Effects of cell growth and a mobile genetic element on propagation of the phages SP16
and SP-beta in Bacillus subtilis
by
Eleina (Helen) M. England
B.A. Biology
Columbia College, 2010
SUBMITTED TO THE DEPARTMENT OF BIOLOGICAL SCIENCES IN PARTIAL
FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
MASTERS OF SCIENCE IN BIOLOGY
AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
FEBRUARY, 2014
© 2014 Eleina (Helen) M. England. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly
paper and electronic copies of this thesis document in whole or in part in any medium
now known or hereafter created.
Signature of Author: ___________________________________________________________
Department of Biology
December 9, 2014
Certified by: __________________________________________________________________
Alan D. Grossman
Professor of Biology
Thesis Supervisor
Accepted by: _________________________________________________________________
Stephen P. Bell
Professor of Biology
Chair of Committee for Graduate Students
1 Effects of cell growth and a mobile genetic element on propagation of the phages SP16
and SP-beta in Bacillus subtilis
by
Eleina (Helen) M. England
Submitted to the Department of Biology
on December, 2014 in Partial Fulfillment of the
Requirements for the Degree of Master of Science in
Biology
ABSTRACT
Two studies were performed on Bacillus subtilis phages SP16 and SP-beta to characterize
the effects of growth and an integrative conjugative element, ICEBs1, respectively. I
found that B. subtilis strains are significantly more susceptible to SP16 infection early in
the growth phase and less susceptible during exponential growth and as cells approach
stationary phase. SP16 makes plaques of different sizes. I found that this variegated
plaque size did not seem to breed true. That is, phage from large plaques gave rise to
both large and small plaques. On the other hand, phage were difficult to recover from
small plaques, indicating that the phage were either not viable or gave rise to small
plaques that were barely visible. A second study analyzed growth of SP-beta and
established the time required for a single round of phage growth. I verified that
presence of ICEBs1 in the B. subtilis chromosome, prevented lytic production of SP-beta.
I also verified that this phenotype was due to a single gene in ICEBs1.
Thesis Supervisor: Alan D. Grossman
Title: Professor of Biology
2 Introduction
Prokaryotes have the ability to transfer genetic material via mechanisms of
horizontal gene transfer: namely transformation (acceptance of naked environmental
DNA), transduction (infection by bacteriophage), and conjugation (cell-to-cell transfer
of DNA [reviewed in Thomas, 2005]. This ability of bacterial species to take up foreign
DNA from the environment can confer incredible advantages for a host population.
New genes, encoding potentially useful functions may provide a bacterial cell with just
the novel capacity it needs to survive in a challenging environment. However, the
introduction of foreign DNA may not always be beneficial to a host—as exemplified by
the bacteriophage, which, upon injecting its genome into a susceptible bacterium, can
initiate a series of events that lead to the hosts’ destruction.
Bacillus subtilis is a common gram positive bacteria found in soil and, more recently,
the human gut [Madigan, 2005; Huynh, 2009]. In these natural settings B. subtilis comes
in contact with many bacteriophages with different physical characteristics, modes of
infection, and lifestyles inside the host. Just like other bacterial species, B. subtilis copes
with dangerous, pathogenic phage by employing life-saving counter-responses to
infections. The descriptions that follow detail preliminary work to characterize putative
defensive strategies used by B. subtilis to deal with two phage: SP16 and SPß.
Bacteriophage SP16
SP16 was first isolated and described in 1972 [Mele, 1972]. Its broad host range—able
to infect a range of Bacilli beyond B. subtilis—made it a subject of interest to those
seeking to develop a generalized transducing agent in B. subtilis with which to conduct
molecular and genetic experiments. This broad host range possibly reflected the unique
conditions under which SP16 was discovered and cultivated (acidic conditions, using
3 strain W23 as a host, rather than the classic 168) compared to other B. subtilis phage
[Dean, 1978]. SP16 is the only known member of the group IV B. subtilis temperate
phages. It has a double stranded linear genome of ~60 kb, whose ends are terminally
redundant and circularly permuted [Parker, 1986]. When investigators realized the SP16
would not be a useful genetic tool, exploration of the biology of SP16 largely came to a
halt.
Bacteriophage SPß
SPß is a temperate phage that is found integrated in the chromosome of many B.
subtilis strains. The phage can be induced to form viable infective particles. Like SP16
and most other B. subtilis phage, the phage DNA is linear and double-stranded [Fink,
2006]. B. subtilis phage are classified into five groups based on immunological, physical,
and host-range characteristics [Dean, 1976; Wilson, 1974; Fink, 2006]. According to this
categorization, SPß is included in group III: Siphoviridae phages with long,
noncontractile tails. The SPß genome is 134,416 bp with a G+C content of 34.6 mol%
[Lazarevic, 1998], significantly less than that of the B. subtilis genome (43.5 mol%). This
difference in GC content is characteristic of many horizontally acquired elements. SPß
has 187 putative open reading frames (ORFs), many encoding short peptides (<100
amino acids long) with unknown functions. These ORFs can be divided into functional
clusters that represent genes likely expressed during the early, mid-, and late points of
the SPß lytic lifecycle [Zuber, 2001]. A study utilizing spontaneously generated, viable
deletion mutants of SPß was able to crudely define these regions and show,
importantly, that transcription of SPß genes during lytic growth follow a regular,
chronological pattern (thus, “early” and “late” genes) [Spancake, 1985]. The attachment
site for SPß in the B. subtilis chromosome falls near the terminus of DNA replication.
4 Interestingly, this region, between ilvA and gltA genes, contains the attachment sites for
several other temperate B. subtilis phage, possibly reflecting the origin of the sequence
as ancient prophage DNA [Zahler, 1977; Regamey, 2000].
ICEBs1
Along with the SPß prophage, many strains of B. subtilis contain the ~20kb
integrative and conjugative element, ICEBs1 (Fig. 1) [Burrus, 2002; Auchtung, 2005;
Goranov, 2006; Auchtung, 2007]. Normally found integrated in the host chromosome,
the global DNA damage response and an intercellular peptide signaling mechanism can
activate excision and transfer of ICEBs1. Induction can be experimentally initiated in
more than 90% of cells in a population. ICEBs1 is easily manipulated and therefore very
experimentally tractable. Additionally, the genes controlling ICEBs1’s regulation and
integration are homologous to systems found in many phages, serving as potential
models for other horizontal gene transfer systems. Until recently, the only phenotype
thought to be conferred by ICEBs1 is a reduced ability to acquire an additional copy of
ICEBs1 [Burrus, 2002; Auchtung, 2007].
Fig 1. (Reproduced from Auchtung, 2005) Organization of ICEBs1 mobile genetic element.
ORFs are indicated as arrows with names above. Black boxes indicate attachment site
sequences. immR encodes the putative repressor; rapI and phrI encode genes involved in
intercellular peptide signaling for excision and transfer. yddK is indicated to the right of rapI.
5 ICEBs1 and SPß
Recently, Christopher Johnson (Grossman lab) identified a new phenotype conferred
by ICEBs1 in the host: namely, suppression of SPß lytic growth in the host. The presence
of ICEBs1 in the host chromosome inhibits lytic infection by exogenous SPß phage, but
does not inhibit lysogenization. Additionally, presence of ICEBs1 in an SPß lysogen
prevents lytic growth following induction of the phage. The ICEBs1 gene yddK is
needed for ICEBs1 to prevent lytic growth of SPß. Moreover, expression of yddK, in the
absence of any other ICEBs1 gene, is sufficient to prevent lytic growth of SPß (C.
Johnson, ADG, unpublished results). yddK is not required for ICEBs1 conjugation, nor
are other functions known for this gene. A psi- BLAST analysis of the yddK sequence
shows that it contains a Toll/interleukin-1 receptor (TIR) domain. Understanding the
function of this domain may be useful for understanding the mechanism of yddK’s
interaction with SPß.
TIR domains
TIR domains (reviewed in Spear, 2009) were initially identified in the cytoplasmic
domains of Toll receptors of Drosophila and the mammalian interleukin-1 receptor [Gay,
1991; Sims, 1988]. In both organisms, the domains function in immune response
signaling receptors [Lemaitre, 1996; Medzhitov, 1997]. These receptors share
homologous TIR domains with the intracellular proteins involved in binding to such
receptors and propagating appropriate signaling cascades [O’Neill, 2007]. TIR domains
have subsequently been found to be widespread in disparate multicellular organisms,
from sea urchins to plants [Rast, 2006; Burch-Smith, 2007]. In plants, as in mammals and
Drosophila, they have been implicated in pathogen-recognition proteins [Burch-Smith,
2007].
Because of the independent evolutionary paths of plants and mammals,
6 however, it is likely that these functions of TIR domains also arose independently
[Rokas, 2008; Spear, 2009].
A bioinformatic study identified >200 bacterial TIR domains [Newman, 2006]. They
also demonstrated that one of these proteins, TlpA, in Salmonella enterica could subvert
immune signaling when expressed in eukaryotic cytoplasm [Newman, 2006]. Other
studies of bacterial TIR proteins from Brucella and E. coli have suggested similar
mechanisms of ‘subversion,’ in which a bacterial cell can secrete TIR-containing
proteins that act as competitive inhibitors for the immune signaling cascade mediated
by TIR-TIR protein-protein interactions in hosts [Cirl, 2008]. However, many questions
about such potential mechanisms for bacterial TIR proteins remain unresolved.
Moreover, bacterial TIR domains have been found in both pathogenic and nonpathogenic bacteria and, within a given pathogenic strain of bacteria (from the same
physical isolate), some cells may possess a TIR protein that others lack [Spear, 2009].
Thus, although there are some clues as to what the function of yddK’s TIR domain might
be (and whether this is important for its interaction with SPß), the full range of
possibilities for this domain’s function is wide.
Abortive Infection Mechanisms in Bacterial Hosts
Bacteria have evolved many different mechanisms to avert destruction by phage
both at the single cell and the population levels [reviewed in Labrie, 2010]. As an
example of the former, bacterial hosts may block phage infection at their surfaces by
mutating receptors used by phage to enter the cell [Beler, 2008]. If a phage succeeds at
injecting its DNA into a cell, a bacterium possessing CRISPR-Cas system, restriction
modification system, or phage-encoded repressor can recognize and neutralize foreign
DNA and thereby survive the attack [Barrangou, 2007]. If the phage is capable of
7 beginning its cycle of lytic infection and replication, the bacterium does not have the
option of clearing the infectious virion without sacrificing itself; however, if a cell can
interrupt the process of phage replication it can minimize the number of phage
released, thereby saving other bacteria in the surrounding population from destruction
[Labrie, 2010]. This last mechanism, known as abortive infection (Abi), is widespread in
bacteria, having been found in a diverse range of species [Snyder, 1995; Forde, 1999;
Hemphill, 1975; Tran, 1999; Smith, 1969; Behnke, 1978; Chowdhury, 1969].
Much work has been done on L. lactis Abi's (for a review: Chopin, 2005) because of
the importance of L. lactis for commercial production of dairy products and the
associated economic threat of phage predation on these bacteria [Chopin, 2005]. Taking
together the information collected from the at least 23 Abi systems found in lactococci,
several patterns emerge about how these systems function. Nearly all lactococcal Abi
systems are encoded by conjugative plasmids. Most Abis function via a single gene,
with several exceptions requiring two or three genes. There is little similarity between
these proteins, and their functions and those of their homologs are, for the most part,
unknown [Chopin, 2005].
The question of how Abi's interfere with phage production is made more confusing
by the fact that phage differ so drastically in DNA sequence and proteins [Chopin,
2001]. Evidence indicates that Abis can target both conserved protein/DNA sequences
in phage or nonhomologous phage proteins that have convergent functions [Chopin,
2005; Curtis, 2005; Bidnenko, 1995; Bouchard, 2004]. Similarly, the number of phages an
Abi is active against varies from highly specific (only one phage) to more broad
[Chopin, 2005]. Looking at broad phenotypic effects on phage production in lactococci,
Abis may delay, arrest, or have no effect on DNA replication [Chopin, 2005]. They may
also cause decay or destabilization of phage transcripts, delay transcription, or block
8 regulation of transcription (disabling early transcripts from being turned off when they
are no longer needed).
The most informative method for identifying phage targets of Abis in lactococci has
been through analyses of spontaneous phage mutants that are resistant to an Abi
[Chopin, 2005; Bouchard, 2002; Bouchard, 2000; Bouchard, 2004; Dinsmore, 1994;
Dinsmore, 1997; Moineau, 1994]. These methods have proven to be more informative
than biochemical experiments because the physiological effects of Abi systems can be
widespread. For example, one can imagine a scenario in which an Abi whose function is
to block the function of an endonuclease may have the primary effect of halting
transcription of phage genes but may subsequently also block replication of the phage
genome. This information should bear on the kind of experiments it makes sense to
conduct in Bacillus Abi systems.
Several Abis have been studied in more mechanistic detail in E. coli [Molineaux,
1991; Snyder, 1995; Kaufmann, 2000; Bingham, 2000]. The Rex Abi is encoded by phage
lambda [Molineaux, 1991; Snyder, 1995]. When a foreign phage infects its host, Rex can
reduce the cell’s membrane potential and deplete the pool of available energy (ATP) for
phage replication. Two other Abis found in E. coli, Lit and PrrC, act by inhibiting
different aspects of translation, thus blocking the production of phage proteins
[Molineaux, 1991; Snyder, 1995; Kaufmann, 2000; Bingham, 2000].
9 RESULTS
I. SP16
Introduction
This study was motivated by observations made by Tyler DeWitt (Grossman lab)
that the B. subtilis integrative conjugative element, ICEBs1, may confer immunity to host
infection by SP16 phage. In preliminary experiments, it appeared that the presence of
ICEBs1 might inhibit the ability of SP16 to form plaques (TDW, ADG, unpublished).
The aim of my experiments was to characterize and optimize conditions for SP16
infection, so as to better determine if there were any differences between strains that did
or did not contain ICEBs1. I found that the efficiency of SP16 infection and growth
depends on the growth state of the host. Controlling for differences in growth state
between hosts revealed that ICEBs1 did not detectably confer any resistance to SP16
infection. Additionally, observed variegation in SP16 plaque morphology and size is
addressed.
Methods
Strains
Strains JMA222 (trpC2, pheA1, ICEBs10) and AG174 (trpC2, pheA1), were used as
isogenic ICEBs10 and ICEBs1+ strains, respectively. These strains are lysogenic for SPß.
PY79, the prototrophic derivative of B. subtilis 168, was used as an SPß-defective ICEhost strain for assays of plaque purified phage sources.
Media
Phage assay (PA) broth and agar were used for all dilutions of phage and indicator
strain (except when spores were being harvested), during phage infection. PA broth
10 consisted of, per liter, 8 g of Difco nutrient broth, 5 g of NaCl, 0.2 g of MgSO4·7H2O, 0.05
of MnSO4·H2O, 2.15 of CaCl2, diluted with distilled water [Vennison, 2009]. To make
1.5% or 0.7% soft agar, the appropriate amount of agar was added to the same broth
base.
Difco sporulation medium (DSM; per liter, 8 g of nutrient broth, 10 ml of 10% KCl,
10 ml of 1.2% MgSO4·7H2O, 0.50 ml of 1 M NaOH, 1.0 ml of 1 M Ca(NO3)2, 1.0 ml of
0.010 M MnCl2, 1.0 ml of 1 mM FeSO4) [Zhou, 2002] was used to generate spores from
starter colonies of B. subtilis strains.
Spizizen minimal salts (per liter, 14.8 g of K2HPO4, 5.4 g of KH2PO4, 2 g of (NH4)2SO4,
1.9 g of tri-sodium citrate, 0.2 g of MgSO4·7H2O) [Anagnostopoulos, 1961] was used to
wash and store pellets of spores after spore generation through overnight incubation in
DSM sporulation medium.
SP16 infection
Plaque forming assays (based on Adams, 1959) were used to assess the number of
viable plaque forming units of phage on a given indicator strain. Indicator strains were
grown in liquid LB cultures from single colonies on freshly streaked LB-agar plates or
light lawns to mid-exponential growth phase. Approximately 2-3 x 108 cells in 300 ul
were combined with various dilutions of 100 ul phage stocks and heated at 37°C to
allow for phage adsorption to cell surfaces. Phage-cell mixtures were plated by adding
molten PA soft agar and overlaying this mixture onto PA-agar plates. These plates were
incubated at 37°C overnight; plaques were counted the following day.
11 Spore generation and purification
In some experiments, germinating spores were used to assay for phage growth and
production. Initiating spore germination in the presence of phage allowed for phage
infection of different strains under virtually identical stages of growth and facilitated
direct comparisons between strains while minimizing possible differences due to
differences in growth stage.
Spores were made by inoculating DSM sporulation
medium with single colonies. Cultures were heated to inactivate surviving vegetative
cells. Sporulation efficiency was calculated by plating cultures before and after heat
treatment. Spores were purified using a Renografin column. Briefly, unpurified spore
cultures were pelleted by centrifugation, resuspended in the column, and spun to pellet
spores. Cell debris was collected from the supernatant and disposed. Final pellets
contained pure spores, which were washed and then stored 1x Spizizin minimal salts.
Plaque purification
In order to determine whether SP16 heterogeneity was an inherent characteristic of
the phage, plaques were purified and used as new phage sources for a subsequent
infection. Briefly, plaques of different sizes were picked from a plate containing SP16
plaques generated on PY79 indicator strain. These agar ‘plugs’ were mixed with single
colonies of PY79 in PA broth, and allowed to grow to stationary phase. Mitomycin C
was added to induce any indicator cells that had been lysogenized by SP16. Cultures
were then centrifuged and pellets discarded. The supernatant, containing the plaquepurified phage stock, was syringe filtered and used the following day as phage sources
to infect PY79 indicator strains in the standard agar overlay assay.
12 Quantification of plaque sizes
Tiff images of similar size and magnification were imported into the NIH ImageJ
software. These images were converted to 8-bit, binary images. The “analyze particles”
tool was used to assess the pixel area of plaques on each of four images. The
distribution of plaque sizes was plotted in Matlab as histograms.
Results
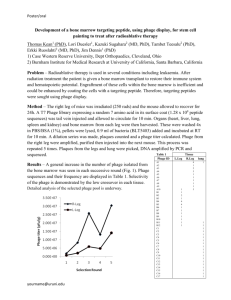
Growth phase of host affects efficiency of plaque formation
My initial experiments comparing infection on a single host (JMA222, ICEBs10) at
various points in growth phase (as measured by OD600; a light scattering measure of
culture density) indicated that fewer plaques would form on hosts isolated later in
growth phase (Fig. 2). Thus, any differences observed between ICEBs1+ and ICEBs10
strains infected with SP16 could have been due to the differences in when the host
strain was isolated in culture, prior to infection. To monitor how the strains varied as
indicator strains for SP16 infections over a range of OD600 values, I did a time-course
infection for isogenic strains: AG174 (ICEBs1+) and JMA222 (ICEBs10), isolating the
cultures at multiple points in growth and performing the standard soft agar overlay
phage infection. The number of indicator cells of either strain was kept constant for each
time point.
The results of this experiment confirmed my hypothesis that infection efficiency
depends highly on the growth phase of either host. Between early and late exponential
growth phases, infection efficiency for either strain varied by over an order of
magnitude. Moreover, the paths of either strain seemed to follow nearly parallel
trajectories. Notably, the ICEBs10 strain, JMA222, appeared moderately sensitized early
in exponential phase, which raised the question of whether differences in immunity
13 might only be seen when cells are just beginning to reach exponential growth (when
plaque formation is most efficient).
OD600
0.234
0.752
1.44
PFU/ml,
JMA222 (ICEBs10)
5.34*105
2.87*104
1.45*104
OD600
0.087
0.35
0.84
1.47
PFU/ml,
AG174 (ICEBs1+)
2.99*105
2.06*105
2.12*104
1.62*104
Fig 2. Effect of host growth phase on plaque formation efficiency. Blue line represents
data for JMA222 (ICEBs10); red line represents data for AG174 (ICEBs1+). Y-axis is on a
logarithmic scale. OD600 reflects light scattering measurement of culture density, a
correlate of growth phase. Exact values shown in table.
This led me to repeat this experiment with B. subtilis spores of the same strains as
host cells for infection, reasoning that spores induced to germinate while subjected to
phage infection would be ‘synchronous’ in terms of their growth state. Moreover,
germinating spores might be even better indicators of phage infection, since the time
course experiment demonstrated that plaques are more efficiently formed on indicator
strains early in growth phase.
14 Spore infection demonstrates no immunity conferred by ICEBs1
Spores were made by growing cultures of JMA222 and AG174 in DSM (sporulation
media) for 24 hours. Cultures were then heat treated to inactivate any surviving
vegetative cells that remained. Cultures were plated on LB before and after heat
treatment to determine sporulation efficiency and spore number. JMA222 sporulated
more efficiently than AG174 (85% versus 75%, respectively). This slight difference may
have resulted in a 2-fold difference observed in my initial test of phage infection on the
two strains. A difference in sporulation efficiency, followed by heat treatment, may
have resulted in an excess of dead cell debris in the AG174 infection, resulting in a
titration of phage away from productive infections. To control for this, I purified the
spores using a Renografin column and multiple centrifugation/wash steps. The final
spore infections were done using the same number of purified spores as cells used for
the standard agar overlay assay. The results of this experiment demonstrate little
difference between the ICE+ and ICE- strains (JMA222: 6.21*106 total PFU/ml; AG174:
5.41*106 total PFU/ml). Notably, the numbers of plaques obtained for either indicator
strain were an order of magnitude higher than those observed at the earliest time point
in the time course infection, corroborating data suggesting that infections earlier in
growth yield a greater number of plaques.
Growth phase of host affects plaque size
While doing the time course experiment, I observed that plaque sizes also varied
with the growth phase of the host. I quantified these differences for strain AG174 (four
time points), using the NIH ImageJ software, and plotted these results as a histogram
(Fig. 3). The greater number of large outliers at early growth stages and the shift
15 towards a greater number of small plaques later in growth phase corroborates these
initial observations.
Fig. 3 Effect of host growth phase on plaque area in strain AG174. Images of plaques
produced from host infections initiated at different host growth phases were taken and
converted to plain images readable by NIH ImageJ software. Plaque areas were calculated;
plot above shows histograms of plaque areas for each growth phase utilized. Y-axis shows
OD600, a light scattering measurement of culture density/growth phase.
Variegation in plaque morphology is a feature of SP16 biology
Another observation made during the course of the previous experiments was that
plaque morphology varies considerably under all infection conditions. Namely,
infections resulted in a mixture of small, clearer plaques and larger, turbid plaques. This
could either result from an inherent characteristic of SP16 biology or from a mixed
infection resulting from a mixed phage stock (i.e., the original lysogenic phage source
strain, when induced by mitomycin C, released multiple, distinct phage species). In
order to test which of these explanations were correct I picked several small, clear and
large, turbid plaques for plaque purification and subsequent infection. Notably, phage
16 sources derived from small plaques did not yield a significant or visible number of
plaques (if any). Infections from larger plaque phage stocks led to mixed plaque
morphologies (Fig. 4), similar to what was observed for all previous infections. This
‘true-breeding’ result suggests that the mixed plaque morphology is an inherent
characteristic of SP16 biology and not due to using a mixed phage stock source. Smaller
‘opaque’ purified plaques that did not yield new plaques may represent smaller phage
populations that did not contain enough phage titer to produce visible plaques upon
secondary infection. Alternatively, these could be common mutant alleles that are
defective for lysis on new hosts.
Fig. 4 Natural variegation of SP16 plaques. Shown are plaques generated during a
standard SP16 infection of PY79. Phage used were obtained from a large (bullseye)
purified plaque picked from a previous infection performed with standard phage stock.
No plaques were observed from the same experiment conducted from small purified
plaques.
17 Conclusions
ICEBs1 does not appear to confer immunity to SP16 infection for the strains tested in
this study. An unexpected finding—that infection by SP16 is more productive earlier in
the host’s growth phase, measured both by number of plaques and area of plaques—
might be suggestive of a more general phenomenon in B. subtilis biology. It is possible
that B. subtilis is generally more susceptible to phage infection at earlier stages in
germination or exponential growth because the proper repressive conditions have not
yet been established. Whether such a model applies to other insults to the organism or
is, like variegation in plaque morphology, an inherent feature of SP16 biology remains
to be understood.
II. SPß
Introduction & Motivation
This work was motivated by the finding that cells containing ICEBs1 are partially
resistant to the phage SPß (Christopher Johnson, unpublished results). This phenotype
is caused by the ICEBs1 gene, yddK. The following preliminary experiments sought to
build tools necessary for mechanistically characterizing the effects of yddK on SPß. The
results detail methods that can be used to quantitatively demonstrate the reduction in
phage produced in the presence of yddK and how to utilize a temperature sensitive
version of SPß, SPßc2, for functional assays, such as for the determination of time
required for one cycle of phage replication.
18 Methods
B. subtilis strains
All the strains used in this study (Table 1) were obtained from Christopher Johnson
(Grossman lab). CMJ28 (thr, leu, metB5, ade) was the parent strain from which all other
strains were derived. It lacks both SPß and ICEBs1 and therefore can act as an indicator
strain for SPß infection. CMJ81 is a derivative of CMJ28 with a kanamycin-resistance
tagged variant of ICEBs1 (ICEBs1::kan), while CMJ82 is a derivative of CMJ28 with a
chloramphenicol-resistance marked variant of yddK in the amyE locus (amyE::{yddK cat}).
CMJ114 is a derivative of CMJ28 with a spectinomycin-resistance marked SPßc2
lysogen, while CMJ116 is a derivative of CMJ28 with a chloramphenicol-resistance yddK
in the amyE locus and a spectinomycin-resistance marked SPßc2 lysogen.
Strain
Genotype
CMJ28
thr, leu, metB5, ade, SPß0, ICEBs10
CMJ81
thr, leu, metB5, ade, ICEBs1::kan SPß0
CMJ82
thr, leu, metB5, ade, amyE::{yddK cat} SPß0
CMJ114
thr, leu, metB5, ade, SPßc2::spc
CMJ116
thr, leu, metB5, ade, SPßc2::spc amyE::{yddK cat}
Table 1. B. subtilis strains used.
SPß strains
Wild type SPß is found in most B. subtilis lab strains and was obtained by inducing
lab strains (i.e. with DNA damaging agents, such as mitomycin C) and collecting
supernatant. The temperature sensitive phage, SPßc2, first characterized by Rosenthal,
19 et al. was also obtained by inducing B. subtilis strains possessing this lysogen (see Table
above) [Rosenthal, 1979]. This variant carries a heat-sensitive repressor allele, such that
upon shift to high temperature, the lysogen is derepressed, causing lytic growth of the
phage.
SPß Plaque forming assay
Plaque forming assays were used to assess the number of viable plaque forming
units of phage on a given indicator strain. Indicator strains were grown in liquid LB
cultures from single colonies on freshly streaked LB-agar plates or light lawns to midexponential growth phase. Approximately 2-3 x 108 cells in 300 ul were combined with
various dilutions of 100 ul phage stocks and heated at 37°C to allow for phage
adsorption to cells. Phage-cell mixes were plated by adding molten LB top agar and
overlaying this mixture onto LB-agar plates. These plates were incubated at 37°C
overnight and PFUs were counted the following day.
Heat induction of SPßc2 lysogens
To enable easier experimental manipulation of phage replication, I induced growth
of the heat sensitive SPß allele, SPßc2, from lysogenic cells. Briefly, fresh single colonies
or light lawns of lysogenic strains were grown in liquid LB culture to mid-exponential
growth phase at low temperature (30°C). Cultures were shifted to high temperature
(52°C) for twenty minutes, after which they were moved to 37°C to allow for phage
growth.
20 One-step Assay
A one-step assay is used to determine the kinetics of a single round of phage
replication. Thus, it essentially involves 1) initiation of a single round of phage growth,
and 2) a quantitative assessment of the number of viable plaques produced at different
time points after phage growth is induced. During an experiment, cultures of strains
lysogenic for SPßc2 were heat induced by the method described above. Small volumes
(100 ul) of culture were collected before and after initiation of heat induction. The
supernatant, containing released phage, was isolated from these volumes and utilized
in the standard plaque forming assay described above.
Results
SPß plaque formation on hosts with and without yddK
Consistent with previous findings, I found that yddK was sufficient to inhibit
production of SPß (Table 2). I measured the ability of SPß to form plaques on a strain
containing ICEBs1 (CMJ81), a strain with yddK in the absence of all other ICEBs1 genes
(CMJ82) and a strain without ICEBs1 (CMJ82). All three strains were cured of SPß. The
presence of yddK or ICEBs1 essentially prevented plaque formation; there were no
detectable plaques (< 3.33*10^3 PFU/ml). In contrast, when grown on the strain with
no ICEBs1 genes, there were 1.9 x 106 plaque forming units (PFU) per ml of phage stock.
These results are consistent with previous findings that demonstrated that yddK is
sufficient to inhibit lytic growth of SPß.
Strain
PFU/mL
CMJ28
1.9*106
CMJ81
< 3.33*103
21 CMJ82
< 3.33*103
Table 2. SPß plaque formation on various B. subtilis strains. Strains
included contained neither ICEBs1 nor yddK (CMJ28), ICEBs1 (CMJ81),
or yddK alone (CMJ82). Note: experiment repeated > 5 times.
Induction of the temperature sensitive SPß lysogen in hosts with and without
yddK
Using a temperature sensitive mutant of SPß, SPßc2 [Rosenthal, 1979], I was able to
determine the rough time required for one cycle of phage replication. The lytic cycle of
SPßc2 is activated when the host lysogen is transferred to high temperature. Lysogens
were grown at 30°C until mid-exponential growth phase, then shifted to 50°C for 20
minutes before shifting cells to 37°C.
Using this protocol the time required for one cycle of phage release to take place was
roughly between 20 and 30 minutes, as demonstrated by the sudden increase in plaque
forming particles following temperature shift (Fig. 5). The number of plaque forming
units produced during growth at 30°C (before temperature shift) by strain CMJ114,
lacking a copy of yddK, was ~10-fold higher than that of CMJ116, possessing a copy of
yddK. Following the temperature shift (phage induction), the number of PFU increased
for both strains (Fig. 5) increased and the difference between CMJ114 (yddK-) and
CMJ116 (yddK+) was approximately 100-fold (Fig. 5). These results are consistent with
previous data indicating that yddK can block lytic growth of the phage.
In a cruder experiment 1 , following optical density and colony forming units
(survival) of the B. subtilis strains similar results were found (Fig. 6). No significant
change in viability was seen for CMJ116 (yddK+), as colony forming units and optical
1 Cells
were grown at 37°C, rather than 30°C, before temperature shift, increasing the baseline number of
excised phage/cured lysogens in both populations. 22 density remained constant. However, CMJ114 (yddK-) cells experienced a drastic
reduction in optical density and an order of magnitude decrease in viable colony
forming units by the end of the experiment. Both one-step experiments again
demonstrate the protective effect of yddK against death by SPß lytic replication. This is
consistent with the results of the plaque forming assays on hosts with and without
yddK, as described above.
Strain CFU, T = 0’ CFU, T = 50’
CMJ116
7.4*107
1.26*107
7
CMJ114
6.8*10
4.0*106
Fig. 5 Plaque forming units per ml of temperature sensitive SPß using B. subtilis strains with and
without yddK. Red line represents data for CMJ116 (yddK+); blue line represents data for CMJ114
(yddK-). Y-axis is displayed in logarithmic scale.
23 Fig. 6 Effect of SPß induction on optical density and colony formation in host. Graph
displays optical density (light scattering measure of culture density) before and after heat
induction of SPß. Table shows colony forming units from the same cultures sampled before
heat induction of phage and at 50’ post-induction.
Conclusions & proposals for future experiments
From the above experiments, it is clear that the previously-observed phenomenon,
namely, that host strains possessing a copy of yddK from ICEBs1 are more resistant to
death by SPß infection, is valid. This study has laid the groundwork for using a heat
sensitive allele of SPß in order to understand more about the mechanism of this
interaction. Several key experiments would greatly inform us about this mechanism in
the near future. Namely, looking at how genome replication and transcription of SPß
are affected by yddK would give us insight into what step is first affected by this
protein; these methods have been successfully employed by previous researchers of Abi
systems in lactococci [Chopin, 2005].
24 References
Adams, MH. Bacteriophages. 1959. Print. pp. 29-31.
Anagnostopoulos C, Spizizen J. (1961). “Requirements for Transformation in Bacillus
subtilis.” 81(5): 741–746.
Auchtung JM, Lee CA, Monson RE, Lehman AP, Grossman AD. (2005). “Regulation of a
Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA
damage response.” Proceedings of the National Academy of Sciences. 102(35): 12554-12559.
Auchtung JM, Lee CA, Garrison KL, Grossman AD. (2007). “Identification and
characterization of the immunity repressor (ImmR) that controls the mobile genetic
element ICEBs1 of Bacillus Subtilis.” Molecular Microbiology. 64(6): 1515-1528.
Behnke D, Malke H. (1978). “Bacteriophage interference in Streptococcus pyogenes L
Characterization of prophage-host systems interfering with the virulent phage A25.”
Virology. 85: 118-128.
Beler D, Gross R. pp 149-160. In Bacterial Signal Transduction: Networks and Drug Targets.
Utsumi R (ed). Springer, New York: 2008.
Bidnenko E, Ehrlich SD, Chopin MC. (1995). “Phage operon involved in sensitivity to
the Lactococcus lactis abortive infection mechanism AbiD1.” Journal of Bacteriolgy.
177:3824-3829.
Bingham R, Ekunwe SIN, Falk S, Snyder L, Kleanthous C. (2000). “The major head
protein of bacteriophages T4 binds specifically to elongation factor Tu.” Journal of
Biological Chemistry. 275, 23219-23226.
Bouchard JD, Moineau S. (2000). “Homologous recombination between a lactococcal
bacteriophage and the chromosome of its host strain.” Virology. 270:65-75.
Bouchard JD, Dion E, Bissonnette F, Moineau S. (2002). “Characterization of the twocomponent abortive phage infection mechanism AbiT from Lactococcus lactis.” Journal
of Bacteriology. 184:6325-6332.
Burch-Smith TM, Dinesh-Kumar SP. (2007). “The functions of plant TIR domains.”
Signal Transduction Knowledge Environment. pe46.
Burrus V, Pavlovic G, Decaris B, Guedon G. (2002). “The ICESt1 element of
Streptococcus thermophilus belongs to a large family of integrative and conjugative
elements that exchange modules and change their specificity of integration.” Plasmid.
48, 77–97.
Chopin A, Bolotin A, Sorokin A, Ehrlich SD, Chopin MC. (2001). “Analysis of six
prophages in Lactococcus lactis IL1403: different genetic structure of temperate and
virulent phage populations.” Nucleic Acids Research. 29:644-651.
Chopin MC, Chopin A, Bidnenko E. (2005) “Phage abortive infection in lactococci:
25 variations on a theme.” Current Opinion in Microbiology. 8:473-479.
Chowdhury R, Biswas SK, Das J. (1969). “Abortive replication of choleraphage f149 in
Vibrio cholera biotype E1 Tor.” Journal of Virology. 63: 392-397.
Cirl C, Wieser A, Yadav M, Duerr S, Schubert S, Fischer H, Stappert D, Wantia N,
Rodriguez N, Wagner H, Svanborg C, Miethke T. (2008). “Subversion of Toll-like
receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor
domaincontaining proteins.” Nature Medicine. 14, 399–406.
Curtis FA, Reed P, Sharples GJ. (2005). “Evolution of a phage RuvC endonuclease for
resolution of both Holliday and branched DNA junctions.” Molecular Microbiology.
55:1332-1345.
Dean DH, Fort CL, Hoch JA. (1978). “Characterization of Temperate Phages in Bacillus
subtilis.” Current Microbiology. 1: 213-217.
Dinsmore PK, Klaenhammer TR. (1994). “Phenotypic consequences of altering the copy
number of abiA, a gene responsible for aborting bacteriophage infections in Lactococcus
lactis.” Applied Environmental Microbiolgy. 60:1129-1136.
Dinsmore PK, Klaenhammer TR. (1997). “Molecular characterization of a genomic
region in a Lactococcus bacteriophage that is involved in its sensitivity to the phage
defense mechanism AbiA.” Journal of Bacteriolgy. 179:2949-2957.
Fink PS, Zahler SA. “Temperate Bacteriophages of Bacillus subtilis,” pp. 557-551. In R
Calendar (ed.) The Bacteriophages, 2nd ed. Plenum Press, New York: 2006.
Forde A, Fitzgerald GF. (1999). “Bacteriophage defense systems in lactic acid bacteria.”
Antonie Van Leeuwenhoek. 76: 89-113.
Gay NJ, Keith FJ. (1991). “Drosophila Toll and IL-1 receptor.” Nature. 351, 355-356.
Goranov AI, Kuester-Schoeck E, Wang JD, Grossman AD. (2006). “Characterization of
the global transcriptional responses to different types of DNA damage and disruption
of replication in Bacillus subtilis.” Journal of Bacteriology. 188(15):5595-5605.
Hemphill HE, Whiteley HR. (1975). “Bacteriophages of Bacillus subtilis.” Bacteriology
Review. 39: 257-315.
Huynh AH, Khaneja R, Tam NMK, Cazzato A, Tan S, Urdaci M, Brisson A, Gasbarrini
A, Barnes I, Cutting SM. (2009). “Bacillus subtilis isolated from the human
gastrointestinal tract.” Research in Microbology. 160 (2): 134-143.
Kaufmann G. (2000). “Anticodon nucleases.” Trends in Biochemical Sciences. 25,70-74.
Labrie SJ, Samson JE, Moineau S. (2010). “Bacteriophage resistance mechanisms.”
Nature Reviews Microbiology. 8(5): 317-27.
26 Lazarevic V, Soldo B, Dusterhoft A, Hilbert CM, Karamata D. (1998). “Introns and intein
coding sequence in the ribonucleotide reductase genes of Bacillus subtilis temperate
bacteriophage SPßc2.” Micobiology. 145:1055-1067.
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. (1996). “The
dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal
response in Drosophila adults.” Cell. 86(6):973-83.
Madigan M, Martinko J, eds. (2005). Brock Biology of Microorganisms. (11th ed.) Prentice
Hall.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. (1997). “A human homologue of the
Drosophila Toll protein signals activation of adaptive immunity.” Nature. 388(6640):
394-7.
Mele, J. (1972). “Biological characterization and prophage mapping of a lysogenizing
bacteriophage for Bacillus subtilis.” Ph.D. thesis, University of Massachusetts, Amherst,
Mass.
Moineau S, Pandian S, Klaenhammer T. (1994). “Evolution of a lytic bacteriophage via
DNA acquisition from the Lactococcus lactis chromosome.” Applied Environmental
Microbiolgy. 60:1832-1841.
Molineaux IJ. (1991). “Host-parasite interactions: recent developments in the genetics of
abortive phage infections.” New Biology. 3, 230-236.
Newman RM, Salunkhe P, Godzik A, Reed JC. (2006). “Identification and
characterization of a novel bacterial virulence factor that shares homology with
mammalian Toll/interleukin-1 receptor family proteins.” Infection and Immunology. 74,
594–601.
O’Neill LA, Bowie AG. (2007). “The family of five: TIR-domain-containing adaptors in
Toll-like receptor signaling.” Nature Reviews Immunology. 7, 353-364.
Parker AP, Dean DH. (1986). “Temperate Bacillus bacteriophage SP16 genome is
circularly permuted and terminally redundant.” 167(2): 719.
Rast JP, Smith LC, Loza-Coll M, Hibino T, Litman GW. (2006). “Genomic insights into
the immune system of the sea urchin.” Science. 314(5801): 952-956.
Regamey A, Lazarevic V, Hauser P, and Karamata D. (2000). “Study of chromosome
rearrangements associated with the trpE26 mutation of Bacillus subtilis. Molecular
Microbiology. 36:1234-1249.
Rokas, A. (2008). “The molecular origins of multicellular transitions.” Current Opinions
in Genetics & Development. 18, 472–478.
Rosenthal R, Toye P, Korman R, Zahler S. (1979). “The Prophage of SPßc2dcitK1, a
Defective Specialized Transducing Phage of Bacillus Subtilis.” Genetics. 92(3):721-39.
27 Sims JE, March CJ, Cosman D, Widmer MB, MacDonald HR, McMahan CJ, Grubin CE,
Wignall JM, Jackson JL, Call SM, Friend D, Alpert AR, Gillis S, Urdal DL, Dower SK.
(1988). “cDNA expression cloning of the IL-1 receptor, a member of the
immunoglobulin superfamily.” Science. 241, 585-589.
Smith HS, Pfizer LL, Pylkas L, Lederberg S. (1969). “Abortive infection of Shigella
dysenteriae P2 by T2 bacteriophage.” Journal of Virology. 4: 162-168.
Snyder L. (1995). “Phage-exclusion enzymes: a bonanza of biochemical and cell biology
reagents?” Molecular Microbiology. 15:415-420.
Spancake GA, Hemphill HE. (1985). “Deletion mutants of Bacillus subtilis
bacteriophage SP beta.” Journal of Virology. 55(1): 39-44.
Spear AM, Loman NJ, Atkins HS, Pallen MJ. (2009). “Microbial TIR domains: not
necessarily agents of subversion?” Trends in Microbiology. 17(9):393-8.
Thomas CM, Nielsen KM. (2005) “Mechanisms of, and Barriers to, Horizontal Gene
Transfer between Bacteria.” Nature Reviews Microbiology. 3: 711-721.
Tran LSP, Szabo L, Ponyi T, Orosz L, Sik T, Holczinger A. (1999). “Phage abortive
infection of Bacillus licheniformis ATCC 9300; identification of its promoter region.”
Applied Microbiology Biotechnology. 52:845-852.
Vennison, SJ. Laboratory Manual for Genetic Engineering. 2009. Print. pp 84.
Wilson GA, Williams MT, Baney HW, Young EE. (1974). “Characterization of temperate
bacteriophages of Bacillus subtilis by the restriction endonuclease EcoRI: evidence for
three different temperate bacteriophages. Journal of Virology. 14:1013-1016.
Zahler SA, Korman RZ, Rosenthal R, Hemphill HE. (1977). “Bacillus subtilis
bacteriophage SPß: localization of the prophage attachment site, and specialized
transduction.” Journal of Bacteriology. 129:556-558.
Zhou B, Wirsching P, Janda KD. (2002). “Human antibodies against spores of the genus
Bacillus: a model study for detection of and protection against anthrax and the
bioterrorist threat.” Proceedings of the National Academy of Sciences USA. 99(8): 5241-5246.
Zuber P. (2001). “A peptide profile of the Bacillus subtilis genome.” Peptide. 22:155515557.
28 Detailed Protocols
SP16 infection
The standard phage agar overlay method [Adams, 1959] was adapted for infection
of B. subtilis strains by SP16. Briefly, host indicator strains (cells or spores) were diluted
to a concentration of 7.75 x 108 per ml (or OD600 = 0.5, for cells). 300 µl of host
cells/spores were mixed with 100 µl of phage stock dilution (obtained from Tyler
DeWitt) in a 1.5 ml Eppendorf tube. A range of several phage dilutions was used for
each infection (between 1 – 10-5) to ensure that the final plates included one that could
easily be counted. Once the host cells/spores were added to the phage dilution, tubes
were briefly vortexed and set on a 37°C heat block for 10 minutes to allow for
adsorption of the phage to host cells. After adsorption, this mixture was transferred to a
glass test tube containing 3 ml of 0.7% PA agar (soft agar). Tubes were inverted 8 times,
and then the mixture was pipetted uniformly onto the surface of a 1.5% PA agar plate.
Once the soft agar was allowed to solidify (approximately 30 minutes), plates were
inverted and transferred to a 37°C room for overnight incubation (~16 hours). The
following day, plaques were manually counted by visual inspection.
Spore generation and purification
Spores were made by inoculating 2 ml of DSM sporulation medium with single
colonies of JMA222 or AG174, growing the cultures for 24 hours. Cultures were then
heat treated at 80°C for 20 minutes to inactivate any surviving vegetative cells. Cultures
were plated on 1.5% LB agar plates before and after heat treatment to determine
sporulation efficiency and spore number. Spores were purified using a Renografin
column. Briefly, unpurified spore cultures were pelleted by centrifugation. These spores
were resuspended in 200 µl of 20% Renografin and layered onto 1 ml 50% Renografin in
29 a 1.5 ml eppendorf tube. Tubes were spun at 14,000 x g for 30 minutes to pellet spores.
Cell debris was collected from the supernatant and disposed. Final pellets contained
pure spores, which were washed 2x with 500 µl 1x Spizizin minimal salts medium.
Purified spores were stored until use in 1x Spizizin minimal salts medium.
Plaque purification
Plaques were picked from a plate containing SP16 plaques generated on PY79
indicator strain. These agar ‘plugs’ were added to a single colony of PY79 in PA broth,
and allowed to grow, shaking at 37°C, until the host reached an OD600 of ~1.0. At this
point, mitomycin C was added to the cultures at a final concentration of 1 ug/ml to
induce any indicator cells that had been lysogenized by SP16. After an additional 20minute incubation, the cultures were isolated and centrifuged at 10,000 x g for 5
minutes to pellet cell debris. The supernatant, containing the plaque-purified phage
stock was syringe filtered through a 0.2 um Acrodisc filter. The following day, these
phage sources were used to infect PY79 indicator strains in the standard agar overlay
assay.
Plaque forming assay: SPß
B. subtilis strain(s) to be used as plaque indicators were plated from frozen stocks
onto fresh LB-based plates the day before setting up the plaque assay. The following
day, single colonies were used to inoculate LB cultures in Erlenmeyer flasks of variable
volumes. Alternatively, instead of using individual colonies to start cultures, light
lawns were made using freshly streaked plates the day before the assay was to be
conducted. The day of the experiment, light lawns were resuspended in LB and diluted
in LB in an Erlenmeyer flask to an OD600 of ~0.01. In either scenario, cultures were
30 grown to mid-exponential growth phase, an OD600 of 0.5 (in the latter case, this
amounted to five doublings). 300 ul of indicator strain was mixed with 100 ul of a phage
stock. Dilutions of the phage stock were typically used to ensure accurate counting of
plaques. 400 ul of indicator strain and phage dilution in a 1.5 ml tube were incubated in
a heat block at 37°C for 10 minutes to allow phage adsorption to cell surfaces. At the
end of adsorption, this mixture was added to 3 ml tube of molten 0.7% agar and
inverted 3 times, before being plated on LB plates with the appropriate selective
antibiotic. These plates were incubated overnight at 37°C, and plaques were counted the
next day.
Heat induction of SPßc2 lysogens
SPßc2 lysogenic strains were streaked onto LB-selective agar plates from frozen
stocks on day 1 and grown at 30°C. On day 2, single colonies of each strain were used to
create light lawns on LB-selective agar plates. On day 3, light lawns were resuspended
in LB and diluted into fresh LB in an Erlenmeyer flask at an OD600 of ~0.01. Cells were
grown to mid-exponential growth phase at 30°C in a shaking water bath. At an OD600 of
~0.5, cells were shifted to a 52°C incubator for twenty minutes. After heat shock, flasks
were moved back to a shaking water bath at 37°C to allow for phage replication and
growth.
One-step Assay
The one-step assay is a modified heat induction followed by a plaque-forming assay,
which allows one to watch the kinetics of one round of phage replication. Rather than
using a phage stock, lysogenic strains are used as a phage source for each timepoint in
the experiment by spinning down 100 uL of cells in a tabletop centrifuge at 8000 rpm for
31 3 minutes and removing supernatant containing phage. Lysogens are grown up from
light lawns, in a 30°C shaking, water bath as described above, and allowed to double
five times until reaching an OD600 of ~0.5. Then, each strain (phage source) is sampled
before being shifted to 52°C for twenty minutes. In some experiments, samples were
taken during the twenty-minute heat treatment. In all experiments, samples were taken
every 5-10 minutes for 30-60 minutes. The exact time for sampling was determined by
carrying out pilot experiments to test roughly how long it took for one phage burst to
occur. Once phage source supernatants were isolated for each sample, they were
combined with an indicator strain grown in parallel at 37°C, and the regular plaque
assay protocol was used for the remainder of the experiment. Plaques were counted the
next day and plotted over the time course of the experiment to determine the kinetics of
replication.
32