Capsid catalysis: de novo enzymes on viral proteins
by
John P. Casey, Jr.
B.S.B.E., Louisiana State University (2009)
Submitted to the Department of Biological Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
I
C=w
Z
(/2L
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2015
Massachusetts Institute of Technology 2015. All rights reserved.
Signature redacted
department of Biolqd$'Ei$gineering
May 11, 2015
Certified by...
Signature redacted
..................
Angela M. Belcher
W.M. Keck Professor of Energy
Professor of Biological Engineering
Professor of Materials Science and Engineering
Thesis Supervisor
___%1 -
Accepted by .............
5
Cy')
at the
A uthor ........................
C:)
I
Signature redacted
Forest White
Professor of Biological Engineering
Chairman, Department Graduate Program Committee
<
2
Capsid catalysis: de novo enzymes on viral proteins
by
John P. Casey, Jr.
Submitted to the Department of Biological Engineering
on May 11, 2015, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy
Abstract
Biocatalysis has grown rapidly in recent decades as a solution to the evolving demands
of industrial chemical processes. Mounting environmental pressures and shifting supply chains underscore the need for novel chemical activities, while rapid biotechnological progress has greatly increased the utility of enzymatic methods. Enzymes, though
capable of high catalytic efficiency and remarkable reaction selectivity, still suffer from
relative instability, high costs of scaling, and functional inflexibility. Herein, M13
bacteriophage libraries are engineered as a biochemical platform for de novo semisynthetic enzymes, functionally modular and widely stable. Carbonic anhydrase-inspired
hydrolytic activity via Zn 2 + co6rdination is first demonstrated. The phage clone identified hydrolyzes a range of carboxylic esters, is active from 25'C to 80*C, and displays
greater catalytic efficacy in DMSO than in water. Reduction-oxidation activity is subsequently developed via heme and copper cofactors. Heme-phage complexes oxidize
multiple peroxidase substrates in a pH-dependent manner. The same phage clone also
binds copper(II) and oxidizes a catechol derivative, di-tert-butylcatechol, using atmospheric oxygen as a terminal oxidant. This clone could be purified from control phage
via Cu-NTA columns, enabling future library selections for phage that co6rdinate
Cu2+ ions. The M13 semisynthetic enzyme platform complements biocatalysts with
characteristics of heterogeneous catalysis, yielding high-surface area, thermostable
biochemical structures readily adaptable to reactions in myriad solvents. As the viral structure ensures semisynthetic enzymes remain linked to the genetic sequences
responsible for catalysis, future work could tailor the biocatalysts to high-demand
synthetic processes by evolving new activities, utilizing high-throughput screening
technology and harnessing M13's multifunctionality.
Thesis Supervisor: Angela M. Belcher
Title: W.M. Keck Professor of Energy
Professor of Biological Engineering
Professor of Materials Science and Engineering
3
Acknowledgments
"Knowledge for its own sake" - that is the last snare of morality: with
that one becomes completely entangled in it once more.
Friedrich Nietzsche, Beyond Good and Evil
First and foremost, I want to thank my family. From the start, my parents have
taught me to work hard, treat people well, and live honestly, come rain or shine. Such
ideals are especially relevant in an increasingly competitive, corporatized research
environment and have guided me during my academic career. My sister, meanwhile,
has always looked out for me and kept me grounded. Throughout the past six years,
all three have provided unwavering support for my plodding research progress.
The two people most responsible for my interest in the field, and for training me
as an engineer, are LSU professors Dr. Todd Monroe and Dr. Marybeth Lima. With
extremely wide-ranging expertise, Dr. Monroe advised me throughout my time at
LSU on numerous projects inside the lab and out. The diversity of the projects in
which I was engaged, as well as his depth of involvement and gracious encouragement
to me in seeking complementary opportunities, have time and again helped me address
challenges during my thesis project in an expeditious manner. Additionally, without
his guidance with respect to coursework, fellowships, and my career in general, I likely
would never have had the opportunity conduct research here at MIT. Dr. Lima,
meanwhile, forced me early on to consider fundamentally what it means to be a
biological engineer, and how to do so with maximum impact for society. She also
made clear the importance of effective communication as an engineer, a lesson that
has only been magnified since.
At MIT, my work is the product of the campus' collegiality and BE's eminent,
erudite faculty. I would of course like to thank my thesis advisor, Angie, for her advice
on my project and trust & encouragement in my pursuit of untraditional ideas. Prof.
Wittrup has imbued a strong dose of skepticism and empirical rigor, from 20.420
onward, that colors how I conduct my own work and review others'. Regardless
the topic, my meetings with Prof. Bhatia have always ended in a wellspring of
exciting ideas and directions. Even after drastically changing my research focus,
my interactions with my committee members have been rich with both constructive
criticism and productive support, for which I am extremely grateful.
The Belcher Lab has been a consistent inspiration and driver of the ideas behind
my project. Drs. Robbie Barbero, Nimrod Heldman, and Paul Widboom conceived
the idea for phage-based biocatalysis and built the original libraries. Robbie and
Nimrod, especially, have been regular sources of guidance and expertise throughout
the subsequent experimentation. Profs. Mark Allen and John Burpo were similarly generous with their time in all manner of troubleshooting. Jackie Ohmura and
Dong Soo Yoon helped me with electron microscopy, and the Swanson Biotechnology Center is responsible for most of the sequencing used to confirm phage identity.
Dr. Dahyun Oh, Dr. Gaelen Hess, Matt Klug, and Griffin Clausen were supportive
4
sounding boards and engaged me when I sought to learn about other biotechnologies.
Additionally, I'd like to thank my UROP Anu Sinha for his help in characterizing the
TENM clone.
More broadly, the MIT community has provided a great environment in which to
work and think. The BE community in particular has been a continuing source of energy and support. My classmates Melissa, Carrie, Alex & Seymour, Aaron, Rebecca,
Adrian, Kara and Deepak made the program here unforgettably fun. The Biological Engineering Communication Lab has resulted in a number of deep friendships
and thoughtful conversations while immensely improving my ability to express my
ideas. Diana Chien has been especially helpful in improving my writing. The MIT
Rugby Football Club provided a home-away-from-home while deeply humbling and
strengthening me. The MIT $100K gave me an immediate, hands-on, simultaneous
education in organizational management, entrepreneurship, and mentorship, which
has become invaluable. The MIT community in aggregate has made my doctoral
progression fascinating and fulfilling.
5
6
Contents
15
1.1
Strategies for Applied Biocatalysis . . . . . . . . . . . . . . . . . . .
16
1.1.1
Engineering enzymes for greater utility . . . . . . . . . . . .
17
1.1.2
De novo enzyme design . . . . . . . . . . . . . . . . . . . . .
18
.
.
Introduction
.
1
1.2
Objective: M13 Bacteriophage as a Platform for Biocatalyst Engineering 21
1.3
Dissertation Overview
. . . . . . . . . . . . . . . . . . . . . . . . . .
23
2 Isolating Enzymatic Bacteriophage From an Engineered p8 Library 25
2.1
Background: Zinc-Co6rdinating Enzymes
2.2
Strategy for M13 Enzyme Engineering
2.3
R esults . . . . . . . . . . . . . . . . . . . .
28
2.3.1
pNPA hydrolysis
28
2.3.2
Minimizing DDAH aggregation
. .
32
2.3.3
Column purification of clone DDAH
32
2.3.4
Structure-function relationships of the DDAH active site
36
. . . . . . . . . . . . . .
26
.
.
26
Sum m ary
. . . . . . . . . . . . . . . . . .
38
2.5
Experimental Methods . . . . . . . . . . .
40
2.5.1
Library Construction . . . . . . . .
40
2.5.2
Bacterial culture and M13 stocks
.
43
2.5.3
Hydrolysis reactions
. . . . . . . .
45
2.5.4
TEM of DDAH with Zn 2
. . . . .
46
2.5.5
Ni-NTA purification of phage clones
46
.
.
.
.
.
.
2.4
.
.
.
.
. . . . . . . . . .
7
3 DDAH Hydrolysis Activity is Versatile
4
47
3.1
Background: Engineering Robust Enzymes ...............
48
3.2
Results . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
50
3.2.1
DDAH activity is robust in nonaqueous solvents . . . . . . . .
50
3.2.2
The DDAH active site can accommodate large substrates . . .
53
3.2.3
DDAH exhibits thermostable catalysis
. . . . . . . . . . . . .
55
3.2.4
Alternate esterolysis substrates
. . . . . . . . . . . . . . . . .
61
3.3
Sum m ary
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
63
3.4
Experimental Methods . . . . . . . . . . . . . . . . . . . . . . . . . .
64
3.4.1
Hydrolysis reactions
64
3.4.2
Bacterial culture and M13 stocks
3.4.3
TEM of DDAH in water and DMSO
. . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . .
. . . . . . . . . . . . . .
67
68
M13 Catalyzes Heme- and Copper-Based Redox Reactions
71
4.1
Background . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
72
4.1.1
Heme peroxidase engineering and applications . . . . . . . . .
72
4.1.2
Copper oxidase engineering and applications . . . . . . . . . .
75
4.2
4.3
Results: Heme Peroxidase Activity
. . . . . . . . . . . . . . . . . . .
76
4.2.1
Identifying and characterizing heme-binding M13 clones . . . .
76
4.2.2
Estimating DDAH p8-heme KD via Soret peaks . . . . . . . .
79
4.2.3
DDAH peroxidase oxidizes TMB
. . . . . . . . . . . . . . . .
81
4.2.4
DDAH peroxidase oxidizes ABTS . . . . . . . . . . . . . . . .
84
4.2.5
Heme-agarose columns retain DDAH phage
. . . . . . . . . .
88
Results: Copper Oxidase Activity . . . . . . . . . . . . . . . . . . . .
89
4.3.1
DDAH co6rdinates Cu 2 +
. . . . . . . . . . . . . . . . . . . .
89
4.3.2
DDAH oxidation of catechol and derivatives . . . . . . . . . .
92
4.3.3
Cu-NTA columns selectively retain Cu2+ -binding phage
. . .
93
4.4
Sum m ary
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
95
4.5
Experimental Methods . . . . . . . . . . . . . . . . . . . . . . . . . .
97
4.5.1
97
Bacterial culture and M13 stocks
8
. . . . . . . . . . . . . . . .
4.5.2
Protocols for assaying phage activity with heme . . . . . . . .
99
4.5.3
Protocols for assaying phage-copper interactions . . . . . . . .
103
9
10
Binary patterning of oz-helices . . . . . . . . . .
. . . . . . . .
20
1-2
M13 Dimensions
. . . . . . . . . . . . . . . . .
. . . . . . . .
22
2-1
Design of semisynthetic active sites for hydrolysis
. . . . . . . .
27
2-2
Activity of tested M13 clones
. . . . . . . . . .
. . . . . . . .
30
2-3
Overview of data regressions . . . . . . . . . . .
. . . . . . . .
31
2-4
DDAH-cation dissociation constants . . . . . . .
. . . . . . . .
33
2-5
Zinc-induced phage aggregation . . . . . . . . .
. . . . . . . .
34
2-6
Ni-NTA purification of DDAH . . . . . . . . . .
. . . . . . . .
35
2-7
Catalytic rates for DDAH and mutants . . . . .
.
. . . . . . . .
37
2-8
Oligonucleotides used for library construction
.
. . . . . . . .
42
3-1
Reaction Rates in DMSO . . . . . . . . . . . . .
. . . . . . . .
51
3-2
DDAH stably hydrolyzes pNPA in DMSO
. . .
. . . . . . . .
52
3-3
TEM of DDAH in water and DMSO
. . . . . .
. . . . . . . .
54
3-4
DDAH catalyzes a range of substrates . . . . . .
. . . . . . . .
56
3-5
Hydrolysis activity after 80'C heat kill . . . . .
. . . . . . . .
58
3-6
DDAH catalysis at raised temperatures . . . . .
. . . . . . . .
59
4-1
Enzymatic cycle for heme
73
4-2
Amplification of cysteine and 4-His clones
.
77
4-3
Soret shift by DDAH . . . . . . . . . . . . .
79
4-4
Soret peaks for 5-50 pM heme with DDAH.
80
4-5
Fitting heme-DDAH KD via Soret peak values
81
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1-1
.
List of Figures
.
.
. . . . . . . . . .
11
DDAH oxidizes TMB ......................
4-7
Inferring
4-8
Squared errors from
4-9
DDAH oxidizes ABTS
82
. . . . . . .
. . . . . . .
83
regressions to TMB oxidation
. . . . . . .
84
. . . . . . . . . . . . . . . . .
. . . . . . .
86
. . . . . . .
87
4-11 Hemin-agarose columns retain and purify DDAH stocks
. . . . . . .
88
4-12 Spectra of phage-copper mixtures . . . . . . . . . . .
. . . . . . .
90
4-13 Oxidation of catechol by DDAH . . . . . . . . . . . .
. . . . . . .
91
4-14 Oxidation of DTBC by DDAH . . . . . . . . . . . . .
. . . . . . .
94
4-15 Spectra of 2-Cys and 4-His phage stocks . . . . . . .
. . . . . . .
100
from TMB oxidation rates
KD
.
KD
.
. . . . . . .
.
4-6
. . . .
.
.
.
.
4-10 DDAH oxidation of ABTS is specific to heme
12
List of Tables
2.1
Clones tested for catalysis . . . . . . . . . . . . . . . . . . . . . . . .
29
2.2
Catalysis by DDAH variants and controls . . . . . . . . . . . . . . . .
38
3.1
Background hydrolysis rates . . . . . . . . . . . . . . . . . . . . . . .
51
3.2
DDAH infectivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
52
3.3
Hydrolysis rates for various substrates
. . . . . . . . . . . . . . . . .
57
3.4
Hydrolysis parameters at various temperatures . . . . . . . . . . . . .
60
3.5
Aqueous phenylethyl acetate hydrolysis characterization . . . . . . . .
62
4.1
Soret peaks of phage clones
. . . . . . . . . . . . . . . . . . . . . . .
78
4.2
Extinction coefficients for phage-Cu2+ complexes . . . . . . . . . . . .
92
4.3
Sequenced plaques from Cu-NTA column . . . . . . . . . . . . . . . .
94
4.4
Infectivity of 2-Cys and 4-His clones . . . . . . . . . . . . . . . . . . .
98
13
14
Chapter 1
Introduction
[.. . I
the organism is capable of performing highly specific chemi-
cal transformations which can never be accomplished with the customary
agents. To equal Nature here, the same means have to be applied, and I
therefore foresee the day when physiological chemistry will not only make
extensive use of the natural enzymes as agents, but when it will also prepare synthetic ferments [enzymes] for its purposes.
-
Emil Fischer's Nobel acceptance speech, 1902
A globular protein modified by introduction of a catalytically active
metal at an appropriate site could, in principle, provide an exceptionally
well-defined steric environment around that metal, and should do so for
considerably smaller effort than would be required to construct a synthetic
substance of comparable stereochemical complexity.
-
M.E. Wilson & G.M. Whitesides, J. Am. Chem. Soc. 19781
Biocatalysts potentiate a wide range of chemical activities and functionality not
practical with abiotic processes. As such, they represent a growing corner of industrial
chemical production. In order to fully utilize enzymatic activity* in industrial settings,
however, new strategies and tools for adaptation will be required. M13 bacteriophage,
*Enzymes are a proteinaceous subset of biocatalysts; for the purpose of this text, though, the
terms "enzyme" and "biocatalyst" (and, for Dr. Fischer, "ferment") are interchangeable.
15
a robust macromolecule previously engineered and evolved for numerous applications,
is a promising candidate for stable, adaptable biocatalysis. In this dissertation, I'll
discuss:
1. applied biocatalysis and where enzymatic bacteriophage could add value;
2. how we have engineered phage-displayed peptides with catalytic potential;
3. the versatility of M13 bacteriophage as an esterolytic supramolecular scaffold;
and
4. the efficacy of M13-displayed enzymes toward reduction-oxidation reactions.
1.1
Strategies for Applied Biocatalysis
Biocatalysis offers many advantages over traditional heterogeneous catalysis for industrial applications. Substrate specificity, reaction selectivity, and catalytic efficiency
drive the accelerating implementation of biocatalysts in reactions that have traditionally employed inorganic catalysts, such as the chiral resolution of alcohols and the
oxidation of alkane precursors.
2,3,4
Enzymes can result in lower process temperatures
for industrial reactions while decreasing the amount of solvent and waste, reducing
energy and material costs.' Biocatalytic products often have no need for purification
or protection /deprotection procedures, greatly simplifying protocols.6 Additionally,
the unique geometries and energy states created by enzyme active sites allow for the
generation of many important chemicals with no effective inorganic means of production
for example, artemisinin 7 and carotenoids.' The inherent modularity of
biomolecules complements such advantages by promising engineers increasing degrees
of control over enzyme outputs: not only is protein structure manipulable by genetic
means, but well-characterized biochemical functional handles allow for catalytically
useful post-translational modifications.
As a result, enzymes have become integral parts of the production of many important medicines and other small molecules 16
penicillins, taxols, and rapamycins,
for instance. Cytochromes cyclize morphine and codeine molecules; esterases, lipases,
and proteases resolve racemic mixtures; oxygenases facilitate bioremediation; hydroxylases catalyze the addition of water molecules to insecticides; thermolysines couple
substrates for food additives such as aspartame.3 ,9 In a growing array of industries,
engineers look to biocatalysis for efficient, scalable transformations.
1.1.1
Engineering enzymes for greater utility
As successful examples of enzymatic approaches proliferate,10"" 1 2 however, challenges
persist. 13 Native enzymes often exhibit short lifetimes, on the scale of hours without modification or immobilization;1 4 outside of native pH and temperature, their
functionality can become less predictable. Similarly, while some enzymes have been
found to have novel activity or even greater thermostability in organic solvents, 4 1, 5
they often become substantially less active when removed from aqueous conditions.16
Moreover, developing and optimizing enzyme-based chemical production systems remains slow and costly, with limited screening tools.' 7 While whole-cell biocatalytic
processes overcome issues related to enzyme half-life and can facilitate pathway evolution, the additional costs of salts and glucose to feed such systems can greatly exceed
even those of equipment and solvents, decreasing their value in industrial settings.18
Opportunities remain, therefore, in improving the stability, versatility, and economic
efficiency of biocatalytic processes. Many strategies have been developed to augment
the utility of enzymes, and usually fall into one of two strategies: modifying pre~xisting enzymes by rational engineering and directed evolution, or the generation and
fine-tuning of de novo catalytic entities.
Rational optimization of existing enzymes can include targeted mutations to improve protein lifetime via insertion of disulfide linkages,1 9 or to modulate nucleophilicity. 20 Bulky residues can be removed to decrease steric interference with larger
substrates.21 Immobilization techniques have been especially successful in improving
some forms of protein stability, and have been implemented broadly for the industrial production of pharmaceuticals and a number of bulk chemicals. However, while
serving to increase the thermochemical stability of enzymes, immobilization often si17
multaneously decreases the catalytic activity and complicates molecular transport in
industrial processes, requiring further optimization (for review, see Zhou and Hartmann22 or Bommarius and Paye 23 ).
Directed evolution approaches have been immensely successful in improving enzyme activity, sampling larger windows of the functional landscape to identify unintuitive active site modificationsi1,24,25
(see Turner 26
for recent review). This strategy
has been implemented in a variety of processes, including Lipitor® production and
the manufacture of chiral amines. 13 Directed evolution can also produce enzymes
which catalyze new, non-natural transformations. 27 While the engineering of existing
enzymes has been successful in improving those enzymes and, in some cases, adapting
them to tangential or novel reactions, 2 8 such approaches are rarely fully generalizable.29 Each attempt at engineering a different protein construct encounters a fresh
set of distinct, independent challenges specific to that structure, and such endeavors
are unlikely to sample beyond the nearby region of fitness landscape occupied by that
enzyme.
1.1.2
De novo enzyme design
An alternate approach to studying and improving enzyme activity involves co6pting
structural domains of unrelated proteins for novel active sites or designing new protein structures entirely. By sampling completely independent domains of the fitness
landscape, such de novo enzymes allow for unique combinations of targeted functional
attributes, especially when function can be uncoupled from structural stability. 30 The
insertion of catalytic chemistries into otherwise non-catalytic proteins dates at least
to the 1970s, when biotin-avidin biochemistry was utilized to fabricate an asymmetric
rhodium biocatalyst. 1 Later, in 1986, the Lerner and Schultz laboratories simultaneously engineered the first generation of enzymatic antibodies. Hypothesizing that
stabilizing transition states would lower the energy barrier for a given reaction, the
authors raised antibodies against transition state analogues and isolated antibodyborne hydrolases with high specificity. 31,32 Hilvert and colleagues subsequently prepared antibodies which catalyzed a chorismate rearrangement 3 3 and the Diels-Alder
18
reaction. 34 This initial work indicated that non-enzymatic proteins could be promising templates for catalytic engineering.
Subsequently, a number of other proteins
, bovine pancreatic
have been utilized to create functional enzymes. Thioredoxin,35 36
polypeptides, 3 7 calmodulin, 38 transcription factors, 39 and zinc fingers
40
have each
been imbued with catalytic activity by inserting foreign active sites.
The Baker Lab has combined this approach with computational methods to create
and optimize numerous functional de novo active sites. By scouring protein databases
for appropriate scaffolds -
e.g. TIM barrels or periplasmic binding proteins -
and
computationally assaying their fit for a desired transition state model, promising
combinations can be identified in a high-throughput manner. 41 When combined with
display and directed evolution tools, this Rosetta software-based strategy has led
to Kemp elimination catalysts; 4 2 hydrolytic cysteine-histidine dyads 43 and histidineserine-aspartate/ glutamate triads; 44 and histidine-coordinated zinc biocatalysts for
organophoshate hydrolysis. 45 While in some cases the resulting proteins have aligned
closely with the intended design, in others the optimization and evolution experiments
led to active sites catalytically effective but quite distinct from original designs. Such
outcomes highlight our functionally incomplete understanding of enzyme mechanisms
and the need for high-throughput experimental validation to identify optimum structures. 4 6
While inserting active sites into a priorithermostable structures provides a useful starting point, engineering protein domains from scratch increases even further
the potential for diverse activity and mechanistic inference from de novo enzymes.
Helix-loop-helix motifs and multimer helical bundles have been the most prominent
de novo structures developed by protein engineers, and have seen remarkable catalytic successes in recent years. Strategically inserting polar and non polar amino
acid residues into a peptide - "binary patterning" - can provide a sufficient free
energy shift to drive the assembly of helical bundles. 47 ,48 Such bundles house a relatively hydrophobic core and present hydrophilic surfaces on their exterior (Figure
1-1). a-Helical bundles have been utilized for redox and peroxidase;
4 95, 0 5, 1 5, 2
boxylation; 5 3 peptide ligase; 5 4 phosphate hydrolysis and phosphoesterase;
19
55,56
decar-
phenol
AA
K
K
KK
K
A
E
A
Figure 1-1: Binary patterning of polar and non-polar residues can stabilize interhelical interactions for parallel (left) and antiparallel (right) helices. Arrows indicate
potential salt bridges. Wheel diagram is of Coil-Ser, adapted from Betz, et al. with
permission.72
oxidase;
57
carbonic anhydrase;58 esterase and lipase; 59 ,60 ,6 1 and nitrate reductase ac-
tivity.6 2,6 3 In addition, other work has sought to stabilize the insertion of active site
65 66
cofactors with catalytic potential, such as chlorins6 4 and iron sulfur clusters, , or to
optimize small molecule binding in general.67 Further inclusion of non-natural amino
acids, 5 5,6 1,68, /-amino acid peptides, 6 9 synthetic nucleic acid polymers, 70 or
-sheet
geometries 71 has also resulted in successful biocatalysts. Such varied approaches are
rapidly broadening the scope of de novo biocatalytic structures in their activity and
applications.
In summary, previous de novo engineered enzymes have recapitulated a range
of enzymatic activities to a remarkable degree, though few have been developed for
the variety of non-native environments common to industrial processes or for facile,
high-throughput adaptation. Computational analysis has greatly increased the field's
fluency in combining a catalytic active site or "theozyme"73 with a stable protein
backbone to produce a functional enzyme. Such predictions still lack information on
conformational flexibility or non-aqueous solvent interactions, however, and must be
combined with considerable mutation and screening experiments to yield high rates
20
of catalysis. The rapidly expanding proficiency in protein engineering now allows
one to build enzymatic structures from the ground up, but the limited number of
molecular interactions often precludes the use of such de novo structures outside of
their originally designed thermal and solvent conditions. For both strategies, high-
throughput evolution is inhibited by their lack of a phenotype-genotype connection
without a genome to amplify and sequence after assays, variants can only be tracked
in an identifiable manner if each is sequestered in its own volume, e.g. a well plate.
1.2
Objective: M13 Bacteriophage as a Platform for
Biocatalyst Engineering
In this work, I seek to develop a robust, versatile biocatalytic platform potentiating
the catalysis of targeted reactions in variable environments. Multiple strategies described above are integrated, mimicking active site geometry from a highly efficient
enzyme within an otherwise structural protein assembly, M13 bacteriophage.
M13 bacteriophage is a rod-shaped Escherichia coli virus 7 nm in diameter and
900 nm in length that is frequently used for protein engineering and nanomaterial
development (Figure 1-2). The phage has 2700 "major" coat proteins - protein
eight or p8 - arrayed helically around its axis. The infectious "proximal" terminus
has about five copies each of p3 and p6 proteins, while the distal terminus expresses
five each of p7 and p9 proteins. The p 3 (406 amino acid residues) and p8 (fifty amino
acid residues) proteins are those most commonly engineered, although researchers
have manipulated other coat proteins as well.74 75' 76 . Structurally, M13's major coat
protein, p8, has its C-terminus facing the interior of the columnar structure, with positively charged lysine residues interacting electrostatically with the negative charges
on its single-stranded DNA loop. The N-terminus of p8, the solvent-exposed exterior
of the capsid, has a bias towards negatively charged aspartate and glutamate residues.
These respective charges facilitate the electrostatic organization of individual helical
p8s during capsid formation at the E. coli membrane. Recent engineering has inserted
21
additional restriction enzyme sites into the M13 genome, increasing its tractability
for protein engineering. 77
M13's genetic pliability has made it a valuable tool for protein engineering. For
example, the Belcher Lab has in recent years used phage to build lithium ion battery
electrodes 78,79 and dye-sensitized solar cells. 80', 81 Phage display has been employed
in the search for tumorigenic biomarkers, as Weissleder and colleagues discovered
phage peptides which successfully bind to the extracellular SPARC protein on prostate
82 8 3
tumor cells and plectin-1 on pancreactic ductal adenocarcinoma. ,
More relevantly, phage display has been applied to the field of biocatalysis. Hunt
and Fierke expressed a library of carbonic anhydrase variants on p3 and selected for
those variants which bound best to a sulfonamide resin, ultimately achieving nearwild type levels of activity and elucidating key aspects of the CAII binding pocket. 84
#-lactamase, 85 ' 86 , glutathione
transferase, 8 7 , and full catalytic antibodies 88 ,89 90 have
been fused to p3 and biochemically characterized.
In 1998 and 1999, the Schultz
and Neri laboratories developed elegant substrate /product-enzyme crosslinking assays
whereby covalent bonding to or cleavage from a solid phase -
"proximity coupling"
could select for catalytically active peptide sequences displayed on p3. 919, 2 More
recently, self-aggregating peptide products were used to isolate p3-displayed proteins
capable of catalyzing amide condensation. 93
While these latter projects utilized M13 to assay the catalytic potential of proteins expressed five-fold on the phage terminus, in this dissertation I develop the
capsid major coat protein p8 for highly multivalent, stable, versatile biocatalysis. By
co6rdinating cofactors between N-terminal eight-residue p8 inserts and single point
900 nm
-4 p7, p9
p3, p6
-2700 copies of pVIlI
Figure 1-2: M13 has a high aspect ratio and is composed of 2700 copies of p8.
22
mutations further C-terminal, the bacteriophage particle is imbued with 2700 enzymatic active sites, resulting in novel biocatalysts. Nested in the p8 a-helices, the
created active sites are analogous to those within de novo helical bundles, but with
the added advantage of the stability evolved into M13's macromolecular structure.
1.3
Dissertation Overview
The dissertation is organized into four chapters, covering the isolation and characterization of a catalytic M13 clone as well as tools for further engineering of enzymatic
phage.
In Chapter 2, I present a strategy for constructing M13 libraries enriched for
catalytic motifs. A clone is isolated which exhibits the target enzymatic activity,
zinc-mediated esterolysis, and characterized for its zinc affinity and structural biochemistry.
In Chapter 3, this clone is shown to hydrolyze a range of esters in myriad environments. Significantly, the clone's activity increases in a chosen nonaqueous solvent,
dimethyl sulfoxide, and at raised temperatures, both of which are critical for the adaptation of enzymes to industrial processes. The clone also hydrolyzes larger substrate
molecules and less reactive esters.
In Chapter 4, the clone is adapted for reduction-oxidation biochemistry. It demonstrates significant interaction with both ferric heme and cupric salts, allowing for
catalysis of peroxidase and oxidase reactions, respectively. These interactions are additionally utilized to develop library selection protocols via affinity chromatography.
N.B.: Figures 2-1, 2-3, 2-7, 3-2, 3-4 and 3-6 are adapted from Casey, et al.,
2014,94 which can be found at http:/doi.org/10.1021/ja506346f.
23
24
Chapter 2
Isolating Enzymatic Bacteriophage
From an Engineered p8 Library
Dr. Stokes insists that d'Herelle and you are right in calling bacteriophage an organism. But what about Bordet's contention that it's an
enzyme?
- Oliver Marchand in Sinclair Lewis' Arrowsmith95 , 1924
In this section, an M13 bacteriophage library incorporating transition metal active site motifs is constructed and functional viral clones studied. The hydrolysis of
para-nitrophenyl acetate (pNPA) is chosen as a representative reaction for the targeted enzyme, carbonic anhydrase. A selected clone, DDAH, mimics the active site of
carbonic anhydrase in its distribution of hydrophilic and hydrophobic residues. The
clone's zinc-binding properties are characterized in order to understand and limit selfaggregating phenomena. The clone is then purified on transition metal-codrdinating
nitrilotriacetic acid (NTA) columns, demonstrating that catalytic activity is a function of the virion alone and not contaminant proteins. The presence of nonpolar amino
acids is important for DDAH's activity, as their deletion significantly decreases catalytic efficiency. The activity is also contingent upon the quaternary structure of
M13's coat proteins. Clone DDAH thus demonstrates simple enzyme activity on an
easily evolvable scaffold, potentiating the development of more efficient and intricate
25
de novo enzyme systems.
2.1
Background: Zinc-Codrdinating Enzymes
Metal ions serve as powerful, versatile catalysts when co6rdinated by proteins, and
approximately one in three enzymes has a metal center. 96 Zinc, in particular, is a
critical ion in hundreds of enzymes,
97
playing many disparate catalytic and struc-
tural roles, even stabilizing quaternary structures in some protein complexes; 98 ,99 it
is the most common metal ion activated for hydrolysis reactions. 100 Zinc ions are predominantly co6rdinated by proton-shuttling histidines in catalytic sites, along with
aspartate and glutamate residues, and a water molecule or hydroxide ion. Cysteine
residues also frequently contribute to zinc codrdination, but mostly when the ion
serves a structural role.101
Carbonic anhydrase, a class IV enzyme, is the classic zinc-based enzyme, with
extra6rdinarily high rates of activity -
kt/KM- 1.5 x 108 M-'s-1 -
more thoroughly studied than any other metalloenzyme.
100
and has been
For its well-understood
biochemistry and catalytic efficiency, carbonic anhydrase's active site (specifically,
that of bovine carbonic anhydrase II, bCAII) was chosen as the model for M13's
displayed enzyme motifs.
2.2
Strategy for M13 Enzyme Engineering
M13's a-helical p8s were engineered such that each could co6rdinate an ion along
with the adjacent helix. CAII's active site has two histidine residues separated by a
phenylalanine in one structure and a third histidine residue in a proximal 43-sheet; the
three residues together bind a Zn2 + ion whose fourth co6rdination site is occupied by a
hydroxyl ion or a substrate molecule. To replicate this geometry, a library of plasmids
were constructed to encode a pair of His residues at fixed positions, surrounded by
randomized amino acids in 8 AA inserts located at the surface-exposed N-terminus of
p8 (Figure 2-1; see experimental section for more details on oligonucleotide libraries).
26
b
a
b
... LVQFH94FH 96WGS...
0
02N
n-A, X 1 X 2 Hi3 Xi 4 Hi5 Xi 6 XiXi8 D 5P 6A7K8 ... l4Hl5A1...
XiAXi 3 Hi 4Xi5 Hi6 Xi7 Xi8
+
Figure 2-1: Design of semisynthetic active sites for hydrolysis. (a) The active site of
10 2
). The
bCAII consists of three histidine residues co6rdinating a Zn2+ ion (1CA2
front-most histidine residue shown comes from an adjacent -sheet. (b) The cloning
strategy involved mutating three specific residues on M13 p8 to histidine (red, sequence at bottom) and randomizing adjacent terminal residues (blue). Histidine pairs
were at i3,i5 or i4,i6; backgone mutations were at Ser13, Gln15, Gly23, or Ala27. Im-
age includes PDB file 2COW. 10 3 (c) The hydrolysis of p-nitrophenyl acetate (pNPA)
yields colorimetric p-nitrophenylate (pNP), which can be detected via absorbance at
94
400 nm, and acetate. Adapted from Casey et al..
A third histidine ("backbone" histidine) was inserted deeper into the p8 further from the N-terminus -
that is,
such that each N-terminal pair of His residues would
be in close proximity to the backbone His of an adjacent p8 protein.
Since the 8 AA inserts are likely not part of the backbone a-helix, which is believed to be broken by Pro-6,* the specific orientation of the eight residues within
each hypothetical insert is not readily inferable. We generated an ensemble of M13
libraries with two variables: the position of the histidine pair in the N-terminus and
the location of the backbone histidine, seeking to maximize the potential for ion co6r10 2
for
dination between three histidine residues on adjacent helices. PDB files 1CA2
CAII and 2C0W1 0 3 for M13 were used for evaluation. Our analyses of the structural
models indicated that the most favorable trios involved pairs i3,i5 and i4,i6 on the
*Positions within the 8 AA insert will be indicated with an 'i' prefix; the other positions will
retain wild-type numbering with no prefix.
27
insert complemented with backbone histidines at the 13th, 15th, 23rd, or 27th residue
from the native N-terminus. Supporting these selections, the positions match closely
with those residues downstream of the N-terminus expected to be most surface exposed in wild-type models.104 See the experimental section for more details on library
construction.
Following bacterial transformation and plating of the libraries, individual plaques
were picked for sequencing, amplification and characterization. Sequenced clones were
surveyed for primary structure similarity to the bCAII active site residues, with an
emphasis on hydrophobicity in the immediate vicinity of the cobrdinating histidines.
Active site hydrophobicity is believed to be critical for effective enzymatic catalysis, serving both to reduce the conformational variability of charged co6rdinating
residues 84 and/or increase the surrounding local dielectric field. 10 5 bCAII's metalbinding histidines are flanked by phenylalanine, tryptophan (FHFHW) and leucine
(LHL). As the N-terminus of M13's p8 preferentially displays negatively charges, and
histidines are mildly basic, many identified sequences were aspartate- and glutamaterich. We nonetheless identified a number of sequences enriched for nonpolar residues
immediately adjacent to the histidine pair and selected ten for experimentation (Table 2.1).
2.3
2.3.1
Results
pNPA hydrolysis
In order to characterize bacteriophage-displayed de novo enzymes, we measured hydrolysis of nitrophenyl esters. The carbonic anhydrase reaction is the reversible conversion below:
CO 2 + H2 0 ++ HCO- + H+
(2.1)
However, as this is an extremely rapid and difficult-to-detect reaction (rates on
28
ID
B
1
2
3
4
5
6
7
8
9
10
11
Catalyst
bCAII
DVHSHGLP, 15H
EGHVHATM, 23H
TENMHGTM, 23H
ESHVHDSE, 27H
DDAHVHWE, 23H
ESHTHGNE, 23H
VSGSSPDS (Ctrl)
DLHSHSEP, 15H
ATHDHGDQ, 23H
ADHDHHSV, 15H
DMHEHNGE, 13H
Activity* (%)
35.5
28.3
25.6
20.1
19.9
16.4
15.5
6.3
5.5
3.1
1.2
-1.9
95% CI
8.1
40.9
44.5
3.0
24.7
10.3
17.5
19.9
3.0
4.2
14.5
19.8
Table 2.1: Clones tested for catalytic activity towards the hydrolysis of pNPA. VSGSSPDS is a control bacteriophage clone from independent gold-binding work.109
*Activity is displayed as percent above the background rate of hydrolysis in 50mM
Tris buffer. Average of n > 3 samples. bCAII was dissolved to 34 nM; phage were dissolved such that the concentration of p8 was 3-4 pM. tConfidence interval presented
as the amount above and below the mean activity that sets a 95% CI.
the order of 105 events per second), the hydrolysis of para-nitrophenyl esterst (Figure
2-1c) is a common proxy reaction for those studying and seeking to replicate the carbonic anhydrase enzyme.
21,84 ,9810
, 6 10
, 7
Esterase activity itself is a common industrial
transformation for the production of cosmetic compounds or paints 108 and for general
enantiospecific deprotection of compounds. 2 Accordingly, the M13 clones discussed
were initially assayed for the hydrolysis of pNPA.
M13 clones demonstrate disparate activity
Only three clones demonstrated significant hydrolysis activity in initial assays: TENMHGTM, 23H ("TENM"); DDAHVHWE, 23H ("DDAH"); and DLHSHSEP, 15H
("DLHS"). While all but one of the tested clones displayed some level of activity,
most samples had too much variance for further characterization (Table 2.1 and
Figure 2-2). Clone TENM was especially interesting, as one of its insert histidine
residues had mutated to arginine during transformation and amplification, and the
methionine was also thought to be potentially catalytically significant. However, in
tFor the substrates evaluated, "esterolysis" and "hydrolysis" are used interchangeably.
29
B
1
2
3
4
5
TENMHGTM,23H
DDAHVHWE, 23H
6
7
8
DLH
9
10
11
50
25
75
Activity (%)
Figure 2-2: Three of the selected clones demonstrate activity significantly above background. Activity is displayed as the percent above the background rate of hydrolysis
in 50 mM Tris buffer. Sample ID on y-axis corresponds to Table 2.1. Average of
n > 3 samples. bCAII (black bar, top) was dissolved to 34 nM (1 pg/mL); phage
were dissolved such that the concentration of p8 was 3-4 pM. Error bars represent
95% CI.
further experiments, its level of activity was too inconsistent and could not be reliably
determined. DLHS had a more repeatable level of activity than TENM, but demonstrated consistently lower levels of activity than DDAH. Clone DDAH was therefore
chosen for more thorough characterization.
Reaction kinetics
The hydrolysis activity of the bacteriophage was quantified by the catalytic efficiency,
kcat/KM, according to Michaelis-Menten kinetics (see experimental section for details):
d[P]
dt
_
kcat [E]
[S]
KM + S]
(2.2)
Briefly, sample Abs 400 was monitored for product (nitrophenolate) formation at
various starting concentrations of pNPA (Figure 2-3a). Initial slopes were measured
30
b
ab 1
-
0.8
0.6
S ---
4mM
2mM
1 mM
500 M
250
125
-
1.6
4
14
x
Background
DDAH
1.2
$1
M
M
cr_ 0.8
0
0.4.
ca
S0.4
0.2
0
500
1000
Time (s)
0
1500
100
200
[pNPA]
0
300
(p M)
400
500
Figure 2-3: Initial slopes are regressed and plotted verses initial substrate concentration. (a) Representative absorbance vs. time plot for a range of pNPA concentrations.
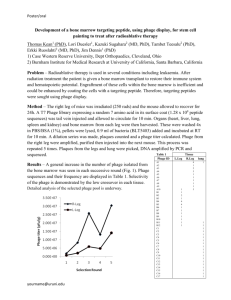
(b) DDAH is significantly more active than background hydrolysis. Adapted from
Casey et al.. 94
and plotted against starting substrate concentration (Figure 2-3b). The kcat/Km
was then determined by Lineweaver-Burk analysis.
Each p8 protein was considered conservatively to be one active site, so that each
bacteriophage represented 2700 catalytic sites. As such, clone DDAH was found to
hydrolyze pNPA with
kcat/K
= 0.535 M-'s- 1 for each p8 subunit (Figure and Ta-
ble) in PBS at pH 7.4; by comparison, wild-type bovine carbonic anhydrase (bCAII,
Worthington Biochemical Corporation #LS001260) hydrolyzed pNPA at a rate of 194
M-'s' and solubilized L-histidine measured 0.021 M-s 1 . The activity of DDAH
thus represents a significant co5rdination between the residues, while remaining far
lower than that of the wild-type enzyme. The inferred
kcat,
0.002 and 1.2 s-1 for
DDAH and bCAII, respectively, implies that a difference in turnover rate is a far
larger contributing factor than substrate binding, though the kcat values themselves
are far less precise calculations from our Lineweaver-Burk plots than
kcat/KM.
It is
hypothesized that the low activity compared to carbonic anhydrase is in part related
to the conformational flexibility of the p8 termini in aqueous solutions. Additionally, the current strategy did not target second-shell interactions between nearby AA
residues and the inserted histidines; such interactions play a large role in catalytic
31
efficiency and their inclusion would likely improve the construct's activity.
2.3.2
Minimizing DDAH aggregation
While ion binding is a critical component of the designed active sites, the positive
charge can also result in charge shielding and aggregation of phage. Using microscale
thermophoresis, DDAH was found to bind Zn2+ with a KD of 6.4 pM, while Ni 2 + and
Cu2+ measured 8.5 and 3.4 puM, respectively (Figure 2-4). During early experiments,
absorbance vs. time plots indicated that saturation of the binding sites with zinc
may be causing the phage to aggregate and precipitate.
TEM experiments were
subsequently conducted at 0, 25, and 50 pM ZnSO 4 with 1 x 1012 /mL DDAH (Figure
2-5). The resulting electron micrographs indicate that as little as 50 AM aqueous zinc
is sufficient to induce heavy aggregation of DDAH viruses. 25 pM, in contrast, led to
only moderate bundling, though still visibly more than with no zinc salts in solution.
25 puM was accordingly chosen as the Zn2+ concentration for all hydrolysis reactions.1
2.3.3
Column purification of clone DDAH
To demonstrate that clone DDAH is itself responsible for observed activity and not a
contaminating protein, 1 0 I developed a column-based purification protocol for clearing away the non-phage bacterial detritus resulting from amplification. While M13
is the predominant protein resulting from DDAH amplification and preparation (see
experimental section for protocols), it is possible that a less prevalent but more active
hydrolytic enzyme is co-precipitating with the phage during preparatory centrifugation.
Nickel nitrilotriacetic acid (Ni-NTA)-conjugated agarose beads were used for separation. Ni-NTA affinity chromatography is most often used with six-histidine peptide
tags, for which reliable protocols are available. Although DDAH has three embedded
histidine residues rather than six consecutive terminally expressed histidines, I hytAt 25 pM ZnSO 4 and 4.5 pM p8, only 3.5 pM of p8-Zn 2+ complex would be formed with a
KD of 6.4 pM; this equates to 2081 p8s per virion. However, to remain conservative, activities were
assumed to be the product of all 2700 p8s per virion for catalytic rate determination.
32
-
995 _
'
A
U
N
1.1
1.1
1.1
1.1
pM
pM
pM
pM
DDAH,
DDAH,
DDAH,
DDAH,
Ni2+, KD = 8.5 pM
Cu 2+, K 0 =3.4 pM
Zn2+, KD = 6.4 pM
Ca 2
I
+
1,000
1 ,1 MfDDAH Mn2+
_
990
985-
980-
975
0.01
0.1
10
1
100
1,000
Ion Concentration (pM)
Figure 2-4: DDAH binds Zn2+ and other soft ions, but not hard ions like Ca2+ and
Data is from a Nanotemper Microscale Thermophoresis instrument, which
Mg 2 +.
measures the thermally induced diffusion of fluorescently labeled proteins across a
range of ligand concentrations to assess bimolecular binding. The low state is unbound, the top of the shoulder is a state where all proteins are bound to the respective
ion, and the midpoint of the curves indicates the KD. For calcium and magnesium,
no binding is detectable.
33
Figure 2-5: Ionic zinc induces aggregation of DDAH phage in aqueous solutions. (a)
With no zinc, DDAH remains mostly separate filaments. (b) At 25 pM ZnSO 4 , some
bundling of DDAH is apparent. (c) DDAH exists mostly as aggregated filaments at
50 pM ZnSO 4 . TEM by Dr. Dongsoo Yun. Scale bars: (a), 200 nm; (b), 500 nm;
(c), 200 nm.
34
b
a
1E+13
-DDAHVHWE,
8E+12
8E+1215
1
2
3
4
5
6
7
8
23H
SCPDCGAE, 15H
#
E 6E+12
4E+12
2E+12
-
0
0
2
4 mL 6
10
8
Figure 2-6: DDAH is retained on Ni-NTA columns with a high efficiency. (a) Two
clones were independently run through Ni-NTA affinity chromatography. Phage concentration was assessed for each aliquot of solvent added to and collected from the
column. DDAH adheres to the beads until elution by imidazole (8 mL mark, red
arrow). (b) PAGE of samples collected from column. Lanes: 1, ladder indicates (top
to bottom) 160, 80, 60, 50, 40, 30, 20, 15, 10 kD; 2, stock DDAH; 3, flow-through of
DDAH after refrigerated shaking (1 mL in (a)); 4 and 5, elutions of DDAH (8.5 and
9 mL in (a)); 6, stock SCPD phage; 7, flow-through of SCPD phage (2 mL in (a)); 8,
elution of SCPD phage (8.5 mL in (a)).
the *-marked band is p8 protein.
#
indicates bands of contamination, while
pothesized that the clone's 3-His motif would provide sufficient affinity to differentiate
it from non-specific proteins. After loading the column with a stock of bacteriophage,
shaking it at 4C for at least an hour, and washing with high salt buffer, the remaining
phage was eluted with 500 mM imidazole.
Two clones were purified by the Ni-NTA columns: DDAH and SCPDCGAE, 15H
(SCPD, a bi-cysteine clone also hypothesized to interact with a column displaying
transition metal ligands).
While SCPD was rather poorly retained on the column,
the vast majority of DDAH adhered (Figure 2-6). Of the 1.75 x 10'3 phage particles
added, 91% (1.6 x 1013) of DDAH and only 22% (3.9 x 1012) of SCPD were retained and
eluted. It is likely that the cysteine residues displayed by SCPD are less strongly interacting with Ni-NTA than histidine, and also that the original SCPD phage preparation had a high proportion of non-phage proteins confounding the spectroscopy-based
quantification of phage particles (compare the p8 bands of SCPD in Figure 2-6b to
35
those of DDAH). DDAH, however, demonstrated relatively strong affinity. The original stock of DDAH had some non-phage proteins present, but these were washed
from the column and were undetectable after elution.
The resulting DDAH elutions were dialyzed to remove imidazole and subsequently
assayed for pNPA hydrolysis. The purified phage was catalytically indistinguishable
from the un-purified stock of DDAH, indicating that the observed hydrolysis is catalyzed by the DDAH virus and not by other bacterial proteins remaining in the stock.
Because the purification process is time-consuming and significantly dilutes phage
stocks, later experiments are from un-purified bacteriophage preparations. Zinc-NTA
columns were also constructed and used to purify DDAH, but the loading capacity
was reduced when compared to Ni-NTA.
2.3.4
Structure-function relationships of the DDAH active site
To more thoroughly understand the contributing factors of the DDAH active site,
I performed an alanine scan on the peptide insert and engineered two additional
histidine substitutions: DDAH with the His residue at position 23 mutated H23G, as
in wild-type M13, and a A3H clone engineered with H4A, H6A, and H23G mutations
so that no histidine residues remain. The results are summarized in Figure 2-7 and
Table 2.2, and demonstrate the importance of the active site design and the phage's
quaternary structure in promoting catalysis.
Each mutant's catalytic activity was significantly lower than the original DDAH
clone (two sample p-value <0.05), though all but the A3H clone had measurable hydrolysis activity above background. Notably, the tryptophan residue in the insert,
which holds the same position relative to the histidine pair as in CAII (sequence: F-HF-H-W, Figure 2-la), was found to be important for hydrolysis activity. Affirming
the hypothesized importance of nonpolar residues near zinc-co6rdinating histidines,
both W7A (p <0.0005) and V5A (p <0.005) mutations substantially lowered measured catalysis rates. Any pair of two histidines was sufficient for hydrolytic activity,
albeit lower than with all three histidine residues, while deletion of all three histidines
fully extinguished activity. The lack of detectable activity from the A3H clone cor36
CU
C,)
A3H
DDAH-8
SDDAH-1 2
**
0.4
0.2
k/KM (M-s
0.6
)
0
Figure 2-7: Alanine mutations in the DDAH active site lower its kcat/KM significantly. A3H: terminal two histidines mutated to alanine and third histidine returned
to glycine (as in wild-type); DDAH-8 and DDAH-12: 8-mer and 12-mer synthetic
peptides, respectively, comprised of the terminal peptide sequence of DDAH p8. Error bars represent 95% confidence intervals for values fit by Lineweaver-Burk analysis.
p-values: *<0.05; **<0.005; ***<0.0005; represents two-sample t test for regressed
coefficients of DDAH and given sample.
37
kcat/KM
Sample
DDAH
H4A
V5A
H6A
W7A
H23G
A3H
DDAH-8
DDAH-12
L-His
bCAII
PBS*
(M-'s-1 )
0.535
0.413
0.352
0.219
0.263
0.346
0.021
0.013
0.021
193.51
6.92E-07
95%
lower
0.033
0.071
0.064
0.034
0.024
0.072
0.008
0.003
0.001
13.667
3.44E-08
CI
upper
0.038
0.108
0.101
0.050
0.029
0.122
0.032
0.004
0.001
15.915
3.44E-08
p-value
0.0158
0.0017
3.86E-07
5.33E-10
0.0040
9.96E-04
7.14E-09
Sequence
DDAHVHWE, 23H
DDAAVHWE, 23H
DDAHAHWE, 23H
DDAHVAWE, 23H
DDAHVHAE, 23H
DDAHVHWE, 23G
DDAAVAWE, 23G
DDAHVHWE
DDAHVHWEDPAK
--
Table 2.2: Catalytic efficiency kcat/KMwas determined for clone DDAH, alanine mutants, synthetic peptides, and controls, each in PBS. Confidence is expressed as lower
and upper intervals to the boundaries of 95% CI region. p-values represent two-sample
t-test between DDAH and a given phage mutant for the regression from n > 8 initial
slopes. *For PBS buffer, parameter shown is kuncat (s- 1).
roborates the data in Section 2.3.3 demonstrating that activity results from DDAH
and not co-precipitating bacterial proteins. In addition to the M13 point mutants,
free-floating synthetic peptides were tested, comprised of the N-terminal 8 and 12
AAs -
that is, DDAHVHWE and DDAHVHWEDPAK, not expressed on the sur-
face of M13. These peptides, referred to as "DDAH-8" and "DDAH-12," demonstrated
negligible activity.
2.4
Summary
In this chapter, a novel strategy for biocatalytic development is explored: the incorporation of a transition metal-activating enzymatic motif into thermostable bacteriophage particles to yield semisynthetic enzymes. After the creation of libraries
expressing numerous combinations of terminal His pairs and backbone His insertions,
hydrolysis assays indicated that the clone DDAHVHWE, 23H was a promising candidate for robust catalysis. The clone binds Zn2+ with a KD of 6.4 PM, along with other
transition metals. Saturating the phage with zinc ions can lead to charge shielding
38
and filament aggregation, but lower (< 25 piM) concentrations of zinc result in the
vast majority of active sites housing an active ion with minimal viral aggregation.
While non-phage proteins were detected in the crude virus preparations after amplification, these could be removed by Ni-NTA affinity chromatography without any
concomitant loss in activity.
The inclusion of nonpolar amino acids in wild-type enzyme active sites is known to
be critical for efficient catalysis, and was found to be important in the DDAH active
site, as well. The activity exhibited by DDAH is additionally contingent upon the
quaternary structure of M13's coat proteins, as soluble peptides exhibited negligible
activity.
In addition, the complete inactivity of the A3H clone in water renders
highly unlikely the possibility that measured catalysis is the result of contaminant
Escherichia coli detritus from our bacteriophage preparation protocols.
While the measured activity remains substantially lower than that measured for
wild-type bCAII, the activity is nonetheless comparable to de novo enzymes reported
by Zastrow et al.58 for the same pH (pH 7.5, kcat/KM = 1.38 M- 1 s-
1 per
three helices)
and Patel et al. 59 (at pH 8, kcat/KM = 9.9 M-'s-1 per four helices). Recent works
by the Kuhlman group99 (kcat/KM at pH 7.5 = 90 M-s-1 for two zinc codrdination
sites) and Rufo et al.9" (at pH 8, kcat/KM = 62 M-'s- 1 per amyloid peptide) have
made substantial improvements in rates for aqueous esterolysis and are informative
for ongoing enzyme design, but neither approach demonstrates solvent /thermostable
activity nor enables genetic screens. In most such de novo hydrolases, the presence
and accessibility of the substrate-binding/ catalytic domain are critical for resulting
reaction rates. DDAH, with a relatively open active site, should allow for significantly higher rates in future work, especially if "second-shell" interactions can also be
engineered into p8 termini. 99
The demonstrated display and organization of multiple metal-co6rdinating residues
on all 2700 p8 proteins is a new capability and will allow for the future insertion of
other cofactors for the replication of additional enzymatic activities. The orthogoThis protein has the highest reported aqueous activity for de novo enzymes, with
630 M 1 s- 1 at pH 9.
39
kcat/KM
nally engineer-able proteins on the phage coat - p3, p8, and p9 - present an exciting
opportunity for the combination of multiple enzymatic activities on one macromolecular structure.
Such multifunctional biocatalysts can simplify multistep processes
and increase overall efficiency. 2 pNPA hydrolysis, a useful representative reaction for
establishing enzymatic activity, will inform future engineering efforts toward more
complex target reactions.
2.5
Experimental Methods
2.5.1
Library Construction
We built M13 p8 libraries using two components: bi-histidine 8 AA N-terminal peptide inserts with a theoretical diversity of 1.664 x 1010
and four distinct M13 vectors
encompassing single histidine mutations. BspHI and BamHI cut sites upstream and
downstream, respectively, from the p8 N-terminus were used for genome engineering.
Vector preparation
Vectors were prepared by mutating a chosen p8 residue to histidine, then amplifying, digesting, and dephosphorylating resulting M13 genomes. p8 backbone mutations -
S13H, Q15H, G23H, A27H -
were made using Agilent Technologies
QuikChange® Lightning Kit and Primer Design Program with the M13SK 77 bacteriophage vector. Resulting vectors were purified from bacterial pellets (see "Bacterial
culture and M13 stocks," below) and 50 pg of DNA was double digested with BamHIHF and BspHI (3 hrs at 37'C),, then purified on a 0.8% agarose gel and extracted
using the QlAquick® Gel Extraction Kit. Digested, purified vector was dephosphorylated using Antarctic phosphatase (37C for 1 hour, 65C for 10 min to heat kill)
to minimize self-ligation, and purified once more on a spin column.
113 x 207, as last codon is limited to NNG. With two histidines at fixed positions, theoretical
diversity is 4.16 x 10 7 , though the practical diversity of assembled phage particles is likely to be
much smaller.
40
Insert preparation
Inserts were designed with two primers, an 89 nt sense strand and a 95 nt antisense
strand (Figure 2-8). The 89 nt sense strand starts 15 nt upstream of p8's BspHI
cut site and ends at the start of the displayed random peptide region. The 95 nt
antisense primer has a 48 bp overlap with the sense primer; 27 nt composing an 8 AA
randomized peptide sequence; and a 20 nt region extending 15 bp past the BamHI cut
site. The first guanine of this cut site, GGATCC, overlaps with the random peptide
region and dictates the last nucleotide therein. After annealing and extension, the
resulting duplex extends from the BspHI cut site upstream of the p8, 118 bp through
the N-terminus and past BamHI cut site that terminates the region coding for the
displayed peptides. The antisense primer's randomized region is designed such that,
after extension, the sense strand codes for two histidine residues, in either the third
and fifth or fourth and sixth positions, flanked by random codons: XXHXHXXZ or
XXXHXHXZ, where X is the mixed base codon NNK11 and Z is the mixed base codon
NNG, G being necessary to maintain the BamHI cut site.
Thusly designed, the primers needed to be annealed, extended, and digested to
be functional inserts. Annealing reactions were performed by mixing 3 pL 100 PM of
each primer (5 pg total for each) and 44 pL TE buffer (10 mM TrisHCl, pH 8.0, 1
mM EDTA) with 100 mM NaCl. These were added to a thermocycler, heated to 95*C
for 8 min, then cooled to 25'C at 0.1 0 C/s. To extend the duplex, the 50 pL reaction
was mixed with 20 pL 1oX Klenow Buffer (NEB Buffer 2), 8 pL 10 mM dNTPs, 8
pL Klenow Fragment (5 U/pL) and 114 pL deionized water. This 200 pL reaction
was heated to 37'C for 20 min to extend, and then heated to 65'C for 20 min to kill
the enzyme. The resulting DNA was purified and concentrated on a QlAquick® spin
column. 10 pg of purified DNA was digested with BamHI-HF and BSPHI and gel
purified with a 3% agarose gel to yield pure duplexes with exposed phosphorylated
cut sites.
1N is any nucleotide, K is guanine or thymine; this sequence preserves all amino acids but prevents
the two most frequent stop codons from being included.
41
Engineered phage vector (M13SK):
5' -ATG AAA AAG TCT TTA GTC CTC AAA GCC TCT GTA GCC GTT GCT ACC CTC GTT CCG ATG CTG TCT
S
L
M
P
V
L
T
A
V
A
V
S
A
K
L
V
L
S
K
K
M
nTTC GCT GCA GAG GGTGAG GAT CCC GCA AAA GCG GCC TTT...-3'
c
F ...
A
A
K
A
P
D
E
G
E
A
A
F
Primers
-- > overlaps with antisense primer
BspHI
7ene .
5'-TTAATGGAAACTTCCTCATGAAAAAGTCTTTAGTCCTCAAAGCCTCTGTAGCCGTTGCTACCCTCGTTCCGATGCTGTCT
In-terminus
TTCGCTGCA-3'
T'tUisense:
I--> overlaps with sense primer
BamHI
His 3,5:
5'-AAGGCCGCTTTTGCGGGATCCNNMNNMNNGTGMNNGTGMNNMNNTGCAGCGAAAGACAGCATCGGAACGAGGGTAGCAAC
GGCTACAGAGGCTTT- '3
I--> overlaps with sense primer
BamHI
His 4,6:
5'-AAGGCCGCTTTTGCGGGATCCNNMNNGTGMNNGTGMNNMNNMNNTGCAGCGAAAGACAGCATCGGAACGAGGGTAGCAAC
GGCTACAGAGGCTTT-'3
Figure 2-8: DNA oligonucleotides used for library construction. Black font color
indicates that the amino acid coded for is expressed on p8; grey is either not expressed
or expressed and cleaved away before virion assembly. Underlined nucleotides are cut
sites. Blue lines indicate coverage of sense primer and red lines antisense; dashed
section indicates randomized inserts.
42
Ligation and transformation
Digested vectors and insert libraries were mixed and ligated, then electroporated into
XL1-Blue electroporation competent cells (Strategene #200228).
For ligation, a
loX
molar ratio of insert DNA to vector DNA was used. The DNA was mixed with T4
Ligase and incubated at 16C for 16 hours, then the enzyme heat-killed 20 min at 65'C.
The reaction product (40-200 pL in 20 pL aliquots) was combined and concentrated
to 20 [tL using a QlAquick® spin column followed by an Amicon Ultra 0.5 mL 30K
spin filter, so as to increase subsequent transformation efficiency.
of ligated
1 pL
1
DNA was used in electroporation transformations with XL1-Blue cells according to
the Stratagene protocol.
N.B. During cloning experiments, our G23H vector also mutated to N12D on p8,
meaning that the resulting p8s were technically
fD
p8 rather than M13; however
the other proteins and genome were all that of the original M13SK vector. Previous
work does show greater permissivity of fD p8 to charge-changing mutations at the
N-terminus.104
Mutational analysis and alanine scan
Mutations were engineered using the Agilent QuikChange Lightning kit and pimer
design program.
The A3H clone was constructed by first creating the backbone
mutant H23G with one site-directed primer, then mutating the His4 and His6 residues
to alanine with a separate QuikChange primer.
2.5.2
Bacterial culture and M13 stocks
E. coli K12 ER2738 (NEB #E4104S) was used for phage amplification as per the
NEB manual on phage display libraries. Bacterial stocks were stored at -80'C in
50% glycerol. Colonies were streaked out monthly on LB/Xgal/IPTG/Tet plates and
kept at 4C. For started cultures, a single colony was picked and grown in 5mL of
LB broth with 20 mg/L tetracycline hydrochloride (Tet) overnight (12-16 hours).
The following day, a single phage plaque was picked and injected into 5 mL fresh
43
LB/Tet with with 50 pL of the bacterial overnight culture (0.O1X), then grown for
6-8 hours. 10 mL of fresh ER2738 overnight culture was prepared and added to 1 L
fresh LB/Tet, followed by 1 mL of the M13-infected culture. This was grown for 8-20
hours, depending on the growth rate of the phage in question (DDAH was grown for
at least 16 hours). The liter culture was then centrifuged (8000 RPM, 30 min) to
remove bacteria. The supernatant was mixed 5:1 with a 25 mM polyethylene glycol
(PEG) 8000, 2.5 M NaCl solution and refrigerated overnight. This was centrifuged
(8000 RPM, 30m) and the pellet redissolved in 20 mL PBS (Omnipur; 137 mM
NaCl, 10 mM phosphate). A second round of bacteria pelleting by centrifugation,
PEG addition to supernatant and incubation, then phage pelleting by centrifugation
was conducted to further purify and concentrate the M13. The final phage pellet
was diluted to 5-10
x
1013 phage/mL in PBS, spun 10 min at 7500 RPM to remove
remaining bacteria, and stored at 4C.
M13 preparations were quantified by spectrophotometer, according to the following equation:
[Phage] - (Abs
2 69
- Abs 3 2 0) x (6 x 1016)
genome length
(2.3)
where [Phage] is in particles per mL, absorbance is for a 1 cm path length, and the
genome length is in base pairs. Titration assays were also used to quantify phage
in plaque-forming units per mL (PFU/mL). However, because titers were commonly
an order of magnitude lower than the concentrations indicated by the spectrophotometer (see Table 3.2) and tended to have greater variation, spectrophotometer
measurements were used in regression analysis. DDAH grows much more slowly than
wild-type M13 bacteriophage in culture, and it is likely that its displayed peptides
can interfere with both infection and assembly, such that titer assays underestimate
the actual virion counts. Were titer counts used for analysis of catalytic experiments,
the regressed kcat and kcat/KMparameters would increase by roughly an order of
magnitude.
For titer assays and sequencing, E. coli XL1-Blue host cells (Strategene #200268)
were used to culture phage. Titering was otherwise carried out as described in the
44
NEB Ph.D. Phage Display manual. Briefly, stocks were diluted ten-fold in water
and 10 pL of a given dilution added to 200 pL overnight XL1-Blue.
After three
minutes, the 210 pL sample was mixed with 3 mL melted top agar and poured onto
Tet/Xgal/IPTG agar plates.
Plates with approximately 100 plaques were sought
for quantification. Sequencing was either prepped from 5 mL overnight cultures of
XL1-Blue cells and M13 or from infected bacterial pellets dissolved in 350 AL PB1.
Subsequent steps were carried out as described in the Qiagen® Miniprep Handbook.
2.5.3
Hydrolysis reactions
Solutions were mixed fresh each day and reactions assayed in 96-well plates on a
Tecan Infinite M200 Pro plate reader.
Substrate (para-nitrophenyl acetate from
Sigma-Aldrich) was stored as solid powder at -20C. Stock solutions were made at
50X, 2.5-40 mM in DMSO. 4 pL substrate solution was mixed on the well plate with
196 pL M13 (or control) in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM phosphate, pH
7.4) with 25 pMZnSO 4 . Final reaction mixtures were 2% DMSO, 98% PBS, with M13
at 1 x 1011-1
x
101 phage/mL (0.45-4.5 pM p8). Synthetic peptide controls DDAH-8
and DDAH-12 were analyzed at 20 pM final concentration. The initial slope of product (p-nitrophenylate, pNP;
E4OOnm =
13000 M- 1 cm 1 at pH 7.4) formation over time
was regressed and plotted for a range of starting substrate concentrations, and background rates subtracted from samples. Lineweaver-Burk analysis was then utilized to
determine the catalytic parameter kcat/KM by fitting all data points with MATLAB's
fit function, weighting the inverted x-values (1/[S] 0 , in M-') by initial substrate concentrations ([S]O, in M) to decrease the regression bias towards lower concentrations
inherent to Lineweaver-Burk analysis. CytoOne 96-well plates (CC7682-7596) with
reservoir space between the wells were used for all reactions, filling the interwell space
with room temperature water to humidify the reaction chamber and minimize well
temperature fluctuations during measurements.
Background rates were fit with simple linear regressions:
45
d[P]
(2.4)
kuncat [S]
dt
from initial slopes with the assumption that IS]
[SIO.
Lineweaver-Burk analysis proceeded via the following regression:
1
1
d[P]Idt
1
1
K
x
+
1(2.5)
kcat[E] X [S]
kcat[E]
TEM of DDAH with Zn 2
+
2.5.4
_KM
TEM samples were prepared on carbon film, 400 square mesh copper grids (Electron
Microscopy Sciences, CF-400-Cu) and imaged with a JEOL 2100 FEG analytical
transmission electron microscope (Figure 2-5). DDAH stocks were diluted 50X into
.
water for a final concentration of 7-8 x 10" phage/mL, with 0, 25, or 50 /pMZnSO4
10 piL of sample was added to the grid and left for 10 min. The sample was then
aspirated off via kimwipe and the grid inverted on a 1 mL drop of water for 10 min
to leach off unbound material. The grid was rinsed by addition and aspiration of
10 pL of water. I'd like to thank Dr. Dongsoo Yun in the Koch Institute Swanson
Biotechnology Center's Nanotechnology Materials core for imaging these grids.
2.5.5
Ni-NTA purification of phage clones
The protocol largely follows that provided by QIAGEN for their Ni-NTA kit, "Batch
purification of 6xHis-tagged proteins from E. coli under native conditions," but with
no imidazole in loading or wash steps. The column is stored with 1 mL QIAGEN
Ni-NTA beads in 50 mM NaH 2 PO 4 , 300 mM NaCl, pH 8 buffer. It was drained and 1
mL of 1.5x 1013 DDAH phage added in imidazole-free loading buffer, then shaken for
2 hr at 4'C. The column was drained ("flowthrough"), rinsed with 8 mL loading buffer,
and then treated with 0.5 mL elution buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 500
mM imidazole). Four 0.5 mL aliquots of elution buffer were added to and collected
from the column at room temperature. Infectivity assays and sequencing were then
conducted as described in Section 2.5.2.
46
Chapter 3
DDAH Hydrolysis Activity is
Versatile
Future biocatalytic processes generally will not be limited by the available technology or the nature of the substrates and the products. Instead,
the feasibility of new biocatalytic processes will often be determined by the
availability of the biocatalyst ...
- A. Schmid, et al. Nature 20013
In this chapter, the versatility of DDAH phage as a catalytic macromolecule is
demonstrated with respect to solvents, substrates and temperature. DDAH activity
is found to increase over an order of magnitude in dimethyl sulfoxide (DMSO), in
which the phage maintains its overall structure but is no longer infective. Solubility
and stability in DMSO allows the phage to catalyze a wider range of substrates, and
its efficiency towards larger molecules is determined. DDAH robustness with respect
to heat is characterized in two ways -
the remaining catalytic activity after heat-
ing to 80C for varying lengths of time before cooling back to room temperature,
and the activity towards substrates while in reaction mixtures at varying temperatures. The stability of DDAH at higher temperatures further increases the breadth
of hydrolyzable substrates, and activities toward less active esters are characterized.
47
3.1
Background: Engineering Robust Enzymes
Industrial catalysis is rarely optimized at room temperature in pH 7.5 saline, wherein
most enzymes evolved and the starting point for many de novo enzyme attempts.
A breadth of functional environments and outputs for a given enzyme is important
for its utility outside the laboratory. Poor catalytic activity in unnatural reaction
environments and instability therein continue to slow enzymatic implementation. 111
Organic solvents, for instance, increase the range of soluble substrates, decrease
deleterious side reactions, can alter substrate specificity, and modulate reaction thermodynamics beneficially. " For enzymes which function in organic solvents, the apolar
environment can minimize conformational flexibility, locking in a protein's structure.
Nonpolar solvents have been used in biocatalytic asymmetric transformation of acids,
alcohols, or diols, for instance, producing pharmaceuticals, herbicides1 1 2 and antifungals.13 Enzymatic polyphenol production, in particular, requires an organic solvent,
as the growing chains become insoluble and the reaction extinguished in aqueous
medium.
114
However, activity from native enzymes drops considerably in organic sol-
vents as opposed to water, if the enzyme functions at all, and the enzymes themselves
are frequently insoluble, requiring stirring of reaction mixtures. Their stability in nonpolar solvents can be inversely correlated with water content, but such lingering water
is often be essential for effective catalysis.15 Directed evolution approaches have been
applied to stabilize esterases and laccases in apolar solvents, and have stabilized the
target enzyme to mixtures with 30% and 50% of organic solvent in water, respectively. The resulting enzymes still see massive drops in activity relative to aqueous
reactions.
241
Immobilization techniques, while contributing their own transport and
chemical complications to a given process, nonetheless have been successfully implemented in stabilizing enzymes for catalysis in organic solvents. 22
Increasingly, de novo enzymes are also developed with an eye to functioning in
organic solvents. For enzyme mimics, the presence of an organic solvent and its corresponding low dielectric field can allow simple catalytic structures to emulate the
hydrophobic active sites of wild-type enyzmes.99 '1 6 Recently, Michael Gagn6 and col48
leagues utilized a bead tagging strategy in dichloromethane (DCM) to isolate reactive
8- and 12-amino acid
-hairpins. Mass spectroscopy identified a number of two- and
three-histidine sequences which subsequently exhibited rate accelerations in the thousands for esterolysis in trifluoroethanol (TFE). 71 A similar strategy was also used with
short helical peptides to find histidine-displaying biocatalysts active in DSM and TFE,
though the helices demonstrated more moderate catalysis and required nonnatural
amino acids to maintain helicity; maintenance of secondary structure was, unsurprisingly, deemed critical for activity.61 Such outcomes demonstrate both the promise of
de novo biocatalysts in apolar solvents and the potential utility of a macromolecular
structure with a robust biochemical structure.
In addition to solvent stability, thermostability can be extremely useful for commercial applications. An enzyme that works at a wide range of temperatures has
access to a broader range of solubilized and active substrates, potentially higher activity when heated, and can be implemented in reactors too hot for bacterial contamination.
17
Sufficient reactivity at raised temperatures can be problematic to ob-
tain, though, resulting in dead-ends for some attempted processes. 2 ' As with solvent
stability, immobilization and directed evolution are common approaches to improve
thermostable functionality.2 3 '11 The former has been used to stabilize enzymes involved in penicillin production at high temperatures, for example, among numerous
other enyzmes.
Directed evolution has facilitated the thermostability of xylanase,
carbamoylase, amylosucrase, and amidase enzymes,"' demonstrating the broad utility of the method. Engineered de novo enzymes generally are more robust in heated
reactions when metallic cations help keep domains together. The zinc ion in the helixloop-helix dimer produced by Der, et al., 99'118 stabilizes the protein to a Tm of 81C,
while a designed three-helix protein demonstrates a 370 C increase in its TM upon
binding iron-sulfur clusters." Sites engineered into naturally stable proteins, such as
Stable Protein One, also demonstrate resilient catalysis at raised temperatures.
119
A broad substrate range for an enzyme is itself useful, both making the enzyme
applicable for more processes and facilitating schemes that involve inhomogeneous
feedstocks. An enzyme's ability to simultaneously process many ester compounds
49
can simplify large-scale reaction schemes for the production of important fragrance or
cosmetic compounds, for instance.108 A wide starting substrate range also facilitates
directed evolution to greater specificity for a desired substrate that may not be the
primary target of an enzyme. When wild-type enzymes are rationally engineered for
substrate versatility, the deletion of bulky active site residues frequently leads to less
steric hindrance and greater promiscuity.21,20 As yet simple systems, de novo enzymes
often lack extreme specificity for one substrate, instead demonstrating measurable
activity for multiple substrates 93 and in some cases effectively replicating the diversity
of activity exhibited by a targeted wild-type enzyme. 58
Here, the diversity of functional environments and substrates compatible with
DDAH is examined, demonstrating activity in a heated apolar solvent and with numerous ester compounds. The relative activity with larger substrates is proportionally
greater than that of wild-type bCAII, which retains no activity outside of aqueous
mixtures, making DDAH a highly versatile biocatalytic macromolecule.
3.2
3.2.1
Results
DDAH activity is robust in nonaqueous solvents
As many synthetic processes involve harsh, abiotic environments, 3 I sought to determine the catalytic efficiency of DDAH in relevant nonaqueous solvents. The pNPA
substrate has excellent solubility and stability in DMSO, a highly useful solvent in,
for instance, the production of pharmaceuticals,
121
so this solvent was chosen for
further investigation. DDAH exhibited a thirty-fold increase in catalytic efficiency
in 98% DMSO, wherein kat/K
-= 16.87 M-'s-', while background levels remained
roughly the same (Table 3.1). The proportion of organic solvent was determined
by the dilution factor of DDAH stocks
(50X,
or 2% final aqueous concentration), but
including a bit of water in otherwise nonpolar reaction mixtures has also been shown
to critically increase resulting biocatalytic activity.
15
The wild-type bCAII enzyme
showed no measurable activity in DMSO (at 67 nM, Figure 3-1). Similarly, another
50
b
+PBS
a 0.15
14
MbCA6A
ATENM
E
10
O DDAH
MbCA11
C
A TENM
6
cc
.0DDAH
o 0.05
-2
0
20
10
Time (min)
300
200
100
pNPA (pM)
0
30
Figure 3-1: DDAH catalyzes pNPA hydrolysis in DMSO. (a) Absorbance vs time
plots for bCAII, TENM (a control phage), DDAH, and background reactions in PBS.
Darker color of marker indicates higher initial concentration of pNPA: 50, 100, 150,
200, 250 pM. (b) Resulting reaction rates calculated from initial slope of reactions
after subtracting background hydrolysis. Only DDAH demonstrates measurable hydrolytic activity. Hashed gray line indicates y = 0 nM/s. Error bars represent 95%
CI from three replicates each.
M13 clone, TENM, has activity in PBS but demonstrates none in DMSO. Such a lack
of activity could be the result of protein denaturation and/or precipitation.
kuncat (s
1)
(95% CI)
Solvent
Substrate
DMSO
pNPA
pNPB
pNPA
pNPP
6.92E-07
1.52E-06
9.50E-07
6.19E-07
3.44E-08
3.84E-08
4.70E-08
3.87E-08
pNPPa
5.69E-08
2.03E-08
Table 3.1: Background hydrolysis rates. Confidence expressed as the interval above
and below kunecat that sets the boundaries for the 95% confidence region.
In order to characterize the durability of the bacteriophage in DMSO, hydrolysis
reactions were run after various lengths of incubation in the solvent.
The activity
is stable for hours of exposure (Figure 3-2), through the length of the experiments
run. The phage assemblies demonstrate a possible physical rearrangement, wherein
both Km and kcat increase modestly such that their ratio kcat/KM remains the same.
This could be a bundling phenomenon, whereinin multiple phage filaments aggregate,
51
2
10
15m
25m
38m
68m
M
X
0
8-
CD
6
+
X
C>
--.
C
m
125m
X
1.5
1.5-
0~
4
(D0.5
0kcat
2-
0
0
KM
kcat/KM
01
0
200
400
[pNPA]0 (IM)
600
800
0
50
100
Incubation Time (min)
150
Figure 3-2: DDAH activity is stable during exposure to DMSO. (left) Hydrolysis
'
rate of pNPA vs. substrate concentration for DDAH clones solubilized in DMSO for
different lengths of time (markers), with regressions (lines). (right) kcat/KMvalues
through 2 hr in DMSO stay > 90% of the initial value. Catalytic parametetrs (at)
are determined from a NLLS fit to the Michaelis-Menten equation and normalized
to their t, = 15 min values (a0 , earliest time point to mix and plate out samples
with substrate). Time points: 15, 25, 38, 68, 90, and 125 min. Error bars are 95%
confidence intervals for kcat and KM, 90% confidence for kcat/Km (0.952). Adapted
from Casey et al.. 9
or a statistically slow rearrangement of active sites. The rates reach a steady-state
within 40 min, and after 125 min the kcat/KM ratio is not significantly different than
at t = 15 min.
PBS
Et(OH) 2
DMSO
Input Phage
Plaques
Factor
4540
91
644
7
7.05
13.00
9.10E07
0
Table 3.2: DDAH is fully resilient in ethane diol, but is rendered non-infectious after
exposure to DMSO. Stock phage, stored in PBS, was diluted 50X into the appropriate
medium and incubated for 80 min at room temperature. Samples were then diluted
into water and plated with XL1-Blue cells.
Two further experiments were conducted to assay the stability of the clone in
DMSO: infectivity assays and transmission electron microscopy.
When stored in
aqueous solution, approximately seven DDAH particles are measured by UV-Vis spectroscopy for every resulting plaque-forming unit (PFU) identified by titering stocks
52
onto XL1-Blue E. coli plates (Table 3.2). This ratio varies by clone (data not shown);
it may indicate that not all filamentous phage structures are successfully assembled
such that they are infective, or that they are aggregating and not infecting cells
individually. This factor, the ratio of spectroscopically measured phage to PFU determined by titers, is nonetheless consistent for a given clone. It decreases moderately
for DDAH stored in ethane diol (e.g., ethylene glycol), to thirteen.* Phage exposed to
98% DMSO is rendered completely noninfectious, with no plaques formed, even when
millions of particles are added to a culture plate. TEM experiments demonstrate that
the filamentous structure remains intact, however (Figure 3-3). This would indicate that the p3 protein responsible for bacterial binding and infection is irreversibly
denatured upon exposure to DMSO, but that the p8 coat proteins maintain their
quaternary structure. The stability of these a-helices allows the sites inserted on p8
to remain active in such a non-aqueous solvent.
3.2.2
The DDAH active site can accommodate large substrates
A critical feature of the M13 semisynthetic enzyme design is the openness of the active
site; situated between the most N-terminal nine amino acids of p8 and an adjacent
p8 a-helix, the site is relatively unimpeded. I therefore sought to characterize the
activity of DDAH with respect to substrates with increasing carbon chain length
(Figure 3-4a):
1. pNPA (n = 2);
2. p-nitrophenyl propionate (pNPP, n = 3);
3. p-nitrophenyl butyrate (pNPB, n
=
4); and
4. p-nitrophenyl palmitate (pNPPa, n = 15, lipase substrate).
Only pNPA and pNPB were soluble in the aqueous reaction mixture, but the
functionality of DDAH in DMSO enabled the study of substrates not soluble in PBS.
*Ethane diol and other solvents were considered for hydrolysis studies, but pNPA was found to
be poorly solubilized by both; see "Hydrolysis of nitrophenyl esters in DMSO" subsection of 3.4.1
Hydrolysis reactions.
53
b
d
c
Figure 3-3: DDAH retains its filamentous structure in DMSO. (a) and (b), DDAH
kept in PBS. (c) and (d), DDAH diluted 50X into DMSO prior to TEM sample prep.
In (d), phage has been incubated with quantum dots for contrast. Scale bars are 1
pM, except for (d), which is 500 nm.
54
While pNPP is one carbon shorter than pNPB, its aqueous solubility is lower, and it
could not be reliably mixed at sufficiently high concentrations for hydrolysis assays.
Solubilized L-histidine was used as a control, as it catalyzes ester hydrolysis at a
measurable level and because histidine residues are critical for the aqueous activity
of DDAH clones (Figure 2-7). In aqueous reactions, DDAH hydrolyzed pNPB at
just over half the efficiency of pNPA, with kcat/KM = 0.282 M-'s-' (Figure 34b). By comparison, wild-type carbonic anhydrase has been found to catalyze pNPB
hydrolysis with only 2% the activity with which it hydrolyzes pNPA.
1
L-histidine
demonstrated measurable but meager hydrolysis for both substrates, far below that
of the DDAH sites (Figure 3-4 and Table 3.3).
The nonaqueous reactions, again, were catalyzed far faster than the water-based
reactions. In 98% DMSO, the kcat/KM -- 16.87 M-'s- 1 for DDAH-catalyzed hydrolysis of pNPA -
dropped to 8.92 M-'s-1 for pNPP (Figure 3-4c). Notably, DDAH
catalyzed the lipase substrate pNPPa with a catalytic efficiency of 5.89 M- 1 s 1 , which
is 35% of the catalytic efficiency of DDAH with the much smaller substrate pNPA. As
pNPPa has a 15-unit hydrocarbon chain, this activity indicates that the substrates
primarily interact with DDAH p8s in an orientation such that the nitrophenyl group
is proximal. The capacity to react with substrates of such varying size makes DDAH
a promising alternative to biocatalysts whose substrate versatility is limited by steric
hindrance.
Interestingly, despite no demonstrable activity in aqueous reactions and no histidines on p8, the A3H clone (Table 2.2) was catalytically active in DMSO. While
not as efficient a catalyst as the full DDAH sequence, the clone showed significant
hydrolysis above background for each of the three substrates tested in DMSO. This
activity may be a result of remaining exposed aspartates (three per p8) and glutamates (one).
3.2.3
DDAH exhibits thermostable catalysis
In order to characterize the effect of temperature on DDAH activity, two experiments
were undertaken. First, DDAH samples were heated to 80'C for varying lengths of
55
a
0
02N
02 N
1-Phenylethyl
Propionate (pNPP)
Acetate (pNPA)
Acetate (PEA)
H
N
C
02N
0
0
O
)2N
0
Palmitate (pNPPa)
Butyrate (pNPB)
b
T
pNPA
pNPB
IndAc, 800 C
(I,
Indoxyl Acetate (IndAc)
20
C
10
13
T_
T
151
100
pNPA
pNPP
pNPPa
'~Q
10
T
10~
5
102
L-His
0
DDAH
L-His
DDAH
A3H
Figure 3-4: DDAH demonstrates versatile catalytic activity in water and DMSO. (a)
Structures of ester hydrolysis substrates assayed. (b) Log-scale plot of L-histidine
and DDAH in the aqueous (98% PBS) hydrolysis of nitrophenyl esters at room temperature and indoxyl acetate at 80'C. (c) Catalytic efficiency of L-histidine, DDAH,
and 3H solubilized in a 98% DMSO reaction mixture with substrates of differing
carbon chain lengths. Error bars in (b) and (c) represent 95% confidence intervals for
values fit by Lineweaver-Burk analysis. In (b), IndAc hydrolysis by L-histidine is indistinguishable from background rate; Lineweaver-Burk upper error bar not available.
Adapted from Casey et al.. 94
56
Solvent
PBS
Substrate Catalyst
L-His
pNPA
DDAH
pNPB
L-His
DDAH
0.036
0.282
IndAc
L-His
DDAH
L-His
DDAH
A3H
L-His
DDAH
A3H
L-His
DDAH
A3H
0.025
3.132
0.229
16.872
6.025
0.268
8.921
4.587
0.029
5.893
2.528
pNPA
DMSO
(M--s-)
0.021
0.535
pNPP
pNPPa
95% CI
upper
lower
0.001
0.001
0.033
0.038
0.018
0.009
0.081
0.190
0.016
1.018
2.905
0.048
0.083
0.620
0.669
2.032
1.124
0.087
0.249
2.720
6.971
1.146
0.764
0.063
0.019
1.412
2.709
0.248
0.309
-
kcat/KM
Table 3.3: Hydrolysis rates for tested substrates in PBS and DMSO. Indoxyl acetate was hydrolyzed at 80'C; all other rates are from room temperature reactions.
Confidence is expressed as lower and upper intervals to the boundaries of 95% CI
region.
time, then cooled off and assayed for pNPA hydrolysis. Second, pNPA hydrolysis was
measured at room temperature (25'C), 40'C and 65'C.
DDAH shows remarkable stability at 80'C as compared to the wild-type enzyme,
bCAII (Figure 3-5). Samples were heated to 80'C for 2, 10, 30, 60, 120, or 240
min and allowed to cool back to room temperature.
bCAII hydrolytic activity is
completely extinguished within minutes of heat treatment, and is afterward not detectable above background rates of hydrolysis (enzyme concentration = 150 nM).
DDAH, while far lower than the starting activity of bCAII, nonetheless maintains
activity substantially above background for hours of heat treatment. The reason for
the dip and recovery in DDAH activity is unclear, but may be related to thermal disaggregation of phage filaments, or insufficient sampling (n = 2 for each time point,
so that all samples could be run on a single well plate).
Many industrial processes are conducted at raised temperatures, both to improve
catalytic performance and to limit bacterial contamination."
57
7
Accordingly, hydrolysis
b
6E-08
6E-08
4hr
4hr
2hr
"- 1hr
10m
bCAII
4E-08
-2hr
'1h
-1hr
4E-08
10m
-DDAH
2E-08
2E-08
0
0
1
0
C
2
pNPA (mM)
1
0
4
3
2
pNPA (mM)
3
4
140%
120%
100%
80%
60%
40%
0
ODDAH
20%
0%
-20%-20%50t
T
-40%
Incubation Time @ 800C (min)
100
150
200
Figure 3-5: DDAH is stable to 80'C heat treatment. (a) bCAII enzyme (150 nM)
hydrolysis versus [S], plots after incubation at 80'C in PBS for varying lengths of
time. (b) DDAH-catalyzed hydrolysis versus [S], after equivalent incubations. All
reactions are conducted at room temperature (25'C) in PBS. The y-axes in (a) and
(b) are equal to facilitate comparison. (c) Resulting activity (kcat/KM) for bCAII
and DDAH, normalized to their respective kcat/KM at t, = 0 min. bCAII activity
collapses to background noise, while DDAH remains active through the four hour
experiment. Error bars represent 95% confidence.
58
5
cc
RT
400C
650C
43
> 2N
0
0
kcat
KM
kcat/KM
kuncat
kcat/kuncat
,
Figure 3-6: DDAH catalytic efficiency increases at raised temperatures. DDAH samples were solubilized in DMSO at the given temperatures and incubated for 30 min before substrate was added. After subtracting background activity from 25 PM ZnSO 4
each catalytic parameter - 0 T- is determined by Lineweaver-Burk analysis (except
for kuncat, which was found by simple linear regression) and normalized to the room
temperature value - aRT. Error bars represent 95% confidence. Adapted from Casey
et al.. 94
experiments were conducted in DMSO at 40'C and 65'C, with the latter representing
a temperature at which bacterial growth is unlikely. Regressed catalytic parameters
are shown in (Figure 3-6), normalized to values at room temperature to facilitate
comparison. The DDAH catalytic rate
kcat
increased significantly for the heated re-
actions, while the constant KN remained close to the room temperature value, such
that the overall catalytic efficiency increased with higher temperatures to twice that
at room temperature. The background rate of hydrolysis, kuncat, also increased with
temperature, dramatically so at 65'C. This background hydrolysis increased proportionally more than DDAH's
kcat,
such that the fold acceleration over background was
lower at 65'C than at room temperature, albeit still substantial (Table 3.4). All
determined kcat/kncat values were in the thousands, indicating significant rate acceleration at each temperature. The stability of the M13 capsid structure therefore leads
to a robust active site capable of catalysis at a wide range of functional temperatures.
59
95% CI
Temp.
Param.
kcat
Km
RT
kcat/KM
kuncat
kcat/kuncat
kcat
Km
40 0C
kcat/KM
kuncat
65 0C
Value
0.00352
5.72E-04
6.16
1.90E-07
18537
0.00694
6.15E-04
11.28
2.59E-07
lower
0.00034
8.63E-05
0.22
5.40E-08
5506
0.00097
1.31E-04
0.57
5.76E-08
upper
0.00042
1.11E-04
0.24
5.40E-08
10490
0.00136
1.93E-04
0.63
5.76E-08
kcat/kuncat
26806
7956
14398
kcat
Km
kcat/Km
0.00514
0.00063
0.00084
kuncat
3.91E-04
13.15
8.56E-07
8.02E-05
0.81
9.27E-08
1.11E-04
0.92
9.27E-08
kcat/kuncat
6007
1252
1824
Table 3.4: Hydrolysis rates at room temperature, 40 0C, and 65'C. DDAH samples
were solubilized in DMSO at the given temperatures and incubated for 30 min before
substrate was added. Initial slopes of reactions were regressed, and parameters derived
via Lineweaver-Burk analysis (except for kuncat which was found by simple linear
regression). Confidence expressed as the interval above and below the parameter
that sets the boundaries for the 95% confidence region.
60
3.2.4
Alternate esterolysis substrates
In addition to larger nitrophenyl esters, less active carboxylic esters were assayed for
reactivity with DDAH. 1-phenylethyl acetate (PEA) is a small molecule lipase substrate often assayed for enantiospecific esterolysis1 2 2 and esterification1 2 3 reactions.
As it has no electron-withdrawing nitrogenous groups on the phenyl ring, and the
ester oxygen is further removed from the phenyl than in nitrophenyl esters, it is less
readily hydrolyzed than the NP substrates. Racemic PEA was mixed with DDAH to
assess, if the phage hydrolyzed the substrate, whether the phage had an enantiomeric
preference. As PEA lacks a clear light spectroscopic signal, its products were analyzed
by a gas chromatography-mass spectroscopy instrument.
While product and substrate peaks could be clearly resolved following solvent
extraction, no hydrolytic activity was detectable in the tested conditions. Toluene,
hexane, acetonitrile and DMSO were all assayed as nonaqueous reaction solvents,
without success. In water, several pHs were assayed and, while higher pHs (8 and
above) showed a measurable level of background hydrolysis, in no samples did DDAH
increase this rate (Table 3.5). If anything, the addition of phage (26 [M p8) impeded
the transformation. Samples were allowed to react at room temperature, 50'C, and
80'C for one hour or overnight, still with no catalytic activity. The reverse action
was also tested, the esterification of 1-phenylethanol by a carboxylic acid, and again
no activity was found. As a positive control for the targeted semisynthetic enzyme
design, bCAII was reacted with PEA, but also exhibited no hydrolytic activity.
PEA is often hydrolyzed by lipase enzymes such as Candida antarctica lipase
B (CaLB), and the enzymatic mechanisms seem to be too different for a carbonic
anhydrase-inspired active site to catalyze the substrate. While a histidine-co6rdinated
zinc ion and hydroxide mediate bCAII catalysis, lipases have a non-metallic active site
which requires a triad of histidine, aspartate and serine. The serine not only interacts
with the histidine residue but also acts to stabilize various reaction intermediates
alongside glutamine and threonine residues in CaLB, for instance.' 21
While such
active site design considerations were not part of the libraries responsible for DDAH,
61
Table 3.5: DDAH does not hydrolyze phenylethyl acetate in aqueous solution. Different pH solutions of 20 mM phosphate buffer were assayed for the conversion of 1phenylethyl acetate (PEA) to 1-phenylethanol (PE). Solutions were incubated at 80'C
for two hours before extracting product in hexane. The addition of 5.75 x 1012 /mL
DDAH (26 pM p 8 ) led to less product. Presented numbers are ion counts from gas
chromatography-mass spectroscopy: "peaks" are the highest ion count on the chromatogram for the given compound, and the sum is integrated across the peak on the
chromatogram. The bottom row of the table is the PE sum divided by the total ion
count sums for PE and PEA of a given sample.
Quantity
PE peak
PE sum
PEA peak
PEA sum
% PE
pH 8
PBS
+DDAH
2585
6179
67330
155436
129019
584821
1458531
6580962
4.41
2.31
pH 8.5
PBS
+DDAH
12563
8144
307526
200918
144982
185662
1667765
2003936
15.57
9.11
pH 9
PBS
+DDAH
16234
8764
399533
219403
159772
152800
1760578
1727485
18.50
11.27
alternate libraries centered on this triad may be considered in the future for identifying
phage clones capable of similar transformations.
Additionally, the ester molecule indoxyl acetate (IndAc) was assayed.
A sub-
strate used to measure the activity of numerous enzymes, indoxyl acetate is better
hydrolyzed by esterases than lipases, phosphatases, or acylases,
2
5
which makes it
a fitting substrate for DDAH assays. Though the ester is more activated than that
of PEA, IndAc is a less reactive molecule than the previously measured nitrophenyl
esters. Upon hydrolysis of the acetate moiety, indoxyl is rapidly converted to indigo
when in the presence of oxygen. 12' Though insoluble in water, aqueous indigo production could be quantified upon dilution into DMSO by absorbance at 618 nm. No
DDAH-catalyzed activity was detectable at room temperature. However, at 80*C,
DDAH hydrolyzed the substrate with kct/KM
3.13 M-'s-
1
(Figure 3-4b and
Table 3.3). Solubilized L-histidine measured 0.025 M-s-'and was statistically in.
distinguishable from background hydrolysis, while bCAII measured 30.47 M-s- 1
In this case, the thermal stability of DDAH activity afforded it catalytic activity
which could not be measured at 25'C, and should promise a broad range of reactive
substrates in future work.
62
3.3
Summary
The environmental robustness inherent to the M13 viral capsid is a key feature in
its use as a biocatalytic platform. In particular, its compatibility with organic solvents and its thermostability are advantageous for numerous applications. Here, the
stability of DDAH in DMSO allowed for the catalysis of substrates and reactions
not practical in aqueous solutions, broadening the potential application space. The
increase in activity observed in DMSO may be a result of conformational rigidity
imparted by hydrophobic solvent interactions with p8 N-terminal residues, 1 5 which
lack known secondary structure in aqueous solutions. The well-characterized a-helix
formed by p8 is likely broken by the proline in position 6, resulting in an unknown
conformation for the most N-terminal ten residues. If these residues, which include
our inserts, are more restrained conformationally in less polar solvents, then this likely
contributes substantially to the increase in catalytic activity observed. Additionally,
as the M13 active site is relatively small, substrates may remain partially exposed to
the surrounding solvent, in which case nonpolar solvents can increase activity. 116 In
addition to DMSO, the phage is compatible with ethane diol, and other solvents will
be characterized in future work. While the lack of infectivity after exposure to DMSO
may impede potential catalytic selections done with libraries in DMSO in the future,
the maintenance of the filamentous quaternary structure will still be extremely useful. Given the number of important feed stocks and precursor compounds insoluble
in water, such diverse solvent compatibility is of immense utility.
In addition, the substrate and temperature range of the active site are broader
than most natural or de novo enzymes. While substrate promiscuity is unfavorable for
some processes, applications like petrochemical refining and bioremediation benefit
from enzymes demonstrating a broader range of working substrates. In this work, pnitrophenyl palmitate, a 15-carbon ester, is in the size range of industrially important,
heterogeneous alkyl acetate esters for fragrance and flavor compounds.1 0 8 ,127 The
output of enzymes used industrially for ester production can be modulated by the
starting feedstocks, and in some cases heterogenous input and output is desirable,
63
which relies upon active site promiscuity. Furthermore, DDAH's hydrolysis at 65'C
and 80'C affords the enzyme more efficient catalysis, a broader range of substrates
both soluble and active, greater diffusivity of substrate molecules, and the ability to
stymie potential bacterial infections.
3.4
Experimental Methods
3.4.1
Hydrolysis reactions
Aqueous hydrolysis of nitrophenyl esters
Solutions were mixed fresh each day and reactions assayed in 96-well plates on a
Tecan Infinite M200 Pro plate reader. Substrates (para-nitrophenyl acetate and butyrate) were stored as solid powder at -20'C. All substrates were purchased from
Sigma-Aldrich Inc. Stock solutions were mixed to 2.5-40 mM in DMSO. 4 PL substrate solution was mixed on the well plate with 196 pL M13 (or control) in PBS
(137 mM NaCl, 2.7 mM KCl, 10 mM phosphate, pH 7.4) with 25 pM ZnSO 4 . Final reaction mixtures were 2% DMSO, 98% PBS, with M13 at 1 x 1011-1
x 1012
phage/mL (0.45-4.5 pM p8). The initial slope of product (p-nitrophenylate, pNP;
64OOnm
=
13000 M- 1 cm-
1
at pH 7.4) formation over time was regressed and plotted
for a range of starting substrate concentrations, and background rates subtracted
from samples. Lineweaver-Burk analysis (Equation 2.5) was then utilized to determine the catalytic parameter kcat/KM by fitting all data points with MATLAB's fit
function, weighting the inverted x-values (1/[SI 0, in M- 1) by initial substrate concentrations ([S]O, in M) to decrease the regression bias towards lower concentrations
inherent to Lineweaver-Burk analysis. CytoOne 96-well plates (CC7682-7596) with
reservoir space between the wells were used for all reactions, filling the interwell space
with fluid of the appropriate temperature to humidify the reaction chamber and minimize well temperature fluctuations during measurements.
64
Aqueous hydrolysis of indoxyl acetate
Substrate and cofactor solutions were mixed fresh each day and reactions assayed by
absorbance. Indoxyl acetate was stored as a dry powder at 4C and mixed to 25200mM as 50X stock solutions. 1 pL 50X substrate was added to 49 pL 1E12/mL
DDAH (4.5 pM p8) in PBS with 25 pM ZnSO 4 . Final reaction mixtures were 2%
DMSO, 98% PBS with 0.5-4 mM indoxyl acetate. Samples were mixed in triplicate
in PCR strips and heated for 30 min, then diluted 5X with DMSO to dissolve indigo
precipitates. Resulting mixtures of solubilized indigo in 80% DMSO were quantified
on the plate reader by absorbance at 618 nm (6618 determined to be 12600 M- 1 cm- 1 ).
After subtracting background rates (25 pM ZnSO 4 in PBS), catalytic parameters were
determined via Lineweaver-Burk analysis (Equation 2.5).
Hydrolysis of nitrophenyl esters in DMSO
Solutions were mixed fresh each day and reactions assayed in 96-well plates. Substrates (para-nitrophenyl acetate, propionate and palmitate) were stored as solid powder at -20'C. Stock solutions were made at 50X, 2.5-40 mM in DMSO. For measuring
rates of hydrolysis with different substrates, 3 yL of M13 sample in PBS was added
to a plate well, followed by 196 pL of a substrate dilution in DMSO and 1 PL of
5 mM ZnSO 4 , for final mixtures of 2% PBS, 98% DMSO, with 25 pIMZnSO 4 and
50-800 pM substrate. Adding a small amount of aqueous solution to the plate first
and dispersing subsequently with substrate in DMSO led to the best mixing of the
two phases, with
kuricat = 1
x 106 s-1 and
kcat/KM
rates at 17 M- 1 s 1 . This was
used for the experiments in Figures 3-1 and 3-4. For incubation experiments, however, this was not possible, and the aqueous solution had to be dissolved in DMSO
first, then later added to the substrate aliquots already on the plate. This led to
kuncat
= 0.2 x 10-6 and kcat/KM = 6 M-s- 1 . This setup was used for the experi-
ments in Figures 3-2 and 3-6. In a 98% DMSO, 2% PBS mixture, it was determined
.
that the absorbance peak red shifts 30 nin, and pNP E430= 22000 M-em- 1
In addition to DMSO, other solvents considered for reactions included acetonitrile,
65
glycerol, and ethane diol. Acetonitrile solubilized the substrate poorly and had substantial background variation. Glycerol didn't solubilize the substrate at all, even at
raised temperatures, and ethane diol had extremely high rates of background hydrolysis of pNPA. Accordingly, none were further explored for nitrophenyl ester hydrolysis,
but all could be considered for future alternative reactions.
Heated hydrolysis of nitrophenyl esters in DMSO
For the heated reactions in Figure 3-6, phage in PBS was diluted into DMSO and
incubated at the appropriate temperature for 30 min before adding to 4 PL of substrate aliquoted on the well plate. The well plate was kept on a heating block at
the appropriate temperature during mixing, and the interwell space filled with 40'C
water or 65'C glycerol to jacket the wells at the intended temperature. The plate
reader itself was heated to 40*C or 42'C (maximum temperature for the instrument).
For 65'C reactions, the plate reader was held at 42'C and slopes were taken during
the first 66 s of the reaction, before substantial cooling could take place.
Glycerol was chosen to jacket the wells for its combination of high specific heat
and low thermal conductivity, which minimize the rate of temperature change during
reactions. The heat exchange was modeled as primarily one-dimensional through the
open (top) surface of the well plate, where the change in thermal energy, 'Q,' would
be proportional to exposed area:
dQ = A x kzXT
kT(3.1)
dt
f
where 'Q' is in Joules, 'A' is area, 'k' is thermal conductivity in W/m-K, 'AT' is the
temperature difference between the reaction mixture and the air in Kelvin, and 'f'
is the length of the temperature gradient in meters. The corresponding change in
temperature is a function of the mass 'in' and specific heat 'cp' in J/g.K:
dQ
dt
dT
d
When rearranged, the rate of temperature change of the jacketing fluid is
66
(3.2)
dT
dt
The term
k
PCP
A
AT
k
f
mcP
(33)
(p, density in g/mL, replacing m) was taken as an intrinsic property
of potential jacketing fluids, with lower values indicating slower temperature change.
For water,
k
PCp
is 0.16, for ethane diol 0.15, and for glycerol 0.02 mL/m-s, each at
65'C. Glycerol was therefore chosen to jacket the wells, as its temperature would be
most stable.
Regression methods for nonaqueous hydrolysis
Two methods of data regression were used for the nonaqueous reactions after subtracting background activity (25 PMZnSO 4 in described solvent). For determining
the kcat/KM ratio, conducting Lineweaver-Burk analysis is most precise (Equation
2.5). However, for the individual kcat or KM parameters emphasized in Figure 3-2,
fitting the initial slopes directly with the Michaelis-Menten equation gave more precise fits and less uncertainty. Accordingly, the values reported in Figures 3-4 and
3-6 were determined by Lineweaver-Burk analysis, while those in Figure 3-2 were
determined by fitting to the equation
dy
dt
_a
xx
b+x
(3.4)
where dy/dt is the initial slope of product over time, a is kcat * [Enzyme] = Vmax,
b is Km, and x is IS],. Background hydrolysis activity was determined by a simple
linear regression (Equation 2.4).
3.4.2
Bacterial culture and M13 stocks
DDAH was amplified as described in Section 2.5.2 and quantified by spectrophotometer, according to Equation 2.3. Briefly, an overnight 10 mL culture of E. coli
K12 ER2738 was added to 1 L of fresh LB with 1 mL 1000X Tet and 1 mL of a previously sequenced DDAH culture. This was grown overnight (16 hours) then centrifuged
to remove E. coli, mixed with PEG-NaCl, refrigerated overnight and centrifuged once
67
more to pellet phage. After dissolving the M13 pellet in PBS, a second round of bacteria pelleting by centrifugation, PEG addition and incubation, then phage pelleting
by centrifugation was conducted to further purify and concentrate the phage.
For titer assays, E. coli XL1-Blue host cells (Strategene #200268) were used to
culture phage. Titering was otherwise carried out as described in the NEB Ph.D.
Phage Display manual. Briefly, phage incubated in a given medium was diluted in
ten-fold series in water and 10 yL of a given dilution added to 200 pL overnight
XL1-Blue. After three minutes, the 210 pL sample was mixed with 3 mL melted top
agar and poured onto Tet/Xgal/IPTG agar plates. Plates with approximately 100
plaques were sought for quantification.
3.4.3
TEM of DDAH in water and DMSO
TEM samples were prepared on carbon film, 400 square mesh copper grids (Electron
Microscopy Sciences, CF-400-Cu) and imaged with a JEOL JEM 200CX transmission
electron microscope.
For aqueous samples (Figure 3-3a-b), DDAH stocks were
diluted 50X into water for a final concentration of 8 x 1011 phage/mL. 10 pL of
sample was added to the grid and left for 8-10 min. The sample was then aspirated
off via kimwipe and the grid inverted on a 1 mL drop of water for 10-15 min to leach
off unbound material. The grid was rinsed twice by addition and aspiration of 10 AL
of water.
Bare phage in DMSO (Figure 3-3c) samples were prepared similarly. DDAH was
diluted 50X into DMSO for a final concentration of 8 x 1011 phage/mL and left for
5 min. 10 pL of sample was then added to a carbon film grid and left for 25 min,
for a total of 30 min in DMSO. The sample was aspirated by kimwipe and the grid
inverted onto 1 mL of water for 10 min to remove unbound material. The grid was
rinsed twice by addition and aspiration of 10 pL of water.
The quantum dot-stained phage was prepared for potentially better contrast of
phage filaments (Figure 3-3d). A 5 x 1013 phage/mL stock was diluted 50OX into
97% DMSO for a final concentration of 1 x 1011
/mL.
The final solution included 2%
PBS and 1% (by solution volume) cadmium-selenium core, zinc sulfide shell quan68
turn dots stabilized with L-histidine in tris/borate/ethylenediaminetetraacetic acid
(EDTA) (TBE buffer). 10 pL of sample was added to a grid and left for 10 min
before wicking away with a kimwipe. The grid was inverted on a 1 mL drop of water
for 15 min before removing, aspirating remaining water, and rinsing with a 10 1L
sample of water.
I'd like to thank Jacqueline Ohmura for imaging the grids shown in Figure 3-3b
and c.
69
70
Chapter 4
M13 Catalyzes Heme- and
Copper-Based Redox Reactions
An ideal peroxidase for industrial applications should be one that is
abundant, can be easily purified, shows broad substrate specificity and has
better stability against high temperature and extreme pH conditions.
- F. Cai, et al., J. Mol. Cat. B: Enzym 2012128
In this chapter, M13 is assayed for cofactor binding and catalytic activity within
reduction-oxidation biochemistry. Two catalytic cofactors are chosen for initial characterization -
heme and copper.
Heme-phage interactions are characterized by
Soret peak analysis, and a successful heme-co6rdinating phage demonstrates oxidation of two peroxidase substrates, tetramethylbenzidine (TMB) and 2,2'-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS). Heme-agarose columns are subsequently developed for the potential selection of heme-co6rdinating phage from libraries. Copper binding is also studied by spectroscopy, and oxidative activity towards catechol and 3,5-di-tert-butylcatechol assayed. Copper-NTA columns are then
developed as a potential selection strategy for use with protein libraries. The activity of M13 towards these disparate redox reactions and separability with simple
chromatography establish a baseline from which future biocatalysts could be evolved.
71
4.1
Background
Following the demonstration of engineered M13 phage as a versatile biocatalytic platform via esterolysis, I sought additional reactions interfacing industrial chemical synthesis with enzymology wherein the platform could add value. Reduction-oxidation
enzymes are particularly suitable targets for phage engineering, as they
* catalyze reactions broadly useful for numerous industries;
" remain under-utilized due to cost (scarcity), stability and substrate breadth
limitations; and
* utilize widely available cofactors with thoroughly understood biochemistry.
Specifically, heme- and copper-based redox enzymes catalyze a diverse array of important reactions. While nickel is also a cofactor in some redox enzymes, such as
nickel superoxide dismutase, its prevalence and potential utility were deemed to be
lower.
4.1.1
Heme peroxidase engineering and applications
Hemoproteins are the most well-studied of the relevant enzymes. Resting in either
ferrous or ferric (Fe 2 + or Fe3+) states, the heme is axially ligated by water or oxygen
on one side and an amino acid on the other. This residue is often histidine but occasionally cysteine. 129 The peroxidase* cycle involves the two electron oxidation of heme
to an oxoferryl radical state, followed by either two one-electron dehydrogenations of
substrates or one two-electron oxygen insertion (Figure 4-1 130).
Heme peroxidases are exciting candidates for chemical synthesis, catalyzing oxidations and dehydrogenations with the potential to replace more costly, wasteful
inorganic means, for instance in the production of phenolic resins. 31 1 4 Already, they
*Formally, enzymes that use hydrogen peroxide as an oxidant are catalases, while peroxidase
activity refers to consumption of an alkyl peroxide. 129 Here, peroxidase is used as a general term
for activity with either oxidant.
72
OH
SH
L
S+H 2 0
/
N
N
Fel/
Compound I
N -I-N
N
N
S.
(a)
N H 2 02
/
N
| N
L
0 OH
N-I-N
Fe"/
| N
L
SH
H20
-*
N
/
0
N-I-N'*
Felv/
I N
L
Compound I
(b)
SO--
S
Figure 4-1: The enzymatic cycle for the heme cofactor has two possible routes for
substrate oxidation after the oxoferryl heme "Compound I" is formed: (a) two oneelectron transfers by dehydrogenation or (b) one two-electron transfer by oxygen insertion. Figure adapted with permission from van de Velde, et al., Trends in Biotechnology 2001.130
have found niche commercial use as antimicrobials in toothpaste and as food additives, 130 and have seen concerted development for detoxification of wastes, 13 1 though
are still of limited scope for industrial catalysis.
Peroxidases catalyze four broad types of reactions: oxidative dehydrogenation,
oxidative halogenation, hydrogen peroxide dismutation, and oxygen transfer.
132
De-
hydrogenation reactions have detoxifying applications and halogenation reactions are
useful in saccharide production, while dismutase activity is of little or no commercial
utility.
Oxygen transfer, however, is a potentially extremely valuable transforma-
tion, as selective atom oxygenation with cheap and nontoxic oxidants has few large
scale non-biological alternatives.
agrochemicals,
132
Such reactions are relevant for pharmaceuticals,
and even petrochemical desulfurization. A particularly convenient
feature of peroxidases is the use of cheaper oxidants, such as hydrogen peroxide or
even atmospheric dioxygen.
Molecular oxygen has been utilized as a terminal oxi-
dant for some pharmaceutical syntheses with peroxidases, which nonetheless required
the addition of stoichiometric acid. 1 33 More generally, oxygen transfer by natural
73
peroxidase enzymes remains impeded because the heme molecule is rather buried,
limiting exposure to aromatic substrates. Currently, peroxidases are also difficult to
implement because they are costly to produce and insufficiently stable.
128,130,132,134
Additionally, the ability to catalyze peroxidase reactions in non-aqueous media is
a consistent hurdle 13 2 and will be necessary for polymerization of compounds not
soluble in water, such as polyphenols.
114
Accordingly, researchers have sought to find more stable peroxidases, engineer
broader substrate suitability, and craft de novo peroxidases. A Jatropha curcas peroxidase was recently found and purified with enhanced thermostability and pH resilience; the wild-type enzyme was even stable under some exposure to organic solvents, but only moderate amounts of solvent, and not DMSO.1 28 Native enzymes
such as horseradish peroxidase (HRP) have been engineered for greater substrate access to the active heme by mutating a nearby phenylalanine to remove steric bulk,
which improved sulfoxidation and epoxidation reactions.135,136,137 Other researchers
have directly evolved a fungal "versatile peroxidase" used for lignin degradation, increasing its Tm by 8'C and improving its overall stability. 138 An early attempt at
peroxidase-inspired de novo active sites inserted selenocysteine into subtilisin, creating a relatively stable semisynthetic peroxidase.
139
In addition, a number of de novo helical bundle proteins have been constructed
to recapitulate heme biochemistry:
" several iterations of the helix-loop-helix dimer "maquette" that binds multiple
heme groups with characteristic redox potentiometry;
49 ,50
* a later version of this maquette which binds many cofactors and displays multifaceted electron transfer; 5 2
* an alternate four-helix protein designed for dioxygen binding as an oxygen transport; 140
* multiple heme-binding proteins isolated from a library of four-helix, binarypatterned de novo structures which have displayed measurable peroxidase activity;51,59,141
and
74
* two- and four-helix bundles covalently bound to heme-like iron porphyrins which
13
4 14 2
display substantial activity in the presence of trifluoroethanol.
These efforts demonstrate the promise of coupling rational biochemical engineering
with biological variation to identify functional synthetic enzymes. The approaches use
histidine or cysteine ligands to activate their respective heme cofactors, often oxidizing
multiple substrates and generally peaking around pH 6-6.5. Activities so far shown
have been impressive, especially that of the latter, Pavone group, whose covalently
bound proteins demonstrate a kat/KM of 8.4 x 103 M-Is-1 with ABTS (as compared
to 5.1 x 105 M-s- 1 for wild-type HRP). However, cost and instability remain as
obstacles,13 4 and higher-throughput adaptability could be useful in identifying even
more active catalysts.
4.1.2
Copper oxidase engineering and applications
While a less frequent target for enzyme engineering, copper-based oxidoreductases are
also potentially very valuable industrial biocatalysts. Enzymes like laccase and copper
oxidase oxidize substrates via molecular oxygen, obviating the cost and complication
of adding stoichiometric oxidants.1 29 Such reactions are useful in the processing of
textiles and paper products, where dyes can be modulated and lignin degradaded,
respectively. A larger opportunity exists in organic syntheses, where the oxidation of
aromatic compounds is in demand.
143
Like heme cofactors, copper centers are often ligated with imidazole and sulfhydryl
groups from histidine and cysteine. In some cases, methionine is also a ligand. Copper enzymes co6rdinate the Cu2 + dication in a square coplanar configuration, the
distortion of which is likely responsible for an observed strong ~615 nm absorption
peak). Many copper enzymes catalyze a two-electron oxidation, such as amine oxidase
and galactose oxidase, both of which reduce dioxygen to hydrogen peroxide while oxidizing their respective targets. Ascorbate oxidase and cytochrome oxidase, however,
reduce dioxygen fully to water in a four-electron oxidation reaction. Tyrosinase is a
copper monooxygenase that can oxidize catechol to o-benzoquinone (Figure), also
75
reducing molecular oxygen to water -
two molecules of catechol for each molecule
of dioxygen. 129 Laccases have four copper ions, each of which is responsible for a
one-electron transfer from a different substrate molecule for a total four-electron oxidation, reducing one molecule of dioxygen to two water molecules in the process.
143
These varied activities represent promising alternatives to inorganic, heterogeneous
means of substrate oxidation.
Recent de novo enzyme strategies with non-heme redox centers have had encouraging results. While not cupric, a helix-loop-helix dimer strategy from the DeGrado
and Lombardi groups yielded a four-helix bundle which bound di-iron to oxidize a
number of phenols, including DTBC (kt/KM = 105 M-'s- 1 )." A tri-helix approach
from the Pecoraro group successfully mimicked a nitrite reductase, co6rdinating copper ions to reduce nitrite in the presence of the reductant ascorbate.6 2 6, 3 These helical
bundle strategies, though difficult to design and adapt, have produced significant catalytic activities and demonstrate the potential of de novo active sites.
4.2
Results: Heme Peroxidase Activity
As heme peroxidase enzymes are well understood and catalyze highly useful transformations, I sought to develop M13 p8 proteins as robust, versatile peroxidase catalysts.
A selection of clones were first assayed for co6rdination of solubilized heme cofactors,
which can be characterized by spectroscopic analysis. A heme-binding clone, DDAH,
is then assayed with two peroxidase substrates to understand the p8-heme interaction
and the pH dependency of resulting peroxidase activity. Lastly, I develop a strategy
to purify heme-binding phage with hemin-agarose beads, which could be used for
library selection or enrichment in the future.
4.2.1
Identifying and characterizing heme-binding M13 clones
In order to identify M13 clones that successfully co6rdinate heme, sequences from the
original triple histidine library (Section 2.2) were examined along with clones from
a similar cysteine-based library. While the heme cofactor in peroxidases is most often
76
b
1.5
-
R9
2Cys13H
-2Cys15H
2Cys27H
e
--
40
ADHD
-
< 0.5
*
10,
200
350
300
250
Wavelength (nm)
400
C
-
+
-
+
-
+
-
+
0
Figure 4-2: Two-cysteine and four-histidine amplifications retain other E. coli proteins. (a) UV-Vis spectroscopy of these phage stocks indicate impure preparations.
Clean preparations have a clear peak at ~265 nm. (b) PAGE of two-cysteine and
four-histidine clones indicates phage is present along with non-phage proteins. The
first lane is a ladder, with specific bands annotated in kD. p8 runs near l0kD marker
(*). Other bands visible are likely contaminating, non-phage proteins. 'C' indicates
whether or not the phage preparation had been further cleaned and purified. Se-
quences as in Table 4.1. ADHD is ADHDHHSV, 15H.
ligated by a histidine residue, as in horseradish peroxidase, chloroperoxidase utilizes
a cysteine sulfhydryl instead and is a rather versatile biocatalyst."' Accordingly,
M13 sequences rich in histidines and cysteines were selected for amplification and
characterization.
After amplifying the selection of clones, several seemed to purify poorly (Figure
4-2).
Specifically, three clones chosen for their display of cysteine (SCPDCGAE,
with various single histidine insertions, Table 4.1) and an additional clone chosen
for its display of four histidines (ADHDHHSV, 15H) lacked the 269 nm peak ill
their absorbance spectra which is standard for clean phage stocks. These conflated
spectra indicated the presence of other proteins. As infectivity plates also indicated
low phage levels (see Experimental Methods), PAGE experiments were conducted
to check that phage proteins were actually present in the stocks.
Purified phage
presents a clear p8 band at 10 kD with little else, as the other phage proteins express
at such lower levels (see Figure 2-6b).
While various non-phage proteins littered
77
Catalyst
None
DDAH
DEHS
DMHE
2Cysl3H
2Cysl5H
2Cys27H
Soret (nm)
390
408
390
390
-
Abs
0.134
0.368
0.329
0.323
0.505
0.521
0.523
Sequence
Empty (PBS)
DDAHVHWE, 23H
DEHSHGLP, 15H
DMHEHNGE, 13H
SCPDCGAE, 13H
SCPDCGAE, 15H
SCPDCGAE, 27H
Table 4.1: Most clones fail to cobrdinate heme. Phage variants were added to 5
pM hemin chloride in PBS with a 45 pM final p8 concentration. Soret peaks determined from the absorbance maximum for A > 370 nm; the cysteine clones had no
maxima in the relevant range (Figure 4-3).
the PAGE samples, a clear p8 band is also visible for the cysteine (asterisked). The
four stocks were subsequently processed with DNAse treatment as well as rounds of
pelleting & centrifugation (C '+' lanes; see Experimental Methods), as the broad
absorbance spectrum may represent both protein and DNA remnants. Non-phage
proteins were diminished, but not eliminated, and the ratio of contaminant proteins
to p8 does not seem to improve.
The clones also bound poorly to a nickel-NTA
column, proving resistant to better purification. Weak protease treatment may be
considered for cleaning future stocks.
The heme co6rdination studies below were
conducted with the as-cleaned phage preparations from Figure 4-2b, though ADHD
was not further characterized due to poor expression and lack of signal with earlier
heme measurements.
Of the clones tested, only DDAH clearly co6rdinates heme (Figure 4-3). A large
(nine-fold) excess of p8 was chosen so that the hemin chloride would be maximally
affected and any shift in the peak more easily detected. The unbound hemin chloride
control had a peak at 390 nm. The spectra for DMHE and DEHS phage were little
changed, only shifted slightly higher. The two-cysteine clones also showed no change
in the peak, and manifested as shoulders rather than peaks due to the other material
remaining in the preparations.
DDAH, however, had a clear 18 nm shift, with a
sharp increase in the peak. This shift indicates functional coardination of the heme
cofactor, and DDAH was chosen for further characterization.
78
b
a
2
Background
2Cysl 3H
2Cysl 5H
2Cys27H
DMHE
DEHS
DDAH
1.5
0)
C.,
-o
0
C,,
1
-o
0.6
0.41}
.0
E.
0
0.2
0.5
0I
0-
200
600
400
Wavelength (nm)
350
800
400
Wavelength (nm)
450
Figure 4-3: DDAH co6rdinates heme, as evidenced by a shift in the Soret peak.
(a) Six clones were assayed for binding of heme, whose 390 nm Soret peak redshifts and sharpens when co6rdinated. Samples were mixed to a final hemin chloride
concentration of 5 pM, final p8 concentration of 45 pM. (b) A magnified view of the
Soret spectra, with the same line colors as in (a). While the absolute values of the
absorbance varied wildly, only DDAH showed a clear red shift and narrowing of the
peak.
4.2.2
Estimating DDAH p8-heme KD via Soret peaks
The Soret peak absorbance was utilized to estimate the binding affinity of DDAH
to heme. By titrating in hemin chloride to a solution of DDAH, a range of Soret
peak curves was obtained (Figure 4-4). Two values were then plotted against the
input concentration of heme, adjusted for dilution effects: the peak absorbance, at
408 nm, and the difference between the 408 absorbance and the 360 absorbance (a
parameter representative of heme co6rdination5 9 ). These were normalized and fit to
the following equation:
y=
[(x + [p 8 ]o + KD) - V/(x + [p8]o + KD) 2
-
4x- [p8]o]
(4.1)
wherein 'y' is an absorbance signal, 'a' is a scaling factor, 'x' is the input heme
concentration, and [p8]o is the starting concentration of phage p8 (here, 45 piM). This
is itself derived from applying the quadratic equation to the formula for dissociation
79
2
1.5
4A
0.5
0
360
400
480
440
520
Wavelength (nm)
Figure 4-4: DDAH-heme Soret peaks were measured increasing hemin chloride concentration with DDAH. Hemin was added in 5 ptM increments from 0 (bottom curve)
to 50 (top peak) pM to a 45 btM solution of phage.
constants, KD:
[A] [B]
[AB]
where [AB] is the concentration of complex formed by free 'A' and 'B' such that
[A]o
=
[A] + [AB] and [B]o = [B] + [AB].
The regressions of Equation 4.1 to
the Soret peaks from 0-50 pM hemin chloride with 45 pM-p8 are shown in Figure
4-5. While the inferred dissociation constants are internally consistent, ultimately
the fits are inconclusive. The KD values obtained for Abs 408 and AAbs 4 08 -3 6 o are,
respectively, 13.2 and 12.1 pM. However, the residuals plot in (b) shows a non-random
pattern of variance from the model, indicating that the model does not adequately
fit the measured absorbance data. It is thus far unclear what source of variation is
driving the inconsistency between measured Soret spectras and the model. Instead,
the heme-DDAH dissociation constant is inferred from catalytic data in Section
4.2.3, below.
80
a
b
015
1.2
1
0.1
0.S0.8
0.05
No
408 Abs
A 408-360
0. 6
50
E 0.4
-0.05
408 Abs
0.2
A 408-360
-0.1
0-'
0
10
20
30
[Hemin] (g M)
40
50
[Hemin] ( M)
Figure 4-5: Regressions for heme-DDAH KD from Soret peaks vs. heme concentrations data results in inadequate fits. (a) Normalized absorbance values for the
Soret peak, at 408 nm, and the absorbance difference Abs 408 - Abs 360 , from Figure
4-4. Data points are plus signs, and the fits from Equation 4.1 are represented as
solid lines. (b) The residuals from regressions to data points in (a); the non-random,
systematic pattern of residuals indicates that the data cannot be properly fit to the
model.
4.2.3
DDAH peroxidase oxidizes TMB
The colorimetric compound 3,3'-5,5'-tetramethylbenzidine (TMB) was chosen for initial assays of DDAH-heme complexes' peroxidase activity. TMB is a commercially
available peroxidase substrate which comes in pre-mixed reaction solutions for ELISA
plates. While the substrate is extremely sensitive to peroxidase, with a strong 652
nm absorbance peak after oxidation, the pre-mixed solutions lack details on extinction coefficients and constituent concentrations. Nonetheless, the compound's ease of
detection made it a useful starting point for DDAH peroxidase measurements.
DDAH was mixed at 0, 1, 2, and 5 pM p8 concentrations with 1, 2, or 5 ptM hemin
chloride to assay TMB activity (Figure 4-6). Unco6rdinated heme exhibited measurable oxidation, but the addition of DDAH drastically increased the rate of catalysis,
as measured by change in Abs 65 2 . Without heme, negligible catalysis was observed,
with or without phage. Background solubilized heme solutions accelerated the reaction 2.3 x
107;
the addition of 5 pM DDAH p8 increased this rate acceleration to
81
b
0.0016
I
H2N
I
0.0012
T
*0 pM p8
I
0.0008
0.0004
NH2
i 1 pM p8
A2 pM p8
HN)
x5
pM p8
0
0
6
4
2
[Hemin] (pM)
NH
Figure 4-6: DDAH-heme complexes oxidize TMB. (a) The addition of DDAH to TMB
reactions substantially increases oxidation of the substrate. Final mixtures were 2%
DMSO (hemin chloride stocks in DMSO diluted 50X), 48% PBS with DDAH, and 50%
Ultra TMB solution. y-axis is the initial slope, A Abs6 2 /At. Error bars represent the
95% confidence interval. (b) Oxidizing TMB (top) results in a brightly colorimetric
product (bottom).
3.5 x 108, or ~15-fold. The substantial rate increase indicates DDAH p8 proteins are
effective peroxidase mimics. However, with no information as to the concentration of
H 2 0 2 or TMB, catalytic rate inferences were limited.
Estimating heme-DDAH KD via TMB oxidation
While TMB oxidation did not generate any standard catalytic parameters, it did
facilitate the estimation of the heme-DDAH dissociation constant. In doing so, two
key assumptions were made:
* the rate of product formation in the presence of catalyst is comprised of (1) a
term linearly proportional to catalyst concentration and (2) a background rate:
dP
dt
dt = ki[E] + k 2
82
(4.3)
a
b
0.03
0.25 r
KD = 20RM
0.02
0.2
CO
0.151
0.01
-8
K D=5p M
-
0.1
I
LI
L
2O M
0
C.,O
0.
.
CO
0.05
-0.01
t30u
M
40g M
C0
0
20
10
[DDAH p8] (g M)
-0.02
30
0.041
Cr
25.6
0.04
K
K =30gM
0.021
0.01
0
-0.01
-0.02'
=
40pM
0.03
0.031
CO,
a)
3.2
[p8] (g M)
d
C
CO
0.4
c,
I LL.I
I 11
0.4
I
CD
Cu
I
0.01
0
'II II
3.2
[p8] (g M)
0.02
-0.01
-0.02
25.6
0.4
3.2
25.6
[p81 ( M)
Figure 4-7: TMB oxidation rates can be regressed to infer KD. (a) Rates of absorbance increase at 652 nm were measured from initial slopes of reactions and plotted against the concentration of DDAH p8 (gray plus signs). p8 concentrations were
100 nM to 25.6 pM, in nine two-fold increments. Solid lines are regressions from
Equation 4.1 with the given KD. (b-d), the residuals from fits with KD
20, 30,
different
is
a
bar
color
each
concentration;
p8
vs.
log-scale
plotted
were
or 40 pM
replicate.
83
where k, could be, for example, the unisubstrate expression
strate expression
9
1+Ka/(5
kcat[S]
KAI+[S]
or the bisub-
and
KB;
the concentration of catalyst is appropriately represented by Equation 4.1.
7.E-09
6.E-09
2
0
5.E-09
0:
4.E-09
Er3.E-09
4-0
00
0
tA
0
2.E-09
1.E-09
0.E+00
0
10
20
30
40
50
Modeled KD (iM)
Figure 4-8: A regression with a KD of 20 puM best approximates the TMB oxidation
data. Differences in model regression and data points, as in Figure 4-7b-d, were
squared and summed with n - 3 at each KD.
TMB solutions were added to 1 pM hemin with a range of DDAH p8 concentrations, 100 nM to 25.6 pM in two-fold steps (Figure 4-7). The resulting data points
were fit with Equation 4.1, where the scaling factor 'a' now corresponds to a rate
(AAbs 652 /At) rather than an absorbance peak. For these regressions, the KD was
The resulting fit was evaluated by residuals analysis.
fixed at 5, 10, 20, 30, or 40 pM.
1
A KD of 20 puM had relatively balanced residuals, with no clear pattern of variance,
and minimized the sum of squared error (SSE) (Figure 4-8).
4.2.4
DDAH peroxidase oxidizes ABTS
DDAH exhibits pH-dependent peroxidase activity
In order to quantify the catalytic activity of DDAH as a peroxidase, ABTS oxidation was measured. 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) is
a sensitive reagent for HRP activity assays and has been used in many previous de
84
novo peroxidase studies.5 9,134 ,142 The substrate is highly colorimetric, with a strong
peak at 660 nm 134 after oxidative dehydrogenation. Reactions with 1 mM ABTS and
micromolar H 2 0 2 demonstrate substantial peroxidase activity from DDAH (Figure
4-9). Little activity is detectable from the uncatalyzed reaction (phosphate buffer,
here at pH 6, with ABTS and H 2 0 2 ), while solubilized heme demonstrates meager
catalysis. Adding DDAH, at a loX ratio of p8 to heme, drastically increases the rate
of substrate oxidation.
In order to better understand the DDAH peroxidase activity, ABTS assays were
conducted from pH 4-9 (Figure 4-9c). The catalytic activity peaked at pH 6, where
kcat/KM
= 86.8 M-'s-for mixtures of DDAH and hemin chloride.
Below pH 6,
catalytic efficiency drops but remains far above (approximately 30-fold) solubilized
heme.
Above pH 7, DDAH shows no significant catalysis over heme in solution.
The pH for peak activity is in accordance with that of previous studies with de
novo proteins 134,142, wherein activity is maximum around pH 6-6.5. At lower pHs,
protonation likely reduces the interaction between histidine and heme, decreasing
activity. At higher pHs, hydroxide ions replace the water molecule occupying the
heme iron's distal co6rdination site, also nullifying activity.
134
Using the DDAH p8-heme KD inferred above, 20 pM, the concentration of p8heme complexes could be estimated, and an adjusted kcat/KM determined in which
p8-co6rdinated heme activity is isolated from unco6rdinated heme in solution. With
this adjustment, the heme-p8 kcat/KM is 257.4 M-'s- 1 at pH 6, fifty-fold higher
than that for unbound heme. While the measure is indirect and not relevant to the
functional output of the clone, this disparity is informative for future development.
It implies that increasing the clone's affinity for heme could double or even triple
the phage activity, before increasing the actual catalytic efficiency of formed heme-p8
complexes. Covalent attachment of heme-like groups to the p8 protein could also
increase the overall catalytic efficacy.
134,142
85
SO 2^
S02-
S
2S
C2H
C2Hs
~
-
S
.02
S
N"
N
\
I
4-N
C 2H 5
C 2Hs
-
300
pH 6
N
.02
C
b
50
N
N
-
N
\I
I
N
.
N"
S
S02.
-
C 2 H5
C 2 H5
R
A
40
(D
*Heme
DDAH
A
A
-- 20
30
A
-2 00
10
2100
+ Heme
DDAH+Hemin
A
(1
A
A
0 Blank
20
AAdj. DDAH
A
A
0
200
400
3.5
600
/N
-R
0
0
5.5
7.5
9.5
pH
[H 20 2] (pM)
Figure 4-9: DDAH catalyzes the oxidation of ABTS in the presence of H 2 0 2 in a
strongly pH-dependent manner. (a) The substrate goes through a two-step oxidation;
reduced compound at left and fully oxidized at right. (b) H 2 0 2 was added to blank 20
mM phosphate buffer, pH 6, buffer with 1 pM hemin chloride, or buffer with hemin
and 10 pM DDAH p8. Measurements made in duplicate. (c) Oxidation of ABTS was
measured at pH 4-9 in 0.5 pH increments and indicates that DDAH-heme peroxidase
activity peaks at pH 6. Final concentrations were 1 uM hemin chloride with 5-10
pM DDAH p8 (1.1-2.2 x 1012 phage/mL) and 25-800 pM H 2 0 2 . Samples were made
in duplicate and each data point presented represents a kcat/KM regression from at
least 10 independent product vs. time curves, according to Michaelis-Menten kinetics.
"DDAH" values represent kcat/KM values per heme (1 pM) once phage is added; "Adj.
DDAH" represents the kcat/KM of heme-p8 complexes after accounting for a KD of
20 pM with given input concentrations heme and p8.
86
10
DDAH
DDAH+H
DDAH+Cu
DDAH+H+Cu
x
8
S+
6
i
E
4
0
CZ
P1
IK
lit
-
1
-2
0
Figure 4-10:
50
200
150
100
[H 20 2 (j M)
250
Only DDAH-heme complexes productively catalyze the oxidation of
ABTS. 16-256 pM H 2 0 2 was mixed in triplicate with DDAH (8 pM), DDAH with
heme (1 pM), DDAH with copper (1 pM), or DDAH with both cofactors. Reactions
mixed in 10 mM phosphate buffer, pH 6.5. Markers are single data points representing
the initial slope of a product vs time curve, and lines are fits to the Michaelis-Menten
equation via Matlab.
DDAH peroxidase activity is specific to heme
Copper ions are also redox-active, and copper-based laccase enzymes can oxidize
ABTS. 1 4 4 Additionally, heme copper oxidases can reduce 02 to oxidize substrates,
145
Accordingly, ABTS
and has been the target of previous de novo enzyme attempts.
2
assays were undertaken in the presence of Cu + to assess DDAH's capacity for bind-
ing other cofactors to accelerate peroxidase reactions (Figure 4-10). Without any
cofactor, DDAH peroxidase activity was undetectable.
Similarly, in the presence of
CuCl 2 (1 pM), DDAH exhibited no activity. With equimolar heme and Cu2+ (1 pM),
DDAH clearly oxidizes the substrate, but still at rates less than DDAH-heme complexes alone. The decrease in activity possibly results from Cu2+ ions competing off
hemin from the p8 binding sites. In any case, DDAH p8s failed to demonstrate any
cupric peroxidase activity, although p8-copper complexes are catalytically active in
other transformations (see Section 4.3, below).
87
b
-0-Cuml Ph
5.E+12
4.E+1 2
wog(
3.E+12
2.E+12
1.E+12
O.E+00
0
5
Volume (mL)
10
Figure 4-11: Hemin-agarose columns retain and purify DDAH stocks. (a) PAGE
analysis of a sample of DDAH run through a hemin agarose column. Lanes: 1), the
unpurified phage stock has a heavy p8 band (asterisk), as well as non-phage proteins
(higher bands); 2) flowthrough, the initial solution collected from the column after
adding phage and shaking for 2 hours, has no visible p8; 3) a rinse with phosphate
buffer, with no p8 protein detectable; 4-5), column elutions with pH 2 citrate buffer
show clear bands of p8. (b) Samples collected were quantified by UV-Vis spectrometry
and plotted both as the phage concentration for that sample and the cumulative phage
collected from the column. Each data marker represents one collection point, and the
red arrow indicates the first elution after addition of citrate buffer.
4.2.5
Heme-agarose columns retain DDAH phage
The methods discussed so far for analyzing phage have been low-throughput, requiring
the independent amplification of each clone of interest for biochemical characterization. A higher-throughput strategy would allow us to assess a greater diversity of
phage clones and potentially identify rarer but more functional sequences. One such
strategy is affinity chromatography, whereby a library of phage could be separated by
strength of interaction with a chosen cofactor covalently bound to a column. The data
shown in Figure 4-9c, in which unbound cofactor undermines catalytic efficiency,
highlights the potential utility of finding stronger heme-binding p8 proteins.
Accordingly, I developed a protocol for the binding and elution of heme-co6rdinating
phage from a column. Hemin agarose beads were washed with a high-salt phosphate
88
buffer, pH 7.5, and treated with a stock solution of DDAH. The column was shaken
for two hours at 4C, rinsed with buffer, and remaining phage eluted with citrate, pH
2. PAGE analysis of the DDAH samples indicates that the eluted protein is rich in p8
and retains little non-phage material from the original stock (Figure 4-11a). While
2 pMol of covalently-bound heme did not retain all of the added phage (~5 x 1012),
experiments with 5 [IMol heme (2.5 mL bead solution with 2 pMol/mL) retained 87%
of DDAH applied to the column (Figure 4-11b). The experiments indicate that 1
piMol of hemin on agarose beads will retain 0.5-1 x 1012 phage particles, or 2.5-5
nMol p8 worth of the DDAH clone.t Hemin agarose affinity chromatography is thus
a promising strategy to enrich libraries for heme-binding bacteriophage.
4.3
Results: Copper Oxidase Activity
Copper oxidase enzymes, though less well-studied than peroxidases, catalyze extremely valuable oxidations with molecular 02 and thus represent a particularly
promising biocatalytic opportunity.
In characterizing the capacity of M13 bacte-
riophage to mimic the activity of these enzymes, I followed a similar path as with
the development of peroxidase activity. First, DDAH was spectroscopically analyzed
for interaction with the Cu2 + ions. DDAH-copper complexes were then used to oxidize a pair of aromatic diol substrates, catechol and 3,5-di-tert-butylcatechol, using
atmospheric oxygen as an oxidant. Finally, an affinity column strategy is used to
separate copper-binding DDAH from a control phage, potentiating library selections
for stronger binders.
DDAH co-rdinates Cu 2
+
4.3.1
Spectroscopic studies were undertaken to characterize the interaction between phage
+
and Cu 2+ ions. Previous experiments with microscale thermophoresis (Figure 2-4)
yielded a DDAH p8-Cu 2+ KD of 3.4 [tM, a stronger affinity than that for Zn2+ or Ni 2
tThe hemin agarose beads are not as stable after removal from the stock solution, and this
retention number decreased after several weeks. A preservative such as thimerosal may be used to
preserve the heme's retention capacity in future work.
89
0.008
0.007
CuCI2
0.006
-
CuNO3
3 0.005
-
0.004
<
0.003
CuSO4
0.002
0.001
0
400
500
600
700
Wavelength (nm)
800
900
Figure 4-12: Phage-bound copper spectra exhibit an absorbance peak at 610-620 nm.
Final concentrations were 79 pM Cu2+ salt with 42 pM DDAH p8 in PBS.
with DDAH. While other triple histidine phage also likely bind Cu2+ ions, DDAH was
chosen for the initial studies presented here.
Copper co6rdination in the active sites of "blue" redox enzymes results in a distinct
1
.
absorption peak around 600 nm with extinction coefficients from 700-10000 M- 1 cm-
Such blue enzymes catalyze the four-electron reduction of oxygen to water, while those
that catalyze a two-electron oxidation to H 2 0 2 show no such peak.
in laccase enzymes, for instance, absorbs with
6615
= 5400 M
1
129
The copper ion
cm 1 . Phage-copper
spectra were measured with three copper salts, CuCl 2 , CuSO 4 , and CuNO 3 , in PBS
(Figure 4-12), to determine peaks and extinction coefficients (Table 4.2).
The
phage-co6rdinated ions exhibited peaks from 610-621 nm, with extinction coefficients
e - 130-163 M- 1 cm'. Thus, while seemingly a weaker interaction than in wild-type
enzymes, the phage-copper spectra indicate potential activation of the copper ions
for catalysis.
90
OH
a
0
0
OH
o-Benzoquinone
Catechol
b
0.4
(
Aqueous PBS) Reactions
Uncat
DDAH
K2 S 2 08
DDAH
K 2 S2 08
0.3
cc
C,
RT
-
-20.2
0
-- DDAH
_K2S20 8
0.1
800C
0
300
500
400
Wavelength (nm)
600
C
Organic (D MSO) Reactions
0.6
Uncat
-Bkgd
-DDAH
0.4
-K2S2C
.0
RT
8
0.2
800C
0
300
500
(nm)
Wavelength
400
600
Figure 4-13: (a) Catechol undergoes a two-electron oxidation to o-benzoquinone. (b)
Spectra and images of reactions in PBS. Potassium persulfate, K 2 S 2 08 , is an oxidant
used as a positive control. Spectra are from 80'C samples after two hours of heating,
with the background absorbance spectrum of substrate in 80C PBS subtracted. Images are of the final mixtures after reacting overnight at room temperature or 80C.
(c) Spectra and images of reactions conducted in DMSO overnight. Spectra shown
.
are 80*C samples for background and DDAH, RT for K 2 S 2 08
91
Peak A (nm)
610
621
619
CUC12
CUS04
CuNO3
EA
(M-cm--1
)
Salt
130
135
163
Table 4.2: Extinction coefficients for DDAH p8-Cu2+ complexes, as determined from
Figure 4-12. It was assumed no more than one complex formed per p8.
4.3.2
DDAH oxidation of catechol and derivatives
Oxidation of catechol
The oxidation of aromatic diols to form carbonyls is a useful reaction for industrial
synthesis, and one which can be performed by copper oxidases without stoichiometric oxidants such as H 2 0 2. Catechol, a simple phenol, is known to be oxidized by
blue enzymes such as tyrosinase and catechol oxidase, and was selected for initial
copper oxidase assays. Reactions were mixed in 98% PBS (Figure 4-13b) or 98%
DMSO (Figure 4-13c), at room temperature and 80'C overnight. Reaction product
o-benzoquinone was unavailable commercially, meaning the expected spectrum after
oxidation was not known. As a positive control, therefore, the oxidant potassium
persulfate (K 2 52 08 ) was added to mixtures. Mixtures included 1 mM catechol; 20
PM CuCl 2 ; and no catalyst,
1x
1012
/mL DDAH (45 pM p8), or 2 mM K 2 S 2 08 . In both
aqueous and nonaqueous solvent, only potassium persulfate oxidized the substrate at
room temperature. At 800 C, all solutions browned, indicating chemical transformation: an aqueous peak formed at ~330 nm, with Abs
Abs 334
=
326 .5
= 0.38 for K 2 S 2 08 and
0.22 for DDAH, after subtracting the background spectrum (these measure-
ments taken after two hours; after overnight incubation, peaks dropped and fattened).
In DMSO, a peak grew closer to 400 nm, and the phage reaction mixture was visibly darker than the uncatalyzed reaction. However, all spectra were multimodal
and, without a definite spectrum for the product, inconclusive. Benzoquinone can
be further oxidized to melanin derivatives, complicating catalytic characterization.
Measurements were attempted on a gas chromatography-mass spectroscopy instrument to confirm the molecular weight of reaction constituents, but peaks could not
be reliably identified. The catechol reactions indicated the phage was catalyzing the
92
oxidation of substrate, but were ultimately non-quantifiable.
Oxidation of 3,5-di-tert-butylcatechol
Instead, a catechol derivative with very clear spectroscopic differences was chosen to
characterize DDAH's oxidase activity. The compound 3,5-di-tert-butylcatechol(DTBC)
undergoes a two-electron oxidation to di-tert-butyl-o-benzoquinone(DTBB), similar to catechol oxidation.1
4OO
Stock DTBB has an absorbance peak at 400 nm, with
= 1500 M-'cm', while substrate DTBC absorbs strongly at 283 nm but not
at 400 nm. Preliminary assays with this substrate indicate significant oxidase activity from DDAH (Figure 4-14). Reactions were conducted in 98% DMSO with 20
PM CuCl 2 and 4.5
pM DDAH p8 at room temperature. Lineweaver-Burk analysis
determined the DDAH kcat/KM to be 50.0 M-'s
1
(95% CI: 34.7-81.7 M-'s-'). With
a kuncat of 2 x 10-6 s-1, DDAH kcat/kuncat = 438. Though more controls and reaction
parameters will need to be tested, these early results nonetheless indicate that the
bacteriophage is successfully codrdinating Cu2+ ions to catalyze the oxidation of diols
by molecular oxygen.
4.3.3
Cu-NTA columns selectively retain Cu2+ -binding phage
While M13 clone DDAH successfully catalyzes copper oxidase activity, a higher
throughput method of assaying potential biocatalytic clones could identify a phage
with greater activity. Accordingly, in a strategy parallel to Section 4.2.5, I sought
to develop an affinity chromatography-based method of finding catalytic candidates.
Ni-NTA beads were flushed with ethylenediaminetetraacetic acid (EDTA) to strip
away the chelated nickel, and then treated with CuCl 2 to form Cu-NTA beads. A
mixture of phage -
DDAH, known to bind Cu2+ ions, and A3H, a negative con-
trol with no histidines (Table 2.2) -
was added to the column in a loading buffer
with 10 mM imidazole. After shaking for an hour at 4'C, the column was washed
with 20 mM imidazole and then eluted in 250 mM imidazole. Each sample along the
tAnd, more conveniently, both substrate and product are commercially available.
93
4.5
x
X
4
3.5
3
0
*
C
a:
~1.
OH0
a: 2.5
cc1
OH0
0
100
400
300
200
[DTBC] 0 (pM)
Figure 4-14: DDAH oxidizes DTBC. 25-400 pM DTBC was mixed with phage (4.5
uM DDAH p8) and CuCl 2 (20 pM) in 98% DMSO at room temperature. Initial
slopes of AAbs 4 oo/At were taken and Lineweaver-Burk analysis used for the regression
50.0 M 1 s-1). Inset, conversion of DTBC to DTBB.
shown (solid line; kat/KM
Mix
Phage
1.0E13
# Seq.
18
#A3H
10
#DDAH
8
Flowthrough
Elution
6.0E12
7.1E11
8
8
8
0
0
8
Sample
Table 4.3:
Cu-NTA specifically retains copper-binding phage DDAH. Amount of
phage shown in column two was determined by UV-Vis spectroscopy.
the amount of plaques grown, miniprepped and sequenced.
# Seq. is
way was characterized by UV-Vis spectroscopy and infectivity assays to quantify the
phage concentration. The presence of imidazole limited non-specific interactions and
ensured that only copper-binding particles were retained through to the elutions; the
majority of phage washed off the column in 10 and 20 mM buffers. Sequencing was
conducted by picking titered plaques from plates for three samples (Table 4.3):
" the initial phage mixture (18 plaques);
* the first sample that came off the column ("flowthrough", 8 plaques); and
" the elution in 250 mM imidazole (8 plaques).
94
While the initial mixture was approximately 1:1 A3H:DDAH, none of the A3H phage
was found in the plaques sequenced from the elution. Most of the DDAH came off
early, as well, presumably in the washes with 20 mM imidazole. As a result, a pure
sample of the DDAH was collected in the elution. The Cu-NTA strategy thus successfully separates out the copper-binding phage, and could be a powerful strategy
for enriching libraries for ion binders in future work.
4.4
Summary
In this chapter, catalytic M13 clone DDAH binds to the redox cofactors heme and
copper to oxidize multiple substrates. Heme peroxidase activity is demonstrated with
tetramethylbenzidine, which undergoes oxidative dehydrogenation, and with ABTS,
which undergoes two one-electron transfers. The activity with ABTS is highly pHdependent, peaking at pH 6, when the histidine-based active site and the heme iron
are optimally ligated. The activity is entirely dependent upon the presence of heme,
and is not reproduced or enhanced by Cu 2+ cofactors. While the catalytic efficiency of
DDAH is approximately two orders of magnitude lower than that previously reported
for a designed four-helix bundle,1 4 2 the thermostability and evolvability of M13 make
it more broadly applicable than most de novo enzymes and should allow for the
identification of clones with improved activity.
Estimates of the DDAH p8-heme
dissociation constant indicate that increasing the affinity for heme could drastically
improve the overall efficacy of the system. Towards this end, a strategy was developed
for purifying heme-binding M13 clones with hemin-agarose beads. Such a strategy
could be used with large libraries of M13 variants to enrich those interacting most
strongly with the heme molecule, though non-heme binding controls will need to
tested first.
In addition to peroxidase activity, DDAH cobrdinates Cu2+ ions to mimic blue copper oxidase enzymes. Microscale thermophoresis measurements show a strong affinity
between DDAH p8 and copper(II), while UV-Vis spectra exhibit an absorption peak
indicative of copper activation. Copper enzymes catalyze many transformations, but
95
the oxidation of aromatic diols is especially valuable and was chosen here for initial assays. While catechol oxidation reactions could not be sufficiently quantified to
infer catalytic parameters, the oxidation of catechol derivative DTBC shows significant catalytic activity with only molecular oxygen as an oxidant. This activity is
demonstrated in DMSO, emphasizing the usefulness of the highly stable M13 coat
proteins. The activity is comparable to that of previous de novo enzymes catalyzing
DTBC oxidation (kt/KM = 105 M-ls-1 )" and effectively equal on a per-amino
acid or per-metal ion basis. In order to identify potentially stronger biocatalytic candidates, an affinity chromatography strategy was developed whereby Cu-NTA could
efficiently select for Cu 2 +-binding phage clones. This approach should enable the
high-throughput enrichment or assessment of M13 libraries and the isolation of more
active de novo enzyme.
More precise characterization of the phage redox activity and substrate range could
be quite valuable for future enzymatic phage development. In particular, measuring
the reduction-oxidation potentials of the heme and copper active sites could guide
library engineering, reaction conditions, and the selection of additional substrates.
For instance, hemeprotein cytochrome c has a relatively low redox potential of +0.2 V,
while copper proteins are often +0.3-0.6 V, such that copper proteins have few natural
terminal electron acceptors besides 02.129 The potential of a given cofactor is in turn
modulated by its affinity for surrounding ligands at a given pH, offering additional
engineering handles for future de novo biocatalyst work.63 In addition to the aromatic
diols discussed above, the oxidations of precursors such as 1-phenylethanol (1-PE) are
useful transformations in the chemical synthesis industry and promising candidates
for future assays.
More broadly, M13's well-understood biology and dense display of peptides make
it appealing for future development. The ease of bacteriophage amplification and purification allows for greater scalability of the catalysts compared to other enzymatic
proteins, while p8's tightly packed, highly multivalent structure leads to higher numbers of reaction centers. With thousands of p8 proteins per biomolecule, M13 can
stably approach millimolar concentrations of active sites with or without immobiliza96
tion. Moreover, the inherent inclusion of the bacteriophage genome with all progeny
makes functional catalytic selections from diverse libraries possible, whereas traditional enzymes lack the requisite genotype-phenotype connection. Accordingly, future engineering of the bacteriophage will harness its high multivalency and genotypephenotype linkage in conjunction with recent developments in high-throughput assays,
such as microengraving and microemulsions,' 46 to search a larger sequence space for
active clones. M13 biology further allows for directed evolution and host rescue assays
to rapidly select functional variants. M13's genome itself is highly engineered, with
numerous restriction enzyme sites in the genes of its various coat proteins, such that
complementary binding or catalytic functions can be incorporated at other positions
on the bacteriophage capsid.
4.5
4.5.1
Experimental Methods
Bacterial culture and M13 stocks
Phage clones were amplified as described in Section 2.5.2 and quantified by spectrophotometer, according to Equation 2.3. Briefly, an overnight 10 mL culture of
E. coli K12 ER2738 was added to 1 L of fresh LB with 1 mL 1000X Tet and 1 mL
of a previously sequenced DDAH culture. This was grown overnight (16 hours) then
centrifuged to remove E. coli , mixed with PEG-NaCl, refrigerated overnight and
centrifuged once more to pellet phage. After dissolving the M13 pellet in PBS, a second round of bacteria pelleting by centrifugation, PEG addition and incubation, then
phage pelleting by centrifugation was conducted to further purify and concentrate
the phage. Final bacteriophage pellets were diluted to 5-10
x
1013 phage/mL in PBS
for storage.
Clones 2Cysl3H, 2Cys15H, 2Cys27H and ADHD were subjected to further cleaning and purification. Each amplification reliably yielded the intended sequence, but
their spectra and preparations indicated the presence of other proteinaceous or nucleic acid E. coli detritus. ADHD, in particular, had a thick, disproportionally large
97
Initial
Clone
2Cys13H
2Cys15H
2Cys27H
ADHD
Titer
5.7E9
1.5E10
7.0E9
8.4E8
Factor
3965
2067
3286
34524
Post-DNAse
Titer
Factor
4.3E9
4053
8.0E9
3537
5.0E9
4481
6.3E8
73898
Final
Titer
2.0E9
2.8E9
1.9E9
1.1E8
Factor
6450
7536
6158
2.5E5
Table 4.4: Cleaning via DNAse and centrifugation fails to improve the absolute or
relative infectivity of two-cysteine and four-histidine phage. The infectivity ("titer")
of the M13 variants was assayed on XL1-Blue E. coli plates at the different stages of
cleaning and purification: after initial amplification and concentration; after DNAse
treatment and resuspension; and after final round of centrifugation and dissolution
at 37C. The "factor" columns represents the amount of phage as determined by
spectroscopy (Equation 2.3) divided by the amount of plaques formed in the "titer"
column.
white precipitate when pelleted and a cloudy appearance after dissolution. 1 mL of
each clone's stock was treated with 5 pg/mL DNAse in the presence of 1 mM CaCl 2
and 5 mM MgCl 2 for 3 hrs. After 3 hrs, 200 pL of 25 mM PEG 8000, 2.5 M NaCl
solution was added and the samples moved to 4'C overnight. The next day, the solutions were spun 10 min at 10,000 RPM to pellet out the phage, which was then
re-dissolved to the original 1 mL volumes. The resulting samples were visibly cloudy,
and so were shaken at 37TC for two hours to facilitate dissolution. These were then
spun an additional 20 min at 6000 RPM to remove un-dissolved material, forming
white pellets. The supernatant was moved to fresh tubes and shaken at 37*C for an
additional 2 hours, after which the samples were visibly clearer.
The infectivity (titer) and absorbance spectra for the clones was assayed after
the initial stock preparation, after DNAse treatment, pelleting and resuspension,
and after the final rounds of centrifugation and incubation at 37'C (Figure 4-15).
Not only did the phage concentration drop and spectra not present a clearer peak,
but the titer factor of spectroscopically measured phage to PFU determined by titers
worsened, indicating a lower ratio of phage "signal" to bacterial detritus "noise" (Table
4.4). Additionally, the ADHD clone showed extremely low titers, which corresponds
to the weak p8 signal in the gel shown in Figure 4-2b. Overall, the processing
failed to purify the phage stocks, and protease or other treatments may be considered
98
in the future for clones that pull down similar amounts of bacterial detritus during
preparations.
For titer assays and sequencing, E. coli XL1-Blue host cells (Strategene #200268)
were used to culture phage. Titering was otherwise carried out as described in the
NEB Ph.D. Phage Display manual.
Briefly, stocks were diluted ten-fold in water
and 10 pL of a given dilution added to 200 [L overnight XL1-Blue.
After three
minutes, the 210 pL sample was mixed with 3 mL melted top agar and poured onto
Tet/Xgal/IPTG agar plates.
for quantification.
Plates with approximately 100 plaques were sought
Sequencing was either prepped from 5 mL overnight cultures of
XL1-Blue cells and M13 or from infected bacterial pellets dissolved in 350 PL PB1.
Subsequent steps were carried out as described in the Qiagen® Miniprep Handbook.
4.5.2
Protocols for assaying phage activity with heme
Measuring Soret spectra
Soret peaks were assayed using hemin chloride (Sigma-Aldrich
# H-5533) on a Beck-
man Coulter DU-800 spectrophotometer. Hemin chloride was stored as a powder at
4 0C, and mixed in DMSO fresh each day. Clones were assayed in 98% PBS (137 mM
NaCl, 2.7 mM KCl, 10 mM phosphate, pH 7.4), with hemin chloride diluted 50X
from a stock in DMSO to a final concentration of 5 pM. The phage was mixed to
1 x 1013
/mL (as measured by UV-Vis spectroscopy), or 45 pMp8. A large excess of
p8 to heme facilitates the detection of any potential phage clone interaction with the
heme. Samples were measured from 200 to 800 nm with a 1 nm resolution and a path
length of 10 mm, and the region from 350 to 450 nm examined for the Soret peak.
The background (PBS) sample was measured before and after the phage samples to
check for any signal drift.
Estimating DDAH-heme dissociation constants
Dissociation constants, KD, were inferred from two data sets: (1) Soret peak values
for varied heme concentration with a fixed phage concentration; and (2) catalytic
99
b
1.5
1.5
2Cysl3H
2Cysl 5H
31
0
0.5
<0.5
0
O
200
350
300
250
Wavelength (nm)
400
200
350
300
250
Wavelength (nm)
400
d
1.5
2
2Cys27H
ADHD
0)
1.5
C.)
1
C
CU
.0
1
.0
.0
0.5
nl
0
MMMMMMMMMML
200
350
300
250
Wavelength (nm)
-
0.5
200
400
350
300
250
Wavelength (nm)
400
Figure 4-15: The spectra of two-cysteine and four-histidine phage stocks remain indefinite. Each panel is a separate clone at the different stages of cleaning and purification:
after initial amplification and concentration (light gray); after DNAse treatment and
resuspension (medium gray); and after final round of centrifugation and dissolution
at 37'C (black). The phage 269 nm peak becomes no clearer after the additional
processing.
100
rates from varied phage concentrations with fixed heme and substrate (TMB) concentrations. For (1), the data was generated by measuring absorbance spectra from a
mixture of 45 pMDDAH p 8 (1 x 101 DDAH/mL) protein with 0-50 pMhemin chloride, in increments of 5 [M. 20 pL of 5 x 1013 /mL DDAH was added to 77.5 PL PBS
in a cuvette with a 10 mm path length for the initial [heme] = 0 puM measurement.
Subsequently, 0.5 pL 1 mM hemin chloride was added, mixed, and measured ten
times, for a final 102.5 piL sample volume. Resulting heme concentrations and Soret
peaks were adjusted for the slight change in fluid volume resulting from each aliquot
and for the signal generated by unbound heme (generated in background measurements). Two sets of values, Abs 408 and AAbs 40 8- 3 6 0 , were normalized and fit with
MATLAB's fit function.
The second method used to estimate DDAH-heme KD was fitting the curve of
TMB oxidation vs p8 concentration.
Slopes were measured for AAbs 6 52 /At with 0
and 0.1-25.6 pM p8, in nine two-fold increments, and 1 pM hemin chloride, all in
triplicate. The resulting data was fit to Equation 4.1 for KD = 5, 10, 20, 30, or 40
pM, so that only the scaling parameter 'a' and an offset constant 'c' were inferred.
The [p8]
12.8 pM data points were outliers and were not included in presented
data. After MATLAB's fit function built a regression, residuals were calculated by
subtracting the measured data points at each p8 concentration from that KD fit.
These were individually squared and summed over all data for a given input KD and
the resulting SSE values presented in Figure 4-8.
TMB assays
3,3'-5,5'-Tetramethylbenzidine (TMB) is a sensitive peroxidase substrate sold as a
pre-mixed solution for ELISA assays (Thermo Scientific
#
34028). TMB solution
was stored at 4C and mixed fresh for each experiment to 50% (by volume) with
catalysts in PBS. Phage and hemin chloride were stored at 4'C and mixed fresh for
each day of experimentation.
Heme stocks were made in DMSO at 50X, so that
final reaction mixtures were 2% DMSO. 100 pL catalyst was added to a CytoOne 96well plate (CC7682-7596) followed by 100pL TMB mix, and the plates immediately
101
analyzed on a Tecan Infinite M200 Pro plate reader. Initial AAbs 6 5 2 /At slopes were
measured and plotted for a range of p8 or heme concentrations.
ABTS assays
2,2 '-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) was procured from SigmaAldrich and stored at room temperature as a powder. Hydrogen peroxide was stored
as a 30% solution at 4'C. Reaction solutions were mixed fresh on the day of experiment: 1 pM hemin chloride (diluted from 50pM stock in DMSO), 10 mM sodium
phosphate buffered at appropriate pH, 5-10 pM DDAH p 8 (1.1-2.2
x10 12
phage/mL),
1-2 mM ABTS, and 25-800 yM H 2 0 2 ; the final mixture was 2% DMSO in 98% water. Reactions were mixed in 96-well plates at 200 pL volumes: 50 pL 4X H 2 0 2 ;
50 pL 4X catalyst mix (ABTS
heme pm phage); 100 pL 2X phosphate buffer.
Plates were immediately analyzed on a Tecan Infinite M200 Pro plate reader. Initial
A Product/At slopes were measured and plotted for a range of H 2 0 2 concentrations,
using
6660
=
14000 M-'s- 1 . kcat/KMvalues were determined by fitting the data di-
rectly to the Michaelis-Menten equation as in Equation 3.4. However, using this
regression, confidence intervals could not be obtained due to unexpected data offsets
from the origin; work is ongoing in identifying reaction conditions so that data curves
properly intersect the origin.
The reactions shown in Figure 4-10 were conducted in 10 mM phosphate buffer,
pH 6.5, with 8 pMDDAH p8, 1 mM ABTS, 0 or 1 pM hemin chloride and 0 or 1
1iM
copper chloride.
Independent assays were conducted to determine that the ABTS reactions are
saturated when [ABTS] > 1 mM and the resulting catalytic rates insensitive to more
substrate. This allows the bisubstrate reaction
d[P]
dt
_
kcat[E]
1+hM[A]+ Kqai / [B]
to simplify to the unisubstrate Michaelis- Menten Equation 2.2.
102
Hemin-agarose columns
Hemin-agarose beads (Sigma-Aldrich H6390) were procured and stored at 4'C as a
2 pMol/mL heme solution. 2.5 mL of bead suspension was added to a 7 mm inner
diameter column, drained, and washed with a 10 mM sodium phosphate, 500 mM
sodium chloride pH 7.5 loading buffer.
5
x 1012
DDAH phage was added to the
column in 0.5 mL loading buffer and shaken for 2 hours at 4'C. The column was
drained ("flowthrough") and rinsed with 4.5 mL buffer.
The phage was eluted by
adding 1 mL 200 mM citrate, pH 2 and leaving at room temperature for two hours,
then collecting. Infectivity assays were conducted as described in Section 2.5.2.
4.5.3
Protocols for assaying phage-copper interactions
Copper spectroscopy
Copper spectra were measured in 10 mm path length cuvettes on a Beckman Coulter
DU-800 spectrophotometer. Roughly equimolar p8 and Cu2+ were mixed so that the
copper was sufficient for detection without crashing out the phage or masking the
p8-Cu2+ signal. Final concentrations were 79 pM Cu2+ salt with 42 pM DDAH p8 in
PBS; the absorbance curve for this concentration of p8 was subtracted from displayed
spectra. Extinction coefficients were determined by dividing the peak absorbance by
the concentration of p8.
Oxidation of catechol
Catechol (1,2-dihydroxybenze, Sigma-Aldrich #135011) was stored as a powder at
room temperature under nitrogen. Several parameters were tested for the reactions:
" temperature: 25'C or 80'C;
" solvent: 98% PBS or 98% DMSO;
* time: 2 hours or overnight (16 hours); and
" catalyst: Cu 2+ in solution, Cu2 + with phage, or potassium persulfate.
103
Final mixtures were 1 mL with 1 mM catechol, 20 pM CuCl 2
DDAH (5 pM p8)
or 2 mM K 2 S 2 0 8 . All images in Figure 4-13 were taken of vials after overnight
incubation. The spectra shown in (b) are from 98% PBS 80'C samples after 2 hours,
baselined with the background 80CuC1 2 spectrum.
Spectra in (c) are from 98%
DMSO samples after overnight incubation; positive control K 2 52 08 spectrum was at
room temperature (the 80'C K 2 S 2 08 spectrum dissipated relative to RT), while the
background and DDAH spectra are from 80'C samples.
Oxidation of DTBC
3,5-di-tert-butylcatechol(Sigma-Aldrich #D45800) and di- tert-butyl- o-benzoquinone(SigmaAldrich #157457)
were stored as powders at room temperature.
The spectra and
extinction coefficients for substrate and product in DMSO were determined experimentally in 96-well plates with 25-400 pMtwo-fold increment concentrations in quadruplicate. DTBC had a peak with 283= 2130 M-cm-1, while product DTBB had
a broader peak with E400
1500 M- 1 cm 1 . Reaction solutions were mixed fresh daily
and assayed in 96-well plates with 200 pL samples. Final concentrations were 25-400
jiM DTBC with 20 pM CuCl 2 and 4.5 pM DDAH p8. Copper chloride was added
last and initial slopes measured immediately after.
Background (20 pM CuCl 2 in
98% DMSO) was subtracted, and the phage data regressed using Lineweaver-Burk
analysis (Equation 2.5).
Cu-NTA columns to purify copper-binding phage
The protocol largely follows that provided by QIAGEN for their Ni-NTA kit, "Batch
purification of 6xHis-tagged proteins from E. coli under native conditions," but with
the column flushed and repopulated with copper ions instead. 1 mL of beads was
flushed with 6M guanidinium chloride, 0.2 M acetic acid, then water, sodium dodecyl
sulfate, ethanol, and EDTA, in sequence. After flushing with more water, the column
was charged with 100 mM CuCl 2 . Another wash of guanidinium chloride was followed
by the loading buffer: 50 mM NaH 2 PO 4 , 300 mM NaCl, 10 mM imidazole.
The
column was drained and 1 mL of 1E13 phage (A3H and DDAH mix) added in loading
104
buffer, then shaken for 1 hr at 4'C. The column was drained ("flowthrough"), rinsed
with 2 mL loading buffer, washed with 4 mL wash buffer (50 mM NaH 2PO 4 , 300
mM NaCl, 20 mM Imidazole), and then treated with 0.5 mL elution buffer (50 mM
NaH 2 PO 4 , 300 mM NaCl, 250 mM imidazole).
All buffers were ph 8. Four 0.5
mL aliquots of elution buffer were added to and collected from the column after 5
min incubation at room temperature. Infectivity assays and sequencing were then
conducted as described in Section 2.5.2.
105
106
Bibliography
[1] M. E. Wilson and G. M. Whitesides. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a
diphosphinerhodium(i) moiety. Journal of the American Chemical Society, 100
(1):306-307, 1978.
121 K. M. Koeller and C. H. Wong. Enzymes for chemical synthesis. Nature, 409
(6817):232-240, 2001.
[3] A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts, and B. Witholt.
Industrial biocatalysis today and tomorrow. Nature, 409(6817):258-268, 2001.
[4] Vicente Gotor-FernAndez, Rosario Brieva, and Vicente Gotor. Lipases: Useful biocatalysts for the preparation of pharmaceuticals. Journal of Molecular
Catalysis B: Enzymatic, 40(3A;4):111-120, 2006.
[5] OECD. The Application of Biotechnology to Industrial Sustainability - A
Primer. OECD Publishing, Paris, France, 2001.
[6] J. D. Keasling. Building with biology. Nature, 492(7428):188-188, 2012.
[7] N. J. White. Qinghaosu (artemisinin): The price of success. Science, 320(5874):
330-334, 2008.
[8] C. Schmidt-Dannert, D. Umeno, and F. H. Arnold. Molecular breeding of
carotenoid biosynthetic pathways. Nature Biotechnology, 18(7):750-753, 2000.
[9] C. Walsh. Enabling the chemistry of life. Nature, 409(6817):226-231, 2001.
[10] F. H. Arnold. Combinatorial and computational challenges for biocatalyst design. Nature, 409(6817):253-257, 2001.
[11] B. Joseph, P. W. Ramteke, and G. Thomas. Cold active microbial lipases:
Some hot issues and recent developments. Biotechnology Advances, 26(5):457470, 2008.
[12] J. Nielsen, M. Fussenegger, J. Keasling, S. Y. Lee, J. C. Liao, K. Prather,
and B. Palsson. Engineering synergy in biotechnology. Nat Chem Biol, 10(5):
319-22, 2014.
107
[13]
M. T. Reetz. Biocatalysis in organic chemistry and biotechnology: Past, present,
and future. Journal of the American Chemical Society, 135(34):12480-12496,
2013.
[14] J. Kim, H. F. Jia, and P. Wang. Challenges in biocatalysis for enzyme-based
biofuel cells. Biotechnology Advances, 24(3):296-308, 2006.
[15]
A. M. Klibanov. Improving enzymes by using them in organic solvents. Nature,
409(6817):241-246, 2001.
[16] A. L. Serdakowski and J. S. Dordick. Enzyme activation for organic solvents
made easy. Trends in Biotechnology, 26(1):48-54, 2008.
[17]
R. Hatti-Kaul, U. Tornvall, L. Gustafsson, and P. Borjesson. Industrial biotechnology for the production of bio-based chemicals - a cradle-to-grave perspective.
Trends in Biotechnology, 25(3):119-124, 2007.
[18] R. G. Mathys, A. Schmid, and B. Witholt. Integrated two-liquid phase bioconversion and product-recovery processes for the oxidation of alkanes: Process
design and economic evaluation. Biotechnology and Bioengineering, 64(4):459477, 1999.
[19]
L. J. Perry and R. Wetzel. Disulfide bond engineered into t4-lysozyme - stabilization of the protein toward thermal inactivation. Science, 226(4674):555-557,
1984.
[20] M. D. Toscano, K. J. Woycechowsky, and D. Hilvert. Minimalist active-site
redesign: Teaching old enzymes new tricks. Angewandte Chemie-International
Edition, 46(18):3212-3236, 2007.
[21] G. Host, L. G. Martensson, and B. H. Jonsson. Redesign of human carbonic
anhydrase ii for increased esterase activity and specificity towards esters with
long acyl chains. Biochimica Et Biophysica Acta-Proteins and Proteomics, 1764
(10):1601-1606, 2006.
[221 Z. Zhou and M. Hartmann. Progress in enzyme immobilization in ordered
mesoporous materials and related applications. Chemical Society Reviews, 42
(9):3894-3912, 2013.
[23] A. S. Bommarius and M. F. Paye. Stabilizing biocatalysts. Chemical Society
Reviews, 42(15):6534-6565, 2013.
[24]
J. C. Moore and F. H. Arnold. Directed evolution of a para-nitrobenzyl esterase
for aqueous-organic solvents. Nature Biotechnology, 14(4):458-467, 1996.
[251 D. Lipovsek, E. Antipov, K. A. Armstrong, M. J. Olsen, A. M. Klibanov,
B. Tidor, and K. D. Wittrup. Selection of horseradish peroxidase variants with
enhanced enantioselectivity by yeast surface display. Chemistry & Biology, 14
(10):1176-1185, 2007.
108
[26]
N. J. Turner. Directed evolution drives the next generation of biocatalysts. Nat
Chem Biol, 5(8):567-73, 2009.
[27] P. S. Coelho, E. M. Brustad, A. Kannan, and F. H. Arnold. Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome p450 enzymes.
Science, 339(6117):307-310, 2013.
[28] H. Jochens, K. Stiba, C. Savile, R. Fujii, J. G. Yu, T. Gerassenkov, R. J.
Kazlauskas, and U. T. Bornscheuer. Converting an esterase into an epoxide
hydrolase. Angewandte Chemie-InternationalEdition, 48(19):3532-3535, 2009.
[29] N. J. Turner and E. O'reilly.
Biology, 9(5):285-288, 2013.
Biocatalytic retrosynthesis.
Nature Chemical
[30] P. Gatti-Lafranconi and F. Hollfelder. Flexibility and reactivity in promiscuous
enzymes. Chembiochem, 14(3):285-292, 2013.
[31] S. J. Pollack, J. W. Jacobs, and P. G. Schultz. Selective chemical catalysis by
an antibody. Science, 234(4783):1570-3, 1986.
[32] A. Tramontano, K. D. Janda, and R. A. Lerner. Catalytic antibodies. Science,
234(4783):1566-70, 1986.
[331 D. Hilvert, S. H. Carpenter, K. D. Nared, and M. T. M. Auditor. Catalysis
of concerted reactions by antibodies - the claisen rearrangement. Proceedings
of the National Academy of Sciences of the United States of America, 85(14):
4953-4955, 1988.
[34] D. Hilvert, K. W. Hill, K. D. Nared, and M. T. M. Auditor. Antibody catalysis
of a diels-alder reaction. Journal of the American Chemical Society, 111(26):
9261-9262, 1989.
[35] D. E. Benson, M. S. Wisz, W. T. Liu, and H. W. Hellinga. Construction of a
novel redox protein by rational design: Conversion of a disulfide bridge into a
mononuclear iron-sulfur center. Biochemistry, 37(20):7070-7076, 1998.
[36] D. E. Benson, M. S. Wisz, and H. W. Hellinga. Rational design of nascent
metalloenzymes. Proceedings of the National Academy of Sciences of the United
States of America, 97(12):6292-6297, 2000.
[37] A. J. Nicoll and R. K. Allemann. Nucleophilic and general acid catalysis at
physiological ph by a designed miniature esterase. Organic & Biomolecular
Chemistry, 2(15):2175-2180, 2004.
[38] I. V. Korendovych, D. W. Kulp, Y. Wu, H. Cheng, H. Roder, and W. F. DeGrado. Design of a switchable eliminase. Proc Natl Acad Sci U S A, 108(17):
6823-7, 2011.
109
[39]
J. Bos, F. Fusetti, A. J. Driessen, and G. Roelfes. Enantioselective artificial
metalloenzymes by creation of a novel active site at the protein dimer interface.
Angew Chem Int Ed Engl, 51(30):7472-5, 2012.
[40] B. Seelig and J. W. Szostak. Selection and evolution of enzymes from a partially
randomized non-catalytic scaffold. Nature, 448(7155):828-U13, 2007.
[41] L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Rothlisberger,
A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, 3rd Barbas, C. F.,
D. Hilvert, K. N. Houk, B. L. Stoddard, and D. Baker. De novo computational
design of retro-aldol enzymes. Science, 319(5868):1387-91, 2008.
[42] D. Rothlisberger, 0. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie,
J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, 0. Dym, S. Albeck, K. N.
Houk, D. S. Tawfik, and D. Baker. Kemp elimination catalysts by computational
enzyme design. Nature, 453(7192):190-U4, 2008.
[43] F. Richter, R. Blomberg, S. D. Khare, G. Kiss, A. P. Kuzin, A. J. T. Smith,
J. Gallaher, Z. Pianowski, R. C. Helgeson, A. Grjasnow, R. Xiao, J. Seetharaman, M. Su, S. Vorobiev, S. Lew, F. Forouhar, G. J. Kornhaber, J. F. Hunt,
G. T. Montelione, L. Tong, K. N. Houk, D. Hilvert, and D. Baker. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. Journal
of the American Chemical Society, 134(39):16197-16206, 2012.
[44] S. Rajagopalan, C. Wang, K. Yu, A. P. Kuzin, F. Richter, S. Lew, A. E. Miklos, M. L. Matthews, J. Seetharaman, M. Su, J. F. Hunt, B. F. Cravatt, and
D. Baker. Design of activated serine-containing catalytic triads with atomiclevel accuracy. Nature Chemical Biology, 10(5):386-U100, 2014.
[451 S. D. Khare, Y. Kipnis, P. J. Greisen, R. Takeuchi, Y. Ashani, M. Goldsmith,
Y. F. Song, J. L. Gallaher, I. Silman, H. Leader, J. L. Sussman, B. L. Stoddard,
D. S. Tawfik, and D. Baker. Computational redesign of a mononuclear zinc
metalloenzyme for organophosphate hydrolysis. Nature Chemical Biology, 8(3):
294-300, 2012.
[46] L. Giger, S. Caner, R. Obexer, P. Kast, D. Baker, N. Ban, and D. Hilvert.
Evolution of a designed retro-aldolase leads to complete active site remodeling.
Nat Chem Biol, 9(8):494-8, 2013.
[47]
L. Regan and W. F. Degrado. Characterization of a helical protein designed
from 1st principles. Science, 241(4868):976-978, 1988.
148]
S. Kamtekar, J. M. Schiffer, H. Y. Xiong, J. M. Babik, and M. H. Hecht. Protein
design by binary patterning of polar and nonpolar amino-acids. Science, 262
(5140):1680-1685, 1993.
[491 D. E. Robertson, R. S. Farid, C. C. Moser, J. L. Urbauer, S. E. Mulholland,
R. Pidikiti, J. D. Lear, A. J. Wand, W. F. Degrado, and P. L. Dutton. Design
and synthesis of multi-heme proteins. Nature, 368(6470):425-431, 1994.
110
[50] S. S. Huang, R. L. Koder, M. Lewis, A. J. Wand, and P. L. Dutton. The hp-i
maquette: from an apoprotein structure to a structured hemoprotein designed
to promote redox-coupled proton exchange. Proc Natl Acad Sci U S A, 101(15):
5536-41, 2004.
[51]
A. Das and M. H. Hecht. Peroxidase activity of de novo heme proteins immobilized on electrodes. Journal of Inorganic Biochemistry, 101(11-12):1820-1826,
2007.
[52] T. A. Farid, G. Kodali, L. A. Solomon, B. R. Lichtenstein, M. M. Sheehan, B. A.
Fry, C. Bialas, N. M. Ennist, J. A. Siedlecki, Z. Y. Zhao, M. A. Stetz, K. G.
Valentine, J. L. R. Anderson, A. J. Wand, B. M. Discher, C. C. Moser, and
P. L. Dutton. Elementary tetrahelical protein design for diverse oxidoreductase
functions. Nat Chem Biol, 9(12):826-+, 2013.
[53] K. Johnsson, R. K. Allemann, H. Widmer, and S. A. Benner. Synthesis, structure and activity of artificial, rationally designed catalytic polypeptides. Nature,
365(6446):530-2, 1993.
[54] K. Severin, D. H. Lee, A. J. Kennan, and M. R. Ghadiri. A synthetic peptide
ligase. Nature, 389(6652):706-9, 1997.
[55]
P. Rossi, P. Tecilla, L. Baltzer, and P. Scrimin. De novo metallonucleases based
on helix-loop-helix motifs. Chemistry, 10(17):4163-70, 2004.
[56] J. Razkin, H. Nilsson, and L. Baltzer. Catalysis of the cleavage of uridine
3'-2,2,2-trichloroethylphosphate by a designed helix-loop-helix motif peptide.
Journal of the American Chemical Society, 129(47):14752-8, 2007.
[57] M. Faiella, C. Andreozzi, R. T. de Rosales, V. Pavone, 0. Maglio, F. Nastri,
W. F. DeGrado, and A. Lombardi. An artificial di-iron oxo-protein with phenol
oxidase activity. Nature Chemical Biology, 5(12):882-4, 2009.
[58] M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey, and V. L. Pecoraro. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nature Chemistry, 4(2):118-123, 2012.
[59] S. C. Patel, L. H. Bradley, S. P. Jinadasa, and M. H. Hecht. Cofactor binding
and enzymatic activity in an unevolved superfamily of de novo designed 4-helix
bundle proteins. Protein Sci, 18(7):1388-400, 2009.
[60] Y. Bai, Y. Ling, W. Shi, L. Cai, Q. Jia, S. Jiang, and K. Liu. Heteromeric
assembled polypeptidic artificial hydrolases with a six-helical bundle scaffold.
ChemBioChem, 12(17):2647-58, 2011.
[61] S. Bezer, M. Matsumoto, M. W. Lodewyk, S. J. Lee, D. J. Tantillo, M. R. Gagne,
and M. L. Waters. Identification and optimization of short helical peptides with
novel reactive functionality as catalysts for acyl transfer by reactive tagging.
Organic & Biomolecular Chemistry, 12(9):1488-1494, 2014.
111
[62] M. Tegoni, F. Yu, M. Bersellini, J. E. Penner-Hahn, and V. L. Pecoraro. Designing a functional type 2 copper center that has nitrite reductase activity
within alpha-helical coiled coils. Proc Natl Acad Sci U S A, 109(52):21234-9,
2012.
163] F. Yu, J. E. Penner-Hahn, and V. L. Pecoraro. De novo-designed metallopeptides with type 2 copper centers: modulation of reduction potentials and nitrite
reductase activities. Journal of the American Chemical Society, 135(48):18096107, 2013.
[641 M. R. Razeghifard and T. Wydrzynski. Binding of zn-chlorin to a synthetic fourhelix bundle peptide through histidine ligation. Biochemistry, 42(4):1024-30,
2003.
[65] A. Roy, I. Sarrou, M. D. Vaughn, A. V. Astashkin, and G. Ghirlanda. De novo
design of an artificial bis[4fe-4s] binding protein. Biochemistry, 52(43):7586-94,
2013.
[66] J. Grzyb, F. Xu, L. Weiner, E. J. Reijerse, W. Lubitz, V. Nanda, and D. Noy.
De novo design of a non-natural fold for an iron-sulfur protein: alpha-helical
coiled-coil with a four-iron four-sulfur cluster binding site in its central core.
Biochim Biophys Acta, 1797(3):406-13, 2010.
[67] C. E. Tinberg, S. D. Khare, J. Dou, L. Doyle, J. W. Nelson, A. Schena,
W. Jankowski, C. G. Kalodimos, K. Johnsson, B. L. Stoddard, and D. Baker.
Computational design of ligand-binding proteins with high affinity and selectivity. Nature, 501(7466):212-6, 2013.
[681 A. Roy, C. Madden, and G. Ghirlanda. Photo-induced hydrogen production
in a helical peptide incorporating a [fefe] hydrogenase active site mimic. Chem
Commun (Camb), 48(79):9816-8, 2012.
[69] M. M. Muller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman, and D. Hilvert.
A rationally designed aldolase foldamer. Angew Chem Int Ed Engl, 48(5):922-5,
2009.
[70] A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew,
C. Cozens, K. M. Weeks, P. Herdewijn, and P. Holliger. Catalysts from synthetic
genetic polymers. Nature, 518(7539):427-30, 2015.
[71]
M. Matsumoto, S. J. Lee, M. L. Waters, and M. R. Gagne. A catalyst selection
protocol that identifies biomimetic motifs from beta-hairpin libraries. Journal
of the American Chemical Society, 136(45):15817-15820, 2014.
[72] S. F. Betz, D. P. Raleigh, and W. F. Degrado. De-novo protein design - from
molten globules to native-like states. Current Opinion in StructuralBiology, 3
(4):601-610, 1993.
112
[731 J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St Clair,
J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E.
Michael, and D. Baker. Computational design of an enzyme catalyst for a stereoselective bimolecular diels-alder reaction. Science, 329(5989):309-13, 2010.
[74] G. P. Smith and V. A. Petrenko.
391-410, 1997.
Phage display.
Chemical Reviews, 97(2):
[75] Carlos F. Barbas III, Denis R. Burton, Jamie K. Scott, and Gregg J. Silverman.
Phage Display - A Laboratory Manual. Cold Spring Harbor Laboratory Press,
Cold Pring Harbor, New York, 2001.
[76] J. W. Kehoe and B. K. Kay. Filamentous phage display in the new millennium.
Chemical Reviews, 105(11):4056-4072, 2005.
[77
S. K. Lee, D. S. Yun, and A. M. Belcher. Cobalt ion mediated self-assembly
of genetically engineered bacteriophage for biomimetic co-pt hybrid material.
Biomacromolecules, 7(1):14-17, 2006.
[78] K. T. Nam, D. W. Kim, P. J. Yoo, C. Y. Chiang, N. Meethong, P. T. Hammond,
Y. M. Chiang, and A. M. Belcher. Virus-enabled synthesis and assembly of
nanowires for lithium ion battery electrodes. Science, 312(5775):885-888, 2006.
[79] Y. J. Lee, Y. Lee, D. Oh, T. Chen, G. Ceder, and A. M. Belcher. Biologically
activated noble metal alloys at the nanoscale: For lithium ion battery anodes.
Nano Letters, 10(7):2433-2440, 2010.
[80] X. N. Dang, H. J. Yi, M. H. Ham, J. F. Qi, D. S. Yun, R. Ladewski, M. S. Strano,
P. T. Hammond, and A. M. Belcher. Virus-templated self-assembled singlewalled carbon nanotubes for highly efficient electron collection in photovoltaic
devices. Nature Nanotechnology, 6(6):377-384, 2011.
[81] J. F. Qi, X. N. Dang, P. T. Hammond, and A. M. Belcher. Highly efficient plasmon-enhanced dye-sensitized solar cells through metaldoxide coreshell nanostructure. Acs Nano, 5(9):7108-7116, 2011.
[82] K. A. Kelly, P. Waterman, and R. Weissleder. In vivo imaging of molecularly
targeted phage. Neoplasia, 8(12):1011-8, 2006.
[831 K. A. Kelly, N. Bardeesy, R. Anbazhagan, S. Gurumurthy, J. Berger, H. Alencar,
R. A. DePinho, U. Mahmood, and R. Weissleder. Targeted nanoparticles for
imaging incipient pancreatic ductal adenocarcinoma. Plos Medicine, 5(4):657668, 2008.
[84] J. A. Hunt and C. A. Fierke. Selection of carbonic anhydrase variants displayed
on phage - aromatic residues in zinc binding site enhance metal affinity and
equilibration kinetics. Journal of Biological Chemistry, 272(33):20364-20372,
1997.
113
[85] P. Soumillion, L. Jespers, M. Bouchet, J. Marchandbrynaert, G. Winter, and
J. Fastrez. Selection of beta-lactamase on filamentous bacteriophage by catalytic activity. Journal of Molecular Biology, 237(4):415-422, 1994.
[86] D. Legendre, P. Soumillion, and J. Fastrez. Engineering a regulatable enzyme
for homogeneous immunoassays. Nature Biotechnology, 17(1):67-72, 1999.
[87]
L. 0. Hansson, M. Widersten, and B. Mannervik. Mechanism-based phage
display selection of active-site mutants of human glutathione transferase al-i
catalyzing snar reactions. Biochemistry, 36(37):11252-11260, 1997.
188] I. Fujii, S. Fukuyama, Y. Iwabuchi, and R. Tanimura. Evolving catalytic antibodies in a phage-displayed combinatorial library. Nature Biotechnology, 16(5):
463-467, 1998.
189] M. Baca, T. S. Scanlan, R. C. Stephenson, and J. A. Wells. Phage display of a
catalytic antibody to optimize affinity for transition-state analog binding. Proceedings of the National Academy of Sciences of the United States of America,
94(19):10063-10068, 1997.
[901 K. D. Janda, L. C. Lo, C. H. L. Lo, M. M. Sim, R. Wang, C. H. Wong, and R. A.
Lerner. Chemical selection for catalysis in combinatorial antibody libraries.
Science, 275(5302):945-948, 1997.
[91] H. Pedersen, S. Holder, D. P. Sutherlin, U. Schwitter, D. S. King, and P. G.
Schultz. A method for directed evolution and functional cloning of enzymes.
Proceedings of the National Academy of Sciences of the United States of America, 95(18):10523-10528, 1998.
[92] J. L. Jestin, P. Kristensen, and G. Winter. A method for the selection of
catalytic activity using phage display and proximity coupling. Angewandte
Chemie-InternationalEdition, 38(8):1124-1127, 1999.
[93] Y. Maeda, N. Javid, K. Duncan, L. Birchall, K. F. Gibson, D. Cannon,
Y. Kanetsuki, C. Knapp, T. Tuttle, R. V. Ulijn, and H. Matsui. Discovery of
catalytic phages by biocatalytic self-assembly. Journal of the American Chemical Society, 136(45):15893-15896, 2014.
[94] J. P. Casey, R. J. Barbero, N. Heldman, and A. M. Belcher. Versatile de
novo enzyme activity in capsid proteins from an engineered m13 bacteriophage
library. Journal of the American Chemical Society, 136(47):16508-16514, 2014.
195]
Sinclair Lewis. Arrowsmith. Harcourt, Brace and Company, New York, 1924.
[96] B. Hocker. Protein design: A metalloenzyme reloaded. Nature Chemical Biology, 8(3):224-5, 2012.
[97] K. A. McCall, C. C. Huang, and C. A. Fierke. Function and mechanism of zinc
metalloenzymes. Journal of Nutrition, 130(5):1437S-1446S, 2000.
114
[981 C. M. Rufo, Y. S. Moroz, 0. V. Moroz, J. Stohr, T. A. Smith, X. Hu, W. F.
DeGrado, and I. V. Korendovych. Short peptides self-assemble to produce
catalytic amyloids. Nat Chem, 6(4):303-9, 2014.
[991
B. S. Der, D. R. Edwards, and B. Kuhlman. Catalysis by a de novo zincmediated protein interface: Implications for natural enzyme evolution and rational enzyme engineering. Biochemistry, 51(18):3933-3940, 2012.
[1001 D. W. Christianson and J. D. Cox. Catalysis by metal-activated hydroxide in
zinc and manganese metalloenzymes. Annual Review of Biochemistry, 68:33-57,
1999.
1101] D. S. Auld. Zinc coordination sphere in biochemical zinc sites. Biometals, 14
(3-4):271-313, 2001.
[102] A. E. Eriksson, T. A. Jones, and A. Liljas. Refined structure of human carbonic
anhydrase-ii at 2.0-a resolution. Proteins-StructureFunction and Genetics, 4
(4):274-282, 1988.
[1031 D. A. Marvin, L. C. Welsh, M. F. Symmons, W. R. P. Scott, and S. K. Straus.
Molecular structure of fd (fl, m13) filamentous bacteriophage refined with respect to x-ray fibre diffraction and solid-state nmr data supports specific models
of phage assembly at the bacterial membrane. Journal of Molecular Biology,
355(2):294-309, 2006.
[104] D. A. Marvin, R. D. Hale, C. Nave, and M. H. Citterich. Molecular-models and
structural comparisons of native and mutant class-i filamentous bacteriophages
ff (fd, fl, m13), ifi and ike. Journal of Molecular Biology, 235(1):260-286, 1994.
11051 M. M. Yamashita, L. Wesson, G. Eisenman, and D. Eisenberg. Where metalions bind in proteins. Proceedings of the National Academy of Sciences of the
United States of America, 87(15):5648-5652, 1990.
[106] W. Hung, C. Hu, and C. Liu. Hydrolysis catalyzed with a resin containing
histidine groups. Journal of Molecular Catalysis A: Chemical, 106:67-73, 1996.
[107] K. S. Broo, L. Brive, P. Ahlberg, and L. Baltzer. Catalysis of hydrolysis and
transesterification reactions of p-nitrophenyl esters by a designed helix-loophelix dimer. Journal of the American Chemical Society, 119(47):11362-11372,
1997.
[108] G. M. Rodriguez, Y. Tashiro, and S. Atsumi. Expanding ester biosynthesis in
escherichia coli. Nature Chemical Biology, 10(4):259-+, 2014.
[109] Y. Huang, C. Y. Chiang, S. K. Lee, Y. Gao, E. L. Hu, J. De Yoreo, and
A. M. Belcher. Programmable assembly of nanoarchitectures using genetically
engineered viruses. Nano Letters, 5(7):1429-34, 2005.
115
[110] M. A. Dwyer, L. L. Looger, and H. W. Hellinga. Computational design of a
biologically active enzyme (retraction of vol 304, pg 1967, 2004). Science, 319
(5863):569-569, 2008.
[111] A. Kumar and S. Singh. Directed evolution: tailoring biocatalysts for industrial
applications. Crit Rev Biotechnol, 33(4):365-78, 2013.
[112] G. Kirchner, M. P. Scollar, and A. M. Klibanov. Resolution of racemic mixtures via lipase catalysis in organic-solvents. Journal of the American Chemical
Society, 107(24):7072-7076, 1985.
[1131 A. Zaks and D. R. Dodds. Application of biocatalysis and biotransformations to
the synthesis of pharmaceuticals. Drug Discovery Today, 2(12):513-531, 1997.
[1141 J. A. Akkara, M. S. R. Ayyagari, and F. F. Bruno. Enzymatic synthesis and
modification of polymers in nonaqueous solvents. Trends in Biotechnology, 17
(2):67-73, 1999.
11151 M. Zumarraga, T. Bulter, S. Shleev, J. Polaina, A. Martinez-Arias, F. J. Plow,
A. Ballesteros, and M. Alcalde. In vitro evolution of a fungal laccase in high
concentrations of organic cosolvents. Chemistry & Biology, 14(9):1052-1064,
2007.
[116] P. Gomez-Tagle, I. Vargas-Zuniga, 0. Taran, and A. K. Yatsimirsky. Solvent
effects and alkali metal ion catalysis in phosphodiester hydrolysis. Journal of
Organic Chemistry, 71(26):9713-9722, 2006.
[117] A. E. Wheals, L. C. Basso, D. M. G. Alves, and H. V. Amorim. Fuel ethanol
after 25 years. Trends in Biotechnology, 17(12):482-487, 1999.
[1181 B. S. Der, M. Machius, M. J. Miley, J. L. Mills, T. Szyperski, and B. Kuhlman.
Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. Journal of the American Chemical Society, 134
(1):375-385, 2012.
[1191 L. Miao, X. Y. Zhang, C. Y. Si, Y. Z. Gao, L. L. Zhao, C. X. Hou, 0. Shoseyov,
Q. Luo, and J. Q. Liu. Construction of a highly stable artificial glutathione
peroxidase on a protein nanoring. Organic & Biomolecular Chemistry, 12(2):
362-369, 2014.
[1201 G. E. Host and B. H. Jonsson. Converting human carbonic anhydrase ii into a
benzoate ester hydrolase through rational redesign. Biochimica Et Biophysica
Acta-Proteins and Proteomics, 1784(5):811-815, 2008.
[121] C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis,
J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman,
and G. J. Hughes. Biocatalytic asymmetric synthesis of chiral amines from
ketones applied to sitagliptin manufacture. Science, 329(5989):305-309, 2010.
116
[122] Y. X. Fan, Z. M. Xie, H. W. Zhang, and J. Q. Qian. Kinetic resolution of both
1-phenylethanol enantiomers produced by hydrolysis of 1-phenylethyl acetate
with candida antarctica lipase b in different solvent systems. Kinetics and
Catalysis, 52(5):686-690, 2011.
[123] C. Solarte, E. Yara-Varon, J. Eras, M. Torres, M. Balcells, and R. CanelaGarayoa. Lipase activity and enantioselectivity of whole cells from a wild-type
aspergillius flavus strain. Journal of Molecular Catalysis B-Enzymatic, 100:
78-83, 2014.
[124] C. H. Kwon, D. Y. Shin, J. H. Lee, S. W. Kim, and J. W. Kang. Molecular modeling and its experimental verification for the catalytic mechanism of
candida antarctica lipase b. Journal of Microbiology and Biotechnology, 17(7):
1098-1105, 2007.
[125] G. G. Guilbault and D. N. Kramer. Resorufin butyrate and indoxyl acetate as
fluorogenic substrates for cholinesterase. Analytical Chemistry, 37(1):120-123,
1965.
[126] J. A. Kiernan. Indigogenic substrates for detection and localization of enzymes.
Biotechnic & Histochemistry, 82(2):73-103, 2007.
[127] B. M. Barney. The sweet smell of biosynthesis. Nature Chemical Biology, 10
(4):246-247, 2014.
[128] F. Cai, 0. Y. Chao, P. P. Duan, S. Gao, Y. Xu, and F. Chen. Purification
and characterization of a novel thermal stable peroxidase from jatropha curcas
leaves. Journal of Molecular Catalysis B-Enzymatic, 77:59-66, 2012.
[129] C. Walsh. Enzymatic Reaction Mechanisms. W. H. Freeman and Company,
San Francisco, 1978.
[130] F. van de Velde, F. van Rantwijk, and R. A. Sheldon. Improving the catalytic
performance of peroxidases in organic synthesis. Trends in Biotechnology, 19
(2):73-80, 2001.
[131] M. Wagner and J. A. Nicell.
Detoxification of phenolic solutions with
horseradish peroxidase and hydrogen peroxide. Water Research, 36(16):40414052, 2002.
[132] S. Colonna, N. Gaggero, C. Richelmi, and P. Pasta. Recent biotechnological
developments in the use of peroxidases. Trends in Biotechnology, 17(4):163-168,
1999.
[133] A. M. Klibanov, Z. Berman, and B. N. Alberti. Preparative hydroxylation of
aromatic-compounds catalyzed by peroxidase. Journal of the American Chemical Society, 103(20):6263-6264, 1981.
117
[1341 M. Faiella, 0. Maglio, F. Nastri, A. Lombardi, L. Lista, W. R. Hagen, and
V. Pavone. De novo design, synthesis and characterisation of mp3, a new catalytic four-helix bundle hemeprotein. Chemistry-a European Journal, 18(50):
15960-15971, 2012.
[135]
S. L. Newmyer and P. R. 0. Demontellano. Horseradish-peroxidase his-42-Iala,
his-42-]val, and phe-41-]ala mutants - histidine catalysis and control of substrate
access to the heme iron. Journal of Biological Chemistry, 270(33):19430-19438,
1995.
[136] S. I. Ozaki and P. R. 0. Demontellano. Molecular engineering of horseradishperoxidase - highly enantioselective sulfoxidation of aryl alkyl sulfides by the
phe-41-Jleu mutant. Journal of the American Chemical Society, 116(10):44874488, 1994.
[1371 S. Ozaki and P. R. 0. Demontellano. Molecular engineering of horseradishperoxidase - thioether sulfoxidation and styrene epoxidation by phe-41 leucine
and threonine mutants. Journal of the American Chemical Society, 117(27):
7056-7064, 1995.
[138] E. Garcia-Ruiz, D. Gonzalez-Perez, F. J. Ruiz-Duenas, A. T. Martinez, and
M. Alcalde. Directed evolution of a temperature-, peroxide- and alkaline phtolerant versatile peroxidase. Biochemical Journal, 441:487-498, 2012.
[139]
Z. P. Wu and D. Hilvert. Selenosubtilisin as a glutathione-peroxidase mimic.
Journal of the American Chemical Society, 112(14):5647-5648, 1990.
[140] R. L. Koder, J. L. R. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser, and
P. L. Dutton. Design and engineering of an o-2 transport protein. Nature, 458
(7236):305-U64, 2009.
[141] S. C. Patel and M. H. Hecht. Directed evolution of the peroxidase activity
of a de novo-designed protein. Protein Engineering Design & Selection, 25(9):
445-451, 2012.
[1421 F. Nastri, L. Lista, P. Ringhieri, R. Vitale, M. Faiella, C. Andreozzi, P. Travascio, 0. Maglio, A. Lombardi, and V. Pavone. A heme-peptide metalloenzyme
mimetic with natural peroxidase-like activity. Chemistry-a European Journal,
17(16):4444-4453, 2011.
[143] S. Riva. Laccases: blue enzymes for green chemistry. Trends in Biotechnology,
24(5):219-226, 2006.
[1441 B. Branchi, C. Galli, and P. Gentili. Kinetics of oxidation of benzyl alcohols by
the dication and radical cation of abts. comparison with laccase-abts oxidations:
an apparent paradox. Organic H Biomolecular Chemistry, 3(14):2604-2614,
2005.
118
[1451 K. D. Miner, A. Mukherjee, Y. G. Gao, E. L. Null, I. D. Petrik, X. Zhao,
N. Yeung, H. Robinson, and Y. Lu. A designed functional metalloenzyme that
reduces o-2 to h2o with over one thousand turnovers. Angewandte ChemieInternationalEdition, 51(23):5589-5592, 2012.
[146] K. R. Love, S. Bagh, J. Choi, and J. C. Love. Microtools for single-cell analysis
in biopharmaceutical development and manufacturing. Trends in Biotechnology,
31(5):16-22, 2013.
119