reactants. Lee and coworkers have de- scribed a similar AuNP probe that utilized Cy5.5-labeled MMP-targeted substrates on the nanoparticle surface and relied on Cy5.5 self-quenching to induce a dark state in the inactivated form [102]. However, the analogous construct in our study, a BHQ2-deficient AuNP probe (50-0-5), demonstrated only 14.9 t 2.4% recovery of fluorescence after trypsin proteolysis, which represents a greater than 50% reduction from the activation ratio observed for the optimal BHQ2-containing probe (50-2-5). Under the experimental conditions described herein, self-quenching interactions between adjacent Quasar 670 dyes on the nanoparticle surface did not provide the lowest inactivated fluorescence signal; a dark quencher, such as BHQ-2, was necessary to further suppress the background fluorescence to produce optimal signal and image contrast. Figure 4-4a illustrates the general trend towards increased signalto-background ratios in trypsin activation of BHQ2-containing probes over BHQ2deficient probes, with the best performing nanoparticles exhibiting a greater than 12-fold signal enhancement. To examine the applicability of these observations in the context of a different protease, we constructed a separate library of uPA-targeted AuNP probes in the same manner as the trypsin- targeted probes, using a core substrate sequence of Gly-Gly-Ser-Gly-Arg-Ser-Ala-Asn-Ala-Lys. The activation fluo- rescence signal after reacting 0.29 pmol AuNP probe with 10 U uPA was compared to the background signal observed for 0.29 pmol AuNP probe without the addition of protease. Similar to the results reported for BHQ-2 influences in the activation of trypsin-targeted probes, uPA-specific probes containing BHQ-2 exhibited a wider dynamic range in activation signal, as illustrated by the expected near unity signal enhancement ratio observed for the 0-2-5 probe as opposed to the 0-0-5 probe, and higher signal-to-background ratios for almost all probe formulations (Figure 4-4b). These data suggest that the advantages of incorporating a dark quencher into the AuNP probe, namely additional background signal reduction and enhanced fluorescence recovery, rather than relying exclusively on self-quenching between near-infrared fluorophores, can be generalized to multiple protease targets. Surface composition analysis of trypsin-targeted AuNP probes revealed several characteristics that correlate with probe performance and explain the effects of re- 150 30 15 6 0670-Peptide reaction concentration (pM) OB+15PEG 90 150 30 15 6 0 0670-Peptide reaction concentration (pM) Figure 4-4: Protease-induced fluorescence enhancement over unactivated AuNP probe. Table 4.1: AuNP Probe Surface Composition as Defined by Reaction Concentrations of Quasar 670-labeled Peptide Substrate No. BHQ2-peptides No. Q670-peptides Q670:BHQ2 per AuNP per AuNP Ratio 0-2-5 ** 5 ** 10-2-5 11 25 2.3 20-2-5 10 26 2.7 30-2-5 4 1 0.33 40-2-5 6 2 0.34 50-2-5 6 2 0.30 Formulation action mixture formulation on surface adsorption. Table 4.1 outlines the number of surface Q670-substrates and BHQ2-substrates found on purified AuNP probes. After determining the importance of BHQ-2 incorporation and increased SHPEG reactant levels to probe performance, we investigated the effects of variable Quasar 670 reactant levels on probe surface formation. The addition of more Q670-substrate to the reaction mixture did not result in a monotonic increase in the amount of surfaceadsorbed species. At an intermediate concentration of Q670-substrate, between the 20-2-5 and 30-2-5 probes, the ratio of BHQ2-containing substrates to Q670-containing substrates reverses to favor more Quasar 670 molecules on the surface. Furthermore, the total number of dye-labeled peptides on the nanoparticle surface drops between the 20-2-5 and 30-2-5 probe formulations. This phenomenon may be attributed to dimer formation between uncomplexed reactant dye-labeled peptides via their thiol groups at high concentrations in the reaction mixture, precluding their availability for AuNP surface adsorption. AuNP probes that exhibited higher signal-to-background ratios included more dye-labeled substrates on their surfaces, whereas probes demonstrating higher activation ratios, or fluorescence recovery, incorporated fewer dyelabeled substrates. Signal-to-background ratio optimization requires the AuNP probe to balance a low signal in the inactivated state with high activated fluorescence, which must be supported with an abundance of surface-adsorbed fluorophores. However, this abundance of fluorophores raises two issues for quantifying fluorescence recovery, namely the creation of a large total fluorescence base and increased steric hindrance to the target protease on the nanoparticle surface. In consideration of these seemingly opposing but important measures of AuNP probe performance, we chose the 50-2-5 probe as the optimal formulation based on its sufficiently high signal-to-background ratio and excellent fluorescence recovery. 4.4.3 Effects of AuNP probe design on substrate kinetics and in vivo biocompatibility 120 - * 100 80 --- , * C' 60 - AuNP probe 0 40 S20 20 Uncomplexed 60 -20 - peptide I 0 01 120 180 240 Time post-injection (min) Figure 4-5: In vivo circulation retention of AuNP probes as compared to the equivalent mole quantity of uncomplexed Quasar 670-labeled and BHQ2labeled peptide substrates. The determined kt : Km for the trypsin-targeted 50-2-5 probe was 1.8 M-s 1 as compared to values on the order of 105 M-'s- 1 reported for uncomplexed peptide substrates [103]. Clearly, the attachment of dye-labeled substrates to a nanoparticle surface at high density greatly impeded proteolytic efficiency, as has been observed by others [104]. Originally designed for imaging PSA activity, these AuNP probes demonstrated such a significant reduction in enzyme reactivity and selectivity as to preclude this application. As determined by Denmeade and coworkers [42], the kcat : Km value for the core substrate as an uncomplexed peptide reacted with PSA is 23.6 M-'s 1 ; any further reduction in enzymatic efficiency would render the reaction nearly undetectable within a physiologic context. However, this same inefficiency in proteolysis represents a significant advantage in reducing non-specific degradation of the AuNP probe in vivo. Another potential benefit of utilizing AuNPs as vehicle and assembly platform is that their delayed clearance from the circulatory system [105] may allow the protease-specific substrates an extended window of access to target tissues. Circulation time in vivo of the 50-2-5 AuNP probe and an equimolar amount of uncomplexed dye-labeled peptide, corresponding to the probe surface composition, was determined by intravenous injection of each species into 6-8 week old athymic nude mice via a lateral tail vein. Serum was collected from each mouse at predetermined time points to measure Quasar 670 fluorescence after DTT treatment of each sample. Rehor and coworkers have suggested that serum levels of intravenously administered nanoparticles within two minutes after injection depict a relatively unperturbed basis for estimating the initial dose [106]. The percentages of initial dose for both probe and uncomplexed peptide retained in serum were calculated by normalizing the fluorescence values at each time point with those determined for serum collected at 1 min post-injection. In the first hour, serum levels of AuNP probe remain high at greater than 88% of initial dose (Figure 4-5). Circulating half-life for the probe was determined to be greater than 4 h. In contrast, more than 90% of the initial administered amount of uncomplexed peptide was removed from serum within the first hour. These data indicate that peptide substrate attachment to a larger vehicle, such as the AuNP, along with PEG incorporation significantly prolongs its circulation time in vivo and support its use via intravenous injection. The utility of the AuNP probe as an imaging agent in vivo was validated using a subcutaneous tumor phantom model in athymic nude mice. Small animal fluores- Uncomplexed peptide + AuNP AuNP probe trypsin Uncomplexed peptide - AuNP AuNP probe + trypsin Bare AuNP Bare AuNP Figure 4-6: In vivo near-infrared fluorescence imaging of subcutaneous tumor phantom model. cence imaging of the trypsin-targeted 50-2-5 AuNP probe was performed by mixing 1.16 pmol of functionalized AuNPs or equimolar amounts of the probe components with either Matrigel or PuraMatrix and injecting the solution subcutaneously to form a gelated mass, comparable to the size and shape of a tumor. Since gold nanoparticles demonstrate strong absorption and fluorescence quenching characteristics, probeequivalent quantities of uncomplexed dye-labeled peptides in the presence and absence of nonfunctionalized AuNPs were imaged subcutaneously to determine the degree to which AuNPs would suppress fluorescence intensity after simulated release of all surface dye components in vivo. Figure 4-6a is a representative fluorescence overlay image of this experiment, illustrating no significant decrease in signal intensity due to the presence of AuNPs. Trypsin-induced activation of the 50-2-5 AuNP probe was also confirmed in this tumor phantom model by exposing 1.16 pmol of probe to 250 U of trypsin (Figure 4-6b). The AuNP probe exhibited no significant fluorescence signal in the absence of trypsin with levels that were comparable to those observed for bare AuNPs. These data demonstrate that the AuNP probe is not non-specifically activated by the in vivo microenvironment nor does it degrade spontaneously but instead, generates a strong fluorescence signal in the presence of the target protease. 4.5 Conclusions In summary, near-infrared fluorogenic AuNP probes have been characterized and optimized for protease-induced fluorescence enhancement based on functional screens of trypsin and uPA-targeted probe libraries. We described a simple one-step reaction method for producing NIRF AuNP probes with variable surface compositions of dye-labeled peptide substrates and PEG and correlated reactant concentrations with the resulting quantities of adsorbed species on the AuNP probe surface and probe performance in vitro. Several design criteria for self-assembled fluorescence quenched AuNP probes emerged from these studies. AuNP probe stability in aqueous solutions was greatly improved by inclusion of 15 pM SH-PEG in the reaction mixture (- 6500 molecules of PEG per AuNP). Moreover, incremental increases in the concentration of reactant Quasar 670-labeled peptide substrate beyond 60 pM (- 26, 000 molecules per AuNP) resulted in a decrease in dye-labeled surface substrates, most likely attributable to dimer formation between uncomplexed reactant substrates. Our most unexpected finding was that the incorporation of a dark quencher, such as BHQ-2, provided additional background signal suppression beyond the fluorescence quenching attributes of the gold nanoparticle and neighboring Quasar 670 fluorophores. Optimized probes designed with the aforementioned criteria were found to exhibit extended circulation time in vivo, with t 1 / 2 > 4 h, and high image contrast in a subcutaneous tumor phantom model. Although many studies have demonstrated successful development and validation of a multitude of nanoparticle, polymer [107], and peptide based protease-activatable NIRF probes, few have attempted to establish design guidelines or generalized principles for constructing optimal imaging agents. To the best of our knowledge, the present study is the first to report on optimization parameters important to probe performance. Future studies will build upon this knowledge and the simple synthesis method introduced in this report to tailor AuNP probes for multiplexed activation and detection as well as therapeutic delivery. Chapter 5 Phage-derived Peptides for Targeting Highly Metastatic Cells 5.1 Abstract The majority of cancer patients succumb to complications related to metastatic disease. Consequently, targeting these lesions for imaging and therapy is important for mitigating patient mortality. Subtractive phage-display screening was performed on prostate cancer cell lines with low and high metastatic potential to identify differential cell surface protein expression between these populations. Using this method, a cell surface protein with M, ~ 130 kDa that exhibits higher expression on the more metastatic cell line was identified. The selected metastasis-homing phage was fluorescently labeled and shown to target the more aggressive human prostate tumor in vivo. 5.2 Introduction The natural history of cancer has traditionally been described as a multistep process that spans tumor initiation and progression within the primary site to metastatic spread and growth. Carcinogens catalyze the cascade of genetic alterations that lead to disruptions in the intrinsic cell regulatory circuits. Normal cells proceed to malignancy along this path by acquiring multiple pro-survival capabilities: unchecked replicative potential, escape from apoptosis, and resistance to anti-growth signals [108]. Rapid tumor growth in situ is supported by the local microenvironment through its secretion of growth factors as well as angiogenesis. The transition from carcinoma in situ to an invasive phenotype whereby cancer cells exhibit high motility and escape past the basement membrane marks the first step towards metastasis. Cells that intravasate into blood vessels and lymphatics are disseminated to distant sites where a subset of these cells can arrest and adhere in organ capillaries and extravasate from the circulation into the tissue. Once embedded in the metastatic site, tumor cells may lay dormant or progress into secondary tumors [109]. Cancers exhibit preferential tropism for specific organs as metastatic sites. Prostate cancer metastases are typically found in bones, whereas breast tumors can spread to bone, lungs, liver, and brain [110]. Traditionally, it was believed that genes involved in tumorigenesis were activated first and separately from those responsible for metastatic spread, which was believed to be a subsequent event. However, recent evidence has altered this view, highlighting the possibility that a subset of tumorigenesis genes may be equally responsible for metastasis and thus, cause tumor cell dissemination much earlier in the cancer progression cascade [111]. Different types of cancers exhibit varied metastatic latency periods where tumor cells exit the proliferative cycle and do not fully colonize the secondary site. A subset of these disseminated tumor cells never expand into clinical metastasis. Organ-confined primary tumors are rarely the cause of death in cancer patients; nearly 90% of cancer-related deaths are due to metastases [112]. However, most current therapies are directed at reducing primary tumor burden in patients; some adjuvant therapies are used to address metastatic disease and cancer recurrence. Consequently, there is a dearth of agents and treatments for diagnosing and eliminating secondary tumors. Metastatic disease is typically detected via anatomic imaging modalities, such as MRI, PET, and X-ray, and occasionally, by screening patient serum for recurrence biomarkers, such as prostate-specific antigen, in the case of prostate cancer. Bone metastases can be found in almost 90% of patients with ad- vanced prostate cancer. The extent of skeletal metastatic tumor burden is generally considered an indicator of survival [113]. However, current methods of identifying these metastases involve a multitiered imaging approach that requires lengthy diagnostic periods and high costs. Bone scintigraphy relies on the dynamic uptake or exclusion of a technetium-99m-labeled ligand, methylene-diphosphonate (MDP), by bone which varies according to changes in bone mineral turnover that can be associated with secondary tumor activity. Although this imaging modality provides a whole body scan of skeletal changes, anatomic detail is lacking. MRI and tracerenhanced PET/CT address these issues surrounding resolution, and the choice of imaging modality for staging prostate cancer based on metastatic spread may be reduced to a comparison of equipment availability and cost. Prostate-specific membrane antigen (PSMA) and androgen receptor expression levels have been targeted as potential in vivo biomarkers for contrast agent enhancement. The value of these contrast agents still remains to be proven as it evolves with the establishment of the biomarkers' clinical relevance. Despite current advances in imaging prostate cancer metastases, preclinical discovery and validation of biomarker targets requires the development of molecular imaging agents. Although many of the clinical contrast agents are based on tracer-labeled antibodies for affinity-based identification or small molecule ligands for functional assays of tumor activity, the use of affinity peptides is becoming increasingly more widespread. Peptides possess the advantages of largescale synthesis, ease of sequence evolution and screening, and controllable modifications. In this study, we sought to identify peptides that bind preferentially to highly metastatic cells as reagents for targeting these lesions in vivo as well as to elucidate differentially expressed cell surface biomarkers. The ability to specifically image and identify metastatic lesions provides an additional index by which to stratify clinical disease and possibly inform the choice of treatment. Peptide sequences are constrained to an alphabet of 20 natural amino acids and also a host of post-synthetic modifications. Libraries of fixed-length peptides can be generated synthetically or via random gene libraries expressed on bacteriophage. Phages are virions that infect and reproduce through bacteria and include several forms, such as the T series of lytic phage containing dsDNA, lambda phage, small DNA phages (spherical and filamentous forms that contain ssDNA), and RNA phages. We utilized a commercially-available phage display library from New England Biolabs that generates combinatorial peptide sequences on M13 filamentous phage. This phage particle contains a single strand of DNA of approximately 6400 bp with a diameter of 6.5 nm [114]. The ssDNA is encapsulated within the core of the filament structure and surrounded by several coat proteins, named pVIII, pIII, pVI, pVII, and pIX. The Ph.D.TM phage display libraries fuse the peptide sequences to the Nterminus of the pIII minor coat protein for pentavalent external display. Several groups have used phage display biopanning to select peptide sequences and motifs that bind specifically to cancer cells [115, 77, 116], endothelial cells associated with atherosclerosis [117], and clotted plasma that can be observed in tumor stroma and wounds [118]. The primary advantages of biopanning for target-specific ligands using phage display libraries over synthetic peptide libraries are the replicative ability of phage in bacterial culture and the ease of sequencing and identifying the selected ligand amino acid composition. As advances in small-scale peptide synthesis and mass spectrometry begin to emerge, the controllability of synthetic libraries may supercede the advantages of phage display libraries. Our study represents one of the first reports of phage display selection of peptides for identifying metastatic lesions. Yang and coworkers have successfully demonstrated a peptide sequence that can be used to image metastatic over nonmetastatic tumors in vivo [119]. The works described herein extend beyond the application of peptides for imaging metastases to methods for identifying the associated biomarker protein(s). Phage display was implemented in a subtractive identification strategy to select phage particles that have higher affinity for high metastatic cancer cells over low metastatic ones. Figure 5-1 illustrates the method by which phages were isolated and enriched through multiple rounds of subtractive display. Since the ultimate goal of finding metastasis-binding peptides is for in vivo applications and the first point of access to the tumor cells is via their cell surface proteins, we focused on identifying plasma membrane associated biomarkers by exposing phages to intact cells. 1. Expose low metastatic cells to phage display lbrary for I h at 37*C 2.Collect unbound phage In the media and clear It of cells by centrifugation 3.Add media to high metastatc cells and Incubate at 37C for 1 h 4. Wash cells 4x with HBSS IFO 5. Colect and lyse cells to ampfy cel srface4bound and Internalized phage Figure 5-1: Schematic diagram of subtractive phage display procedure. Phages that remained in the culture media after the negative selection round with low metastatic cells were then incubated with high metastatic cells. After rigorous washes with a salt solution, phages bound to or internalized by the more aggressive cells were isolated by cell lysis. Cell lysates were then added to bacterial cultures to amplify phages that were still associated with the cells. Multiple repetitions of this procedural sequence ensures an enrichment for these selected ligands. We used a modified version of Jacobson's pellicle method to isolate plasma membrane proteins [120, 121]. Rahbar and coworkers have implemented this method along with mass spectrometry to determine subcellular distribution of proteins enriched by Jacobson's pellicle isolation [122] and plasma proteins associated with drug resistance in cancer cells [123]. 5.3 5.3.1 Experimental Methods Cell culture The human prostate cancer cell lines, PC3M and PC3M-LN4, were a gift from Dr. Isaiah J. Fidler (M. D. Anderson Cancer Center, The University of Texas, Houston, TX). PC3M cells were originally derived from gross liver metastases from an intrasplenic injection of the parental PC-3 cells [124]. Subsequent orthotopic inoculation of PC3M cells and resulting lymph node metastases through four cycles of orthotopic inoculation yielded the PC3M-LN4 cell line [125]. Both cell lines were maintained in RPMI media (Invitrogen, Carlsbad, CA) supplemented to a final composition containing 10% fetal bovine serum (FBS) and 1% L-glutamine and grown under 37'C and 5% CO 2 atmosphere. 5.3.2 Phage display selection The Ph.D.TM_12 phage display peptide library (New England Biolabs, Beverly, MA), comprised of linear dodecapeptides was used to conduct the ligand selection process. PC3M and PC3M-LN4 cells were seeded in 10-cm tissue culture dishes at a density such that the cells would reach 90-100% confluence after approximately 48 h. Phage stock was diluted in both serum-containing and serum-free RPMI culture media to a final concentration of 1 x 1011 PFU/ml. Negative selection was performed on near confluent monolayers of PC3M cells by first washing each dish of cells twice with either serum-containing or serum-free RPMI media followed by addition of 2 ml of diluted phage solution in media such that each dish contained 2 x 10" PFU of phage. PC3M cells with addition of phage were incubated in the tissue culture incubator (37 C under 5% CO 2 atmosphere) for 1 h. The culture media was then collected and centrifuged at ~ 900 x g for 5 min to pellet cellular material. The resulting supernatant, containing phage that did not bind to PC3M cells, was added to PC3M-LN4 cells that had been washed twice with the corresponding media. PC3M-LN4 cells in phage-containing media were incubated under tissue culture conditions for 1 h. The cells were then washed three times with Hanks' Balanced Salt Solution (HBSS) supplemented with Ca++ and Mg++ (HBSS++) followed by three washes with HBSS without Ca++ and Mg++ (HBSS--). PC3M-LN4 cells were then scraped into 300 pl of lysis buffer (1% Triton X-100 in PBS supplemented with cOmplete mini protease inhibitor cocktail). Lysates were set on ice for 30-60 min. Cell surface and internalized phage contained in the PC3M-LN4 lysate was amplified per manufacturer's instructions. Three rounds of negative selection in PC3M cells followed by positive selection in PC3M-LN4 cells were conducted prior to clone selection, DNA isolation and sequencing. 5.3.3 Modified Jacobson's pellicle method for plasma membrane isolation Materials All materials were purchased from Sigma-Aldrich (St Louis, MO) unless otherwise indicated. Phosphate-buffered saline (Invitrogen, Carlsbad, CA) containing 1 mM MgCl 2 and 1 mM CaCl 2 is denoted by PBS++. Plasma membrane coating buffer (PMCBB) was prepared to contain 0.5 mM CaCl 2 , 1 mM MgCl 2 , 20 mM MES, 135 mM NaCl, and adjusted to pH 5.3. The colloidal silica solution (5%w/v) was prepared by diluting LUDOX® (30% wt) with PMCBB solution. Poly(acrylic acid) (PAA) solution contains 10 mg/ml PAA in PMCBB adjusted to pH 6-6.5. Lysis buffer refers to 2.5 mM imidazole pH 7 and is differentiated in this protocol as that with and without one cOmplete mini protease inhibitor cocktail tablet (Roche Applied Science, Indianapolis, IN) per 10 ml buffer. Plasma membrane isolation Cells were seeded at 2 x 106 cells in 10-cm tissue culture plates approximately 48 h prior to membrane isolation, such that on the day of the experiment, cells were 90-100% confluent. Each dish was washed twice with PBS++ followed by one wash with PMCBB. Each 10-cm dish of cells was then coated with 5 ml of colloidal silica solution and left on ice for 1 min. The silica suspension was removed, and the cells were washed once with PMCBB to remove excess PAA. Cells were washed quickly once with lysis buffer without protease inhibitors followed by addition of 5 ml lysis buffer with protease inhibitors. The dishes were left on ice for 1 h to swell the cells. Dishes were then allowed to adjust to room temperature by placing them on the benchtop for 15-30 min. Apical membranes were isolated by pipetting the lysis buffer from each dish over the cells at an angle to shear open each cell with a P1000 Pipetman and collected into BD Falcon centrifuge tubes (BD Biosciences, San Jose, CA). The lysate was then centrifuged at 950 x g for 30 min at room temperature to sediment nuclei and silica-coated plasma membranes. The resulting pellet was resuspended in 1 ml of lysis buffer and diluted with 1 ml of 100% (w/v) HistodenZTM solution. A density gradient was formed by layering the following solutions in the indicated order in a clean ultracentrifuge tube (Beckman-Coulter, Fullerton, CA): (1) 3 ml of 70% HistodenzTM solution, (2) 50% HistodenzTM solution containing the cell pellet, and (3) lysis buffer to near the top of the tube. The tubes were spun at 60, 000 x g (- 21, 000 rpm in a Beckman LK-90 ultracentrifuge using a SW28 rotor) for 30 min at 10'C. The supernatant containing nuclei suspended at the 50-70% HistodenzTM interface was discarded; the plasma membrane pellet at the tube bottom was resuspended in 1 ml lysis buffer. Excess HistodenzTM was removed by centrifuging the suspension at 14,000 rpm in a tabletop Eppendorf microfuge at 4'C for 5 min. The plasma membrane pellet was washed twice more with lysis buffer before resuspension in 30 ml of 100mM Na 2 CO 3 pH 11.4 in a 50 ml Falcon centrifuge tube. The membrane sheet suspension was sonicated for 30 min with vortexing every 5-10 min followed by centrifugation at 12,000 rpm (~ 17, 211 x g for SS-34 rotor) for 30 min in a Sorvall 50 ml tube. Pellets were then resuspended in 1 ml of Na 2 CO 3 solution followed by centrifugation at maximum speed for 15 min in a tabletop microfuge. Isolated proteins were recovered from the silica coating by direct solubilization in 2% SDS solution and incubation in a 60'C water bath for 15 min. Solubilized samples were sonicated for 1 min and spun down at max speed in a microfuge for 15 min at room temperature to pellet the silica coating. The supernatant was collected as the plasma membrane protein containing component and either used immediately or stored at -20 C for further analysis. 5.3.4 Gel electrophoresis and immunoblotting Protein samples were quantified for total protein content by the bicinchoninic acid method using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL) and reduced in Laemmli sample buffer containing #-mercaptoethanol by boiling the mixture for 5 min. Proteins were resolved on either 4-20% linear gradient or 7.5% Tris-HCl precast polyacrylamide gels (Bio-Rad, Hercules, CA) by one-dimensional sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE). Molecular weights were determined by comparison to the Precision Plus Protein Kaleidoscope standards (Bio-Rad, Hercules, CA). Proteins resolved electrophoretically were transferred to Immobilon-P polyvinylidene fluoride (PVDF) membranes (0.45 pm pore size; Millipore, Billerica, MA) using a Bio-Rad electroblotting transfer apparatus. Membranes were blocked with 5% (w/v) nonfat dry milk in PBS for 1 h followed by five washes with PBS supplemented to contain 0.1% Tween 20 (PBS-T). Primary antibodies were prepared to the specified dilutions in PBS containing 2% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO) and 0.1% sodium azide. Membranes were incubated in primary antibody solution for 1 h at room temperature followed by three washes of PBS-T for 5 min each. Horseradish peroxidase-conjugated secondary antibodies were diluted in 5% milk in PBS and added to the membrane for 1 h incubation at room temperature. Membranes were then washed six times with PBS-T for 5 min each and developed with chemiluminescent substrates as indicated by manufacturer. 5.3.5 Phage-facilitated immunoprecipitation PC3M and PC3M-LN4 cells were seeded at 2 x 106 and 3 x 106 cells per 10-cm tissue culture dish approximately 48 h prior to experiment to achieve near confluence. SulfoN-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido benzamido)-hexanoamido) ethyl1,3'-dithioproprionate (Sulfo-SBED; Thermo Fisher Scientific, Rockford, IL) labeled phage was prepared by PEG precipitation of M13KE and target phage. The phage pellet was then resuspended in 490 pl of 0.3 M sodium bicarbonate buffer. Pre- aliquoted sulfo-SBED (1 mg per vial) was reconstituted in 100 pl of DMSO and protected from light. SBED was added to each vial of phage solution at 10 [l per sample. The labeling reactions were vortexed briefly and left at room temperature and protected from light for 30 min. Labeled phage was then re-precipitated using PEG/NaCl on ice for 30-45 min and reususpended in serum-free RPMI (with L-glutamine) media at 2 x 1013 PFU/ml. Each 10-cm dish of cells was washed twice with 5 ml cold serumfree RPMI media. SBED-phage was added to each dish at 2 x 1013 PFU per dish and incubated at 40 C for 1 h with occasional rotation to disperse media. Each dish was then washed twice with 5 ml Mg++ and Ca++-free cold PBS and immersed in 10 ml of PBS. Cells were exposed to 365 nm UV light at 8 W power output (Thermo Fisher Scientific, Rockford, IL) for 30 min at room temperature to induce biotin label transfer between labeled phage and cell surface proteins. PBS was then suctioned off each dish and replaced with 100 pl lysis buffer (1% Triton X-100 in PBS supplemented with cOmplete mini protease inhibitor cocktail without EDTA). Cells were then scraped from the dish surface and collected into clean microcentrifuge tubes. Lysates were isolated by incubating the mixture on ice for 10 min to 1 h followed by centrifugation at 10, 000 x g for 5 min at 4C. Total protein content was assayed on a small aliquot of lysate using the BCA assay; the remainder of the lysate was mixed with 50 pl of streptavidin-agarose beads (Thermo Fisher Scientific, Rockford, IL) and rotated end-over-end overnight at 40C. Bound resin was pelleted by centrifugation at ~ 2, 500 x g for 2 min at room temperature. Resin was washed at least three times with lysis buffer or PBS. Resin-bound proteins were eluted by boiling resin pellet in SDS-PAGE Laemmli sample buffer for 5 min. Resin was discarded by centrifuging the resulting solution at 10,000 x g for 5 min at room temperature. Denatured proteins in the supernatant were assayed immediately or stored at -20 C. 5.3.6 Mass spectrometry Isolated plasma membrane protein samples were subjected to SDS-PAGE in duplicate for simultaneous immunoblotting and gel band identification. After gel electrophoretic separation, gels were washed three times in 100 ml water for 5 min each. Gels were then incubated in SimplyBlue TM SafeStain (Invitrogen, Carlsbad, CA) for 1-3 h followed by addition of 2 ml of 20% NaCl (w/v) in water for every 20 ml of stain. Gel destaining was performed by washing the gels with 100 ml of water. Coomassie-stained protein bands were compared to the duplicate gel transferred and immunoblotted for phage-binding protein bands to determine target bands. These bands were cut from the gel and analyzed at the Harvard Medical School Taplin Mass Spectrometry Facility (Boston, MA) by microcapillary LC/MS/MS techniques. 5.3.7 FACS analysis PC3M and PC3M-LN4 cells were seeded in 60-mm tissue culture dishes such that they would reach 90-100% confluence on day of experiment. Culture media was suctioned off and incubated with 0.75 ml of phage solution (5 x 10" PFU/ml diluted in serumcontaining RPMI media) under tissue culture conditions (37 C and 5% CO 2 ) for 1 h. Unbound phage was then suctioned off the cells; cells were then washed twice with 4 ml of PBS++. Cells were fixed by adding 1 ml of freshly diluted 4% paraformaldehyde in PBS++ to each 60-mm dish at room temperature for 10 min. Each dish was then washed twice with 4 ml PBS. Cells were labeled in succession with 1 ml of 1:50 dilution of mouse anti-M13 monoclonal antibody (GE Healthcare Biosciences, Piscataway, NJ) diluted in serum-containing media for 1 h at room temperature followed by 1 ml of 1:200 dilution of Alexa Fluor 488-labeled goat anti-mouse secondary antibody diluted in serum-containing media for 1 h at room temperature while protected from light. Each dish was washed twice with 4 ml PBS between labeling reactions. After final labeling step, cells were washed three times with 4 ml PBS followed by addition of 1.5 ml of FACS buffer (0.1% BSA in PBS containing 0.02% NaN 3 ) to each dish. Cells were scraped into the FACS buffer solution and dissociated into individual cells with repetitive pipetting. Cell suspensions were then set on ice. FACS analysis was performed on a BD FACSCaliburTM flow cytometer. 5.3.8 Confocal microscopy Ethanol-sterilized coverslips (18 mm diameter) were coated with human fibronectin (10 pg/ml in sterile PBS) for 1 h at 370 C in a 12-well tissue culture plate. Coverslips were then washed three times with sterile PBS and incubated in serum-containing RPMI media. PC3M and PC3M-LN4 cells were then seeded into each well at varying densities and grown to approximately 60-70% confluence. Cells were fixed and stained as described under FA CS analysis. Coverslips were then mounted on microscope slides using VECTASHIELD® mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Optical sections were scanned using a Leica TCS SP2 AOBS spectral confocal scanner mounted on a Leica DM IRE2 inverted microscope with a 40x objective and 488 nm argon, 543 nm HeNe, 633 nm HeNe, and 405 nm diode lasers. 5.3.9 Cy5.5-labeled phage FACS analysis Phage was isolated at 1 x 1013 PFU per conjugation reaction mixture and suspended in the reaction buffer (0.3 M NaHCO 3 pH 8.5). Cy5.5-NHS (GE Healthcare Bio- sciences, Piscataway, NJ) stock solution was prepared in the reaction buffer at 2 mg/ml concentration. The conjugation reaction was prepared by mixing the Cy5.5 solution with the phage suspension at varying final volumes but at 1 mg/ml final Cy5.5-NHS concentration. The reaction was allowed to proceed at room temperature for 1 hr followed by PEG/NaCl precipitation of labeled phage per usual protocol. The resulting labeled phage pellet was washed with Tris-buffered saline (TBS) and re-precipitated twice to remove any remaining uncomplexed dye. The purified Cy5.5labeled phage pellet was resuspended in 0.2% NaN3 in TBS and stored at 4'C. Labeled phage was titered per usual protocol; Cy5.5 content was quantified by absorbance measurements at 675 nm (e = 250, 000 M-1 cm- 1 ). Fluorescently-labeled phage was diluted in serum-containing RPMI media at 1 nmol dye per 750 pl. Cell monolayers approaching confluence in 60-mm dishes were incubated with 750 pl phage solution in the tissue culture incubator for 1 h while protected from light. Each dish was then washed twice with 4 ml of PBS and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were again washed twice with 4 ml of PBS and scraped into 1.5 ml FACS buffer. The cell suspension was collected into 5-ml round bottom tubes and set on ice for analysis. 5.4 5.4.1 Results Phage peptide selection converges on a common sequence Phage peptide selection was performed by successive rounds of negative selection in PC3M cells followed by positive selection in PC3M-LN4 cells to enrich for peptide sequences that preferentially bind the surface of cell lines with higher metastatic potential. Parental PC-3 cells were originally isolated from a bone metastasis of a grade IV prostatic adenocarcinoma from a 62-year-old male Caucasian. After these parental cells were inoculated intrasplenically in nude mice, liver metastases were cultured to result in PC3M cells. Orthotopic re-introduction of PC3M cells resulted in lymph node metastases that were isolated, propagated, and re-inoculated over four cycles to result in the establishment of PC3M-LN4 cells. PC3M-LN4 cells are thus considered to possess higher metastatic potential and aggressiveness than PC3M cells. Pettaway and coworkers characterized the tumorigenicity, prostate weight, mean weight of regional lymph nodes, and presence of distant metastasis after orthotopic inoculation of athymic nude mice using these cells lines. Regional lymph nodes in mice with PC3M-LN4 tumors were weighed significantly more than those in mice with PC3M lesions, suggesting that there was more or larger lymph node metastases. In addition, the incidence of distant metastasis was much higher in PC3M-LN4 inoculated mice as compared to mice with PC3M tumors [125]. Consequently, PC3M/PC3M-LN4 cell line pairing was considered to be the appropriate match of a less aggressive/low metastatic potential versus a more aggressive/higher metastatic potential cell line, as both lines were derived after passage in mice of a similar genetic and physiologic background. We chose to use an unbiased peptide selection process by exposing the phage display library to intact living cells to detect any macromolecular differences within and on the plasma membrane. The unbiased approach does not assume the presence of any particular protein, as is typically the case in in vitro biopanning against known biomarkers for affinity peptides, and thus, allows for the discovery of novel biomolecules. Moreover, the use of intact living cells for the panning process opens up the possibility of identifying multiprotein complexes as well as individual proteins. We compared phage peptide selection in serum-free media to that in serumcontaining media as a measure of peptide affinity dependence on serum proteins. Large proteins have been used in the research literature as carriers for drugs and other smaller ligands and could potentially act as non-specific blocking agents for cell surface receptors. Consequently, it was of great significance to dissect the differences in phage affinity in the presence and absence of serum proteins. After three rounds of panning, peptide sequences converged to a nearly complete 12-amino acid motif that could be found under both media conditions. Table 5.1 outlines the peptide sequences and frequencies of occurrence for clones selected after phage display selection in serum-free and serum-containing media. Clones whose DNA sequencing results did not pass quality control and/or contained incorrect pIII leader sequences were eliminated from further evaluation, yielding 15 total clones between the two media formulations. Eight of the 15 clones exhibited the same peptide sequence, Glu-TrpLeu-Trp-Glu-Phe-Pro-Ser-Asp-Thr-Arg-Ser, which we termed LN4P-1. Phage clones selected under serum-containing media conditions converged to this sequence much more quickly than did those diluted in serum-free media, as illustrated by the diversity of peptide sequences found in the third panning round in the absence of serum. One possible explanation for this dichotomy may be that the absence of serum induces artifactual changes in the cell surface composition in both PC3M and PC3M-LN4 cell lines. Serum contains growth factors, hormones, and other proteins that maintain cell viability, growth, and substrate attachment. Moreover, the inclusion of serum proteins more accurately recapitulates physiologic conditions in which cancer cells are bathed in a complex mixture of secreted proteins. The presence of clones selected through serum-free media conditions displaying the LN4P-1 peptide sequence is evidence that the cell surface association and potential internalization of this specific phage is independent of serum proteins, such as albumin. Peptide sequence searches based on the LN4P-1 sequence was performed using the NIH BLASTP engine in a database comprised of expressed human proteins. The top protein matches are outlined in Table 5.2. The BLASTP search revealed no putative conserved domains and no conclusive matches to known human proteins. It is of interest to note that several of the identified proteins were plasma membrane associated while others were DNA-interacting proteins. Table 5.1: Peptide Sequences from Subtractive Phage Display Selection for Ligands with Preferential Affinity for Highly Metastatic Prostate Cancer Cells Amino Acid Position and Sequence (N No. of Clones C) Media w/o Media w/ Serum Serum P1 P 2 P3 P4 P5 P 6 P7 Ps P9 Pio P11 P 12 3/8 5/7 E W L W E F P S D T R S * 1/7 D W Q Y T F P S V T R S * 1/7 K W T W E Y S P F L N A 1/8 * A S I P Y Q M N N A V P 1/8 * G W 1I E Y P I A N H P 1/8 * L N P F H A G Y L K K P 1/8 * ID S I G H K L R G T K 1/8 * A D S Y D L R K P A R T Table 5.2: LN4P-1 Peptide Sequence Matches to Expressed Human Proteins using BLASTP Engine E-Value Gene Name Reported Protein Function 20 Hypothetical protein FLJ13236 Integral membrane and HSP-binding protein 27 mutY homolog A/G-specific adenine DNA glycosylase 37 Fibroblast growth factor receptor 4 Growth factor signaling 49 BBX protein Member of the HMG-box superfamily of DNA-binding proteins 49 Solute carrier family 29, member 1 Nucleoside transporters 119 KLRD1/CD94 Cell signaling in natural killer cells 5.4.2 LN4P-1 phage preferentially binds PC3M-LN4 cell surface Selection and identification of a a convergent phage peptide sequence led to a process of validating the specificity of phage interaction and localization. Fluorescence activated cell sorting (FACS) analysis was performed on PC3M and PC3M-LN4 cells labeled with LN4P-1 phage as compared to those reacted with a control insertless M13KE phage. After phage labeling, a fluorochrome-conjugated secondary antibody was used to quantify the degree of phage binding to each cell followed by flow analysis to determine the percentage of total cells that exhibited phage-associated cell surface fluorescence above a background threshold. In the comparison between LN4P-1 phage binding affinities to PC3M and PC3M-LN4 cells, the background fluorescence cutoff values were determined by analyzing the maximum fluorescence threshold exhibited by M13KE-labeled cells. Nonspecific antibody binding to both cell lines was observed after anti-M13 mouse monoclonal primary antibody and Alexa Fluor@ 488-labeled rabbit anti-mouse secondary antibody reactions (Figure 5-2). For both cell lines, there is a bimodal distribution of fluorescence intensities after only antibody labeling without initial phage incubation. However, with the addition of phage, differences in LN4P-1 phage affinity to each cell line emerged. Independent experiments demonstrated that 83.7 ± 1.0% of PC3M-LN4 cells were labeled by LN4P-1 phage as compared to 10.2 ± 2.9% of PC3M cells (Figure 5-3). Despite a distinct peak separation between PC3M and PC3M-LN4 fluorescence intensity distributions, there is a slight overlap in their labeled cell populations. The broader fluorescence distributions and overlap are indicative of the heterogeneity of the cancer cell populations, particularly between two cell lines with such close lineage. It would be of great interest to further fractionate the PC3M cell population into low, average, and high LN4P-1 binding groups to determine the association between LN4P-1 affinity and metastatic ability as compared to PC3M-LN4 cells. As much of the field's research efforts have been directed at elucidating molecular and epigenetic mechanisms involved in metastasis, it is interesting to note the possibility of differences in cell surface proteins and/or protein complexes that may contribute to metastasis rather than purely genetic alterations. Further validation of LN4P-1 affinity to PC3M-LN4 cells over PC3M cells was performed by confocal microscopy of immunofluorescently labeled cells. Figure 5-4 illustrates the higher LN4P-1 staining of PC3M-LN4 cells over that of PC3M cells. After phalloidin staining for actin, phage staining can be observed almost exclusively at the cell periphery or surface in a punctate pattern. Confocal microscopy confirmed the targeting preference of LN4P-1 to PC3M-LN4 cells and localization of LN4P-1 phage binding to the cell surface, as staining permeabilized cells did not result in detectable intracellular LN4P- 1-associated fluorescence. LN4P-1 Phage Primary & Secondary Antibody V. PC3M 20.8% PC3M-LN4000407.002 PcaM-LN4.0407.001 PC3M-LN4 io 10 71.6% --- - 0 - 101j _102 FLI-H 13 1 0 10 --10 10 FLI-H 0 0 Figure 5-2: Background fluorescence as a result of nonspecific antibody binding during FACS. % of Total Cell Population above Gated Background Fluorescence FL1IH FU1-H 'I Figure 5-3: FACS analysis of LN4P-1 phage binding to PC3M and PC3MLN4 cells as quantified by fluorescence distribution. M ... .... ........ ........... PC3M-LN4 PC3M + DAPI + DAPI + Phallokfdn Figure 5-4: Confocal microscopy images of PC3M and PC3M-LN4 cells labeled with LN4P-1 phage. LN4P-1 (green) and actin (red) are shown. 100 WMMMWM 5.4.3 Development of imaging probe from LN4P-1 phage peptide Based on the identified LN4P-1 phage peptide sequence and validation of its preferentially binding affinity for PC3M-LN4, a prostate cancer cell line with high metastatic potential, fluorescently-labeled synthetic peptides were prepared for imaging applications. A cognate peptide comprised of the LN4P-1 peptide sequence as well as a scrambled version of the same sequence were synthesized (Anaspec, Inc., Fremont, CA) and labeled with fluorescein to result in the following: (1) Glu-Trp-Leu-Trp-GluPhe-Pro-Ser-Asp-Thr-Arg-Ser-Lys(5-FAM-LC)-NH2 and (2) Phe-Ser-Asp-Trp-Glu-ProLeu-Trp-Ser-Arg-Thr-Glu-Lys (5-FAM-LC)-NH2. Each peptide was incubated with the cells at different concentrations and under varying serum conditions to determine its cell binding characteristics. FACS analysis was employed to measure relative cell fluorescence intensities after labeling reactions. Figure 5-5 illustrates the labeled cell populations using 20 pM and 100 pM of cognate and control scrambled peptides under serum-containing media conditions. Under the two peptide concentrations assayed in these experiments, more cells in both PC3M and PC3M-LN4 cell line populations were labeled with scrambled peptide over the cognate sequence. Morever, the labeled fraction of the cell populations was much lower than previously observed for cells labeled with LN4P-1 phage. We extended this assay to serum-free conditions to ascertain the contribution of serum proteins to peptide binding. Figure 5-6 demonstrates the same trend observed for serum-containing conditions. Scrambled peptide labels more cells than cognate peptide. Several phenomena may have contributed to the low binding efficiency of the synthetic peptides to the target prostate cancer cell line. The primary cause for low peptide affinity may be the monovalent nature of the uncomplexed peptides. The selected LN4P-1 phage displays five copies of its peptide sequence and may benefit from multivalent binding coordination and kinetics. Morever, the failure of a highstringency control as the scrambled peptide to demonstrate a differential binding effect from cognate peptide would indicate a lack of sequence order specificity in cell 101 surface binding but not necessarily of peptide composition specificity. Preferential phage binding to highly metastatic cells is proposed to occur through hydrophobic or electrostatic interactions between the peptide amino acids and the target protein. Consequently, the order of the amino acids within the peptide may not be of primary importance in this interaction. As a result of these insights, we pursued the development of Cy5.5 labeled LN4P-1 phage as a potential in vivo imaging agent. Cy5.5-NHS, a near-infrared fluorophore suitable for in vivo imaging applications, was used to label free amines on the surface of phage particles. Both insertless M13KE and cognate LN4P-1 phages were labeled with Cy5.5 to differentiate between phage binding to cells due to coat protein interactions and diplayed peptide interactions with the cell surface. As demonstrated in Figure 5-7, fluorescent M13KE phage bound to PC3M and PC3M-LN4 cells to similar proportions. A significant differentiation between the two cell lines is exhibited after incubating cells with Cy5.5-LN4P-1 phage. A much higher fraction of PC3MLN4 cells (9.94%) demonstrated Cy5.5 surface binding than PC3M cells (1.39%). Although the degree of phage binding was much lower in this experiment over prior analyses using a three-step immunofluorescence labeling protocol, involving the succession of phage, anti-M13 primary antibody, and fluorochrome conjugated secondary antibody, there is a distinct difference in binding efficiency of Cy5.5-LN4P-1 phage to each cell line. The presence of multiple Cy5.5 dye molecules on the phage particles may interfere with the overall phage binding kinetics. These observations may also indicate a possible role for the coat proteins in coordinating the pentavalent peptide binding to the cell surface. Furthermore, the FACS machine used in the analysis may have limited dynamic range in the detection of such long wavelength dyes, thus, creating an artificially compressed fluorescence distribution and an inability to distinguish positive from background signal intensities. Since the amine labeling process is nonspecific, optimization of Cy5.5 content on the phage particle surface was performed by varying the stoichiometry of labeling reactions. The final reaction concentration of Cy5.5-NHS was maintained at 1 mg/ml across all reactions with variable concentrations of phage. Results of the labeling reactions are summarized in Table 5.3. All 102 reactions produced relatively high levels of Cy5.5 conjugation, ranging from - 2, 000 to ~ 10, 000 molecules of dye per phage particle. Regardless of the degree of Cy5.5 labeling, there was a greater proportion of PC3M-LN4 cells with bound phage over PC3M cells. However, the fraction of positively labeled cells was still much lower than that observed with antibody-based detection. Evidence of cell line distinction based on labeling with Cy5.5-phage led to in vivo application of these phages as imaging agents. Male Nu/nu mice (6-8 weeks old) were inoculated subcutaneously with PC3M cells on the left flank and PC3M-LN4 cells on the right flank for bilateral tumors. Approximately four weeks after inoculation, mice with size-matched tumors were selected for the imaging experiment. Each mouse was intravenously injected with 500 pl of PBS, Cy5.5-M13KE, or Cy5.5-LN4P-1; the volume of injected phage corresponded to 750 pmol of Cy5.5 dye as determined by absorption measurements. Fluorescence images were then recorded over time while the mice were under anesthesia. Figure 5-8 illustrates the time sequence of fluorescence images. The region of interest (ROI), as highlighted in yellow, demarcates the tumor region from areas where nonspecific intestinal fluorescence due to phytoestrogen content in the feed. Photographs of the mice confirmed subcutaneous PC3M tumors on the left flank (orange arrow) and PC3M-LN4 tumors on the right flank (green arrow). Tumors prior to injection exhibited extremely low background fluorescence. After administration of PBS only, PC3M-LN4 tumor fluorescence increased slightly without a concomitant increase in the PC3M tumor fluorescence. This selective enhancement of PC3M-LN4 tumor fluorescence persisted through six hours after injection and could not be explained, as PBS is not fluorescent. In evaluating LN4P-1 phage targeting, we only considered the M13KE phage control. Selective fluorescence enhancement of the PC3M-LN4 tumor was observed at 3.5 h after Cy5.5-LN4P-1 injection, whereas PC3M and PC3M-LN4 tumors exhibited similar fluorescence intensities after Cy5.5M13KE phage administration. The preferential binding and fluorescence contrast provided by LN4P-1 phage as compared to control M13KE phage persisted through 6 h post-injection. All tumor fluorescence signals had been reduced to background levels at 20 h after initial injection. 103 a3 20 uM Peptide in Complete Media PC3M PC3M-LN4 Scramble LN4P-1 Peptide - Cognate LN4P-1 Peptide 0.3% - 0.9% FL11 FL" b 100 uM Peptide in Complete Media PoCX&= .* Scramble LN4P-1 Peptide 0- Cal O 1 1&5% *, Cognate LN4P-1 Peptide 4A% FLl IM ~ ; Figure 5-5: LN4P-1 peptide binding to PC3M and PC3M-LN4 cells 104 aI 20 uM Peptide in SF Media PC3M-LN4 PC3M w Po3M.04 I. Scramble LN4P-1 Peptide C3M-uK&Os 4.1% '100 01 t -14-H to' o 1to' PC3.WI Cognate LN4P-1 Peptide 3EVI 70 0.2% 0.8% 0 17*~ 2 FL144 PLI44 to" o uM Peptide b 100 in SF Media POM-N009 Scramble LN4P-1 Peptide i *7.6% le to, 2 j0 F1L. a to Cognate LN4P-1 Peptide Figure 5-6: LN4P-1 peptide binding to PC3M and PC3M-LN4 cells in serum-free media 105 PC3M-LN4 PC3M 0.11% Unlabeled Celia at I * 0 g t S0.00% 1 0.07% R"t ga 1 eI11310 0.26% M13KE-labeled Cells FL"1~ I*0.18% LN4P-1-labeled Cells 0.09% -.- 0.24% 0 o P 9 9 9.94% *. i. tl 1.39%2 o~10 3 -1121 _L --- 1--.1 i.~ to' to; to04 FL" Figure 5-7: FACS analysis of PC3M and PC3M-LN4 cells using Cy5.5-labeled LN4P-1 phage Table 5.3: Optimization of Cy5.5 loading on phage for maximal distinction in phage labeling of cells % PC3M Cells % PC3M-LN4 Dye per Labeled Cells Labeled Phage Unlabeled 0.15 0.11 0 5.0 x 10" 100 pg dye 0.33 0.87 2136 2.8 x 1011 300 pg dye 0.36 0.84 9824 6.1 x 1011 600 pg dye 0.25 1.49 3053 2.0 x 1011 107 Phage Titer PBS LN4P-1 M13KE Before Injections After PBS Injection Before LN4P3-1 & M13KE Injection 3.5 h after Injections 4h 6h 20h Figure 5-8: In vivo fluorescence imaging of intravenously administered Cy5.5-labeled phage 5.4.4 Validation of Jacobson's pellicle method Demonstration of LN4P-1 phage utility as an imaging agent led us to pursue the identity of any potential biomarkers that are specifically bound by LN4P-1 on the PC3M-LN4 cell surface. In order to reduce the protein complexity inherent in lysing open whole cells for more efficient biomarker identification, we sought a method to provide plasma membrane enrichment over proteins from other organelles. We had demonstrated previously that LN4P-1 binding is localized to the plasma membrane. Consequently, we utilized Jacobson's pellicle method to dissociate and isolate the plasma membrane from the rest of the cell. The pellicle method relies on silica colloid and polymer binding to the apical membrane to exogenously increase its density; the apical membrane can then be dissociated mechanically from the cell substrate and isolated by density gradient centrifugation. Basolateral (BL) membrane pro- teins can also be isolated by washing the substrate-adherent monolayer followed by scraping to collect the proteins. Comparisons in the purity of collected plasma membrane fractions were conducted between samples collected by the pellicle method and those isolated by a commercially available eukaryotic membrane protein extraction kit (Mem-PER from Pierce Biotechnology/Thermo Fisher Scientific). The main disadvantage of many commercial membrane extraction kits is that they do not sufficiently distinguish between all the different membranous organelles. Consequently, we compared organelle-specific protein expression levels between several extraction methods. Cell lysate samples obtained by the pellicle method, Mem-PER extraction kit, and 1% Triton X-100 (TX100) in PBS lysis buffer, were collected from PC3M and PC3MLN4 cell culture monolayers. PC3M protein samples collected by the pellicle method were enriched in Na/K ATPase, a marker for plasma membrane, and showed negligible expression of GS28, calnexin, lamin b, and VDAC1/porin, which are markers for the golgi apparatus, endoplasmic reticulum, nuclear envelope, and mitochondria, respectively, as shown by immunoblotting (Figure 5-9 and Figure 5-10a). PC3M- LN4 protein samples collected by the pellicle method also demonstrated the same expression patterns with the exception of some GS28 expression in the basolateral 109 membrane component. Most of the membranous organelle marker proteins could be found at moderate to high levels in the whole cell lysates prepared by 1%TX100 lysis buffer and the hydrophobic portion of the Mem-PER extraction sample. The data thus indicate that plasma membrane protein samples isolated by the pellicle method are largely devoid of contaminating proteins from other cellular membranes as compared to protein samples collected by a commercial extraction kit which appear to contain many different membranes. After establishing the minimal non-plasma membrane protein contamination in samples collected by the pellicle method, we pursued the level of plasma membrane protein enrichment achieved by the aforementioned sample isolation methods. Several plasma membrane proteins, such as E-cadherin, STEAP, EpCAM, and PSMA, were found in isolates from pellicle preparations but not in whole cell lysates nor MemPER derived samples (Figure 5-10b). The presence of E-cadherin in PC3M samples and absence in PC3M-LN4 samples, regardless of isolation method, concurs with other reports of E-cadherin expression patterns in these cells. Furthermore, STEAP, six-transmembrane epithelial antigen of the prostate, which was originally characterized as a cell-surface antigen that is highly upregulated in human prostate tumors [126], was found only on immunoblots of pellicle method derived protein samples and not in whole cell lysate or Mem-PER samples. EpCAM expression was detected in pellicle-derived samples from PC3M-LN4 cells but not in samples prepared by other methods. This observation highlights the extreme dependence of protein detection on cell lysate preparation methodology. Based solely on whole cell lysate analysis, one may conclude that neither PC3M nor PC3M-LN4 cells express EpCAM protein. It was also interesting to note that under reducing conditions, E-Cadherin, EpCAM, and PSMA protein epitopes were identified at multiple molecular weights depending on lysate preparation method. Multiple protein bands on Western blots may indicate the presence of degradation products or different post-translationally modified proteins, which are important data in protein characterization. The pellicle method also leads to enrichment of protein levels within each band as observed through increased chemiluminescence intensities when compared to other isolation methods. 110 444 4.!!.!.222 R~iil''i'ii'ii" 2!!2 222~i~~i!!!!iiliiliil"M 022 22 !!!99 """ ""."."""."""" """.""" !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!1|il|!| .!|d !!!!|d uusu !!!!,!!,!,,!,,!,,!,!!!!d!! MemPER Pellicle m << m m g - Plasma Membrane Na/K ATPase Golgi GS28 Calnexin ER Lamin B Nuclear Envelope Plasma Membrane Na/K ATPase Golgi GS28 Calnexin ER Lamin B Nuclear Envelope Figure 5-9: Western blot analysis of membrane content in lysates 111 ............. .- VDAC1/PorIn Immunoblot MemPER Pelilie m m I Ii I PC3M PC3M-LN4 -j 0 C7 MemPER z z-j z z LIp) WOL BL Apical V) E-Cadhertn STEAP EpCAM I I PSMA I | Figure 5-10: Western blot analysis of membrane content in lysates 112 LN4P-1 phage binds to ~ 130 kDa plasma membrane- 5.4.5 associated protein Validation of plasma membrane protein enrichment by Jacobson's pellicle method allowed us to pursue the ligand or ligand complex that binds LN4P-1 phage on the PC3M-LN4 cell surface. We had confirmed preferential binding of LN4P-1 phage to whole PC3M-LN4 cells through FACS analysis and immunofluorescence visualization. Therefore, In subsequent experiments, efforts were focused on elucidating phage interactions at the protein level. One-dimensional SDS-PAGE of protein samples followed by electrophoretic transfer onto PVDF membranes were performed per usual protocol; modifications were introduced in the reagents used during immunoblotting to probe LN4P-1 phage interaction with membrane confined proteins. LN4P-1 phage at an optimal concentration was used in the first immunoblotting step followed by HRP-conjugated anti-M13 antibody as the signal amplification reagent. Insertless M13KE phage was used to immunoblot replicate membranes to determine nonspecific bands that interact with the phage coat proteins rather than the displayed peptide sequence. Figure 5-11 demonstrates the successful use of phage in an immunoblotting application and identifies multiple nonspecific protein bands at - 15, 30, 50, and 100 kDa. The primary difference between the control phage and LN4P-1 immunoblots is an apical membrane-associated protein overexpressed in PC3M-LN4 cells at - 130 kDa. No bands at 130 kDa molecular weight was observed in the con- trol phage immunoblot. However, faint expression was observed in PC3M cells under long exposure. These immunoblot comparisons confirm the presence of a LN4P-1 binding plasma membrane-associated protein of M. - 130 kDa that is expressed more highly in PC3M-LN4 plasma membrane than PC3M plasma membrane. Since all immunoblots were conducted with reduced protein samples, the identity of the LN4P-1 phage binding protein is most likely a single protein rather than a multiprotein complex. Immunoprecipitation (IP) efforts to selectively pull down the LN4P-1 phage binding protein began with optimization of lysis conditions and capture antibodies. Initial 113 pull-down experiments using a fd-tet capture antibody revealed a candidate protein band recognized by LN4P-1 phage on immunoblot (Figure 5-12). By titrating the amount of capture antibody in the immunoprecipitation reaction, we found that 1 pg of fd-tet antibody yielded the highest levels of phage binding protein at ~ 130 kDa. Control IP reactions with just phage and capture antibody or antibody only produced bands with molecular weights at 50 kDa and below, providing evidence that the candidate protein band was not an artifact of nonspecific immunoblotting interactions. The addition of further control IP reactions involving insertless M13KE phage to eliminate proteins associated with just the phage coat proteins and rabbit IgG to preclude nonspecific capture antibody interactions yet again revealed a 130 kDa protein band captured during IP and recognized by LN4P-1 during immunoblotting (Figure 5-13). PC3M-LN4 IP reactions with capture antibody but no LN4P-1 phage demonstrated no LN4P-1 immunoblot bands above 75 kDa, further supporting the specificity of the 130 kDa band pulled down with LN4P-1 phage. Coomassie-stained gel bands corresponding to the approximate molecular weight region of PC3M-LN4 pellicle method isolated plasma membrane proteins were analyzed by mass spectrometry to determine potential LN4P-1 phage binding ligands. Tables 5.4 and 5.5 list the resulting peptide fragment matches to a human protein database. From this list, we attempted to identify those proteins that were plasma membrane associated and had molecular weights around 130 kDa. Cub domaincontaining protein 1 (CDCP1) was of primary interest, as it fulfilled both requirements and had been documented to be potentially related to cancer aggressiveness [127]. Immunoprecipitation of CDCP1 from PC3M and PC3M-LN4 lysates was performed followed by immunoblotting for LN4P-1 binding as well as confirmation of CDCP1 identity. Figure 5-14 demonstrates that the co-IP did not isolate the LN4P1 ligand, despite strong expression of CDCP1 in pellicle samples from PC3M-LN4. The extreme abundance of CDCP1 in the pellicle plasma membrane preparation, as shown on immunoblot, led us to pursue an alternative LN4P-1 ligand isolation strategy to enrich the collected sample not just for plasma membrane proteins but more specifically, cell surface proteins within proximal range of LN4P-1 phage interaction. 114 SBED reagents were employed to tag LN4P-1 phage with a biotin moiety that could be photochemically transferred from the tagged 'bait' protein (LN4P-1 phage) to the target protein, which may presumably be the ligand, via UV exposure. After biotin transfer from phage to proximal interacting protein, avidin-based agarose beads were used to pull down biotin-tagged proteins for further resolution and analysis on Western blot. Figure 5-15 demonstrates the successful pull down of a 130 kDa band that associates with LN4P-1 phage on immunoblotting. Under M13KE control phage and no phage exposure, no LN4P-1 phage interacting band at 130 kDa was detected. UV exposure of at least 15 min in duration could induce sufficient biotin transfer to pull down the LN4P-1 interacting protein. Coomassie staining and subsequent mass spectrometry analysis of a replicate gel band resulted in a list of possible ligand proteins (Tables 5.6 and 5.7). Pyruvate carboxylase appeared as the highest protein match between the 130 kDa and 75 kDa bands from SBED-mediated immunoprecipitation of LN4P-1 interacting proteins on PC3M-LN4 cell surfaces. However, both this protein and acetyl-CoA carboxyalse 1, another protein hit, involve biotin cofactors in their endogenous functions. Consequently, it is difficult to dissect avidin-based pull down due to the endogenous biotin association versus exogenous biotin tagging due to phage interaction. Other protein hits found in the 75 kDa band which had been highlighted on LN4P-1 phage immunoblot included heat shock protein 70, grp 78, hnrnp m, and ezrin. Hsp70 presentation on the plasma membrane appears to be a tumor-specific phenomenon and is not observed for normal cells [128]. Ezrin is also a known mediator associated with cancer metastasis. No conclusive LN4P-1 phage ligand was found using these methods, but these studies highlighted several existing and emerging proteins associated with metastasis. A major weakness of traditional mass spectrometry is the lack of quantitation available for characterized protein matches. We also suspect that despite positive immunoblot identification of a phage interacting protein, excised gel bands corresponding to this band did not contain sufficient amounts of protein for robust MS analysis, which requires nanogram levels of protein. 115 .................................... .. ....... Insertless Phage LN4P-1 Phage WCL Apical Apical WCL -250- -150- 100 - 75 - - 50- - 37 - 1 min exposure -25 - -20 - - 15 - I I I I -250- 150- -100- 3 min exposure - 75 - - 50- - 37 - -25 -20 - I I 15 - Figure 5-11: Indirect phage Western blot of pellicle and WCL 116 CD 0 (D I-i 5 ug fd-tet Ab 0.25 ug fd-tet Ab 1El 0 PFU LN4P-1 + 5 ug fd-tet Ab 1El 0 PFU LN4P-1 +1 ug fd-tet Ab 1El 0 PFU LN4P-1 + 0.5 ug fd-tet Ab 1El 0 PFU LN4P-1 + 0.25 ug fd-tet Ab PC3M-LN4 + LN4P-1 + 5 ug fd-tet Ab PC3M-LN4 + LN4P-1 +1 ug fd-tet Ab PC3M-LN4 + LN4P-1 + 0.25 ug fd-tet Ab PC3M-LN4 Pellicle (15 ug) PC3M Pellicle (15 ug) 2E1 0 PFU LN4P-1 + fd-tet Ab 2E10 PFU M13KE + fd-tet Ab PC3M-LN4 - LN4P-1 + fd-tet Ab PC3M-LN4 + LN4P-1 + rabbit IgG PC3M-LN4 + LN4P-1 + fd-tet Ab PC3M-LN4 + M13KE + fd-tet Ab Table 5.4: Mass spectrometry identified proteins from 100-150 kDa protein bands from pellicle isolation Protein Matches IProtein Name M. 11 cub domain-containing protein 1 precursor 92,875 7 cadherin-2 precursor (neural-cadherin) 99851 4 neuropilin-1 precursor (cd304 antigen) (VEGF-165 receptor) 103,120 2 neutral amino acid transporter 56,598 2 4f2 cell-surface antigen heavy chain (cd98) 57,945 2 thyroid hormone receptor-associated protein 3 108,666 2 hepatocyte growth factor receptor precursor 155,527 8 integrin alpha-2 precursor (cd49b antigen) 129,295 1 slit-robo rho gtpase-activating protein 2 120,881 1 potassium-transporting atpase alpha chain 2 115,899 1 150 kda oxygen-regulated protein precursor 111,335 1 kinesin-like protein kifl4 186,492 1 cd44 antigen precursor (phagocytic glycoprotein 1) 81,554 1 macrophage-stimulating protein receptor precursor 152,227 1 ephrin type-a receptor 2 precursor 108,254 1 monocarboxylate transporter 1 53,958 Table 5.5: Mass spectrometry identified proteins from 100-150 kDa protein bands from pellicle isolation Protein Matches Protein Name M. 1 cd151 antigen 28,295 1 sodium-driven chloride bicarbonate exchanger 125,946 1 myosin 6 145,016 1 integrin beta-4 precursor (gp150) (cd104 antigen) 202,151 1 thyrotropin-releasing hormone-degrading ectoenzyme 117,000 1 cdna flj43793 157,962 1 integrin alpha-5 precursor 114,536 1 bcl-2 associated transcription factor 1 106,122 1 nucleolin (protein c23) 76,483 1 tyrosine-protein kinase-like 7 precursor (cck-4) 118,260 z.A 0 0 I PC3M-LN4 Pellicle (20 ug) PC3M-LN4 CDCP1 IP PC3M CDCPI IP PC3M-LN4 Pellicle (20 ug) PC3M-LN4 CDCP1 IP PC3M CDCP1 IP _j z PC3M PC3M-LN4 -. Phage 30m M13KE No Phage - 15m - - - 30m - 30m - - - 30m - 30m - 30m Figure 5-15: Indirect phage Western blot of SBED cross-linking immunoprecipitation 122 Table 5.6: Mass spectrometry identified proteins from 130 kDa protein bands from phage immunoprecipitation Protein Matches 47 Protein Name M. pyruvate carboxylase, mitochondrial precursor 129,634 6 acetyl-coa carboxylase 1 265,040 2 acetyl-coenzyme a carboxylase alpha isoform 2 variant 114,618 1 acetyl-coa carboxylase 2 279,694 1 clathrin heavy chain 1 191,483 Table 5.7: Mass spectrometry identified proteins from 75 kDa protein bands from phage immunoprecipitation Protein Matches Protein Na m e 1M 32 propionyl-coa carboxylase alpha chain, mitochondrial precursor 77,354 26 methylcrotonoyl-coa carboxylase subunit alpha 80,473 14 heat shock cognate 71 kda protein 70,898 13 pyruvate carboxylase, mitochondrial precursor 129,634 11 stress-70 protein, mitochondrial precursor (grp 75) (mortalin) 73,680 5 heat shock protein 70 kda protein 11 70,375 5 heat shock-related 70 kda protein 2 70,021 4 replication protein a 70 kda dna-binding subunit 68,138 3 78 kda glucose-regulated protein precursor 72,333 3 acetyl-coa carboxylase 1 265,040 3 heterogeneous nuclear ribonucleoprotein m (hnrnp m) 77,384 2 heat shock 70 kda protein 1 70,052 1 acetyl-coenzyme a carboxylase alpha isoform 2 variant 114,618 1 heat shock 70 kda protein 6 71,028 1 lysyl-trna synthetase 68,048 1 atp-dependent dna helicase 2 subunit 1 (ku70) 69,712 1 ezrin (p81) (cytovillin) 69,268 5.5 Conclusions Metastatic potential of primary tumors is often a hallmark of cancer aggressiveness and a direct contributor to patient prognosis. Recent evidence has underscored the ability of tumor cells to disseminate from the primary site at a much earlier stage during tumorigenesis and progression than previously believed. Consequently, there is a great need to locate secondary sites of tumor cell seeding for monitoring as well as to identify novel biomarkers that may elucidate metastatic progression pathways. We performed subtractive phage display biopanning on two human prostate cancer cell lines with differing metastatic potential to determine peptide sequence(s) with preferential binding to cells with higher metastatic ability. One peptide sequence, Glu-Trp-Leu-Trp-Glu-Phe-Pro-Ser-Asp-Thr-Arg-Ser, which we termed LN4P-1, was selected through multiple rounds of panning under both serum-deficient and serumcontaining media conditions. LN4P-1 phage specifically bound to a protein localized to the plasma membrane, as evidenced by FACS analysis and confocal microscopy. However, monovalent peptide targeting of PC3M-LN4 cells resulted in low binding and suggested the need for either phage coat protein coordination and/or multivalent peptide presentation for efficient labeling of PC3M-LN4 cells. The development of an in vivo imaging probe led us to label LN4P-1 phage with a near-infrared dye, Cy5.5. Although Cy5.5-LN4P-1 had reduced binding affinity to PC3M-LN4 cells, in vivo applicability was successfully demonstrated in mouse models with bilateral subcutaneous PC3M and PC3M-LN4 tumors. Using Jacobson's pellicle method for plasma membrane isolation, we showed that LN4P-1 phage specifically bound the reduced form of a - 130, 000 kDa protein asso- ciated with the cell surface. Efforts to isolate the binding ligand resulted in successful immunoprecipitation and immunoblot evidence of a LN4P-1-associating protein. However, mass spectrometry and subsequent validation experiments did not conclusively result in the identification of the LN4P-1 ligand but instead, highlighted several membrane proteins that have been previously shown or are currently being investigated for their involvement in metastasis. 125 This study represents one of the first investigations into targetable cell surface-associated proteins for identifying highly metastatic cells. Yang and coworkers have used a similar subtractive peptide display system to select for metastasis-binding peptides and demonstrated its utility in imaging metastases from several different cancers [119]. However, they did not attempt to identify the peptide binding partner, as this step seems to be the more difficult task but one with potentially great value to basic understanding of the metastatic process. 126 Chapter 6 Conclusion 6.1 Summary The work described in this thesis has addressed the need for improved prostate cancer diagnosis through the use of biomarker-targeted molecular imaging. Current diagnostic methods suffer from poor resolution and accuracy and involve invasive procedures with variable outcomes. Biomarker research has focused mainly on correlating disease state with expression levels of specific proteins. However, biochemical cascades important to cancer progression are often catalyzed by the level at which a protein biomarker fulfills its functional role rather than its sheer quantity. Molecular imaging supported by target-specific probes has emerged to fill this gap in knowledge by providing a means of visualizing biochemical function. In the first portion of this thesis, prostate cancer biomarkers that demonstrated significant correlation between their expression levels and disease progression in the literature were characterized with respect to their proteolytic activity. Results suggested that the in vivo environment provides additional cues that control active uPA secretion and are not recapitulated in vitro with prostate cancer cell lines of varying metastatic potential. Furthermore, total PSA secretion was negatively correlated with the cell line aggressiveness, with undetectable levels of enzymatically active PSA. Attempts to engineer proteolytically active PSA-secreting cell lines for future use as small animal models of clinically relevant disease succeeded in the form of human prostate cancer cells exhibiting co127 expression of full-length PSA and its upstream activator, hK2. These results highlight the sensitive balance and stable control of protease activity by a combination of cancer cells and its tumor microenvironment, as in vitro experiments rarely capture the same phenomena and trends observed in experimental animal models as well as clinical patient samples. These observations also emphasize the importance of the protease activity of known cancer biomarkers, rather than total protein levels, as cells resist external manipulation in attempts to increase activity levels. Given the importance of biomarker protease activity in clinical disease progression and newly-gained knowledge of activity levels in various human cancer cell lines, protease-activatable molecular imaging probes were developed as a potential supplemental tool for diagnosing cancer presence and stage. Optimal gold nanoparticle probes targeted to trypsin and urokinase-type plasminogen activator required the incorporation of a dark quencher to achieve 5 to 8-fold signal amplification. These probes exhibited extended circulation time in vivo and high image contrast in a mouse tumor model. This platform system is easily adaptable for targeting a wide array of cancer-associated biomarker proteases. As much of the clinical cancer research literature addresses the general detection and imaging of neoplastic lesions regardless of clinical stage and progression, I sought to develop a molecular imaging probe for the preferential detection of cells with higher metastatic potential based on cell surface protein biomarkers. Typically, metastases are identified on imaging scans where multi-organ lesions are seen or when patients experience a biochemical failure after medical or surgical interventions to treat the primary tumor. Identification of metastatic cells in a non-invasive and highly sensitive setting would be greatly advantageous towards tailoring patient treatment based on disease aggressiveness. Subtractive phage display screening yielded a peptide sequence, termed LN4P-1, that preferentially binds to a -130 kDa protein found on the cell surface of highly metastatic PC3M-LN4 cells. The LN4P-1 containing phage, when labeled with a fluorescent dye, selectively target and bind subcutaneous PC3MLN4 tumors in mice, as shown on whole animal images. The possibility of identifying a novel metastasis-associated biomarker protein that can be targeted for imaging as 128 well as therapeutic applications represents an exciting opportunity to advance patient care before what is commonly considered the end stages of cancer. 6.2 Future Directions The emergence of molecular imaging as a field has drawn great interest from both translational medicine researchers as well as basic scientists who seek to understand disease pathologies in vivo. As knowledge of molecular medicine and diseaseassociated biochemical pathways expands, the possibilities for tailoring molecular probes for specific detection become boundless. New insights gained from using these molecular imaging tools then create new avenues of research, opening the doors to even more advanced imaging probes and reagents. With the aim of establishing new technology platforms and biomolecular targets for imaging cancer through its many stages, the work described in this thesis establishes a foundation for expanding into more advanced imaging probes and therapeutic delivery constructs as well as increasing basic understanding of end stage disease. The development of molecular imaging probes represents the first step towards the realization of a general targeted delivery vehicle. A nanomaterial-based vehicle is appealing as a means of targeting specific sites within the body, imaging its location, and delivering a therapeutic payload to eradicate lesions. The gold nanoparticle probe system described in this thesis can be further expanded to include a targeting molecule, such as a peptide or oligonucleotide, to selectively concentrate the probe and its payload in a desired location. Furthermore, multiple adaptations can be made to the AuNP surface to render it suitable for other forms of imaging, such as surfaceenhanced Raman scattering (SERS), or as a conduit for photothermal heating and destruction of select tissue regions. The additional insight gained from identifying a metastasis-targeting peptide sequence opens new avenues of research. Efforts to uncover the identity of the LN4P-1 ligand may shed light on novel biochemical and signaling pathways that are critical for metastatic spread. Future experiments may involve adapting the LN4P-1 to the 129 AuNP probe as a targeting moiety with which to locate metastatic sites and expand into potential therapeutic applications. Since metastatic cancers tend to exhibit drug resistance, the LN4P-1 modified AuNP may be a beneficial tool with which to further explore this phenomenon. 130 Bibliography [1] American Cancer Society. Cancer facts & figures 2005. Technical report, American Cancer Society, 2005. [2] T. F. Massoud and S. S. Gambhir. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev, 17:545-580, 2003. [3] A. Moore, L. Josephson, R. M. Bhorade, J. P. Basilion, and R. Weissleder. Human transferrin receptor gene as a marker gene for MR imaging. Radiology, 221:244-250, 2001. [4] T. A. Stamey, N. Yang, A. R. Hay, J. E. McNeal, F. S. Freiha, and E. Redwine. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N. Engl. J. Med., 317:909-916, 1987. [5] P.R. Hubler, Y. Schnell, F. Hering, and G. Rutishauser. Prostate specific antigen. experimental and clinical observations. Scand. J. Urol. Nephrol. Suppl., 104:33-39, 1987. [6] J. W. Colberg, D. S. Smith, and W. J. Catalona. Prevalence and pathological extent of prostate cancer in men with prostate specific antigen levels of 2.9 to 4.0 ng/ml. J. Urol., 149:507-509, 1993. [7] W. J. Catalona, J. P. Richie, F. R. Ahmann, M. A. Hudson, P. T. Scardino, R.C. Flanigan, J.B. deKernion, T.L. Ratliff, L.R. Kavoussi, and B.L. Dalkin et al. Comparison of digital rectal examination and serum prostate specific antigen 131 in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J. Urol., 151:1283-1290, 1994. [8] J. S. Krumholtz, G. F. Carvalhal, C. G. Ramos, D. S. Smith, P. Thorson, Y. Yan, P. A. Humphrey, K. A. Roehl, and W. J. Catalona. Prostate-specific antigen cutoff of 2.6 ng/ml for prostate cancer screening is associated with favorable pathologic tumor features. Urology, 60:469-473, 2002. [9] M. K. Brawer, M. A. Rennels, R. B. Nagle, R. Schifman, and J. A. Gaines. Serum prostate-specific antigen and prostate pathology in men having simple prostatectomy. Am. J. Clin. Pathol., 92:760-764, 1989. [10] P. Cohen, H. C. Graves, D. M. Peehl, M. Kamarei, L. C. Guidice, and R. G. Rosenfeld. Prostate-specific antigen (psa) is an insulin-like growth factor binding protein-3 protease found in seminal plasma. J. Clin. Endocrinol. Metab., 75:1046-1053, 1992. [11] P. Cohen, D. M. Peehl, H. C. Graves, and R. G. Rosenfeld. Biological effects of prostate specific antigen as an insulin-like growth factor binding protein-3 protease. J. Endocrinol., 142:407-415, 1994. [12] M. M. Webber, A. Waghray, and D. Bello. Prostate-specific antigen, a serine protease, facilitates human prostate cancer cell invasion. Clin. Cancer Res., 1:1089-1094, 1995. [13] S.R. Denmeade, L. Litvinov, L.J. Sokoll, H. Lilja, and J.T. Isaacs. Prostatespecific antigen (PSA) protein does not affect growth of prostate cancer cells in vitro or prostate cancer xenografts in vivo. Prostate,56:45-53, 2003. [14] E.T. Keller and J. Brown. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J. Cell Biochem., 91:718-729, 2004. [15] V.I. Romanov, T. Whyard, H.L. Adler, W.C. Waltzer, and S. Zucker. Prostate cancer cell adhesion to bone marrow endothelium: the role of prostate-specific antigen. Cancer Res., 64:2083-2089, 2004. 132 [16] N. Blaszczyk, B. A. Masri, N. R. Mawji, T. Ueda, G. McAlinden, C. P. Duncan, N. Bruchovsky, H. U. Schweikert, D. Schnabel, E. C. Jones, and M. D. Sadar. Osteoblast-derived factors induce androgen-independent proliferation and expression of prostate-specific antigen in human prostate cancer cells. Clin. Cancer Res., 10:1860-1869, 2004. [17] H. H. Heidtmann, D. M. Nettelbeck, A. Mingels, R. Jager, H. G. Welker, and R. E. Kontermann. Generation of angiostatin-like fragments from plasminogen by prostate-specific antigen. Br. J. Cancer,81:1269-1273, 1999. [18] A. H. Fortier, J. W. Holaday, H. Liang, C. Dey, D. K. Grella, J. Holland-Linn, H. Vu, S. M. Plum, and B. J. Nelson. Recombinant prostate specific antigen inhibits angiogenesis in vitro and in vivo. Prostate,56:212-219, 2003. [19] A. Kumar, S. D. Mikolajczyk, A. S. Goel, L. S. Millar, and M. S. Saedi. Expression of pro form of prostate-specific antigen by mammalian cells and its conversion to mature, active form by human kallikrein 2. Cancer Res., 57:3111-3114, 1997. [20] A. Lundwall and H. Lilja. Molecular cloning of human prostate specific antigen cDNA. FEBS Lett., 214:317-322, 1987. [21] T. K. Takayama, K. Fujikawa, and E. W. Davie. Characterization of the precursor of prostate-specific antigen: activation by trypsin and by human glandular kallikrein. J. Biol. Chem., 272:21582-21588, 1997. [22] T. K. Takayama, C. A. Carter, and T. Deng. Activation of prostate-specific antigen precursor (pro-PSA) by prostin, a novel human prostatic serine protease identified by degenerate pcr. Biochemistry, 40:1679-1687, 2001. [23] M. C. Wang, L. A. Valenzuela, G. P. Murphy, and T. M. Chu. Purification of a human prostate specific antigen. Invest. Urol., 17:159-163, 1979. [24] Y. Qian, J. A. Sensibar, D. J. Zelner, A. J. Schaeffer, J. A. Finlay, H. G. Rittenhouse, and C. Lee. Two-dimensional gel electrophoresis detects prostate-specific 133 antigen-a 1 -antichymotrypsin complex in serum but not in prostatic fluid. Clin. Chem., 43:352-359, 1997. [25] F. Espafia, J. Sanchez-Cuenca, A. Estelles, J. Gilabert, J. H. Griffin, and M. J. Heeb. Quantitative immunoassay for complexes of prostate-specific antigen with a 2-macroglobulin. Clin. Chem., 42:545-550, 1996. [26] U. H. Stenman, J. Leinonen, H. Alfthan, and 0. Alfthan. S. Rannikko, K. Tuhkanen, A complex between prostate-specific antigen and ai- antichymotrypsin is the major form of prostate-specific antigen in serum of patients with prosatic cancer: assay of the complex improves clinical sensitivity for cancer. Cancer Res., 51:222-226, 1991. [27] M. Martinez, F. Espana, M. Royo, J. M. Alapont, S. Navarro, A. Estelles, J. Aznar, C. D. Vera, and J. F. Jimenez-Cruz. The proportion of prostate-specific antigen (psa) complexed to ai-antichymotrypsin improves the discrimination between prostate cancer and benign prostatic hyperplasia in men with a total psa of 10 to 30 pg/l. Clin. Chem., 48:1251-1256, 2002. [28] T. Saika, T. Tsushima, Y. Nasu, N. Kusaka, Y. Miyaji, H. Takamoto, K. Takeda, S. Uno, and H. Kumon. Prostate specific antigen complexed to aiantichymotrypsin in patients with intermediate prostate specific antigen levels. Cancer,94:1685-1691, 2002. [29] R. W. Veltri and M. C. Miller. Free/total PSA ratio improves differentiation of benign and malignant disease of the prostate: critical analysis of two different test populations. Urology, 53:736-745, 1999. [30] L. F. Wymenga, F. J. Duisterwinkel, K. Groenier, P. Visser van Brummen, J. Marrink, and H. J. Mensink. Clinical implications of free-to-total immunoreactive prostate-specific antigen ratios. Scand. J. Urol. Nephrol., 34:181-187, 2000. 134 [31] D. Weckermann, C. Maassen, F. Wawroschek, and R. Harzmann. Improved discrimination of prostate cancer and benign prostatic hyperplasia by means of the quotient of free and total PSA. Int. Urol. Nephrol., 31:351-359, 1999. [32] P. J. Van Cangh, P. De Nayer, L. De Vischer, P. Sauvage, B. Tombal, F. Lorge, F. X. Wese, and R. Opsomer. Free to total prostate-specific antigen (PSA) ratio improves the discrimination between prostate cancer and benign prostatic hyperplasia (BPH) in the diagnostic gray zone of 1.8 to 10 ng/ml total PSA. Urology, 48:67-70, 1996. [33] K. Jung, C. Stephan, U. Elgeti, M. Lein, B. Brux, G. Kristiansen, B. Rudolph, S. Hauptmann, D. Schnorr, S. A. Loening, and P. Sinha. Molecular forms of prostate-specific antigen in serum with concentrations of total prostate-specific antigen i4 pg/l: are they useful tools for early detection and screening of prostate cancer? Int. J. Cancer,93:759-765, 2001. [34] H. Lilja. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J. Clin. Invest., 76:1899-1903, 1985. [35] H. Lilja, J. Oldbring, G. Rannevik, and C. B. Laurell. Seminal vesicle-secreted proteins and their reactions during gelation and liquefaction of human semen. J. Clin. Invest., 80:281-285, 1987. [36] A. Christensson, C. B. Laurell, and H. Lilja. Enzymatic activity of prostatespecific antigen and its reactions with extracellular serine proteinase inhibitors. Euro. J. Biochem., 27:755-763, 1990. [37] J. Leinonen, W. M. Zhang, and U. H. Stenman. Complex formation between PSA isoenzymes and protease inhibitors. J. Urol., 155:1099-1103, 1996. [38] S. R. Denmeade, L. J. Sokoll, D. W. Chan, S. R. Khan, and J. T. Isaacs. Concentration of enzymatically active prostate-specific antigen (psa) in the extracellular fluid of primary human prostate cancers and human prostate cancer xenograft models. Prostate,48:1-6, 2001. 135 [39] M. Robert, B. F. Gibbs, E. Jacobson, and C. Gagnon. Characterization of prostate-specific antigen proteolytic activity on its major physiological substrate, the sperm motility inhibitor precursor/semenogelin i. Biochemistry, 36:3811-3819, 1997. [40] J. Malm, J. Hellman, P. Hogg, and H. Lilja. Enzymatic action of prostatespecific antigen (PSA or hK3): substrate specificity and regulation by Zn2+, a tight-binding inhibitor. Prostate,45:132-139, 2000. [41] M. Jonsson, S. Linse, B. Frohm, A. Lundwall, and J. Malm. Semenogelins I and II bind zinc and regulate the activity of prostate-specific antigen. Biochem. J., 387:447-453, 2005. [42] S. R. Denmeade, W. Lou, J. L6vgren, J. Malm, H. Lilja, and J. T. Isaacs. Specific and efficient peptide substrates for assaying the proteolytic activity of prostate-specific antigen. Cancer Res., 57(21):4924-4930, 1997. [43] C. F. Yang, E. S. Porter, J. Boths, D. Kanyi, M. Hsieh, and B. S. Cooperman. Design of synthetic hexapeptide substrates for assaying the proteolytic activity of prostate-specific antigen. Peptide Res., 54:444-448, 1999. [44] G. S. Coombs, R. C. Bergstrom, J. L. Pellequer, S. I. Baker, M. Navre, M. M. Smith, J. A. Tainer, E. L. Madison, and D. R. Corey. Substrate specificity of prostate-specific antigen (psa). Chem. Biol., 5:475-488, 1998. [45] S. Rehault, M. Brillard-Bourdet, L. Bourgeois, G. Frenette, L. Juliano, F. Gauthier, and T. Moreau. Design of new and sensitive fluorogenic substrates for human kallikrein hK3 (prostate-specific antigen) derived from semenogelin sequences. Biochim. Biophys. Acta., 1596:55-62, 2002. [46] P. Wu, J. Leinonen, E. Koivunen, H. Lankinen, and U. H. Stenman. Identification of novel prostate-specific antigen-binding peptides modulating its enzyme activity. Eur. J. Biochem., 267:6212-6220, 2000. 136 [47] S. R. Denmeade, A. Nagy, J. Gao, H. Lilja, A. V. Schally, and J. T. Isaacs. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res., 58:2537-2540, 1998. [48] S. R. Khan and S. R. Denmeade. In vivo activity of a psa-activated doxorubicin prodrug against PSA-producing human prostate cancer xenografts. Prostate, 45:80-83, 2000. [49] D. DeFeo-Jones, S. F. Brady, D. M. Feng, B. K. Wong, T. Bolyar, K. Haskell, D. M. Kiefer, K. Leander, E. McAvoy, P. Lumma, J. M. Pawluczyk, J. Wai, S. L. Motzel, K. Keenan, M. Van Zwieten, J. H. Lin, V. M. Garsky, R. Freidinger, A. Oliff, and R. E. Jones. A prostate-specific antigen (psa)-activated vinblastine prodrug selectively kills PSA-secreting cells in vivo. Mol. Cancer Ther., 1:451459, 2002. [50] C. M. Jakobsen, S. R. Denmeade, J. T. Isaacs, A. Gady, C. E. Olsen, and S. B. Christensen. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J. Med. Chem., 44:4696-4703, 2001. [51] V. M. Garsky, P. K. Lumma, D. M. Feng, J. Wai, H. G. Ramjit, M. K. Sardana, A. Oliff, R. E. Jones, D. DeFeo-Jones, and R. M. Freidinger. The synthesis of a prodrug of doxorubicin designed to provide reduced systemic toxicity and greater target efficacy. J. Med. Chem., 44:4216-4224, 2001. [52] S. P. Leytus, K. R. Loeb, F. S. Hagen, K. Kurachi, and E. W. Davie. A novel trypsin-like serine protease (hepsin) with a putative transmembrane domain expressed by human-liver and hepatoma-cells. Biochemistry, 27(3):1067-1074, 1988. [53] A. Tsuji, A. Torres-Rosado, T. Arai, M. M. Le Beau, R. S. Lemons, S.-H. Chou, and K. Kurachi. Hepsin, a cell membrane-associated protease. J. Biol. Chem., 266(25):16948-16953, 1991. 137 [54] A. Torres-Rosado, K. S. O'Shea, A. Tsuji, S.-H. Chou, and K. Kurachi. Hepsin, a putative cell-surface serine protease, is required for mammalian cell growth. Proc. Natl. Acad. Sci., 90:7181-7185, 1993. [55] Y. Kazama, T. Hamamoto, D. C. Foster, and W. Kisiel. Hepsin, a putative membrane-associated serine-protease, activates human factor-VII and initiates a pathway of blood-coagulation on the cell-surface leading to thrombin formation. J. Biol. Chem., 270(1):66-72, 1995. [56] P. Moran, W. Li, B. Fan, R. Vij, C. Eigenbrot, and D. Kirchhofer. Pro- urokinase-type plasminogen activator is a substrate for hepsin. J. Biol. Chem., 281(41):30439-30446, 2006. [57] S. Herter, D. E. Piper, W. Aaron, T. Gabriele, G. Cutler, P. Cao, A. S. Bhatt, Y. Choe, C. S. Craik, N. Walker, D. Meininger, T. Hoey, and R. J. Austin. Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers. Biochem. J., 390:125-136, 2005. [58] M. Tripathi, S. Nandana, H. Yamashita, R. Ganesan, D. Kirchhofer, and V. Quaranta. Laminin-332 is a substrate for hepsin, a protease associated with prostate cancer progression. J. Biol. Chem., 283(45):30576-30584, 2008. [59] Q. Wu, D. Yu, J. Post, M. Halks-Miller, J. E. Sadler, and J. Morser. Generation and characterization of mice deficient in hepsin, a hepatic transmembrane serine protease. J. Chin. Invest., 101(2):321-326, 1998. [60] R. T. Aimes, A. Zijlstra, J. D. Hooper, S. M. Ogbourne, M.-L. Sit, S. Fuchs, D. C. Gotley, J. P. Quigley, and T. M. Antalis. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb. Haemostasis, 89:561-572, 2003. [61] C. Morrissey, L. D. True, M. P. Roudier, I. M. Coleman, S. Hawley, P. S. Nelson, R. Coleman, Y.-C. Wang, E. Corey, P. H. Lange, C. S. Higano, and R. L. 138 Vessella. Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clin. Exp. Metastas., 25:377388, 2008. [62] J. A. Magee, T. Araki, S. Patil, T. Ehrig, L. True, P. A. Humphrey, W. J. Catalona, M. A. Watson, and J. Milbrandt. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res., 61(15):5692-5696, 2001. [63] S. M. Dhanasekaran, T. R. Barrette, D. Ghosh, R. Shah, S. Varambally, K. Kurachi, K. J. Pienta, M. A. Rubin, and A. M. Chinnaiyan. Delineation of prognostic biomarkers in prostate cancer. Nature, 412:822-826, 2001. [64] 0. Klezovitch, J. Chevillet, J. Mirosevich, R. L. Roberts, R. J. Matusik, and V. Vasioukhin. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell, 6:185-195, 2004. [65] F. Blasi, J. D. Vassalli, and K. Dano. Urokinase-type plasminogen activator: proenzyme, receptor, and inhibitors. J. Cell Biol., 104(4):801-804, 1987. [66] S. Sheng. The urokinase-type plasminogen activator system in prostate cancer metastasis. Cancer Metast. Rev., 20(3-4):287-296, 2001. [67] K. Miyazawa, Y. M. Wang, S. Minoshima, N. Shimizu, and N. Kitamura. Structural organization and chromosomal localization of the human hepatocyte growth factor activator gene - phylogenetic and functional relationship with blood coagulation factor XII, urokinase, and tissue-type plasminogen activator. Eur. J. Biochem., 258(2):355-361, 1998. [68] R. McGowen, H. Biliran, R. Sager, and S. Sheng. The surface of prostate carcinoma DU145 cells mediates the inhibition of urokinase-type plasminogen activator by maspin. Cancer Res., 60(17):4771-4778, 2000. [69] S. A. Rabbani, P. Harakidas, D. J. Davidson, J. Henkin, and A. P. Mazar. Prevention of prostate-cancer metastasis in vivo by a novel synthetic inhibitor 139 of urokinase-type plasminogen activator (uPA). Int. J. Cancer, 63(6):840-845, 1995. [70] A. Billstrom, I. Lecander, F. Dagnaeshansen, B. Dahllof, U. Stenram, and B. Hartley-Asp. Differential expression uPA in an aggressive (DU-145) and a nonaggressive (1013L) human prostate-cancer xenograft. Prostate,26(2):94104, 1995. [71] Z. L. Gokaslan, S. K. Chintala, J. E. York, V. Boyapati, S. Jasti, R. Sawaya, G. Fuller, D. M. Wildrick, G. L. Nicolson, and J. S. Rao. Expression and localization of urokinase-type plasminogen activator in human spinal column tumors. Clin. Exp. Metastas., 16(8):713-719, 1998. [72] H. Miyake, I. Hara, K. Yamanaka, K. Gohji, S. Arakawa, and S. Kamidono. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate, 39(2):123-129, 1999. [73] M. Yebra, L. Goretzki, M. Pfeifer, and B. M. Mueller. Urokinase-type plasminogen activator binding to its receptor stimulates tumor cell migration by enhancing integrin-mediated signal transduction. Exp. Cell Res., 250(1):231240, 1999. [74] A. Angelucci, S. D'Ascenzo, C. Festuccia, G. L. Gravina, M. Bologna, V. Dolo, and A. Pavan. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin. Exp. Metastas., 18, 2000. [75] Z. Dong, A. D. Saliganan, H. Meng, S. M. Nagha, A. L. Sabbota, S. J. Sheng, R. D. Bonfil, and M. L. Cher. Prostate cancer cell-derived urokinase-type plasminogen activator contributes to intraosseous tumor growth and bone turnover. Neoplasia, 10(5):439-449, 2008. [76] M. Gutova, J. Najbauer, R. T. Frank, S. E. Kendall, A. Gevorgyan, M. Z. Metz, M. Guevorkian, M. Edmiston, D. Zhao, C. A. Glackin, S. U. Kim, and K. S. 140 Aboody. Urokinase plasminogen activator and urokinase plasminogen activator receptor mediate human stem cell tropism to malignant solid tumors. Stem Cells, 26(6):1406-1413, 2008. [77] K. A. Kelly, S. R. Setlur, R. Ross, R. Anbazhagan, P. Waterman, M. A. Rubin, and R. Weissleder. Detection of early prostate cancer using a hepsin-targeted imaging agent. Cancer Res., 68(7):2286-2291, 2008. [78] B. Ballou, G. W. Fisher, T. R. Hakala, and D. L. Farkas. Tumor detection and visualization using cyanine fluorochrome-labeled antibodies. Biotechnol. Prog., 13(5):649-658, 1997. [79] A. Becker, C. Hessenius, K. Licha, B. Ebert, U. Sukowski, W. Semmler, B. Wiedenmann, and C. Gr6tzinger. Receptor-targeted optical imaging of tumors with near-infrared fluorescent ligands. Nat. Biotechnol., 19(4):327-331, 2001. [80] S. Achilefu, R. B. Dorshow, J. E. Bugaj, and R. Rajagopalan. Novel receptortargeted fluorescent contrast agents for in vivo tumor imaging. Invest. Radiol., 35(8):479-485, 2000. [81] W. K. Moon, Y. H. Lin, T. O'Loughlin, Y. Tang, D. E. Kim, R. Weissleder, and C. H. Tung. Enhanced tumor detection using a folate receptor-targeted nearinfrared fluorochrome conjugate. Bioconjugate Chem., 14(3):539-545, 2003. [82] S. Ke, X. X. Wen, M. Gurfinkel, C. Charnsangavej, S. Wallace, E. M. SevickMuraca, and C. Li. Near-infrared optical imaging of epidermal growth factor receptor in breast cancer xenografts. Cancer Res., 63(22):7870-7875, 2003. [83] Y. Koyama, T. Barrett, Y. Hama, G. Ravizzini, P. L. Choyke, and H. Kobayashi. In vivo molecular imaging to diagnose and subtype tumors through receptortargeted optically labeled monoclonal antibodies. Neoplasia, 9(12):1021-1029, 2007. 141 [84] S. B. Lee, M. Hassan, R. Fisher, 0. Chertov, V. Chernomordik, G. KramerMarek, A. Gandjbakhche, and J. Capala. Affibody molecules for in vivo characterization of HER2-positive tumors by near-infrared imaging. Clin. Cancer Res., 14(12):3840-3849, 2008. [85] C. H. Tung, S. Bredow, U. Mahmood, and R. Weissleder. Preparation of a cathepsin D sensitive near-infrared fluorescence probe for imaging. Bioconjugate Chem., 10(5):892-896, 1999. [86] C. H. Tung, U. Mahmood, S. Bredow, and R. Weissleder. In vivo imaging of proteolytic enzyme activity using a novel molecular reporter. Cancer Res., 60(17):4953-4958, 2000. [87] R. Weissleder, C. H. Tung, U. Mahmood, and A. Bogdanov Jr. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat. Biotechnol., 17(4):375-378, 1999. [88] S. M. Messerli, S. Prabhakar, Y. Tang, K. Shah, M. L. Cortes, V. Murthy, R. Weissleder, X. 0. Breakefield, and C. H. Tung. A novel method for imaging apoptosis using a caspase-1 near-infrared fluorescent probe. Neoplasia, 6(2):95105, 2004. [89] F. A. Jaffer, D. E. Kim, L. Quinti, C. H. Tung, E. Aikawa, A. N. Pande, R. H. Kohler, G. P. Shi, P. Libby, and R. Weissleder. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation,115(17):2292-2298, 2007. [90] W. Pham, Y. Choi, R. Weissleder, and C. H. Tung. Developing a peptidebased near-infrared molecular probe for protease sensing. Bioconjugate Chem., 15(6):1403-1407, 2004. [91] D. Maxwell, Q. Chang, X. Zhang, E. M. Barnett, and D. Piwnica-Worms. An improved cell-penetrating, caspase-activatable, near-infrared fluorescent peptide for apoptosis imaging. Bioconjugate Chem., 20(4):702-709, 2009. 142 [92] L. Shang, J. Y. Yin, J. Li, L. H. Jin, and S. J. Dong. Gold nanoparticle- based near-infrared fluorescent detection of biological thiols in human plasma. Biosens. Bioelectron., 25(2):269-274, 2009. [93] A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane, and C. A. Mirkin. Nano-flares for mRNA regulation and detection. A CS Nano, 3(8):2147-2152, 2009. [94] C. C. You, 0. R. Miranda, B. Gider, P. S. Ghosh, I. B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Bunz, and V. M. Rotello. Detection and identification of proteins using nanoparticle-fluorescent polymer 'chemical nose' sensors. Nat. Nanotechnol., 2(5):318-323, 2007. [95] A. Bajaj, 0. R. Miranda, I. B. Kim, R. L. Phillips, D. J. Jerry, U. H. F. Bunz, and V. M. Rotello. Detection and differentiation of normal, cancerous, and metastatic cells using nanoparticle-polymer sensor arrays. Proc. Nati. Acad. Sci., 106(27):10912-10916, 2009. [96] D. J. Maxwell, J. R. Taylor, and S. M. Nie. Self-assembled nanoparticle probes for recognition and detection of biomolecules. J. Am. Chem. Soc., 124(32):96069612, 2002. [97] E. Dulkeith, M. Ringler, T. A. Klar, J. Feldmann, A. M. Javier, and W. J. Parak. Gold nanoparticles quench fluorescence by phase induced radiative rate suppression. Nano Lett., 5(4):585-589, 2005. [98] E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Moller, and D. I. Gittins. Fluorescence quenching of dye molecules near gold nanoparticles: radiative and nonradiative effects. Phys. Rev. Lett., 89(20):203002-1-4, 2002. [99] K. Aslan and V. H. Perez-Luna. Quenched emission of fluorescence by ligand functionalized gold nanoparticles. J. Fluoresc., 14(4):401-405, 2004. 143 [100] P. Anger, P. Bharadwaj, and L. Novotny. Enhancement and quenching of singlemolecule fluorescence. Phys. Rev. Lett., 96(11):113002-1-4, 2006. [101] F. Tam, G. P. Goodrich, B. R. Johnson, and N. J. Halas. Plasmonic enhancement of molecular fluorescence. Nano Lett., 7(2):496-501, 2007. [102] S. Lee, E.-J. Cha, K. Park, S.-Y. Lee, J.-K. Hong, I.-C. Sun, S. Y. Kim, K. Choi, I. C. Kwon, K. Kim, and C.-H. Ahn. A near-infrared-fluorescence-quenched gold-nanoparticle imaging probe for in vivo drug screening and protease activity determination. Angew. Chem., Int. Ed., 47(15):2804-2807, 2008. [103] T. Tanaka, B. J. McRae, K. Cho, R. Cook, J. E. Fraki, D. A. Johnson, and J. C. Powers. Mammalian tissue trypsin-like enzymes-comparative reactivities of human-skin tryptase, human-lung tryptase, and bovine trypsin with peptide 4-nitroanilide and thioester substrates. J. Biol. Chem., 258(22):3552-3557, 1983. [104] N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han, and C. A. Mirkin. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science, 312:1027-1030, 2006. [105] D. Kim, S. Park, J. H. Lee, Y. Y. Jeong, and S. Jon. Antibiofouling polymercoated gold nanoparticles as a contrast agent for in vivo X-ray computed tomography imaging. J. Am. Chem. Soc., 129(24):7661-7665, 2007. [106] A. Rehor, H. Schmoekel, N. Tirelli, and J. A. Hubbell. Functionalization of polysulfide nanoparticles and their performance as circulating carriers. Biomaterials, 29(12):1958-1966, 2008. [107] S. Lee, J. H. Ryu, K. Park, A. Lee, S. Y. Lee, I. C. Youn, C. H. Ahn, S. M. Yoon, S. J. Myung, D. H. Moon, X. Chen, K. Choi, I. C. Kwon, and K. Kim. Polymeric nanoparticle-based activatable near-infrared nanosensor for protease determination in vivo. Nano Lett., 10.1021/n1902709m [doi]:http://pubs.acs.org (accessed October 22, 2009), 2009. 144 [108] D. Hanahan and R. A. Weinberg. The hallmarks of cancer. Cell, 100:57-70, 2000. [109] G. Poste and I. J. Fidler. The pathogenesis of cancer metastasis. Nature, 283:139-145, 1980. [110] D. X. Nguyen, P. D. Bos, and J. Massague. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer, 9:274-284, 2009. [111] M. J. Duffy, P. M. McGowan, and W. M. Gallagher. Cancer invasion and metastasis: changing views. J. Pathol., 214:283-293, 2008. [112] G. P. Gupta and J. Massagu6. Cancer metastasis: building a framework. Cell, 127:679-695, 2006. [113] C. Messiou, G. Cook, and N. M. deSouza. Imaging metastatic bone disease from carcinoma of the prostate. Br. J. Cancer, 101(8):1225-1232, 2009. [114] S. Cabilly. The basic structure of filamentous phage and its use in the display of combinatorial peptide libraries. Mol. Biotechnol., 12:143-148, 1999. [115] J. R. Newton, K. A. Kelly, U. Mahmood, R. Weissleder, and S. L. Deutscher. In vivo selection of phage for the optical imaging of PC-3 human prostate carcinoma in mice. Neoplasia, 8(9):772-780, 2006. [116] K. A. Kelly, N. Bardeesy, R. Anbazhagan, S. Gurumurthy, J. Berger, H. Alencar, R. A. DePinho, U. Mahmood, and R. Weissleder. Targeted nanoparticles for imaging incipient pancreatic ductal adenocarcinoma. PLoS Med., 5(4):657-668, 2008. [117] M. Nahrendorf, F. A. Jaffer, K. A. Kelly, D. E. Sosnovik, E. Aikawa, P. Libby, and R. Weissleder. Noninvasive vascular cell adhesion molecule-1 imaging identifies inflammatory activation of cells in atherosclerosis. 114(14):1504-1511, 2006. 145 Circulation, [118] J. Pilch, D. M. Brown, M. Komatsu, T. A. H. Jarvinen, M. Yang, D. Peters, R. M. Hoffman, and E. Ruoslahti. Peptides selected for binding to clotted plasma accumulate in tumor stroma and wounds. Proc. Natl. Acad. Sci., 103(8):2800-2804, 2006. [119] W. Yang, D. Luo, S. Wang, R. Wang, R. Chen, Y. Liu, T. Zhu, X. Ma, R. Liu, G. Xu, L. Meng, Y. Lu, J. Zhou, and D. Ma. TMTP1, a novel tumor-homing peptide specifically targeting metastasis. Clin. Cancer Res., 14(17):5494-5502, 2008. [120] L. K. Chaney and B. S. Jacobson. Coating cells with colloidal silica for high yield isolation of plasma membrane sheets and identification of transmembrane proteins. J. Biol. Chem., 258(16):10062-10072, 1983. [121] D. B. Stolz and B. S. Jacobson. Examination of transcellular membrane protein polarity of bovine aortic endothelial cells in vitro using the cationic colloidal silica microbead membrane-isolation procedure. J. Cell Sci., 103:39-51, 1992. [122] A. M. Rahbar and C. Fenselau. Integration of Jacobson's pellicle method into proteomic strategies for plasma membrane proteins. J. Proteome Res., 3(6):1267-1277, 2004. [123] A. M. Rahbar and C. Fenselau. Unbiased examination of changes in plasma membrane proteins in drug resistant cancer cells. J. Proteome Res., 4(6):21482153, 2005. [124] J. M. Kozlowski, I. J. Fidler, D. Campbell, Z. L. Xu, M. E. Kaighn, and I. R. Hart. Metastatic behavior of human tumor cell lines grown in the nude mouse. Cancer Res., 44(8):3522-3529, 1984. [125] C. A. Pettaway, S. Pathak, G. Greene, E. Ramirez, M. R. Wilson, J. J. Killion, and I. J. Fidler. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin. Cancer Res., 2(9):1627-1636, 1996. 146 [126] R. S. Hubert, I. Vivanco, E. Chen, S. Rastegar, K. Leong, S. C. Mitchell, R. Madraswala, Y. Zhou, J. Kuo, A. B. Raitano, A. Jakobovits, D. C. Saffran, and D. E. H. Afar. STEAP: a prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Nat. A cad. Sci., 96(25):14523-14528, 1999. [127] J. D. Hooper, A. Zijlstra, R. T. Aimes, H. Liang, G. F. Claassen, D. Tarin, J. E. Testa, and J. P. Quigley. Subtractive immunization using highly metastatic human tumor cells identifies sima135/cdcp1, a 135 kda cell surface phosphorylated glycoprotein antigen. Oncogene, 22(12):1783-1794, 2003. [128] M. Gehrmann, G. Liebisch, G. Schmitz, R. Anderson, C. Steinem, A. De Maio, G. Pockley, and G. Multhoff. Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS One, 3(4):e1925, 2008. 147