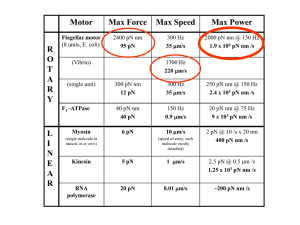
L~oE by Single-molecule studies of protein degradation ARCHE
advertisement

Single-molecule studies of protein degradation ARCHE and kinesin-8 motility by MASS^C APR 15 2015 B.S., Seoul National University (2007) S.M., Massachusetts Institute of Technology (2009) LIBRARIES Submitted to the Department of Mechanical Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mechanical Engineering at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2015 Massachusetts Institute of Technology 2015. All rights reserved. Signature redacted Author ..................... Department of Mechanical Engineering October 6, 2014 Matthew J. Lang Associate Professor of Chemical and Biomolecular Engineering, Vanderbilt University Thesis Supervisor Signature redacted C ertified by ....... ................................. Roger D. Kamm Professor of Mechanical Engineering Signature redacted Chair, Thesis Committee ......................... David E. Hardt Professor of Mechanical Engineering Chairman, Department Committee on Graduate Studies Accepted by ........... STTUTE L~oE by Yongdae Shin Certified by.redacted I I Single-molecule studies of protein degradation and kinesin-8 motility by Yongdae Shin Submitted to the Department of Mechanical Engineering on October 6, 2014, in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mechanical Engineering Abstract Molecular machines drive living organisms out of equilibrium and perform critical functions in almost every aspect of cellular processes. They are mechanical enzymes transducing free energy stored in chemical forms to generate motions and forces useful in cell. How these tiny machines operate in the presence of thermal agitation has been studied for a few model systems but still largely elusive. Especially, mechanisms of how molecular machines that were evolved from common ancestors diversified their machine actions to fit specific cellular requirements need to be answered. This thesis explores mechanisms of molecular machines in an effort to reveal roles of thermal fluctuations on machine functions, using two biological systems: ClpXP proteases and kinesin-8 Kif18B. As members of bigger protein families, these two fascinating nanomachines perform important cellular tasks in protein quality control and spindle formation, respectively. AAA+ ClpX unfoldases use energy from ATP binding and hydrolysis to drive mechanical unfolding and translocation of target proteins to associated peptidase ClpP. Previous ensemble biochemical and structural studies uncovered many aspects of degradation activity of ClpXP but the mechanistic understanding of ClpXP function is currently still lacking. We employed single-molecule biophysical techniques including optical trapping and single-molecule fluorescence to directly monitor unfolding and translocation activity of single ClpX hexamer as well as conformational dynamics of single subunit of ClpX. Statistical kinetic analyses on unfolding and translocation uncovered that unfolding kinetics were dominated by futile ATP hydrolysis and translocation steps contained more than one rate-limiting process for each physical stepping. Single-molecule fluorescence resonance energy transfer (smFRET) assay revealed dynamic switching of ClpX subunit conformations between multiple states. In the absence of nucleotide, ClpX explored available conformational spaces thermally in an erratic manner. Nucleotide binding to ClpX hexamer leads to ring contraction as well as defined hexameric conformation arrangements. Conformational transitions were not directly coupled to ATP hydrolysis, suggesting an important role of thermal fluctuation in ClpX machine function. 3 The Kinesin-8s are plus-end directed motors that negatively regulate microtubule length. The canonical members of this kinesin sub-family showed ultra-processivity which enables Kinesin-8s to enrich preferentially at the plus-ends of microtubules to alter microtubule dynamics. Kif18B is an understudied human Kinesin-8 that also limits MT growth during mitosis. Using single-molecule assays, we found that Kif18B was only modestly processive, and that the motor switched frequently between plusend directed and diffusive modes of motility. Measurements with truncated motors showed diffusion was promoted by a second MT-binding site located in the Kifl8B tail. Our model accounting for motility switching is consistent with autoinhibition mechanism of Kinesin-1, implying that kinesins may share common regulatory mechanisms to drive varying functional consequences. Thesis Supervisor: Matthew J. Lang Title: Associate Professor of Chemical and Biomolecular Engineering, Vanderbilt University 4 Acknowledgments Looking back on past years of graduate school, many things have happened in my life. It was not only an academic journey but also a time to look into myself. Facing the end of the graduate life, I am happy that I feel like I know what I want to do and what I should do better than I did 7 years ago. I would like to thank all people who gave me huge support and help and made my graduate life productive and enjoyable. I want to thank my advisor, Matt Lang for having me in his laboratory. When I first came to MIT, I was a naive college graduate and rather a theory-minded person. He trained and guided me to think how to precisely measure things and to build high-end instruments to accomplish that. Now I always think how better measurements can be made to tackle problems facing me. In addition to being a fantastic research advisor, Matt has been a mentor for personal stuffs as well as my career. I really appreciate his support and patience during my stay in his lab and it is just impossible to list here all his role and contribution during my graduate study. I also want to thank Bob Sauer, Tania Baker and Ryoma (Puck) Ohi for giving me a chance to work on ClpXP and Kif18B. Bob and Tania provided insightful advices on ClpXP, without them it would have been impossible to continuously make progress in my ClpX study. Puck introduced me a world of cell biology. His support and encouragement helped me to keep moving on. I am also grateful to my thesis committee, Roger Kamm and Peter So, for their commitment and critical advice during my graduate study. I was very fortunate enough to work with many talented collaborators. Joey Davis and Andreas Martin taught me how to work with ClpXP and made critical contributions to make the first single-molecule fluorescence assay for ClpXP. Adrian Olivares's invaluable contribution made it possible for us to propose the mechanochemical cycles of ClpX. I also want to thank Ben Stinson, Andrew Nager and Karl Schmitz for their significant help in developing the fluorescence assay for ClpXP conformational dynamics. Sun Taek Kim introduced me to the exciting world of immunology. I am also thankful to Yaqing Du who provided most kinesins I used in my thesis and taught 5 me biology of the cell division. It has been a great pleasure to interact and share memory with the former and current Lang Lab members; Carlos Castro, David Appleyard, Ricardo Brau, Mo Khalil, Jorge Ferrer, Hyungsuk Lee, Marie Aubin-Tam, Hoi Siew Kit, Bill Hesse, Ted Feldman, Juan Carlos Cordova, Sonia Brady, Harris Manning, Yinnian Feng, Nikki Reinemann, Taishi Zhang, James Smith and Dibyendu Das. Everyday life was enjoyable due to their presence in the lab and it has been great fun to exchange scientific idea with them. I also want to thank my friends in KGSAME for their invaluable support. Finally, and most importantly, I am thankful to my family for their countless encouragement and support; many thanks to Mom, Dad and Nuna. This work is dedicated to my wife, Mihee. It was only possible with her support and love. I cannot finish without mentioning my little boy and girl; Jeongmu and Jeongwon. Their smile made me to forget all hard works in the lab and to realize what is important in my life. 6 Contents 1 Introduction 15 1.1 Molecular Machines . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16 1.1.1 ClpXP Protease . . . . . . . . . . . . . . . . . . . . . . . . . . 18 1.1.2 K inesin . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21 Single-Molecule Techniques . . . . . . . . . . . . . . . . . . . .. . . . . 23 1.2.1 Single-Molecule Fluorescence . . . . . . . . . . . . . . . . . . . 24 1.2.2 Optical Trapping . . . . . . . . . . . . . . . . . . . . . . . . . 28 1.2 2 3 Monitoring conformational dynamics of single ClpX subunits 31 2.1 Sum m ary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31 2.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32 2.3 Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 2.3.1 Single-Molecule FRET assay for ClpXP . . . . . . . . . . . . . 33 2.3.2 Nucleotide dependence of ClpX conformation . . . . . . . . . 35 2.3.3 Substrate dependence . . . . . . . . . . . . . . . . . . . . . . . 39 2.3.4 K inetics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39 2.4 D iscussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 2.5 Materials and Methods . . . . . . . . . . . . . . . . . . . . . . . . . . 45 2.5.1 Single-molecule fluorescence assay . . . . . . . . . . . . . . . . 45 2.5.2 Data analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . 46 Single-molecule kinetics of ClpXP-mediated protein unfolding and 47 translocation 7 . . 48 . 49 3.3.1 Single-molecule optical trapping assay of ClpXP . . . . . . . 49 3.3.2 Single-molecule kinetics . . . . . . . . . . . . . . . . . . . 51 3.3.3 Multiple ATP hydrolysis cycles yet single rate limiting step 56 3.3.4 Translocation kinetics . . . . . . . . . . . . . . . . . . . . 59 Materials and Methods . . . . . . . . . . . . . . . . . . . . . . . . 64 3.4.1 Protein constructs 64 3.4.2 Single-molecule mechanical measurements . . . . . . . . . 64 3.4.3 Kinetic analysis . . . . . . . . . . . . . . . . . . . . . . . . 65 Summary 3.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.3 Results and Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.4 Non-canonical Motility of Kif18B Sum m ary . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67 4.2 Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68 4.3 R esults . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 4.3.1 Distinct motile properties of Kif18B . . . . . . . . . . . . . 70 4.3.2 Dual mode of motility: diffusion and directed motion . . . . 74 4.3.3 The tail domain of Kif18B : regulator of motility . . . . . 77 4.3.4 Motility of Kif18B under load . . . . . . . . . . . . . . . . 79 4.4 D iscussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81 4.5 Materials and Methods . . . . . . . . . . . . . . . . . . . . . . . . 83 4.5.1 Protein constructs . . . . . . . . . . . . . . . . . . . . . . 83 4.5.2 Single-molecule fluorescence assay . . . . . . . . . . . . . . 83 4.5.3 Video tracking assay and optical trapping assay . . . . . . 84 . . . . . . . . . 4.1 . 67 . 4 47 3.1 A ClpXP Protocols 85 85 A.2 Single-Molecule Fluorescence Assay . . . . . . . . . . . . . . . . . . 87 . . A.1 Suface Passivation Using PEG . . . . . . . . . . . . . . . . . . . . . 8 B Kif18B Protocols 89 B.1 Tubulin Polymerization . . . . . . . . . . . . . . . . . . . . . . . . . . 89 B.2 Single-Molecule Fluorescence Motility Assay . . . . . . . . . . . . . . 91 B.3 Kinesin Bead Assay . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93 9 10 List of Figures 1.1 M olecular m achines ............................ 1.2 ClpXP, protein degradation machine . . . . . . . . . . . . . . . . . . 19 1.3 General domain organization of the kinesin . . . . . . . . . . . . . . . 22 1.4 Diverse motility of kinesin . . . . . . . . . . . . . . . . . . . . . . . . 23 1.5 Jablonski diagram for fluorescence and FRET . . . . . . . . . . . . . 25 1.6 Qualitative depiction of the optical trapping . . . . . . . . . . . . . . 29 2.1 Single-molecule FRET assay for ClpXP . . . . . . . . . . . . . . . . . 34 2.2 Nucleotide binding leads to contraction . . . . . . . . . . . . . . . . . 36 2.3 Correlation analysis for varying nucleotide conditions . . . . . . . . . 37 2.4 The effect of substrates on ClpX conformations 40 2.5 State identification using hidden Markov Modeling . . . . . . . . . . 42 2.6 Dwell time distributions for U and L conformations . . . . . . . . . . 43 3.1 Optical trapping assay of ClpXP . . . . . . . . . . . . . . . . . . . . 50 3.2 Example traces of ClpXP unfolding and translocation . . . . . . . . . 52 3.3 Completion time distributions report the number of rate limiting steps 53 3.4 Distributions of randomness parameters for three kinetic schemes . . 56 3.5 Unfolding completion time distributions . . . . . . . . . . . . . . . . 57 3.6 Fluctuation analysis reveals hidden kinetic information . . . . . . . . 60 3.7 Fluctuation analysis on protein translocation by ClpXP . . . . . . . . 62 4.1 K if18B is dim eric. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71 4.2 Distinct motile properties of Kif18B. . . . . . . . . . . . . . . . . . . 72 11 17 . . . . . . . . . . . . Kifl8B dwells shortly at the microtobule end. . 73 4.4 Kif18B has dual mode of motility. . . . . . . . . . 75 4.5 Kif18B spends the majority of time in diffusion. . 76 4.6 Tailless motors show only directed motion. . . . . 78 4.7 Two proposed mechanisms of motility switching . 79 4.8 Diffusional behaviors of the tailless motor and the tail domain 80 4.9 Kif18B is a low-force motor 81 . 4.3 . . . . . . . . . . . . 12 List of Tables 4.1 Summary of motile parameters from MSD analysis 13 . . . . . . . . . . 80 14 Chapter 1 Introduction A hallmark of life is that living matter is out of equilibrium 11]. Any biological matter from bacteria to humans continuously exchanges energy and materials with their surroundings. Moreover, internal dynamic gradients in temperature, mechanical force and molecular species are maintained as long as they are alive. Ultimately, biological non-equilibrium states originate from the level of constituent molecules. In living organisms, there exist numerous biomolecules which operate in unidirectional manner, forbidden at thermodynamic equilibrium according to the principle of detailed balance, using energy from chemical, electrochemical or photonic sources [2, 3]. These biomolecules, termed molecular machines, often form higher-order multiprotein complexes and are involved in almost every aspect of cellular processes such as cell signaling, growth, metabolism, movement, adhesion, and division. The thermal energy, kT, does not depend on an object's size unlike other forms of energy. At the length scale of biomolecules, it becomes comparable to those that maintain structures of biomolecules such as van der waals energy or hydrogen bonds 14]. This leads to inevitable fluctuations in every molecular processes. Living matter ranging from molecular machines to cells have evolved to deal with thermal fluctuations and sometimes even utilize them for their functions [5, 6, 7, 8]. This thesis is devoted to studying mechanisms of molecular machines with emphasis on roles of thermal fluctuations on machine functions. Two biological systems are chosen as model systems: ClpXP protease and Kinesin-8 Kif18B. Both proteins 15 are diversified from each protein family sharing similar architectures in energy transduction. A broader understanding of linkage between specific motile properties of molecular machines and their specialized cellular function is clearly necessary and pursued to answer in this thesis. To address these, collective approaches are taken using state-of-the-art biophysical techniques, statistical analysis as well as numerical simulations. The system manipulation aspects of this research were done in collaboration with the Sauer Lab and Baker Lab at Department of Biology, MIT for the ClpXP work and the Ohi Lab at Department of Cell and Developmental Biology, Vanderbilt for the Kif18B work. Chapter Two describes a single-molecule assay to monitor conformational dynamics occurring in single subunit of ClpXP. Next, the enzymatic activity of ClpXP is directly measured using optical trapping and analyzed with statistical kinetics in Chapter Three. Finally, Chapter Four presents unusual motile behaviors of Kif18B as well as the underlying molecular mechanism of motility. 1.1 Molecular Machines The first motor protein identified was skeletal muscle myosin, which is also called myosin II, which is responsible for generating forces during muscle contraction 191. Typically, the motor proteins refer to those that interact with cellular cytoskeleton such as actin and microtubule, and include myosin, kinesin and dynein. However, molecular machines can be more broadly defined as a collection of molecules that generate mechanical motions, like macroscopic machines, by converting energy from other sources, mostly chemical sources. They include a series of proteins involved in the central dogma of molecular biology which generate unidirectional mechanical motions along nucleic acid tracks such as DNA polymerase, RNA polymerase, ribosome, helicase, topoisomerase and so on. ATP synthases and bacterial flagellum are examples of rotary molecular machines. The molecular machines can also incorporate synthetic molecules recently designed and demonstrated [10, 11, 12]. In cells, the most prevalent source of energy storage is a nucleotide such as adeno16 a b *\Cago binding N Multimedzation Lover Traniocalon ftrns&"cr F Track Figure 1.1: Molecular machines. (A) Cartoon diagram for a general translocating motor, illustrating its mechanical components. Small conformational changes occuring at the fuel processor are transduced into the force generator where mechanical work is produced. Typically, molecular machines possess levers whose action amplify mechanical motions. Typically, multimerization is necessary for processive motion of the motor along its partner track. (Figure adapted from 1131). (B) Actin protrusion as an example molecular machine. Actin filament is a dynamic biopolymer which constitutes important family of the cytoskeleton. Cycles of ATP binding and hydrolysis in actin monomers (G-actin) drive polymerization of actin filament against the cell membrane, generating protrusive forces by rectifying Brownian motion [8]. Blue circles are Arp2/3 complexes responsible for generating branched networks of actin filaments. sine triphosphate (ATP). An ATPase domain in molecular machines acts like a automobile engine converting chemical energy stored in the form of ATP to mechanical motions. Typically, structural rearrangements near ATP binding pocket in the ATPase domain during ATP hydrolysis cycle are tiny (on the order of A). These small conformational changes near the ATP binding site are transduced and magnified through a series of structural elements in the molecular machines to ultimately produce nanometer scale motions along their partner tracks (Fig. 1.1a). How this coupling between chemical cycle of ATP hydrolysis is linked to the mechanical motion is called mechanochemical (or chemomechanical) coupling and it is a major theme in studies of molecular machines. Some subcellular organelles can be also regarded as molecular machines. For example, acto-myosin networks in the leading-edge of motile cells as well as the spindle complex of dividing cells are dynamic ensemble of 17 molecules where nucleotide hydrolysis impart force generation and motility to drive complex cellular behaviors (Fig. 1.1b). 1.1.1 C1pXP Protease The AAA+ protein family is a large and functionally diverse group of enzymes present in all kingdoms that are able to induce conformational changes in a wide range of target substrates using energy from ATP hydrolysis. The functional repertoire of the AAA+ enzymes range from protein degradation and DNA replication to membrane fusion and the movement of microtubule motors [14]. From bacteria to humans, intracellular degradation driven by a series of AAA+ proteases plays vital roles not only in general protein-quality control but also in the regulation of a variety of cellular processes such as the response to environmental stress/DNA damage and adaptation to changing growth conditions. [151. The work herein is focused on ClpXP, a relatively simple and well-characterized AAA+ protease, which serves as a model system for other ATP-dependent proteases, including ClpAP, HslUV, Lon, FtsH, and the 26S proteasome. ClpXP is an ATP-dependent protease that consists of ClpX, a homohexameric AAA+ ATPase, and ClpP, a tetradecameric peptidase (Fig. 1.2). The active proteolysis sites in ClpP are sequestered in a hollow interior chamber which is only accessible through axial portals too narrow to admit folded native proteins. This restriction prevents the unregulated destruction of most cellular proteins but requires the assembly of ClpXP complex in which ClpX performs recognition, denaturation and translocation of correct substrates to achieve proper degradation [161. Irreversible protein degradation is tightly regulated at the substrate recognition stage by ClpX. Several types of short unstructured peptide sequences at both C and N terminus are known to serve as degradation tags for ClpX including the ssrA tag (AANDENYALAA-COOH) [16]. When ribosomes stall during protein synthesis in eubacteria, the ssrA tag is added to the incomplete nascent peptide by the tmRNA system, ensuring degradation of these molecules [17]. Appending the ssrA tag to any proteins generates substrates for ClpXP, which allows biochemical and biophysical 18 tagV unfokdlas trsnekoce peptidase ADP ADP fm substrate and enzyme recognition uannpendentg demdation Figure 1.2: Cartoon model of ClpXP mediated protein degradation activity. The ClpXP protease is composed of a unfoldase ClpX and a peptidase ClpP. ClpXPdriven protein degradation consists of four distinct steps: recognition, unfolding, translocation and proteolysis. In an initial substrate recognition step, a peptide tag in the substrate binds in the axial pore of the ClpX hexamer. In subsequent ATP-dependent steps, ClpX mechanically unfolds the substrate and translocates the unfolded polypeptide into the degradation chamber of ClpP, where it is cleaved into small peptide fragments. (Figure adapted from [161) studies of the ClpXP operation. Several mutational studies on ClpX core residues, located at the Walker B and Sensor II motifs, demonstrated that ATP binding but not hydrolysis is required for productive binding of ClpX to both ClpP and substrates [18]. Substrate degradation, however, depends on the rate of ATP hydrolysis generally in a linear manner, yet the unfolding of GFP by ClpXP displayed nonlinear dependence, suggesting a complex interplay between the ClpXP machinery and the energy landscape of substrates 1191. Three types of loops, RKH, pore-1 and pore-2 loop, located along a central pore of ClpX are essential in mediating translocation and mechanical unfolding of substrates as well as binding to ClpP [19, 20]. Several studies suggest ClpXP successfully unfolds stable substrates only after numerous failed trials: 1) ClpXP maintains a similar ATP turnover rate to degrade substrates ranging in stability but simply spends more time and ATPs to degrade more stable ones 1211. 2) Recent single-molecule optical trap assays showed no detectable 19 movement during long dwells between the end of translocation and the next unfolding events [22]. Encountering an ultra stable protein that stubbornly resists unfolding, ClpX prevents being stalled by partitioning between frequent release and infrequent denaturation of the substrate [231. ClpXP can even process multiple polypeptide chains simultaneously [24]. The molecular mechanism of ClpXP mediated protein degradadtion is currently under investigation. Recent application of single-molecule techniques to ClpXP studies allowed for direct observation of individual unfolding and translocation events by ClpXP [22, 25] and showed that ClpXP could work against forces of 20 pN or higher. They further demonstrated that the smallest translocation steps were - 1 nm and revealed physical steps that were multiples of this value, resulting from two, three, or four power strokes happening simultaneously. Recently, a phosphate release was proposed to be a force-generating step in the ATP hydrolysis cycle [26]. Based on the observation that dwell times before translocation steps were constant across varying concentrations of ATP, they proposed a mechanochemical cycle where size of steps were dictated by the numbers of ATP binding during a constant time set by 'internal clock'. However, the propose model allows ATP hydrolysis and mechanical action even from ClpX hexamers with two ATPs bound, which conflicts with the biochemical observation that ATP binding to subunits with weak-affinity drives conformational changes required for the ClpX ring to hydrolyze ATP and perform mechanical work [27j. The nature of rate-limiting kinetic steps in ClpX translocation is also a mystery. Translocation velocity of ClpXP is insensitive to small force, indicating that the force-generating step is not rate-limiting in the unloaded translocation kinetics [22, 25]. Since - 1 mM ATP concentration is well above saturation levels in these measurement, other steps in ATP hydrolysis cycles such as hydrolysis and ADP release or some kind of conformational changes can be rate-determining steps. Thus, it will be highly advantageous to study details of degradation kinetics in varying conditions and to monitor conformational changes of ClpX in action. 20 1.1.2 Kinesin Kinesin is also a molecular machine that converts the chemical energy from ATP hydrolysis to mechanical motions along microtubule tracks. Kinesin was discovered in search of a molecular factor responsible for axonal transport [28]. In the human body, axons can be up to one meter in length and the active transport of organelles by kinesin is necessary to speed up molecular exchange between the neuronal cell body and the synapse. Since the discovery of this conventional kinesin (also called kinesin-1 or KIF5), molecular, genetic, and biochemical analyses of various organisms have identified a superfamily of proteins that share a kinesin motor domain ( 350 amino acids). On the basis of homology between motor domains, the kinesin related proteins have been classified into 14 families (kinesin-1 to kinesin-14) [29]. The kinesin motor protein is modular and generally consists of a kinesin motor domain, a coiled coil stalk and a tail domain (Fig. 1.3). The motor domain binds to the microtobule and generates force during the ATP hydrolysis cycle. The stalk is mostly an oz- helical coiled coil which is important for oligomerization but sometime harbors hinge segments which enable the folding of the tail region and interaction with the motor head for regulatory purposes [30]. The tail domain is responsible for kinesin binding to cargo, adaptors or scaffold proteins. Kinesin walks on the microtubule hand-over-hand manner [31, 32]. As far as the conventional kinesin was concerned, the mechanochemical coupling has been highly studied and reasonably well established [33, 13]. Essentially, nucleotide dependent changes of the motor head affinity to the microtubule as well as coordination between two motor domains turned out to be key components for high processivity of the conventional kinesin. Since the microtubule has polarity, specific interaction between the motor head and its corresponding track generates unidirectional motion. Typical N-kinesin (kinesin with N-terminal motor domains) walks toward the plus end of microtubules (Fig. 1.4a). However, C-kinesin (kinesin with C-terminal motor domains) provides minus-end directed motility. Interestingly, M-kinesin (kinesins with motor domain in the middle) does not show directional motility yet performs diffusion on 21 Motor domain Coiled coil Oligomerization Tail domain - Regulation - Targeting " ATP hydrolysis " Microtubule binding Figure 1.3: General domain organization of the kinesin. Most kinesin are comprised of an N-terminal motor domain, coiled coil and C-terminal tail domain. Exceptions from this domain organization include Kinesin-14 with C-terminal motor head and Kinesin-13 with the motor head in the middle of the protein. The motor domain is responsible for ATP hydrolysis and microtubule binding, where the coupling between them as well as coordination between two motor heads result in processive motion of kinesin along the microtubule. The length as well as function of the tail domain vary highly between different types of kinesins. The tails contain recognition sequences for co-proteins, regulatory kinases and cargo. the microtubule. The mechanisms of directionality in the kinesin motility have been studied and a neck region linking the motor head to the coiled-coil stalk is shown to be important in determining the motor directionality [34, 35, 36, 37]. The prototypical Kinesin-1 moves along a single protofilament of the microtubule [38, 391 (Fig. 1.4). However, several studies showed that some kinesins such as Kinesin-2, Eg5 and Kinesin-8 kip3 switched their profilament tracks in a biased way thereby generating helical motions on the microtubule lattice [40, 41, 421. The stability of the neck region was thought to be the origin of this torque generation. The side stepping could be advantageous in motility on the potentially highly crowded surface of microtubules to evade macromolecular obstacles encountered in the linear path. Recently, dynein is also shown to have helical motility but unlike kinesins dynein displayed bidirectional helical motion 1431. More work is necessary to determine the significance of the propensity of molecular transporters to side step. 22 n h N-terminal motor: plus-end directed Move along a single protofilament C-terminal motor: minus-end directed Central motor: diffusion Move along a helical path - __ _ _ _ _ + Figure 1.4: Diverse motility of kinesin. (A) Generally speaking, a location of the motor domain in the polypeptide sequence dictates the direction of motility of the protein. Kinesins with an N-terminal motor domain show the plus end directed motility, whereas kinesins with a C-terminal motor (such as Kinesin-14) undergo motility to the minus end. Kinesin motors having a central motor domain (such as Kinesin-13) do not undergo directed motility but instead perform diffusion. (B) The prototypical kinesin-1 moves along a single protofilament on the microtubule. However, recently several kinesins are shown to spiral around microtubules at varing degrees, suggesting functional advantages of helical motions on the microtubule. 1.2 Single-Molecule Techniques Recent advances in single-molecule techniques have allowed for probing motions of individual molecules with nm accuracy and manipulating them with pN force resolution [44, 45, 461. Notably, these length and force ranges exactly cover biomolecular scales, which is one of reasons why single-molecule tools have been so powerful in studying biomolecules. Single-molecule approaches have allowed for tracking dynamic behaviors of individual proteins over microsecond to minute time scales, detecting rare but important events which are averaged out in ensemble studies, and observing inherent stochastic fluctuation of biological processes. Among single-molecule manipulation methods, optical tweezers have been used extensively to measure displacements and forces generated by individual motor proteins such as kinesin [47] and to perturb their free energy to understand underlying mechanisms. Single-molecule localization and fluorescence resonance energy transfer (FRET) methods using total internal reflec23 tion fluorescence (TIRF) microscopy has allowed for measuring the activity of single enzymes [481 and monitoring conformational changes of proteins and nucleotides in action [49, 50]. These two branches of powerful single-molecule measurement tools have been combined to demonstrate the capability of providing high-resolution mechanical control over molecular conformations with fluorescence-based structural reporting [51, 521. Here, I briefly describes basic principles of single-molecule fluorescences as well as optical trapping. 1.2.1 Single-Molecule Fluorescence Fluorescence is a luminescent process in which susceptible molecules at an electronically excited state return to the ground energy level by emitting light (Fig. 1.5) 153]. Fluorescent molecules absorb light at a particular wavelength determined by their electronic energy levels and subsequently emit light of longer wavelength after a brief interval, called the fluorescence lifetime (typically - ns). The difference in wave- lengths between the absorbed and emitted light is known as Stokes shift and implies the existence of energy loss during the fluorescence cycle. In fact, excited electrons in the excited singlet state, Si, an energy level too high to be thermally populated at room temperature, undergo fast vibronic relaxation to the lowest vibrational level of the excited single state before returning to the ground state by fluorescence. Due to the energy loss in this vibronic relaxation process, emitted light has less energy and longer wavelength compared to the absorbed light. Because the time scale of the relaxation process is typically several orders of magnitude faster than the fluorescence lifetime, vibronic relaxation is complete prior to emission, resulting in an emission spectra independent of the excitation wavelength. Electrons in an excited singlet state can also undergo intersystem crossing to reach an excited triplet state (Fig. 1.5). A transition with emission of light from the excited triplet state to the ground energy level is also possible yet much slower than typical fluorescence and is termed phosphorescence. Long-lived dark triplet state is also problematic since the molecule can interact with oxygen resolved in solution to generate reactive singlet oxygen species. Molecular oxygen is a primary source of 24 ksc S, k,hva k ,hv, kr so b kFRET SD1 S 0A ;A S0 D S 0^ -_ Figure 1.5: Jablonski diagram for fluorescence and FRET. (A) A molecule in the ground singlet state So is excited to one of vibrational sublevels of the first excited singlet state S1 upon absorption of a photon of energy hva. The excited molecule will then rapidly undergo vibrational relaxation to the lowest energy level of state Si before returning to the ground state via multiple possible pathways. Fluorescence occurs when the molecule returns by emission of a photon of energy hve (< hva; red-shifted). Alternatively, excited molecules undergo intersystem crossing to the first excited triplet state T 1 , where they can return to the ground state via slow phosphorescence. Multiple relaxation pathways compete kinetically each other. In the diagram, k denote rate constants for each conversion. (B) FRET takes place when 1) the emission spectrum of a donor D and the absorptoin spectrum of an acceptor A overlaps and 2) two dyes are in close proximal (typically < 10nm). Under these conditions, donor fluorescence emission is quenched by nonradiative energy transfer to the acceptor by dipole-dipole interaction. This energy transfer rate depends on the distance between the dye pair, thus providing a rationale of using FRET as a nanoscopic ruler. (Figure adapted from [541) 25 early photobleaching so it is essential to remove molecular oxygen for achieving longer observation times. A enzymatic oxygen scavenging system based on glucose, glucose oxidase and catalase has been widely to effectively remove oxygen during fluorescence measurements. Since oxygen also has a positive role as a triplet quencher, removing oxygen should be accompanied by employing an alternative triplet quencher [55]. 0mercaptoethanol has long been used for this purpose, but recently Trolox was shown to have superior performance in terms of stabilizing fluorescent molecules [56J. Fluorescence resonance energy transfer (FRET) is yet another way of relaxing excited energy (Fig. 1.5b). FRET occurs when another molecule (called an acceptor) with its absorption spectra overlapped with the emission of original molecule locates in vicinity (typically < 10 nm). It is due to nonradiative dipole-dipole interactions and the energy transfer rate depends on the distance between two molecules, R as: kFRET _1(Ro6 (1 1 -o R where To is the donor fluorescence lifetime in the absence of acceptor and RO is the Forster radius. RO is proportional to the orientation factor K2, the donor-acceptor spectral overlap J (in M'cm-'nm 4 ) and the quantum yield of the donor bD RO = 0.21 (I 2 4DJn - 4 ) 1 /6(in A) (1.2) where n is a refractive index of medium. The orientation factor equals to 2/3 for isotropic rotation. Generally, K2 is defined as = where 0 T (2 sT - 3cosODCOsOA) 2 (1.3) is the angle between the donor emission dipole and the acceptor absorption dipole and OD (repectively, OA) is the angle between the donor-acceptor connection line and the donor emission dipole (repectively, acceptor absorption dipole). The 26 donor-acceptor spectral overlap J (in M - J = where fD j cm- nm4 ) is (1.4) fD(A)E(A) A 4 dA is normalized emission spectrum of the donor and CA (in M-'cm- 1 ) is the molar extinction coefficient of the acceptor. The wavelength A is in nm. The FRET efficiency E is defined as EkFRET kr + knr + kFRET where kFRET, kr and knr [ (R Ro )6 _ T O (1.5) indicate FRET, radiative and nonradiative rate respectively. The FRET efficiency can also be measured ratiometrically from the donor and acceptor fluorescence intensities IA and E ID : (1.6) = 1A - iYID where -y is a correction factor incorporating the donor and acceptor quantum yield (41) and the detection efficiencies of both channel (y) TIDD (1.7) Experimentally, 'y factors are computed as the ratio of change in average acceptor intensity (AIA) to change in average donor intensity (AID) upon acceptor photo- bleaching [57] AIA AITD (1.8) All fluorescence measurements presented in this thesis were performed using a custom-built instrument based on an inverted microscope with objective-side total internal reflection (TIR) fluorescence capabilities. The instrument is equipped with three fiber-coupled excitation lasers (488 nm, 532 nm and 642nm, all from Blue Sky Research, Milpitas, CA) whose output power can be controlled by an NI board (National Instruments, Austin, TX) and a custom program written in Labview software 27 (National Instruments). All lasers are collimated from fiber outputs and then expanded before being focused at the back focal plane of the objective (100X, 1.45 NA, Nikon, Tokyo, Japan) for TIR. Emitted fluorescence signals are collected with the same objective, split into the donor and acceptor emissions, and imaged side by side on the EMCCD camera (Andor Technology, Belfast, UK). For single color imaging (for example, ClpX labeled with TAMRA or Cy3 or GFP-Kifl8B), simply only the donor side of the CCD is used with appropriate dichroic filter sets. 1.2.2 Optical rapping An optical trap, also known as an optical tweezer, was first demonstrated by Arthur Ashkin where he showed microscopic dielectric particles can be trapped using radiation pressure alone [581. A three dimensional potential well forming the optical trap is the result of gradient restoring forces that are best described by geometric optics in the ray optics (Mie) regime (i.e., when the object's dimension d is much larger than the wavelength of trapping light: d << A) and by electromagnetic dipoles in the Rayleigh regime (i.e., d << A) [59]. In practice, most applications involving micrometer-sized particles and near-infrared lasers require an approximate combination of the two regimes. Nonetheless, it is useful and informative to provide a short description of optical forces generated from both regimes. In the ray optics regime, the gradient forces that collectively make up the optical trap arise from momentum exchange between refracted light rays and dielectric particles. The net effect of the gradient forces is to generate restoring forces toward the focus of a trapping laser, as depicted in Fig. 1.6. Since there are some photons reflected and absorbed by the particle, there is also a small amount of net force, called a scattering force, acting on the particle in the direction of light propagation. Thus, successful trapping is only achieved when the gradient force along the optical axis overcome the scattering force, which necessitates the very steep optical gradient achieved by using a high NA objective and slightly overfilling the back aperture of the objective. In the Rayleigh regime, the particle is treated as a point dipole in an inhomogenous 28 a LASER SEAM CAMCOPE LES ik bb 0 b bb f C F FS a b Figure 1.6: Qualitative depiction of the optical trapping. Two rays of light refract when entering into a dielectric sphere with a refractive index higher than the surrounding. Refraction of light requires transfer of momentum from the sphere to light. By the principle of momentum conservation, the same amount of momentum is delivered from light to the sphere which always pushes the sphere towards the focus of the trapping beam. (Figure adapted from [60]) 29 electromagnetic field. The gradient force is given as Fgrad 2a V0 cnm2 (1.9) where a = nm2a3(r 1) 22 (1.10) where 1o is the intensity of the incident light, c is the speed of light in vacuum, m is the ratio of the index of refraction of the particle to the index of the medium, nm is the index of refraction of the medium and a is a radius of the particle. The gradient force is proportional to the intensity of gradient, and points up the gradient toward the focus of light when m > 1. The experiments in this thesis were performed using custom-built instruments, described previously [22, 511. Briefly, optical trapping is achieved by tightly focusing a 1064 nm laser with a high numerical aperture objective (10OX, 1.40 NA, oil IR; Nikon, Tokyo, Japan). The trap location is computer-controlled with a pair of orthogonally oriented acousto-optic deflectors (AODs; intra-Action, Bellwood, IL) and the sample position is manipulated using a nanometer-resolution piezo-stage (Polytech PI, Auburn, MA). A separate 975 nm laser is employed in conjunction with a position sensitive device (PSD; Pacific Silicon, West Lake Village, CA) to monitor position of beads using back-focal-plane interferometry [61]. The second pair of the optical trap and detection laser are employed if necessary by splitting each laser with polarizing beam splitters. The output volages from the PSD are collected with an A/D board (National Instruments, Austin, TX) and custom programs coded in Labview software (National Instruments) are used to control experimental runs and data acquisition. 30 Chapter 2 Monitoring conformational dynamics of single ClpX subunits 2.1 Summary AAA+ ClpX unfoldases utilize energy from ATP binding and hydrolysis to drive mechanical unfolding and translocation of target proteins to a degradation chamber. Crystal structures of the ClpXP hexamer showed two distinct classes of subunit conformations where only one type of conformation allows nucleotide binding. A recent ensemble measurement suggested conversions between subunit classes can occur, yet the exact nature of transitions as well as their mechanisms are not known. Here, we study conformational dynamics of single ClpX subunits that are critical for mechanical action of the molecular machine. Single-molecule FRET assay allows direct observation of ClpX subunit conformation in real time and reveals dynamic switching between multiple conformational states. In the absence of nucleotide, ClpX explores available conformational space thermally in an erratic manner. Nucleotide binding to ClpX hexamer leads to ring contraction as well as defined hexamer arrangements. Conformational changes are not directly related to ATP hydrolysis, suggesting an important role of thermal energy in the machine function. 31 2.2 Introduction AAA+ molecular machines use the chemical energy of ATP hydrolysis to power the degradation, remodeling, disassembly, or molecular transports in a wide range of cellular processes [14]. ClpXP is an ATP-dependent protease that consists of ClpX, a hexameric-ring AAA+ unfoldase and ClpP, a peptidase. Controlled protein degradation occurs via ClpX-mediated specific recognition of target substrates, followed by mechanical denaturation and translocation of unfolded polypeptides into the ClpP chamber for proteolysis [161. Each subunit of the ClpX hexamer is identical in sequence and consists of large and small AAA+ domains and a family-specific N domain that is not required for degradation of native ssrA-tagged proteins [62]. A series of pore loops along a central channel of the ClpX ring was shown to be important for efficient substrate recognition and unfolding, and thought to be linked to ATP hydrolysis cycles. Recent single-molecule studies on ClpXP began to uncover mechanochemical coupling of ATP hydrolysis and degradation activity [22, 63, 25, 26]. In current models, ClpX fires bound ATPs to produce multiples of 1 nm steps where the step size is dictated by the number of ATPs hydrolyzed concomitantly. Kinetics of translocation were required to have either an internal clock 126] or conformational changes activating the ClpX ring [63] but the exact nature of this critical kinetic step is still elusive. Several previous biochemical and structural studies showed asymmetry in organization and function of ClpX subunits [64, 65, 271. Crystal structures of the ClpX hexamer revealed two distinct classes of subunits (nucleotide loadable L subunits and unloadable U subunits) that are arranged in an L/U/L/L/U/L pattern (Fig. 2.1b). For all subunits, the large AAA+ domain forms a rigid-body unit with the small AAA+ domain of the counterclockwise subunit. U and L subunits differed in relative orientation of the small domain to large one. Nucleotide binding pockets were located at the hinge region connecting two AAA+ domains(Fig. 2.1a), thus ATP binding and hydrolysis were thought to alter the orientation between the domains, ultimately giving rise to allosteric effects across subunits via rigid-body interfaces. In fact, a recent 32 ensemble study using novel fluorescence assays showed that initial nucleotide loading to the ClpX enzyme drives staged allosteric changes, setting a ring ready for ATP hydrolysis and mechanical work [27]. They also used a mutant ClpX with low nu- cleotide affinity to show all subunits go through conformational changes between U and L. However, the driving force of this conformational switching as well as kinetics of U-L switching has been unknown. Moreover, monitoring conformational changes of single subunits in multi-protein complexes has been technically challenging. 2.3 2.3.1 Results Single-Molecule FRET assay for ClpXP To address these questions, we used single-molecule fluorescence resonance energy transfer (smFRET) and total internal reflection fluorescence (TIRF) microscopy to monitor conformational dynamics of single ClpX subunit within the hexamer. For the specific labeling of a FRET pair to designed locations, we constructed a sortaselinked single-chain ClpX hexamer. First, two engineered trimers with single reactive cysteines, X-X-Q174C and D170C-X-X, were labeled with an acceptor (Alexa 647) and a donor (Cy3), respectively. Then, dye-labeled trimers were linked using the sortase reaction to generate single-chain ClpX hexamers with a single FRET pair. In the assembled ClpX hexamer, the dye pair was positioned in the large domains of neighboring subunits (Fig. 2.1c), resulting in inter-dye distances between - 4 nm and ~ 6 nm for L and U subunit, respectively. Due to the rigid-body interface between the small domain of the acceptor labeled subunit and the large domain of the donor labeled subunit, this FRET construct was designed to report intra-subunit motions of the ClpX enzyme. Based on crystal structures [27, 65], the FRET efficiency was expected to be higher in the L conformation than U due to shorter inter-dye distances. If subunit conformations of the labeled ClpX switch between U and L, this smFRET assay would allow real-time observation of the dynamics (Fig. 2.1d). 33 a Large domain small U Hinge c Small domain barge e L Nuc eotide colored by rigid-body unit colored by subunit d C Non-switching >1 Non-switching C: U subunit .. 31 L subunit W W W L subunit (High FRET) Time Time Switching between L and U L C, W) U U_ ime U subunit (Low FRET) Figure 2.1: Single-molecule FRET assay of ClpXP. (A) In the ClpX monomer, a nucleotide binding pocket resides at the hinge region linking the small and large domains (3HWS). ATP hydrolysis cycles occurring in the hinge presumably lead to structural rearrangement between the small and large domains. (B) Most crystal structures of ClpX consist of nucleotide unloadable U subunits and loadable L subunits. A small domain forms a rigid-body unit with a large domain of the neighboring subunit. (Figure adapted from [161) (C) To utilize a lever-arm effect and label two trimers separately with different dyes, a donor (subunit D: D170C-Cy3) and an acceptor (subunit C: Q174C-Alexa647) are labeled at two neighboring large domains. Inter-dye distances span - 4 nm (for L subunit) to - 6 nm (for U subunit). (D) If a subunit conformation does not change, a FRET trace from individual ClpX molecules will be stationary. In the switching case, the single-molecule FRET assay allows for direct observation of switching and kinetic analysis 34 2.3.2 Nucleotide dependence of C1pX conformation The biotinlyated FRET ClpXP construct was immobilized on a PEG-passivated glass surface via streptavidin at the single-molecule limit and FRET signals from individual molecules were monitored using TIRF microscopy equipped with a dual view EMCCD [48]. Using individual FRET traces, we first constructed and compared FRET efficiency histograms in four different nucleotide conditions; 1 mM ATP, 1 mM ATPyS, 1 mM ADP and no-ATP (Fig. 2.2a-d). Clearly, the nucleotide binding to ClpX, regardless of the exact nature of nucleotides (ATP, ADP, ATP 'S), resulted in an increase of ClpX monomer populations with high FRET, indicating that the association of nucleotides in the binding pocket of ClpX subunit brings the small and large domain closer. The asymmetric shape of FRET histograms suggests the presence of multiple FRET states. To see the minimal number of states that can fit FRET histograms, Gaussian functions representing distinct populations of ClpX conformations were fitted to the histograms. Two different populations were adequate to fit histograms except for the no-ATP condition where triple Gaussian functions were necessary. In 1 mM ATP, a high-FRET population (FRET efficiency E ~ 0.8), tentatively assigned to the L conformation, represented 75 % of all molecules. A low-FRET population (FRET efficiency E r-zd 0.65) was tentatively assigned to the U conformation and accounted for the remaining 25 %. Thus, ClpX monomers preferably adopt L con- formation, consistent with previous crystal structures [65, 27]. Similar to the results for 1 mM ATP, two distinct populations sufficiently describe FRET histograms for 1 mM ATP-yS and 1 mM ADP, with 60 - 83 % high-FRET and 17-40 % low-FRET populations. Notably, Gaussian fits to the FRET histogram for 1 mM ADP (a-= 0.05) had smaller widths than 1 mM ATP (o- = 0.07), suggesting that there might be sub-classes in U and L conformations (we return to this point below). In the absence of nucleotide, ClpX conformations are rather broadly distributed over E r 0.2 - 1 with a minimum three conformational states. We next asked if ClpX conformations dynamically change between U and L con35 a f bh 5 ~1mM 1mM ATP ATPgS C3 - Alexa 647 4000 K 4 3 - 8000 2000 3 0 10 20 0.. 0 0.2 0.4 06 0.8 0 1 1 0.4 0.2 5 4 1I U LL 0 10 30 20 S21mM ATPgS 0.80 f 40 (s) 1Time No ATP 2 01 40 0 4 ImM ADP 30 1 0.8 d -5 -- 0.6 FRET FRET C 20 Time (s) 1 1 10 30 20 Time (s) 6 08 1 0 0 02 0.8 0.6 0.4 1 M M FRET FRET 0.4 U FRET L FRET ATP 0.65 0.80 75 ATPgS 0.60 0.80 83 0.67 0.79 60 ADPI L (L + U) ) Populatilon e 0 NoAP10 20 30 40 Time (s) 02 0 1mMADP 0 10 Time (s) Figure 2.2: Nucleotide binding leads to contraction. (A-D) FRET histograms of the ClpX in different nucleotide conditions (a: 1 mM ATP, b: 1 mM ATP -yS, c: 1 mM ADP and d: no nucleotide). For a-c, a double Gaussian distribution with equal standard deviation was fitted for each histogram. For d, a triple Gaussian distribution with fixed standard deviation (a - 0.07) was fitted. (E) Summary of FRET histogram analysis. The peak FRET values for U and L conformation as well as population percentages of L conformation are indicated. (F) Example time trace of fluorescence intensity of the ClpX labeled with a single FRET pair in the presence of 1 mM ATP. (G) Representative smFRET trajectories under each nucleotide condition. Blue, FRET efficiency; red, HMM of the FRET trace. For 1 mM ATP, the FRET trace is from the same ClpX molecule as one in (f). At the end of all traces, acceptors bleached, leading to abrupt drops in FRET efficiencies to near zero. A trace in the absence of nucleotide exhibits low FRET and frequent transitions. In ADP, the trace is stationary. 36 a C 0 CU 0) 8 C 0 00 0 0.5 0(000 0 0 0 ATP 00 C 0 0.4 ADP No ATP 0.3 O)oO0 C - 0 0.2 0 0 -0.1 ATP 0 0.1 -0.2 ADP No ATP -0.3 0c 0 0 0 00 00 0__0008 2 1 0 3 2 1 3 Time (s) Time (s) b d Amplitude Time constant (s) ATP -0.16 1.76 1.75 ATPgS - 0.08 1.25 4.21 0.04 ADP - 1300 0.01 0.44 0.58 No ATP 0.28 0.45 Amplitude Time constant (s) ATP 0.35 1.36 ATPgS 0.19 ADP No ATP - Figure 2.3: Correlation analysis for varying nucleotide conditions. (A) The autocorrelation of FRET efficiency traces for four different nucleotide conditions (red: ATP, green: ATP 7 S, black: ADP, violet: No ATP). Circles are data and lines are single exponential fits. In ADP, the autocorrelation immediately drops to zero similar to stable signal with white noise. This indicates that there is no detectable transition in ADP. A decay of the autocorrelation in the absence of nucleotide is faster than in ATP or ATPyS. (B) Summary of fitting parameters for auto-correlation analysis in (A). (C) The cross-correlation of donor and acceptor signals. The color codes are the same as (A). (D) Summary of fitting parameters for cross-correlation analysis in (C). 37 formations observed in the FRET histogram. Examining individual traces revealed that most smFRET traces over time showed transitions between different FRET levels yet at varying degrees depending on nucleotide conditions (Fig. 2.2f). Without ATP, the ClpX enyzme changed its conformation frequently to multiple states in an apparently erratic manner. Together with the broad FRET histogram, this suggests that in the absence of bound nucleotide ClpX subunits explore available conformational spaces by thermally hopping between states. In saturating ATP, ClpX spent most of time in the L conformation but briefly visited the U state. Thus, the ClpX subunit indeed dynamically switches its conformation between U and L, which is consistent with a previous ensemble study which used mutant CpX enzymes with low nucleotide affinity 1271 and rules out the nonswitching model of ClpX conformations. Traces in 1 mM ATP7S tend to have similar types of transitions as ATP yet at slower rates. In contrast, traces from ADP-bound ClpX appeared largely static within the observation timescale (- 15 s). To better quantify rates of overall transitions, we performed correlation analyses for smFRET signals. The correlation analyses provide overall time scale of transitions present in signals without the necessity of identifying individual states. For a two-state system with representing signals hopping between two states, a rate constant from a single exponential fit to the auto-correlation of the signal is theoretically equal to the sum of the two transition rate constants for each state [661. We first determined the auto-correlations of FRET efficiency traces for each nucleotide condition (Fig. 2.3a). In ADP, the autocorrelation immediately vanished to zero (Fig. 2.3a). This "Dirac-dela-function" like behavior of autocorrelation is a distinct feature of white noise with a constant mean. Thus, the correlation analysis further sup- ports that ClpX conformation is stationary in ADP at least during our observation timescale. The decay of the autocorrelation was more than twice as fast in the absence of nucleotide than in ATP, implying that thermal wobbling of ClpX subunits without bound ATP is faster than conformational changes occurring in the ATP-bound one. Fits to cross-correlations of donor and acceptor signals showed similar time scales (Fig. 2.3). However, more importantly, they showed negative correlation in short 38 time lags, reflecting the existence of anti-correlated emission from a dye pair with fluctuating FRET coupling. 2.3.3 Substrate dependence The recent ensemble study showed that U-L conformational changes were critical for ClpX-mediated substrate degradation [271. A ClpX mutant with two subunits locked in the L conformation with disulfide bonds hydrolyzed ATP at faster rate than WT ClpX, but failed to degrade either folded or unfolded substrates. Thus, U-L conformational transitions were thought to couple ATP hydrolysis with substrate degradations. To examine the effect of substrates on conformations of ClpX molecules, we performed the smFRET assay in the presence of saturating concentrations of either folded or unfolded titin 2 7-ssrA in solution. At the population level, conformations which ClpX adopted in the presence of unfolded substrates were similarly distributed to those observed without any substrate (Fig. 2.4a). However, when folded substrates were present, the low-FRET population corresponding to the U conformation increased (Fig. 2.4b). Global fits to FRET histograms of no-substrate, folded titin and unfolded titin resulted in identification of FRET states as well as relative populations between them. FRET values corresponding to U and L conformations were identical to those obtained in the individual fit to the no-substrate condition (Fig. 2.2e; ATP). In the presence of folded substrates, the population of the L conformation decreased to 65 %, but this value is likely underestimated due to the presence of populations (E ~ 0.4 - 0.5) not included in the fitted profile (black arrow in Fig. 2.4b). Thus, during substrate unfolding, the number of subunits with U conformations is on average increased in the ClpX hexamer, however, for translocation, the number remains similar to the case without any substrate present. 2.3.4 Kinetics Altered steady state distributions of FRET values of ClpX subunits suggests kinetics between ClpX conformations are changed in the presence of substrate. To test if this 39 a C 5 U FRET unfolded titin-ssrA Population LI(L+ U)(%) ATP 4 76 cm-titin W, L FRET 3 0.65 78 0.80 titin 65 C 2 d -0 2~ 1 0 b 0 0.8 0.2 0.4 0.6 FRET 0.8 h LA iikJ R -A -i 1 0.4 0.2 5 unfolded titin-ssrA 0 folded titin-ssrA' 0 4 10 20 30 lime (s) 3 - 0.8 I- 0.6 wL 0.4 0.2 0 2 Ca M Q- 1 0 0 0.2 0.4 0.6 FRET 0.8 0 1 folded titin-ssrA 10 20 Time (s) 30 Figure 2.4: The effect of substrates on ClpX conformations. (A-B) FRET histograms of ClpXP in the presence of 15 pM unfolded titin1 2 7-ssrA (a) and native titin 2 7-ssrA (b). Double Gaussian distributions with equal standard deviations were globally fitted to three FRET histograms of no substrate (Fig. 2.2a), unfolded titin1 27 and native titin 27. A black arrow indicates a population not incorporated in the global fit. In the FRET histogram measured with the folded titin, there is an increase in population with U conformations. (C) Summary of FRET histogram analysis. The peak FRET values for U and L conformations as well as population percentages of L conformations are indicated. (D) Representative smFRET trajectories for unfolded and folded titin. Blue, FRET efficiency; red, HMM of the FRET trace. In the presence of unfolded substrates, ClpX often exhibits rapid back and forth transitions between U and L conformations. 40 is the case, we performed the Hidden Markov Modeling (HMM) to identify conformational states present in FRET trajectories (Red lines in all FRET time traces in this thesis are identified from HMM). The FRET transitions obtained from HMM were plotted in the form of a transition density plot (TDP) reflecting the frequency of transitions between the various FRET states (Fig. 2.5a-c). Peak locations in the TDP were well aligned vertically as well as horizontally across three distinct substrate conditions tested and were classified into four different FRET states. Together with steady-state FRET histograms, this reveals that U and L conformations consist of two sub-states respectively, resulting in total 4 conformational states (E ~ 0.52 (U 1 ), 0.65 (U 2 ), 0.72 (L 1 ) and 0.8 (L 2 )). Transition frequencies varied heavily depending on the presence of substrates (Fig. 2.5g). In the absence of substrates, conformational switching between U and L was as common as transitions between L sub-states (L 1 and L 2 ). However, the presence of substrates increased switching frequency between L and U conformations by 70 % while decreasing the frequency of transitions between L sub-states, implying important roles of U and L switching in substrate processing. Comparing folded and unfolded substrates, frequent fast transitions between L 2 and U 1 were noticeably observed in the presence of unfolded substrates (Fig. 2.5b, e and 2.4d), suggesting that ClpX exhibits different types of conformational changes in each stage of the degradation process. As mentioned above, the increased ClpX subunit population with U conformations in the presence of folded substrates (Fig. 2.4b) must accompany the increase in the ratio of U dwell times to L dwells. To verify this, dwell times between U and L conformations were measured and collected for comparison (Fig. 2.6). Here, we defined either U or L dwells as the total time ClpX spent, regardless switching between substates, either in an U or L conformation before transitioning to the other state. In Fig. 2.5h, an example U dwell time is equal to the sum of dwell times in U 2 and U 1 before switching to L 1 occurs. As expected, dwell times were much shorter in U conformations compared to L in all cases, consistent with the observation that the subunit population with L conformations was more prevalent (Fig. 2.2a). For U dwells, at 41 a b No substrate C Unfolded titin-ssrA 0.9 0.9 C 0 0.8 0.8 0.7 0.7 0.6 0.6 0.5 0.5 0.4 0.4 Native titin-ssrA 7 . 8 0. M (U 0.3 0.3' 0.3 0.4 0.5 0.6 0.7 0.8 0.9 0.3 1 0.4 0.5 0.6 0.7 0.8 0.9 1 FRET before transition d 0 (D Native titin-ssrA V 10 0 E - Unfolded titin-ssrA No substrate L2 55 10 10 0 0 --- 10 5 0 0.6 0.5 U C 0.8 0.7 U2 10 10 5 25 U2 10 5 0 _ L 10 10 5 5 0.9 0.6 0.5 0.7 0,8 09 0.5 0.6 0.7 0.9 0.8 L2 Initial FRET h Cr 0.07 Unfolded titin Native titin 0.06 0.6 0.04[ C L2 L 0.8 0.051 U- 0.03 0.4 UI 0.02' 0.2 0.01 F U dwell time 0I 0 U U U +L L U L -L 0 10 20 40 30 50 60 70 time (s) Figure 2.5: State identification using Hidden Markov Modeling (HMM). (A-C) Transition density plots in the absence of substrate (a), in the presence of unfolded titin-ssrA (b) native titin-ssrA (c). There are four FRET states (E - 0.52, 0.65, 0.72 and 0.8 corresponding to U 1 , U 2 , L 1 and L 2 respectively) present through all three conditions. (D-F) Mean transition times between each FRET state. Each row corresponds to the same final FRET state. Errors are SEM. (G) Transition frequency normalized with trace lengths (green: no-substrate, red: unfolded titin, violet: folded titin). Transition frequencies of sub-states such as U 1 and U 2 are combined to represent either U or L conformations. (H) Example FRET trace with four FRET states. Dwell times for U or L conformations are defined as the total time ClpX spends, regardless of transition between sub-states, before making a transition to the other state. An example dwell time for U conformation is shown. 42 a b No substrate 1 0 C 0.8 21 -. U 0.4 E0.2 L L E U 1 0 5 10 15 20 25 0.8 0.8 0 2 0.4 U -+L 0.2 L 0 3( 1 0 5 10 15 20 0.6 U 0.4 E U __2_ 25 30 0 og 0.2 Singleexponential Time constant (s) 0 5 15 20 25 30 Double-exponential Amplitude (%) Time constant (s) Amplitude (%) Time constant (s) U- L L -+ U 52 0.2 47 2.7 6.2 76 5.3 24 12.5 Unfolded titin-ssrA U L L U 92 0.3 11 34.6 3.2 28 0.8 73 6.8 Native titin-ssrA U -+ L 71 0.33 31 5.6 1.2 77 6.0 3.8 10 Dwell time (s) Nosubstrate LU L L -+U 0 Dwell time (s) Dwell time (s) d Native titin-ssrA or9v 0.6 0.6 0 C Unfolded titin-ssrA 22 1 Figure 2.6: Dwell time distributions for U and L conformations. Plots of cumulative frequency versus dwell times of U (blue) and L (red) conformations are shown for nosubstrate (a), unfolded titin (b), and native titin (c). At least a double exponential function (black dashed lines) was necessary to fit U dwell times. For L dwell times, although single exponentials fit the data reasonably well (black solid lines), double exponential fits (black dashed lines) the data better except for the no-substrate case where the double exponential fit overlapped with the single exponential one. (D) Summary of parameters used for fits in (a-c). least a double-exponential fit was necessary to generally capture the distribution, implying the presence of two distinct populations with an order of magnitude different transition rates. When the U dwell times were split into two populations based on initial FRET states before making a transition (U 1 and U 2 ), double-exponential like distributions still existed for each sub-state (data not shown). Thus, the conformations of fast-converting and slow-converting species were indistinguishable at least in our experimental conditions. For L dwells, dwell times were well fitted with a single-exponential function decaying with a time constant of ~ 6 s in the absence of substrate but the second fast converting population with a time constant of ~1 s appeared in the presence 43 of substrate that accounts for 22 - 28 % of the total population. This accompa- nied an increase in amplitudes of fast converting species in U dwells, consistent with the observation that frequent fast switching between U and L were present in traces (Fig. 2.4d). Dwell-time analyses showed that, in the presence of folded substrates, shortened dwell times in L conformations mainly contribute the increase in the ClpX subunit population with U conformation found in steady-state FRET distributions (Fig. 2.4b). 2.4 Discussion Our result support a model where U-L conformational switching in the ClpX hexamer occurs in a probabilistically defined subunit at rates close to every translocation steps. In a sequential switching model, once a given subunit switched from U to L, it needs to wait until its next turn arrives. Depending on the allowed number of U conformations in the hexamer (< 2), more than 2 kinetic steps are required to occur before the given subunit can switch back to U, resulting in L dwell time distributions similar to the Gamma distribution. However, in our result, dwell times for L conformations were distributed in a single-exponential manner, indicating that switching from L to U is a single-rate limiting kinetic step. U dwells consist of two distinct populations; fast and slow converting U. The amplitudes of fast converting U were increased in the presence of substrates (from 52 % to either 92 or 71 %). Notably, the time scale of fast converting species was 0.3 s which is close to the measured dwell times for individual physical steps during substrate translocation 163, 26]. Then, what drives conformational switching of ClpX subunits between U and L? Even in the presence of ATP-yS which ClpX hydrolyzes ~ 90 times slower than ATP [26], the rate of conformational changes was still similar to ATP (Fig. 2.3), suggesting that it is not ATP hydrolysis that drives U-L conformational transitions. This is also consistent with the previous study which showed intact ATP hydrolysis of ClpX even when U-L switching was prevented by disulfide locking [27]. Our result showed, in the absence of any nucleotide, ClpX explored different conformations frequently (Fig. 44 2.2), implying energy barriers between conformational states can be readily overcome using thermal energy. It is possible that nucleotide binding to ClpX raised energy barriers between U and L yet to the extent for thermal energy to allow occasional switching. Interestingly, ADP-bound ClpX showed little U-L transitions during our observation time. A difference in dissociation rates between ATP-yS and ADP could be a possible explanation. The important role of thermal energy on ClpX was also evidenced in the recent study which showed the fraction of active ClpX supporting substrate degradation decreased dramatically as temperature dropped 1631. It is possible that at a lower temperature, the available thermal energy is not high enough for ClpX conformations to be exchanged readily. Together with the observation that U-L conformational changes were critical for protein degradation activity [27], ClpX is likely to utilize thermal fluctuation to drive its conformational transitions suitable for its mechanical actions. 2.5 2.5.1 Materials and Methods Single-molecule fluorescence assay Single-molecule FRET assays were performed using a custom built objective-side TIRF microscope equipped with a 100X 1.49 NA TIRF objective, an EMCCD camera (Andor) and a 532/640 nm two-color illumination system. Cy3 and Alexa647 emissions were split by a 640 nm longpass dichroic mirror (Chroma) and simultaneously focused onto the EMCCD camera. PEGylated coverslips were prepared based on a published protocol [67]. In brief, glass coverslips were sonicated sequentially in deionized water, methanol and 1 M KOH. The cleaned coverslipes were then sonicated with a solution of 1 % methoxypolyethyleneglycol-silane (1 % mixture of biotin-PEG-silane) (mPEG-silane-5000 and biotin-PEG-silane-5000, Laysan Bio) in anhydrous toluene in the presence of 0.8 mM triethylamine. The reacted coverslips were washed with toluene and deionized water, 45 - and finally dried under Argon gas. PEGylated coverslips were stored in vacuum at 20 0 C and assembled into flow chambers before use. Flow cells were made from double-sided sticky tape gaskets sandwiched between a glass slide and the PEGylated coverslip. While flow cells were treated with 0.01 mg/ml streptavidin, the ClpX labeled with the FRET pair (20 nM) were incubated with ClpP (0.83 MM), 1 mM ATP and ATP-regeneration system (5 mM creatine phosphate and 0.03 mg/ml creatine kinase) in PD buffer(25 mM Hepes (pH 7.8), 100 mM KCl, 10 mM MgCl 2 , 10 % glycerol (vol/vol)) to form the ClpXP complex. After unbound streptavidin were washed out, assembled ClpXP (diluted to ~ 0.7 nM) were introduced into the flow cell and incubated for 20 min to allow binding of the FRET ClpXP to the biotin-PEG surface. The samples were then washed with imaging buffer (PD buffer supplemented with ClpP (0.5 pM), appropriate nucleotides/ substrates and oxygen scavenging system; 0.8 % D(+)-glucose, 165 units/mL glucose oxidase, 2,170 units/mL catalase, and 2 mM Trolox; all from Sigma) and incubated for 5 min before imaging. Movies were recorded at 127 ms time resolution. 2.5.2 Data analysis Single-molecule data were processed using custom-made MATLAB scripts. In brief, fluorescent peaks in the images were identified and traced throughout the trajectory. Traces that showed a single-donor bleaching event were used for data analysis. The FRET efficiency, E, was calculated from donor and acceptor intensities (ID, IA) by using Equation 1.6 where 'y is the ratio of change in average acceptor intensity (AIA) to change in average donor intensity (AID) upon acceptor photobleaching [571. FRET histograms were built using averages of every 3 points up to initial 10 points of each FRET trace. MATLAB embedded functions were used to calculated auto-correlation and cross-correlation of signals. Correlations were calculated using individual traces and then weight-averaged with lengths of traces. Hidden Markov modeling (HMM) to FRET traces was performed using the HaMMy program 1681. To prevent underestimation of kinetic rates, only traces lasting longer than 5 s were used for HMM. 46 Chapter 3 Single-molecule kinetics of ClpXP-mediated protein unfolding and translocation 3.1 Summary The development of single-molecule biophysical techniques such as optical trapping and single-molecule fluorescence allows one to obtain valuable biological information which was previously inaccessible using ensemble measurements. Especially, singlemolecule assays provide whole distributions of observables as well as averages, presenting an unprecedented opportunity to perform statistical analysis using moments of distributions. The completion time distribution for reactions is specifically shaped by an underlying kinetic scheme. Thus, statistical measures of fluctuations provides insight into the reaction mechanism. In this chapter, single-molecule statistical analyses were performed on the unfolding and translocation kinetics of the ClpXP protease. Unfolding pre-dwells were dominated by a single-rate limiting process, providing a direct evidence of futile ATP hydrolysis during unfolding dwells. Fluctuation analysis on ClpXP translocation was also presented as well as its limitation. This chapter is partially reproduced from [63]. 47 3.2 Introduction AAA+ proteases (ATPases associated with diverse cellular activities) maintain protein quality control in the cell by converting the energy derived from ATP binding and hydrolysis into work that powers mechanical protein unfolding, translocation, and ultimately degradation [151. How these destructive enzymes degrade proteins with widely varying sequences, structures, and stabilities is only beginning to be understood. ClpXP, one of the best-characterized members of this family of degradation machines, consists of ClpX, a hexameric AAA+ ATPase, and ClpP, a barrel-shaped peptidase 116]. Degradation is initiated when the ClpX ring binds a substrate via an unstructured degron, such as the ssrA tag, and attempts to translocate this peptide through its narrow axial pore. For native substrates, degron translocation by ClpX pulls on the folded portion of the protein, driving mechanical denaturation that allows subsequent translocation steps to spool the unfolded polypeptide into ClpP for degradation. Single-molecule studies, using optical tweezers to monitor ClpXP unfolding and translocation of multidomain substrates, establish that ClpXP can work against forces of 20 pN or higher, demonstrate that the smallest translocation steps are - 1 nm (ap- proximately four to eight amino acids), and reveal physical steps that are multiples of this value, resulting from kinetic bursts of two, three, or four power strokes [22, 25, 261. Studies of variants containing inactive subunits support a probabilistic mechanism of ATP hydrolysis and mechanical function by ClpXP 169], but this model is not firmly established and a related AAA+ protease has been proposed to operate by a sequential mechanism 170]. At present, it is not known how the physical properties of native and unfolded substrates affect the kinetics of single-molecule ClpXP unfolding and translocation or if these reactions account for solution-degradation rates. Moreover, no current model satisfactorily explains how the ClpX ring generates translocation steps of different sizes, accounts for the kinetics of unfolding and translocation, or explains the linkage between ATP consumption and these mechanical reactions. Any deep understanding of AAA+ proteases and related remodeling machines requires 48 answers to these questions. In a recent published work [63], we used optical trapping to assay single-molecule ClpXP unfolding and translocation of substrates consisting of domains with varying stabilities and sequences. ClpXP unfolds most domains by a single pathway, with kinetics that depend on the native fold and structural stability. Subsequent translocation or pausing occurs at rates that vary with the sequence of the unfolded substrate. During translocation, ClpXP does not exhibit a sequential pattern of step sizes, supporting a fundamentally stochastic reaction, but a mechanism of enzymatic memory results in short physical steps being more probable after short steps and longer physical steps being more likely after longer steps, allowing the enzyme to run at different speeds. Based on the experimental result in the work, we presented a mechanochemical model in which initial stochastic ATP hydrolysis in the AAA+ ring can be followed by a cascade of coordinated power strokes. In this thesis, I focus on kinetic aspects of ClpXP-mediated unfolding and translocation. A brief review on single-molecule statistical kinetics was introduced and then kinetic theories were applied to unfolding and translocation data of ClpXP. Additional unpublished materials were also presented. 3.3 3.3.1 Results and Discussion Single-molecule optical trapping assay of ClpXP ClpXP degrades ssrA-tagged variants of the titin 127 domain at different rates [21]. For example, the V13P and V15P mutations disrupt or eliminate hydrogen bonds close in space to the C-terminal ssrA tag (Fig. 3.1A and B); reduce thermody- namic, kinetic, and mechanical stability; and accelerate ClpXP degradation, with the wild-type (WT) domain being most stable and degradation resistant, V15P having intermediate stability and degradation rates, and V13P being least stable and most rapidly degraded 171, 211. For single-molecule studies, we constructed Halo-WT-WT-WT-WT-ssrA, Halo-V13P-V13P-V13P-V13P-ssrA, Halo-Vi5P-V15P49 V15P-V15P-ssrA, and Halo-WT-V13P-V13P-V13P-ssrA substrates, in which Halo is an N-termiinal HaloTag domain that allows covalent attachment to a biotinylated DNA spacer. For optical-trapping (Fig. 3.1C), multidomain substrates were at- tached via the Halo domain and DNA spacer to one streptavidin-coated bead and a biotinylated variant of ClpXP was attached to a second streptavidin-coated bead [22]. In all substrates, the Halo domain was connected to the adjacent titin domain by a 22-residue linker, whereas the remaining titin domains were connected by four-residue linkers. A B 14 15 H-bon Z H-b:ynd rsnited in V15P - disrupled in V131P 86 88 87 C four titin domains Halo DNA Figure 3.1: Optical trapping assay of ClpXP. (A) Cartoon structure of titin 127 (Protein Data Bank code iTIT), colored from the N terminus (blue) to the C terminus (red). Spheres show a carbons for residues 13, 15, and 87. ClpXP pulling on a Cterminal ssrA tag is resisted by local structure, including / sheet hydrogen bonding between the C-terminal / strand and the /3 strand with residues 13 and 15. (B) The V13P and V15P mutations disrupt hydrogen bonds that directly or indirectly stabilize the titin 127 domain. (C) Experimental setup for single-molecule assays of ClpXP unfolding and translocation. ClpXP is attached to one laser-trapped bead and has engaged the ssrA tag of a multidomain substrate consisting of four titin domains and a Halo domain, which is attached to a second laser-trapped bead via a DNA linker. (Figure adapted from [631) 50 Optical-trapping measurements under constant force 122] were used to visualize single-molecule ClpXP unfolding and translocation. Individual traces displayed three signatures of ClpXP mechanical function as shown in Fig. 3.2. First, abrupt increases in bead-to-bead distance occurred upon unfolding, with the size of the transition being smaller for titin domains than for the Halo domain. Second, bead-to-bead distance decreased following unfolding, as C1pXP translocated the unfolded polypeptide, with the total decrease depending upon the size of the denatured domain and the length of the linker to the next domain. Third, between completed translocation of one unfolded domain and denaturation of the next native domain, there was a preunfolding dwell with little change in bead-to-bead distance. 3.3.2 Single-molecule kinetics Single-molecule studies allow for measurement of not only averages of observables but also the whole distributions. In fact, this extra information contained within the shape of the distribution can be highly informative about underlying processes. Let us consider a simple kinetic pathway shown in Fig. 3.3a, where the system begins at State A and the reaction goes through several potential intermediate States ay before being completed at State B. Assume that we can measure State A and B directly using single-molecule tools but any intermediate states are inaccessible. Further assume that each reaction step is Markovian. For the substrate unfolding by ClpX, State A can be a state when ClpX just finishes translocation of an unfolded polypeptide and encounters next folded domain and State B can be a state when the domain finally becomes denatured by ClpX. For translocation, State A and B can be a beginning and end of dwells between each physical stepping. In this case, intermediate states can be any biochemical events such as ATP binding, hydrolysis, and product release or any conformational changes of ClpX not directly linked to mechanical motions. Again, all these State A and B in unfolding and translocation are directly measurable as shown in Fig. 3.2. Then, completion times for each reaction can be pooled to generate completion time distributions. The shape of the completion time distribution provides information on underlying 51 V13P V1 3Pip Halo V13P e V15P V15P Unfolding dwell time \A- V1 5P Halo Translocation -oo 10 s Figure 3.2: Trajectories for ClpXP unfolding and translocation of multidomain substrates. Unfolding of individual domains increases bead-bead distance (upward movement), whereas translocation decreases bead-bead distance (downward movement). After completed translocation of one domain, there is a variable unfolding dwell time before ClpXP unfolds the next domain. The dwell baselines before and after titinunfolding events are spaced as expected for the end-to-end distance of a native titin domain (4.4 nm) or native titin plus the linker to the Halo domain. kinetics of the reaction 1721. Fig. 3.3 shows completion time distributions generated using Monte Carlo simulations based on three distinct simple kinetic schemes between State A and B (See Materials and Methods). It must be noted that all three reactions occur on average at the same rate. However, there are huge fluctuations in terms of when individual reactions occur in the given kinetic scheme. Moreover, it is clear that different kinetics generate distinct shapes of fluctuations. The first reaction scheme involves a single Markovian step (no intermediates, Fig. 3.3b) between States A and B. Since each kinetic step has no memory of when the 52 b a Intermediate steps z A -a a1 -+> Reaction I 0.9 0.8 a2 -+----+ B e 0.7 1s 0.6 0.5 A -- B 0.4 2 a. Completion time (T) 0.3 0.2 0.1 0 0 C 0.9 0.5s 0.5s 0.7 0.5 s 1 A -+- al -> B 0.6 0.5 0.8 62% A -> B 38% 2s A' -> 0.6 0.4 . 0.3 0.2 0.1 01 1 1 LhW~ 2 3 0.4 B 0.2 fI 4 5 6 0 - - - 0 Reaction III 1.2 0.8 6 Completion time (s) d Reaction II 5 4 3 2 1 1 2 3 4 5 6 Completion time (s) Completion time (s) Figure 3.3: Completion time distributions report the number of rate limiting steps. (A) A biochemical reaction from State A to State B with multiple intermediate steps, a n. Both State A and B are experimentally measurable but intermediates are hidden in measurements. Measuring completion times of the reaction from A to B and examining their distributions allow for determination of hidden intermediate kinetics. (B-D) Completion time distributions of a single-rate limiting reaction (b), double-rate limiting reaction (c) and single-rate limiting reaction with two distinct populations or pathways (d). All completion times are generated using a Monte Carlo simulation (see Materials and methods). Average completion times for all three conditions are identical (- 1 s) yet their distributions are very distinct depending on the underlying kinetic schemes. Red lines are fits to data: (b) single exponential (r 1.97, Fr = 0.51 s), (d) double exponentials 0.97 s), (c) gamma distribution (N 1.81 s (40 % population)). Total number of (Ti = 0.51 s (60 % population) and T2 completion times simulated are n ~ 2000. 53 system arrived or from where it arrived, the rate at which the system leaves the State A should be constant, and therefore, the probability of finding the system in State A should decrease exponentially in time. Thus, the probability density of completion times, 0(t) should be O(T)= kexp(-kT) (3.1) where k is a rate constant from State A to B and T is a completion time. For the simulation, k =1 s is used. When there are two rate-limiting steps in series (reaction II, Fig. 3.3c), the completion time distribution becomes the convolution of two exponential distributions [731. If it takes time t for the first step (A to al), then it takes T-t for the second one (al to B). Thus, the completion time, T, for the total reaction should be distributed as = where p 1 (t) and # 2(t) j()#1(t)#2(T - t) dt (3.2) are completion time distributions of each kinetic step. It is clear that the distribution from two rate-limiting steps is more narrowly distributed (i.e. fluctuation is less and the reaction occurs more regularly) compared to the completion times from single rate-liming step. Larger fluctuations can occur when there are alternative pathways from State A to B. For a simple case with two parallel kinetic pathways (Fig. 3.3d), the completion time distribution adopts a double exponential shape with time constants from each pathway. The presence of off-pathway kinetic steps can increase fluctuations further since visits to the off-pathway states are stochastic and do not occur for every reaction. To objectively judge the shape of the completion time distributions, a useful value, called randomness, can be defined [74]. The randomness of completion times is defined as <T >-<T> <T_ (3.3) >2 where <T> is the mean of the completion time distribution and <T 2 >_<T> 2 is the variance. Thus, for cases with equal mean completion times like three reactions in 54 the previous example (Fig. 3.3), the randomness parameter increases as variance increases. The exponential distribution is unique in that the square of mean is the same as variance, therefore the kinetic reaction with a single-rate limiting step (reaction I) has randomness equal to 1. For a general case where there are N rate-limiting steps with equal kinetic time constants, A k a k >a 2 - . .aN- - k 1 - (3.4) B The completion time distribution is the Gamma distribution of q(T) = F(N) (3.5) exp(-kr) where F(N) is the Gamma function (for a positive integer N, F(N) = (N-1)!). Variance of the Gamma distribution is Nk 2 , while the mean is Nk. Therefore, the randomness parameter, r = 1/N. For the reaction 1I with two rate-limiting steps of equal rates, r = 1/2. If there are infinite numbers of rate limiting steps, the randomness parameter becomes close to 0. This means that the reaction from A to B occurs in a completely periodic manner like a clock. It should be noted that the randomness parameter calculated by equation 3.3 was theoretically proved to provide a strict lower bound on the number of kinetic states that compose the underlying kinetic model r= 1 1751: (3.6) nmin In reality, the finite size of data lead to deviation of randomness parameters from values calculated above. To test this, a total 130 or 260 numbers of completion times are generated based on three reaction schemes in Fig. 3.3 and randomness parameters for each data set was computed. This process was iterated by 1000 times to generate 1000 randomness parameters for each condition and their distribution was plotted in Fig. 3.4. Peaks of reaction I and II are located at randomness 1 and 0.5, respectively, which is consistent with theory. As mentioned above, reaction scheme III shows randomness parameters larger than 1. 55 250- reaction I n = 130 n = 260 200 150 reaction I 0 0 100 reaction50I 0 0 0.5 1 1.5 2 2.5 Randomness value Figure 3.4: Distributions of randomness parameters for three kinetic schemes. Either 130 or 260 dwell times are generated for each reaction scheme, and randomness parameters are calculated for each set of dwell times. This procedure is repeated 1000 times to generate 1000 randomness parameters for each reaction. The distribution of these randomness values are plotted. Peak locations are consistent with theory and as data size increases, randomness distributions become narrower. 3.3.3 Multiple ATP hydrolysis cycles yet single rate limiting step Completion times for unfolding titin V13P and V15P domain by ClpXP were directly measured using the optical trapping assay (Fig. 3.2). A completion time distribution of V13P is best fit by double exponential functions (Fig. 3.5). To better access shapes of distributions, the randomness parameters are computed. The randomness parameters for unfolding completion time were 1.50 (V13P) and 0.95 (V15P). To estimate likely errors, we performed trials in which half of the completion times from each data set were randomly removed, calculated r values, and then determined an average + 1 SD for a set of 10 independent trials, yielding values of 1.44 56 0.16 (V13P) and 0.91 0.19 (V15P). For V13P unfolding, the lower error bound of the randomness parameter was substantially above 1, as expected for a reaction with two populations, in agreement with the better fit of these data by exponential processes operating on two populations (Fig. 3.5). For ClpXP unfolding of V15P, the fits and randomness values indicate that a single predominant kinetic step is rate limiting. ClpXP unfolding of V13P fit better to exponential processes acting on less-stable and more-stable populations of similar size (Fig. 3.5), with enough events (n = 262) to make sampling error unlikely. This result is consistent with the existence of two unfolding pathways, which could depend upon which parts of the V13P domain are stochastically destabilized. For example, the N-terminal portion of V13P might be transiently frayed in the more-stable population and the C-terminal region transiently frayed in the less-stable population. b a 1 - - 1 C) Cr cc) V15P V13P E E 0. , . , M 0-.01h I double single S0 r D 01 single double "WON& -0.1 80 pre-unfolding dwell time (s) 0 0"0 V, A' . . 0 1 20 40 60 0 20 40 60 80 100 pre-unfolding dwell time (s) % Figure 3.5: Plots of cumulative frequency versus ClpXP unfolding completion times for titin V13P (a) and titin V15P domains (b). Although single exponentials fit the data reasonably well (black solid lines, top panels), a double exponential function, y = Ai.(1-exp(-t/Tunf1) + A 2 -(1-exp(-t/rnf2) (gray dashed lines, top panels), fits the 0.2 s 3.6 data better (residual plots shown in bottom panels). For V13P, Trnfl 1.1 s (52 % A 2 ). For V15P, Tunf, = 5.3 + 0.9 s (16 (48 % A 1 ) and Tunf2 = 13.1 1.3 s (84 % A 2 ). (Figure adapted from [631) A 1 ) and Tunf2 = 24.4 57 At first glance, it seems odd that unfolding proceeds with a single-rate limiting kinetic step since it has been previously known that ClpXP hydrolyze ~ 150 ATP per min at 30 'C during substrate denaturation [21]. Is the fact that there are futile ATP hydrolysis events sufficient enough to explain the presence of a single-rate limiting step during substrate unfolding? To answer this question, let us think about an arbitrary kinetic cycle with a completion time distribution of 0m(F) [761. Assume that there is very low probability of finishing a reaction after each cycle, E. Under this condition, the final completion time distribution, Of (T) = where f E0m(T) + E(1 - E)#/m(T) *m(T) + c(1 - 6) 2 (0 off(T), m should be (T) *om(T)) * #,(T) +- (3.7) * g means convolution of f and g. The first term on the right hand side corresponds to the completion times from reactions finished in the first cycle and the second term is from reactions finished in the second cycle (failed to finish in the first round). Using the Laplace transform, the above equation can be simplified as f (S) (38) m(S) 1 - (1 - E)#m(S) where of(s) and om(s) are Laplace transformed version of off(T) and om(T), respec- tively. All moments of #ff(T) can be found through differentiation, < T' >= (1'd dSn (sS=0 (3.9) resulting in the randomness of the final completion time with small success rate, E, r = (1 - E) + Ero where ro is a randomness of single kinetic cycle from (3.10) #m(T). Thus, any kinetic scheme, regardless of its detail, will result in a single-rate limiting kinetics if it has very small probability of finishing the reaction (when c ~ 0, r - 1) at the end. This suggests that any stable substrates requiring multiple ClpX power strokes for unfolding would 58 have single-rate limiting unfolding kinetics and at the limit of weak substrate whose unfolding energy barrier can be lowered significantly by ClpX single power stroke, the unfolding kinetics would start to show the kinetics occurring inside the motor. 3.3.4 Translocation kinetics In theory, analyses of the translocation kinetics can be performed in an identical manner. As long as individual physical steps are well identifiable without being missed, dwell times between steps can be directly measured and the minimum number of RLS per physical steps can be decided using the randomness parameters defined in equation 3.3. Defining the minimum number of RLS is important for the investigation of the mechanochemical coupling of molecular machines. A general ATP hydrolysis cycle of molecular motors consists of motor + ATP +-< motor-ATP - motor-ADP-Pi ( motor-ADP - motor (3.11) Molecular motors produce mechanical motions using energy from chemical sources such as ATP, so at least one of reaction step in the ATP hydrolysis cycle needs to be coupled to mechanical motion. Altering concentrations of reactants (ATP) or products (ADP or Pi) affects transition rates of involved steps and applying different loads changes the rate constant of force-generating kinetic step. Thus, a series of kinetic analyses in varying conditions of chemical species as well as mechanical loading is highly effective in studying mechanochemical cycles of molecular motors. In the published original article, we were able to identify individual steps in a sub-fraction of our data, and kinetic analyses for ClpX translocation were performed based on dwell times between physical steps [63]. In this thesis, some of the initial effort in identifying the number of RLS in ClpX translocation are summarized as well as limitations in the analysis presented. Historically, randomness parameters were originally introduced using trajectories 59 1 RLS 2 RLS 5 RLS 40 40 40 20 20 0 0 a.0 (L 00 a.0 0 -20 -20 -200 2 4 Time (s) 6 0 8 2 4 Time (s) 6 0 8 50 50 40 40 40 0 30 E 30 0 0 20 ,20. 50 0 4 ime (s) 6 8 6 8 30 20 Q_ 0 10 10 10 00 0 0 0 CL 2 2 4 Time (s) 0 6 4 Time (s) 6 12 c 8 4 2 4 Time (s) 20 16 E S12 16 E S12 16 0 8 20 f 20 0 2 .cu 8 8 4 0 0.5 0 1 Time (s) 1.5 0 1 0.5 Time (s) 1.5 00 0.5 1 1.5 Time (s) Figure 3.6: Fluctuation analysis reveals hidden kinetic information. Simple simulations are performed to generate a series of traces based on different numbers of rate limiting steps (RLS) per physical step (See Materials and Methods) : single RLS (leftmost column), double RLS (center column) and quintuple RLS (rightmost column). The first row shows single simulated traces (raw data in gray and noise-free steps in red) for each condition. As the number of RLS increases, physical steps occur in more periodic manner and the motor speed is temporally unchanged. The second row depicts overlaid plots of total 20 traces. At the population level, traces with smaller number of kinetic rate limiting steps tend to spread more. The last row shows an increase of position variance over time for each simulation condition (averaged variance in black and variances of individual traces in all other colors). The variance increases more rapidly for smaller number of RLS. Slopes of variance increase are divided by d-<-v>, where d is a 2 nm step size and <v> is a average velocity of 4 nm/s, to obtain the randomness parameters for each condition. The inverse of calculated randomness parameters (1.12, 1.82 and 5.3 for 1, 2 and 5 RLS, respectively) are very close to the number of RLS. 60 of individual motor runs in the following form rtrajectory [771. limX(x(t) <x(t) >) > =--t-++00 (3.12) d < x(t) > where d is a step size of the motor protein. Like the previous definition of the randomness parameter (equation 3.3), r becomes 1/N for an enzyme with N rateliming kinetic steps with equal kinetic rates. High signal-to-noise is not required for the purpose of this analysis since the variance in position due to motor kinetics grow without bound with time, whereas the instrument noise remains constant. However, the major limitation of the method is its requirement of knowledge of step size, d. Thus, this method is useful in a case where a rough idea on the step size is already known but signal-to-noise is not sufficient to detect all individual steps reliably. In theory, missing short steps can give rise to nonexponential dwell time distributions based on which one can falsely interpret it as a sign of multiple kinetic RLS. To illustrate the method, traces based on varying number of RLS were simulated (Fig. 3.6) and fluctuation analysis was performed on traces to test its ability to report the underlying kinetics. Inspecting simulated traces, it is clear that fluctuations of motor stepping shaped by underlying kinetic schemes generate different levels of variations in time as well as across populations. For example, traces from 5 RLS showed highly regular stepping and less spread across different runs. Variance of positions increased linearly with varying slopes which correlate with an inverse of the number of RLS, demonstrating the fluctuation analysis is robust to report underlying kinetics. Translocation traces of ClpX were analyzed using the same method and the variance increase over time was plotted for V13P and V15P (Fig. 3.7). Using a step size of 2 nm, the computed randomness parameters were r = 0.63 0.26 and 0.62 0.35 for V13P and V15P, respectively. Since V13P and V15P are point-mutations made on WT-titin, it was expected that randomness parameters from two substrates were similar. The randomness values suggest the presence of a minimum two rate limiting processes per translocation stepping. However, some care should be taken in 61 a 305 25- E 20V13P c15 >10 51 0 0.2 0.6 0.4 0.8 1.2 1 time interval (s) b - 35 - 30 C) C 20 V1 5P - .0 CU 15 0 0.2 0.6 0.4 0.8 1 time interval (s) Figure 3.7: Fluctuation analysis on protein translocation by ClpXP. Variance of translocation traces, averaged over 403 runs for V13P (a) and 187 runs for V15P (b) and line fits (red) over the interval 0.3 s to 0.8 s. Position data were decimated every 10 points and then moving-averaged every 5 points before being fed into fluctuation analysis. Either decimation or moving averaging do not affect slopes of variance increase yet change the y-intercept. The rapid rise in variance at short times reflects the brownian correlation time for bead position. 62 interpreting these values. As mentioned above, the randomness parameter calculated using completion times of reactions, equation 3.3, reports a lower bound of the RLS in underlying kinetics. However, the equality of this randomness parameter to the one based on trajectories, equation 3.12, is not always guaranteed [78, 79, 801. For example, when step sizes vary for each completion of kinetic cycle, fluctuations in the step size give rise to additional variations in trajectories [76]. In a case where step sizes and completion times are uncorrelated, the randomness parameter from the trajectories is equal to the sum of two terms Ttrajectory < d 2 >- < d>2 + 2 = < d>2 T2 < 2 >-_T < (.3 T >2(3.13) >2 where rtrajectory refers to the definition given in equation 3.12. The first term on the right hand side corresponds to fluctuations due to the varying step sizes and the second term is from fluctuations in the motor stepping kinetics. Thus, this relationship provides a way, for the uncorrelated case, to remove the effect of varying step sizes on variances of trajectories and to recover the fluctuation due to the underlying kinetic cycles of motor stepping. The presence of correlation between the completion time and step size decreases the amount of additional contribution from varying step sizes. However, the randomness from the raw trajectories (a term in the left hand side of equation 3.13) is still larger than the randomness from stepping kinetics (the second term in the right hand side of equation 3.13), providing the lower bound of the number of kinetic RLS. It turned out that during ClpXP translocation an interesting correlation between step sizes and stepping dwell times was observed [631. ClpXP spent a longer time before taking bigger steps. Moreover, another layer of complexity, some levels of 'memory' in step sizes, was also present in the ClpX stepping. In this complicated case, an ideal way of performing kinetic analysis for motor stepping is to use dwell times for each step as we did in the original article [63]. Notably, kinetic analyses based on the pre-stepping dwell times also suggested the presence of at least two kinetic RLS per physical step [26, 63], consistent with the result presented in this chapter. In summary, although some limitations mentioned in this chapter need 63 to be cautiously considered in interpreting results, the fluctuation analysis using the raw motility trace provides a simple way to study the underlying kinetics of motor stepping. 3.4 3.4.1 Materials and Methods Protein constructs SsrA-tagged protein substrates, E. coli ClpP, and single-chain hexamers of wild-type ClpXAN or RWERWE ClpXAN with a C-terminal biotinylation site were cloned, expressed, and purified as described [81, 21, 69, 22]. In multi-domain substrates, the linker between the Halo domain and the adjacent titin domain had the sequence ISGEPTTEDLYFQSDNAIAPRM; all additional titin domains were connected by the sequence GTRM. The C-terminal sequence of each multi-domain substrate was KVKELGH6GAANDENYALAA, where the ssrA tag that targets the substrate to ClpXP is underlined. 3.4.2 Single-molecule mechanical measurements Complexes of ClpXP with multi-domain substrates containing an N-terminal Halo domain, which was covalently linked to biotinylated double-stranded DNA, were tethered between two laser-trapped beads as described [22]. Briefly, DNA-linked substrates were tethered to a 1 pm streptavidin-coated polystyrene bead that was loosely bound to the surface of a glass cover slip via a DNA-tethered glass-binding peptide aptamer. Biotinylated ClpXP was attached to a 1.26 Pum streptavidin-coated polystyrene bead, which was trapped and brought into the vicinity of the bead containing the DNA-linked substrate. Upon substrate recognition by ClpXP, as determined by inter-bead tension, the laser trap for the substrate bead was turned on and the cover slip was moved to rupture the aptamer-glass attachment, resulting in tethering the ClpXP-substrate complex between two laser-trapped beads (Fig. 3.1c). Experiments were performed at room temperature (18-22 'C), using 2 mM ATP and 64 ATP-regeneration and oxygen-scavenging systems [221. Data acquisition was carried out as described (Aubin-Tam et al., 2011). Custom MATLAB scripts were used to calculate inter-bead distances, measure the magnitude of unfolding distances, and measure the time elapsed from the end of one translocation event to the next unfolding event, which represents the pre-unfolding dwell time. Translocation events in each trace were separated and fit with a linear equation to determine the average translocation velocity. We developed a pause-detecting MATLAB script in which the translocation data is smoothed to decrease environmental noise, and then differentiated to determine the instantaneous velocity. Pauses were identified as time periods in which this velocity remained at or below zero for longer than 2.5 s for ClpXP translocation or 7.5 s for RWERWE ClpXP translocation. 3.4.3 Kinetic analysis Monte Carlo simulation was performed to simulate completion times of three distinct kinetic schemes in Fig. 3.3. For a Markovian process, a probability for the event to occur is constant, k, in time. At time t = 0, a system starts at State A and for each time increment, 6t, a probability of the reaction to occur, k 6t, was compared with a randomness number x generated with uniform probability between 0 and 1. If k 6t is larger than x, the reaction occurred and the completion time was collected. If k 6t is smaller than x, the reaction did not occur and let the time increase by 6t for next run. Simulated completion times were combined to generate probability density distribution of completion times. Fluctuation analysis was performed as previously described [821. Briefly, the slopes of line-fits to displacement data vs. time for each run were calculated to obtain the mean velocity <v>. To calculate the randomness, pairwise distances between all measured positions in a given record were used to generate a running plot of variance, [y(t + At) - (y(t) + <v> At)1 2, vs. At. The variance increased linearly and the slope of the line between 0.3 s and 0.8 s was then divided by d - <v>, where d is the step size, to obtain the r parameter for the record. 65 66 Chapter 4 Non-canonical Motility of Kif18B 4.1 Summary The kinesin-8s are plus-end directed motors that negatively regulate microtubule (MT) length. The canonical members of this kinesin sub-family, are "ultra-processive", a property enabled by a second MT-binding site that tethers these motors to the MT track. Ultra-processivity causes kinesin-8s to enrich preferentially at the plus-ends of long MTs, where they promote MT catastrophes or pausing. Kif18B is an understudied human kinesin-8 that also limits MT growth during mitosis. In contrast to archetypal kinesin-8s, localization of Kif18B to plus-ends relies on binding to the plus-end tracking protein (+TIP) EB1, obfuscating the relationship between its potential plus-end directed motility and plus-end accumulation. Using single molecule assays, we show that Kif18B is only modestly processive, and that the motor switches frequently between plus-end directed and diffusive modes of motility. Diffusion is promoted by a second MT-binding site located in the Kif18B tail. In cells, Kif18B concentrates at the extreme tip of a subset of MTs, superseding EB1. While the speed of ATP-driven plus-end motility of Kif18B is well below the velocity of growing MT plus-ends, computer simulations suggest that a combination of directed motility and diffusion allows Kif18B to outpace a growing MT plus-end. At MT plus-ends, we demonstrate that Kif18B promotes catastrophe through a pause intermediate in a manner that is potentiated by EB1. Our data are consistent with a widespread 67 function for kinesin-8s in regulating MT dynamics, but demonstrate that they use diverse design principles to accomplish this task. 4.2 Introduction Microtubules (MTs) are polar cylindrical polymers comprised of head-to-tail stacked strands of a,3-tubulin that are vital for a diverse range of physiological processes [83, 84]. During mitosis, for example, the interphase MT array is remodeled into a fusiform structure, the mitotic spindle, which segregates chromosomes between two daughter cells. A key property that underlies the functional versatility of MTs is their ability to assemble with non-equilibrium kinetics, a behavior termed dynamic instability [85, 86]. MTs grow and shorten by subunit addition and loss at their plus-ends and switch randomly between these states in a manner independent of neighboring MTs. Although most parameters of MT dynamics do not vary grossly through the cell cycle, dividing cells increase the frequency at which growing MTs convert to shortening (catastrophe; [87]). This alteration allows MTs to efficiently explore cytoplasmic space, until their plus-ends are captured and stabilized by specialized chromosomal sites called kinetochores [88]. The best understood factor that increases MT catastrophes is the kinesin-13 MCAK. Kinesin-13s are immotile motors that have adopted their abilities to hydrolyze ATP to cause catastrophe-promoting conformational changes at MT ends [89, 90, 91]. Instead of using directed motility, these motors target MT ends by diffusing along the lattice [7]. MCAK is largely responsible for the 10-fold elevation in the catastrophe frequency in Xenopus egg extracts [92], and its depletion from this system prevents spindle assembly because MTs overgrow in its absence [93]. Consistent with a major role for MCAK in shaping MT dynamics, it can cause MTs assembled from pure tubulin to exhibit physiological MT dynamics when combined with XMAP215, a pro-growth MT-associated protein (MAP; [94]). Kinesin-8s have emerged as important MT length suppressors, but they do so through complex mechanisms that vary by family member. Similar to MCAK, yeast 68 Kip3 and human Kifl9 are MT depolymerases [95, 96]. Human Kif18A, in contrast, does not depolymerize MTs but instead causes MT plus-ends to pause, a state where tubulin subunits neither add to or release from the plus-end [97, 98]. In contrast to their differential effects on MT dynamics, an invariant feature of all kinesin-8s studied to date is that they are plus-end directed motors that are capable of walking unusually long distances ( - 15 pm) on a MT track before dissociating [99, 100]. This "ultra-processivity" depends on the ability of the non-motor C-terminal tail domains of Kif18A and Kip3 to bind MTs, an activity which tethers the motors to the MT track [99]. Enhanced processivity enriches kinesin-8 motors on long, stable MTs and consistently, a major function of Kif18A is to promote chromosome congression by regulating the plus-end dynamics of kinetochore-MTs 11011. Whether the paradigm set by Kip3 and Kif18A will apply to all kinesin-8 motors is unclear. Kif18B, a third human kinesin-8, localizes to and suppresses the overgrowth of non-kinetochore-MT plus ends but does so in a manner that requires the plusend tracking protein (+TIP) EB1 [1021. In principle, Kif18B would not require high processivity to target MT plus-ends. In addition, Kif18B has been suggested to transport MCAK to the MT tip, raising the possibility that it may not regulate MT dynamics directly [103]. To shed light on how Kif18B prevents hyper-elongation of non-kinetochore-MTs, we used high-resolution single molecule assays to show that Kif18B is not ultra-processive like other kinesin-8s, and that it uses a hybrid form of motility involving diffusion and plus-end directed motility to target the extreme MT plus-end. Interestingly, diffusion is promoted by a second MT-binding site located in the tail of Kif18B, a function distinct from the processivity promoting MT-binding tail of Kif18A. Kifl8B thus uses a suite of biophysical properties unique to kinesin-8s to navigate around crowded MT ends for its function. 69 4.3 4.3.1 Results Distinct motile properties of Kif18B To investigate motile properties of individual Kif18B on the microtubule, we first constructed and purified GFP-tagged Kif18B. The oligomeric nature of GFP-Kif18B was first examined by comparing intensity distributions of individual fluorescent spots of GFP-Kifl8B, dimeric GFP-Kif18A and single GFP molecules (4.1A, Fig. 4.1B). The mean intensity of surface-bound GFP-Kif18B was similar to that of GFP-Kif18A and twice of single GFP, confirming that GFP-Kif18B was dimeric. For single-molecule measurements of Kif18B motions on the microtubule, highly diluted GFP-Kif18B was introduced into a flow cell with surface-immobilized GMPCPP-microtubules and imaged with total internal reflection fluorescence (TIRF) microscopy (Fig. 4.2A). In the presence of 1 mM ATP, multiple fluorescent spots appeared on the microtubule lattices and moved along them before disappearing (Fig. 4.21B). Fluorescence excitation was modulated at 1 Hz to extend the longevity of GFP. In a small number of long binding events, intensities of fluorescent spots showed double fluorescence drops to the background level as expected for dimeric proteins (Fig. 4.1C). We found that Kif18B had distinct motile properties from other Kinesin-8 such as Kip3p and Kif18A in terms of processivity and dwell times at the microtubule end. Interestingly, Kif18B displayed only modest levels of processivity, 1.19 pm (mean 0.07 SEM; N = 106), (Fig. 4.2B and Fig. 4.2C), contrary to the previously discovered ultra-processivity of other Kinesin-8 [104, 991. Among randomly bound Kif18B along the microtubule, only a small fraction of molecules reached the microtubule end. After arriving at the end, Kif18B was not stationary but exhibited brief back-and-forth movements (Fig. 4.2D). This is quite distinct from Kip3p which stayed at the microtubule end without showing any noticeable backward movements 1104]. Given that the localization accuracy of individual fluorescent spots is around ~ 20 nm in our experimental condition, we speculated that Kif18B could move backward more frequently by distances smaller than the localization limit. To observe movements of Kif18B with high spatial and temporal resolution, we 70 a X104 I 2 Kif18B Kif1 8A I a-o I 1 0 single GFP I 0 1 0.5 . lu in111I1II 2 1.5 - 3 2.5 3.5 -, 4 x 104 Initial fluorescence intensity (a.u.) b x10 C 12000 3 2 10000 C (D C (D C', C 8000 Cu C 6000 o 4000 C 2000 0 x 104 50 100 150 100 150 Cu 0 2 . - C (D 1 I- 0 '0 C"* 'q 0 0 50 Time (s) Figure 4.1: Kif18B is dimeric. (A) Intensity histograms of GFP-Kif18B (n = 682; red), GFP-Kif18A (n = 991; black), and single GFP (n = 103; blue) respectively. The mean intensities of initial three time points for each isolated fluorescent spot were used to generate histograms. (B) The mean intensity of GFP-Kif18B is similar to that of dimeric GFP-Kif18A and twice of single-GFP, suggesting GFP-Kif18B is also a dimer. (C) Representative intensity profiles with two-step fluorescence drops for GFP-Kif18B moving on the microtubules. Arrows indicate points of fluorescence drops. The second fluorescence drop can be either dissociation of Kif18B from the microtubule or the photobleaching of the remained GFP. The excitation laser was modulated at 1 Hz to extend the longevity of GFP, resulting in the fraction of fluorescence spots with two-step fluorescence drops is minimal. 71 8 Microtubule a- GFP-Kifl8B TIRF Streptavidin Biotin - PEG Coverslip b C~ c 30 50 d 40 30 E U 20s Run Length (pm) Figure 4.2: Distinct motile properties of Kif18B. (A) Single-molecule TIRF assay of Kifl8B. Diluted GFP-Kifl8B in solution is allowed to interact with a MT specifically anchored to a glass coverslip via streptavidin-biotin linkage. TIRF microscopy is used to track individual GFP-Kifl8B. (B) Sequential frames of a GFP-Kifl8B-FL (green) images showing moderate processivity. Motility of single GFP-Kifl8B-FL molecules on a MT (red) is marked by white arrows. Elapsed time is reported in seconds. (C) Run length distribution of GFP-Kifl8B-FL in 1 mM ATP. The red curve is a single exponential fit to the data. n = 106. (D) Examples of kymographs showing backand-forth movements of Kifl8B at the MT end. The regions of the movements are marked by white arrows. 72 attached a 440-nm-diameter polystyrene bead to the tail of Kif18B and imaged its motion with differential interference contrast (DIC) microscopy (Fig. 4.3A) [391. Using video tracking, positions of the Kifl8B-bound beads were localized with a spatial resolution of about 6 nm at a time resolution of 33 ms. Similar back-and-forth motions were observed in the video-tracking assay when the Kif18B bead reached the microtubule end (Fig. 4.3B). Short pauses at the microtubule end were flanked by frequent back-and-forth movements spanning from tens of nm to several hundred nm. The average dwell time at the very end of the microtubule was 1.84 + 0.48 s (mean SEM; N = 38; Fig. 4.3C). a b Y axis Bead XExsCoesi - 200 Microtubule end -200 0 0 0-400 Coverslip to -600 -800 W-1000 -1200 GFP-KI1 8B Microtubule 0 10 20 30 40 50 60 70 80 Time (s) C 15 10 0 0 5 0 0 1 2 3 4 5 6 Dwell time at MT tip (s) Figure 4.3: Kif18B dwells shortly at the microtobule end. (A) Video-tracking assay of Kif18B. GFP-Kifl8B is linked to a bead via biotinylated pentahistidine antibody and the position of the bead is monitored using cross-correlation method. (B) Example trace of the Kif18B coated bead near the MT end. Frequent back-and-forth movements are evident. To compute dwell times at the microtubule end, positions up to 50 nm away from the maximum value are defined as the microtubule end. (C) Dwell time distribution of Kif18 at the MT end. The red curve is a single exponential fit to the data. n = 38 73 4.3.2 Dual mode of motility: diffusion and directed motion We next inspected motility of Kif18B on the microtubule lattice. Trajectories of individual Kif18B molecules showed overall directionality but, notably, they exhibited frequent backward motions (Fig. 4.4A), suggesting that Kif18B possesses two distinct modes of motility; diffusion and directed stepping. The mean squared displacement (MSD) analysis was performed on the fluorescently tracked positions of Kif18B in the presence of ATP, resulting in a diffusion constant, 0.01 0.001 Pm 2 sec-1 and a velocity, v, of 0.052 + 0.003 [tm sec- 1 (Fig. 4.4B). These values were used to compute a randomness parameter, r, which describes the extent of stochasticity in the stepping of motor proteins [77]. For a motor whose motility is tightly coupled to stochastic ATP hydrolysis, r approaches near 1; for example, kinesin-1/Kif5C has an r of 0.77 [1051 whereas Cin8 and Eg5, which show both directional stepping and diffusion, exhibit an r of 73 and 13.7, respectively 1106, 1071. Our data indicate that for Kifl8B, r = 50 consistent with the dual modes in the Kif18B motility. Single-molecule fluorescence tracking measurements in the presence of ADP showed that ATP hydrolysis was required for directional movements of Kif18B yet diffusive motility was intact (Fig. 4.4C and Fig. 4.4D). To determine temporal fractions for each mode of motility in Kifl8B, we further examined motions of Kif18B on the microtubule lattice acquired from the bead-based video tracking assay. Consistent with results from single-molecule fluorescence tracking, Kifl8B-bound beads showed transient directional motions flanked by diffusive phases (Fig. 4.5B). A control measurement with Kinesin-1 exhibited only unidi- rectional motion (Fig. 4.5A). To objectively identify temporal periods of transient directed stepping from diffusion, we employed an automated algorithm based on the fact that velocity vectors, for directed motions, are temporally highly correlated (Fig. 4.5D) [108]. Since stochastic fluctuations of Brownian diffusion can occasionally generate motions appeared to be directed during short time periods, we determined regions of directed motions so that the probability of any detected events being due to pure diffusion is less than 5 %. Detected directional phases had much faster in74 stantaneous velocities than the overall velocity based on the whole trajectories (Fig. 4.5C), suggesting that Kif18B moves during directional phases much faster than calculated from the overall MSD calculation in Fig. 4.4B. A model with only directional motion fits well the MSD from detected directed phases, resulting in the instantaneous velocity during the directional stepping of 0.183 t 0.002 ptm sec-1 (Fig. 4.5E). Difference between the instantaneous velocity and the overall one is expected when the motor switches between directional stepping and diffusion without any net directionality. Taking the ratio of overall velocity to the instantaneous velocity provides that Kif18B spends only 28 % temporally in the directed phase during its association with the microtubule (Fig. 4.5F). a 0.8 b 20s 1mM ATP E -~ CN C 1mM ATP 1mMADP 0.6 0.4 40s E 0.2 - 0 2 4 6 8 10 12 Time interval (s) 1mM ADP 0.6 =L _____________________ ________________ 0.4 20 0.2___________ CO, 0 0 2 8 10 12 4 6 Time interval (s) + Figure 4.4: Kif18B has dual mode of motility. (A) Representative kymograph of GFP-Kifl8B-FL depicting two modes of motility (diffusive and directed motion) in the presence of 1 mM ATP. Examples of backward motion are indicated by white arrows. (B) Mean-squared displacement (MSD) of GFP-Kifl8B-FL in 1 mM ATP. The red curve is a fit to MSD = v 2 2 + 2DT+ 2c2 with tracking precision e = 37 nm. Error bars represent the SEM of the squared displacement values. n = 98. (C) Representative kymographs of GFP-Kifl8B-FL showing diffusive movements in 1 mM ADP. (D) MSD of GFP-Kifl8B in 1 mM ADP. The red line is a fit to MSD = 2DT 2c2 with tracking precision c = 62 nm. Error bars represent the SEM of the squared displacement values. n = 78. All fitted parameters are listed in table 4.1. b a E C UC) + CO I d C 02 .: -' <cos 0 > 0 t 2 <cosO>~1 3 2 1 Kinesin-1 3 10S Kifl8B e f 1 0.3 0.8 . 0.2 0.6 0 U, 0.1 E 0.4 IT 0.2 0 0 0.5 1 1.5 2 2.5 Diffusion Directed 0 Time interval (s) Figure 4.5: Kifl8B spends the majority of time in diffusion. (A) Example 2D trajectories of Kinesin-1 on MT (right). Traces are based on top-down view and the motility is upward (left). (B) Example 2D trajectories of Kif18B. Different colors denote distinct modes of motility (red : diffusion, black : directed motion) detected using a computer algorithm (see Materials and Methods). (C) Position versus time curve for the trace 3 in (b). The instant velocity is much higher than the average one. (D) Velocity vectors are highly correlated for directed motion (down, black) but uncorrelated for diffusion (top, red). (E) MSD calculated with transient directed phases. The red curve is a fit to MSD = v 2 2 + 262. (F) Temporal fraction of Kif18B in each motility mode (72 % in diffusion and 28 % in directed motion). All fitted parameters are listed in table 4.1. 76 4.3.3 The tail domain of Kif18B : regulator of motility How does Kif18B switch between two different modes of motility? To access potential roles of the tail region of Kif18B on its motile properties, we constructed tailless version of GFP-Kif18B (Kifl8B-N574) as well as the tail domain fused to mCherry (Kifl8B-C268) (Fig. 4.6A). Interestingly, single-molecule fluorescence measurements with the tailless Kif18B showed only unidirectional motions (Fig. 4.6B). To address the possibility of unresolved short periods of diffusion due to low spatial and temporal resolution in fluorescence imaging, we examined the motility of tailless Kif18B-bound beads in the video tracking assay. Two-dimensional trajectories of beads on the microtubule did not show any sign of diffusion (Fig. 4.6C), confirming that the motor head domain of Kif18B steps directionally along the microtubule. Positional changes of Kif18B beads along the microtubule showed not only the lack of diffusive phases but also faster overall velocity in the tailless Kif18B motility compared with the full length motor (Fig. 4.6D). Indeed, MSD analysis to the fluorescence data of the tailless motor revealed that the velocity of the tailless Kif18B, 0.166 1 sec- 0.001 Pm , is similar to that of the instantaneous velocity of the full length motor (Fig. 4.6E, Fig. 4.5E and Fig. 4.1). This data suggest that the motor head domain of the Kif18B steps directionally along the microtubule and the tail domain gives rise to diffusing behaviors. What is happening at the molecular level during the switching of the motility mode in Kif18B? One possibility is that the tail domain stochastically interacts with the motor head to directly switch its mode of motility (Fig. 4.7). The second possibility is when the motor head detaches from the microtubule the molecule remains bound to the microtubule via the tail domain which diffuses along the microtubule (Fig. 4.7). Two models differ in terms of which domain executes diffusive phases. To discriminate between these possibilities, we analyzed diffusive motilities of individual tail domain and tailless motor in the presence of ADP using a single-molecule TIRF assay. Both proteins briefly interacted with the microtubule and their motility was only detectable with a high acquisition rate (17 Hz). The diffusion constant of the 77 b a 30 s """ Motor Coiled coil Tail Kifl8B-FL Kifl8B-N574 Kif18B-C268 C d e 2500 E 5 2000 4 1500 C i~A E C: 0 E3 1000 2 500 ( 0 Tailless in ATP A/V~4kiA 0 ~fr 1 2 3 -500 0 00 - ~UU 5 10 15 20 30 35 2 4 6 8 10 12 14 Time interval (s) Time (s) Tailless-Kif18B 25 Figure 4.6: Tailless motors show only directed motion. (A) Schematic diagrams of three Kif18B constructs used in this study. (B) Representative kymographs of GFP-Kifl8B-N574 showing the absence of diffusion in 1 mM ATP. (C) Example 2D trajectories of GFP-Kifl8B-N574 from the video tracking assay show only directed motions. (D) Example distance versus time plots of GFP-Kifl8B-N574 (black) and GFP-Kifl8B-FL (other color) from video tracking. The tailless motor lacks diffusion and moves faster than the full length motor. (E) MSD of GFP-Kifl8B-N574 in 1 mM ATP calculated from single-molecule fluorescence tracking (blue square) and video tracking (black line). The red curve is a fit to MSD from the single-molecule fluorescence data with MSD = v 2 2 + 262. All fitted parameters are listed in table 4.1. tail, 0.598 0.020 pm 2 sec 1, was much larger than that of the tailless motor, 0.014 + 0.001 Pm 2 sec- 1 (Fig. 4.8A-c). Notably, the diffusion constant of the tailless motor is similar to that of the full length motor in ADP (Table 4.1), suggesting that the interaction between the motor head and the microtubule limits diffusion speed of the full length motor. With our observation that the velocity of tailless motor in ATP is similar to the instantaneous velocity of the full length motor during the directed 78 Model I Diffusion Directed motion Model 11 Diffusion Directed motion Figure 4.7: Two proposed mechanisms of motility switching. In model I, the motor domain is responsible not only for directed motion but also for diffusion. An intramolecular interaction between the motor domain and the tail modulates switching between two motile modes. In model II, diffusion of the tail domain along the microtubule becomes evident upon unbinding of the motor domain from the microtubule. Reattachment of the motor head results in reversal of motility to directed phases. stepping phases (Table 4.1), our data collectively favors the model that the motor head executes both directional stepping and diffusion and the stochastic interaction between the motor head and the tail mediates switching in motility modes. 4.3.4 Motility of Kif18B under load To access how much mechanical forces Kif18B can exert, we performed optical trapping assays for both the full length and tailless constructs (Fig. 4.9A). Under the fixed trap configuration, Kif18B beads showed minimal force generation, typically below 1 pN, before snapping back to the trap center (Fig. 4.9B). Motility of the motor immediately before detaching from the microtubule did not show typical stalling behaviors as shown in Kinesin-1 motility [109]. Thus, we termed a peak force defined less stringently compared to the stalling criteria of Kinesin-1 (See Materials and Methods, Fig. 4.9C). In some runs, beads moving away from the trap center returned back to the 79 a s Tailless in ADP b E C Tailless in ADP 0.016 0.012 E a 0.008 Tail 0 C,) 0.004 0 Tail 0.6 0.4 0.2 0- 0.1 0.2 0.3 Time interval (s) 0 0.1 0.2 0.3 Time interval (s) Figure 4.8: Diffusional behaviors of the tailless motor and the tail domain. (A) Representative kymographs of GFP-Kifl8B-N574 (top) in 1 mM ADP and GFPKif18B-C268 (bottom) showing diffusion. The diffusion of the tail domain is much faster than the tailless motor. (B-C) MSD of GFP-Kifl8B-N574 in 1 mM ADP (b) 2D + 2c2. All fitted and GFP-Kifl8B-C268 (c). The red curves are fits to MSD parameters are listed in table 4.1. Table 4.1: Summary of motile parameters from MSD analysis [rm/s| ATP FT Tailless ATP AD Kif18B-FL 0.015 - 0.166 - ATP ATP(T.D.P) 0.001 0.045 0.0002 0.183 t 0.002 Relevent figure 4.4B 4.4D 4.6E 0.001 ADP- Tail VT 0.003 0.052 ADP Diffusion constant (D) [pm2 /si 0.001 0.010 0.014 0.001 4.8B 0.598 0.020 4.8C 0.0105 0.0004 - Velocity (V) 4.5E FT Single-molecule fluorescence tracking. VT : Video tracking. T.D.P : Transient directed phase detected using the computer algorithm. All errors are 95% confidence bounds of fitted parameters baseline not abruptly but continuously. This tendency was decreased for the tailless Kif18B (data not shown), suggesting the presence of immature stalling potentially due to the diffusive mode in the full length Kif18B motility. Taken together with a previous work 11101, our result indicates that Kinesin-8 motors are generally evolved to exert only small amount forces, consistent with their roles as microtubule dynamics regulators. Close inspection of Kif18B traces from optical trapping assay revealed that the 80 a b1 0.5 Y axis Optical twee z X axis 0 z -0.5 V P-Kif18B 0 i -1 -1.5 -2 0 C 0.5 1 0 0.5 1 1.5 0 0.5 1 1.5 2 Time (s) 40 d e 30 8 nm 3 2 20 0 m0 ** * * ** 0 0 0.2 * 10 0.4 0.6 0.8 Peak force (pN) 1 Figure 4.9: Kif18B is a low-force motor. (A) Schematic diagram of an optical trap assay for Kifl8B. The optial trap is stationary. As Kifl8B walks along the microtubule, the force exerted on the motor increases. (B) Representative records of Kifl8B-FL coated beads held in the optical trap along the microtubule axis (top, black) and lateral axis (bottom, blue). Kif18B moves sideway frequently (rightmost). (C) Peak force distribution of Kif18B-FL. n - (D-E) Example 2D trajectories (black) of Kif18B coated beads in the optical trap assay. Potential binding sites on the microtubule are shown as green dots. motor frequently stepped sideway (Fig. 4.9b rightmost). This is even clearer when two-dimensional movements of Kif18B beads held by the trap were viewed from the top of the microtubule (Fig. 4.9d and e). 4.4 Discussion We propose a Kif18B motility switching model where the tail domain of Kif18B stochastically interacts with its motor head to lock the nucleotide state of it in ADP-bound form, resulting in diffusion of the motor head. This model is supported by our multiple observations: 1) the Kif18B motor head diffuses in the presence of ADP, 2) Tailless Kif18B showed only directed motions, and 3) During transient di81 rected phases, the full-length Kif18B showed similar instantaneous velocities as tailless Kif18B. The tail-mediated autoinhibition mechanism has been observed in multiple kinesins including Kinesin-1, 2 3, and 7, where the unstructured hinges in the coiled coil stalk enable the tail to fold back on itself, resulting in contact inhibition of the motor head [111, 112, 113, 114]. For these kinesins, autoinhibition can be an effective mechanism to prevent futile ATP hydrolysis in the absence of cargo. The molecular mechanism of Kinesin-1 inhibition is recently studied [115]. In fact, they showed that the tail domain of Kinesin-1 directly contacts the enzymatically important Switch I helix in the motor domain and prevents ADP release from the nucleotide binding pocket. Thus, in Kif18B, a similar molecular mechanism could lead to a change in motile mode from directed to diffusion instead of inhibition of the motor head. Kif18B requires EBi to target MT plus-ends and are likely to dictate the rate of Kif18B accumulation at the extreme MT tip. In particular, since diffusional movements outcompete directed motions in short time/distance scales, targeting of Kif18B proximal to MT plus-ends by EBI could provide a reasonable mechanism for occasional capture of a growing MT plus-end by Kif18B via fast diffusion, even in the case where the growth rates of MTs are faster than the average stepping velocity of Kif18B. MT plus ends are likely to be densely populated by various MT regulators. Thus, diffusion as well as sideway steps observed in Kifl8B motility could be highly advantageous in navigating at the MT tip. Our results showed that human Kif18B possesses unique motile properties distinct from other Kinesin-8 motors. Previously studied yeast Kip3 and human Kif18A exhibit ultra-processivity which allows them to reach microtubule plus ends without dissociation and ultimately result in length-dependent regulation of microtubule dynamics. Like this, the motile properties of different kinesins are generally tuned for distinct functions of each motor. Kif18B shows moderate levels of processivity, short dwell times at the very end of the microtubule and motility switching between diffusion and directed motion, implying its biological roles distinct from other studied Kinesin-8 motors. Our biophysical study of Kif18B motility will be useful in revealing cellular functions of this novel Kinesin-8. 82 4.5 4.5.1 Materials and Methods Protein constructs GFP-Kifl8B-FL-HislO and GFP-Kif18B-N574-His10 were expressed in Sf-9 cells and purified using methods described previously [971. His6-mCherry-Kif18B-C268 was expressed in BL21 cells. 4.5.2 Single-molecule fluorescence assay Motility of single Kif18B molecules was measured using a heavily modified inverted microscope equipped with a 1OOX 1.49 NA TIRF objective, an EMCCD camera and a 488/532nm two-color TIRF illumination system (see Chapter 1.2.1). Single molecule experiments were performed in flow cells with biotinylated GMPCPP MTs, grown from purified bovine brain tubulin labeled with X-Rhodamine, linked to biotin-PEGcoated coverslips via streptavidin. TIRF experiments were carried out in BRB80 containing 0.1 mM DTT, 1 mg/ml casein, 1 mM ATP (1 mM ADP to monitor the diffusional behavior only) and an oxygen scavenging system (5 mg/ml -D-glucose, 0.25 mg/ml glucose oxidase and 0.03 mg/ml catalase). For measurements of motility, 1 nM GFP-Kifl8B was added to the assay buffer and frames were collected at 1 frame per second. 100 ms exposures were used in all recordings. Images were analyzed using custom MATLAB software and the validity of each track was confirmed by visual inspection. The mean squared displacement (MSD) of each single-molecule trace was computed and then averaged over all traces. The diffusion coefficient and drift velocity were then determined by performing weighted fits with appropriate models described in the main text to the first 5-8 points of the MSD because in general the error of the MSD quickly increases when r becomes large [1161. Kymographs and dual color images were generated from representative GFP-Kifl8B motors in ImageJ 1.44p (NIH). 83 4.5.3 Video tracking assay and optical trapping assay Streptavidin conjugated beads (0.44 pm diameter, Spherotech) were coated with biotinylated pentahistidine antibody (Qiagen) through 1-hour incubation at 4 'C. Excess antibody was removed by centrifuging the bead solution and resuspending it in PBS four times. The appropriate Kif18B construct was diluted and mixed with beads in BRB80 with 1 mM DTT, 1 mg/ml casein, 20 pM Taxol (paclitaxel), and 1 mM ATP and incubated for 1 h at 4 'C. After the oxygen scavenging system was added to the Kif18B coated bead solution, the beads were introduced into the flow cell with Taxol-stabilized MTs immobilized on the poly(L-lysine)-coated etched coverslip. To ensure the single-molecule limit, the protein-bead ratio was adjusted so that fewer than half of the beads captured by the optical trap and tested on microtubules showed binding. The video-tracking assay was performed by trapping a freely diffusing bead and placing it on a microtubule. The trap was immediately shuttered upon binding of the bead and the bead motion was recorded at 30 Hz using video-enhanced differential interference contrast (DIC) microscopy. Custom built MATLAB scripts based on a cross-correlation method [391 were used to track positions of beads. The tracking programs were tested using immobilized beads on a glass surface and moving a piezo stage at predetermined step sizes. Detection of transient directed phases from diffusion were carried out based on a speed correlation index (SCI) [108]. The optical trapping measurements were performed in a stationary optical tweezers intrument with separate trapping and detection systems. The setup and calibration procedures are described in detail elsewhere [47]. Briefly, a 1.5 W Nd:YVO4 laser (1064 nm) was expanded and coupled into an inverted microscope with a lOOX/1.3 NA oil-immersion objective. Displacements from the trap center of beads were recorded at 3 kHz, antialias-filtered at 1.5 kHz. Trap stiffnesses ranging 0.01 - 0.02 pN/nm were used for all experiments. Motility data were analyzed using custom built MATLAB scripts. The peak force were detected using criteria : stall force > 0.2 pN, snapback velocity > 1000 nm/s, run lasting longer than 33 ms and a snapback to baseline. 84 Appendix A ClpXP Protocols A.1 Suface Passivation Using PEG Materials 1. PEG: mPEG-Silane, MW 5000 and Biotin-PEG-Silane MW 5000 (Laysan Bio) 2. Triethylamine : Sigma 90335 3. Toluene : Sigma 244511 4. Methanol : Sigma 179337 5. KOH : Sigma 221473 6. glass coverslip : Fisher 12-544-C (24 x 40, thickness 1.5) Protocol 1. Remove triethylamine from the 4 'C refrigerator and the PEG-silane jar (contains mPEG-SIL and Bio-PEG-SIL) from the -20 0C freezer. Allow them to thaw for a couple of hours. These reagents are moisture sensitive and must equilibrate to room temperature before use. 2. Rinse the glass-staining jar (reaction container) multiple times with DI water. Fill it with 1 M KOH and sonicate for 20 minutes. Rinse again multiple times with DI water. 85 3. Place glass coverslips in the glass staining jar. Rinse twice and sonicate for 10 minutes with DI water. Repeat the process with methanol. 4. Sonicate the coverslips in IM KOH for 20 minutes then rinse them multiple times with DI water. 5. While the coverslips are sonicating in KOH, remove mPEG-SIL and Bio-PEGSIL from the PEG jar. Place approximately 297 mg of mPEG and 3 mg of Bio-PEG-SIL (1 %) in the falcon tube. 6. Rinse the KOH-etched coverslips three times with Toluene. Pour 30 mL of Toluene into a small beaker. Add 3.38 piL of Triethylamine and the falcon tube of PEG to the beaker of Toluene. Mix thoroughly and pour into the jar of coverslips. 7. Sonicate the reaction mixture for 30 minutes at 35 'C. 8. Wash twice with Toluene, then multiple times with DI water until coverslips are clean. 9. Dry the coverslips with Nitrogen, place in a sealed dessicator and store at -20 'C. PEG coverslips are best if used within 2 weeks. 86 A.2 Single-Molecule Fluorescence Assay Materials 1. PEGylated coverslips 2. Streptavidin (Rockland, SOOO-01, 1 mg/ml in PBT) 3. ATP (Sigma A7699, 100 mM in PD, stored in - 80 'C) 4. Proteins (Biotinylated ClpX with a labeled FRET pair, ClpP, ssrA-tagged substrates) 5. ATP regeneration system (prepared in 20 x; 100 mM creatine phosphate (EMD, 2380) and 0.6 mg/ml creatine kinase (EMD, 238395) in PD) 6. Oxygen scavenging system (prepared in 100 x; 16500 U/ml glucose oxidase (Sigma, G2133) and 217000 U/ml catalase (Sigma, C100) in PD, 2 mM Trolox (Sigma, 238813) in PD) 7. PD buffer (25 mM HEPES, pH 7.8, 100 mM KC1, 10 mM MgCl 2 , 10% Glycerol) 8. PBT buffer (100mM phosphate buffer, pH 7.5) 9. flow cell (KOH-etched coverslips, double sticky tape, microscope slides) Protocol 1. Take out a dessicator with PEG coverslips from - 20 'C and let it to be equilibrated with room temperature. 2. Assemble a flow cell with double stick tape flanked by a glass slide and a PEGcoated coverslip. 3. Dilute streptavidin to 0.01 mg/mL in PBT, flow 20 pL into the flow cell and incubate for 10 min. 87 4. During streptavidin incubation, mix ClpX, ClpP, 1 mM ATP in PD to assemble ClpXP complexes. 5. Wash out unbound streptavidin with 100 pL PD. 6. Dilute ClpXP complexes further to reach the single-molecule level and flow 20 pL of the diluted solution into flow cells and incubate for 20 min. 7. Prepare a washing buffer and an imaging buffer. The washing buffer consists of 0.5 pM ClpP and oxygen scavenging system with appropriate nucleotide/substrate conditions. 8. Wash out unbound ClpXP with the washing buffer. Right before data collection, add glucose to the washing buffer before flowing into the flow cell. 88 Appendix B Kif18B Protocols B.1 Tubulin Polymerization Materials 1. PEM80 (80mM PIPES, 1mM EGTA, 4mM MgCl 2 , pH adjusted to 6.9 with KOH) 6.048 g Pipes (Sigma P1851), 95.1 mg EGTA (Sigma E4378), 204.1 PL of 4.9 M MgCl 2 (Mallinckrodt H590) stock into 250 mL final volume 2. PEM104 (103.6 mM PIPES, 1.3 mM EGTA, 6.3 mM MgCl 2 , pH adjusted to 6.9 with KOH) 3.133 g Pipes (Sigma P1851),49.452 mg EGTA (Sigma E4378), 128.57 pL of 4.9 M MgC1 2 (Mallinckrodt H590) stock into 100 mL final volume. 3. STAB 38.6 pL PEM80, 0.5 pL 100 mM GTP (Cytoskeleton BST06), 4.7 PL 65 g/L NaN 3 (Sigma S8032), 1.2 pL 10 mM Taxol (Cytoskeleton TXD01, reconstituted in 20pL DMSO to make 10mM), 5.0 pL DMSO (Sigma D5879). 4. Tubulins tubulin (TL238), biotin-tubulin(T333P) and rhodamine labeled tubulin (TL 590m) all from Cytoskeleton Inc. Protocol 1. Combine 13 pL PEM104 + 2.2 ML 10 mM GTP (dilute 100mM GTP with PEM104) to make PEM/GTP solution. 89 2. Combine 15.2 pL PEM/GTP + 2.2 pL DMSO, vortex mixture, then add - Nonlabeled microtubule : 4.8 pL of 10 mg/mL tubulin - Biotinylated microtubule: 4.5 pL of 10 mg/mL tubulin + 0.3 pL of 10 mg/mL biotinylated tubulin - Biotinylated and dimly fluorescently labeled microtubule : 4.5 PL of 10 mg/mL tubulin + 0.3 pL of 10mg/mL biotinylated tubulin + 0.25 pL of 1 mg/mL fluorescently labeled tubulin (diluted from stock with PEM 80 + 1mM GTP) 3. Place TUB solution in water bath at 37 'C for 30 min. 4. Remove TUB from water bath and add 2 pL of STAB. (For biotinylated MT, centrifuge 7 min at 21 x 103 g at room temp) 5. Store microtubules at room temperature. 90 B.2 Single-Molecule Fluorescence Motility Assay Materials 1. PEGylated coverslips (30 % Biotin-PEG) 2. Tubulins tubulin (TL238), biotin-tubulin(T333P) and rhodamine labeled tubulin (TL 590m) all from Cytoskeleton Inc. 3. GMPCPP (Jena Bioscience, NU405S) 4. Buffers (PEM80, PBS) 5. Streptavidin (Rockland, SOOO-01, 1 mg/ml in PBT) 6. Casein (Sigma, C8654) 7. DTT (0.5 M in 10 mM K-acetate, stored at - 20 0 C) 8. ATP (Sigma, A7699, 100 mM in PEM80, pH 7, stored at - 80 'C) 9. 100 x Oxygen scavenging system (Glucose oxidase (EMD, 345386, 25 mg/mL in PBS), -D-glucose (EMD, 346351, 500 mg/mL in PBS), Catalase (EMD, 219261, 3 mg/mL in PBS, all stored in - 80 C)) Protocol 1. (if GMPCPP stabilized microtubules are not ready) Polymerize GMPCPPmicrotubule (a) For Biotinylated and dimly fluorescently labeled microtubule : 17.2 PL of 10 mg/mL tubulin + 1.6 pL of 10 mg/mL biotinylated tubulin + 1 pL of 1 mg/mL fluorescently labeled tubulin (diluted from stock with PEM 80 + 1 mM GTP) + 87.4 ptL PEM80 + 12 pL GMPCPP (10 mM) + 0.25 pL DTT (0.5 M) (b) The reaction mix can be stored at - 80 'C. (c) For immediate usage, incubate the reaction mixture for 1 hour at 37 0 C. (d) (To remove short MTs and tubulin monomers) Centrifuge for 7 min at 14000 rpm and remove supernatant. Resuspend the pellet in PEM 80. 91 2. Prepare flow channels using PEG coated coverslips. 3. Flow in 20 pL of streptavidin (diluted 4 times with PBS) and incubate for 10 min. 4. Make assay buffer (a) Label a tube AB, and add: i. 1329 pL PEM80 ii. 3 pL DTT (0.5 M in 10 mM K-acetate, aliquots in -20 C) iii. 15 pL ATP (100 mM in PEM80, aliquots in - 80 'C) iv. 150 pL 10 mg/mL Casein in PBS (made fresh once per week and stored at 4 0 C) (b) AB final concentrations: 1 mM DTT, 1 mg/mL Casein, 1 mM ATP (c) Store on ice 5. Wash out unbound streptavidin with 100 pL PEM 80. 6. Flow in 20 pL of GMPCPP-microtubules (diluted 10 times with PEM 80) and % let them bind for 20 min. (When the binding is rare, incubating with 0.2 methylcellulose helps MTs bind to the surface). 7. Start warming AB to RT (do not leave it longer than 5 min at room temp) 8. Wash in 50 pL PEM 80 and 80 pL AB sequentially. 9. Take out a tube of Kinesin from - 80 'C freezer and dilute with AB to appropriate concentrations. Add oxygen scavenging system to the final diluted volume before washing into the flow channel. 92 B.3 Kinesin Bead Assay Materials 1. Biotinylated penta-His antibody (Qiagen, 34440, 125 pL, 0.2 mg/mL) 2. Streptavidin coated polystyrene bead (Spherotech, SVP-05-10, 0.44 pm in diameter, 1% w/v) 3. PBS (100 mM phosphate buffer, pH 7.5, 1860 pL IM NaH 2PO 4 , 8140 pL IM Na 2HPO 4 , 90 mL dH 20) 4. DTT (0.5 M in 10 mM K-acetate, stored at - 20 C) 5. Taxol (Cytoskeleton, TXD01, 10 mM in DMSO, stored at - 20 'C) 6. Poly-l-lysine (Sigma, P8920) 7. ATP (Sigma, A7699, 100 mM in PEM80, pH 7, stored at - 80 'C) 8. 100 x Oxygen scavenging system (Glucose oxidase (EMD, 345386, 25 mg/mL in PBS), #-D-glucose (EMD, 346351, 500 mg/mL in PBS), Catalase (EMD, 219261, 3 mg/mL in PBS, all stored in - 80 C)) Protocol 1. (if streptavidin-coated beads are not ready) Wash streptavidin coated beads (a) Dilute 20 ptL 0.44 pm Streptavidin-coated beads into 80 pL PBS (b) Wash 4 times at 10,000 rpm for 6 min, reconstituting in 100 pL PBS (c) Sonicate for 2 min at 40% (d) Store washed streptavidin-coated beads in a rotator at 4 'C 2. Make bead coated with biotinylated penta His antibody (a) Take 20 pL of cleaned beads, and add i. 50 pL PBS ii. 30 pL 0.2 mg/mL biotinylated Penta-His antibody (b) Incubate at 4 0 C for 1 h in a rotator (during this incubation, proceeds to make buffers below) 93 (c) Add 500 pL of PBS and wash anti-His beads 4 times for 6 min at 10,000 rpm, 4 'C, reconstituting in 500 pL PBS. For last wash, reconstitute in 100 AL AB. (d) Sonicate for 2 min at 30 %. Label HisB 3. Make PemTax (a) Label a tube PemTax, and add: i. 1000 pL Pem80 (stored in 4 'C) ii. 2 piL Taxol (10 mM in DMSO, aliquots in -20 'C) (b) Store at room temperature 4. Make assay buffer (a) Label a tube AB, and add: i. 1329 pLL PEM80 ii. 3 pL DTT (0.5 M in 10 mM K-acetate, aliquots in -20 'C) iii. 3 pL Taxol iv. 15 pL ATP (100 mM in PEM80, aliquots in -80 'C) v. 150 ILL 10 mg/mL Casein in PBT (made fresh once per week and stored at 4 0 C) (b) AB final concentrations: 1 mM DTT, 20 pM Taxol, 1 mg/mL Casein, 1 mM ATP (c) Store on ice 5. Make C-Tax (a) Label a tube C-Tax and add i. 80 pL PemTax ii. 20 pL 10 mg/mL casein (b) store on ice 6. Make Kinesin dilutions (a) K/100: 2 pL Kinesin into 98 pL AB (b) K/1000: 10 pL Kinesin/100 into 90 pL AB 94 . (c) .. 7. Make Kinesin/bead dilution (KDB) (a) Label a tube KDB/ , and add: i. 20 ML HisB ii. 20 pL K/ (b) Incubate for 1 hour on the rotator at 4 'C 8. Prepare Poly-l-lysine coated coverslips (a) Dilute 100 1L Poly-l-lysine into 30 mL EtOH in a falcon tube. (b) Place two KOH cleaned slips into the solution, and let sit for 15 min (c) Dry for 15min in the oven 9. Prepare flow channels using Poly-l-lysine coated coverslips (a) Start warming C-Tax to RT (b) Flow in 15 [pL MT/100 (diluted in PemTax) and let bind for 10 min (c) Wash in 20 pL PemTax with high velocity to RT (do not leave these longer than 5 (d) Start warming AB and KDB/ min at room temp) (e) Wash in 15 pL C-Tax and let coat for 5 min (f) Wash in 50 ALL PemTax (g) Wash in 80 ALL AB (h) Add oxygen scavenging system to each KDB incubation (40 pL) i. Add 0.4 pL of Glucose oxidase, (i) Wash in 20 pL KDB/ 95 -D-glucose and Catalase each 96 Bibliography [11 Erwin Schr6dinger. What is life?: With mind and matter and autobiographical sketches. Cambridge University Press, 1992. [2] Timothy Elston, Hongyun Wang, and George Oster. Energy transduction in atp synthase. Nature, 391(6666):510-513, 1998. [31 Sriram Subramaniam and Richard Henderson. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. 2000. Nature, 406(6796):653-657, [41 Rob Phillips and Stephen R Quake. The biological frontier of physics. Physics Today, 59(5):38-43, 2006. [5] Murat Acar, Jerome T Mettetal, and Alexander van Oudenaarden. Stochastic switching as a survival strategy in fluctuating environments. Nature genetics, 40(4):471-475, 2008. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell, 135(2):216-226, 2008. [6] Arjun Raj and Alexander van Oudenaarden. [71 Jonne Helenius, Gary Brouhard, Yannis Kalaidzidis, Stefan Diez, and Jonathon Howard. The depolymerizing kinesin mcak uses lattice diffusion to rapidly target microtubule ends. Nature, 441(7089):115-119, 2006. 18] Charles S Peskin, Garrett M Odell, and George F Oster. Cellular motions and thermal fluctuations: the brownian ratchet. Biophysical journal, 65(1):316-324, 1993. 191 A Szent-Gy6rgyi. The contraction of myosin threads. Stud. Inst. Med. Chem. Univ. Szeged. I, pages 17-26, 1942. [101 Bartosz Lewandowski, Guillaume De Bo, John W Ward, Marcus Papmeyer, Sonja Kuschel, Maria J Aldegunde, Philipp ME Gramlich, Dominik Heckmann, Stephen M Goldup, Daniel M DSouza, et al. Sequence-specific peptide synthesis by an artificial small-molecule machine. Science, 339(6116):189-193, 2013. 1111 Perrine Lussis, Tiziana Svaldo-Lanero, Andrea Bertocco, Charles-Andre Fustin, David A Leigh, and Anne-Sophie Duwez. A single synthetic small molecule that generates force against a load. Nature nanotechnology, 6(9):553-557, 2011. 97 [12] Jose Berna, David A Leigh, Monika Lubomska, Sandra M Mendoza, Emilio M P6rez, Petra Rudolf, Gilberto Teobaldi, and Francesco Zerbetto. Macroscopic transport by synthetic molecular machines. Nature materials, 4(9):704-710, 2005. [13] Wonmuk Hwang and Matthew J Lang. Mechanical design of translocating motor proteins. Cell biochemistry and biophysics, 54(1-3):11-22, 2009. [141 Phyllis I Hanson and Sidney W Whiteheart. Aaa+ proteins: have engine, will work. Nature Reviews Molecular Cell Biology, 6(7):519-529, 2005. [15] Robert T Sauer and Tania A Baker. Aaa+ proteases: Atp-fueled machines of protein destruction. Annual review of biochemistry, 80:587-612, 2011. [161 Tania A Baker and Robert T Sauer. Clpxp, an atp-powered unfolding and protein-degradation machine. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1823(1):15-28, 2012. [17] Kenneth C Keiler, Patrick RH Waller, and Robert T Sauer. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger rna. SCIENCE-NEW YORK THEN WASHINGTON-, pages 990-993, 1996. [18] Shilpa A Joshi, Greg L Hersch, Tania A Baker, and Robert T Sauer. Communication between clpx and clpp during substrate processing and degradation. Nature structural & molecular biology, 11(5):404-411, 2004. 1191 Andreas Martin, Tania A Baker, and Robert T Sauer. Protein unfolding by a aaa+ protease is dependent on atp-hydrolysis rates and substrate energy landscapes. Nature structural & molecular biology, 15(2):139-145, 2008. [201 Andreas Martin, Tania A Baker, and Robert T Sauer. Diverse pore loops of the aaa+ clpx machine mediate unassisted and adaptor-dependent recognition of ssra-tagged substrates. Molecular cell, 29(4):441-450, 2008. 1211 Jon A Kenniston, Tania A Baker, Julio M Fernandez, and Robert T Sauer. Linkage between atp consumption and mechanical unfolding during the protein processing reactions of an aaa+ degradation machine. Cell, 114(4):511-520, 2003. 1221 Marie-Eve Aubin-Tam, Adrian 0 Olivares, Robert T Sauer, Tania A Baker, and Matthew J Lang. Single-molecule protein unfolding and translocation by an atp-fueled proteolytic machine. Cell, 145(2):257-267, 2011. [231 Jon A Kenniston, Tania A Baker, and Robert T Sauer. Partitioning between unfolding and release of native domains during clpxp degradation determines substrate selectivity and partial processing. Proceedings of the National Academy of Sciences of the United States of America, 102(5):1390-1395, 2005. 98 [241 Randall E Burton, Samia M Siddiqui, Yong-In Kim, Tania A Baker, and Robert T Sauer. Effects of protein stability and structure on substrate processing by the clpxp unfolding and degradation machine. The EMBO journal, 20(12):3092-3100, 2001. 125] Rodrigo A Maillard, Gheorghe Chistol, Maya Sen, Maurizio Righini, Jiongyi Tan, Christian M Kaiser, Courtney Hodges, Andreas Martin, and Carlos Bustamante. Clpx (p) generates mechanical force to unfold and translocate its protein substrates. Cell, 145(3):459-469, 2011. [261 Maya Sen, Rodrigo A Maillard, Kristofor Nyquist, Piere Rodriguez-Aliaga, Steve Presse, Andreas Martin, and Carlos Bustamante. The clpxp protease unfolds substrates using a constant rate of pulling but different gears. Cell, 155(3):636-646, 2013. [27] Benjamin M Stinson, Andrew R Nager, Steven E Glynn, Karl R Schmitz, Tania A Baker, and Robert T Sauer. Nucleotide binding and conformational switching in the hexameric ring of a aaa+ machine. Cell, 1523(31):628-637-9, 20131998. [28] Ronald D Vale, Thomas S Reese, and Michael P Sheetz. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell, 42(1):39-50, 1985. [29] Nobutaka Hirokawa, Yasuko Noda, Yosuke Tanaka, and Shinsuke Niwa. Kinesin superfamily motor proteins and intracellular transport. Nature reviews Molecular cell biology, 10(10):682-696, 2009. [301 Kristen J Verhey, Neha Kaul, and Virupakshi Soppina. Kinesin assembly and movement in cells. Annual review of biophysics, 40:267-288, 2011. 131] Ahmet Yildiz, Michio Tomishige, Ronald D Vale, and Paul R Selvin. Kinesin walks hand-over-hand. Science, 303(5658):676-678, 2004. [32] Charles L Asbury, Adrian N Fehr, and Steven M Block. Kinesin moves by an asymmetric hand-over-hand mechanism. Science, 302(5653):2130-2134, 2003. [331 Bason E Clancy, William M Behnke-Parks, Johan OL Andreasson, Steven S Rosenfeld, and Steven M Block. A universal pathway for kinesin stepping. Nature structural & molecular biology, 18(9):1020 1027, 2011. [341 Sharyn A Endow and Kimberly W Waligora. Determinants of kinesin motor polarity. Science, 281(5380):1200-1202, 1998. [351 Richard H Wade and Frank Kozielski. Structural links to kinesin directionality and movement. Nature Structural 6 Molecular Biology, 7(6):456-460, 2000. 99 [361 Nicholas F Endres, Craig Yoshioka, Ronald A Milligan, and Ronald D Vale. A lever-arm rotation drives motility of the minus-end-directed kinesin ncd. Nature, 439(7078):875-878, 2005. [37] Sirish Kaushik Lakkaraju and Wonmuk Hwang. Hysteresis-based mechanism for the directed motility of the ncd motor. Biophysical journal, 101(5):1105-1113, 2011. [381 Sanghamitra Ray, Edgar Meyh6fer, Ronald A Milligan, and Jonathon Howard. Kinesin follows the microtubule's protofilament axis. The Journal of cell biology, 121(5):1083-1093, 1993. [39] Jeff Gelles, Bruce J Schnapp, and Michael P Sheetz. Tracking kinesin-driven movements with nanometre-scale precision. Nature, 331(6155):450-453, 1988. [40] Volker Bormuth, Bert Nitzsche, Felix Ruhnow, Aniruddha Mitra, Marko Storch, Burkhard Rammner, Jonathon Howard, and Stefan Diez. The highly processive kinesin-8, kip3, switches microtubule protofilaments with a bias toward the left. Biophysical journal, 103(1):L4-L6, 2012. [411 Melanie Brunnbauer, Renate Dombi, Thi-Hieu Ho, Manfred Schliwa, Matthias Rief, and Zeynep Okten. Torque generation of kinesin motors is governed by the stability of the neck domain. Molecular cell, 46(2):147-158, 2012. 1421 Junichiro Yajima, Kana Mizutani, and Takayuki Nishizaka. A torque component present in mitotic kinesin eg5 revealed by three-dimensional tracking. Nature structural & molecular biology, 15(10):1119-1121, 2008. [43] Sinan Can, Mark A Dewitt, and Ahmet Yildiz. Bidirectional helical motility of cytoplasmic dynein around microtubules. eLife, 3:e03205, 2014. [441 William J Greenleaf, Michael T Woodside, and Steven M Block. Highresolution, single-molecule measurements of biomolecular motion. Annual review of biophysics and biomolecular structure, 36:171, 2007. [45 Carlos Bustamante, Wei Cheng, and Yara X Mejia. Revisiting the central dogma one molecule at a time. Cell, 144(4):480-497, 2011. 146] Keir C Neuman and Attila Nagy. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nature methods, 5(6):491, 2008. [471 Ahmad S Khalil, David C Appleyard, Anna K Labno, Adrien Georges, Martin Karplus, Angela M Belcher, Wonmuk Hwang, and Matthew J Lang. Kinesin's cover-neck bundle folds forward to generate force. Proceedings of the National Academy of Sciences, 105(49):19247-19252, 2008. 100 [48] Yongdae Shin, Joseph H Davis, Ricardo R Brau, Andreas Martin, Jon A Kenniston, Tania A Baker, Robert T Sauer, and Matthew J Lang. Single-molecule denaturation and degradation of proteins by the aaa+ clpxp protease. Proceedings of the National Academy of Sciences, 106(46):19340-19345, 2009. [49] Ruobo Zhou, Alexander G Kozlov, Rahul Roy, Jichuan Zhang, Sergey Korolev, Timothy M Lohman, and Taekjip Ha. Ssb functions as a sliding platform that migrates on dna via reptation. Cell, 146(2):222-232, 2011. [50] Guus B Erkens, Inga Hdnelt, Joris MH Goudsmits, Dirk Jan Slotboom, and Antoine M van Oijen. Unsynchronised subunit motion in single trimeric sodiumcoupled aspartate transporters. Nature, 502(7469):119-123, 2013. 151] Ricardo R Brau, Peter B Tarsa, Jorge M Ferrer, Peter Lee, and Matthew J Lang. Interlaced optical force-fluorescence measurements for single molecule biophysics. Biophysical journal, 91(3):1069-1077, 2006. [52] Peter B Tarsa, Ricardo R Brau, Mariya Barch, Jorge M Ferrer, Yelena Freyzon, Paul Matsudaira, and Matthew J Lang. Detecting force-induced molecular transitions with fluorescence resonant energy transfer. Angewandte Chemie InternationalEdition, 46(12):1999-2001, 2007. [53] Joseph R Lakowicz. Principles of Fluorescence Spectroscopy. Springer, 2006. [541 Xavier Michalet, Shimon Weiss, and Marcus Jdger. Single-molecule fluorescence studies of protein folding and conformational dynamics. Chemical reviews, 106(5):1785-1813, 2006. [551 Taekjip Ha and Philip Tinnefeld. Photophysics of fluorescence probes for single molecule biophysics and super-resolution imaging. Annual review of physical chemistry, 63:595, 2012. [56] Ivan Rasnik, Sean A McKinney, and Taekjip Ha. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature methods, 3(11):891-893, 2006. [57] Taekjip Ha, Alice Y Ting, Joy Liang, W Brett Caldwell, Ashok A Deniz, Daniel S Chemla, Peter G Schultz, and Shimon Weiss. Single-molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proceedings of the National Academy of Sciences, 96(3):893-898, 1999. [58] Arthur Ashkin. Acceleration and trapping of particles by radiation pressure. Physical review letters, 24(4):156, 1970. 1591 Keir C Neuman and Steven M Block. Optical trapping. Review of scientific instruments, 75(9):2787-2809, 2004. [60] Arthur Ashkin. Forces of a single-beam gradient laser trap on a dielectric sphere in the ray optics regime. Biophysical journal, 61(2):569-582, 1992. 101 [61] Frederick Gittes and Christoph F Schmidt. Interference model for back-focalplane displacement detection in optical tweezers. Optics letters, 23(1):7-9, 1998. [62] Satyendra K Singh, Jan Rozycki, Joaquin Ortega, Takashi Ishikawa, John Lo, Alasdair C Steven, and Michael R Maurizi. Functional domains of the clpa and clpx molecular chaperones identified by limited proteolysis and deletion analysis. Journal of biological chemistry, 276(31):29420-29429, 2001. [63] Juan Carlos Cordova, Adrian 0 Olivares, Yongdae Shin, Benjamin M Stinson, Stephane Calmat, Karl R Schmitz, Marie-Eve Aubin-Tain, Tania A Baker, Matthew J Lang, and Robert T Sauer. Stochastic but highly coordinated protein unfolding and translocation by the clpxp proteolytic machine. Cell, 158(3):647-658, 2014. 164] Greg L Hersch, Randall E Burton, Daniel N Bolon, Tania A Baker, and Robert T Sauer. Asymmetric interactions of atp with the aaa+ clpx6 unfoldase: Allosteric control of a protein machine. Cell, 121(7):1017-1027, 2005. [65] Steven E Glynn, Andreas Martin, Andrew R Nager, Tania A Baker, and Robert T Sauer. Structures of asymmetric clpx hexamers reveal nucleotidedependent motions in a aaa+ protein-unfolding machine. Cell, 139(4):744-756, 2009. [661 Harold D Kim, G Ulrich Nienhaus, Taekjip Ha, Jeffrey W Orr, James R Williamson, and Steven Chu. Mg2+-dependent conformational change of rna studied by fluorescence correlation and fret on immobilized single molecules. Proceedings of the National Academy of Sciences, 99(7):4284-4289, 2002. 1671 Kyung-Jin Jang and Jwa-Min Nam. Direct-write nanoparticle microarrays for cell assays. small, 4(11):1930-1935, 2008. [68] Sean A McKinney, Chirlmin Joo, and Taekjip Ha. Analysis of singlemolecule fret trajectories using hidden markov modeling. Biophysical journal, 91(5):1941-1951, 2006. 1691 Andreas Martin, Tania A Baker, and Robert T Sauer. Rebuilt aaa+ motors reveal operating principles for atp-fuelled machines. Nature, 437(7062):11151120, 2005. 170] David M Smith, Hugo Fraga, Christian Reis, Galit Kafri, and Alfred L Goldberg. Atp binds to proteasomal atpases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell, 144(4):526-538, 2011. [711 Hongbin Li, Mariano Carrion-Vazquez, Andres F Oberhauser, Piotr E Marszalek, and Julio M Fernandez. Point mutations alter the mechanical stability of immunoglobulin modules. Nature Structural & Molecular Biology, 7(12):1117 1120, 2000. 102 [721 Daniel L Floyd, Stephen C Harrison, and Antoine M Van Oijen. Analysis of kinetic intermediates in single-particle dwell-time distributions. Biophysical journal, 99(2):360-366, 2010. [731 Sunney Xie. Single-molecule approach to enzymology. 2(4):229-236, 2001. Single Molecules, 174] MJ Schnitzer and SM Block. Statistical kinetics of processive enzymes. In Cold Spring Harborsymposia on quantitative biology, volume 60, pages 793-802. Cold Spring Harbor Laboratory Press, 1995. [751 Aldous David and Shepp Larry. The least variable phase type distribution is erlang. Stochastic Models, 3(3):467-473, 1987. [761 Joshua W Shaevitz, Steven M Block, and Mark J Schnitzer. Statistical kinetics of macromolecular dynamics. Biophysical journal, 89(4):2277-2285, 2005. [77] Karel Svoboda, Partha P Mitra, and Steven M Block. Fluctuation analysis of motor protein movement and single enzyme kinetics. Proceedings of the National Academy of Sciences, 9174(25):11782-11786A183-A183, 19948. [78] Hongyun Wang. A new derivation of the randomness parameter. Journal of Mathematical Physics, 48(10):103301, 2007. [791 Jeffrey R Moffitt, Yann R Chemla, and Carlos Bustamante. Chapter tenmethods in statistical kinetics. Methods in enzymology, 475:221-257, 2010. [80] Jeffrey R Moffitt and Carlos Bustamante. Extracting signal from noise: kinetic mechanisms from a michaelis-menten-like expression for enzymatic fluctuations. FEBS Journal, 281(2):498-517, 2014. [811 Yong-In Kim, Randall E Burton, Briana M Burton, Robert T Sauer, and Tania A Baker. Dynamics of substrate denaturation and translocation by the clpxp degradation machine. Molecular cell, 5(4):639-648, 2000. [82] Steven M Block, Charles L Asbury, Joshua W Shaevitz, and Matthew J Lang. Probing the kinesin reaction cycle with a 2d optical force clamp. Proceedings of the National Academy of Sciences, 100(5):2351-2356, 2003. [83] Arshad Desai and Timothy J Mitchison. Microtubule polymerization dynamics. Annual review of cell and developmental biology, 13(1):83-117, 1997. [841 Eva Nogales. Structural insights into microtubule function. Annual review of biochemistry, 69(1):277-302, 2000. 1851 Tim Mitchison, Marc Kirschner, et al. Dynamic instability of microtubule growth. Nature, 312(5991):237-242, 1984. 103 [86] Kazuhisa Kinoshita, Bianca Habermann, and Anthony A Hyman. Xmap215: a key component of the dynamic microtubule cytoskeleton. Trends in cell biology, 12(6):267-273, 2002. [87] Nasser M Rusan, Carey J Fagerstrom, Anne-Marie C Yvon, and Patricia Wadsworth. Cell cycle-dependent changes in microtubule dynamics in living cells expressing green fluorescent protein-a tubulin. Molecular biology of the cell, 12(4):971-980, 2001. [88] lain M Cheeseman and Arshad Desai. Molecular architecture of the kinetochore-microtubule interface. Nature Reviews Molecular Cell Biology, 9(1):33-46, 2008. [891 Carolyn A Moores, Ming Yu, Jun Guo, Christophe Beraud, Roman Sakowicz, and Ronald A Milligan. A mechanism for microtubule depolymerization by kini kinesins. Molecular cell, 9(4):903-909, 2002. [901 Dongyan Tan, William J Rice, and Hernando Sosa. Structure of the kinesin13microtubule ring complex. Structure, 16(11):1732-1739, 2008. [91] Claire E Walczak, Sophia Gayek, and Ryoma Ohi. Microtubule-depolymerizing kinesins. Annual review of cell and developmental biology, 29:417-441, 2013. [92] R6gis Tournebize, Andrei Popov, Kazuhisa Kinoshita, Anthony J Ashford, Sonja Rybina, Andrei Pozniakovsky, Thomas U Mayer, Claire E Walczak, Eric Karsenti, and Anthony A Hyman. Control of microtubule dynamics by the antagonistic activities of xmap215 and xkcml in xenopus egg extracts. Nature cell biology, 2(1):13-19, 1999. 193] Claire E Walczak, Timothy J Mitchison, and Arshad Desai. Xkcml: a xenopus kinesin-related protein that regulates microtubule dynamics during mitotic spindle assembly. Cell, 84(1):37-47, 1996. [941 Kazuhisa Kinoshita, Isabelle Arnal, Arshad Desai, David N Drechsel, and Anthony A Hyman. Reconstitution of physiological microtubule dynamics using purified components. Science, 294(5545):1340-1343, 2001. [95] Mohan L Gupta, Pedro Carvalho, David M Roof, and David Pellman. Plus end-specific depolymerase activity of kip3, a kinesin-8 protein, explains its role in positioning the yeast mitotic spindle. Nature cell biology, 8(9):913--923, 2006. [96] Shinsuke Niwa, Kazuo Nakajima, Harukata Miki, Yusuke Minato, Doudou Wang, and Nobutaka Hirokawa. Kifl9a is a microtubule-depolymerizing kinesin for ciliary length control. Developmental cell, 23(6):1167-1175, 2012. [971 Yaqing Du, Chauca A English, and Ryoma Ohi. The kinesin-8 kifl8a dampens microtubule plus-end dynamics. Current Biology, 20(4):374-380, 2010. 104 [98] Jason Stumpff, Michael Wagenbach, Andrew Franck, Charles L Asbury, and Linda Wordeman. Kifl8a and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension. Developmental cell, 22(5):1017-1029, 2012. 199] Jason Stumpff, Yaqing Du, Chauca A English, Zoltan Maliga, Michael Wagenbach, Charles L Asbury, Linda Wordeman, and Ryoma Ohi. A tethering mechanism controls the processivity and kinetochore-microtubule plus-end enrichment of the kinesin-8 kifl8a. Molecular cell, 43(5):764-775, 2011. 1100] Vladimir Varga, Jonne Helenius, Kozo Tanaka, Anthony A Hyman, Tomoyuki U Tanaka, and Jonathon Howard. Yeast kinesin-8 depolymerizes microtubules in a length-dependent manner. Nature cell biology, 8(9):957-962, 2006. [101] Monika I Mayr, Stefan HUmmer, Jenny Bormann, Tamara Griiner, Sarah Adio, Guenther Woehlke, and Thomas U Mayer. The human kinesin kifl8a is a motile microtubule depolymerase essential for chromosome congression. Current biology, 17(6):488-498, 2007. [102] Jane R Stout, Amber L Yount, James A Powers, Chantal LeBlanc, Stephanie C Ems-McClung, and Claire E Walczak. Kifl8b interacts with ebi and controls astral microtubule length during mitosis. Molecular biology of the cell, 22(17):3070-3080, 2011. [103] Marvin E Tanenbaum, Libor Macurek, Babet van der Vaart, Matilde Galli, Anna Akhmanova, and Rene H Medema. A complex of kifl8b and mcak promotes microtubule depolymerization and is negatively regulated by aurora kinases. Current Biology, 21(16):1356-1365, 2011. [104] Vladimir Varga, Cecile Leduc, Volker Bormuth, Stefan Diez, and Jonathon Howard. Kinesin-8 motors act cooperatively to mediate length-dependent microtubule depolymerization. Cell, 138(6):1174 - 1183, 2009. [105] Yasushi Okada and Nobutaka Hirokawa. A processive single-headed motor: kinesin superfamily protein kifla. Science, 283(5405):1152-1157, 1999. [106] Johanna Roostalu, Christian Hentrich, Peter Bieling, Ivo A Telley, Elmar Schiebel, and Thomas Surrey. Directional switching of the kinesin cin8 through motor coupling. Science, 332(6025):94-99, 2011. [107] Benjamin H Kwok, Lukas C Kapitein, Jeffrey H Kim, Erwin JG Peterman, Christoph F Schmidt, and Tarun M Kapoor. Allosteric inhibition of kinesin-5 modulates its processive directional motility. Nature chemical biology, 2(9):480485, 2006. [1081 Cedric Bouzigues and Maxime Dahan. Transient directed motions of gabaa receptors in growth cones detected by a speed correlation index. Biophysical journal, 92(2):654-660, 2007. 105 [1091 Karel Svoboda and Steven M Block. Force and velocity measured for single kinesin molecules. Cell, 77(5):773-784, 1994. [1101 Anita Jannasch, Volker Bormuth, Marko Storch, Jonathon Howard, and Erik Schdffer. Kinesin-8 is a low-force motor protein with a weakly bound slip state. Biophysical journal, 104(11):2456-2464, 2013. [1111 Miki Imanishi, Nicholas F Endres, Arne Gennerich, and Ronald D Vale. Autoinhibition regulates the motility of the c. elegans intraflagellar transport motor osm-3. The Journal of cell biology, 174(7):931-937, 2006. [112] David L Coy, William 0 Hancock, Michael Wagenbach, and Jonathon Howard. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nature Cell Biology, 1(5):288-292, 1999. [1131 Julien Espeut, Amaury Gaussen, Peter Bieling, Violeta Morin, Susana Prieto, Didier Fesquet, Thomas Surrey, and Ariane Abrieu. Phosphorylation relieves autoinhibition of the kinetochore motor cenp-e. Molecular cell, 29(5):637-643, 2008. [114] Jennetta W Hammond, Dawen Cai, T Lynne Blasius, Zhe Li, Yuyang Jiang, Gloria T Jih, Edgar Meyhofer, and Kristen J Verhey. Mammalian kinesin-3 motors are dimeric in vivo and move by processive motility upon release of autoinhibition. PLoS biology, 7(3):e1000072, 2009. [115] Kristen A Dietrich, Charles V Sindelar, Paul D Brewer, Kenneth H Downing, Christine R Cremo, and Sarah E Rice. The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proceedings of the National Academy of Sciences, 105(26):8938-8943, 2008. 1116] Cedric Bouzigues, Sabine Levi, Antoine Triller, and Maxime Dahan. Single quantum dot tracking of membrane receptors. In Quantum Dots, pages 81-91. Springer, 2007. 106