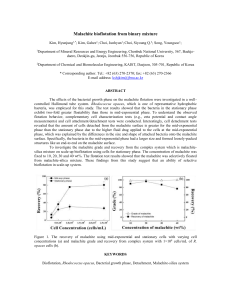
Document 10947897
advertisement

REFLECTANCE SPECTROSCOPY AS A REMOTE SENSING TECHNIQUE FOR THE IDENTIFICATION OF PORPHYRY COPPER DEPOSITS by KRISTINE LOUISE ANDERSEN S.B., Massachusetts Institute of Technology (1974) SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY July, 1978 Signature of Author . Department of Earth and Planetary Sciences, July, 1978 .,r Certified by ....... Accepted by . ... , s 1 4W 4 - .-- . ............... ........................ Thesis Supervisor ........ ... ........ . Chairperson, Departmental Committee on Graduate Students Wif"93IUWN MAS OF iL9,M t NSTTUTE akES LIBRARIES REFLECTANCE SPECTROSCOPY AS A REMOTE SENSING TECHNIQUE FOR THE IDENTIFICATION OF PORPHYRY COPPER DEPOSITS by KRISTINE LOUISE ANDERSEN Submitted to the Department of Earth and Planetary Sciences, July, 1978, in partial fulfillment of the requirements for the Degree of Doctor of Philosophy ABSTRACT Reflectance spectroscopy has been used successfully as a remote sensing technique to study the mineralogy and composition of the surfaces of planetary objects. This thesis attempts to develop reflectance spectroscopy to locate and identify copper bearing regions on the earth. Development of the technique required background laboratory studies involving measurement and interpretation of reflectance spectra of minerals--oxidized copper minerals, ferric oxides, clays, MnO2--occurring in copper bearing soils. Absorption features appearing in the oxidized copper phases, malachite, azurite, and chrysocolla, were assigned on the basis of ligand field theory and theories of molecular vibrations. These studies showed that malachite, the most common oxidized copper phase, could be detected in surface soils from analysis of ratio spectra generated by dividing the spectra of areas containing different malachite proportions. In the laboratory, reflectance spectra of simulated soils were measured, scaled to unity at 0.87 p, and divided by each other, generating a set of ratio spectra. Intensity ratios, 1(0.75 0)/I(1.20 p) versus 1(0.65 y)/I(0.75 P), calculated from the ratio spectra, were found to be sensitive indicators to the presence of malachite. The sensitivity of the technique, determined from the graph of the ratioed intensity values, was found to be on the order of 2000-5000 ppm copper. This technique was tested in the laboratory using natural soil samples collected from the Silver Bell and Mineral Park copper districts since there currently does not exist a spectrometer with the required detection capabilities. Reflectance spectra of these soils were measured in the laboratory, scaled to unity at 0.87 p, and representative samples were ratioed. Intensity ratios were calculated from the ratio spectra and plotted on graphs similar to those generated from the laboratory simulated soils. Relative copper concentrations contained in the natural soils, deduced from the intensity ratio graphs, were found to be too low (<2000 ppm as analyzed from XRF) to be detected by this technique. The laboratory studies determined the instrumental parameters required for the detection of copper in the field. A spectrometer such as the one proposed for geochemical orbiting missions of planetary bodies possesses instrumental capabilities for accurate measurement of field spectra. Problems arising in field measurements of spectra include vegetation, minerals which possess absorption features near the diagnostic copper wavelengths, phase angle variations, and atmospheric effects. Vegetation not only covers the surface, thereby masking the spectral properties of the soil, it also produces a chlorophyll absorption feature in the 0.65-0.70 y region. The effect of this band is to decrease the 0.65 yj/0.75 p intensity value which could lead to erroneous interpretations at high relative amounts of vegetation. Some clinopyroxenes (diopside, augite, salite) possess absorption features near the bands diagnostic of copper which could be misinterpreted. However, these minerals are not expected to be present in abundance in surface soils characteristic of copper bearing regions. The relative absorption band strengths in the clinopyroxene spectra are different from those in the malachite spectrum, so that intensity ratios calculated from the spectra of clinopyroxenes plot outside of the region diagnostic of malachite. Atmospheric effects are minimized by scheduling reconnaissance missions during clear, stable weather conditions. Phase angle variations are minimized by dividing spectra measured at similar phase angles. Reflectance spectroscopy may be used for other earth applications, including mineral exploration. Identification of chromium deposits is a potential candidate for application of this technique. iii ACKNOWLEDGMENTS I wish to acknowledge the Smithsonian Museum of Natural History in Washington, D.C. for providing the many mineral samples described in Appendix I. Gratitude is extended to the following people who provided their time and equipment for sample preparation and analyses: Dr. Michael Rhodes, for XRF sample preparation; Dr. Ramon Barnes, for the XRF machine and for helpful discussions regarding sample analyses; and Dr. Graham Hunt, for generously measuring the infrared spectra of azurite and malachite and for his very useful and helpful discussion regarding the assignment of the vibrational features. I am very grateful to Dr. John Adams for providing the time and the use of his reflectance spectrometer, and to Messrs. Keith Ronnholm and William Thomson, who both very kindly spent much time and energy helping me reduce and manipulate the spectral data. Mr. James Galey of ASARCO generously provided the soil and rock chip samples from the Silver Bell district, as well as many helpful comments and suggestions. I wish to acknowledge ASARCO for releasing the topographic map of the North Silver Bell area for publication in this thesis. My thanks to Mr. Clancy Wendt of the Duval Corporation for iv providing the Mineral Park soil samples. My sincere thanks to Dr. Edward Solomon, who patiently shared his knowledge and insight into the chemistry of copper complexes and whose discussions and suggestions were invaluable to this thesis. I am grateful to Dr. Roger Burns for the use of his X-ray diffractometer, but mostly I am deeply grateful for his interest, discussions, and kind encouragements throughout my graduate research. The existence of this thesis is due totally to Dr. Thomas McCord, who had enough faith (or chutzpah) to admit me into the graduate program. His past concern and guidance is most deeply appreciated. I wish I knew how to express my thanks to Dr. Robert Huguenin. He suggested the idea of applying reflectance spectroscopy to the identification of copper deposits. Without his unbounded enthusiasm, encouragement, direct guidance, and sense of humor, this thesis could never have been possible. My warmest thanks to Mr. Robert Scott and to my family and friends for their support and enthusiasm during my graduate career. I also wish to thank Diane Plosia for her graphic services, and thanks to those individuals whom I may have inadvertently omitted. Finally, I must acknowledge the cosmic soul of the v universes whose interactive forces allow the transient existence of all things material and immaterial, including NASA grant NSG-7405 (through the University of Massachusetts, R.L. Huguenin, principal investigator) which supported this research. In fond and loving memory of my late grandmother, Irene K. Sladek vii TABLE OF CONTENTS Page TITLE PAGE ABSTRACT . . . . . . . . . . . . . . . . - .- - ACKNOWLEDGMENTS DEDICATION . . . . . . . . TABLE OF CONTENTS . . . - i1 - - . iv . . . vii . . . viii xii LIST OF FIGURES AND TABLES LIST OF APPENDICES - xv . Chapter I. INTRODUCTION The Technique of Reflectance . . . . . . . . . . . Spectroscopy . . . . . Ore Exploration Techniques Advantages of the Technique of . . . . . Reflectance Spectroscopy Reflectance Spectroscopy as a Remote Sensing Technique to Identify Porphyry Copper Deposits II. DESCRIPTION OF PORPHYRY COPPER DEPOSITS Definition of a Porphyry Copper . . . Deposit Occurrence and Distribution . . Geology of Porphyry Copper Deposits Nature of Intrusive and Host Rocks . . Structural Controls . . . . Alteration Processes . . . . . . Mineralogy Ore . . . . . . Supergene Enrichment . . . Leached Ore Cappings Genesis of Porphyry Copper Deposits viii 6 6 10 10 10 11 11 12 14 15 Page Chapter III. DIFFUSE REFLECTANCE SPECTRA OF AZURITE, MALACHITE, AND CHRYSOCOLLA . . - - . . . . Introduction The Crystal Structures of Malachite . . . . . . . and Azurite Reflectance Spectra of Malachite and . . . . . . . . - - - - Azurite . . . . . . Spectrum Malachite The . . Spectrum The Azurite . Features Analysis of Vibrational . Reflectance Spectra of Chrysocolla . . . . . . . . Green Chrysocolla . . . . . . . Blue Chrysocolla IV. 23 . . 23 . . 29 . . 30 . . 36 36 36 . . 45 45 SPECTRA OF LABORATORY SIMULATED COPPER. . . . . . . . . . . . BEARING SOILS 49 . . . . . . . . . . . . - Soil Formation Reflectance Spectra of Soil Con. . . . . . . . . . . . stituents . . . . . . . . . . . . . Ferric Oxides . . . . . . . . . . . . . . . . . Clays . . . Desert Varnish and Organic Matter of Mixtures of Reflectance Spectra . . . . . . . . Soil Constituents . . . . . . Malachite and Ferric Oxides Malachite, Ferric Oxides, and . . . . . . . . . . . . . . Kaolinite Malachite, Ferric Oxides, Kaolinite, . . . . - - - - - and MnO 2 . . . Ratio Spectra of Simulated Soils . Presentation of the Ratio Spectra The Graph of Intensity Ratios ix . 40 40 40 41 Definition of the Term "Ferric Oxide" . . . . . . . Crystal Structures Ferric Oxides of Spectra Reflectance TransiElectronic Assignment of . . . . . . . tions Ferric of Spectra of Mixtures . . . . . . . . Oxides V. 19 20 . THE OPTICAL PROPERTIES OF FERRIC OXIDES 19 . . . 49 51 52 52 54 56 56 56 57 57 58 59 Page Chapter VI. VII. SPECTRA OF NATURALLY OCCURRING COPPER. . . . . . . . . . . . . BEARING SOILS . 63 . Description of Soil Samples Mineral Park Samples Silver Bell Samples Reflectance Spectra of the Soil . . . . . . . . . . . . . . Samples Comparison with Laboratory Spectra Effect of Particle Size . . . . . . The Spectra of Large Areas . . . . Ratio Spectra of Representative Soil Samples . . . . Presentation of Spectra . . . . . Predictions of Relative Copper Concentrations from Intensity Ratios The Determination of Copper Concentrations . . . . . . . . . . . . . Graphs of Intensity Ratios Absolute Concentration Versus . . . . Relative Concentration Soil Mineralogy versus Sample Distance . . . . . . . . . . . . Conclusions from Intensity Ratio Graphs . . .. . .. . . . . . 63 64 64 78 78 78 81 81 81 84 87 . 88 Instrumental Requirements.... .*.... .. . . . . . . . . . . Available Instruments 88 89 INSTRUMENTATION . . . . . . The Geochemical Orbiter Spectrometer Instrumental Specifications for Copper Reconnaissance . . . . . . . . . . . . . . . Instrumental Parameters . . . . Calibration Instrumental . . Location of the Ground Track Data Collection . . . . . . . . . Obstacles and Uncertainties Involved in Field Measurements . . . . . . Vegetation . . . . . . . . . . . Phase Angle Effects . . . . . . . Flight Variations . . . . . . . . VIII. 70 70 72 75 .. . . . . . . . . . . . . . . . . .93 91 91 92 92 . . . . . . . . 93 93 94 95 . . . . . DISCUSSION Summary 90 96 .. 96 Page Chapter Potential Problems in Field . . . . . . . . . Application of ClinoSpectra Reflectance . . . . . . . . . . . . . pyroxenes . . . . Occurrence of Clinopyroxenes Clinopyroxene Discrimination from . . . . . . . . . Intensity Ratios Fe 3 +-rich Clinopyroxenes: Salite Reflectance Spectra of Representative . . . . . Desert Vegetation Effects of Vegetation of Soil . . . . . . . . . . Spectra the Technique of Sensitivity Advantages of the Technique . . . . . Potential Applications REFERENCES . . . . . . . . . APPENDIX I . . . . . . . . . APPENDIX II . . . . . APPENDIX III . . . . . . . . . . . . - 97 . 97 100 . . 101 101 . 104 . . . . 107 107 109 109 111 115 120 . 155 APPENDIX IV 204 APPENDIX V 212 LIST OF FIGURES AND TABLES Page I. 1. 2. 3. 4. 5. FIGURES Map of porphyry copper deposits occurring in the southwestern United States (after Titley . . . . . . . . . . . . ... and Hicks, 1966) 8 Map of granitic rocks in the western United States (after Titley and Hicks, 1966) 9 The process of secondary enrichment (after . . . . . . . . . Whitten and Brooks, 1973) . . Cross section of a typical porphyry copper . . . . . . . . system (after Sillitoe, 1973) . 16 Porphyry copper deposits in relation to plate boundaries (after Sillitoe, 1972) . . . . . . . 18 6. Malachite site structures according to Sisse .*.. . . . .21 . . .. . . . . . . .. (1967) . 7. Azurite site structures according to Gattow . . . . . . . . . . . . . . and Zemann (1958) 8. Reflectance spectra of malachite.... 9. Reflectance spectra of azurite. 10. 13 .... . ..... . 22 24 . .. 25 Splitting of the one-electron energy levels of Cu2 + in crystal fields of axial symmetry . . . . . . (after Hathaway and Billing, 1970) 27 spectra of malachite (from Sadtler Labs, 1943) spectra of azurite (from Sadtler .. . . . .. . . . .. Labs, 1943) . 31 12a. XRD pattern of azurite 5000 (CoKa radiation used: X = 1.790A) b. XRD pattern of azurite 5001 . . . . . . . . . . 35 . 37 lla. Infrared Research b. Infrared Research 13. Reflectance spectra of blue and green chrysocolla . . . . . .. . . . . . . xii . . . . Page I. 14. FIGURES (cont.) Reflectance spectra of goethite and limonite . o. . . . .. .. 42 . . . . . . -.-.-. . . . . . . . . 15. Reflectance spectra of hematite 16. Reflectance spectra of maghemite and lepidocrocite . . . . . . . . . - . . . . . . -. 17. 18. 19. 20. 21. 22. Reflectance spectra of mixtures of hematite . . - - . . . . . . ... and goethite -. .- Reflectance spectra of kaolinite and montmorillonite (after Adams, 1975) . . . . . . . 43 44 . 47 . 53 . . 55 Intensity ratio graph calculated from laboratory . . . . . simulated soils . . . . . . . . . .. 60 Soil collection grid of the Mineral Park samples . .. . .. (courtesy C. Wendt of Duval Corp.) 65 Topographic map of the Mineral Park district showing soil sample location (after Eidel .. . . . . . . . . . . .. . . . et al., 1968) 66 Reflectance spectra of desert varnishes (courtesy John Adams) . . . .. . . . . . . 23. Map showing the approximate location of the North Silver Bell district (after Titley and . . . .67 .. . . . . . . .. . .. Hicks, 1966) 24. Topographic map of the North Silver Bell district depicting alteration zones and soil and rock chip sample locations (courtesy . .. . .... .. . . . . ... ASARCO, 1978) 69 Reflectance spectra of a soil sample at four . . ... . .. .. . different particle sizes 73 25. 26. 27. 28. Ratio spectrum of a soil sample showing the effect of particle size . . . . . . . . .. . . 74 Averaged spectra representing large sample .. ....... .. . . . . . areas............. 76 . . . ... 77 Ratio spectra of large sample areas xiii Page I. FIGURES (cont.) 29. Intensity ratio graph generated from the soil . . . . . . . .. . . . . sample ratio spectra 79 30. Intensity ratio graph comparing absolute ..... versus relative copper concentrations 82 31. Intensity ratio graph comparing soil mineralogy versus relative sample .- . - -.-.- -.-. . . . . . . . . .. . distances 85 Plot of centers of absorption bands appearing in the reflectance spectra of common rock. . . . forming minerals (after Adams, 1975) 98 32. . 33. Reflectance spectra of clinopyroxenes (after . . . . . . . . . . - - - - Adams, 1975) 34. Intensity ratio graph of laboratory simulated . . . . . . . soils including clinopyroxenes . 102 Absorption spectra of plant pigments (after . . . . . . . . . . . . . Gates et al., 1965) . 105 36a. Spectra of desert succulent plants (after Gates et al., 1965) b. Spectra of typical lichens (after Gates et al., . . - - - -. - - -. 1965) . . . . . . . . .. 106 Reflectance spectra of chromite and chrome. . . . . . diopside (after Adams, 1975). 110 35. 37. II. 1. 2. .. TABLES Energies (cm~ ) of the fundamental ligand vibrations in malachite and azurite . . . . . . 33 . . 34 Energies (cm 1) of electronic transitions in ferric oxides . . . . . . . . . . . . . . . . . 46 Vibrational features appearing in the reflectance spectra of malachite and azurite 3. 99 xiv . . LIST OF APPENDICES Page . 115 . . . . 115 117 117 117 118 APPENDIX II: REFLECTANCE SPECTRA OF LABORATORY . . . . . . . . . . . . SIMULATED SOILS 120 APPENDIX I: SAMPLE DESCRIPTION AND ANALYSES . Sample Description . . . Analyses Sample Reflectance Spectra . Infrared Spectra X-ray Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . Figure 1: Malachite and ferric oxides Figures 2-4: Malachite, ferric oxides, and . . . . . . . . . . . . . . . . .. kaolinite oxides, ferric Figures 5-32: Malachite, - - - - - - - - - - - kaolinite, and MnO 2 . APPENDIX III: RATIO SPECTRA OF LABORATORY . . . . .. SIMULATED SOILS Figures 1-16: MnO2, malachite, kaolinite, . . . . . . . . . . . .. and goethite Figures 17-32: MnO 2 , malachite, kaolinite, . . . . . . . . . . . . . and hematite Figures 33-48: MnO 2 , malachite, kaolinite, . . .. (50% hematite + 50% goethite) Figure 7: Silver Bell rock chip samples . . . . . 156 . . . . and . . .. 172 . . o . . . ..... . . . . APPENDIX V: RATIO SPECTRA OF NATURALLY OCCURRING . . . o . . . . .... .. SOIL SAMPLES Figures Figures 1-4: Mineral Park soil sample ratios 5-28: Silver Bell soil sample ratios xv 125 155 . APPENDIX IV: REFLECTANCE SPECTRA OF NATURALLY . . . . . .. OCCURRING SOIL SAMPLES Figure 1: Mineral Park soil samples. . . Figures 2- 6: Silver Bell soil samples 122 . . . 121 188 204 205 206 211 212 . . . . 213 217 C H A P T E R I INTRODUCTION The Technique of Reflectance Spectroscopy Reflectance spectroscopy has been used successfully as a technique for remotely sensing the mineralogy and composition of the surfaces of some solar system objects using ground based telescopes, cf. McCord et al. (1977); Head et al. (1978). This thesis attempts to develop reflectance spec- troscopy for earth applications; in particular, to locate and identify copper bearing regions. Once this technique is de- veloped for copper, it may be applied to include remote sensing identification of other economically important ore deposits and other applications such as pollution and crop monitoring, geological surveying, etc. The theory of reflectance spectroscopy has been established in the literature (Ballhausen, 1962; Burns, 1970; Adams, 1975). Briefly, spectral reflectance measures the fraction of incident solar radiation that is reflected from a surface as a function of wavelength. Sufficient spectral resolution and intensity'precision can resolve absorption bands which are diagnostic of surface composition and miner1 alogy. Mineralogical interpretation depends primarily on the wavelength dependence of reflectance and to a lesser extent on the albedo and the angular dependence of the reflected radiation. Material on the surfaces of planetary bodies including the earth occurs as randomly oriented fragments. When observed in reflected light, the surface particulate material returns two components of radiation: (1) a specular component consisting of first surface reflections described by Fresnel's laws for absorbing dielectrics; and (2) a diffuse component which is composed of light that has entered at least one grain and has been scattered back toward the observer. It is the diffuse component that contains the most compositional information. Most of the absorption features appearing in reflectance spectra arise from electronic transitions and charge transfers by valence shell electrons in transition metal ions 2+ 2+ .4+ (Cu , Fe , Ti , etc.) and from overtones and combinations of molecular vibrations. The wavelength positions of the ab- sorption band centers depend on the types of ions present and on the dimensions and symmetry of the sites in which the ions are situated. To a large extent, these two factors define the mineralogy of a sample. Reflectance spectra are interpreted using laboratory and theoretical techniques. Laboratory spectra of samples of known composition are studied as functions of particle size, phase angle, mixing ratios, and mixing heterogeneity. Interpretations of absorption bands are based on ligand field theory, molecular orbital theory, and theories of molecular vibrations. Relative modal abundances are analytically de- termined by comparing the relative strengths of bands contributed by the constituent minerals using assumptions about homogeneity and relative grain sizes. Ore Exploration Techniques There are many techniques being used in mineral exploration, most of them involving indirect identification of ore deposits. Geophysical detection techniques include studies of magnetic susceptibilities, gravitational anomalies, electromagnetic induction, and induced polarization. Geochemical techniques include the study of surface color anomalies, and soil, plant, and ground water sampling analyses. Remote identification techniques have also been used. Dykstra (1975) used ERTS imaging in an attempt to delineate copper bearing regions according to rock facies associated. with disseminated deposits. In his study of the Silver Bell porphyry copper district, he found that the spectral contrast among individual rock types was too subtle to be diagnostic -since surface weathering processes obscured the characteristics of the underlying rocks. Advantages of the Technique of Reflectance Spectroscopy The advantage of reflectance spectroscopy as a remote sensing technique compared with ERTS broad band imaging and most of the other techniques is that it is able to directly detect the presence of copper bearing minerals. It is able to scan areas that may be remote and inaccessible to in situ exploration regimes. With this technique, a wide region may be surveyed during a single reconnaissance, and subtle changes in surface composition can be detected where they may not be discerned by visual inspection. Reflectance Spectroscopy as a Remote Sensing Technique to Identify Porphyry Copper Deposits Identification of disseminated copper deposits was chosen as the initial earth application of the technique of reflectance spectroscopy for the following reasons. Most of the porphyry type copper deposits found in the United States occur in semi arid environments (Arizona and New Mexico) which support minimal ground vegetation thus making surface soils accessible for spectral measurement. The surface ex- pressions of copper.deposits contain oxidized copper minerals (azurite, malachite, chrysocolla) which possess absorption bands at spectrally diagnostic wavelengths. In this thesis, those wavelengths diagnostic of oxidized copper minerals 5 will be determined. The sensitivity of the technique will also be determined from laboratory and field studies. C H A P T E R I I DESCRIPTION OF PORPHYRY COPPER DEPOSITS Definition of a Porphyry Copper Deposit The term "porphyry copper deposits" refers to disseminated ore of low grade usually mined by large scale/low cost techniques such as open pit versus bulk or vein-type mining. It is now mostly a mining-engineering term used to designate a large, low grade deposit regardless of genesis and wall rock type. However, there are features inherent to this type of deposit as will be revealed. It is the pur- pose of this section to give a brief description of these deposits and to discuss their genesis in relation to occurrence. Most of the information in this section was obtained from the Geology of the Porphyry Copper Deposits by Titley and Hides (1966), and references to this book of collected papers will be made by quoting the author and page number. Occurrence and Distribution The United States is the largest copper producer in the world with about 1,411,000 tons mined in 1975 (World Mining Copper Map, 1976). Most of the ore came from deposits in the porphyry copper region of the southwest shown in 6 Figure 1. The dominant physiographic feature here is the Colorado Plateau which extends through Utah, Colorado, Arizona, and New Mexico. It is bounded on the east by the Southern Rocky Mountains and on the south and west by the Basin and Range province which consists of mountain blocks bounded by faults. The Colorado Plateau is well known for its rich uranium deposits, but it is the Basin and Range province surrounding it which hosts porphyry copper (Figure 1) as well as lead, silver, gold, and molybdenum deposits. This seemingly preferred distribution of deposits has been related to the local crustal history (Gilluly, 1963). The porphyry copper deposits formed before the active uplift of the Colorado Plateau and the development of the Basin and Range province. They are a part of what Anderson (p. 13) calls an orogenic-epeirogenic cycle which includes chronologically: (1) Cretaceous sedimentation due to a west- ward advancing sea; (2) Rocky Mountain orogeny from the Cretaceous to early Tertiary resulting in intense deformation and thrust faulting; (3) associated plutonic and vol- canic activity; and (4) Cenozoic development of the Colorado Plateau and the Basin and Range province. torially summarized in Figure 2. This cycle is pic- Recent studies (Sillitoe, 1972 and 1973) have given strong evidence that porphyry copper deposits are indeed related to crustal history particularly in orogenic areas associated with consuming plate margins. The genesis of these deposits will be discussed at MILES M E X I C 0 *CANANEA Figure 1. Map of the southwestern United States showing the principal physiographic subdivisions and the location of porphyry copper deposits. (After Titley and Hicks, 1966). 0 200 MILES EXPLANATION Middle T.rtiary x + Lawer Tertiary and Upper Cretaceoum Middle Cretaceous A Lower Jurassic and Upper Taimc . Facies boundary' Major Cretaceous thrust belt Figure 2. Granitic rocks of Mesozoic and Cenozoic age inmte western United States. The age assignments are based in part on all radiometric ages published to July 1962; the ages of many small plutons are arbitrary. Facies boundary in Washington, Idaho, and Nevada: eugeosvclinal rocks to the west, miogeosynclinal rocks to the east. From Gilluly (21, fig. 22). Porphyry copper deposits: 1. Chino (Santa Rita); 2. Tyrone; 3. Morenci; 4. Bisbee; 5. Cananea; 6. Miami-Inspiration; 7. Ray; 8. San Marpel; 9. Silver Bell; 10. Pima and Mission; 11. Esperanza; 12. Bagdad; 13. Ajo; 14. Bingham Canyon; 15. Ely; 16. Yerington. (After Titley and Hicks, 1966). 10 the end of this section. Geology of Porphyry Copper Deposits Although the term "porphyry copper" has become largely a mining term (Schmitt, p. 18) referring to the size and grade of the deposit, there are geologic features which are characteristic of this type of deposit. be discussed briefly in this section. These features will In general, dissemin- ated copper deposits are closely related to igneous intrusions (usually monzonitic porphyries), intense hydrothermal alteration is usually found, and supergene enrichment has operated to concentrate copper over the primary sulfide zone. Nature of intrusive and host rocks. Stringham (pp. 35-40) discussed the nature of the intrusive and host rocks associated with porphyry copper deposits. He found that the in- trusives are of intermediate stock with a preference for quartz monzonite-quartz latite associations, and that intrusive porphyry is an absolutely necessary feature. The wall rock may be of all lithologic types, ages, and thicknesses. The most favorable host rocks for ore emplacement are intrusive or highly siliceous metamorphic and sedimentary rocks. Structural controls. Locally, porphyry copper districts are found to be concentrated at orogenic and fault-zone intersections, usually triple or more complex intersections (Schmitt, p. 30). In the desert Southwest, most of the cop- 11 per deposits occur along the central rift of the WasatchJerome orogen, and more than half of the total U.S. copper production comes from the intersection of this copper with the Texas structural zone (consisting of many thrust-fault areas) (Schmitt, p. 30). The structural controls favoring ore emplacement arise from the tectonics of consuming plate margins (see Figure 5) and will be included in the discussion of ore genesis. Alteration processes. Intense hydrothermal alteration is a characteristic feature of porphyry deposits. The alteration is complex and varied due to differences in the prevailing thermodynamic and compositional factors. The principal al- teration minerals are alunite, antigorite, micas, and clays. Sericitization is the single most important alteration process which can attack almost every mineral in the primary assemblage except for pyrite. Under favorable conditions, a porphyry deposit may end up essentially as a quartzsericite-pyrite aggregate (Schwartz, p. 44). Ore mineralogy. Although the mineralogy of a typical por- phyry copper deposit is made up of a complex group of primary, hydrothermal, and supergene minerals, the ore minerals form a relatively simple assemblage. The primary ore minerals are sulfides--dominantly chalcopyrite and bornite with minor amounts of molybdenite, sphalerite, and ubiquitous pyrite. Chalcocite and covellite are the predominate supergene sulfides. In the oxidized zone, malachite, azur- ite, cuprite, and chrysocolla are more or less abundant depending on the individual deposit. The leached ore cappings are relatively depleted in copper minerals, and the concentration of copper naturally increases downward. The primary sulfides occur as discrete grains throughout the host rock and also as fillings of small veins. Pyrite is the most abundant sulfide in the disseminated deposits; it usually is the first sulfide to be deposited, but it may also continue to form throughout the period of metallization (Schwartz, p. 46). The primary copper sulfides are deposited more or less simultaneously and display mutual boundary relations (Schwartz, p. 46). Supergene enrichment. The process of supergene or secondary enrichment serves to concentrate the copper ore below the level of the water table. Massive blankets of chalcocite are formed in this highly reducing environment. Replacement of primary sulfides is selective; chalcocite replaces bornite in preference to chalcopyrite and then to pyrite. Figure 3 depicts the process of supergene enrichment. Upon oxidation, the sulfides of a porphyry copper deposit form sulfuric acid and copper and iron sulfates. If the con- centration of carbonate and silicate ions is high, copper becomes fixed as an oxidized mineral (malachite, azurite, chrysocolla). Otherwise, copper remains soluble and migrates 13 Figure 3 COSSAN Secondary enrichment. Gossan is the residuum of iron oxides (limonite) from which mobile elements have been removed, e.g. copper, sulphur, as sulphates, etc. In the leacbed zone the sulphides are oxidised to sulphates and transported in solution. In the zone of oxidised enrichment, the reaction of the sulphate solution with the original ore .in an oxygen-rich environment (carbon dioxide is also usually present) results in the formation of carbonates and o:ides, native metals, and (rarely) silicates. Below the water table, secondary sulphide enrichment occurs in an oxygen-free environment. (After Whitten and Brooks,1973). downward. Under reducing conditions at depth, it replaces sulfides low or lacking in copper. Since the porphyries are only mildly neutralizing, they provide ideal conditions for the process of secondary enrichment by allowing sulfuric acid to exist long enough to oxidize the primary sulfides. Leached ore cappings. The leached capping of the typical disseminated copper deposit is a highly iron-stained, porous, siliceous mass in which the original characteristics of the deposit are obscured. Oxidation of pyrite and chalcopyrite finally results in limonite which is mostly a mixture of ferric oxides. According to Locke (1926), the leached cap- pings normally contain copper, though not necessarily evident to the naked eye. Limonitic characteristics (gossan) have been used by geologists as a guide to the location of copper deposits. The outcrops of these deposits display a deep maroon or reddish-brown color in contrast to the brick red color over pyritic rocks. Locke (1926) and Blanchard (1968) both relate texture of the limonitic capping to the relative amounts and types of primary sulfides from which it was derived. 1 Lovering (1950) found that soils taken from a chrysocolla-rich outcrop near the San Manuel copper deposit contained copper contents ranging from 1,000-10,000 ppm and that the highest Cu concentrations occurred in the finer soil fractions. Clarke (1952) collected soil samples near the Ray copper deposit and found copper values from 150-2,000 ppm with an average value of 650 ppm above the ore body. Genesis of Porphyry Copper Deposits The genesis of porphyry copper deposits has long been the subject of much interest. Schmitt (pp. 17-33) proposes that deep seated tectonic activity such as that found in orogenic zones associated with consuming plate margins remobilizes copper and associated metals. More recent studies (Sillitoe, 1972; 1973) confirm this "orthomagmatic" model of ore genesis. system. Sillitoe defines the typical porphyry copper It is originally emplaced 1.5 to 3 km beneath the summit of a strato volcano and can possess a vertical extent up to 8 km. During ascent to higher crustal levels, bodies of calc-alkaline magma lose fluid during retrograde boiling. Alteration, mineralization, and brecciation in the early consolidated hood of the intrusive and the immediate wall rocks are accomplished by these escaping fluids. Fumarolic activ- ity accompanies porphyry copper formation suggesting that the magma bodies that host the porphyry copper may represent magma chambers that had not erupted but evolved passively. Figure 4 summarizes the typical porphyry copper system. Porphyry copper deposits are intimately genetically related to igneous calc-alkaline activity. This restricts ore formation to orogenic areas associated with subduction zones since it is this type of magmatism that is produced at consuming plate margins. The temporal and spatial dis- tribution of deposits depends upon the level of erosion and Fig . . Idealized cross section of a typical, simple porphyry copper deposit showing its position at the boundary between plutonic and volcanic environtbents. (After Sillitoe, Vertical and horizontal dimcnsions arerneant to be only approximate. 1973). 17 the amount of copper available in the underlying subduction zone. Figure 5 is a powerful statement in support of this theory, and it suggests regions for future porphyry copper prospecting. 18 ------- - - Accretnq platemargins - Active transform faults plate margins Consuming - Plate margins of indeterunate nature Mesozoc-Cenozoic Mountain bells Reqions with Dorphyry copper and molybdenum deposits Plate boundories token from Dewey and bir d, 170 Fig. 5. The western Americats. southwest Pacific and Alpide porphyry belts in relation to Mesozoic-cenozoic orogenic belts and accreting and consuming plate boundaries. (After Sillitoe, 1972). C H A P T E R I I I REFLECTANCE SPECTRA OF MALACHITE, AZURITE, AND CHRYSOCOLLA Introduction The copper carbonates azurite, Cu3 (CO3 ) 2 (OH)2 , and malachite, Cu2 CO 3 (OH) 2 ' are formed in the oxidized zone of copper deposits and usually occur together. Although they are chemically similar, the fact that their crystal structures are different is apparent from analysis of their spectra. Hunt and Salisbury (1970b) recorded the diffuse re- flectance spectra of both minerals, and Lakshman and Reddy (1973) recorded the optical absorption spectrum of malachite. It is the intention of this chapter to refine the previous work by assigning all of the optical and near infrared features appearing in the reflectance spectra of a suite of both minerals. Features arising in the reflectance spectra are electronic transitions due to Cu2+ and ligand vibrations. Electronic transitions arising from Cu2+ in tetragonally distorted octahedral sites are assigned on the basis of ligand field theory. The near infrared vibrational features arise from combinations and overtones of ligand CO 3 and OH 19 fundamentals whose energies are determined from analysis of their infrared spectra. The copper silicate chrysocolla also occurs in association with azurite and malachite in the oxidized zone of copper deposits, and features appearing in its reflectance spectrum are also discussed at the end of the chapter. The Crystal Structures of Malachite and Azurite The malachite crystal structure was determined by Wells (1951) and refined by Ssse (1967). The crystal be5 longs to the space group P2 /a = C2h with four formula units 2+2 per unit cell. Cu2+ occupies two different sites as shown in Figure 6, both of which are axially elongated octahedra with the ligands in trans-positions. 2+ populated by Cu Both sites are equally Gattow and Zemann (1958) determined the azurite crystal structure as shown in Figure 7. The crystal belongs to 5 the space group P2 /c = C2 h with two formula units in the 2+2 unit cell. Cu2+ occupies two different sites, one of which is essentially square planar with the ligands in trans-positions and the other site which is a square-based pyramid with the ligands in cis-positions. This square pyramid site oc- curs twice as often as the other site in the crystal structure. OH 03C03 OH to OH ' 1.915 e 1. 03 1.899 Cu OH 003 C 2.116 Cu 2.042 C \ \-p (0 /c' nw C03 SITE CN OH I SITE 2 Figure 6. Malachite site structures according to SUsse (1967). Both sites are axially elongated octahedra (0 h + D4h distortion). 3 003 C0 3 C03 OH 1"98 1.98 / Cu OH OH 2.04 / Cu 2.01 C03 OH C03 SITE I SITE 2 Figure 7. Azurite site structures according to Gattow and Zemann (1958). The extent of axial elongation in Site 1 is so great that i.t is essentially square planar (D4h). Site 2 is nearly a square pyramad (C4v). Reflectance Spectra of Malachite and Azurite ) reflectance Visible-near infrared (4000+ 29,000 cm spectra of suites of malachite and azurite samples are shown in Figures 8 and 9 respectively. The sharp, weak absorption features in the infrared region arise from carbonate and hydroxyl ligand vibrations, and the broad, strong absorption 2+. electronic features in the visible region are due to Cu transitions. The reflectance peaks near 20,000 cm~ give azurite its blue color and malachite its green color. The electron configuration of Cu2+ is 3d9 which means that only one electronic transition, 2 Eg+ T2g is possible for Cu2+ in an octahedral (0 h) ligand environment. However, 2+ complexes are found in it is well known that very few Cu perfect octahedral coordination (CuSiF6*6H 20 is the one exception; Pappalardo, 1961) but rather they are found in tetragonally (D4 h) distorted 0h environments. Such is the case in azurite and malachite where inspection of their 2+. occupies cation sites (Figures 6 and 7) shows that Cu axially elongated (tetragonally distorted (D4 h)) octahedral sites in both minerals, although the extent of axial elongation is greater in the azurite sites. Figures 8 and 9 show the occurrence of more than one ligand field transition in 2+ .. is not in the spectra of each mineral which means that Cu perfect octahedral coordination. The malachite spectrum. The malachite spectra (Figure 8) Figure 8. Reflectance spectra of malachite. 24 0.8 1 - Malachite 6000 0.7 2 - Malachite 6001 0.6 3 - Malachite 6002 4 - Malachite 6003 5 - Malachite 6004 0.5 0.4 0.3 0.2 0.I 0.1 0.1 0.1 0.1 0.0 L 3.0 - 2.5 2.0 1.5 C M~1 x 1.0 iO~4 0.5 Figure 9. Reflectance spectra of azurite. 1.0 1 - Azurite 5000L 0.9 2 - Azurite 5001 - 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 0.0 -- 3.0 2.5 2.0 1.5 CM~' x I.0 I0-4 0.5 show three electronic transitions centered near 9200 cm 13,000 cm~ (1.08 p), (0.77 p), and 22,400 cm~ 1 (0.45 y); the azurite spectra show two such transitions centered near 13,000 cm~ (0.77 p) and 16,100 cm 1 (0.62 y). The bands may be tentatively assigned on the basis of Figure 10, bearing in mind that band assignments based on spectral studies alone are rarely definitive in the case of copper complexes (Hathaway and Billing, 1970). In malachite, the extent of axial elongation is not great enough for the sites to be considered square planar (the ratio of the average copper-ligand band length along the z-axis/the average copper-ligand band length in the x-y plane is approximately 5/4). Therefore, the 2B + 2A transition should be the least energetic and may be assigned to the band at 9,200 cm 1. (dxz' d z) Figure 10 shows that the b 2 g (dxy) and e orbitals lie close in energy, even when the extent of axial elongation is great. Thus, the band at 13,000 cm~1, which is more broad and intense than the 9,200 cm 1 band, may 2. 2 2 2 transitions. + E and B + B be assigned to both the B g lg 2g lg This 13,000 cm 1 band has an asymmetric band width which may be due to a small energy separation between the b2g and eg orbitals. The assignment of these transitions compares well with band assignments of other similarly coordinated Cu 2 + complexes (Hathaway and Billing, 1970, p. 176). The value of 10 D equals the energy of the transi2 2 which depends on the actual site symmetry, tion B + B lg 2g A&-41 (bit) .lIS, N Nek~~iL - - 4'~ ~ N , N c~q LbL1J ____ j a Y1 Il Coepmrcaf d~;,d1 L N ~ z Ctaz (01) oc~k,'e&tc SgtroJe C00v - -1 - ?( ran LDjy6% Figure 10. The splitting of the one electron energy levels of Cul* in crystal fields of axial symmetry. (After Hathaway and Billing, 1970). C~) but not on the extent of axial elongation (Lever, 1968, p. -l Thus, in malachite, D = -1300 cm which compares well q with other similarly coordinated Cu2+ complexes (Burns, 1970; 78). Pappalardo, 1961; Hathaway and Billing, 1970). The origin of the band at 22,400 cm is uncertain, but it may be due to some low-lying change transfer transition. A metal to metal charge transfer transition from Cu 2 + to Cul+ is probably not the reason for this band as it is found to generally occur in the 1-2 p region (E.I. Solomon, personal communication). However, if a ligand has a rela- tively high energy filled orbital, and the metal has a relatively low lying empty orbital, then a charge transfer transition will be observed either in the visible or near-ultraviolet region (Lever, 1968, p. 228). This may be the case in malachite where such a transition is possible from a ligand -bonding orbital to a copper eg (7*) orbital. Origin of this transition may arise from the hydroxyl ligand as it is more reducible than carbonate (E.I. Solomon, personal communication). Few studies have been made concerning the na- ture of charge transfer transitions arising in transition metal complexes, and further studies are needed in order to confirm the above hypothesis. The origin of this transition could also be due to 3+ the presence of an impurity such as Fe . However, the spectrum of reagent grade malachite (spectrum #5, Figure 8), whose maximum limit of iron impurities is less than 0.01%, exhibits this 22,400 cm feature. Thus, this feature must be intrinsic to the malachite structure. The azurite spectrum. The ligand field transitions in azurite may also be tentatively assigned on the basis of Figure 10. All three transitions occur in the 13,000-16,000 cm-1 region, and considerable overlap can occur since electronic transitions have half-widths on the order of 500-2000 cm~ (Lever, 1968). This could account for the higher inten- sity of these bands relative to those in malachite. The extent of axial elongation in the azurite sites is much greater than in the malachite sites (compare Figures 6 and 7) which means an increase in the extent of tetragonal (D4 h) distortion. This would account for the shift to higher energies relative to malachite of the 2B + 2E transition lg g 1 (from 13,000 cm- to 16,100 cm ) with the alg orbital shifting into the e orbital (refer to Figure 10). The b2g or- bital is not much affected by the extent of axial elongation so that the 2B + 2B transition energy should be approxilg 2.g mately the same as it is in malachite (13,000 cm ). The ligand field transitions in azurite are therefore assigned to 2 2 the B + B transition (= 10 D ) at 13,000 cm -i and the lg 2g qi 2 2 2 B + 2A , E transitions at 16,100 cm -l1. The occurrence of two transitions near 16,100 cm- would account for this band's greater intensity and broadness relative to the single transition at 13,000 cm-1 . The absorption feature found at 22,400 cm in mala- chite may also occur in the azurite spectrum but at an energy higher than can be recorded by reflectance measurements ( 29,000 cm 1 ). The strong fall-off in reflectivity at the high energy end of both the azurite and the malachite spectra (2 25,000 cm~ ) most likely arises from ligand to metal charge transfer transitions of the type discussed previously. Analysis of Vibrational Features The absorption features in the infrared region (7700 to 4000 cm- ) of the reflectance spectra arise from combinations and overtones of the fundamental vibrations of the hydroxyl and carbonate ligands. The hydroxyl ligand pos- sesses only one fundamental vibration which is IR active. Carbonate, governed by its intrinsic D 3 h point group symmetry, has four fundamental vibrations, two of which are doubly degenerate (v3 and v4 ) and one IR inactive mode (v). However, carbonate is found to occupy a general lattice position (C1 symmetry) in both azurite and malachite (Halford, 1946) thus removing all selection rules and degeneracies under its intrinsic D3 h symmetry. IR spectra (4000-250 cm~ N In Figure 11, representative ) of both minerals suggest that the vibration becomes IR active, and that the v4 degeneracy is removed (the degenerate component.s arising from v3 are not resolvable). The frequencies of the fundamental vibrations in a Figure 11. Infrared spectra (4000-4 250 cm 1 ) of (A) malachite (smithsonian sample B1044 2), and (B) azurite (Smithsonian sample R15412) (Source: Sadtler Research Laboratories, Inc., 1943). 100 80 60 40 20 0 L4.0 3.5 3.0 2.5 2.0 100 - 1.6 1.8 1.4 1.2 1.0 0.8 0.6 0.4 0.2 1.2 1.0 0.8 0.6 0.4 0.2 x IO- 3 CM~ B 80 604020 04.0 3.5 3.0 2.5 2.0 1.8 1.6 1.4 v x 10-3CM-1 32 suite of both minerals are listed in Table 1. The reason for the appearance of two sets of frequencies for each fundamental is that the ligands occupy two different positions in both crystal structures, viz. the average copper-ligand distance in the x-y plane is much shorter than that along the z-axis of the site (refer to Figures 6 and 7). In azurite, the Cu-OH bond lengths are all approximately equal (~2.OOA) so that only one frequency for VOH is observed. Only one value of the o. fundamental appears in both minerals: the frequency of this node may be invariant with respect to ligand position or one vibration may be of very low intensity. Also, the 1025 cm 1 V 1 frequency does not appear in the IR spectrum of azurite 5000. The IR and reflectance spectra as well as the x-ray diffraction pattern of this sample (Figure 12) all possess sharper and more well defined peaks than the other azurite sample indicating a more ordered crystal structure. This would affect ligand orientation to some degree and consequently destroy the symmetry coupling needed to produce the (IR forbidden) v -1 cm vibration at 1025 Table 2 lists the observed frequencies of the vibrational features appearing in the malachite and azurite reflectance spectra. The calculated energies are found to agree well with the observed values, but the fact that each fundamental possesses two frequencies due to unequivalent structural positions leads to some rather broad bands where Table 1. Energies (cm 1) of the fundamental liyand vibrations appearing in the infrared spectra (4000-250 cm~ ) of malachite and azurite. OH CO 3 V 1030-1045 V 2 875 V3 1375-1390 4 4 700-705 810-820 Malachite 1080-1095 Azurite (1025) 1080 1 L3395-2405. 1475-1495. _735-745] 945 1410 730 800-805 _1460] 760 815-820 This feature does not appear in sample 5000. 3300-3320 3415-3420 Table 2. Observed energies (cm ) of ligand vibrational features appearing in the reflectance spectra (29,000-4000 cm- 1 ) of malachite and azurite. Observed Energy Malachite (cm-1 ) Assignment 5500 5450-4720 V + 3v3 2v + 2v3 3v 1+ 2v4 V1 + 2v3 + \4 4480 3v 3 4420 Azurite 7690 3v 6755 + 3v 3 2v OH 6290-6210 2v + 3v3 6175-5585 3v + 2v3 2v + 3v3 V 5375 V 4975 4855-4610 + 2v 2 + 2vo3 + 3v 3 2v1 + 2v3 S+ 2v3 + V 3v1 + 2v4 4320 3v3 * All vibrations are due to carbonate unless otherwise noted. A (D H B 50 45 Figure 12. Powder X-ray diffraction pattern of azurite using Co Ka radiation (X = 1.790 A). (A) Azurite 5000 (B) Azurite 5001. several features overlap. The hydroxyl overtone at 6755 cm 1 appears in the azurite but not the malachite spectrum since the overtone position would be obscured by the low energy electronic transition at 9200 cm 1 . Reflectance Spectra of Chrysocolla Green chrysocolla. Chrysocolla, CuSiO3-nH20, is a blue to green mineral found in the oxidized zone of copper deposits associated with azurite and malachite. Blue and green ap- pearing chrysocollas have been thought to be structurally the same, but inspection of Figure 13, showing reflectance of representative blue and green chrysocollas, reveals that this is not the case. Intense infrared features occur in the spectrum of blue chrysocolla near 7150 cm 1 , 5260 cm 1 , and 4440 cm 1 due to water vibrations, while the infrared bands in green chrysocolla are very similar to the carbonate and hydroxyl bands found in the malachite spectrum. Further, the ligand field transitions in green chrysocolla occur at the same energies as those in the malachite spectrum, viz. 9200 cm and 13,000 cm Blue chrysocolla. Green chrysocolla, therefore, seems to be an intimate mixture of malachite and silica. Blue chryso- colla, however, is apparently a hydrous copper silicate which does possess a well-defined crystal structure (although it has not yet been reported in the literature) since its re- Figure 13. Reflectance spectra of chrysocolla. 0.7 A - Blue chrysocolla (7002) B - Green chrysocolla (7005) 0.6 8 0.5 0.4 I 0.3 0. 2 0.1 -B A 0.I 2.5 2.0 1.5 cm 1 x 10~4 1.0 0.5 flectance spectrum (Figure 13) is unique compared with the spectra of other copper minerals. Ligand field bands in blue chrysocolla occur near 11,800 cm and 14,000 cm . These transitions may be tentatively assigned on the basis of Figure 10 since the effective ligand environment in blue chrysocolla is similar to those found in azurite and malachite. (Indeed, it is well known that almost every Cu2+ com- plex similar to the ones in this discussion possesses a tetragonally distorted octahedral ligand environment (Hathaway and Billing, 1970).) The energies of the ligand field transitions in blue chrysocolla occur between the energies of the corresponding transitions in azurite and malachite (between 9000 cm 1 and 16,000 cm~). In blue chrysocolla, the energy separation between the bands (~2000 cm than that found in azurite (~3000 cm~ -l ) is less ) which means that the extent of tetragonal distortion in blue chrysocolla is probably not as great as it is in azurite where the a is expected to occur at the same energy as the e The low energy transition at 11,800 cm orbital orbital. in blue chrysocolla is more intense relative to the intensity of the corresponding feature in azurite and malachite, suggesting that the a g orbital may occur near the same energy as the b2g orbital. This analysis leads to the assignment of the 2B +2 A , 2B2g transitions at 11,800 cm 1 and the 2 gB+ 2 Eg transition at -gl 1400 cm~ , and implies that the extent of tetragonal distortion in blue chrysocolla is greater than in malachite but 39 less than in azurite. The reason for this cannot be ascer- tained until the chrysocolla structure is determined. A relatively weak feature is noted at 23,800 cm1 in blue chrysocolla similar to the one at 22,400 cm~ chite. in mala- The mechanism for this transition is probably similar to the one discussed in malachite. C H A P T E R I V THE OPTICAL PROPERTIES OF FERRIC OXIDES Definition of the Term "Ferric Oxide" Ferric (Fe +) oxides are common and widespread in occurrence since they are weathering products of metallic iron and iron bearing minerals. In arid regions, they impart a characteristic brick red to ochre color to surface rocks and soils, and they tend to be enriched in areas associated with ore depoxits due to the weathering of iron bearing sulfides (mostly pyrite). The term "ferric oxide" is used here to encompass the polymorphs of Fe 2 0 3 and the polymorphs of FeO(OH). By far the most abundant ferric oxides are those with the hexagonal close-packed oxygen structure--hematite (a-Fe20 3) and goethite (a-FeO(OH)). The cubic close-packed oxygen poly- morphs are maghemite (y-Fe 2 O 3 ) and lepidocrocite (y-FeO(OH)) which occur naturally but to a much lesser extent than the a structures. Crystal Structures Bernal et al. (1959) found the crystal structures of these ferric oxides to be composed of different stackings of 40 41 close-packed oxygen/hydroxyl ions with various arrangements of Fe + ions in the interstices. Fe + occupies octahedral sites in all of these structures, and it also occupies tetrahedral sites in maghemite which has a spinel-like structure very similar to that of magnetite (y-Fe 3 04 ). The degree of cation ordering can vary greatly in the maghemite structure, but the octahedral sites are always enriched relative to the tetrahedral sites. Reflectance Spectra of Ferric Oxides The electron configuration of Fe gives rise to a 6A field. is (Ar)3d5 which ground state in a weak octahedral ligand Since all of the electrons are unpaired, any elec- tronic transition involves spin pairing and thus all transitions are spin forbidden. tra (29,000 cm 1 Representative reflectance spec- - 4000 cm-1 ) of goethite, hematite, mag- hemite, and lepidocrocite are shown in Figures 14-16; Figure 14 contains spectra of limonite, an omnibus term implying a mixture of x-ray amorphous iron hydroxides and hydrated iron oxides. X-ray diffraction analyses of these samples showed that they contain mostly goethite. The spectra of all of the ferric oxides are very similar in that the electronic transitions occur at similar energies and intensities. The mag- hemite specimen was obtained by oxidizing magnetite, and its electronic transitions occur at higher energies than in the other ferric oxides. This may be due to contributions from Figure 14. Reflectance spectra of limanite and goethite. 1.0 1 - Limcnite 2001L 0.9- 2 - Limonite 2000L 3 - Goethite 3004 4 - Limonite 2003 0.80.70.60.5- 4 3 0.4 0.3 u 2 4 Ld : 0.1 O0 0 0.1 -3 0.1 -2 0.1 0.0I 2.5 2.0 1.5 c m~' x 1.0 I0- 4 0.5 0.0 1.0 Figure 15. Reflectance spectra of hematite. 43 1 - Hematite 1003 0.9 2 - Hematite 1001 3 - Hematite 1000 4 - Hematite 1005 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 -4 -2 0.1 -3 0.1 0.1 0.0 II I 2.5 I 2.0 I 1.5 cm x I0~4 1.0 I 0.5 0.0 44 Figure 16. Reflectance of maghemite and lepidocrocite. 1.0 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.I 0.I 0.0 2.5 2.0 1.5 cm' x i0~4 1.0 0.5 0.0 45 Fe in the tetrahedral sites (Dq would be smaller meaning a higher energy separation between levels in the weak field d5 case). Assignment of electronic transitions. The bands are assigned according to those given in the literature (Jorgensen, 1963, p. 92; Ballhausen, 1962, p. 253; Mao and Bell, 1973-74; Hunt et al., 1971a) for other high spin ferric oxide complexes. The lowest energy transition near 11,000 cm~1 (0.90 P) is as4 + T transition; the transition near lg lg -16 4 16,000 cm 1 (0.63 p) is assigned to the A +4 T transilg 2g tion; and the high energy transitions near 21,000 cm~ signed to the (0.50 y) to the 6 A and 23,000 cm 6 A 4 46 +4 Agr 4 E 1 (0.44 p) are assigned respectively and A + 4 T (D) transitions. lg lg g lg 2 3 lists the band assignments and energies for the ferric Table oxides studied here as well as for some others reported in the literature. Spectra of mixtures of ferric oxides. When several phases of iron oxides are present, the band positions are found to vary linearly with relative amount. Figure 17 shows how the band positions change with varying amounts of hematite and goethite. The success of reflectance spectroscopy as a technique to identify copper deposits depends on the ability to spectrally identify the weathering phases of copper, viz. azurite, malachite, chrysocolla. In arid regions and es- Table 3. Energies (cm 1) and assignments of electronic transitions appearing in the reflectance spectra of ferric oxides. (cm 1) 6A -+ 4T T2g IA 4E 4T 2g(D) aFe(H0) 3+ H2 0 6 12,600 18,500 24,600 27,500 a-FeO(OH) 11,200 16,000 22,000 23,700 b(s) 10,930 15,500 b(y) 11,210 15,340 y-FeO(OH) 11,800 15,600 21,250 23,200 b(H) 10,140 13,700 15,300 b (Y) 10,890 14,950 a-Fe 2 0 3 11,900 16,000 18,850 20,000 y-Fe 2 0 3 12,100 16,300 20,000 20,650 aData from Jorgensen, 1963. bData from Mao and Bell, 1973-74. Figure 17. Reflectance spectra of mixtures of hematite 47 (reagent) and goethite (limonite 2001). Mixtures are in wt.%. 0.9 I I 1 2- 2% hematite + 78% goethite 0.8 32% hematite + 68% goethite 3 - 59% hematite + 41% goethite 2 -- 0.7 - 4 - -- 4 78% hematite + 22% goethite 0.6- 3 0.5 0.4- 2 LU i z0.3 FU U 0.2 4 0.1 -3 0 .1I --2 0.0 0.0 2.5 2.0 2.0 1.5 cm- 1 x 10-4 .. 1.0 0.5 0.0 48 pecially over mineral deposits, the surface concentration of ferric oxides will be much higher relative to the concentration of copper phases. However, the electronic transi- tions in the copper phases--malachite in particular--occur at different energies than in ferric oxides. A spectrum measured above a copper deposit will be dominated by ferric oxide, so the presence of copper will be detected from analyzing the subtle changes in the ferric oxide spectrum. C H A P T E R V SPECTRA OF LABORATORY SIMULATED COPPER-BEARING SOILS The success of reflectance spectroscopy as an exploration technique to identify copper deposits depends upon the ability to detect the presence of copper bearing minerals in soils above copper deposits. Development of this tech- nique requires that the composition of copper-bearing soils be known so that the presence of copper may be determined from the soil spectra. It is the purpose of this chapter to present laboratory spectra of samples containing representative amounts of natural soil constituents including various amounts of malachite, the most common weathering phase of copper. The spectra are studied to determine how the pres- ence of malachite can be detected in the representative soil spectra. Finally, the spectra of natural samples taken from copper bearing regions are analyzed, and the results are correlated with their laboratory determined copper concentrations. Soil Formation The majority of porphyry copper deposits in the United States are situated in the desert Southwestern states 49 (Arizona and New Mexico). The semi-arid climate of this re- gion supports little extensive ground vegetation so that the surface is mainly composed or rocks and soils. The soils contain minor amounts of decayed organic matter mixed with the weathered mineral phases derived from the surrounding surface rocks. The geology of porphyry copper deposits was discussed in a previous section where it was noted that hydrothermal alteration is an important and necessary process in the formation of a porphyry deposit. Creasey (1966, in Geol. of the Porphyry Copper Deposits) notes three types of hydrothermal alteration processes characteristic of porphyry copper deposits--potassic, argillic (phyllic), and propylitic. Al- teration of granodiorite rocks associated with porphyry copper deposits produces clays (chiefly kaolinite and montmorillonite), micas (sericite and minor biotite), potassic feldspar, and to a lesser extent, chlorite, carbonates, and epidote which are formed mostly by propylitic alteration. Surface exposure of these altered rocks results in subaerial weathering of their surfaces--iron bearing minerals are . 2+ oxidized to Fe oxides (limonite, hematite) and feldspars are altered to clays. Soils are formed by mechanical weathering of the rock surfaces, and thus their compositions will reflect the composition of the rock surfaces from which derived. Soils from the porphyry copper region should be composed mostly of Fe + oxides, clays, and some decayed organic matter. Reflec- tance spectra of soils obtained from the region of porphyry copper deposits show that this is indeed the case (see Figures 1- 7, Appendix IV). These spectra will be discussed in a later section. Huff (1952) found that copper tends to accumulate in soils with high concentrations of clay or organic matter, and that leaching does not affect the concentration of copper in soils nearly as much as it affects the rocks from the supergene zone of a copper deposit. A copper anomaly is formed by the weathering of exposed rock surfaces, and the shape and size of the anomaly are influenced by topography which controls soil creep. Although the extent of surface vegetation is minimal in the porphyry copper district, decayed organic matter and desert varnish are. present in soils and on rock surfaces. Reflectance Spectra of Soil Constituents In the following sections, reflectance spectra of samples modeled after soils typical of copper anomalies are presented, and spectral features are correlated with the constituent minerals. The addition of various amounts of malachite to these representative soil samples is studied for the effects on the spectra. Finally, ratio spectra are generated, and the regions of the spectrum which are diagnostic of malachite are determined. Ferric oxides. Soils from copper bearing regions generally contain ferric oxides, clays, and decayed organic matter. Understanding the spectrum of a mixture of these minerals involves determination of the spectral contributions arising from each constituent mineral. Reflectance spectra of com- monly occurring ferric oxides and hydroxides were presented and discussed in the previous chapter (refer to Figures 1416). Limonitic goethite and hematite, the predominate weathering phases of iron bearing minerals, form on the surfaces of exposed rocks and are thus released to the soil as a result of mechanical weathering. Clays. Clays are formed by hydrothermal alteration or weathering of silicates, and they generally make up a large percentage of many soils. Kaolinite and montmorillonite are expected to be the primary clay constituents found in copper bearing soils. Reflectance spectra (0.35-2.50 p) of representative samples of kaolinite and montmorillonite are shown in Figure 18. Salisbury and Hunt (1970) and Adams (1975) discussed the spectra of these clays, and their findings are summarized here. The major spectral features exhibited by these clays are several strong hydroxyl bands in the infrared region, centered near 1.4 p and 2.2 p. Compared to the montmoril- lonite spectrum, kaolinite exhibits a weak band at 1.9 p in- Figure 18 0.5 1.0 15 WAVELENGTH (;pm) 2.0 2.5 Diffuse reflectance spectra of micas and clays. Muscovite and the clays (kao- linite and montmorillonite) show strong hydroxyl and water bands, respectively. Phlogopite and biotite in contrast have Fe2 + and Fe+-Fel+ absorptions. (After Adams, 1975). dicating the lack of appreciable bound H 2 0. The features near 0.95 p and 1.15 p are also due to vibrations of the water molecule, but they are much weaker (by about a factor of ten) than the bands at 1.4 p and 1.9 p. Desert varnish and organic matter. Although the extent of surface vegetation is minimal in the porphyry copper district, decayed organic matter and desert varnish are present in soils and on rock surfaces. Spectral features arising from these materials are expected to be minimal; however, they act as darkening agents which would reduce the overall albedo thus affecting the shape of the entire soil spectrum. Desert varnish forms a dark coating on exposed rock surfaces. Engel and Sharp (1958) and Potter and Rossman (1977) both studied desert varnishes and showed that most of them are composed of iron and manganese oxides, and that they tend to be enriched in trace elements, especially copper. Reflectance spectra (0.35-2.50 p) of desert varnish coating two different types of rocks are shown in Figure 19 along with the spectrum of finely ground MnO 2 for comparison. Spectral features arising from the rock-forming minerals are strongly obscured by the varnish. The low albedo and fea- tureless MnO 2 spectrum is probably due to intense charge transfer bands arising from metal-metal and metal-ligand electron transfer processes. 60 50 uJ C 40 - 30 z .J u.20 0 10 \0 \0 0 0.5 1.0 1.5 WAVELENGTH Figure 19. 2.0 (/L) Reflectance spectra of desert varnishes. (Courtesy J. Adams) . 2.5 Reflectance Spectra of Mixtures of Soil Constituents Samples containing predetermined amounts of malachite, hematite, goethite, kaolinite, and manganese dioxide (MnO 2 ) were prepared in the laboratory, and their reflectance spectra (0.35-2.50 y) were measured. The minerals used were made up of very small sized particles ( 5 50 p diameter), although they were not sorted. Malachite and ferric oxides. Figure 1 in Appendix II shows shows reflectance spectra of samples containing selected weight percentages of malachite combined with an equal weight percent mixture of hematite and goethite. Increasing the amount of malachite depresses the ferric oxide reflectance peak at 0.75 p and changes the slope of the spectrum between 1.0 p and 1.5 p. This is due to the electronic transitions in malachite centered at 0.77 p and 1.08 p. (Azurite and chrysocolla (see Chapter III) would likewise depress the ferric oxide reflectance peak due to their electronic transitions at 0.72 p and 0.76 p, respectively.) Car- bonate vibrational features at 2.25 p and 2.40 p gain intensity at higher malachite percentages. Malachite, ferric oxides, and kaolinite. Reflectance spectra of mixtures of malachite, an equal weight percent mixture of hematite and goethite, and kaolinite are presented in Figures 2-4 in Appendix II. The addition of kaolinite raises the 57 overall spectral albedo and adds characteristic H2 0 vibrational bands to the spectrum at 1.4 p, 1.9 p, and 2.2 p. In- creasing the weight percentage of malachite relative to ferric oxides also has the effect here of flattening the ferric oxide peak at 0.75 P and lowering the spectral albedo between 1.0 and 1.5 p. Malachite, ferric oxides, kaolinite, and MnO Selected weight percentages of MnO 2 were added to mixtures of malachite, ferric oxides, and kaolinite, and their reflectance spectra are shown in Figures 5-34 in Appendix II. Increasing the weight percentage of MnO 2 correspondingly lowers the total spectral albedo by approximately the same percentage, e.g., increasing the amount of MnO 2 from 5% to 50% lowers the overall curve albedo by about 40%. The spectral contrast is also lowered at higher percentages of MnO 2* At the same amount of MnO 2 ' malachite affects the ferric oxide spectrum as described in the paragraph above. Ratio Spectra of Mixtures of Simulated Soils In the laboratory, a reflectance spectrum is measured against a standard reference source, usually MgO or halon. However, in the field, a reference standard will not be available. To eliminate spectral contributions arising from the atmosphere and instrumentation, an individual spectrum may be ratioed by another. This residual ratio spectrum will depict any relative differences between the two spectra involved. It is hoped that differences in copper concentra- tions between two samples will diagnostically affect the ratioed spectrum. Presentation of the ratio spectra. Reflectance spectra (0.35 p + 2.50 y) of laboratory prepared samples involving mixtures of malachite, ferric oxides, kaolinite, and MnO 2 were scaled to unity at 0.87 p and divided by each other to generate a set of ratio spectra shown in Figures 1-48 in Appendix III. Albedo differences among the spectra are mini- mized by normalizing each spectrum by the 0.87 y intensity value. This wavelength was chosen because the spectral al- bedo in this region is essentially unvarying due to characteristic ferric oxide absorptions (the 0.85-0.89 p electronic transitions). Since the albedos of naturally occurring soils are not expected to vary greatly over a small range (on the order of 1/2 km), each ratio spectrum was generated at a constant amount of MnO 2 with either the amount of malachite or ferric oxide being varied. This was done so that effects arising from the variation of one constituent could be easily analyzed from the ratio spectrum. In the interest of conserving space, the ratio spectra presented here were chosen so that the intensity values at wavelengths shorter than 0.5 p are less than 1.0. In most of the ratioed spectra, this criterion is met by placing the sample with the greater concentration of ferric oxides in the numerator. This criterion breaks down at high concen- trations (-50%) of hematite (see Figures 17, 22, and 29 in Appendix III) because the 64 A + T electronic transition lg 2gelcrnctasio at 0.63 p gains intensity relative to the 6A g 4 A g 4 E transition at 0.53 y with increasing hematite concentrations. Thus, a ratio spectrum containing 50% hematite in the numerator and 20% hematite in the denominator will generally have ratioed intensity values greater than unity below 0.63 p. The graph of intensity ratios. The determination of relative copper differences from natural soil sample spectra may require visual comparison with representative laboratory ratio spectra. However, the copper signature sought by visual in- spection may be quantified and a method devised which will determine the relative copper concentration and will hopefully be at least as effective as visual inspection. Figure 20 is a graph of ratioed intensity values-1(0.75 p)/I(l.20 p) versus 1(0.65 p)/I(0.75 p)--calculated from the normalized ratioed reflectance spectra of mixtures containing kaolinite, ferric oxides, and malachite. The spectra were ratioed at a constant amount of MnO 2 ranging from 0% to 50% by weight. The 0.65 i/0.75 p ratio was chosen to measure the depression of the 0.75 p ferric oxide reflectance peak by the 0.77 p electronic transition in malachite; the value at 0.65 p was chosen because the intensity values 60 Figure 20. Graph of intensity ratios, 1(0.75 y)/I(l.20 p) versus 1(0.65 p)/I(O.75 -f), calculated from ratio spectra of laboratory simulated soils. Region I depicts relative malachite differences when there is more malachite in the numerator spectrum. Region II depicts relative malachite differences when there is more malachite in the denominator spectrum. Ferric oxide differences were calculated from ratio spectra containing more ferric oxide in the numerator, except for the points denoted by "Z" which contain more hematite in the denominator. See text for full discussion. Graph Key Relative malachite concentrations* Region I Region II 5%/l% 10%/l% 10%/5% 15%/10% 20%/5%,15%/5% 20%/10% 20%/15% 50%/20% 50%/5% Relative ferric oxide concentrations* Graph Code 10%/5% 20%/10% 20%/5% 50%/5% 50%/20% 20%/50% (hematite) wt. % in numerator spectrum/wt. % in denominator spectrum. Special Points Points Special Symbol_ Represented Symbol Represented M,R h M,0 D,H i D,F N,0 U,vW O,P k KY F,G 1 v,Y C,F,I m VX L,0,S n I,J nnuiluinninmuuin II.~niionmioninnni Fn-niininnon Iminoigion niinionininniginninin~~~~ I-.=i~~~an==i. lignininnjg~mniggg-Mnin~gpli~nnnillinin nnu. miuimlli m.I~nimnlllmieln ------------ Tie.m.-uinjirIiis -v-q-n 4 Nl -4nvw -pTmls-=I:.:=-n.I UnonXprnnliinn mnnnn-noi7nninni+NII ==I=.-~~ uminileninemnimiimumonen5m I.im ====|====.====::==::::::===7=:1::::::7=:::=1-=::::=::: iniinimininninini....M~~~~~~~InIn~numn~niiinininnimniini iltjliiingginpnnnlmniijiinimncniliilnil -- gmIIILun .. IlleennuenioinininonnnninminnrS- geliimuliinillnillumaninnemingl~I .i~~~~~~~~ Ann^ f%4- lin xm it u I:=:L n-n-----in .. -=ml|=- Ininnlin I1=:iimrm=:-en~~irIlr mj"++lin+ ninlpnirnnnio.Nn. ir--inn--iin+ n-jAjn-=N - - -in-nn--n-P-- r-== at shorter wavelengths may vary greatly due to the relatively 6 4 4 strong A g A g E transition occurring at 0.53 y in hematite and 0.45 y in goethite. The 0.75 p/1.20 p ratio was chosen to reflect the slope change between 1.0 and 1.5 y due to the 1.08 p electronic transition in malachite. It can be seen from inspection of Figure 20 that points reflecting relative malachite differences cluster within a diagnostic area apart from the points reflecting ferric oxide differences. Increasing relative concentrations of malachite systematically affect the intensity ratio values. The ratio of two spectra containing much smaller relative amounts of copper than are depicted in Figure 20 is expected to show a corresponding lack of spectral contrast, i.e., the two intensity ratios would tend to cluster closer to unity. C H A P T E R V I SPECTRA OF NATURALLY OCCURRING COPPER-BEARING SOILS Success of the remote-sensing regime developed here is determined from field testing. Currently, there does not exist a spectrometer capable of testing this method in the field, but a simulated field test can be modeled in the laboratory. Spectra of surface regions are simulated by measuring the spectra in the laboratory of soil samples collected from two copper bearing districts. Ratio spectra, generated by dividing the spectra of representative soil samples, simulate the spectra that would be obtained from ratioing the spectra of adjacent surface regions. The ratio spectra are analyzed for their relative copper contents. The results are correlated with the measured copper concentrations of the soil samples, thus testing the sensitivity of this technique in practical applications. Description of Soil Samples Soil samples were collected from the regions of two porphyry copper deposits--the Mineral Park (Ithaca Peak) and Silver Bell mining districts. A brief description of these deposits follows, and their locations are shown in Figure 1. 63 Mineral Park samples. Ithaca Peak is a mining district situated on the Mineral Park property owned by the Duval Corporation. Thirty-six soil samples were collected at 1.5 meter intervals in a 6x6 grid pattern as shown in Figure 21. The samples were obtained near the Ithaca Peak stock, and the exact location is shown in Figure 22. Assayed copper and molybdenum values in this area are on the order of 0.5% and 0.03-0.05%, respectively (Clancey Wendt, personal communication). The predominate rock type is a quartz-feldspar gneiss, and the Ithaca Peak stock is pervasively sericitized or argillized. The entire Mineral Park area is surrounded by a zone of propylitic alteration. A complete description of this deposit is given by Eidel et al. (1968). Since the soils were collected at only 1.5 meter intervals, one sample representative of a 3x3 grid portion of the entire area was made by combining the nine samples contained in this area into one representative sample (see Figure 21). Four samples, each representative of one quadrant of the grid, were sieved to contain particle sizes less than 125 my. in diameter. Silver Bell samples. Twenty soil samples and five rock chip samples were collected from the North Silver Bell district, owned by the American Smelting and Refining Company (ASARCO), shown in Figure 23. Copper mineralization occurs in hydro- thermally altered volcanics--alaskite, dacite, and dacite and AS6 10 86 0 D6 C6 0 0 A C46. 1 I A4 84 F6 0 0 DF46 C5 I E6 I C4' D5 0 E5 0 FS 04 14 e- ~1 ---- A3 0 83 0 C3 D F 13 A C13 A2 I At0 82 81 e C2 F2 .0 I I I I I CI e ) I It Dl El 0 Ft O. SCALE 5 FEET Figure 21. Soil collection grid of the Mineral Park samples (courtesy C. Wendt, Duval Corp.). Nine soil samples were combined to give the sample representative of a 10' x 10' (3 x 3 m) area. The soil collection site is shown in Fig. 22. -- 66 AA r AR TIN RIDGE U PE AK- in Feet \Scale TUROUOIsE Figure 22. Topographic map of the Mineral Park district (Arizona) depicting the soil collection site (e) and the present mining pit (After perimeter. Eidel, et al, 1968). 67 0WW 4 CC* -o o W a- w * *i I w1 a ..- o Lo - x a - ,J o0- - - z . --... -... Figure 23. Map of the Silver Bell district (Arizona) showing the approximate area (shaded) of the soil and rock chip collection sites (see Fig. 24). (After Titley and Hicks, 1966). 68 quartz-monzonite porphyries. Potassic, phyllic, and propy- litic alteration zones are all present at Silver Bell, but the copper tends to be concentrated mostly in the phyllic zone. Mining operations have been confined to the El Tiro and Oxide pits which consist of tabular chalcocite blankets formed by twofold to threefold enrichment of copper contained in the primary mineralization. Richard and Courtright (1966, in Geology of the Porphyry Copper Deposits) have described the geology of Silver Bell. Figure 24 is a topographic map (courtesy of ASARCO) showing the sites where the soils and rock chips were collected. (Soil samples are prefaced by "SS"; rock chips by "GOC.") The map also delineates the zones of hydrothermal alteration. For each sample site, ten small samples at an estimated weight of 10-15 grams were collected within a 15 meter radius and at a depth of a centimeter into the soil. The small samples were combined to give the sample for that site, the sites being situated at least one hundred meters apart. Some samples contain dark organic matter, most of it being roots of "resurrection moss" munication). (J. Galey, personal com- The sieving process separated most of this or- ganic matter into the larger particle fractions (125 mp to 1000 mp diameters), especially in the 125-500 my size range. Five rock chip samples were collected near the soil sample sites indicated in Figure 24. Each sample was ob- tained by collecting 10-20 rock chips of approximately 50-100 p K0 I 4 to p -'-Figure 24. Topograghic map of !the North Silver Bell area depicting alteration zones and Athe soil and rock chip sample sites (courtesy ASARCO). Soil samples are denoted by "SS"; samples are denoted by "GOC". Location: Vaca Hills Quad. Sections 33 & 34 Township 11 South Range 8 East SScale: s( // jj 1/ rock chip - ..... Air P- --- Nt I A lk -h N lip f 0 m FT wr 00#v JIM %A Jv N 1 ; Lox f~N\ - mat 4I~j %, ,00 / METT 00o 7100, TGO G$1GO0 'N'I 4 8 S "N'~ -- SS5 (I1 (##3 c P'T4 7A j 4' > si ....... J s 7 s 20 1 I 'I \ 1001, ,- Jr K ~ 1/',, 4"T ~ £KA - 0 P .0000 grams within a 15 meter radius. The combined sample was finely pulverized for assay (performed by ASARCO). Since the copper concentrations in these samples were predetermined, they were used here as standards in the XRF analyses of the soil samples (Appendix I). Reflectance Spectra of the Soil Samples Reflectance spectra (0.35-2.50 p) of soil and rock chip samples collected from the Mineral Park and Silver Bell porphyry copper districts are presented in Figures 1-7 in Appendix IV. The smallest particle sizes were used to generate the spectra: the Silver Bell samples were all less than 45 p, and the Mineral Park samples were less than 125 y in size. Comparison with laboratory spectra. Laboratory samples (de- scribed in Chapter V) chosen to model natural soils contained mixtures of ferric oxides, clays, and a substance to lower the spectral albedo (MnO2 ). Examination of the natural soil spectra shows that the laboratory generated spectra (Figures 5-34 in Appendix II) accurately represents them. The pres- ence of clay is inferred from the natural soil spectra by the intense OH and H20 vibrational features at 1.4, 1.9, and 2.2 p. The intensity of the 1.9 p feature indicates a water retaining clay, such as a montmorillonite. A mixture of fer- ric oxides is indicated by characteristic electronic transi- tions near 0.5, 0.6, and 0.85 p." The exact positions of these bands indicate a more goethite rich, rather than hematite rich, mixture (refer to Figure 14), especially in the Mineral Park samples. The albedos of the soil samples measured at the highest reflectance value in each spectrum ranges between 45% and 50%, indicating the presence of at least one albedo lowering substance such as the organic matter noted during sample preparation, desert varnish, or an opaque mineral. If an opaque is responsible for the lowering of the albedo, then comparison with laboratory spectra (Figures 5-32 in Appendix II) containing MnO 2 (an opaque) indicates that it is present at 30-35% by weight in the natural soil samples. The rock chip spectra (Figure 7 in Appendix IV) show compositions similar to the soil samples, although their albedos are generally higher. The rock chip samples were pulverized, and their particle sizes were estimated by inspection to be smaller than the 0-45 p particle size fraction of the soil samples. Smaller particle sizes enhance scattering effects which lower the spectral contrast and increase albedo (Adams and Filice, 1967). The spectrum of rock chip sample G0C43 (Figure 7 in Appendix IV) is significantly lower throughout the visible region compared with the other rock chip spectra. The assayed value for copper in this sample (3200 ppm) is about ten times greater on the average than the other rock samples. Copper in this sample may occur in opaque oxides such as cuprite, Cu 2 0, or tenorite, CuO. Although the reflectance spectrum of tenorite has not been measured, Salisbury and Hunt (1971a) showed that cuprite is a trans-opaque mineral, i.e., it is reflecting in the infrared region but highly opaque in the visible, which could account for the spectral behavior of QOC43. Effect of particle size. It was previously mentioned that the smallest sized particles control the albedo and band contrast of a spectrum. The effect of particle size is demonstrated in Figure 25, which shows reflectance spectra of a representative soil sample (SS2) at four different particle sizes. Note that the albedo increases with de- creasing particle size due to enhanced scattering. The spectral contrast, however, decreases with decreasing grain size. The H 2 0 vibrational features near 1.4 y and 1.9 p are more intense in the spectra of the larger sized particles, which may be due to the presence of small water inclusions. Figure 26 represents the spectrum of a mixture containing equal amounts of the three smallest particle sizes (0-500 y) divided by the spectrum of the smallest particle size (0-45 p). Ratioed intensity values tend close to unity across the entire wavelength region. This is probably due to the fact that the smallest sized particles tend to coat the larger particles, but it shows that the spectral 50 40 30 20 10 I0 I0 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (L) Figure 25. Reflectance spectra of a soil (SS2) showing the effect of particle size. See text for discussion. 74 cd 1.50- LCD 1.00- -P .H 0 - 050- I 050 I I 1.00 I I 1.50 I I 200 Wavelength (y-) Figure 26. The effect of particle size as demonstrated by the ratio spectrum of a soil sample (SS2). The numerator spectrum contains 0-500p sized particles; the denominator spectrum contains 0-451, sized particles. properties are controlled by the smaller sized grains. The spectra of large areas. The spectra of several adjacent soil samples taken from the Silver Bell district were averaged together to form a spectrum representative of a larger single area containing those samples. The averaged spectrum should approximate the spectrum of the large area since the soil spectra themselves are very similar. (If they were very different, then this would not be the case as some spectral bands, especially electronic transitions, are stronger than others--thus, a spectrum weighted according to the optical depth/grain size ratio would be more accurate.) The averaged spectra, each representing a 1/4 - 1/2 km area, were generated by averaging the spectra of the following soil samples: SSAV1 = SS1, SS2, SS9 SSAV2 = SS3-SS8, SS10 SSAV3 = SS12, SS14, SS15, SS17, SS18 SSAV4 = SS19, SS20. The averaged area spectra are shown in Figure 27. The fact that these spectra are very similar is shown by their ratio spectra presented in Figure 28. Thus, over a large area, the reflectance spectra lose individuality. This means that in practical application, any reconnaissance technique would require a high spatial resolution, probably on the order of the soil sample dimensions (-15 meters). 76 - 1 - SSAV1 (Average of SS1, SS2, SS9) 2 - SSAV2 (Average of SS3 - SS8, SS10) 3 - SSAV3 (Average of SS12, SS14, SS15, SS17, SS18) 4 - SSAV4 (Average of SS19, SS20) 0.25b- 0.25 0.25- 0.2 5i- , 0.50 I 0.75 I 1.00 I I -I 1.75 1.50 1.25 2.00 I 2.25 Wavelength (y) Figure 27. Spectra of the average of several soil samples representing the spectra of large sample areas. 7 __ ~ 1 - SSAVl/SSAV2 2 - SSAV3/SSAV2 3 - SSAV4/SSAV3 4 - SSAV3/ SSAV1 5 - SSAV4/sSAV1 ,00 1OOH- 2 00 3 1.OOFH 4 1.0OH- I 0.50 I 0.75 I 1.00 I 1.25 I 1.50 1 1.75 | 2.00 2.25 Wavelength (P) Figure 28. Ratio spectra of the averaged spectra representing large sample areas (shown in Fig. 27). Ratio Spectra of Representative Soil Samples The reflectance spectra of the Presentation of spectra. soil samples were normalized to the intensity value at 0.87 p, and then representative samples were divided, generating a set of ratio spectra. Twenty-four ratio spectra, shown in Figures 5-28 in Appendix V, were generated from the Silver Bell soil spectra, with most of the samples involved in each ratio being less than 1/3 km apart. Figures 1-4 in Appendix V show four ratio spectra generated from the Mineral Park soil spectra. All of the samples here lie next to each other, the mean sample distance being 5-6 meters. Predictions of relative copper concentrations from intensity ratios. The ratio spectra were visually inspected, but no copper bands were plainly evident. 1(0.75 y)/I(l.20 p) Intensity ratios, and 1(0.65 p)/I(O. 7 5 y), were calculated for each ratioed spectrum and plotted as shown in Figure 29. This graph was compared to the same graph generated from the laboratory ratio spectra (Figure 20) in an attempt to predict the relative copper concentrations of the soil samples before analyzing for their copper contents. Comparison of the graphs show that thirteen samples plot within the laboratory designated regions showing copper differences; the other samples may contain relatively lower amounts of copper which would yield undiagnostic ratios. Figure 29. Graph of intensity ratios generated from the soil sample ratio spectra. The outlined regions I and II are diagnostic of malachite differences as explained in the caption to Figure 20. Graph Key Silver Bell soil sample ratios SS1/SS2 SS5/SS8 SS8/SS7 SS6/SS3 SS6/SS4 SS8/SS6 SS5/SS6 SSll/SS10 SS14/SS12 SSll/SS12 SS18/SS17 SS19/SS16 SS17/SS15 SS16/SS15 SS19/SS20 SS10/SS7 SS11/SS2 SS18/SS12 SS15/SS2 SS16/SS2 SS20/SS2 SS9/SS2 SS13/SS9 Graph Code Copper concentrations 370/285 1585/305 305/710 395/270 395/710 305/395 1585/395 290/760 175/95 290/95 135/100 145/55 100/275 55/275 145/105 760/710 290/285 145/95 275/285 55/285 105/285 245/285 125/245 Duval soil sample ratios ACl3/DF46 ACl 3/AC4 6 DF13/DF46 DF1 3/AC4 6 930/1140 930/1250 1075/1140 1075/1250 (ppm) Figure 29 80 gensoiimimiinllisiisilntilannislilinniiAisli IIIIIIIII"IIIIIIIIII-- - II----IIIIIIII IIIIII -- ......'-- --------- ---M---IIIII-IIIII-I-i1-III-l--III--Iiii IIIIII - ------------EEMMEEMMMMMMMEMMMMMMMMM ----- -| i n n n n n +Ifin inMM -- i----MM MM EM EM ME M-------------MM il~ n n---i -------------- --ni . inn4n MINIIII!I!II~JIII!IIIIIII n li4I MMMM EME MMM MMMMMMM EMEM MMM -- ----------.----------- il~ ili H~n MMM EMEM MMM i n lliiiiiiill I--IIIIIIIIIIIIII MMM MMMLMM MM BE MM MM liinn--n---- MM---MM ---------M MMM niM ini-n---ni--------------iliiliil inn !!IIIIIIIIIII IIMMIM-n------------I-I--IIIIIII EMEErJ 4 MMMMEMMMEMIMEIMMMMEMLM AMMEMI.,MMME iiiiin iinnilisinininnilig- IViiiiiniininnsii M~ninilinnEMIIINMI"IIIIIIIII~niIITI MMMMMMMEMMEMMMMMEMMMMEMEMMMM-4 IMEMME^MM IiniliniiniliinnnifiEII~liligligliglgiigigr MqMEMM MMEraMMMMMMMEMEMEMMMMMMEEMMEMMM---MMMMMMM-M--M--M ----- n------------ ---------- ---- --- ----- l M M III n------n--------i.-------iH i I I .. ---- ------!IIIIIIM!II EMMMMMEEMBEEMMMEMMMMMEMESEAMMMM-MM------EMMM-------MMM I~li-n--n----n---n---il i+44#niiiilii-li---n-in-n-. llil---.-- ininN~onMMIIIIIMIMMMIMIIIIII----II----g--g-- i-n llrVnMI -- -- - -- The determination of copper concentrations. Standard XRF analysis, described in Appendix I, was used to determine the copper content of each soil sample. The mean copper concentration of each sample is listed in Table 2 in Appendix I. Most of the Silver Bell samples have copper con- centrations onthe order of 100-500 ppm; the Mineral Park samples have higher copper concentrations, on the order of 1000-1200 ppm. Of the thirteen samples that showed relative copper differences in Figure 29, seven were correctly predicted, meaning that the copper concentrations in the soil samples are too low to be diagnostic. Graphs of Intensity Ratios Graphs of intensity ratios are presented in the following sections. In the interest of conserving space and avoiding repetitive explanations, the format of the graphs will be described here. Each graph consists of ratioed intensity values, 1(0.75 p)/I(l.20 y) and 1(0.65 y)/I(0.76 p), each taken from the ratioed spectrum of normalized (at 0.87 y) spectra. soil The 0.75 y/l.20 p intensity ratio is plotted against the 0.65 p/1 .2 0 p ratio. The ratioed spectra were chosen to contain the sample with the greater copper concentration in the numerator. Absolute concentration versus relative concentration. Figure 30 is a graph of intensity ratios which compares the absolute 82 Figure 30. Intensity ratio graph comparing relative and absolute copper concentrations. The intensity ratios are calculated from soil sample ratio spectra which contain more copper in the numerator sample spectrum. See text for discussion. Graph Key Absolute concentration (ppm) Relative concentration 0-200 200-500 1.0-1.5 V 1.5-2.0 w 2.0-3.0 3.0-5.0 >5.0 a b c d e >1000 U -1.0 Special Symbol_ 500-1000 Points Represented D,G,I B,J,S B,H,Y B,0,Z H,I Figure 30 ILnoInIi MooinnnII 8 IoI nInnlinin III~nlinninP"..M!H!MM!IMI+ I IIMP.MHII Iinnin iiilinntniininn"t" ME!MEN!~~~~~I I=IMPIn"".I..L"H~gnM ===n==P===on====.n==.==== EI hnni.~innnniliinnnonMI Ininiinlinnoiliinno~IT TUI EIM! !in!MMI inlPillnninnininilnnoP.IninliMIM.Innninili MMMMMME EMMM.EMMMMMEIMI.M~non= !==E!!!n!!E=!!!EP.=.i.. I U = Ii!!= WnnnI!Mni.WgiMMPIMIMIIM !!IIMM~nP.M~inIME..noininniEEP!EE f M~niIM~nniiP~nliInlll=I MINIMP.IIMI1171n11 M!IM~.9 iiliCnii innin iliinninniinn rnil 1 1nnnn11iin lPIrnii _II~inInin HE~iinlinininlnnNI~nPInnniP~liiilliin®ri vInniME M~oi.~in!MMM concentration of copper to the relative copper concentration in some 60 Silver Bell soil sample ratio spectra. The samples involved in each ratio were chosen to lie at relative distances less than 1/2 km. The absolute concentration of a sample ratio is defined to be the copper concentration of the sample involved in the numerator of each ratio. The graph shows no correlation between the absolute and the relative concentrations at the levelof copper detectedin the soil samples. Soil mineralogy versus sample distance. A graph of some 70 Silver Bell soil sample intensity ratios is shown in Figure 31. This graph compares soil mineralogy with relative sample distance, i.e., the distance between the two samples used to generate a ratio spectrum. The graph shows that intensity ratios made from samples contained within the same zone of alteration (refer to the Silver Bell topographic map, Figure 24) all cluster near unity. Samples taken from different al- teration zones have intensity ratios which are quite different from unity, reflecting intrinsic differences in soil mineralogy rather than differences in copper concentrations. Thus, in practical application, sample areas should all be ratioed within the same zone of alteration. Since the size of an altera- tion zone depends on the individual deposit, a reasonable approach that would insure meaningful results would be to ratio sample areas that lie fairly close to each other (on the order of 1/2 km in the case of the Silver Bell samples). 85 Figure 31. Intensity ratio graph comparing soil mineralogy and relative distances between soil samples. The intensity ratios are calculated from soil sample ratio spectra which contain more copper in the numerator spectrum. See text for discussion. Graph Key Sample distance (km) Soil mineralogy 1/4 1/4-1/2 1/2-1 1-2 Potassic A B C D Phyllic E F G H Propylitic I J K L Potassic-Phyllic M N 0 P Potassic-Propylitic R S T U Phyllic-Propylitic W X Y Z Special Symbol a b c d e f' g h i Points Represented B,J,K F,I,J B,E,I N,X BMX B,Y A,T AM A,N Figure 31 HinlilI MI~~nnininn~I En inilii == =i===I IE Ii L I I I IInJCnili" I IIMI I IM 8 lilinnlnMIIniin I I 11in1nnn1j1 .. =P I======.1= MM !III!E!M!!!EininE!I!!!!HiR!nnE.I...nn!! innnl .. IIMM giiig n.. InInIniiliiniPgni Iiiniinl IP.Miinnilln~nnn.nlMMnniiin Immnneni minenomenmiP-mm VnnnIn!. !IE~ninni!!!nnE~ii!!Enin.-IF, Pli""EM iniini~~~ininilinilliii~~LIiin iin EIIMMM~~nn!IIMEMME~INEEI~n!Mnn.!M E!inEE!!~n!!!E!=in~nnia!!!!n!!+! E!!IMEINEE!!IonininnMM~nLIn!IE!IM MEMEMMEMIEMP.IMEIIMI~noINinEIMi niiniinniinnnininiiiniiniioninP.niini1i E!M~nP"EMMEM!!IEninEI~nEMIIn!Minho ====Eig==========ll'====== II IT 111M.... ME=IMTnilnienFMMI innioinnnM!!n.!EIIn!!MIE!MMEMIIMM!5IM inliniilliinl iI ~I IlIra-%Iinin InIIIM mem=omm~n~mnminiufill I======imm=I ==PHil=== iin iniiini njnM I Conclusions from intensity ratio graphs. Analysis of the intensity ratio plots (Figures 30 and 31) show that, in practical application, samples taken from relatively far apart should not be ratioed because the mineralogy of the soil is not constant over a large distance. Since there was no correlation between the absolute and relative copper concentrations, the amount of copper analyzed in the soil samples (<2000 ppm) is probably below the detection limit of this technique. Inspection of Figure 20 shows that points diagnostic of copper cluster within a certain area, as delineated on the graph. A relative copper difference of at least 2:1 is needed to produce diagnostic intensity ratios. The minimum absolute concentration of malachite needed to be detectable ranges between -5 - 10%, depending on the overall spectral albedo. (At lower albedos, higher concentrations of malachite are needed because of the corresponding decrease in spectral contrast.) This means that copper concentrations on the order of at least 2000-5000 ppm are needed to be detectable by this technique. C H A P T E R V I I INSTRUMENTATION Instrumental Requirements In developing the technique of reflectance spectroscopy for the detection of copper deposits, laboratory studies are necessary to define the instrumental parameters needed to delineate copper bearing regions. The laboratory studies presented in previous chapters of this thesis adequately define the important instrumental parameters which are intensity precision, spectral range and resolution, and spatial resolution. Determination of these parameters sug- gests the method of reconnaissance and the type of instrument needed to detect surface copper anomalies. The required wavelength range is determined from the optical properties of the weathered (oxidized) phases of copper which most commonly occur in surface anomalies, e.g., malachite, chrysocolla, azurite. Electronic transi- tions diagnostic of these phases occur in the 0.4-1.20 y region. (Infrared transitions due to ligand (CO3 and OH) vibrations are non-unique.) The amount of information regarding mineralogy and modal abundances obtainable from a spectrum depends on the extent to which absorption band positions, strengths, and 88 asymmetries can be resolved. A spectral resolution of 5 to 10 nm is required between 0.4 y and 1.50 p. The intensity precision should be in the neighborhood of a few tenths of a percent (relative reflectance) across the entire wavelength region. Greater spectral resolution or intensity precision would not much improve the interpretation, but a degradation of either parameter would significantly limit the interpretation. The spatial resolution depends on the size of the typical copper anomaly. The ASARCO soil samples collected from the Silver Bell area used in this thesis are representative of a circular region thirty meters in diameter. This value would be a reasonable upper limit to the spatial extent since spectra taken of larger areas tend to lose spectral individuality. Available Instruments Possible instruments available at the moment for remote sensing of the earth include the Skylab S-191 spectrometer and a variety of spectral imagers (ERTS-1, Mariner, etc.). None of these, however, may be used to detect copper by the remote sensing regime proposed in this thesis. The spectral imagers all employ broad band filters (~100 nm) which certainly do not give the required spectral resolution. The S-191 spectrometer had problems in instrument calibration as the data was found to be shifted along the wavelength axis. 90 Nonetheless, thereis a proposed instrument system that does meet the required specifications. The Geochemical Orbiter Spectrometer The reflectance spectrometer designed for geochemical orbiting missions of planetary objects (McCord et al., 1975 (NASA proposal) designed a spectrometer for lunar orbiting) has instrumental specifications that meet (or exceed) those required here for the detection of copper deposits. The spectrometer is designed according to the above mentioned proposal to measure the spectrum of light reflected from the lunar surface along a ground track. The spectrum would then be interpreted in terms of the mineralogy and composition of the regolith, and would be comparable in quality to the presently available laboratory spectra of lunar samples. The geochemical orbiter spectrometer is designed to obtain high resolution spectra of the surface in 220 channels between 0.35 p and 2.50 p with intensity precision of 0.1 to 0.5 percent. The spectral resolution ranges between 5 nm at the high energy end of the spectrum (-0.35 p) to 20 nm near the low energy end of the spectrum (-2.50 y). spatial resolution of 200:1 is attainable (0.5 tion from an altitude of 100 km). A km resolu- In order to obtain a high signal-to-noise ratio (103 ), a 0.5 second integration time is used so that the instrument will not be photon limited at an altitude of 100 km. The ground track rate is 1.7 km/sec which yields a nominal spatial element of 1.35 km x 0.1 km at 0.5 second integration time. At a 100 km altitude, a circular aperture 1.0 mm in diameter situated at the focal point is required to give 0.5 km spatial resolution. Instrumental Specifications for Copper Reconnaissance Instrumental parameters. A high spatial resolution should be used to locate small highly rich copperareas. bearing To attain 10 m resolution using the design specifi- cations for the geochemical orbiter reflectance spectrometer requires an instrument flying altitude of 2 km. Hence, aircraft reconnaissance is the most feasible method to attain the required spatial resolution of 200:1. The spectral resolution and intensity precision of the geochemical orbiter spectrometer meets the specifications for this application. The spectral range in this case may be limited to 0.35 p to 1.50 p (instead of 2.50 p) since the infrared end of the spectrum does not contain information necessary for this application. If the average speed of a low-flying aircraft is about 140 mph (62.5 m/sec), then an integration time of 0.16 seconds or less is obtained. To attain an integration time of 0.1 seconds (which is one-fifth of the integration time for the geochemical orbiter spectrometer) means that the 92 light reflected from the earth's surface should be roughly five times as great as that which is reflected from the moon. The Bond albedo of the moon is on the order of 6-12% and that of the earth ranges between 30-50%. Thus, 0.1 second is a reasonable integration time for use here to give a high signal-to-noise count. The nominal spatial element as measured by the spectrometer, then, would be an area 16.25 m x 10 m and would require an aperture 20 y in diameter for 10 m resolution. Instrumental calibration. Preflight instrument calibration should include defining maximum (100%) reflectance on high albedo soils at low phase angles along the noon meridian (high sun angles). Inflight calibration will not be neces- sary as ratioing the spectra will eliminate instrumental as well as most of the atmospheric contributions. Location of the ground track. During flight, a camera may be used to precisely locate the ground track. Its optic axis should be bore sighted with that of the spectrometer. The camera's field of view should be larger than that of the spectrometer's (10 m) in order to put the spatial element in perspective with the surrounding area. A camera with a field of view on the order of 200 meters is sufficient for this purpose, and would require that a picture be taken every one second to provide sufficient frame overlap at the expense of a minimum amount of film. Data collection. Inflight data collection should consist of uncalibrated reflectance spectra of contiguous 16.25 m x 10 m segments along the ground track. Each spectrum will be accompanied by its time of measurement and phase angle, and the flight altitude and ground speed may be recorded, say, every tenth frame. The surface location should be recorded on photographic film by the camera attached to the spectrometer. The data may be ratioed immediately during flight according to a prescribed ratio pattern, i.e., contiguous spectra may be ratioed, but ratioing every fifth or tenth spectrum will produce greater spectral contrast. Candidate spectra may be ratioed later on the ground after visual inspection of the ground track segments. Obstacles and Uncertainties Involved in Field Measurements Vegetation. There are some obstacles and uncertainties in- volved in the field measurement of spectra. The major ob- stacle is the effect of vegetation on reflectance. The porphyry copper deposits in the United States are situated in desert environments so that the extent of surface vegetation is minimal. However, local areas may be rather heavily vegetated with cactus and sagebrush, and such areas should be avoided. For those areas not heavily covered by cactus and brush, desert varnish ean cover a large part of the surface of many desert floors. Desert varnish is a dark material which can coat up to the entire surface of an exposed rock, and it is mainly composed of manganese and iron oxides (Engeland Sharp, 1958; Potter and Rossman, 1977). The presence of desert varnish would lower the overall albedo of a reflectance curve and thus decrease spectral contrast. The concentration of desert varnish does not vary greatly on a local scale, and so its effects may be minimized by surveying areas of low concentrations (525%). Lichens and moss may also abound in certain regions, and highly lichenous areas should be avoided. There are several types of lichens which are able to assimilate copper and have been reported to change color dramatically according to the amount of copper assimilated (Czehura, 1977). A study of the spectral properties of these indicator lichens may provide a useful and worthwhile addition to this proposed remote sensing regime. Representative spectra of selected desert plants and lichens are presented in the discussion chapter (refer to Figure 36). Absorption fea- tures appearing in the plant spectra are discussed in relation to their contributions to soil spectra. Phase angle effects. An uncertainty in the measurement of spectra involves the effect of phase angle variations on reflectance. Laboratory spectra measures the overall Bond albedo of the reflected material (using an integrating sphere), while inflight spectra are measurements of the 95 geometric albedo, that is, the albedo measured at a certain phase angle. Variations in phase angle affect the extent of shadowing and the overall slope of the spectrum. Band centers and symmetries are also slightly affected by this. The effects of phase angle variations on reflectance would be minimized by flying at low phase angles. A 10* phase angle will permit observations of the least shadowed portions of the surface while not encountering opposition (anomalously high specular reflection which occurs near 0* phase angle). Useful data may be acquired between 100 and 60* (the albedo may vary by a factor of 50 between 0* and 60*), and only those spectra taken at similar phase angles should be ratioed. The effect of phase angle may also be minimized by changing the viewing angle of the spectrometer. This would be useful to survey strongly sloping objects such as the sides of mountains and deep trenches. Flight variations. Other obstacles involved in the measure- ment of spectra include variations in flight speed, altitude, and pitch, and variations in atmospheric conditions. The effect of flight variations should be negligible over 1 to 5 seconds (10-50 measurements). Atmospheric variations will be minimized by flying during relatively clear conditions. C H A P T E R V I I I DISCUSSION Summary Reflectance spectroscopy has been used successfully to determine the composition of solar system objects as was discussed in the Introduction. In this thesis, the technique is used to determine copper bearing regions on the earth. Development of the technique for this purpose re- quired understanding the spectral properties of minerals which are contained in surface soils and rocks. This was accomplished from background laboratory studies which not only determined the wavelength regions diagnostic of copper bearing minerals and other representative soil constituents such as clays, ferric oxides, and vegetation, but also determined the limits of copper concentration needed for detection. Since there is not a field spectrometer which currently possesses sufficient detection capabilities, a field test was simulated in the laboratory using naturally occurring soil samples taken from the regions of copper deposits. Reflectance spectra of these samples were used to generate representative ratio spectra simulating those that would be taken in the field. These laboratory ratio spectra were analyzed for their relative copper concentrations and 96 the results defined the sensitivity limit of the technique. Development of a spectrometer with the required instrumental parameters (such as the geochemical orbiter spectrometer described in the chapter on Instrumentation) is needed before this technique can be applied in the field. Potential Problems in Field Application There are some potential problems which can arise when reflectance spectroscopy is applied in the field. Obstacles involved in the measurement of spectra such as the effect of phase angle, atmospheric conditions, and flight variations were discussed in the Instrumentation Chapter. However, a potential problem involved in spectral interpretation may arise from the presence of other minerals possessing absorption features near the wavelengths diagnostic of copper. Reflectance spectra of clinopyroxenes. Figure 32 is a plot (fromAdams, 1975) of centers of absorption bands appearing in the reflectance spectra of common rock-forming minerals. Inspection of the graph shows that a few minerals possess absorption bands near the diagnostic copper (malachite) wavelengths at 0.77 p and 1.08 p. These minerals are the clinopyroxenes diopside, augite, and salite. Representative reflectance spectra of these minerals are shown in Figure 33, and features appearing in the spectra have been discussed by 98 Figure 32 I I I I I I I I ALMANDINE ~dsPYROXENE . 2 ABSORPTION BANDS ONLY WER . b 3 ABSORPTION - - IflO'F BANDS METALS, LOWER OXIDES . OLIVINE SCALE FELDSPARI GLASS PHLOG-SAL O0LO, . SEPID * EPID 090 CMICA S90- KR K 1P- DOP- -SulAL RCDIOP- gSAL 00 Q70 cr-iNST F8T0- Cr00~P 050 CraOs * JCr-DIOP i . 04 I 0 iCr-EST I 0.6 Q8 W FlOiFt 08 I I I LO 12 1.0 I L4 I I I II I I I I L6 L8 20 22 24 L2 L4 L6 12 WAVELENGTH, MICRONS 20 Plot of centers of absorption bands in the diffuse reflectance spectra of several common minerals. Minerals having only one main band are plotted on the lower scale. Minerals with two or more bands in their spectra are designated by points in the main upper field. Hydroxyl and water bands at 1.4 and 1.9 sm are not used. Bands for minerals species and for glasses plot in separate fields. See text for discussion. Diop-sal - disosidesalite; bio-phlog - biotite-phlogopite; aug - augite; pige - pigeonite; opx -= orthopyroxene; dolo - dolomite; epid - epidote; actin-hornb - actinolite-hornblende; trem-ged-kaer - tremolite-gedrite-kaersutite; amph - amphibole; enst - enstatite; chlor - chlorite; Fo - forsterite; Fa - fayalite; Ab - albite; Olig - oligoclase; And andesine; Lab - labradorite; Byt - bytownite; An - anorthite. (After Adams, 1975). Figure .33 I .1 a' 0 4I- I 0.5 1.0 1.5 WAVELENGTH (pm) 2.0 2.5 Diffuse reflectance spectra of clinopyroxenes. Iron-rich caleie pyroxenes typically have more complex Fe'+ and Fe'-Fes+ band structure than do the orthopyroenes and pigeonites. (After Adams, 9 1975). 100 several authors (Burns, 1970; Adams, 1975; Hunt and Salisbury, 1970a). Briefly, the absorption feature in the 0.75- 0.80 ypregion is attributed to Fe 2 -Fe + change transfers whose absorption intensity varies with the amount of iron substitution. The absorption strength of this feature is not greater than the 1.1 p feature except in the spectra of salite, which is more iron rich than diopside. The presence 2+ of the 1.1 y band is due to Fe in the pyroxene Ml site, and the presence of a fairly weak feature near 0.95 y in the salite-diopside spectra arises from Fe + substitution in the M2 site (Burns, 1970). Occurrence of clinopyroxenes. Although clinopyroxenes have absorption bands in the regions of the malachite bands, the presence of these minerals in the spectra of naturally occurring soils may not be much of a problem. Disseminated copper deposits are formed mostly in acidic-intermediate rock facies, usually monzonite, whereas the clinopyroxenes are formed mostly in basic igneous rock facies and diopside is commonly found in contact metamorphosed limestone (skarn). Areas chosen for the reconnaissance of copper will be those containing rocks of principally acidic-intermediate composition associated with subduction zones, and so the occurrence of mafic rock facies which are more likely to contain the clinopyroxenes should be limited in these areas. 101 Clinopyroxene discrimination from intensity ratios. However, should clinopyroxenes be encountered during reconnaissance, the fact that the 1.1 y band is generally more intense than the one near 0.75 y in the spectra of these minerals will be evident when the 0.65 y/0.75 y and 0.75 y/1.20 y intensity ratios are calculated. In malachite, the 0.75 p feature is always more intense than the one at 1.1 y which results in the 0.75 p/l.20 y intensity ratio generally being less than or equal to unity. However, in clinopyroxenes, this inten- sity ratio will generally be greater than unity since the 1.1 y band is more intense than the 0.75 y band. The pyrox- ene spectra (Figure 33) also show that the 0.65 y/0.75 y intensity ratio is greater than unity. A tentative pyroxene field generated from estimates of the 0.65 y/0.75 y and 0.75 y/1.20 y intensity ratios is outlined on the graph of the malachite and ferric oxide intensity ratios in Figure 34, which shows that pyroxene will probably not be confused with malachite. Fe +-rich clinopyroxenes: salite. The only possible case when a clinopyroxene may be confused with malachite is when the pyroxene is relatively rich in Fe3+ compared to Fe 2 + as in salite. The spectrum of salite (Figure 33) shows that the 0.75 P band becomes more intense than the 1.1 p band due to Fe + enrichment. Olivines and pyroxenes with high rela- tive abundances of Fe3 + show exsolved lamallic intergrowths 102 Figure 34. Intensity ratio graph of laboratory simulated soils (Figure 20) including ratios calculated from the spectra of clinopyroxenes. The effect of vegetation is also shown. See text for full discussion. Figure 34 103 103 44- -T-t- Im I s4$ A 44 1 w V IA If IA All I V 11.1 v I /I F ol%1 ri liT V 1611 If I I/ .4, 1 All V 3111. r IF I IN I L L/L Y I J v V- -ely I -- I YIf + V of I ri or tj I 0,1 VT -Op 4pvv T-T (X, 11A 104 of hematite (a-Fe 2 03 ) within the crystal structure (Deer, Howie, and Zussman, 1962a,b). exposed at the surface, When these minerals are the presence of Fe 3+ tends to break up the crystal structure in favor of formation of ferric oxides (Deer, Howie, Zussman, 1962a). enriched in Fe Thus, pyroxenes are not expected to be common at the sur- face since weathering processes would probably have destroyed them. Reflectance spectra of representative desert vegetation. The presence of vegetation may also lead to erroneous spectral interpretation. Although highly vegetated areas should be avoided during field measurements, some vegetation will invariably be encountered. UV-visible-IR absorption spectra of chlorophyll and other essential plant pigments are shown in Figure 35. The predominant plant pigments absorb in the 0.44 p region (22,500 cm1 ), but only chlorophyll absorbs in the vicinity of 0.65 y (15,000 cm~ ) (Gates et al., 1965). Vibrational bands due to water occur throughout the infrared region. The materials comprising leaves are moderately reflecting in the green, near 0.54 y (18,500 cm 1) highly reflecting in the 0.70-2.0 y region. and are The reflectance and absorptance spectra of typical desert succulent plants and typical lichens are shown in Figures 36a and 36b respectively. The prominent feature in the spectra of both types of plants is the chlorophyll absorption in the 0.65-0.70 y region. 105 55 44 is 2- ir re'0 3 s) 40r VW^J41.MICRO me1MMI ars.1 Figure 35. Specific absorption versus frequency of the principal plant pigments and liquid water. (After Gates, et al., 1965). 106 94 #1 -ULTRAVO.ET VIStOLE a - ------ 05 as it sl WAVELENGTH, MICRONS - is 0 (G400 INRARE0 -0 CEREUS GIGANTEUS AGAVAMERICANA (LOWR M A E £AMvi SWRACE) O"MTI LAEVIS ''-''' WA/EMAER% cm 1 &w9 I A SAVELENGTH. MILLlNCA B Fig re16. (A) The spectral absorptance and reflectance of the stems of desert succulent plantt. (B) Reflectance spectra of selected lichens. (After Gates, et al., 1965). 107 Effects of vegetation on soil spectra. This chlorophyll feature does not occur at the wavelengths diagnostic of malachite (0.77 y and 1.08 y), but it will affect the 0.65 p/0.75 p intensity ratio calculated from the spectra of soils containing vegetation. The effect of plants will be to lower the reflectance at 0.65 p relative to the reflectance at 0.75 p which will cause a decrease in the 0.65 p/0.75 p intensity ratio. Intensity ratios calculated from spectra containing a relatively high proportion of vegetation will fall shortward of those values calculated from spectra containing little vegetation on the 0.65 p/0.75 p axis. Assuming that malachite is present in diagnostic amounts, a high proportion of vegetation in the spectra would yield a 0.65 p/0.75 p ratio outside of the field diagnostic of malachite. The relative proportion of vege- tation required to produce an undiagnostic 0.65 p/0.75 p intensity ratio has not been studied, and further laboratory work is needed in order to determine the effects of vegetation. Sensitivity of the Technique The detection limit of the technique was determined from Figure 20 which was discussed in Chapter VI. It was found that at least a 2:1 relative copper difference needed to produce diagnostic intensity ratios. is The minimum absolute concentration of malachite needed to be detected was '108 found to be on the order ,of 5-10% (2,000-5,000 ppm Cu) depending on the overall spectral albedo. In application, ad- jacent spectra are ratioed in order to minimize instrumental and atmospheric variations. The ratio spectrum of two areas both containing a high absolute malachite concentration, but with less than a 2:1 relative malachite concentration would yield undiagnostic to unity). intensity ratios (they would both be close Therefore, there is a trade-off between the sen- sitivity of the technique and minimizing the atmospheric and instrumental effects. These effects, however, may not vary greatly during a reconnaissance mission so that spectra taken from rather widely separated areas which contain different relative malachite concentrations would produce diagnostic intensity ratios thus increasing the sensitivity of the technique. Higher concentrations of malachite may be needed if a relatively high proportion of vegetation is present in the spectra. Although this technique is not sensitive to soils containing approximately less than 2,000 ppm Cu such as were found in the natural soil samples studied here, it should still be useful to identify copper deposits. Small, localized surface areas can contain malachite and other oxidized copper minerals at levels high enough to be detected. al., (Lovering et 1950, found that some soils collected around the San Manual porphyry deposit contained copper concentrations on the order of 10,000 ppm.) A 10 m spatial resolution for the 109 spectrometer proposed in the Instrumentation chapter should be high enough to detect small areas that are enriched in malachite. Advantages of the Technique Reflectance spectroscopy has advantages as an exploration technique over other geophysical, geochemical, and remote sensing techniques. It is capable of directly detecting the presence of copper in surface soils, it can survey a wide area during a single reconnaissance, and it can scan areas that may be inaccessible to in situ measurements. Potential Applications The technique of reflectance spectroscopy may be used for other applications. The remote sensing regime developed in this thesis for copper exploration may be extended to other economically important transition metals. Chromium is a good candidate since it possesses absorption features at diagnostic wavelengths. Figure 37 shows reflectance spectra of a chrome- diopside containing 1.2% Cr2 0 3 and chromite (Cr2 3 ). Strong, relatively sharp absorption features arising from electronic transitions in Cr 3 + occur near 0.45p and 0.60p. Inspection of Figure 32 shows that the 0.6Op feature is unique only to Cr 3 +bearing minerals, and hence chromium would be a good choice towards application of this technique. 110 Cr DDPMDE U. 0* 1.0 . ~ Figure 37. Reflectance spectra of chrome bearing minerals. (After Adams, 1975). REFERENCES Adams, J. (1975). Interpretation of visible and near-infrared diffuse reflectance spectra of pyroxenes and other rock-forming minerals. In: Karr, C. (Editor), Infrared and Raman Spectroscopy of Lunar and Terrestrial Minerals, Academic Press, New York, 91-116. Argall, G. and Wyllie, R. (1976). World Mining Copper Map. Miller Freeman Publications, Belgium. Ballhausen, C. (1962). Introduction to Ligand Field Theory. McGraw-Hill, New York. Bernal, J. et al. (1959). The oxides and hydroxides of iron and their structural inter-relationships. Clay Min. Bull. 4, 15-31. Blanchard, R. (1968). Interpretation of Leached Outcrops. Nevada Bureau of Mines, Reno. Burns, R. (1970). Mineralogical Applications of Crystal Field Theory. Cambridge University Press, Cambridge. Chaffee, M. (1976). The zonal distribution of selected elements above the Kalamazoo porphyry copper deposit. J. Geochem. Exploration 5, 145-165. Clarke, 0. (1952). Geochemical prospecting for copper at Ray, Arizona. Econ. Geol. 48, 39-45. Creasey, S. (1966). Hydrothermal alteration. In: Titley, S. and Hicks, C. (Editors), Geology of the Porphyry Copper Deposits, Univ. of Arizona Press, 51-74. Czehura, S. (1977). A lichen indicator of copper mineralization, Lights Creek District, Plumas County, California. Econ. Geol. 42, 796-803. Deer, W., Howie, R. and Zussman, J. (1962a). Rock Forming Minerals: Vol. I: Ortho and Ring Silicates. Longmans, Green and Co., London. . (1962b). Rock Forming Minerals: Vol. 2: Chain Silicates. Longmans, Green and Co., London. 111 112 Dykstra, J. (1975). Detection of Porphyry Copper Alteration by Computer Processing of ERTS-1 Digital Data. M.A. Thesis, Dartmouth College, Hanover, New Hampshire. Eidel, J., Frost, J. and Clippinger, D. (1968). Coppermolybdenum mineralization at Mineral Park, Mohave County, Arizona. In: J.D. Ridge (Editor), Ore Deposits of the United States, 1933-1967 (GratonSales Vol.), SPE/AIMIE, New York, 1258-1281. Engel, C. and Sharp, R. (1958). Chemical data on desert varnish. Geol. Soc. Am. Bull. 69, 487-518. Galey, J. (personal communication). Gates, D. et al. (1965). Spectral properties of plants. Applied Optics 4, 11-20. Gattow, V. and Zemann, J. (1958). Neubestimung der Kristallstruktur von Azurit. Acta Cryst. 11, 866-872. Gilluly, J. (1963). The tectonic evolution of the western United States. Geol. Soc. London Quart. Jour. 119, 133-174. Halford, R. (1946). Motions of molecules in condensed systems: I. Selection rules, relative intensities and orientation effects for Raman and infrared spectra. J. Chem. Phys. 14, 8-15. Hathaway, B. and Billing, D. (1970). The electronic properties and stereochemistry of mono-nuclear complexes of the copper (II) ion. Coordin. Chem. Rev. 5, 143-207. Head, J. et al. (1978). Definition and detailed characterization of lunar surface units using remote observations. Icarus 33, 145-172. Huff, L. (1952). Abnormal copper, lead, and zinc content of soils near metalliferous veins. Econ. Geol. 47, 517542. Hunt, G. and Salisbury, J. (1970a). Visible and near infrared spectra of minerals and rocks: I. Silicate minerals. Mod. Geol. 1, 283-300. . (1970b). Visible and near infrared spectra of minerals and rocks: II. Carbonates. Mod. Geol. 2, 23-30. 113 , and Lenhoff, C. (1971a). Visible and near infra- red spectra of minerals and rocks: III. Oxides and hydroxides. Mod. Geol. 2, 195-205. . (1973a). Visible and near infrared spectra of minerals and rocks: VI. Additional silicates. Mod. Geol. 4, 85-106. Jorgensen, C. (1963). Spectroscopy of transition group complexes. Advances in Chemical Physics 5, 33-147. Lakshman, S. and Reddy, B. (1973). Optical absorption spectra of Cu 2 + in chalcanthite and malachite. Can. Min. 12, 207-210. Lever, A. (1968). Inorganic Electronic Spectroscopy. Elsevier Publishing Co., New York. Liehr, A. (1969). Reciprocation of electrostatic and electromagnetic forces in ligand field theory. J. Phys. Chem. 64, 43-51. Locke, A. (1926). Leached Outcrops as Guides to Copper Ore. Baltimore, Williams, and Wilkins Co. Lovering, T., Huff, L., and Almond, H. (1950). Dispersion of copper from the San Manual copper deposit, Pinal County, Arizona. Econ. Geol. 45, 493-514. Lowell, J. and Guilbert, J. (1970). Lateral and vertical alteration-mineralization zoning in porphyry ore deposits. Econ. Geol. 65, 373-408. Mao, H. and Bell, P. (1973-74). Crystal field effects of ferric iron in goethite and lepidocrocite: band assignment and geochemical applications at high pressure. Annual Report of the Director, Geophysical Laboratory, Carnegie Institution, Washington, D.C. McCord, T. (principal investigator)(1975). Surface Mineralogy Experiment for the Lunar Polar Orbiter. NASA proposal. . et al. (1977). Spectral reflectance of Martian areas during the 1973 opposition: photoelectric filter photometry 0.33-1.10 p. Icarus 31, 25-39. Pappalardo, R. (1961). Absorption spectra of Cu2+ in different crystal coordinations. J. Mol. Spectroscopy 6, 554573. 114 Potter, R. and Rossman, G. (1977). Desert varnish: the importance of clay minerals. Science 196, 1446-1448. Sillitoe, R. (1972). A plate tectonic model for the origin of porphyry copper deposits. Econ. Geol. 67, 184197. . (1973). The tops and bottoms of porphyry copper deposits. Econ. Geol. 68, 799-815. Silman, J. (1958). The stabilities of some oxidized copper minerals in aqueous solutions at 25*C and 1 atmosphere pressure. Ph.D. Thesis, Harvard University, Cambridge, Massachusetts. Solomon, E.I. (personal communication). Ssse, V. (1967). Verfeinerung der Kristallstruktur des Malachits. Acta Cryst. 22, 146-151. Titley, S. and Hicks, C. (1966). Geology of the Porphyry Copper Deposits. Univ. of Arizona Press, Tucson. Wells, A. (1951). Malachite: re-examination of the crystal structure. Acta Cryst. 4, 200-204. Whitten, P. and Brooks, J. (1972). A Dictionary of Geology. Penguin Books, England. A P P E N D I X I SAMPLE DESCRIPTIONS AND ANALYSES Sample Description Mineral samples used to generate the laboratory reflectance spectra presented in this study were obtained from the Smithsonian Museum of Natural History in Washington, D.C. as well as from mineral and chemical supply companies. The origins of the samples are listed in Table 1 along with their respective Smithsonian identification numbers, where applicable. The samples used to generate the laboratory mixtures of ferric oxides, clays, malachite, and MnO 2 are the following: Limonite 2001 (Cartersville) Hematite, reagent Malachite 6004 (reagent) Kaolinite, England MnO 2 , reagent Samples that needed to be ground were done so with a porcelain mortar and pestle. Most samples contain unsorted par- ticles sized less than 90 p, and a few samples noted in the text were dry sieved using standard Tyler sieves according to the particle sizes: J = 63-90 p; K = 45-63 p; L = <45 p. Standard x-ray diffraction analysis was used to identify the azurite, malachite, and ferric oxide samples. 115 116 Table 1. Sample descriptions and identification numbers. Mineral M.I.T. Sample # Smithsonian Sample # Sample Origin Azurite 5000 5001 118913 117707 Tsumeb, S.W. Africa Hermosillo, Mexico Malachite 6000 6001 6002 6003 6004 R13881 10323 122455 C5823 Bisbee, Arizona Kennecott, Alaska Broken Hill, Rhodesia Apex Mine, Utah Reagent (Mallinckrodt) Chrysocolla (blue) (blue) (blue) (green) (green) (green) 7000 7001 7002 7003 7004 7005 104956 120272 R4844 B17050 R16369 R4859 Bisbee, Arizona Arizona Gila County, Arizona Herrengrund, Hungary Tagilsk, U.S.S.R. Tagilsk, U.S.S.R. Goethite 3004 C1736 Minas Gerais, Brazil Limonite 2000 2001 2003 118532 1000 1001 1003 1005 46422 128254 64655 2002 104895-1 Hematite Lepidocrocite Tuscaloosa, Ga. (Wards) Cartersville, Ga. (Wards) Bisbee, Arizona Ironton, Minn. (Wards) Camerata, Africa Mt. Newman, Australia Missouri Reagent (Matheson, Coleman, and Bell) (Limonite) Bisbee, Az. Maghemite Steam oxidized magnetite Kaolinite England (Mallinckrodt) Manganese dioxide 8002 Reagent (Baker) 117 Sample Analyses Reflectance spectra. The theoretical basis for interpreta- tionof visible-near infrared spectra of minerals has been established in the literature 1962). (Burns, 1970; Ballhausen, Adams (1975) presents references describing the application of crystal field theory to the interpretation of optical spectra of natural minerals. Laboratory diffuse reflectance spectra of powdered samples were measured in the range 0.35-2.50 y with a Beckman DK-2A ratio-recording spectrophotometer belonging to John Adams at the University of Washington. MgO was used as a reference surface except for those spectra measured of the natural soil and rock chip samples, where halon was used as a reference. Spectra were recorded simultaneously on chart paper and on magnetic tape except for the azurite and malachite spectra, which were recorded on digital papertape. Infrared spectra. Infrared spectra of a suite of azurite and malachite samples were measured by Graham Hunt at the Air Force Cambridge Research Laboratories in the 2.50-40 y range with a Perkin-Elmer Model 180 spectrophotometer. conventional KBr pellet technique was used as well as a reflection technique, microreflectance which employed a modified standard attachment. The 118 X-ray fluorescence. Soil samples collected from the Silver Bell and Mineral Park districts were analyzed for their copper concentrations using X-ray fluorescence. The smallest sized particle samples (<45 y for the Silver Bell samples and <125 y for the Mineral Park samples) were pressed into pellets and then measured on a Diano (General Electric XRD-5) X-ray fluorescent instrument operated by the Department of Chemistry at the University of Massachusetts. A tungsten tube pro- vided the X-ray source operating at 40 kV and 35 mA. The op- tical path was air,and the X-rays were dispersed using a LiF crystal. A proportional detector operating at 1.4 kV was used to convert the X-ray photons to electron pulses. Copper was analyzed at the CuKa peak at 45.03*, and the background was measured at 44.20* and 48.200 for each sample. Counts were recorded at a fixed time of 10 seconds per measurement, and 10 measurements were used to calculate the average number of counts at each angle setting. Since the copper concentra- tions of the Silver Bell rock chip samples were previously determined by ASARCO, they were used as reference standards with copper concentrations ranging between 70 and 3200 ppm. The estimated error in the copper concentrations of the soil samples ranged between 15 and 45 ppm, with the average error on the order of 25 ppm, although the error in the rock chip analyses could not be included, since it was not given by ASARCO. The average copper concentration of the soil and rock chip samples are given in Table 2. 119 Table 2. Copper concentrations of the Silver Bell and Mineral Park samples as determined from XRF. The concentrations of the rock chip samples was provided through the courtesy of ASARCO. Silver Bell soil sample Cu (ppm) Silver bell rock chip Cu (ppm) SS1 370 GOC43 3200 SS2 285 GOC44 310 SS3 270 GOC46 100 SS4 465 GOC49 SS5 1585 GOC78 750 SS6 395 SS7 710 SS8 305 Mineral Park soil sample Cu (ppm) SS9 245 ACl 3 930 SS10 760 DFl3 1075 SSll 290 AC4 6 1250 DF46 1140 SS12 SS13 125 SS14 175 SS15 275 SS16 SSl7 100 SS18 135 SS19 145 SS20 105 A P P E N D I X I I REFLECTANCE SPECTRA OF LABORATORY SIMULATED SOILS Figure 1: Malachite and ferric oxide Figures 2-4: Malachite, ferric oxides, and kaolinite Figures 5-32: Malachite, ferric oxides, kaolinite, and MnO 2 120 121 80 70 A 60- B W 50 Z 40 W 30 - D I 20 - od 10 A - 5% malachite + 95% ferric oxides 10 B - 25% malachite + 75% ferric oxides C - 50% malachite + 50% ferric oxides 10 D - 75% malachite + 25% ferric oxides 10 0 0.5 1.0 1.5 2.0 WAVELENGTH 2.5 (p) Figure 1. Reflectance spectra of laboratory prepared mixtures of malachite and an equal mixture (wt.%) of hematite and goethite. 3.0 122 100 9,0 80- 10 z U <60 C z ~ L, 50-~ ;- 4,0 A - 5%malachite + 5% ferric oxides + 90% kaolinite B - 5% malachite + 20% ferric oxides + 75% kaolinite 30~ A C - 5% malachite + 50% ferric oxides + 45% kaolinite B 10 C 0.5 10 1.5 2.5 Z.0 WAVELENGTH 3.0 (/.) Figure 2. Reflectance spectra of laboratory prepared mixtures of malachite, and equal mixture (wt.%) of hematite and goethite, and kaolinite. 123 90 80 70 W Q Z W -j LW 60 A 50 50 B C 40 30 20 10 A - 20% malachite + 5% ferric oxides + 75% kaolinite B - 20% malachite + 20% ferric oxides + 60% kaolinite C - 20% malachite + 50% ferric oxides + 30% kaolinite 10 10 t 0 L 0.5 1.0 1.5 2.5 2.0 WAVELENGTH 3.0 (p4 Figure 3. Reflectance spectra of laboratory prepared mixtures of malachite, and equal mixture (wt.%) of hematite and goethite, and kaolinite. 124 80 70 60 A 50 B 40 30 20 A - 50% malachite + 5% ferric oxides + 45% kaolinite B - 50% malachite + 20% ferric oxides + 30% kaolinite 10 - 0 0.5 1.0 1.5 2.0 2,5 WAVELENGTH Figure 4. Reflectance spectra of laboratory prepared mixtures of malachite, an equal mixture (wt.%) of hematite and goethite, and kaolinite. 3.0 125 90 80 70 w A 60 50 B W 40 I C w 30 0 $ 20 A - 5% malachite + 5% goethite + 85% kaolinite + 5% MnO B -5% malachite + 20% goethite + 70% kaollinite + 5% MnO2 10 C- 5%malachite + 50% goethite + 40% kaolinite + 5% MnO 2 - 10 0 0.5 1 .0 2.0 1.5 WAVELENGTH (,L) Figure 5.Reflectance spectra of laboratory prepared mixtures (wt.%). 2.5 126 70 60 W U Z 50 A 40 B C w 03 0 -J LI- W 20 o 10 A - 5% MnO 2 + 20% malachite + 5% goethite + 70% kaolinite B - 5% MnO 2 + 20% malachite + 20% goethite + 55% kaolinits. C - 5% Mn0 2 + 20% malachite + 50% goethite + 25% kaolinite 10 10 ~ 0 0.5 1.0 1.5 2.0 2.5 3.0 WAVELENGTH (p.) Figure 6. Reflectance spebtra of laboratory prepared mixtures (wt.%). 127 80 70 w 60 U Z 50 W 40 40 W 30 A B 20 A - 5% MnO2 + 50% malachite + 5% goethite + 40% kaolinite 10 B - 5% MnO 2 + 50% malachite + 20% goethite + 25% kaolinite 10 0 0.5 1.0 1.5 2.5 2.0 WAVELENGTH 3.0 ( Figure 7. Reflectance spectra of laboratory prepared mixtures (wt.%). 128 70 60 A 50 Z U 40 B C W 30 30 -j IL W 20 O IO A - 15% MnO + 5% malachite + 5% goethite + 75% kaoiinite B - 15% MnO 5% malachite + 20% goethite + 60% kaoiinite C - 15% Mno + 5% malachite + 50% goethite + 30% kaoIinite 10 10 0 0.5 1.0 1.5 2.0 WAVELENGTH (p) Figure 8. Reflectance spectra of laboratory prepared mixtures (wt.%). 2.5 129 70 60 A 50 0 z 40 W30 C - 20 $ \0 10 10 10 -+ A - 15% Mno + 20% malachite + + 60% k olinite 5% goethite B - 15% MnO, + 20% malachite + 20% goethite + 45% kaolinite C - 15% Mno + 20% malachite + 50% goethite 15% k olinite 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 A (p. Figure 9. Reflectance spectra of laboratory prepared mixtures (wt.%). 130 80 70 W60 z 5,0 I- 40 W4 LL Cr 30 0 20 -. A - -0 10 10 A- 15% Mato 5% goethite + B - 15% MnO + 50% malachite + 20% goethite + 50% .malachite + 30% kao~inite 2 15% kaolinite 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 10. Reflectance spectra of laboratory prepared mixtures (wt.%). 131 70 60 w 50 0 z 4 A W30 C W 20 o* O 5% malachite + 5% goethite + 65% kaoiinite A B - 10 + 25% MnO + 5% malachite + 20% goethite + 50% kaoiinite C 10 10 25% MnO - 25% MnO + 5% malachite + 50% goethite + 20% kao~inite - II 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p) Figure 11. Reflectance spectra of laboratory prepared mixtures (wt.%). 132 70 60 W 50 z p. 4 0 130 C Li X: 20 $ 10 A - 215% Mno + 20% malachite + 5% goethite + 50% k olinite B - 25% MnO + 20% malachite + 20% goethite + 35% k alinite C - 25% MnO + 20% malachite + 50% goethite + 5% ka1inite 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 12. Reflectance spectra of laboratory prepared mixtures (wt.%). 133 70 60 w 050 zA 40 0 w -3 20 0 A 25% MnO + 50% malachite + 5% goethite + 20% kaoiinite B - 25% MnO + 50% malachite + 20% goethite + 5% ka linite 10 0 0.5 - 1.0 1.5 WAVELENGTH 2.0 2.5 (p) Figure 13. Reflectance spectra of laboratory prepared mixtures (wt.%). 134 70 A - 50% MnO + 5% malachite + 20% goethite + 25% kaoiinite 60 w 0 Z 5% goethite + B - 50% MnO + 20% malachite + C - 50% MnO + 20% malachite + 20% goethite + - 25% kaolinite 10% kaolinite 50 40 [ A LL B 30 -J IL W 20 o 10 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (.L) Figure 14. Reflectance spectra of laboratory prepared mixtures (wt.%). 135 80 70 A 60 z 50 w 40 U. 30 w B C 20 0 10 A - 5% MnO + 5% malachite + 5% hematite + 85% Naolinite B - 5% MnO + 5% malachite + 20% hematite + 70% iaolinite 10 C- 5% MnO + 5% malachite + 50% hematite + 40% aolinite 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (l.) Figure 15. Reflectance spectra of laboratory prepared mixtures (wt.%). 136 80 70 60 W 50 0 z H 0 40 w LL 3 0 w r.20 0 10 A a 5% MnO + 20% malachite + + 70% aolinite 10 B C 10 - - 5% MnO 5% hematite + 20% malachite + 20% hematite - + 55% 5% MnO + 20% malachite + 50% hematite + 25% aolinite aolinite 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p) Figure 16. Reflectance spectra of laboratory prepared mixtures (wt.%). 137 80 70 W 60 0 z 4 B 50 0 IL Oc 0 W40 30 20 A - 5% MnO + 50% malachite + + 40% Raolinite 5% hematite B - 5% MnO + 50% malachite + 20% hematite + 25% aolinite 10 - 10~ 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 17. Reflectance spectra of laboratory prepared mixtures (wt.%). 138 70 60 Li 0B z 50 40 C 1- W 30 W 20 0* 10 Q A - 15% MnO + 5% malachite + 5% hematite + 75% k.olinite B - 10 10 15% MnO + 5% malachite + 20% hematite + 60% k linite C - 15% MnO + 5 % malachite + 50% hematite + 30% k olinite - - 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (piL) Figure 18. Reflectance spectra of laboratory prepared mixtures (wt.%). 139 70 60 50 B U Z 40 A 0 C w w 30 LL W a: 20 9 10 A - 15% MnO + 20% malachite + 5% hematite + 60% kaolinite B - 15% MnO + 20% malachite + 20% hematite +45% kaglinite 10 C - 1O 15% MnO + 20% malachite + 50% hematite +15% kalinite 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (.) Figure 19. Reflectance spectra of laboratory prepared mixtures (wt.%). 140 80 + 30% kiolinite B + 50% malachite + 20% hematite 15% MnO + 15% kiolinite 70 w - 60 0A Z 50 W 40 IL W 30 $ 20 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 20. Reflectance spectra of laboratory prepared mixtures (wt.%). 141 70 60 A 50 w Z 40 W30 - C w20 0 10 10 0.5 A - 25% MnO + 5% malachite + + 65% kaolinite B - C - 25% MnO + 5% malachite + 20% hematite + 50% kaolinite 25% MnO + 5% malachite + 50% hematite + 20% k olinite 1.0 1.5 WAVELENGTH 5% hematite 2.0 2.5 (L) Figure 21. Reflectance spectra of laboratory prepared mixtures (wt.%). 142 70 60 W z 50 0 40 A -B W 30 -J LLC X 20 %% 0 10 A - B - t0 25% MnO + 20% malachite + + 50% k olinite 25% MnO 5% hematite + 20% malachite + 20% hematite + 35% kaolinite C - 25% MnO + 20% malachite + 50% hematite + 5F kaglinite 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (/p) Figure 22. Reflectance spectra of laboratory prepared mixtures (wt.%). 143 80 70 A - 25% MnO + 50% malachite + + 20% k~olinite B - 5% hematite + 50% malachite + 20% hematite 25% MnO + 5% kaglinite w60 0 z 50 (U laa -J 40- L w 30 o 20 B 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p. ) Figure 23. Reflectance spectra of laboratory prepared mixtures (wt.%). 144 70 60 w Q z 50 - + 25% kaolinite B - 50% MnO + 20% malachite + 5% hematite + 25% kiolinite C - 50% MnO + 20% malachite + 20% hematite + 10% k~olinite - - 40 A - W 30 ..J LL B X20 IS 10 - 10 10 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 24. Reflectance spectra of laboratory prepared mixtures (wt.%). 145 80 V 70 A 60 B 050 z - 40 --J30 w0 20 A - 5% MnO + 5% malachite + 5% (hematite + goethite) + 85% kaolinite B - 5% MnO + 5% malachite + 20% (hematite + goethi~e) + 70% kaolinite I0 C - + 5% malachite + 50% (hematite + goethife) + 40% kaolinite 10 10 5% MnO - 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p) Figure 25. Reflectance spectra of laboratory prepared mixtures (wt.%). 146 70 A 60 w 0 z 50 40 C I W30 LiL W 20 - 5% MnO + 20% malachite + 5% (hematite + goet ite) + 70% kaolinite 10 B - 5% MnO + 20% + goet ite) + C - 5% MnO + 20% + goethite) + 10 malachite + 20% (hematite 55% kaolinite malachite + 50% (hematite 25% kaolinite 10 0.5 1.0 1 .5 WAVELENGTH 2.0 2.5 (p.) Figure 26. Reflectance spectra of laboratory prepared mixtures (wt.%). 147 80 70 w 60 z 50 - A 0 - W 40 -J w 30 0f%20 -- A - 5% MnO + 50% malachite + 5% (hematite + goet~ite) + 40% kaolinite B - 5% MnO + 50% malachite + 20% (hematite + goet ite) + 25% kaolinite 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 27. Reflectance spectra of laboratory prepared mixtures (wt.%). 148 70 60 A 50 B z40 FUC W30 LiL w 20 A - 15% MnO + 5% malachite + 5% (hematite + goethite) + 75% kaolinite B - 15% MnO + 5% malachite + 20% (hematite + goethite) + 60% kaolinite C - 15% MnO + 5% malachite + 50% (hematite + goethite) + 30% kaolinite 10 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (/p.) Figure 28. Reflectance spectra of laboratory prepared mixtures (wt.%). 149 70 60 A w 50 C. z<40 B 0 C . w 0 o 20 10 A - 15% MnO + 20% malachite + 5% (hematite~ + goethite) + 60% kaolinite 10 B - 15% MnO C 15% MnO + 20% malachite + 20% (hematite + goethite) + 15% kaolinite 10 0 -- 0.5 - + 20% malachite + 20% (hematite + goethite) + 45% kaolinite 1.0 1 .5 WAVELENGTH 2.0 2.5 (p4 Figure 29. Reflectance spectra of laboratory prepared mixtures (wt.%). 150 80 B - goethit6) + 30% kaolinite 15% MnO + 50% malachite + 20% (hematite + goethit ) + 15% kaolinite 70 60 0A z 50 W 040 -J IL LLJ3 W 30o 20 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p.) Figure 30. Reflectance spectra of laboratory prepared mixtures (wt.%). 151 70 60 50 A z 040 B W30 1 U C 20 A - 10 B - 5% MnO + 5% malachite + 5% (hematite + goethite) + 65% kaolinite 25% MnO + 5% malachite + 20% (hematite + goethite) + 50% kaolinite 10 C - 25% MnO + 5% malachite + 50% (hematite + goethite) + 20% kaolinite 10 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p. Figure 31. Reflectance spectra of laboratory prepared mixtures (wt.%). 152 70 60 50 Z - B 40 W30 - 0 -oJ o1010 B - + goethite) + 50% kaolinite 25% MnO + 20% malachite + 20% (hematite + goethite) + 35% kaolinite C - 25% MnO + 20% malachite + 50% (hematite _ + goethite) + 5% kaolinite 10 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 ( p.) Figure 32. Reflectance spectra of laboratory prepared mixtures (wt.). 153 70 goethit6) + 20% kaolinite B 60 -- - 25% MnO + 50% malachite + 20% goethit ) + (hematite + 5% kaolinite W 50 z -40 B 0w LL 3 0 w 20 10 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (p) Figure 33. Reflectance spectra of laboratory prepared mixtures (wt.%). 154 60 50 w 0 40 - z H30 0 w L.L20 w 10 10 A - 50% MnO + 5% malachite + 20% (hematite + goethite) + 25% kaolinite B - 50% MnO + 20% malachite + 5% (hematite + goethite) + 25% kaolinite C - 50% MnO + 20% malachite + 20% (hematite kaolinite + go th~te) + 10f 10 L 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (.) Figure 34. Reflectance spectra of laboratory prepared mixtures (wt.%). A P P E N D I X I I I RATIO SPECTRA OF LABORATORY SIMULATED SOILS Figures 1-16: MnO , malachite, kaolinite, and goethite ra io spectra Figures 17-32: MnO 2 , malachite, kaolinite, and hematite ratio spectra Figures 33-48: MnO 2 , malachite, kaolinite, and (50% hematite + 50% goethite) ratio spectra Note: Each spectrum was scaled to unity at 0.87 p before ratioing. 155 5% MnO2 + 5% malachite + 40% kaolinite + 50% goethite 5% MnO 2 + 5% malachite + 70% kaolinite + 20% goethite 09 -I - (D ~* Os WAVELENGTH - 5% MnO 2 + 20% malachite + 25% kaolinite + 50% goethite 5% MnO 2 + 20% malachite + 55% ksolinite + 20% goethite - L1 -JJ (1 (D -GT In .- . -. . 5 2 3 05 -7 M S WAVELEN- H" e1 5% MnO2 + 5% malachite + 70% kaolinit.e. + 20% goethite 5% MnO 2 + 20% malachite + 55% kaolinite + 20% goethite Ln FJ. ~1 09 (D LA Is(A .150 CJ75 1A 15 WAVELENGTH - 1w MICRONS Ij7T5 eJoW "2 5% MnO 2 + 5% malachite + 40% kaolinite + 50% goethite 5% MnO 2 + 20% malachite + 25% kaolinite + 50% goethite r2 -4 Ufl . fia .r W.. WAVELENGTH - MICRONS e 's ' lse 1A 15% MnO 2 + 5% malachite + 60% kaolinite + 20% goethite 15% MnO2 + 5% malachite + 75% kaolinite + 5% goethite 0 (D Lu -5 'ise so0 ()7 ti75 ii5s iles WAVELENGTH - MICRONS 2)00 2)25 2250 15% MnO 2 + 5% malachite + 30% kaolinite + 50% goethite 15% MnO 2 + 5% malachite + 75% kaolinite + 5% goethite IC I . Z s Q1$0 t2TJ0 1i2 WAVELENGTH - ij MICRONS E) 1175~r a 50 us Mls N ps- 15% MnO 2 + 5% malachite + 30 % kaolinite + 50% goethite 15% MnO 2 + 5% malachite + 60 % kaolinite + 20% goethite 70 n VS7S o2 O tICe WAVELENGTH - MICRONS I7 Qles Las .' 50n 15% MnO 2 + 20% malachite + 15% kaolinite + 50% goethite 15% MnO 2 + 20% malachite + 45% kaolinite + 20% goethite IU, (ft M ast I'-S (D 00 -I. 0 -1 :~i )~s 9 9 se 075 0AE LE - I50 WAVELENGTH - MICRONS 1ji~J W ~ceCc. '50 15% MnO 2 + 5% malachite + 60% kaolinite + 20% goethite 15% MnO 2 + 20% malachite + 45% kaolinite + 20% goethite Ln P-e I4 om -- UjSe Q'75 tow0 ije5 WAVELENGTH - 1jbw MICRONS 1 a? I5:D Cifw I;e a. 2 15% MnO 2 + 20% malachite + 15% kaolinite + 50% goethite 15% MnO 2 + 5% malachite + 30% kaolinite + 50% goethite vi Zn (/ " S e Oise U7/5 1000 25 WAVELENGTH I.5 - MICRONS c) e w 7 es c5oA 50 ii 25% MnO 2 + 5% malachite + 50% kaolinite + 20% goethite 25% MnO 2 + 5% malachite + 65% kaolinite + 5% goethite U.' /1 afse es G)-75 0 u.25 WAVELENGTH - i3S J MICRONS - 2)J0 27e 2ise 25% MnO 2 + 5% malachite + 20% kaolinite. + 50% goethite -J 25% MnO 2 + 5% malachite + 65% kaolinite + 5% goethite ru UI u0 eI- sU, is am -1.1 61e U350 0175 100 125 WAVELENGTH - 1.150 MICRONS iJ75 270e j2es 2J50 25% MnO 2 + 5% malachite + 20% kaolinite + 50% goethite 25% MnO 2 + 5% malachite. + 50% kaolinite + 20% goethite Lfl U' Iljjjjrl 111 -4' ''111111till PolII! M6 MJ U' L, Lfl4 sS 61 Gi Qf'75 t 00 U bw U25 WAVELENGTH - MICRONS c caca ewe 1 J to .o 150 SI S -J 25% MnO 2 + 20% malachite + 5% kaolinite + 50% goethite I 25% MnO 2 + 20% malachite + 35% kaolinite + 20% goethite 'Ji I U1 Ul I U725 iWo ie25 150 WAVELENGTH - MICRONS .-- 1 ~ ~ ~ec ~ '53 c C fAewe 25% MnO + 5% malachite + 50% kaolinite + 20% goethite 2* 25% MnO + 20% malachite + 35% kaolinite + 20% goethite 2 -I. -J ~DISW Djso 0)75 1,J75 1)50 1)00 iNTHW7 i)~5 ies iCR0ON 0)7s WAVELENGTH - MICRONS - - a)0s~eia~~a~50 ejoe ej25 215so 25% MnO 2 + 20% malachite + 5% kaolinite + 50% goethite 25% MnO 2 + 5% Lfl malachite + 20% kaolinite + 50% goethite Lfl M 00 I(I) H a' t % Mno 2 + 5% malachite + 40% kaolinite + 50% hematite 5% MnO 2 + 5% malachite + 70% kaolinite + 20% hematite -4.. In L'I $ZWAVELENGTH - MICRONS 1w 6fQ 5% MnO 2 + 20% malachite + 55% kaolinite + 2G% hematite 5% MnO 2 + 50% malachite + 25% kaolinite + 50% hematite LI! -I. CD co L1 ga (Gj7S t00 1)25 WAVELENGTH - L50 MICRONS J.75 LJWW c) ears 50 owl 5% MnO 2 + 5% malachite + 70% kaolinite + 20% hematite 5% MnO2 + 20% malachite + 55% kaolinite + 20% hematite !1 4- ru Ln - aI do UYse 0P7S t0 tie5 I050 WAVELENGTH - MICRONS i7s 2Je 25es 21s 5% MnO 2 + 5% malachite + 40% kaolinite + 50% hematite 5% MnO 2 +20% malachite. + 25% kaolinite + 50% hematite ~J1 -4. S dl S - U' S S S ) -m U)s@ 0. 75 ' 1)00 1)2J WAVELENGTH - 1e5 MICRONS 1 /5 eI w4eD 22b 2.5S 15% MnO 2 + 5% malachite + 60f kaolinite + 20% hematite 15% MnO 2 + 5% malachite + 75% kaolinite + 5% hematite Lfr Lu &I 0 50 0 -75 i) 1MCt!S 1N25 WAIVELENG'fH - M'ICIRN3 I1 /b 2 2J25 1 15% MnO 2 + 5% malachite + 30% kaolinite + 50% hematite 15% MnO 2 + 5% malachite + 75% kaolinite + 5% hematite Q~q -4. 09 )S O2-7S 00 T.ON 7n ) )I$~ 11w 11 15% MnO 2 + 5% malachite + 60% kaolinite + 20% hematite 15% MnO 2 + 5% malachit-e + 30% kaolinite + 50% hematite n Pu L' 4. a& Sn - W25 ~ i:~~~~~ WAVELENGTH - MICRONS irAW.L iJ - 15% MnO + 20% malachite + 15% kaolinite + 50% hematite 15% MnO + 20% malachite + 45% kaolinite + 20% hematite I-s CD R) 4:::- C-4WAVELENG'TH Wres 0[15L - 02~7C --MICRONS Z 500 15% MnO, + 5% malachite + 60% kaolinite + 20% hematite 15% MnO 2 + 20% malachite + 45% kaolinite + 20% hematite LU U (b) so 0)-75 10 L25 MICbN 5 WAVELENGTH - MICRONS IZ .we eC s eCI 1f/i se 15% MnO2 + 20% malachite 15% kaolinite + 50% hematite -J 15% MnO2 + 5% malachite + 30% kaolinite + 50% hematite In C!' CD R) 0)SG &'7S 1)75 I)~0 I)~S 1)00 WA VEL5 t15 s o t. 25 i j 75 WAIVELENGTH - MICRONS 2)00 2joe 2)25 2j25 2.~50 2.'50 25% MnO 2 + 5% malachite + 50% kaolinite + 20% hematite 's Lfl2 25% MnO 2 + 5% malachite + 65% kaolinite + 5% hematite ru l'! ~wv~'V~ IWAVE0LEN7GS WAJVELENG'TH EwJ - M)IoN -j~s MICRONS Id~~~~~~~ .W ;..J L. A. 25% MnO 2 + 5% malachite + 20% kaolinite + 50% hematite ' 25% MnO 2 + 5% malachit.e + 65% kaolinite + 5% hematite 00 r\M WAVE.LENGTH - MIICRONS 25% MnO 2 + 5% malachite + 20% kaolinite + 50% hematite 25% MnO 2 + 5% malachite + 50% kaolinite + 20% hematite rx) -j )sO no i)es WAVELENGTH - -Jsbi MICRONS ls i l e e -- - 'es e - se 25% MnO 2 + 20% malachite + 35% kaolinite + 20% hematite 25% Mn O 2 + 20% malachite + 5% kaolinite + 50% hematite W ru U1 l U1 ru Ul 25 25 0.150 /b e025 ow 1V .TH - 1ICRON WAVELENGTH - MICRONS 1 1i.1 53 C i 2 0 W 2 ! 2-C 2In W 50 25% MnO 2 + 20% malachite + 35% kaolinite + 20% hematite 25% MnO 2 + 5 % malachite + 50% kaolirite + 20% hematite , . lv"V , v 111, 0 ,JFA l,'111,1111, ,, 111111 111"111,11,11111 CD w Le*i ,_j eYse e2s i)00 tjes i0se WAVELENGTH - MICRONS 13/b i.,ws e1 e-e Z> .s ' W 25% MnO 2 + 5% malachite + 20% kaolinite + 50% hematite 25% MnO2 + 20% malachi-te + 5% kaolinite + 50% hematite en Ln (n IS- Lfl -A 0.So 0)25 1j00 iies WAVELENGTH - iise MICRONS t 12/b0 e2es5 els.,e i.,b 5% MnO2 + 5% malachite + 40% kaolinite + 50% ' (hematite + goethite) 5% MnOn2 + 5% malachite + 70% kaolinite + 20% (hematite + goethite) U9 11 0 ru -4 Lt U,1 WAVELENGTH - MICRONS i)7S' 2je eis 2 5% MnO2 + 20% malachite + 25% kaolinite + 50% (hematite + goethite) 5% MnO2 + 20% malachite + 55% kaolinite + 20% (hematite + goethite) -4 LI E; U, 49 V S2 1 Hes t 150 i AV0L0(DO Sj25 WAVELENGTH - MICRONS 2joo 2ej)2?5 250 5% MnO2 + 20% malachite + 55% kaolinite + 20% (hematite + goethite) 5% MnO2 + -J 5% malachite + 70% kaolinite + 20% (hematite + goethite) (-1 Ul ....... ....................... ............ LI. QI U1 S ,I f- 0)54 0)2 S1i25 1*7 MICo 5 i j275 WAVELENGTH - MICRONS 23 2j25 2150 5% MnO2 + 20% malachite + 25% kaolinite + 50% (hematite + goethite) 5% MnO 2 + 5% malachite + 40% kaolinite + 50% (hematite + goethite) Ul A (A -J S in U1 S I S Tys 7T3W5 i25 WAVELENGTH - i5s MICRONS ' ee i~~~~ 3 5 5 M 2 53 15% MnO2 + 2 15% NnO 2+ 5% malachite + 60% kaolinite + 20% 5% malachite + 75% kaolinite + (hematite + goethite) 5% (hematite + goettehite) Lfl LI, 1j7s WAVELENG'fH - NICRONS 2) 00 2j2s 2.150 15% MnO2 + 5% malachite + 30% kaolinite + 50% (hematite + goethite) 15% MnO 2 + 5% malachite + 75% kaolinite + 5% (hematite + goethite) rk) 1-'09 (D w 4.' co L9~ (YR )$ s -)2S i)Vo 5 1,IIis I25 WAnVELENGTrH - MICRONS in2 27o 2j25 2.'s 15% MnO 2 + 5% malachite + 30% kaolinite + 50% (hematite + goeth te) 15% MnO2 + 5% malachite + 60% kaolinite + 20% (hematite + goethite) EQ 4 Li, *1 2-S--5- 5 1"S 12 50 I j15O e WAVELENGTH - MICRONS i j75 e1o 2)2s 2.50 15% MnO 2 + 20% malachite + 15% kaolinite + 50% (hematite + goethite) 15% MnO 2 + 20% malachite + 45% kaolinite + 20% (hematite + goethite) Ui C) U' S 0 -~7 t)SO 12S WAVELENGrH - MICRONS i7S e)2 o 2j2S 2si (n 15% MnO 2 15% NnO2 15% MnO 2 5% malachite + 60% kaolinite + 20% (hematite + goethite) 5% malachite + 60% kaolinite + 20% (hematite + goethite) + 20% malachite + 45% kaolinite + 20% (hematite + goethite) r- LI Lii ,) so 25 1.100 1 WAVELENGTH 1 .5 J25 i - MICRONS 1-' 1aw /1 C ei c. C50 15% MnO2 + 5% malachite + 30% kaolinite + 50% (hematite + goethite) i 15% MnO 2 + 20% malachite + 15% kaolinite + 50% (hematite + goethite) Lin Ul Ln H. CD PU - 1 w WAVELENGTH - MICRONS ~ ~ ~ aas ~ E5 Z vi5E~e 25% MnO 2 + 5% malachite + 50% kaolinite + 20% (hematite + goethite) 25% 'MnO2 + 5% malachite + 65% kaolinite + 5% (hematite + goethite) 4 i 0, 250 Q WAVELENGTH - M 25% MnO2 + 5% malachite + 20% kaolinite + 50% 25% MnO 2 + 5% malachite + 65% kaolinite + (hematite + goethite) 5% (hematite + goethite) L9 ro) 09 (D L-4 Ln F-Y75s 1 EG(H125 r-1IC WAVELENGTH - MICRONS I. I /s w. C *m ' eww c CA 25% MnO + 5% malachite + 20% kaolinite + 50% (hematite + goethite) 25% MnO 2 + 5% malachite + 50% kaolinite + 20% (hematite + goethite) L-e ~:ij I-i. CD '31 -- j V1 1/2 .e:: 0t25 0)5 iL 115C O Ei25 WAVELENGTH - MICRONS ' i El j m5 i:JeCA 0 ful 25% MnO + 20% malachite + 2 5% kaolinite + 50% (hematite + goet ite) 25% MnO + 20% malachite + 35% kaolinite + 20% 2 (hematite + goethite) CD 0~\ Ln )nSO s r62 fb '7S Ges tiso WfVELENGTH -,MICRONS if2s 2J)O ej2S 2.'so 25% MnO2 + - 5% malachite + 50% kaolinite + 20% 25% MnO2 + 20% malachite + 5% kaolinite + 20% (hematite + goethite) (hematite + goethite) (.n U51 -4 CD U5ln :D ... ... ~ Gy)5 Q)-75 ij00 tj25 L!)5Q WfWELENGTH - MICRONS - -L I75 e wa a -- 50 25% MnO2 + 20% malachite + 25% MnO 2 + 5% kaolinite + 50% (hematite + goethite) 5% malachite + 20% kaolinite + 50% (henatite + goethite) , "111", ICD X=: Co (il - is (21b) IiV(bbHjes jICo i)2S WAVELENGTH - MICRONS 2JO0 2)ls 2lse A P P E N D I X I V REFLECTANCE SPECTRA OF NATURALLY OCCURRING SOIL SAMPLES Figure 1 : Mineral Park soil sample spectra Figures 2-6: Silver Bell soil sample spectra Figure 7 : Silver Bell rock chip sample spectra 204 205 50 40 LU 30 Z 20 LL. I0 I0 \0 |0 0 0.5 1.0 1.5 2.0 2.5 WAVELENGTH (L) Figure 1. samples. Reflectance spectra of the MIneral Park soil 206 50 40 2 LU 3 0 3 20 4 uio -J 10 LL -Ljj 1 - SS 1 10 2 - SS 2 3 - SS 3 0* 4 10 10 - SS 4 - 0 0.5 1.0 1 .5 WAVELENGTH Figure 2. Reflectance of Silver Bell 2.0 (a) soils. 2.5 207 50 5 40 6 zS30|020 -J LL 8 1 O' 10 5 - SS 5 - SS 6 _6 - SS 7 -07 8 - ss 8 10 - 10 0 0.5 1 .0 1.5 WAVELENGTH Figure 3. 2.0 (p) Reflectance spectra of Silver Bell soils. 2.5 208 50 40 z 030 20 uL 10 10 9---9 1-20 10 10 - SS 10 11 - SS 11 12 SS 12 - 200- -0.5 1.0 1.5 WAVELENGTH Fifgure 4. 2.0 (p) Reflectance spectra of Silver Bell soils. 2.5 209 50 40 U Z 30 20 U_j) 10 10 13 - SS 13 0 14 - SS 14 10 15 - SS 15 16 - SS 16 10 0 0.5 1.0 1.5 WAVELENGTH 2.0 2.5 (pL) Figure_5. Reflectance spectra of Silver Bell soils. 210 50 -40 18 - UJ 30 2 <.. 20 -9 - 20 LU LU 10 17 - SS 17 10 18 - SS 18 19 - SS 19 20 SS -- 20 I0 - 0 0.5 1.0 1.5 WAVELENGTH Figure 6. 2.0 2.5 (u) Reflectance spectra of Silver Bell soils. 211 60 50 40 LUJ Q z 30 -2 LU LOft 10 A - GOC 43 B - GOC 78 C - GOC 44 IN0 E - GOC 49 10 10 0 0.5 1 .0 O.5 WAVELENGTH Figr 2.0 2.5 (L) . Reflectance spectra of Silver Bell rock chips. A P P E N D I X V RATIO SPECTRA OF NATURALLY OCCURRING SOIL SAMPLES Figures 1-4: Mineral Park soil sample ratio spectra Figures 5-28: Silver Bell soil sample ratio spectra Note: Each spectrum was scaled to unity at 0.87 p before ratioing. 212 AC13 / AC46 I'll - Q0 0)25 1)1010 i25 WAVELENGTH i )50 - MICRONS i )-7S 2)00 2)25 2.150 DF13 / DF46 1 1,10 111, 0 ru Gaes t)7S WAVELENGTH - MICRONS 2 0 ees 2Jso DF13 / 12 0.Se s AC46 U)-75 l)eo 0)~7) 12 WAVELENGTH - MICRONS 200 2)27 2j50 AC13 / DF46 UDE S- f3 '/ l wbw -41r 0.2-b t w 1. j: Z) 1 0 I WAVELENGTH - MICRONS I I2 c- Ijw 2)251 !5 50 SS1 / SS2 I.~~1. I-s CD f- ro L - A5 VE25NG) 2 WAVELENGTH MRN -MC O 1125 2109 2j25 2.)50 II i~. ~ I 1.25 I 1.50 I 1.25 Figure 6 I 1.00 RATIO 0.75 0.50 9.25 218 to z r- 0 Lf9 L..J Rit 8S5 / ss8 (ll s.eb ka. lb 1 je -L g WAVELENGTH - I MICRONS 4- tA p h Pips 2s so se SS8 / SS6 -J LFI u Ln ;co -4. -J - t 1 WAVELENGTH - MICRONS 75 .'Es )5 fU S SS11 / .. SS10 01 U1 00 \D OS SU, S U1 --- -j - - - 0)500S25 0100 t 25 WAVELENGTH - t se MICRONS I. Z i /s ww G~ Q t c A 5 t0f SS11 / SS12 -". Ul U1 L, .F S IS - o)25 Oj i) tj5 WAVELENGTH - 1iJs IS0 1 )25 MICRONS w e-. 2/25 2 5b0.,Z ful ;Q IS3 SS13 / SS9 S- Ul Ln Q.sbibL2G 100 U 25s t1 b50 WAVELENGTH - MICRONS I J 1 /5 2.00 2.125 2.150 SS14 / SS12 U1 ru - 61 - ru (A gjg Us -soS f 1GTes 00) WAVELENGTH - MICRONS ins e ej )eis 2es 8S19 / ss16 S ~ 225 0)50 0)5 I., e i5 so WAVELENGTH - MICRONS U /b 2j75.1 SS19 / SS20 -J U, U, U, H' 2' CD 4:::- -J U, S - 'I, S ,-I-- SO -00 IC5o V25 1.25 WAVELENGTH - MICRONS I, - / Cw ) 2)5 C,2 SS18 / SS12 m4~ lo11 11,"All 11,111 116 1,11111110 , 4l,lilill 3>1 MIVELENG TH - MICRONS SS11 / SS2 (D 0\ l G- cn U1' II 0s5 (b2 U00 V-7Stjes 1iiCo 1I7 WAVELENGTH - MICRONS 2y0 2 25 -2'50 SS16 / SS2 -I (ft Li~ (ft -4 W6VELENGTH - MICRONS I P7S 2joo 2j~s ~2.is SS20 / SS2 Ul Ln I Ln 4v "All''P, 11F'll 101111,1%111 Ln 4- ro WAVELENGTH - MICRONS ins eles 25 e.es se Ss3 / Ss6 -J ru L S Ul -.- U1 Ul ru J2s e~uso 0.75 1)00 1.M3s WAVELENGTH - iise MICRONS i j-s easo 2.12s e.so SS4 / SS6 r- <n L1q CD -450 ED a U, -J L9 isojso 0)7S 100J tjs t-so NS0 iMCRO WAVELENGTH - MICRONS 2 e.so SS7 / SS6 01 Cr' Ul, L2 Ul ru .25 s I25 02~55 t) 5 NS M25 9!;;lWAVELENGTH - MICRONS s SS7 / SS8 U -i 'A i f-f, ru Uo p- I1 A . 1 )CI iz L 0 AI lj/b WAVELENGTH I. /b - MICRONS 2.100 2j~s -2.1s Vn SS7 / SS10 D- U' !i, S - da Su 0 WAVELENGTH - 1)so ----MICRIONS 7 S 2)oo 2j2S .S SS15 / SS16 -j U, U, S -4.. (D -Jq UX= IQ 1 i WAVELENGTH - MICRONS )?5 Ejs ~gg E25 S SS15 / SS17 Ul m (Jl - ru n pls qi A 1 1 . 0 1 1 )5 WAVELENGTH - 15 MICRONS - --- -------- SS18 / SS17 S U1 U' 1-1PL 015 02S 1 go i.2s WAVELENGTH - I MICRONS 1Utb i:'Jiow 2125 ~250 2150 SS2 / SS9 (f 09 (D fr> Lo ru - s s ibeIn -- i !es MICRONS WAVELENGTH - MICRONS I.sns eJOT 2s 2 eEs SS2 / SS15 -L c0 00 (f? SI .!e 0j Sib a !-7c: 4i ICA~ WAVELENGTH "A 41c - MICRONS n