Affinity Flow Fractionation For Label-Free
Cell Sorting
By
Suman Bose
B.Tech., Indian Institute of Technology Kharagpur, India (2007)
M.Tech., Indian Institute of Technology Kharagpur, India (2007)
S.M., Massachusetts Institute of Technology, USA (2009)
Submitted to the Department of Mechanical Engineering in Partial Fulfillment of the
Requirements for the Degree of
Doctor of Philosophy in Mechanical Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
February 2014
© 2014 Massachusetts Institute of Technology. All rights reserved
Signature of Author
Department of Mechanical Engineering
September 27th, 2013
Certified by
Rohit Karnik
Associate Professor of Mechanical Engineering
Thesis Supervisor
Accepted by
David E. Hardt
Professor of Mechanical Engineering
Graduate Office
This page is intentionally left blank.
Affinity Flow Fractionation For Label-Free Cell Sorting
by
Suman Bose
Submitted to the Department of Mechanical Engineering on September 27th, 2013 in
partial fulfillment of the requirements for the Degree of Doctor of Philosophy in
Mechanical Engineering.
ABSTRACT
Capture and isolation of flowing cells from body fluids such as peripheral blood, bone
marrow or pleural effusion has enormous implications in diagnosis, disease monitoring,
and drug testing. However, in many situations the conventional methods of cell sorting
are of limited use due to complex sample preparation steps, high costs, or low sensitivity.
Drawing inspiration from nature, a novel platform technology for cell separation known
as Affinity Flow Fractionation (AFF) was developed. AFF relies on interaction of cells
with asymmetric patterns of weak adhesive molecules allowing for continuous sorting of
cells with high purity without irreversible capture of cells. Cells are sorted in a single
step, which is a significant advance over conventional immunocapture methods,
especially for point-of-care and point-of-use applications.
In this work, first, the interaction of cells under shear flow with asymmetric
patterns of weak adhesive molecules was studied systematically to highlight the
underlying mechanism of AFF at a phenomenological level. Next, an optimized
separation device was fabricated and its performance was characterized using model cell
lines. A detailed predictive mathematical model, which accounts for the major transport
processes involved in cell separation by AFF, was developed and the results validated
using experiments.
Finally, AFF was applied for rapid isolation of neutrophils from blood, which is
important for several applications where conventional procedures involve multiple steps
and time-intense manual skills. It was demonstrated that asymmetric patterns of Pselectin, a weak adhesive molecule involved in cell trafficking, can directly draw
neutrophils out of a continuously flowing stream of blood, with high purity (92%). As
cells exhibiting non-specific adhesion are not drawn out of the flowing stream, an
ultrahigh 400,000-fold enrichment of leukocytes over erythrocytes is achieved.
Moreover, the sorted neutrophils remain viable, unaltered, and functionally intact. The
lack of background erythrocytes enabled direct enumeration of neutrophils by a
downstream detector, which could distinguish the activation state of neutrophils in blood.
This method is compatible with capillary microfluidics and may find use in isolation of
neutrophils for diagnosis of sepsis, genetic analysis, HLA typing, assessment of
chemoreadiness, and other applications.
Weak molecular interactions govern a large number of important physiological
processes such as stem cell homing, inflammation, immune modulation and cancer
metastasis. Yet, currently there are no effective technologies that can separate cells based
on weak interactions alone. We believe, AFF would fulfill this un-met need in the area of
cell sorting and enabling new discoveries.
Keywords: Microfluidics, Cell sorting, cell rolling, selectin, blood, point-of-care,
neutrophils.
Thesis Committee:
Prof. Rohit Karnik, Associate Professor of Mechanical Engineering, MIT. (Thesis Supervisor)
Prof. Roger D. Kamm, Professor of Mechanical and Biological Engineering, MIT
Prof. Jeffrey M. Karp, Associate Professor of Medicine, Harvard Medical School, Brigham and
Women’s Hospital.
To my Mother, Father and my Teachers, who have shown me the light….
This page is intentionally left blank.
Table of Contents
Acknowledgements………………………………………………………………………..…..……………..9
List of Abbreviations………………………………………………………………………..……………...10
List of Symbols…………………………………………………………………………….…………….…..11
List of Figures and Tables……………………………………………………………….…………...……12
1. Introduction………………………………………………………….…………………….….15
1.1 Principles of cell sorting…………………………………….………………………..…..16
1.2 Microfluidic cell sorting and clinical relevance……………………………………….....17
1.3 Affinity based separation: use of moderate to low affinity molecules…………………...20
1.4 Asymmetric weak adhesive interactions: Towards AFF……………………………..…..21
2. Studying Interaction Of Cells With Asymmetric Receptor Patterns………………….…..25
2.1 Introduction
…………………………………………………………………….……25
2.2 Materials and Methods………………………………………………………….…….….28
2.2.1 Materials………………………………………………………………………………….…28
2.2.2 Fabrication of Patterned Substrates…………………………………….…………….…29
2.2.3 Cell Rolling Experiments in a Flow Chamber…………………………………….……30
2.2.4 Data Analysis……………………………………………………………………….………30
2.2.5 Simulation of Cell Rolling Trajectories…………………………………………………35
2.3 Results and Discussion………………………………………………………………..….35
2.3.1 Effect of Edge Angle on the Rolling Behavior of HL60 Cells ………………………..37
2.3.2 Effect of Shear Stress on Rolling Behavior of HL60 Cells …………………………...39
2.3.3 Effect of P-selectin Density on Rolling Behavior of HL60 Cells…………………….40
2.3.4 Detachment of cells rolling along an edge modeled as a Poisson process......…….41
2.3.5 Prediction of cell trajectories on a receptor-patterned substrate…………………...46
2.4 Conclusion…………………………………………………………………..……………49
Acknowledgements…………………………………………………………..………………50
3. Creating functional surfaces…………………………………………………….…………...51
3.1 Introduction………………………………………………………………....……………51
3.2 Design considerations………………………………………………………….……...…52
3.3 Background…………………………………………………………………..………..…52
3.4 Microfluidic device design ………………………………………………….……….…..56
3.5 Screening of surface functionalization protocol ……………………………...………….57
3.6 Immobilization of P-select………..…………………………………………….….…… 60
3.7 Final fabrication protocol …………………………………………………………..……65
3.8 Characterization of substrate ………………………………………………………..…...67
3.9 Conclusion…………………………………………………………………………..……68
Acknowledgements…………………………………………………………………..… …...68
4. Separation of model cell lines……………………………………………………….……… 69
4.1 Introduction………………………………………..……………………………..………69
4.2 Experimental setup for cell separation………………………….…………………..……70
4.3 Separation of HL60 cells from K562 cells……………………………..…………...……70
4.4 Separation metrics…………………………………………………………………......…73
4.5 Mathematical Modeling of separation process …………………….…………….……...76
4.5.2 Estimation of the model parameters…………………………..…………...………77
4.5.3 Monte Carlo simulation……………………………………………………………80
4.5.4 Scaling model………………… ………………………………...…………………81
4.5 Conclusion…………………………………………………………………………..……83
5. Application of AFF in Sorting of Neutrophils from blood …………………………….…..85
5.1 Introduction…………………………………………………..…………………..………85
5.2 Sorting of neutrophils from blood …………………………………………..………...…86
5.3 Purity and efficiency …………………………………………………………..…...……88
5.4 Characterization of sorted cells …………………………………………….....…………90
5.4.1 Hematological analysis …………………………………………………..….……90
5.4.2 P-selectin binding …………………………………………………………...….…91
5.4.3 Activation assay ……………………..…………….………….……………...……92
5.4.4 Phagocytosis assay ……………………………………………………….…….…94
5.5 Label-free detection of neutrophil activation in blood ……………………………....…..95
5.6 Conclusion ……………………………………………….………………………..…..…98
6. Conclusions and Discussion ………………………………………………………....…..…101
References ……………………………………………………………………………….……..107
Acknowledgements
It gives me great pleasure to thank the people who have made this work possible.
I feel very fortunate to have the opportunity to work closely with Prof. Rohit
Karnik. Rohit is an extraordinary teacher, adviser, mentor and a friend. Rohit allowed
immense creative freedom in my research and let me follow my path, while at the same
time providing necessary guidance and support, which ultimately helped me develop
confidence, passion and conviction in my work. Rohit’s passion for science, commitment
to his students, principles and life vision has been truly inspiring for me, and hopefully I
can carry his teachings into my career ahead.
Next, I would like to thank Prof. Jeff Karp who has been like a second adviser to
me. Jeff had been instrumental in shaping up the project right from its conception and
especially in realizing the clinical aspect of the work. Jeff’s enthusiasm about
translational research was one of the key factors that motivated me to pursue a career in
translational medicine. Also, I would like to thank Prof. Roger Kamm who has given
many valuable suggestions and insightful comments on the work. Despite his busy
schedule, Roger has been very generous with his time and has always provided great
career advices.I would also like to thank Dr. Ulrich von Adrian and Dr. David Sloane
who provided a number of great suggestions about the project.
Next, I would like to thank my colleagues whom I have worked closely in my
grad school on various projects - Chia-Hua Lee, Chong Shen, Rishi Singh, Mikhail
Hanewich Hollatz, Cheryl Cui, Weian Zhao, Minhee and Lim. Working together with
Chia was a great experience, which I will miss. She has been an amazing colleague and a
great friend. I feel privileged to have had the opportunity to mentor Chong, Rishi and
Mikhail. This work would not have been successful without their hard work and
dedication. While mentoring them was a great experience, I have probably learned more
than I have taught my students.
I owe a special thanks to all my friends whom I have met over the years at MIT.
These were probably some of the best minds I have come across. They have been
motivating, inspiring, and all the amazing time I spend with these people made graduate
life memorable. Thanks Marco –for all the late night beer, life philosophies and for
cheering me up after failed experiments, Jongho – for your friendship, dinner and coffees,
Sankha –for the evening strolls, ‘adda’ and your support, Ioannis – for pushing me when
I would give up. I would also like to thank – Sean, Tarun, Sung-Young, Jason, Mike,
Sunandini, Jong-Min, Maria, Tom and Sumeet. It was my pleasure to know you guys and
thank you for all your support.
Last, but not the least, I would like to specially thank my mom, dad and my
family in India. Today, I have reached this position because of what they have taught me,
values they instilled in me, sacrifices they made, their love and support.
List of Abbreviations
AFF
AFM
APC
BSA
CD
CDF
CTC
DC
DPBS
DSP
E.R
EC
EDTA
ELISA
ESL-1
FACS
FBS
FC
FITC
HBSS
HIV
MACS
MHC
MSC
NHS
NSAID
PDF
PDMS
PE
PEG
PNAd
POC
PSGL-1
RBC
SAM
SD
SEM
VCAM
WBC
Affinity flow fractionation
Atomic force microscope
Allophycocyanin
Bovine serum albumin
Cluster of differentiation
Cumulative distribution function
Circulating tumor cell
Dendritic cells
Dulbecco’s Phosphate Buffered Saline
Dithiobis succinimidyl propionate
Enrichment ratio
Endothelial cells
Ethylenediaminetetraacetic acid
Enzyme linked immunosorbent assay
E-selectin ligand 1
Fluorescence activated cell sorting
Fetal bovine serum
Flow cytometry
Fluorescence isothiocyanate
Hank’s Balanced Salt Solution
Human Immunodeficiency Virus
Magnetic activated cell sorting
Major Histocompatibility Complex
Mesenchymal stem cells
N-hydroxysuccinimide
Non-steroidal anti-inflammatory drug
Probability distribution function
Polydimethylsiloxane
Phycoerythrin
Polyethylene glycol
Peripherial node addressin
Point of care
P-selectin glycoprotein ligand 1
Red blood cells
Self assembled monolayer
Standard deviation
Scanning electron microscope
Vascular cell adhesion molecule
White blood cells
List of Symbols
English symbols
b
d
D
D*
g
h
L
L*
le
Neff
P∞
P0
Pattach
Peff
pi
ps
pw
pnt, sorted
pnt,input
pt,input
pt,sorted
Q
r
ve,x
ve,y
ve
Vfluid,x
vp
w
wc
wp
z
distance between patterns
lateral displacement of cell
Mean displacement of the cell population
Normalized D
gravitational acceleration
channel height
Length of travel for a cell
Normalized L
edge tracking length
Number of effective bands on which the cells rolled on
Probability of attachment from free stream
Probability of reattachment of a rolling cell on the next downstream band
Net attachment probability of a cell flowing over a number of bands
Net average probability of attachment of a cell on a single band
purity of input sample
purity of sorted sample
purity of waste sample
purity of non-target cells in sorted sample
purity of non-target cells in input sample
purity of target cells in input sample
purity of target cells in sorted sample
flow rate
recovery of target cells
component of edge velocity along fluid flow
component of edge velocity normal to fluid flow
velocity of rolling cells on pattern edge
velocity of the fluid
velocity of rolling cells on plain region
channel width
width of cell stream
width of patterned region
height of cell from channel floor
Greek symbols
α
λ
µ
σ
τ
edge inclination angle
mean of Poisson distribution
viscosity
Standard deviation of displacements of cells
wall shear stress
List of Figures and Tables
Figures
Figure 1.1: Cell sorting: Principles and applications.
Figure 1.2: Procedure of performing diagnostic tests with a centralized laboratory.
Figure 1.3: Affinity Flow Fractionation of cells.
Figure 2.1. Studying interaction of cells with asymmetric receptor patterns
Figure 2.2. Schematic diagram for patterning of P-selectin on a gold substrate
Figure 2.3. Flow chart describing the cell tracking and analysis algorithm.
Figure 2.4. Characterization of P-selectin patterned substrates for cell rolling.
Figure 2.5. Tracks of HL60 cells rolling on P-selectin lines.
Figure 2.6. Effect of edge inclination angle α on rolling behavior of HL60 cells
Figure 2.7. Effect of shear stress τ on rolling behavior of HL60 cells
Figure 2.8. Effect of P-selectin incubation concentration on rolling behavior of HL60
cells
Figure 2.9. Detachment of cells rolling along an edge is well described by a Poisson
distribution.
Figure 2.10. Probability distribution of net lateral displacement of HL-60 cells after
rolling on consecutive bands of P-selectin patterns as obtained from Monte Carlo
simulations and experimental observations.
Figure 3.1. Schematic showing the different mechanisms though which proteins can
interact with solid substrates.
Figure 3.2. Design of the separation channel.
Figure 3.3. Rolling velocity of HL60 cells at different shear stress on substrates
functionalized with P-selectin using different functional groups.
Figure 3.4. Passivation of glass.
Figure 3.5. Immobilizing P-selectin with proper orientation on the substrate
Figure 3.6. Testing immunogenicity of rolling surfaces prepared using different
immobilization protocols.
Figure 3.7. Schematic for fabrication and assembly of separation devices
Figure 4.1. Schematic of the experimental setup for cell separation
Figure 4.2. Separation characterization using HL60 and K562 cells.
Figure 4.3. Performance of separation
Figure 4.4. Schematic description of the key processes governing the transport of cells
inside the separation device.
Figure 4.5. Schematic of parameters used in mathematical model.
Figure 4.6. Edge tracking length of HL60 cells and neutrophils on P-selectin patterns
Figure 4.7. Theoretical modeling of cell separation in the device.
Figure 5.1. Direct isolation of neutrophils from blood using AFF.
Figure 5.2. Purity of sorted neutrophils.
Figure 5.3. Purity of the samples determined by flow cytometric analysis.
Figure 5.4. P-selectin binding affinity of the input and sorted fractions.
Figure 5.5. Activation assay of sorted neutrophils.
Figure 5.6. Phagocytosis activity of neutrophils in whole blood and sorted fraction.
Figure 5.7. Detecting neutrophil activation using activation-dependent cell sorting.
13
Figure 6.1. Applications of Affinity Flow Fractionation of cells
Tables
Table 1.1: Clinical applications of cell sorting
Table 2.1. Comparison of the experimental and Poisson average value of λ.
Table 5.1. Comparison of AFF with other blood cell separation methods
14
This page is intentionally left blank.
1
Introduction
Isolation of cells from complex mixtures is of immense importance in disease
diagnosis1,2, stem cell therapeutics4, genetic analysis5, and biological research. The
samples of interests vary in composition and complexity, and can range from simple cell
suspensions, complex mixtures such as - blood, peritoneal fluids, cerebrospinal fluids and
bronchial aspirate or even tissue samples such as bone marrow. Blood, which is a
common clinical sample, is composed of a wide variety of cells of different shapes and
sizes. The abundance of cells also vary over a wide range, with RBCs present in the order
of 5 billion per mL, leukocytes at 10 million per mL and rare cells such as antigen
specific T cells, circulating tumor cells and stem cells at 1 or less per mL of blood6. Thus
a variety of techniques have been developed to address the needs for different
applications.
In clinical diagnostics, RBC and WBC counts and leukocyte differentials are routinely
used to diagnose infections, anemia, parasitimea, hematological diseases, stress and
variety of other disorders2. Cell based therapies also require isolation and purification of
the donor cells before transferring them to the recipient. Enrichment of CD34+ cells and
depletion of T-cells from umbilical chord blood7 and bone marrow, followed by ex vivo
expansion increases efficacy of stem cell graft treatment8-11. Similarly, mesenchymal
Chapter 1: Introduction
16
stem cells (MSCs) are usually rare in adult tissue and needs to be isolated from donor
tissue (marrow and adipose tissue) and expanded ex vivo before transferring to
recipient12-15. With many new cell based therapies being recently approved by FDA16 or
in the pipeline, cell isolation methods are certainly going to take center stage in the
therapeutics market17-24. Table 1.1 summarizes the major clinical applications of cell
sorting.
Table 1.1. Clinical applications of cell sorting
Therapeutics
Diagnostics
Application
Cells
Source
Disease
RBC
Blood
Anemia, Sickle cell anemia, Malaria
Neutrophils
Blood
Infection, immune system functioning, sepsis,
2,25
genotyping, genetic analysis
Lymphocyte
Blood
Viral infection, genotyping, genetic analysis
Eosinophil
Blood
Allergy, parasitic infections
Basophils
Blood
Hypersensitivity, allergy
CD4 T cells
Blood
HIV infection
Neoplastic
hematopoietic cells
Blood or
Marrow
Leukemia and lymphoma
Circulating Tumor Cells
Blood
Cancer
RBC
Blood
Thalassemia, Sickle cell anemia
Leukocytes
Blood
Adoptive immune therapy,
Extracorporeal photopheresis for treatment of
GVHD, cutaneous T cell lymphoma and
2
Autoimmune diseases
Platelets
Blood
Thrombocytopenia
Hematopoietic stem cells
(CD34+)
Marrow
Leukemia, stem cell transplant
Mesenchymal stem cells
Marrow,
adipose
tissue
Regeneration therapy
Dendritic cells
Blood
Immune therapy
2
2
2
2
2,26
2
2
33-36
27-30
19,31,32
, DC Vaccination
16,37
1.1 Principles of cells sorting
A number of parameters such as – shape, size, density, deformability, membrane
electrical impedance and intracellular and extracellular protein expression have been used
to differentiate and isolate cells38. Physical parameters such as shape, size, density,
stiffness and membrane conductance provide only a moderate degree of specificity to cell
type, and correlation to clinical condition is often weak. On the other hand, isolation
based on expression of proteins provides a more specific approach to identification and
Chapter 1: Introduction
17
isolation of cell populations and if often more widely used in clinics and laboratories2.
Cluster of differentiation (CD) proteins, which are expressed on cell surface, are used as
surface markers to immunophenotype cells into different sub groups, and are often the
target for specific cell isolation techniques39.
Specific cell surface molecules are often detected though molecules (ligands, antibody,
aptamer), which specifically bind to the target marker. The antibody can be ‘tagged’ with
a - fluorescent molecules that enable fluorescent based sorting (FACS), magnetic
nanoparticle to pull the target cells out of the solution (MASC) or just used to capture
cells on a solid surface (Affinity based separation). These methods are shown in Fig 1.1.
While a number of variations on this theme are possible, it is important to note that all of
these methods rely upon specific receptor-ligand interactions on the cell surface.
Figure 1.1. Cell sorting: Principles and applications. Sorting of cells based on specific
surface marker relies on using antibody-antigen interactions to produce a signal.
Popular methods of separation are fluorescence based sorting (FACS), magnetic based
sorting (MACS) and affinity capture.
1.2 Microfluidic cell sorting and clinical relevance
Chapter 1: Introduction
18
Cell sorting and counting is performed as a routine process in the clinics and forms a
major process in disease diagnosis. Most commonly, blood counts are performed using
automated counters, which use a combination of Coulter principle and light scattering
pattern to identify major blood cell types i.e Red blood cells (erythrocytes), granulocytes,
monocytes and lymphocytes2. However, further immunotyping of blood cells into further
subgroups requires identification of specific CD surface marker and is commonly done
though FACS2.
Figure 1.2. Procedure of performing diagnostic tests with a centralized laboratory
involves transportation, storage and sample preparation which adding to the time and
cost of performing tests. Microfluidic chips can pack several functionalities from sample
collection, processing and analysis into a single disposable and inexpensive unit allowing
for diagnostic tests to be performed near the patient.
The workflow of clinical samples from the patient to the hematology labs usually
involves multiple transportation and sample preparation steps that are time consuming
and labor intensive, adding to the cost and time required preforming these tests. Because
of the physical distance between the patient and diagnostic instrument especially in
resource limited settings, obtaining a simple blood count may take anywhere between a
day upto a week, delaying diagnosis and treatment. Point-of-care (POC) devices which
perform diagnostic test on the patient’s bed side offers a solution to this problem40-42. An
ideal POC device would be resource independent, fast, require less or little manual
Chapter 1: Introduction
19
intervention and inexpensive. Microfluidics offers a great alternative to the traditional
diagnostic methods since they have the potential to pack the functionalities of sample
preparation and analysis into a single unit (see Fig 1.2), consumes very little reagents and
are fast and inexpensive43-46.
While a large number of microfluidics POC devices have been developed and
commercially adopted for performing clinical chemistry and immunodiagnostics47-54,
development of devices for hematological tests remains limited55. The Toner group
developed a CD4+ T-cell counter though capture of CD4+ cells on antibody coated
microfluidic channel56,57 and developed methods to quantify the levels of such cells in
blood though electrical impedance measurement though the channel58. Daktari Diagnostic
Inc has commercialized the CD4 chip for monitoring HIV treatment in developing
countries. The same group also developed chips based on immunocapture for isolation of
circulating tumor cells (CTCs) for monitoring of cancer metastatis59 and isolation of
granulocyte from trauma and burn patients for genomic and proteomic studies5. A
number of other studies have looked at immunocapture based sorting of cells within
microchannels with the focus on modifying the channel geometry60,61 or surface
topographies62 to enhance cell-surface interaction increasing efficiency. In an alternative
approaches to immunocapture, cells pre-labeled with magnetic particles63 or dielectric
particles64 have been sorted from a cell mixture inside a microfluidic chip with high
efficiency and purity. Microfluidics MACS have been used to isolate viable CTCs for
single cell genomics65 and could potentially be used to filter bacteria from blood of septic
patients66. However, the requirement for labeling and high cost of the magnetic beads
makes MACS based techniques more suitable for laboratory scale cell processing and
analysis tool, impacting its use as a POC instrument.
A number of techniques have also been developed for label-free sorting of cells based on
non-specific characteristics67. On-chip lysis of RBC though osmotic shock has been
reported to isolate leukocyte suitable for genomic analysis68. Several groups have
developed microfluidic filters that perform size-based separation of cells allowing sorting
of different cellular component of blood - RBC, leukocytes and platelets6. On similar
lines, interaction of cells with microstructures inside channel have been leveraged to
isolate cells based on deformability69 and detect diseases such as sickle cell anemia70.
Chapter 1: Introduction
20
Recently, spiral microchannel with trapezoidal cross section have been used to isolate
leukocytes71 and CTCs72 from diluted blood using differences in density between the cell
types. Differences in electric and magnetic properties of RBCs and WBCs have been
used to attempt at separating the cell types using dielectrophoresis73 and MACS74. For a
more comprehensive review of different cell sorting techniques, the readers are directed
elsewhere75,76. A review of the current microfluidic devices that have been
commercialized can be found here77.
1.3 Affinity based separation: use of moderate to low affinity molecules
Affinity based separation has been widely used for achieving label-free and surface
marker specific sorting of cells. However, the use of high-affinity antibodies as affinity
molecules limits the usability of these devices since retrieval of the sorted sample is at
best difficult if not impossible. Counting of cells trapped inside microfluidic channels
poses a problem in obtaining easy reliable counts and might require uses of microscope
or other equipment. For example, the CD4 cell counting device developed by the Toner
group and Daktari requires successive rinses with several buffers actuated in sequence by
an external instrument to enumerate the captured cells. In contrast to antibodies, other
molecules having low affinity have also been investigated for affinity based fractionation
of cells.
Lectins – which are a class of carbohydrate binding proteins, have been used for sorting
cells though affinity columns78. Binding of lectins is relatively non-specific to cell types
and depends on a number of factors like metabolic state, stage of cell division and
differentiation and surface protein glycosylation. Lectins affinity columns have been thus
used to isolate stem cells based on differentiation or homing potential79-82.
Physiological weak affinity adhesive interactions have also been exploited in an
interesting manner in vivo for cell separation. Nature has evolved a number of molecules
that exhibit weak, yet relatively specific adhesive interactions including bacterial
adhesion molecules83, selectins involved in homing of circulating cells84, and MHC-II
molecules on antigen presenting cells that exhibit weak affinity towards the T-cell
receptor85. Selectins – glycoproteins involved in cell trafficking, have been particularly
Chapter 1: Introduction
21
studied extensively and used for cell separation purpose. A brief review of the
phenomenon of cell trafficking along with different ligands involved is presented below.
Selectin coated surfaces have been proposed for separation of leukemia cells based on
their differential rolling velocity86. In another approach, P-selectin coated microtubes
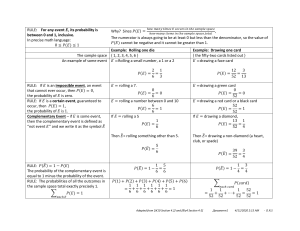
Cell rolling: A brief synopsis The immune system serves as to protect the body against harmful foreign objects and is critical for our survival. A major role of the immune system is to traffic different immune cells present in the blood to their target sites in the organ. This is achieved though a process called ‘cell rolling’, wherein the endothelial cells express weak affinity ligands, which interacts with the receptors on 3
the surface of the leukocytes allowing them to latch on and stick to the endothelium . Due to the weak nature of the interactions, the drag force on the cell due to blood flow break the bonds at the trailing edge of the cell allowing the cell to ‘roll’ forward such that new bonds can be formed at the leading edge. Next, specific signaling molecules called ‘chemokines’ acts upon the rolling cells initiating activation of other high-­‐affinity surface adhesive molecules that allows for arrest of the cell on the endothelium. Then, the cell transmigrates though the endothelium and extravasates into the tissue. Figure. Rolling of leukocytes on endothelium mediated through selectins. Source: Janeway’s th
Immunobiology 8 ed, GS. Cell rolling is mediated by glycoproteins known as selectins, primarily expressed on endothelial cells, which interacts with a variety of ligands on leukocyte surface. Apart from leukocytes, stem cell and cancer cells also exhibit cell rolling. A list of selectins and corresponding ligands are listed below. Selectin Expression Function Leukocytes Constitutive on all Leuk, Lym homing, L-­‐selectin except effector/ PSGL-­‐1 inflammation memory T c ells Inflamed EC, hemangioblast, E-­‐selectin constitutive in skin and bone marrow x
a
Ligands EC Other Mannose PNAd receptor, (CD34) versican, sulfatide s-­‐Le , s-­‐Le , inflammation PSGL-­‐1, ESL-­‐1 x
s-­‐Le , glycolipids Inflammation
PSGL-­‐1 PNAd CD24 , hemostasis Leuk: leukocyte, EC: endothelial cell, Lym: lymphocyte, PSGL-­‐1: P-­‐selectin glycoprotein ligand 1, PNAd: Peripheral node addresin, s-­‐Le: sialyl lewis, ESL-­‐1: E-­‐selectin ligand-­‐1. Source: IM701 Lecture notes 2009 (Uli Von Adrian). P-­‐selectin EC, platelets have been used to capture CD45+ cells and CTCs from whole blood87,88 and selectin co-
Chapter 1: Introduction
22
patterned with antibody has shown enhanced cell capture efficiency than antibody
alone89. Lee and coworkers developed a microfluidic device with selectin coated
micropillars and demonstrated the feasibility of using such devices for cell separation
application90. Sorting is based on the fact that cells that interact strongly with the selectin
are retained longer in the device while the weakly interacting cells would be washed out
faster. However, the above-mentioned devices perform batch processing requiring
multiple wash steps. Karnik and coworkers recently developed a microfluidic device that
combined the principles of hydrophoresis with selectin based affinity separation91. Cells
that interacted with selectin coated channel were transported into microgrooves, where
the direction of the fluid stream pushes the cells away from the original stream. This
technique overcomes the previous challenge of batch processing since cells can now be
processed in a continuous manner. However, the hydrophoretic flow causes substantial
size based dispersion of cells within the channel92 and hence these devices cannot be used
for separation directly from blood.
1.4 Asymmetric weak adhesive interactions: Towards Affinity Flow
Fractionation
Affinity based fractionation allows for separation of cells based on a specific surface
marker in a label-free method. Using weak affinity molecules allows for retrieval of
sorted cells, but so far most of the reported affinity devices work in a batch operation
mode restricting their use in building simple POC devices. On the opposite end of the
spectrum, flow fractionation of cells - where flowing target cells are pulled across
streamlines resulting in separation of cells in a continuous manner, have been limited to
long-range physical forces arising from dielectrophoresis, acoustophoresis, gravitational,
magnetic, or inertial effects. The non-specific action of these long-range force fields
limits the use of flow fractionation of cells to a few applications, while its extension to
sorting based on molecular recognition requires pre-labeling of cells with magnetic or
dielectric beads93.
Karnik and coworkers in an earlier study94 demonstrated that transient interactions of
cells with asymmetric patterns of weak affinity adhesive molecules exert forces on the
cell to deflect it perpendicular to the direction of fluid flow, without capture. Specifically,
Chapter 1: Introduction
23
they patterned P-selectin – a molecule involved in trafficking of neutrophils during early
phase of inflammation, at an angle to the flow direction and reported the deflection of
rolling HL60 cells (promyelocytic leukemia cell line that demonstrates rolling on Pselectin) as they encountered the edges of the pattern (See Fig 1.3). Subsequently, other
groups have also investigated the phenomenon theoretically95 and experimentally96. This
effect provides a new paradigm for label-free flow fractionation of cells based on specific
molecular interaction, and is called affinity flow fractionation (AFF).
Figure 1.3. Affinity Flow Fractionation of cells. a) Patterns of weak affinity receptors
introduce a displacement in the trajectory of a rolling cell as it follows the edge of the
patterns. b) Tracks of rolling HL60 cells on a P-selectin patterned edge (P-selectin
region shown in pink) could be seen to follow the edge. c) Quantification of angle of the
tracks demonstrates that cells tend to follow the pattern direction. d) Schematics of a
AFF based microfluidic separation device which uses a series of inclined receptor
patterns to separate the target cells from the sample stream. Figure a-d are reproduced
from Karnik et al. Nano Lett, 2008 94.
Chapter 1: Introduction
24
This work aims at mechanistic understanding the phenomenon of how the cells interact
with asymmetric receptor patterns, and developing microfluidic devices based on AFF to
achieve label-free surface marker specific fractionation of cells. Our aim is finally to
develop AFF microfluidics devices such as the one shown in Fig1.3d, where a stream of
cells is flowed over a patterned substrate, which draws a stream of purified target cells
out of the original cell stream into the parallel buffer stream. Such devices should find
wide usage in separating cells for diagnostics and therapeutic application.
The thesis has been divided into six chapters each covering investigation, design and
fabrication, characterization, demonstration of application and finally conclusion. The
interaction of cells with asymmetric patterns has been investigated in the second chapter,
which gives mechanistic insights into the process of cell rolling on the edge. In Chapter
3, an optimized design for the AFF device has been developed based on the findings of
the mechanistic study. A fabrication protocol was also developed. Next, in Chapter 4, we
demonstrate the operational characteristics of the AFF devices using model cell lines
(HL60 and K562) and develop a mathematical model to accurately predict the behavior
of AFF devices. Then, in Chapter 5, we demonstrate application of AFF for sorting
neutrophils from blood in a single step with high purity. Finally, in Chapter 6, we discuss
the potentials for AFF as a general cell sorting tool and some of the applications where
AFF might be useful.
2
Studying
Interaction
Of
Cells
With
Asymmetric Receptor Patterns
Note: This chapter is a modified version of the paper “ Examining the Lateral Displacement of
HL60 Cells Rolling on Asymmetric P-Selectin Patterns” Lee et al., Langmuir, 2011, 27 (1), pp
240–249.
2.1 Introduction
Cell separation devices employ a host of different mechanisms to enable sorting of target
cells from a mixed population6,67,97. However, at a fundamental level all of these
techniques reply upon recognition of specific cell surface molecules by cognate ligand,
which are linked to effector mechanisms that pulls the cells away from the general
population. In affinity-based separations, the ligands are immobilized on surfaces or
beads, which allows the target cells to adhere to the surface while the non-target
contaminating cells can be washed off98. Understanding the transport of cells over these
affinity surfaces and their capture is critical for design, engineering and optimization of
the affinity based separation process.
Two major transport processes mainly govern the binding of cells to an affinity surface.
First, the cells need to arrive in close proximity to the surface through the action of long-
Chapter 2: Cells on asymmetric receptor patterns
26
range forces such as fluid flows and gravity. Second, the chemical kinetics of the reaction
between the ligands on the surface to receptors on the cell dictates the adhesion process
of the cells to the affinity surface99. However, in cases where the chemical affinity of the
receptor-ligand interaction is weak, a third transport process becomes important. In such
a scenario, bonds between the cell and surface breaks under the mechanical stress of the
fluid flow resulting in transient interaction of the cell with the surface, allowing the cell
to move along the direction of the force (fluid flow)100. This phenomenon, known as cell
rolling
101
, occurs due to continuous breakage of bonds due to fluid shear at the trailing
edge while new bonds form at the leading edge resulting in a ‘rolling’ motion of the cell
on the substrate. Rolling of leukocytes, lymphocytes and stem cells has been studied
extensively in vivo101 and in vitro102 and many interesting observations about the
dynamics of cells on selectin coated surfaces been reported102-107. For example, it was
observed that the rolling of cells is fundamentally stochastic with periods of slow velocity
followed by fast hopping of cells where the cells often return to free stream
velocity102,105. The role of cellular structures such as the microvillus and kinetics of the
receptor ligand interaction have been shown to play a crucial role on stability of rolling
and its resistance to fluid shear stress108. Cell rolling has also been studies at a
fundamental nano- as well as macro- scale though a number of analytical109, semianalytical110,111 and computational models112-114. However most of these studies examined
the motion of cells on unpatterned selectin coated surfaces. Karnik et.al94 in their
pioneering study showed that patterned edges of selectin can deflect the motion of rolling
cells along the edge direction. Based on that idea, a new class of separation devices is
envisioned where the effect of displacement by asymmetric edges is amplified though
interaction over multiple edges, resulting in separation of the target cells as shown in
Figure 2.1(a). Since the mechanism of rolling on the receptor pattern edge is very
different from that over plain surface, the results from the studies on the later form of
rolling cannot be directly extrapolated to understand rolling on edge. Hence, fresh studies
on quantitative understanding of the nature of cell rolling trajectories and lateral
displacement on receptor-patterned substrates is warranted. Figure 2.1(b) shows the
trajectories of cells rolling within such a patterned device substrate. Studies by Karnik
et.al demonstrated that HL60 cells could track along P-selectin edges94; however, the
Chapter 2: Cells on asymmetric receptor patterns
27
effect of flow conditions and pattern inclination angle on cell rolling trajectories was not
studied. Furthermore, the distribution of edge tracking lengths and lateral displacements
was not analyzed due to insufficient data obtained from a single patterned edge.
Therefore, how the parameters of cell rolling relate to cell rolling trajectories and lateral
displacements for such asymmetric patterns is poorly understood. For example, it is
unknown to what extent cell rolling is affected by shear stress magnitudes or pattern
inclination angles. Further, the nature of the detachment of cells after tracking along an
edge is not known, and is it not yet established whether detachment of cells from such
patterns is a random process that is unaltered by interaction history with the pattern itself.
Systematic study of cell rolling trajectories along such well-defined receptor patterns is
therefore prerequisite for addressing these issues and enabling the development of labelfree devices for separation or analysis of cells such as the device envisioned in Figure
2.1.
In this chapter, the cell rolling trajectories have been quantified and the effect of pattern
geometry, shear stress, and P-selectin incubation concentration on rolling of HL60 cells,
which are widely used as a model to study leukocyte rolling90,101,115-117, have been
studied. HL60 cell surfaces express a specific ligand termed P-selectin glycoprotein
ligand-1 (PSGL-1)101 , which binds reversibly to the receptor P-selectin to enable rolling
in vivo and in vitro. A technique based on microcontact printing (µCP) to pattern
alternating µm-scale lines of adhesive P-selectin regions with passivating poly(ethylene
glycol) regions on a gold substrate was developed. The edge tracking lengths and rolling
velocities of HL60 cells along these patterned substrates within a flow chamber at
different edge inclination angles and shear stress magnitudes were quantified. The
distribution of edge tracking lengths and observation of re-attachment of cells were
incorporated into a computational simulation tool to predict the trajectories of cells on a
patterned substrate.
Chapter 2: Cells on asymmetric receptor patterns
28
Figure 2.1. (a) Schematic diagram of a device for separation of cells. Cells rolling along
patterned edges are laterally displaced into the adjacent buffer stream, resulting in
separation. Pink lines indicate receptor-functionalized regions. Red and blue circles are
cells that do and do not, respectively, express ligands that bind specifically to those
receptors. (b) Illustration of a typical cell rolling trajectory along the receptor pattern
inclined an angle α to the fluid flow direction: The cell binds within the receptor line, and
rolls in the direction of shear flow toward the pattern edge. The cell then tracks the edge
to define an edge tracking length le, resulting in a net lateral displacement d, before
detaching to continue along the direction of fluid flow before possible reattachment and
rolling along a new receptor line. Cell rolling velocity vp along within the receptorfunctionalized line in the x-direction of fluid flow can be distinguished from the velocity
ve along the line edge, where ve,y is lateral velocity (the vertical component perpendicular
to the streamlines and parallel to the lateral displacement, d).
2.2 Materials and Methods
2.2.1 Materials
Gold-coated glass slides were purchased from EMF Corp. All slides were cleaned with
piranha solution prior to use (3:1 mixture of sulfuric acid (Sigma-Aldrich) to 30%
hydrogen peroxide (Sigma-Aldrich)). (1-Mercaptoundec-11-yl)tetra(ethylene glycol)
(PEG alkanethiol; Sigma-Aldrich) was diluted in absolute ethanol (Pharmco-AAPER) at
a concentration of 5 mM for microcontact printing. Recombinant human P-selectin (R&D
Chapter 2: Cells on asymmetric receptor patterns
29
Systems Inc.) and bovine serum albumin (BSA, Rockland Immunochemicals, Inc.) were
diluted in 150 mM NaCl Dulbecco's phosphate buffered saline (DPBS, Mediatech Inc.).
All materials employed in this study were used without further purification unless
specified.
Figure 2.2 . Schematic diagram for patterning of P-selectin on a gold substrate involving
microcontact printing. Step 1: Selective deposition of PEG molecules on the gold surface.
Step 2: Filling in of the uncoated surface with P-selectin.
PDMS Stamps. Microcontact printing stamps that defined the receptor pattern were
fabricated in polydimethylsiloxane (PDMS) using an SU-8 molding process. The small
line-patterned stamp (SS) (15 µm line width and 10 µm spacing between adjacent lines)
and the large line-patterned stamp (LS) (70 µm stamping regions spaced 50 µm apart)
were used to characterize the patterning process and cell rolling behavior, respectively.
2.2.2 Fabrication of Patterned Substrates.
A schematic diagram of the patterning process is shown in Figure 2.2.
Step 1: Microcontact printing (µCP) was used to form alternating self-assembled
monolayers (SAMs) of PEG molecules on the gold substrate. The PDMS stamp was first
inked with PEG solution in ethanol (5 mM), dried, and pressed onto the surface to be
patterned for 40 s. The surface was then rinsed with ethanol and dried under a stream of
N2.
Step 2: After selective deposition of PEG molecules, the substrates were incubated in Pselectin solution (15 µg/mL in DPBS, unless stated otherwise) using a perfusion chamber
(Electron Microscopy Sciences) for 3 hours at room temperature to pattern the remaining
areas with P-selectin. The surfaces were then backfilled with BSA (1 mg/mL in DPBS)
for 1 h to block non-specific interactions.
Chapter 2: Cells on asymmetric receptor patterns
30
Substrate Characterization. Atomic force microscopy (Veeco Dimension 3100; Tapping
mode; scan rate: 1 Hz) and scanning electron microscopy (JEOL 6700; acceleration
voltage 3.5 kV) were used to characterize the patterned surface geometry. All substrates
for AFM and SEM characterization were placed in a vacuum chamber overnight before
imaging to minimize residual solvent on the surface; no further coating was employed for
SEM imaging.
2.2.3 Cell Rolling Experiments in a Flow Chamber.
A suspension of HL60 cells (~105 cells/mL) was flowed over the patterned surfaces in a
rectangular flow chamber (Glycotech, Inc; width w = 1.0 cm; length = 6 cm; height h =
0.005”) with inclination angles of the receptor pattern of either α = 5º, 10º, 15º or 20º at
room temperature of 24.5°C. A syringe pump (World Precision Instruments (WPI),
SP230IW) was used to generate different flow rates between 75 and 300 µL/min, with
corresponding shear stresses of 0.5 – 2.0 dynes/cm2 (~0.05 to 0.2 Pa). Flow was laminar
(Reynold’s number Re ~ 0.1-3) and shear stress τ was calculated using the plane
Poiseuille flow equation τ = 6µQ/wh2, where µ is the kinematic viscosity, Q is volumetric
flow rate, w is width of the flow chamber, and h is height of the flow chamber. An
inverted microscope (Nikon TE2000-U) with a mounted camera (Andor iXon 885) was
used to record HL60 rolling interactions with adhesive P-selectin substrates using a 4×
objective, typically at a rate of 1 frame per second for durations of 300 s. For each shear
stress magnitude and pattern inclination angle, three independent experiments were
performed. Data are presented as mean and standard deviation of the average values
obtained from each experiment.
2.2.4 Data Analysis.
The experiments in flow chamber produces a large volume of data in form of time
sequenced images, which then needs to be analyzed to fish out the relevant data. We
developed a custom in-lab program, coded in Matlab (Mathworks, Inc.) program that
utilized a particle tracking freeware118 to detect the cells and generate tracks along the
patterned line edges. We defined various filters to remove spurious tacks and then used
Chapter 2: Cells on asymmetric receptor patterns
31
optimized fitting programs to analyze the tracks. The algorithm and the methodology is
detailed below.
Tracking of cells: The general scheme of the algorithm is shown in Figure 2.3. The image
sequences were analyzed using a customized Matlab (Mathworks, Inc.) program that
utilized a particle tracking freeware118 to detect the cells and generate tracks along the
patterned line edges. The procedure used to generate tracks has been described in earlier
work94. Typical rolling velocity of cells are in the range of 1-10 µm/sec while free
flowing cells move close to fluid stream velocity typically >100 µm/sec. We set a
tracking criterion that required the cell displacement between consecutive frames to be
less than 10 µm. This criterion successfully filtered out the free flowing cells, while
conservatively tracking rolling cells successfully. Note that while setting this criterion at
a higher value could potentially increase the number of cells tracked (as cells moving
with higher velocity will also be tracked), it also increases the chances of spurious tracks
as the probability of proximal tracks being getting connected increases. Through a
manual trial method we found that setting the search radius of 10 µm gave best result. It
was also observed that cells which jumped for a short distance while rolling resulted in
the tracks being broken into a number of smaller segments. Thus a second filter was used
wherein tracks with total length <40 µm were not included in analysis of the distribution
of cell responses, as these short tracks predominantly represented unlinked fragments of a
single track and in some cases artifacts on the substrate such as pinholes. Tracks
generated by the software were randomly selected and inspected manually by comparing
with the images to ascertain their accuracy.
Sorting of Tracks: The patterned edges were identified using the difference in contrast
between the PEG and P-selectin regions as imaged using optical microscopy. The
positions of the edges were calculated based on the information of position of one edge,
and the geometry of the pattern. Following this, the starting and end point of each track
was compared with the edge list and tracks with endpoints within 10 µm of the nearest
edge were identified as having encountered an edge. This way, the tracks were sorted into
three major classes.
Chapter 2: Cells on asymmetric receptor patterns
32
Type ‘A’: Tracks that does not reach the edge.
Type ‘B’: Tracks that reach the edge and also roll on it.
Type ‘C’: Tracks of cells only on edge.
Tracks of type ‘A’ and ‘C’ are used only for velocity calculation on the plain surface and
edge, while tracks ‘B’ are used for calculating the edge tracking length le , along with
calculating the velocities.
Identification of the portion of a track representing cell rolling along the edge: Two
intersecting straight lines were fitted to every selected track – one aligned with the flow
direction and the other aligned with the edge. A constrained error minimization scheme
with the slopes of the two lines and the intersection point (Pi) as the fitting parameter was
used. While the slopes of the two lines were allowed to vary ±3o from the flow direction
and the angle of the pattern, respectively, their intersection point was confined such that
the x- and y- coordinates of the intersection point were within the limits of the minimum
and maximum values of the x- and y- coordinates of the points on the track. Thus the
tracks were subdivided into two segments – one that represented rolling inside the Pselectin line, and another representing rolling on the edge (Figure 2.5, inset).
Extraction of edge tracking length, le: The length travelled along the patterned line edge
was calculated from the distance between Pi and the end of the track. In order to avoid
biasing the population, tracks which were restricted by space (field of view of the
camera) or time (tracks that existed before or continued after the image sequence) or
tracks which only rolled on the edge without a segment of track on the band (distance
between the intersection point Pi and beginning of track <10 µm) were excluded from the
calculation of the average edge tracking length.
Extraction of rolling velocities on the edge and within the P-selectin lines: Velocity was
calculated by choosing two points on the track and dividing the distance between the
points by the time taken to traverse them. We observed that the rolling velocity was
smaller around Pi when a cell transitioned from rolling within the P-selectin line to
Chapter 2: Cells on asymmetric receptor patterns
33
rolling along the edge. We therefore excluded the portion of the track which was within a
distance of 10 µm from the predicted Pi for calculation of rolling velocities. Thus, the
starting point and the point located 10 µm before Pi, were used for calculating the rolling
velocity vp within the patterned lines, while the point located 10 µm ahead of Pi and the
end point of the track were used for calculating the velocity ve along the edge of the
patterned lines. Tracks that did not encounter the patterned edge (Type ‘A’ tracks) were
used only to calculate vp, while the tracks which were confined only to the edge (Type
‘C’ tracks) were used only for calculating ve, each by taking the ratio of total track length
to the total elapsed time. Thus for each experiment an array of edge tracking length,
velocity on edge and velocity on plain region was generated and used for further analysis.
Chapter 2: Cells on asymmetric receptor patterns
Figure 2.3. Flow chart describing the cell tracking and analysis algorithm.
34
Chapter 2: Cells on asymmetric receptor patterns
35
2.2.5 Simulation of Cell Rolling Trajectories.
A Monte Carlo simulation of rolling of cells on a substrate patterned with P-selectin lines
with edge inclination angle α was performed by assuming that the cell detachment from
the edge followed a Poisson distribution. The value of λ for the Poisson distribution
corresponding to the edge inclination angle α was obtained as described in Restults and
Discussion. The direction of fluid flow was along the positive x-direction and the cells
were assumed to roll on all P-selectin lines that they encountered. For each cell, the edge
tracking length was calculated by generating a random number based on the Poisson
distribution, and the position of the cell was correspondingly updated. The cell was then
assumed to detach and begin rolling on the next downstream edge at the same ycoordinate. This process was repeated with the cell starting at (0, 0) on the first edge,
until the x-coordinate of the cell was equal to the travel distance, which yielded a final ycoordinate (net lateral displacement). The above sequence was iterated for 105 cells and
the final y-positions of all cells were used to calculate the probability density.
2.3 Results and Discussion
Direct microcontact printing (µCP) of proteins has been used widely to control the
geometry of protein patterns on various planar surfaces119-124, including printing of Pselectin to study neutrophil rolling
125
. However, it is possible that the protein will
denature or lose bioactivity during PDMS stamping steps126. Additionally, transfer of the
stamp material (PDMS) from the stamp to the surface during µCP can contaminate the
patterned areas127. In the present method, after selective deposition of PEG molecules, the
gold substrate is patterned with P-selectin in the solution phase so the possibility of
denaturation due to protein drying can be ruled out. AFM images of P-selectin patterned
using the small line-patterned stamp (SS) show clearly defined 10 µm-wide lines of Pselectin with well-resolved, straight edges (Figure 2.4(a), (b)). The large line-patterned
stamp (LS) was used to prepare surfaces for cell rolling experiments. The sharp contrast
between P-selectin regions and PEG regions confirms that the resulting patterns had welldefined edges over large areas as revealed by SEM (Figure 2.4(c), (d)). In addition to this
Chapter 2: Cells on asymmetric receptor patterns
36
surface characterization, we observed that HL60 cells exhibited rolling specifically in the
P-selectin patterned regions with velocities in a similar range as those reported in other
studies94. These results confirm that the PEG-functionalized regions on either side of Pselectin lines could block P-selectin adsorption (as expected from the non-fouling
property of PEG) and that P-selectin molecules retained their activity after being
adsorbed to the exposed gold. In addition, we performed experiments to characterize cell
adhesion on PEG surfaces and surfaces coated with BSA, and did not observe any cellsurface interactions, further confirming that the observed interactions were due to Pselectin.
Figure 2.4. Characterization of P-selectin patterned substrates for cell rolling. AFM
images of 10 µm wide P-selectin lines separated by 15 µm wide PEG bands (after step 2),
displaying the contrast between P-selectin and PEG regions in (a) height and (b) phase,
respectively. The phase image indicates a difference between the mechanical properties
of the surface in the two regions. SEM images of surfaces after PEG printing (c) (step 1)
and after P-selectin adsorption (d) (step 2), respectively, showing uniformity of
patterning (brighter areas correspond to PEG regions). Scale bars are 5 µm in (a) and
(b) and 100 µm in (c) and (d).
Chapter 2: Cells on asymmetric receptor patterns
37
The P-selectin patterned substrates were incorporated into a flow chamber for studying
the rolling behavior of HL60 cells at different edge inclination angles and shear stress
magnitudes. Figure 2.5 shows an example of tracks obtained from the automated tracking
software for an experiments where HL60 cells were flowed over patterns at an edge angle
of
10°
and
shear
stress
of
0.5
dyn/cm2
(Movie
available
at
http://pubs.acs.org/doi/suppl/10.1021/la102871m/suppl_file/la102871m_si_001.qt).
2.3.1 Effect of Edge Angle on the Rolling Behavior of HL60 Cells.
We first examined the effect of the edge inclination angle α subtended by edges of the Pselectin lines with respect to the direction of fluid flow on the edge tracking length le, the
lateral displacement d, and the rolling velocities vp an ve, respectively. At α=5º, HL60
cells rolled an average distance of more than 135 µm along the edges before detachment
at a shear stress of 0.5 dyn/cm2. Figure 2.6(a) shows that, as the edge angle was increased
from 5º to 20º in 5º increments, the average edge tracking length le decreased
significantly (ANOVA, F = 18.403, p = 0.001). In other words, the ability of the cells to
roll along the edges was reduced with increasing edge inclination angle (and increasing
component of the fluid force on the cell directed away from the edge). Comparison of
data pairs (post-hoc t-test) demonstrated statistically significant differences in le for every
5º increase in α. In contrast, Figure 2.6(b) shows that the lateral displacement d = le sinα,
did not show a significant trend with increasing α (ANOVA, F = 3.075, p = 0.091), and
varied from 7.0 to 12.5 µm over this range of α. However, a statistically significant
difference was observed between the lateral displacements at α=10º and 20º (post hoc ttest). This behavior can be understood in that although sinα increases with increasing
edge inclination angle, edge tracking length le concomitantly decreases. It is the
magnitude of this lateral displacement that is more relevant to separation of cells by
rolling on such a patterned substrate.
Chapter 2: Cells on asymmetric receptor patterns
38
Figure 2.5. Tracks of HL60 cells rolling
(blue lines) on P-selectin lines (pink) were
obtained
by
analyzing
300
images
acquired at 1 fps using a customized
Matlab code. The edge inclination angle
was 10° and the shear stress was 0.5
dyn/cm2.
Inset
shows
a
track
corresponding to a cell that first rolled
inside the P-selectin line (green) in the
direction of fluid flow and then tracked
along the edge (black).
Figure 2.6. Effect of edge inclination angle α on rolling behavior of HL60 cells at a fluid
shear stress magnitude of 0.5 dyn/cm2. Variation of (a) edge tracking length, le; (b)
lateral displacement, d; (c) rolling velocities vp and ve within the P-selectin lines and on
the edge, respectively; and (d) lateral velocity, ve,y (component of the edge rolling velocity
in the direction of lateral displacement). Error bars represent one standard deviation,
where n = 3 replicate experiments for each condition.
Chapter 2: Cells on asymmetric receptor patterns
39
In addition to altering the direction of cell rolling, asymmetric receptor patterns can also
alter the rolling velocity of the cells94. To examine the effect of α on rolling velocity, we
quantified the average rolling velocity of cells within and along the edges of the Pselectin lines as a function of α at a fixed shear stress magnitude of 0.5 dyn/cm2 (Figure
2.6(c)). The rolling velocity within the P-selectin line was in the range of vp = 2.9 - 3.6
µm/s, and was always less than that along the edge regions (vp = 4.4 - 6.0 µm/s). This
can be understood in terms of the expectation that, as α increases, surface area of
interaction and adhesion resistance to rolling between the cell and the surface decreases,
leading to an increase in rolling velocity. Pairwise (t-test) statistical analyses show a
significant increase in ve at large edge angles (15º and 20º) compared to vp, consistent
with previous observations94. The average rolling velocity on the edge ve increased from
4.4 µm/s for an edge inclination angle of 5º to 6.0 µm/s at an edge inclination angle of
20º, though this trend did not reach a degree of statistical significance (ANOVA, F =
3.55, p=0.067). In contrast, ve,y (lateral velocity, defined previously as the edge velocity
component in the direction of lateral displacement d) increased significantly from 0.4
µm/s to 2.1 µm/s as α increased from 5º to 20º. Thus, receptor patterns characterized by
large edge inclination angles (α=15º, 20º) led to greater lateral displacement of cells over
a given rolling duration.
2.3.2 Effect of Shear Stress on Rolling Behavior of HL60 Cells.
We next examined the effect of shear stress (τ= 0.5 dyn/cm2 to 2.0 dyn/cm2) on rolling
behavior of HL 60 cells. Figure 6 summarizes the edge tracking length, lateral
displacement, and rolling velocity of as a function of the τ at a fixed edge angle of 5º.
The edge tracking length le varied in the range of 118.6 - 173.1 µm over τ = 0.5 to 2.0
dyn/cm2. However, there was no statistically significant effect of shear stress on le
(ANOVA, F=2.119, p=0.176) (Figure 2.7(a)). The lateral displacement d varied between
10.3 - 15.1 µm, again with no statistically significant dependence on the shear stress
(Figure 2.7(b)). Similarly, Figure 2.7(c) shows that the rolling velocity within the Pselectin lines and on the edge also did not vary significantly with shear stress (ANOVA,
p=0.917 and p=0.165, respectively). This lack of dependence on shear stress is not
Chapter 2: Cells on asymmetric receptor patterns
40
surprising, given that cell rolling involves mechanisms at the cellular and molecular
levels to regulate the rolling response over a range of shear stresses128. Similar
independence of rolling velocity with shear stress has been observed before in the case of
HL60 cells rolling on E-selectin129: the rolling velocity of HL60 cells has been observed
to increase with shear stress at low shear stress (τ<0.5 dyn/cm2) and reach a plateau at
higher shear stresses. Our experiments indicate that similar to rolling within the Pselectin line, shear stress also does not have a significant effect on the rolling behavior of
HL60 cells on asymmetrically patterned edges within τ = 0.5 to 2.0 dyn/cm2.
Figure 2.7. Effect of shear stress τ on rolling behavior of HL60 cells at an edge
inclination angle of 5º. Variation of (a) edge tracking length, le; (b) lateral displacement,
d; (c) rolling velocities vp and ve within the P-selectin lines and on the edge, respectively;
and (d) the lateral rolling velocity, ve,y. Error bars represent one standard deviation,
where n = 3 replicate experiments for each condition.
Chapter 2: Cells on asymmetric receptor patterns
41
2.3.3 Effect of P-selectin Incubation Density on Rolling Behavior of HL60 Cells.
Apart from shear stress and edge inclination angle, the P-selectin density on the surface
may be expected to affect the trajectories of cells rolling on the inclined edges. Increasing
P-selectin density on microslided surface under high shear stresses (~20 dyn/cm2)
resulting in decreasing rolling velocity of HL60 cells has been observed, whereas
receptor density has less effect on rolling velocity under low shear stress (< 2
dyn/cm2)116. Particle rolling velocity decreases with increasing E-selectin density at low
relative site density and reach a plateau at high relative site density and low shear stress
(0.6 dyn/cm2), which suggests that the sensitivity of particle rolling velocity occurs when
the site density of E-selectin is low.
100
. When the P-selectin incubation density was
varied in the range of 5 to 30 µg/mL while maintaining an incubation time of 3 h, we
observed a change from no rolling adhesion to robust rolling adhesion around a P-selectin
concentration of 15 µg/mL, and then a change from rolling adhesion to slow rolling /firm
adhesion around a P-selectin concentration of 30 µg/mL, at a shear stress of 0.5 dyn/cm2.
Thus, the useful range of P-selectin incubation concentrations that resulted in a useful
rolling response was 15 to 25 µg/mL. We therefore characterized the edge tracking length
and cell rolling velocities in this range of P-selectin concentrations. Interestingly, we did
not observe a significant change in rolling velocity with change of P-selectin incubation
concentration: the average rolling velocities were 3.15±0.23, 2.69±1.16, and 3.14±0.23
for P-selectin concentrations of 15, 20, and 25 µg/mL at edge inclination angle of 10° and
4.79±0.8, 4.07±0.99, and 4.42±0.86 for P-selectin concentrations of 15, 20, and 25
µg/mL at edge inclination angle of 20°, respectively. Similarly, we did not observe
significant change in the cell behavior along the edge including edge tracking length le,
lateral deflection d, edge rolling velocity Ve, and lateral velocity Ve.y. These results are
in agreement with previous observation of less change in rolling variation with a
variation of ligand density under low shear stress (< 2 dyn/cm2) done by Dong et al116.
These results indicate that when P-selectin is directly immobilized on a gold substrate,
the rolling behavior along the edge cannot be controlled as easily as that by changing the
edge inclination angle.
Chapter 2: Cells on asymmetric receptor patterns
42
Figure 2.8. Effect of P-selectin incubation concentration on rolling behavior of HL60
cells at a shear stress of 0.5 dyn/cm2. Variation of (a) edge tracking length, (b) lateral
deflection, (c) edge rolling velocity, and (d) lateral velocity with P-selectin incubation
concentration at edge inclination angles α= 10º and 20º.
2.3.4 Detachment of cells rolling along an edge can be described by a Poisson
process.
While the average edge tracking length and lateral displacement are useful to elucidate
the effect of edge angle and shear stress on cell rolling, knowledge of the distributions of
the distance rolled along the edge and the lateral deflection is important for predicting the
eventual distribution of a population of cells in a separation device. Similarly, this
knowledge is required to elucidate the number of edge tracking events that must be
observed to distinguish between cell phenotypes by observing rolling on the patterned
substrates. We therefore examined the distribution of the edge tracking lengths, with the
aim of developing a model that would serve as a tool to predict cell rolling trajectories
and their spread, with direct implications on analysis resolution and separation efficiency.
Chapter 2: Cells on asymmetric receptor patterns
43
Figure 2.9. Detachment of cells rolling along an edge is well described by a Poisson
distribution. (a) Cumulative distribution function of edge tracking lengths le (filled
triangles) was fitted to a Poisson distribution described by Eq. 1 (solid lines). Insets show
the frequency distribution of the experimentally measured edge tracking lengths, along
with that predicted by the Poisson distribution fit to the CDF (solid lines). Colors
indicate different inclination angles α of the receptor pattern. Representative results are
shown for only one experiment for each α. (b) Variation of the average value of λ with
edge inclination angle is well described by a linear fit on a semi-log plot (solid line). (c)
Variation with the edge inclination angle of the average value of the lateral displacement
(solid line) obtained from the empirical fit shown in (b) along with the experimental
results (open circles). Error bars in (b) and (c) represent one standard deviation. Shear
stress is 0.5 dyn/cm2.
Chapter 2: Cells on asymmetric receptor patterns
44
We observed that for all the experiments conducted, distributions of the edge tracking
length exhibited a decaying exponential characteristic similar to that of a Poisson process
(Figure 2.9). Poisson process occurs when individual events within the process are
random in nature with uniform probability of occurrence independent of history. It is well
known that cell rolling is a stochastic process involving discrete adhesive interactions
between the cell and the surface109,112,114,130. Rolling cells continuously form adhesive
contacts with the surface with receptors localized at the tips of extensible structures
known as microvilli131. In the case of cells rolling along an edge, the region of overlap
between the cell’s contact area and the receptor-coated region is the site where new cellsurface adhesions are formed. As the cell tracks along an edge, new cell-surface
adhesions are continuously formed in the region of overlap between the cell’s contact
area and the receptor-coated region. The cell will detach from the surface when a new
adhesive interaction fails to form before the last adhesive interaction is broken under
shear flow. If the probability of formation of new adhesive interactions in a given period
is constant (i.e., independent of the past rolling history of each cell and/or uncorrelated
among cells within a population), detachment is expected to be a random event and the
distance traveled by each cell along the edge is expected to follow a Poisson distribution.
To test this hypothesis, we calculated the cumulative distribution function (CDF) from
the data and fit it with a Poisson distribution (Figure 2.8) given by:
C (le ) = 1 − exp(−le / λ )
(2.1)
Here, λ is the mean value of the Poisson distribution. In the ideal case where detachment
is a random process, λ will approach the mean value of the edge tracking length. The
CDF does not involve any arbitrary bin widths and therefore allows for an objective
comparison of the actual distribution of the edge tracking lengths with that predicted for a
Poisson process (Eq. 2.1). Note that the CDFs displayed in Figure 2.8 do not begin at the
origin because edge tracking lengths <10 µm could not be resolved by the present video
frame rate and magnification; these unresolved lengths were used for calculation of the
CDF itself but were not used for fitting the Poisson function. The Poisson distribution
described by Eq. 2.1 well fit the CDF for all edge inclination angles considered, and the
Chapter 2: Cells on asymmetric receptor patterns
45
mean value of λ obtained by fitting to the CDF also accurately predicted the observed
edge tracking length histograms (Figure 2.9(a), inset) at different edge inclination angles.
Additionally, to confirm that the process of detachment of cells from the edge was indeed
a Poisson process for a given cell, we calculated λ under different experimental
conditions and compared it to the measured average edge tracking lengths (Table 1). We
found strong correspondence between λ and the average edge tracking length from the
experiments; even at different shear stress magnitudes, we found the maximum difference
to be within 7% (data not shown). As an additional confirmation, we also manually
compared the edge tracking lengths for several cells that exhibited multiple rolling and
rebinding events within a single experiment; we found no correlation between the initial
and subsequent le for such cells, as would be expected for a history-independent
detachment process (data not shown). These data strongly suggest the fact that
detachment of these HL60 cells rolling along an edge is indeed a random process.
Interestingly, we observed that the average value of λ obtained by fitting the CDF to the
Poisson distribution showed an exponential decrease with increasing edge inclination
angle (Figure 2.9(b)). This observation provides an empirical relation between the edge
angle and the average edge tracking length, while the distribution of the lengths itself can
be modeled using a Poisson process. Thus, this exponential dependence combined with
modeling cell detachment as a Poisson process can be used to interpolate the average
edge tracking length as well as its distribution for any edge angle between 5o and 20o. We
also predict the variation of lateral displacement with edge inclination angle based on this
exponential fit which agreed well with the experimental data (Figure 2.9(c)).
Interestingly, the curve predicts an optimal edge inclination angle between 5º and 10º that
maximizes the lateral displacement for rolling along a single edge. This prediction
explains why a statistically significant difference was not observed between lateral
displacements at edge inclination angles of 5º and 10º (Figure 2.6). The average lateral
displacement exhibits a maximal value at the edge inclination angle between 5º and 10º.
Chapter 2: Cells on asymmetric receptor patterns
46
Table 2.1. Comparison of the experimental and Poisson average value of λ.
Edge angle
Poisson mean
Experimental average
%Error
λ±SD (µm)
le±SD (µm)
100.(λ-le/)le (µm)
5
134.1±33.2
135.5±36.3
-0.98%
10
66.2±21.7
72.2±17.2
-8.35%
15
34.7±9.4
37.2±7.6
-6.60%
20
20.3±3.9
20.3±3.7
0.11%
* n = 3 replicate experiments. Data were obtained by fitting to a Poisson distribution with
experimentally measured average edge tracking length le. Shear stress is 0.5 dyn/cm2.
2.3.5 Prediction of cell trajectories on a receptor-patterned substrate
The experimental results obtained in this study enable us to develop a simulation tool for
predicting the trajectories and distribution of cells rolling across a substrate patterned
with asymmetric P-selectin lines. Here we incorporate the experimental observations to
simulate the downstream distribution of HL60 cells that are injected at a particular
location (0, 0) on a surface (similar to Figure 2.1(a)). In the simulation study, the device
was assumed to be patterned with parallel P-selectin lines having a width of 50 µm and a
gap of 70 µm between adjacent lines (similar to the pattern used in experiments). In
addition to rolling behavior of cells on the P-selectin regions, re-attachment of cells that
have detached is an important parameter for predicting the trajectories of cells that
encounter several parallel edges. From our experiments we observed that after
detachment from the edge >80% of the cells reattached at the next available downstream
P-selectin line at a shear stress of 0.5 dyn/cm2. Hence, in our simulation we assumed that
cells rolled on all the P-selectin lines that they encountered with the edge tracking lengths
described by a Poisson distribution with the mean λ as obtained earlier.
We performed a Monte Carlo simulation for 105 cells, each entering the pattern
sequentially from a single point source and predicted their net lateral displacement and
resulting distributions at a longitudinal distance of 1 cm downstream of the origin. Figure
2.10(b) summarizes the probability density functions of the net lateral displacement,
defined here as the sum of all displacements (analogous to the experimental lesinα in
Chapter 2: Cells on asymmetric receptor patterns
47
Figure 2.1(a)) that a given cell acquired over the longitudinal travel distance of 1 cm. The
net lateral displacement increased with increasing edge inclination angle α from 5º to
15º, with little change as α increased from 15º to 20º. The net lateral displacement
showed maximum sensitivity to the edge inclination angles between 5o to 10o. This αdependence and sensitivity can be understood as follows. The net lateral displacement
was the result of displacements on each edge encountered by the cells. Increasing the
edge inclination angle α increases the total number of edges encountered by the cells,
but simultaneously decreases the mean lateral displacement on each edge (λsinα)
(Figure 2.9(c)). These opposing effects resulted in the net lateral displacement exhibiting
an increase till α = 15º, and little change between 15º and 20º. Interestingly, if we
assume that the logarithmic dependence of the edge tracking length on α (Figure 2.9(b))
is valid at α = 25º, the net lateral displacement decreases to 152±24 µm, indicating that
the effect of decreasing edge tracking length offsets the effect of the increased edge
inclination angle. With increasing number of edges encountered (i.e. at larger a), the
distribution of cells is seen to approach a Gaussian distribution, consistent with the
central limit theorem. Interestingly, although α=10°˚ and 20º yield similar net lateral
displacements, it is noteworthy that the Gaussian spread is smaller for α=20°˚. In fact, the
ratio of the mean displacement to the standard deviation increases monotonically from
2.72 to 5.68 as the inclination angle increases from 5º to 20º. This result can again be
explained by the fact that the cells undergo more tracking events with increasing α due
to the higher number of edges per unit length in the direction of flow. It is noteworthy
that although the distribution of cells approaches a Gaussian form, it is not exactly
Gaussian as some of the cells in the distribution are rolling along the edge; these cells
create “spikes” in the distribution at specific positions where the edges intersects the
axis (at a longitudinal travel distance of 1 cm). Simulations for longitudinal travel lengths
up to 10 cm confirmed that the mean lateral displacement remained proportional to the
longitudinal distance traveled by the cells; although the spread of the distribution
increased, being approximately proportional to the square root of the downstream
distance as is typical of chromatographic separation (data not shown). These results
demonstrate that a microfluidic device based on asymmetric receptors patterns can be
Chapter 2: Cells on asymmetric receptor patterns
48
used for efficient modulation of cell rolling trajectories, as required for flow-based
separation of cells. From the distributions in Figure 2.10, it can be predicted that a 100
µm wide stream of a heterogeneous mixture of receptor-binding and non-binding cells
can be separated for a receptor pattern of α=20o and device of length 1 cm provided the
solution is dilute enough to minimize dispersion due to cell-cell interactions.
Figure 2.10. (a) Probability distribution of net lateral displacement of HL-60 cells after
rolling on three consecutive bands of P-selectin patterns as obtained from Monte Carlo
simulations (shaded area) and experimental observations. α = 20º (shear stress 0.5
dyn/cm2). (b) Prediction of the downstream distribution of HL60 cells rolling on
patterned P-selectin in a separation device. HL60 cells are introduced at a single
location and travel a distance of 1 cm in the downstream direction, interacting with a
substrate patterned with P-selectin lines of 50 µm width separated by 70 µm PEG gaps.
Chapter 2: Cells on asymmetric receptor patterns
49
Net lateral displacement of 105 cells was calculated assuming the detachment on edge
was governed by a Poisson process with parameters as obtained earlier by fitting to
experimental data at a shear stress of 0.5 dyn/cm2. The numbers in parentheses denote
net lateral displacement and standard deviation. The “spikes” appear at the position
where the edges intersect the 1 cm downstream position (full extent of the spikes is not
shown).
2.4 Conclusion
In summary, we have designed substrates with multiple P-selectin patterns by a facile,
versatile method utilizing microcontact printing. These substrates were incorporated in a
flow chamber for studying HL60 cell rolling behavior including quantification of edge
tracking lengths and cell rolling velocities. Among the parameters we considered, the
pattern edge inclination angle modulated the cell rolling trajectory most strongly; in fact,
the edge tracking length decreased exponentially with increasing edge inclination angle.
In addition, the nature of cell rolling and detachment along the edge was consistent with
the Poisson process. This correlation suggests that detachment of rolling cells from
receptor-functionalized edges is a random process that is not disrupted measurably by
cell-surface interactions (e.g., pattern biofouling or transmembrane ligand redistributions)
over the device timescales considered. Experimental characterization of cell rolling
enabled the development of a computational Monte Carlo tool to simulate the trajectories
of cells. These simulations indicated that cell separation may be achieved within a short
(cm-scale) separation channel and also provided a potentially useful approach to optimize
pattern geometry to ultimately maximize cell separation efficiency.
The increasing
resolution obtained by multiple tracking events on parallel edges indicates that small
differences in rolling behavior can be amplified and resolved by increasing the
longitudinal traveling distance or the number of edges encountered by thes cells,
analogous to other analytical processes. As cell rolling behavior depends on the ligand
density on the cell (among several other parameters), we hypothesize that different cell
phenotypes can be separated or identified based on difference in their characteristic
lateral displacements. While the present work does not directly address the feasibility of
Chapter 2: Cells on asymmetric receptor patterns
50
separating different types of cells that interact with a given selectin receptor, it lays the
groundwork for future studies in this direction by elucidating the statistical nature and
effect of edge inclination angle on cell rolling along patterned edges. The ultimate goal is
to develop devices that incorporate asymmetric receptor patterns for point-of-care
diagnostic and therapeutic applications where rapid cell sorting or analysis with minimal
cell processing would be beneficial. Our work indicates the feasibility of realizing such
devices, and also provides quantitative tools for future device design.
Acknowledgement
I would like to acknowledge the contribution of Ms. Chia-Hua Lee who is an equal
author contributor to the Langmuir paper, where this work is published. Chia-Hua led the
development of the microcontact printing technique and performed the experiments with
HL60 cells. I lead the developed of the automated image analysis software and data
analysis. I would also like to acknowledge the contribution of Ms. Minhee Sung and Mr.
Lim Wanapahoon who helped with the experiments and data analysis.
3
Creating functional surfaces
3.1 Introduction
Realization of the separation devices envisioned in Chapter 1 primarily depends on
designing and fabricating protein patterns that are the key to the separation process. The
phenomenological studies outlined in Chapter 2 guide the designing process of these
separation surfaces. However, fabricating these protein patterns, especially in the context
of a separation device is a challenging task. Micro-contact printing, which was used in
our last study132 (Chapter 2), is probably one of the simplest methods for protein
patterning but was found to be unsuitable for use in separation devices for a number of
reasons which will become clear in the later sections in this chapter. Hence, developing a
new strategy for patterning protein that is in-line with the design requirements was
necessary. Here we review the different approaches to immobilize proteins onto surfaces
and also common techniques for creating two-dimensional patterns of proteins. Then, we
outline the design requirements for creating affinity-flow-fractionation (AFF) surfaces
and present the rationale for choosing a particular strategy over the others.
Chapter 3: Creating functional surfaces
52
3.2 Design considerations
The main design considerations are:
1. Maximizing displacement of cells over each edge, and minimize device length
2. Fast transit time of cells though the device
3. Robust rolling of cells and high efficiency of separation
4. Robust passivation to prevent biofouling
3.3 Background
Protein immobilization is a long-standing problem that is important in a wide range of
application such as protein assays, biosensors, affinity surfaces to name a few133,134.
While there isn’t a single best universal method for immobilizing proteins, a number of
different approaches and chemistries have been developed depending on the protein and
the application. Interaction of proteins with solid surfaces is a complex problem and a
number of different interactions can be simultaneously involved. Proteins can interact
with solid surfaces though hydrophobic interactions, van der walls forces or through less
common hydrogen bonding with the surface135-138. All of the above results in noncovalent immobilization of the proteins onto the surface. While non-covalent methods are
the easiest to implement, they are very non-specific, less controllable and due to the low
bond energy of the non-covalent interactions are short lived on the surface with half-lives
as low as a few hours. On the other hand, functional groups on proteins can be made to
chemically react with reactive groups on the substrate resulting in formation of133,134,139141
covalent bond between the two. Such interactions are significantly stronger than non-
covalent interactions and can result in a long lasting immobilization of proteins with halflives on the order of days. The differences in kinetics of the reaction of the reactive group
on the surface with different functional groups on the protein can be harnessed to
introduce a moderate to high degree of specificity in the immobilization reaction. For
example, the group N-hydroxysuccinimide is susceptible to nucleophilic attack and hence
in principle reacts with hydroxyl (-OH), sulfhydryl (-SH) and amine (-NH2) groups.
However, because the reaction kinetics favors reaction to primary amines, it can be
successfully used to link proteins even in aqueous solutions although hydrolysis occurs as
Chapter 3: Creating functional surfaces
53
a side reaction142. The last and probably the most specific and controlled method of
immobilizing proteins is a hybrid strategy wherein a linker protein is immobilized on the
substrate, which in turn captures and binds the target protein directly or a specific ‘tag’
attached to the target142. Reaction kinetics between the linker and the ‘tag’ dictates the
kinetics of binding and are often chosen to be strong such that the interaction is long
lasting.
For example, the biotin-avidin binding - the strongest non-covalent interaction known
(Ka≈1015 M-1) in nature – is used to immobilized biotinylated proteins onto avidin-coated
surfaces. The advantage of this hybrid system is that the reactions occurs in physiological
buffer under benign condition, requires less amount of target protein and maintains
protein activity. Target proteins can be ‘tagged’ though chemical modification, which
introduces the tag at random positions. Often times, chimeric proteins with the ‘tag’ fused
into the protein can be generated which allows for the protein to be specifically oriented
on the surface. A list of schemes for immobilizing proteins on surfaces is described in
Figure 3.1. The different reactions and interactions are detailed in Figure 3.1(b). Note that
this list is not exhaustive, and the readers are directed elsewhere142,143 for a through
review of different strategies and protocols.
Chapter 3: Creating functional surfaces
54
Figure 3.1. Schematic showing the different mechanisms though which proteins can
interact with solid substrates. The chemical and ligand-specific interactions that are
commonly employed to immobilize proteins are illustrated142.
Chapter 3: Creating functional surfaces
55
Creating protein patterns poses an additional level of complexity since in order to create
2D patterns, protein immobilization must now be restricted to specific area on the
surface. The readers are directed to some excellent reviews on different techniques of
protein patterning for in-depth understanding13. The different approaches to achieve this
can be divided into two broad categories. First, where the chemical makeup of the surface
is homogenous but the contact of the protein to the substrate is restricted to specific areas.
Methods under this category include direct micro-contact printing of proteins, dip-pen
lithography, microfluidic patterning and using photolithography to mask the substrate17.
The above-mentioned methods are relatively straightforward, but the harsh treatment
required in some of the protocols tends to denature the protein. The second approach is
to create a chemical pattern on the substrate such that when exposed to the protein
solution, immobilization occurs only in the desired areas. A number of different strategies
can be devised to create patterns of chemical groups on surfaces; some examples include
microcontact printing and photolithography. Creating chemical patterns prior to protein
immobilization allows the protein to be immobilized in relatively benign conditions
preserving protein activity. However, since the protein comes in contact with whole
surface, successful patterning requires creation both protein repellent ‘passivated’ regions
along with ‘activated’ regions capable of immobilizing the protein. While common
methods to ‘activate’ a surface are shown in Figure 3.1, passivation of surfaces against
non-specific adsorbtion of proteins is itself challenging. Amongst the different
approaches, making surfaces hydrophilic, engraftment of molecules such as albumin,
polyethylene glycol (PEG) and glycosaminoglycan or coating with perfluorocarbons has
been explored144,145. However, PEG coatings have so far outperformed most of the other
methods and are broadly accepted as superior passivation molecule145. The protein
repellant performance of PEG coatings depends on the molecular weight of the protein
and the PEG146, the density of PEG engraftment147 and the age of the coating since PEG
is easily prone to oxidation in air. For further information on PEG coatings, the readers
are directed elsewhere145.
Chapter 3: Creating functional surfaces
56
3.4 Microfluidic device design
The studies conducted in Chapter 2 provided the guidelines to design the separation
devices. It was found that an edge inclination angle between 15°-20° provided the highest
lateral displacement of cells per unit horizontal length traversed, and accordingly the
inclination angle was chosen to be 15°. In our early experiments with HL60 cells we
observed that the width of the bands didn’t have an effect on efficiency of capture of cell
or their rolling behavior as long as they were wider then the cell diameter, specifically
10 µm and 100 µm wide bands performed similarly at least qualitatively (data not
shown). Since diameters of HL60 cells and K562 cells vary from 10-16 µm while
leukocytes are between 8-12 µm, we decided to keep the width of the patterns to 15 µm.
Since cell rolling velocity is order of magnitude slower than the free stream velocity of
cells – 1-5 µm /s compared to >100 µm /s – it was required that the longitudinal rolling of
cells be minimized since it does not contribute towards separation. We achieved this by
having a non-patterned gutter region that collects separated cells allowing their quick
elution out of the device. Since the pattern is at the bottom of the channel, the cells need
to settle under action of gravity before interacting with the patterns. Assuming a channel
height of 100 µm, back of the envelope calculations fixed the value for settling distance
of the cells range 3 – 5 cm (wall shear stress 0.5-2 dyn/cm2). Assuming that the inlet
stream of cells is 100 µm wide, results of section 2.3.5 demonstrates that at least 1 cm
length is required for resolution of the separated cells from original stream. Note that, this
estimates ignores the dispersion of the non-target cells, and assumes that once settled the
cells don’t skip any bands – something we found to be not true in our experiments
especially over large number of patterns. Thus assuming a factor of safety of ~3 we
decided to have the channel of 20 cm in length. The channels were designed to be 1 mm
wide and 100 µm high. The final design is shown in Figure 3.2. Our studies indicated, as
discussed in later sections, that the best method of creating the protein patterns was on
plain glass substrate without having the microfluidic device attached. A vacuum suction
cup was designed into the microfluidic manifold that was used to attach the device to the
substrate after protein patterning.
Chapter 3: Creating functional surfaces
57
Figure 3.2. Design of the separation channel. Design of the substrate (yellow) is shown
aligned with the microfluidic
3.5 Screening of surface functionalization protocol
The design of the substrate required aligning of the pattern with the microfluidic device.
Thus it was necessary for the pattern to be easily visible so that it could be manually
aligned. Since, creating self assembled monolayes (SAM) on gold and glass using thiols
and organosilanes respectively is well established, we decided to etch the pattern in gold
on glass substrate and use gold/glass selective chemistry to functionalize one region
while blocking the other region. Although different functional groups can be used to
immobilize the protein (P-selectin in our case) it has been reported earlier148 that different
functionalization protocols alters the behavior of rolling of cells. Thus we screened for
different functionalization protocols in search for the optimal method. We created
Chapter 3: Creating functional surfaces
58
substrates with different end functional groups as described below and incubated them
with 15µg/ml of P-selectin (R&D Systems, Catalog# ADP3) for 1 hr followed by
blocking with 1% BSA for 1 hr after which HL60 cells were flowed over them and the
rolling velocity measured at different shear stress. The specific protocol for each
functionalization is described below:
Glass: Glass slide (VWR), piranha cleaned (1:3 H2O2 to H2SO4)
Gold: 100nm gold coated slides on glass substrate (obtained from EMF Corp, NY),
piranha cleaned.
PEG: Gold slides (EMF) were piranha cleaned, dried and flooded with 5 mM solution of
PEG-alkanethiol (1-Mercaptoundec-11-yl)tetra(ethylene glycol), Sigma) in ethanol. The
slides were washed with ethanol followed by DI water after 15 min of incubation with
PEG solution.
Amine: Glass slides were piranha cleaned, dried and immersed in 1% solution of 3aminopropyltrimethoxysilane (Gelest) in anhydrous toluene for 12 hr. The slides were
washed in acetone, ethanol and dried.
Thiol: Glass slides were piranha cleaned, dried and immersed in 1% solution of MPTS
(3-mercaptopropyltrimethoxysilane (Gelest)) in anhydrous toluene for 12 hr. The slides
were washed in acetone, ethanol and dried.
Hydrophobic: Glass slides were piranha cleaned, dried and immersed in 1% solution of
Octadecyltrimethoxysilane (Gelest) in anhydrous toluene for 12 hr. The slides were
washed in acetone, ethanol and dried.
NHS: Thiol modified glass slides were taken and treated with 10mM of GMBS (N(Gamma-Maleimidobutyryloxy) Succinimide, Sigma) in ethanol for 30 min. The slides
were washed in ethanol, dried and used immediately.
The results for the study are summarized in Figure 3.3, which shows the rolling velocity
for different functionalization protocols. Rolling on hydrophobic and amine surfaces
showed highest sensitivity to shear stress than the other methods, while rolling on NHS
immobilization seemed to be most resistant against shear stress. Thus we decided to use
Chapter 3: Creating functional surfaces
59
NHS functionalization to attach to the surface. Also, the PEG surfaces exhibited excellent
passivation since no rolling or cell attachment was seen.
Figure 3.3. Rolling velocity of HL60 cells at different shear stress on substrates
functionalized with P-selectin using different functional groups.
Now that the immobilization technique was selected, there are two options – either,
activate the glass with NHS and passivate the gold using PEG or, vice versa. In our
preliminary tests we found that simultaneous activation of glass using MPTS damages the
passivation of the gold region even when the PEG-alkanethiols have been grafted prior to
exposure to the MPTS. We believe that this might be due to the replacement of the PEGalkanethiols with MPTS resulting in degradation of passivation layer. Moreover, NHSalkane-silanes are expensive and unstable. Hence we decided to choose the alternate
strategy i.e. to activate the gold and passivate glass. Activation of gold was performed
using Dithiobis (succinimidyl propionate) (DSP) which being a di-thiol forms a SAM
with NHS functional end groups on gold surface. We also developed a protocol for
grafting PEG-trimethoxysilane (8-12 PEG units, Gelest) on glass. Piranha cleaned glass
slides were immersed in 1% PEG-silane solution in anhydrous toluene overnight. The
sides were then removed form the PEG solution, washed with acetone followed by
Chapter 3: Creating functional surfaces
60
ethanol and the dried under nitrogen and used for experiments immediately. For
measuring protein repellant property of the PEG surfaces, the PEG grafted glass slides
were incubation with FITC labeled BSA (100µg/ml) for 1 hr after which the excess
protein was washed with DI water and dried before fluorescence signal measurements
were performed using an epifluorescent microscope. The PEG surfaces showed excellent
protein repellant property as seen in Figure 3.4. Later, tests with incubation of P-selectin
also demonstrated that cells did not attach to the PEG region on glass.
Figure 3.4. Passivation of glass. Plain glass, piranha cleaned and PEG-grafted glass was
incubated with 100µg/ml of FITC labeled BSA for 1 hr and washed after which
fluorescence measurement was done to estimate the amount of protein adsorbed. The
fluorescence measurement was performed on epi-fluoresnece microscope with CCD
camera. The values have been corrected for background noise. Error bars represent SD
of n>3 measurements.
3.6 Immobilization of P-selectin
The extracellular domain of P-selectin contains there main regions – starting from the Nterminus there’s the EGF domain, the C-type lectin domain and the consensus repeat
units (CRR). Thus proper orientation of P-selectin is critical for effective binding with
PSGL-1. While direct chemical immobilization is an effective method to immobilize Pselectin, as noted earlier, the orientation of the grafted protein is random. Hence we
attempted to try and orient the P-selectin on the surface close to how it is displayed
physiologically.
Chapter 3: Creating functional surfaces
61
Recombinant P-selectin (from R&D systems) was available in two forms – as a truncated
protein from Tryptophan at the 42nd position till Alanine at 771st position, or as a fusion
protein which contain a IEGRDMD (sensitive to Factor Xa and Thrombin cleavage) and
human IgG1 Fc domain fused to the P-selectin segment (see Figure 3.5a). We used the Pselectin-Fc fusion protein to orient the P-selectin on the surface as shown in Figure 3.5b.
Protein A (binds Fc domain of immunoglobins) was first immobilized on NHS
functionalized slides by incubation with 1mg/ml of Protein A for 1 hr, after which the
surface was blocked using 1% BSA (2 hr incubation). Next, P-selectin-Fc was incubated
at varying concentration (1-10µg/ml) for 1 hr, slides were washed and rolling experiment
was performed inside flow chambers with HL60 cells at a shear stress of 1dyn/cm2. We
had observed earlier in unrelated experiments that although HL60 cells did roll on Pselectin-Fc coated substrates, there would be a substantial number of cell that would be
stuck on the surface. In fact, HL60 cells express Fc gamma receptors (FcγR) which bids
human Fc domain with high avidity149,150, which explains for the sticking behavior
specifically on P-selectin-Fc and not on P-selectin. We hypothesized that, if the Pselectin-Fc was properly oriented, then the Fc domain will not be accessible to FcγR and
hence there should reduced sticking of cells. However, we observed substantial amount
of sticking (~25%) on surfaces prepared by Protein A immobilization (as described above
and shown in Figure 3.5b) indicating that the P-selectin-Fc failed to be properly oriented.
In order to investigate further, we performed controls where BSA replaced Protein A in
one of the incubation steps and still observed sticking of cells (Figure 3.5c) indicating
that BSA does not provide proper passivation against P-selectin. Hence, we believe that
although the Protein A could successfully orient the P-selectin, there were substantial
number of P-selectin molecules which would attach to BSA resulting in display of Fc to
the HL60 cells.
Although P-selectin-Fc caused substantial sticking, we did observed more stable rolling
of cells with lower rolling velocity on these surfaces compared to sP-selectin (without Fc
tag). Improvising on our experience, we wanted to test whether P-selectin-Fc can be
grafted with the Fc domain blocked. Note that, we in the following experiments we did
not attempt to orient the protein anymore. In order to test the blocking of Fc, we reacted
P-selectin-Fc with Protein A or anti human Fc mouse IgG at a ratio of 1:50 (P-selectin-Fc
Chapter 3: Creating functional surfaces
62
to blocking agent). We then incubated NHS substrates with per-reacted solutions having
15µg/ml of P-selectin-Fc for 2 hr and blocked using 1% BSA solution or the commercial
blocking solution Superblock (Pierce). We found that while IgG binding failed to block
the Fc binding to FcγR on cells, Protein A successfully blocked all such interaction and
there was very little to no sticking observed in all experiments (see Figure 3.5d).
There are more then four different types of Fc receptors on human leukocytes with
different immune functions151,152. After demonstrating successful blocking of Fc
interaction with HL60 cells though Protein A, we wanted to test if this protocol would
work for human leukocytes. We prepared substrates immobilized with P-selectin-Fc
(blocked with Protein A) or with sP-selectin (no Fc tag) and flowed whole blood at 1
dyn/cm2 over them. While we observed rolling of leukocytes (mostly neutrophils) on
both the substrates, the rolling cells on P-selectin-Fc quickly transitioned to permanent
adhesion and then spread out within 10 min indicating activation of these cells (see
Figure 3.6a). On the other hand, cells rolling on sP-selectin continued to roll with
moderate velocities and maintained their round shape even after 30 min – 1 hr after
infusion of blood. Hence we concluded that Protein A failed to block the Fc from
interacting with FcγRI on neutrophils which resulted in their activation. Thus we decided
to use sP-selectin for separation experiments since the immunogenicity of the P-selectinFc could not be avoided.
Chapter 3: Creating functional surfaces
63
Chapter 3: Creating functional surfaces
64
Figure 3.5. Immobilizing P-selectin with proper orientation on the substrate. A) Two
commercial forms of P-selectin (R&D Systems) with or without the Fc tag. B) Protocol
adopted for orienting P-selectin-Fc though the Protein A. Protein A is immobilized on
NHS functionalized substrate following BSA blocking. When P-selectin-Fc is exposed to
the surface, binding of Fc domain with Protein A orients the P-selectin. C) Microscope
images of interaction of HL60 cells with three different substrates - BSA coated, BSA
coating followed by P-selectin-Fc exposure and P-selectin-Fc immobilized using the
protocol shown in B. The images were taken with a CCD camera with 0.5 s exposure,
such that only rolling or suck cells can be seen. Adhesion and rolling could be seen in all
surfaces exposed to P-selectin-Fc irrespective of presence of Protein A, indicating that
BSA could not effectively block non-specific adsorption of P-slectin-Fc. Scale bar 100
µm. D) Percentage of stuck cells in rolling experiments on substrates prepared using
different protocols. Either P-selectin-Fc was immobilized using Protein A as shown in B,
or was pre-reacted with Protein A or anti-Fc IgG in order to block the Fc domain, and
then directly immobilized on NHS functionalized surfaces. Both %BSA and Superblock
(Pierce) were tested as blocking agent. Error bars show SD of n=3 experiments.
Figure 3.6. Testing immunogenicity of rolling surfaces prepared using different
immobilization protocols. P-selectin-Fc pre-reacted with Protein A (1:50 ratio) or sPselectin (with Fc) was immobilize on NHS functionalized surfaces followed by blocking
by 1% BSA. Whole human blood was flowed at 1 dyn/cm2 and cell rolling was observed.
Chapter 3: Creating functional surfaces
65
Rolling cells on P-selectin-Fc surface rapidly transitioned to permanent adhesion
followed by morphological changes and cell spreading, indicating cell activation. In
comparison, cells on sP-selectin rolled without sticking and maintained their cellular
morphology even after 1 hr of infusion. The images were taken with CCD at 0.5 s
exposure, where by only rolling and stick cells are imaged. Inset shows magnified view of
the cells highlighting the morphology of rolling cells. Scale bar 100 µm (inset 15 µm).
3.7 Final fabrication protocol
Fabrication of receptor-patterned substrate
Gold coated slides (gold thickness 100 nm for HL60 experiments and 20 nm for blood
experiments, 5 nm chromium adhesion layer) (EMF Corp, NY) were coated with
OCG825 photoresist and patterned using photolithography (Figure 3.2) using Gold
etchant and Chromium etchant (Sigma) followed by photoresist stripping in acetone. The
patterned slides were then washed successively in acetone and ethanol, dried and stored
for future use.
Surface functionalization of the patterned gold slides was performed using a previously
established method
153
with a slight modification. The patterned slides were cleaned by
immersing in piranha bath (3:1 H2SO4 to H2O2) maintained at 80°C for 10 min, washed
with DI water and ethanol, and dried thoroughly. The slides were then immersed in a
solution containing 1% (v/v) Polyethyleneglycol (10-12 units) trimethoxysilane (Gelest,
PA) and 2% (v/v) Triethylamine (Sigma) in anhydrous Toluene (Sigma). The slides were
incubated for more than 6 h at room temperature in a closed container after which they
were removed, washed with acetone, and dried. Next, the slides were flooded with 5 mM
solution of 3,3’–Dithiopropionic acid di(N-succinimidyl ester) (Sigma) in anhydrous
Dimethylformamide (Sigma), placed on orbital shaker and incubated for 1 h. The
functionalized slides were sonicated in an ethanol bath for 10 min (to remove unbound
DSP), washed with ethanol, dried, and immediately used for protein immobilization.
The protein incubation was done in customized incubation chambers. Thin strips (~1 mm
width) of Secureseal Adhesive sheet (250 µm thick) (Electron Microscopy Science, PA)
were cut and pasted on the border of the functionalized slide. Next, inlet and outlet ports
Chapter 3: Creating functional surfaces
66
were drilled on a Hybrislip (Electron Microscopy Science, PA) before sticking on the
adhesive strips previously placed on the slide. Human recombinant P-selectin (R&D
Systems, MN) solution of desired concentration was made in Dulbecco’s Phosphate
Buffered Saline (DPBS) and was used to fill the chamber using a micropipette. The
whole setup was placed in a humidified enclosure to prevent drying of the solution. For
HL-60 separation experiments, 5 µg/mL of P-selectin solution was incubated for 3 h
while for blood separation experiments 15 µg/mL of P-selectin was incubated for 1 h.
The above protocol achieved a P-selectin site density of ~500 sites/µm2 for HL-60
separation substrates and ~1100 sites/µm2 for blood separation substrates measured as
described in the next section. After incubating for the desired time, the incubation
chamber was removed from the slides using tweezers, and the slides were washed in a
stream of DPBS for 1 min, placed in sterile 1% BSA solution (Teknova, CA), and stored
at 4°C. The slides were used within 48 h of protein immobilization.
Fabrication of microfluidic channels
The microfluidic channel was made using soft lithography. The design of the channel
used in separation experiments is shown in Figure 3.2. The master mold of the channel
was created on a silicon wafer using SU-8 photoresist by photolithography. The thickness
of the SU-8 layer measured using a profilometer (Dektak) was found to be 95 µm with a
maximum
deviation
of
±4
µm
across
the
wafer.
The
silicone
elastomer
Polydimethoxysilane (PDMS) (Sylgard 184, Dow Corning Corp, MI) and curing agent
were mixed in 1:10 ratio and poured on the master to form a layer 5-8 mm thick,
degassed to remove trapped air, and cured at 80°C. The mold dimensions included a
1.2% shrinkage allowance in order to account for the thermal shrinkage of the cured
PDMS. After curing, the PDMS was removed from the master, cut to size, and punched
to create inlet and outlet ports. A vacuum port was also punched over the vacuum suction
area.
Chapter 3: Creating functional surfaces
67
Figure 3.7. Schematic for fabrication and assembly of separation devices.
3.8 Characterization of substrate
The density of P-selectin immobilized on the gold substrate was estimated using indirect
ELISA according to a published protocol
154
. The protein-immobilized substrates were
placed on an orbital shaker, flooded with 15 µg/mL of anti-human P-selectin mouse IgG1
antibody (clone AK4, Biolegend, CA), and incubated for 6 h. Next, the slides were
washed thoroughly with DPBS, flooded with 50 µg/mL Goat anti-mouse IgG1
Horseradish Peroxidase conjugate (SantaCruz Biotech, CA), and incubated for 4 h with
constant shaking. The slides were then washed again, placed on a clean pertidish flooded
with 300 µL of UltraTMB (Thermo Fisher Scientific, IL), and incubated for another 30
min with slow shaking on orbital shaker. After the completion of the reaction, 100 µL of
the stop buffer (2 N H2SO4) was added to the slide, the solution was transferred to a well
Chapter 3: Creating functional surfaces
68
plate, and the absorbance was measured using a plate reader (Tecan 200Pro). Standard
calibration curve was generated by mixing known amounts of HRP conjugated secondary
antibody to 300 µL of UltraTMB in a well plate and allowing the reaction to proceed for
30 min before adding the stop buffer and measuring the absorbance in a plate reader. The
P-selectin density was found to be ~500 sites/µm2 for HL-60 separation substrates and
~1100 sites/µm2 for blood separation substrates.
3.9 Conclusion
In this work we systematically investigated the different approaches of creating proteinpatterned substrate that would enable AFF. Amongst the different strategies investigated,
we found NHS functionalization for protein immobilization and PEG functionalization
for protein blocking to be most optimal. We also attempted to control the orientation of
the engrafted P-selectin on the surface using Fc tagged fusion recombinant P-selectin, but
blocking Fc against interaction with different FcR on leukocytes was unsuccessful, which
led to immunogenicity of P-selectin-Fc. Thus sP-selectin was found to be most suitable
for separation of cells. Finally a successful protocol for creating the protein patterns
based on photolithograhy and then using selective activation of gold was developed. The
protocol uses NHS functionalization and hence this method could be easily adapted to
immobilize any protein of choice. Using Nickel patterns instead of gold might also allow
engraftment of His tagged proteins with desired orientation, provided such recombinant
proteins could be produced. In general, this method should be useful in a broad range of
applications.
Acknowledgement
I would like to acknowledge the contribution of Mikhail Hanewich Hollatz and Rishi
Singh in developing the protocol for fabricating AFF surfaces. Mikhail was involved in
screening functional groups and development of a working protocol for passivation of
glass though using PEG silane. Rishi was involved in studies attempting to orient Pselectin on the surface and selecting the best method to immobilize the P-selectin.
4
Separation of model cell lines
Note: This chapter has been published in the paper “Affinity flow fractionation of cells via
transient interactions with asymmetric molecular patterns”, Bose et al., Scientific Reports
3, 2013.
4.1 Introduction
In the last chapter, we developed a protocol to fabricate substrates to enable affinity flow
fractionation of cells. Through testing different functionalization strategies we choose a
protocol that demonstrated best rolling behavior and had minimal immunogenicity. While
we had looked at the optimized rolling behavior of cells for different functionalization,
actual separation of cells was not demonstrated and measurement of performance of was
not performed. The performance of the device depends on a number of factors including
the patterns design, quality of the patterned substrate and operating conditions. A through
characterization of the device is essential to optimize operating conditions, enhance
performance and in order to compare with existing technologies. In this chapter, as a first
step, we used model cell lines that mimic the behavior of leukocytes, to demonstrate
separation capabilities of the AFF surfaces. This approach ensured that there is no sample
variability – something, which is common with using complex mixtures such as whole
blood. Then we quantify the separation in terms of recovery and purity and study the
Chapter 4: Separation of model cell lines
70
operational parameters of the device. Lastly, we identify the main transport phenomenon
that governs separation and model the separation process.
4.2 Experimental setup for cell separation
The experimental apparatus is shown as a schematic in Figure 4.1. The cells were
injected and collected in polypropylene pressure vessels (Sigma) while the buffer was
injected using a syringe pump (PhD Ultra, Harvard Apparatus) and glass syringes
(Hamilton Co). PTFE tubing (Cole Parmer) was used for interfacing with the device. The
ratio of buffer to cell sample flow rate was adjusted by controlling the inlet pressure,
while the fraction of the outlet flow that is collected as sorted stream was adjusted by
controlling the outlet pressure.
Figure 4.1. Schematic of the experimental setup for cell separation.
4.3 Separation of HL60 cells from K562 cells
To investigate the transport of cells in the device, we studied the separation of HL60 cells
from K562 cells, both of which are myeloid in origin. HL60 cells express PSGL-1 (the
major ligand for P-selectin)155 and exhibits ‘cell rolling’ on P-selectin surfaces, which is a
process involving continuous formation and breakage of bonds between the cell and the
surface under the influence of hydrodynamic shear.155 K562 cells do not exhibit strong
interactions with P-selectin surfaces156. HL60 cells and K562 cells were stained using
Cell Tracker Red and Cell Tracker Green (Invitrogen) respectively and mixed in
Chapter 4: Separation of model cell lines
71
approximately 1:1 ratio to a final concentration of 106 cells/mL in cell culture media. The
mixture was injected alongside a Dublecco’s Phosphate Buffered Saline (DPBS)
(containing Ca2+ and Mg2+) buffer stream into the assembled separation device. The inlet
pressure was adjusted such that the cell stream occupied about 10% of the channel width
(ratio of the flow rates of cell to buffer streams around 1:9). The wall shear stress inside
the channel is a critical parameter that modulates attachment rate and rolling velocities
and was calculated (assuming plain Poiseuille flow) from the equation
Q=
τ wh3
6µ
(4.1)
where τ is the wall shear stress, w and h are channel width and height respectively and µ
is viscosity of the buffer. We performed experiments at different shear stresses ranging
from 0.2 dyn/cm2 up to 1.5 dyn/cm2 and found that lower shear stresses promoted a
tendency of the cells to get stuck on the substrate, while higher shear stresses
significantly decreased the cell-surface attachment rate, reducing the separation
efficiency. A shear stress of 0.5 dyn/cm2 was found to be optimal for overall device
performance and was used in the reported experiments. We found that the HL60 attached
to the P-selectin patterns, rolled over them and got separated from the K562 cells. Over
the 20 cm length of the separation channel, the mean position of HL60 cells was
displaced laterally by ~800 µm, and about 80% of the HL60 cells were found to be
concentrated in a narrow band on the sorted side (Figure 4.2a).
The flow of cells within the channel was recorded by taking time lapse images of the
cells at particular channel locations without P-selectin patterns in different fluorescence
channels. Positions of at least 200 flowing cells of each type were recorded and analyzed
using the software ImageJ (NIH). The number of cells flowing per unit time at different
positions across the channel width was converted to frequency histograms, which were
normalized using the total area under the curve to obtain the probability distribution
function (PDF) representing the flux of the cells. The resolution of the HL60 cells from
the non-rolling K562 was found to be dependent on the length travelled inside the
channel, similar to that in chromatography. Control experiments were used to verify that
the cell separation was due to P-selectin / PSGL-1 interactions. HL60 cells were not
Chapter 4: Separation of model cell lines
72
laterally displaced when BSA was immobilized on the gold patterns instead of P-selectin
(Figure 4.2b), or when PSGL-1 on the cells was blocked with an antibody (Figure 4.2b).
Furthermore, in the absence of P-selectin, there was no significant difference between
distribution of cells on the BSA passivated substrates with or without the gold patterns
(Figure 4.2b), indicating that the gold pattern did not significantly affect the cell
distribution. The observed broadening of the cell distribution during separation on Pselectin may be attributed to weak interactions between the P-selectin and K562 cells.
Chapter 4: Separation of model cell lines
73
Figure 4.2. Separation characterization using HL60 and K562 cells. A) Overlaid
fluorescence images (false colored) showing the distribution of the two cell types at
different locations along the microchannel. Images were acquired via a continuous 10 s
exposure for each fluorescence channel. Scale bar is 100 µm. B) Evolution of distribution
of the flux of the two cell types along the channel length shown as a dimensionless
probability density function (PDF) against channel position normalized by the channel
width. The active substrate containing the P-selectin patterns is shown against a control
where BSA replaced P-selectin, P-selectin patterned surface with cells treated with antiPSGL-1 antibody (clone # KPL-1, Biolegend Inc.) and BSA passivated glass substrate
(no gold patterns).
4.4 Separation metrics
Performance of separation is measured by two primary quantities – 1) Purity, which gives
the percentage of target cells in sorted sample and 2) Recovery, which is the percentage
of total target cells sorted, as defined below.
# of HL60 cells in sorted sample
Total # of cells in sorted sample
# of HL60 cells in sorted sample
Recovery =
Total # of HL60 cells in injected in the device
Purity =
(4.2)
While the purity can be measured directly from flow cytometry (FC) of the sorted
samples, calculating the recovery by directly measuring the absolute cell counts in the
input, sorted and waste streams is prone to pipetting errors and cell loss during sample
preparation for flow cytometry. Here we used an indirect method to obtain the cell
recovery from purity of the three samples. Below, we detail the mathematical derivation.
For a given system, if the fractions of the target cell in the input, sorted, and waste
streams are pi , ps and pw (%) respectively, then for every 100 cells injected in the
device, there are pi target cells and assuming a recovery of r (%), the number of target
cells in the sorted stream is r.pi/100 while the number of target cells in the waste stream
Chapter 4: Separation of model cell lines
74
is (1– r/100)pi . If the purity of the cells in the sorted stream (measured by flow
cytometry) is ps, the total number of cells (both target and non-target) in the sorted
sample is r.pi/ps. In a similar manner, the total number of cells in the waste stream is (100
– r)pi/pw. Thus using mass balance we get: (100 – r)pi/pw+ r.pi/ps=100 which on
simplification gives:
r=
ps ( pw − pi )
pi ( pw − ps )
100
(4.3)
In one experiment, we collected 25% of the flow into the sorted stream which yielded a
purity of 94.6%, which using Equation 4.3 estimates a recovery of 83% (Figure 4.3a). It
can be easily noted that the recovery and purity depends on the location in the channel
and the fraction of flow collected in the sorted stream. We calculated the purities and
recover for different fraction of flow collected at 20 cm channel length from the
probability densities (Figure 4.3b). As a higher fraction of flow is collected as the sorted
stream, the recovery increases, while the purity monotonically decreases. Tuning the
fraction of the flow collected as the sorted stream determines the trade-off between purity
and recovery, depending on the needs of the application.
Chapter 4: Separation of model cell lines
75
Figure 4.3. Performance of separation. A) Flow cytometric analysis of the samples in an
experiment where the sorted stream comprised 25% of the total flow. B) The purity (red)
and recovery (blue) of HL60 cells at the channel exit (L = 20 cm) for different fractions
of the total flow collected into the sorted stream as calculated from the PDF. Mean and
standard deviation for n = 3 experiments are shown.
4.5 Mathematical Modeling of separation process
Quantitative understanding of the transport mechanisms of cell separation is essential for
optimizing the device performance and guiding future development of AFF for other
applications. Through our observation of the behaviors of cells inside the AFF devices we
identified six processes which, we believe, primarily determines the transport of cells and
hence the separation performance. We developed a phenomenological mathematical
model that accounts for these major transport processes within the device and can be used
to predict the separation performance (Figure 4.4). Chapter 4: Separation of model cell lines
76
4.5.1 Model description
The cells entering the device settle to the bottom of the channel under the action of
gravity, with the settling length depending on geometry, flow velocity, and initial
distribution of the cells. We observed that HL60 cells that settled to the bottom of the
channel attached to the P-selectin stripes and tracked along their edges for a certain
distance
before
detaching
and
reattaching
on
a
downstream
stripe
(http://www.nature.com/srep/2013/130731/srep02329/extref/srep02329-s2.mov).
The
cells tracked along the edges of several consecutive P-selectin stripes, but then detached
and traveled a long distance before re-attaching and tracking along the edges of another
set of P-selectin stripes. These observations suggest that two different capture
probabilities are at play during the repeated rolling and detachment of cells. The cells in
the free stream flowing close to the surface can initiate rolling on the patterns with a
relatively low probability, P∞99. These cells track the patterned edges for a certain
distance (le) before detaching and reattaching to an immediately downstream stripe with a
significantly higher probability, P0. For HL60 cells on P-selectin patterns, we determined
the values of P0 and le from experiments (See section 4.5.2), and combined the Davis &
Giddings model for near wall particle setting under gravity157 with the Goldman’s model
for particle convection158 to describe the gravitational settling of the cells within the
microchannel. Assuming that rolling along the edge is a Poisson process132 with the mean
edge tracking length le depending the angle of inclination (α) of the patterns, we
performed a Monte Carlo simulation to obtain the distribution of cells at different
locations in the separation channel.
Chapter 4: Separation of model cell lines
77
Figure 4.4. Schematic description of the key processes governing the transport of cells
inside the separation device
4.5.2 Estimation of the model parameters
Figure 4.5. Schematic of parameters used in mathematical model. The x-axis is along the
device length while the y-axis is the lateral direction. The perpendicular distance
between the edges of the pattern is b, while the patterns are inclined at an angle α to the
flow direction. wc is the width of the cell stream, wp is the width of the patterned region
while w is the channel width.
The coordinate convention and the nomenclatures used in the model are shown in Figure
4.5. There are two primary length scales associated with the transport of rolling cells in
the microchannel. First is the length scale associated with the settling of the cell to the
Chapter 4: Separation of model cell lines
78
bottom of the channel under the action of gravitational force which is estimated using a
formulation similar to Zang & Neelamegam 99. The settling velocity for a cell of radius r
is given by 157:
2
r2
ρc − ρm ) g
(
dz
9
µ
=−
r
dt
1+
z−r
(4.4)
where ρm and ρc are fluid and cell densities, g is gravitational acceleration and µ is the
viscosity of medium. The convection velocity of the cell is affected by the presence of the
wall - an effect which becomes dominant in small confinements. To account for it, we
assumed that the convection velocity is equal to the local flow velocity if the separation
from the wall is greater than 0.2h (h is channel height), while the convection velocity of
the cells closer to the wall than 0.2h is estimated using the equation derived by Goldman
et.al. 158. For a channel of height h, the cell convection velocity is given by:
⎧ 12 ⎛⎜ r ⎞⎟ F rT s − F sT r
dx ⎪ ⎝ z ⎠ t r
(γ z )
= ⎨ F T − F rT t
dt ⎪
⎩V fluid , x
if min [(h − z ), z ] < 0.2h
(4.5)
if min [(h − z ), z ] ≥ 0.2h
F r , F s , F t , T r , T s , T t are non-dimensional parameters that depend on the distance of
separation of the cell from the wall and the readers are directed elsewhere
158
for their
values. γ is the local shear rate of the fluid near the cell which is given by the partial
derivative of the flow velocity in z-direction. The flow velocity inside a rectangular
channel for a flow rate Q is given by 159 :
(
)
y−w 2
1 ⎡ cosh nπ h ⎤
⎢
⎥ sin nπ
1
−
∑ 3
cosh nπ 2wh ⎥⎦
36Q n ,odd n ⎢⎣
V fluid , x ( y, z ) =
∞
192h
whπ 3
1 − ∑ 5 5 tanh nπ 2wh
n
π w
n , odd
∞
(
)
(
(
z
h
)
(4.6)
)
The second length scale in the process is associated with the actual separation of the
settled cells and is affected both by the probability of attachment to the patterns and
deflection by the edges. We learnt from the experiments that cells flow freely with the
fluid till they get captured on a ligand patterned band after which they track the edge
getting displaced laterally and eventually detaching, usually reattaching to the immediate
pattern downstream. Interestingly, this process of detachment and reattachment continues
until after a detachment event the cell fails to attach to the downstream band and re-enters
Chapter 4: Separation of model cell lines
79
the free stream, after which the whole cycle may be repeated. More than 120 HL60 cells
were tracked and it was found that on an average 90% of the detached cells attached to
the next immediate pattern, 5% attached to the second pattern while the rest did not attach
in the image frame (>5 stripes after the detachment) and were assumed to have returned
to free flowing velocity. For simplicity, we assume that the reattachment probability P0
refers to the attachment only to the next immediate pattern and is 0.95 for the HL60
experiments.
Unlike P0, it is difficult to experimentally determine the free stream attachment
probability P∞ since very few free flowing cells attached in a given image frame. Using
the current microfluidic device, we performed experiments where free flowing HL60
cells (gravitationally settled inside the channel) were introduced to a series of P-selectin
stripes at different positions in the microchannel and time lapse images were acquired
continuously. The images were manually analyzed and the probability of attachment of
cells (Pattach) was obtained as the fraction of cells which attached in the given image area.
Since the attachment of the cells follows a binomial probability, the total probability that
a cell attaches to at least one stripe out of ‘n’ stripes it encounters is given by:
Pattach = 1− (1− P∞ ) n . The above formula is used to determine P∞ from the values of Pattach
and ‘n’ obtained from image analysis. Our experiments yielded an average value of P∞ =
0.027 with 95% confidence bounds between 0.015 – 0.038. We ran the Monte Carlo
simulation (described later) with different values of P∞ and obtained an excellent match
with the experimental result for P∞ = 0.007, which is only in modest deviation from the
range of values determined experimentally. The experiments for determining P∞ were
performed with lower number of cells unlike the separation devices where a large number
of cells compete for relatively limited rolling area, which might explain the lower value
of P∞ at which the simulations matched experimental results.
Rolling of HL60 cells on asymmetric receptor patterns have been characterized by us
previously 132, and it was found that the tracking length on a single pattern edge exhibited
a exponential distribution which was a strong function of the inclination angle of the
pattern (α), amongst other parameters ( see Chapter 2). We measured the edge tracking
lengths in our current system from independent experiments and confirmed that the
Chapter 4: Separation of model cell lines
80
events were exponentially distributed as expected (Figure 4.5); the mean of the fitted
curve (average edge tracking length le) was found to be 23.96 µm for HL60 cells (95%
confidence bound between 22.13 – 25.79 µm) compared to 67.32 µm for neutrophils
(95% confidence bound between 65.32 – 69.32 µm).
Figure 4.6. Edge tracking length of HL60 cells and neutrophils on P-selectin patterns.
Distributions of the edge tracking length of (a) HL60 cells and (b) neutrophils were fitted
with an exponential function (red) to obtain the mean edge tracking length le. The fits
have an R-square value above 0.9 in each case.
4.5.3 Monte Carlo simulation
The numerical scheme was implemented using a custom code in the commercial software
MATLAB (Mathworks, Inc.). The values of the parameters used for the simulation were
g = 9.8 m/s2, ρc-ρm = 77 kg/m3, µ = 0.0009 N-s/m2, h = 95 µm, w = 1 mm, b = 30 µm, α
= 15o, wc = 100 µm, wp = 850 µm. The cells were assumed to be uniformly distributed at
the entrance of the device and the starting position of a cell was calculated by random
sampling of the cross section of the cell stream. Trajectory of the settling cell was
calculated by solving the coupled differential equations Eq. (4.4, 4.5, 4.6) using a
variable order solver. Once the cell settled, its x-position in the channel was incremented
iteratively. As the cell passed over each band, the attachment state was decided by
generating a random number and comparing it with the attachment probability (P0 or P∞
depending on the previous attachment history of the cell). The tracking length of a rolling
Chapter 4: Separation of model cell lines
81
cell on an edge was calculated by generating a random number based on exponential
distribution by inverse transform sampling. The above steps were repeated until the xposition of the cell exceeded the upper limit (the desired length of separation) or the cell
travelled into the non-patterned region. The position of the cell was recorded at every
step. The simulation was performed for 10,000 cells to calculate the cell distribution and
mean displacement. The simulation results showed excellent agreement with the
experimental results (Figure 4.7a) for P∞ =0.007, which is only in modest deviation from
the range of P∞ determined experimentally (0.015 – 0.038).
4.5.4 Scaling model
To obtain scaling relations between the fundamental separation parameters, we developed
a simple analytical model that estimates the lateral displacement of the cells. We assume
that the cells are already settled when they enter the separation channel and thus neglect
the gravitational settling length. For the given free stream attachment probability P∞ , on
an average a cell is expected to cross 1/P∞ bands before getting attached. Similarly for a
reattachment probability of P0, the cell will roll on 1/(1- P0) bands before returning to
free flow. Thus, the effective attachment probability Peff - defined as the ratio of the
number of bands to which the cell attached to the number of bands the cell crossed - can
be derived as:
Peff =
1 (1 − P0 )
P∞
=
1 (1 − P0 ) + 1 P∞ P∞ + (1 − P0 )
(4.7)
If at given length L of the channel the mean displacement of the population of cells is D,
then the total number of bands on which the cell attached is Neff = D (le sin α ) . Using the
effective attachment probability, L can be estimated as: L=
N eff
Peff
b
D
+
sin α tan α
(4.8)
where b is the perpendicular distance between the edges of the bands (Fig. 4.5). The first
term represents the actual number of bands that the cells cross, while the second term is
the added length in x-direction that the cells travel while tracking the edge. The above
expression can be rearranged to get:
Chapter 4: Separation of model cell lines
D* = L*
82
1
1+ b
(4.9)
Peff le sin α cos α
where D* and L* are the non-dimensional mean displacements and length given by
D* = D ( cos α b ) and L* = L (sin α b ) . Since the length travelled along each edge by the
cells follows an exponential distribution, the sum of all the edge tracking lengths for a
cell is expected to follow a gamma distribution for which the ratio of the standard
deviation to the mean of the distribution is expected to be:
σ
D
=
1
N eff
(4.10)
For large values of Neff (or large values of L), the second term in Eq. (4.8) is small
compared to the first term and hence Neff can be approximated as Neff ≈ (L.Peff .sinα)/b,
which when combined with Eq. (4.10), gives the relation:
σ
D
≈
1
L Peff
*
(4.11)
This scaling holds true when P∞ is comparatively small i.e. when the length scale for
separation dominates over the settling length; while the simulation results deviate
modestly from the theoretical scaling predictions as the two length scales became
comparable at larger values of P∞ (Figure 4.7b). The ratio of standard deviation (σ) of the
lateral displacement to the mean displacement (D) is indicative of the separation
resolution, which may be expected to depend on the number of stripes that the cells
interact with (given by L*.Peff once the cells settle). It exhibits an initial peak in magnitude
with increasing L*.Peff during the gravitational settling phase before transitioning to an
inverse power law relation (Figure 4.7c). The decreasing value of σ/D with increasing the
number of stripes that the cells interact with is in accordance with theoretical scaling
predictions (Equation 4.11), and is analogous to the increased resolution observed in
chromatographic separations for longer columns.
Chapter 4: Separation of model cell lines
83
Figure 4.7. Theoretical modeling of cell separation in the device. A) Comparison of the
experimental distribution of the flux of HL60 cells for the same conditions described
previously with that predicted by the model (P∞ = 0.007, P0 = 0.95, le = 23.96 µm).
Channel position is normalized by the channel width. (B-C) Scaling relations between
different operational parameters. Mean normalized lateral displacement (D*) is
proportional to the normalized length of travel (L*) scaled by the non-dimensional
parameter θ (B), while the ratio of standard deviation to mean lateral displacement (σ/D)
scales with the number of patterns on which the cells roll (L*Peff) (C). Plots of simulated
results are shown for different values of P∞ (0.001 –
(10 µm –
×
, 100 µm –
×
, 0.01 – × ×, 0.9 –
) and le
).
4.5 Conclusion
The performance of the AFF devices was measured through sorting of model cell lines.
Observation of behavior of cells on the AFF surface led to development of a novel
Chapter 4: Separation of model cell lines
84
mathematical model, which successfully predicted the gross separation characteristic of
the devices. A simplified scaling model was also developed which indicated that the
attachment of free flowing cells to the pattern was the rate limiting step to the separation
process. New channel designs, which enhance attachment of cells to the surface, should
prove beneficial in the future to enhance performance. Another interesting point worth
noting is the fact that the very weak interaction of the K562 cells with P-selectin was
sufficient to alter the cell distribution in the channel beyond passive diffusion. Thus it
might be possible that stable rolling in not a prerequisite to separation though AFF, which
should allow AFF surfaces to be used in a large number of applications involving other
physiological weak interactions such as those between lectins and glycoproteins.
5
Application of AFF in Sorting of
Neutrophils from blood
Note: This chapter has been published in the paper “Affinity flow fractionation of cells via
transient interactions with asymmetric molecular patterns”, Bose et al., Scientific Reports
3, 2013.
5.1 Introduction
Neutrophils constitute the majority of leukocytes in blood and are the first line of defense
against infection, playing a key role in shaping of the immune response
160,161
. They are
involved in pathophysiology of many diseases and are highly activated in a number of
diseases such as sepsis
162
, venous disease
163
, and sickle cell disease
164
. Neutrophil
counts are therefore commonly used to access the state of the immune system, detecting
bacterial infections and inflammatory diseases 2, and in some special cases such as
determining chemo-readiness of cancer patients. Inappropriate positioning of activated
neutrophils in the tissue is believed to cause multiple organ failure during sepsis
162
detection of activation of neutrophil has shown to be highly prognostic in neonates
and
165
.
Separation of neutrophils or leukocytes from other blood components that interfere with
DNA amplification can also provide samples suitable for genetic analysis 166.
Chapter 5: AFF for sorting neutrophils from blood
86
The conventional method for neutrophil isolation based on Ficoll-Hypaque density
gradient centrifugation followed by dextran
sedimentation takes ~3 h and requires
hypotonic lysis of contaminating erythrocytes to achieve purity > 95%
97
. The standard
methods for neutrophil enumeration are microscopic examination of blood films
following staining, or use of one of the several multichannel automatic counters such as
Coulter and Seimens 2. Detection of neutrophil activation is typically performed using
flow cytometry by assessing the expression levels of markers that are up- or downregulated upon activation
167
. While these techniques work very well using laboratory
equipment, their complexity precludes easy implementation in a disposable device or in
point-of-care settings.
AFF provides an excellent alternative to existing technologies for sorting neutrophils, and
is uniquely suited to be a simple and effective method that can be used at the point-ofcare. The performance of the separation devices with HL-60 cells, which are similar to
neutrophils in several aspects, gave us confidence to use the AFF devices in sorting
neutrophils form blood. In this chapter, we detail the step by step approach taken in order
to overcome the challenges of using blood as the sample, and demonstrate separation of
neutrophils from whole blood with high purity and efficiency in a single step.
5.2 Sorting of neutrophils from blood
All experiments involving human samples were approved by the Committee On Use of
Humans as Experimental Subject (COUHES) at MIT and the BWH Institutional review
board. Blood was collected from consenting adult healthy donors who had not taken
aspirin or other NSAIDs within 48 h prior to blood withdrawal. Blood was collected in
1.5 mL citrate vacutainers (3.2% buffered sodium citrate)
168
and was immediately used
for experiments. It should be noted that, amongst the various anticoagulant screened
(Heparin, Low molecular weight heparin, Hirudin fragment, PPACK, citrate, EDTA),
citrate worked best in terms of preventing coagulation without interfering with cell
rolling.
The buffer were used in this experiment was made by supplementing DPBS with 30 µM
CaCl2 (Sigma) and 200 units/mL of Polymxin B. This Ca2+ concentration was sufficient
to enable P-selectin bond formation without causing blood coagulation. The experiment
Chapter 5: AFF for sorting neutrophils from blood
87
was set up as described before (section 4.2) using sterile collection vials. The input
sample which consisted of either whole anticoagulated blood or diluted blood (whole
blood mixed with buffer in 1:1 ratio) was injected parallel to the buffer stream at a total
wall shear stress of 0.5 dyn/cm2. This wall shear corresponds to a 5.18 µL/min of buffer
flow rate and 0.25 µL/min blood flow rate, with a width of the input (blood) stream at
~10% of the channel width. The sorted neutrophils were either collected in HBSS (-/-) on
ice (for the viability, activation and phagocytosis assay) or in a 1% formaldehyde solution
(for determining purity through FC). The outlet pressure was adjusted so as to collect
20% of the net flow into the sorted stream.
Within the separation channel, leukocytes rolled on the P-selectin patterns and were
displaced from the blood stream into the parallel buffer stream, while the erythrocytes
exhibited only a small lateral dispersion (leukocytes can be fairly easily distinguished
from erythrocytes by their morphology). By the end of the separation channel almost all
of the erythrocytes remained on the blood input side and could be rejected, while a highly
pure stream of leukocytes was obtained on the other side (see Fig 5.1).
Figure 5.1. Direct isolation of neutrophils from blood using AFF. Citrate anticoagulated
whole blood was diluted 1:1 in the running buffer (supplemented with 30 µM Ca++) and
injected parallel to a stream of running buffer at a shear stress of 0.5 dyn/cm2 as shown
in the top left panel. Neutrophils were seen to adhere and roll on the P-selectin
immobilized gold stripes, follow the edge and separate from the blood stream (shown in
right panel). At the channel exit, a pure stream of neutrophils was separated from the
Chapter 5: AFF for sorting neutrophils from blood
88
blood stream which were collected in different outlet channels (left bottom). The scale bar
on left panel is 50 µm while that in the right panel is 30 µm.
5.3 Purity and efficiency
For the purpose of purity determination, 20% of the flow was collected in vials
containing 1% formaldehyde. The samples were then washed and re-suspended in cell
staining buffer (Biolegend, CA) and stained using the antibody cocktail - anti CD14V450 and anti CD66b- PE from BD Pharmigen, anti CD45-FITC from Biolegend , and
anti CD235a-APC (GlycophorinA) from eBioscience Inc., CA following manufacturer’s
protocols. Erythrocytes were lysed only in the input and waste sample and each sample
was analyzed using BD LSR Fortessa flow cytometer. The gates were defined using a
positive control which was prepared by taking whole blood, depleting the erythrocytes
though lysis followed by addition of 106 erythrocytes per milliliter of original blood
sample; the reduction of erythrocyte concentration ensures that the antibody staining
intensities are similar to those expected in the sorted sample and was hence important for
defining the proper gates. Appropriate isotype matched controls were used for defining
negative gates (Figure 5.2).
The populations were identified as - erythrocytes (GlycophorinA+ CD45-), leukocytes
(CD45+ GlycophorinA-), granulocytes (CD66+ CD14low), monocytes (CD14+ CD66-),
lymphocytes (CD66- CD14-) in the input, sorted, and waste streams (Fig. 5.2 a,b). We
found that the sorted stream consisted of highly pure population of leukocytes 99.6 ± 0.3
% (up to 99.84%) (Figure 5.3). The enrichment ratio (E.R.) as defined in previously5, was
calculated as follows:
E.R. =
(
)
pt ,sorted pnt ,sorted pt ,sorted 1− pt ,input
=
pt ,input pnt ,input
pt ,input (1− pt ,sorted )
(0.1)
where pt,input and pt,sorted are purities of the target cells in input and sorted fractions
respectively, and pnt,input and pnt,sorted are purities of the non-target cells in input and sorted
fractions respectively (measured by flow cytometry). E.R. of leukocytes (which is same
as rejection ratio of RBCs) is calculated considering leukocytes as target cells and RBC
as non-target cells and was found to be 0.4×106 fold. This enrichment ratio was three
Chapter 5: AFF for sorting neutrophils from blood
89
orders of magnitude higher than that typically obtained in other chip-based continuous
flow fractionation methods.5 The sorted leukocyte population was highly enriched in
neutrophils with purity 92.1 ± 0.2 % (Fig 5.3b), representing a neutrophil enrichment
ratio of 18,760 with respect to other cells, while the waste stream was significantly
depleted of neutrophils. The neutrophil purity is comparable to existing methods of
neutrophil separation by antibody capture4 and to the best of our knowledge is the first
demonstration of using transient weak adhesive interactions to attain such high purities
and enrichment ratios in a continuous flow system.
Figure 5.2. Purity of sorted neutrophils. (a,b) Phenotyping via flow cytometry analysis
of the (a) input and sorted sample, and (b) waste samples. Input is depleted of
erythrocytes to enable cytometry. For this experiment, 25% of the flow was collected as
sorted sample. The sorted sample could be seen enriched in neutrophils while the waste is
depleted. (e) Histological staining of the sorted sample showing polymorphonuclear
neutrophils. Scale bars are 20 µm.
In neutrophil separation experiments using diluted blood, based on the percentage of
neutrophils in the samples and using the above formula we found the recovery of
neutrophils to be ~67% (Input: 62.2%, Sorted: 91.9%, Waste: 37.75%). Neutrophil
recovery was also independently measured to corroborate the finding from the purity
Chapter 5: AFF for sorting neutrophils from blood
90
data. We obtained time lapse images at the channel exit at regular intervals (between 40 90 min of injection) that were manually analyzed to determine the average flux of the
sorted neutrophils. The neutrophil recovery was determined to be 68.5% based on the
flux of the sorted neutrophils and flux of input neutrophils (obtained from the measured
concentration of neutrophils in the input sample and the flow rate of blood). Thus we
estimate the recovery of neutrophils to be between 65-70%. Although the device also
worked with whole blood, the neutrophil recovery was 4-fold lower than with 1:1 diluted
blood possibly due to increased steric hindrance by the erythrocytes or the presence of
PSGL-1 in plasma.25
Figure 5.3. Purity of the samples determined by flow cytometric analysis. (a) The ratios
of erythrocytes and leukocytes in the samples before and after sorting demonstrate high
erythrocyte rejection. (b) Composition of the leukocyte population in the sample before
and after sorting demonstrates high degree of neutrophil enrichment. Error bars
represent SD of n = 3 independent experiments.
5.4 Characterization of sorted cells
Cells are sensitive to the treatment undertaken and several separation methods have been
known to affect the biological activity of separated cells. Using the trypan blue dye
exclusion assay we found the viability of the sorted cells to be 98.8 ± 0.7%. In addition,
we performed a battery of tests on the sorted cells to establish effect of sorting on the
cellular activity.
5.4.1 Hematological analysis
Chapter 5: AFF for sorting neutrophils from blood
91
The surface marker CD66b belongs to the carcino-embryonic antigen family and is
broadly expressed on all granulocytes, i.e – neutrophils, eosinophils and basophils. Since
Neutrophils usually out number the later two subtypes by 30:1 ratio, we classified all
CD66b+ cells as neutrophils. Therefore, we performed hematological staining on the
sorted cells in order to subtype the sorted granulocytes. We added 100 µL of the sorted
sample on a poly-L-Lysine cover slip and allowed the cells to attach to the glass surface.
The cover slips were then air dried, fixed in methanol and stained using eosin and azure
B (VWR LLC) followed by washing in pH 6.8 buffer. The stained cells were analyzed on
an inverted microscope in bright field. Hematological staining revealed that 95% of the
sorted granulocytes had segmented nuclei (41 out of 43 analyzed) typical of mature
polymorphonuclear neutrophils (Fig. 5.2c)
5.4.2 P-selectin binding
PSGL-1 is the major ligand for P-selectin and is known to be broadly expressed on all
leukocytes
169
. However not all forms of expressed PSGL-1 are functional and hence
there is variability between the myleiod and lymphoid population in terms of rolling on
P-selectin surfaces
170
. Hence, instead of using anti-PSGL-1 antibody, we directly tested
the affinity of the cells toward P-selectin binding by using fluorescent P-selectin
complexes. The staining solution was made by mixing P-selectin-Fc (R&D systems),
biotinylated anti-Human Fc (Sigma) and Streptavidin-PE (Biolegend) at a final
concentration of 10µg/mL, 40 µg/mL, and 40 µg/mL, respectively in 1% BSA solution in
DPBS containing 1mM CaCl2 (Fig. 5.4a). The samples (whole blood and sorted cells)
were washed and resuspended in 100 µL of cell staining solution. A nuclear staining dye
Hoechst 33342 (Invitrogen) was then added to the solution at 5µM final concentration
and the sample was kept on ice for 30 min in dark. Next, the samples were washed,
resuspended in the staining buffer and analyzed within 30 min on a flow cytometer.
Leukocytes were gated using Hoechst fluorescence (Pacific blue channel) and forward
scatter. Whole blood stained with a mixture of streptavidin-PE and biotinylated antihuman Fc was used as negative control to determine non-specific binding of streptavidin
to the cells. In whole blood two clear populations of leukocytes emerged – one that bind
Chapter 5: AFF for sorting neutrophils from blood
92
to P-selectin avidly (~47%) and the non-binders (~53%). However majority of the sorted
fraction (~91%) consisted of the strong P-selectin binding cells (Fig. 5.4b).
Figure 5.4. P-selectin binding affinity of the input and sorted fractions. a) Schematic
showing the steps involved in making the fluorescent P-selectin complexes. b) Flow
cytometry was used to measure the binding of P-selectin complexes to the leukocytes in
input and sorted fractions. Leukocytes were gated using nuclear staining by Hoechst
33342. Whole blood stained with streptavidin-PE and antibody complex (without Pselectin) was used as negative control.
5.4.3 Activation assay
Neutrophils are extremely sensitive to stimulus and can undergo rapid activation resulting
in several phenotypic and morphological changes.26-28 To detect whether the sorted
neutrophils were activated, we used the classical markers – L-selectin and Mac-1 – that
are shed and up-regulated respectively upon activation.29 For measuring the expression
level of L-selectin and Mac-1, the sorted cells were collected in HBSS(-/-) on ice after
which they were washed and stained using anti-CD66-FITC, anti-CD62L-V450 and antiCD11b-PE antibody (Biolegend) in cell staining buffer for 20min . Then, formaldehyde
solution was directly added to the tubes until the final concentration was 4%
Chapter 5: AFF for sorting neutrophils from blood
93
formaldehyde, and the cells were left to fix in dark for 30 min. Then the cells were
washed and immediately analyzed in a BD LSR Fortessa flow cytometer. In parallel
experiment, the same cocktail of antibody was added to either whole blood or whole
blood previously incubated with HBSS, dextran (Sigma), Mono-poly resolving media
(Cederlane Labs), Histopaque 1077 (Sigma) and Percoll (GE Healthcare). All samples
preparation, centrifugation and staining were done on ice or at 4oC in order to avoid cell
activation. Neutrophils were gated from the FCS and FITC fluorescence and the V450
(Pacific Blue channel) and PE fluorescence of each sample was recorded. Whole blood
incubated with LPS (1 µg/mL for 30 min at 37oC) was used as positive control (activated
neutrophils).
When compared to fresh whole blood and activated control, the sorted neutrophils
exhibited minimal change in expression of L-selectin and Mac-1, indicating that they
were not activated (Fig. 5.5a). Variability of activation levels between different AFF
experiments on the same sample was also absent confirming the reproducibility of the
method (Fig 5.5b). In contrast, all of the separation media affected L-selectin expression
(most notable with Histopaque), although little change in Mac-1 expression was observed
Figure 5.5. Activation assay of sorted neutrophils. (a) Expression level of activation
markers L-selectin and Mac-1 on neutrophils in sorted cells and fresh whole blood.
Isotype control (negative control) and activated neutrophils (positive control) are shown
for reference. (b) Effects of different neutrophil isolation methods on activation. Whole
blood was collected from a healthy donor and neutrophils were isolated using affinity
flow fractionation (AFF, labeled as ‘Neutrophil -1,2,3’) in three separated experiments.
Chapter 5: AFF for sorting neutrophils from blood
94
At the same time, aliquots of whole blood was mixed with buffer, dextran, histopaque1077, mono-poly resolving media and percoll, incubated for 30 min and washed.
Expression level of L-selectin and Mac-1 were measured using flow cytometry and the
results are shown as mean fluorescence intensity (MFI) with the error bar showing SD
for each distribution. Blood incubated with LPS (1µg/ml for 30 min) was used as positive
control for activation.
5.4.4 Phagocytosis assay
A major physiological function of neutrophils is to phagocytose pathogens and opsonized
foreign particles in blood and tissue. This is a major defense mechanism of the body
against bacterial infections. Mature neutrophils are also very efficient in killing of the
phagocytosed pathogens though lysosome-phagosome fusion which leads to acidification
and killing of its contents. Thus the bactericidal effects of the neutrophils depends on the
two step process of successful phagocytosis followed by its acidification. We used
pHRoho® E.Coli bioparticles (Invitrogen) in order to determine bactericidal activity of
the sorted neutrophils. The assay consists of E.Coli particles tagged with pHRodo - a pHsensitive dye that becomes fluorescent at low pH environments such as those found
inside lysosomes, which helps to distinguish between particles attached to the cell from
those that are internalized.
The bioparticles were reconstituted in buffer at a concentration of 1mg/mL and the
particle count was determined using a hemacytometer. For the phagocytosis assay the
sorted cells (collected in HBSS on ice) were resuspended in autologous plasma (prepared
beforehand by removing the cells in whole blood through centrifugation). The
bioparticles were added in excess (greater than 1000 particles per neutrophil) to 200 µL
samples of whole blood or sorted cells and incubated at 37oC for 1 h. Whole blood
without the bioparticles kept at 37oC and whole blood with the bioparticles kept on ice (to
measure baseline fluorescence of attached but non-internalized particles) served as
negative control. At the end of 1 h, 200 µL of ice cold stop solution 97 (1% FBS in HBSS
with 2 mM of NaF) was added to each sample followed by washing to remove unbound
particles. All further washing and resuspension steps were performed with the stop
Chapter 5: AFF for sorting neutrophils from blood
95
solution at 4oC. Next, the samples were stained with CD66-FITC, washed and
immediately analyzed using a flow cytometer. Neutrophils were gated using forward
scatter and FITC fluorescence and phagocytosis was measured using the fluorescence in
PE channel. Whole blood with bioparticles kept on ice was used to define the extent of
positive gates (Fig. 5.6a,b). We found that level of phagocytosis was similar in whole
blood and sorted cells – 90.2% and 92% respectively (Fig. 5.6b).
Figure 5.6. Phagocytosis activity of neutrophils in whole blood and sorted fraction. (a)
Brightfeild, fluorescent and overlaid images of the sorted neutrophils demonstrating
successful phagocytosis of E.coli particles tagged with a pH-sensitive fluorescent dye.
Scale bars are 20 µm. (b) Flow cytometric analysis of the cells to quantify level of
phagocytosis.
5.5 Label-free detection of neutrophil activation in blood
The ability to sort cells in continuous flow without extensive sample preparation is useful
for point-of-care analysis as it greatly simplifies the device design, potentially enabling
Chapter 5: AFF for sorting neutrophils from blood
96
instrument-free disposable devices. Neonatal sepsis accounts for a large fraction of
childhood deaths in developing countries, but diagnosis is challenging due to non-specific
clinical symptoms.15 While detection of neutrophil activation by up-regulation of CD64
has high diagnostic value,30 it requires flow cytometry that is typically not available in
resource-limited settings. Given that activation of neutrophils reduces their efficiency of
rolling on P-selectin,31 we investigated whether the present method could be used to
detect neutrophil activation in blood. A concept of such a device is shown in Fig. 5.7a,
where a downstream detector counts the number of cells that are sorted out. Since
background cells are rejected, the detector does not need to discriminate between the
types of cells being sorted, analogous to a detector at the end of a chromatography
column. If neutrophils are activated, we expect a decrease in the flux of sorted cells.
To detect activation of blood, the microfluidic device design was slightly modified to
include four independent 10 cm long channel on a single chip. Blood was collected in
citrate vaccuitainer as before and four aliquots of 250 µL each were immediately made.
Next, 25 µL of buffer (DPBS), Lipopolysaccharide (Sigma) (10 µg/ml in DPBS), Tumor
Necrosis Factor – α (Biolegend) (10 µg/mL in DPBS each) and Platelet Activating Factor
(Sigma) (1 µg/mL in DPBS) were added in each tube and incubated for 30 min at room
temperature. Then the four samples were diluted (1:1) in running buffer and injected in
each separated channel. The experimental setup and flow parameters were similar to that
in separation experiment, except the outlets of the channels were connected to a waste
collection tube. Time lapse images were taken at the exit of each channel at regular
intervals and were manually analyzed to obtain unbiased counts of cells separated from
the blood stream (i.e. all cells separated from the blood stream were counted). The
experiment was performed in triplicate with independent samples.
In case of normal blood (incubated with buffer), neutrophils rolled on the edges and were
separated from the blood stream as early as 10 min after injection. However, in the case
of activated blood there was very little or no attachment of neutrophils to the P-selectin
patterns and separation of cells from the blood stream did not occur (Figure 5.7b). We
also did not observe sticking of neutrophils or platelets inside the device. We quantified
the average flux of the sorted cells between 15 and 30 min after injection was started in a
Chapter 5: AFF for sorting neutrophils from blood
97
region 10 cm downstream of the inlet defined as shown in Fig. 5.7a. Using blood samples
from different donors, we found that the flux of sorted cells in activated blood was
dramatically diminished as compared to a normal blood sample (Fig. 5.4b,c).
Figure 5.7. Detecting neutrophil activation using activation-dependent cell sorting. (a) A
cartoon showing the concept for detecting activation of neutrophils in blood. A narrow
stream of blood is flowed parallel to a buffer stream on the asymmetric P-selectin
patterned surface and an unbiased cell counter (that counts the flux of all cells separated
from the blood stream) enumerates the sorted cells downstream of the inlet as shown.
While neutrophils in normal blood separate from the blood stream at a high rate,
activated neutrophils fail to separate due to lack of interaction with the P-selectin
patterns leading to very low flux that correlates with the activated state of the blood. (b,c)
Anticoagulated whole blood was activated ex vivo by incubation with LPS, TNF-α and
PAF or buffer as control, and infused into the device after dilution (1:1) parallel to the
buffer stream following the same protocol for separation. (b) The flux of sorted cells
measured 10 cm downstream at different time points after injection. While the flux of cells
in normal blood seemed to peak at 30 min, the flux of activated blood remained low
Chapter 5: AFF for sorting neutrophils from blood
98
throughout. (c) The average flux of sorted cells between 15-30 min post infusion. Blood
activated using LPS, TNF-α and PAF as agonist resulted in significantly lower (** p <
0.038) flux of sorted cells when compared to time-matched samples of normal blood
(incubated with buffer). Error bars show the SD of n = 3 independent experiments.
5.6 Conclusion
There are multiple technologies that enable isolation of cells from blood. While the
method of affinity capture, seemed to provide with the highest purity, density gradient
and inertial separation methods allow for higher throughput and hence processing of
larger sample volumes. Table 5.1 summarizes the performance of some of the state-of-art
devices for sorting blood cells in terms of purity and recovery.
Table 5.1. Comparison of AFF with other blood cell separation methods
Principle
Molecule
Cells
Performance
Ref
Affinity Flow Fractionation
P-selectin
Neutrophils
form blood
>92% purity | 70% efficiency
18,500x neutrophil enrichment
400,000x leukocyte enrichment
Dean flow separation
-
Leukocytes
from blood
20x leukocyte enrichment
>95% efficiency
172
Micropost array
-
Leukocytes
from blood
110x leukocyte enrichment
173
Microfluidic Leukapheresis
-
Leukocytes
from blood
2x leukocyte enrichment
174
Density gradient separation
-
Granulocytes,
lymphocytes
60-70% efficiency, >95% purity
(after lysing RBC)
97
Antibody capture
anti-CD66
Neutrophils
>95% purity
5
Antibody capture
anti-CD4
T-cells
Efficiency >85%
Purity>95%
1
Affinity capture
α6-integrin
Cervical
cancer cells
Efficiency >30%,
Purity >95%
175
171
Neutrophils were isolated within 30 min using AFF, which is significantly faster than the
conventional density gradient isolation that requires ~1 h and involves multiple wash
steps.28 While the purity of 92% with AFF is comparable to previous antibody based
methods4 and density-gradient methods,28 the RBC rejection ratio of AFF is highest
Chapter 5: AFF for sorting neutrophils from blood
99
reported amongst current continuous flow fractionation systems, for example - 400,000fold with AFF compared to ~20-fold in microfluidic centrifugal sorting,37 110-fold in
micropost assay,38 ~2-fold (at optimum flow rate of 5 µL/min) in leukophesesis.39 We
believe AFF may provide a method for rapid extraction of leukocytes and neutrophils
from unprocessed blood to yield cells ready for biological assays or genetic analysis
without requiring washing or other preparatory steps.40
This page is intentionally left blank.
6
Conclusions and Discussion
In this thesis, we developed Affinity Flow Fractionation of cells - a new method to isolate
cells in a continuous manner, based on interaction of specific ligands on cell surface with
an asymmetric receptor pattern. This method overcomes the limitations of previous labelfree cell separation techniques where specific isolation was limited to permanent capture
of cells. The specific contributions of the thesis are:
•
First demonstration of continuous sorting of cells based on weak interactions.
•
Developed a standard approach to characterize weak interactions and design
optimized devices to enable separation.
•
First demonstration of sorting leukocytes directly from blood using selectins in
continuous flow.
•
Highest RBC rejection amongst similar continuous flow systems, purity
comparable to antibody based methods.
•
First microfluidic device enabling activation dependent sorting of neutrophils.
Weak interactions play an important role in many biological and physiological processes
such as bacterial adhesion83, cell homing176, immune surveillance160, hemostasis177,
embryogenesis178, neural circuit formation179 etc. Selecins and their interaction with sialyl
Lewis180,181 residues on other glycoproteins, perhaps is has been the most widely studied
Chapter 6: Conclusions and Discussion
102
weak affinity interaction. Hence, in this work we have demonstrated AFF using Pselectin as a model weak-affinity ligand. Apart from neutrophil separations, selectins
have been shown to have potential to separate hematopoietic stem cells from bone
marrow88, CD34+CD38+ from CD34+CD38-
182
, and cancer cells from blood87. Selectin-
based separation of these cells has been limited to batch process requiring separate
capture and release that yields relatively low purity87,88. Devices with selectin-coated
microstructures have also been employed for sorting90,183, but have failed to sort cells
from blood due to mixing of the flows leading to dispersion of the red blood cells, which
does not occur in AFF due to the absence of lateral flows. Our results open new avenues
for selectin-based sorting that overcome the limitations of batch processing in a format
suitable for direct isolation in a flow-through device. The high purity separation enabled
by AFF is due to the absence of lateral fluid flows and the multiple molecular recognition
events that gradually lead to lateral displacement, in contrast to approaches that utilize a
single recognition event such as capture by high-affinity antibodies where non-specific
adhesion directly impacts the purity.
Neutrophils were isolated within 30 min using AFF, which is significantly faster than the
conventional density gradient isolation that requires ~3 hrs and involves multiple wash
steps97. While the purity of 92% with AFF is comparable to previous antibody based
methods5 and density-gradient methods97, the RBC rejection ratio of AFF is highest
reported amongst current continuous flow systems, for example - 400,000-fold with AFF
compared to ~20-fold in microfluidc centrifugal sorting184, 110-fold in micropost
assay185, ~2-fold (at optimum flow rate of 5 µL/min) in leukophesesis186. We believe
AFF may provide a method for rapid extraction of leukocytes and neutrophils from
unprocessed blood to yield cells ready for biological assays or genetic analysis without
requiring washing or other preparatory steps187. The current device can process blood at
~1 µL/min and is ideally suitable for analytical applications such as obtaining neutrophils
counts and isolation of cells for genetic analysis. Given that most common blood cells
exist at high concentrations (>10 to104 µL-1) and standard automated counters typically
analyze very small volumes (~10-50 µL)2, similar results can be obtained in 20-30 min in
our device. The system can potentially be massively parallelized to enhance the
Chapter 6: Conclusions and Discussion
103
throughput which would enable processing large sample volumes extending to
applications such as isolation and fractionation of stem cells for therapeutics.
The fact that this technique relies on the functional ability of cells to roll can be harnessed
to detect alterations in the rolling behavior of cells that can potentially be used to
diagnose disease states such as neonatal sepsis188. The low driving pressures (< 1 kPa)
and small sample volumes (<10 µL of blood and 200 µL of buffer) make the cell sorting
method potentially compatible with use of capillary forces for device operation. While
optical detection was used in the present work, we envision that cells may also be
counted in a variety of ways ranging from collection of the sorted cells over a
downstream filter for crude manual counting, to integrating simple capacitive sensors.
Figure 6.1. Applications of Affinity Flow Fractionation of cells
In general, the applications of AFF can be categorized into two broad areas – cell sorting
and cell analysis, as describe in Figure 6.1.
Cell sorting using AFF overcomes a number of challenges of previous methods,
and hence is uniquely suited for use in disposable hematology counters for diagnostic
applications. A number of physiological ligands can be used to sort a variety of cells. For
example – VCAM32,189 and MHCII190 can be used to sort lymphocytes and specifically
CD4 T cells respectively, RBCs infected specifically with plasmodium falciparum are
Chapter 6: Conclusions and Discussion
104
known roll on CD36191 amongst other ligands which might be useful for detecting
malaria and the difference in interactions of different subtypes of leukocytes with
selectins can be leveraged to fractionate the leukocyte population106. AFF of cells can
potentially be used to for a number of therapeutic applications. The advantage of labelfree isolation of cells without capture allows for minimal modification of cells,
preserving biological activity ultimately reducing risk of complications. Leukoreduction
of transfusion blood is one of the key areas where AFF might be useful. Activation of
neutrophils in transfusion blood is a major cause of TRALI (Transfusion Related Acute
Lung Injury) which might be prevented by pre-processing blood samples to isolate
neutrophils192. But, it is imperative that high throughput devices based on AFF must be
developed for application in therapeutic sorting. In order to extend AFF as a generic cellsorting tool, synthetic weak affinity ligands must be engineered and produced. Synthetic
antibodies193 with customized affinities, weak affinity aptamers194 and yeast-displayed
peptides195 are some of the methods that can be used to develop AFF ligands against
desired targets. Existing theoretical studies on effects of ligand affinity110,113 on nature of
cell adhesion and rolling will be useful for guiding development of such synthetic weak
affinity ligands.
Cell analysis is another area where AFF might shape up to be a powerful tool.
Presently, quantitative analysis of expression levels of surface makers is possible though
flow cytometry but is limited to strong interactions and only for known cell surface
markers. While a number of important physiological ligands are weak affinity, the
transient nature of these interactions makes it difficult to quantify them. AFF is one of the
only techniques that provide a direct method to measure, albeit in a semi-quantitative
manner. Weak interaction between the cell and the asymmetrically receptor patterned
surface produces displacement in the cell trajectory which might be used to quantify the
degree of affinity to the surface. We had demonstrated that between K562 and HL60
cells, the former owing to only weak interaction with P-selectin is deflected (more than
control cells) less than the later which shows robust rolling on P-selectin surfaces. Since
knowledge of the ligands on cell surface participating in the process is not required, it
open up possibility for a number of applications such as - analyzing differentiation of
stem cell196 and measuring changes in adhesion signature of cancer cells197,198.
Chapter 6: Conclusions and Discussion
105
One might envision a number of exciting extensions of AFF to engineer novel separation
substrates. For example, separation channels could potentially be arranged in series to
sort cells based on more than one surface marker, or even different patterns could be
superimposed on each other to control trajectory of different cell populations. As such,
the ability to sort cells with high purity in continuous flow stream offers new
opportunities in analysis of cells, and at the point-of-care.
This page is intentionally left bank.
References
1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. Cheng, X., et al. A microfluidic device for practical label-­‐free CD4(+) T cell counting of HIV-­‐infected subjects. Lab Chip 7, 170-­‐178 (2007). McPherson, R.A., Pincus, M.R. & Henry, J.B. Henry's clinical diagnosis and management by laboratory methods, (Saunders Elsevier, Philadelphia, 2007). Ley, K., Laudanna, C., Cybulsky, M.I. & Nourshargh, S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7, 678-­‐689 (2007). Bosio, A., et al. Isolation and enrichment of stem cells. Adv Biochem Eng Biotechnol 114, 23-­‐72 (2009). Kotz, K.T., et al. Clinical microfluidics for neutrophil genomics and proteomics. Nat Med 16, 1042-­‐U1142 (2010). Toner, M. & Irimia, D. Blood-­‐on-­‐a-­‐chip. Annual Review of Biomedical Engineering 7, 77-­‐103 (2005). Habibi, N., et al. Self-­‐assembly and recrystallization of bacterial S-­‐layer proteins of Bacillus sphaericus and Bacillus thuringiensis on silicone, mica and quartz crystal supports. Conf Proc IEEE Eng Med Biol Soc 2010, 3739-­‐
3742 (2010). Herrmann, A., Fleischer, K., Czajkowska, H., Muller-­‐Newen, G. & Becker, W. Characterization of cyclin L1 as an immobile component of the splicing factor compartment. Faseb J 21, 3142-­‐3152 (2007). Dandulakis, G., Herr, J.C. & Kirwan, D.J. Cell growth and monoclonal antibody production in the presence of antigen and serum. Biotechnol Prog 11, 518-­‐
524 (1995). Bierer, B.E., Herrmann, S.H., Brown, C.S., Burakoff, S.J. & Golan, D.E. Lateral mobility of class I histocompatibility antigens in B lymphoblastoid cell membranes: modulation by cross-­‐linking and effect of cell density. J Cell Biol 105, 1147-­‐1152 (1987). Meyers, M.H. & Herron, M. A fibrin adhesive seal for the repair of osteochondral fracture fragments. Clin Orthop Relat Res, 258-­‐263 (1984). Bioconjugate Techniques 2nd Edition Preface to the Second Edition. Bioconjugate Techniques, 2nd Edition, Xxiii-­‐Xxv (2008). Blawas, A.S. & Reichert, W.M. Protein patterning. Biomaterials 19, 595-­‐609 (1998). Erdman, C.P., et al. Enrichment of adipose-­‐derived mesenchymal stem cells using resveratrol. Tissue Engineering Part A 14, 730-­‐730 (2008). Mooney, J.F., Rogers, C.T., Hunt, A.J. & McIntosh, J.R. A general technique for patterning of functional proteins with photolithography of silane monolayers. Biophys J 70, Tu216-­‐Tu216 (1996). Thio, T., et al. Hybrid Capillary-­‐Flap Valve for Vapor Control in Point-­‐of-­‐Care Microfluidic CD. Ifmbe Proc 35, 578-­‐581 (2011). References
17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 108
Mooney, J.F., et al. Patterning of functional antibodies and other proteins by photolithography of silane monolayers. Proc Natl Acad Sci U S A 93, 12287-­‐
12291 (1996). Ramaswamy, B., Yeh, Y.T.T. & Zheng, S.Y. Microfluidic device and system for point-­‐of-­‐care blood coagulation measurement based on electrical impedance sensing. Sensor Actuat B-­‐Chem 180, 21-­‐27 (2013). Kim, U., Ravikumar, A., Seubert, J. & Figueira, S. Detection of Bacterial Pathogens through Microfluidic DNA Sensors and Mobile Interface toward Rapid, Affordable, and Point-­‐of-­‐Care Water Monitoring. 2013 Ieee Point-­‐of-­‐
Care Healthcare Technologies (Pht), 1-­‐4 (2013). Dean, D., et al. A Multiplexed Microfluidic PCR Assay for Sensitive and Specific Point-­‐of-­‐Care Detection of Chlamydia trachomatis. Plos One 7(2012). Govindarajan, A.V., Ramachandran, S., Vigil, G.D., Yager, P. & Bohringer, K.F. A low cost point-­‐of-­‐care viscous sample preparation device for molecular diagnosis in the developing world; an example of microfluidic origami. Lab Chip 12, 174-­‐181 (2012). Iwai, K., Sochol, R.D., Lee, L.P. & Lin, L. Finger-­‐Powered Bead-­‐in-­‐Droplet Microfluidic System for Point-­‐of-­‐Care Diagnostics. 2012 Ieee 25th International Conference on Micro Electro Mechanical Systems (Mems) (2012). Li, P., Sherry, A.J., Cortes, J.A., Anagnostopoulos, C. & Faghri, M. A blocking-­‐
free microfluidic fluorescence heterogeneous immunoassay for point-­‐of-­‐care diagnostics. Biomed Microdevices 13, 475-­‐483 (2011). Samorajski, J., Wang, J., Sutherland, A.M. & Heath, J. Magnetically activated valves for point-­‐of-­‐care microfluidic "lab-­‐on-­‐a-­‐chip" devices. Abstr Pap Am Chem S 241(2011). Al-­‐Gwaiz, L.A. & Babay, H.H. The diagnostic value of absolute neutrophil count, band count and morphologic changes of neutrophils in predicting bacterial infections. Medical Principles and Practice 16, 344-­‐347 (2007). Anidi, I.U., et al. CD36 and Fyn Kinase Mediate Malaria-­‐Induced Lung Endothelial Barrier Dysfunction in Mice Infected with Plasmodium berghei. Plos One 8(2013). Omi, K., et al. CD36 as a candidate gene predisposing to severe malaria: polymorphism screening and association with cerebral malaria. Am J Hum Genet 71, 367-­‐367 (2002). Tandon, N.N., Chen, W., Greco, N.J., Ockenhouse, C.F. & Jamieson, G.A. Characterization of anti-­‐platelet CD36 monoclonal antibodies and their effects on platelet activation and cytoadherence of malaria-­‐infected red blood cells. Tissue Antigens 48, Pl203-­‐Pl203 (1996). Asch, A.S., Silbiger, S., Heimer, E. & Nachman, R.L. Cd36-­‐Transfected Jurkat Cells Bind Thrombospondin Via a Malaria-­‐Homologous Domain. Clin Res 39, A338-­‐A338 (1991). Khan, S.Y., et al. Lysophosphatidylcholines activate G2A inducing G(alpha i-­‐
1)-­‐/G(alpha q/11)-­‐ Ca2+ flux, G(beta gamma)-­‐Hck activation and clathrin/beta-­‐arrestin-­‐1/GRK6 recruitment in PMNs. Biochemical Journal 432, 35-­‐45 (2010). References
31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 109
Tantra, R. & van Heeren, H. Product qualification: a barrier to point-­‐of-­‐care microfluidic-­‐based diagnostics? Lab Chip 13, 2199-­‐2201 (2013). Thankamony, S.P. & Sackstein, R. Enforced hematopoietic cell E-­‐ and L-­‐
selectin ligand (HCELL) expression primes transendothelial migration of human mesenchymal stem cells. P Natl Acad Sci USA 108, 2258-­‐2263 (2011). Olagnier, D., et al. Nrf2, a PPAR gamma Alternative Pathway to Promote CD36 Expression on Inflammatory Macrophages: Implication for Malaria. Plos Pathog 7(2011). Olagnier, D., et al. Nrf2-­‐Mediated Cd36 over Expression Independently of Ppar Gamma in Inflammatory Macrophages Improves the Outcome of Severe Malaria. Inflamm Res 60, 38-­‐38 (2011). Patel, S.N., Lu, Z.Y., Ayi, K.J., Katz, K.C. & Kain, K.C. Disruption of CD36 impairs cytokine response to Plasmodium falciparum GPI and confers susceptibility to severe and fatal malaria in vivo. Am J Trop Med Hyg 75, 257-­‐257 (2006). Serghides, L., Smith, T.G., Patel, S.N. & Kain, K.C. CD36 and malaria: friends or foes? Trends Parasitol 19, 461-­‐469 (2003). Franke-­‐Fayard, B., et al. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. P Natl Acad Sci USA 102, 11468-­‐11473 (2005). Tomlinson, M.J., Tomlinson, S., Yang, X.B. & Kirkham, J. Cell separation: Terminology and practical considerations. Journal of tissue engineering 4, 2041731412472690 (2013). Zola, H., et al. CD molecules 2006 -­‐ Human cell differentiation molecules. J Immunol Methods 319, 1-­‐5 (2007). Ye, M., et al. Generating Aptamers by Cell-­‐SELEX for Applications in Molecular Medicine. Int J Mol Sci 13, 3341-­‐3353 (2012). Lauridsen, L.H., Rothnagel, J.A. & Veedu, R.N. Enzymatic Recognition of 2 '-­‐
Modified Ribonucleoside 5 '-­‐Triphosphates: Towards the Evolution of Versatile Aptamers. Chembiochem 13, 19-­‐25 (2012). Song, K.M., Lee, S. & Ban, C. Aptamers and Their Biological Applications. Sensors-­‐Basel 12, 612-­‐631 (2012). Wyman, T.H., et al. A two-­‐insult in vitro model of PMN-­‐mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol-­‐Cell Ph 283, C1592-­‐C1603 (2002). Yang, X.B., Li, N. & Gorenstein, D.G. Strategies for the discovery of therapeutic aptamers. Expert Opin Drug Dis 6, 75-­‐87 (2011). Topp, S. & Gallivan, J.P. Emerging Applications of Riboswitches in Chemical Biology. Acs Chem Biol 5, 139-­‐148 (2010). Inganas, M., et al. Integrated microfluidic compact disc device with potential use in both centralized and point-­‐of-­‐care laboratory settings. Clin Chem 51, 1985-­‐1987 (2005). Mayer, G., Rohrbach, F., Potzsch, B. & Muller, J. Aptamer-­‐based modulation of blood coagulation. Hamostaseologie 31, 258-­‐263 (2011). Harris, L.F., Rainey, P., Castro-­‐Lopez, V., O'Donnell, J.S. & Killard, A.J. A microfluidic anti-­‐Factor Xa assay device for point of care monitoring of anticoagulation therapy. Analyst 138, 4769-­‐4776 (2013). References
49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 110
Liu, J., et al. Recent Developments in Protein and Cell-­‐Targeted Aptamer Selection and Applications. Curr Med Chem 18, 4117-­‐4125 (2011). Zhou, J.H. & Rossi, J.J. Cell-­‐Specific Aptamer-­‐Mediated Targeted Drug Delivery. Oligonucleotides 21, 1-­‐10 (2011). Sun, W., Du, L.P. & Li, M.Y. Advances and Perspectives in Cell-­‐Specific Aptamers. Curr Pharm Design 17, 80-­‐91 (2011). Shaw, R.A., et al. Toward point-­‐of-­‐care diagnostic metabolic fingerprinting: quantification of plasma creatinine by infrared spectroscopy of microfluidic-­‐
preprocessed samples. Analyst 134, 1224-­‐1231 (2009). Huang, Y.F., Kim, Y., Meng, L. & Tan, W.H. Assembly of aptamer conjugates as molecular tools in therapeutics. Chim Oggi 27, 52-­‐54 (2009). Ulrich, H. & Wrenger, C. Disease-­‐Specific Biomarker Discovery by Aptamers. Cytom Part A 75A, 727-­‐733 (2009). Peyrin, E. Nucleic acid aptamer molecular recognition principles and application in liquid chromatography and capillary electrophoresis. J Sep Sci 32, 1531-­‐1536 (2009). Gatto, B., Palumbo, M. & Sissi, C. Nucleic Acid Aptamers Based on the G-­‐
Quadruplex Structure: Therapeutic and Diagnostic Potential. Curr Med Chem 16, 1248-­‐1265 (2009). Wang, W. & Jia, L.Y. Progress in Aptamer Screening Methods. Chinese J Anal Chem 37, 454-­‐460 (2009). Zheng, J., He, P.G. & Fang, Y.Z. Aptamer-­‐Based Biosensor. Prog Chem 21, 732-­‐
738 (2009). Kulbachinskiy, A.V. Methods for selection of aptamers to protein targets. Biochemistry-­‐Moscow+ 72, 1505-­‐1518 (2007). Tan, Y. & Zhang, X.M. Aptamer Mediated Delivery of Small Interfering RNAs. Prog Biochem Biophys 39, 410-­‐415 (2012). Yang, Y., Ren, X., Schluesener, H.J. & Zhang, Z. Aptamers: Selection, Modification and Application to Nervous System Diseases. Curr Med Chem 18, 4159-­‐4168 (2011). Eniola, A.O., Willcox, P.J. & Hammer, D.A. Interplay between rolling and firm adhesion elucidated with a cell-­‐free system engineered with two distinct receptor-­‐ligand pairs. Biophys J 85, 2720-­‐2731 (2003). Bhatia, S.K., King, M.R. & Hammer, D.A. The state diagram for cell adhesion mediated by two receptors. Biophys J 84, 2671-­‐2690 (2003). Tino, P. & Hammer, B. Architectural bias in recurrent neural networks: Fractal analysis. Neural Comput 15, 1931-­‐1957 (2003). Tannenberg, R.K., et al. Nucleic Acid Aptamers as Novel Class of Therapeutics to Mitigate Alzheimer's Disease Pathology. Curr Alzheimer Res 10, 442-­‐448 (2013). Bhatia, S.K. & Hammer, D.A. Dynamic simulations of inflammatory cell recruitment: The state diagram for cell adhesion mediated by two receptors. Proceedings of the Ieee 29th Annual Northeast Bioengineering Conference, 158-­‐159 (2003). Gossett, D.R., et al. Label-­‐free cell separation and sorting in microfluidic systems. Anal Bioanal Chem 397, 3249-­‐3267 (2010). References
68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 111
Obrecht, D., et al. Recent Progress in the Discovery of Macrocyclic Compounds as Potential Anti-­‐Infective Therapeutics. Curr Med Chem 16, 42-­‐
65 (2009). Baytas, S.N. & Linhardt, R.J. Combinatorial carbohydrate synthesis. Mini-­‐Rev Org Chem 1, 27-­‐39 (2004). Bertucci, C., Bartolini, M., Gotti, R. & Andrisano, V. Drug affinity to immobilized target bio-­‐polymers by high-­‐performance liquid chromatography and capillary electrophoresis. Journal of Chromatography B-­‐
Analytical Technologies in the Biomedical and Life Sciences 797, 111-­‐129 (2003). Wang, G.D., Reed, E. & Li, Q.Q. Molecular basis of cellular response to cisplatin chemotherapy in non-­‐small cell lung cancer (Review). Oncol Rep 12, 955-­‐965 (2004). Greene, M.E., Kinser, C.R., Kramer, D.E., Pingree, L.S.C. & Hersam, M.C. Application of scanning probe microscopy to the characterization and fabrication of hybrid nanomaterials. Microsc Res Techniq 64, 415-­‐434 (2004). Seidel, R. & Engelhard, M. Chemical Biology of Prion Protein: Tools to Bridge the In Vitro/Vivo Interface. Top Curr Chem 305, 199-­‐223 (2011). Di Bella, G., Mascia, F., Gualano, L. & Di Bella, L. Melatonin Anticancer Effects: Review. Int J Mol Sci 14, 2410-­‐2430 (2013). Fischer, P.M. The design, synthesis and application of stereochemical and directional peptide isomers: A critical review. Curr Protein Pept Sc 4, 339-­‐356 (2003). Kidoaki, S. & Matsuda, T. Mechanistic aspects of protein/material interactions probed by atomic force microscopy. Colloid Surface B 23, 153-­‐
163 (2002). Chin, C.D., Linder, V. & Sia, S.K. Commercialization of microfluidic point-­‐of-­‐
care diagnostic devices. Lab Chip 12, 2118-­‐2134 (2012). Janshoff, A. & Steinem, C. Scanning force microscopy of artificial membranes. Chembiochem 2, 799-­‐808 (2001). Yuan, Y., Oberholzer, M.R. & Lenhoff, A.M. Size does matter: electrostatically determined surface coverage trends in protein and colloid adsorption. Colloid Surface A 165, 125-­‐141 (2000). Shah, M.A. & Schwartz, G.K. The relevance of drug sequence in combination chemotherapy. Drug Resist Update 3, 335-­‐356 (2000). Simon, A., Nagel, D., Kitchen, B.J. & Bode, U. Clinical relevance of etoposide (VP-­‐16) pharmacokinetics in children and adults. Int J Pediat Hem Onc 4, 401-­‐
414 (1997). Ruardy, T.G., Schakenraad, J.M., vanderMei, H.C. & Busscher, H.J. Preparation and characterization of chemical gradient surfaces and their application for the study of cellular interaction phenomena. Surf Sci Rep 29, 3-­‐30 (1997). Anderson, B.N., et al. Weak rolling adhesion enhances bacterial surface colonization. J Bacteriol 189, 1794-­‐1802 (2007). Tedder, T.F., Steeber, D.A., Chen, A. & Engel, P. The Selectins -­‐ Vascular Adhesion Molecules. Faseb J 9, 866-­‐873 (1995). References
85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 112
Stone, J.D., Chervin, A.S. & Kranz, D.M. T-­‐cell receptor binding affinities and kinetics: impact on T-­‐cell activity and specificity. Immunology 126, 165-­‐176 (2009). Greenberg, A.W. & Hammer, D.A. Cell separation mediated by differential rolling adhesion. Biotechnol Bioeng 73, 111-­‐124 (2001). Hughes, A.D., et al. Microtube device for selectin-­‐mediated capture of viable circulating tumor cells from blood. Clin Chem 58, 846-­‐853. Narasipura, S.D. & King, M.R. P-­‐selectin-­‐coated microtube for the purification of CD45+ hematopoietic cells directly from human peripheral blood. Blood Cells, Molecules, and Diseases 42, 136-­‐139 (2009). Launiere, C., et al. Channel Surface Patterning of Alternating Biomimetic Protein Combinations for Enhanced Microfluidic Tumor Cell Isolation. Anal Chem 84, 4022-­‐4028 (2012). Chang, W.C., Lee, L.P. & Liepmann, D. Biomimetic technique for adhesion-­‐
based collection and separation of cells in a microfluidic channel. Lab Chip 5, 64-­‐73 (2005). Choi, S.Y., Karp, J.M. & Karnik, R. Cell sorting by deterministic cell rolling. Lab Chip 12, 1427-­‐1430 (2012). Choi, S., Song, S., Choi, C. & Park, J.K. Continuous blood cell separation by hydrophoretic filtration. Lab Chip 7, 1532-­‐1538 (2007). Hu, X.Y., et al. Marker-­‐specific sorting of rare cells using dielectrophoresis. P Natl Acad Sci USA 102, 15757-­‐15761 (2005). Karnik, R., et al. Nanomechanical control of cell rolling in two dimensions through surface Patterning of receptors. Nano Lett 8, 1153-­‐1158 (2008). Usta, O.B., Alexeev, A. & Balazs, A.C. Fork in the road: Patterned surfaces direct microcapsules to make a decision. Langmuir 23, 10887-­‐10890 (2007). Edington, C., et al. Tailoring the Trajectory of Cell Rolling with Cytotactic Surfaces. Langmuir 27, 15345-­‐15351 (2011). Clark, R.A. & Nauseef, W.M. Isolation and functional analysis of neutrophils. Curr Protoc Immunol Chapter 7, Unit 7 23 (2001). Nordon, R.E., Milthorpe, B.K., Schindhelm, K. & Slowiaczek, P.R. An Experimental-­‐Model of Affinity Cell-­‐Separation. Cytometry 16, 25-­‐33 (1994). Zhang, Y. & Neelamegham, S. Estimating the efficiency of cell capture and arrest in flow chambers: Study of neutrophil binding via E-­‐selectin and ICAM-­‐
1. Biophys J 83, 1934-­‐1952 (2002). Brunk, D.K. & Hammer, D.A. Quantifying Rolling Adhesion with a Cell-­‐Free Assay-­‐ E-­‐Selectin and Its Carbohydrate Ligands. Biophys J 72, 2820-­‐2833 (1997). Norman, K.E., Moore, K.L., Mcever, R.P. & Ley, K. Leukocyte Rolling in-­‐Vivo Is Mediated by P-­‐Selectin Glycoprotein Ligand-­‐1. Blood 86, 4417-­‐4421 (1995). Ramachandran, V., Williams, M., Yago, T., Schmidtke, D.W. & McEver, R.P. Dynamic alterations of membrane tethers stabilize leukocyte rolling on P-­‐
selectin. P Natl Acad Sci USA 101, 13519-­‐13524 (2004). Puri, K.D., Finger, E.B. & Springer, T.A. The faster kinetics of L-­‐selectin than of E-­‐selectin and P-­‐selectin rolling at comparable binding strength. J Immunol 158, 405-­‐413 (1997). References
113
104. Rainger, G.E., Fisher, A.C. & Nash, G.B. Endothelial-­‐borne platelet-­‐activating factor and interleukin-­‐8 rapidly immobilize rolling neutrophils. Am J Physiol-­‐
Heart C 272, H114-­‐H122 (1997). 105. Ramachandran, V., et al. PSGL-­‐1 dimerization stabilizes cell rolling on P-­‐
selectin in shear flow. Arterioscl Throm Vas 21, 709-­‐709 (2001). 106. Reinhardt, P.H. & Kubes, P. Differential leukocyte recruitment from whole blood via endothelial adhesion molecules under shear conditions. Blood 92, 4691-­‐4699 (1998). 107. Shao, J.Y., Ting-­‐Beall, H.P. & Hochmuth, R.M. Static and dynamic lengths of neutrophil microvilli. P Natl Acad Sci USA 95, 6797-­‐6802 (1998). 108. Alon, R., Chen, S.Q., Puri, K.D., Finger, E.B. & Springer, T.A. The kinetics of L-­‐
selectin tethers and the mechanics of selectin-­‐mediated rolling. J Cell Biol 138, 1169-­‐1180 (1997). 109. Korn, C.B. & Schwarz, U.S. Dynamic states of cells adhering in shear flow: From slipping to rolling. Phys Rev E 77, -­‐ (2008). 110. Bose, S., Das, S.K., Karp, J.M. & Karnik, R. A Semianalytical Model to Study the Effect of Cortical Tension on Cell Rolling. Biophys J 99, 3870-­‐3879 (2010). 111. Krasik, E.F. & Hammer, D.A. A semianalytic model of leukocyte rolling. Biophys J 87, 2919-­‐2930 (2004). 112. Chang, K.C. & Hammer, D.A. Adhesive dynamics simulations of sialyl-­‐
Lewis(x)/E-­‐selectin-­‐mediated rolling in a cell-­‐free system. Biophys J 79, 1891-­‐1902 (2000). 113. Chang, K.C., Tees, D.F.J. & Hammer, D.A. The state diagram for cell adhesion under flow: Leukocyte rolling and firm adhesion. P Natl Acad Sci USA 97, 11262-­‐11267 (2000). 114. Jadhav, S., Eggleton, C.D. & Konstantopoulos, K. A 3-­‐D computational model predicts that cell deformation affects selectin-­‐mediated leukocyte rolling. Biophys J 88, 96-­‐104 (2005). 115. Lawrence, M.B., Kansas, G.S., Kunkel, E.J. & Ley, K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E). J Cell Biol 136, 717-­‐727 (1997). 116. Dong, C. & Lei, X.X. Biomechanics of cell rolling: shear flow, cell-­‐surface adhesion, and cell deformability. J Biomech 33, 35-­‐43 (2000). 117. Wu, L., et al. Impact of carrier stiffness and microtopology on two-­‐
dimensional kinetics of P-­‐selectin and P-­‐selectin glycoprotein ligand-­‐1 (PSGL-­‐1) interactions. Journal of Biological Chemistry 282, 9846-­‐9854 (2007). 118. http://physics.georgetown.edu/matlab/. 119. James, C.D., et al. Patterned protein layers on solid substrates by thin stamp microcontact printing. Langmuir 14, 741-­‐744 (1998). 120. Bernard, A., et al. Printing patterns of proteins. Langmuir 14, 2225-­‐2229 (1998). 121. Tan, J.L., Tien, J. & Chen, C.S. Microcontact printing of proteins on mixed self-­‐
assembled monolayers. Langmuir 18, 519-­‐523 (2002). References
114
122. Graber, D.J., Zieziulewicz, T.J., Lawrence, D.A., Shain, W. & Turner, J.N. Antigen binding specificity of antibodies patterned by microcontact printing. Langmuir 19, 5431-­‐5434 (2003). 123. Foley, J.O., Fu, E., Gamble, L.J. & Yager, P. Microcontact printed antibodies on gold surfaces: Function, uniformity, and silicone contamination. Langmuir 24, 3628-­‐3635 (2008). 124. Ghosh, M., et al. Multifunctional surfaces with discrete functionalized regions for biological applications. Langmuir 24, 8134-­‐8142 (2008). 125. Lee, D. & King, M.R. Microcontact Printing of P-­‐Selectin Increases the Rate of Neutrophil Recruitment Under Shear Flow. Biotechnol Progr 24, 1052-­‐1059 (2008). 126. Folch, A. & Toner, M. Microengineering of cellular interactions. Annual Review of Biomedical Engineering 2, 227-­‐256 (2000). 127. Glasmastar, K., Gold, J., Andersson, A.S., Sutherland, D.S. & Kasemo, B. Silicone transfer during microcontact printing. Langmuir 19, 5475-­‐5483 (2003). 128. Yago, T., et al. Distinct molecular and cellular contributions to stabilizing selectin-­‐mediated rolling under flow. J Cell Biol 158, 787-­‐799 (2002). 129. Chen, S.Q., Alon, R., Fuhlbrigge, R.C. & Springer, T.A. Rolling and transient tethering of leukocytes on antibodies reveal specializations of selectins. P Natl Acad Sci USA 94, 3172-­‐3177 (1997). 130. Smith, M.J., Berg, E.L. & Lawrence, M.B. A direct comparison of selectin-­‐
mediated transient, adhesive events using high temporal resolution. Biophys J 77, 3371-­‐3383 (1999). 131. Shao, J.Y. & Hochmuth, R.M. Micropipette suction for measuring piconewton forces of adhesion and tether formation from neutrophil membranes. Biophys J 71, 2892-­‐2901 (1996). 132. Lee, C.H., Bose, S., Van Vliet, K.J., Karp, J.M. & Karnik, R. Examining the Lateral Displacement of HL60 Cells Rolling on Asymmetric P-­‐Selectin Patterns. Langmuir 27, 240-­‐249 (2011). 133. Williams, R.A. & Blanch, H.W. Covalent Immobilization of Protein Monolayers for Biosensor Applications. Biosens Bioelectron 9, 159-­‐167 (1994). 134. Wong, L.S., Khan, F. & Micklefield, J. Selective Covalent Protein Immobilization: Strategies and Applications. Chem Rev 109, 4025-­‐4053 (2009). 135. Gray, J.J. The interaction of proteins with solid surfaces. Curr Opin Struc Biol 14, 110-­‐115 (2004). 136. Hlady, V. & Buijs, J. Protein adsorption on solid surfaces. Curr Opin Biotech 7, 72-­‐77 (1996). 137. Elwing, H., et al. Protein Adsorption on Solid-­‐Surfaces -­‐ Physical Studies and Biological Model Reactions. Acs Sym Ser 343, 468-­‐488 (1987). 138. Lundstrom, I. Models of Protein Adsorption on Solid-­‐Surfaces. Progress in Colloid and Polymer Science 70, 76-­‐82 (1985). 139. Wasserberg, D., et al. Oriented Protein Immobilization using Covalent and Noncovalent Chemistry on a Thiol-­‐Reactive Self-­‐Reporting Surface. J Am Chem Soc 135, 3104-­‐3111 (2013). References
115
140. Smith, M.T., Wu, J.C., Varner, C.T. & Bundy, B.C. Enhanced protein stability through minimally invasive, direct, covalent, and site-­‐specific immobilization. Biotechnol Progr 29, 247-­‐254 (2013). 141. Frost, R.G., Monthony, J.F., Engelhorn, S.C. & Siebert, C.J. Covalent Immobilization of Proteins to N-­‐Hydroxysuccinimide Ester Derivatives of Agarose -­‐ Effect of Protein Charge on Immobilization. Biochim Biophys Acta 670, 163-­‐169 (1981). 142. Hermanson, G.T. Bioconjugate Techniques, 2nd Edition. Bioconjugate Techniques, 2nd Edition, 1-­‐1202 (2008). 143. Rusmini, F., Zhong, Z.Y. & Feijen, J. Protein immobilization strategies for protein biochips. Biomacromolecules 8, 1775-­‐1789 (2007). 144. Andrade, J.D. & Hlady, V. Protein Adsorption and Materials Biocompatibility -­‐ a Tutorial Review and Suggested Hypotheses. Adv Polym Sci 79, 1-­‐63 (1986). 145. Park, K. Protein-­‐ and cell-­‐repellant surfaces. Colloid Surface B 18, 167-­‐167 (2000). 146. Archambault, J.G. & Brash, J.L. Protein repellent polyurethane-­‐urea surfaces by chemical grafting of hydroxyl-­‐terminated poly(ethylene oxide): effects of protein size and charge. Colloid Surface B 33, 111-­‐120 (2004). 147. Unsworth, L.D., Sheardown, H. & Brash, J.L. Protein resistance of surfaces prepared by sorption of end-­‐thiolated poly(ethylene glycol) to gold: Effect of surface chain density. Langmuir 21, 1036-­‐1041 (2005). 148. Hong, S., et al. Covalent immobilization of P-­‐selectin enhances cell rolling. Langmuir 23, 12261-­‐12268 (2007). 149. Mccool, D., Birshtein, B.K. & Painter, R.H. Structural Requirements of Immunoglobulin-­‐G for Binding to the Fc-­‐Gamma Receptors of the Human-­‐
Tumor Cell-­‐Lines U937, Hl-­‐60, Ml-­‐1, and K562. J Immunol 135, 1975-­‐1980 (1985). 150. Raychaudhuri, G., Mccool, D. & Painter, R.H. Human Igg1 and Its Fc Fragment Bind with Different Affinities to the Fc-­‐Receptors on the Human U937, Hl-­‐60 and Ml-­‐1 Cell-­‐Lines. Mol Immunol 22, 1009-­‐1019 (1985). 151. Bancu, A.C., Sulica, A. & Moraru, I. Characterization of Fc-­‐Receptors Present on Human Polymorphonuclear Leukocytes. Rev Roum Biochim 18, 163-­‐174 (1981). 152. Murphy, K., Travers, P., Walport, M. & Janeway, C. Janeway's immunobiology, (Garland Science, New York, 2012). 153. Veiseh, M., Zareie, M.H. & Zhang, M.Q. Highly selective protein patterning on gold-­‐silicon substrates for biosensor applications. Langmuir 18, 6671-­‐6678 (2002). 154. Edington, C., et al. Tailoring the trajectory of cell rolling with cytotactic surfaces. Langmuir 27, 15345-­‐15351. 155. Moore, K.L., et al. P-­‐Selectin Glycoprotein Ligand-­‐1 Mediates Rolling of Human Neutrophils on P-­‐Selectin. J Cell Biol 128, 661-­‐671 (1995). 156. Snapp, K.R., Wagers, A.J., Craig, R., Stoolman, L.M. & Kansas, G.S. P-­‐selectin glycoprotein ligand-­‐1 is essential for adhesion to P-­‐selectin but not E-­‐selectin in stably transfected hematopoietic cell lines. Blood 89, 896-­‐901 (1997). References
116
157. Davis, J.M. & Giddings, J.C. Influence of Wall-­‐Retarded Transport on Retention and Plate Height in Field-­‐Flow Fractionation. Separ Sci Technol 20, 699-­‐724 (1985). 158. Goldman, A.J., Cox, R.G. & Brenner, H. Slow Viscous Motion of a Sphere Parallel to a Plane Wall .2. Couette Flow. Chemical Engineering Science 22, 653-­‐& (1967). 159. Bruus, H. Theoretical microfluidics, (Oxford University Press, Oxford ; New York, 2008). 160. Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 33, 657-­‐670 (2010). 161. Mantovani, A., Cassatella, M.A., Costantini, C. & Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11, 519-­‐531 (2011). 162. Brown, K.A., et al. Neutrophils in development of multiple organ failure in sepsis. Lancet 368, 157-­‐169 (2006). 163. Whiston, R.J., Hallett, M.B., Davies, E.V., Harding, K.G. & Lane, I.F. Inappropriate neutrophil activation in venous disease. Br J Surg 81, 695-­‐698 (1994). 164. Lard, L.R., Mul, F.P., de Haas, M., Roos, D. & Duits, A.J. Neutrophil activation in sickle cell disease. J Leukoc Biol 66, 411-­‐415 (1999). 165. Bhandari, V., Wang, C., Rinder, C. & Rinder, H. Hematologic profile of sepsis in neonates: neutrophil CD64 as a diagnostic marker. Pediatrics 121, 129-­‐134 (2008). 166. Nordvag, B.Y., Husby, G. & Raafat el-­‐Gewely, M. Direct PCR of washed blood cells. Biotechniques 12, 490-­‐493 (1992). 167. Kuijpers, T.W., et al. Membrane surface antigen expression on neutrophils: a reappraisal of the use of surface markers for neutrophil activation. Blood 78, 1105-­‐1111 (1991). 168. Abbitt, K.B. & Nash, G.B. Characteristics of leucocyte adhesion directly observed in flowing whole blood in vitro. Br J Haematol 112, 55-­‐63 (2001). 169. Laszik, Z., et al. P-­‐selectin glycoprotein ligand-­‐1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 88, 3010-­‐3021 (1996). 170. Vachino, G., et al. P-­‐Selectin Glycoprotein Ligand-­‐1 Is the Major Counter-­‐
Receptor for P-­‐Selectin on Stimulated T-­‐Cells and Is Widely Distributed in Nonfunctional Form on Many Lymphocytic Cells. Journal of Biological Chemistry 270, 21966-­‐21974 (1995). 171. Bose, S., et al. Affinity flow fractionation of cells via transient interactions with asymmetric molecular patterns. Scientific reports 3, 2329 (2013). 172. Wu, L., Guan, G., Hou, H.W., Bhagat, A.A. & Han, J. Separation of leukocytes from blood using spiral channel with trapezoid cross-­‐section. Anal Chem 84, 9324-­‐9331 (2012). 173. Davis, J.A., et al. Deterministic hydrodynamics: taking blood apart. Proc Natl Acad Sci U S A 103, 14779-­‐14784 (2006). 174. Sethu, P., Sin, A. & Toner, M. Microfluidic diffusive filter for apheresis (leukapheresis). Lab Chip 6, 83-­‐89 (2006). References
117
175. Du, Z., Colls, N., Cheng, K.H., Vaughn, M.W. & Gollahon, L. Microfluidic-­‐based diagnostics for cervical cancer cells. Biosens Bioelectron 21, 1991-­‐1995 (2006). 176. Quesenberry, P.J. & Becker, P.S. Stem cell homing: Rolling, crawling, and nesting. P Natl Acad Sci USA 95, 15155-­‐15157 (1998). 177. Jackson, S.P., Mistry, N. & Yuan, Y.P. Platelets and the injured vessel Wall -­‐ "Rolling into action" -­‐ Focus on glycoprotein Ib/V/IX and the platelet cytoskeleton. Trends Cardiovas Med 10, 192-­‐197 (2000). 178. Pincet, F., et al. Ultraweak sugar-­‐sugar interactions for transient cell adhesion. Biophys J 80, 1354-­‐1358 (2001). 179. Chao, D.L., Ma, L. & Shen, K. Transient cell-­‐cell interactions in neural circuit formation. Nat Rev Neurosci 10, 262-­‐271 (2009). 180. Taylor, M.E. & Drickamer, K. Paradigms for glycan-­‐binding receptors in cell adhesion. Current opinion in cell biology 19, 572-­‐577 (2007). 181. Hakomori, S. Le(X) and related structures as adhesion molecules. The Histochemical journal 24, 771-­‐776 (1992). 182. Greenberg, A.W., Kerr, W.G. & Hammer, D.A. Relationship between selectin-­‐
mediated rolling of hematopoietic stem and progenitor cells and progression in hematopoietic development. Blood 95, 478-­‐486 (2000). 183. Choi, S.Y., Karp, J.M. & Karnik, R. Cell sorting by deterministic cell rolling. Lab on Chip (2012). 184. Wu, L.D., Guan, G.F., Hou, H.W., Bhagat, A.A.S. & Han, J. Separation of Leukocytes from Blood Using Spiral Channel with Trapezoid Cross-­‐Section. Anal Chem 84, 9324-­‐9331 (2012). 185. Davis, J.A., et al. Deterministic hydrodynamics: Taking blood apart. P Natl Acad Sci USA 103, 14779-­‐14784 (2006). 186. Sethu, P., Sin, A. & Toner, M. Microfluidic diffusive filter for apheresis (leukapheresis). Lab Chip 6, 83-­‐89 (2006). 187. Abu al-­‐Soud, W. & Radstrom, P. Purification and characterization of PCR-­‐
inhibitory components in blood cells. J Clin Microbiol 39, 485-­‐493 (2001). 188. Chiesa, C., Panero, A., Osborn, J.F., Simonetti, A.F. & Pacifico, L. Diagnosis of neonatal sepsis: a clinical and laboratory challenge. Clin Chem 50, 279-­‐287 (2004). 189. Tedder, T.F., Chen, A.J. & Engel, P. L-­‐Selectin Regulation of Lymphocyte Homing and Leukocyte Rolling and Migration. Cardiovascular Disease 2, 173-­‐
184 (1995). 190. Artyomov, M.N., Lis, M., Devadas, S., Davis, M.M. & Chakraborty, A.K. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. P Natl Acad Sci USA 107, 16916-­‐16921 (2010). 191. Ockenhouse, C.F., Tandon, N.N., Magowan, C., Jamieson, G.A. & Chulay, J.D. Platelet Membrane Glycoprotein-­‐Iv (Cd36) as Binding-­‐Receptor for Malaria-­‐
Infected Erythrocytes. Thromb Haemostasis 62, 506-­‐506 (1989). 192. Shaz, B.H., Stowell, S.R. & Hillyer, C.D. Transfusion-­‐related acute lung injury: from bedside to bench and back. Blood 117, 1463-­‐1471 (2011). 193. Miersch, S. & Sidhu, S.S. Synthetic antibodies: concepts, potential and practical considerations. Methods 57, 486-­‐498. References
118
194. Meyer, M., Scheper, T. & Walter, J.G. Aptamers: versatile probes for flow cytometry. Appl Microbiol Biot 97, 7097-­‐7109 (2013). 195. Boder, E.T. & Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15, 553-­‐557 (1997). 196. Karp, J.M. & Teol, G.S.L. Mesenchymal Stem Cell Homing: The Devil Is in the Details. Cell Stem Cell 4, 206-­‐216 (2009). 197. Reticker-­‐Flynn, N.E., et al. A combinatorial extracellular matrix platform identifies cell-­‐extracellular matrix interactions that correlate with metastasis. Nat Commun 3(2012). 198. Kim, G., et al. Adhesion molecule protein signature in ovarian cancer effusions is prognostic of patient outcome. Cancer-­‐Am Cancer Soc 118, 1543-­‐
1553 (2012).