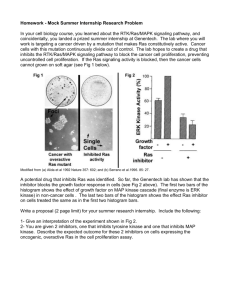
Cloning and Characterization of RIP1, ...
advertisement

Cloning and Characterization of RIP1, an Effector of Ral by Sang-Ho Park B. S., Yonsei University 1985 Submitted to the Department of Biology in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY at the Massachusetts Institute of Technology May 1996 0 1996 by Sang-Ho Park. All rights reserved. The author hereby grants to MIT permission to reproduce and to distribute publicly copies of this thesis document in whole or in part. I1 Signature of Author I' I Department of Biology May 29, 1996 Certified by N6 Robert A. Weinberg Thesis Supervisor Accepted by .... 77.INS;: ,Ui'L ,ASAC- -~ETTS OF TECHNOLOGY JUL 0 81996 Science LIBRARIES .. ,, F-rank Solomon Chairman Graduate Committee To Winnie, You started me off on this path of research, questioning what is, and why. I will never forget you, for every day I am reminded of the things you taught me, what you meant to me. I only regret not having realized it sooner. Enduring pain, agony and hopelessness, showing only perseverance, hope and love throughout. An inspiration always..... ACKNOWLEDGEMENTS First and foremost, I would like to thank Mina and Chan-Jun for their love, support, and understanding. I also give thanks to my parents and parents-in-law for their unwavering support. I would also like to thank all the members of the Weinberg lab, past and present, not only for their help in scientific matters, but also for their companionship and camaraderie, without which my stay in the Weinberg lab would not have been as fun and enjoyable. I give special thanks to Snezna Rogelj, who was always there for me when I needed help or advice; to Dennis Templeton, who taught me sequencing, as well as many other fine points in experimentation; to Steve Friend, who took me under his wing when I first joined the lab; to Bart Giddings and other members of the now defunct Ras group, who helped me start afresh when I returned from the Army; to past baymates Roman Herrera and Qiang Yu, who were there to commiserate and encourage me when things weren't going just right; to the sheik, the monk, the wire, and, of course, the chairman, may you never be called before a congressional hearing. I thank Bob for letting me join his lab, twice; for his support, encouragement and endurance for tolerating me for the eight years I have been in his lab; and especially for his support during the period I was off being all that I could be. I also thank Phil Sharp, David Housman for their helpful discussions and scientific supervision, as well as for past WIBR vs. CCR softball games, Peter Kim for his interest and encouragement and Andre Bernards for his insight, willingness and especially for his generous gift, without which this work would have been much delayed. I also thank Suzanne Brill for reading this manuscript and for her many helpful comments. I have very fond memories of the Whitehead and M.I.T. The Pit (yes, good things happen in the Pit, too), Tang Hall, project lab (yes, I enjoyed that too), softball games, Eastgate, Summer barbecues, the Retreats. But my fondest memories are of the third floor of the Whitehead, from back in the days when it was cram-packed with absolutely no extra space to now where we are practically the only lab left on the floor. There was always an excitement about doing Science and, again, I thank Bob for his part in creating and maintaining such an atmosphere. If I leave this place with nothing else, let me take that excitement with me. TABLE OF CONTENTS DEDICATION i ACKNOWLEDGEMENT ii TABLE OF CONTENTS iv FIGURES vii ABSTRACT 1 CHAPTER 1. RAS-RELATED GTPases AND THEIR EFFECTORS 3 GTPases 5 Ras, the oncogene 7 GTPase Activating Proteins 8 Guanine Nucleotide Exchange Factors 10 Down-stream signaling from Ras 12 Ras-related 17 proteins The Rho subfamily 18 Effectors and regulators of Rho-related GTPases 21 FIGURES 27 CHAPTER 2 CLONING AND MOLECULAR ANALYSIS INTRODUCTION 32 RESULTS Isolation of the RIP1 clone 34 Sequence analysis 36 Northern Analysis 36 Nucleotide specificity 37 Interacting domain mapping 38 DISCUSSION 38 MATERIALS AND METHODS 42 ACKNOWLEDGEMENT 44 FIGURES 45 CHAPTER 3 ANALYSIS OF RIP1 PROTEIN 58 INTRODUCTION RESULTS Expression of RIP1 60 Covalent Modification of RIP1 62 Ral-RIP1 64 interaction GAP assays 65 Immunofluorescence 67 DISCUSSION 69 MATERIALS AND METHODS 74 ACKNOWLEDGEMENT 81 FIGURES 82 CHAPTER 4. CONCLUSIONS AND PROSPECTS Effector proteins 101 RIP1 as an effector 104 Covalent modification of RIP1 105 GTPase cascade 106 FIGURES 109 CHAPTER 5. REFERENCES 112 FIGURES Figure 1-1. The basic GTPase cycle. Figure 1-2. Family tree of Ras-related proteins. Figure 2-1. Nucleotide and deduced amino acid sequence of RIP1. Figure 2-2. 29 45 Amino acid sequence comparison of proteins containing Rho/Rac GAP activity. Figure 2-3. Tissue distribution of RIP1 transcript. Figure 2-4. Preferential binding of 19-6 to KH-ral preloaded with GTP vs. GDP. Figure 2-5. 27 Mapping of the Ral-binding region. 47 49 51 53 Figure 2-6. Amino acid sequence comparison of RIP1 homologs. 55 Figure 3-1. Western analysis of RIP1. 82 Figure 3-2. Effect of cycloheximide treatment on RIP1. 84 Figure 3-3. Effect of phosphatase on RIP1. 86 Figure 3-4. Effect of serum on RIP1 modification. 88 Figure 3-5. Ral-RIP1 complexes in Cos-1 transient transfections. 90 Figure 3-6. The effect of RIP1 on the GTPase activity of several GTPases. Figure 3-7. The effect of RIP1 on guanine nucleotides bound to GTPases. Figure 3-8. Immunofluorescence of RIP1. Figure 3-9. Co-localization of RIP1 and vinculin. Figure 4-1. 92 Models of Ral-RIP1 interaction. 95 96 98 109 The Cloning and Characterization of RIP1, an Effector of Ral by Sang-Ho Park Submitted to the Department of Biology on May 29, 1996 in partial fulfillment of the requirements for the Degree of Doctor of Philosophy ABSTRACT I report here the cloning of a gene encoding a novel Ralinteracting protein (RIP1) from a cDNA expression library using radiolabeled Ral as probe. manner. RIP1 binds to Ral in a GTP-dependent The 4.1 kb transcript of the RIP] gene is present in all tissues analyzed and encodes for a protein product of 648 residues. RIP1 shares sequence similarity with GTPase activating proteins that are capable of activating the GTPase activity of members of the Rho/Rac family of GTPases. When tested, RIP1 could activate the GTPase activity of CDC42 and, to a lesser extent, Racl but not RhoA, Ras, or Ral. Activated Ral had no direct effect on the GTPase-activating ability of RIP1, in vitro. RIP1 can be covalently modified by phosphorylation as a result of serum stimulation in serum starved Balb/3T3 cells. RIP1 is found in a complex with Ral within five minutes after serum stimulation of serum starved cells. RIP1 co-localizes with vinculin in focal adhesion complexes. Thesis Advisor: Robert A. Weinberg, Ph.D. Professor of Biology, M.I.T. Member, Whitehead Institute for Biomedical Research Chapter 1. Ras-related GTPases and Their Effectors Ral is a member of a large family of small molecular weight GTPases, the most well-known of which is Ras. Like other members of this family of proteins, Ras functions as a molecular switch, oscillating between a GTP bound 'on' state and a GDP-bound 'off' state. The GTPase cycle (Figure 1-1) is regulated by two families of proteins. The guanine nucleotide exchange factors (GEFs) function in response to upstream signals to increase the rate of dissociation of the guanine nucleotide from the GTPase. The empty guanine nucleotide binding pocket is then rapidly occupied by GTP due to the high intracellular GTP concentration compared to GDP, switching the GTPase to the 'on' state. thereby The rate at which the GTPase hydrolyzes GTP to GDP varies widely among the different GTPases and determines the length of time in which the GTPase remains in an activated state. Many GTPases are then downregulated by specific GTPase-activating proteins (GAPs) that greatly increase the rate of hydrolysis by factors of 102-105. There is a third class of proteins involved in the regulation of some of the GTPases called guanine nucleotide dissociation inhibitors (GDIs) which function by blocking the dissociation of GDP from the GTPase. Recently, the role of Ras in the transduction of growth regulatory signals has been elucidated at the molecular level. As a result of activation, Ras undergoes a conformational change as a result of GTP binding and is able to interact with and cause the activation of its 'effector', Raf, the protein through which the effect of Ras activation is mediated. Raf activation in turn leads to the activation of a kinase cascade that has been shown to be important in signaling growth. Ever since Ras was identified as a gene that was capable of transforming cells (Reddy et al., 1982; Tabin et al., 1982; Taparowsky et al., 1982), much effort has been exerted into identifying the role that Ras plays in normal cells and the mutations that allow Ras to become transforming, as well as identifying novel genes that might mimic Ras in structure or function. To date, in excess of 60 Ras-related proteins have been isolated from a wide variety of species, ranging from yeast and slugs to frogs, fruit flies and mammals, with some estimates predicting the existence of at least 60 Ras-related proteins in mammals, at least 30 of which are expected to be Rab proteins (Chardin, 1993). Recent work in this laboratory has identified a protein that is involved in the regulation of Ral, a protein related to Ras. In this thesis, I will present work done in an attempt to elucidate the role of Ral in signaling by identifying a candidate effector protein for it. As an introduction to the world of GTPases, I will start off with a brief summary of GTPase in general and the conserved structure of Rasrelated proteins. I will then introduce Ras and its regulatory proteins, and the role of Ras in signal transduction. Finally, I will introduce other Ras-related proteins with emphasis on the Rhorelated protein for reasons that will become apparent later. GTPases. GTPases play a role in a wide variety of cellular functions, ranging from protein synthesis and vision to hormone action and cancer. These GTPases function primarily as switches oscillating between an activated, GTP-bound form to an inactive GDP-bound form. There are three families of GTPases that share a common design, in that all members of these families contain five regions of homology whose function is to either participate in guanine nucleotide binding or undergo conformational change in response to the nucleotide bound (Bourne et al., 1991). These families are the alpha subunits of heterotrimeric G proteins, the Ras-related small molecular weight GTPases, and EF-Tu, an elongation factor involved in bacterial protein synthesis. The heterotrimeric G-proteins are involved in hormonal or sensory signaling as a response to stimulation of heptahelical receptors. Representative of heterotrimeric G-proteins are GS, the stimulatory regulator of adenylyl cyclase, and GT (transducin), which mediates phototransduction. EF-Tu performs a proofreading function that ensures that the mRNA codon matches the anticodon of the aminoacyl-tRNA. The Ras-related GTPases are involved in a wide variety of functions ranging from signaling proliferation and differentiation to regulation of the actin cytoskeleton and vesicular trafficking. Although the amino acid sequence of Ras and EF-Tu guanine nucleotide binding domains are only 16% identical, their three dimensional structures are almost superimposable (Valencia et al., 1991). All members of the Ras family possess three conserved regions, all of which are critical for the proper function of Ras. The first region (residues 10-18, 57-63, 116-119, 143-146) is involved in GTP-binding. Mutations in this region, such as those frequently found in residues 12 or 61, result in a slowing down of the intrinsic GTP hydrolysis activity. The second region (residues 32-40) is commonly referred to as the 'effector' domain. Mutations in this region render oncogenic mutants of Ras no longer able to transform, without having any effect on the localization, GTPase activity or stability of the protein. It is thought that this is the region that binds to the cellular effector of Ras. Interestingly, this also overlaps nicely with one of the two domains (residues 30-37 and 60-76) shown to undergo a conformational switch when GTP replaces GDP in the protein (Kim et al., 1988). The third region is the C-terminal end which contains sites for isoprenylation, either a CAAX motif, or a CXC or CC motif where C is cysteine, A is an aliphatic amino acid, and X in any amino acid. The isoprenylation helps anchor the C-terminal end to the membrane. Ras, the oncogene. In 1964, Jennifer Harvey first reported a virus that was capable of inducing sarcomas and splenomegaly in infected rats (Harvey, 1964). Several years later, Kirsten and Mayer reported a mouse lymphoma virus that was capable of inducing sarcomas after passage through rats (Kirsten et al., 1967). It was later shown that both viruses carried closely related genes that encoded a 21 kD protein, namely, Ha-ras and Ki-ras (Shih et al., 1979). Further, it was soon demonstrated that these Ras proteins were capable of not only binding guanine nucleotides (Shih et al., 1980), but also of hydrolyzing GTP to GDP (McGrath et al., 1984; Sweet et al., 1984). Sequence analysis of the viral oncogene and the proto-oncogene revealed point mutations in residues 12 and 59. It was later shown that a single mutation at the position 12 residue was sufficient to cause normal Ras to become transforming (Reddy et al., 1982; Tabin et al., 1982; Taparowsky et al., 1982). This was due to the fact that although the residue 12 mutated form of Ras was capable of binding either GTP or GDP, its ability to hydrolyze GTP to GDP was greatly reduced compared to the wild type Ras (Gibbs et al., 1984). This was the first indication that the GTP-bound form of Ras was able to transduce a signal to a downstream target that allowed the cell to become transformed, and that the ability to regulate the guanine nucleotide binding and GTPase activity properly was crucial for normal cell function. GTPase Activating Proteins. Additional mutations in residues 13, 61 and 63, discovered in other human tumors, were shown to possess a reduced intrinsic rate of GTP hydrolysis (Colby et al., 1986; Gibbs et al., 1985; McGrath et al., 1984; Sweet et al., 1984). These activating mutations in Ras were long thought to endow Ras with a more aggressive growth potential as a direct consequence of reduced GTPase activity. But upon further investigation, a direct correlation between reduction in intrinsic GTPase activity and transforming potency did not hold up (Der et al., 1986; Lacal et al., 1986; Trahey et al., 1987). For example, although the T59 mutant form of Ras, where residue 59 was mutated to threonine, possesses GTPase activity comparable to that of wild type, it has a transforming potency comparable to the L12 mutant, indicating that activation of the transforming property of Ras can occur by mechanisms not involving just a reduction in in vitro GTPase activity (Lacal et al., 1986). In another study, two different mutants in position 12 that possessed a 4-fold difference in GTPase activity were shown to be equally potent in their transforming ability, suggesting that position 12 mutations might affect some other aspect of Ras function (Trahey et al., 1987). In experiments in Xenopus oocytes using the induction of maturation as an assay for Ras activity two activated forms of Ras, D12 and V12, could induce maturation while the wild type Ras, G12, was found to be relatively inactive. While both mutant forms were found to be GTP-bound, the wild type Ras was found to be predominately GDP-bound, indicating the presence of an activity capable of inducing the GTPase activity of Ras and, thereby, rendering it inactive. A cytoplasmic protein was subsequently purified that was capable of activating the GTPase activity of wild type Ras by more than 200-fold while having no effect on the mutant forms. A similar activity was detected in mammalian cells (Trahey and McCormick, 1987) and later purified as pl20GAP (Gibbs et al., 1988; Vogel et al., 1988). These data provide strong support for the model that the role of the position 12 mutants is to render these molecules unresponsive to GAP, allowing them to remain in their active state for a protracted period of time. Overexpression of pl20GAP can inhibit the function of wild type Ras by blocking the accumulation of its GTP-bound form (Zhang et al., 1990). pl20GAP was later shown to interact with Ras through its effector domain, which provided initial support for its role not only as a downregulator but also as a potential effector for Ras (Adari et al., 1988). Other GAPs for Ras have since been identified. Neurofibromin- 1 (NF1) is another mammalian protein that possesses GAP activity. Originally identified as a gene disrupted in neurofibromatosis patients (Cawthon et al., 1990; Viskochil et al., 1990; Wallace et al., 1990), when cloned, NF1 was found to have 26% identity to the Cterminal third of human pl20GAP (Xu et al., 1990). Although cell lines derived from these tumors have normal levels of p120GAP, Ras is predominately in the GTP-bound form, supporting the role of NF1 as a down regulator of Ras (Basu et al., 1992; DeClue et al., 1992). Two yeast proteins, IRA1 and IRA2, have also been identified as Ras GAPs (Tanaka et al., 1990). NF1 expressed in yeast is capable of replacing IRA (Ballester et al., 1990; Martin et al., 1990; Tanaka et al., 1990; Xu et al., 1990). Guanine Nucleotide Exchange Factors. Much effort has been invested in identifying the factor responsible for the activation of Ras. Although several groups had identified exchange activity in mammalian cell extracts (Downward et al., 1990; West et al., 1990; Wolfman and Macara, 1990), the protein responsible remained elusive. S. cerevisiae have two Ras homologs, RAS1 and RAS2, that function in the activation of adenylyl cyclase. Through genetic methods, CDC25 had been identified as functioning upstream of RAS (Broek et al., 1987; Robinson et al., 1987). Disruption of the CDC25 gene is lethal but its function can be replaced by an activated RAS2 10 gene. Wild type RAS2 cannot replace CDC25 even at very high levels, indicating that the CDC25 gene product is required for the activation of RAS2. The CDC25 gene product was subsequently shown to be able to promote guanine nucleotide exchange on mammalian Ras. A second related protein, SDC25, was also shown to function as an exchange factor when activated by truncating its C-terminal third (Crechet et al., 1990). In fission yeast, the ste6 gene product acts as an exchange factor for Ras and was found to share substantial homology with CDC25 (Hughes et al., 1990). A mammalian homologue of CDC25, CDC25Mm, was cloned from a mouse brain cDNA library by functional complementation in yeast (Martegani et al., 1992). In Drosophila, the sev gene, a gene found to be required for the proper development of the R7 photoreceptor cell, was shown to be structurally similar to mammalian receptor tyrosine kinases (Hafen et al., 1987; Simon et al., 1989). One of the genes identified in a screen looking for downstream elements was the Drosophilaras] gene, indicating that Drasl was involved the signaling from Sev. Indeed, activated mammalian Ras was able to rescue the formation of the R7 cell in sev null mutants (Fortini et al., 1992). Another gene, sos, that was identified in that screen was capable of suppressing a weak loss of function mutant of sev and, interestingly, contained homology to the yeast CDC25 gene (Bonfini et al., 1992; Simon et al., 1991). Not so surprisingly, Sos turned out to be the exchange factor for Drasl. A widely expressed human homologue of the Drosophila sos gene was soon isolated and found to able to stimulated guanine nucleotide exchange specifically on mammalian Ras proteins in vitro (Chardin et al., 1993). Also, mammalian cells overexpressing the full length human Sos were found to have increased guanine nucleotide exchange activity on Ras, indicating that the human Sos was indeed the exchange factor for Ras. Downstream signaling from Ras. Analysis of mutations in Ras identified regions that were indispensable for function, such as those required for guanine nucleotide binding and the C-terminal residues (Willumsen et al., 1986). Some of these mutations, such as those on positions 35, 36, 38, and 40, did not have an obvious function in that they did not have any effect on the biochemical properties of the protein. Mutations at these positions greatly reduced the ability of Ras to transform cells, and it had been postulated that these mutations defined the effector domain of Ras, a region that was important for transmitting the downstream signal (Sigal et al., 1986). Efforts to identify cellular responses to mitogen stimulation of mammalian cells led to the discovery that following an initial burst of receptor-mediated phosphorylation on tyrosines came a more widespread phosphorylation on serine/threonine residues, and that the prominent targets that were phosphorylated in response to a variety of different growth factors seemed to overlap (Avruch, 1985). One of the targets of phosphorylation turned out to be the S6 ribosomal subunit (Blenis and Erikson, 1986; Maller et al., 1986; Pelech et al., 1986; Stefanovic et al., 1986). The kinase responsible for the phosphorylation, Rsk, was purified (Blenis et al., 1987; Erikson and Maller, 1986) and its gene was cloned (Alcorta et al., 12 1989; Jones et al., 1988). Soon afterward, a second type of mitogen- activated protein kinase, the p42/p44 MAPKs, was identified that was shown to be able to phosphorylate and activate the Xenopus S6 kinase (Sturgill et al., 1988). While the crucial targets of Rsk remain uncertain, the p42/p44 MAPKs was found to be able to regulate a number of other protein kinases as well as a variety of protooncogenic transcription factors, such as c-Jun and c-Myc (Davis, 1993). MAPKs were then shown to be regulated by growth factor- stimulated tyrosine phosphorylation (Anderson et al., 1990), although the regulation turned out not to be a direct regulation by the receptor tyrosine kinases, but through yet another kinase, the MAPK kinase, that was able to activate the p42/p44 MAPs by phosphorylation on tyrosine as well as serine/threonine residues (Ahn et al., 1992). Thus, the regulation of proteins involved in the response to mitogens relied on a cascade of protein phosphorylation events (Ahn and Krebs, 1990; Ahn et al., 1990). MEK, a MAPK kinase, was found to be directly phosphorylated, and thus activated, by c-Raf, a cytoplasmic serine/threonine kinase whose oncogenic counterpart, v-Raf, was identified in mouse sarcoma virus 3611 (Rapp et al., 1988). Although other kinases capable of activating MEK in vitro have been identified (Lange-Carter et al., 1993), genetic evidence suggests that Raf is the primary element through which receptor tyrosine kinases exert their regulation of MEK (Tsuda et al., 1993). The initial indication that Ras and Raf might be on the same signaling pathway came from experiments in which the Y13-259 13 antibody was microinjected into cells. Y13-259, a rat monoclonal antibody against Ras (Furth et al., 1982), was critical in the functional studies of Ras in that it was able to neutralize Ras in the context of inhibiting morphological transformation induced by mutated Ras as well as being able to block the effect of serum induced mitogenic responses in certain cell lines (Kung et al., 1986; Mulcahy et al., 1985). Interestingly, this antibody could not inhibit the mitogenic effects of the v-raf gene product, an activated serine/threonine kinase, or an activated mutant Ras that was not recognized by the antibody. Later, it was found that a dominant negative Raf could block the mitogenic signal of Ras (Kolch et al., 1991). Conversely, while the activation of several serine/threonine kinases implicated in mitogenesis, including Raf, has been shown to require Ras, the stimulation of these kinases as a response to receptor tyrosine kinase activation can be inhibited by the N17 dominant negative Ras, while oncogenic Ras can activate their kinase activities (Wood et al., 1992). Soon after Raf had been identified as the activator of MEK and the primary element through which receptor tyrosine kinases regulate MEK, examination of the possible regulation of Raf by Ras led to a number of reports showing direct interaction between the two proteins. This interaction was shown to be dependent on Ras activation, in that Raf could interact only with the GTP-bound form of Ras. Also, these interactions were mediated through domains of the two proteins known to be crucial for their functions, the effector domain in the case of Ras and the N-terminal, presumably regulatory, domain of Raf (Koide et al., 1993; Moodie et al., 1993; Van Aelst et al., 1993; Vojtek et al., 1993; Warne et al., 1993; Zhang et al., 14 1993). The emerging model was that activation of receptor tyrosine kinases led to the activation of Ras through the translocation/activation of Sos, which in turn led to the activation of Ras and the subsequent recruitment of Raf to the plasma membrane where it is activated. Studies in which the CAAX box of Ras is fused to Raf further validated this model by demonstrating that by allowing Raf to be localized to the membrane independent of Ras through its own membrane localization domain, Ras was no longer necessary for, nor was the dominant negative N17 Ras able to inhibit, the activation of Raf (Leevers et al., 1994; Stokoe et al., 1994). Although the Raf-CAAX possessed an eight- to ten- fold higher specific activity compared to that of wild type Raf in the absence of Ras activation, treatment by EGF further activates Raf-CAAX by ten fold indicating that membrane localization is not sufficient for full activation of Raf and that other events are necessary. Activation of Raf is most likely not the only consequence of Ras activation. Ras activation is able to activate multiple signaling events that regulate cell growth and differentiation. In quiescent fibroblasts, the expression of ectopic H-Ras activated by the V12 mutation can induce membrane ruffling, activation of the MAP kinase and stimulation of DNA synthesis. Mutations in the effector domain of Ras in conjunction with the V12 activating mutation can further dissect the effector domain. The V12C40 mutant (where residue 40 is mutated to cysteine in the presence of the V12 mutation) can stimulate membrane ruffling but was defective in MAP kinase activation and stimulation of DNA synthesis. The V12S35 mutation, on the other hand, could activate MAP kinase but 15 was defective in stimulation of both DNA synthesis and membrane ruffling. The expression of both V12C40 and V12S35 could restore the stimulation of DNA synthesis to a level comparable to V12 Ras. These data suggest that Ras has multiple effectors, and that membrane ruffling and MAP kinase activation require the activation of distinct effector pathways while input from both are required for the stimulation of DNA synthesis (Joneson et al., 1996). A possible alternative effector for Ras is the phosphatidylinositol-3-OH kinase (PI3K) (Rodriguez-Viciana et al., 1994). Ras can interact directly with the catalytic subunit of PI3K in a GTP-dependent manner through its effector domain. The expression of the N17 dominant negative mutant of Ras can inhibit the production of 3' phosphorylated phosphoinositides in PC12 cells, while in COS cells the transfection of Ras, but not Raf, leads to a large elevation in the level of these lipids, supporting the existence of an effector pathway distinct from the Raf pathway. Also, by yeast two hybrid screens, RalGDS, a guanine nucleotide exchange factor specific for Ral was shown to be able to interact with the effector domain of Ras and with Rap (Hofer et al., 1994; Kikuchi et al., 1994; Spaargaren and Bischoff, 1994). Indeed, these screens were able to identify a family of related proteins that have RalGDS homology and that can interact with Ras (Ikeda et al., 1995), supporting not only the existence of multiple Ras signaling pathways, but also the existence of other Ral-like proteins. 16 Ras-related proteins. Soon after the H-ras and K-ras genes were discovered, other genes that shared homology with these genes were identified; the ypt gene in yeast (Papageorge et al., 1984) and the rho gene in Aplysia (Madaule and Axel, 1985), each of which share about 30% sequence similarity with the ras gene, indicating that Ras belonged to a family of related proteins. Since then, through the use of degenerate oligonucleotide probes against conserved sequences, through low stringency hybridization, or by biochemical purification of proteins based on their ability to bind guanine nucleotides, more than 60 Ras-related proteins have been identified (Chardin, 1993). The Ras family of proteins can be divided into four subfamilies according to their sequences: Ras, Rho/Rac, Rab, and Ran. (Figure 1-2) This division delineates function as well as structural conservation. Members of the Rho/Rac subfamily are involved in the regulation of the cytoskeleton, and members of the Rab family are involved in vesicular transport, while Ran proteins are involved in nuclear transport. Rap and Ral are the other members of the Ras subfamily. These GTPases share approximately 50% homology with Ras (Kitayama et al., 1989; Pizon et al., 1988). The Rap proteins are enigmatic, in that one of the ways in which it was isolated was by its ability to cause reversion of the transformed phenotype in DT fibroblasts transformed by v-K-ras. The effector domain of Rapl is identical to Ras, leading to a model in which Rapl competes for downstream targets with Ras (Frech et al., 1990; Hata et al., 1990). Interestingly, although Rapl has a much higher affinity for pl20GAP 17 than does Ras, Rap1 GTPase hydrolysis is not stimulated as a result of interaction (Quilliam et al., 1990). To complicate matters, Rap2, along with R-Ras, also has an identical effector domain with Ras, although it is incapable of suppressing the ability of Ras to transform cells (Schweighoffer et al., 1990), indicating that structural components outside of the effector domain are important for the ability of Rapl to antagonize Ras. A GAP protein for Rapl has been purified (Polakis et al., 1991), and its gene subsequently cloned (Rubinfeld et al., 1991). Interestingly, the Rapl GAP shows no homology to the pl20GAP or NF1. A guanine nucleotide exchange factor for Rapl has also been identified (Kaibuchi et al., 1991), and was shown to have limited homology to CDC25 and SDC25 proteins. Despite its high degree of homology with Ras, little is known about its exact function. Rapla has been shown to associate with the NADPH oxidase cytochrome b component in human neutrophils (Bokoch et al., 1991), but in light of its ubiquitous expression, a regulatory role for a phagocyte specific enzyme is unlikely to be its only function. The other member of the Ras subfamily is the Ral GTPase which was identified through its sequence homology with Ras (Chardin and Tavitian, 1986). As it is the focus of this thesis, a detailed introduction will be given in Chapter 2. The Rho subfamily. The Rho subfamily is comprised of RhoA, RhoB, and RhoC; Racl and Rac2; G25K/Cdc42Hs; TC10O; RhoG. Members of this subfamily are involved in rearrangement of the actin cytoskeleton, such as the 18 formation of stress fibers, filopodia and lamellipodia. The signals that cause these different events are the result of the stimulation of a wide variety of different receptors, but it seems that these divergent signals converge on one or more the Rho-related GTPases (Zigmond, 1996). Microinjection of constitutively active Rho into serum-starved Swiss 3T3 cells will cause the formation of stress fibers and focal adhesion plaques (Ridley and Hall, 1992). Constitutively active Rac will induce the formation of membrane ruffles and lamellipodia (Ridley et al., 1992), and Cdc42 will induce filopodia (Kozma et al., 1995; Nobes and Hall, 1995). Although Rac and Cdc42 are not able to induce the formation of focal adhesion plaques, they are capable of inducing the formation of vinculin-containing focal complexes. The factor that will induce only stress fibers, and cause the activation of only Rho, has been shown to be lysophosphatidic acid (LPA). PDGF or bombesin can induce the activation of Rac and will result in the formation of membrane ruffling and subsequently the formation of stress fibers through the activation of Rho. Bradykinin-induced activation of Cdc42 will also result in the seemingly stepwise activation of Rac followed by Rho. The activation of these Rhorelated GTPases seem both necessary and sufficient for the induction of actin rearrangement, in that dominant negative forms of these GTPases or specific inhibitors can block the corresponding rearrangement from occurring. For example, in the presence of C3 transferase, a specific inhibitor of Rho, PDGF treatment will result in the formation of lamellipodia but not of stress fibers. 19 A wide range of cellular functions in response to the interaction of a cell with its surrounding extracellular matrix is mediated through integrin adhesion receptors. Contact with extracellular matrix induces integrins to cluster and form complexes with cytoplasmic proteins and the actin cytoskeleton. It is thought that the signaling by integrins is mediated by the focal adhesion kinase (FAK) and that after recruitment to the focal adhesion complex FAK is then able to activate the ERK1/2 MAP kinase pathway. Rho- related GTPases are important in the formation of focal adhesion complexes in that neither the interaction of extracellular matrix and integrins alone nor the activation of Rho or Rac by extracellular growth factors in the absence of extracellular matrix is capable of inducing focal complex formation. Also, the interaction of integrins with the matrix in the absence of activated Rho or Rac does not lead to the activation of ERK1/2 kinases in Swiss 3T3 fibroblasts, indicating that both extracellular matrix and activation of Rhorelated GTPases are necessary for integrin-dependent signal transduction (Hotchin and Hall, 1995). In lymphocytes, the leukocyte integrin LFA-1 is involved in aggregation. While the actin polymerization inhibitor cytochalasin B has no effect on integrin-mediated aggregation, C3 transferase, an inhibitor of Rho, completely blocked phorbol ester induced aggregation (Tominaga et al., 1993). This strongly suggests that, while actin polymerization is not required for aggregation, Rho is involved in the activation of integrin complexes, and that the changes in actin polymerization observed in fibroblasts might be a secondary consequence of Rho activation. 20 A number of other cellular events have also been shown to result from, or require, the activation Rho-related proteins. The expression of a constitutively activated Racl has be shown to be sufficient for causing malignant transformation (Qiu et al., 1995). Also, although Rho itself cannot transform cells, the expression of the constitutively activated V14-RhoA strongly cooperates with RafCAAX in focus-formation assays in NIH 3T3 cells (Qiu et al., 1995). Furthermore, dominant-negative N19 RhoA inhibits focus formation by V12-H-Ras and Raf-CAAX in NIH 3T3 cells. The Rho-related GTPases have also been shown to stimulate cell cycle progression through G1 and subsequent DNA synthesis when microinjected into quiescent fibroblasts (Olson et al., 1995), while the microinjection of dominant negative forms of Rac and Cdc42 or of the Rho inhibitor C3 transferase blocked serum-induced DNA synthesis. None of the Rho GTPases activated the mitogen- activated protein kinase (MAPK) cascade. Instead, Rac and Cdc42, but not Rho, could stimulated the c-Jun kinase JNK/SAPK (Jun NH2terminal kinase or stress-activated protein kinase), a distinct MAP kinase (Coso et al., 1995; Minden et al., 1995; Olson et al., 1995; Vojtek and Cooper, 1995). Effectors and regulators of Rho-related GTPases. The first demonstration of a direct role for a Rho-related GTPase came in phagocytic cells where Rac was found to be involved in NADPH oxidase through its interaction with its effector, p67phOx (Abo et al., 1991). Later, in a screen for proteins capable of interacting with the Rho-related GTPases, Cdc42 and Racl were 21 found to directly interact with and cause the activation of p65PAK (Manser et al., 1994) as well as other members of the PAK65 and STE20 family serine/threonine kinases (Martin et al., 1995). However, activation of p65PAK does not seem to be required for the formation of membrane ruffles (Knaus et al., 1995). In budding yeast, the actin cytoskeleton plays a role in bud construction by polarizing the machinery necessary in providing the building materials for cell wall assembly to the bud tip. Rholp has been co-localized with cortical actin at the inner surface of the plasma membrane at the bud tip and at the mother-daughter neck (Yamochi et al., 1994). Recently, Rholp has been shown to interact with and activate the regulatory subunit of 1,3-0-glucon synthase, an enzyme involved in cell wall biogenesis (Drgonova et al., 1996; Qadota et al., 1996), thus directing glucon synthesis to sites of cell wall growth. Rholp has also been shown to interact with and activate Pkclp, the yeast homolog of protein kinase C (Kamada et al., 1996; Nonaka et al., 1995). Mutants in pkcl displayed no defect in glucon synthesis while overexpression of Pkcl was not able to rescue glucon synthesis in rhol mutants indicating that Rholp functions in two distinct regulatory roles. Recently, protein kinase N and the protein kinase N-related protein rhophilin have been identified as targets of mammalian Rho. Activated Rho has been shown to directly interact and activate protein kinase N. LPA treatment of Swiss 3T3 cells can cause the phosphorylation of protein kinase N, while treating these cells with C3 transferase can block this phosphorylation (Amano et al., 1996; Watanabe et al., 1996). The p70S6Kinase has also been shown to 22 interact directly with Rac and can be activated by Rac and Cdc42 but not Rho in Cos transient transfection assays (Chou et al., 1996). Although a number of kinase cascades can be activated by the stimulation of Rho-related GTPases, none of these yet provide a direct link to actin rearrangement. By probing immobilized protein with radiolabeled Racl, Racl has been shown to bind tubulin (Best et al., 1996). occurs only in the GTP-bound state. This binding While the dominant negative mutants did not significantly affect the ability of Racl to interact with tubulin, the effector domain mutant D38A prevented interaction. These results suggest that the Racl-tubulin interaction may play a role in Racl function. Sequence analysis of Myr 5, a novel type of widely expressed myosin from rat tissue, also exhibited sequence similarity to the GTPase activating domain for Rho-related proteins. A fusion protein containing the GAP domain of Myr 5 was shown to stimulate the GTPase activity of RhoA and Cdc42, but only mildly that of Racl. Myr 5 provides not only a link between Rho-related proteins and the actin cytoskeleton, but also implies the importance of myosins and the actin cytoskeleton in signal transduction (Reinhard et al., 1995). Another link between a Rho family member and the actin cytoskeleton can be found in Cdc42. Cdc42 has been recently shown to bind WASP, a protein that is defective in Wiskott-Aldrich syndrome, in a GTP-dependent manner. While overexpression of WASP induces clusters of WASP that are highly rich in polymerized actin, co-expression of dominant negative Cdc42 prevents this clustering (Symons et al., 1996). 23 A number of guanine nucleotide exchange factors have been identified for the Rho family members. Most of these contain the Dbl homology domain, which has the exchange activity, along with a pleckstrin homology domain, which can bind phosphatidylinositol 4,5-bisphosphate. The Dbl oncoprotein was initially identified in transfection experiments from B-cells, in which it was found to be activated by truncation. Later it was found to be homologous to the CDC24 of S. cerevisiae, which was genetically the upstream regulator of CDC42 (Ron et al., 1991). CDC24 was shown to be an exchange factor for CDC42 as was Dbl (Hart et al., 1991). Dbl showed no homology to the Ras-specific guanine nucleotide exchange factor CDC25, although sequence similarity was identified between Dbl, Bcr and Vav. Since then a number of oncoproteins have been identified that show homology to Dbl. Ost, identified from osteosarcoma cDNA library, shares sequence similarity with both Dbl and pleckstrin. Ost was shown to be able to activate the guanine nucleotide exchange activity of RhoA and Cdc42 but not Racl (Horii et al., 1994). Tiam-1 is a gene that affects the invasiveness of T lymphomas. Sequence analysis indicated that the protein product shared sequence similarity with Dbl and pleckstrin (Habets et al., 1994). In fibroblasts, Tiam-1 was able to induce the same phenotypes as activated Rac (Michiels et al., 1995) suggesting guanine exchange activity for Rac. This exchange activity was confirmed in vitro. Ectopic expression of activated Tiam-1 was able to transform fibroblasts and was dependent on the presence of the Dbl-homology 24 domain. The transformed phenotype was similar to cell transformed with activated Rac, suggesting that the oncogenic transformation of Tiam-l was mediated through Rac (van Leeuwen et al., 1995). Lbc, another oncoprotein that shares homology with Dbl, has been shown to associate specifically with Rho (Zheng et al., 1995). The recombinant, affinity-purified Lbc specifically catalyze guaninenucleotide exchange on Rho in vitro. Also, microinjected onco-Lbc potently induces actin stress fiber formation in quiescent Swiss 3T3 fibroblasts indistinguishable from that induced by Rho. Lbc-induced NIH 3T3 focus formation is inhibited by the co-transfection with the dominant negative Rho mutant, indicating that Lbc is a specific guanine nucleotide exchange factor for Rho and causes cellular transformation through activation of the Rho signaling pathway. Recently a protein that interacts with the phosphotyrosine phosphatase LAR has been shown to contain two separate Dbl-like domains. Each domain was shown to activate the GTPase activity of a specific Rho-related GTPase, one for Rho and the other for Rac (Debant et al., 1996). A growing number of proteins that can act as GAPs for Rhorelated proteins have also been identified. During the purification of a protein that was able to activate the GTPase activity of Rho but not Ras, a 30 amino acid peptide was found to have homology with Bcr (Diekmann et al., 1991). The C-terminal domain of Bcr and of a related protein, n-chimerin, has been shown to act as GAPs on Rac. Later, a homolog of Bcr, Abr, was also shown to share homology with Dbl, and have GAP activity towards Rac (Heisterkamp et al., 1993). 25 Intriguingly, p190, a protein shown to interact with p120GAP of Ras, has been shown to function as a GAP for not only Rac, but also for Cdc42 and Rho as well (Settleman et al., 1992). This was the first direct indication of cross-regulation between members of the Ras subfamily and Rho subfamily members. While RhoGAP can also act as a GAP for Rac, Cdc42 and Rho in vitro (Lancaster et al., 1994), in vivo experiment show that p190 and, Bcr and RhoGAP have distinct specificities for GTPases (Ridley et al., 1993). p122-RhoGAP, a protein involved in the activation of phospholipase C-51, has also been found to share sequence similarity with Bcr, and has been shown to activate the GTPase activity of RhoA, but not RacI (Homma et al., 1995). A protein that binds to the C-terminal domain of focal adhesion kinase (FAK) has recently been shown to function as a GAP for Rho and Cdc42, but not Rac. Overexpression of this protein, Graf, greatly reduces the number of stress fibers, consistent with in vivo Rho GAP activity (Hildebrand et al., 1996). A search of proteins sharing homology with these proteins shown to have GAP activity to Rho-related proteins shows that the p85a regulatory subunit of PI3-kinase also shares homology with the domain implicated in GAP function, although GAP activity has not yet been demonstrated. 26 Figure 1-1. The basic GTPase cycle. Dissociation of GDP from the GTPase-GDP complex is accelerated by guanine nucleotide exchange factors (GEFs). transient empty state of the GTPase. GTP binds to the The intrinsic GTPase activity of the GTPase hydrolyses GTP to GDP, and GTPase activating proteins (GAP) greatly enhances this activity. 27 (4EF GDP GTP GAP Figure 1-2. Family tree of Ras-related proteins. Mammalian proteins are on the outer part, Drosophila,Aplysia, or plant proteins underneath, while yeast proteins, indicated in capital letters, and Dictyostelium proteins are closest to the center. Chardin (1993). 29 From -4' ar ), U, 0 O0 ~ N,b cr g 4 S> r -Y m .3 .0 " - 93 ar P ) 000 Chapter 2 Cloning and Molecular Analysis 31 INTRODUCTION The gene encoding Ral was first isolated from a simian B cell library using a synthetic oligonucleotide mix corresponding to amino acids 57 to 63 in Ras, a region implicated nucleotide binding (Chardin and Tavitian, 1986). It codes for a 206 amino acid protein that shares greater than 50% sequence identity with Ras. Like Ras, Ral can bind guanine nucleotides and possesses an intrinsic GTPase activity. Nuclear magnetic resonance studies have shown that Ras and Ral possess a similar three dimensional environment surrounding the bound nucleotide (Frech et al., 1990), indicating a similar structure. Moreover, mutant forms of Ral that contain mutations comparable to those activating Ras exhibit the identical, albeit less pronounced changes in function. Thus, Gly 1 2 to Val and Gln 6 1 to Leu mutations decrease the GTPase activity by factors of 10 and 2, respectively. A factor of 10 is usually observed in the corresponding mutants of Ras. Like Ras, the carboxyl terminal of Ral is isoprenylated. (Kinsella et al., 1991), but unlike Ras which is farnesylated, Ral is modified by 20-carbon isoprenyl groups, most likely a geranylgeranyl moiety. distribution as well. Ral and Ras differ in their subcellular While Ras is found exclusively associated with the inner surface of the plasma membrane, Ral has been found in association with endo- and exocytic vesicles along with the plasma membrane (Bielinski et al., 1993; Urano et al., 1996; Volknandt et al., 1993). Also, Ral contains an extra 11 amino acids on the N-terminus that do not have corresponding residues in Ras. 32 Ral transcripts are expressed in a wide variety of tissues, with the highest levels detected in testis and ovaries. Somewhat less was found in brain, adrenal and pituitary glands, kidney and ovary and the lowest levels in muscle tissue (Wildey et al., 1993) (Olofsson et al., 1988). The Ral proteins have been identified as being a major GTP binding protein in human platelets (Bhullar et al., 1990; Polakis et al., 1989) as well as being a major protein in the supernatant fraction of rabbit and bovine brains (Bhullar, 1992). A GAP activity for Ral has been demonstrated in cytosolic fractions of brain and testis (Emkey et al., 1991). The activity sediments between 150 and 443 kDa and fails to activate the GTPase activity in mutant forms of Ral that contain amino acid substitutions corresponding to those rendering Ras insensitive to interaction with its corresponding GAP. The Ral-GAP protein has yet to be identified. RalGDS, the guanine nucleotide exchange factor specific for only RalA and RalB, has been identified in this lab (Albright et al., 1993), and has been implicated as a possible effector for Ras (Hofer et al., 1994; Kikuchi et al., 1994; Spaargaren and Bischoff, 1994) through its association with activated ras in yeast two hybrid systems. Furthermore, a recent report suggests that activated H-Ras, but not R-Ras or Rapl, can induce RalGDS to stimulate Ral activation and that although RalA activation cannot induce oncogenic transformation independently it can enhance the transforming abilities of both HRas and Raf (Urano et al., 1996). Ral has recently been implicated in the activation of phospholipase D (PLD) (Jiang et al., 1996). 33 Although it is unable to activate PLD alone, Ral has been reported to constitutively associate with PLD through its N-terminal tail. In an attempt to elucidate the role of Ral in signaling, I set out to identify proteins that would interact with Ral in a GTP-specific manner. To this end, I set up a screen to search for proteins that were capable of physically interacting with Ral. In this chapter, I describe the cloning of a gene encoding such a protein and the preliminary characterization of this protein. RESULTS Isolation of the RIP1 clone. In order to detect proteins that were capable of physical interaction with Ral, a radiolabeled Ral protein was used as probe to screen a cDNA expression library. This method was chosen instead of the commonly used yeast two-hybrid system because the use of a protein probe made it possible to preload the probe with either GDP or GTP; this in turn made it possible to screen for interacting proteins that bound specifically to the GTP form of the Ral protein. The probe used in this screen was a modified version of human RalB. In order to facilitate radiolabelling of this protein in vitro, an arg-arg-asp-ser-val (K-domain) sequence, a substrate for cAMPdependent protein kinase, was fused to the N-terminus of Ral (Hateboer et al., 1995). In addition, a poly-histidine tag (Janknecht et al., 1991) was added to the C-terminus for purification purposes. The resulting chimeric protein, termed KH-Ral, was expressed in bacteria and purified over a nickel column (Hochuli et al., 1987). 34 The guanine nucleotide-binding property of the chimeric KH-Ral protein was found to be similar to that of previously purified, bacterially produced, human RalB (data not shown; Albright et al., 1993). I wished to confirm that the effector domain of KH-Ral also remained unaffected by the modifications undertaken to construct this probe. The effector domain was originally defined in the related Ras protein as the region in which mutations rendered oncogenic mutants of Ras no longer able to transform; at the same time, such mutations had no effect on the localization, GTPase activity or stability of the protein. Previously, we have found (B. Giddings and R. A Weinberg, unpublished) that the intactness of the corresponding region of the Ral protein, which we presume serves as the effector domain of Ral, was required in order for RalGDS to stimulate guanine nucleotide exchange on Ral. As a test of whether the effector region of KH-Ral remained functionally intact, nucleotide exchange reactions using purified RalGDS were conducted on this protein. Neither the presence of the extra N- and C-terminal residues sequence nor the phosphorylation of the K-domain caused a substantial difference in the ability of KH-Ral to be recognized by RalGDS, indicating that the presence of the K-domain and the His-tag do not alter the structure of the effector domain of Ral (data not shown). KH-Ral was used to screen a cDNA expression library generated from day 10 mouse embryo cells. 2 x 105 plaques were screened with the radiolabeled, GTPyS-loaded KH-Ral. were identified. Three positive plaques One of the positive plaques, termed 19-6, was purified and its DNA insert of 1331 bases pairs was sequenced on 35 both strands. This clone was named RIP1 (for Ral interacting protein). A 342 base pair EcoRI-PstI fragment subcloned from this cDNA clone was used as a DNA probe to screen the library for full length clones. Fourteen plaques that reacted with this probe were identified and divided into six groups by restriction mapping. Although none of the detected clones contained the entire sequence, overlapping sequences between them made it possible to deduce the full length sequence (Figure 2-1), which encompassed 3661 base pairs in which I found an open reading frame of 1944 bases encoding a protein of 648 amino acids. Approximately 3 kilobases including the open reading frame was sequenced on both strands, while the majority of the 3' untranslated region was sequenced only on one strand. Sequence analysis. Analysis of the imputed reading frame revealed a striking sequence similarity shared with other proteins known to possess GTPase activating (GAP) activity toward members of the Rho/Rac sub-family of GTPases. (Figure 2-2) 147 amino acids. The homology extended over No significant homologies were identified in the domains flanking the rho GAP homology domain. The C-terminal domain was found to be rich in glutamic acid residues while the Nterminal domain was rich in both glutamic acid and lysine residues. 36 Northern Analysis. I wished to determine the range of expression of the RIP1 gene in various tissues. To this end, I used a 342 base pair EcoRI-PstI fragment of RIP1 to probe two panels of Northern blots containing RNA from a total of 14 different human tissues. (Figure 2-3) Although the levels of expression varied substantially, a 4.1 kilobase transcript was evident in all of the tissues examined. The highest level of expression was observed in ovaries and skeletal muscle, whereas the lowest was found in spleen, liver and peripheral blood leukocytes. Interestingly, these expression patterns did not resemble those of the Ral mRNA previously reported for these various tissues (Olofsson et al., 1988; Wildey et al., 1993). Nucleotide specificity. To test whether the binding of Ral to the RIP1 protein was dependent on the specific guanine nucleotide bound by Ral, we tested the relative affinities of the GDP- and GTP-loaded forms of the KH-Ral probe for RIP1. Using the initially isolated phage, filters were prepared for the protein screen that contained induced 19-6 protein (19-6 protein refers to the fusion protein product expressed in phage 19-6). Each filter was then cut in half and incubated with radiolabeled KH-Ral that was preloaded with either GTPyS or GDP. As shown in Figure 2-4, 19-6 bound KH-Ral strongly when it was preloaded with GTPyS, while only background level binding was detected when the KH-Ral probe was preloaded with GDP. This indicated that the interaction between RIP1 and Ral was GTP- 37 dependent, and thus provided strong support for the notion that RIP1 functions as an effector of Ral. Interacting domain mapping. I wished to localize the region in RIP1 involved in its interaction with Ral. To do so, derivatives of the pEXloxl9-6 plasmid encoding fragments of RIP1 were generated. Two series of nested deletions were constructed, one beginning from the C-terminus, the other from an internal Eco47 III site. The truncated RIPI proteins encoded by these deleted forms of pEXloxl9-6 were then synthesized by in vitro translation and incubated with HA (hemagglutinin antigen)-tagged human RalB protein that had been preloaded with GTPyS. Anti-HA antibody was used to immunoprecipitate any Ral-19-6 complexes that formed. (Figure 2-5) It was clear that ability of RIP1 to bind Ral was critically dependent upon a domain that lay between 390 and 445. This segment is 34 amino acids C-terminal of the domain containing homology to Rho GAPs. I concluded that the observed binding was attributable to the interaction of Ral with a specific, well-defined domain of RIP1. DISCUSSION Almost a decade has past since the isolation of the gene encoding Ral was first described (Chardin et al., 1986). Although much work has been done in characterizing the biochemistry of the Ral protein and in cataloguing the various tissues in which it is 38 We expressed, the physiologic function of Ral has yet to be revealed. describe here an attempt to elucidate the function of Ral through the identification of a putative effector protein. A protein probe was used to screen directly a cDNA expression library for proteins that associated physically with Ral. A clone was identified encoding a protein, termed RIP1, that would interact with GTPyS-loaded Ral, which we presume to be the activated form of this protein. Importantly, the RIP1 protein clone would not interact with Ral when Ral was preloaded with GDP, underscoring the specificity of this interaction. Of interest is the fact that RIP1 shares substantial sequence similarity with proteins known to exhibit GAP activity toward Members of the Rho/Rac family proteins in the Rho/Rac subfamily. have been implicated in cytoskeletal regulation. Thus, a strong induction of stress fibers was demonstrated when activated Rho protein was microinjected into serum-starved Swiss 3T3 fibroblasts (Ridley et al., 1992). Rac activation, in contrast, leads to the formation of lamellipodia, while CDC42 leads to the formation of filopodia in the same system (Nobes and Hall, 1995; Ridley et al., 1992). RIP1 was found to be ubiquitously expressed and is highly conserved between species. Since the identification and cloning of RIP1 in mouse, two other groups reported the cloning of a protein that was capable of interacting with Ral from a Rat brain cDNA library (Cantor et al., 1995) and from a Jurkat cDNA library (JullienFlores et al., 1995). (Figure 2-6) Both of these group used the yeast two hybrid system to identify RalBP-1 and RLIP, respectively. 39 They were shown to bind activated Ral, but incapable of interacting with effector domain mutants of Ral. The amino acid sequence shows a 95.2 % identity between the mouse and rat proteins, with a single residue deletion and 23 of the 31 substitutions being conservative. Between the mouse and human proteins 92.3 % of the amino acid sequence is identical with 25 of the 51 substitutions being conserved. There is also a 7 amino acid insertion near the C-terminus. The Ral binding domain is absolutely conserved between mouse and human, and there is a single leucine to phenylalanine substitution between mouse and rat. Interestingly, in the 4 out of 128 acidic residues that are substituted, the substitution is to the other acidic residue. (99 out of 100 glutamic acid residues are identical.) Similarly, in the 6 out 110 basic amino acids that are substituted, 5 are conserved while the remaining arginine to tryptophan substitution observed between mouse and rat might be due to a single base pair error in sequencing. (The human protein also has an arginine at that position.) The triple KEEKH repeat, along with numerous other multi-K stretches, is absolutely conserved among the three species, suggesting an important structural or functional role for these repeats. The question remains as to whether the region in RIP1 that shares sequence similarity with other Rho GAPs is, indeed, capable of activating the GTPase activity of one or more of the Rho-related GTPases, and if so how RIP1 activity might be regulated. In the next chapter, I will describe GAP assays using purified RIP1 protein designed to answer this question. I also look into possible regulation of RIP1 through post-translational modification, as well as 40 intracellular localization of RIPI. 41 MATERIALS AND METHODS Preparation and labeling of KH-Ral. The human Ralb coding region was cloned into the KpnI-SacI site of pKH-Ela (Hateboer et al., 1995), replacing Ela with Ral resulting in the expression of Ral upstream of six histidine codons and downstream of arginine-arginine-aspartate-serine-valine, a cAMP-dependent protein kinase phosphorylation site. The plasmid was transformed into the BL21(DE3) strain of E. coli (Novagen). Following a 1 hour induction with 1 mM IPTG (Sigma) the bacteria were pelleted and resuspended in lysis buffer (50 mM TrisHCl pH 8.0, 20% sucrose, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM beta-mercaptoethanol, 0.2 mM sodium-metabisulfite) plus protease inhibitors (1 mM PMSF, 5 gg/ml aprotinin, 5 ptg/ml leupeptin) and sonicated. An equal volume of high salt buffer (50 mM TrisHC1 pH 8.0, 600 mM KC1, 10 mM beta-mercaptoethanol, 0.2 mM sodiummetabisulfite) was added to the sonicate and centrifuged. Ni-NTA resin (Qiagen) was added to the supernatant and washed with a 1:1 mix of lysis buffer and high salt buffer plus 1 mM imidazole (Sigma), then with PBS plus 16 mM imidazole. The protein was eluted in PBS plus 100 mM imidazole and dialyzed against PBS to remove the imidazole. The purified protein was labeled in vitro by combining 100 gCi [y- 3 2 P]ATP (ICN), 30 units cAMP-dependent protein kinase (Sigma), and 1-2 p g protein to a final volume of 30 gl in protein kinase buffer (20 mM Tris pH 7.5, 100 mM NaC1, 12 mM MgC12, 1 mM DTT). Following incubation at 40 C for 30 min the probe was purified over a 42 Sephadex G25 column prewashed with Tris/NaC1 (100 mM Tris pH 8.0, 120 mM NaCi) by eluting with Tris/NaCi. KH-Ral was then incubated in exchange buffer (50 mM Tris 7.5, 10 mM EDTA, 5 mM MgC12, 1 mg/ml BSA) with GTPyS (Sigma) for 20 min at 370 C. The reaction was stopped by adding MgC12 to a final concentration of 10 mM. Isolation of RIP] cDNA. A 10-day whole mouse embryo cDNA expression library (,EXlox, Novagen) was plated on BL21(DE3)pLysE (Novagen) at 104 plaques/ plate. A total of 2x10 5 plaques were plated. The plates were then incubated at 370 C until plaques are about 0.5 - 1 mm in size and then overlaid with nitrocellulose filters (Schleicher & Schuell) previously saturated in 10 mM IPTG and incubated for an additional 3-5 hours at 370 C. The filters were blocked at 40 C in HBB (25 mM HEPES-KOH pH 7.7, 25 mM NaC1, 50 mM MgC12, 5% milk) 410 hours and hybridized in Hyb 75 (20 mM HEPES pH 7.7, 75 mM KC1, 0.1 mM EDTA, 2.5 mM MgCl2, 0.05% NP-40) plus 1% dried nonfat milk with 2 - 7x10 5 cpm probe per ml of hybridization solution, overnight with rocking at 40 C. The filters were then washed in Hyb 75 and exposed at -80 0 C for 1 - 3 hrs. Phage from positive plaques were used to infect the BM25.8 strain of E. coli to allow the cre-lox mediated excision of the insert containing plasmid, pEXlox, from the ,EXlox vector. was done using a Sequenase kit (Amersham). sequencing was from Amersham DNA sequencing [3 5 S]dATP for Oligonucleotides were synthesized on a Applied Biosystem model 391 DNA synthesizer. 43 A 342 base pair EcoRI-PstI fragment was used as a DNA probe to rescreen the library for full length cDNAs. Mapping of the interacting domain. Nested deletions were created using Erase-a-base (Promega) and verified by sequencing. 1 jig of plasmid was used per 25 tl in vitro translation reaction (TNT coupled Reticulocyte Lysate System, Promega; [3 5 S]-methionine, NEN DuPont). 5 pl were diluted into 1 ml of ELB (150 mM NaC1, 0.1% NP-40, 50 mM Hepes, pH 7.4, 5 mM EDTA, 0.5 mM DTT, 1 mM PMSF) and incubated with HA(hemagglutinin antigen)-tagged human Ralb preloaded with GTPyS. The complexes were then immunoprecipitated with anti-HA antibody, washed with ELB, electophoresed on an SDS/10% polyacrylamide gel and exposed overnight at -70'C. Northern analysis. A 342 base pair EcoRI-PstI fragment was used to probe (Oligolabelling Kit, Pharmacia; [a- 32P]dCTP, NEN DuPont) Multiple Tissue Northern Blots (Clontech). ACKNOWLEDGEMENTS I would like to gratefully acknowledge Dr. Ren6 Bernard for the pKH-Ela plasmid and many helpful comments, and Dr. Andr6 Bernards for an aliquot of the XEXlox library used in this study. 44 Figure 2-1. Nucleotide and deduced amino acid sequence of RIP1. The Rho/Rac GAP homology domain is underlined. the Ral-binding region. The box indicates The stop codon is indicated by an asterisk. 45 ft P p,00p0ftPJo 0 (0Of-t (q0 ftrt seeo~~onoooe rt QcOt P a0prO p) o Q p) o ftO H r 0 Pir t f0 t t o 0 (0, ~oj~' o( 0 ft0ftP)p@fp Q 4 0C) >K ) r) ) PSOQPS t Oc O(D)O9 C C) 0 C) >) ) 0 40LQOGQcttOW R rt Q 0 0p Q00 P 0) (Qr P 0Q 4WR oLQO otp0 it0 0 rr 0 0 >CQp 0 C 0 E"ympgp 0Q mjP> O gggaaaoogggg po 0) (a t rr o ) trt 0 oP o(0 OPSQ LSP 0) ftO( p 0 rt 0000t ( 0 0OPO (0 ( 0 0 L ft 0ftOf (PQftDtt to Di(t 0 Pt 0 ft St0 Q0ft o t0 t O o O 00 (0)0)t 0t pif PO(OfOftO0 o H -3 > >0 0 m rt Ct (-) (t0 (0 0 0 Q 0Q ( o 0 > PS IQ 0C q 0rtt~lH 0 H7) - /0 - 0 > C) C) 0( 0 C 0) O C O 0) H 0q H -C >t C n 0 H CS >r C C) C) C) 8H C-3 0 a so 4 0 rt. ( t sft(p Lfo(OPOft oPp oPSa A ftf0P tftQPSP1OOOPJ0 Pftfto (0ftQ rt0ftfOPP r QC3 t( 0ftP(10(0f-r tOP(f OPft S w oO pn rt r ft 0ft W P0 J o o r00 0 0C) C)C ) 0 ( I(0 ft Poo 4 0 H ft, 0 ftr10n C) t Qf 0 P wt 1o rttOQrt 0 rt 0 w 00 rt Wt o C) C 0 C H C 0 Ct( ) C Hr ) ý3H 0 0 C) 0 H 0 pi L) 0f0ftO w tfQ o StQO (0((0((Q ftPS0t 4 c Pi (w ft(oftP 4 0 fttPPrc t(QL D( rtPS(0tP W (00r)o(P 0) S )o rttt rt.(t 0 ftQSrr PwP Q (0ft ftPSP 0 ft Q (tft 00 (0 Q 0 C Q (7) H 0C -3 C) 0C) > :CCH qH H0PC) (0fr P t pt(0ptP P ft ft 0 r rrtt(0 Pt ft w( OL ft(t 0 LI (t cr0f rft0( o 0 f( P rQ Q0i( (0)O fQ(0 0) rO t)L (Q (-r 0Pt o p0r-t Pr rS( 0 0o Pr rt rt 0 p)fQ (0 ft rI (0 0 0 C)O rjP (t ýi H C)H 0)ft Q fT(0(0Qt0(0tft0 ri (0QfO D)p)(0tft0 0 Q o ft(tpioftft ftf tP)ift(0 r 00 C) <HG) 0 HH r) 0 ct 0t 0 Q Q QrPtr( PL ftftt oP o ft(0 ( r t0 ct w 4 t to (-t Q 0w 0 f tftftftP( r o0ftO(fP 0) tp rrff0 0 rrQt(0)S0(fP( Q f tftQ (0ftP P) 0LQ (Q Qo (00 0G 0 C 0 000- P C) C) H C 0 C ) H C) 0 C) 0 0 C > -iQ (0 0(c PS(0 PS prt P0)S f )fftfp QtP 0 f(Q0 ( PS r p(t(0(0(0o rr SQ tftPW 0 PS rfQOPSOO)0(0Q0Ort (r-S o 0) Qtftftfto ft o0 o fPS LQ~t 0 PS OPQ(0Qf oofptf( (0Q ft < 0 0 CQ ) Q ft > 0 03 C) Q r0 0 ~ C)> Q C PS0 0t f O(Q0ftQ 0) ft 0) r H H C 4 ft p, Q wft(O frt srr (0 ftPSS0 rr0S 0 SP QftW fr 0 Qt ftft0fft Prftft(f(000t fta(0 t(0 ctP ftft tPS(0 ft ctpftofr tp((p 00, rOpprtp94 ft rrp,ofts p ftPaft(0ft pS(00(OPS w 0)mlýt)000m H Nw ti wwwwww )t~it &t)00 N ft P PS PS Ift pC) 0 09 tJ ) )t) P. & Otl) 0w C O Hr C) 0 G 60 t' n 0 H C) Hx~0 C) C) C 0 r C C) (C) C) C) 3 0H 0 > n0 > H a) toýd0 C) 0 3 0H 1- 0 C) H q t.4 > > > < P C C) 0 C)0 )n0C) C ) >1- 10 Hf C) nC) C( H (-) LO)C H H C) - q 0 - 0 >(0 C)0 ~0 0 Hq n( 0 C) >0Q C) 'i) t'r C) Q 0 (Q 00( 0C) 0 nOft n CnG) ) nC) gnP0 C) (LQ ~(Q 7) a C)3 C) H C 00 o n g C)3 ) p H a C) ) 0 0 )0 >Hp, n >( 0H 1- (00(0 0 CH <to rJ 0 C) > H z C 1n> C ýOt C) >00t 0 0 0q > C G) H 0C O H O C ý &1 tiC) >n (D C) C) 0Q q 0 C D 0(0 0 F-3 C) C) H > 08 0 0 C) a n rtq Qe Q nQ o HO H0 P 0g ýG) 0(0 0 0 (Q P(Q 0 QH0 o( q0 Qp q Q o o n 6t r a C a 10q C C)pC C) m m C< > C 0 C) 0 C) C)3 C) C) m0-C) oý HC(QO P a C) PS 0 D 0 C) n ) 0C)HO C 0@ 0 0 00 psg 0) 0 nC) 2 C H(n CP( C)> PO0 0 qS C) "r C) w S 0 ~(Q r) n 0- 0 H C) Fq H a Cg C) CH 0 0 >r) C) C pq H n0 H 0 0 0 i) > Q 13 C) m 0 0 r C)3 <) 3 ) r H 3 0 H ?0 > 1-H C) > H C) a C) C) 7) H F H H 00 ONMN O i mSoo ON 0t) ý3 >HC C)0a H aOPCp(0 0C r o qH r (0(0 C) HQ C) n 0(0 0 ; C0 ( C) n0 t o > u naH rT(Q a~< r):P HO n 5DC0 0 C) nP 000 C) 0 C) ) 0 nC CC <Hl H iC C) 0 C) C) HýPS C) C) > t-H 0 ~- 0 )L'H ~ C n q co-a <H0 coP ) 0 a n LC (00 >C E C) C) 0 C H qC)00 r 0 > C C) t2*) 7PS 00 > C) ýaCC H 0 C)n 0 C)HI H > > ) ) 0 C) C C) 3 n 0> ' C) 0 H H 0-3 r~ > > C > H 0 C)q ro) oC) C C) C ) 0 0 C H3 C) > 0 >) ~ q 0 En C (q >j (QC)0o 0 0w ( C )(0( a t -3 co<OF )a 0 ;1 ON 00 ON wO 1 ON) f > 0( C) 0 r) > C) M 009 C) 0 PS 0t C) H9 r C) C)c >s H Hl w i o C) > H qH x H F3 H Cý3 ý- Hr~t ) > )C3C) C) P 0t >1 (7 C C) H 0 (7) 0 C) q 0 He n t C) sC) > 4 S4 7 S0 00 H C) C C)3 C) r C0 0 0r w ; LtJ O t) 0 C a HC)3 g(7) Be" <g<ag 13 a- 0a > 1C) c C)(S yc 0C pi P (7) 0 C) C) 0 >7) 0 0 >H n r 0H 0 3 >HH C) 0 Q nO g 0 (0 C) (0 Q 0q 0 0) QfQt> >H n X C C)ýg a C)-3)C n G) H PSP ft(Q( Qr 0 St ftft0(Q S(0 (0(0t0 PSP)Jr pSo(o (T0(0(0SQfP) Q(0P 0)tf t (p0(0) (0OfrLQto t 0 n H ) S H C) H <H tzi S0 0C) C) Cn)H 0 H C)ZC C) CK) C)) S 0 l H 0lF3 C) C) > 0 00 Q C) C C L, H C) 0 - C) H HtC) ýd H 0 0 C) H 0 t C C > n n C)e r) Q 0 0 p-PD (t 0 C) ft C) (0 0 C) ftQ N C) 0 r) ft C) 0 C C) H0 H C) C) C) > C) 0 0 0 C) 0 H 2) 0tftftpft 0fQ P)O(00(0ftPs(0ft(0 QO Q(0OPS0 D)ft0 ftP (0fD)tOL rQ0t P P rPrtft rft(0t((00rt (0(0O SP 0 2)0 w O0) Q tp 0OOPS ftPSPQ ftoftw(0 0PS (PQQ (00 rt ftPW(P 0 0 (0OQ 0t(P4(0(Q ftft 0) 2)0)Q0 rt rf rt Q rt 0f 0 Q i Pt0 rt 0 D) (0f t rt OtQ tft)(Q( rt rr (q rr (0 0) ) 8 t:O C) n > S H 0 C) C) )0 H i 0C c it ( C >0() td H S 0 -0 Q OftO Hn ~1- 0 -3 H (() H ) r) - 0 C) 0H 0 H C0 C) 0 0 0 0 0 PS aC C) 0 0 0 >ag f P) 0 rt f0PQ > r) (r Q0 W f rP 0 0a Om0G Q Q rQ orrr(Qft0LQ )t ) z0 C) 0Pr Q S(0 o 0 0 0 LQ C rr P) 000tPSJ0) WOt 0S0P P) t O P) (Q 00a >rG Z CQ pC 0(7) rtrt ftrt p) Q tQ rri(Q 0> ti C>) Qtr) H C) 8 0 (Q 0t ft rt rr rtrn 0 0 00" 00 0 10 8 0ti 8>t HHQ I Lrt0 0r L rt nLQ 0 rt Pr rrt o f t wtf 0P rt ) >a·1~ SS o> a C) H HC ) C) a ()3 C) C ra XPý1 K)1 'aj~C t H -s 0a, C) H C C) C C H S 0 0 C) S p PS > Mo > > C W0 agag0 0 H H1P t-'Hý ) C) 18 - (0 00(00P0) LQ 0 0t0)C) crf() 0) f O)PJ)ftt 0 00Q 0 w 000 QQP pltasarcarctpSr pW rt(00 ft t r(0 PW ft 0 PH 0 04 0(0(00 (tOO0)OOOiO00) 0 W) 0 0500 QPs rt rt0 o g(Q0)f(Q rt (Q(QLQrt LO0 2) t 0 Ct 0 Pc-t'ftOO0 p,Ofr fr(-tOPJPJ(0OQ t 1)0) rO OLcrt( W D0 P 0() rtf 0 Qt 9OOt o0 t00H0 0f 0Q Pi) r,0fr portt QO o 0 Q 0)0 Q ((000f00ft Q t 0 )00P o rtOPr o) 0)rt CS QC) cao (a) 'i Ot0w co & 0t)O' H w co OS)& S ON )N) O.J ) ON 0 0 ON) Dlý ON) )00 ONN t) N) Figure 2-2. Amino acid sequence comparison of proteins containing Rho/Rac GAP activity. The sequence of RIP1 is compared to the sequence of n-Chimerin (Hall et al., 1990), Bcr (Heisterkamp et al., 1985), p190 (Settleman et al., 1992), Myr 5 (Reinhard et al., 1995), Graf (Hildebrand et al., 1996), RhoGAP (Lancaster et al., 1994), 3BP-1 (Cicchetti et al., 1995), p122-RhoGAP (Homma et al. 1995), and p85 alpha subunit of PI3-K (Otsu et al., 1991). Identical residues are indicated with black boxes. Amino acids present in 7/9 sequences are indicated in the consensus sequence (amino acids present in all sequences are indicated in bold), while conservative substitutions present in 8/9 sequences are indicated in the consensus sequence as a (acidic residues: D, E), b (basic residues: K, R, H), f (hydrophobic residues: A, F, L, M, P, V, W), and p (polar residues: C, G, N, Q, S, T, Y). 47 DA ER MMYDGVR------ cMEV - ----- SR R S --------- - - - TT - - - - P :K - - - - - - - - - KNPEQE-------DKA- ---------S IGV F SI KDNQSEGTAQLDS S Q F A PP -- -- ----D .. GV . f .. . .. . . . - RhoGAP PL AI Myr 5 gH VL Mgg HAL L L QVCS-T AO MM QQAM L R N RS QFAPP --------- INAgPL LI - - - - - A L QvCS-IT KK YNMGL --- DFDQYN--E QTNPA--T K D AE--MDPKTATETET MA D -- f YR . S G . P . L. ..... Graf 3BP-1 Y NY GQS --p122 p85 alpha . . . . . . . - . f . . fA . fL K . Y Consensus T -S . . . . . . C GKTT IA S IN IN - LNI- -I VE RhoGAP Myr S ICAEWE PH - - -Sc- NEC E D S- G ID- - - - - - - iPE QS S ....... MQ Myr 5 Graf 3BP-1 p122 p85 alpha - EN -N . fp .. f f RhoGAP - PE - K EPY Q Y PLfT. RIP1 Bcr n-Chim p19 0 - S fRaLP. - L P . . N Consensus RIP1 Bcr n-Chim pl90 RhoGAP Myr 5 Graf 3BP-1 p122 p85 alpha WV C L AIASIF H L N T L K MLNARVMSEIESEMEFEFSAM .. . L . . L f . H L . . V .. .... Consensus RIP1 Bcr n-Chim p190 I VN Y D - T --LAEKD GEKA HN-L Graf 3BP-1 p122 p85 alpha P . ff . . . f . . fE . . G f . . . -G S P --DY PN VMEM -S RE NK N V F F R FEQ RIP1 Bcr n-Chim p190 N .Mp . . N f . . . F . PpL f ...... S Consensus Figure 2-3. Tissue distribution of RIP1 transcript. A 342 base pair EcoRI-PstI fragment was used to probe multiple tissue Northern blots. 2 micrograms of poly-(A) selected RNA from the indicated tissue was run in each lane. in kilobases. 49 The number indicates size 60 eb; $ 2!5&; e %•O 4.1 -- CPSr 4.1 - Figure 2-4. Preferential binding of 19-6 to KH-Ral preloaded with GTP vs. GDP. Filters were prepared as described in Material and Methods and cut in half. The left half was hybridized to KH-Ral probe preloaded with GDP, the right half was hybridized to KH-Ral probe preloaded with GTPyS. 17 is a negative control. 51 19-6 GDP : GTP GDP GTP Figure 2-5. Mapping of the Ral-binding region. Two series of deletions were constructed and translated in vitro. The translates were incubated with HA-tagged human RalB preloaded GTPyS and immunoprecipitated with anti-HA antibody. indicate the position of the deletion. gene 10 fusion protein of pEXlox. 53 The numbers The hashed bar indicates the T7 + + I I + 4 + w ND co Sco w I 0..J 0D 0 U 0 I- C13 • * I C0 .o I + 0CAD u + rl U l .o Figure 2-6. Amino acid sequence comparison of RIP1 homologs. The sequence of RIP1 is compared to RalBP-1 (Cantor et al., 1995), a rat homolog, and RLIP (Jullien-Flores et al., 1995), a human homolog. Conserved residues are indicated with black boxes. The GAP homology domain and the Ral-interacting domain (RID) are indicated. 55 ripi raflhp nip nipi ralbpl nlip nipi ralbpl nlip ripi rallhpl1 nlip F ripi rJlip I -_ a . i- a nipi raflbp1 nlip GAP domain I| ripi ralbpl nlip m................. ml II ii i I II I I III I I .. •. I nipi ralbhrplnlip L F [ I Kl N10 i : nipi ralbpl nlip ~-I RID domain ripi ralbpl nlip ripi flip F nipi EL I I L E D R MIý7E1,E'TK 1ýMH 1 C)6 I AV l,"1 ElA 1 11I'lI"? Q raftpl 0 Am H17ER LAI I E!L RV0 L R1,L 0 M RAKýnlip, _ID _E1 WRGu E. L R A E 1AVQ L a~aer~ PKAGKEP R ripi railpi nlip nipi ralbpl nlip Chapter 3. Analysis of RIP1 Protein 57 INTRODUCTION In the previous chapter, I described the cloning of the RIP1 gene along with initial analysis of its expression in different tissues and the guanine nucleotide specificity of the Ral-RIP1 interaction. Analysis of the amino acid sequence predicted a region in RIP1 that likely possesses GAP activity toward members of the Rho subfamily of GTPases. An activity that was capable of activating GTPase activity of Rho but not of Ras was attributed to a 29 kD protein (Garrett et al., 1991), that was subsequently purified and sequenced. Analysis of the sequence indicated that it contained a domain that was 50% identical to one in Bcr and n-chimerin (Diekmann et al., 1991), both of which were shown to be GTPase activating proteins (GAPs) for Rac but not Rho. Since then, other proteins have been identified that also share homology with this region. They include the p190 (Settleman et al., 1992) that was identified as a protein capable of binding to the pl20GAP; Abr (Heisterkamp et al., 1993), a homolog of Bcr, that, like Bcr, also contains a Dbl-like domain; 3BP-1 (Cicchetti et al., 1995), an SH3 domain containing protein capable of binding the Abl protein; Myr 5, a novel type of myosin (Reinhard et al., 1995); p122-RhoGAP, a regulatory protein of phospholipase C-81 (Homma et al., 1995); Graf, a protein that associates with p125FAK (Hildebrand et al., 1996); p85a, the regulatory subunit of PI3K (Otsu et al., 1991). All but p85a has been shown to possess GAP activity toward one or more of the Rho subfamily members. While Rho-GAP and p190 exhibit activity toward Rho, Rac and Cdc42, Bcr and Abr 58 are only active toward Rac and Cdc42 as is N-chimerin, while 3BP-1 is active only toward Rac. The key function for Ras-related GTPases is to transduce upstream signals through their respective downstream effectors to elicit a cellular response. A number of proteins have been identified whose enzymatic activity has been shown to be modified as a result of their interaction with their respective GTPase. In most cases, this modification is due to covalent modification of the effector protein, usually phosphorylation. For example, following translocation to the membrane as a result of Ras activation, Raf-1 becomes phosphorylated and it becomes catalytically activated as a kinase. The same is true for p65PAK, a serine/threonine kinase that gets activated through phosphorylation as a result of its interaction with Rac or Cdc42 (Manser et al., 1994). More recently, protein kinase N (PKN) has been shown to autophosphorylate and thereby activate itself as a result of Rho interaction (Watanabe et al., 1996). In this chapter, I will describe studies looking at possible covalent modification of RIP1 as a result of Ral activation, analogous to those found in other GTPase-effector interactions. I will also describe Ral-RIP1 interaction in a transient transfection system. I will then describe results from GAP assays using RIP1 to ascertain whether the Rho GAP homology domain found within RIP1 is catalytically active. Lastly, I will describe immunofluorescence studies examining intracellular localization of RIP1 protein. 59 RESULTS Expression of RIP1. To examine the protein expression of RIP1, the C-terminal portion (see Material and Methods) of RIP1 was expressed in an E. coli expression system and then purified; the resulting protein was used to generate anti-RIP1 antibodies in 2rabbits. The resulting unfractionated polyclonal sera failed to detect specific protein species in both immunoprecipitation and Western blot experiments. However, affinity purification against full length RIP1 protein yielded an antibody which, when used as a probe in Western blot analysis, detected a 110 kilodalton species in lysates of Balb/3T3 cells. (Figure 3-1) We compared this size to that seen following direct expression of the cloned RIP1 gene. Introduction of the RIP1 coding segment into the pGEX-4T plasmid resulted in expression of a protein composed of RIP1 fused to glutathione S-transferase; this fusion protein could subsequently be cleaved with thrombin, releasing full length RIP1 protein. As an alternative, a segment containing the RIP reading frame was introduced into the pVL1393 baculovirus vector which allows expression in transfected insect cells. While the amino acid sequence predicted a 75 kilodalton protein, a protein of approximately 110 kilodaltons was observed when bacterially produced protein was cleaved with thrombin. A protein of similar size was observed in the insect cells transfected with pVL1393. These sizes were virtually identical to those seen upon Western blotting of Balb/3T3 lysates, indicating that the cloned 60 cDNA contains the entire reading frame of RIP1. The slight size discrepancy between the bacterially produced protein and the protein detected in Balb/3T3 lysates might be due to cleavage of RIP1 by thrombin at a potential cleavage site near the C-terminus or to the lack of proper post-translational modification. (Figure 3-1) Subsequently, the full length protein produced in insect cells was also used to generate antiserum. The polyclonal antiserum generated from a rabbit injected with the full length protein was able to detect endogenous RIP1 protein by Western blot and ectopically RIP1 by immunoprecipitation. Because the endogenous RIP1 in Balb/3T3 cells could not be detected by immunoprecipitation, the metabolic stability of RIP1 could not be determined by pulse labeling. For that reason, cells were treated with cycloheximide to inhibit protein synthesis and the subsequent degradation of RIP1 was followed by Western blotting. (Figure 3-2) The metabolic stability of proteins following cycloheximide treatment has been shown to mimic the results obtained from pulse labeling in a number of cases (Hasegawa et al., 1995; Seely et al., 1982). RIP1 was found to be relative stable since no decrease in protein levels were detected within 10 hours of cycloheximide treatment. Covalent Modification of RIP1. When endogenous RIP1 was examined, multiple bands could be detected by Western blot, indicating that RIP1 might undergo posttranslational modification or alternative forms of RIP1 might be synthesized within the cell. To examine this further, RIP1 was 61 immunoprecipitated from COS-1 cells transiently transfected with an expression vector designed to overexpress wild type mouse RIP1. The protein was then treated with alkaline phosphatase. While the immunoprecipitated RIP1 not treated with alkaline phosphatase retains the modified forms, treatment with this enzyme resulted in the multiple bands being reduced to a single band, indicating that the multiple bands were due to post-translational phosphorylation of RIP1. (Figure 3-3) To examine further the nature of the phosphorylation, the RIP1 immunoprecipitated from COS-1 cells was resolved on a SDS-PAGE gel, transferred to nitrocellulose, and probed with an antiphosphotyrosine antibody. Although an EGF-treated control lysate demonstrated that the antibody could indeed recognize phosphotyrosine residues present in EGF-receptor as a result of EGF treatment, this antibody did not recognize RIP1. This is a strong indication that the phosphorylation of RIP1 in likely due to a serine/threonine kinase, although definitive proof can only be obtained through phosphoamino acid analysis. Previous work on RalGDS, an exchange factor specific for the RalA and RalB GTPases, demonstrated that the protein levels of the exchange factor changed dramatically in response to serum starvation/stimulation. To examine whether the modification of RIP1 could also respond to serum starvation/stimulation, Balb/3T3 cells were serum-starved for 60 hours and then stimulated with 10% calf serum. (Figure 3-4) Under conditions of serum starvation, the modifications of RIP1 gradually disappeared, resulting in a single 62 RIP1 band in Western blots. Following serum stimulation, although the first indication of protein phosphorylation became evident within 1 hour, the full range of phosphorylation was not achieved until 24 hours of serum stimulation. 63 Ral-RIP1 interaction. One of the difficulties in studying signal transduction through Ral is the inability to gauge whether Ral is activated hence identify signals that can activate Ral. In the case of Ras, the activation of Ras can be examined directly by looking at the guanine nucleotide bound to Ras following immunoprecipitation thereby allowing the factors capable of activating Ras. The unavailability of anti-Ral antibodies capable of immunoprecipitating Ral made it difficult to ascertain conditions in which Ral is activated. In light of the fact that the interaction of RIP1 and Ral is dependent on the activation of Ral (Fig 2-4) , this provides an indirect method of gauging Ral activation. The COS-1 transient transfections applied previously were used to this end. Cos-1 cells were co-transfected with the vector that expressed the full length wild type RIP1 along with a vector that expressed the wild type RalB protein with the Myc epitope fused to its N-terminus. Following transfection, the cells were starved in 0.2% calf serum for 24 hours and then stimulated with serum for 5, 15, and 30 minutes. The cells were lysed, the Ral protein immunoprecipitated using the 9E10 anti-Myc monoclonal antibody, and the immunoprecipitates were resolved on an SDS-PAGE gel. This gel was then transferred to a nitrocellulose filter and probed with anti-RIP1 antiserum. Under conditions in which Ral is activated and in its GTP-bound state, I expected to see RIP1 co-immunoprecipitate with Ral. indeed the case, as shown in Figure 3-5a. This was As early as five minutes following serum stimulation, RIP1 could be seen coimmunoprecipitating with Ral while none could be detected in 64 serum-starved cells. Interestingly, the level of co- immunoprecipitating RIP1 seems to decline with time suggesting that Ral activation is transient and subject to downregulation and/or RIP1 is modified such that it is no longer able to interact with Ral. To test whether the inability of RIP1 in serum-starved cells to bind to Ral was due to a modification of RIP1 in those cells rather than a change in Ral, the Ral-binding ability of RIP1 present in serum-starved lysate to that of RIP1 from serum-stimulated were compared. lysate As shown in Figure 3-5b, GTPyS-bound Ral was able to interact with RIP1 from either lysate equally well, indicating that the interaction between Ral and RIP1 observed in serum-stimulated cells was most likely due to a change in Ral rather than RIP1, specifically the activation of Ral through guanine nucleotide exchange. GAP assays. To ascertain whether the Rho/Rac GAP homology domain found in RIP1 was indeed capable of activating the GTPase activity of the Rho, Rac, or related proteins, full length, recombinant RIP1 protein was used in GAP assays with either Racl, CDC42Hs, RhoA, c-H-Ras, or RalB. The assay consists of loading the GTPase with [y32 P]GTP and incubation in the presence of the protein being studied. The reaction is then stopped by the addition of stop buffer, filtered through a nitrocellulose filter to retain the GTPase and any associated nucleotide, and then washed to remove non-specific radioactivity. An activation of the GTPase activity will result in the loss of the yphosphate hence a decrease in the amount of radioactivity retained. 65 Initially 20 ng of each GTPase were incubated in the presence of 0.5 gxg of purified RIP1 protein (a molar ratio of approximately 1 to 10) for 10 minutes at 25 0 C. RIP1 succeeded in stimulating the GTPase activity of Racl and CDC42. Under these conditions, 22% more of the GTP was hydrolyzed to GDP by Racl in the presence of RIP1, while as much as 35% was hydrolyzed by CDC42 in the presence of RIP1. However, no increase in GTPase activity could be detected when Ras or Ral were used in this assay, and very little, if any, could be detected when RhoA was used. (Figure 3-6) While Ral interacts with RIP1 in a GTP-dependent manner, as shown above, this interaction had no effect on the intrinsic GTPase activity of Ral. This indicates that although RIP1 can interact with Ral, the interaction is such that it does not affect on the GTPase activity of Ral, rather RIP1 is able to activate the GTPase activity of Cdc42 and Racl. This correlates nicely with the mapping of the Ral-binding domain of RIP1 since the GAP homology region and Ral-binding region does not overlap. To test whether the effects observed were indeed an activation of specific GTPase activity and not due to the simple dissociation of the nucleotide from the GTPase or due to protease activity, these assays were repeated using [a- 32 P]GTP. Since the a-phosphate is not lost as a result of GTPase activation, a loss of radioactivity would indicate that the nucleotide was not retained on the filter due to either dissociation of the nucleotide from the protein or its degradation. As illustrated in Figure 3-7, no radioactivity was lost when the GTPases were loaded with [a- 32 P]GTP. To exaggerate any effect RIP1 might have on the GTPases, the amount of purified RIP1 66 used in these assays were increased to 5 tg while the GTPase was decreased to 10 ng per reaction giving a Ral to RIP1 molar ratio of approximately 1 to 200. I also tested whether the binding of GTP-activated Ral to RIP1 affected the GAP activity of RIP1 toward Racl, Cdc42Hs or RhoA. To do this, RalB preloaded with either GTPyS or GDP was added to these GAP assays. Neither of these forms of Ral had an observable effect on the GAP activity of RIP1 toward these various GTPases (data not shown). Therefore, although Ral has the ability to bind RIP1 in a GTP-dependent manner, that interaction itself does not have a regulatory role for the GAP domain. Immunofluorescence. To study the intracellular localization and possible translocation of RIP1 as a result of Ral activation immunofluorescence microscopy was performed using anti-RIP antibody. Initially, polyclonal rabbit antiserum generated against the full length RIP1 protein was used. Several different fixation methods were tested, including the organic solvents methanol or acetone and formaldehyde or glutaraldehyde fixation followed by permeablization by detergent or organic solvent. The most striking pattern was a punctate pattern seen most convincingly following fixation by organic solvents although still visible when cells were fixed with formaldehyde. (Figure 3-8a) Glutaraldehyde fixation was problematic since the fixation process itself generated a fluorescence that was difficult to eliminate consistently. 67 Interestingly, following formaldehyde fixation, another pattern is also discernible. Aside from the punctate pattern observed when organic solvents were used to fix cells, a pattern reminiscent of vinculin or focal adhesion plaques was observed. I recognized that these patterns of staining might be due to non-specific crossreactivity of the polyclonal antiserum used in these experiments. Therefore, in an attempt to determine which, if any, of the observed patterns were indeed due to RIPl-specific staining, a blocking strategy was attempted. Thus, the polyclonal serum was preincubated with purified RIP1 protein prior to staining in an attempt to block the antiserum from recognizing RIP1-specific epitopes while allowing it to still recognize nonspecific proteins. As a result, the pattern reminiscent of vinculin staining disappeared while the punctate pattern remained. (Figure 3-8b) This suggested that the punctate pattern observed was likely due to non-specific staining. To ascertain whether the focal adhesion plaque suggestive staining pattern was indeed RIPl-specific, purified RIP1 protein was coupled to Affigel-15 (Bio-Rad) and used to affinity purify RIP1specific antibodies. The results of the immunofluorescence using the affinity purified antibody corroborated the blocking results in that, although the punctate pattern was no longer observed, the pattern reminiscent of vinculin staining remained. (Figure 3-8c) To test whether the pattern observed actually coincided with vinculin staining, a mouse monoclonal antibody against the human vinculin protein was used to do a double staining with the RIP1 specific antibody. As shown in Figure 3-9, the major staining 68 observed using the affinity purified did indeed co-localize with vinculin staining, indicating that RIP1 was present in focal adhesion plaques. To test whether RIP1 undergoes a translocation as a result of Ral activation, the staining patterns of serum-starved and The bulk of the staining pattern restimulated cells were compared. remained constant, indicating that RIP1 did not undergo a drastic translocalization as a result of Ral activation. DISCUSSION When antibodies generated against the full length RIP1 was used for Western analysis, RIP was found to be a protein running at about 110 kD on an SDS-PAGE gel. Using lower percent acrylamide gels (6% to 7.5%), the RIP1 band could be resolved as multiple bands, indicating that either the RIP1 protein might undergo covalent modification resulting in proteins with altered mobility or that the antibody generated against the full length RIP1 protein might be recognizing possible RIPl-related proteins. Upon further analysis, the multiple bands were found to be attributable to phosphorylation, as RIP1 immunoprecipitated and treated with calf alkaline phosphatase resulted in the multiple bands collapsing into a single lower molecular weight band. Interestingly, the single lower molecular weight band resulting from phosphatase treatment of RIP1 is slightly faster in migration than the RIP1 immunoprecipitated from cells, indicating that all RIP1 probably undergoes a basal 69 phosphorylation and the multiple bands observed in Western blots were the result of additional phosphorylation events. This would nicely explain the discrepancy observed between the bacterially produced protein and endogenous RIP1 found in mammalian cells, in that the basal phosphorylation seen in the latter might be dependent on kinases not present in E. coli. In Balb/3T3 cells, I could resolve three distinct forms of RIP1 upon acrylamide gel electrophoresis of lysate prepared from exponentially growing cells. Following serum starvation in 0.2% calf serum, the two upper bands disappeared while the lowest band remained. Within 1 hour of serum stimulation, an extra RIP1 band appeared, indicating a phosphorylation of RIP1 in response to serum stimulation. 24 hours. A second phosphorylated band became apparent after In the transient transfections where Cos-1 cells received expression vectors for RIP1 and myc-tagged Ral, a Ral-RIP coimmunoprecipitating complex was detectable within five minutes of serum stimulation following 24 hours of serum starvation, indicating that a component within calf serum was capable of activating Ral, allowing it to form a complex with RIP. An attractive mechanistic model of RIP1 action would be one in which, in analogy to the behavior of Ras, Ral serves to recruit RIP1 to the membrane where RIP1 gets activated. As a result of its interaction with Ral, RIP1 gets phosphorylated, possibly activating the catalytic activity of RIP1. While in Figure 3-3 very little, if any, phosphorylated RIP1 is detectable by Western after five minutes of serum stimulation, it may be enough to function properly. 70 Obviously, a more detailed study of RIP1 phosphorylation within one hour after serum stimulation is required. Of course, it is also possible that phosphorylation of RIP1 has nothing to do with its interaction with Ral and is the result of an independent regulatory mechanism, and Ral serves to recruit RIP1 to a compartment in which it can function, or alternatively, to sequester RIP1, removing it from a compartment in which it serves as a GAP toward proteins like CDC42 or Rac. When recombinant RIP1 protein was purified and used in GAP assays, it was shown that this protein could activate the GTPase activity of CDC42 and, to a lesser extent, Racl but not that of RhoA, Ral, or Ras. While RIP1 interacts specifically with the GTP-bound form of Ral, this binding had no effect on the GTPase activity of Ral. Moreover, when the Ral-binding domain of RIP1 was mapped, it was found to be in a region C-terminal to, and not overlapping with, the GTPase activating domain of RIP1. serve as a regulator of Ral. This suggests that RIP1 does not Rather, this indicates that RIP1 is the target of activated Ral and thus serves as an effector of Ral. The degree of GAP activity of RIP1 toward CDC42 and Racl is much lower than has been associated with other previously identified Rho/Rac GAP-containing proteins. This might be explained by the fact that the RIP1 protein may be labile, resulting in its substantial inactivation in vitro. Although several independent bacterial and insect cell preparations exhibited the same low specific GAP activity, it is possible nevertheless that one of the steps used in all of the purifications caused the inactivation of much of the protein. 71 Alternatively, it is possible that the RIP1 protein requires functional activation, either through covalent modification or the presence of an associated, distinct regulatory protein. Finally, it is possible that the bona fide target of RIP1 GAP activity has not yet been identified. I considered the possibility that Ral might function as an allosteric regulator of RIP1, such that the binding of activated Ral to RIP1 would modulate the GAP activity of RIP1 toward its target GTPases. To test this possibility, GAP assays were done in the presence of excess GTPyS or GDP-loaded Ral. No modulation of RIP1 GAP activity toward RhoA, Racl or CDC42 was observed. This failure to observe a modulation of RIP1 GAP activity in vitro is not definitive. Immunofluorescence studies indicate that a substantial fraction of RIP1 is present in focal adhesion complexes. This was demonstrated by using antibodies affinity-purified against RIP1 which showed that the observed staining pattern overlapped with that of the anti-vinculin antibody. Vinculin is a component of, and is found almost exclusively in, focal adhesion complexes. This is provocative in that one of the effects of Rac or Cdc42 activation is the formation of such complexes. Focal adhesions are sites through which fibroblasts adhere to the extracellular matrix and are the sites of integrin-mediated signaling. Thus, RIP1 might function in the regulation of signals through these complexes by regulating Rac or Cdc42. Interestingly, other regulatory proteins for Rho-related GTPases have been found in focal adhesion plaques. 72 Graf is a recently identified member of the Rho GAP family that has GTPase activating activity toward Rho and Cdc42. Graf been shown to bind the C-terminal domain of FAK, a protein tyrosine kinase associated with focal adhesions (Hildebrand et al., 1996). Lar is a transmembrane phosphotyrosine phosphatase that co-localizes at the ends of focal adhesion (Serra-Pages et al., 1995). Trio, a recently identified protein that associates with LAR, has been shown to contain two Dbl-like domains, one of which has been shown to have guanine nucleotide exchange activity specific for Rac, the other for Rho (Debant et al., 1996). This indicates that Rho-related GTPases play a major role in integrin-mediated signaling and that RIP1 probably functions in the regulation such signaling. 73 MATERIALS AND METHODS Expression of recombinant RIP1. The coding region of RIP1 was cloned into pGEX-4T (Pharmacia). Transformed bacteria were induced with 0.1 mM IPTG for 4 hours, centrifuged and washed. The pellet was resuspended in STE (10 mM Tris, pH 8.0, 150 mM NaC1, 1 mM EDTA) containing 100 tg/ml of lysozyme and incubated on ice. DTT was added to final concentration of 5 mM. The bacteria were sonicated and centrifuged. Triton X-100 was added to the supernatant to a final concentration of 2%. Glutathione Sepharose 4B (Pharmacia) beads were added and incubated at 40 C for 30 minutes. The beads were washed in PBS. RIP1 was cleaved off by adding thrombin in PBS, incubating overnight. Alternatively, the coding region of RIP1 was also cloned into pVL1393 (Pharmingen). A high titer baculoviral stock was prepared by using the BaculoGold System (Pharmingen) in Sf9 insect cells and used to infect High Five insect cells (Invitrogen). The cells were collected, washed with PBS and resuspended in buffer A (10 mM Tris, pH 7.5, 1.5 mM MgC12, 10 mM KC1, 5 mM P-mercaptoethanol, 1 mM EDTA) + protease inhibitors. The cells were lysed in a Dounce homogenizer and centrifuged to pellet nuclei. 0.11 volumes of buffer B (100 mM Tris, pH 7.5, 1,500 mM NaC1, 15 mM MgCl2, 100 mM KC1, 50 mM P-mercaptoethanol, 10 mM EDTA, 1% Triton X-100) was added to the supernatant. Following a second centrifugation to pellet cellular debris, Ni-NTA resin (Qiagen) was added to the supernatant and incubated for 1 hour. 74 The beads were then washed with 0.1x buffer B + 1 mM imidazole, loaded onto a column and washed with buffer B-(0.1 x buffer B minus Triton X-100) plus 16 mM imidazole and 10% glycerol. The protein was eluted in a 10 ml gradient of 16 - 150 mM imidazole in buffer B-. Positive fractions were dialyzed against buffer B- to remove the imidazole. Antibody Preparation. The EcoRI-HindIII insert from pEXloxl9-6 was cloned into pET29a (Novagen). Protein was expressed, purified (S-Tag Purification System, Novagen) and used to immunize rabbits. The resulting antiserum was affinity purified against full length GST-RIP1 protein by binding the protein to Glutathione Sepharose 4B (Pharmacia) and washing with 0.1 M borate buffer (pH 8.0), 0.2 M triethanolamine (pH 8.2). The protein was cross-linked to the column by incubation in 40 mM dimethylpimelimidate (DMP) in 0.2 M triethanolamine. The column was then washed in 40 mM ethanolamine (pH 8.2), 0.1 M borate (pH 8.0). The antiserum was passed over the column. Following washes in PBS and PBS + 2 M KCl the antibody was eluted in 5 M Nal and 1 mM sodium thiosulfate in PBS. For affinity purification of antibody using Affi-gel 15 beads (Bio-Rad), 10 micrograms of RIP1 protein purified from insect cells infected with the RIP1 expressing virus described above was crosslinked to 2 ml of Affi-Gel 15 beads according to the protocol supplied by the manufacturer. passed over the beads. Five milliliters of anti-RIP1 antisera was Following washes in PBS the antibody was eluted in 0.2 M glycine, pH 2.5 and neutralized with 1/10 volumes of 75 1 M Tris, pH 8.0. The affinity purified antibody was then dialyzed against PBS and concentrated. Western analysis. Lysates from Balb/3T3 cells and purified RIP1 protein were run on a SDS/7.5% acrylamide gel and transferred to nitrocellulose. The filter was blocked in PBS plus 10% dried non-fat milk for 1 hour, incubated with affinity purified anti-RIP1 antibody in PBST (PBS plus 0.1% Tween-20) for 1 hour, washed with PBST and incubated with horseradish peroxidase conjugated donkey anti-rabbit IgG antibody (Jackson ImmunoResearch) in PBST. The filter was washed in PBST and visualized used ECL (Amersham). For cycloheximide treatment, cells were grown in the presence of 45 gg/ml of cycloheximide. GTPases. H-ras and Ral were prepared as previously described (Albright et al., 1993). HA-Ral was expressed by cloning the coding region of human RalB downstream of the HA epitope and upstream of 6 histidine codons into pET-15 and purified as described above for KHRal. Racl, CDC42, and RhoA were kindly provided by Dr. Marc Symons (Malcolm et al., 1994). GAP Assay. 1 gg GTPase was combined with 10 gCi [y- 32P]GTP (NEN DuPont) in 100 p•l exchange buffer (50 mM Tris, pH 7.5, 50 mM NaC1, 5 mM EDTA, 1 mg/ml BSA, 1 mM DTT), 10 min, 25'C. The exchange 76 reaction was stopped by adding 100 p.l stop exchange buffer (50 mM Tris, pH 7.5, 50 mM NaC1, 20 mM MgC12, 5 mM EDTA, 1 mg/ml BSA, 1 mM DTT), diluted into reaction buffer (50 mM Tris, pH 7.5, 10 mM MgC12, 1 mg/ml BSA, 1 mM DTT), and aliquoted into 100 p.l reactions (10 ng of GTPase per reaction). 0.5 micrograms of RIP1 was added per reaction and incubated 10 min, 25°C. The reaction was diluted in 500 pl of wash buffer (50 mM Tris, pH 7.5, 10 mM MgC12, 1 mM DTT), filtered through nitrocellulose and washed with additional 10 ml of wash buffer. For testing the RIP1 protein for guanine nucleotide dissociation activity or protease activity, the assays were done identically as describe above except that [O- 32 P]GTP (NEN DuPont) was used instead of [y- 32 P]GTP and 10 ng of labeled GTPase was incubated in the presence of 5 micrograms of RIP1 for 15 minutes at 25°C instead. For GAP assays in the presence of Ral, 5 micrograms of GTPyS or GDP preloaded Ral was incubated with 0.5 micrograms of RIP1 on ice for 30 min prior to addition to the GAP assay. Construction of expression vectors for transfection. pCIN-RIP1 and pCIN-mRal are based on pCI-Neo (Promega). ACGCGTTCTAGAATGGCTGAGTGCTTCCTGCC was ligated onto the 5' Ava I site of RIP1 and GACGCCCATCTCCGGATGAGCGGCCGC ligated onto the 3' BsaHI site. of pCI-Neo. This was then cut and ligated into the XbaI-NotI sites pCIN-mRal was constructed by PCR cloning the Myc- epitope (GGCGAACAAAAGCTGATTTCTGAAGAAGACTTG) directly 5' of the starting ATG of human RalB and cloning into the XbaI-NotI sites of pCI-Neo. The sequence was confirmed by sequencing. 77 Transient transfections. Cos-1 cells were split 1:4 the day before transfection to approximately 2x106 cells per 10 cm plate and grown in 10% calf serum in DME. The plates were washed with PBS (137 mM NaC1, 2.7 mM KC1, 4.3 mM Na 2 HPO 4 -7H 20, 1.4 mM KH 2PO 4 , pH 7.2) and the growth media was changed to 4 mls 10% Nu serum in DME 1 hr prior to transfection. 8 gg of DNA per plate was brought up to 40 pl in TE and mixed with 80 pl of 10 mg/ml DEAE dextran in water. This mixture is added to the plate, followed by 16 p1 of 5 mM chloroquine diphosphate to a final concentration of 20 gM. The cells were incubated at 370 C for 4 hours and then shocked with 5 mls 10% glycerol in PBS for 60 seconds, washed twice with PBS and fed with 10 ml 10% calf serum in DME. Immunoprecipitations. For phosphatase treatment, cells transfected with pCIN-RIP1 were harvested 36 to 48 hours after transfection and lysed in 1 ml PLB (PBS + 0.1% NP-40, 10 mM sodium fluoride, 1 mM sodium vanadate,10 mM 03-glycerophosphate) plus protease inhibitors. Cellular debris was removed by centrifugation. One microliter of anti-RIP1 antiserum was added per ml of lysate and incubated on ice for 1 hour. Ten microliters of Protein A sepharose (Bio-Rad) was added and incubated for 1 hour at 4VC on a rotator. The beads was then washed two times with PBS, once with a 1:1 mixture of PBS and alkaline phosphatase reaction buffer, and two times in alkaline phosphatase reaction buffer (50 mM Tris-C1, pH 7.5, 1 mM MgCl2). 78 The beads were then resuspended in 50 microliters of alkaline phosphatase reaction buffer and incubated at 300 C for 10 minutes. 20 units of calf intestinal phosphatase (New England Biolabs) was added per sample and each sample was incubated an additional 30 minutes. The reaction was stopped by addition of ten microliters of 6x protein sample buffer (0.35 M Tris-C1, pH 6.8, 30% glycerol, 1.28% SDS, 6mM DTT, 0.012% bromophenol blue), the sample was then boiled for 10 minutes and resolved on a 7.5% SDS-PAGE gel. RIP1 was visualized by Western blot analysis as described above. For co-immunoprecipitation of RIP1 and Ral, Cos-1 cells were transfected with three micrograms of pCIN-RIP1 and five micrograms of pCIN-mRal per 10 cm plate. 36 to 48 hours after transfection, the cells were serum starved for 24 hours and lysed in 1 ml of PLB+ (PLB + 15 mM MgCl2) per plate. Cellular debris was removed by centrifugation and one microliter of anti-Myc ascites fluid was added per ml of lysate. Following one hour of incubation on ice ten microliters of Protein G agarose (Sigma) was added to each sample and incubated for 1 hour at 40 C on a rotator. The beads was then washed four times with PLB+ and boiled in 50 gl lx protein sample buffer. Protein was resolved on a 10% SDS-PAGE gel and RIP1 was visualized by Western analysis as described above. For KH-Ral co-precipitation, the lysates described above were incubated with one microgram KH-Ral preloaded with GTPyS as described previously, and then incubated for 1 hour on ice. Ten microliters of Ni-NTA resin (Qiagen) were added and the samples were rotated at 4°C for one hour. The samples were then processed identically as described for co-immunoprecipitation of RIP1 and Ral. 79 Immunofluorescence. Balb/3T3 cells were grown on 12 mm coverslips (Fisher) in 24 well tissue culture plates. For organic solvent fixation, the coverslips were submerged in either methanol or acetone for two minutes at room temperature. For formaldehyde, coverslips were submerged in a 1:5 dilution of 16% EM-grade formaldehyde (Ted Pella) in PBS for 15 minutes at room temperature, rinsed once in PBS and permeabilized by submerging in acetone for 2 minutes at room temperature. The coverslips were rinsed twice in PBS and placed cell-side up on parafilm on top of wet Whatman paper in a culture plate. The coverslips were blocked with 50 microliters of 3% BSA in PBS for 30 minutes. The block solution was removed by aspiration and replaced with 50 microliters of primary antibody diluted 1:100 in 3% BSA in PBS for 1 hour. The coverslips were washed three times fifteen minutes and then replaced in the incubation chamber with 50 microliters of secondary antibody diluted 1:100 to 1:250 in 3% BSA in PBS for 1 hour. The primary antibodies used were a mouse anti-human vinculin monoclonal antibody (Sigma) and affinity purified and neat rabbit anti-mouse RIP1 antiserum described above. The secondary antibodies were rhodamine- or fluorescein- conjugated donkey anti-mouse or rabbit IgG antibodies from Jackson ImmunoResearch. 80 ACKNOWLEDGEMENT I would like to gratefully acknowledge Dr. Marc Symons for the GTPases. Figure 3-1. Western analysis of RIP1. Purified RIP1 cleaved from a GS T fusion was run parallel to lysate prepared from Balb/3T3 cells. purified anti-RIP1 rabbit serum. RIP1 was detected using affinity The numbers indicate molecular weight in kilodaltons. 82 -112 53.2 34.9 Figure 3-2. Effect of cycloheximide treatment on RIP1. Balb/3T3 cells were treated with 45 tg of cycloheximide per ml of media for the indicated period of time. Cells were lysed and proteins separated on a 7.5% polyacrylamide gel, transferred to nitrocellulose. RIP1 was visualized with anti-RIP1 antibodies and bands are indicated by the arrows. treatment is indicated in hours. 84 the multiple The length of cycloheximide Cycloheximide treatment (hrs) I I 1 1 3 3 6 6 12 12 24 24 im a 1 RIP1 I Figure 3-3. Effect of phosphatase on RIPi. RIP1 from Cos-1 cells transiently transfected with RIP1 was immunoprecipitated, incubated with or without calf intestinal phosphatase, resolved on a 7.5% polyacrylamide gel, transferred to nitrocellulose and detected with anti-RIP1 antibody. 86 Alkaline Phosphatase - RIP1 Figure 3-4. Effect of serum on RIP1 modification. Balb/3T3 cells were grown in 10% calf serum, transferred to 0.2% serum for 60 hours, and stimulated with 10% serum. RIP1 was visualized by Western blot after separating whole cell lysate on a 7.5% polyacrylamide gel. Length of serum starvation or stimulation is indicated in hours, except for the five minute time point (5'). 88 Serum starved Serum stimulated (hr) % f 0 8 13 24 45 60 0 5' 1 4 12 18.5 24 -I RIP1 Figure 3-5. A. Ral-RIP1 complexes in Cos-1 transient transfections. Expression vector for RIP1 and Myc-tagged Ral were transiently transfected into Cos-1 cells. The cells were serum starved in 0.2% calf serum for 24 hours and stimulated with 10% calf serum for the indicated time. Ral was immunoprecipitated using anti-Myc antibody and co-immunoprecipitating RIP1 was visualized by Western blot of a 10% polyacrylamide gel. B. The lysate described above was incubated for 1 hour in the presence of GTP-loaded KHral. Ral-RIP1 complexes were precipitated with Ni-NTA resin. RIP1 was visualized by Western blot of a 7.5% polyacrylamide gel. 90 serum stimulation : 0 (min) 15 5 D 30 mWSW -- RIP1 IP: anti-myc Western blot: anti-RIP1 Transfection: pCIN-RIP1 + pCIN-mycRal 10% polyacrylamide gel B KH-Ral GDP GTPyS serum stimulation + + ++ w-- RIP1 ppt: Ni-agarose Western Blot: anti-RIP1 7.5% polyacrylamide gel Figure 3-6. The effect of RIP1 on the GTPase activity of several GTPases. 20 ng of each GTPase was preloaded with [y- 3 2 P]GTP and incubated with or without 500 ng RIP1 for 10 minutes at 25°C. The products were separated by filtration, the filter-bound radioactivity was determined and is indicated as percent radioactivity retained. sample without RIP1 addition is depicted as 100 percent. 92 The M - RIP1 El + RIP1 I I T ~I T 75- Z A= 0 IC 0-- 50 50- 25 - 01 . II H-Ras RalB Racl Cdc42 RhoA Figure 3-7. The effect of RIP1 on guanine nucleotides bound to GTPases. 10 ng of each GTPase was preloaded with [iX- 3 2 P]GTP or [y- 3 2 P]GTP and incubated with or without 5 gg RIP1 for 10 minutes at 25oC. The products were separated by filtration, the filter-bound radioactivity was determined and is indicated as percent radioactivity retained. The sample without RIP1 addition is depicted as 100 percent. 94 S[a-32p] GTP- RIP1 [0Ia-32P] GTP+ RIP1 S[y-3 2 P] GTP- RIP1 E] [y. 32 P] GTP+ RIP1 Racl CDC42 RhoA Figure 3-8. Immunofluorescence of RIP1. Balb/3T3 cells were seeded on glass cover slips, fixed with formaldehyde, permeabilized with acetone, and incubated with A. anti-RIP1 antiserum neat, B. anti-RIP1 antiserum preincubated with purified RIP1 protein, C. affinity purified anti-RIP1 antibody. The pattern reminiscent of vinculin localization is indicated by the arrows. Rhodamine-conjugated secondary antibody was used in A and B, while fluorescein-conjugated secondary was used in C. 96 Figure 3-9. Co-localization of RIP1 and vinculin. Balb/3T3 cells were seeded on glass cover slips, fixed with formaldehyde, permeabilized with acetone, and incubated with affinity purified anti-RIP1 antibody and a mouse monoclonal antibody against human vinculin, and visualized with A. fluoresceinconjugated anti-rabbit antibody or B. rhodamine-conjugated antimouse antibody, or C. doubly exposed. 98 Chapter 4. Conclusions and Prospects 100 In the previous two chapters, I described the cloning of the RIP1 gene and the characterization of this putative effector of Ral. The RIP1 gene was cloned by virtue of the ability of its encoded protein to bind GTP-bound Ral, and was shown to be incapable of binding GDP-bound Ral. One domain in RIP1 shares homology with that of a family of known proteins in the domain capable of activating the GTPase activity of members of the Rho-related GTPases. RIP1 was shown to have limited but nonetheless significant ability to activate the GTPase activity of Cdc42 and, to a lesser extent, that of Racl, but not that of RhoA. RIP1 is phosphorylated, presumably on serine/threonine residues, in response to serum stimulation of serum-starved Cos-1 cells transiently transfected with a RIP expression vector. In Cos-1 cells transiently transfected with RIP and Myc-tagged Ral expression vectors, starved and restimulated with serum, RIP1 can coimmunoprecipitate with Ral within five minutes after stimulation. This indicates that as a response to activation by an, as yet, unidentified factor present in serum Ral is activated and binds to RIP1. By immunofluorescence, RIP1 has been shown to co-localize with vinculin in focal adhesion plaques. Effector proteins. For a protein to be a bona fide effector of a GTPase, two criteria must be met. The first of these is the ability to bind specifically to the GTP-bound form, but not the GDP-bound form, of the GTPase. I have shown in Chapter 2 that this is indeed the case. A corollary of this would be that the effector protein be able to bind 101 activating mutants of the GTPase but not effector mutants. This was shown nicely by two other groups using yeast two hybrid system (Cantor et al., 1995; Jullien-Flores et al., 1995). Both groups show that the rat and human homolog of RIP1 can interact with the activated V23 Ral but not with the N49 or A46 effector domain mutants of Ral. The second criterion of an effector is a change in its activity as a result of the interaction. There are several models by which one could envision such an activation to take place (Marshall, 1996). The first is one in which the effector is directly altered through its interaction with the GTPase. For example, the binding of the GTPase to the effector may induce a conformational change in the effector that increases its activity. In this model, one wouldn't expect that membrane localization of the GTPase is required, but it is formally possible that the plasma membrane contributes by constraining the interaction between the effector and GTPase to one favorable for activation of the effector. A second model is one in which recruitment to a membrane is required for activation, either to allow activation by a component present only at the plasma membrane or to allow association with a substrate present only at the plasma membrane. An example of the first model can be found in PAK activation by Cdc42 (Manser et al., 1994). Another example is B-Raf activation by Ras (Yamamori et al., 1995). The addition of activated Ras to complexes containing B-Raf and 14-3-3 can lead to the activation of B-Raf. Although this activation can occur in the absence of plasma membrane, it requires the isoprenylation of Ras, indicating that 102 although Ras might be able to activate B-Raf through a mechanism involving conformational change, an additional role is played by the isoprenylation, possibly through the stabilization of the activating configuration. It is also possible that the requirement for 14-3-3 for the activation can only be fulfilled at the membrane. Although 14-3- 3 is distributed throughout the cell, the isoform required for activation might be present only at the membrane, or possible modification on 14-3-3 required to allow its participation in the activation might occur only at the membrane. Raf-1 activation is an example of the second model in which interaction with the activated GTPase induces translocation to the membrane where subsequent activation of the Raf-1 effector can occur. Many groups have reported the direct interaction between Raf-1 and Ras as a result of Ras activation (Moodie et al., 1993; Morrison et al., 1993; Warne et al., 1993; Zhang et al., 1993). Indeed, the requirement for Ras in Raf-1 activation can be bypassed by directly targeting Raf-1 to the membrane by the addition of the membrane-targeting CAAX-box of K-Ras to the C-terminus of Raf-1, allowing Raf-1 to then be activated in the absence of activated Ras. (Leevers et al., 1994; Stokoe et al., 1994) The interaction between RalGDS and activated Ras (Hofer et al., 1994; Kikuchi et al., 1994; Spaargaren and Bischoff, 1994), might be an example of membrane localization that allows association to a substrate present only at the membrane, since translocation of RalGDS to the membrane as a result of Ras activation would increase the probability of RalGDS to interact with its substrate Ral. Whether 103 the guanine exchange activity of RalGDS undergoes activation as a result of Ras interaction is not known. In the case of phosphatidylinositol-3-OH kinase (PI3K), it is not yet known whether the interaction between the GTPase and its effector leads to a direct activation of the kinase activity or whether the translocation of PI3K to the plasma membrane as a result of Ras activation merely increases the proximity of the kinase to its substrate (Rodriguez-Viciana et al., 1994). RIP1 as an effector protein. Experiments in which GTPyS-bound Ral was incubated with RIP1 prior to GAP assays showed no elevation of GAP activity as a result of Ral-RIP1 interaction, suggesting that the role of Ral in RIP1 activity is not one in which it can induce a direct activation of the GAP activity of RIP1. This would lead to the speculation that recruitment of RIP1 to the membrane by Ral is important in some aspect of RIP1 regulation, perhaps by allowing activation of RIP1 by other factors present at the membrane and/or by recruiting RIP1 to the membrane where it is now in close proximity to its substrate. (Figure 4-la) In Chapter Three, I have presented data indicating that some component present in calf serum is capable of activating Ral and that, within five minutes of serum stimulation of serum starved cells, a complexing of RIP1 and Ral can be observed, indicating an immediate activation of Ral and, presumably, an immediate role for RIP1 following serum stimulation. There were also repeated observations of the level of Ral-RIP1 interaction 104 decreasing over time, suggesting that Ral activation is transient, and subject to negative regulation by a Ral-specific GAP. Covalent modification of RIP1. Covalent modification of RIP1 can occur as a result of serum stimulation. I have demonstrated that RIP1 can be phosphorylated within one hour of stimulation. An attractive model for RIP1 action is one in which, as a result of Ral activation, RIP1 is recruited to the membrane where it gets phosphorylated as a result of the Ral-RIP1 interaction. Experiments to determine the role of Ral in the phosphorylation of RIP1 are currently underway using activated and dominant-negative mutants of Ral. If Ral-RIP1 interaction is a direct cause of RIP1 phosphorylation, cells transfected with activated mutants would be expected to have a higher proportion of phosphorylated RIP1, while cells transfected with dominant negative Ral would be expected to be inhibited in their ability to phosphorylate RIP1. This experiment does have the caveat that the kinase responsible for the phosphorylation of RIP1 might itself be regulated. It is also possible that the phosphorylation of RIP1 is independent of Ral-RIP1 interaction and is the result of a separate mechanism that regulates the activity of RIP1. (Figure 4-1b) The function of Ral in this model would be one in which Ral mediates the translocation of RIP1, allowing it to function at a new site and the duration of the signal is determined by the length of Ral activation or by the regulation of RIP1 through phosphorylation. Attempts are being made to directly address the role of RIP1 phosphorylation by 105 immunoprecipitating RIP1 from cells at different growth states to correlate phosphorylation with either activation or inactivation of its GAP activity. It is possible that Ral-RIP1 interaction allows the activity of RIP1 by bringing it to close proximity to its target, while the subsequent phosphorylation of RIP1 inactivates its GAP function rendering any RIP1 still complexed with Ral inactive. (Figure 4-la) Alternatively, it is possible that the RIP1 is activated through phosphorylation and that the amount phosphorylated is not detected at five minutes after serum stimulation. The secondary phosphorylation observed on RIP1 would support a model in which RIP1 is activated by the initial phosphorylation and that it remains active until the secondary inactivating phosphorylation occurring around 24 hours after serum stimulation. GTPase cascade. In the majority of cases where a protein has been shown to interact with a GTPase in a GTP-dependent manner, the interacting proteins have turned out to be kinases, able to regulate downstream elements directly through covalent modification. Examples of these are Raf-1, PI3K, PAK, and PKN. However, emerging data from a variety of different organisms suggest that some GTPases might be involved in a quite different mode of signaling, through a GTPase cascade. For example, in S. cerevisiae, Cdc24 has previously been shown to act as a guanine nucleotide exchange factor for Cdc42. (Zheng et al., 1994) More recently, Cdc24 has been implied to be a putative effector for Rsrl, a Ras-related GTPase, through its ability to 106 inhibit GAP-stimulated GTPase activation of Rsrl and its preferential binding to the activated form of Rsrl (Zheng et al., 1995). Data suggest that Rsrl does not directly control the guanine nucleotide exchange activity of Cdc24, but instead exerts its control on Cdc42 by positioning Cdc24 such that Cdc24 is then able to regulate Cdc42. A similar situation is observed in S. pombe between Cdc42sp, Rasl1 and Scd1, an S. pombe homolog of Cdc24 (Chang et al., 1994). The interaction between RalGDS and activated Ras suggests yet another GTPase cascade in which activated Ras regulates the activity of members of the Rho subfamily through Ral activation, which would then lead to the inactivation of Cdc42 or Racl. This simple- minded GTPase regulatory cascade is raised to a higher level of complexity when Ras regulatory proteins and p190 is included since p 190, a Rho subfamily regulatory protein shown to have GAP activity on Rho, Rac and Cdc42, can bind directly to pl20GAP, a Ras regulatory protein shown to have Ras GAP activity. A recent report indeed indicates that an increase in GTP-bound Ral is observed in response to H-Ras activation (Urano et al., 1996). Although R-Ras and Rapl were also shown to be capable of interacting with RalGDS, it seems that the interaction of RalGDS with H-Ras is the only productive one. These results, however, are in contrast to results obtained previously from in vitro experiments in which GTPyS-bound H-Ras was incubated with RalGDS prior to guanine nucleotide exchange assays on Ral. Exchange reactions conducted in the presence of activated Ras were found to be indistinguishable from those done in the presence of GDP-bound Ras. Since these assays were utilizing purified proteins, it is possible that 107 another component necessary for promoting the dissociation of the guanine nucleotide from Ral was missing from the reaction. It is also possible that the mere mixing of proteins is not sufficient to activate RalGDS, and that certain membrane constraints that allow the proper juxtaposition of Ras, RalGDS and Ral must be met in order to observe an increase in exchange activity. Using transient transfections, I have shown that a component present in serum had the ability to activate Ral. Using this as an assay, it should be possible to identify factors that are capable of activating Ral and subsequently identify the consequences of Ral activation. The proof that RIP1 is indeed a true effector for Ral would be to demonstrate that these downstream events of Ral signaling are mediated through a function of RIP1 and that a Ralindependent activation of RIP1 is capable of bypassing the need for Ral activation. In this thesis, I have described the cloning and characterization of RIP1. Although the experiments described in this thesis do not yet answer the question of what kind of signaling Ral is involved in, it does bring us one step closer in elucidating the Ral-dependent signal transduction pathway. The consequence of Ral signaling is not yet clear, but it is likely that Ral activation leads to the regulation of other GTPases through its effector RIP1. It is also becoming clear that the signaling through Ras-related GTPases is not a simple unidirectional event but rather a complex network of signaling events that involve cross-talking and multiple regulatory mechanisms. 108 Models of Ral-RIP1 interaction. Figure 4-1. a. Ral-dependent activation of RIP1. is dependent on the activation of Ral. In this model, RIP1 activation The Ral activation leads to the binding of RIP1 to Ral which allows the activation RIP1 by other proteins. Once activated, RIP1 may or may not be required to maintain its interaction with Ral for full activity. Subsequently, RIP1 may be inactivated by a Ral-independent phosphorylation of RIP1 either free or still complexed with Ral. b. Ral-independent activation of RIP1. In this model, the activation of the catalytic activity of RIP1 is independent of Ral activation, and activated RIP1 can function properly only when interacting with activated Ral. An independent signal leads to the activation of the catalytic activity of RIP1 through phosphorylation, thus priming RIP1 for action. Upon Ral activation, activated RIP1 can bind to Ral and form an active complex. The activity of RIP1 in this complex is dependent on the maintenance of activated Ral. Subsequently RIP1 can then be inactivated by an additional phosphorylation on RIP1. The interaction of inactive RIP1 (either the unphosphorylated or doubly phosphorylated RIPI) with Ral would not lead to the formation of an active complex. 109 RIP1 (inactive) 1 (active) RIP1 (active) 1 (inactive) (inactive)P (inactive) p (inactive) RIP1 activation Ral activation "*V (catalytically active) \RIP inactivation RIP1 RIP1 inactivation Active complex (inactive) Ral inactivation (inactive) (inactive) Chapter 5. References 112 Abo, A., Pick, E., Hall, A., Totty, N., Teahan, C. G., and Segal, A. W. (1991). Activation of the NADPH oxidase involves the small GTPbinding protein p21racl. Nature 353, 668-70. Adari, H., Lowy, D. R., Willumsen, B. M., Der, C. J., and McCormick, F. (1988). Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science 240, 518-21. Ahn, N. G., and Krebs, E. G. (1990). Evidence for an epidermal growth factor-stimulated protein kinase cascade in Swiss 3T3 cells. Activation of serine peptide kinase activity by myelin basic protein kinases in vitro. J Biol Chem 265, 11495-501. Ahn, N. G., Seger, R., and Krebs, E. G. (1992). The mitogen-activated protein kinase activator. Curr Opin Cell Biol 4, 992-9. Ahn, N. G., Weiel, J. E., Chan, C. P., and Krebs, E. G. (1990). Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J Biol Chem 265, 11487-94. Albright, C. F., Giddings, B. W., Liu, J., Vito, M., and Weinberg, R. A. (1993). Characterization of a guanine nucleotide dissociation stimulator for a ras-related GTPase. Embo J 12, 339-47. Alcorta, D. A., Crews, C. M., Sweet, L. J., Bankston, L., Jones, S. W., and Erikson, R. L. (1989). Sequence and expression of chicken and mouse 113 rsk: homologs of Xenopus laevis ribosomal S6 kinase. Mol Cell Biol 9, 3850-9. Amano, M., Mukai, H., Ono, Y., Chihara, K., Matsui, T., Hamajima, Y., Okawa, K., Iwamatsu, A., and Kaibuchi, K. (1996). Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271, 648-50. Anderson, N. G., Mailer, J. L., Tonks, N. K., and Sturgill, T. W. (1990). Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 343, 651-3. Avruch, J. e. a. (1985). . In Molecular Basis of Insulin Action, M. Czech, ed.: Plenum), pp. 263-296. Ballester, R., Marchuk, D., Boguski, M., Saulino, A., Letcher, R., Wigler, M., and Collins, F. (1990). The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63, 851-9. Basu, T. N., Gutmann, D. H., Fletcher, J. A., Glover, T. W., Collins, F. S., and Downward, J. (1992). Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients [see comments]. Nature 356, 713-5. 114 Best, A., Ahmed, S., Kozma, R., and Lim, L. (1996). The Ras-related GTPase Racl binds tubulin. J Biol Chem 271, 3756-3762. Bhullar, R. P. (1992). Identification of some of the brain Gn27 as the ral gene product. Comparison between the brain and platelet Gnproteins. Febs Lett 298, 61-4. Bhullar, R. P., Chardin, P., and Haslam, R. J. (1990). Identification of multiple ral gene products in human platelets that account for some but not all of the platelet Gn-proteins. Febs Lett 260, 48-52. Bielinski, D. F., Pyun, H. Y., Linko-Stentz, K., Macara, I. G., and Fine, R. E. (1993). Ral and Rab3a are major GTP-binding proteins of axonal rapid transport and synaptic vesicles and do not redistribute following depolarization stimulated synaptosomal exocytosis. Biochim Biophys Acta 1151, 246-56. Blenis, J., and Erikson, R. L. (1986). Stimulation of ribosomal protein S6 kinase activity by pp60v-src or by serum: dissociation from phorbol ester-stimulated activity. Proc Natl Acad Sci U S A 83, 17337. Blenis, J., Kuo, C. J., and Erikson, R. L. (1987). Identification of a ribosomal protein S6 kinase regulated by transformation and growth-promoting stimuli. J Biol Chem 262, 14373-6. 115 Bokoch, G. M., Quilliam, L. A., Bohl, B. P., Jesaitis, A. J., and Quinn, M. T. (1991). Inhibition of RaplA binding to cytochrome b558 of NADPH oxidase by phosphorylation of RaplA. Science 254, 1794-6. Bonfini, L., Karlovich, C. A., Dasgupta, C., and Banerjee, U. (1992). The Son of sevenless gene product: a putative activator of Ras. Science 255, 603-6. Bourne, H. R., Sanders, D. A., and McCormick, F. (1991). The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117-27. Broek, D., Toda, T., Michaeli, T., Levin, L., Birchmeier, C., Zoller, M., Powers, S., and Wigler, M. (1987). The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 48, 78999. Cantor, S. B., Urano, T., and Feig, L. A. (1995). Identification and characterization of Ral-binding Protein 1, a potential downstream target of Ral GTPases. Molecular and Cellular Biology 15, 4578-4584. Cawthon, R. M., Weiss, R., Xu, G. F., Viskochil, D., Culver, M., Stevens, J., Robertson, M., Dunn, D., Gesteland, R., O'Connell, P., and et, a. (1990). A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations [published erratum appears in Cell 1990 Aug 10; 62(3):following 608]. Cell 62, 193-201. 116 Chang, E. C., Barr, M., Wang, Y., Jung, V., Xu, H. P., and Wigler, M. H. (1994). Cooperative interaction of S. pombe proteins required for mating and morphogenesis. Cell 79, 131-41. Chardin, P. (1993). Structural conservation of Ras-related proteins and its functional implications. In GTPases in Biology, B. Dickey and L. Birnbaumer, eds.: Springer-Verlag), pp. 159-176. Chardin, P., Camonis, J. H., Gale, N. W., van Aelst, L., Schlessinger, J., Wigler, M. H., and Bar-Sagi, D. (1993). Human Sosl: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338-43. Chardin, P., and Tavitian, A. (1986). The ral gene: a new ras related gene isolated by the use of a synthetic probe. Embo J 5, 2203-8. Chou, M., Blenis, J. (1996). The 70 kDa S6 kinase complexes with and is activated by the Rho family of G proteins Cdc42 and Racl. Cell 85, 753-584. Cicchetti, P., Ridley, A. J., Zheng, Y., Cerione, R. A., and Baltimore, D. (1995). 3BP-1, an SH3 domain binding protein, has GAP activity for Rac and inhibits growth factor-induced membrane ruffling in fibroblasts. Embo J 14, 3127-35. Colby, W., Hayflick, J., Clark, S., and Levinson, A. (1986). Mol Cell Biol 11, 202-212. 117 Coso, O. A., Chiariello, M., Yu, J. C., Teramoto, H., Crespo, P., Xu, N., Miki, T., and Gutkind, J. S. (1995). The small GTP-binding proteins Racl and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81, 1137-46. Crechet, J. B., Poullet, P., Mistou, M. Y., Parmeggiani, A., Camonis, J., Boy, M. E., Damak, F., and Jacquet, M. (1990). Enhancement of the GDP-GTP exchange of RAS proteins by the carboxyl-terminal domain of SCD25 [see comments]. Science 248, 866-8. Davis, R. J. (1993). The mitogen-activated protein kinase signal transduction pathway. J Biol Chem 268, 14553-6. Debant, A., Serra-Pages, C., Seipel, K., O'Brian, S., Tang, M., Park, S.-H., and Streuli, M. (1996). The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA in press. DeClue, J., Papageorge, A. G., Fletcher, J. A., Diehl, S. R., Ratner, N., Vass, W. C., and Lowy, D. R. (1992). Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69, 265-73. 118 Der, C. J., Finkel, T., and Cooper, G. M. (1986). Biological and biochemical properties of human rasH genes mutated at codon 61. Cell 44, 167-76. Diekmann, D., Brill, S., Garrett, M. D., Totty, N., Hsuan, J., Monfries, C., Hall, C., Lim, L., and Hall, A. (1991). Bcr encodes a GTPase-activating protein for p21rac. Nature 351, 400-2. Downward, J., Riehl, R., Wu, L., and Weinberg, R. A. (1990). Identification of a nucleotide exchange-promoting activity for p21ras. Proc Natl Acad Sci U S A 87, 5998-6002. Drgonova, J., Drgon, T., Tanaka, K., Kollar, R., Chen, G.-C., Ford, R. A., Chan, C. S. M., Takai, Y., and Cabib, E. (1996). Rholp, a yeast protein at the interface between cell polarization and morphogenesis. Science 272, 277-279. Emkey, R., Freedman, S., and Feig, L. A. (1991). Characterization of a GTPase-activating protein for the Ras-related Ral protein. J Biol Chem 266, 9703-6. Erikson, E., and Maller, J. L. (1986). Purification and characterization of a protein kinase from Xenopus eggs highly specific for ribosomal protein S6. J Biol Chem 261, 350-5. 119 Fortini, M. E., Simon, M. A., and Rubin, G. M. (1992). Signalling by the sevenless protein tyrosine kinase is mimicked by Rasl activation [see comments]. Nature 355, 559-61. Frech, M., Schlichting, I., Wittinghofer, A., and Chardin, P. (1990). Guanine nucleotide binding properties of the mammalian RalA protein produced in Escherichia coli. J Biol Chem 265, 6353-9. Furth, M. E., Davis, L. J., Fleurdelys, B., and Scolnick, E. M. (1982). Monoclonal antibodies to the p21 products of the transforming gene of Harvey murine sarcoma virus and of the cellular ras gene family. J Virol 43, 294-304. Garrett, M. D., Major, G. N., Totty, N., and Hall, A. (1991). Purification and N-terminal sequence of the p21rho GTPase-activating protein, rho GAP. Biochem J 276, 833-6. Gibbs, J., Sigal, IS, Poe, M, and Scolnick, EM (1984). Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci USA 81, 5704-5708. Gibbs, J., Sigal, I., and Scolnick, E. (1985). TIBS 10, 350-353. Gibbs, J. B., Schaber, M. D., Allard, W. J., Sigal, I. S., and Scolnick, E. M. (1988). Purification of ras GTPase activating protein from bovine brain. Proc Natl Acad Sci U S A 85, 5026-30. 120 Habets, G. G., Scholtes, E. H., Zuydgeest, D., van der Kammen, R. A., Stam, J. C., Berns, A., and Collard, J. G. (1994). Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell 77, 53749. Hafen, E., Basler, K., Edstroem, J. E., and Rubin, G. M. (1987). Sevenless, a cell-specific homeotic gene of Drosophila, encodes a putative transmembrane receptor with a tyrosine kinase domain. Science 236, 55-63. Hall, C., Monfries, C., Smith, P., Lim, H. H., Kozma, R., Ahmed, S., Vanniasingham, V., Leung, T., Lim, L. (1990). Novel human brain cDNA encoding a 34,000 Mr protein n-chimerin, related to both the regulatory domain of protein kinase C and BCR, the product of the breakpoint cluster region gene. J. Mol. Biol. 221, 11-16 Hall, A. (1992). Signal transduction through small GTPases--a tale of two GAPs. Cell 69, 389-91. Hart, M. J., Eva, A., Evans, T., Aaronson, S. A., and Cerione, R. A. (1991). Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature 354, 311-4. Harvey, J. J. (1964). An unidentified virus which causes the rapid production of tumors in mice. Nature 204, 1104-1105. 121 Hasegawa, H., Kojima, M., Oguro, K., and Nakanishi, N. (1995). Rapid turnover of tryptophan hydroxylase in serotonin producing cells: demonstration of ATP-dependent proteolytic degradation. Febs Lett 368, 151-4. Hata, Y., Kikuchi, A., Sasaki, T., Schaber, M. D., Gibbs, J. B., and Takai, Y. (1990). Inhibition of the ras p21 GTPase-activating proteinstimulated GTPase activity of c-Ha-ras p21 by smg p21 having the same putative effector domain as ras p21s. J Biol Chem 265, 7104-7. Hateboer, G., Gennissen, A., Ramos, Y. F. M., Kerkhoven, R. M., Sonntag-Buck, V., Stunnenberg, H. G., and Bernards, R. (1995). BS69, a novel adenovirus E1A-associated protein that inhibits E1A transactivation. EMBO J., 3159-3169. Heisterkamp, N., Stam, K., Groffen, J., De Klein, A., Grosveld, G. (1985). Structural organization of the bcr gene and its role in Ph' translocation. Nature 315, 758-761. Heisterkamp, N., Kaartinen, V., van Soest, S., Bokoch, G. M., and Groffen, J. (1993). Human ABR encodes a protein with GAP r ac activity and homology to the DBL nucleotide exchange factor domain. J Biol Chem 268, 16903-6. Hildebrand, J. H., Taylor, J. M., and Parsons, J.T. (1996). An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol. Cell Biol. 16, 3169-3178. 122 Hochuli, E., D6beli, H., and Schacher, A. (1987). New metal chelate adsorbents selective for proteins and peptide containing neighbouring histidine residues. J. Chromatography 411, 177-184. Hofer, F., Fields, S., Schneider, C., and Martin, G. S. (1994). Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc Natl Acad Sci U S A 91, 11089-93. Homma, Y., and Emori, Y. (1995). A dual functional signal mediator showing RhoGAP and phospholipase C-8 stimulating activities. Embo J. 14, 286-291. Horii, Y., Beeler, J. F., Sakaguchi, K., Tachibana, M., and Miki, T. (1994). A novel oncogene, ost, encodes a guanine nucleotide exchange factor that potentially links Rho and Rac signaling pathways. Embo J 13, 4776-86. Hotchin, N. A., and Hall, A. (1995). The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol 131, 1857-65. Hughes, D. A., Fukui, Y., and Yamamoto, M. (1990). Homologous activators of ras in fission and budding yeast. Nature 344, 355-7. 123 Ikeda, M., Koyama, S., Okazaki, M., Dohi, K., and Kikuchi, A. (1995). rapl p21 regulates the interaction of ras p21 with RGL, a new effector protein of ras p21. Febs Lett 375, 37-40. Janknecht, R., Martynoff, G. d., Lou, J., Hipskind, R. A., Nordheim, A., and Stunnenberg, H. G. (1991). Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. PNAS 88, 8972-8976. Jiang, H., Luo, J.-Q., Urano, T., Frankel, P., Lu, Z., Foster, D. A., and Feig, L. A. (1996). Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature, 409-412. Jones, S. W., Erikson, E., Blenis, J., Maller, J. L., and Erikson, R. L. (1988). A Xenopus ribosomal protein S6 kinase has two apparent kinase domains that are each similar to distinct protein kinases. Proc Natl Acad Sci U S A 85, 3377-81. Joneson, T., White, M. A., Wigler, M. H., and Bar-Sagi, D. (1996). Stimulation of membrane ruffling and MAP kinase activation by distint effectors of RAS. Science 271, 810-812. Jullien-Flores, V., Dorseuil, O., Romero, F., Letourneur, F., Saragosti, S., Berger, R., Tavitian, A., Gacon, G., and Camonis, J. H. (1995). Bridging Ral GTPase to Rho pathways. RLIP76, a Ral effector with CDC42/Rac GTPase-activating protein activity. J Biol Chem 270, 22473-7. 124 Kaibuchi, K., Mizuno, T., Fujioka, H., Yamamoto, T., Kishi, K., Fukumoto, Y., Hori, Y., and Takai, Y. (1991). Molecular cloning of the cDNA for stimulatory GDP/GTP exchange protein for smg p21s (ras p21-like small GTP-binding proteins) and characterization of stimulatory GDP/GTP exchange protein. Mol Cell Biol 11, 2873-80. Kamada, Y., Qadota, H., Python, C. P., Anraku, Y., Ohya, Y., and Levin, D. E. (1996). Activation of yeast protein kinase C by Rhol GTPase. J. Biol. Chem. 271, 9193-9196. Kikuchi, A., Demo, S. D., Ye, Z. H., Chen, Y. W., and Williams, L. T. (1994). ralGDS family members interact with the effector loop of ras p21. Mol Cell Biol 14, 7483-91. Kim, S. H., de Vos, A. M., Tong, L., Milburn, M. V., Matias, P. M., Jancarik, J., Ohtsuka, E., and Nishimura, S. (1988). ras oncogene proteins: three-dimensional structures, functional implications, and a model for signal transducer. Cold Spring Harb Symp Quant Biol 1, 273-81. Kinsella, B. T., Erdman, R. A., and Maltese, W. A. (1991). Carboxylterminal isoprenylation of ras-related GTP-binding proteins encoded by racl, rac2, and ralA. J Biol Chem 266, 9786-94. Kirsten, W., and Mayer, LA (1967). Morphological responses to a murine erythroblastosis virus. J. Natl Cancer Inst 39. 125 Kitayama, H., Sugimoto, Y., Matsuzaki, T., Ikawa, Y., and Noda, M. (1989). A ras-related gene with transformation suppressor activity. Cell 56, 77-84. Knaus, U. G., Morris, S., Dong, H. J., Chernoff, J., and Bokoch, G. M. (1995). Regulation of human leukocyte p21-activated kinases through G protein-coupled receptors. Science 269, 221-3. Koide, H., Satoh, T., Nakafuku, M., and Kaziro, Y. (1993). GTPdependent association of Raf-1 with Ha-Ras: identification of Raf as a target downstream of Ras in mammalian cells. Proc Natl Acad Sci U S A 90, 8683-6. Kolch, W., Heidecker, G., Lloyd, P., and Rapp, U. R. (1991). Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature 349, 426-8. Kozma, R., Ahmed, S., Best, A., and Lim, L. (1995). The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol 15, 1942-52. Kung, H. F., Smith, M. R., Bekesi, E., Manne, V., and Stacey, D. W. (1986). Reversal of transformed phenotype by monoclonal antibodies against Ha-ras p21 proteins. Exp Cell Res 162, 363-71. 126 Lacal, J. C., Srivastava, S. K., Anderson, P. S., and Aaronson, S. A. (1986). Ras p21 proteins with high or low GTPase activity can efficiently transform NIH/3T3 cells. Cell 44, 609-17. Lancaster, C. A., Taylor-Harris, P. M., Self, A. J., Brill, S., van Erp, H. E., and Hall, A. (1994). Characterization of rhoGAP. A GTPase-activating protein for rho-related small GTPases. J Biol Chem 269, 1137-42. Lange-Carter, C. A., Pleiman, C. M., Gardner, A. M., Blumer, K. J., and Johnson, G. L. (1993). A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260, 315-9. Leevers, S. J., Paterson, H. F., and Marshall, C. J. (1994). Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369, 411-4. Madaule, P., and Axel, R. (1985). A novel ras-related gene family. Cell 41, 31-40. Malcolm, K. C., Ross, A. H., Qiu, R. G., Symons, M., and Exton, J. H. (1994). Activation of rat liver phospholipase D by the small GTPbinding protein RhoA. J Biol Chem 269, 25951-4. Maller, J. L., Pike, L. J., Freidenberg, G. R., Cordera, R., Stith, B. J., Olefsky, J. M., and Krebs, E. G. (1986). Increased phosphorylation of ribosomal protein S6 following microinjection of insulin receptorkinase into Xenopus oocytes. Nature 320, 459-61. 127 Manser, E., Leung, T., Salihuddin, H., Zhao, Z. S., and Lim, L. (1994). A brain serine/threonine protein kinase activated by Cdc42 and Racl. Nature 367, 40-6. Marshall, C. J. (1996). Ras Effectors. Curr. Opin. Cell Biol. 8, 197-204. Martegani, E., Vanoni, M., Zippel, R., Coccetti, P., Brambilla, R., Ferrari, C., Sturani, E., and Alberghina, L. (1992). Cloning by functional complementation of a mouse cDNA encoding a homologue of CDC25, a Saccharomyces cerevisiae RAS activator. Embo J 11, 2151-7. Martin, G. A., Bollag, G., McCormick, F., and Abo, A. (1995). A novel serine kinase activated by racl /CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. Embo J 14, 1970-8. Martin, G. A., Viskochil, D., Bollag, G., McCabe, P. C., Crosier, W. J., Haubruck, H., Conroy, L., Clark, R., O'Connell, P., Cawthon, R. M., and et, a. (1990). The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63, 843-9. McGrath, J. P., Capon, D. J., Goeddel, D. V., and Levinson, A. D. (1984). Comparative biochemical properties of normal and activated human ras p21 protein. Nature 310, 644-9. 128 Michiels, F., Habets, G. G., Stam, J. C., van der Kammen, R. A., and Collard, J. G. (1995). A role for Rac in Tiaml-induced membrane ruffling and invasion. Nature 375, 338-40. Minden, A., Lin, A., Claret, F. X., Abo, A., and Karin, M. (1995). Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 81, 1147-57. Moodie, S. A., Willumsen, B. M., Weber, M. J., and Wolfman, A. (1993). Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase [see comments]. Science 260, 1658-61. Morrison, D. K., Heidecker, G., Rapp, U. R., and Copeland, T. D. (1993). Identification of the major phosphorylation sites of the Raf-1 kinase. J Biol Chem 268, 17309-16. Mulcahy, L. S., Smith, M. R., and Stacey, D. W. (1985). Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 313, 241-3. Nobes, C. D., and Hall, A. (1995). Rho, Rac, and Cdc42 GTPase regulate the assemble of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53-62. Nonaka, H., Tanaka, K., Hirano, H., Fujiwara, T., Kohno, H., Umikawa, M., Mino, A., and Takai, Y. (1995). A downstream target of RHO1 129 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J. 14, 5931-5938. Olofsson, B., Chardin, P., Touchot, N., Zahraoui, A., and Tavitian, A. (1988). Expression of the ras-related ralA, rhol2 and rab genes in adult mouse tissues. Oncogene 3, 231-4. Olson, M. F., Ashworth, A., and Hall, A. (1995). An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through Gl. Science 269, 1270-2. Otsu, M., Hiles, I., Gout, I., Fry, M. J., Ruiz-Larrea, F., Panayotou, G., Thompson, A., Dhand, R., Hsuan, J., Totty, N., Smith, A. D., Morgan, S. J., Courtneidge, S. A., Parker, P., and Waterfield, M. D. (1991). Characterization of two 85kD proteins that associate with receptor tyrosine kinases, middle T/pp60 c -src complexes, and PI3-kinase. Cell 65, 91-104. Papageorge, A. G., Defeo-Jones, D., Robinson, P., Temeles, G., and Scolnick, E. M. (1984). Saccharomyces cerevisiae synthesizes proteins related to the p21 gene product of ras genes found in mammals. Mol Cell Biol 4, 23-9. Parsons, J. T. (1996). Integrin mediated signallng: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr. Opin. Cell Biol. 8, 146-152. 130 Pelech, S. L., Olwin, B. B., and Krebs, E. G. (1986). Fibroblast growth factor treatment of Swiss 3T3 cells activates a subunit S6 kinase that phosphorylates a synthetic peptide substrate. Proc Natl Acad Sci U S A 83, 5968-72. Pizon, V., Lerosey, I., Chardin, P., and Tavitian, A. (1988). Nucleotide sequence of a human cDNA encoding a ras-related protein (raplB). Nucleic Acids Res 16, 7719. Polakis, P. G., Rubinfeld, B., Evans, T., and McCormick, F. (1991). Purification of a plasma membrane-associated GTPase-activating protein specific for rapl/Krev-1 from HL60 cells. Proc Natl Acad Sci U S A 88, 239-43. Polakis, P. G., Weber, R. F., Nevins, B., Didsbury, J. R., Evans, T., and Snyderman, R. (1989). Identification of the ral and racl gene products, low molecular mass GTP-binding proteins from human platelets. J Biol Chem 264, 16383-9. Qadota, H., Python, C. P., Inoue, S. B., Arisawa, M., Anraku, Y., Zheng, Y., Watanabe, T., Levin, D. E., and Ohya, Y. (1996). Identification of yeast Rholp GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science 272, 279-281. Qiu, R. G., Chen, J., Kirn, D., McCormick, F., and Symons, M. (1995). An essential role for Rac in Ras transformation. Nature 374, 457-9. 131 Qiu, R. G., Chen, J., McCormick, F., and Symons, M. (1995). A role for Rho in Ras transformation. Proc Natl Acad Sci U S A 92, 11781-5. Quilliam, L. A., Der, C. J., Clark, R., O'Rourke, E. C., Zhang, K., McCormick, F., and Bokoch, G. M. (1990). Biochemical characterization of baculovirus-expressed raplA/Krev-1 and its regulation by GTPaseactivating proteins. Mol Cell Biol 10, 2901-8. Rapp, U. R., Heidecker, G., Huleihel, M., Cleveland, J. L., Choi, W. C., Pawson, T., Ihle, J. N., and Anderson, W. B. (1988). raf family serine/threonine protein kinases in mitogen signal transduction. Cold Spring Harb Symp Quant Biol 1, 173-84. Reddy, E., Reynolds, RK, Santos, E, and Barbacid, M (1982). A point mutation is responsible for the acquistition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149-152. Reinhard, J., Scheel, A. A., Diekmann, D., Hall, A., Ruppert, C., and Bahler, M. (1995). A novel type of myosin implicated in signalling by rho family GTPases. Embo J 14, 697-704. Ridley, A. J., and Hall, A. (1992a). The small GTP-binding protein rho regulates the assemble of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389-399. 132 Ridley, A. J., Paterson, H. F., Johnston, C. L., Diekmann, D., and Hall, A. (1992b). The small GTP-binding protein rac regulates growth factorinduced membrane ruffling. Cell 70, 401-10. Ridley, A. J., Self, A. J., Kasmi, F., Paterson, H. F., Hall, A., Marshall, C. J., and Ellis, C. (1993). rho family GTPase activating proteins p190, ber and rhoGAP show distinct specificities in vitro and in vivo. Embo J 12, 5151-60. Robinson, L. C., Gibbs, J. B., Marshall, M. S., Sigal, I. S., and Tatchell, K. (1987). CDC25: a component of the RAS-adenylate cyclase pathway in Saccharomyces cerevisiae. Science 235, 1218-21. Rodriguez-Viciana, P., Warne, P. H., Dhand, R., Vanhaesebroeck, B., Gout, I., Fry, M. J., Waterfield, M. D., and Downward, J. (1994). Phosphatidylinositol-3-OH kinase as a direct target of Ras [see comments]. Nature 370, 527-32. Ron, D., Zannini, M., Lewis, M., Wickner, R. B., Hunt, L. T., Graziani, G., Tronick, S. R., Aaronson, S. A., and Eva, A. (1991). A region of protodbl essential for its transforming activity shows sequence similarity to a yeast cell cycle gene, CDC24, and the human breakpoint cluster gene, bcr. New Biol 3, 372-9. Rubinfeld, B., Munemitsu, S., Clark, R., Conroy, L., Watt, K., Crosier, W. J., McCormick, F., and Polakis, P. (1991). Molecular cloning of a 133 GTPase activating protein specific for the Krev-1 protein p21rap1. Cell 65, 1033-42. Schweighoffer, F., Rey, I., Barlot, I., Soubigou, P., Mayaux, J. F., and Tocque, B. (1990). Rap gene products mobilize a different metabolic pathway than p21 ras proteins. In The biology and medicine of signal transduction, Y. e. a. Nishizuka, ed. (New York: Raven), pp. 329-334. Seely, J. E., Poso, H., and Pegg, A. E. (1982). Effect of androgens on turnover of ornithine decarboxylase in mouse kidney. Studies using labeling of the enzyme by reaction with [14C] alphadifluoromethylornithine. J Biol Chem 257, 7549-53. Serra-Pages, C., Kedersha, N. K., Fazikas, L., Medley, Q., Debant, A., and Streuli, M. (1995). The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. EMBO J 14, 2827-2838. Settleman, J., Albright, C. F., Foster, L. C., and Weinberg, R. A. (1992). Association between GTPase activators for Rho and Ras families. Nature 359, 153-4. Shih, T., Papageorge, AG, Stokes, PE, Weeks, MO, and Scolnick, EM (1980). Guanine nucleotide-binding and autophosphorylating activities associated with the p21src protein of Harvey murine sarcoma virus. Nature 287, 686-691. 134 Shih, T., Weeks, MO, Young, HA, and Scolnick, EM (1979). Identification of a sarcoma virus-coded phosphoprotein in nonproducer cells transformed by Kirsten or Harvey murine sarcoma virus. Virology 96, 64-79. Sigal, I. S., Gibbs, J. B., D'Alonzo, J. S., and Scolnick, E. M. (1986). Identification of effector residues and a neutralizing epitope of Haras-encoded p21. Proc Natl Acad Sci U S A 83, 4725-9. Simon, M. A., Bowtell, D. D., Dodson, G. S., Laverty, T. R., and Rubin, G. M. (1991). Rasl and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701-16. Simon, M. A., Bowtell, D. D., and Rubin, G. M. (1989). Structure and activity of the sevenless protein: a protein tyrosine kinase receptor required for photoreceptor development in Drosophila. Proc Natl Acad Sci U S A 86, 8333-7. Spaargaren, M., and Bischoff, J. R. (1994). Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-ras, K-ras, and Rap. Proc Natl Acad Sci U S A 91, 12609-13. Stefanovic, D., Erikson, E., Pike, L. J., and Maller, J. L. (1986). Activation of a ribosomal protein S6 protein kinase in Xenopus oocytes by insulin and insulin-receptor kinase. Embo J 5, 157-60. 135 Stokoe, D., Macdonald, S. G., Cadwallader, K., Symons, M., and Hancock, J. F. (1994). Activation of Raf as a result of recruitment to the plasma membrane [see comments]. Science 264, 1463-7. Sturgill, T. W., Ray, L. B., Erikson, E., and Maller, J. L. (1988). Insulinstimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 334, 715-8. Sweet, R. W., Yokoyama, S., Kamata, T., Feramisco, J. R., Rosenberg, M., and Gross, M. (1984). The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 311, 273-5. Symons, M., Derry, J. M. J., Karkak, B., Jiang, S., Lemanhieu, V., McCormick, F., Franke, U., and Abo, A. (1996). Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84, 723-734. Tabin, C., Bradley, SM, Bargmann, CI, Weinberg, RA, Papageorge, AG, Scolnick, EM, Dhar, R, Lowy, DR, and Chang, EH (1982). Mechanism of activation of a human oncogene. Nature 300, 142-149. Tanaka, K., Nakafuku, M., Tamanoi, F., Kaziro, Y., Matsumoto, K., and Toh-e, A. (1990). IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Mol Cell Biol 10, 4303-13. 136 Taparowsky, E., Suard, Y, Fasano, O, Shimizu, K, Goldfarb, M, and Wigler, M (1982). Activation of the T24 bladder carcinoma transforming gene is linked to a single amino acid change. Nature 300, 762-765. Tominaga, T., Sugie, K., Hirata, M., Morii, N., Fukata, J., Uchida, A., Imura, H., and Narumiya, S. (1993). Inhibition of PMA-induced, LFA1-dependent lymphocyte aggregation by ADP ribosylation of the small molecular weight GTP binding protein, rho. J Cell Biol 120, 1529-37. Trahey, M., and McCormick, F. (1987). A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 238, 542-5. Trahey, M., Milley, R. J., Cole, G. E., Innis, M., Paterson, H., Marshall, C. J., Hall, A., and McCormick, F. (1987). Biochemical and biological properties of the human N-ras p21 protein. Mol Cell Biol 7, 541-4. Tsuda, L., Inoue, Y. H., Yoo, M. A., Mizuno, M., Hata, M., Lim, Y. M., Adachi-Yamada, T., Ryo, H., Masamune, Y., and Nishida, Y. (1993). A protein kinase similar to MAP kinase activator acts downstream of the raf kinase in Drosophila. Cell 72, 407-14. Urano, T., Emkey, R., and Feig, L. A. (1996). Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular tranformation. EMBO Journal 15, 810-816. 137 Valencia, A., Kjeldgaard, M., Pai, E. F., and Sander, C. (1991). GTPase domains of ras p21 oncogene protein and elongation factor Tu: analysis of three-dimensional structures, sequence families, and functional sites. Proc Natl Acad Sci U S A 88, 5443-7. Van Aelst, L., Barr, M., Marcus, S., Polverino, A., and Wigler, M. (1993). Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci U S A 90, 6213-7. van Leeuwen, F. N., van der Kammen, R. A., Habets, G. G., and Collard, J. G. (1995). Oncogenic activity of Tiaml and Racl in NIH3T3 cells. Oncogene 11, 2215-21. Viskochil, D., Buchberg, A. M., Xu, G., Cawthon, R. M., Stevens, J., Wolff, R. K., Culver, M., Carey, J. C., Copeland, N. G., Jenkins, N. A., and et, a. (1990). Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62, 187-92. Vogel, U. S., Dixon, R. A., Schaber, M. D., Diehl, R. E., Marshall, M. S., Scolnick, E. M., Sigal, I. S., and Gibbs, J. B. (1988). Cloning of bovine GAP and its interaction with oncogenic ras p21. Nature 335, 90-3. Vojtek, A. B., and Cooper, J. A. (1995). Rho family members: activators of MAP kinase cascades. Cell 82, 527-9. 138 Vojtek, A. B., Hollenberg, S. M., and Cooper, J. A. (1993). Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74, 205-14. Volknandt, W., Pevsner, J., Elferink, L. A., and Scheller, R. H. (1993). Association of three small GTP-binding proteins with cholinergic synaptic vesicles. Febs Lett 317, 53-6. Wallace, M. R., Marchuk, D. A., Andersen, L. B., Letcher, R., Odeh, H. M., Saulino, A. M., Fountain, J. W., Brereton, A., Nicholson, J., Mitchell, A. L., and et, a. (1990). Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients [published erratum appears in Science 1990 Dec 21; 250(4988):1749]. Science 249, 181-6. Warne, P. H., Viciana, P. R., and Downward, J. (1993). Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 364, 352-5. Watanabe, G., Saito, Y., Madaule, P., Ishizaki, T., Fujisawa, K., Morii, N., Mukai, H., Ono, Y., Kakizuka, A., and Narumiya, S. (1996). Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 271, 645-8. West, M., Kung, H. F., and Kamata, T. (1990). A novel membrane factor stimulates guanine nucleotide exchange reaction of ras proteins. Febs Lett 259, 245-8. 139 Wildey, G. M., Viggeswarapu, M., Rim, S., and Denker, J. K. (1993). Isolation of cDNA clones and tissue expression of rat ral A and ral B GTP-binding proteins. Biochem Biophys Res Commun 194, 552-9. Willumsen, B. M., Papageorge, A. G., Kung, H. F., Bekesi, E., Robins, T., Johnsen, M., Vass, W. C., and Lowy, D. R. (1986). Mutational analysis of a ras catalytic domain. Mol Cell Biol 6, 2646-54. Wolfman, A., and Macara, I. G. (1990). A cytosolic protein catalyzes the release of GDP from p21ras. Science 248, 67-9. Wood, K. W., Sarnecki, C., Roberts, T. M., and Blenis, J. (1992). ras mediates nerve growth factor receptor modulation of three signaltransducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 68, 1041-50. Xu, G. F., Lin, B., Tanaka, K., Dunn, D., Wood, D., Gesteland, R., White, R., Weiss, R., and Tamanoi, F. (1990). The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63, 835-41. Xu, G. F., O'Connell, P., Viskochil, D., Cawthon, R., Robertson, M., Culver, M., Dunn, D., Stevens, J., Gesteland, R., White, R., and et, a. (1990). The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62, 599-608. 140 Yamamori, B., Kuroda, S., Shimizu, K., Fukui, K., Ohtsuka, T., and Takai, Y. (1995). Purification of a Ras-dependent mitogen-activated protein kinase kinase kinase from bovine brain cytosol and its identification as a complex of B-Raf and 14-3-3 proteins. J. Biol. Chem. 270, 1172311726. Yamochi, W., Tanaka, K., Nonaka, H., Maeda, A., Musha, T., and Takai, Y. (1994). Growth site localization of Rhol small GTP-binding protein and its involvement in bud formation in Saccharomyces cerevisiae. J Cell Biol 125, 1077-93. Zhang, K., De Clue, J. E., Vass, W. C., Papageorge, A. G., McCormick, F., and Lowy, D. R. (1990). Suppression of c-ras transformation by GTPase-activating protein [see comments]. Nature 346, 754-6. Zhang, X. F., Settleman, J., Kyriakis, J. M., Takeuchi-Suzuki, E., Elledge, S. J., Marshall, M. S., Bruder, J. T., Rapp, U. R., and Avruch, J. (1993). Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364, 308-13. Zheng, Y., Bender, A., and Cerione, R. A. (1995). Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. J Biol Chem 270, 626-30. Zheng, Y., Cerione, R., and Bender, A. (1994). Control of the yeast budsite assembly GTPase Cdc42. Catalysis of guanine nucleotide exchange 141 by Cdc24 and stimulation of GTPase activity by Bem3. J Biol Chem 269, 2369-72. Zheng, Y., Olson, M. F., Hall, A., Cerione, R. A., and Toksoz, D. (1995). Direct involvement of the small GTP-binding protein Rho in lbc oncogene function. J Biol Chem 270, 9031-4. Zigmond, H. S. (1996). Signal transduction and actin filament organization. Curr Opin Cell Biol 8, 66-73. 142