High-throughput Approaches to Sourcing of Human Hepatocytes ... Cell-based Therapies 18
advertisement

High-throughput Approaches to Sourcing of Human Hepatocytes for
Cell-based Therapies
By
MASSACHUSETT INS TE
OF TECHNOLOGY
Jing (Meghan) Shan
JUN 18 2014
B.S. Biomedical Engineering,
Columbia University, New York, New York 2007
LIBRARI ES
Submitted to the Harvard-MIT Division of Health Sciences and Technology
in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY IN HEALTH SCIENCES AND TECHNOLOGY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
May 2014
C 2014 Massachusetts Institute of Technology. All rights reserved.
Signature redacted
Signature of Author
Harvard-MIT Program in Health Sciences and Technology
May 19, 2014
A
Certified by
I
-
.
_Signature redacted
6'r
Sangeeta N. Bhatia, MD, PhD
Professor of Health Sciences and Technology & Electrical Engineering and Computer Science
Thesis Supervisor
Accepted by
Signature redacted
Emery N. Brown, MD, PhD
Director, Harvard-MIT Program in Health Sciences and Technology
Professor of Computation Neuroscience & Health Sciences and Technology
High-throughput Approaches to Sourcing of Human Hepatocytes
for Cell-based Therapies
By
Jing (Meghan) Shan
B.S. Biomedical Engineering,
Columbia University, New York, New York 2007
Submitted to the Harvard-MIT Division of Health Sciences and Technology in Partial
Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY IN HEALTH SCIENCES AND TECHNOLOGY
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
May 2014
Thesis Supervisor: Sangeeta N. Bhatia
1
Abstract
Chronic liver disease affects more than 500 million people worldwide. The only therapy
shown to directly prevent mortality is organ transplantation. However, there is growing
discrepancy between supply and demand of transplant-grade organs and transplant recipients are
subject to a lifetime of immunosuppressive regimens. Therefore, the overall aim of this thesis is
to advance alternatives to whole organ transplantation for liver diseases.
Treating the failure of organs serving a multitude of biochemical functions, such as the
liver, requires cell-based therapies. Such therapies should ideally employ human cells due to
immunological concerns and the substantial differences between animal and human
hepatocellular functions. Human cell lines, while renewable, lack the full functional capacity of
primary adult hepatocytes, and for clinical applications there is a tumorigenic risk. Primary
human hepatocytes have exhibited therapeutic potential; however, limited sourcing has been a
bottleneck for many fields of research and clinical therapies. Pluripotent human stem cells are an
attractive cell source, but to date, the hepatocyte-like cells obtain by directed differentiation
continue to exhibit an immature hepatic phenotype, which resembles fetal hepatocytes more than
adult hepatocytes. Motivated by these limitations, we report in the first section of this thesis, the
development of a 384-well liver platform that incorporates cell-cell interactions to stabilize the
hepatocyte phenotype, and enable high-throughput screening using cryopreserved primaly
human hepatocytes. We also developed attendant assays to assess cell fates in this platform
through an automated image-based proliferation assay and an ELISA-based functional assay.
In the second section of this thesis, we use the high-throughput liver platform to conduct
a chemical screen of 12,480 small molecules. We identified 12 bioactive factors in three classes:
small molecules that enhanced proliferation (PH), or function (FH) or both proliferation and
function (FPH) of human primary hepatocytes. 2 FPHs expanded primary human hepatocytes in
vitro, resulting in up to 10 fold more cells over 7 days. This proliferation rate is consistent with
in vivo liver regeneration kinetics and similar to PGE2-induced Wnt-mediated human hepatocyte
proliferation in vitro. To date, we have tested 10 different donors of primary human hepatocytes
and found all to expand upon FPH treatment, though kinetics and degrees of response vary.
Additionally, FPH1 and FHl were shown to differentiate hepatocyte-like cells derived from
induced pluripotent stem cells (iPS) toward a phenotype more mature than what was previously
obtainable, causing upregulation of previously low or absent adult markers such as CYP3A4,
CYP2A6, PXR and BSEP, concomitant with downregulation of persistent fetal marker AFP.
In the final section of this thesis, we explore the therapeutic potential of identified small
molecules in vivo. Using zebrafish models from the Goessling/North lab, we observed that both
PHIl and FHl enhanced liver sizes in lfabp:GFP reporter zebrafish embryos. In another model of
acute liver failure induced by overdose of acetaminophen (APAP), PH1 and FHl acted as
hepatoprotectants that increased liver size, and in the case of FHi, rescued zebrafish survival. At
sub-lethal concentrations (5mM APAP), the respective therapeutic windows of FHl and PHI
both exceed that of N-acetylcystein, the only antidote in use clinically, by at least 12 hours. At
lethal concentrations (IOmM APAP), FHl therapy further improved zebrafish survival by up to
63% in both embryonic and adult models of APAP toxicity.
The high-throughput liver platform developed in this thesis will enable studies of
previously inaccessible aspects of liver biology and small-molecule bioactivity, and have led to
the identification of first generation small molecules that have the potential to address cell
sourcing challenge impacting many facets of liver disease.
2
Acknowledgements
I am an extremely fortunate beneficiary of what has proven to be a phenomenal academic upbringing. It was
made possible by an unimaginably brilliant, capable, kind and generous group of people. I am very certain that
no collection of words I can string together will be sufficient to express my gratitude, but I nevertheless wish
to try.
First and foremost, I'd like to thank my PhD advisor Sangeeta Bhatia. Sangeeta has been a constant source of
amazing mentorship during my 6 years here at MIT. She patiently coached me through my initial unsure steps
as a fledgling graduate student, then gave me the freedom to grow but immediately appeared beside me again
if I stumbled. Through it all, she reassured me when I had doubts, inspired me when I felt stagnant, protected
me from irrelevant hardships and gently re-routed me when I started down the wrong paths. Sangeeta
personifies everything I had hoped for in a mentor, and more. For all that, I will be forever thankful.
I am also thankful for a number of other truly excellent mentors. Dr. Wolfram Goessling has been a brilliant
scientific advisor, an ever generous collaborator and an inspiring life-mentor. He has shown me feats of
accomplishments and kindness that I didn't think were possible but now see were made possible by his
extraordinary strength and ability. My thesis committee members Dr. Mehmet Toner and Dr. Gregory Verdine
have both offered many hours of input and support that have made this thesis possible and expanded my
perspectives. Thank you all for shaping my science, my career and my views on life.
Within the Laboratory for Multiscale Regenerative Techmologies (LMRT), I have made friends who, much like
their ability to multitask with superhuman efficiency, are at once my mentors, playmates, and consultants,
among other identities. They have done everything from showing me lab techniques to interrogating me about
my life plan in a way that I thought only a concerned parent could. Thank you Sandra March-Riera, Neetu
Singh, Salman Khetani, Shengyong Ng, Nathan Reticker-Flynn, Kartik Trehan, Cheri Yingjie Li and everyone
else in LMRT for making lab a home away from home.
Within the MIT and Harvard community, there are countless people who have made this PhD possible. These
include collaborators like Andy Cox, Nathan Ross, David Logan, and Robert Schwartz, as well as staff like
Sue Kangiser and Heather Fleming. The HST program has been a truly special home department, full of
curious, bright and dedicated scientists and physicians who have challenged and motivated me to attempt
seemingly insurmountable problems. Thank you all for pushing me to grow.
Last, but definitely not least, I would like to thank my family and friends-who-are-family. Mom and dad, I
cannot imagine the challenges you volunteered for and the sacrifices you made so that I can have what I have
today. Thank you for your unconditional love, which has empowered me to dream, then to go beyond even the
wildest of those dreams. To my grandparents, who raised me until I was 10 while my parents studied in
Germany, thank you for giving me a wonderful childhood and great values; they remain the foundation that
firmly reinforces who I am today. To Lin Jia, thank you for sticking by me through the good times, the hard
times and the really hard times, and for being my greatest believer. And to Stephen Gillanders, thank you for
being the textbook definition of a perfect best friend.
This one page acknowledgement is grossly inadequate in light of all that the aforementioned people have done
for me and inspired in me. Fortunately, most of them seem able to read minds and have seen and fixed or
forgiven much worse faults. So I'd like to conclude with just a simple thank you. I could not have done any of
the things I am proud of today without your unfailing guidance and support. I would not be me without you.
3
Biographical Information
EDUCATION
Massachusetts Institute of Technology
Ph.D.
Health Sciences and Technology (Harvard-MIT); GPA: 4.9/5.0
Expected June 2014
Cambridge, Massachusetts
Columbia University, Fu Foundation School of Engineering and Applied Science
B.S.
May 2007
Biomedical Engineering, Summa cum laude; GPA: 4.0/4.0
New York, New York
International Baccalaureate (IB) Organization
June, 2003
Full IB Diploma
Geneva, Switzerland
SELECTED HONORS
Sponsored Delegate
Womensphere - 5th Emerging Leaders Global Summit, 2014
Poitras Pre-Doctoral Fellowship
Massachusetts Institute of Technology, 2012
Robert E. And Claire S. Reiss Award in Biomedical Engineering (given to graduating
seniors judged by the faculty most likely to contribute substantially to the field)
Columbia University, 2007
Canby Robinson Award (for leadership and scholarship) [Declined]
Vanderbilt University, 2007
(two times) MacLaren Scholar
Columbia University, 2005-2006 and 2006-2007
Steamboat Summer Scholar Finalist
Steamboat Foundation and The Hospital for Special Surgery, 2006
(two times) Extraordinary Teaching Assistant Award
Columbia University, 2005 and 2006
(two times) Academic Scholarship
University of Lethbridge Faculty Association, 2005 and 2006
Member of Tau Beta Pi Engineering Honor Society
Columbia University, 2005-Present
Member of Golden Key Honor Society
Columbia University, 2005-Present
Invited Delegate
China Synergy Program for Outstanding youth, 2004
The Governor General's Academic Medal (given to the valedictorian)
The Government of Canada, 2003
National Champion / Gold Medallist in Fermat Mathematics Competition (Perfect Paper)
CMC-Centre for Mathematics and Computing, Canada, 2002
s
Place
in Logical Thinking Competition
1
4
Canadian Centre for Behavioural Neuroscience, 2002
RESEARCH EXPERIENCES
01/2008-Present
Harvard-MIT Division of Health Sciences and Technology
Graduate Research Assistant
Laboratory for Multiscale Regenerative Technologies
Principle Investigator: Dr. Sangeeta Bhatia
" Developed high-throughput tools to enable studies of previously inaccessible aspects
of liver biology and drug bioactivity
* Identified the first chemical factors to enable human hepatocyte growth in vitro
- Demonstrated the first functional maturation of any stem cell-derived cell, addressing
a major question in the field as to their therapeutic potential
* Identified genetic factors important for human hepatocyte maintenance in vitro
- Studied mechanisms of hepatocyte survival and function
* Participated in grant writing, mentorship, and outreach
05/2006-08/2006
Mayo Clinic/ Mayo Graduate School
Summer Undergraduate Research Fellow
Enteric Neuroscience Program
Principle Investigator: Dr. Gianrico Farrugia
" Examined the role of Telethonin in the gastrointestinal tract
e
Aided research on exploring H2 S in the gastrointestinal tract as a signaling molecule
11/2003-05/2007
Columbia University
Undergraduate Research Assistant, and Summer Undergraduate Research Fellow
Biomaterials and Interface Tissue Engineering Laboratory
Principle Investigator: Dr. Helen H. Lu
- Examined fibroblast-osteoblast interactions and their role in modulating cell
phenotypes through paracrine and autocrine regulations
- Explored osteoblast and fibroblast paracrine regulations of human mesenchymal stem
cell fate
e
Aided various other research projects on functional engineering of ligament-bone
interface
07/2002-08/2002
Canadian Centre for Behavioural Neuroscience
Alberta Heritage Youth Researcher
PrincipleInvestigator:Dr.Robert Sutherland
e
Determined the effects of fluoxetine on adult neurogenesis in rodents
- Studied the effects of aging on the Hippocampus in rodents
PUBLICATIONS
[1] Shan, J., Schwartz, R.E., Ross, N.T., Logan, D.J., Duncan, S.A., North, T.E., Goessling, W.,
Carpenter, A.E., Bhatia, S.N. (2013) High-throughput identification of small molecules for
human hepatocyte expansion and iPS differentiation. Nature ChemicalBiology, 9: 514-520.
[2] March, S, Ng, Shengyong, Velmurugan, S, Galstian, A, Shan, J, Logan, D, Carpenter, AE,
Thomas, D, Sim, BKL, Mota, MM, Hoffman, SL, and Bhatia, SN (2013) A microscale
human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax,
Cell Host Microbe. 14: 104-115.
5
[3] Shan, J., Logan, D.J., Carpenter, A.E., Bhatia, S.N. (2014) High-throughput identification of
molecular factors that promote phenotypic stabilization of primary human hepatocytes in
vitro., [In preparation]
[4] Stevens, K.R., Schwartz, R.E., Ng, S., Shan, J., and Bhatia, S.N. (2013). Hepatic tissue
engineering, in Principles of Tissue Engineering. R. Lanza, R. Langer, J.P. Vacanti (ed.),
Elsevier / Academic Press, 951-986
[5] Shan, J, Stevens, KR, Trehan, K, Underhill, GH, Chen, AA, and Bhatia, SN (2011). Hepatic
tissue engineering, in Molecular Pathology of Liver Diseases. S.P.S. Monga (ed.), Springer
Science, 321-342
[6] Wang, I.E., Shan, J., Choi, R., Oh, S., Kepler, C.K., Chen, F.H., Lu, H.H. (2007) Role of
osteoblast-fibroblast interactions in the formation of the ligament-to-bone interface. Journal
of OrthopedicResearch, 25(12): 1609-1620
PATENTS
[1] International Patent Application No. PCT/ US2014/028219, "Systems and Methods for
Culturing Epithelial Cells", filed March 14, 2014.
[2] International Patent Application No. PCT/US2014/028408, "Compounds for Inducing
Proliferation and Differentiation of Cells, and Methods of Use Thereof', filed March 14,
2014
PEER REVIEWED CONFERENCES
[1] Shan, J., Schwartz, R.E., Logan, D.J., Ross, N.T., Duncan, S.A., North, T.E., Goessling, W.,
Carpenter, A.E., Bhatia, S.N. (2013) Identification of small molecules for maturation of iPSderived hepatocyte-like cells. InternationalSocietyfor Stem Cell Research. Boston, Ma,
USA; Podium Presentation
[2] Shan, J., Schwartz, R.E., Logan, D.J., Ross, N.T., Duncan, S.A., North, T.E., Goessling, W.,
Carpenter, A.E., Bliatia, S.N. (2012) High-throughput identification of small molecules for
human hepatocyte expansion and iPS differentiation. FASEB Summer Research Conferences.
Snowmass Village, CO, USA; Poster
[3] Shan, J., Logan, D.J., Ross, N.T., Carpenter, A.E., and Bhatia, S.N. (2010) High-throughput
identification of small molecules for in vitro human hepatocyte expansion. FASEB Summer
Research Conferences. Snowmass Village, CO, USA; Poster
[4] Logan, D.J., Hartwell, K., Miller P., Shan, J., Stewart, A., Golub, T., Ebert B., Schreiber S.,
Bhatia S., and Carpenter A. (2009) Identifying chemical regulators of hematopoeisis and
hepatocyte proliferation via image-based screens of co-cultured cells. Nature Chemical
Biology Symposium. Cambridge, MA, USA; Poster
[5] Shan, J., Khetani, S., Ploss, A., Syder, A., Rice, C., and Bhatia, S. (2008) Engineering
microscale human liver models for applications in drug discovery and development. FASEB
Summer Research Conferences. Snowmass Village, CO, USA; Poster
[6] Shan, J., Wang, I.E., and Lu, H.H. (2006) Osteoblast-Fibroblast Interactions Modulate Cell
Phenotypes through Paracrine and Autocrine Regulations. OrthopedicResearch Society
Annual Meeting. San Diego, CA, USA; Podium Presentation
[7] Shan, J., and Lu, H.H. (2005) Effects of Conditioned Media on ACL Fibroblast and
Osteoblast Growth and Differentiation in Vitro. Columbia University SEAS Symposium. NY,
USA; Podium Presentation
6
[8] Shan, J., Wang, I.E., and Lu, H.H. (2005) Paracrine Regulation of Human BMSC Growth
and Differentiation by Osteoblasts and Fibroblasts. Biomedical EngineeringSociety Annual
Meeting. Baltimore, MD, USA; Poster
[9] Shan, J., Wang, I.E., and Lu, H.H. (2004) Effects of Conditioned Media on Osteoblast and
Ligament Fibroblast Growth and Differentiation. Biomedical EngineeringSociety Annual
Meeting. Philadelphia, PA, USA; Podium Presentation
TEACHING EXPERIENCES
01/2010-05/2011
Massachusetts Institute of Technology
(Two times) Graduate TeachingAssistant
HST. 500 Frontiersin BioMedicalEngineeringand Physics
Harvard-MITDivision of Health Sciences and Technology
" Provided iterative scientific feedback on -40 graduate research proposals
- Updated lecture materials
- Assisted faculty in preparing and running grant-writing workshops
- Evaluated students
- Helped plan course schedule
03/2009-05/2012
Massachusetts Institute of Technology
Research Mentor
Laboratoryfor Multiscale Regenerative Technologies
Harvard-MITDivision of Health Sciences and Technology
Mentored undergraduate researchers, one of whom was named an Amgen Research
Scholar
09/2006-12/2006
Columbia University
Head Teaching Assistant
DepartmentofBiomedical Engineering
e
Trained and mentored other teaching assistants
- Planned and delivered review sessions and weekly recitations
- Graded midterm, final exam and weekly written reports
09/2004-12 /2005
Columbia University
(Two times) UndergraduateTeaching Assistant
Departmentof BiomedicalEngineering
e
Planned and delivered review sessions and weekly recitations
* Graded midterms, final exams, and weekly written reports
11/2005-05 /2007
Columbia University
Research Mentor
Biomaterials and interface Tissue EngineeringLaboratory
Departmentof Biomedical Engineering
- Mentored undergraduate and master's students, one of whom won the McNair
Research Award
7
Table of Contents
Abstract................................................................................
. ..........-2
... 3
Acknowledgements........................................................................
Biographical Information..............................................................................4
.... 8
Table of Contents..............................................................................
List of figures and tables.................................................................................10
Chapter 1. Introduction............................................................................-16
1.1
Liver Diseases and Current Treatments............................................16
1.2
Alternative Therapies for Liver Diseases...........................................18
1.3
Cell Sources for Liver Therapies......................................................21
1.4
Maintenance of Hepatocytes ex vivo...............................................28
1.5
High-throughput Screening and Small Molecule Modulation of Complex
Cellular Phenotypes...................................................................31
Chapter 2. High-throughput Liver Platform for the Development of Novel Therapeutics.33
2.1
Introduction.............................................................................33
2.2
Results and Discussions..............................................................35
2.3
Conclusions.............................................................................52
2.4
Materials and Methods................................................................53
Chapter 3. High-throughput Identification of Small Molecules for Inducing in vitro
Proliferation and Function in Primary Human Hepatocytes..................................57
3.1
Introduction.............................................................................57
3.2
Results and Discussions..............................................................60
3.3
Conclusions.............................................................................
3.4
Materials and Methods...........................
8
69
............. 70
Chapter 4. Small Molecules for Inducing Hepatocyte Expansion and iPS Differentiation in
vitro............................................................................................................87
4.1
Introduction.............................................................................87
4.2
Results and Discussions..............................................................88
4.3
Conclusions...............................................................................106
4.4
Materials and Methods.................................................................106
Chapter 5. Small Molecules for Enhancing Liver Development and Treating Acute Liver
Failure in vivo..............................................................................................111
5.1
Introduction.............................................................................111
5.2
Results and Discussions...............................................................112
5.3
Conclusions..............................................................................119
5.4
Materials and Methods................................................................120
Chapter 6. Perspectives and Future Directions....................................................121
6.1
Understanding Mechanisms of Small Molecule Bioactivity....................122
6.2
Small Molecules as Enabling Research Tools for Understanding Human
Biology.....................................................125
6.3
Using Small Molecules to Develop Molecular and Cellular Therapeutics ..127
6.4
Maintenance of human hepatocytes in vitro using coatings of recombinant
proteins and soluble factors...........................................................129
References...................
......................
.....................
9
139
List of figures and tables
Figure 1.1 Cell sourcing of human hepatocytes. Approaches have focused on modulating in
vitro culture conditions of primary hepatocytes, including the addition of soluble factors,
extracellular matrix proteins, and heterotypic cell-cell-interactions. Various immortalization
techniques have also been attempted, such as spontaneous transformation through long-term
culture in collagen gel and targeted mutations through viral transduction. Other sources include
tumor-derived cell lines, stem-cell derived cell lines and in vivo expansion in mice models
21
providing regenerative stimuli.........................................................................
Table 2.1. Donors of cryopreserved primary human hepatocytes. Eight different donor lots
were tested for suitability for high-throughput screening through examination of plate-ability and
baseline functions such as albumin secretion, urea production and cytochrome P450 activity.. .36
Figure 2.1 High-throughput liver platform. Cryopreserved primary human hepatocytes (green)
are maintained in vitro through co-cultivation upon a feeder layer of J2-3T3 fibroblasts (red) in
384-well formats. Line graph shows representative rate of albumin secretion in screening cocultures and hepatocyte-only cultures (green) over time. Phase contrast imaging shows
morphology of feeder-layer co-cultures (scale bar = 100um). All data presented mean ± standard
38
dev iation .......................................................................................................
Figure 2.2 Distinctive nuclei morphology. Hepatocytes in co-culture with J2-3T3 fibroblasts
can be separated based on nuclei morphology. Hepatocyte nuclei (left) are smaller, rounder and
more uniform in texture while fibroblast nuclei (right) are punctate.................................40
Figure 2.3 Image-based assay workflow. Nuclei are visualized with Hoechst stain, imaged
using a high-content screening microscope, identified, characterized and counted through a
custom image-based proliferation assay. The user interface window of the classification
software, CellProfiler Analyst, is shown. It allows manual classification of randomly presented
nuclei and error correction of machine-classified nuclei................................................41
Figure 2.4 Uniformity of image intensity throughout screen. Permeabilization treatment is not
necessary for traditional Hoechst staining but helped normalize Hoechst 33258 staining
intensities throughout screening. Upper panel shows heatmap of image intensities for each 384well plate; arrows indicate location of brightest and dimmest images. Bottom panel shows
42
acquired im ages...........................................................................................
Figure 2.5 Schematic of Automated Image Acquisition. Treated sample plates are robotically
loaded into high-throughput screening microscope....................................................43
Figure 2.6 Nuclei Identification. Feederlayer co-culture led to overlapping objects in Hoechst
images that proved challenging to segment. Final algorithin was able to correct identify nuclei
locations and borders.....................................................................................45
Figure 2.7 Identification of sub-nuclear structures. Punctate sub-nuclear structures were
identified as objects and associated with their parent nucleus. Yellow circles indicate hepatocyte
islands. Yellow square surrounds one region of fibroblast cluster....................................47
10
Figure 2.8 Classification Accuracy. Screening images were classified without (left) and with
(right) the identification of punctate sub-nuclear structures. Yellow squares indicate fibroblast
nuclei that were erroneously identified as hepatocyte nuclei..........................................48
Figure 2.9 Mitotic nuclei morphology. Left gray square marks a nuclei with morphology
consistent with metaphase; right gray square marks a nuclei with morphology consistent with
anaphase......................................................................................................50
Figure 2.10. Biochemical functional assays. Bar graph displays albumin secretion (left), urea
production (middle) and cytochrome P450 activity (right) as a function of hepatocyte density in
screening cultures. All data presented as mean ± standard deviation.................................51
Figure 2.11 Schematic of competitive ELISA.....................................................52
Figure 3.1 Overview of High-throughput Screening. A, Primary human hepatocytes (green)
were seeded on a feeder layer of confluent J2-3T3 fibroblasts (red) in 384-well plates. B, Cells
were seeded onto a collagen matrix, cultured for 7 days, and treated with small molecules for 48
hrs before analyses through image-based proliferation assay and competitive-ELISA-based
functional assay. C, hit validation. D, classes of confirmed hits. Two classes of hits were selected
for further characterization. Functional proliferation hits were examined for their ability to
expand mature human hepatocytes. Functional hits were explored as inducers of iHeps
59
m aturation..................................................................................................
Figure 3.2 Chemical Library Composition. Categories of screened (white) and primary
screening hit (black) com pounds...........................................................................60
Table 3.1 Small molecule screening data from high content co-culture imaging assay......62
Table 3.2 Small molecule screening data from co-culture albumin secretion ELISA assay.63
Figure 3.3 Primary screening results. Scatterplots display replicates of the screen of 12,480
small molecules, shown separately for the image-based proliferation and competitive-ELISA
functional readouts. Blue and red data points represent DMSO and experimental small molecules
respectively. Boxed regions indicate hit zones.......................................................65
Figure 3.4 Dose curves of confirmed hits and PGE2. Proliferation hits have increasing curves
of hepatocyte and/or metaphase nuclei count. Functional hits have decreasing curves of
competitive [Albumin] or increasing curves of fold change in [Albumin]. Control cultures were
treated with empty vehicle (DMSO). PGE2 was tested as a putative positive control since it was
previously reported by Goessling and colleagues to promote liver regeneration in zebrafish.....66
Table 3.3 Classification of 12 Confirmed Hits. Functional proliferation hits (FPHs) were
selected as hits by both the image-based proliferation assay and the ELISA-based functional
assay. Proliferation hits (PHs) enhanced hepatocyte proliferation only during primary screening.
Functional hits (FHs) enhanced hepatocyte functions only..........................................67
Figure 3.5 Structure Activity Relationship of FPH1. Structure of the functional proliferation
hit FPH 1 and a series of analogs (Compounds 4-10) depicting the structure-activity relationships
of this series of compounds. Both a 5-chloro-2-methyl substituted sulfonamido phenyl ring and
11
a phenylamide ring without significant steric bulk at the para position are required for compound
69
activity .......................................................................................................
Supplementary Note 3.1. FPH1, FPH2 and FH1 chemical characterization................77
Figure 4.1 Primary screening data for FPHl and FPH2. Data presented as mean
dev iation ......................................................................................................
+
standard
89
Figure 4.2 Morphology and colony size of FPH-treated human hepatocytes. Cells were
cultured in 12-well plates over time. Untreated (day 1) hepatocytes are shown for comparison,
90
far left (scale bar = I 00um )..............................................................................
Figure 4.3. Ki67 and albumin staining of FPH treated hepatocyte cultures. Primary human
hepatocytes were cultured in 24-well formats on top of a confluent feederlayer of growtharrested J2-3T3 fibroblasts and exposed to small molecules as described. After six days in
culture, samples were fixed and stained with for albumin (green) and Ki67 (red). Bar graphs
represent quantifications of displayed images. Scale bar = 1 00um................................91
Figure 4.4. Automated Cell Counter analysis. Fibroblasts were labeled with CM-DiI prior to
initiation of culture in order to allow identification of hepatocytes via negative selection. FACS
cell counting was further enabled by fluorescent counting beads. Control cultures were treated
with empty vehicle (DMSO). Data presented as mean ± SEM.....................................92
Figure 4.5 FPH-induced expansion of multiple different donors of primary human
hepatocytes. Six additional sources of primary human hepatocytes were treated with FPHs and
A) stained for albumin (green) and Ki67 (red) after 6 days in culture. Scale bars represent
300um. B) Day 1 untreated control was added for reference (top). On day 7, we quantified the
number of hepatocytes in culture using FACS analysis...............................................93
Figure 4.6. Functional analysis of FPH-treated primary human hepatocytes. A, gene
expression profiling of FPH-treated human hepatocytes. A panel of 83 liver-specific genes were
analyzed via Luminex. Columns of the heatmap are averaged values of replicate (n=3) loadings
of mRNA extracted from various populations of human hepatocytes (250ng total RNA per
replicate). mRNA expression was determined relative to the average of control gene transferrin,
and heat maps are row-normalized. Bar graphs are select gene sets comparing the relative mRNA
expression of FPH-treated hepatocytes (patterned bars) and HepG2 (solid red bar), normalized to
primary human hepatocytes (solid black bar) for nuclear receptors, phase I, phase II, and phase
III drug metabolism genes. Data represent the mean ± SEM of Luminex-loaded replicates. B,
phase 3 transporter activity. Cultures were incubated with 5-(and-6)-carboxy-2',7'dichlorofluorescein diacetate, which is internalized by hepatocytes, cleaved by intracellular
esterases and excreted into the bile canaliculi between hepatocytes by transporters (scale bar =
50pm). C, biochemical characterization of key hepatocyte functions. Albumin secretion reflects
protein synthesis capability; urea content is a surrogate marker for protein metabolism;
detoxification functions were measured via processing of substrate BFC into fluorescent
products. For all analyses, primary and FPH-treated hepatocytes were cultured for 7 days in 12well plates (n=3). Control cultures were treated with empty vehicles (DMSO). All data presented
as m ean ± standard deviation..............................................................................95
12
Figure 4.7. iHeps generation. A) Undifferentiated iPS (top) and hepatic progenitor cells (iHEP)
generated from iPS (bottom). B) Immunostaining of hepatic lineage markers, not present in iPS
(top), but expressed by iHEP (bottom). C) FACS analysis illustrating expression of iPS markers
and hepatic progenitor markers in undifferentiated iPS (top) and iHEP (bottom). Scale bars =
50 jm ...................................................................................................
. . .. 96
Figure 4.8 Functional enhancement of human primary hepatocytes and iHeps. A, primary
screening data and dose curve of FHl. Control cultures were treated with empty vehicle
(DMSO). All data presented as mean ± s.d. C, morphology and colony size of FPH1- and FH1treated iHeps in 6-well plates 9 days post-treatment. Untreated iHeps are shown for comparison
(scale bar = 100pm). D, Albumin (green), CYP3A (turquoise) and AFP (red) staining of iHeps
after 9 days of culture (scale bar = 10O m). Bar graphs represent quantifications of displayed
imag es.........................................................................................................97
Figure 4.9 Maturation of human iHeps. Gene expression profiling of FHl-treated (A) and
FPHl-treated (B) iHeps. Heat map displays of Luminex analysis for 83 liver-specific genes,
shown separately for independent experiments. Columns of the heatmap are averaged values of
replicate (n>4) loadings of mRNA extracted from various populations of iPS cells (250ng total
RNA per replicate). mRNA expression was determined relative to the average of control gene
transferrin, and heat maps are row-normalized. Bar graphs are select gene sets comparing the
relative mRNA expression of small molecule-treated hepatocytes (solid colored bars) to iPS
(solid white bars), untreated iHeps (solid gray bars) and fetal human hepatocytes (patterned
white bars), normalized to control (patterned gray bar) for nuclear receptors, phase 1, phase 11,
and phase III drug metabolism genes. Control refers to adult cryopreserved human primary
hepatocytes stabilized by micropatterned co-culture (MPCC, more details in supplementary note
1). Data represent the mean z SEM of Luminex-loaded replicates. C, Quantifications of
Albumin, CYP3A and AFP staining of iHeps after 9 days of culture. D, biochemical
characterization of key hepatocyte functions. Albumin and AFP secretion are measured as a liver
marker and a fetal marker respectively; detoxification functions were measured via processing of
substrates with fluorescent or luminescent products. Specific activities of CYP2A6 and CYP3A4
were measured using coumarin and luciferin-IPA respectively. For all analyses, iHeps were
cultured for 9 days post-differentiation, in 6-well plates (n=3). All data presented as mean -
SE M ......................................................................................................
. .99
Figure 4.10 Stability of Mature iHep Phenotype. iHeps were treated with small molecules
once on day 20. Cultures were then maintained in nonnal basal media containing OSM (without
small molecule addition) for 9 days prior to A) phase contrast imaging (scale bars represent
100im) and immunofluorescent staining (scale bars represent 50pm) for Albumin, CYP3A and
AFP. B) ELISA assay for secreted albumin and AFP, and C) quantitative CYP3A4 and CYP2A6
activity assays, benchmarked against cryopreserved human hepatocytes that have been stabilized
in culture (MPCC, more details in supplementary note 1)............................................101
Figure 4.11. Hepatocyte induction kinetics. Hepatocytes were induced with 250pm Pnaphthoflavone (BNF) at various times during culture. Black arrows indicate addition of BNF;
white arrows indicate removal. CYP450 In all cases, elevations in CYP450 activity secondary to
induction are mostly reverted by 24hrs post removal of inducer.....................................102
13
Figure 5.1 Small molecule enhancement of in vivo liver development. Zebrafish embryos
were allowed to develop normally for 24 hours post fertilization prior to exposure to small
molecules as previously described until 72 hours post fertilization. Embryo liver sizes were then
3
measured via fluorescent microscopy of transgenic Tg(-2.8fabpJ:EGFP)s (lfabp:GFP)
zebrafish and in situ hybridization.......................................................................114
Figure 5.2 Effect of small molecule treatment on early zebrafish survival.
Zebrafish embryos were allowed to develop normally for 48 hours before co-administration of a
small molecule hit and a lethal dose of APAP. % survival was measured at 80 hours post
116
fertilization ................................................................................................
Figure 5.3. FH1 increases zebrafish embryo survival post lethal doses of APAP.
Zebrafish embryos were allowed to develop normally for 72 hours before co-administration of
10mM APAP and various doses of FHl. % survival was measured through 168 hours post
117
fertilization ..................................................................................................
Figure 5.4. Hepatoprotective effects of PH1 and FHI following delayed administration.
Ifabp:GFPzebrafish embryos were allowed to develop normally for 48 hours before exposure to
5mM APAP alone for 24 hours. Small molecule treatment was given another 24 hours later, at
72 hours post fertilization. At 96 hours post fertilization, liver sizes were examined via
fluorescent m icroscopy....................................................................................118
Figure 5.5. Small molecule treatments are hepatoprotective against acute liver injury in
adult zebrafishes. Adult zebrafishes ranging in age from 3 months to 1 year were coadministered 10mM APAP and FH 1. % survival was measured 72 hours post exposure........119
Figure 6.1. High-throughput liver platform for validation of stromal factors hypothesized
to mediate the co-culture effect. A, -50 3T3 genes were hypothesized to be important for coculture mediated maintenance of primary hepatocytes based on gene expression profiling. B,
vector design of shRNA library. C, screening workflow. D, platform and assay validation. Left
panel shows that Alamar Blue assay is able to accurately reflect fibroblast cell numbers. Middle
panel shows kill curve of various infection conditions. Right panel shows infection efficiency
during screening.............................................................................................131
Table 6.1. Negative regulators of hepatocyte functions............................................131
Table 6.2. Positive regulators of hepatocyte functions.............................................132
Table 6.3. 12 genes validated by custom shRNA screening.......................................134
Figure 6.2 High-throughput identification of gene products important for J2-3T3-mediated
stabilization of primary hepatocytes in culture. A, primary screening data. Hits were selected
based on decreased total albumin secretion, decreased total number of hepatocyte nuclei and
decreased albumin output on a per cell basis. B, selected hits.......................................135
Figure 6.3. Schematic of combinatorial shRNA screen.
12 modified lines of J2-3T3s will be generated through shRNA-mediated knockdown of
previously validated genes involved in the hepatocyte-fibroblast co-culture effect. Each line will
14
be used to screen a custom set of ~450 genes annotated as cell surface factors, secreted factors or
factors involved in cell-cell signaling...................................................................136
Figure 6.4. Schematic of integrating chemical and genetic screening to generate a
renewable source of functional human primary hepatocyte in vitro. Our hypothesis is that
molecular signals from the stroma provide inductive cues, enabling hepatocyte maturation and
replication in vitro in response to small molecule exposure, and that these stromal signals can be
isolated and used to replace the fibroblasts in stabilizing hepatocytes. The platform described in
chapter 2 of this thesis can be used to identify small molecules and stromal factors involved in
the regeneration, maturation and phenotype maintenance of human hepatocytes in vitro.
Together, these studies can provide key insights into leveraging HTS technologies to provide
human hepatocytes needed for advancing cell-based therapies and furthering our understanding
of liver development, regeneration, and maintenance.................................................137
15
Chapter 1. Introduction
1.1 Liver Diseases and Current Treatments
Liver disease afflicts over 500 million people', 30 million in the United States alone,
leading to over 40,000 deaths annually.
Liver failure can generally be divided into two
categories: fulminant or acute liver failure, and chronic hepatic failure caused by chronic
disorders.
Acute liver failure is relatively rare but exhibits a high mortality rate of -28%1. Common
causes include acetaminophen overdose, infections such as hepatitis A and B, and idiosyncratic
drug reactions'. Fulminant hepatic failure is characterized by hepatic encephalopathy and
impaired liver synthetic functions within 26 weeks of initial onset of jaundice. Encephalopathy
is a neuropsychiatric disorder whose symptoms can range from mild confusion and sleep
disruption to deep coma.
Accompanying clinical symptoms can further include microbial
infections as well as metabolic and cardio-respiratory abnormalities. Thanks to the phenomenal
regenerative capacity of the liver, spontaneous recovery from acute liver failure is possible, but
difficult to predict. Additionally, some etiologies, such as acetaminophen overdose and hepatitis
B cause such extensive liver damage that regeneration is often disabled. For these patients, liver
transplantation is the only therapy to directly offer survival benefits.
Chronic liver failure is more widespread and a leading source of death for the United
States in 20102. Common causes include hepatitis C virus (HCV) and fatty liver diseases, both
alcohol-induced and nonalcoholic (NAFLD) 3. Collectively, chronic liver disorders can lead to
the development of decompensated cirrhosis, resulting in clinical symptoms such as ascites,
portal hypertension, variceal bleeding, and hepatic encephalopathy.
16
Liver cirrhosis resulting
from HCV infection is the #1
cause of liver transplantation and accounts for 40-50% of
transplant candidates 4. Long-term inflammation precipitated by chronic HCV infections also
increase risks for the development of hepatocellular carcinoma. Medical management of chronic
liver disorders can minimize the impact of ensuing consequences on patient quality of life but
transplantation is the only effective therapy currently available to patients.
Unlike other major causes of mortality, death rates from liver diseases are rising instead
of declining, leading to widening discrepancy between supply and demand of liver transplants.
As a result, several surgical options have been examined to maximize usage of available organs.
These include the use of non-heart-beating donors or split liver transplants from both cadaveric
and living sources 5 . Split liver transplants are based around the significant regenerative capacity
possessed by the liver. Liver regeneration has been extensively studied, mostly through the use
of rodent models, which have shown that replacement of lost liver mass following surgical
removal of liver lobes or chemical injury is enabled by the expansion of existing mature cell
populations within the liver, led by hepatocytes, and followed by others including bile duct
epithelial cells. More recently, an in vivo RNAi screen identified MKK4 as a key regulator of
liver regeneration.
While powerful, liver regeneration is difficult to control clinically; thus
despite some effectiveness, biliary and vascular complications are major concerns in these
procedures5.
The risks of split liver transplants, which have resulted in several donor deaths,
have raised significant ethical barriers.
Furthermore, the wide gap between the number of
patients on transplant wait lists and the number of available organs is unlikely to be met by liver
transplantation procedures alone.
Alternative approaches are therefore highly desired and
actively being pursued.
17
1.2 Alternative Therapies for Liver Diseases
ExtracorporealBioartificialLiver Devices
Extracorporeal support devices process the blood or plasma of liver failure patients.
These devices primarily aim to provide transient support for patients suffering from acute or
acute-on-chronic liver failure, providing time to allow for innate liver regeneration or serving as
a bridge to transplantation. Early device designs mostly employed nonbiological mechanisms
including plasma exchange, plasmapheresis, hemodialysis, molecular adsorbents recirculation
system, or hemoperfusion over charcoal or various resins . Hemoperfusion involves the passage
of blood or plasma through a charcoal column in order to remove toxins and capture other useful
metabolites.
Charcoal-based systems are the most extensively examined embodiment of
extracorporeal support devices and have been evaluated clinically in patients with acute liver
failure, although no clear survival benefits have been observed 8 . This is likely due to the limited
range of functions served by each of these devices, particularly considering the complex array of
functions performed by a healthy liver.
In order to supply a more complete array of synthetic, metabolic, and detoxification
functions, cell-based bioartificial liver (BAL) devices have been extensively explored. Central to
the clinical success of any BAL device is the ability to scale to levels that provide effective
therapy. Different types of liver diseases (e.g., acute liver failure, end-stage cirrhosis, genetic
metabolic disorders) will likely have different demands, but it is estimated that the minimum
cellular requirement is approximately 10% of total liver weight, or I x 1010 hepatocytes 9 .
Ultimately, to achieve clinical efficacy, BAL devices will require a) renewable sources of
functional human hepatocytes and b) scaleable systems capable of maintaining a large number of
functional human hepatocytes ex vivo. Overall, the development of therapeutic BAL devices is a
18
major challenge and substantial efforts have been placed towards continued improvements in the
capacity and efficiency of these systems.
Implantable Technologiesfor Liver Therapies
In addition to temporary extracorporeal support, the development of in vivo therapies for
liver treatment aimed at the eventual cell-based replacement of damaged or diseased tissue is an
active area of investigation.
Cell Transplantation.
Direct injection of hepatocytes into animals via the spleen or splenic artery,
intraperitoneal space, peripheral veins, or portal vein have been shown to improve host
survival in models of both acute and chronic liver failure as well as disease models of
genetic metabolic defects10
14
.
Animal models of acute liver failure can be induced
chemically through toxic doses of acetaminophen or carbon tetrachloride, or generated
surgically through the resection or partial hepatectomy of 2/3 of the liver 5 . Chronic liver
16
disorders can be modeled through administration of CCL4 or bile duct ligation' .
Metabolic liver diseases are studied through the use of transgenic mice strains, such as
the fumarylacetoacetate hydrolase (FAH)-deficient mouse for modeling of familial
tyrosinemia '.
Clinically, cellular transplantation appears to offer the most therapeutic
benefits to primarily pediatric patients with liver-based metabolic diseases. Certain
disorders such as urea cycle defects exhibit better responses than others, but even in best
case scenarios, transplanted hepatocytes deteriorate over time and liver transplantation is
required by six months'.
A major condition driving the success of cell transplantation in animal models of
liver diseases is the presence of regenerative stimuli provided by hepatotoxins, surgical
19
alterations, and/or transgenic injury. Such stimuli provide the donor cells with a
repopulation advantage, promoting expansion of transplanted cells to enable more
extensive engraftment of healthy hepatocytes to compromised host tissues. However,
these strategies are difficult to translate to the clinics. Major limitations of cell
transplantation include the lack of a renewable source of functional and safe human
hepatocytes. This difficulty is further compounded by extremely inefficient engraftment
and survival of transplanted cells in host tissue, collectively reported at only 10-30% of
injected cells 19 . Consequently, while hepatocyte transplantation therapy has been shown
to possess long-term safety, poor cell engraftment and inadequate survival benefits limit
its effectiveness as a clinical therapy2 0
.
Tissue EngineeredHepatocellularConstructs.
One approach for mitigating the challenges faced by direct cell transplantation is
0 20
the development of implantable tissue engineered hepatocellular constructs , . Such
constructs typically encapsulate cells in biomaterial scaffolds that provide physical
support to normally adhesive cells such as hepatocytes. Additionally, various strategies
have been developed to enhance hepatocyte survival and function in such constructs,
involving but not limited to the partial re-creation of the complex microenvironment that
supports hepatocytes in vivo 2 3 -66 . The ability to introduce cell-cell, cell-matrix and cellsoluble factor interactions into such artificial constructs allows the generation of liverlike tissues in vitro prior to in vivo implantation.
Overall, studies that improve cell
delivery, survival, and integration with host can greatly improve the effectiveness of cellbased therapies, and will need the support of a renewable source of functional human
hepatocytes.
20
1.3 Cell Sources for Liver Therapies
Studies into cell-based therapies suggest great promise but progress has been hindered by
the propensity of hepatocytes to lose both phenotypic functions and the ability to proliferate in
vitro67' 68 . Thus, the continued elucidation of molecular mediators that regulate hepatocyte
function and proliferation will be critical for the advancement of cell-based therapies and their
routine use in clinics to treat compromised liver functions. In addition, the potential of alternative
cell sourcing approaches, based on stem cell differentiation and reprogramming, are active areas
of investigation.
Soluble Factors
Viral Transduction
Extracellular Matrix
Tumor-Derived Cell Lines
Co-Cultivation
Stem-cell Derived
Spontaneous Immortalization
Genetically Altered Mouse Strains
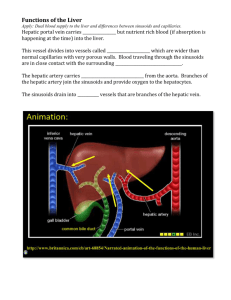
Figure 1.1 Cell sourcing of human hepatocytes. Approaches have focused on modulating in
vitro culture conditions of primary hepatocytes, including the addition of soluble factors,
extracellular matrix proteins, and heterotypic cell-cell-interactions. Various immortalization
techniques have also been attempted, such as spontaneous transformation through long-term
culture in collagen gel and targeted mutations through viral transduction. Other sources include
tumor-derived cell lines, stem-cell derived cell lines and in vivo expansion in mice models
providing regenerative stimuli.
Mature Hepatocytes
Primary human hepatocytes are functionally the most robust cell type for cell-based
therapies for liver diseases69 7 0 . Within their native microenvironments
21
in vivo, human
hepatocytes have phenomenal proliferative capability. Following resection of two-thirds of the
liver through a surgical procedure known as partial hepatectomy (PHx), the residual mature cell
populations, comprised mainly of hepatocytes, are able to proliferate and restore lost liver
mass' 5 . This full regenerative response can be seen after each of at least 12 sequential PHx's.
To demonstrate the clonogenic potential of the hepatocyte itself, mouse models were generated
in which livers were rendered incapable of supporting animal life through experimentally
induced defects. Healthy hepatocytes injected into these compromised livers can proliferate,
72
generate nodules of normal hepatocytes, and rescue the animals . As low as 1000 normal
hepatocytes were found to be sufficiently therapeutic. Furthermore, cells from newly formed
nodules of normal hepatocytes can be isolated and serially transplanted, through as many as four
generations, to rescue other animals. Mathematical calculations based on this model predict that
a single hepatocyte can undergo at least 34 cell divisions to give rise to 1.7 X 1010 cells,
suggesting that a single rat hepatocyte can generate 50 rat livers of 300 million hepatocytes
each.
Various attempts have been made in the last several decades to harness ex vivo this
tremendous replication potential of mature human hepatocytes (Figure 2). It is recognized that
proliferating hepatocytes in vivo are presented a complex and dynamic mixture of soluble factors
via the blood while maintained within an interactive support system of extracellular matrix
(ECM) and non-parenchymal cells. Thus, early studies focused on providing select key
components to in vitro culture systems, including humoral and nutritional supplements as well as
ECM and supportive cell types7 4. To specifically promote hepatocyte expansion in vitro, primary
75
cultures have been treated with serum and cytosol collected from livers that underwent PHx
76
76 77
and with more defined soluble factors including various growth factors ' , sugars , amino
22
acids 76, hormones 7778, vitamins 76,
serum proteins76'80, and trace metals
76 80
,
. The effect of any
individual supplement on hepatocyte proliferation can be difficult to directly determine, as the
effect depends on the state of the hepatocyte, which is synergistically determined by the
combination of all culture components 74. Nevertheless, investigations have yielded a multi-factor
media formulation, which can be used for moderate expansion of rat hepatocytes through a
dedifferentiated bi-potential intermediate76 . Non-soluble culture components such as different
ECM 7 68, 1 and supportive cell types8 1-84 have also been examined for mitogenic effects on
hepatocytes. These include physiologic liver ECM proteins, and non-physiologic tumor-secreted
protein mixtures in different configurations, in addition to co-cultures of hepatocytes with
various intrahepatic and extrahepatic cell types, both live and dead. Many different combinations
of culture components have been shown to support moderate expansion of rat hepatocytes
although translation of these findings to human cultures has not been reported.
Human cells are critical for cell-based therapies due to substantial species-specific
differences between animal and human hepatocellular functions including apolipoprotein
expression, metabolic regulation of cholesterol, and phase I detoxification enzymes
85-87
-
. To
overcome the growth limitations of primary human cells, investigations are underway to develop
highly functional human hepatocyte cell lines. A common approach is to introduce oncogenes
through retroviral transduction. The simian virus 40 tumor antigen gene (SV40 Tag) is a
common immortalization agent, whose product binds to cell cycle regulator proteins Rb and
p53"'8. Cell lines have also resulted from spontaneous immortalization of hepatocytes in cocultures or collagen gel sandwich cultures", and additionally can be derived from liver tumors,
as in the case of the HepG2 hepatoma cell line 90 . Although these cell lines are growth-competent,
they introduce new safety concerns and typically underperform primary cells in terms of liver
23
functions 9 1,92 . The principal safety concern is the transmission of oncogenic agents to the host,
especially in the case of implanted cells. To address this, researchers have developed
mechanisms to inactivate transduced oncogenes through temperature-sensitive SV40 Tag93, CreloxP-mediated oncogene excision9 4 , and suicide genes such as herpes simplex virus thymidine
kinase (HSV-tk)9 5 .
Another intriguing approach for human hepatocyte expansion, particularly as a model
system, is the transplantation of human hepatocytes into genetically-altered mouse strains17,96-98
This strategy takes advantage of the in vivo mitogenic environment, known to orchestrate many
rounds of hepatocyte replication and can be generated through experimentally induced defects to
host livers. Such defects can be produced by large amounts of urokinase, which can be
abnormally over-expressed under the influence of the albumin promoter in hepatocytes'
99 100
.
While effective as a hepatic xeno-repopulation system, these mice are fragile and present only a
limited time window for transplantation. Alternatively, Grompe and colleagues have produced
regeneration-inducing liver defects through an experimentally introduced deficiency in the
catabolic enzyme fumarylacetoacetate hydrolase (Fah). After pretreatment with a urokinaseexpressing adenovirus, Fah-deficient mice can be very receptive hosts to human hepatocytes17 .
Findings from these animal studies suggest that human hepatocytes do retain their considerable
proliferation potential upon isolation and can expand given the appropriate stimuli. However,
similar to the use of hepatocyte cell lines, the therapeutic utility of hepatocytes expanded in
animal models is limited by safety concerns such as the transmission of pathogenic agents and
the incorporation followed by expression of animal glycoproteins on human hepatocyte cell
surfaces.
24
Ultimately, sustainable proliferation of highly functional human hepatocytes could
generate patient-specific cell populations. These cells can be used to provide sufficient
autologous
materials
for
cell-based
treatments,
thus
circumventing
post-surgical
immunosuppressive regimens. In vitro, the ability to expand human hepatocytes can enable drug
therapies to be selected according to the characteristics of individual patients, thus minimizing
adverse drug reactions.
Stem Cells and ProgenitorPopulations
Due to limitations in mature hepatocyte expansion in vitro, alternative cell sources are
being pursued. These include various stem cell populations, which can self-renew in vitro and
exhibit pluripotency or multipotency and thereby serve as a possible source of hepatocytes, as
well as other non-parenchymal liver cells.
Studies have shown that embryonic stem cells can be induced to differentiate down the
hepatic lineage in culture through the carefully orchestrated addition of various growth factors,
and when supported by the appropriate ECM10103 More recently, studies are also exploring in
more scope and detail the functional capacity of these differentiated populations, both in vitro
and in vivo
4 106
. Such endeavors are being guided by improved insight into how different cell
types are specified in embryonic development. This insight is typically gained through
observations of cellular responses to individual inductive signals. Zaret and colleagues have
further investigated how different inductive signals interrelate and have reported complex,
dynamic signaling networks that could help explain incomplete cell programming in stem cell
differentiation protocols'0 7 .
In addition to embryonic stem cells, a wide range of fetal and adult progenitor cell types
have been explored. Continuing investigations are focused on determining the differentiation
25
potential and lineage relationships of these populations. Fetal hepatoblasts are liver precursor
cells present during development that exhibit a bipotential differentiation capacity, defined by
1 08
the capability to generate both hepatocytes and bile duct epithelial cells . Furthermore, within
the adult liver, a rare percentage of resident cells have been demonstrated to exhibit properties
consistent with their designation as adult hepatic stem cells''9,10.
It has been suggested that
these cells represent precursors to adult progenitor cells, termed oval cells, which share
phenotypic markers and functional properties with fetal hepatoblasts. In adult livers suffering
certain types of severe and chronic injury, oval cells can mediate liver repair through a program
similar to hepatic development'
2
Various cell lines exhibiting characteristics comparable to
fetal hepatoblasts and oval cells have been developed, for example, lines derived from mouse
E14 embryos by Weiss and colleagues. These bipotential mouse embryonic liver (BMEL) cells
are proliferative, can be induced to be hepatocyte-like or bile duct epithelial-like in vitro"13,
and
can home to the liver to undergo bipotential differentiation in vivo within a regenerative
environment'
4
.
Outside the liver, there may also exist multipotent stem/progenitor-like cells that are of
therapeutic and biomedical interest
15
. For example, multipotent adult progenitor cells (MAPCs)
116
derived from the bone marrow have been shown to generate hepatocyte-like cells in vitro
Similarly, various mesenchymal stem cell preparations have been reported to give rise to cells
exhibiting many characteristics of mature liver cells117-120, including the ability to engraft in vivo;
5
however, the extent of functional liver repopulation has been modest" . Other sources of
extrahepatic liver cell progenitors include human amniotic fluid and membranes, which may
contain cells capable of hepatic differentiation121-125
ReprogrammedAdult Cells
26
Fully differentiated adult cells, such as skin cells, were recently demonstrated to be
reprogrammable
to an
undifferentiated, pluripotent
state through
forced expression
of
reprogramming factors Oct3/4 and Sox2 along with either Klf412 6- 12 9 or Nanog and Lin28"3 .
These reprogrammed cells are termed induced pluripotent stem (iPS) cells and highly resemble
embryonic stem (ES) cells, sharing many characteristics such as significant self-renewal
capabilities in vitro and pluripotent differentiation potential. However, iPS cells offer an
additional advantage of sourcing from adult somatic cells for the generation of patient-specific
cell populations, potentially enabling therapies to be developed according to the characteristics
of an individual patient.
Work
that
researchers,demonstrated
done by
Duncan
as well
as other
a subsequent
multistep
and colleagues,
through iPS reprogramming
and
differentiation protocol, skin cells can give rise to hepatocyte-like cells, which not only exhibit a
variety of hepatocyte-specific functions in vitro, but can also be induced to generate intact fetal
livers in mice in vivo
.
As a parallel strategy, work done by Melton and colleagues has demonstrated that it is
also possible to directly reprogram one adult cell type into another, without an undifferentiated
pluripotent intermediate. Similar to the use of master transcriptional regulators in the
reprogramming to iPS cells, the expression of a key set of transcription factors in pancreatic
exocrine cells in vivo induced conversion into cells that highly resemble
P-cells13 4 . These
findings raise future possibilities for deriving hepatocytes directly from another adult cell type.
Ultimately, understanding the mechanisms governing the fates of stem and progenitor cell
populations can empower the development of cell-based therapies. However, many challenges
remain, including the ability to program differentiation completely. Furthermore, regardless of
the cell source, phenotypic stabilization of hepatocytes ex vivo remains a primary issue.
27
Accordingly, the development of robust in vitro liver models is an essential stepping-stone
towards a thorough understanding of hepatocyte biology and improved effectiveness of cellbased therapies for liver disease and failure.
1.4 Maintenance of Hepatocytes ex vivo
In order to engineer an optimal system for the maintenance of hepatocytes in vitro, one
can utilize as a guide the complex architecture of the liver, in which hepatocytes interact with
diverse extracellular
matrix molecules, nonparenchymal cells,
and soluble factors (i.e.,
hormones, oxygen). The liver is organized into functional units known as lobules, which contain
mostly hepatocytes (70% of total liver cells), aligned into cords and surrounded by different
types of stromal cells. At the interface between hepatocytes are gap junctions, tight junctions,
and bile canaliculi, where they contribute to homotypic cell-cell interactions and coordinate the
excretion of bile to the gall bladder via bile ducts. These hepatocyte cords are flanked by the
Space of Disse, which are layers of extracellular matrix between heaptocytes and the liver
sinusoids. Within this space reside hepatic stellate cells, a type of pericytes implicated in liver
fibrosis. The liver sinusoids are lined with fenestrated endothelium; Kupffer cells (macrophages)
and Pitt cells (Natural Killers) are free to roam the blood and tissue compartments; and toward
the end of the sinusoid, biliary ductal cells (cholangiocytes) also interact with hepatocytes.
The liver is supplied by two major blood vessels through its right lobe: oxygen-rich blood
enters via the hepatic artery and comprise one-third of the afferent blood supply while the
remaining two-thirds enter via the portal vein. The portal vein brings nutrient- and hormone-rich
venous blood from the digestive system to the liver for processing before entry into systemic
28
circulation. The efferent vessel, hepatic vein, drains directly into the inferior vena cava posterior
to the liver.
Within the liver lobule, hepatocytes are divided into three zones along the length of the
sinusoid, each exhibiting a unique morphology and function. Zonal differences can be observed
in virtually all hepatocyte
functions.
For instance, gene expression is thought to be
compartmentalized to enable the liver to function as a "glucostat". Other differences in
cytochrome P450 enzymes have been implicated in zonal hepatotoxicity of certain xenobiotics
13S.
It is not quite clear how zonation is established or maintained but possible mediators include
blood-borne hormones, oxygen tension, pH levels, extracellular matrix composition, and
innervations
136
. It is believed that a precisely orchestrated microarchitecture, coupled with cell-
cell, cell-matrix and cell-soluble factor interactions allows the liver to perform its many diverse
functions, which can be broadly categorized into protein synthesis (e.g., of blood-borne proteins
including albumin and clotting factors), energy and cholesterol metabolism, bile production, and
detoxification of both endogenous (e.g., bilirubin, ammonia) and exogenous (e.g., drugs and
environmental toxins) compounds.
Heterotypic interactions between parenchymal cells (hepatocytes) and their stromal
neighbors in particular are known to be important in the liver in vivo. During development, the
formation of liver from endodermal foregut and mesenchymal vascular structures is believed to
be mediated by heterotypic interactionsm 1
. In the adult liver, stromal cells modulate
hepatocyte phenotype under both physiologic and pathologic conditions99,139. For instance,
stellate cells activated by TGFP-1 produce excessive amounts of extracellular matrix proteins
(e.g., collagen), leading to liver fibrosis, which can progress to cirrhosis, portal hypertension and
ultimately liver failure.
29
In vitro, the viability and liver-specific functions of hepatocytes from multiple species
have been shown, through extensive studies, to stabilize for several weeks upon co-cultivation
with stromal cell types. This co-culture effect can be observed using a wide variety of stromal
cell types, both primary and immortalized, from intra-hepatic and extra-hepatic sources, crossing
even species barriers 14 1-143. Hepatocytes in co-cultures, particularly with murine embryonic J23T3 fibroblasts, maintain for weeks the distinct nuclei, polygonal morphology, well-demarcated
cell-cell borders, and visible bile canaliculi network displayed by cells in vivo1 44 145 . This is in
stark contrast to hepatocytes in pure mono-layers, where most cells rapidly (hours) lose viability,
while surviving cells lose liver-specific functions and adopt a fibroblastic morphology1 43
Hepatocyte phenotype should ideally be maintained for the lifetime of the clinical
intervention (-weeks for extracorporeal devices and ~years for in vivo therapies).
To address
this problem, various approaches have manipulated the culture environment (e.g., seeding
hepatocytes onto matrigel substrates or in between collagen gels, assembling hepatocytes into 3D spheroids or maintaining hepatocytes in bioreactors)' 4 6 . While many of these methods are
useful for capturing liver-specific functions in vitro, they are difficult to implement for clinical
applications due to solute transport or scaling limitations.
Co-culture does not suffer such
limitations and have been utilized to investigate various physiologic and pathologic processes
including host response to sepsis, mutagenesis, xenobiotic toxicity, response to oxidative stress,
lipid metabolism, induction of the acute phase response
47 157
-
, and more recently in the
development of in vitro models for pharmaceutical drug screening and engineered hepatic
tissues1'4.
30
1.5 High-throughput Screening and Small Molecule
Modulation of Complex Cellular
Phenotypes
Small molecules have been shown to modulate a wide range of complex cellular
processes, including stem cell self-renewal and differentiation, and the proliferation of normally
quiescent adult cells such as pancreatic
P-cells and cardiomyocytes 5 8 - 60 . Compounds can act
through a variety of mechanisms to induce cell division, including activation of developmental
signaling pathways such as Wnt 16
or recruitment of GEFs to the plasma membrane for
RAS/MAPK pathway activation160.
Traditionally, drug discovery in the pharmaceutical industry is conducted through targetbased screening.
Such screens aim to identify small molecule modulators of specific protein
activities, often inhibitors or activators of rate-limiting enzyme of the biochemical process of
interest. By providing the isolated targets during initial large-scale screening, understanding the
mechanism of action of the resultant drug candidates is relatively straightforward'62,163
However, there is significant attrition rate when progressing through the drug development
pipeline, initially when natural transport barriers, such as the plasma membrane, are introduced
and again when moving from animal physiology into clinical trials.
While target-based
screening has allowed many major advances in modem drug development, a prerequisite for a
target-based screen, however, is a reasonably well-characterized target, which is somewhat
limiting when exploring new phenotypes and new areas of biology.
More recently, whole-cell or phenotypic screens have evolved to offer the ability to target
any protein (or other entity, such as a lipid or nucleic acid) in its biological context, without the a
priori need to know the target.
This means that, in addition to either enzyme inhibitors or
receptor agonists or antagonists, small molecule hits from phenotypic screens could, for example,
31
act as allosteric inhibitors or could ablate protein-protein interactions
164,161
. Phenotypic screens
are also key in providing tools to study entire signaling pathways or networks, which are
important features in modem chemical and systems biology.
Although the potential of
phenotypic screens for the discovery and characterization of active compounds is high, target
identification remains a major challenge and is the main reason why phenotypic screens are not
widely used in the pharmaceutical industry1 66' 167.
Recent advances in target identification
strategies, including chemical proteomics and computational methods, are making this problem
more tractable16 3
32
Chapter 2.
High-throughput Liver Platform for the Development of Novel
Therapeutics
2.1 Introduction
Liver disease is a major healthcare burden and its lack of efficacious treatments presents
a critical unmet medical need, which is further compounded by the absence of an in vitro model
of human hepatocyte biology capable of predicting clinical outcome. The liver has a central role
in the metabolism of all molecular therapeutics, thus an accurate model of human liver
physiology is a critical tool for research and development of novel therapeutics, not just for liver
diseases, but for other organ systems as well.
Drug development is a costly endeavor, due largely in part to the high attrition rates of
candidate compounds that proceed through preclinical testing only to fail in humans. A third of
drug withdrawals from the market and most Phase I clinical trials fail due to unforeseen liver
toxicity and/or altered bioavailability of drug candidates in patients, indicating that existing in
vitro liver models are not fully predictive of in vivo liver biology. The primary cause of this high
attrition rate is the species-specific variations in liver metabolic pathways, particularly
cytochrome P450 isoforms and activities, which limit the translatability of results from animal
studies to the clinics16 1. Thus, this chapter reports the development of a scale-able model of
human liver physiology for high-throughput identification of small molecules with clinically
relevant bioactivity.
Cellular functions in the liver are supported by a complex microenvironment, consisting
of various cell-cell, cell-matrix and cell-soluble factor interactions organized in hierarchical
structures ranging from single cells to functional subunits.
33
Conventional tissue cultures of
primary cells lack such multi-faceted cellular stimuli and primary human hepatocytes cultured
alone rapidly lose viability and liver phenotype in vitro. Liver microsomes are currently used in
high-throughput identification of detoxification enzymes, but their lack of cytoarchitecture and
functional cellular machinery including dynamic gene expression systems limits their use in the
study of other aspects of liver biology. Liver slices do contain intact cells, but have extremely
limited viability (~I day) and are not compatible with high-throughput screening.
Similarly,
hepatic spheroids, and models that manipulate the extracellular matrix (ECM) microenvironment
of hepatocytes with Matrigel and/or collagen require a high degree of hepatocyte confluency for
long-term survival, thus limiting their use in the exploration of many cellular process, such as the
proliferation of functional hepatocytes, whose expansion is contact-inhibited.
Such 3D models
are also difficult to miniaturize into a standard and high-throughput format.
Bhatia et. al. had previously reported that co-cultivation of non-parenchymal cells with
hepatocytes can stabilize primary human hepatocytes in vitro for an extended period of several
weeks23',142514,168,169.
Such systems provide sufficient time for phenotypic screens and are also
amenable to automation. However the inclusion of a second, supportive cell type introduces new
challenges.
When probing cellular phenotypes, it is now necessary to develop assays that can
separate the hepatocytes and non-parenchymal cells that co-exist within the platform. Functional
assays can be designed to measure hepatocytes-specific outputs such as albumin and urea;
proliferation assays, however, have less innate ability to distinguish different cell types.
In this chapter, we report the development of a human liver platform and various highthroughput assays for examination of cellular phenotype including cell proliferation and death as
well as protein synthesis, detoxification and amino acid metabolism functions.
34
2.2 Results and Discussions
Cell Sources
Sourcing of hepatic cells for liver platforms is a fundamental challenge for many fields of
liver research. Many are forced to employ xenogeneic sources such as rodent and porcine, or
immortalized human hepatocyte cell lines 170-173 . Animal hepatocytes are well studied and easily
sourced, but exhibit significant species-specific differences in hepatocellular functions including
apolipoprotein expression, metabolic regulation of cholesterol, and most importantly, phase I
detoxification enzymes.
For clinical applications, use of animal cells are further limited by
safety concerns, such as the possibility of transmitting pathogens like porcine endogenous
retrovirus (PERV), across species.
Human hepatocyte cell lines, while expandable, contain
mutations and exhibit an abnormal repertoire of liver functions, which limits their clinical impact.
Primary human hepatocytes present the best functional output and are thus ideal for modeling
human liver physiology. However, to date, chemical screening on primary human hepatocytes
has been prohibited by their limited availability in large quantities, and their precipitous decline
in viability and liver-specific functions in vitro.
Human livers become available for research purposes only after they are disqualified for
transplantation, thus the vast majority of donor organs suffer from significant liver-related
conditions such as chronic alcoholism, hepatitis infections, fibrosis, cirrhosis, and cancer.
Fortunately, recent advances in cryopreservation technologies enabled the storage of entire livers
of human hepatocytes from the rare donors that can still represent normal physiology. A human
liver can yield hundreds of vials of cryopreserved hepatocytes, each containing 5 to 10 million
cells. A single donor would allow multiple batches of phenotypic screening, ultimately testing
hundreds of thousands of small molecules on a constant genetic background over a period of
35
several months.
However, while extensive previous characterizations have shown that some
cryopreserved primary human hepatocytes exhibit phenotypes that approach fresh hepatocytes
and are thus very useful for in vitro liver studies23 1 74 ,175 , not all donors were suitable for
screening (Table 2.1).
During our initial testing of eight different donors of cryopreserved
human hepatocytes, three were non-plateable, thus incompatible with phenotypic screening.
While the remaining five donors all yielded hepatocytes that adhered to rigid collagen in culture,
one donor was too young (0.1 years) to exhibit a full repertoire of mature hepatocyte functions
while another two donors had poor functions at baseline. Ultimately, we chose donor GHA, who
was a one-year-old Caucasian female with a cause of death of dry drowning. GHA hepatocytes
attached well to rigid collagen and demonstrated good synthetic, detoxification and metabolic
functions.
Donor
Age
Plate-able?
Function
HU0845
HU4122
H4U4088
HU4100
KQG
SCT
47 yrs
19 yrs
2 yrs
40 yrs
38 yrs
38 yrs
No
No
No
Yes
Yes
Yes
N/A
N/A
N/A
N/A
Low
Low
RQO
0.1 yrs
Yes
Too young
GHA (donor a)
1 yr
Yes
Good
Table 2.1. Donors of cryopreserved primary human hepatocytes. Eight different donor lots
were tested for suitability for high-throughput screening through examination of plate-ability and
baseline functions such as albumin secretion, urea production and cytochrome P450 activity.
Platform Design
To maintain primary human hepatocytes in culture, we co-cultivated them with murine
embryonic J2-3T3 fibroblasts, which have been shown to transiently stabilize hepatocytes in
vitro. Earlier studies suggest that this co-culture effect is mediated by heterotypic cell-cell
36
contact between primary hepatocytes and stromal cells as well as continuous stimulation with
stromal-derived, short-range signaling molecules
There exist multiple configurations of co-cultures of primary hepatocytes and J2-3T3
fibroblasts, with varying degrees of architectural organization.
The simplest implementation
consists of a co-planar distribution of randomly mixed hepatocytes and J2-3T3 fibroblasts on a
matrix of rigid collagen type I.
More sophisticated designs comprise the application of
semiconductor-driven microtechnology to organize primary hepatocytes into in vitro colonies of
empirically optimized island sizes, subsequently surrounded by J2-3T3 fibroblasts; this particular
configuration is termed micro-patterned co-culture (MPCC). All configurations of hepatocyte-J2
co-cultures were found to maintain primary human hepatocyte functions in vitro for at least 9
days but none recapitulates their innate potential for substantial proliferation.
In general,
increased architectural organization of cells in culture leads to longer-term stabilization of
hepatocyte functions, with MPCC being the most optimal configuration, enabling maintenance
of hepatocyte functions in vitro for 4-6 weeks. However, while the packing of hepatocytes onto
pockets of circular islands tightly surrounded by J2-3T3 fibroblasts maximizes the homotypic
cell-cell interactions that enhance long-term hepatocyte survival and function, the confluent cellcell contact may prevent expansion of any cell type whose proliferation is normally contactinhibited. Additionally, MPCCs are difficult to miniaturize beyond 96-well platforms and in any
case, such prolonged periods of hepatocyte functions are neither necessary nor practical for most
whole-cell screens.
We designed our high-throughput liver platform to assume a feeder layer co-culture
configuration in order to provide both time and space for hepatocyte expansion. The platform
contains a sparse population of hepatocytes on top of a confluent layer of J2-3T3s within 384-
37
well plates (Figure 2.1 top panel).
This design enabled fibroblast-mediated
hepatocyte
stabilization for at least 9 days (Figure 2.1 left panel) without hepatocyte crowding and is
amenable to 384-well and smaller formats.
S16Co-culture
IE14.
M 12-
E 6
4
2
0
Hep
0
2
4
6
8 10 12 14
Culturing Time (Days)
Figure 2.1 High-throughput liver platform. Cryopreserved primary human hepatocytes
(green) are maintained in vitro through co-cultivation upon a feeder layer of J2-3T3 fibroblasts
(red) in 384-well formats. Line graph shows representative rate of albumin secretion in screening
co-cultures and hepatocyte-only cultures (green) over time. Phase contrast imaging shows
morphology of feeder-layer co-cultures (scale bar = 100pm). All data presented mean ± standard
deviation
The number of fibroblasts per well was empirically optimized to 8,000 cells/well in order
to establish a confluent feeder layer without contraction and aggregation of overcrowded J2-3T3
fibroblasts. The number of hepatocytes per well was empirically optimized to 2,000 cells/well,
minimizing the number of hepatocytes in culture to allow room for expansion while balancing
the need for sufficient albumin output detectable via ELISA (details under functional assay).
Similarly, the amount of media used per well was empirically optimized to balance opposing
needs of oxygen transport and nutrient supply - too much media presented a transport barrier for
gas diffusion, causing observable steatosis in cultured hepatocytes; too little media caused
38
nutrient deprivation. There was an additional restriction that all fluids handled robotically must
be dispensed in volumes that are multiples of 10ul; while the robots can be programmed to
dispense in single microliter gradients, only multiples of I Oul offered sufficient accuracy. All
cells were robotically seeded at the lowest possible speed setting in order to minimize physical
stresses. During pilot testing, it was observed that fibroblasts had difficulty remaining attached
to plain tissue culture plastic in 384-well formats, thus a matrix coating of Collagen type I was
added at a concentration of I OOgg/mL. To assess cell fates in this platform, we developed two
separate high-throughput readouts: an image-based proliferation assay and three biochemical
functional assays.
ProliferationAssay
The co-cultivation of hepatocytes with J2-3T3 fibroblasts allows long-term maintenance
of primary human hepatocytes in culture but renders the measurement of hepatocyte proliferation
challenging.
The co-existence of two different cell types in each well requires a proliferation
assay that is specific to hepatocytes.
However, all existing measurements
of cellular
proliferation and number, such as Alamar Blue, Cell Titer Glow and cell cycle stains including
Ki67, BrdU and PNA, all reflect the proliferation state of the whole well, which allows behavior
of the more populous J2-3T3 fibroblasts to mask moderate hepatocyte expansions in culture.
Therefore, we developed a custom image-based readout to specifically measure hepatocyte
proliferation in our high-throughput liver platform.
Hepatocytes in culture can be distinguished from underlying J2-3T3 fibroblasts via a
variety of methods, including phase-contrast microscopy, staining for hepatocyte-specific
markers such as Albumin and CD44, and striking differences in nuclear morphology. Brightfield
images, while easy to acquire, are difficult to quantify, particularly in a high-throughput manner.
39
Immunofluorescent staining of particular antigens, while easy to measure in an automated
fashion, are difficult to execute in 384-well and smaller formats. Therefore,, we chose to develop
an image-based proliferation assay that uses nuclear morphology to quantify hepatocyte nuclei
numbers in culture. When visualized with Hoechst stain, hepatocyte nuclei are more uniform in
texture while fibroblast nuclei are punctate (Figure 2.2). The assay thus visualizes all cell nuclei
in culture using a simple Hoechst stain, specifically identifies hepatocytes based on nuclear
morphology and provides a count of the number of hepatocyte nuclei in culture (Figure 2.3).
Fibroblast Nuclei
Hepatocyte Nuclei
Figure 2.2 Distinctive nuclei morphology. Hepatocytes in co-culture with J2-3T3 fibroblasts
can be separated based on nuclei morphology. Hepatocyte nuclei (left) are smaller, rounder and
more uniform in texture while fibroblast nuclei (right) are punctate.
40
1. High-throughput
2. Automated Nuclei Profile Construction
Image AcquisitionTexture
Spotslntensity-O)
Texture SpotsCount-0)
TextureRadialDistribution=0.5)
IntensityNMaxintensityEdge=0.1)
AreasapePerimeter=64)
AreaShapeMajorAxisLength =12)1
AreaShapeEccentricity =0.1)
Construct Nuclear Profile
using 300 Measured Properties
3. Nuclei Classifier Training
Fetch.3O
random
cells from
#pritSenl
IF (HepNuCLoQTextureCorrelation CorrNuclei_10 > O.25663000000000002.0t.79518227119604368'
Max number of rules:
(
d
s
ore Ai (.score imge)
unclass fled (100>
_____________
Iterative
manual training
ofthe nuclei classifier
----
Caust 6e rt rained vat
sW
uIes I n s.
(Ad
new las
4. Automated Nuclei Classification and Counting
..
F (Texture SpotsMeanintensity > 4.57)
F(TextureSpotsCount > 3)
F (Texture RadialDistribution > 1.04)
F (intensityMaxintensityEdge > 0.50)
.+.....
F (AreaShapePerlmeter > 79.70)
F (AreaShapeMajorAxisLength > 46.24)
Rules F(AreaShapeEccentricity > 0.38)
Hep
Fb
Figure 2.3 Image-based assay workflow. Nuclei are visualized with Hoechst stain, imaged
using a high-content screening microscope, identified, characterized and counted through a
custom image-based proliferation assay. The user interface window of the classification
software, CellProfiler Analyst, is shown. It allows manual classification of randomly presented
nuclei and error correction of machine-classified nuclei.
41
It should be noted that primary hepatocytes have been known to exist in multi-nucleated
states and/or initiate cell cycle without completing cytokinesis.
Thus we recognize that this
assay is not strictly a measure of hepatocyte expansion. However, cellular proliferation cannot
occur without DNA synthesis. We designed the image-based assay to minimize the loss of
active molecules to false negative errors during primary screening as they cannot be recovered
later on; On the other hand, false positive molecules that induce DNA synthesis and/or multinucleation without cellular expansion can be filtered away during secondary screening, in vitro
or in vivo hit validation.
Image Acquisition
Image Intensities - No Treatment
Image Intensities - Permeabilized
1-12
Dimmest
Dimmest
Brie test
Figure 2.4 Uniformity of image intensity throughout screen. Permeabilization
treatment is not necessary for traditional Hoechst staining but helped normalize Hoechst
33258 staining intensities throughout screening. Upper panel shows heatmap of image
intensities for each 384-well plate; arrows indicate location of brightest and dimmest
images. Bottom panel shows acquired images.
Cultures of hepatocytes and J2-3T3 fibroblasts were fixed using 4%
paraformaldehyde (PFA) in black-walled, clear and flat-bottomed 384-well plates
42
(Coming). Fixed samples were then stained with Hoechst 33342. It is important to note
that the cell membrane is much more permeable to Hoechst 33342 than Hoechst 33258;
thus an additional permeabilization step using 0.1% Triton-X for 30 min is necessary if
visualizing nuclei with Hoechst 33258.
Without membrane permeabilization, Hoechst
33258 leads to heterogenous staining intensities, which will lower the accuracy of
subsequent image analyses (Figure 2.4).
Images of fluorescently labeled nuclei are acquired and digitized using a highthroughput screening microscope (Molecular Devices IXM) coupled to a barcode reader
and robotic arm (Thermo) for automated plate loading (Figure 2.4). The microscope is
configured to self-focus, first using lasers to identify the bottom of wells via differences
in the refractive index of plastic and fluids, then using image-based focusing algorithms
that scan through a z stack of -200 um in -50um steps in search of the plane with the
sharpest images.
IX Micro HTS microscope
Thermo robot with plate stack
Figure 2.5 Schematic of Automated Image Acquisition. Treated sample plates are
robotically loaded into high-throughput screening microscope.
In earlier implementations of this image assay, we explored the distinction of hepatocytes
from fibroblasts using z-position alone, but the differences between the two layers of the
co-culture was too minute (-5pm).
The greater number of fibroblast nuclei (8,000
fibroblasts vs. 2,000 hepatocytes) and their punctuate nature ensured automated focusing
43
on the fibroblast plane, further enhancing the morphological differences between
image
hepatocytes
and fibroblasts to facilitate subsequent
mechanisms
do fail occasionally, burying populations
analyses.
Self-focus
of blurred images among
successful acquisitions, depressing the accuracy of hepatocyte nuclei counts. To address
this, we developed subsequent analyses pipelines to flag the occurrence of these normally
rare failures using blurry nuclear morphologies.
Accurate examination of nuclear morphology required image acquisition at 20x
magnification. Given the large volume of images required to cover 100% of well area at
this high magnification, we chose to sample 50% of the well area in a checkerboard
fashion, imaging a total of 21 sites per well. To speed up image acquisition, laser-based
focusing occurred once per plate and image-based focusing executed once per well.
Image Processingand Nuclei Identification
We developed automated image analysis pipelines to identify every nucleus in
every Hoechst-stained image of the screening cultures, and to measure various
characteristics (e.g., shape, size, intensity, texture) of each nucleus using the open-source
CellProfiler software
177
An important
first
step
in image processing
is illumination
correction.
Illumination can vary by more than 1.5-fold across a field of view, despite the use of fiber
optic light sources. This adds an unacceptable level of noise, and can compromise the
accuracy of subsequent analyses involving object intensity, including nuclei identification
and classification. Thus for our proliferation assay, we configured CellProfiler to stack
all acquired images from a single experiment to identify and normalize consistent
discrepancies in the staining intensities across the field of view.
44
Nuclei identification or segmentation is challenging when source images are
crowded or, in our case, also contain overlapping objects. The accuracy of this step plays
a central role in determining the accuracy of the resulting nuclei counts. We tested a
variety of object identification modules offered within CellProfiler, starting with a
modular strategy that first identifies object edges based on intensity, then separates
clumped objects based on their measurements such as shape or size. This module offered
a great degree of versatility, allowing customization of a number of parameters such as
expected object size and intensity thresholds.
While we were able to configure this
module to successfully identify crowded nuclei, we were unable to get accurate
segmentation of overlapping objects. Therefore, we opted instead to assemble a custom
module of nuclei segmentation.
Original Algorithm:
Final Algorithm:
Figure 2.6. Nuclei Identification. Feederlayer co-culture led to overlapping objects in
Hoechst images that proved challenging to segment. Final algorithm was able to correct
identify nuclei locations and borders.
The segmentation pipeline implemented in our proliferation assay first uses
relative peaks in intensity to pinpoint positions of potential nuclei, so that overlapping
45
nuclei can be correctly identified as separate objects. After the locations of nuclei are
found, their edges can then be outlined more accurately using Propagation algorithms.
We implemented test modules to compare several algorithms side by side in order and
incorporated into the pipeline the most accurate segmentation algorithm (Figure 2.6).
For each identified nucleus, we measured a large number of features to construct a
nuclear profile, including nuclear size, shape and texture. This profile will subsequently
be used to train machine learning algorithms to automatically classify nuclei as
hepatocytes or fibroblasts.
During assay development, we observed that the single
feature of nuclear morphology that most effectively distinguished fibroblast nuclei from
hepatocyte nuclei was the punctate sub-nuclear structures present in fibroblasts but absent
in hepatocytes. Unfortunately, these punctate structures, while numerous, are very small,
thus occupying only a minute percentage of the nuclei area; consequently, their impact on
the measurements at the whole-nucleus level was too dilute for effective machine training.
To address this, we added an additional segmentation module to identify the punctate
sub-nuclear structures and measured how many and what type (e.g., big or small, bright
or dull) of punctuates are associated with each nucleus (Figure 2.7).
46
Original Measurements
Final Measurements
Figure 2.7 Identification of sub-nuclear structures. Punctate sub-nuclear structures
were identified as objects and associated with their parent nucleus. Yellow circles
indicate hepatocyte islands. Yellow square surrounds one region of fibroblast cluster.
Nuclei Classificationand Quantification.
The
nuclear
profiles
generated
by
CellProfiler
are
inputted
into
CellProfilerAnalyst17 8 for training of machine learning algorithms to distinguish and
count hepatocyte nuclei. We manually initiate the training phase by identifying a few
hepatocytes and a few fibroblasts. To avoid over-fitting the machine learning algorithm
to a few particular samples, the initial training sets are populated with -50 hepatocytes
and ~ 50 fibroblasts taken randomly from the general population without references to
specific wells or plates. Using this initial training set, a machine learning algorithm is
used to generate a preliminary set of rules for nuclei classification, using the
GentleBossting algorithm applied to regression stumps.
This rule set is used by
CellProfilerAnalyst to classify a new batch of nuclei, outputting the results for manual
error correction.
The corrections are then used to refine the rule set in an iterative
47
process until an accuracy plateau is reached. Once finalized, the rule set is applied to the
nuclear profiles of every nucleus of every image in the experiment to classify each object
as a hepatocyte or fibroblast before outputting a count of each nucleus type per well.
Initial training was conducted without info on sub-nuclear structures and required
approximately 1 day to complete a training set containing -5000 manually classified
objects with an accuracy plateau of -75% using a total of 300 rules. With the assistance
of punctate sub-nuclear structures, this assay now requires only a few hours to generate a
training set of -500 hundred objects with an accuracy plateau of at least 90%, often 95%,
using a total of 100 rules (Figure 2.8).
Final Classification Accuracy:
>90%
Original Classification Accuracy:
-70%-75%
Figure 2.8 Classification Accuracy. Screening images were classified without (left) and
with (right) the identification of punctate sub-nuclear structures. Yellow squares indicate
fibroblast nuclei that were erroneously identified as hepatocyte nuclei.
Assay Validation.
Assay readiness for high-throughput screening is most often assessed via
statistical parameters such as z'-factor17 9 , which reflects both assay signal dynamic range
and variation, and is mathematically defined:
Z'
1
(3a-.+ + 3o-)
IL+ - Mc-
where "c+"=positive control, "c-"=negative control, "a"=standard deviation and
"p"=average. Assuming normal distribution, assays with positive z'-factors can separate
48
99.8% of the negative and positive control populations (i.e.,
the two populations, as
defined by mean signal ± 3 standard deviations, do not overlap), essentially separating
signal from noise.
Imaging of multiple 384-well plates containing untreated hepatocyte-fibroblast
co-cultures showed that the image-based readout can confidently (Z'>O) detect doublings
in hepatocyte nuclei numbers with low variance (CV<20%) and good reproducibility. It
should be noted, however, that the highly textured nature of fibroblast nuclei rendered
their segmentation difficult, often leading to the breakup of a single nucleus into multiple
nuclei. Therefore, while the assay does report numbers of fibroblast nuclei as well as
hepatocyte nuclei, it is optimized for accurate detection of hepatocyte nuclei only.
Detection ofMitotic Bodies
In addition to quantifying hepatocyte nuclei in interphase, we also developed two
additional analyses pipelines to quantify the number of nuclei in the process of mitosis.
These pipelines were built to detect nuclear morphologies consistent with cells
undergoing metaphase and anaphase (Figure 2.9). While CellProfilerAnalyst is capable
of simultaneously identifying more than 2 morphologies, its classification accuracy
becomes significantly impaired with each additional category, thus we opted to generate
separate training sets for each morphology of interest (i.e., metaphase nuclei, anaphase
nuclei and hepatocyte nuclei in interphase).
Cells undergoing metaphase have very
distinctive nuclear morphologies and were easily quantifiable using the image assay
outlined earlier in this section. Cells undergoing anaphase unfortunately assumed very
similar morphologies to staining/camera artifacts, with just one distinction: anaphase
49
nuclei always appear in closely positioned pairs. Therefore, we made minor adjustments
to the measurements of these objects to include neighbor relationships.
Figure 2.9 Mitotic nuclei morphology. Left gray square marks a nuclei with
morphology consistent with metaphase; right gray square marks a nuclei with
morphology consistent with anaphase.
Training
of automated nuclei classification was
also altered
slightly to
accommodate the rare nature of these mitotic bodies. Instead of populating the training
set extensively with randomly selected objects, which would result in a severely
imbalanced training set containing thousands of negative examples but only a few
positive items, we focused on iterative error correction.
Validation of mitotic body detection was conducted manually through visual
inspection of raw images due to the lack of a positive control that can induce proliferation
of primary human hepatocytes. In general, program reported counts of mitotic bodies
were in good agreement with manually obtained values.
FunctionalAssays
In addition to the above image-based proliferation assay, we equipped the highthroughput liver platform with several functional assays in order to probe whether hepatocytes in
the platform retain their liver identity. Due to the diverse repertoire of the 500+ documented and
yet unidentified biochemical functions of the liver, there does not exist a single all-inclusive,
gold-standard assay for measuring hepatocyte functions. We thus opted to sample 3 major types
50
of liver functions: 1) ELISA-based quantification of albumin output as a surrogate marker for
protein synthesis functions of the liver, 2) colorimetric assay quantifying urea generation as a
surrogate marker for amino acid metabolism functions of the liver, 3) enzyme activity assay
measuring cytochrome P450 activity as a surrogate marker for detoxification functions of the
liver.
For all 3 assays, we optimized parameters such as reagent type, concentration and volume
to develop them into biochemical assays compatible with high-throughput screening, with Z'>0
and wide dynamic ranges of detection (Figure 2.10). Ultimately, for screening purposes, it is
neither necessary nor practical to implement all 3 assays, thus we chose 1, the ELISA-based
albumin quantification, as the functional assay for the human liver platform.
6
0.7,
Z'=0.2
Z'= O.5
i cv<
10%
P05$
P2
ir
C <%
P4
Ps6
r
P
19
9"
P2
P4
PS
Psht
Z'=0.4
CV<1 6%
2
4
0
8
Figure 2.10. Biochemical functional assays. Bar graph displays albumin secretion (left), urea
production (middle) and cytochrome P450 activity (right) as a function of hepatocyte density in
screening cultures. All data presented as mean ± standard deviation.
The most common form of the ELISA assay is a sandwich ELISA that captures the
antigen of interest in between 2 layers of antibodies. This assay is difficult to adapt to highthroughput screening due to the long protocol, which limits through-put, and the extensive
washes, which are difficult to program robotically.
Therefore, for our liver platform, we
employed a competitive ELISA assay, which reduces the length of workflow by approximately a
third. A saturating amount of human albumin is first coated onto the walls of adsorptive 384well plates.
Sample supernatant is then introduced and competes with coated albumin for
binding to HRP-conjugated antibodies. The amount of bound antibodies is then quantified via a
51
colorimetric substrate. Automation of the ELISA assay necessitated a few adjustments to the
platform. Volume of media used to maintain cultures were increased to 30ul/well in order to
allow withdrawal of 20ul of sample without disturbing the cell layer. An adhesive breathable
membrane was added to the top of culture plates to minimize edge effects arising from fluid
evaporation. The visualization agent was changed from TMB to an ultra-sensitive luminescent
Ultimately, validation data showed that this
substrate to increase sensitivity (Figure 2.11).
biochemical functional assay can confidently (Z'>O) detect doublings in hepatocyte populations
with low variance (CV<10%) and good reproducibility.
4RP-Conjugated
Antibody
0
Albumin
Robotically Coated
Albumin Plate
Automated
Plate
Wash
Iruminogenic
Substrate
Figure 2.11 Schematic of competitive ELISA.
2.3 Conclusions
Predictive high-throughput liver models are a critical tool for research and development
of novel therapeutics as well as for the study of liver biology. Co-cultivation of hepatocytes with
J2-3T3 fibroblasts represents a scale-able platform that can maintain primary human hepatocytes
52
in culture for at least 9 days, providing both time and space for a wide range of cellular activities.
This chapter described a miniature feeder layer co-culture platform for primary human
hepatocytes and attendant assays for probing multiple hepatocyte phenotypes including cellular
proliferation, cell death, protein synthesis functions, detoxification functions and amino acid
metabolism.
2.4 Materials and Methods
HepatocyteMedium Composition
1X DMEM
10% fetal bovine serum (FBS)
15.6 pig/ml insulin
7.5 pg/ml hydrocortisone
16 ng/ml glucagon
1% penicillin-streptomycin
FibroblastMedium Composition
1X DMEM
10% bovine serum (BS)
1% penicillin-streptomycin
Cell Culture Conditions
53
J2-3T3 Culture. Passage 2 J2-3T3 fibroblasts were obtained from Howard Green (Harvard) and
kept in liquid nitrogen until use. Cells were maintained under standard tissue culture conditions,
in DMEM media containing 10% BS and 1% Penicillin-streptomycin. Fibroblasts were grown in
T- 150 tissue culture flasks and passaged 1:10 using 0.25% Trypsin-EDTA when cells reached
confluency. Experiments used J2-3T3s ranging in passage numbers from P9 to P12.
Automated Cell Seeding. Cells suspensions were diluted to the desired densities and kept in
suspension using a magnetic stir bar. Thermo Combi robot was used to dispense cells into 384well formats using speed setting low and standard cassette.
Assay Conditions
Biochemical assays. Urea concentration was quantified using a colorimetric assay that employs
diacetylmonoxime with acid and heat (Stanbio Labs). Albumin content was measured using
ELISA assays (MP Biomedicals) with horseradish peroxidase detection and TMB (Fitzgerald
Industries) substrate.
Cytochrome-P450induction. 7-benzyloxy-4-trifluoromethylcoumarin (BFC, BDGentest) was
added to cultures at 50uM and incubated for 1 hr at 37C in phenol-red free media. Many
different CYP450 isoforms process BFC into its fluorescent product of 7-hydroxy-4trifluoromethylcoumarin (7-HFC), which is then quantified fluorometrically.
Automated Plate Washing. Washing for plates containing cells were done manually to prevent
cell loss. Plate washing for ELISA was performed on the BioTek ELx-405 HT, using the
following optimized settings:
e
Prime: Prime_200 using DI water
Wash: Named program HEPELISA
54
o
Method
-
Number of cycles = 02
-
Wash Format = Plate
-
Soak/Shake
"
Soak Duration = 010 sec
-
Shake before soak = yes
=
Yes
" Shake Duration = 005 sec
*
Shake Intensity = 4 (18 cycles/sec)
-
Prime after soak = No
o Disp
-
Dispense volume = 100 t/well
-
Dispense flow rate = 05
" Dispense height = 120 (15.240 mm)
" Horizontal X disp pos = 25 (1.143mm)
" Horizontal Y disp pos = 20 (0.914mm)
o
=
Bottom wash Ist = no
"
Prime before start = no
Aspir
* Aspiration Height = 020 (2.540 mm)
" Horizontal X Aspiration Position = 00
" Horizontal Y Aspiration Position = 00
-
Asp rate = 05 (6.4 mm/sec)
"
Asp delay = 0000 msec
55
=
Cross-wise aspir = yes
-
Cross-wise on = all
=
Cross-wise height = 020 (2.540mm)
=
Cross-wise X horiz. Pos = 00
-
Cross-wise Y horiz. Pos = 00
"
Final asp = Yes
"
Final asp. Delay = 0000 msec
Automated Plate Reading. PerkinElmer EnVision 2102 Multilabel Reader was used to quantify
ELISA signal. Program named ShanMeghan Chemillum and contains integration duration of 0.1
sec, luminescence mirror, luminescence 700 emission filter and measurement height of 6.5mm.
Note: High-throughput screening protocols are provided in detail in Chapter 3.
56
Chapter 3. High-throughput Identification of Small Molecules for Inducing in
vitro Proliferation and Function in Primary Human Hepatocytes
3.1 Introduction
The repertoire of liver functions exhibited by primary human hepatocytes is the most
faithful to in vivo human physiology of all available cell sources. Therefore, primary human
hepatocytes are the ideal cell type for in vitro liver models and liver cell-based therapies.
However, development of these platforms and therapies has been hindered by the lack of a
renewable source of functional human hepatocytes.
Currently, human hepatocytes allocated for research purposes are derived from an
extremely limited source of human livers deemed unsuitable for transplantation. Most of these
livers suffer from liver diseases such as HCV infection and hepatocellular carcinoma and thus
exhibit abnormal functions. As a result, alternative sources for hepatic cells are being
investigated. Various stem cell populations, for example, are capable of virtually unlimited selfrenewal in vitro and exhibit either pluripotency or multipotency, thereby representing a possible
source of hepatocytes as well as other liver cell types',1-82
While such cell populations offer
powerful possibilities for personalized medicine, their application to the clinics is premature.
There are entire axes of epigenetic modifications that the field has yet to explore; even the
cellular reprogramming mechanisms that are being widely employed are not fully understood. In
fact, it is not yet possible to completely dictate differentiation, particularly for in vitro
applications. Therefore, substantial efforts continue to be undertaken to maximize the usage of
human hepatocyte isolations.
57
Notably, despite their rapid and extensive innate expansion capabilities in vivo, mature
hepatocyte proliferation in culture is insignificant, particularly for human hepatocytes
'.
In
response, researchers have created genetically-altered mouse strains to serve as an in vivo
incubator for the expansion of primary human hepatocytes17' 96-98 . Their findings suggest that
isolated human hepatocytes, both fresh and cryopreserved preparations, retain their capacity for
considerable proliferation
and will self-renew ex vivo if given the appropriate stimuli.
Elucidation of these stimuli and their application towards the development of in vitro expansion
platforms would dramatically enhance the utility of human hepatocytes, generating new
opportunities for a wide spectrum of liver-related fields including drug discovery and
Unfortunately, traditional gene-by-gene and
development and hepatic tissue engineering.
protein-by-protein approaches attempted thus far have not been able to harness in vitro the
tremendous replication potential of human hepatocytes. We hypothesize that a major cause of
this is the declining liver phenotype in conventional culture systems, which lack critical stromal
support.
Co-cultivation of primary hepatocytes with stromal cells has resulted in modest
proliferation of rodent hepatocytes. Michalopoulous and colleagues have further reported a list of
mitogenic factors 17,96-98,158 capable of inducing transient expansion of rat primary hepatocytes
through a de-differentiated, bipotential intermediate. However, translation of these findings to
human cultures has not been straightforward.
In this chapter, we report the utilization of a large-scale small molecule screening
approach (12,480 molecules) to search for factors that can induce human hepatocyte proliferation
and function within the context of a stromal microenvironment (Figure 3.1).
We hypothesize
that the presence of supportive stromal cells will, in addition to functional stabilization, maintain
58
or promote the responsiveness of human hepatocytes to molecular stimuli responsible for
significant replication in vitro. Ultimately, we hope our findings will help assemble a more
complete understanding
of the mechanisms underlying hepatocyte replication and liver
regeneration.
A
High-throughput Platform
and Assay Development
B
Primary Screening of
12,480 Small Molecules
Co
I
4tCYtes
ats
Fib
7 days&
C
tf
Hit Validation
Proliferation Assay Functional Assay:
ELISA
Imaging
(Hep. Nuclei Coun 0)(Albumin Content)
Retest 400 compounds in 8-point dose
ELISA counter assay
2 donors of primary human hepatocytes
Primary Screening
12,48 d
compounds
Donor
+
Smal
Moecle
L
Retest
12
compounds
D
Functional
>roliferation
FPH1
FPH2
FH1
Jt
ir .
1.
FPHs
Expand Primary Human Hepatocytes
2.
FH
Induce Maturation of iHeps
Aaft
Sam
Figure 3.1 Overview of High-throughput Screening. A, Primary human hepatocytes (green)
were seeded on a feeder layer of confluent J2-3T3 fibroblasts (red) in 384-well plates. B, Cells
were seeded onto a collagen matrix, cultured for 7 days, and treated with small molecules for 48
hrs before analyses through image-based proliferation assay and competitive-ELISA-based
functional assay. C, hit validation. D, classes of confirmed hits. Two classes of hits were selected
59
for further characterization. Functional proliferation hits were examined for their ability to
expand mature human hepatocytes. Functional hits were explored as inducers of iHeps
maturation.
3.2 Results and Discussions
Chemical Library
Using the high-throughput liver platform, and its attendant proliferation and functional
readouts, we screened 12,480 small molecules, supplied by the Broad Institute. The collection
comprised
960
chromatin-biased
compounds
(CHRM),
3,520
commercially
available
compounds (ComA), 2,560 products of diversity-oriented synthesis (DOS), 3,200 kinase-biased
compounds (KinA), 1,920 known bioactives (BioA) and 320 natural products (NatP, figure 3.2).
Selection of this library was based on factors including compound purity and quality, historical
hit rates and ease of re-synthesis of potential small molecule hits. Other considerations included
the nature of this work as a phenotypic whole-cell screen, instead of a target-driven protein
screen.
Sceene
400-
Hit
350300250-
o 200-
E
o
o
150
100-1
E50-
R
II
4i
CHRM
ComA
DOS
II
I
nII
KinA
BboA
NatP
Type of Compound
Figure 3.2 Chemical Library Composition. Categories of screened (white) and primary
screening hit (black) compounds.
60
Within this library are proprietary compounds designed and synthesized at the Broad
Institute. Most of these novel chemicals are aggregated in the DOS collection, comprising ~20%
of the 12,480 small molecules tested. While traditional screening libraries typically consist of
structurally similar compounds all designed to interact with a specific target protein, DOS
collections are assembled to be structurally diverse
185
. In fact, their complexity rival that of
natural products, which is desirable because many small molecules known to affect biological
processes are structurally complex. Structural diversity is also desirable for this particular screen
because we are looking for a novel in vitro cell fate, thus every macromolecule in the cellular
machinery is a potential target. Having a library that occupies a large volume in the abstract
space whose dimensions represent different physical and chemical properties maximizes the
likelihood of finding active chemicals exhibiting the perfect combination of characteristics
necessary for eliciting the desired biological response.
PrimaryScreening and Hit Selection
The workflow of primary screening is summarized in figure 3.1 B.
In order to avoid
donor-to-donor variability, human primary hepatocytes from a single donor (donor a) were preconditioned for seven days by J2-3T3 fibroblasts in collagen-coated 384-well plates. During cell
seeding, the stock solution was kept under constant agitation through magnetic stirring in order
to prevent gravity-mediated cell aggregation.
Upon hepatocyte stabilization in vitro, cultures
were treated with individual molecules from the chemical library for 48 hours, during which time
the sample plates were kept in metal stacks with uniform air buffers between each plate in order
to provide uniform gas and heat exchange. Additionally, breathable-membranes and extra water
reservoirs were employed to minimize edge effects arising from fluid evaporation.
61
All compounds were tested in duplicate, at a single concentration of -1 5uM, in weekly
batches of -3,000 compounds each. This single concentration was selected via a pilot screen of
~2,000 small molecules, in which ~15 RM yielded a toxicity rate typical for most primary
screens conducted with the Broad chemical library.
Following compound treatment, media
supernatants were collected for functional analyses via competitive ELISA and cultures were
fixed in 4% paraformaldehyde for proliferation analyses via imaging (Tables 3.1 and 3.2).
Category
Parameter
Description
Assay
Type of assay
High content imaging
Target
Primary human hepatocytes
Primary measurement
Hepatocyte nuclei count
Key reagents
Primary human hepatocyte co-culture system
Assay protocol
Primary human hepatocytes were seeded onto a
confluent monolayer of fibroblasts in 384-well clear
bottom plates at 2,000 hepatocytes/well. 7 days
later, compounds were added and incubated for 48
days. Supematants were then removed for albumin
ELISA and cultures were fixed and stained with
Hoechst stain. Images were collected on an IX
Micro microscope and were analyzed using
CellProfiler.
Additional comments
None
Library size
12,480
Library composition
Small molecules
Source
Broad Institute
Additional comments
None
Format
384-well high content imaging
Concentration(s) tested
8-point dose curve at 2x dilutions, starting -15pM
Plate controls
No treatment, DMSO
Reagent/ compound dispensing system
Cybio pin tool, Thermo Combi, Cybio Vario
Detection instrument and software
IX Micro, Cell Profiler
Assay validation/QC
CV < 20% with DMSO treatment
Correction factors
None
Normalization
DMSO control
Additional comments
None
Hit criteria
Z 3
Library
Screen
Post-HTS analysis
62
Hit rate
0.07%
Additional assay(s)
Phase contrast imaging, albumin staining, Ki67
staining, cytometer counting, FACS counting, gene
expression analysis, MRP2 activity assay, urea
assay, CYP450 activity assay, CYP3A staining, AFP
staining.
Confirmation of hit purity and structure
Hits retested and then purchased as dry powders
Additional comments
None
Table 3.1. Small molecule screening data from high content co-culture imaging assay
Category
Parameter
Description
Assay
Type of assay
ELISA
Target
Albumin secreted from primary human hepatocytes
Primary measurement
Secreted albumin
Key reagents
Primary human hepatocyte co-culture system
Assay protocol
Primary human hepatocytes were seeded onto a
confluent monolayer of fibroblasts in 384-well clear
bottom plates at 2,000 hepatocytes/well. 7 days
later, compounds were added and incubated for 48
days. Supematants were transferred into ELISA
plates for albumin detection. Plates were read out
on a Perkin Elmer Envision and ELISA data was
analyzed using GeneData.
Additional comments
None
Library size
12,480
Library composition
Small molecules
Source
Broad Institute
Additional comments
None
Format
384-well ELISA assay
Concentration(s) tested
8-point dose curve at 2x dilutions, starting -15pM
Plate controls
No treatment, DMSO
Reagent/ compound dispensing system
Cybio pin tool, Thermo Combi, Cybio Vario
Detection instrument and software
Perkin Elmer Envision, GeneData
Assay validation/QC
ELISA spiked with purified albumin
Correction factors
None
Normalization
DMSO control
Additional comments
None
Hit criteria
IZ| 4
Library
Screen
Post-HTS analysis
63
Hit rate
0.06%
Additional assay(s)
Phase contrast imaging, albumin staining, Ki67
staining, cytometer counting, FACS counting, gene
expression analysis, MRP2 activity assay, urea
assay, CYP450 activity assay, CYP3A staining, AFP
staining.
Confirmation of hit purity and structure
Hits retested and then purchased as dry powders
Additional comments
None
Table 3.2. Small molecule screening data from co-culture albumin secretion ELISA assay
To identify proliferation hits, we integrated the three image-based readouts (the number
of nuclei in metaphase, in anaphase, and hepatocyte nuclei in interphase).
z scores were
converted into p values in order to have statistical measurements that correlated linearly with the
extent of observed effect. p values were then used to generate ranked lists of compounds based
on the efficacy and consistency of effects across the different proliferation readouts. Efficacy is
assessed by the product of p values
(pprod = Pinter X Pmeta X Pana);
consistency is evaluated by the
maximum of p values (pmax = max (pinter, Pmeta, Pana). Compounds were considered proliferation
hits if Pprod < IX 10-6 and Pmax < 0.25.
Functional hits were separately identified based on the
ELISA readout. Functional hits were selected by an ELISA p<-0.05. Compounds with ELISA
z>3.0 were eliminated as toxic (figure 3.3).
64
E...
200-
..
...
~200tE
I
I
b.
N
N
M
-100
-100
-100
0
100
200
fj~
-100
z Normalized HepCount 1
0
100
200
Z Normalized [Albumin] 1
Figure 3.3 Primary screening results. Scatterplots display replicates of the screen of 12,480
small molecules, shown separately for the image-based proliferation and competitive-ELISA
functional readouts. Blue and red data points represent DMSO and experimental small molecules
respectively. Boxed regions indicate hit zones.
93 compounds met all hit selection criteria, qualifying as functional proliferation hits
(FPH); figure x shows the types of compounds that constituted this set of hits. In addition to
these 93 FPHs, we also identified proliferation-only (PH) and function-only (FH) hits.
Secondary Validation and Hit Prioritization
A total of 400 primary hits from the proliferation readout and/or functional readout were
retested in eight-point dose-response curves using a different donor of cryopreserved hepatocytes
(donor b) for biological diversity (Fig. 3.1 Panel C). The two donors differ greatly in age (1 year
old for donor a vs 54 years old for donor b). Remaining hits were further tested using a cell-free
ELISA assay to eliminate compounds that interfered with the ELISA process chemically, instead
of eliciting real biological responses from the hepatocytes.
confirmed hits (Figure 3.4).
65
Ultimately, we obtained 12
i
A
B
FPH3
40
FH2
40
0
U
z
VIOU
2]
C:
-
FH3
E0
FPH4
E
-1:0
D
-25-
-25
log Concer tration [Ml
Iog Concentration [Ml
C
D
PH2
PH1
PGE2
1.50-
2
20
E
0) =
C
:3
0
U
'0
~0
2
1.00-
LL
PH4
PH3
z
0r
C
-C
2
-20
1.25-
20-
0.75-
4-.
U
Low
High
1.75-
.2H
C
0
Cti
Concentration
4-,
1.50.
PH-5
300
1.25-
z
.c 1.00-
CL)
4-
oL'-0.75
log toncentration [M]
Ctri
Low
High
Concentration
Figure 3.4 Dose curves of confirmed hits and PGE2. Proliferation hits have increasing curves
of hepatocyte and/or metaphase nuclei count. Functional hits have decreasing curves of
competitive [Albumin] or increasing curves of fold change in [Albumin]. Control cultures were
treated with empty vehicle (DMSO). PGE2 was tested as a putative positive control since it was
previously reported by Goessling and colleagues to promote liver regeneration in zebrafish.
66
We divided these confirmed hits into three separate classes of compounds: proliferation
hits, functional hits and functional proliferation hits (Table 3.3). Each of the three classes may
enable a distinct approach
towards generating renewable
sources of functional human
hepatocytes, and may synergize when used in combination. For in vitro applications, we focused
on candidates within the functional proliferation hit (FPH) and functional hit (FH) categories
only (Figure 3.1). FPHs induced functional proliferation of hepatocytes in vitro (e.g., FPH1,
FPH2), and thus may be useful for expanding mature human primary hepatocytes. FHs enhance
the functions of cultured hepatocytes (FH1), prompting us to hypothesize that these molecules
may promote the differentiation of iPS-derived hepatocytes toward a phenotype more mature
than what has been obtainable to date.
Confirmed Hits
Functional Proliferation Hit (FPH)
4
Proliferation Hit (PH)
5
Functional Hit (FH)
3
Table 3.3 Classification of 12 Confirmed Hits. Functional proliferation hits (FPHs) were
selected as hits by both the image-based proliferation assay and the ELISA-based functional
assay. Proliferation hits (PHs) enhanced hepatocyte proliferation only during primary screening.
Functional hits (FHs) enhanced hepatocyte functions only.
We prioritized follow-up characterization of 3 particular hits: FPHI, FPH2 and FHl.
FPH1 and FHl were selected for their strong activities during primary and secondary screening
as well as their ease of synthesis. FPH2 was selected because it is the only hit shared with a
separate screen examining Hematopoietic Stem Cell homeostasis. HSC homeostasis and liver
regeneration both involve Wnt signaling pathways. This intersection had previously led to the
67
identification of PGE2 by Goessling and colleagues as an in vivo enhancer of liver regeneration
in zebrafish; thus it is possible that FPH1 also acts through a mechanism involving Wnt signaling.
Chemical Characterizationand Structure-Activity Relationships
1
We characterized top hits through Liquid Chromatography / Mass Spectrometry, H-
NMR and
13 C-NMR
in order confirm compound purity and identity (Supplementary Note 3.1).
In an effort to determine if any structure-activity relationships (SAR) were present for the
three strongest hits (FH1, FPH1, and FPH2), we searched the remaining 12,477 compounds
screened for analogs (similarity score > 0.8). Using this criterion, FHI and FPH2 were singletons,
while 21 analogs of FPH1 were found. These analogs all contained the same N-phenyl-2-(Nphenylmethylsulfonamido) acetamide core as FPH1, but had varying substitutions around the
sulfonamide and amide phenyl rings (Figure 3.).
The key driver for compound activity in the
FPH1 series was the presence of a 5-chloro-2-methyl substitution on the sulfonamido phenyl ring
and a small functional group at the para position of the phenylamide ring. This is depicted in
Supplementary Figure 4, where, FPH1 and compound 5 were proliferation hits, compound 6 a
weak hit, and compounds 7 and 8 were inactive. Compounds 9 and 10 illustrate that low steric
bulk at the para position of the phenylamide ring alone is not sufficient to drive compound
activity of the FPH1 series.
68
CI
F
0
N
N
90
S
H
N
H
4
FPH9
DI
CI
CI
CI
F
F
0
O ,
N
H
16
5
N
0
j
N
H
HH
0
F
6
N
N
0
H
8
7
0
CI
CI
0F
0N
S
N)
H
0
CI:O
9'
F
9
N
AN-_-N
0
10
F
0
F
Figure 3.5 Structure Activity Relationship of FPH1. Structure of the functional proliferation
hit FPHI and a series of analogs (Compounds 4-10) depicting the structure-activity relationships
of this series of compounds. Both a 5-chloro-2-methyl substituted sulfonamido phenyl ring and
a phenylamide ring without significant steric bulk at the para position are required for compound
activity.
3.3 Conclusions
Utilizing the high-throughput liver platform described in chapter 2, we screened 12,480
small molecules using primary human hepatocytes and identified 12 confirmed hits in 3 classes:
functional proliferation hits, functional hits and proliferation hits. Functional proliferation hits
enhanced both hepatocyte nuclei numbers and albumin output during primary screening and will
be examined in subsequent chapters as candidate inducers of primary human hepatocyte
expansion in vitro. Similarly, functional hits, which enhanced the albumin output of culture
69
hepatocytes, will be studied as potential inducers of hepatocyte differentiation. Both will be
explored as strategies for generating a renewable source of functional human hepatocytes.
3.4 Materials and Methods
Hepatocyte Medium Composition
IX DMEM
10% fetal bovine serum (FBS)
15.6 ptg/ml insulin
7.5 ptg/ml hydrocortisone
16 ng/ml glucagon
1% penicillin-streptomycin
FibroblastMedium Composition
IX DMEM
10% bovine serum (BS)
1% penicillin-streptomycin
Cell Culture Conditions
J2-3T3 Culture. Passage 2 J2-3T3 fibroblasts were obtained from Howard Green (Harvard) and
kept in liquid nitrogen until use. Cells were maintained under standard tissue culture conditions,
in DMEM media containing 10% BS and 1% Penicillin-streptomycin. Fibroblasts were grown in
70
T-1 50 tissue culture flasks and passaged 1:10 using 0.25% Trypsin-EDTA when cells reached
confluency. Experiments used J2-3T3s ranging in passage numbers from P9 to P12.
High-throughputscreen. 384-well screening plates (Coming) were incubated with a solution of
type-I collagen in water (100 mg/ml, BD Biosciences) for 1 h at 37 0 C. A feeder layer of J2-3T3
fibroblasts (gift from Howard Green) were robotically plated onto the collagen at a density of
8,000 cells/well (designated as day -2), and allowed to reach confluency over 48 hours, when
their growth became contact inhibited.
Primary human hepatocytes were plated onto the
fibroblasts on day 0 at a density of 2,000 cells/well and maintained under standard culture
conditions with daily replacement of hepatocyte medium for 7 days. Primary human hepatocytes
were purchased in cryopreserved suspension from Celsis In vitro Technologies (donor a) and
Invitrogen (donor b), and pelleted by centrifugation at 50g for 10 min. The supernatant was
discarded before re-suspension of cells in hepatocyte culture medium.
A library of 12,480
compounds was added on day 7 at a final concentration of -15pM, and allowed to incubate for
48 hours. On day 9, culture supernatants were collected for automated ELISA analysis, and cells
were fixed in 4% PFA for imaging analysis.
competitive
ELISA
(MP
Biomedicals)
using
Hepatocyte functions were determined via
horseradish
peroxidase
detection
and
chemiluminescent luminol (Pierce) as a substrate. The cell-free counter assay involved ELISAs
on fresh media incubated for 48 hrs with compounds of interest. Hepatocyte proliferation was
assessed through customized, automated high-content imaging protocol.
Fixed cells were
permeabilized with 0.1% Triton-X, nuclei visualized with Hoechst stain (invitrogen) and
robotically imaged (Thenno, Molecular Devices) at 13 dispersed sites per well.
Images were
digitized and analyzed using CellProfiler and CellProfiler Analyst (Broad Institute). Details of
robotic protocols are provided below.
71
Screening Protocols
Cell Culture:
1.
Coat tissue culture plates with rat Collagen I:
a. Load Collagen I solution:
i.
Volume: 20ul/well
ii. Concentration: 100pg/mL
iii. Equipment: Thermo Combi, using standard cassette on high speed
b. Wash plates:
i.
Washing solution: DI water
ii. Volume: 30pl/well
iii. Cycle: 2X
iv. Equipment: Thermo Combi, using standard cassette on high speed
v. Manually bang out liquids for each wash
c.
Air dry plates and sterilize with UV for 20 minutes
2. Seed mouse embryonic fibroblasts (J2-3T3):
a. Harvest fibroblasts:
i. Enzyme: 0.25% Trypsin
ii. Volume: 8mL / T150 flask
iii. Duration: 5min
iv. Neutralize with serum media and spin down at 1000 rpm for 5min
v. Resuspend in serum media pre-warmed to 37'C
vi. Add sterile stir bar
72
b. Load fibroblasts:
i.
Keep cells in suspension and heated
ii. Volume: 10pl/well
iii. Concentration: 80,000 cells/mL
iv. Equipment: Thermo combi, using standard cassette on high speed
3.
Add 40ul of serum media to fibroblasts 24 hrs after
a. Equipment: Thermo combi, using standard cassette on low speed
4. Seed cryopreserved primary human hepatocytes:
a. Thaw cells:
i.
Place vial in water bath for 1.5 min
ii. Pipet vial contents to 50 mL centrifuge tube
iii. Slowly add 50mL of serum media pre-warmed to 37'C
iv. Spin down at 800rpm for 5 min
v. Resuspend in serum media pre-warmed to 37'C
vi.
Add sterile stir bar
b. Load hepatocytes:
i. Keep cells in suspension and heated
ii. Volume: 30pi/well
iii. Concentration: 66,667 cells/mL
iv. Equipment: Thermo combi, using standard cassette on low speed
5.
Maintain hepatocytes daily:
a.
Manually dump out old media
b. Add Media:
73
i.
Volume: 30pi
ii. Equipment: Thermo combi, using standard cassette on low speed
c.
After compound pinning, seal plates with breathable membrane (Nunc 249720) to
prevent evaporation and subsequently edge effects.
ELISA:
1.
Coat white MaxiSorp plates (Cat#: NUNC 460372) with human albumin:
-
Load human albumin (MP biomedical cat# 55912) solution:
i. Volume: 40pl/mL
ii. Concentration: 50ug/mL in coating buffer
iii. Equipment: Thermo Combi, using standard cassette on high speed
*
Seal plates and Spin down at 1000rpm
*
Re-seal plates and Incubate:
i.
Temperature: r.t.
ii. Duration: overnight
iii. Equipment: Thermo plate mixer set to level 6
2. Wash albumin-coated plates:
3.
e
Wash Buffer: 0.1% PBST
-
Equipment: BioTek ELX405 (96 pins)
-
Program: Prime_200 for priming, HEPEELISA for washing
*
Manually bang plates on paper towel after robotic washing
Load samples:
-
Volume: 20ptl/well
e
Equipment: Cy-Bi Well Vario, using 384 head and sterile tips
74
-
Program: Meghan's fixing with PBS - PJ
4. Load HRP-conjugated antibodies (MPBio Cat #55235)
5.
-
Volume: I Opl/well
-
Concentration: 0.125 ptg/mL of 0.1% PBST
-
Equipment: Combi, using standard cassette on low speed
Seal plates and spin down at 1000 rpm
6. Re-seal plates and Incubate:
a.
Temperature: r.t.
b.
Duration: 3hrs
c.
Equipment: Thermo plate mixer set to level 6
7. Wash plates
a. Wash Buffer: 0.1% PBST
b. Equipment: BioTek ELX405 (96 pins)
c. Program: Prime_200 for priming, HEPEELISA for washing
d. Manually bang plates on paper towel after robotic washing
8.
Load developer
a. Volume: 20pl/well
b. Equipment: Combi, using standard cassette on high speed
9. Seal plates and spin down at 1000rpm
10. Read plates
a. Equipment: Perkin Elmer Envision 2102 Multilabel Reader
b. Program: ShanMeghan Chemilum
Imaging:
75
1.
Wash cell culture plates
e
Wash Buffer: IX PBS
e
Volume: 25pL/well
* Cycle: 2X
-
Equipment: Cy-Bi Well Vario, using 384 head and sterile tips
-
Program: Meghan's Fixing with PBS - PJ
2. Fix cells with PFA
*
Volume: 25pLI/well
e
Concentration: 4%
-
Duration: 15min
"
Equipment: Cy-Bi Well Vario, using 384 head and sterile tips
* Program: Meghan's Fixing with PBS - PJ
3.
Wash cell culture plates
a. Wash Buffer: IX PBS
b. Equipment: BioTek ELX405 (96 pins)
c. Program: MAGSPRIME for priming, MEG for washing
4. Stain cells with Hoechst
5.
*
Manually dump out IX PBS
-
Volume: 30pl/well
*
Concentration: 0.001% in DI water
e
Duration: 10min
e
Equipment: Combi, using standard cassette on low speed
Wash cell culture plates
76
a. Manually dump out staining solution
*
Washing solution: DI water
-
Volume: 50 pl/well
-
Cycle: IX
-
Equipment: Combi, using standard cassette on low speed
6. Immerse samples in 1XPBS
a. Manually dump out washing solution
b. Volume: 50 pl/well
c. Equipment: Combi, using standard cassette on low speed
7.
Seal plates and store covered with foil at 4C.
Supplementary Note 3.1. FPHJ, FPH2 andFHJ chemical characterization.
Lead Compounds
FPHI (BRD-6125) was purchased as a dry powder from ChemBridge Corporation.
FPH2 (BRD-9424) was purchased as a dry powder from Molport.
FHI (BRD-K4477) was purchased as a dry powder from Molport.
Characterizationof Lead Compounds
Compound 1 (BRD-6125)
F
1
77
LCMS: Expected [M+H]+ 389.0533, Determined [M+H]+ 389.0534; HPLC: 98% (@ 210nm)
(Rt;0.66); 'H NMR (300 MHz, DMSO-d 6 ) 6 9.93 (s, 1H), 7.61 (s, 1H), 7.36 (m, 3H), 7.16 (t, J=
8.2 Hz, 2H), 3.34 (s, 2H), 3.21 (s, 3H), 2.33 (s, 3H);
13C
NMR (75 MHz, DMSO-d 6 ) 6 167.15,
159.13, 139.97, 138.04, 132.50, 130.28, 129.30, 128.62, 128.29, 113.66, 112.04, 111.73, 52.95,
17.50.
LCMS for Compound 1(BRD-6125):
3
DW Detector: 210
Ra.r0
3*9.52
387.39
1. OUG-2
0..
0.30
0.40
0.50
0 4*
0.70
78
0.10
&.90
1.00
1.10
1.10
2. 312*-l
0 4flh-
1H
NMR for Compound 1(BRD-6125):
I
i
p
(I
.1; 1
_
,0
_W-WI
11.5
11.0
10.5
10.0
IX95
9.0
8.5
8.0
7.5
7.0
6.5
.0
fi (PPM')
79
5.5
5.0
45
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0
13C
NMR for Compound 1(BRD-6125):
W
1!
Ii
I
I.
to0
200
1;0
180
' 170 '160
A-
Ii
'150
140
13
±10
'2 110
fl (Ppm)
90
80
70
4
'05030
20
10
Compound 2 (BRD-9424)
Os
Cl
CI
N
N
H
2
N
~
N
H
H2N
N
N
0
LCMS: Expected [M+H]+ 354.0786, Determined [M+H]+ 354.0793; HPLC: 94% (@ 210nm)
(R,;0.60); 'H NMR (300 MHz, DMSO-d 6 ) 8 10.32 (s, 1H), 9.99 (s, 1H), 8.85 (s, 1H), 7.63 (s,
1H), 7.46 (s, 2H), 7.31 (dd, J= 8.8, 2.4 Hz, 1H), 7.12 (d, J= 8.9 Hz, 1H), 4.17 (q, J= 7.2 Hz,
2H), 3.77 (s, 3H), 3.34 (s, 2H), 1.37 (t, J= 7.2 Hz, 3H); "C NMR (75 MHz, DMSO-d 6 ) 8 177.14,
80
165.26, 152.74, 132.23, 127.58, 127.29, 127.11, 124.73, 123.52, 121.47, 113.76, 55.86, 47.10,
15.26.
LCMS for Compound 2 (BRD-9424):
3
vv Datoctor:
2
Ranga
1it 42
352.40
0,40
320.54
31829
0.4
-1.0,-i
0.30
0.40
G.50
0.60
0.70
81
0.80
0.90
1 00
1,10
1,20
5.94.-1
t.004a-i
1
H NMR for Compound 2 (BRD-9424):
iR
9 a
I
L~4~
I,
f)
I-
j
Its
itO
1O~S
iO~O
9.5
F
I
I
~Qy~JK
___
~.O
J
(
910
8.5
8.0
7
7.0
6.5 6.0 5-5
fi (ppm)
82
S
4.3
4.0
;.5 '.0
2.5
2.0
.5
10
0.
0.
13
C NMR for Compound 2 (BRD-9424):
~:g S
WX
1
, a
io0
20'
1;0 '1;v0
I'
1
I-
170 '160
1110a
--
' LW
~dL I
WMINS,
''
140
130
0
0
120
10
~
I
oil
I
f1 (PPM)
~iz~
Ii ~
i P
WON
100
I10
I
90
s0
70
60
s0
40
30
20
10
Compound 3 (BRD-K4477)
H
N
H
N
3
LCMS: Expected [M+H]+ 283.1441, Determined [M+H]+ 283.1440; HPLC: 99% (@ 210nm)
(RI;0.50); 1H NMR (300 MHz, DMSO-d 6) 8 9.86 (s, 2H), 7.46 (d, J = 8.5, 4H), 7.10 (d, J = 8.4,
4H), 3.80 (s, 2H), 2.01 (s, 6H); "C NMR (75 MHz, DMSO-d6 ) 6 168.01, 137.26, 135.98, 128.73,
119.10, 39.92, 23.88.
83
LCMS for Compound 3 (BRD-K4477):
I, W atsctor 210
n94
28 357
281 49
0 50
a
o
6,
4 0-Ia-IA
2.0.-i
0. &
0.40
0.50
0.60
0.70
84
0.80
0.90
L.*
1.10
120
'H NMR for Compound 3 (BRD-K4477):
7
BV
BREjK44777625_001_01_9
(
ii
I,
---
A
10.5
10.0
t
JLI
T I rl
TA
.0
jI.
9.5
9;0
8.5
8.0
7.5
7.0
65
6.0
5.5
fl (ppm)
85
5.0
4.5
4.0
3.5
T
3.0
2.5
2.0
1.5
1.0
0.5
"C NMR for Compound 3 (BRD-K4477):
U
7
77
i
i
J0
190
160
170
164
150
140
13)
120
110
10)
fi (Ppm)
86
90
80
70
60
50
4)
30
20
10
Chapter 4.
Small Molecules for Inducing Hepatocyte Expansion and iPS
Differentiation in vitro
4.1 Introduction
Chronic liver disease affects more than 500 million people worldwide'86 .
Most
treatments are palliative; the only therapy shown to directly alter outcome and prevent mortality
87
is organ transplantation, but its utility is limited by a persistent shortage of donor organs' . Cell-
based therapies, such as cell transplantation, engineered hepatocellular tissue constructs and
bioartificial liver devices, have long held promise as alternatives to whole organ transplantation'.
Such therapies require human hepatocytes due to substantial species-specific differences between
animal and human hepatocellular functions including apolipoprotein expression, metabolic
6 87
regulation of cholesterol, and phase I detoxification enzymes ' .
Human hepatocytes, within the stromal context of the liver in vivo, are capable of
extensive expansion.
Following 2/3 partial hepatectomy (PHx), the residual mature cell
populations comprised mainly of hepatocytes are able to proliferate and replace lost liver mass.
In vitro, hepatocytes, particularly human ones, lose this phenomenal proliferative ability'.
Various attempts have been made in the last several decades to harness the innate replication
potential of hepatocytes ex vivo.
Investigations have yielded a number of different culture
conditions that can support moderate expansion of rodent hepatocytes76,78',79, '82,84, including a
multi-factor media formulation that expands rat hepatocytes through a dedifferentiated bipotential intermediate7 6 . However, translation of these findings to human cultures has not been
reported.
To overcome the growth limitations of primary human cells, human hepatocyte cell
lines have been developed, derived from tumor cells or generated through introduction of the
87
SV40 immortalizing gene'188.
Although these cell lines are growth-competent, their use is
limited by safety concerns and their abnormal levels and repertoire of hepatic functions,
including notable divergences in cytochrome P450 activity9 2, 88 .
Human stem cells are an attractive alternative cell source to primary human hepatocytes
and immortalized cell lines, holding great promise due to their ability to self-renew without limit
and to differentiate along many lineages, including hepatocytes. Induced pluripotent stem cells
(iPS cells) additionally create the possibility of establishing patient-specific cell types, thus
facilitating in vitro modeling of rare diseases, and enabling personalized medicine. Human iPS
cells are generated from somatic cells via forced expression of reprogramming factors, and can
be differentiated towards hepatocyte-like cells (iHeps) in a step-wise manner, using defined
However, the resulting iHeps exhibit an immature hepatic phenotype, which
factors""3.
resembles fetal hepatocytes more than adult hepatocytes.
Notably, iHeps persistently express
fetal markers like alpha fetoprotein (AFP) and lack key mature hepatocyte functions, as reflected
by drastically reduced activity (0.1%) of many detoxification enzymes (e.g.,
CYP3A4)
1 3
-
CYP2A6,
. These key differences between iHeps and adult hepatocytes have limited the use
of stem cells as a renewable source of functional human hepatocytes, especially for in vitro
applications.
For decades, human hepatocyte sourcing has been a bottleneck for many fields of
research and clinical therapies.
To overcome this limitation, we utilized a small molecule
screening approach and identified factors that can either induce proliferation of mature primary
human hepatocytes or induce maturation of human iPS-derived hepatocyte-like cells.
4.2 Results and Discussions
88
Expansion of humanprimary hepatocytes
We assayed the ability of our prioritized FPHs from chapter 3 to expand human primary
hepatocytes in vitro. As a first step, we tested Prostaglandin E2 (PGE2) as a putative positive
control as it has been shown to promote liver regeneration in zebrafish and mouse via wnt
signaling..'"...
We tested PGE2 on human primary hepatocytes in our high-throughput liver
platform and found it to indeed perform as a FPH in this system. Two other strong FPHs (FPHl
and FPH2), identified through unbiased screening, were also tested. Both induced a 1.5-fold
increase in hepatocyte nuclei count during primary screening (Figure 4.1 left), elevated the
number of nuclei undergoing mitosis (Figure 4.1 middle), and these effects on hepatocytes were
concentration dependent (Figure 4.1 right). Cells treated with these FPHs also maintained their
liver-specific functions.
e 250FH
-E
ZP~~toeo~ hi
Rltp4~
P
FP
PH2. RI FPC42 R
Figure 4.1 Primary screening data for FPH1 and FPH2. Data presented as mean
deviation.
standard
To further characterize the effects of FPH1 and FPH2, we stained for proliferation marker
Ki67, counted hepatocyte cell numbers and evaluated the morphology, gene expression and
functions of treated hepatocytes. Human primary hepatocytes were cultured in standard 12-well
tissue culture plates on top of a feeder layer of growth-arrested J2-3T3 fibroblasts. A single FPH
was supplemented into the culture media on days 1 and 5 at a concentration of 20 [tM for FPH1
and 40uM for FPH2.
Treated hepatocyte colonies increased in area over time, with more
hepatocytes populating each colony (Figure 4.2).
89
Figure 4.2 Morphology and colony size of FPH-treated human hepatocytes. Cells were
cultured in 12-well plates over time. Untreated (day 1) hepatocytes are shown for comparison,
far left (scale bar = I OOjm).
Immunofluorescent staining showed that FPH treatment increased Ki67 staining, which
co-localized with Hoechst stains for cell nuclei, and also with human albumin stains for
hepatocytes (Figure 4.3). Quantitative image analysis showed an up to 6.6-fold increase in the
area of albumin-positive colonies upon small molecule treatment. The vast majority of Ki67positive nuclei exhibited hepatocyte nuclear morphologies, which is consistent with the lack of
proliferating cells in fibroblast-only cultures treated with FPHs. These results strongly indicate
that human primary hepatocytes can be induced to proliferate in vitro using FPHs.
90
04
9 --
Figure 4.3. Ki67 and albumin staining of FPH treated hepatocyte cultures. Primary human
hepatocytes were cultured in 24-well formats on top of a confluent feeder layer of growtharrested J2-3T3 fibroblasts and exposed to small molecules as described. After six days in
culture, samples were fixed and stained with for albumin (green) and Ki67 (red). Bar graphs
represent quantifications of displayed images. Scale bar = 100p~m
To characterize the degree and kinetics of proliferation, we quantified the number of
hepatocytes in culture, using both an automated cell counter, and FACS analysis.
Results
showed a dramatic, up to 10-fold increase in the number of hepatocytes when treated with
various FPHs (Figure 4.4). The strongest proliferation inducer was FPH1. Over 7 days, FPH1
induced hepatocyte doublings at a rate that is consistent with reported liver regeneration kinetics
in vivo99.
91
j0
~o4
0
103
Hepatoyte
60
1
to-I
10
10,
I
1
0
01
ted C
Atom
01040.
fr03
a yes
0
si
0
Cos
adtoa
to
e
0atys
Fto
suea
we
10
th
ao
FPH2.
th
10
50
toI0101010
obts
0
00
20
P
j
16, o1'i01010
10010110
10 10
to
to
1.
Figure 4.4. Automated Cell Counter analysis. Fibroblasts were labeled with CM-Di pnor to
initiation of culture in order to allow identification of hepatocytes via negative selection. FACS
cell counting was further enabled by fluorescent counting beads. Control cultures were treated
with empty vehicle (DMS0). Data presented as mean ± SEM.
To generalize our findings across multiple donors, we obtained primary human
hepatocytes from six additional cell sources and treated them with FPH1 and FPH2.
Immunofluorescent staining for Ki67 and albumin, along with FACS-mediated cell counting
revealed that hepatocytes from every source examined expanded upon small molecule exposure
(Figure 4.5). These results, together with the primary screening and validation data obtained
using cells from donors a and b, suggest that the FPHs are active across a wide range of
genetically diverse individuals.
92
A
No Tx
FPH2
FPH1
B
FACS
U-_
0
0
0
0
0
00
Figure 4.5 FPH-induced expansion of multiple different donors of primary human
hepatocytes. Six additional sources of primary human hepatocytes were treated with FPHs and
A) stained for albumin (green) and Ki67 (red) after 6 days in culture. Scale bars represent
300um. B) Day 1 untreated control was added for reference (top). On day 7, we quantified the
number of hepatocytes in culture using FACS analysis.
To assess the phenotype of the treated hepatocytes, we performed phase-contrast imaging,
gene expression profiling and biochemical analyses. Imaging showed that normal hepatocyte
morphology was maintained throughout the treatment period (Figure 4.2).
93
Gene expression
profiling demonstrated that there are no significant differences between FPH-treated and
untreated primary human hepatocytes (Figure 4.6 A).
Urea synthesis, a surrogate marker of
nitrogen metabolism, and albumin secretion were both stable throughout FPH treatment (Figure
4.6 C).
Metabolic functions were assessed via examinations of cytochrome P450 (CYP450)
activity and canalicular transport. Results showed active transport of a fluorescent substrate into
the bile canaliculi between hepatocytes (Figure 4.6 B), and that CYP450 activity was not
compromised by small molecule treatment (Fig 4.6 C). These data agree with published findings
of sustained liver functions throughout liver regeneration".
94
Ar
MControl MPGE2 0FPH2 MFPH1 MHEPG2
CYP2AS
CYP2B6
CES2
Nuclear Receptors
FXR
SLCOIA2
CYP3A7
CYP7A1
CYPI 182
CYPIA2
CYP2C8
CAR
CYP2C9
CYP2CI9
CYP2D$
CYP3A4
CYP3A5
RXRS
CYP2EI
CYPIAI
CYPlel
CYP2A%13
CYP2FI
FMO3
FXR
FMO4
MAOA
MAOS
EPHXI
EPHX2
CESI
NOWI
N002
CORI
0CXR
0*IRS2
DHRS4
AKtt1AI
AhR
LTB4DH
ALOHIAI
ALOH2
UGT1AI
UGT1A3
IJGTIAG
UGTIA9
SULTIA1
StJLT1A2
SULTIAWI
SUL72AI
COMT
TMVT
HNUT
NNUT
NATI
GSTAI
GSTA4
081)11
081)1
GSTPI
oSTT
MOSTi
MGST2
ABCA2
ASOM
PXR
RR
AS=0
AS=8
ASCSI
AS=C
SLC22AI
SLCO103
MF A2
ABGG
AOS
ASB11l
Abuffdn
PXR
HNFg
1.0
1.G
1.0
OZ
0
05
0.0
O.G 9
0.010
Phase I Enzymes
CYP2C9
C
FM03
to
1.0
1,o
0.
0.5
0.5
0.0
- 0.0
16] T
.
12-
OX
8-
ALDH1A1
AKR1A1
EPHX1
20
MAOB
4.
0_1
120
100
Phase 11 Enzymes
UGT1A9
UGT1A3
T
II
E2
FPH2
FP'H1
80
SULTIA2
80
40
1.04MO
1.0
1o
2001
CtrI
PGE2 FPH2 FPH1
D
1.2-
Phase III Enzymes
ABCA2
0.9-
ABCA6
ABC3
0.8
0.G
1.0
1.0 .
0,3.
0.5
050.5
0-A.
0.0
0.0
M
0.0.
Ctrd PGE2 FPH2 FPH1
Figure 4.6. Functional analysis of FPH-treated primary human hepatocytes. A, gene
expression profiling of FPH-treated human hepatocytes. A panel of 83 liver-specific genes were
analyzed via Luminex. Columns of the heatmap are averaged values of replicate (n=3) loadings
of mRNA extracted from various populations of human hepatocytes (250ng total RNA per
replicate). mRNA expression was determined relative to the average of control gene transferrin,
and heat maps are row-normalized. Bar graphs are select gene sets comparing the relative mRNA
expression of FPH-treated hepatocytes (patterned bars) and HepG2 (solid red bar), normalized to
primary human hepatocytes (solid black bar) for nuclear receptors, phase I, phase II, and phase
III drug metabolism genes. Data represent the mean 1 SEM of Luminex-loaded replicates. B,
phase 3 transporter activity. Cultures were incubated with 5-(and-6)-carboxy-2',7'-
95
dichlorofluorescein diacetate, which is internalized by hepatocytes, cleaved by intracellular
esterases and excreted into the bile canaliculi between hepatocytes by transporters (scale bar =
50um). C, biochemical characterization of key hepatocyte functions. Albumin secretion reflects
protein synthesis capability; urea content is a surrogate marker for protein metabolism;
detoxification functions were measured via processing of substrate BFC into fluorescent
products. For all analyses, primary and FPH-treated hepatocytes were cultured for 7 days in 12well plates (n=3). Control cultures were treated with empty vehicles (DMSO). All data presented
as mean ± standard deviation.
Maturationof human iHeps
We tested the ability of hit molecules to promote the differentiation of iPS cells towards a
hepatic lineage and to induce the maturation of iHeps towards more adult-like liver phenotype.
Undifferentiated iPS cells were cultured on Matrigel, supported by conditioned media from
primary mouse embryonic fibroblasts and differentiated into iHeps as previously described.
In
brief, iPS cells were cultured in differentiation media, with sequential addition of growth factors
(Activin A, BMP-4, bFGF, HGF, and OSM) to guide differentiation, first into endoderm, then
into hepatic specified endoderm, then into hepatic progenitor cells and finally into iHeps (Figure
4.7).
Phase
a-1-Antit
111 1
DLK
w/ Rob Schwartz
E UDLX
SSEA1
5SZAI
Figure 4.7. iHeps generation. A) Undifferentiated iPS (top) and hepatic progenitor cells (iHEP)
generated from iPS (bottom). B) Immunostaining of hepatic lineage markers, not present in iPS
(top), but expressed by iHEP (bottom). C) FACS analysis illustrating expression of iPS markers
and hepatic progenitor markers in undifferentiated iPS (top) and iHEP (bottom). Scale bars
50pim.
Small molecules were added to basal media (without OSM in Figures 4.8 and Figure 4.9;
with OSM in figure 4.10) on day 21 post initiation of differentiation and acted over a period of 9
96
days. One FH (FH1) and one FPH (FPH1) were used to treat iHeps. FHl doubled albumin
secretion during primary screening (Figure 4.8 A) and the effects were dose-responsive (Figure
4.8 A). FPH1 had a similar effect on hepatocyte functions, which interestingly, was much more
pronounced in the younger donor (a, age 1).
A
B
2.5.
DMSORep I Rep 2
~04C
i
FHI
FP
00g concentroon M
Merged
AFP
Albumin
Merged
CYP3A
Albumin
.
U
0
C
a
Albumn
s
gsa
d
(gruen C
OIL
f
t
(a
b
=
A
p.
RI
Bar
sMMered)
A
P2
s
400
gr
p
represent
A, primary
.eps.of human primary hepatocytes and
Figure 4.8 Functional enhancement
vehicle
empty
with
treated
were
cultures
Control
FH1
of
curve
dose
and
screening data
size of
colony
and
morphology
C,
deviation.
(DMSO). All data presented as mean ± standard
shown
are
iHeps
Untreated
FPH I-and F il-treated iieps in 6-well plates 9 days post-treatment.
for comparison (scale bar = 100pm). D, Albumin (green), CYP3A (turquoise) and AFP (red)
staining of iileps after 9 days of culture (scale bar = 100pm). Bar graphs represent
quantifications of displayed images.
Cultures treated with FHl and FPH1 contained larger colonies of illeps, which exhibited
more pronounced hepatocyte morphologies, including polygonal cell shapes, visible nuclei, and
more noticeable bile cannaliculi between hepatocytes (appearing as phase bright demarcations
97
between hepatocytes, Figure 4.8 B). We subsequently examined the maturity of treated iHeps via
gene expression profiling, immunofluorescent staining, and various biochemical assays probing
protein secretion and enzyme activity.
Gene expression profiles showed that treated iHeps more closely resemble mature hepatocytes
than untreated cells (Figure 4.9 A and 4.9 B). Euclidian clustering analyses grouped untreated
cells with fetal hepatocytes, and treated cells with adult hepatocytes. Of particular interest are the
expression levels of various ABC transporters and GSTPL. ABC transporters are known to
mature after birth. ABCB1 1, also known as bile-salt export pump (BSEP), increased -4 fold in
expression levels with FH1 treatment, reaching 100% of adult hepatocyte levels. In contrast,
GSTP1 expression, whose levels decrease with maturity, remained low upon small molecule
treatment.
98
OiPS MiHeps EFPH2
U Fetal h. Hepatocyte
U Adult h. Hepatocyte
OIPS miHeps *FH1
E Fetal h. Hepatocyte
EAdult h. Hepatocyte
B 80
0
40
Nuclear Receptors
CAR
25
2
2.6
...
Nuclear Receptors
XR
..
CAR -
)
-~2.0
PXR
.
ips
FPH2
FHI
Wlkp
FPH2
FH1
io
FPH2
FHI
Hope~ FPH2
Fl-t
1.4
12
Phase I Enzymes
Phase I Enzymes
CYP2A6
CYP2A13
2,1CYP2A6
IL
<- 0.8
" CYP2A13
0.46
02
0.2.
2.C
CYP2B6
201
FMO3
2CYP2B6
-
FMO3
45-
Os
Phase 11 Enzymes
: -
GSTP1
-
Phase 11 Enzymes
GSTP1
0
HNMT
~270.
2
18-
HNMT1
(0
0
Phase IIl Enzymes
2ABCB1
1
2
Phase III Enzymes
ABCC4
2
IABCB11
to.0
o:s1 lf
2E
1Al
tOA
.Is ~jjf.5 IA -es
SLC2 2A1
ABCG5
uJABCC4
Zs
2011
..
0.
.. 0
Figure 4.9 Maturation of human iHeps. Gene expression profiling of FHl-treated (A) and
FPH1-treated (B) iHeps. Heat map displays of Luminex analysis for 83 liver-specific genes,
shown separately for independent experiments. Columns of the heatmap are averaged values of
replicate (n>4) loadings of mRNA extracted from various populations of iPS cells (250ng total
RNA per replicate). mRNA expression was determined relative to the average of control gene
transferrin, and heat maps are row-normalized. Bar graphs are select gene sets comparing the
relative mRNA expression of small molecule-treated hepatocytes (solid colored bars) to iPS
(solid white bars), untreated iHeps (solid gray bars) and fetal human hepatocytes (patterned
white bars), normalized to control (patterned gray bar) for nuclear receptors, phase I, phase II,
and phase III drug metabolism genes. Control refers to adult cryopreserved human primary
hepatocytes stabilized by micropatterned co-culture (MPCC, more details in supplementary note
1). Data represent the mean ± SEM of Luminex-loaded replicates. C, Quantifications of
Albumin, CYP3A and AFP staining of iHeps after 9 days of culture. D, biochemical
characterization of key hepatocyte functions. Albumin and AFP secretion are measured as a liver
marker and a fetal marker respectively; detoxification functions were measured via processing of
99
substrates with fluorescent or luminescent products. Specific activities of CYP2A6 and CYP3A4
were measured using coumarin and luciferin-IPA respectively. For all analyses, iHeps were
cultured for 9 days post-differentiation, in 6-well plates (n=3). All data presented as mean
SEM.
To examine the effects of FH1 and FPHl at the protein level in iHeps, we visualized AFP,
albumin and CYP3A4 levels via immunofluorescent staining (Figure 4.8 C and Figure 4.10).
Images showed dramatic increases in albumin upon both FHI and FPH1 treatment, although the
effects of FH1 are more pronounced. This pattern is in agreement with morphological findings.
Consistent with prior work13 1 ~1 , untreated cultures contained islands that were strongly positive
for both albumin and the fetal marker AFP, but showed minimal staining for the mature marker
CYP3A4. In contrast, treated islands double stain strongly for albumin and CYP3A4, with AFP
largely absent. This more mature phenotype was stable for at least 1 week after removal of small
molecule treatment (Figure 4.10).
100
A
Alb/Hoechst
CYP3A/Hoechst
Merge
AFP/Hoechst
Phase
>4t)
C:
0
ON
B
E
4101
12~g
0
-31
0
DOp 20 Noh010P*1
Day 29
PHI
Day 20 NoSa 0*0PHI4
020 NOTXDWO00PH0I PHI
FHI
Day 29
Day 29
C"l
--
2
NotxUSOPHIl PHI
CtA
Day 29
Figure 4.10 Stability of Mature iHep Phenotype. iHeps were treated with small molecules
once on day 20. Cultures were then maintained in normal basal media containing OSM (without
small molecule addition) for 9 days prior to A) phase contrast imaging (scale bars represent
100um) and immunofluorescent staining (scale bars represent 50pm) for Albumin, CYP3A and
AFP. B) ELISA assay for secreted albumin and AFP, and C) quantitative CYP3A4 and CYP2A6
activity assays, benchmarked against cryopreserved human hepatocytes that have been stabilized
in culture (MPCC, more details in supplementary note 1).
To confirm the staining results, we measured secreted levels of albumin and AFP via
ELISA, and CYP450 activities through isoenzyme-specific
substrates with fluorescent or
luminescent metabolites (Figures 4.9, 4.10). ELISA results verified that small molecule
101
treatment both increased albumin secretion and decreased AFP secretion. CYP3A4 activity was
found to increase by 16 and 45 times upon treatment with FHl and FPH1, respectively. CYP2A6,
another mature CYP450, was also found to increase significantly upon small molecule treatment.
We considered enzyme induction as a possible explanation for these elevations in CYP450
activity. However, this is unlikely. While human hepatocytes treated with specific inducers do
exhibit elevated CYP450 activity, such elevations are typically reverted by 24 hrs after removal
of the inducer (Figure 4.11). A period of at least 48 hrs separates small molecule treatment and
the measurement of CYP450 activity, thus the iHeps are expected to have recovered from any
general elevations in CYP450 activity.
1.41.2
wo
1.4
-r-Induced
-+-Uninduced
1.
1-0
0.8
0.8
0.6-
0.6
0.4
o
E 0.2
0.0
0
100
200
300
400
-
z 0.4
E 0.2
0.0
500
-Induced
-+-Uninduced
1.2
-
0
100
200
300
400
500
Culture Time (Hrs)
Culture Time (Hrs)
Figure 4.11. Hepatocyte induction kinetics. Hepatocytes were induced with 2501M pnaphthoflavone (BNF) at various times during culture. Black arrows indicate addition of BNF;
white arrows indicate removal. CYP450 In all cases, elevations in CYP450 activity secondary to
induction are mostly reverted by 24hrs post removal of inducer.
Discussions
Developing novel cell-based therapies for liver disease requires human hepatocytes due
8 6 87
to substantial species-specific differences ' . Human hepatocytes, in their native environment,
have phenomenal regenerative capabilities 1s,71,72,99,100, which is lost ex vivo. Consequently, for
102
decades, human hepatocyte sourcing has been a bottleneck for many fields of research and
clinical therapies.
In traditional cultures of other cells, small molecules have shown promising roles in
modulating a wide range of complex cell phenotypes. These outcomes include stem cell selfrenewal and differentiation, and the proliferation of normally quiescent mature adult cells such as
pancreatic P-cells and cardiomyocytes'5
160 .
Compounds can act through a variety of
mechanisms to induce cell division, including activation of developmental signaling pathways
such as Wnt16 1 or recruitment of GEFs to the plasma membrane for RAS/MAPK pathway
activation 160.
However, such systems have limited applicability towards primary human
hepatocytes due to the rapid loss of hepatocyte phenotype and viability in vitro. To date, highthroughput screens related to the liver have been restricted to hepatocyte cell lines or hepatocyte
extracts, neither of which offers the full repertoire of hepatocellular functions and responses
exhibited by primary human hepatocytes. Such deviation from clinically-relevant biology has
been a major cause of the high attrition rates currently troubling drug discovery. The highthroughput platform developed here leveraged stromal interactions, known to be important in
vivo, to enable screening using primary human hepatocytes. Such a platform enables studies of
many previously inaccessible aspects of liver biology and small-molecule bioactivity. In this
work, we used the platform to address two long-standing issues: lack of a renewable human
hepatocyte source, and incomplete differentiation of human stem cells.
Early work on hepatocyte expansion focused on in vitro addition of growth factors,
hormones, serum and vitamins75 ' 76 78' 79 , and has led to a media formulation for the expansion of
rodent hepatocytes76. Our study contributed to this progress through the identification of small
molecules that can induce proliferation of human hepatocytes in vitro. With this advance, we
103
foresee the potential to extend our findings to generate renewable materials (i.e., functional
human hepatocytes) for various cell-based therapies, and in vitro liver models and assays,
including tissue engineered liver constructs, cell transplantation, ADME/Tox screening, and
humanized mice for in vivo disease modeling. Our future work will determine the maximum
expansion potential and functionality of treated hepatocytes using animal rescue experiments,
and explore use of hepatocytes expanded from patient biopsies for cell transplantation therapy
and other forms of personalized medicine. Such outcomes can contribute to our understanding of
liver regeneration, and carry significant implications for regenerative medicine.
An alternative cell source to primary human hepatocytes is human iPS cells, which can
be differentiated towards hepatocyte-like cells (iHeps) 3 1 132 1, 9 1 . Among the factors used to drive
differentiation is Oncostatin M. It has been observed that continued supplementation of
Oncostatin M beyond the first 20 days of differentiation promotes survival of the resulting
hepatocyte-like cells but suppresses the late stages of hepatocyte maturation. Thus, in order to
isolate the effect of hit molecules on maturation (rather than differentiation), oncostatin M was
excluded beyond the 20 day point for the differentiations depicted in Figures 4.8 and 4.9. Under
these conditions, a small percentage of hepatocyte-like cells were derived; however, the few that
were obtained exhibited dramatic improvements in maturation.
When oncostatin M was
maintained throughout the treatment period (Figure 4.10), high efficiencies of differentiation to
hepatocyte-like cells were observed. Interestingly, and contrary to our expectations, the hit
molecules were also able to mature these hepatocyte-like cells; however, as expected, the levels
of maturation as judged by CYP2A6 and 3A4 activity were not as robust as in the absence of
oncostatin M.
104
The efficiency of differentiation is generally stochastic yielding some highly efficient
differentiations and others far less efficient-even in the same laboratory. The variables that
underly this stochasticity are yet poorly understood but may include the variability in other cell
types that emerge over time during differentiation, effectively creating a variable co-culture
environment. Even the most robust cultures of differentiated iHeps persistently express fetal
markers (e.g., AFP) and lack key mature hepatocyte flmctions (e.g., CYP2A6, CYP3A4)
131-133,
a
trait which presents a significant hurdle to the application of these cells in both therapeutic and
basic research assays. Our iPS results suggest that FH1 and FPH1 are able to promote the
maturation of well-differentiated cultures of iHeps beyond what has been obtainable to date, thus
potentially alleviating a major obstacle to the use of iPS cells as a renewable source of functional
human hepatocytes. We found that this effect generalized across two pluripotent cell lines;
nonetheless, we did observe stochasticity in the efficiency of maturation akin to the variability
observed in response to existing directed differentiation protocols.
Based on tetraploid complementation experiments with mice, it is clear that murine iPSderived progeny are capable of differentiating into mature, adult liver cells, when provided with
the correct in vivo context and developmental cues",193. Given that it is not possible to conduct
the parallel experiment with human cells, it remains to be seen whether similar conditioning is
sufficient to promote complete maturation to a comparable, fully mature hepatocyte. The
evidence that human iHeps mature when xenografted in a 'gold standard' adult, in vivo
environment is mixed 71'
94;
the remaining donor cells still appear to exhibit low function, on a
per cell basis I',191,195-197. This apparent defect could indicate that the cells are truly blocked from
late-stages of tissue-specific maturation, or that the pre-conditioning protocols provided in vitro
are deficient in some way - perhaps lacking specific cues that the human embryo experiences in
105
the third trimester of development, for example. Our data suggest that human iPS cells are
indeed capable of further maturation in vitro, consistent with their potential observed in the
mouse system. In light of similar challenges in other cell types 198 , our platform and its resulting
compounds offer a roadmap for testing the generality of this finding in other tissues, and for
identification of regulatory networks that are influenced by small molecule exposure.
4.3 Conclusions
Through a high-throughput screen of 12,480 small molecules on primary human
hepatocytes, we identified compounds in two separate classes that can be used to establish
renewable sources of functional human hepatocytes.
One class, FPH, induces proliferation of
mature human primary hepatocytes. The second class, FH, helps differentiate human iPS cells
towards more mature, adult hepatocytes. The identification of these small molecules may impact
several areas of research including maturation of other iPS-derived cell types, expansion of other
'terminally' differentiated cell types, and the translational potential of the resultant hepatocytes
and other cell types.
4.4 Materials and Methods
Hepatocyte Medium Composition
1X DMEM
10% fetal bovine serum (FBS)
15.6 ptg/ml insulin
7.5 gg/ml hydrocortisone
106
16 ng/ml glucagon
1% penicillin-streptomycin
FibroblastMedium Composition
IX DMEM
10% bovine serum (BS)
1 % penicillin-streptomycin
Cell Culture Conditions
J2-3T3 Culture. Passage 2 J2-3T3 fibroblasts were obtained from Howard Green (Harvard) and
kept in liquid nitrogen until use. Cells were maintained under standard tissue culture conditions,
in DMEM media containing 10% BS and 1% Penicillin-streptomycin. Fibroblasts were grown in
T- 150 tissue culture flasks and passaged 1:10 using 0.25% Trypsin-EDTA when cells reached
confluency. Experiments used J2-3T3s ranging in passage numbers from P9 to P12.
Expansion ofMature Hepatocytes. J2-3T3 fibroblasts were growth-arrested using 8ug/mL
mitomycin-C (sigma) for 3hrs before trypsinization and seeding into the desired plate format
(24-well or 12-well or 6-well) at a density of ~400,000 cells / cm 2 . The following day,
cryopreserved human primary hepatocytes were thawed following standard protocol (with
centrifugation at 50g for 5min) and seeded onto the confluent fibroblast feederlayer at a density
of ~50,000 cells/cm 2 . Small molecules were supplemented to the culture media on days 1 and 5
at 40uM for FPH1 and 20uM for FPH2 and FH1. Cultures were maintained in 200ul/well of
hepatocyte media if in 24-well tissue culture plates, 400ul/well if in 12-well plates and 1 mL/well
107
if in 6-well plates, with media changes every other day. Culture supernatants were collected
during media change for functional analyses and kept at -20C until analysis.
Differentiationof PS cells. Validation experiments examining the maturation effects of FHI and
FPH1 were conducted in standard tissue culture vessels (24 or 6-wells) using human iPS-derived
hepatocyte-like cells (iHeps). iHeps were cultured in hepatocyte basal media, I mL/well if in 6well tissue culture plate format and 200ul/well if in 24-well format. Media change and
supernatant collection occurred every other day. Small molecules were added to cultures every
other day at a concentration of 1 ORM. Experiments examining maturation (rather than
differentiation) of iHeps were conducted without OSM supplementation in the late stages (after
day 20) of cultures while experiments reported in fig. x were conducted in the presence of OSM.
Phenotype of treated iHeps were benchmarked to cryopreserved primary human hepatocytes
stabilized by a micropatterned co-culture system (MPCC), previously reported to allow for
functional recovery of primary human hepatocytes following the trauma of isolation and
disruption from the native microenvironment, and is thus more representative of in vivo liver
physiology.
Proliferationand FunctionalAssays
Immunofluorescent Staining. Samples were fixed in 4% PFA for 15 min at room temperature,
permeabilized with 0.1% Triton-X for 30 min at room temperature, and blocked with 3% FBSPBS for 1 hr at room temperature. Primary antibodies for Ki67 (Millipore), Albumin (Abcam,
Bethyl), CYP3A (Santa Cruz), and/or AFP (Fitzgerald) were added to cultures at manufacturerrecommended concentrations and incubated at 4*C over night on rocker. Secondary antibodies
108
(BD Biosciences) were added to cultures at 1:400 dilution and incubated at room temperature for
45 min. Nuclei were visualized through Hoechst Staining.
Biochemical Assays. Culture media were collected and frozen at -20'C until analysis. Albumin
content was measured through sandwich ELISA assays (MP Biomedicals, Fitzgerald, Bethyl
Laboratories) using horseradish peroxidase detection and 3,3',5,5'-tetramethylbenzidine (TMB,
Fitzgerald Industries) as a substrate. Urea concentration was determined colorimetrically using
diacetylmonoxime with acid and heat (Stanbio Labs). To quantify CYP450 activity, co-cultures
were incubated with 7-benzyloxy-4-trifluoromethylcoumarin (BFC, BDGentest) at 50KM for 1
hr at 37'C in phenol-red free media. iHep cultures were incubated with various substrates
(coumarin from Sigma for CYP2A6, luciferin-IPA from Promega for CYP3A4) for 4 hours at
37'C.
Incubation medium was
collected and metabolite
concentration
quantified via
luminescence, or fluorescence after hydrolization of potential metabolite conjugates by Bglucuronidase/arylsulfatase (Roche, IN).
Bile CanaliculiStaining.Live co-cultures were washed to remove phenol-red, then incubated
with 2 gg/mL CDF [5-(and-6)-carboxy-2',7'-dichlorofluorescein diacetate, Molecular Probes] for
10 minutes at 37'C. Excess stain was removed prior to examination with fluorescence
microscopy (excitation/emission wavelengths: 495/520 nm).
Cell Counting. J2-3T3 fibroblasts were covalently labeled with Vybrant CM-Dil (Invitrogen)
before initiation of co-culture.
While J2-3T3s are in suspension, CM-Dil stain was added at
manufacturer-recommended concentration of 5ul per lmL of cell suspension (1 million cells /
mL) and allowed to incubate at 37'C for 15min. Cell suspension was washed 3 times prior to
seeding. For FACS analysis, co-cultures were treated with collagenase, then accutase, and
suspended in PBS-0.2%FBS.
Cell suspensions were supplemented with 50,000 fluorescent
109
counting beads (CountBright, Invitrogen) per sample. Data were acquired with a 4-color flow
cytometer (FACSCalibur, BD Biosciences) and analyzed with CellQuest (BD Biosciences). For
Cellometer analysis, cells were trypsinized and placed on cell counting chambers (Nexcelom) for
automated cell counting.
Luminex analysis. Cells were lysed using RLT buffer (Qiagen) or Trizol (Invitrogen) and
purified using the Mini-RNeasy kit (Qiagen). Gene expression was determined using Luminex
analysis, as previously described 46.
Briefly, total RNA was immobilized on a Qiagen turbo
capture 384-well plate, and reverse-transcribed using oligo dT priming. A biotinylated FlexMap
tag sequence unique to each gene of interest and a phosphorylated downstream probe were then
added to resulting cDNAs to generate biotinylated FlexMap-tagged amplicons. Universal PCR
was then performed for 35 cycles using a biotinylated T7 forward primer and T3 reverse primer
in buffer with dNTPs and Taq polymerase. FlexMap microsphere beads conjugated with antitag
oligonucleotides were then added and allowed to hybridize.
Amplicons were captured by
streptavidin-phycoerythrin, and 100 events per bead were analyzed for internal bead color and
phycoerythrin reporter fluorescence on a Luminex FlexMap 3D analyzer.
Data for replicate
loadings, expressed in mean fluorescent intensity of at least 100 beads per sample, were scaled to
the human transferrin gene and row-normalized for heat map representation using GeneE open
software (Broad Institute).
110
Chapter 5. Small Molecules for Enhancing Liver Development and Treating
Acute Liver Failure in vivo
5.1 Introduction
The growing discrepancy between supply and demand of transplant-grade livers for endstage liver diseases necessitates the need for alternative liver therapies.
Approaches being
actively pursued include cell-based strategies such as bioartificial liver devices, cell
transplantation, and implantation of hepatocellular constructs, as well as molecular inducers of in
vivo liver regeneration. In earlier chapters, we had reported the high-throughput identification of
small molecules for hepatocyte expansion and iPS differentiation in vitro. Here, we explore the
therapeutic potential of these small molecules and further report their bioactivity in vivo as
enhancers of liver development and protectors against acute liver failure in zebrafish.
Zebrafish (Danio rerio) has a lengthy and successful history as a critical tool in
developmental biology
99 .
Its fast and lucrative reproductive cycles, coupled to its desirable
optical properties and external development enable easy visualization of complex organ
developments in real time. More recently, it has also gained traction as a powerful platform for
genetic and chemical screening' 9
16 1
,s9 ,2oo. Its genome and body plan are similar to those of
other vertebrates and there is well-documented conservation between zebrafish and mammalian
pathways involved in liver development, regeneration and pathogenesis' 89 .
Zebrafish models of several human diseases have been developed, including a clinically
relevant model of acetaminophen overdose induced acute liver injury, recently reported by
Goessling and colleagues' 8 9 .
Acetaminophen (N-acetyl-p-aminophenol,
IlI
or APAP) is an
analgesic and antipyretic available over the counter. Accidental or suicidal overdose of APAP is
the leading cause of acute liver failure and results in more than 300 deaths each year in the
United States alone20 1 . APAP toxicity results from the accumulation of the toxic metabolite Nacetyl-p-benzoquinone
imine
(NAPQI),
which
is
normally
neutralized
by
glutathione
conjugation in the liver but accumulates in contexts of APAP overdose when glutathione stores
become depleted by excess production of NAPQI.
Unconjugated NAPQI causes oxidative
stress, leading to liver necrosis and dysfunction. The only clinical therapy available is Nacetylcysteine (NAC), which inhibits the mechanism of toxicity by directly inactivating NAPQI.
NAC can thus improve clinical outcome and survival if administered before the onset of
extensive liver damage (<12 hrs post ingestion)202 .
Here, we utilize previously characterized zebrafish models of liver development and
acute liver failure to demonstrate in vivo bioactivity of small molecule inducers of hepatocyte
proliferation (PHI) and function (FHl). Data show that PHI and FHi are molecular enhancers of
in vivo liver development and that FHl can further serve as a hepatoprotectant against APAP
overdose induced liver toxicity. These results serve not only as in vivo validation of the highthroughput liver platform and subsequent chemical screening described in earlier chapters, but
also represent important first steps towards the development of clinically relevant liver
therapeutics.
5.2 Results and Discussions
PHI and FH1 Enhance Liver Development in Zebrafish
112
Previously, we had identified small molecule inducers of in vitro hepatocyte proliferation
and function through a chemical screen of 12,480 small molecules using the high-throughput
liver platform we developed for primary human hepatocytes. Here, we examine the ability of 4
prioritized hits, FPH1, FPH2, FH1 and PHl to modulate liver development in zebrafish embryos.
3
(lfabp:GFP)
Transgenic liver fatty acid binding-protein reporter Tg(-2.8fabp1O:EGFP)as
zebrafish embryos were allowed to develop normally for 24 hours post fertilization (hpf).
Embryos were then transferred into 6-well plates and maintained in 5mL of fluids each
containing a small molecule treatment (50-75 embryos/group) until 72 hpf. At 72 hpf, the in vivo
liver size of embryos were examined by fluorescent microscopy and confirmed by in situ
hybridization for the hepatocyte-specific marker lfabp.
Data showed that PHI and FHI increased liver size in a dose dependent manner. Both
compounds were examined at 2.5uM, 5uM or lOuM doses. Maximum effect was observed at
1 OuM for PHI, with no evidence of toxicity, suggesting that greater effects may be possible at
higher concentrations. Embryo exposure to IOuM of PHI shifted liver size distribution from 17%
small-size livers, 66% normal-size livers and 17% large-size livers to I % small liver, 59%
normal-size livers and 40% large-size livers (Figure 5.1).
113
0
O
f
M75pf
mygp
MSmalliver
N = 50-75 embryos/group
V0
FH1 2.5uM
*LargeUver
so
DMSO
DMSO
vNormalLiver
100
0
PH1 2.5uM
0
5
2.5
IFHI]J(uM)
*SmsllLiver
UNormal
Liver
ELargeLver
100......
PH1 5uM
.2
FH1 lOuM
0-
FH11w
M
PH1 1OuM
7
50
0
5
2.5
[PHIJ uM)
10
w/ Andrew Cox
Figure 5.1 Small molecule enhancement of in vivo liver development. Zebrafish embryos
were allowed to develop normally for 24 hours post fertilization prior to exposure to small
molecules as previously described until 72 hours post fertilization. Embryo liver sizes were then
3
measured via fluorescent microscopy of transgenic Tg(-2.8fabplO:EGFP)=
(lfabp:GFP)
hybridization.
situ
in
zebrafish and
Similar effects were observed with FH1, with peak activity at 5pM. There is a decrease in
embryo liver size at 10pM of FHI, suggesting toxicity at higher doses of FH1. This is consistent
with medicinal chemistry predictions of FH1 bioactivity. Terminal hydrolyzation of FH1 can
generate aniline, which is hepatotoxic, thus unmodified FH1 will exhibit a narrow concentration
window of therapeutic efficacy.
FPHl, tested at 2.5p.M to lOuM, and FPH2, tested at 5pRM to 20pM, did not enhance liver
development and appeared toxic at higher doses. This may signify the lack of in vivo bioactivity
in zebrafish, but it is also possible that immature livers lack the adult metabolic functions
114
necessary to activate these molecules. Further examination into the mechanisms of action,
particularly the identification of active chemical components, will be beneficial.
FH1 Protects Against APAP-induced Acute Liver Toxicity
APAP exposure in zebrafish leads to biological changes reflective of human physiology,
including elevated ALT levels signifying liver injury, widespread necrosis and sinusoidal
hemorrhage, hepatocyte and ultimately zebrafish death. Thus we examined whether small
molecules hits identified using human primary hepatocytes can protect against acute in vivo liver
injury in zebrafish.
Zebrafish embryos were allowed to develop normally for 48 hrs in order to allow
metabolism of APAP and other small molecules. They were then simultaneously exposed to
10mM APAP and a small molecule, PHI, or FH1 from 48 hours post exposure (hpe) to 80 hpe.
At this early time point, APAP overdose results in only 23% loss in survival, which is alleviated
to 7% upon 5ptM FHI treatment (Figure 5.2). Surprisingly, PHI
had no protective effect. It is
possible that the acute injuries caused by APAP overdose disabled the liver's ability to activate
PHI.
115
1n mMAPAP
I
80 hpf
48 hpf
0 hpf
100
I
N
-
=
150 embryos/group
80
6040
20
0
-- ,
DMSO
APAP
APAP+FH1
APAP+ PHi
w/ Andrew Cox
Figure 5.2. Effect of small molecule treatment on early zebrafish survival. Zebrafish
embryos were allowed to develop normally for 48 hours before co-administration of a small
molecule hit and a lethal dose of APAP. % survival was measured at 80 hours post fertilization.
Next, we examined the effects of PHI and FHl on longer-term zebrafish embryo survival.
Embryos developed normally for 72 hrs, then were exposed to concurrent treatments of 10mM
APAP and 2.5pM, 5pM or 10pM of either PHI or FHl. Consistent with results from earlier time
points, PHI offered no protection against acute liver injury, while FHl was able to increase
survival from 16% to 71% at its optimum dose (Figure 5.3). Interestingly, the optimal dose of
FH1 decreased from 5pM for enhancing liver development to 2.5pM for protecting against acute
liver injury.
116
Mai
0 hpf
r
72hpf
168 hpf
N = 55-75 embryos/group
100
p
S
752
*w
mU.APAP
50
atrwAPAP + 2.5uM F H 1
25
SuM FH1
-APAP+
--
PAP+ 1OuM FH1
0
0
1
2
Time (dpe)
3
4
w/Andrew Cox
Figure 5.3. FHl increases zebrafish embryo survival post lethal doses of APAP. Zebrafish
embryos were allowed to develop normally for 72 hours before co-administration of 10mM
APAP and various doses of FHl. % survival was measured through 168 hours post fertilization.
Given the hepatoprotective effects of FHI and liver enhancing effects of PHI, we
explored whether they can address the key shortcoming of NAC therapy, which is that NAC
must be administered within 12 hours post ingestion in order to offer significant therapeutic
benefits. Zebrafish embryos developed normally for 48 hrs before they were treated with 5mM
APAP for 24 hours. At 72 hpf, embryos were administered 10pM of PHI or FHl. Fluorescent
117
microscopy showed increases in liver size upon both PHI and FHl treatment, suggesting that
both molecules can offer therapeutic benefits beyond the clinical standard for treating APAPoverdose (Figure 5.4). It was noted that 5pM of PHI offered therapeutic benefits for liver injury
caused by 5mM APAP but not 10mM APAP, which supports the hypothesis that PHI needs to
be metabolically altered by the liver in order to be active. This property limits the use of
unmodified PHI molecule for liver diseases but this limitation may be circumvented by
identification and direct administration of the active derivatives of PHI.
O hpf
DMSO
72 hpf
48 hpf
FH1 10sM
96hpf
PH 110sM
w/ Andrew Cox
Figure 5.4. Hepatoprotective effects of PHI and FHl following delayed administration.
Ifabp:GFPzebrafish embryos were allowed to develop normally for 48 hours before exposure to
5mM APAP alone for 24 hours. Small molecule treatment was given another 24 hours later, at
72 hours post fertilization. At 96 hours post fertilization, liver sizes were examined via
fluorescent microscopy.
Finally, we explored whether the hepatoprotective effects of FH1 is conserved in adult
zebrafish. Using 30 zebrafishes aged 3 to 12 months, we found that FHI enhances survival for all
ages. Survival benefit can be up to 63% in zebrafishes aged 3-6 months (Figure 5.5 left), with an
average (Figure 5.5 right) increase in survival of 40% (17.8% survival with APAP exposure only,
58.5% with FH1 rescue).
118
0hpf
Adultfish
N = 30 zebrafishes aged 3-12 months
zebrafishes aged 3-6 months
100
100
75
50
25
---
APAP
-
APAP+ FHI I uM
0
6
0
12
25
IS 24 30 36 42 48 54 60 66
Time (hpe)
-APAP
-
72
0
APAP+ FHI I uM
20
40
Time (hpe)
60
80
w/ Andrew Cox
Figure 5.5. Small molecule treatments are hepatoprotective against acute liver injury in
adult zebrafishes. Adult zebrafishes ranging in age from 3 months to 1 year were coadministered 10mM APAP and FH1. % survival was measured 72 hours post exposure.
5.3 Conclusions
Using the high-throughput liver platform described in earlier chapters, we had previously
identified several small molecule inducers of in vitro human primary hepatocyte expansion and
function. In this chapter, we further provide evidence of in vivo bioactivity of these small
molecules. Using well-established zebrafish models from Goessling and colleagues for studying
liver development and acute liver injury, we found Proliferation Hit 1 (PHI) and Functional Hit
1 (FH1) to both enhance liver development and protect against APAP overdose induced liver
failure. These results not only validate our previous in vitro findings but also represent important
first steps towards the development of these compounds as novel molecular therapeutics for liver
diseases.
119
5.4 Materials and Methods
Zebrafish
Zebrafish transgenic lines were developed and maintained as previously described.
Small Molecule Exposure
PHI was purchased in powder form from Enzo Life Sciences; FHl was initially
purchased in powder form from Maybridge and subsequently custom synthesized by Fisher
Scientific. Both molecules were reconstituted in DMSO at 20mM and kept at -80'C until use.
Zebrafish were exposed to DMSO, APAP, PHI and FHl at doses and times as described. Post
exposure, fishes were transferred to and maintained in fresh water until analysis.
In Situ Hybridization
Zebrafish embryos were fixed in paraformaldehyde and processed for lfabp in situ
hybridization as previously described (http://zfin.org/ZFIN/Methods/ThisseProtocol.html).
FluorescenceImaging
Zebrafish were anesthetized with 0.04 mg/mL Tricaine-S prior to microscopy.
120
Chapter 6. Perspectives and Future Directions
The overall goal of this thesis work is to identify and develop novel molecular and
cellular therapies for organ regeneration. Organ transplantation is the gold standard treatment for
the failure of many organ systems, including heart, liver and kidneys. However, transplant
recipients are subject to a lifetime of immunosuppressive regimens, and there is growing
discrepancy between supply and demand of transplant-grade organs. Therefore, this research
aimed to advance alternatives to whole organ transplantation, specifically molecular inducers of
in vivo organ regeneration, and cell-based therapies such as cell transplantation and engineered
tissue constructs.
Treating the failure of organs serving a multitude of biochemical functions, such as the
liver requires cell-based therapies, particularly when not all functions are well understood. Such
therapies should ideally employ human cells due to substantial species-specific functional
differences, as well as safety considerations. Ex vivo, human primary cells often lose their
phenotype, and for hepatocytes, their innate ability to replicate extensively. Consequently, for
decades, human cell sourcing has been a bottleneck for many fields of research and clinical
therapies.
Accordingly, this work engineered a high-throughput liver platform for primary human
hepatocytes to enable large scale chemical screening for small molecules that can used to
generate renewable source of functional human hepatocytes in vitro. Towards this end, we found
2 classes of molecules, FPH and FH. FPH compounds enhanced both hepatocyte proliferation
and function during primary screening and were shown to induce expansion of functional
primary human hepatocytes under standard tissue culture conditions. FH molecules increased
hepatocyte functions during primary screening and were subsequently found to induce
121
differentiation of iPS cells towards an adult hepatic phenotype more mature than what was
previously obtainable. In addition to FPH and FH, we identified a third class of prioritized hits
from the small molecule screen termed PH. All three classes were tested in well-characterized
zebrafish models to assess their potential as molecular therapeutics for liver diseases. Both PH
and FH were found to enhance liver size during development and protect against APAP
overdose-induced acute liver failure.
Ultimately, the identification of these small molecules may impact several areas of
research including differentiation and maturation of other iPS-derived cell types, expansion of
other 'terminally'
differentiated cell types, and the translational potential of the resultant
hepatocytes and other cell types as well as the design and development of molecular therapies for
acute liver failure. In this chapter, we outline future applications of these small molecules and
provide preliminary data for promising areas of investigation; additionally, we also discuss ongoing work towards the improvement of in vitro liver models and offer mechanistic insight on ex
vivo maintenance of primary human cells.
6.5 Understanding Mechanisms of Small Molecule Bioactivity
Traditionally,
screening-based
discovery
of bioactive
small molecules
has been
overwhelmingly target-based, involving purified proteins with known mechanisms of action.
With modern technological advances in cell culture and cell fate assays, there has been a recent
shift towards phenotypic screens that benefit from intact cellular machineries and programs to
better mimic in vivo physiology. Such whole-cell screens additionally offer the possibility of
discovering previously unknown therapeutic targets. The cost paid for these benefits is the loss
of mechanistic insight. In order for FPH, PH and FH molecules to reach their full potential as in
122
vitro research tools and particularly as therapeutics for liver diseases, we need to first understand
their mechanisms of action. Such knowledge would not only provide a better understanding of
the biology being modulated by these small molecules but it is also a prerequisite for medicinal
chemistry efforts toward improving the specificity and potency of the small molecule hits.
An important first step for understanding the mechanism of action is target identification.
There now exist several methods for target deconvolution, including direct biochemical methods,
genetic interaction methods and computational inference methods.
Direct biochemical methods
Direct methods employ stable isotope labeling of amino acids in culture (SILAC)
and quantitative mass spectrometry to identify direct and indirect binding partners of
small molecules of interest. Chemical hits are immobilized on beads and allowed to
interact with lysates from two different cell populations, distinguished by either heavy or
light isotope labels. One population interacts with bead-immobilized small molecules in
the presence of competitive inhibitors while the other is allowed to interact without
interference. Following processing through mixing, washing and electrophoresis, samples
are treated with trypsin and peptides are analyzed by quantitative mass spectrometry.
Proteins that are detected in equal amounts in both heavy and light isotope labeled
samples are non-specific binding partners and may represent mediators of off-target
effects; minimizing binding to these proteins may improve the specificity of the small
molecule. Peptides that are differentially detected in heavy versus light isotope labeled
samples represent specific binding partners, which can be either the direct target or
members of complexes including the direct target.
123
An important pre-requisite for direct biochemical target identification is an
understanding of structure-activity relationships (SAR) for the small molecules being
investigated. Without such knowledge, one risks alteration of small molecule bioactivity
through accidental tethering of beads to active sites. In chapter 3, we offered some SAR
for FPHl, which was obtained through analyses of structural analogs included in primary
screening. Among the remaining 11 prioritized hits were 3 products of diversity oriented
synthesis (DOS), with ~60 additional structural analogs that were not included in the
original screen. We have since initiated testing on all analogs in our human liver platform
and expect to gain insight on the SAR of these 3 DOS products. For other hits without
structural analogs, one can either commission custom synthesis of related structures or
employ other target deconvolution methods.
Genetic interactionmethods
In mammalian cells, genetic interaction methods often encompass genome-wide
RNAi modulation of cellular phenotypes. Full or partial phenocopy of the small molecule
effect by RNAi treatment suggests that the gene product is a small molecule target. When
specific pathways are implicated, RNAi tools can further help generate mechanistic
hypotheses through the activation and/or suppression of pathway activators and/or
inhibitors.
The use RNAi technology to understand cellular processes can be used in
conjunction with chemical modulators or independently. Later in this chapter, we will
discuss on-going work that utilizes genetic screening to study mechanisms that mediate
the co-culture effect.
Computationalinference methods
124
Computational methods can infer protein targets of small molecules mostly
through pattern recognition of profiling data. For example, by contrasting gene
expression levels of small molecule treated and untreated cells, one may be able to
identify proteins and possibly entire signaling pathways perturbed by chemical exposure.
Additionally, it is believed that small molecules acting through the same pathways will
elicit similar behaviors in different biological assays. Thus new compounds can be
compared with landmark molecules with known mechanisms of action in reference
databases to establish connections and generate target hypotheses.
In addition to the above systematic approaches, we can also gather mechanistic insight
through hypothesis-driven investigations. FPH2, for example, is a small molecule hit shared with
an independent phenotypic screen examining hematopoietic stem cell (HSC) self-renewal. HSC
homeostasis shares well-documented signaling pathways with liver regeneration, most notably
Wnt signaling. Thus it may be worthwhile to examine the effects of FPH2 on Wnt pathways in
vitro through techniques such as Western blotting of downstream proteins including
P-catenin,
and in vivo through reporter zebrafish lines.
Ultimately, we hope to utilize mechanistic understandings of small molecule bioactivity
to design and develop improved derivates of FPH, PH and FH molecules towards the generation
of more effective in vitro inducers of hepatocyte proliferation and differentiation as well as in
vivo therapeutics for liver diseases.
6.6 Small Molecules as Enabling Research Tools for Understanding Human Biology
Advances in the development cell culture systems can provide important insights into
human-specific biological processes that were not previously accessible.
125
Specifically, the
identification of small molecule inducers of hepatocyte proliferation and differentiation can
enable novel studies into a broad spectrum of human biology including liver regeneration, liver
development and more generally, cell fate control of both "terminally" differentiated cell types
as well as pluripotent stem cells.
Liver regeneration has been the subject of extensive research dating back to the early
1900s. Anderson and Higgins developed in 1931 what remains to this day the gold standard
model for studying liver regeneration - 2/3 partial hepatectomy. Over several decades, the
community has gained much insight into the underlying biology of a powerful regenerative
process; however, many questions remain. Through parabiotic studies, we learned about the
existence of a systemic soluble signal that enables liver growth in one animal upon liver injury in
its connected peer. Extensive investigations have been launched over the years looking to
identify and exploit these factors for in vitro expansion of primary hepatocytes. While
researchers have found lists of both direct mitogens such as hepatocyte growth factor and
epithelial growth factor and indirect mitogens such as norepinephrine, these molecules have
demonstrated limited in vitro bioactivity. This suggests that there may be currently unidentified
processes and pathways. Towards this end, FPHs and PHs may prove to be valuable research
tools.
In addition to studying the proliferative aspects of liver regeneration, FPHs and PHs may
also help answer questions regarding the source of liver functional output during hepatocyte
expansion. Literature reports that all essential liver functions (e.g., glucose regulation, synthesis
of blood proteins including albumin and coagulation proteins, secretion of bile, biodegradation of
toxic compounds) are intact even when 90% of resident hepatocytes are undergoing proliferation.
It is not clear how this functional output is maintained.
126
It is possible that all hepatocytes,
including those undergoing active cell cycle continue
normal functional output during
regeneration. However, traditional cell biology principles state that proliferative cellular
programs and functional ones are mutually exclusive states. In this case clinical observations of
intact liver functions during regeneration can only be explained by a temporary transformation of
the 10% of quiescent hepatocytes into a super-functional cell type capable of compensating for
the 90% of its peers undergoing proliferation. FPH and PH now enable investigation of such
questions in vitro.
Moving beyond the liver, identification of these small molecules may also empower the
study of cell fate control in other organ systems. We are currently investigating whether FPH and
PH molecules can induce in vitro proliferation of other normally quiescent primary cells,
particularly those intimately related to the liver throughout development. FPH2 was found to
enhance self-renewal of hematopoietic stem cells and we will look for bioactivity in primary
pancreatic beta cell systems.
In addition to understanding adult cellular processes, FH1 can also be applied towards
stem cell biology. While stem cells represent extremely attractive alternative cell sources, their
use is currently limited by our inability to fully dictate complete differentiation. FHl has been
shown to promote differentiation and maturation of iPS-derived human hepatocytes, and may
thus be helpful in the elucidation of cellular pathways involved in cell reprogramming. We also
have preliminary data showing that FHl promotes maturation of iPS-derived human endothelial
cells. The joint effects of FHl on both endodermally- and mesodermally-derived cell lineages
suggest that the reprogramming pathways accessed by FH 1 may be universal.
6.7 Using Small Molecules to Develop Molecular and Cellular Therapeutics
127
The bioactivity of FPH, PH and FH molecules both in vitro and in vivo engenders a wide
variety of novel applications worth further investigations:
"
Cells treated with these small molecules can be used for research and development of
novel therapeutics, including tissue engineered constructs, bioartificial liver devices, and
cell transplantation.
-
Small molecules can be used to expand patient biopsies or differentiate patient iPS cells
towards personalized medicine, which may include auto-transplantation of expanded
functional cells either directly or delivered within implantable hepatocellular constructs.
Furthermore, treated cells can be applied towards in vitro disease modeling and/or
ADME-toxicity testing for patient-specific drug selection and dose optimization.
-
Small molecules can be administered
systemically or locally to promote organ
regeneration, rescue failing and/or diseased organs, or serve as a bridge-to-transplant.
Compounds may act alone or in conjunction with other therapies in order to elicit
synergistic protection through the activation of multiple disease-prevention mechanisms.
-
Small molecules can enable smart implantable systems. FPH, PH and/or FH compounds
can be encapsulated in bio-responsive nanoparticles to trigger distinct cell fates based on
the type of therapy needed. Different classes of small molecules and nanoparticles can be
combined to generate a system that responds to multiple stimuli and can output multiple
different therapies.
-
Small molecules can be used to generate humanized mouse models for research and
therapy development.
-
Small molecules can be used to elucidate
cellular growth mechanisms, towards
identification of novel targets for cancer therapies.
128
Small molecules can be used to generate genotype-specific cells for disease modeling and
generation of new therapies customized to different genotypes. Such customization can
reduce adverse drug effects and help identify therapies appropriate to the patient's
genotype.
*
Small molecules may serve as adjunct therapy during organ regeneration following
partial liver transplantation.
It should be noted that the use of new therapies, while deserving of great excitement and
significant research efforts, should be implemented with due caution. Yamanaka, who first
reported the generation of iPS cells, has warned against their premature application to the clinics.
There are entire axes of stem cell biology that we do not yet understand and buried in those axes
could be cellular programs that will accidentally harm the patients we are trying so hard to help.
6.8 Maintenance of human hepatocytes in vitro using coatings of recombinant proteins and
soluble factors
While stromal cell co-cultures enable hepatocyte maintenance ex vivo, the use of a second
cell type of animal origins poses several key challenges including overgrowth of stromal cells
leading to nutrient and oxygen depletion and safety concerns. Thus it is desirable to replace
stromal cells with acellular biomaterials and/or recombinant protein products.
Previous work conducted by Bhatia and colleagues examining mechanisms involved in
hepatocyte-fibroblast co-culture had shown that heterotypic cell-cell interactions are required for
18-24 hrs before stromal-derived short range soluble factors alone can maintain hepatocyte
functions in vitro. This co-culture effect is independent of reciprocal hepatocyte-fibroblast
interactions, suggesting that it is mediated by constitutive expression of critical soluble factors by
129
fibroblasts. Using a gene expression profiling approach, Khetani et. al. had identified -50
candidate fibroblast genes that may play a role in stabilizing liver-specific functions, including a
novel candidate, truncated-cadherin (T-cadherin). As with other molecules previously implicated
in the co-culture effect (e.g., LRP, E-cadherin, TGF-betal, decorin), T-cadherin can induce
functions in rat hepatocytes for a limited time in vitro, but it cannot rescue the hepatic phenotype,
nor is it expressed by all cell types capable of inducing the co-culture effect. These findings
suggest that several stromal molecules may coordinate to stabilize hepatocyte functions in vitro.
Although these findings are promising, a complete picture of the mechanisms underlying the
stabilizing effect of fibroblasts remains elusive. This includes the unknown identity of a single
factor or a cocktail of factors that can adequately support hepatocytes in single culture,
emphasizing the need to apply objective, genome-wide approaches to these studies.
Currently, we are implementing high-throughput RNAi-based screens to identify the
most critical stromal-derived factors involved in the stabilization of primary human hepatocytes
in vitro. Such lentiviral-based RNAi methods have been shown to be compatible with high
content screening and valuable for identifying novel regulators of cellular processes. To date, we
have assembled and screened a custom set of -50 fibroblast genes previously implicated in the
hepatocyte-fibroblast co-culture effect through gene expression profiling studies (Table 6.1,
Table 6.2). These studies identified genes whose differential expression by three different
fibroblast cell lines correlated with their ability to rescue hepatocytes in culture (Figure 6.1 A).
The screening workflow is outlined in figure 6.1 C. Wild type J2-3T3 fibroblasts are seeded in
each well of a 384 well plate near confluency (4000 cells/well), and allowed 24 hours for
attachment, spreading, and adaptation to culture. These fibroblast monolayers are then infected
with lentivirus carrying shRNAs of interest (5-10 shRNA per gene) under optimized transduction
130
conditions. Infected fibroblasts were selected using puromycin for 48 hrs prior to introduction of
primary human hepatocytes. Fibroblast viability before and after puromycin selection is
quantified via Alamar Blue assay and used to determine infection efficiency (InfE). Wells with
InfE < 20% are discarded during analysis. Primary human hepatocytes are conditioned by
shRNA-modified fibroblasts for seven days, prior to functional analyses via ELISA and toxicity
analysis via image-based quantification of hepatocyte nuclei.
28 Positive Regulators of
Hepatocyte Functions
Gene
s
Prfn
}
B
LLJ -{+Lm J
Nio
egative Regulators of
Hepatocyte Functions
121K
10
D
lee
(Hep.
vh* vd
M
40M -
C
2000-
0 1000
12600 1600
8O
400
0
Fibroblast Density (cells/well)
.2
- -4
0L
0
2
4
6
[Puromycin] (ug/mL)
EEmpty Vectors
Pury Resistant
z
0
200-
1000
P
I
sr
Count)Atundn
0.
--
5
45000
-3000
ide
L Cortnn
Pr11tillnAsmW. Fu5.
A-
0
U
8
0
0.4 0.8 1.2
0
Infection Efficiency
Figure 6.1. High-throughput liver platform for validation of stromal factors hypothesized
to mediate the co-culture effect. A, ~50 3T3 genes were hypothesized to be important for coculture mediated maintenance of primary hepatocytes based on gene expression profiling. B,
vector design of shRNA library. C, screening workflow. D, platform and assay validation. Left
panel shows that Alamar Blue assay is able to accurately reflect fibroblast cell numbers. Middle
panel shows kill curve of various infection conditions. Right panel shows infection efficiency
during screening.
Gene
Symbol
Cd44
Igfbp2
U ( Jup
Ptprf
NCBI
Gene
NCB1Description
Gene ID
12505 CD44 antigen
16008 insulin-like growth factor binding protein 2
16480
junction plakoglobin
19268
protein tyrosine phosphatase, receptor type, F
131
3
Pkp2
67451
plakophilin 2
Adm
11535
adrenomedullin
Col8al
12837
collagen, type VIII, alpha I
Ctgf
14219
connective tissue growth factor
Inhba
16323
inhibin beta-A
Timp2
21858
tissue inhibitor of metalloproteinase 2
Dkk3
50781
dickkopf homolog 3 (Xenopus laevis)
Actal
11459
actin, alpha 1, skeletal muscle
Cdknla
Cyba
12575
13057
cyclin-dependent kinase inhibitor IA (P21)
cytochrome b-245, alpha polypeptide
F2r
14062
coagulation factor II (thrombin) receptor
Fhll
14199
four and a half LIM domains 1
Gtf2hl
14884
general transcription factor II H, polypeptide I
Laspi
Ndn
16796 LIM and SH3 protein 1
17984 necdin
Rock2
19878
Rho-associated coiled-coil containing protein kinase 2
Sec23a
20334
SEC23A (S. cerevisiae)
Sorbsl
Shcl
20411
20416
sorbin and SH3 domain containing 1
src homology 2 domain-containing transforming protein CI
Tpd52
Dpysl3
Ddx3y
21985
22240
26900
tumor protein D52
dihydropyrimidinase-like 3
DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, Y-linked
Tsnax
53424
translin-associated factor X
Pdliml
54132
PDZ and LIM domain 1 (elfin)
Htral
56213
HtrA serine peptidase 1
Prrc 1
73137 proline-rich coiled-coil 1
110596 Rho guanine nucleotide exchange factor (GEF) 28
Rgnef
Table 6.1. Negative regulators of hepatocyte functions.
o
Gene
NCBI
Symbol
Cdhl3
Dlkl
H2-K1
Gene ID
12554
13386
14972
12870
13179
14205
18812
20308
Cp
IT
Dcn
Figf
Prl2c3
Ccl9
0 d Dhfr
13361
Description
cadherin 13
delta-like 1 homolog (Drosophila)
histocompatibility 2, K1, K region
ceruloplasmin
decorin
c-fos induced growth factor
prolactin family 2, subfamily c, member 3
chemokine (C-C motif) ligand 9
dihydrofolate reductase
132
HmgbI
15289
high mobility group box 1
Ifi204
Irfl
15951
16362
interferon activated gene 204
interferon regulatory factor 1
Mlfl
Myolb
17349 myeloid leukemia factor 1
17912 myosin IB
Pold2
Srsf3
Ssb
18972
20383
20823
polymerase (DNA directed), delta 2, regulatory subunit
serine/arginine-rich splicing factor 3
Sjogren syndrome antigen B
Ttk
22137
Ttk protein kinase
Racgapl
Timm8al
Hnrpdl
26934
30058
50926
Rac GTPase-activating protein I
translocase of inner mitochondrial membrane 8A1
heterogeneous nuclear ribonucleoprotein D-like
Ifi2711
Dtdl
52668
66044
interferon, alpha-inducible protein 27 like 1
D-tyrosyl-tRNA deacylase 1
SnxlO
Cwc22
Hnrnpa3
Tardbp
N/A
Xlr
71982
80744
229279
230908
22441
sorting nexin 10
CWC22 spliceosome-associated protein homolog (S. cerevisiae)
heterogeneous nuclear ribonucleoprotein A3
TAR DNA binding protein
X-linked lymphocyte-regulated complex
Table 6.2. Positive regulators of hepatocyte functions.
Through this screen, we confirmed that at least 12 of the 50 genes identified via gene
expression profiling plays important roles in the co-culture effect (Table 6.3, Figure 6.2). These
12 genes were identified as hits via a competitive ELISA z score greater than 3.0, for at least two
shRNAs tested. While not all shRNAs can achieve successful knock-down, this requirement for
activity from multiple shRNAs of any given gene minimizes the risk of observing false positives
secondary to off-target effects. Among these 12 validated genes are T-cadherin and decorin.
Both molecules had previously been shown to help stabilize rat hepatocytes for a limited time in
vitro, but they cannot rescue the hepatic phenotype. Consistent with these literature findings, the
knock-down of any one of these 12 validated genes significantly compromised hepatocyte
stability in culture but did not fully abrogate the co-culture effect, strongly suggesting that
stromal molecules coordinate to maintain hepatic phenotype.
133
Table 6.3. 12 genes validated by custom shRNA screening.
134
A1
Empty Vector
Control Vector
Experimental Vector
Ud
B
Z 1.Z
-
-I
Ci
-......
..
-.
Hit Region
3:
-1 0 0102
10 2
-10 0
[Albumin] Z
--
20-
.
........
-C
301
Hepatocyte Toxicity Hepatocyte Function per Cel
Hepatocyte Function
-
1
........ ...
-- 8420
...
0 24
Hop Count Z
202
4
30
-4
4
2
.
Hep Count Z
10-
S -1to-
-
0
-10
CD
1k.
.
.
2.2.
0
-2
4- a
-..
30
20
10
U)
[Albumin] Z Score 1
~0
0d
2.
M
-6
-6
-C4
-4-2--4-2 Z
4
!
t2-
-
~-
-4
4
cC
Score 1
Hep Count Z
10 1
0. ..........
......
04
.
CL
. J......
4
S
N
A e
10
0 - -- .
Hap Count Z
Hap Count Z
[Albumin] Z
1.2 .
...........
o-
+0-
-A1
Hop Count Z
Hop Count Z
[Albumin] Z
.
....
0.6
CI 1
0 0 .4
0.2
-
- ,
0 10 20
[Albuminj Z
-10
-
0
10
-
--t.......... ..... ..... .
10
.
2 0 2
Hap Count Z
R-8-
2
30
4 Hap Count Z
4-
30
20
[Albumin] Z
W08 .
0.4
-C
-
2
--
C
-c
0
02
0420 2 4
Hep Count Z
....
....
cc
.2
-10
1 2-
1.2-
10
...........
4
------
-10
0
[A
-
30
10 20
bumin] Z
C2n
2
- 4 -2 0
Hap Count Z
-8
-4
0
-2
Hop Count Z
2
4
|-
N
20[VJL
..
..
....
12
3 10
..
. ^....
121.
08
-
..
.... .
-
-
..
. -.. - .. . . .
Q &~- -i4
1.Z
-
--
1~ -
1300 -
~
S.
.10l
6
4
-2
0
2
Hep Count Z
4
to
[Adbuminj Z
-1o0010
to
-4
-2
4 024
Hop Count Z
.842
A024
Hop Count Z
Figure 6.2 High-throughput identification of gene products important for J2-3T3-mediated
stabilization of primary hepatocytes in culture. A, primary screening data. Hits were selected
135
.. ...2.
based on decreased total albumin secretion, decreased total number of hepatocyte nuclei and
decreased albumin output on a per cell basis. B, selected hits.
Moving forward, we intend to examine all possible two-way combinations of the 12
validated genes with a new set of -450 genes, cherry-picked to include fibroblast secreted factors
and signaling molecules. In total, we will knockdown approximately 5,500 combinations of
fibroblast genes and assess their effects on hepatocyte function and survival. To this end, we will
generate 12 modified J2 fibroblast cell lines, each selected via blasticidin to harbor vectors
containing a validated shRNA. These modified J2 fibroblasts will then be used in genetic
screening of the new set of -450 custom genes (Figure .6.3), following the same workflow
previously outlined above.
Combinatorial Screen of
12 validated genes x 450 new genes
[~J
~
2
24 ?'t
4
W_
W
FN
t.
x12
~1
udoba
Figure 6.3. Schematic of combinatorial shRNA screen. 12 modified lines of J2-3T3s will be
generated through shRNA-mediated knockdown of previously validated genes involved in the
hepatocyte-fibroblast co-culture effect. Each line will be used to screen a custom set of -450
genes annotated as cell surface factors, secreted factors or factors involved in cell-cell signaling.
We expect that the knock down of multiple stromal factors will synergize to perturb coculture stabilization of primary human hepatocytes, possibly negating hepatocyte maintenance
completely. Promising shRNAs from this genetic screen will be used to perturb co-culture
stabilization of primary human hepatocytes in knock-down and over-expression studies using
136
fibroblasts. We expect the results to help elucidate mechanisms instrumental in the functional
maintenance of hepatocytes in vitro. This new insight will guide the assembly of a cocktail of
recombinant proteins containing hits encoding membrane-bound and secreted factors, towards
eventual replacement of stromal cells by acellular stromal products for maintenance of human
hepatocytes in vitro. Towards this end, various cocktails will be tested on pure primary human
hepatocytes. Lastly, select genes of interest will also be investigated in combination with small
molecule hits, to specifically identify factors that enable replication and phenotypic stabilization
of primary human hepatocytes. Through these investigations, we aim not only to elucidate the
importance of key secreted/membrane factors but also to replace stromal cells with their gene
products in engineered liver tissues (Figure 6.4).
Establish Renewable Source of
Human Hepatocytes
Prmary Screen of
12,480 Small Molecules
rHigh-throughput Platform
M
,L
2.wm~twje
am
and Assay Development
m proide
oom ice
500 genes
ce
7Replace
TIO1.
2ft"2.
Stabilize Human Hepatocytes
in vitro Using Stromal Factors
Non-parenchymal Cells with:
Secreted Factors *
Cell Surface Proteins
wm
Figure 6.4. Schematic of integrating chemical and genetic screening to generate a
renewable source of functional human primary hepatocyte in vitro. Our hypothesis is that
molecular signals from the stroma provide inductive cues, enabling hepatocyte maturation and
replication in vitro in response to small molecule exposure, and that these stromal signals can be
137
isolated and used to replace the fibroblasts in stabilizing hepatocytes. The platform described in
chapter 2 of this thesis can be used to identify small molecules and stromal factors involved in
the regeneration, maturation and phenotype maintenance of human hepatocytes in vitro.
Together, these studies can provide key insights into leveraging HTS technologies to provide
human hepatocytes needed for advancing cell-based therapies and furthering our understanding
of liver development, regeneration, and maintenance.
138
References
1
2
3
4
5
6
Lee, W. M. Acute liver failure in the United States. Semin Liver Dis 23, 217-226, doi:10.1055/s2003-42641 [doi] (2003).
Hoyert, D. L. & Xu, J. Deaths: Preliminary Data for 2011. National Vital Statistics Reports 61
(2012).
Kim, W. R., Brown, R. S., Terrault, N. A. & EI-Serag, H. Burden of liver disease in the United States:
Summary of a workshop. Hepatology 36, 227-242, doi:10.1053/jhep.2002.34734 (2002).
Brown, R. S. Hepatitis C and liver transplantation. Nature 436, 973-978,
doi:10.1038/nature04083 (2005).
Brown, K. A. Liver transplantation. Current Opinion in Gastroenterology 21, 331-336 (2005).
Wuestefeld, T. et al. A Direct In Vivo RNAi Screen Identifies MKK4 as a Key Regulator of Liver
Regeneration. Cell 153, 389-401, doi:S0092-8674(13)00348-6 [pii]
10.1016/j.cell.2013.03.026 [doi] (2013).
7
8
9
Allen, J. W., Hassanein, T. & Bhatia, S. N. Advances in bioartificial liver devices. Hepatology 34,
447-455 (2001).
Yarmush, M. L., Dunn, J. C. & Tompkins, R. G. Assessment of artificial liver support technology.
Cell Transplant 1, 323-341 (1992).
Chamuleau, R. A. Future of bioartificial liver support. World./ GastrointestSurg 1, 21-25,
doi:10.4240/wjgs.vl.il.21 [doi] (2009).
10
11
12
13
14
15
16
17
Lee, S. W., Wang, X., Chowdhury, N. R. & Roy-Chowdhury, J. Hepatocyte transplantation: state
of the art and strategies for overcoming existing hurdles. Ann Hepatol 3, 48-53, doi:8164 [pii]
(2004).
Kobayashi, N. et al. Prevention of acute liver failure in rats with reversibly immortalized human
hepatocytes. Science 287, 1258-1262 (2000).
Matas, A. J. et al. Hepatocellular transplantation for metabolic deficiencies: decrease of plasms
bilirubin in Gunn rats. Science 192, 892-894 (1976).
Bernardi, M., Tacconi, C., Somaroli, M., Gasbarrini, G. & Mazziotti, A. Hyperammonemic,
ammonia-independent coma in experimental acute liver failure induced in the pig.
Gastroenterology 81, 191-192 (1981).
Demetriou, A. A. et al. Transplantation of microcarrier-attached hepatocytes into 90% partially
hepatectomized rats. Hepatology 8, 1006-1009, doi:S0270913988001442 [pii] (1988).
Higgins G.M. and Anderson, R. M. Experimental pathology of liver: restoration of liver in white
rat following partial surgical removal. Arch. Pathol. 12, 186-202 (1931).
Iredale, J. P. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in
a solid organ. J Clin Invest 117, 539-548, doi:10.1172/JCl30542 [doi] (2007).
Azuma, H. et al. Robust expansion of human hepatocytes in Fah(-/-)/Rag2(-/-)/l2rg(-/-) mice.
Nature Biotechnology 25, 903-910 (2007).
Hughes, R. D., Mitry, R. R. & Dhawan, A. Hepatocyte transplantation in the treatment of liver
diseases - future seems bright after all. Pediatr Transplant 12, 4-5, doi:PTR853 [pii]
10.1111/j.1399-3046.2007.00853.x [doi] (2008).
Gupta, S., Gorla, G. R. & Irani, A. N. Hepatocyte transplantation: emerging insights into
19
mechanisms of liver repopulation and their relevance to potential therapies. Journal of
Hepatology 30, 162-170 (1999).
18
139
20
Dhawan, A., Puppi, J., Hughes, R. D. & Mitry, R. R. Human hepatocyte transplantation: current
experience and future challenges. Nat Rev Gastroenterol Hepatol 7, 288-298,
doi:nrgastro.2010.44 [pii]
10.1038/nrgastro.2010.44 [doi] (2010).
21
22
Fox, I. J. et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation.
N Engl J Med 338, 1422-1426, doi:10.1056/NEJM 199805143382004 [doi] (1998).
Puppi, J. et al. Hepatocyte transplantation followed by auxiliary liver transplantation--a novel
treatment for ornithine transcarbamylase deficiency. Am J Transplant 8, 452-457, doi:AJT2058
[pii]
10.1111/j.1600-6143.2007.02058.x [doi] (2008).
23
Chen, A. A. et al. Humanized mice with ectopic artificial liver tissues. Proceedings of the National
Academy of Sciences of the United States of America 108, 11842-11847,
doi:10.1073/pnas.1101791108 (2011).
24
Glicklis, R., Shapiro, L., Agbaria, R., Merchuk, J. C. & Cohen, S. Hepatocyte behavior within three-
dimensional porous alginate scaffolds. Biotechnol Bioeng 67, 344-353, doi:10.1002/(SICI)10970290(20000205)67:3<344::AID-BIT11>3.0.CO;2-2 [pii] (2000).
Chua, K. N. et al. Stable immobilization of rat hepatocyte spheroids on galactosylated nanofiber
scaffold. Biomaterials 26, 2537-2547, doi:S0142-9612(04)00681-7 [pii]
10.1016/j.biomaterials.2004.07.040 [doi] (2005).
Ranucci, C. S., Kumar, A., Batra, S. P. & Moghe, P. V. Control of hepatocyte function on collagen
26
foams: sizing matrix pores toward selective induction of 2-D and 3-D cellular morphogenesis.
Biomaterials 21, 783-793, doi:S0142961299002380 [pii] (2000).
Semino, C. E., Merok, J. R., Crane, G. G., Panagiotakos, G. & Zhang, S. Functional differentiation
27
of hepatocyte-like spheroid structures from putative liver progenitor cells in three-dimensional
peptide scaffolds. Differentiation 71, 262-270, doi:S0301-4681(09)60357-1 [pii]
10.1046/j.1432-0436.2003.7104503.x [doi] (2003).
Bruns, H. et al. Injectable liver: a novel approach using fibrin gel as a matrix for culture and
28
intrahepatic transplantation of hepatocytes. Tissue Eng 11, 1718-1726,
doi:10.1089/ten.2005.11.1718 [doi] (2005).
Fan, J., Shang, Y., Yuan, Y. & Yang, J. Preparation and characterization of chitosan/galactosylated
29
hyaluronic acid scaffolds for primary hepatocytes culture. J Mater Sci Mater Med 21, 319-327,
25
doi:10.1007/s10856-009-3833-y [doi] (2010).
30
31
Kasai, S. et al. Cellulose microcarrier for high-density culture of hepatocytes. Transplant Proc 24,
2933-2934 (1992).
Uygun, B. E. et aL. Organ reengineering through development of a transplantable recellularized
liver graft using decellularized liver matrix. Nat Med 16, 814-820, doi:nm.2170 [pii]
10.1038/nm.2170 [doil (2010).
32
33
Demetriou, A. A. et al. Replacement of liver function in rats by transplantation of microcarrierattached hepatocytes. Science 233, 1190-1192 (1986).
Li, K. et al. Chitosan/gelatin composite microcarrier for hepatocyte culture. Biotechnol Lett 26,
879-883, doi:5274932 [pii] (2004).
34
35
36
Baptista, P. M. et al. The use of whole organ decellularization for the generation of a
vascularized liver organoid. Hepatology 53, 604-617, doi:10.1002/hep.24067 [doi] (2011).
Mooney, D. J. et al. Biodegradable sponges for hepatocyte transplantation. J Biomed Mater Res
29, 959-965, doi:10.1002/jbm.820290807 [doi (1995).
Mooney, D. J. et al. Long-term engraftment of hepatocytes transplanted on biodegradable
polymer sponges. J Biomed Mater Res 37, 413-420, doi:10.1002/(SICI)10974636(19971205)37:3<413::AID-JBM12>3.0.CO;2-C [pii] (1997).
140
37
38
Itle, L. J., Koh, W. G. & Pishko, M. V. Hepatocyte viability and protein expression within hydrogel
microstructures. Biotechnol Prog 21, 926-932, doi:10.1021/bp049681i [doi] (2005).
Liu Tsang, V. et ci. Fabrication of 3D hepatic tissues by additive photopatterning of cellular
hydrogels. FASEB J 21, 790-801, doi:fj.06-7117com [pii]
10.1096/fj.06-7117com [doi] (2007).
Underhill, G. H., Chen, A. A., Albrecht, D. R. & Bhatia, S. N. Assessment of hepatocellular function
within PEG hydrogels. Biomaterials 28, 256-270, doi:S0142-9612(06)00760-5 [pii]
10.1016/j.biomaterials.2006.08.043 [doi] (2007).
Smith, M. K., Peters, M. C., Richardson, T. P., Garbern, J. C. & Mooney, D. J. Locally enhanced
40
angiogenesis promotes transplanted cell survival. Tissue Eng 10, 63-71,
doi:10.1089/107632704322791709 [doi] (2004).
Hasirci, V. et al. Expression of liver-specific functions by rat hepatocytes seeded in treated
41
poly(lactic-co-glycolic) acid biodegradable foams. Tissue Eng 7, 385-394,
39
doi:10.1089/10763270152436445 [doi] (2001).
42
43
Karamuk, E., Mayer, J., Wintermantel, E. & Akaike, T. Partially degradable film/fabric composites:
textile scaffolds for liver cell culture. Artif Organs 23, 881-884, doi:aor6308 [pii] (1999).
Lee, J. S., Kim, S. H., Kim, Y. J., Akaike, T. & Kim, S. C. Hepatocyte adhesion on a poly[N-pvinylbenzyl-4-0-beta-D-galactopyranosyl-D-glucoamide]-coated poly(L-lactic acid) surface.
Biomacromolecules 6, 1906-1911, doi:10.1021/bm049430y Idoi] (2005).
44
45
46
47
Catapano, G., Di Lorenzo, M. C., Della Volpe, C., De Bartolo, L. & Migliaresi, C. Polymeric
membranes for hybrid liver support devices: the effect of membrane surface wettability on
hepatocyte viability and functions. J Biomater Sci Polym Ed 7, 1017-1027 (1996).
Fiegel, H. C. et al. Influence of flow conditions and matrix coatings on growth and differentiation
of three-dimensionally cultured rat hepatocytes. Tissue Eng 10, 165-174,
doi:10.1089/107632704322791817 [doi] (2004).
Gutsche, A. T., Lo, H., Zurlo, J., Yager, J. & Leong, K. W. Engineering of a sugar-derivatized porous
network for hepatocyte culture. Biomaterials 17, 387-393, doi:0142961296855773 [pii] (1996).
Kim, M., Lee, J. Y., Jones, C. N., Revzin, A. & Tae, G. Heparin-based hydrogel as a matrix for
encapsulation and cultivation of primary hepatocytes. Biomaterials 31, 3596-3603, doi:S0142-
9612(10)00100-6 [pii]
10.1016/j.biomaterials.2010.01.068 [doi] (2010).
48
49
50
51
52
Park, T. G. Perfusion culture of hepatocytes within galactose-derivatized biodegradable
poly(lactide-co-glycolide) scaffolds prepared by gas foaming of effervescent salts. J Biomed
Mater Res 59, 127-135, doi:10.1002/jbm.1224 [pii] (2002).
Li, J., Pan, J., Zhang, L. & Yu, Y. Culture of hepatocytes on fructose-modified chitosan scaffolds.
Biomaterials 24, 2317-2322, doi:S0142961203000486 [pii] (2003).
Mayer, J., Karamuk, E., Akaike, T. & Wintermantel, E. Matrices for tissue engineering-scaffold
structure for a bioartificial liver support system. J Control Release 64, 81-90,
doi:S0168365999001364 [pii] (2000).
Carlisle, E. S. et al. Enhancing hepatocyte adhesion by pulsed plasma deposition and
polyethylene glycol coupling. Tissue Eng 6, 45-52, doi:10.1089/107632700320883 [doi] (2000).
Lutolf, M. P. et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of
tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A 100, 5413-
5418, doi:10.1073/pnas.0737381100 [doi]
0737381100 [pii] (2003).
53
Seliktar, D., Zisch, A. H., Lutolf, M. P., Wrana, J. L. & Hubbell, J. A. MMP-2 sensitive, VEGFbearing bioactive hydrogels for promotion of vascular healing. J Biomed Mater Res A 68, 704-
716, doi:10.1002/jbm.a.20091 [doi] (2004).
141
54
Mann, B. K., Gobin, A. S., Tsai, A. T., Schmedlen, R. H. & West, J. L. Smooth muscle cell growth in
photopolymerized hydrogels with cell adhesive and proteolytically degradable domains:
synthetic ECM analogs for tissue engineering. Biomaterials 22, 3045-3051,
doi:S0142961201000515 [pii] (2001).
55
56
57
58
Kim, S. S. et al. Survival and function of hepatocytes on a novel three-dimensional synthetic
biodegradable polymer scaffold with an intrinsic network of channels. Ann Surg 228, 8-13 (1998).
Petronis, S., Eckert, K. L., Gold, J. & Wintermantel, E. Microstructuring ceramic scaffolds for
hepatocyte cell culture. J Mater Sci Mater Med 12, 523-528, doi:353887 [pii] (2001).
Kaihara, S. et al. Silicon micromachining to tissue engineer branched vascular channels for liver
fabrication. Tissue Eng 6, 105-117, doi:10.1089/107632700320739 [doi] (2000).
Ogawa, K., Ochoa, E. R., Borenstein, J., Tanaka, K. & Vacanti, J. P. The generation of functionally
differentiated, three-dimensional hepatic tissue from two-dimensional sheets of progenitor
small hepatocytes and nonparenchymal cells. Transplantation 77, 1783-1789, doi:00007890-
200406270-00002 [pii] (2004).
59
60
61
62
Vozzi, G., Flaim, C., Ahluwalia, A. & Bhatia, S. Fabrication of PLGA scaffolds using soft lithography
and microsyringe deposition. Biomaterials 24, 2533-2540, doi:S0142961203000528 [pii] (2003).
Tan, W. & Desai, T. A. Microfluidic patterning of cells in extracellular matrix biopolymers: effects
of channel size, cell type, and matrix composition on pattern integrity. Tissue Eng 9, 255-267,
doi:10.1089/107632703764664729 [doil (2003).
Wang, X. et al. Generation of three-dimensional hepatocyte/gelatin structures with rapid
prototyping system. Tissue Eng 12, 83-90, doi:10.1089/ten.2006.12.83 [doi] (2006).
Hahn, M. S. et al. Photolithographic patterning of polyethylene glycol hydrogels. Biomaterials 27,
2519-2524, doi:S0142-9612(05)01087-2 [pii]
10.1016/j.biomaterials.2005.11.045 [doi] (2006).
Revzin, A. et aL. Fabrication of poly(ethylene glycol) hydrogel microstructures using
63
photolithography. Langmuir 17, 5440-5447 (2001).
Beebe, D. J. et al. Functional hydrogel structures for autonomous flow control inside microfluidic
64
channels. Nature 404, 588-590, doi:10.1038/35007047 [doi] (2000).
Liu, V. A. & Bhatia, S. N. Three-dimensional photopatterning of hydrogels containing living cells.
65
Biomedical Microdevices 4, 257-266, doi:10.1023/a:1020932105236 (2002).
Tan, W. & Desai, T. A. Layer-by-layer microfluidics for biomimetic three-dimensional structures.
66
Biomaterials 25, 1355-1364, doi:S0142961203006756 [pii] (2004).
Strain, A. J. Ex vivo liver cell morphogenesis: One step nearer to the bioartificial liver?
67
Hepatology 29, 288-290 (1999).
Dunn, J. C. Y., Yarmush, M. L., Koebe, H. G. & Tompkins, R. G. Hepatocyte Function and
68
69
70
71
72
Extracellular-Matrix Geometry - Long-Term Culture in a Sandwich Configuration. Faseb Journal 3,
174-177 (1989).
Allen, J. W. & Bhatia, S. N. Engineering liver therapies for the future. Tissue Engineering 8, 725737 (2002).
Hewitt, N. J. et al. Primary hepatocytes: Current understanding of the regulation of metabolic
enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in
metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug
Metabolism Reviews 39, 159-234 (2007).
Stocker E., W. H. K., Brau G. Capacity of regeneration in liver epithelia of juvenile, repeated
partially hepatectomized rats. Autoradiographic studies after continous infusion of 3Hthymidine. Virchows Arch. B Cell Pathol. 14 (1973).
Overturf, K. et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model
of hereditary tyrosinaemia type 1.Nature Genetics 12, 266-273 (1996).
142
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
Grompe, M., Overturf, K., Al-Dhalimy, M. & Finegold, M. Serial transplantation reveals stem cell
like regenerative potential in parenchymal mouse hepatocytes. Hepatology 24, 256A (1996).
Edwards, A. M., Michalopoulos, G. K. in The Hepatocyte Review (ed M. N. Berry, Edwards, A. M.)
73-96 (kluwer Academic Publishers, 2000).
Michalopoulos, G. et al. LIVER-REGENERATION STUDIES WITH RAT HEPATOCYTES IN PRIMARY
CULTURE. Cancer Research 42, 4673-4682 (1982).
Block, G. D. et al. Population expansion, clonal growth, and specific differentiation patterns in
primary cultures of hepatocytes induced by HGF/SF, EGF and TGF alpha in a chemically defined
(HGM) medium. Journal of Cell Biology 132, 1133-1149 (1996).
Ismail, T. et al. GROWTH OF NORMAL HUMAN HEPATOCYTES IN PRIMARY CULTURE - EFFECT OF
HORMONES AND GROWTH-FACTORS ON DNA-SYNTHESIS. Hepatology 14, 1076-1082 (1991).
Richman, R. A., Claus, T. H., Pilkis, S. J. & Friedman, D. L. HORMONAL-STIMULATION OF DNASYNTHESIS IN PRIMARY CULTURES OF ADULT RAT HEPATOCYTES. Proceedings of the National
Academy of Sciences of the United States of America 73, 3589-3593 (1976).
Mitaka, T., Sattler, C. A., Sattler, G. L., Sargent, L. M. & Pitot, H. C. MULTIPLE CELL-CYCLES OCCUR
IN RAT HEPATOCYTES CULTURED IN THE PRESENCE OF NICOTINAMIDE AND EPIDERMAL
GROWTH-FACTOR. Hepatology 13, 21-30 (1991).
Cable, E. E. & Isom, H. C. Exposure of primary rat hepatocytes in long-term DMSO culture to
selected transition metals induces hepatocyte proliferation and formation of duct-like structures.
Hepatology 26, 1444-1457 (1997).
Uyama, N. et al. Regulation of cultured rat hepatocyte proliferation by stellate cells. Journal of
Hepatology 36, 590-599 (2002).
Mizuguchi, T. et al. Enhanced proliferation and differentiation of rat hepatocytes cultured with
bone marrow stromal cells. Journal of Cellular Physiology 189, 106-119 (2001).
Cho, C. H., Berthiaume, F., Tilles, A. W. & Yarmush, M. L. A new technique for primary
hepatocyte expansion in vitro. Biotechnology and Bioengineering 101, 345-356 (2008).
Shimaoka, S., Nakamura, T. & Ichihara, A. STIMULATION OF GROWTH OF PRIMARY CULTURED
ADULT-RAT HEPATOCYTES WITHOUT GROWTH-FACTORS BY COCULTURE WITH
NON PARENCHYMAL LIVER-CELLS. Experimental Cell Research 172, 228-242 (1987).
Clayton, T. A. et al. Pharmaco-metabonomic phenotyping and personalized drug treatment.
Nature 440, 1073-1077 (2006).
Nelson, D. R. Cytochrome P450 and the individuality of species. Archives of Biochemistry and
Biophysics 369, 1-10 (1999).
Gibbs, R. A. et al. Genome sequence of the Brown Norway rat yields insights into mammalian
evolution. Nature 428, 493-521 (2004).
Werner, A. et al. Cultivation of immortalized human hepatocytes HepZ on macroporous
CultiSpher G microcarriers. Biotechnology and Bioengineering 68, 59-70 (2000).
Kono, Y., Yang, S. Y., Letarte, M. & Roberts, E. A. ESTABLISHMENT OF A HUMAN HEPATOCYTE
LINE DERIVED FROM PRIMARY CULTURE IN A COLLAGEN GEL SANDWICH CULTURE SYSTEM.
Experimental Cell Research 221, 478-485 (1995).
Kelly, J. H. & Darlington, G. J. MODULATION OF THE LIVER SPECIFIC PHENOTYPE IN THE HUMAN
HEPATOBLASTOMA LINE HEP-G2. In Vitro Cellular & Developmental Biology 25, 217-222 (1989).
Jauregui, H. 0. Cellular component of bioartificial liver support systems. Artificial Organs 23,
889-893 (1999).
Nyberg, S. L. et al. PRIMARY HEPATOCYTES OUTPERFORM HEP G2 CELLS AS THE SOURCE OF
BIOTRANSFORMATION FUNCTIONS IN A BIOARTIFICIAL LIVER. Annals of Surgery 220, 59-67
(1994).
143
93
94
95
Yanai, N., Suzuki, M. & Obinata, M. HEPATOCYTE CELL-LINES ESTABLISHED FROM TRANSGENIC
MICE HARBORING TEMPERATURE-SENSITIVE SIMIAN VIRUS-40 LARGE T-ANTIGEN GENE.
Experimental Cell Research 197, 50-56 (1991).
Kobayashi, N., Noguchi, H., Fujiwara, T. & Tanaka, N. Establishment of a reversibly immortalized
human hepatocyte cell line by using Cre/LoxP site-specific recombination. Transplantation
Proceedings 32, 1121-1122 (2000).
Cai, J. et al. Construction of a non-tumorigenic rat hepatocyte cell line for transplantation:
reversal of hepatocyte immortalization by site-specific excision of the SV40 T antigen. Journal of
Hepatology 33, 701-708 (2000).
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
Tateno, C. et al. Near completely humanized liver in mice shows human-type metabolic
responses to drugs. American Journal of Pathology 165, 901-912 (2004).
Katoh, M. et al. In vivo drug metabolism model for human cytochrome P450 enzyme using
chimeric mice with humanized liver. Journal of Pharmaceutical Sciences 96, 428-437 (2007).
Turrini, P. et al. Development of humanized mice for the study of hepatitis C virus infection.
Transplantation Proceedings 38, 1181-1184 (2006).
Michalopoulos, G. K. & DeFrances, M. C. Liver regeneration. Science 276, 60-66 (1997).
Rhim, J. A., Sandgren, E. P., Degen, J. L., Palmiter, R. D. & Brinster, R. L. Replacement of Diseased
Mouse-Liver by Hepatic Cell Transplantation. Science 263, 1149-1152 (1994).
Hamazaki, T. et al. Hepatic maturation in differentiating embryonic stem cells in vitro. Febs
Letters 497, 15-19 (2001).
Chinzei, R. et al. Embryoid-body cells derived from a mouse embryonic stem cell line show
differentiation into functional hepatocytes. Hepatology 36, 22-29, doi:10.1053/jhep.2002.34136
(2002).
Yamada, T. et al. In vitro differentiation of embryonic stem cells into hepatocyte-like cells
identified by cellular uptake of indocyanine green. Stem Cells 20, 146-154 (2002).
Cho, C. H. et al. Homogeneous differentiation of hepatocyte-like cells from embryonic stem cells:
applications for the treatment of liver failure. Faseb Journal 22, 898-909, doi:10.1096/fj.067764com (2008).
Gouon-Evans, V. et al. BMP-4 is required for hepatic specification of mouse embryonic stem cellderived definitive endoderm. Nature Biotechnology 24, 1402-1411, doi:10.1038/nbt1258 (2006).
Soto-Gutierrez, A. et al. Reversal of mouse hepatic failure using an implanted liver-assist device
containing ES cell-derived hepatocytes. Nature Biotechnology 24, 1412-1419,
doi:10.1038/nbt1257 (2006).
Wandzioch, E. & Zaret, K. S. Dynamic Signaling Network for the Specification of Embryonic
Pancreas and Liver Progenitors. Science 324, 1707-1710, doi:10.1126/science.1174497 (2009).
Lemaigre, F. & Zaret, K. S. Liver development update: new embryo models, cell lineage control,
and morphogenesis. Current Opinion in Genetics & Development 14, 582-590,
doi:10.1016/j.gde.2004.08.004 (2004).
Schmelzer, E. et al. Effect of Human Patient Plasma Ex Vivo Treatment on Gene Expression and
Progenitor Cell Activation of Primary Human Liver Cells in Multi-Compartment 3D Perfusion
Bioreactors for Extra-Corporeal Liver Support. Biotechnology and Bioengineering 103, 817-827,
doi:10.1002/bit.22283 (2009).
Zhang, L., Theise, N., Chua, M. & Reid, L. M. The stem cell niche of human livers: symmetry
between development and regeneration. Hepatology 48, 1598-1607, doi:10.1002/hep.22516
[doi] (2008).
111
Oh, S. H., Hatch, H. M. & Petersen, B. E. Hepatic oval 'stem' cell in liver regeneration. Semin. Cell
Dev. Biol. 13, 405-409, doi:10.1016/s1084-9521(02)00127-1 (2002).
144
112
113
Sell, S. The role of progenitor cells in repair of liver injury and in liver transplantation. Wound
Repair and Regeneration 9, 467-482 (2001).
Strick-Marchand, H., Morosan, S., Charneau, P., Kremsdorf, D. & Weiss, M. C. Bipotential mouse
embryonic liver stem cell lines contribute to liver regeneration and differentiate as bile ducts
and hepatocytes. Proceedings of the National Academy of Sciences of the United States of
America 101, 8360-8365, doi:10.1073/pnas.0401092101 (2004).
114
115
Strick-Marchand, H. & Weiss, M. C. Inducible differentiation and morphogenesis of bipotential
liver cell lines from wild-type mouse embryos. Hepatology 36, 794-804,
doi:10.1053/jhep.2002.36123 (2002).
Duncan, A. W., Dorrell, C. & Grompe, M. Stem cells and liver regeneration. Gastroenterology 137,
466-481, doi:S0016-5085(09)00818-X [pii]
10.1053/j.gastro.2009.05.044 [doi] (2009).
116
117
118
119
120
Schwartz, R. E. et a/. Multipotent adult progenitor cells from bone marrow differentiate into
functional hepatocyte-like cells. Journal of Clinical Investigation 109, 1291-1302 (2002).
Hong, S. H. et al. In vitro differentiation of human umbilical cord blood-derived mesenchymal
stem cells'into hepatocyte-like cells. Biochemical and Biophysical Research Communications 330,
1153-1161 (2005).
Ong, S. Y., Dai, H. & Leong, K. W. Hepatic differentiation potential of commercially available
human mesenchymal stem cells. Tissue Engineering 12, 3477-3485 (2006).
Sato, Y. et al. Human mesenchymal stem cells xenografted directly to rat liver are differentiated
into human hepatocytes without fusion. Blood 106, 756-763, doi:10.1182/blood-2005-02-0572
(2005).
Aurich, H. et al. Hepatocyte differentiation of mesenchymal stem cells from human adipose
tissue in vitro promotes hepatic integration in vivo. Gut 58, 570-581, doi:gut.2008.154880 [pii]
10.1136/gut.2008.154880 [doi] (2009).
121
122
123
124
125
126
127
128
129
130
De Coppi, P. et al. Isolation of amniotic stem cell lines with potential for therapy. Nature
Biotechnology 25, 100-106, doi:10.1038/nbt1274 (2007).
in 'tAnker, P. S. et al. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic
transplantation. Blood 102, 1548-1549 (2003).
Miki, T., Lehmann, T., Cai, H., Stolz, D. B. & Strom, S. C. Stem cell characteristics of amniotic
epithelial cells. Stem Cells 23, 1549-1559 (2005).
Miki, T. et al. Production of hepatocyte-like cells from human amnion. Methods Mol Biol 481,
155-168, doi:10.1007/978-1-59745-201-4_13 [doi] (2009).
Tsai, M. S., Lee, J. L., Chang, Y. J. & Hwang, S. M. Isolation of human multipotent mesenchymal
stem cells from second-trimester amniotic fluid using a novel two-stage culture protocol. Human
Reproduction 19, 1450-1456, doi:10.1093/humrep/deh279 (2004).
Lowry, W. E. et al. Generation of human induced pluripotent stem cells from dermal fibroblasts.
Proceedings of the National Academy of Sciences of the United States of America 105, 28832888, doi:10.1073/pnas.0711983105 (2008).
Park, I. H. et al. Reprogramming of human somatic cells to pluripotency with defined factors.
Nature 451, 141-U141, doi:10.1038/nature06534 (2008).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined
factors. Cell 131, 861-872, doi:10.1016/j.cell.2007.11.019 (2007).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and
adult fibroblast cultures by defined factors. Cell 126, 663-676 (2006).
Yu, J. Y. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318,
1917-1920, doi:10.1126/science.1151526 (2007).
145
131
132
133
134
135
136
137
138
139
140
Si-Tayeb, K. et al. Highly Efficient Generation of Human Hepatocyte-Like Cells from Induced
Pluripotent Stem Cells. Hepatology 51, 297-305, doi:10.1002/hep.23354 (2010).
Song, Z. et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem
cells. Cell Research 19, 1233-1242, doi:10.1038/cr.2009.107 (2009).
Sullivan, G. J. et al. Generation of Functional Human Hepatic Endoderm from Human Induced
Pluripotent Stem Cells. Hepatology 51, 329-335, doi:10.1002/hep.23335 (2010).
Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J. & Melton, D. A. In vivo reprogramming of adult
pancreatic exocrine cells to beta-cells. Nature 455, 627-632 (2008).
Lindros, K. 0. Zonation of cytochrome P450 expression, drug metabolism and toxicity in liver.
Gen Pharmacol 28, 191-196, doi:S0306362396001838 [pii] (1997).
Kietzmann, T. & Jungermann, K. Modulation by oxygen of zonal gene expression in liver studied
in primary rat hepatocyte cultures. Cell Biol Toxicol 13, 243-255 (1997).
Douarin, N. M. An experimental analysis of liver development. Medical Biology 53, 427-455
(1975).
Houssaint, E. Differentiation of the mouse hepatic primordium. 1.An analysis of tissue
interactions in hepatocyte differentiation. . Cell Differentiation 9, 269-279 (1980).
Michael J. Olson, M. A. M., Manjeri A. Venkatachalam, Arun K. Roy. in Cell Intercommunications
(ed W. De Mello) 71-92 (CRC press, Florida, 1990).
Bataller, R. & Brenner, D. A. Liver fibrosis. Journal of Clinical Investigation 115, 1100-1100,
doi:10.1172/jci200524282c1 (2005).
141
Corlu, A. et al. The coculture: A system for studying the regulation of liver
differentiation/proliferation activity and its control. Cell Biology and Toxicology 13, 235-242
(1997).
142
143
144
Bhatia, S. N., Balis, U. J., Yarmush, M. L. & Toner, M. Effect of cell-cell interactions in
preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells.
Faseb Journal 13, 1883-1900 (1999).
Guguenguillouzo, C. et al. Maintenance and Reversibility of Active Albumin Secretion by AdultRat Hepatocytes Co-Cultured with Another Liver Epithelial-Cell Type. Experimental Cell Research
143, 47-54 (1983).
Khetani, S. R., Szulgit, G., Del Rio, J. A., Barlow, C. & Bhatia, S. N. Exploring interactions between
rat hepatocytes and nonparenchymal cells using gene expression profiling. Hepatology 40, 545-
554 (2004).
145
146
147
Khetani, S. & Bhatia, S. N. Development and characterization of microscale models of rat and
human livers. Hepatology 46, 773A-773A (2007).
Allen, J. W. & Bhatia, S. N. Improving the next generation of bioartificial liver devices. Semin. Cell
Dev. Biol. 13, 447-454 (2002).
Delavega, F. M. & Mendozafigueroa, T. DIMETHYL-SULFOXIDE ENHANCES LIPID-SYNTHESIS AND
SECRETION BY LONG-TERM CULTURES OF ADULT-RAT HEPATOCYTES. Biochimie 73, 621-624,
doi:10.1016/0300-9084(91)90033-w (1991).
148
149
Guillouzo, A., Delers, F., Clement, B., Bernard, N. & Engler, R. LONG-TERM PRODUCTION OF
ACUTE-PHASE PROTEINS BY ADULT-RAT HEPATOCYTES CO-CULTURED WITH ANOTHER LIVERCELL TYPE IN SERUM-FREE MEDIUM. Biochemical and Biophysical Research Communications 120,
311-317, doi:10.1016/0006-291x(84)91255-5 (1984).
Guillouzo, A., Morel, F., Fardel, 0. & Meunier, B. USE OF HUMAN HEPATOCYTE CULTURES FOR
DRUG-METABOLISM STUDIES. Toxicology 82, 209-219, doi:10.1016/0300-483x(93)90065-z
(1993).
146
150
151
152
153
Langenbach, R., Freed, H. J. & Huberman, E. LIVER CELL-MEDIATED MUTAGENESIS OF
MAMMALIAN-CELLS BY LIVER CARCINOGENS. Proceedings of the National Academy of Sciences
of the United States of America 75, 2864-2867, doi:10.1073/pnas.75.6.2864 (1978).
Lebreton, J. P. et al. LONG-TERM BIOSYNTHESIS OF COMPLEMENT COMPONENT C-3 AND
ALPHA-1 ACID GLYCOPROTEIN BY ADULT-RAT HEPATOCYTES IN A COCULTURE SYSTEM WITH AN
EPITHELIAL LIVER CELL-TYPE. BiochemicalJournal 235, 421-427 (1986).
Mendozafigueroa, T., Hernandez, A. & Lopez, M. D. TRIGLYCERIDE ACCUMULATION IN LONGTERM CULTURES OF ADULT-RAT HEPATOCYTES BY CHRONIC EXPOSURE TO AROCLOR-1254.
Journal of Toxicology and Environmental Health 26, 293-308 (1989).
Mertens, K., Rogiers, V. & Vercruysse, A. GLUTATHIONE-DEPENDENT DETOXICATION IN ADULTRAT HEPATOCYTES UNDER VARIOUS CULTURE CONDITIONS. Archives of Toxicology 67, 680-685,
doi:10.1007/bf01973691 (1993).
154
155
156
157
Monden, K. et al. ENHANCEMENT OF HEPATIC MACROPHAGES IN SEPTIC RATS AND THEIR
INHIBITORY EFFECT ON HEPATOCYTE FUNCTION. Journal of Surgical Research 50, 72-76 (1991).
Rinconsanchez, A. R., Hernandez, A., Lopez, M. D. & Mendozafigueroa, T. SYNTHESIS AND
SECRETION OF LIPIDS BY LONG-TERM CULTURES OF FEMALE RAT HEPATOCYTES. Biology of the
Cell 76, 131-138, doi:10.1016/0248-4900(92)90205-f (1992).
Strom, S., Kligerman, A. D. & Michalopoulos, G. COMPARISONS OF THE EFFECTS OF CHEMICAL
CARCINOGENS IN MIXED CULTURES OF RAT HEPATOCYTES AND HUMAN-FIBROBLASTS.
Carcinogenesis 2, 709-715, doi:10.1093/carcin/2.8.709 (1981).
West, M. A., Manthei, R., Bubrick, M. P., Lanser, M. E. & Moore, E. E. AUTOREGULATION OF
HEPATIC MACROPHAGE ACTIVATION IN SEPSIS. Journal of Trauma-Injury Infection and Critical
Care 34, 473-480, doi:10.1097/00005373-199304000-00001 (1993).
158
159
160
161
Chen, S. B., Hilcove, S. & Ding, S. Exploring stem cell biology with small molecules. Molecular
Biosystems 2, 18-24 (2006).
North, T. E. et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis.
Nature 447, 1007-U1007, doi:10.1038/nature05883 (2007).
Wang, W. D. et al. Identification of small-molecule inducers of pancreatic beta-cell expansion.
Proceedings of the National Academy of Sciences of the United States of America 106, 14271432 (2009).
Goessling, W. et al. Genetic Interaction of PGE2 and Wnt Signaling Regulates Developmental
Specification of Stem Cells and Regeneration. Cell 136, 1136-1147,
doi:10.1016/j.cell.2009.01.015 (2009).
162
Eggert, U. S. The why and how of phenotypic small-molecule screens. Nat Chem Biol 9, 206-209,
doi:nchembio.1206 [pii]
10.1038/nchembio.1206 [doi] (2013).
163
Schenone, M., Dancik, V., Wagner, B. K. & Clemons, P. A. Target identification and mechanism of
action in chemical biology and drug discovery. Nat Chem Biol 9, 232-240, doi:nchembio.1199 [pii]
10.1038/nchembio.1199 [doi] (2013).
164
Swinney, D. C. & Anthony, J. How were new medicines discovered? Nat Rev Drug Discov 10, 507519, doi:nrd3480 [pii]
10.1038/nrd3480 [doi] (2011).
165
Castoreno, A. B. & Eggert, U. S. Small molecule probes of cellular pathways and networks. ACS
Chem Biol 6, 86-94, doi:10.1021/cb1002976 [doi] (2011).
166
167
McNamara, C. & Winzeler, E. A. Target identification and validation of novel antimalarials.
Future Microbiol 6, 693-704, doi:10.2217/fmb.11.45 [doi] (2011).
Knight, Z. A., Lin, H. & Shokat, K. M. Targeting the cancer kinome through polypharmacology.
Nat Rev Cancer 10, 130-137, doi:nrc2787 [pii]
147
10.1038/nrc2787 [doij (2010).
168
169
Khetani, S. R. & Bhatia, S. N. Microscale culture of human liver cells for drug development.
Nature Biotechnology 26, 120-126 (2008).
Bhatia, S. N., Yarmush, M. L. & Toner, M. Controlling cell interactions by micropatterning in cocultures: Hepatocytes and 3T3 fibroblasts. Journal of Biomedical Materials Research 34, 189-199
(1997).
170
171
Millis, J. M. et al. Initial experience with the modified extracorporeal liver-assist device for
patients with fulminant hepatic failure: system modifications and clinical impact.
Transplantation 74, 1735-1746, doi:10.1097/01.TP.0000038483.93833.21 [doi] (2002).
van de Kerkhove, M. P., Hoekstra, R., Chamuleau, R. A. & van Gulik, T. M. Clinical application of
bioartificial liver support systems. Ann Surg 240, 216-230, doi:00000658-200408000-00005 [pii]
172
173
174
(2004).
Sauer, I. M. et al. Primary human liver cells as source for modular extracorporeal liver support--a
preliminary report. IntiJArtif Organs 25, 1001-1005 (2002).
Demetriou, A. A. et al. Prospective, randomized, multicenter, controlled trial of a bioartificial
liver in treating acute liver failure. Annals of Surgery 239, 660-667,
doi:10.1097/01.sla.0000124298.74199.e5 (2004).
Ploss, A. et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte
cultures. Proceedings of the National Academy of Sciences of the United States of America 107,
3141-3145, doi:10.1073/pnas.0915130107 (2010).
Schwartz, R. E. et al. Modeling hepatitis C virus infection using human induced pluripotent stem
cells. Proceedings of the National Academy of Sciences of the United States of America 109,
2544-2548, doi:10.1073/pnas.1121400109 (2012).
Hui, E. E. & Bhatia, S. N. Micromechanical control of cell-cell interactions. Proceedings of the
176
National Academy of Sciences of the United States of America 104, 5722-5726 (2007).
Carpenter, A. E. et al. CellProfiler: image analysis software for identifying and quantifying cell
177
phenotypes. Genome Biology 7, doi:R100
10.1186/gb-2006-7-10-rOO (2006).
Jones, T. R. et al. CellProfiler Analyst: data exploration and analysis software for complex image178
based screens. Bmc Bioinformatics 9, doi:482
10.1186/1471-2105-9-482 (2008).
Zhang, J. H., Chung, T. D. Y. & Oldenburg, K. R. A simple statistical parameter for use in
179
evaluation and validation of high throughput screening assays. Journal of Biomolecular
Screening 4, 67-73 (1999).
Dabeva, M. D. & Shafritz, D. A. Hepatic stem cells and liver repopulation. Seminars in Liver
180
Disease 23, 349-361 (2003).
S. S. & Grisham, J. W. Overview of recent experimental studies on liver stem cells.
Thorgeirsson,
181
Seminars in Liver Disease 23, 303-312 (2003).
Jiang, Y. H. et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature
182
175
183
184
185
418, 41-49 (2002).
Mitaka, T. The current status of primary hepatocyte culture. International Journal of
Experimental Pathology 79, 393-409 (1998).
Runge, D., Michalopoulos, G. K., Strom, S. C. & Runge, D. M. Recent advances in human
hepatocyte culture systems. Biochemical and Biophysical Research Communications 274, 1-3
(2000).
Tan, D. S. Diversity-oriented synthesis: exploring the intersections between chemistry and
biology. Nat Chem Biol 1, 74-84, doi:nchembio0705-74 [pii]
10.1038/nchembio0705-74 [doi] (2005).
148
186
187
188
189
Shepard, C. W., Finelli, L. & Alter, M. Global epidemiology of hepatitis C virus infection. Lancet
Infectious Diseases 5, 558-567, doi:10.1016/s1473-3099(05)70216-4 (2005).
Lin, H. M. et al. Center-specific graft and patient survival rates - 1997 United Network for Organ
Sharing (UNOS) Report. Jama-Journal of the American Medical Association 280, 1153-1160
(1998).
Castell, J. V., Jover, R., Martinez-Jimenez, C. P. & Gomez-Lechon, M. J. Hepatocyte cell lines:
their use, scope and limitations in drug metabolism studies. Expert Opinion on Drug Metabolism
& Toxicology 2, 183-212, doi:10.1517/17425255.2.2.183 (2006).
North, T. E. et al. PGE2-regulated wnt signaling and N-acetylcysteine are synergistically
hepatoprotective in zebrafish acetaminophen injury. Proceedings of the National Academy of
Sciences of the United States of America 107, 17315-17320, doi:10.1073/pnas.1008209107
(2010).
190
191
Michalopoulos, G. K. Liver regeneration. Journal of Cellular Physiology 213, 286-300 (2007).
Touboul, T., Vallier, L. & Weber, A. Robust differentiation of fetal hepatocytes from human
embryonic stem cells and iPS. M S-Medecine Sciences 26, 1061-1066,
doi:10.1051/medsci/201026121061 (2010).
192
Kang, L., Wang, J., Zhang, Y., Kou, Z. & Gao, S. iPS Cells Can Support Full-Term Development of
Tetraploid Blastocyst-Complemented Embryos. Cell Stem Cell 5, 135-138,
doi:10.1016/j.stem.2009.07.001 (2009).
Zhao, X.-y. et al. iPS cells produce viable mice through tetraploid complementation. Nature 461,
86-U88, doi:10.1038/nature08267 (2009).
Payne, C. M. et al. Persistence of functional hepatocyte-like cells in immune-compromised mice.
194
Liver International 31, 254-262, doi:10.1111/j.1478-3231.2010.02414.x (2011).
195
Liu, H., Kim, Y., Sharkis, S., Marchionni, L. & Jang, Y.-Y. In Vivo Liver Regeneration Potential of
Human Induced Pluripotent Stem Cells from Diverse Origins. Science Translational Medicine 3,
doi:82ra39
10.1126/scitranslmed.3002376 (2011).
Jozefczuk, J., Prigione, A., Chavez, L. & Adjaye, J. Comparative Analysis of Human Embryonic
196
Stem Cell and Induced Pluripotent Stem Cell-Derived Hepatocyte-Like Cells Reveals Current
Drawbacks and Possible Strategies for Improved Differentiation. Stem Cells and Development 20,
1259-1275, doi:10.1089/scd.2010.0361 (2011).
Behbahan, I. S. et al. New Approaches in the Differentiation of Human Embryonic Stem Cells and
197
Induced Pluripotent Stem Cells toward Hepatocytes. Stem Cell Reviews and Reports 7, 748-759,
193
doi:10.1007/s12015-010-9216-4 (2011).
198
199
Kroon, E. et al. Pancreatic endoderm derived from human embryonic stem cells generates
glucose-responsive insulin-secreting cells in vivo. Nature Biotechnology 26, 443-452,
doi:10.1038/nbt1393 (2008).
Chu, J. & Sadler, K. C. New school in liver development: lessons from zebrafish. Hepatology 50,
1656-1663, doi:10.1002/hep.23157 [doi] (2009).
200
Peterson, R. T. & Macrae, C. A. Systematic approaches to toxicology in the zebrafish. Annu Rev
Pharmacol Toxicol 52, 433-453 (2012).
201
Lai, M. W. et al. 2005 Annual Report of the American Association of Poison Control Centers'
national poisoning and exposure database. Clin Toxicol (Phila) 44, 803-932,
doi:G33005368TP30667 [pii]
10.1080/15563650600907165 [doi] (2006).
202
Whyte, 1.M., Francis, B. & Dawson, A. H. Safety and efficacy of intravenous N-acetylcysteine for
acetaminophen overdose: analysis of the Hunter Area Toxicology Service (HATS) database. Curr
Med Res Opin 23, 2359-2368, doi:10.1185/030079907X219715 [doil (2007).
149
203
Her, G. M., Chiang, C. C., Chen, W. Y. & Wu, J. L. In vivo studies of liver-type fatty acid binding
protein (L-FABP) gene expression in liver of transgenic zebrafish (Danio rerio). FEBS Lett 538,
125-133, doi:S0014579303001571 [pii] (2003).
Parts of this thesis are adapted from publications number [1] and [5] listed in the Biographical
Information Section.
150