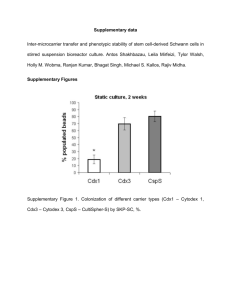
September 2015
advertisement

Vol. September 2015 Vol.12, 12, Supplement Supplement 55 A S p e c i A l S u p p l e m e n t to B i o p r o c e S S i n t e r n At i o n A l Enabling Cell Therapy Manufacturing Enabling Cell Therapy Manufacturing in association with in ASSociAtion with “ Together we can advance cell therapies worldwide.” Pall Life Sciences Pall Life Sciences delivers the expertise and experience to help our customers realize efficient, effective development and commercialization of cell therapies. Working with Pall, you’ll benefit from our industry-leading cell manufacturing technologies and services – from our unique single-use bioreactor systems to our innovative SoloHill® microcarriers. You’ll be fully equipped to overcome the challenges of manufacturing live cells for therapeutics, making your path to industrialization smoother than ever. Pall’s business philosophy is about collaboration and helping you achieve your goals. Together, we can advance the development of cutting-edge cell therapies... and dramatically improve the lives of patients worldwide. Pall Life Sciences Your vision. Our expertise. Their future. www.pall.com/celltherapy © 2015 Pall Corporation. Pall, and SoloHill are trademarks of Pall Corporation. ® indicates a trademark registered in the USA. GN15.9698 September 2015 Expansion and Characterization of Mesenchymal Stem Cells on Pall SoloHill® Microcarriers . . . . . . . . . . . . . . 22 Heather Woolls, Dave Splan, and Mark Szczypka Strategies for Microcarrier Culture Optimization . . . . . . . 28 Mark Szczypka and Alain Fairbank Section 3: Cell Therapy Manufacturing Enabling Cell Therapy Manufacturing . . . . . . . . . . . . . . . . . . . 2 Alain Fairbank Pall’s innovative products and capabilities for production of cellular therapies Designing the Most Cost-Effective Manufacturing Strategy for Allogeneic Cell-Based Therapies . . . . . . . . . . . . . . . . . . . . 36 Section 1: Pall in Cell Therapy Introducing Pall’s commitment to support of cell therapy manufacturing and commercialization Positioning for Success: An Interview with Mario Philips . . 4 S. Anne Montgomery and Brian Caine Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10 Jon Rowley, Eytan Abraham, Andrew Campbell, Harvey Brandwein, and Steve Oh Section 2: Process Development Tools and technologies from Pall Life Sciences designed to facilitate process development and scale-up T-Cell Suspension Culture in a 24-Well Microbioreactor: High-Throughput Screening of Operating Conditions . . 14 Kenny Choi, Jason N. Carstens, and Shelly Heimfeld Thierry Bovy, Alain Fairbank, and Suzanne Farid Production of Viral Vectors Using the iCELLis® Fixed-Bed Bioreactor System: Beyond Mesenchymal Stem Cells — Gene-Modified Cell Therapy, Gene Therapy, and Exosomes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 Matt Kremer Single-Use Bioreactors and Microcarriers: Scalable Technology for Cell-Based Therapies . . . . . . . . . . 48 Mark Szczypka, David Splan, Heather Woolls, and Harvey Brandwein Section 4: Perspectives Experts at Pall Life Sciences discuss cell therapy industrialization solutions and future directions Ask the Experts: Core Technologies Expand Opportunities for Cell Therapy Manufacturing . . . . . . . . . . . . . . . . . . . . . . . . 54 Scaling Up Stem Cells: Moving from Laboratory to Commercial Productions with a Single-Use Multiplate Bioreactor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18 S. Anne Montgomery Matthieu Egloff and Jose Castillo, with Thierry Bovy S. Anne Montgomery and Brian Caine Positioning Tools, Technologies, and Talents for Cell Therapies: An Interview with Harvey Brandwein . . . . . . . 60 Featured Products and Services . . . . . . . . . . . . 21, 43, 47 Editorial Offices Sales and Administrative Offices PO Box 70, Dexter, OR 97431 Editor in Chief S. Anne Montgomery amontgomery@bioprocessintl.com (article and supplement queries, editorial policies) Senior Technical Editor Cheryl Scott cscott@bioprocessintl.com (press releases, art submissions, design) One Research Drive, Suite 400A Westborough, MA 01581 (sales inquiries, media kits, advertising) Production and Creative Manager Genevieve McCarthy 1-212-520-2752 genevieve.mccarthy@informausa.com Publisher Brian Caine 1-508-614-1443 bcaine@bioprocessintl.com Director of Audience Development and Manufacturing Nora Pastenkos 1-212-520-2733 nora.pastenkos@informausa.com Strategic Marketing Consultant (Eastern) Christopher Johnson 1-508-614-1273 cjohnson@bioprocessintl.com Marketing and Digital Content Strategist Leah Rosin 1-508-614-1167 lrosin@bioprocessintl.com Managing Editor Maribel Rios mrios@bioprocessintl.com (article queries, special projects) Strategic Marketing Consultant (Western) Mike Kelly 1- 646-957-8974 mkelly@bioprocessintl.com Editorial Assistant Alison Center acenter@bioprocessintl.com Strategic Marketing Consultant (European) Joanna Taylor 44-(0)-20-7551-9392 joanna.taylor@informa.com Reprints Rhonda Brown 1-866-879-9144 x194 rhondab@fosterprinting.com Sales and Marketing Coordinator Kim Rafferty 1-508-614-1226 krafferty@bioprocessintl.com For subscription inquiries, call 1-847-763-4930, toll free 1-877-232-2399, or email bioprocess@ halldata.com. Find BPI citations online in the Chemical Abstracts Database (www.cas.org). List Rental Amy Miller 1-508-614-1251 amiller@ibcusa.com ©2015 BioProcess International (USPS 0022-044, ISSN 1542-6319) is published eleven times a year by Informa Life Sciences Group at 52 Vanderbilt Ave., New York, NY 10017, Phone: 1-212-520-2777, fax 1-212-661-5052, www.bioprocessintl.com. Periodicals postage is paid in Westborough, MA and additional mailing offices. POSTMASTER: Send address changes to BioProcess International, PO Box 1170, Skokie, IL 60076. Canadian publication agreement No. 41067503. Canadian return address DPGM 7496 Bath Road Unit 2, Mississauga, ON L4T 1L2. Electronic subscriptions are available online. Articles are abstracted by the Chemical Abstracts database at www.cas.org. 1 BioProcess International 13(8)sp S eptember 2015 Sponsored Supplement Introduction Enabling Cell Therapy Manufacturing by Alain Fairbank A s this special Pall supplement of BioProcess International issue goes to press, progress continues in the field of cell therapy research. The revival of cell (and gene) therapy has been driven by some positive achievements that have occurred over the past decade. Cell therapy products differ in many ways from traditional small-molecular and biologic products. The main difference is that, contrary to traditional biopharmaceutical applications in which cells secrete the product of interest, in cell therapy applications cells are the product. That brings a multitude of challenges as manufacturers navigate the industrialization pathway to determine an optimal manufacturing strategy for their therapy. Despite those challenges — or perhaps because of them — the demand for manufacturing tools to support this industry continues to grow. Some key drivers include the following: Scalability — adherent cells such as mesenchymal stem cells (MSCs) present a manufacturing challenge as lot sizes increase from billions to trillions of cells (1) Cost — achieving lot sizes of several hundred billion to trillions of cells efficiently and cost effectively will be imperative for commercial success (1). Flexibility — predicting market demand is never easy, particularly for emerging technologies that lack historic precedent, so manufacturing flexibility (both in scheduling and capacity for rapid production scale-up) is vital to the commercial success of the cell therapy industry (2). Closed systems — one of the most challenging aspects in cell therapy manufacturing is conversion of research 2 BioProcess International 13(8)sp processes to manufacturing campaigns, which is most readily achieved by converting to closed systems (3). The general purpose of this supplement is to discuss many industrialization challenges facing developers of cellular therapies today, while highlighting how Pall’s platform of products and services has been developed to address those challenges and consequently enable its customers on their pathway to cell therapy commercialization. Pall in Cell Therapy This special issue commences with an interview with Mario Philips, president of Pall’s single-use technologies division. He discusses Pall Life Sciences’ commitment to the cell therapy industry and how Pall has positioned itself with a platform of unique and innovative cell expansion technologies and services. Pall Life Sciences recognizes that the advancement of cell therapies — and, ultimately, that the commercial success of such products — can be achieved through the combined strengths of our customers and the Pall team. Long recognized for its expertise in processing and filtration equipment for the biopharmaceutical industry, Pall Life Sciences has S eptember 2015 broadened its offering in upstream manufacturing in recent years. It has done so by expanding its core capabilities in the single-use, bioreactor, and microcarrier arenas, with unique and innovative technologies for cultivation of cells for therapeutic applications. “Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy” is an abridged article originally published in BioProcess International in March, 2012. Readers of this article will learn from industry experts, including Pall’s own Harvey Brandwein, about some hurdles, challenges, and bottlenecks associated with bringing a cell-based therapy to market. Process Development and Cell Therapy Manufacturing Developing the right industrialization strategy is critical to supporting a sustainable cell therapy development and commercialization program. “Designing the Most Cost-Effective Manufacturing Strategy for Allogeneic Cell-Based Therapies” was generated from a webinar presented in March of 2015. In their presentations, Thierry Bovy of Pall Life Sciences and Professor Suzanne Farid from University College London (UCL) discussed advantages Sponsored Supplement and limitations of current technologies available for the commercialization of large-scale allogeneic therapies. Farid also provided some key insights from an advanced bioprocess economics model designed by her team. Also discussed in this article are how the economic aspects of cell therapy products need to be addressed from the early phases of development to enable a viable life cycle. Some of Pall’s key technologies for the production of cell-based therapies — the Micro-24 Microreactor, Xpansion® Multiplate Bioreactor, and Pall SoloHill® Microcarriers — are highlighted in the process development section of this supplement. In the article, “T-Cell Suspension Culture in a 24-Well Microbioreactor” (originally published in BioProcess International in April of 2013), Choi et al. present results from a study using the Micro-24 MicroReactor from Pall Life Sciences, a 24-well microbioreactor with the capability for agitated suspension culture and continuous monitoring and control of pH, temperature, and dissolved oxygen (DO) in each individual well. The team valuated its usefulness as a process development tool to develop and optimize suspension cell culture for manufacturing therapeutic T cells, and they demonstrated the effectiveness of the Micro-24 MicroReactor as an accurate, high-throughput experimental system that properly represents production-scale systems. Designed for shear-sensitive, adherent-cell applications such as stem cell cultivation, the Xpansion multiplate bioreactor is part of the Pall Life Sciences’ single-use bioreactor family. The article, “Scaling Up Stem Cells” was first published in May of 2012. In it, the authors discuss the development of this unique bioreactor system for use in scale-up and expansion of stem cells for therapeutic applications. The authors highlight three key advantages of implementing this technology: speed, ease of adaptation, and reduced footprint. Several clinical trials have been initiated using stem cells in cell therapy treatments. Fast emerging as a premier technology for the (very) large-scale expansion of adherent cells for therapies Sponsored Supplement is the microcarrier production platform used with stirred-tank bioreactors. “Expansion and Characterization of Mesenchymal Stem Cells on Pall SoloHill Microcarriers” was originally published as an application note. In it, the authors discuss some additional benefits of using microcarriers to expand mesenchymal stem cells and demonstrate that such cells can in fact be expanded on a number of microcarrier types while maintaining their ability (post expansion) to differentiate into adipocytes and osteocytes. As Oh, et al. discuss in “Meeting Lot Size Challenges” the ability to scale up to very large volumes of cells has been a challenge that the cell therapy industry has been working to overcome. Professor Farid and her team’s work has identified microcarriers as the most costeffective platform to produce the high billions to trillions of cells required for some cellular therapeutics. In the “Strategies for Microcarrier Culture Optimization” article, Mark Szczypka and I outline key considerations that need to be taken into account when developing an optimized microcarrier platform. We also highlight the strategies necessary to enable a successful process development and commercialization using a microcarrier platform. In recent years, gene therapies have rapidly progressed as products move from academic research laboratories into commercial development. Many such therapies in development require large amounts of viral vectors for use in modification of cells to be used as therapeutics. As Matt Kremer reports in his article, “Production of Viral Vectors Using the iCELLis® Fixed-Bed Bioreactor System — Beyond Mesenchymal Stem Cells: GeneModified Cell Therapy, Gene Therapy, and Exosomes,” Pall’s iCELLis fixedbed bioreactor has been successfully applied to this challenge in a number of academic and industrial institutions. They have demonstrated scalable production of viral vectors including retrovirus, adenovirus, recombinant adenoassociated virus, and lentivirus in the iCELLis system. Kremer presents findings from several experiments demonstrating the effectiveness of that system for such applications. He also discusses its utility in providing a longterm, continuous process environment for cultivation of stem cells and harvesting of exosomes — an up and coming area of research. Perspectives Pall Life Sciences has been long recognized as a leader in processing and filtration systems for the biopharmaceutical industry. In recent years, the company has expanded its product offerings by moving upstream with core technology development and acquisitions in single-use bioreactors and microcarrier technologies. For this special supplement, we’ve invited three of our subject-matter experts — Fabien Moncaubeig, Thierry Bovy, and Mark Szczypka — to discuss some of these technologies. They also provide an overview of the process development capabilities now offered at four of Pall’s sites. This special issue concludes with an interview in which Harvey Brandwein (vice president of business development at Pall Life Sciences) provides his perspectives on the cell therapy industry today, where he sees it going, and how he sees Pall’s role in helping to advance cell-based therapies through the industrialization pathway to clinical success and the ultimate reward: commercialization. We hope that you will find these articles both constructive and informative as you navigate your development journey in this very exciting therapeutic area. References 1 Rowley J, et al. Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy. BioProcess Int. 10(3) 2012: S16–S22. 2 Davie NL, et al. Streamlining Cell Therapy Manufacture. BioProcess Int. 10(3) 2012: S24–S28, S49. 3 Sargent B. Best Practices in Cell Therapy Manufacturing. The Cell Culture Dish 6 February 2013. • Alain Fairbank is director of cell therapy marketing for Pall Life Sciences, alain_fairbank@pall.com. S eptember 2015 13(8)sp BioProcess International 3 S e c t i o n O n e PALL IN CELL THERAPY Positioning for Success An Interview with Mario Philips O n 12 March 2015, BPI met with Mario Philips, president, single-use technologies, at Pall’s Port Washington, NY, facility, to learn about Pall’s reasons for entering the cell therapy market. Also participating in the discussion was Alain Fairbank, director of marketing for cell therapies at Pall Life Sciences. The discussion began on a personal note, asking Philips what it was that launched his interest in single-use technologies and cell therapies. Philips: My background is as a chemical engineer. I had worked for a couple of years in the biopharmaceutical industry, more in the analytical space, and then started my own business. We targeted the chemical market for inline and online process analysis. We were picked up by the semiconductor industry and then were acquired by ATMI in 2002. At that time ATMI had a materials division that dealt with chemicals and packaging of liquids. What not a lot of people know is that ATMI already had a relatively large business in single-use liquid packaging. The semiconductor industry was facing the challenge of molecular and particle contamination when cleaning the stainless steel containers used for transportation; and they had a lot of yield and cleaning issues. So we developed a bag-in-a-bottle and a bagin-a-container system. It’s a very different business model from what you find in life sciences. It was very standardized. We were shipping dangerous chemicals, and there was not a lot of customization. The film materials were, in 70% of the cases, also very different — more 20154 BioProcess International 13(8)sp like working with PTFA because of the very aggressive chemicals. ATMI by then had a $40–$50 million business. Meanwhile, I was staying in touch with my friends in the life sciences industry, where people were beginning to use bags. A friend asked me, “Isn’t that what you do in microelectronics?” So we started to look at that. By then, HyClone and Stedim were already becoming established in the life sciences. We knew that we needed to enter that market with adequate credibility. So we asked customers what was not yet addressed by current technology. Two big answers came out of that: mixing and bioreactors. At that time in our corporation we didn’t have a lot of cell culture competencies. We said: “Let’s try to do one thing very well and become the leader in SUT (singleuse technology) mixing.” Then later, the company evolved its bioreactors business. So that is how I ended up in the single-use technology space. My personal interest in cell therapy began with my exposure to the biotechnology industry in Belgium. That country is quite innovative in biotech for its size. The big player there was GSK Biologics, with more than 10,000 people on site, and a number of those people were launching start-ups. A number of those scientists went into cell therapy. Not many equipment manufacturers saw investment in cell therapy companies as a promising business model. But we did. At first we got into cell therapies more to accelerate our learning curve than as an investment. But investing gave us access to CEOs, business plans, and information about cost of ownerships. The big lesson was that S eptember Brenner Photo Productions (www.brennerphoto.com) by S. Anne Montgomery and Brian Caine Mario Philips, President, Single-Use Technologies at Pall Life Sciences they were all using static devices — CellSTACK® culture chambers, typically. Using multiple containers for each patient works well when your target is to make clinical material for 20 patients. But if you want to treat 2,000 patients, that isn’t going to get you there. That was a problem that we recognized early on and that we are still very focused on today, at Pall. The cell therapy industry is still largely at laboratory and R&D scales. But we want to help companies get more out of their R&D-related technologies and help them either scale out (for autologous therapies) or scale up (for allogeneic therapies). That was a fundamental problem to address. Some of the CEOs acknowledged that if they had to scale up using static devices, they would need a square kilometer of clean room space before they could even think about going to the FDA. For example, the first customer we met needed 20 CellSTACK CF10 units for one patient. His therapy required 725 manual interventions. We were able to simplify the process and streamline Sponsored Supplement that company’s capital investment to make its industrial plan realistic. Cell therapy is exciting in that scientists are doing almost unbelievably innovative work that can have a significant impact on us as patients. We can all be exposed to diseases requiring such treatments. Bringing the initial idea to market is a high-risk project with many challenges. The role we like to play at Pall is to provide innovative, cost-effective technology. That’s a big challenge in all of biopharma, of course, but definitely in cell therapy. And then Pall also brings 60 years of experience in bioprocessing to these smaller start-ups — because that’s what these cell therapy companies typically are. They are, very often, very scientific, but they do not have much expertise in bioprocess engineering. The Need for Innovative Technologies Caine: How much existing proteinbased technology can be used in the cell therapy market? What changes or innovations need to be made for largescale cell therapy manufacturing to become affordable? Philips: There are two types of therapies. Scaling up autologous therapies is quite different from what biotechnology typically does. In the autologous model of personalized medicine, a company deals with small volumes. But if it wants to treat thousands of patients, it needs to find a way to scale that process out. Innovation has to happen. At Pall we are doing some of that — developing replacements for traditional static devices, such as by using expansion bioreactors. The goal is to close the process and develop industrial-scale automation around it. In the case of allogeneic therapies, I think the biotechnology and vaccine models are much more leverageable. We can give a customer two options. If a customer is tackling a treatment for an orphan disease for which there may be fewer than 10,000 patients, that company probably can use some type of expansion reactor. Again, the goals are to close up the process, reduce the risk, and bring in some Sponsored Supplement controls. For allogeneic therapies, much of that technology is already available. We use microcarriers and single-use bioreactors in vaccine development. But the biggest difference between biotechnology and cell therapy is that the cells themselves are the product in cell therapy. That introduces some challenges. One area where innovation is still needed is in segregating the cells from the carriers safely - plastic material must not end up in a patient. I believe also that the volumereduction step requires more attention. We are trying to approach it with centrifuge technology and tangential flow filtration (TFF). But that is still early stage work for us. Allogeneic development uses basic cell culture technologies — bioreactors, microcarriers, and so on. The main difference is how to work with these very fragile cells. Again, the fundamental difference is that you need the cells not to segregate as in the other biotechnology applications. All in all, however, some dedicated discrete products are available. Our vision at Pall is that we want to turn them into an industrial manufacturing platform so that people can produce cells safely in a closed environment. The demand in the market is still low, with maybe four, five, or six companies that are really ready for prime time and have to flesh out their manufacturing. But if you look at the clinical trials, the pipeline is huge. Some won’t make it, but many will. Our focus is on leveraging existing technologies from the biotech space and then trying to close gaps where needed. The goal is to create an industrial solution. When to Enter the Market Caine: There are other suppliers to the cell therapy market. Some of them want to play in this area, but many (if not most) of them are waiting. They are waiting for the market, they are waiting for a return, and they are waiting for a bit more clarity in the market. But Pall doesn’t seem to be one of those companies. Pall is making a significant investment and a significant push into cell therapy now. Why is this the right time? Cell therapy companies are more and more thinking about what their CORE competencies are — where they excel versus where they need partners. Philips: That’s a very good question. If you look back on the cell therapy industry, five or seven years ago a lot of people thought it was breakthrough time, right? You could see that at the Cell Therapy Congress among many vendors and customers. And then we had a bit of a dip when the financial crisis prevented some cell therapy start-ups from obtaining funding. Why we feel that now the chances of a breakthrough are much better are 1), that there is much better funding and 2), that we have a couple of commercial therapies on the market. But it is still fairly early. I think our industry needs a couple of success stories now. Otherwise you could look at it very pessimistically. The two or three approvals of 2014 didn’t really take off. All of them are having problems. But that also helps others learn how to do it better. One of the players did think too late about how to manufacture huge quantities. That sounds like a positive problem: We will deal with it when it’s there. But your investors don’t care. If you are not delivering on your demand, you are destroying value, and they will pull out. At Pall we believe it is still a risk, but we believe it is a more calculated risk, and we want to be first-to-market with this platform. So we are investing ahead of the potential. We want to be associated with the top four, five, or six that come through now, and we hope that will give us the long-term benefit. The Importance of Managing Long-Term Expectations Caine: Your prior example was of a scientific success that was a business failure. Was it a necessary failure in International S eptember 2015 13(8)sp BioProcess 5 20156 BioProcess International 13(8)sp Brenner Photo Productions (www.brennerphoto.com) the evolution of cell therapy companies to understand what they need to do to be successful? Philips: Probably. Yes. If you dig into other industries, they all have such early examples. Montgomery: So it’s still an investment in the future at this point. Do you feel that Pall’s experience in the general biopharmaceutical industry prepares you to wait out the cell therapy industry — to follow it for a couple of years before you see more commercial successes? Philips: Yes. Of course, we are very focused on the short-term returns as well, but we know we also have to place some bets on the long-term. If we look at our strategic plans for our core business in filtration with the vaccines and the biotech market, three years is a good strategic plan to look at. But in cell therapy, we are really looking more at five to seven years. Caine: Do you see a change in behavior and understanding among cell therapy companies toward earlier creation of a commercial business plan? How has that changed in the past few years? Philips: I think cell therapy companies have one big challenge in finding the right sort of partnerships to allow them to advance. In the past, many believed that because the cell is the product, they had to manufacture completely in-house. So although some companies may still set up their businesses in that way, other are starting to look at partners. If you are an investor, you hope that the cell therapy company will use your money for reaching important clinical results. Then you will attract more investors and be able to fund your scale-up. So cell therapy companies are more and more thinking about what their core competencies are — where they excel versus where they need partners. For example, I know that some of them start to do some in-house work and make some clinical materials, but also immediately involve CMOs. They recognize that they need to drive down the needed capital and not put a lot of money into clean rooms and manufacturing infrastructure. That makes any investor quite nervous. The lesson is that some of the work has to be done in-house, but more and more there are specialized CMOs who are helping with the clinical stages. The Role of Contract Manufacturing Organizations Montgomery: Are you seeing more dedicated CMOs coming into this space? Philips: Yes, and pretty much everywhere. In the United States we have Progenitor Cell Therapy and of course Lonza. In Europe, in Belgium, a company there is MaSTherCell. I have met a company in Asia that wants to dedicate itself only to cell therapy. It’s a little bit of a different animal. A lot of the CMOs today have large stainless steel reactors, so they do not necessarily have the right infrastructure to serve a cell therapy customer. They also understand some of the time issues involved, too, especially with autologous therapies, and that the timing differs from any classic model. Caine: Your target customer is the end-user and the CMO. From a technology development standpoint, is one a better partner than the other? If you want to understand the allogeneic need to produce billions, trillions of cells, who do you find to be a more involved or leading partner? Philips: Well, it is definitely always both. But the immediate focus is definitely on the end-user customers because I think in most cases they will prescribe to the CMO what to use. Caine: But whose responsibility is it to develop new technologies? We S eptember talked earlier about gathering information and doing trend analysis regarding what’s needed and where. Would a CMO be more of a partner with you because it is actually manufacturing a cell therapy and can tell you what is lacking, what is needed? Do you have that interaction with PCT and some of these other CMO partners? Philips: Sure. But they bring the same messages to us as the end-users. They are facing the same problems. Fairbank: The challenges exist whether the end-user or CMO is scaling up. There are gaps and challenges that need to be overcome. The CMO is somewhat, I would say, the voice of their customer. Technology Transfer Expectations Montgomery: Are there unique technology transfer issues that people are grappling with? Philips: I think not unique, but it’s a very good question. It’s quite challenging from being at a couple of milliliters, then going to large types of reactors. If you look at the clinical market today, many people today are in phase 1 and in CellSTACKs. So going from a CellSTACK to a microcarrier type of process is quite disruptive. It will be challenging. These cells will be more sensitive than the ones we used to deal with and, honestly, the scale-up and the transfer later on is highly complicated. That is one of the major reasons why we introduced our Xpansion® bioreactor technology first rather than using microcarriers with a bioreactor, because in the Xpansion system, we are really not changing the environment for the cell. So although it’s a big change, with all the benefits of scalability and closed systems, at the end the cells feel completely the same. We have been very successful in tech transfer from static devices into what I could call a huge redesigned CellSTACK. That’s a big benefit because otherwise the technology transfer gets very complicated. Now customers that want to serve markets of ten thousand or hundreds of thousands of patients will have to go to Sponsored Supplement To Manufacture or Be Acquired? Caine: Do you see a trend overall in the strategy of cell therapy companies right now toward being acquired rather than planning to manufacture? Philips: I think we are going to see a lot more therapy companies that only want to bring their product to phase 2 and then try to sell off, which will largely be to big pharma. One of the trends you also see in that respect is that big pharma is starting to get into these start-up companies through its venture capital groups. That’s with only one intention: to have a preferred position to one day acquire the company. So I think the tendency will be that more people will just create value at the point of that investment. Also, bringing a therapy to market requires understanding the logistics and the dynamics, and for that, of course, big pharma has all the experience. I think we’ll see a lot of companies change ownership after initial therapeutic proof that a therapy works. So although some companies will want to go to the end, I think we’ll see more of them being acquired and merged into large pharma. Caine: Do you also see cell therapy–cell therapy mergers and acquisitions? Philips: There are one or two that have happened, but it is still quite exceptional. Sponsored Supplement Caine: Pharma does bring greater financial resources and manufacturing capabilities to the table, however. Montgomery: And established distribution networks. Fairbank: And related logistics. Philips: That’s huge for a standalone cell therapy company to undertake logistics. The one other tendency that I see is that big pharma may be waiting a little bit longer to acquire an early stage cell therapy company. They used to buy companies in phase 1. It was cheaper, despite the risk. Now the trend is to say, “Let it be more expensive, but more proven.” So that’s another thing that we’ll see in cell therapy. Investors don’t want to necessarily get in too early. It’s still cheaper, but there is far more uncertainty. The Role of Quality by Design Brenner Photo Productions (www.brennerphoto.com) something like a reactor, and they will have to go through that change. But it’s always a challenge in our industry. People get into phase 2 and start thinking about how to manufacture product for a hundred thousand patients — and everybody gets nervous about changing the process and explaining it to the FDA. You’re probably going to have a setback and have to go back a phase and re-do a lot of work. Clearly for cell therapy companies it is very important at the very early (stages) to figure out what their end game is. I think some of them are facing issues today because their tech transfer will be highly complicated and explaining the technology to the regulators will be almost impossible. Brian Caine, Publisher of BPI Pall’s Technologies — Present and Future Montgomery: To what extent will current risk-based approaches apply to cell therapies? Does QbD have a place? Philips: For manufacturing allogeneic therapies, it’s quite the same sort of process, and cell-therapy CEOs realize that the need to bring in bioprocess expertise. More and more of these company leaders come from large biotech. They will start using the same risk-based approaches because they bring their expertise into this field. I have seen many, many examples of this already. People who have been with Amgen, Biogen or from vaccine companies are implementing the same type of approaches to risk. I think bringing that expertise into the cell therapy space will definitely give us better chances that this industry will prosper because these people have lived it. Fairbank: This was really validated just yesterday, when I attended a presentation by the FDA. They were discussing the importance of identifying critical process parameters. Applying quality by design and the whole risk assessment approach for cell therapy is just as important. Down the road, we know that there were be more regulations to come. Caine: Can you tell us about Pall’s technologies, their current application to this market segment, and your unique position? Where do you see yourself as a leader in the cell therapy market, and what products does Pall have to support that? Philips: Our focus is currently on industrial manufacturing, from smallto large-scale devices. I think we have a strong position in what I call the expansion step of the cells. We have more product development to do in what I call the volume reduction step. Then we want to leverage expertise from our current business — one such area is process development. We have four process development hubs across the world: one in Brussels, one in Portsmouth in the United Kingdom, one in Ann Arbor, Michigan, and in Westborough, Massachusetts. A lot of our customers are cell therapy people. They ask us, for example, to start the expansion process in a small-scale Xpansion reactor, scale it up, and then either transfer it back to them or transfer it to a CMO. The second area that we want to leverage is our internal biopharm automation and process group. We build large chromatography skids. We set up complete single-use suites with the bioreactor in the middle and the mixers around it. We are building on that expertise to connect these great products into a real platform solution. So I would say our focus is currently on industrial manufacturing; helping customers get out of a relative International S eptember 2015 13(8)sp BioProcess 7 crunch that they are in. In looking at future strategies, one desire for us is to be able to offer cell culture media. We are also dedicated to bringing technology solutions to the downstream process for volume reduction. We have a centrifuge technology in development. We have single-use TFF. These are strengths in our vaccine and the biotechnology businesses, so we hope to close the gaps related to cell therapies. We can do that by combining existing expertise looking into mergers and acquisitions. Because our mission is to really have a total manufacturing solution for future cell therapy companies. We will be less involved in analytical aspects, cell isolation and all that. So even in cell therapy, we will try to focus on our core capabilities. Caine: Not too long ago when protein titers went from one to 10+ grams, there was concern about the downstream bottleneck and not being able to clarify or purify at those volumes. Is the challenge for cell therapies going to be dealing with the very large volumes needed for trillions of cells, or is it about the gentleness with which you handle the cells? What do you see as being perhaps a solution to the current challenges on the downstream side? Fairbank: We know that the cells for cell therapy are the product and they need to be treated more gently, and so I agree that that’s an issue. We’ve got some things in development as Mario said, but scaling up to deal with processing lot sizes of several hundred billion or up to a trillion cells is still a gap that needs to be addressed. Philips: Probably the biggest challenge of the customers is more in the downstream volume reduction step. You see presentations in which people say that have expanded cells, and somebody asks them: “Okay, now what?” Caine: You can’t have a process if you have half a process. Fairbank: The good news is nobody is really at that “now what?” stage where it’s so critical, it’s going to stop the works. Everybody has been talking about it enough that people who are 20158 BioProcess International 13(8)sp Bringing QUALITY BY DESIGN EXPERTISE into the cell therapy space will definitely give us better chances for this industry to prosper. indeed listening. Issues of scale are clearly identified as an unmet need. There is still a window of opportunity to develop the solution before it’s necessary or, worse, becomes an impediment Montgomery: Thinking about these steps so early in the game is still kind of a hard sell for some people, it seems. Fairbank: You know, it is. I was at a conference in Brussels in December and the question was put out to the people who were attending the conference: “How many are in 2D?” Just about everybody in the room raised their hands. “How many are in 3D?” and a few raised their hands. Then the question was posed: “How many people are thinking about the transition from 2D to 3D when you scale?” Only one responded to that question. I found it quite surprising to find that so few people were thinking about it yet. Driving that point home, especially if you know you are developing a treatment for a large population, you need to concurrently — in phase 1, before your process is locked in — develop side-by-side what your commercial process is going to be. Montgomery: Including looking at eventual reimbursement issues? Fairbank: Yes, reimbursement issues — which will help to drive the entire manufacturing strategy. Montgomery: Is this message getting across? Fairbank: It’s starting to more and more. We certainly have been making some noise about it or the past year since I joined Pall. The noise level has certainly been elevated, and the message is getting across. S eptember Bridging the Commercial Gaps — Who Are the Leaders? Caine: I’ll come at it from a publication standpoint. This will be our fifth year with cell therapy and trying to bridge that gap between research and commercialization and the technologies. Three or four years ago, even two years ago, you heard very little about phase 3 commercial scale — understanding the financial business dynamics. You just heard: “Check this out. We can do this. Look how cool this is.” The whole tone has changed in the marketplace. I take it this is also one reason why you’re committing to this market — that you see a transition toward a maturing market. What geographic area do you see as being a leader in development, either from a regulatory standpoint or a scientific standpoint? Philips: I would say the United Stated is definitely leading; that’s where most activity is. There is activity in Europe, but from a leading perspective, it’s the United States, and then Europe, and now Asia. The Japanese industry is receiving quite a bit of support from its government. That’s another element leading toward a breakthrough in the industry: Many governments are starting to incentivize and give subsidies to cell therapy developers, as well as offering big manufacturing initiatives. One example is the Catapult Initiative in the United Kingdom. Its vision is to build a manufacturing hotel where a small cell therapy company can come in to produce its cells. Initially maybe it needs the building for three or four months to do its clinical work, and then it can go out and say, “For my next phase, now I have all the clinical results, and I’m going to find an investor.” That’s another area where you can see some governments becoming more supportive in their regulatory climates, as in Japan recently. If you were to ask me what one big risk remains for this industry, I think the most uncertainty rests in the reimbursement strategies of local governments. Personal medicine is always going to be quite expensive as a therapy. That’s a reality. Allogeneic Sponsored Supplement Sponsored Supplement Philips: As I mentioned earlier, our industry desperately needs a couple of success stories that will attract more investors, more big pharma. They will be seen as providing a proven business model. In the short term it’s all about that. Five years from now, if we have these success stories, I am confident that cell therapy will be a wellaccepted industry already largely dominated by and enhanced by biopharma. It will be almost a condition for cell therapy to succeed that the biotechnology industry will invest into this space and bring in expertise in logistics and infrastructure — as well as experience managing the regulatory piece. So big pharma stepping in will probably increase the chances of success for the industry. Some Final Words Caine: Do you have an opinion on where you think Pall’s role will be in this market if you are, again, to look forward five years? Philips: I hope to be recognized as a leader in industrial manufacturing for cell therapy. Everybody goes to work and does that typically for money. But one thing I’ve learned with my employees is that cell therapy is a very motivating area. Our people like to be successful with the company because everybody can see the impact of our work on the lives of patients. The Pall retrospective in five years will be to say that we have helped world-class scientific people live their dream by offering them cost-effective, innovative technologies. That would be awesome. I think helping them get to an economical, viable manufacturing strategy has to be our role. That’s where we can make an impact. Drive cost out of it, drive risk out of it. We will benefit, of course, from that. But this is the key: Besides reimbursement, making manufacturing economical will also drive cost down the cost of current therapies. Caine: It’s a challenging but exciting place to be. How often you get to be at the early stages of a developing industry? Fairbank: It’s a good reason to get up and go into the office in the morning. It is a very exciting industry, and it’s motivating to be part of it. Philips: When people ask me, “So, Mario. What are you doing?” I very often talk about cell therapy because of the passion that these people bring to their work. They are very entrepreneurial and very scientific, and know what a significant impact their success can have on the lives of other human beings. c S. Anne Montgomery is cofounder and editor in chief, and Brian Caine is cofounder and publisher of BioProcess International, amontgomery@bioprocessintl.com; bcaine@bioprocessintl.com. Brenner Photo Productions (www.brennerphoto.com) therapies might be more affordable, but there are investors that want a return. Only therapies that really make significant impacts on the lives of patients will get reimbursed. It will depend, that is, on how we treat the disease today and what the cost is — and the cost of ownership. If you have a patient with heart disease and poor quality of life and who has to go into the hospital a lot, he might cost the healthcare system $100,000 per year. If you then come in with a fundamental fix that has a one-time cost of $100,000, that’s an easy reimbursement discussion. But there are also going to be therapies where the impact is perhaps quite good, but there is not much benefit to the healthcare system. Will those therapies be reimbursed? That is an ethical question, and there is still uncertainty in our space. But, again, really disruptive therapies will get reimbursed. Montgomery: Do you see the startup companies doing a pretty good job examining their competitive position with regard to existing therapies? Is it something they have to get better at? Philips: I think they are getting better at it. But I think that in the past, our very scientific industry was not paying much attention to market potential. The pipeline definitely has companies targeting the same diseases. But I think newer companies do scan the market to validate the areas where they can play. It’s a more mature industry. Each cell therapy needs easily $60 million to get through the clinical phases. You’re going to have to raise that capital. You’re going to have to show investors what your target markets are, where the competitors are — even among alternative competitive treatments today. The companies that we have invested in have very solid business plans including that type of analysis. Caine: If you had a crystal ball and wanted to flash forward two, three, or five years, where do you think the cell therapy market will be then? What lessons do you think you’ll look back on and say: “These were important lessons or important steps that we had to go through.” (Left to Right) Brian Caine (Publisher, BPI); Mario Philips (President, Single-Use Technologies); Alain Fairbank (Director of Marketing, Cell Therapies); Harvey Brandwein (Chief Technology Officer, Senior Vice President of Research); and Anne Montgomery (Editor in Chief, BPI) International S eptember 2015 13(8)sp BioProcess 9 S e c t i o n O n e PALL IN CELL THERAPY Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy by Jon Rowley, Eytan Abraham, Andrew Campbell, Harvey Brandwein, and Steve Oh A dherent cells such as adult primary cell lines and human multipotent (MSCs) and pluripotent stem cells (hPSCs) present a manufacturing challenge as lot sizes increase from 109 (billions) to 1012 (trillions) cells (1). Typically, manufacturing platforms are good for one log of expansion. So new methods will be required to achieve commercially relevant lot sizes. Traditional two-dimensional culture methods have been used to grow anchorage-dependent cell types. Although such methods are reliable and well defined, they are very labor intensive and limited in scale-up production potential by the available growth surface area (Table 1). Allogeneic “off-the-shelf ” therapies based on adherent-cell platforms may require manufactured lot sizes from 100 billion to a trillion cells depending on a given indication’s market size (2). Here, we examine the three platforms available for producing adherent cells — planar technologies, packed-bed systems, and suspension platforms such as microcarriers and aggregate cultures — for their potential of meeting lot requirements at different scales. As new production methods are introduced, we propose addressing downstream processing bottlenecks before they occur and introduce some large-volume downstream process technologies. Scaling of Planar Flask Cultures Adherent therapeutic cells (e.g., dermal fibroblasts, chondrocytes, and MSCs) 10 BioProcess International 13(8)sp Figure 1: Closed system manufacturing of therapeutic adherent cells in 10-layer vessels using bagged media are typically produced using planar technologies (flasks). Ten-layer vessels (Figure 1) have been used to progress several allogeneic cell therapy products into mid- to late-stage clinical development. By some estimations, planar technologies will reach lot-size limitations of 3–5 million cm2 per lot (Table 1) — capping lot sizes in the 100–400 billion cell range for most adult primary cells. Scaling up traditional flask-based culture processes from laboratory scale usually involves commercially available stacked-plate systems such as Nunc Cell Factories and Corning CellStacks. These multilayer vessels have been used for over 30 years for large-scale cell culture (3). They were first published for therapeutic dendritic cells (DCs) (4) and large-scale culture of MSCs (5) in the early 2000s. More S eptember 2015 recently, Millipore and Becton Dickinson (BD) have brought smallscale parallel-plate vessels to market. Traditional 10-layer vessels have been adopted for closed-system processing and are being used as a platform in the good manufacturing practice (GMP) production of allogeneic therapeutic adherent cells (6). The main strategy for maximizing lot size in planar vessels is to scale up the total surface area manipulated per unit operation and then scale out multiple units. Scaleup can be achieved by increasing the size and number of layers per vessel and then scaling out that unit operation to a manageable size for manufacturing. Traditional 10-layer vessels with ~6,300 cm 2 of surface area have successfully been scaled out to lot sizes of 50–70 vessels for a Sponsored Supplement Table 1: Projected lot sizes by production platform in number or unit operations, total surface area per harvest, and total cell numbers, with robotic manipulation of four large vessels in a single unit operation; cell density at harvest used in the calculations represent harvest densities at confluence for multipotent stem cells, fibroblasts, and pluripotent stem cells. Types of Planar Methods Vessels/Unit Operations Total Surface Areas (cm2) Harvest Density Cell Types (cells/cm2) MSCs 25,000 HDFs 80,000 hPSCs 160,000 Manual 10/12 Layers Low 60/60 High 100/100 372,000 620,000 9 30 60 16 50 99 total of ~400,000 cm 2 (6). These vessels were designed to be scaled-up to 40 layers (3). Robotic instrumentation has been used to manipulate four 40-layer vessels in a single manipulation — or 16-fold greater surface area per unit operation than a single 10-layer vessel. Recent innovations in Hyper technology (Corning) has tripled the surface area per unit volume of traditional multilayer vessels (7). Applying robotics to that technology could allow the manipulation of four 120-layers to achieve >240,000 cm2 per unit operation, with the potential to scale out to several million square centimeters per harvest (Table 1). Table 1 shows approximate surface area per harvest that is achievable for various flask-based vessels, with total harvest yields for typical cell types at estimated harvest densities. Harvest densities of GMP processes can vary greatly depending on media composition, cell type, and confluency at harvest. The two main variables dictating harvest size in planar culture methods are total surface area harvested per lot and cell density at harvest. Increases in lot size require maximizing both variables. Because different cell types can achieve different cell densities at harvest, we estimated lot sizes in billions of cells per harvest over various culture systems (Table 1). For adult primary cells such as MSCs, lot sizes >100 billion cells are not readily achievable with planar technologies outside of massive automation and parallel processing. Suspension technologies are required to achieve scales of 1 trillion cells per lot. Human dermal fibroblasts (HDFs) can achieve much higher densities at harvest, and lot sizes may approach 500 billion Sponsored Supplement Manual 36/40 Layers Low 40/40 High 60/60 1 million 1.5 million Robotic 36/40 Layers Low 64/16 High 80/20 1.6 million 2 million Estimated Billions of Cells Produced 25 38 40 50 80 120 128 160 160 240 256 320 using planar technologies. Human pluripotent stem cells (hPSCs) are very small and grow as tight clusters. So if more robust cell lines are developed, they may achieve 1 trillion cells per lot more readily than adult primary cells. Researchers have made several attempts to apply bioreactor control to planar culture systems, including the RepliCell (Aastrom) system (8), CellCube (Corning) cell culture unit (9), and Xpansion (Pall Life Sciences) bioreactor (10). Those systems automate many operations and monitor and control many traditional culture variables such as dO2, dCO2 , and pH. Suspension Microcarrier and Aggregate Cultures Achieving lot sizes of several hundred billion to trillions cells efficiently and cost effectively will be imperative for commercial success. Suspension culture of therapeutic cells in existing single-use bioreactor manufacturing platforms is likely to be the only way to accomplish that. Nonetheless, other strategies for adherent therapeutic cell scale-up have been investigated. One such method uses microcarriers in a bioreactor-based system. A potential benefit of using microcarriers for large-scale production is that the surface-area-to-volume ratio is greatly increased over traditional static culture processes. So cell density may be increased and the required footprint reduced. Many different types of microcarriers are commercially available. Microcarriers can be made from polystyrene such as the Hillex brand (Pall Life Sciences) or made from other materials such as collagen or dextran. Although most Robotic 120 Layers Low 64/16 High 80/20 3.84 million 4.8 million 120 384 768 150 480 960 microcarriers are spherical and smooth, others have macroporous surfaces and alternatives such as rod-shaped carriers (10). Additional technological advances include infusion of magnetic particles that may help in cell separation from beads (GEM particles from Global Cell Solutions) and chip-based microcarriers such as the µHex product (Nunc) that provide a traditional flat surface for cell growth while maintaining the high SA:V ratio of traditional microcarriers. Selecting a microcarrier for cell expansion is not a trivial task. Different properties of microcarriers may significantly affect expansion rates and cell multi- or pluripotency (11). Some surface chemistry modifications can improve cell adhesion. Such methods include applying positive or negative charges and coating with extracellular matrix proteins such as laminin or vitronectin (12). One advantage of using microcarriers is increased control of a culture’s environment in a bioreactorbased system. Technology borrowed from the development of single-use bioreactors for biopharmaceutical processes can be applied to growing therapeutic adherent cells (13). MSCs typically achieve <1 × 106 cells/mL, whereas ESCs can achieve 1 × 106 to 3 × 106 cells/mL. Using similar bioreactor-based systems of >1,000-L volume for scaling up stem cells may result in lot sizes approaching or surpassing 1 trillion cells (Table 2). Bioreactor technology offers the ability to precisely control process parameters such as gas exchange, nutrient feeding, and pH. In smallerscale systems, however, factors such as shear stress must be controlled closely because stem cells are susceptible to S eptember 2015 13(8)sp BioProcess International 11 Table 2: Achievable cell densities projected to commercial-scale lot sizes (total volumes and cell harvests) for bioreactors Methods for Producing One Lot of Stem Cells Suspension (microcarriers/aggregates) Low High Cell Density (cells/mL) 0.5 million 5 million Volumes (L) 1,000 1,000 Total Cell Harvest 500 billion 5 trillion systems can achieve homogenous cultures of high densities, but that will require fine control to maintain stem cell function as the volumes increase. Downstream Processing Table 3: Estimated harvest volumes and number of product doses per lot produced by planar and bioreactor technologies Number of Doses per Lot Harvested Volume (L) 50 million/dose 250 million/dose 30 200 40 Bioreactor Types 10-layer trays Scale 60 vessels 40 layers per rack 20 racks, 80 vessels 200 1,000 200 120 layers per rack 20 racks, 80 vessels 600 3,000 600 Bioreactor* 250 L 250 5,000 1,000 Bioreactor* 1,000 L 1,000 20,000 4,000 * Assuming a cell density of 1 million cells/mL spontaneous differentiation in an unoptimized system (14, 15). An alternative to a traditional impellerdriven bioreactor system is the XRS20 (Pall Life Sciences) that uses a single-use biocontainer and a rocking motion to agitate cell suspension. As a result, shear stress is potentially reduced. The system chosen, microcarrier, and cell culture medium will influence cell proliferation. Maintaining cell phenotype and differentiation potential is critical. Harvesting: For the biotherapeutic and vaccine markets, in which a supernatant contains the product, there is no need to separate cells from a microcarrier. In cell therapy processes, cells are the product and must be harvested. Cell harvesting and yield of microcarrier-based methods depends on efficiency of cell dissociation and separation from beads. Enzymatic treatment using commercially available recombinant animal-origin–free proteases is commonly used to remove cells from microcarriers. Using new surface chemistries that allow nonenzymatic removal of cells may increase a system’s effective yield. Tangential flow filtration (TFF) and sequential differential centrifugation techniques are options for cell harvesting but require extensive optimization and validation for processing large lot sizes (>1,000 L) to ensure that all microcarriers or particulates are removed from a cell suspension. 12 BioProcess International 13(8)sp Because many cell therapies will be administered intravenously, carryover of particulates or intact microcarriers into final products poses a serious safety risk. Using magnetic-particle infused beads can facilitate cell separation, and incorporating biodegradable and thermosensitive materials may help reduce that concern (16). An alternative approach is culturing stem cell as aggregates. The advantage is that it does not need a carrier or extracellular matrix. The key to ensuring success of this technique is using single-cell seeding and maintaining high viability (e.g., using a ROCK inhibitor) (17). Aggregate size, however, is more difficult to control, and large aggregates will suffer transport limitations. To overcome that problem, aggregates will have to be broken up or passaged every few days. But high cell-density limitations will pose a greater challenge. Studies have demonstrated suspension aggregate cultures for both pluripotent hESCs and iPSCs. Other studies have shown the same for MSCs — albeit at relatively low cell densities — and sometimes slower doubling times result (18–20). Similar to microcarrier cultures, agitation can cause cell differentiation and unstable cell growth in aggregate cultures (17). Regardless of the method used for harvest and cell concentration, robust quality control (QC) assays will be necessary to demonstrate product consistency and efficacy. So suspension S eptember 2015 As therapeutic-cell lot size is scaled from several billions to hundreds of billions (Table 1), manufacturing bottlenecks will shift to the downstream processing (DSP) areas. The cell therapy industry will need to proactively address DSP requirements so that technology is in place to accept larger lot sizes as new culture technology is implemented. Process bottlenecks will shift to two DSP process steps: volume reduction and wash, and final product filling. Volume reduction and washing process requirements will be driven by harvest volume, which is dictated by the culture platform and volumes used during harvest (Table 3). Volumes >5–10 L cannot be easily reduced using laboratory centrifugation or blood processing equipment, and scale-out is cumbersome. Scalable single-use technologies have been adapted from bioprocessing to enable presterilized, closed systems. Other technologies include process automation such as therapeutic cell TFF (21) and continuous centrifugation (e.g., from kSep Systems). Both TFF and continuous centrifugation processes are scalable from tens of liters to hundreds of liters. The kSep technology has the potential to scale up to 1,000 L processing volume. Larger lot sizes may shift bottlenecks to the final-product dosefilling step, which is driven by lot size and the number of cells per dose. Lot sizes will range from several hundred to several thousand doses per lot, requiring a shift from traditional blood bags to pharmaceutical vials and compatible filling automation. New plastic vials from West (22) and Aseptic Technologies (23) coupled with traditional pharmaceutical fill line automation will enable lot sizes in the several hundred to several thousand doses per lot. Holding times for cells in dimethyl sulfoxide will dictate process timing, Sponsored Supplement so the ability to fill several thousand vials per hour should enable lot sizes of at least three to five thousand vials per lot. The DSP manufacturing bottleneck is likely to shift depending on manufacturing platform, harvest process volumes, and product dose size. Final fill will be the manufacturing bottleneck of greatest concern at the highest culture volumes and lowest product doses where fill time may become unmanageable. Challenges Ahead As lot sizes increase from tens of billions to trillions of cells, alternatives to planar technologies will have to be considered. The two main variables dictating harvest size in planar culture methods are total surface area harvested per lot and the density of the cells at harvest. Increases in lot size will require maximizing both. Manipulation of multiple units, however, will limit the scale of this technology to one trillion cells. Packed-bed reactors can achieve very high densities (108/mL), up to 200× more than planar surface densities. The challenge is to increase volumes beyond 40 L with good process control to achieve a trillion cells. Suspension technologies such as microcarriers and aggregate cultures can already achieve densities >106/mL and potentially can scale to thousands of liters. So they are the most promising approaches for meeting lot sizes of trillions of cells. As those commercial-scale lot sizes are reached in the upstream portion of a process, bottlenecks downstream should be addressed proactively so that technology is in place to accommodate large product doses. References 1 Brandenberger R, et al. Cell Therapy Bioprocessing: Integrating Process and Product Development for the Next Generation of Biotherapeutics. BioProcess Intl. 9(3) 2011: S30– S37. 2 Kirouac D, Zandstra P. The Systematic Production of Cells for Cell Therapies. Cell Stem Cell 9 October 2008: 369–381. 3 Davis J. Medicines from Animal Cell Culture. Glyn Stacey and John Davis, Ed. John Wiley & Sons: New York, NY, 2007; 145–172. 4 Tuyaerts S, et al. Generation of Large Numbers of Dendritic Cells in a Closed System Sponsored Supplement using Cell Factories. J. Immuno. Methods 264, 2002: 135– 151. 5 Colter DC, et al. Rapid Expansion of Recycling Stem Cells in Cultures of PlasticAdherent Cells from Human Bone Marrow. PNAS 97(7) 2000: 3213–3218. 6 Rowley JA. Developing Cell Therapy Biomanufacturing Processes. Chem. Eng. Prog. November 2010, S50–S55. 7 Titus K, et al. Closed System Cell Culture Protocol Using HYPERStack Vessels with Gas Permeable Material Technology. J. Vis. Exp. (45) 2010: e2499. 8 Goltry KL, et al. Adult Stem Cell Therapies for Tissue Regeneration: Ex Vivo Expansion in an Automated System. Stem Cell Research and Therapeutics. Yanhong Shi and Dennis O. Clegg, Eds. Springer Science: New York, NY, 2008; 251–274. 9 Aunins JG, et al. Fluid Mechanics, Cell Distribution and Environment in CellCube Bioreactors. Biotechnol. Prog. 19, 2003: 2–8. 10 Oh SK, et al. Long-Term Microcarrier Suspension Cultures of Human Embryonic Stem Cells. Stem Cell Res. 2(3) 2009: 219–230. 11 Chen A, et al. Critical Microcarrier Properties Affecting the Expansion of Undifferentiated Human Embryonic Stem Cells. Stem Cell Res. 7(2) 2011: 97–111. 12 Heng BC, et al. Translating Human Embryonic Stem Cells from 2D to 3D Cultures in a Defined Medium on Lamininand Vitronectin-Coated Surfaces. Stem Cell Dev. 23 December 2011 (epublication). 13 Kehoe D, et al. Scalable StirredSuspension Bioreactor Culture of Human Pluripotent Stem Cells. Tissue Eng., Part A 16 (2) 2010: 405–421. 14 Santos FD, et al. Toward a ClinicalGrade Expansion of Mesenchymal Stem Cells from Human Sources: A Microcarrier-Based Culture System under Xeno-Free Conditions. Tissue Eng. Part C 17(12) 2011: 1201–1210. 15 Leung HW, et al. Agitation Can Induce Differentiation of Pluripotent Stem Cells in Microcarrier Cultures. Tissue Eng. Part C 17(2) 2011: 165–172. 16 Yang HS, et al. Suspension Culture of Mammalian Cells Using Thermosensitive Microcarrier That Allows Cell Detachment without Proteolytic Enzyme Treatment. Cell Transplant 19(9) 2010: 1123–1132. 17 Zweigerdt R, et al. Scalable Expansion of Human Pluripotent Stem Cells in Suspension Culture. Nat. Protoc. 6(5) 2011: 689–700. 18 Larijani MR, et al. Long-Term Maintenance of Undifferentiated Human Embryonic and Induced Pluripotent Stem Cells in Suspension. Stem Cells Dev. 20(11) 2011: 1911–1923. 19 Amit M, et al. Dynamic Suspension Culture for Scalable Expansion of Undifferentiated Human Pluripotent Stem Cells. Nat Protoc. 6(5) 2011: 572–579. 20 Bartosh TJ, et al. Aggregation of Human Mesenchymal Stromal Cells (MSCs) into 3D Spheroids Enhances Their Antiinflammatory Properties. Proc. Natl. Acad. Sci. 107(31) 2010: 13724–1379. 21 Pattasseril J, Rowley JA. High Shear Rates Negatively Affect Cell Viability and Final Product Quality in TFF Processing (poster). Society for Biological Engineering Biannual Meeting on Stem Cell Engineering, Boston, MA, 2010. 22 Woods EJ, et al. Container System for Enabling Commercial Production of Cryopreserved Cell Therapy Products. Regen. Med. 5(4) 2010: 659–667. 23 Thilly J, Conrad D, Vandecasserie C. Aseptic Filling of Closed, Ready to Fill Containers. Pharm. Eng. 26(2) 2006: 1–6. c At the time of this article’s original publication, Jon Rowley, PhD, was innovation director, cell processing technologies, at Lonza Walkersville. Eytan Abraham, PhD was 3D cell culture research and development manager at Pluristem Therapeutics. Andrew Campbell was senior manager of PD direct services at Life Technologies. Harvey Brandwein was vice president at Pall Life Sciences. Corresponding author Steve Oh is associate director and principal scientist at Bioprocessing Technology Institute; steve_ oh@bti.a-star.edu.sg. This article was first published in BioProcess International 10(3)s 2012: 16–22. S eptember 2015 13(8)sp BioProcess International 13 S e c t i o n T w o PROCESS DEVELOPMENT T-Cell Suspension Culture in a 24-Well Microbioreactor High-Throughput Screening of Operating Conditions by Kenny Choi, Jason N. Carstens, and Shelly Heimfeld C ell therapy promises revolutionary new therapeutic treatments for cancer and other serious diseases and injuries. For example, T-cell therapy response rates of >50% and durable complete response rates of 20% have been reported in patients with metastatic melanoma who had failed other therapies (1). In another example, sustained remissions of up to a year were achieved among a small group of advanced chronic lymphocytic leukemia patients upon treatment with autologous T-cells expressing an anti-CD19 chimeric antigen receptor (2). Numerous other examples use cell therapy for cardiac repair, bone or cartilage regeneration, organ repair (pancreas or liver), neurological repair (spinal cord or brain injury), correcting genetic defects, and treating infectious diseases such as human immunodeficiency virus (HIV) (3). Such therapies involve the ex vivo expansion and manipulation of different cell types including stem cells and T-cells. Because many are still in early clinical research phases, often their manufacturing processes are based on research-scale methods using static flasks and bags, with very limited process monitoring and control. Although such methods have been manageable for enabling phase 1 and 2 clinical trials treat a small number of patients per month, the need to produce tens of billions of cells per subject significantly diminishes the practicality of static processes. This is especially true when you take into 14 BioProcess International 13(8)sp Figure 1: The Micro-24 microbioreactor is a 24-well agitated reactor system; each well has individual pH, DO, and temperature control. Controller Microbioreactor Sterile membrane pH sensing material Temp. Thermal sensor conductor Optical pH sensor consideration the future prospects of a manufacturing facility that will provide treatments for hundreds of patients at any given time. Static cellculture processes often require a vast array of small-volume vessels, which are both labor intensive and present risks in aseptic fluid handling. Furthermore, static systems are prone S eptember 2015 DO sensing material Heater Dilute CO2 lowers pH Thermal conductor Purge Gas O2/Air to raise DO Optical DO sensor to mass-transfer limitations due to their heterogeneity. They do not offer the capacity to easily monitor and control physical system parameters such as dissolved oxygen (DO), pH, and temperature — variables that almost certainly influence cell culture expansion and the critical quality attributes of a cellular product. Sponsored Supplement Figure 2: Comparing theoretical DO concentration delivered to the microbioreactor with the average steady state on-line value; the off-line values were determined by running samples on a blood-gas analyzer. Figure 3: Comparing off-line and on-line pH measurements Off-line pH (GEM 3000) 8.0 Measured DO (% air saturation) 100 80 60 40 Average on-line Micro-24 DO 20 0 Off-line DO Theoretical DO 0 20 40 60 The current state of cell therapy manufacturing is similar to that of therapeutic protein manufacturing of 30 years ago. That industry also commonly relied on static culture systems and lacked bioprocess knowhow, well-defined raw materials, sensitive analytical methods, and sophisticated equipment that would permit precise control over the process and therapeutic product. Over decades, the therapeutic protein manufacturing industry evolved, particularly with advances eliminating the need for static culture and instead permitting manufacture of product in suspension culture using fed-batch or perfusion stirred-tank bioreactors. The body of knowledge around bioreactor scaling issues, media composition, and feed strategy has grown considerably over the past few decades, allowing for development of more productive and efficient cell culture manufacturing processes. In addition, improved monitoring and process control capabilities of bioreactors have continually helped to progress the development of increasingly robust and reproducible processes. Finally, the introduction of disposable bioreactors and associated components has reduced costs without compromising aseptic processing and final-product quality. The cell therapy industry faces unique challenges unlike those in protein manufacturing. Nonetheless, many tools originally developed to support therapeutic protein development and manufacturing can be adapted to cell therapy applications. One such technology is the Micro-24 microbioreactor from Sponsored Supplement 80 Theoretical DO (% air saturation) y = 0.9811x + 0.1853 R2 = 0.8834 7.8 7.6 7.4 7.2 7.0 6.8 6.8 100 7.0 Pall Life Sciences, a 24-well bioreactor with the capability for agitated suspension culture and continuous monitoring and control of pH, temperature, and DO in each individual well (Figure 1). The small operating volume (3–7 mL) is well suited to cell therapy applications, in which patient-specific starting cells are in short supply and culture media may contain very expensive growth factors. Finally, the system already has a successful history as a scaledown model for culturing mammalian cells in stirred-tank bioreactors. Because this microbioreactor system had never before been used in a cell therapy application, our laboratory set out to evaluate its usefulness as a process development tool supporting efforts to develop and optimize suspension cell culture for manufacturing therapeutic T-cells. As a first step, we qualified the equipment for use by demonstrating both its ability to accurately measure and control process parameters and its reproducibility in cell growth. Then we evaluated the system for its ability to serve as a scale-down model compared with a 3-L stirred-tank bioreactor. 7.2 7.4 On-line pH (Micro-24) 7.6 7.8 Materials and Methods To validate the precision of on-line DO readings, a range of known oxygen concentrations (from 0 to 100%) were delivered by purging to each bioreactor well with a rotometergoverned blend of oxygen, carbon dioxide, and nitrogen. Using the onboard Micro-24 controls, we set the purge flow rate at 5.0 sccm, the platform agitation at 800 rpm, and the temperature at 37.0 °C. Each well received 5.0 mL of a cell culture medium based on the familiar Roswell Park Memorial Institute (RPMI) medium. For comparative analysis, we took off-line DO readings with a GEM 3000 blood-gas analyzer from Instrumentation Laboratory. That involved removing samples from the microbioreactor using a syringe and needle to puncture the cap, then introducing them to the gas analyzer as rapidly as possible in hopes of minimizing changes in the dissolvedgas concentration. For qualification of on-line pH readings, we performed a retrospective analysis by taking samples during the course of several T-cell culture experiments. We measured the offline pH using the GEM 3000 bloodgas analyzer and compared the Table 1: An example of the instantaneous on-line dissolved oxygen (DO) reading reported in each microbioreactor well; theoretical oxygen concentration is 11.2%. Online DO Readings of Cassette (%) A 1 2 3 4 5 6 16 15 15 15 14 14 B 14 14 16 15 15 13 C 16 16 16 16 16 15 D 13 13 14 13 15 15 Average DO reading of cassette 14.75%, standard deviation 1.07% S eptember 2015 13(8)sp BioProcess International 15 Figure 4: Viable cell density (VCD) over time under the same growth conditions in each well VCD (106/mL) 10 9 8 7 6 5 4 3 2 1 0 8 11 13 Day It is sometimes desirable to maintain certain cell types under hypoxic conditions or otherwise ensure that conditions remain CONSTANT while the microbioreactor is manipulated. resulting values with the Micro-24 on-line readings. We’d set the temperature, agitation, and purge rates at the same values as in our dissolved oxygen qualification experiments. We qualified the cell culture growth performance capabilities of the Micro-24 system using a polyclonal antigen-specific CD8 T-cell line. The T-cells were stimulated under static conditions using irradiated PBMC feeder cells for four days before we transferred them to suspension culture in the microbioreactor. In experiments to assess well-to-well reproducibility, we inoculated all 24 wells identically with a pH set point of 7.0, a DO setting of 25% air saturation, a temperature of 37 °C, and an agitation rate of 500 rpm. When comparing the Micro-24 system with a 3-L stirredtank reactor, we inoculated both from the same starting culture and operated them under similar conditions. Results and Conclusions DO Qualification: From preliminary experiments using a nitrogen purge, we observed that with a constant purge-gas flow rate of 1.0–2.0 sccm it 16 BioProcess International 13(8)sp Table 2: Means and standard deviations from Figure 4 Day 8 11 13 Number 24 24 24 Mean 1.91625 4.83750 6.74792 Standard Deviation 0.34810 0.71843 1.22061 was impossible to drive the DO reading to 0% in any of the wells. In fact, the lowest DO levels we achieved were 10–15%. Presumably that was due to oxygen leaking into the system through the bottom seal or elsewhere in the gas-delivery system. However, as we increased the purge-gas flow rate to >3.0 sccm, the system reached a 0% DO reading in all wells. So we chose a standard operating condition of 5.0 sccm for purge gas. Next we wanted to confirm that the microbioreactor would be capable of maintaining integrity under closed conditions. First, to demonstrate how the system would respond when purposefully breached, we showed that under low-oxygen conditions it would rapidly reabsorb oxygen when exposed to open atmosphere. For example, after 800-rpm agitation under a pure nitrogen purge (with all wells reading 0% oxygen), when agitation was stopped and a well cap was removed for 30 seconds (and then replaced), the DO reading increased by 15–20% almost immediately. When we conducted the same experiment at a DO setting of about 50%, the reading increased by about 5% — not a surprising result when you consider the reduced driving force for oxygen mass transfer. We then demonstrated that the closed system could maintain its integrity when left sealed and closed. Under the same starting conditions — 800 rpm and pure nitrogen purge with all wells uniformly reading 0% DO — the capped plate was unclamped from its platform and placed on the laboratory bench for 120 seconds. After the plate was reclamped and returned to normal agitated operating conditions, all wells had increased their dissolved oxygen levels by only 2–3%, indicating that this microbioreactor could generally maintain a closed, leak-tight system. This feature is important because it is sometimes S eptember 2015 Lower 95% 1.7693 4.5341 6.2325 Upper 95% 2.0632 5.1409 7.2633 desirable to maintain certain cell types under hypoxic conditions or otherwise ensure that conditions remain constant while the microbioreactor is manipulated. Next, we evaluated the precision and accuracy of oxygen readings using a rotometer manifold that delivered a gas mixture of known oxygen composition (0–100%) by purging. We allowed the system to achieve a steady-state operation condition over a period of at least 30 minutes. In an effort to identify performance changes that might occur over time, we performed this evaluation over two weeks. At each setting, we recorded the on-line DO in each well and calculated the average and standard deviation. In all cases, the well-to-well deviation was <3%. Table 1 provides example data collected from each well at a DO setting of 11.2%. Figure 2 compares the known DO concentration delivered to the microbioreactor with the average on-line DO reading in each well. In general, there was excellent agreement between the on-line DO reading and the theoretical value of the known DO. In addition, we also analyzed offline samples with the blood-gas analyzer. Despite our efforts to place samples on the analyzer as rapidly as possible and minimize their atmospheric exposure, reabsorption of ambient oxygen into the samples produced artificially high DO measurements (a more prominent effect at lower DO conditions). These results demonstrated acceptable accuracy and precision of the DO readings. In fact, the on-line instrument readings should be viewed with greater confidence than off-line samples tested with the bloodgas analyzer. pH Qualification: During a threemonth period, we performed several cell culture experiments using T-cells in an RPMI-based medium. At intervals, we pulled samples for offline measurement, including for pH Sponsored Supplement Sponsored Supplement pH 7.1, DO 40% pH 7.0, DO 10% 5 4 3 2 1 0 pH 6.8, DO 40% 30 Fold Expansion (×100) Cell Density (106/mL) 6 pH 7.0, DO 40% pH 6.8, DO 10% 0 5 Run Days 10 25 20 15 10 5 0 15 0 5 Run Days 10 15 Figure 6: Lactate and glucose concentration profiles 2.0 1.8 1.6 1.4 1.2 1.0 0.8 0.6 0.4 0.2 0.0 2.5 pH 6.8 3-L bioreactor pH 7.1 pH 7.0 pH 6.8 2.0 pH 7.1 pH 7.0 3-L bioreactor 0 5 Run Days 10 15 be aimed at optimizing the media composition and feed strategy. More important to the purposes of equipment qualification, the information provides confidence in the scalability of results from the microbioreactor to the stirredtank bioreactor. A Process Development Tool Glucose (g/L) using the GEM analyzer. Whenever a sample was pulled, we also recorded the instantaneous on-line pH measurement for that particular well. From a retrospective analysis, Figure 3 compares on-line and off-line pH measurements. The 0.98 slope shows excellent agreement between those measurements at the given pH values. Cell Culture Qualification: We initiated a CD8 T-cell culture in the microbioreactor by inoculating and operating each well under identical conditions. As Figure 4 shows, cells successfully expanded over a 13-day period with a reasonably comparable cell density in each well (day 13 mean of 6.75 million cells/mL, standard deviation of 1.2 million cells/mL). The total cell number expanded an average of 1,850-fold and exhibited exponential growth through day 11, with an average doubling time of 24.2 hours. Those results were consistent with what we observed in larger-scale bioreactors. Figure 5 illustrates a second cell culture experiment, in which we inoculated a 3-L stirred-tank bioreactor and the Micro-24 system using the same seed culture. Here we operated the microbioreactor in a matrix with three pH set points (6.8, 7.0, and 7.1) and two DO set points (10% and 40%). Observed cell densities and foldexpansion values were reasonably similar in all cases, with data suggesting that perhaps the pH 7.0 condition might be best. Occasionally we sampled the cultures for their glucose and lacticacid concentrations (Figure 6), with data demonstrating that glucose was becoming depleted. Those results suggest that future experiments should 3-L bioreactor pH 7.1, DO 10% Lactate (g/L) More important to the purposes of equipment qualification, the information provides CONFIDENCE in the scalability of results from the microbioreactor to the stirred-tank bioreactor. Figure 5: Comparing cell growth in a 3-L stirred-tank bioreactor and in the microbioreactor at three different pH set points and two DO set points Our instrument qualification results show that the Micro-24 microbioreactor can be used as an accurate, high-throughput experimental system that properly represents production-scale systems. The system offers a sufficient degree of accuracy and precision in its on-line DO and pH measurements and demonstrates a reasonable degree of well-to-well reproducibility. Furthermore, cell growth rates and metabolic profiles are similar to what we observed with a 3-L stirred-tank bioreactor. Our results suggest that the Micro-24 system is suitable as a scale-down model for high-throughput evaluation of operating parameters for therapeutic cell culture. 1.5 1.0 0.5 0.0 0 5 10 Run Days 15 donation from the Bezos Family Foundation and NIH grants P30 DK56465, P01 CA18029, and P30 CA15704. References 1 Rosenberg SA. Durable Complete Response in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 17(13) 2011: 4550–4557. 2 June CH. Chimeric Antigen ReceptorModified T Cells in Chronic Lymphoid Leukemia. NEJM 365, 2011: 725–733. 3 Trounson A. Clinical Trials for Stem Cell Therapies. BMC Medicine 9, 2011: 52. c Kenny Choi is a research technician; corresponding author Jason N. Carstens, PhD, is director of process development; and Shelly Heimfeld, PhD, is scientific director of the cellular therapy and cell processing facilities — all in the clinical research division at Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, MS D5-100, Seattle, WA 98109-1024; 1-206-667-6131; jcarsten@ fhcrc.org. This article first appeared in BPI’s April 2013 issue. Acknowledgments This work was supported by a generous S eptember 2015 13(8)sp BioProcess International 17 S e c t i o n T w o PROCESS DEVELOPMENT Scaling Up Stem Cells Moving from Laboratory to Commercial Production with a Single-Use Multiplate Bioreactor by Matthieu Egloff and Jose Castillo, updated by Thierry Bovy C ell-based products are becoming increasingly important as potential biotherapies. Cell therapy is predicted to have a huge impact on the healthcare sector over the coming decades. Stem cells, in particular, are investigated as potential treatments for a diverse range of applications (such as heart disease and metabolic and inflammatory disorders) in which they might be used to restore lost biological functions. The cell therapy industry is starting to mature. Several emerging companies are now supporting latestage clinical trials, and stem cellbased products should soon appear on the market. However, potential commercial success for such products is linked to the ability of sponsor organizations to industrialize manufacturing processes for ensuring cell supply and managing costs. Successful transition from laboratory scale, which is suitable for producing just a few batches per year, to an efficient and robust good manufacturing practice (GMP) process will be essential. If the economic model is to be viable, and if health authorities and insurers are to reimburse these products, then prices must be controlled. That translates to scaling out for autologous therapies and scaling up for allogeneics. The research and development (R&D) process is based on currently available laboratory-scale technology, in which multiple-tray stacks are used to culture cells. This method is not practical for large-scale production, 18 BioProcess International 13(8)sp however, because it requires multiple manual aseptic operations, which can present problems for consistency, reproducibility, and safety as well as quality assurance and control (QA and QC). The multitray option is also expensive — perhaps prohibitively so. Although the cost of purchasing a stack is not always prohibitive, the cost of running it can be expensive, costly both financially and in labor. That was the problem facing a biotech company working on a cellbased therapy to protect patients’ hearts during myocardial injuries. The developmental autologous therapy uses cardiopoietic cells made from a patient’s bone marrow stem cells, then injected into the patient’s heart. Following successful phase 2 trials, the company then planned to advance into phase 3 studies. This required an effective, efficient, and practical method for growing batches of autologous stem cells for more patients. ATMI, later acquired by Pall Corporation, was approached to develop a single-container solution S eptember 2015 Figure 1: An Xpansion plate with 614 cm2 available for cell growth that might solve its scale-up problem. The company previously used a multitray stack to grow cells in a laboratory. For each patient, 20 such devices were required, with operations during the cell-production process carried out under laminar flow. That made scaling up/out the process very Sponsored Supplement difficult because the company’s objective was to treat approximately 3,000 patients per year in the early stages of commercialization. Using the laboratory process, that would have required some 200 aseptic operations per batch (2,000 aseptic operations per day). Achieving a current GMP (cGMP) process also would have been difficult. Coupled with the sheer space and number of operators required, this made it nearly impossible to create a viable and efficient business model. Simulations demonstrated that this company would have needed a 5,000‑m 2 facility equipped with 500 incubators and 160 biocabinets, employing 300 operators in numerous culture rooms. What our customer needed was a cost-efficient process that cultured high-quality cells to GMP standards on the necessary scale. For safety reasons, a closed system was required to guarantee sterility. The process also needed to be easily controlled to ensure reproducibility. Cost efficiency could be achieved by simplifying and reducing the number of operations (and operators) involved. Figure 2: (left) multitray stack 10; (right) 10 Xpansion plates Figure 3: (left) 20-multitray stack 10; (right) 200-plate Xpansion unit A Lack of Suitable Solutions Single-use bioreactors are well established in biopharmaceutical production, for which they offer many benefits. Not only do they simplify QA and QC functions, but they also reduce overall costs and provide a flexible solution for manufacturing. However, most existing disposable bioreactors are designed for viral or protein production. Fragile adherent cells (such as stem cells) are highly sensitive to the physical parameters of their microenvironment. Factors such as the surface material, pH, dissolved oxygen DO), and shear stress all affect the way these cells grow and differentiate. And harvesting the cells is not trivial. It might be possible to use a standard bioreactor in combination with microcarriers to provide a surface area on which such cells need to grow. But those have not yet been optimized for stem cell culture. Moreover, shear stress might affect stem cell cultures and require intensive process development. A three-dimensional Sponsored Supplement (3D) scaffold might be a potential alternative, but its configuration could affect stem cell behavior (and differentiation) by modifying the niche microenvironment. There is no guarantee that cells grown in any 3D bioreactor would be the same as those grown on a laboratory plate. Developing a Specific Solution All of these problems could be prevented with a new multiplate design approach. This would mimic the cell-growth environment of a multitray stack and minimize risks encountered during process development. Cells would still grow on two-dimensional (2D) plates with similar physical characteristics to those used in traditional multiplate systems. The Xpansion® bioreactor was designed with plates made from the same plastic (polystyrene) as is used in multitray stacks. After a common plasma treatment used to hydrophilize the plastic, studies show that the surface (Figure 1) was very similar to that of a comparator. It was also important to reduce the size of the device. A compact design was created by removing the gas-phase layer that allows gas exchange into media in which cells are growing. Gas exchange is essential, but the Xpansion bioreactor allows it to take place within a central column in which medium circulates (rather than between the plates). That reduces the gap between plates from 15 mm to just 1.6 mm, enabling ≤200 plates to fit in a single device about 80 cm tall (Figure 2 and 3). The surface of each Xpansion plate is ~614 cm 2, roughly equivalent to each plate of a multitray stack. So one 200-plate bioreactor has the same cellgrowing capacity as 20 stacks (122,400 cm 2) with a much smaller footprint than they would require. These circular plates contain 16 radial channels to circulate media. Liquid moves up through the first channel, flows horizontally over a plate, then rises to reach a second plate, and so on through all plates until it reaches the top of the reactor. It is then recirculated and returned to the bottom of the stack — a design optimized to minimize shear stress on cells. The culture operates similarly to multitray stack cultures without their need for complex manual manipulation. And there is only one device to operate, not many. Other technological improvements involved regulation and control of cell S eptember 2015 13(8)sp BioProcess International 19 culture parameters. An Xpansion bioreactor can be operated as a fully closed system, guaranteeing safety and sterility during a cell culture process. In addition to temperature monitoring, pH and dissolved oxygen patches were added to each plate. Combined with sensors on a unit’s head plate, they enable monitoring and control of gas exchange within the bioreactor and maintenance of correct pH and DO. Throughout a process, the precise environment in which cells are growing can be regulated and controlled, which is particularly important for fragile stem cells that are very sensitive to their environment. This will greatly increase process reproducibility. For autologous cells, it is important to remember that each batch will be different. Stem cells harvested from one patient will react differently from those harvested from another, so environmental control is a necessity. Another important feature of the Xpansion system allows operators to observe cells as they grow within the bioreactor. Cell density can be calculated automatically, and cell morphology can be checked. Specialized light microscopy developed by Ovizio enables cells to be observed on multiple layers of the bioreactor with very high image quality. This is also important for autologous cells. Those from one patient might take four days to reach confluence, whereas cells from another could take a week — and that is unpredictable. Proper monitoring of their growth will ensure that cells are harvested at the optimum time (Figure 4). Scale-Up Advantages The biggest advantage of this new single-use bioreactor is that switching to it from multilayer stacks does not affect the quality or nature of the cells. That makes it possible to speed up the scaling process while decreasing risks. It eliminates the need to start from scratch and develop a new 3D manufacturing process in a traditional cell culture bioreactor, which would not be guaranteed to give 20 BioProcess International 13(8)sp Figure 4: Bone-marrow mesenchymal stem cells cultured on an Xpansion plate (shown by an Ovizio microscope) the correct cell morphology. No aseptic operations are required (compared with 2,000 such procedures each day using previous technologies), and both space and operator requirements are cut by >60%. Only a class C cleanroom is required for transfer of cells into the bioreactor because no open handling takes place. And that is much less expensive to install than the class B room needed for multilayer stacks. Reducing bioreactor and facility footprint enables economically feasible commercial-scale production. Scale-up from the multitray stack process to one supporting several thousand patients would have been impractical in space and operator requirements. The Xpansion system makes that possible with a significant reduction in both those parameters. A quick calculation of the number of batches and patients indicates that 300 operators would have been required for a multitray stack scale-up; a validated cost simulation performed with two different customers showed that the number could be halved with an Xpansion system. The potential cost savings are dramatic. We calculate that for an autologous cell therapy to treat 3,000 patients per year, the annual operational expenses would be reduced by 40%. There are also benefits in capital investment. The number of operations that must to be carried out under laminar flow is significantly reduced. With a smaller footprint, fewer incubators are required. These cost savings are important because cellbased therapies are predominantly the domain of small biotech companies with limited access to capital. They S eptember 2015 can’t risk investing several million dollars in constructing new manufacturing facilities. And commercialization becomes a more realistic prospect when capital expenditure requirements are reduced by 50%. Using a close, compact, single-use, multiplate bioreactor provides a realistic solution to the problem of scaling up a fragile, adherent-cell manufacturing process — guaranteeing that stem cells retain the quality and morphology of those grown in an R&D laboratory. This would not otherwise be possible at a commercial scale without prohibitive investment and running costs. Further Reading Placzek MR, et al. Stem Cell Bioprocessing: Fundamentals and Principles. J. R. Soc. Interface 6, 2009: 209–232. Rowley JA. Developing Cell Therapy Biomanufacturing Processes. Stem Cell Eng. 106(11) 2010: S50–S55; www.aiche.org/ uploadedFiles/SBE/Restricted/ SBEOnlyNew/111050.pdf. Rowley JA, et al. Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy. BioProcess Int. 10(3) 2012: S16–S22. At the time of this writing, Matthieu Egloff was product manager, and Jose Castillo was director of cell culture at Pall Life Sciences. Thierry Bovy is global product manager, Xpansion multiplate bioreactor systems, Pall Life Sciences, Brussels, Belgium. Xpansion is a trademark or registered trademark in the United States, other countries, or both. Ovizio is a trademark of Ovizio Imaging Systems SA. Other names are trademarks of their respective companies. An earlier version of this article was published in BPI’s May 2012 Cell Therapy supplement. Sponsored Supplement Featured Products and Services SoloHill® Microcarrier Training Courses Accelerator Development Services Applications: Bioprocess consulting, process development, and qualification Features: Pall Life Sciences supports customers with an experienced team of scientists and engineers — from on-site guidance to larger projects in its own process development laboratories. Speed-to-clinic and -market are primary drivers of research timelines. Often, quickly developed but suboptimal processes are commercialized, placing long-term burdens on future campaigns. Modernization opportunities are lost when researchers are pressured to develop processes using dated methods. Even experienced scientists can feel the impact of learning curves when presented with new technologies or methods. Unknowns can discourage innovative approaches in process development. Incorporating a learning curve into a project plan reduces resources available for optimization and other necessary tasks. Pall process development services can demystify the adoption of new technologies. This removes the burden of learning curve and provides a streamlined path to the most modern bioreactors and methods available. Clear timelines, targeted expertise, and process transfer support help clients to successfully meet aggressive timelines. Contact Pall Life Sciences Accelerator@pall.com PadReactor® Single-Use Bioreactors Features: SoloHill microcarrier training courses are offered at Pall’s sites in Ann Arbor, MI (USA), and Portsmouth, England (UK). Each course can be customized to your specific needs, focusing on the following: Applications: Flexible for suspensionadapted and adherent cell cultures Features: The PadReactor family of single-use bioreactors is designed for cell culture applications. These systems are linearly scalable (5–1,000 L) and suited from laboratory-scale process development to large-scale GMP manufacturing. They are proven to be effective for cultivation of suspended cells and adherent cells on microcarriers (e.g., Pall SoloHill® microcarriers). • Adapting flatware and roller bottle processes to microcarriers • Handling microcarriers and optimizing attachment conditions • Small-scale microcarrier processes (spinner flasks) A PadReactor unit incorporates a cubeshaped biocontainer design with a highly efficient mixing paddle and dynamic sparger for efficient low-speed mixing to minimize shear stress. High O2 supply and CO2 stripping through dynamic sparging supports high cell densities. Paddle and sparger are enclosed in a sleeve made from the same medical-grade ultralow-density polyethylene (ULDPE) material as the biocontainer itself. The flexible drive unit is compatible with different biocontainer sizes and a mobile retaining tank supports the biocontainer and provides mobility. A user-friendly design allows short set-up times. The fully transparent front cover ensures easy cell culture observation during cultivation and harvest. An available controller provides for a turnkey system, but an openarchitecture option allows use with any type of controller. Contact Pall Life Sciences bioreactors@pall.com Sponsored Supplement Applications: Promotes efficient user acquisition of microcarrier operational skills • Glass bioreactor microcarrier processes (2–15 L) • Single-use microcarrier processes (2–20 L) • Process optimization strategies • Microscopic imaging techniques (fluorescent and phase-contrast) • Sterile handling techniques. Pall’s training team works with you to determine your desired areas of focus as well as the appropriate level of instruction required. The trainers’ dedication to curriculum design ensures that experienced cell culture scientists and novices alike will come away from each training course with new confidence to optimize microcarrier culture conditions. Courses are led by experienced PhD- and MS-level scientists trained to discuss your specific process challenges and potential solutions. Contact Pall Life Sciences microcarriers@pall.com S eptember 2015 13(8)sp BioProcess International 21 S e c t i o n T w o PROCESS DEVELOPMENT Expansion and Characterization of Mesenchymal Stem Cells on Pall SoloHill® Microcarriers by Heather Woolls, Dave Splan, and Mark Szczypka M esenchymal stem cells (MSCs) are self-renewing cells that differentiate into several terminally differentiated cell types. These cells have been isolated from multiple sources such as bone marrow, adipose tissue, peripheral blood, and other adult tissues(1-6). The interest in these cells is that they hold the potential to cure disease and are being pursued in clinical trials. Three emerging fields of interest for stem cells are cell therapy, regenerative medicine and screening of candidate drugs. In many cases, poor correlation between efficacy of candidate drugs in animal models and humans is observed. This leads to high attrition rates of candidate drugs from the developmental pipeline and also contributes to large losses in revenue spent on animal model testing. The ability to isolate, expand, and differentiate human stem cells in vitro will streamline drug testing by allowing candidate drug testing on human cells at early stages thereby better predicting how human populations may react to new and developing drugs. It is hoped that the ability to reproducibly isolate and expand these cell types will facilitate 22 BioProcess International 13(8)sp the identification of candidate drugs earlier in the development process. In addition, the ability to differentiate stem cells into various cell lines should allow for more relevant toxicity testing. These achievements should ultimately lead to overall cost savings and decreased health risks in the future. In addition to drug product testing, several clinical trials have been initiated using stem cells in cell therapy treatments. Research has shown stem cell characteristics such as differentiation potential, angiogenic potential, immunosuppression, or immune-privilege may be effective in the treatment of many diseases. Clinical trials using stem cells for the treatment of osteoarthritis, spinal cord injuries, Parkinson’s disease, ischemia due to stroke, cardiac arrests, or diabetes, are seeing promising results. However, for toxicology screening and cell therapy applications, large numbers of cells are needed. Expansion of adult stem cells is difficult since they have a finite life span and pluripotency can be lost. Two-dimensional (2D) culture systems such as T-flasks, cell cubes/factories, and roller bottles are common production platforms for vaccine and S eptember 2015 Mesenchymal stem cells were seeded at low density and expanded for eight days on Pall SoloHill’s Plastic microcarrier. Cells were stained with DAPI (blue) and FITC-labeled phalloidin (green) for visualization. biologics manufacturing as well as cell therapy. These systems are typically used for expansion of cells to seed large bioreactors. Although wellestablished, these formats occupy a large footprint, are labor intensive, and are susceptible to contamination problems due to numerous open handling steps. Microcarriers offer a large surface area for growth of anchorage-dependent cell types, and could thereby facilitate use of bioreactors for stem cell expansion in fewer passages. Herein we characterized MSC expansion on flatware and five Pall SoloHill microcarriers (Collagen, C102-1521; Plastic, P102-1521; Plastic Sponsored Supplement Plus, PP102-1521; Pronectin F, PF102-1521; and Hillex II®, H112‑170) in stirred vessels. Retention of multipotency of the MSCs expanded in stirred culture was verified by immunostaining with stem cell specific antibodies and by assessing their ability to differentiate into osteocytes and adipocytes. Materials and Methods Culturing of MSCs: Human bone marrow-derived MSCs (Passage 1) were purchased from EMD Millipore (SCR108) and expanded in spinners or on flatware in DMEM (GIBCO 11054) supplemented with 10% fetal bovine serum (FBS) (HyClone SH30071.03), 2 mM l-glutamine (HyClone SH30034.02), penicillin/ streptomycin (ATCC 30-2300), and basic Fibroblast Growth Factor (bFGF) (EMD Millipore GF003). Unless otherwise noted, medium refers to this complete formulation. For growth experiments on flatware, MSCs were cultured on Corning T-flasks (430825, 430639, and 430641). To subculture cells, medium was decanted and cells were rinsed once with Dulbecco’s phosphate buffered saline (DPBS; HyClone SH30028.03). The DPBS was immediately decanted and 1–3 mL of TrypLE Select enzyme (Life Technologies 12563) was added (depending on T-flask size). Flasks were incubated at 37 °C until cells detached (5–8 minutes). The cells were resuspended with medium and then centrifuged at 300g for five minutes to pellet the cells. Medium and trypsin were decanted. Cells were resuspended in 2–5 mL plus 10% FBS but without bFGF (volume depends on T-flask size and number) and counted using trypan blue stain and a Nexcelom Cellometer with associated software. Fresh T-flasks were seeded at 3 × 103 cells/ cm2. Fresh bFGF was added (8 ng/mL) to seeded T-flasks, which were then incubated at 37 °C ± 0.5 with 5% CO2 in complete medium. 100% media exchanges (with fresh bFGF) were performed every other day beginning on the second day of culture. Incubation: For initial microcarrier attachment studies 200,000 cells were Sponsored Supplement MICROCARRIERS offer a large surface area for growth of anchorage-dependent cell types, and could thereby facilitate use of bioreactors for stem cell expansion in fewer passages seeded onto the equivalent of seven cm 2 of each microcarrier type. Although this seeding density of ~3 × 104 cells/cm 2 was higher than anything used subsequently, this density was used to provide enough cells for counting and visualization on the microcarriers. Cells were incubated with microcarriers in 1 mL of medium (either ± FBS) in 1.5 mL Eppendorf tubes at 37 °C ± 0.5 with 5% CO2. At various time points, tubes were removed from the incubator and microcarriers were allowed to settle. Cell counting: 20-µL samples of the supernatant were taken for counting on the Nexcelom counter. Time courses for percent cells attached versus unattached were determined for each condition and plotted (Figure 2). For growth experiments on the various Pall SoloHill microcarriers, 0.5 g of microcarriers were used per 50 mL of medium in each Corning brand 125 mL spinner vessel (Fisher Scientific 10-203B). All microcarriers were prepared according to manufacturer’s instructions by autoclaving at 121 °C in deionized water. Spinner cultures were essentially performed as described in a previous microcarrier protocol (7). Briefly, spinners were seeded in complete medium low protein concentration (less than 0.25% FBS and no bFGF) for 30 minutes for initial attachment. After 30 minutes >85% of cells had attached to the microcarriers. Final protein concentrations of 10% FBS and 8ng/μL bFGF were added slowly to prevent any osmotic shock from the serum. The spinners were incubated at 37 °C ± 0.5 with 5% CO2. Cell counts were performed using standard assays to quantify cell numbers and determine viability. Spinners containing cells on Hillex II microcarriers were grown at 60 rpm, whereas all other microcarrier spinners were kept at 40 rpm. Media exchanges of 25 mL (50% volume) were performed every other day beginning on the second day of culture. Samples were retrieved daily for nuclei counts using the citric acid/ crystal violet method. Nuclei were counted using the Nexcelom counter. The number of nuclei per cm 2 surface area was calculated for each sample. Trypsinization: For trypsinization of cells from microcarriers, cells/ microcarriers were allowed to settle and medium was removed. Cells and microcarriers were washed with DPBS for five minutes at room temperature with occasional rocking back and forth by hand to resuspend microcarriers. After five minutes, the microcarriers were allowed to settle, and the DPBS was removed. Five mL of TrypLE Select was added. The spinners were gently pipetted once or twice to thoroughly mix and then incubated at 37 °C for 10–15 minutes (with occasional rocking by hand). Cells and microcarriers were pipetted after five minutes and again after ten minutes to achieve a single-cell suspension that could be used to reseed fresh microcarriers. Visualization: To visualize the expression of several stem cell markers on MSCs expanded on Pall SoloHill microcarriers, samples were transferred from the spinners into 15-mL tubes. Once the microcarriers settled, medium was removed and cells/microcarriers were carefully washed with DPBS for five minutes at room temperature. Once cells and microcarriers settled, DPBS was removed and cells and microcarriers were fixed in 4% paraformaldehyde for ten minutes at room temperature. The paraformaldehyde was removed and cells were washed in DPBS and stored at 4 °C until use. To visualize stem cell markers, 250 µL of each sample was transferred to a 1.5 mL tube, microcarriers settled and DPBS removed. Nonspecific binding was blocked by incubation with 5% FBS in S eptember 2015 13(8)sp BioProcess International 23 Figure 1: MSC growth curve on flatware; cells seeded at 3 × 103 cells/cm2 were grown for 10 days. Data are presented here as means ± SEM (n = 3). 4.5 4.0 Determining Differentiation Potential: Samples were then incubated Cell Density (×104 cells/cm2) 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 0 1 2 3 4 5 6 7 8 Days Figure 2: MSC attachment studies; removing FBS increased attachment rate to all SoloHill microcarriers in semistatic conditions. A B 100 80 Collagen 60 Plastic 40 Plastic Plus Pronectin F 20 Hillex II Percent Attachment Percent Attachment 100 DPBS for one hour at room temperature. Samples were washed in 500 μL DPBS three times for five minutes at room temperature. 1 0 30 60 90 120 150 180 210 240 80 Collagen 60 Plastic 40 Plastic Plus Pronectin F 20 1 Attachment Time (minutes) Hillex II 0 5 10 15 20 25 30 35 40 Attachment Time (minutes) Figure 3: After 30 minutes of attachment at 37 °C, high percentages of cells remained unbound to microcarriers (left column). Decreasing FBS concentrations in medium increased attachment rate, and almost no cells were visible in medium after 30 minutes (right column). ProNectin F Plastic Plus Plastic Collagen – FBS +FBS Hillex II Figure 4: MSC attachment to microcarriers in spinner cultures Hillex II Plastic Plus ProNectin F Plastic Collagen in 250 µL of the dye/antibody solutions. All antibodies were used at 1:1000 except for Stro-1 (1:500). Dyes and antibodies used were DAPI (Life Technologies, D3571), phalloidinFITC (Life Technologies, A12379), FITC antihuman CD44 (BioLegend 338803), APC antihuman CD90 (BioLegend 328113), Alexa Fluor 647 antihuman Stro-1 (BioLegend 340103), FITC antihuman CD18 (BioLegend 302105), FITC antihuman CD19 (BioLegend 302205), Alexa Fluor 647 antihuman CD14 (BioLegend 325611), and Alexa Fluor 647 antihuman CD146 (BioLegend 342005). To determine differentiation potential of MSCs expanded on Pall SoloHill microcarriers, spinners were seeded at 3 × 103 cells/cm2 and grown for eight days. Spinners were subsequently passaged into new spinners or flatware (24 well plate) at 3 × 103 cells/cm2. Spinners at passage 2 on microcarriers were allowed to expand until near-confluency (2–3 × 104 cells/cm2). Samples from the spinners were transferred to a 24-well plate to determine differentiation capabilities on microcarriers compared to cells grown on flatware. Growth/expansion medium was removed, and 1 mL of either osteogenesis induction medium (EMD Millipore SCR028) or adipogenesis induction medium (EMD Millipore SCR020) was added. Induction and maintenance media were changed according to EMD Millipore’s protocol (as recommended by supplier). Osteocyte differentiation was determined by Alazarin Red S staining, and adipocyte differentiation was determined by Oil Red O staining (protocols with EMD Millipore kits). Results Characterization on Flatware: To characterize MSCs on flatware, T-25s were seeded at 3 × 103 cells/cm2 and incubated for up to ten days to generate growth curves (Figure 1). As shown in 24 BioProcess International 13(8)sp S eptember 2015 Sponsored Supplement Figure 5: Nuclei counts for MSC spinner cultures; counts show maximal confluent densities 6 and 10 × 104 nuclei/cm2. Data are presented as means ± SEM (n = 3). 14 12 Cell Density (× 104/cells/cm2 Figure 1, MSCs seeded at 3 × 103 cells/ cm2 MSCs reached a maximum confluent density of ~4 × 104 cells/cm2. Over the course of the ten day growth curve, cells had an average doubling time of about 48 hours. Attachment Studies: To determine initial attachment conditions for MSCs to the Pall SoloHill microcarriers, attachment studies in which FBS was removed from the attachment media were performed as described earlier. As shown in Figure 2A and 2B, MSCs were 70–80% bound to all microcarriers after 15 minute incubations at 37 °C. However, with 10% FBS present in the medium, attachment ranged from 30% to 80% after two hours and from 50% to 90% after four hours. Observations under light microscopy after 30 minutes of incubation at 37 °C supported these cell counts (Figure 3). Since these experiments were performed under semistatic conditions, the faster attachment rates in the conditions with low FBS were chosen for future spinner cultures. To determine the growth capabilities on Pall SoloHill microcarriers, spinner cultures were seeded at 3 × 103 cells/cm 2. The attachment was done in low serum concentration conditions for 30 minutes. As shown in Figure 4, after this 30 minute attachment period in the spinner flasks, very few cells remain unbound. The low seeding density of 3 × 103 cells/cm 2 is approximately 2–3 cells/bead. Some microcarriers were observed to have more than three cells and some had no cells attached. 10 Collagen 8 Plastic 6 Plastic Plus ProNectin F 4 Hillex II 2 0 0 2 4 Days Figure 6: MSC expansion in spinner culture; MSCs tended to clump once higher densities were reached in microcarrier spinner cultures (days 5–0). 6 8 10 Figure 7: Uniform attachment to microcarriers; using low FBS concentration and no bFGF medium during attachment increased efficiency from ~75% to >95%. Figure 8: (a) Stem cell marker expression of microcarrier-expanded MSCs; MSCs expanded on Pall SoloHill microcarriers (collagen shown) were incubated with anti-CD44 (green), anti-CD90 (red), and DAPI (blue). Populations of CD44 expressing cells are indicated by green arrows. Populations of CD90 expressing cells are indicated by red arrows. Populations of cells expressing both markers are indicated by yellow arrows. (b) MSCs were incubated with anti-CD44 (green), anti-Stro1 (red), and DAPI (blue). Populations of CD44 expressing cells are indicated by green arrows. Populations of Stro‑1 expressing cells are indicated by red arrows. Populations of cells expressing both markers are indicated by yellow arrows. MSC Attachment to Microcarriers in Spinner Cultures: When seeded at low densities, low serum attachment conditions led to quick attachment, although not completely uniform attachment. At this low seeding density, uniform attachment was not possible in medium with 10% FBS. However, cells attached to approximately 70% of the microcarriers allowing good expansion over the 10-day growth period. Nuclei Counts: Figure 5 shows the nuclei counts for spinner samples over the ten day growth periods. Nuclei density reached 6–10 × 104 nuclei/cm 2. Sponsored Supplement MSC density on microcarriers appeared to reach a higher maximal confluent density than what was seen in T-flask growth. As shown in Figure 6, cells grown on all microcarriers tended to stretch across two or three microcarriers (with Hillex II as the exception). By stretching between multiple microcarriers, cells could effectively have a larger available threedimensional volume in which to grow, which is not possible on 2D surfaces. S eptember 2015 13(8)sp BioProcess International 25 Figure 9: Potentiality of microcarrier-expanded MSCs; (a) MSCs grown on T-flasks and Plastic microcarriers remain in an undifferentiated state; (b) MSCs grown on T–flasks and plastic microcarriers underwent adipogenesis and osteogenesis. Cells expanded on microcarriers appeared to have similar differentiation capabilities compared with cells expanded on T-flasks only. Plastic Adipogenesis Osteogenesis Undifferentiated A T-Flask Plastic T-Flask B Figure 10: Differentiation; MSCs grown on plastic microcarriers for multiple passages are shown undifferentiated in the left two panels and differentiated into adipocytes and osteocytes in the two right panels. To increase initial attachment, bFGF was removed from the medium during attachment (complete medium with low FBS and without bFGF). After 30 minutes all cells appeared attached to greater than 90 percent of microcarriers. Expansion for seven days verified uniform attachment and excellent growth, shown in Figure 7. To verify the stem cell-like character of MSCs grown on Pall SoloHill microcarriers, the expression of MSC-associated cell surface markers was determined as well as 26 BioProcess International 13(8)sp the ability of these cells to differentiate after being expanded on microcarriers. To check for the expression of cell surface markers, samples were incubated with f luorophore-conjugated antibodies. Shown in Figure 8, imaging on a Nikon (Ti65) determined these cells to be CD44+, CD90+, Stro1+ and CD146+ when grown on all five Pall SoloHill microcarriers. The hematopoetic cell markers CD14 and CD19 were not expressed in MSCs (data not shown). S eptember 2015 Stem Cell Marker Expression: (Figure 8). Expansion on Microcarriers: To determine the potentiality of MSCs when expanded on microcarriers, cells were grown on Plastic microcarriers for multiple passages. MSCs were seeded at 3 × 103 cells/cm 2 and expanded in spinner flasks for eight days. Cells were trypsinized from microcarriers and seeded onto flatware for differentiation into adipocytes and osteocytes. After 21 days cells grown on plastic microcarriers were able to differentiate into adipocytes and osteocytes at a level comparable to cells grown on T-flasks alone (Figures 9a and 9b). Investigating Differentiation: To determine if several passages on microcarriers affected the differentiation ability of MSCs, cells were passaged multiple times on plastic microcarriers. After six passages on microcarriers, cells were seeded onto flatware for differentiation. Table 1 shows that over the six passages on microcarriers, the harvesting density was consistently above 3 × 104 cells/cm 2 with a doubling time of 48–51 hours when seeded at 3 × 103 cells/cm 2, showing that multiple passages on microcarriers did not decrease maximal confluent density or doubling rates. The ability of cells grown on microcarriers for six passages is shown in Figure 10. The cells grown on plastic microcarriers appear undifferentiated (top panels). Additionally, these cells were able to differentiate into both adipocytes and osteocytes (bottom panels) similar to levels seen previously on earlier passages and on flatware (Figure 9, Table 1) Several passages on microcarriers demonstrate the ability to expand MSCs continuously on microcarriers without decreasing maximal confluent density or doubling time when seeded at 3 × 103 cells/cm 2. Conclusions Current obstacles limiting the use of stem cells for therapeutic benefits include a limited number of cell divisions and the potential loss of pluripotency. Due to the restricted Sponsored Supplement number of population doublings, achieving maximal possible expansion in the fewest passages is vital. We have shown here that MSCs can be expanded on various types of Pall SoloHill microcarriers. The benefit of MSC expansion on microcarriers is two-fold. First, expansion on microcarriers allows growth on large surface areas within single containers, and second microcarrier expansion increases the ratio of apparent surface area to medium volume due to the fact that MSC growth on microcarriers outpaces growth on flatware. This is particularly important with stem cells grown in medium that contains expensive supplements. Therefore, the use of microcarriers allows minimal passages for expansion of cells while decreasing the overall cost required to grow enough cells for a therapeutic dose in clinical trial treatments. We have shown that multiple passages on microcarriers do not affect the ability of MSCs to differentiate into adipocytes and osteocytes. The ability to maintain pluripotency while expanding MSCs on microcarriers for five or six passages allows for the isolation of cells from bone marrow onto a T-150 flask. Cells expanded in this fashion can subsequently seed a small scale spinner culture which could be used to seed a small bioreactor. For example, the maximal confluent cell density in a T-150 results in 2.5–3 × 106 cells and 3.5–4 × 104 cells/cm 2 on microcarriers. To seed a 200-mL spinner volume requires 3.1 × 10 6 cells using 5,150 cm 2/L. The maximal densities on spinner cultures achieved here would result in enough cells for a minimum 10-fold expansion into a 2 L bioreactor, by the third passage after isolation and the second passage on microcarriers. Considering the recoverable cell numbers presented here, a 6.7 L bioreactor volume (at 5,150 cm 2/L) would result in enough cells for one therapeutic dose (~1 × 109 cells). Assuming similar growth between small scale spinners and bioreactors, a 2-L bioreactor could be used to seed a 20-L bioreactor, which would result in enough cells for three doses from a single T-150 and multiple passages on Sponsored Supplement Table 1: Continuous passage on microcarriers in spinner cultures; several passages on microcarriers demonstrate the ability to expand MSCs continuously on microcarriers without decreasing maximal confluent density or doubling time when seeded at 3 × 103 cells/cm2. Plastic Spinners Seeding Density1 Harvest Density1 Days of Growth Number of Doublings 1 P1 0.3 4.6 8 4 P2 2 3.3 5 <1 P3 1 2.7 5 1.3 P4 0.3 3.6 7 3.6 P5 0.3 4.3 8 3.8 P6 0.3 4.5 8 3.9 × 104 cells/cm2 Table 2: Extrapolated liters needed for therapeutic doses for various concentrations of plastic microcarrier cultures Microcarrier Concentration (plastic) 10 g/L 20 g/L Surface Area 0.3 Maximum Confluent Density (cells/cm2) 2 Total Cells/L 1 L/Therapeutic Dose 0.3 4.6 3.3 2.7 3.6 microcarriers. Additionally, increasing the microcarrier concentration beyond 5,150 cm 2/L would decrease bioreactor volume required for a large number of recoverable MSCs. Table 2 extrapolates the expected number of cells if the Plastic microcarrier concentrations used were 10 g/L and 20 g/L. The resulting liters needed for therapeutic doses of stems cells would be 7–8 liters and 3–4 liters, for concentrations of 10 g/L and 20 g/L, respectively. Work will continue to further characterize stem cells grown on Pall SoloHill microcarriers, such as defining various populations within bone marrow-derived MSCs, any fluctuations in these populations when grown on microcarriers for multiple passages, and additional stem characteristics such as the ability of microcarrier-expanded cells to produce angiogenesis signals such as VEGF. Future work will also expand to include other stem cells such as induced-pluripotent stem cells, mouse embryonic stem cells, and mesenchymal stem cells from other sources such as placental-derived or adipose-derived. Additionally, work will continue to define growth and expansion conditions under animal component free environments, as well as direct expansion on microcarriers of isolated stem cells, thereby bypassing flatware tissue culture and increasing the number of available passages before senescence. References 1 Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast Precursors in Normal and Irradiated Mouse Hematopoietic Organs. Exp. Hematol. 4, 1976: 267–274. 2 Fraser et al. Fat Tissue: An Underappreciated Source of Stem Cells for Biotechnology. Trends Biotechnol. 2, 2006: 150– 154. 3 Cao C, Dong Y. Study on Culture and In Vitro Osteogenesis of Blood-Derived Human Mesenchymal Stem Cells. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 19, 2005: 642–647. 4 Griffiths M, Bonnet D, Janes SM. Stem Cells of the Aveolar Epithelium. Lancet. 366, 2005: 249–260. 5 Beltrami et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell. 114, 2003: 763–776. 6 Pittenger et al. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science. 284, 1999: 143–147. c Corresponding author Mark Szczypka, PhD, is senior director of applications and new product development, mark_szczypka@pall.com. David Splan is senior research scientist; and Heather Woolls, PhD, is a research scientist, all at Pall Life Sciences. SoloHill and Hillex are registered trademarks of Pall Corporation. S eptember 2015 13(8)sp BioProcess International 27 S e c t i o n T w o PROCESS DEVELOPMENT Strategies for Microcarrier Culture Optimization by Mark Szczypka and Alain Fairbank T he process of delivering an allogeneic stem-cell therapy to patients requires isolation and expansion of rare tissue-specific stem cells, which are subsequently delivered to individual patients for treatment. One type of cell used for such therapies is commonly known as human mesenchymal stem cells (hMSCs). They have been isolated from a number of tissues: e.g., bone marrow, heart, brain, placenta, and umbilical cord. And they have been shown to be immune-privileged in that hMSCs elicit no graft-versus-host (GvH) response such as those observed with allogeneic organ transplants. Thought to provide therapeutic benefits for a range of diseases, hMSCs have hypothesized mechanism of actions of promoting intrinsic organ repair and performing immunomodulatory activities (1). Those functions are postulated to be temporally and site specific. In vitro, hMSCs secrete growth factors and other molecules that may regulate intrinsic repair mechanisms in vivo. Those molecules may represent the therapeutic effectors when they reach sites of damage. It is important to note that hMSCs do not integrate into tissues and “transdifferentiate” into adult-cell types at appreciable frequencies, and their residence time within a patient’s body is relatively short (1–3). That provides a challenge for achieving an effective therapy, making it necessary for therapeutic cells to rapidly reach their site of action to perform their necessary functions 28 BioProcess International 13(8)sp Figure 1a: Monitoring kinetics of cell attachment to microcarriers; samples retrieved from dynamic culture are fixed and stained with DAPI — nuclei can be visualized and counted using a microscope equipped for fluorescence Suboptimal (few cells attached) Optimal (10–12 cells/bead) before they are cleared. Early results from clinical trials indicate that relatively large numbers of cells — in the range of tens of millions to billions — are needed to produce a therapeutic effect. Furthermore, some indications may require multiple treatments to guide and promote curative processes (4). If those observations hold true, then allogeneic hMSC therapies will require implementation of large-scale manufacturing processes similar those performed for many years in mammalian-cell–based manufacturing of biologics and vaccines (5). That requirement is complicated by the fact that hMSCs expanded in vitro must retain their therapeutic potential throughout the entire manufacturing process so that healthy cells can be efficiently harvested for delivery to patients. However, when cells are products, special challenges arise in developing a manufacturing platform that are not found in more traditional biologics S eptember 2015 manufacturing. The theoretical basis and practical demonstrations of the concepts for using adult stem cells to treat disease have come from academic institutions, research institutes, and medical schools. Methods used by their investigators to generate cells for seminal studies were similar to traditional cell culture techniques used in research laboratories: static twodimensional (2D) platforms consisting of cell culture dishes and T-flasks. In many cases, such platforms have been sufficient for small-scale animal studies and some phase 1 clinical trials requiring limited numbers of cells. However, as cell therapies transition to larger clinical trials for multiple indications in humans, developers need to identify more user-friendly manufacturing platforms for cell expansion. The methods are too onerous for handling large numbers of dishes or T-flasks needed to generate enough cells for such purposes. Fortunately, procedures for isolation, in vitro expansion, Sponsored Supplement formulation of hMSCs into a final product, and ultimate delivery to patients have undergone continual process improvements over the past 10–15 years (6–8). Multiplate units are commonly used to expand hMSCs; however, this platform provides only a short-term solution for the expansive surface area (SA) requirements needed for hMSC expansion. Different iterations of multiplate technologies provide sufficient SA to generate tens of billions cells. But even the most sophisticated devices require an unmanageable number of units to generate the trillions of cells required to treat many predicted patient populations, with the required multiple manual manipulations increasing the risk of culture contamination (9). Hollow-fiber units also provide a solution for generating millions to billions of cells. However, when cell number requirements even modestly increase, an unmanageable number of units become required. These limitations have spurred efforts to identify simple, cost-effective, and efficient solutions for generating much larger numbers of cells. Implementing a microcarrier-based production platform has become an obvious option for filling this need (10). Microcarriers for Expansion of hMSCs Microcarriers provide a large SA-tovolume ratio for propagation of adherent cells, making expansion to trillions of cells possible in a single culture vessel. Implementation of this technology greatly reduces the number of production lots needed per year as well as lowering risk and cost while at the same time increasing process control (5, 10, 11). All these improvements lead to product consistency and quality. Microcarriers used for propagation of mammalian cells are micron-sized particles. Manufactured at relative densities that promote their suspension in vessels using gentle stirring, they can take different shapes and sizes. Different core materials include synthetic compounds such as polystyrene and biologic materials such as extracellular matrix molecules. The Sponsored Supplement Table 1: Properties of Pall SoloHill microcarriers Microcarrier Core Material Collagen Cross-linked polystyrene Fact III Cross-linked polystyrene Plastic Cross-linked polystyrene Plastic Plus Cross-linked polystyrene ProNectin F Cross-linked polystyrene Hillex II Cross-linked modified polystyrene Relative Density 1.02–1.04 Animal Protein Free? No 1.02–1.04 No 1.02–1.04 Yes Charged surface 1.02–1.04 Yes Recombinant protein 1.02–1.04 Yes 1.10 Yes Surface Treatment Type I porcine gelatin coating Charged surface with type I porcine gelatin coating Uncharged Cationic amine surfaces of microcarriers also can be chemically modified or coated with different molecules to facilitate efficient cell attachment, spreading, and growth (Table 1). Availability of a broad range of microcarriers provides an opportunity to select an optimal substrate that promotes robust and efficient cell growth while allowing use of simple methods for harvest and separation of cells from the microcarriers after expansion. Unique, innovative approaches to accomplishing harvest across the range of manufacturing scales continue to be developed to ensure that highly viable, healthy, and therapeutically active hMSCs can be efficiently expanded on microcarriers and isolated from bioreactors for delivery into patients. Growth of Adherent Cells on Microcarriers For cell culture processes, bioreactor vessels are assembled, and culture media and microcarriers are added to them. Environmental conditions such as pH and gas concentrations are allowed to stabilize at desired set points prior to cell addition, then adjusted and maintained at preferred levels. Cells are then added at an appropriate concentration for attachment to the microcarriers. Cells are harvested from a smaller bioreactor or static platform (e.g., an Xpansion multiplate bioreactor from Pall Life Sciences) and introduced into a large bioreactor containing microcarriers. Cell densities that promote occupation of all microcarriers in the vessel are determined empirically and used for seeding. Seeding density influences overall culture efficacy because cells must come into contact with a microcarrier in suspension and subsequently attach to its surface. Once attached, cells must spread over the microcarrier through cell division while remaining attached. Dynamic microcarrier culture can be described as a series of discrete, sequential events. Several important physical parameters regulate this process. Kinetics of cell attachment to microcarriers, distribution of cells across the microcarrier population within a vessel, cell spreading, and maintenance of adherence to microcarriers during cell division are therefore all important processes to characterize. Those events can be monitored using qualitative and quantitative methods in small-scale studies that help culturists identify conditions that lead to efficient microcarrier culture. Microcarrier concentration, cell seeding density, microcarrier surface characteristics, medium formulation, mixing dynamics, and environmental controls are process parameters that directly or indirectly influence culture efficiency. Cell Attachment and Distribution: The kinetics of cell attachment to microcarriers is of primary importance because adherent cells that remain in suspension for extended periods can form aggregates and will eventually perish. To measure the kinetics of attachment, representative samples retrieved at predetermined time points after cell seeding are formaldehyde fixed, then stained with a fluorescent dye for visualization of nuclei using S eptember 2015 13(8)sp BioProcess International 29 fluorescence microscopy. The number of cells attached to microcarriers then can be quantitated (Figure 1a). The resulting measure helps culturists assess the quality of cell attachment and reveals the distribution of cells on microcarriers suspended in solution over time. Ideally, cells will attach in a predictable time frame, and all microcarriers will have the same number of cells attached to their surfaces. In practice, however, attachment times depend on cell type and medium. A calculation using cell seeding density (cells/cm2) and the physical properties of the microcarriers can predict the number of cells that will occupy each microcarrier in a culture (Figure 1b). The observed distribution of cell-containing microcarriers in suspension — and the number of cells attached to each microcarrier within a sample — can be compared with the theoretical number of cells expected to attach to each one. Performing that comparison helps culturists determine the attachment efficiency and microcarrier occupancy under different environmental conditions and facilitates identification of conditions that lead to rapid and efficient cell attachment. An efficient culture should have a roughly equivalent number of cells on each microcarrier, and every microcarrier in suspension should have at least one cell bound to its surface. Such a scenario increases the efficiency of culture by facilitating synchronous cell growth and promoting coverage of the entire available SA within a vessel. Environmental conditions such as protein concentration in the medium, pH level, cell-seeding density, mixing dynamics, and microcarrier concentration can be modified to promote cell attachment. Optimal set points for those parameters should be identified and refined in small-scale cultures before further optimization with larger bioreactors. Cell spreading onto microcarriers is indicative of a favorable cell-substrate interaction and thus another critical parameter to assess. Monitoring this process can be subjective because there are no simple and robust methods to 30 BioProcess International 13(8)sp Figure 1b: Monitoring kinetics of cell attachment to microcarriers; calculation to determine theoretical cell numbers attached to microcarriers using seeding density and physical characteristics of microcarriers Cs(SAg) Bg Cells/microcarrier = Cs = seeding density (cells/cm2) } SAg = surface area (cm2) per gram of microcarriers Bg = number of beads quantify the phenomenon on microcarriers. With some guidance and practice, however, this measure can be used to effectively gauge microcarrier performance in early screening efforts. Cells initially attach to substrates with a rounded appearance. If the substrate is favorable, they will rapidly begin to spread across the microcarrier surface and adopt a gumdrop-shaped morphology. As cells continue to spread, they flatten to their typical in vitro morphology — which (depending on cell type) can make them more difficult to visualize. For example, hMSCs adopt a flattened fibroblast-like morphology that makes visualization more challenging. Use of fluorescent dyes and microscopy can help analysts visualize those cells once they have spread onto the surface of nontranslucent microcarriers. Adherence and Growth: Successful propagation of cells on microcarriers in dynamic culture is more complex than on flatware because cells are constantly exposed to forces they do not encounter in static culture. Shear forces make cell division more difficult. Many cell types have specific requirements for attachment and growth, and a number of parameters control their ability to grow effectively in dynamic cultures. As mentioned above for other cellular processes, identification of the optimal combination of these variables for a specific cell type is best performed in small-scale cultures. This can be difficult sometimes because representative scaled-down models that mimic larger-scale vessels are not always available. But this approach can provide initial insight into important process-control variables for specific cells in a selected S eptember 2015 medium type and also identify starting conditions for a particular larger (production-sized) vessel. Cell seeding density can greatly influence the implementation and efficiency of microcarrier cultures (12). Fine tuning this parameter leads to efficient use of the SA within a vessel. Total SA in a bioreactor depends on the mass of microcarriers used, and that SA is distributed among the number of beads within each gram of microcarriers. Using the physical characteristics of microcarriers, culturists can calculate the number of cells expected to occupy individual microcarriers within a bioreactor. This establishes a lower theoretical limit for cell seeding. It is preferable to seed with enough cells to have at least one cell attached to each microcarrier in suspension. Seeding densities used in static cultures should be used as a starting point for smallscale studies. The lowest density that allows for population of all microcarriers should be tested along with several other (higher) seeding densities. Microcarrier Concentration: Total available SA is determined by the concentration of microcarriers added to a bioreactor. Microcarrier properties (e.g., size and shape) influence the maximum SA that can be occupied by cells. For example, the SA of spherical, nonporous microcarriers is dictated by the diameter of those particles. Individual microcarriers with smaller radii have a smaller SA than that of larger microcarriers, but the SA equivalent of their entire population is greater because the number of microcarriers of a given density in a single gram is much larger. That larger number of individual spheres will inverse-exponentially increase the overall surface so that solid microcarriers >300 µm have a very low overall SA per gram. It is important to note that increasing microcarrier concentration is not directly linked to culture performance. Some investigators use concentration of cells (cells/mL) to describe microcarrier culture efficiency, but that measure does not accurately assess the actual SA use. Sponsored Supplement Figure 2: In hMSC attachment studies, removing fetal bovine serum (FBS) increased attachment rate to all SoloHill microcarriers in semi-static conditions. After 30 minutes attachment at 37 °C, high percentages of cells remained unbound to microcarriers (bottom panel). Decreasing FBS concentration in media to 0.5% increased attachment rate after 30 minutes. Low Serum (0.5%) 100 80 Attachment (%) Attachment (%) 80 60 40 Collagen 60 Plastic Plastic Plus 40 Pronectin F 20 20 0 Standard Serum (10.0%) 100 0 5 10 15 20 25 Time (minutes) Increasing microcarrier concentration will expand SA within a vessel, but if the available substrate is not colonized by cells, then the system is inefficient. Microcarrier concentration can influence other culture parameters as well: e.g., the formation of cell microcarrier aggregates and possible increases in shear forces encountered by cell-laden microcarriers. It therefore becomes very important to identify the optimal microcarrier concentration for a given combination of cells and media that will support the highest concentration of cells. Environmental Control: Standard environmental-control methods used in dynamic suspension cultures of mammalian cells are appropriate for implementation with adherent-cell microcarrier cultures. Parameters such as pH and dissolved oxygen (DO) levels, nutrient supplementation, and metabolite removal must be tightly controlled because of their influence on culture health (13). Changes to pH result from cellular metabolic activities, including release of carbon dioxide (CO2), lactic acid, and other metabolites. Those shifts can affect cell adherence to microcarriers. Culture pH can be controlled with the same CO2bicarbonate buffering systems used in flatware along with infusions of CO2, oxygen (O2), nitrogen (N2), and air mixtures. Other buffering systems are available; however, the bicarbonate buffering system is most commonly Sponsored Supplement 30 35 40 0 Hillex II 0 5 10 15 20 25 Time (minutes) used for many microcarrier-based processes. Gases can be infused into a vessel using either a gas overlay or sparging. Overlay entails infusing a gas mixture into the vessel as a constant stream over the surface of the liquid–air interface to promote gas exchange. Sparging a vessel with gas occurs through an apparatus that contains differently sized holes, and is immersed into the culture medium. Although the latter increases gas exchange, the former helps to prevent some of its shortfalls (e.g., bubble formation and foaming, both which can lead to shear stress and loss of microcarriers from solution). Base addition can also be used to maintain pH; however, care should be taken to not expose cells directly to highly concentrated alkaline solutions, which can cause membrane damage and cell death. Batch feeding, perfusion, and nutrient supplementation all can be implemented in microcarrier cultures. Bioreactors should be outfitted with spin filters or dip tubes that can be covered with mesh screens that facilitate removal of spent medium without removing microcarriers from the vessel. Medium can be replenished through additional vessel ports. The amount of medium removed and replaced is regulated through the use of level probes. Feed strategies for large cultures (including nutrient or growth factor supplementation) along 30 35 40 with metabolite monitoring should be optimized first in small-scale cultures to save time and cost. Microcarrier Surface Characteristics: Commercially available microcarrier types are composed of a range of materials and possess unique surfaces. Unique combinations of core materials and surface modifications control physical characteristics of microcarriers, including relative density, rigidity, size, surface porosity, and charge density. Some microcarriers feature a coating with recombinant or animal-sourced extracellular matrix proteins and/or synthetic peptides. Such molecules promote attachment and adherence of cells. The types of molecules used and the manner in which they are attached to the microcarrier influences how effective the substrate is at promoting cell attachment and growth (14–17). Plasma membranes of specific cell types have unique assortments of attachment molecules and distinct molecular properties. The membrane composition influences how cells attach to a surface and also governs whether one substrate is preferred over another. It is therefore important to identify an ideal surface chemistry for specific cells to achieve optimal culture efficiency. Using charged moieties to modify microcarrier surface chemistry also has proven to be useful and can sometimes mitigate the need for protein coatings. Polymeric microcarriers can be chemically charged by modulating monomeric composition using well- S eptember 2015 13(8)sp BioProcess International 31 known chemistries or charged-molecule coatings. Charges can also be imparted onto the surface of microcarriers using plasma-emission coating, a technology commonly used to generate tissueculture plastic. It’s not surprising that some microcarriers combine charges and proteins and have proven to be useful in a number of mammalian cell culture processes. Commercially available Pall SoloHill microcarriers present a number of available surface modifications. Microcarriers containing positive charges, animal-derived attachment proteins, recombinant proteins, and combinations of all those substrates are offered. Many other types of surface modifications have been applied to microcarriers. Application of temperature-responsive molecules to microcarriers has been performed in the past, but new technologies and thermal-responsive molecules thought to improve utility are being explored. The potential benefits of such technology are that cells can be removed from microcarrier surfaces without enzymatic treatment. Media Formulations: It is well known that culture media formulations significantly influence cell growth. Therefore, it follows that the composition of a medium (including pH, osmolality, and protein concentrations) also has a major influence on cell attachment, adherence, and growth in microcarrier cultures. Media components such as serum, recombinant proteins, and synthetic peptides can passively absorb onto microcarrier surfaces during acclimation in reactors, or they can attach to cells and directly or indirectly influence their attachment to microcarriers. Attachment kinetics and cell distribution have been shown to change according to media components and concentrations (18–20). For example, the commonly used culture supplement fetal bovine serum (FBS) contains many proteins and other molecules such as bovine serum albumin (BSA), fibronectin, vitronectin, and galectin. It is wellknown that those and other molecules can affect cell attachment, spreading, and adhesion (21). Some such 32 BioProcess International 13(8)sp molecules can facilitate cell attachment when they are affixed to surfaces, but modulation of protein or serum concentration in culture media also can influence cell attachment and distribution on microcarriers. We have shown that decreasing concentrations of those media components can improve cell attachment and facilitate even cell attachment to microcarriers (Figure 2). Optimization Roadmap and Time Considerations It is paramount that a simple and straightforward experimental strategy be adopted when developing and implementing microcarrier cultures, requiring a high degree of attention to detail. Careful planning and timely execution of experimental plans and implementing multiparameter experiments (where applicable) will expedite developmental timelines and save money. The first step in the process is to fully characterize the selected cell type in flatware cultures. Parameters that should be characterized and optimized include doubling time, population doubling levels, expression of cellspecific markers, cell behavior in functional/potency assays, attachment factor requirements, and conditions for cell harvesting. Those cell characteristics and behaviors in static culture will be used to guide selection of initial conditions used for microcarrier culture. They also can help culturists assess potential changes in cell function that could be attributable to expansion in a dynamic microcarrier culture. Cells must retain their critical phenotypic characteristics when grown in a suspension microcarrier culture. For example, adult hMSCs should retain their differentiation capacity, expression of characteristic markers, and functionality. However, minor differences in some parameters sometimes can be seen. Doubling times might be slightly longer for some cell types, and expression levels of some “house-keeping” proteins can be altered by exposure to dynamic conditions. Attachment kinetics and efficiency are the first parameters to optimize. S eptember 2015 Examination of cell attachment to different microcarrier types and growth in static culture can be used as a method to identify microcarriers that are compatible with a given cell type and medium combination. Using this method, cells and microcarriers combined in plates that have not been treated for tissue culture are gently mixed at defined intervals over the first few hours of culture. Attachment and growth are monitored over two to three days. This is a rapid and economic way to obtain an indication of productive cell–microcarrier interaction. Although useful, this technique does not expose cells to dynamic conditions, so it is not always fully predictive of their behavior on specific microcarriers in stirred-tank vessels. Characterization of attachment and binding in dynamic culture should closely follow. These studies can be performed using small-scale formats such as multiwell bioreactors or smallscale spinners of 50–200 mL. Such formats are convenient for multifactorial studies and can be used to identify and optimize growth conditions in dynamic culture. Parameters such as cell seeding, microcarrier concentration, and medium formulation optimization (including feeding and/or supplementation) can be optimized effectively, expeditiously, and costeffectively using this type of format. This is especially useful for optimization when expensive medium formulations are required. Cell harvest techniques should be optimized at every stage of development, beginning with optimization of conditions for enzymatic treatment (e.g., trypsin, chymotrypsin) in flatware (22–24). Investigators must establish temperature limits and determine minimum and maximum exposure times that do not affect cell health. Harvesting of cells from microcarriers is a multistep process that includes enzymatic treatment to initiate cell detachment, dislodging cells from microcarriers using some type of physical disruption, and then separating the cells from the microcarriers. Durable, rigid polystyrene microcarriers made of materials that are similar to standard Sponsored Supplement culture flatware are ideally suited for harvesting cells. Additionally, the properties of such microcarriers allow for implementation of a range of techniques to dislodge cells from their surfaces with simple processes for capturing cells from the resulting slurry. Although small-scale formats are informative, further optimization in larger formats is required. Transitioning to small (2-L to 5-L) bioreactors will allow for further optimization of conditions required to increase cell numbers and further optimize feed strategy. The intrinsic environmental and process-control options available in such platforms provide an opportunity to further optimize conditions that support superior cell growth. Physical conditions present in these platforms are closer to those observed in larger systems. But for most such platforms, each increase in scale presents specific challenges that must be addressed for optimization of environmental conditions. Fortunately, a large amount of historical information and expertise have accumulated from suspensionculture manufacturing. Such literature can provide starting points for optimization of microcarrier celltherapy cultures. Conclusion Until recently, a common paradigm in the biopharmaceutical industry has been that development of a microcarrier-based production platform is a lengthy and onerous process when compared with the effort needed to scale out static platforms. But new microcarriers have emerged to present a reliable and accessible technology for generating large numbers of cells for allogeneic cell therapies. Although slight differences may arise with transition from static 2D systems to dynamic microcarrierbased technology, the cost benefits and process efficiencies achieved with microcarrier culture far outweigh the time and effort required for bioreactor process development. To facilitate successful production at commercial scale, process development efforts should be initiated as early as possible in product development, which also helps Sponsored Supplement products transition to clinical trials at the earliest practical time point. Implementing the strategies of careful planning, understanding the critical process parameters, and timely execution of experimental designs will expedite developmental timelines; save money; and facilitate development of robust, reproducible, and cost-effective manufacturing platforms for cellbased therapies (Table 1). References 1 Phinney DG, Prockop DJ. Concise Review: Mesenchymal Stem/Multipotent Stromal Cells: The State of Transdifferentiation and Modes of Tissue Repair — Current Views. Stem Cells 25(11) 2007: 2896–2902. 2 Kean TJ, et al. MSCs: Delivery Routes and Engraftment, Cell-Targeting Strategies, and Immune Modulation. Stem Cells Int. 13 August 2013. 3 Chamberlain G, et al. Concise Review: Mesenchymal Stem Cells: Their Phenotype, Differentiation Capacity, Immunological Features, and Potential for Homing. Stem Cells 25, 2007: 2739–2749. 4 Van't Hof W, et al. Direct Delivery of Syngeneic and Allogeneic Large-Scale Expanded Multipotent Adult Progenitor Cells Improves Cardiac Function After Myocardial Infarct. Cytother. 9(5) 2007: 477–487. 5 Rowley J, et al. Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy. BioProcess Int. 10(3) 2012: 16–22. 6 Want AJ, et al. Large-Scale Expansion and Exploitation of Pluripotent Stem Cells for Regenerative Medicine Purposes: Beyond the T Flask. Regen. Med. 7(1) 2012: 71–84. 7 Kehoe D, et al. Scalable StirredSuspension Bioreactor Culture of Human Pluripotent Stem Cells. Tiss. Eng. Part A 16, 2010: 405–421. 8 Santos F, et al. Toward a ClinicalGrade Expansion of Mesenchymal Stem Cells from Human Sources: A Microcarrier-Based Culture System Under Xeno-Free Conditions. Tiss. Eng. Part C Meth. 17, 2011: 1201–1210. 9 Davie NL, et al. Streamlining Cell Therapy Manufacture: From Clinical to Commercial Scale. BioProcess Int. 10(3) 2012: S24–S27. 10 Simaria AS, et al. Allogeneic Cell Therapy Bioprocess Economics and Optimization: Single-Use Cell Expansion Technologies. Biotechnol. Bioeng. 111(1) 2014: 69–83. 11 Szczypka MS, et al. Single-Use Bioreactors and Microcarriers: Scalable Technology for Cell-Based Therapies. BioProcess Int. 12(3) 2014: 54. 12 Hu WS, Meier J, Wang DIC. A Mechanistic Analysis of the Inoculum Requirement for the Cultivation of Mammalian Cells on Microcarriers. Biotechnol. Bioeng. 27, 1985: 585–595. 13 Wlaschin KF, Hu WS. Fed-Batch Culture and Dynamic Nutrient Feeding. Adv. Biochem. Eng. Biotechnol. 101, 2006: 43–74. 14 Varani J, et al. Growth of Three Established Cell Lines on Glass Microcarriers. Biotechnol. Bioeng. 25, 1983: 1359–1372. 15 Giard DJ, et al. Virus Production with a Newly Developed Microcarrier System. Appl. Environ. Microbiol. 34, 1977: 668–672. 16 Varani J, et al. Substrate-Dependent Differences in Growth and Biological Properties of Fibroblasts and Epithelial Cells Grown in Microcarrier Culture. J. Biol. Stand. 13, 1985: 67–76. 17 Varani J, et al. Cell Growth on Microcarriers: Comparison of Proliferation on and Recovery from Various Substrates. J. Biol. Stand. 14, 1986: 331–336. 18 Blüml G. Microcarrier Cell Culture Technology. Animal Cell Biotechnology. Pörtner R, Ed. Humana Press: New York, NY, 2007: 149–78. 19 Butler M, et al. High Yields from Microcarrier Cultures By Medium Perfusion. J. Cell Science 61, 1983: 351–363. 20 Serra M, et al. Improving Expansion of Pluripotent Human Embryonic Stem Cells in Perfused Bioreactors Through Oxygen Control. J. Biotechnol. 148(4) 2010: 208–215. 21 Grinnell F. Cellular Adhesiveness and Extracellular Substrata. Int. Reve. Cytol. 1978: 67–129. 22 Schriebl K, et al. Stem Cell Separation: A Bottleneck in Stem Cell Therapy Biotechnol. J. 5, 2010: 50–61. 23 Heathman TR, et al. Expansion, Harvest and Cryopreservation of Human Mesenchymal Stem Cells in a Serum-Free Microcarrier Process. Biotechnol Bioeng. 112(8) 2015: 1696–1707. 24 Weber C, et al. Expansion and Harvesting of hMSC-TERT. Biomed. Eng. J. 7(1) 2007: 38–46. • Corresponding author Mark Szczypka is senior director of applications and new product development at Pall Life Sciences in Ann Arbor, MI 48108; 1-734-973-2956; mark_szczypka@pall.com. Alain Fairbank is director of cell therapy marketing at Pall Life Sciences in Port Washington, NY; alain_fairbank@pall.com. S eptember 2015 13(8)sp BioProcess International 33 “ We’ve been finding solutions to complex challenges for over 60 years. Just imagine what we can achieve together.” Pall Life Sciences Your vision. Our expertise. Their future. www.pall.com/celltherapy © 2015 Pall Corporation. Pall, , Xpansion, iCELLis and PadReactor are trademarks of Pall Corporation. ® indicates a trademark registered in the USA. S e c t i o n T h r e e CELL THERAPY MANUFACTURING Designing the Most Cost-Effective Manufacturing Strategy for Allogeneic Cell-Based Therapies by Thierry Bovy, Alain Fairbank, and Suzanne S. Farid R apid progress is occurring in the field of stem cell therapy research, and increasing numbers of products will begin reaching the market in the near future. But new cell therapy treatments must fit into a competitive and highly regulated healthcare environment. Succeeding in that environment requires alignment between a company’s business model and its manufacturing strategy. Cell therapy products are different in many respects from traditional small-molecule and even biologic drugs. So developers may need to reconsider preconceptions about the manufacturing process to accommodate the unique characteristics of these therapeutics. Economic aspects should be addressed from early phases of development to enable a viable product life cycle. Identifying cost drivers early on will help companies develop the right manufacturing strategies and determine necessary batch sizes. Commercial strategy helps to identify available resources — and risks — for bioprocess development and full-scale manufacturing. Some key drivers of the rapidly expanding demand for manufacturing tools include cost, scalability, and flexibility. Achieving lot sizes of several hundred billion to trillions of cells both efficiently and cost effectively will be imperative for commercial success. Scaling the culture of adherent cells, such as mesenchymal stem cells, has presented manufacturing challenges as 36 BioProcess International 13(8)sp Implementing Technology Options in Cell Therapy Development lot sizes increase from billions to trillions of cells. Predicting market demand is never easy, particularly for emerging technologies that lack historic precedent. Manufacturing flexibility, in both scheduling and capacity for rapid production scale-up, is therefore vital to commercial success of a cell therapy. On 25 March 2015, Pall Life Sciences presented a webcast on designing cost-effective manufacturing strategy for allogeneic cell-based therapies. The speakers were Thierry Bovy (Pall Life Sciences’ global product manager of Xpansion multiplayer bioreactor systems) and Suzanne Farid (professor in bioprocess systems engineering at University College London’s department of biochemical engineering). Below, Bovy discusses industrialization challenges and the advantages and limitations of implementing different technology options across the productdevelopment pathway. S eptember 2015 Industrialization of a cell therapy process can be challenging. The product is, of course, our center of attention. But some aspects of increasing importance will have to be considered when moving through drug development. First, the scale of a manufacturing process must match market needs for increasing quantities of cellular doses. Raw materials and other supplies need to be carefully managed, as well. No one wants to see a therapy’s progress halted by a shortage in raw materials or consumables. And cost of the final product must allow the sponsor company to make proper benefit while marketing product that the patient/ payer community can afford to buy. Finally, facility design and size will need to match the production process and manufacturing volumes. The quality system will have to be robust enough to cope with increasing workloads associated with intensification of manufacturing and associated validation efforts. Scale of adherent cell culture is associated with the extent of surface available for cells to colonize. The vertical axis of Figure 1 represents the surface area per unit, and the horizontal axis plots the different currently available technologies for adherent cell cultures. Most people are familiar with open, multilayered stacks. But Pall proposes the Xpansion bioreactor as a compact and closed alternative for lateral-scale Sponsored Supplement Figure 2: Upstream options — adherent cells for therapeutic applications >10 6 122,400 25,000 Ho Bi llow or ea Fi ct be o r M rs ul til St ay ac er k M Bi u s or lti ea p l c a M torste i St cro irr ca ed rr Ta iers nk 18,000 manufacturing with up to >122,000 cm 2 per single unit. And microcarrier storage tanks are currently under investigation at most advanced cell therapy companies working on allogeneic treatments. But the Xpansion bioreactor technology is currently the only one able to reach millions of square centimeters per unit. Developers must select the best manufacturing strategy with their end target in mind, taking into account the amount of cells that will need to be produced in the commercial phase. Figure 2 shows different technical options for adherent-cell culture, considering the number of patients to treat each year and the amount of cells per patient. The dose size for autologous treatment is generally smaller than that of allogeneic therapies. Open T-flasks or multilayer stacks are typically used for manufacturing lowdose products. Increasing the production capacity involves scaling out rather than up. Use of bioreactors and/or automation will contribute to reducing cost of goods (CoG). Allogeneic treatment doses can go up to a billion cells per patient (or per injection). When multiplied by the number of patients treated per year, that requires a huge amount of cells to be produced. Existing planar technologies are unable to achieve more than 5 × 1011 cells per lot. Pall’s Xpansion system is currently the only single-use bioreactor providing >100,000 cm2/ unit. Threedimensional (3D) systems such as microcarriers in stirred-tank bioreactors could generate up to 1013 cells per lot. The Xpansion multiplate bioreactor was developed for largeSponsored Supplement Allogeneic (scale-up) Millions of cells/patient Autologous (scale-out) Surface Area (cm2/unit) Figure 1: Available upstream technologies 3D Stirred-tank bioreactors with microcarriers 500 Xpansion (multilayer), automated multilayer 2D Xpansion (multilayer) 250 Tangential-flow filtration (multilayer) 10s 50s Phases 1−3 100s 500s ... 5,000s Commercial 10,000s ... Patients treated/year scale production of traditional 2D cell cultures. Each plate provides 612 cm 2 of surface for adherent cell growth. Stacking 10–200 of those plates provides increasing surface from 6,000 up to >122,000 cm 2 per single unit without modification to the bioreactor’s footprint. Only its volume and weight change. The smallest version (Xpansion 10 bioreactor) is normally used for technical evaluations, whereas the others (Xpansion 50, 100, and 200 bioreactors) are designed for manufacturing purposes. The model numbers correspond to the number of plates in each unit. This bioreactor was designed to provide the same microenvironment as multitray stacks to ease transition from traditional open models to closed and controlled systems. Some tweaking is necessary, mainly optimizing parameters such as pH and dissolved oxygen (DO) as well harvest protocols. The systems scale up linearly from 10 to 200 plates. The same hardware is used across the bioreactor range. So settings contribute to maintaining the culture condition consistent. Parameters such as stirring speed are adapted according to bioreactor size for maintaining linear speed of the culture medium over the monolayer. You change stirring speed to scale out. All Xpansion bioreactors have the same footprint, and they contribute to reducing manual operations. Microcarriers: Transitioning a cell culture system from 2D to 3D and scaling it up requires time and expertise. Special attention must be paid to the selection of microcarriers. Their density, surface area, concentration (g/L) in a bioreactor, and surface coatings all influence mixing requirements, the amount of cells obtained per milliliter of culture, and ultimately product quality. To increase quantities, cells need to colonize more microcarriers. To achieve that goal, you can rely either on bead-to-bead transfer or on traditional subcultivation techniques (e.g., trypsinization). During growth in a bioreactor, microcarriers must predominantly remain in suspension. The vessel design must ensure proper mixing with minimal shear stress on cells regardless of the tank size. Increasing volumes also need to be managed, with an ultimate goal of guaranteeing product quality and its comparability with cells obtained on a planar surfaces. All these steps require process-development efforts that should not be underestimated. The timeline has to be carefully considered. However, microcarriers are scalable to very high-volume manufacturing and do represent the lowest-cost alternative for large production of cells per lot. Processing: Once billions of cells have been generated, they must be harvested, concentrated, and washed, then formulated and finally filled into their final containers — all under time pressure. Upstream manufacturing S eptember 2015 13(8)sp BioProcess International 37 Figure 3: Pall approach — product life cycle T-Flask, Multilayers Cost of Goods (CoG) Microcarriers Process Development Costs Multilayer Bioreactor — Xpansion Product Comparability R&D Phase 1 Phase 2 Phase 3 Commercial ! Commercial ! (high volume) Process Development Table 1: Downstream process scale-up — available technologies Continuous Fluidized (e.g., Ksep) 400–9,600 100–1,600 + Parameter Flow rate (mL/min) Hold-up volume (mL) Process development effort and downstream clarification operations thus need to be aligned. The main consideration in large-scale harvesting include detaching cells from their substrate (either a planar surface or 3-D microcarriers). After that, the cells must be separated from the microcarriers before concentrating, washing, formulating, and inline filling the cell therapy product. Table 1 lists a few common technologies used to concentrate cells along with their indicative flow rates and outputs. Continuous centrifuge systems are the closest thing to laboratory batch centrifuges and thus require the least effort in process development. Along with tangentialflow filtration (TFF) systems, they feature greater flow rates than the others. Clinical Considerations: Figure 3 shows different technologies available for large-scale culture of allogeneic adherent cells. Throughout clinical development, an increasing number of patients will receive cellular doses, with a significant increase beginning in phase 3. Development costs will rise and plateau until phase 1 scale-up is complete. Not only must a marketed 38 BioProcess International 13(8)sp Continuous Pelleted (e.g., Unifuge) 3,000 1,700 – Tangential-Flow Filtration 20–12,500 100s to 1,000s +++ product demonstrate comparability, but it also should be manufactured in an optimized culture system. Progressing through clinical phases require time and effort. Pall supports its customers in this endeavor by providing innovative single-use technologies, teams of specialists for process development assistance, automation and validation, and a global manufacturing presence to secure the supply. When developing an industrial process from research and development (R&D) to good manufacturing process (GMP) production, a company should focus on market approval. In an iterative approach to implement fundamental changes, both upstream and downstream processes should be considered as a whole. Three primary reasons why cell therapy companies fail are inappropriate business plans, overestimated market penetration, and underestimated costs. Making the informed and correct process design decisions will help to align technology and product roadmaps to bring a highquality product to market at the right moment — and at the right price. S eptember 2015 Professor Suzanne Farid leads research into novel, computer-based decision-support tools that provide systematic foundations to help companies make better decisions with inevitable uncertainty in process performance, market projections, and clinical success rates. Her team’s research focuses on establishing and integrating modules on bioprocess economics, manufacturing logistics, dynamic simulation, uncertainty analysis, multiobjective decision making, and combinatorial optimization. Below, she presents insights from the advanced bioprocess economics model designed by her team. An Advanced Bioprocess Economics Model Over the past 15 years, cell therapies have begun to reach the market. Some companies have faced challenges to achieving scalable, cost-effective, and robust cell therapy manufacturing — leading to notable failures caused by manufacturing concerns, such as high CoG. Now the industry is asking, “How can cell therapies achieve the manufacturing success of biopharmaceuticals?” A significant proportion of cell therapy products in development are allogeneic. Obtaining cells from universal donors is closer to a traditional, product-driven biopharmaceutical business model than patient-specific therapies. “Offthe-shelf ” products and processes can benefit from scaling up. Yet key differences remain between biopharmaceuticals and cell therapies, creating unique manufacturing challenges for the latter. Protein-based biopharmaceuticals have benefited from the availability of large-scale technologies and associated economies of scale. That is not the case of cell therapies because of their relative novelty and the inherent complexities of manufacturing living cells as products. A further challenge is the adherent nature of the types of cells used for allogeneic therapies. They come from healthy donors and have limited expansion potential, complicating matters with large Sponsored Supplement Figure 4: Decisional tool for cell therapy manufacturing Demand Technology Options Process/Facility/Cost Parameters Demand (103 doses/year) Cell type Figure 5: Lot size and market demand Lot Size (doses/lot) Decisional Tool Decisional tool integrated: C# • Process economics • Optimization • Visualization Case study scope: • Allogeneic manufacture • Optimal USP & DSP kits • Current technology gaps • Performance targets Bioprocess Economic Model Optimization Algorithm Origin MS Access Visualization Tools MS Excel Data Analysis Tools Database Graphical User Interface Visual C# Optimal upstream and downstream process strategy for each demand Cost of goods per dose (CoG/dose) and CoG breakdowns Sponsored Supplement database of process facility and cost parameters. Key outputs describe the optimal technologies to use for upstream and downstream processing, with details of their CoG structure. Using a step-by-step approach, we first looked at the economic competitiveness of cell culture technologies and then at volumereduction decisions. Finally, we explored cost implications of making process changes at different times throughout a product’s life cycle. Case Study — Cell Expansion (1): We explored multiple scenarios with different demands, lot sizes, and 50 100 20 10 500 1,000 2,500 10,000 <10 lots/year 5 100 50 10 10 200 100 20 10 100 50 20 >200 200 100 lots/year 40 10 200 50 50 100 500 doses, considering million- to a billion-cell doses. Figure 5 shows lots per year for each combination of lot size and demand. For each of practical combination of lot size and demand, we ran an optimization to pick out the most cost-effective technology. We considered planar technologies (e.g., 10-layer vessels and multilayer bioreactors) and microcarriers in singleuse bioreactors. The tool identified that, for low-dose scenarios, planar technologies are feasible (blue in Figure 6). We extended our analysis across all dosages (106 –109 cells). The figure shows where 2D technologies cease to be feasible and it’s better to switch to 3D culture: at the high doses of 108 and 109 cells with four large lot sizes (pink in Figure 6). The gray area in the bottom right-hand corner of the figure for very high-demand, high-dose scenarios indicate production scenarios Figure 6: Allogeneic cell expansion decisions — optimal technologies across demand/lot-size matrix and dosage requirements; blue indicates where planar technologies are feasible, pink where microcarriers in bioreactors are the only option, and gray where no upstream technologies exist. Blue indicates where planar technologies are feasible, pink where microcarriers in bioreactors are the only option, and grey where no technologies exist. Lot Size (doses/lot) Demand (103 doses/year) 50 Demand (103 doses/year) commercial demand. Another key difference is that single-use technologies are considered essential for cell therapies because of sterility concerns, putting further constraint on technology options available. At University College London (UCL), we investigated production processes used for cell therapies in both clinical and commercial processes and found that they tend to rely on T-flask or 10-layer trays. However, based on typical dosages and market demand projections, we estimated that commercial therapies could require lot sizes of over a trillion cells. To meet a maximum demand of 10-trillion–cell lot sizes, you would need 100,000 10-layer vessels. That is a very large number, but operators can handle only about 50–100 vessels per lot. This points to a need for alternative technologies that can support sufficient cell numbers for commercial lot sizes. We created a decisional tool for identifying the most cost-effective technologies for manufacturing commercial allogeneic cell therapies. We tested it across a range of different scales and identified technology gaps and technical innovations required to fill them. Our tool combines bioprocess economics with optimization and visualization (Figure 4). Key costs captured in the tool cover materials (e.g., media and single-use components), labor, quality control (lot release testing), and fixed overhead. Key inputs include cell type and demand, technology options, and a 1 1 100 Lot Size (doses/lot) 500 1,000 2,500 10,000 50 100 500 1,000 2,500 10,000 Dose = 10 cells Dose = 107 cells Dose = 108 cells Dose = 109 cells 6 5 10 50 100 500 1 5 10 50 100 500 S eptember 2015 13(8)sp BioProcess International 39 Figure 7: Allogeneic cell expansion decisions — technology S-curve for cell therapy manufacture Target: 10,000 billion cells/lot (e.g., lot size = 10,000 doses, dose = 109 cells) 10,000 5,000 Performance (109 cells/lot) 1,000 500 Microcarriers Automated multilayers Multilayered bioreactors 100 50 Compact flasks and multilayers 10 5 1 0.5 commercial lot sizes, where even microcarrier technologies would need to double their performance. The team then looked at how this could be achieved. Doubling the cells/lot can be achieved through different combinations of cell concentrations and number of single-use bioreactors (Figure 8, left). The right side of Figure 8 shows combinations of microcarrier densities and surface areas required to achieve the desired cell concentration. Multilayers Case Study — Volume Reduction (2, 3): Trillion-cell lot sizes pose challenges for volume reduction and downstream processing. The current approach is to use bench-top centrifuges. But to meet the maximum demand for very high lot sizes would require a ridiculous number of those instruments (25,000). So we explored the potential of more scalable technologies, determining that they would also need to be single use. The choices were TFF and single-use, fluidized-bed centrifugation. Again, our goal was to identify the most costeffective technology and look for bottlenecks. Figure 9 overlays downstreamprocessing bottlenecks with the optimal cell culture technology matrices from the previous case study. T flasks 0.1 R&D Effort and Investment that cannot be met by any existing technologies because the number of units required per lot exceeds the maximum allowable. We created a technology “S” curve to visualize performance trajectories and limits in cell output for each technology (Figure 7). For each one, we plotted the maximum number of cells per lot against the R&D effort. Each technology covers one log of performance (in billions of cells per lot) before it must be replaced by a newer technology. For example, T-Flasks will allow users to reach a billion-cell lot size, then 10-layer vessels will reach up to 10-billion–cell lot sizes. The output of all planar technologies caps at ~500 billion cells per lot, above which microcarriers become the only feasible option. The target was to reach 10 trillion cells per lot. But a technology gap exists for meeting the largest Figure 8: Allogeneic cell expansion decisions — future performance targets for microcarrier applications 8 Cell harvest density 20,000 cells/cm2 6 Microcarrier surface area 5 Microcarrier density 4 3 10,000 Production target 10,000 A 2 B 1 0 1 2 3 4 5 6 Number of Single-Use Bioreactors 7 LEFT: Base Case: 0.5M cells/mL Production target can be achieved through different combinations of cells/ml and #SUBs: (A) 2.6 million cells/mL with three SUBs per lot; (B) 1.3 million cells/mL with six SUBs per lot 8 Microcarrier Surface Area (cm2/g) Million Cells/mL Characteristics of point X: Billion cells/lot 7 8,000 cm2/g 16 g/L it y 2 20,000 ns ) de /cm s t e lls e r v (c Ha nge 25,000 ra 30,000 9,000 8,000 y si t 2 ) en m 20,000 t d lls/c s e e r v (c 25,000 Ha nge ra 30,000 1.3 7,000 6,000 5,000 But currently: 360–5,500 cm2/g (literature) X 2.6 4,000 3,000 2,000 Million cells/mL 5 RIGHT: Windows of Operation: 10 15 Microcarrier Density (g/L) 20 Desired number of cells/mL can be achieved through different combinations of microcarrier density, surface area, and harvest density. 40 BioProcess International 13(8)sp S eptember 2015 Sponsored Supplement Sponsored Supplement Figure 9: Allogeneic cell downstream process decisions — downstream processing bottleneck identification across demand/lot-size matrix and dosage requirements; blue indicates where planar technologies are feasible, pink where microcarriers in bioreactors are the only option, and gray where no upstream technologies exist. Lot Size (doses/lot) Demand (103 doses/year) Demand (103 doses/year) 50 100 Lot Size (doses/lot) 500 1,000 2,500 10,000 1 50 100 500 1,000 2,500 10,000 Dose = 10 cells Dose = 107 cells Dose = 108 cells Dose = 109 cells 6 5 10 50 100 >200 500 lots/year 1 5 m ea tr n g ns si ck w ce s n e D o pro t tle bo A bottleneck occurs downstream in the pink region, as you move to large, single-use bioreactors with microcarriers (often >1,000 L in volume). Those large, single-use bioreactors would need to be staggered, and only fluidized-bed centrifugation would be an option. We also explored whether CoG is dominated by upstream or downstreamprocessing costs. It is also important to consider cells/lot because the ratio of upstream to downstream processing costs varies with scale. For planar technologies, as scales increase, the ratio of upstream to downstream costs moves from being a 50–50 split to one that is dominated by upstream processing costs. By contrast, for microcarrier processes, the cost of goods is dominated by downstream processing. This type of analysis can help identify where to focus R&D efforts. We then investigated CoG targets as a percent of sales (in relation to reimbursement values) by plotting CoG/dose for different multilayer vessels and microcarrier processes against typical selling prices. For the high-dose scenario of 109 cells/dose, we assumed that allogeneic therapies would have gross margins in line with those of other biologics. If that target is for a CoG as 15% of sales, only microcarrier processes would meet the target and allow for a successful business model. If the selling price is even lower, as some reports suggest, then not even microcarriers could help a company meet that target gross margin without process improvement. More Work to Be Done: In the examples above, results predict that for the cell therapy industry to be sustainable in high-demand cases, technical innovation and optimization are still required to make costeffective and scalable manufacturing processes possible. However, recent research and offerings from some vendors to bridge the gaps that are currently restraining large-scale commercialization suggest that this will be achievable. Our model is being extended in on-going collaborations: e.g., how uncertainties affect robustness of manufacturing processes, as well as 10 50 100 500 their CoG and reimbursement economics. We’re also working to provide a framework for reconciling conflicts between financial and operational benefits for different technologies. And in parallel, the UCL team is exploring how to design feasible business models for autologous manufacture and providing a detailed economic evaluation of centralized and decentralized facility configurations. Questions and Answers The industry faces many challenges to the manufacture of allogeneic cell therapies. With the end goal of market approval in mind, a stepwise approach to fundamental changes is critical. Both upstream and downstream processes should be considered as parts of the whole manufacturing process. Cost, scalability, and flexibility are some key considerations to take into account when choosing the best manufacturing platform for a cell therapy. Dose, patient population, desired scale, and number of lots required are critical determining factors in technology selection. The UCL decisional tool can help you identify optimal technologies for commercial cell therapy bioprocesses and technical innovations required to move these therapies forward. Here are some questions posed by clients interested in these topics. What about porous microcarriers as an alternative to surface-only microcarriers? (TB) Porous microcarriers increase the surface available for cell growth, but they also create physical gaps in some portions where cells can grow but the microenvironment is modified, which could affect cell quality. So although it may be good that you have more cells, their quality may not remain consistent and comparable to that of early cell cultures. What factors do companies need to consider when switching from planar to microcarrier technologies? (SF) Microcarriers can offer >50% CoG savings at commercial scale. Consider whether the CoG savings at commercial scale would outweigh the higher cost of development. Also consider the best time to switch, whether early or late in development or after product approval. Analyses have been extended to capture the consequences of switching from planar to microcarrier technologies on drug development, clinical and commercial manufacture, and clinical trials. Considerations included their effects on process development, tech transfer, comparability studies, and process S eptember 2015 13(8)sp BioProcess International 41 validation or qualification batches. And from a clinical trial perspective, it’s important to consider extra bridging studies and whether you may have parallel arms in your clinical trial studies. So the outcome depends on whether you take a drug-development or product life cycle perspective. That will affect the weightings you assign to costs (of drug development and commercial CoG savings). Go–no-go decisions are also influenced by the potential market size, dose, and therapeutic selling price. What cell types have been successfully grown in the Xpansion bioreactor? (TB) The bioreactor was developed after we were contacted by a potential user who wanted to grow stem cells and maintain them in the same microenvironment as in a multilayer stack. So the default use is for stem cells of different origins. But there have been some successful attempts to grow mammalian cell lines (e.g., VERO) and others for gene therapy, as well. How would you sum up the current state of cell therapy manufacturing? (SF) There are bottlenecks in both the upstream and downstream processes, particularly for the high-demand, high-dose scenarios. This offers innovation potential for technology providers to adapt existing technologies for cell therapies and help bridge the gaps constraining large-scale production. Existing characterization methods make comparability difficult and can act as a disincentive to process changes once human trials have begun. So choosing the best technology early in development can be critical to success. And to pick the right technology early on, you need to have an outlook toward the potential commercial scale and consider the whole life cycle of your drug. That will help ensure success of cell therapies overall, to push them through development to market. What is the primary driver for choosing planar or 3D culture platforms in cell therapy applications? (TB) That would be the ultimate production scale. Current planar systems can achieve only 5 × 1011 cells 42 BioProcess International 13(8)sp at maximum. If you need to go higher than that, then you need to use a 3-D system. Some people are discussing microfibers and hydrogels, but they present some significant problems and have their own limitations of scale. So cost and scale of manufacturing are the main drivers and space as well. With classical multitray, you would need a huge space and many technicians to manage so many units. That’s becoming really problematic. What is the best time to begin process development for scaling up to a microcarrier bioreactor platform to volumes required in phase 3 and beyond? (TB) You have to look at the number of cells you need to produce for the market. You should know how many cells will be needed per patient during or at the end of phase 2. Some people focus on getting to market as quickly as possible with as few process changes as possible just because they are running short of cash and want to move on and raise additional funding. They would have to switch to 3-D a bit later if they need to scale it up. Others to consider going 3-D immediately or as early as possible and really scale up during clinical testing, thus avoiding the switch and maintaining product comparability. It’s always the client’s choice, and our recommendation is to transition whenever you can, knowing that it will represent hard work and eventually additional clinical trials unless you start as early as possible using microcarriers — if that will be necessary in the end. Quality control is critical. You need to ensure that the comparability of the cells remains consistent across the different technologies that you use. So quality control really needs to be cast in stone. What are your general observations regarding cell culture media requirements for the different approaches to cell expansion? (TB) The Xpansion bioreactor was designed to consume the same amount of medium as in a regular cell stack or multitray stack. If you use ~100 mL/layer, then for 20 10-layers you would use 20–30 L. When it comes to microcarriers, you’re going to get more cells per S eptember 2015 volume, so you may consume more medium per batch. But if you consider the amount of medium consumed by each cell, then it remains roughly the same as the planar technologies. Costwise, as you scale up processes and move toward higher demand, you’ll see that material cost becomes a much more significant proportion of CoG. With planar technologies, consumables dominate over media costs; in 3D microcarrier processes, media costs are more dominant than consumables. In all cases, industry needs to move towards a less expensive medium free of animalsourced ingredients, which will follow the media development that we’ve seen for mammalian cell culture. What key questions does the sector need to address for successful commercialization of cell therapies? (SF) It will be underpinned by a number of factors. First, we need a cost-effective and robust manufacturing process at large scale — and in particular, downstream technologies must catch up to the upstream. In addition, transportation and logistics infrastructure will be critical to cell therapy success. Companies need to ensure that they have feasible business models early on to help them decide whether a process is cost effective or requires innovation. They also need to think about that in relation to reimbursement levels. Successful companies will have the know-how related to manufacturing logistics, reimbursement, management of clinical trials, and marketing to create that successful business model. Many people are still using traditional systems such as multiplate stacks for development and early stage clinical trials. What do you believe is the biggest challenge to using those systems as cell therapies move through the clinical pathway to commercialization? (TB) First, they are very labor-intensive just to manipulate large numbers of multitray stacks. And you can imagine the space needed to incubate hundreds of multitray systems. You Sponsored Supplement have to manipulate them and protect them in cleanrooms, validate them, and so on. The volumes are critical. Traditional systems are still very open and not well controlled. Basically, you put the trays under an incubator and rely on that to control the environment in which cells grow. GMPs are about control, so moving toward bioreactors is a major step forward and a critical process improvement. Finally, multitray systems will not be able to support large-scale production at industrial levels. Producing some allogeneic therapies would require the equivalent of one or two Empire State buildings just for incubation space, which is pretty unrealistic. Acknowledgments UCL’s cost modeling outputs shown here were part of a project funded by the UK Technology Strategy Board with Lonza as the lead partner. Financial support from the Technology Strategy Board (UK) and Lonza is gratefully acknowledged. Constructive feedback and technical advice from industrial experts at Lonza and vendors is gratefully acknowledged. References 1 Simaria S, et al. Allogeneic Single-Use Cell Expansion Decisions. Biotechnol. Bioeng. 111(1) 2014: 69–83. 2 Hassan S, et al. Allogeneic Single-Use Volume Reduction Decisions. Regen. Med. 2015 (in press). 3 Hassan S, et al. Process Change Evaluation Framework for Allogeneic Cell Therapies (in preparation). • Thierry Bovy is global product manager of Xpansion multiplayer bioreactor systems (thierry_bovy@pall.com), and Alain Fairbank is director of marketing for cell therapies at Pall Life Sciences (alain_ fairbank@pall.com). Suzanne Farid is a professor in bioprocess systems engineering at University College London’s department of biochemical engineering; s.farid@ucl. ac.uk. Sponsored Supplement Featured Products SoloHill® Microcarriers Applications: Large-scale culture of anchorage-dependent cells Features: SoloHill microcarriers provide relief from the limitations of two-dimensional (2D) flatware culture, offering the advantage of a larger ratio of surface area to volume that supports high adherentcell densities in suspension culture systems. This technology is based on a solid polystyrene core that prevents absorption of serum, cell culture products, and dissociation enzymes. Available microcarrier types include charged/coated carriers designed to foster cell attachment and come in animal-protein–free formats. Xpansion® Single-Use Bioreactors Applications: Cell therapy manufacturing without changing cell environment Features: The Xpansion single-use bioreactor was developed for largescale, two-dimensional (2D) cell culture of fragile adherent cells such as stem cells. The modular design uses stacked polystyrene plates, which offer the same cell environment as conventional multiple-tray stacks to enable straightforward process transfer from traditional technologies. The polystyrene core provides for easy handling and preparation, immediate solubility, and multiple sterility options, including gamma irradiation. These microcarriers support cell growth and recovery at a range of scales: spinner flasks, small- to large-scale stirred-tank bioreactors, and rocking platforms. Pall’s newest SoloHill product (Hillex® CT microcarriers) has the lowest particulate content of any microcarrier available today, providing an ideal substrate for expansion and recovery of stem cells in research and cellular therapy applications. An Xpansion bioreactor has a cellgrowth surface area of ≤122,400 cm2, so a single bioreactor can replace ≤200 stacked trays. It offers real-time monitoring and control of cell culture parameters (pH, dissolved oxygen, and temperature) to ensure reproducibility and data recording. Cell density and morphology both can be monitored with a holographic microscope. A closed system design protects cells from potential contamination. The Xpansion bioreactor represents an excellent solution for adherent cell cultures to drive cell therapies to market faster while reducing cost of goods (CoG) and maximizing safety by lowering the number of operations needed. Contact Pall Life Sciences microcarriers@pall.com Contact Pall Life Sciences bioreactors@pall.com S eptember 2015 13(8)sp BioProcess International 43 S e c t i o n T h r e e CELL THERAPY MANUFACTURING Production of Viral Vectors Using the iCELLis® Fixed-Bed Bioreactor System Beyond Mesenchymal Stem Cells: Gene-Modified Cell Therapy, Gene Therapy, and Exosomes by Matt Kremer I n the past few years, the resurgence of cell-based immunotherapies — and, by extension, gene therapies — has accelerated as products move rapidly from academic research laboratories into commercial development. A successful clinical trial by St. Jude Children’s Research and the University College London of a gene therapy for hemophilia B was a seminal translational event (1), as was the licensing of the University of Pennsylvania’s gene-modified chimeric antigen receptor T-cell technology (CAR-T) for leukemia by Novartis (2). CAR-T cells and gene therapies are now among the most dynamic areas for biotechnology investment and development. Both gene therapies and gene-modified cell therapies require large amounts of viral vectors, either for direct delivery to patients or for ex vivo modification of cells that are later dosed to patients. Another exciting area of research and development is that of exosomes, by which stem cells are cultured with the intent of generating secreted factors that can be administered for a therapeutic effect. Instead of administering cells that will provide growth and trophic factors in situ, the potential exists for those factors to be produced ex vivo, then harvested and subsequently administered for therapeutic purposes. 44 BioProcess International 13(8)sp Figure 1: The iCELLis bioreactor (Pall Life Sciences) is a scalable, single-use, fixed-bed bioreactor. Many processes for the production of viral vectors require the large-scale culture of adherent cells. Although more traditional two-dimensional (2D) culture technologies are sufficient for producing phase 1 and 2 materials, they may not be sufficiently scalable when companies move down the industrialization pathway toward phase 3 clinical trials and commercial production. Single-Use, Fixed-Bed Bioreactors Some companies are attempting to develop suspension culture processes, but fixed-bed bioreactors have emerged as an ideal technology to support large- S eptember 2015 scale production of viral vectors — e.g., >1016 viral particles (vp) per batch. One primary advantage that comes with the use of fixed-bed bioreactor technology is the large surface area to which adherent cells can attach, expand their numbers, and then be infected or transfected for viral-vector production. Other major advantages include the ability to achieve higher density cultures; real-time control of parameters such as pH, dissolved oxygen (DO), temperature, perfusion, and agitation; and low shear stress for shear-sensitive cells (3). Fixed-bed technologies are currently used for commercial Sponsored Supplement biologics production. Until recently, what has been lacking is a large-scale, single-use system. The iCELLis bioreactor (Figure 1) is a fully integrated, high–cell-density bioreactor designed to simplify adherent-cell culture processes by combining the advantages of singleuse technologies with the benefits of a fixed-bed system. Designed for rapid implementation and ease of use, this compact system represents a new generation of single-use bioreactors. The fixed bed in an iCELLis system is based on 0.62-cm × 2.5-cm macrocarriers made of hydrophilized, nonwoven, medical-grade polyethylene terephthalate (PET) microfibers, ensuring excellent cell growth in a 3-D environment (Figure 2, Figure 3). Contained in a housing through which media flows from bottom to top, the macrocarriers remain fixed in place. The iCELLis system comes in two formats: the benchtop iCELLis Nano system and the large-scale iCELLis 500 bioreactor. The height of their fixed beds can vary (2, 4, and 10 cm), as can the density of carrier packing. Table 1 lists available configurations for both models. Customers can choose among three bed heights and two compaction densities for a total of six fixed-bed configurations. So the iCELLis Nano ranges from 0.53 m 2 to 4.0 m 2 of surface area for cell growth; the commercial scale ranges 66–500 m 2 of surface area. Table 2 compares iCELLis surface areas with those of other commercial systems. The scaling factor between iCELLis Nano and iCELLis 500 systems is 125×. Both are fully functional bioreactor systems that include monitoring and control of pH, DO, temperature, and agitation. Notable is the constant bed height and compaction density for scaling. Each small-scale iCELLis Nano bioreactor has a corresponding largescale iCELLis 500 unit with the same bed height and compaction. The cellular microenvironment is the same in an iCELLis 500 as in an iCELLis Nano system, so a process developed in the latter can be transferred directly to the corresponding iCELLis 500. Additionally, significant biomass Sponsored Supplement Figure 2: The iCELLis fixed-bed macrocarrier substrate: polyethylene terephthalate (PET) fiber carriers Table 1: iCELLis bioreactor configurations at small and manufacturing scales Bioreactor iCELLis Nano iCELLis Nano iCELLis Nano iCELLis 500/100 iCELLis 500/200 iCELLis 500/500 Fixed-Bed Bioreactor Diameter Height Volume Volume 110 mm 20 mm 0.04 L 1L 110 mm 40 mm 0.08 L 1L 110 mm 100 mm 0.20 L 1L 860 mm 20 mm 5.00 L 70 L 860 mm 40 mm 10.00 L 70 L 860 mm 100 mm 20.00 L 70 L Surface Area Low High Compaction Compaction 0.53 m2 0.8 m2 2 1.06 m 1.6 m2 2 2.65 m 4.0 m2 2 66.00 m 100.0 m2 2 133.00 m 200.0 m2 2 333.00 m 500.0 m2 multiplication can occur in the fixed bed, which simplifies seed-train development. It is feasible to inoculate an iCELLis bioreactor at low cell density (e.g., 5,000 cells/cm 2 or even lower) and then amplify cells in the fixed bed to easily reach up to 500,000 cells/cm 2 or higher. iCELLis System for Viral Vector Production Both gene therapies and genemodified cell therapies require viral vectors to introduce a gene of interest into target cells. Gene therapy viral vectors are administered to patients. For gene-modified cell therapies (e.g., CAR-T cells), a patient’s own target cells are isolated, then modified by a gene introduced by a viral vector, expanded in vitro, and finally returned to the same patient. Several viral vector types have been used for such applications: e.g., adenovirus, adenoassociated virus, retrovirus, and lentivirus. Some of those are produced in stable producer cell lines; others require transient transfection. The iCELLis system has emerged as a premier platform for viral vector production, with successful academic Figure 3: Micrograph of cells growing on iCELLis Microfiber substrate. and commercial operations using such standard adherent cell lines as human embryonic kidney (HEK 293) cells, adenocarcinomic human alveolar basal epithelial (A549) cells, and mouse leukemia (PG13) cells. Retrovirus: A recent issue of the Journal of Immunotherapy included an article by researchers at Memorial Sloan Kettering Cancer Center (MSKCC) (4). They reported results from comparing retrovirus production by stable packaging PG13 and HEK 293Vec cell lines in an iCELLis Nano system and a Nunc Cell Factory S eptember 2015 13(8)sp BioProcess International 45 Table 2: Comparing surface areas for iCELLis Nano and 500 bioreactors with those of other systems for adherent cell culture; scaling factor between iCELLis Nano and iCELLis 500 systems is 125×. Bed Height (Compaction) 2 cm (C1.0) 2 cm (C1.5) 4 cm (C1.0) 4 cm (C1.5) 10 cm (C1.0) 10 cm (C1.5) iCELLis Nano 850-cm2 Roller 10-Layer Cell Bottles Stacks (CS10) 6.2 0.8 9.4 1.3 12.4 1.7 18.8 2.5 31.8 4.3 47.1 6.3 Surface Area 0.53 m2 0.80 m2 1.06 m2 1.60 m2 2.65 m2 4.00 m2 system (Thermo Scientific), which is a familiar 2D multiplate system. High-titer vector stocks were harvested over 10 days, representing a much broader harvest window than the three-day harvest window afforded by cell factories. For PG13 and 293Vec packaging cell lines, the average vector titer and the vector stocks’ yield in the bioreactor were higher by 3.2- to 7.3-fold and 5.6- to 13.1-fold, respectively than those obtained in cell factories. The vector production was 10.4 and 18.6 times more efficient than in cell factories for PG13 and 293Vec cells, respectively. Furthermore, the vectors produced from the fixed-bed bioreactors passed the release test assays for clinical applications. Therefore, a single vector lot derived from the 293Vec is suitable to transduce up to 500 patients cell doses in the context of large clinical trials using chimeric antigen receptors or T-cell receptors. These findings demonstrate for the first time that a robust fixed-bed bioreactor process can be used to produce γ-retroviral vector stocks scalable up to the commercialization phase. (4) Total vector production by 293Vec cells was reported at 2.53 × 1012 in an iCELLis Nano 200-mL bioreactor, which features a surface area of iCELLis 500 HyperStack 36 0.3 0.4 0.6 0.9 1.5 2.2 Surface Area 66 m2 100 m2 133 m2 200 m2 333 m2 500 m2 850-cm2 Roller Bottles 776 1176 1565 2353 3918 5882 26,500 cm2 (2.65 m2), compared with production at 1.93 × 1011 in a 6CF10 with a surface area of 38,160 cm2 (3.816 m2). If scaled to the corresponding iCELLis 500 bioreactor with 333 m2 of available surface area, a theoretical vector yield could reach 3.2 × 1014/batch. Reported vector production in PG13 cells was 2.9 × 1010 for the iCELLis bioreactor and 3.96 × 109 in a Cell Factory system. At the corresponding iCELLis 500 scale, vector production could reach 3.6 × 1012/batch. Yields could be further increased 1.5× by scaling to higher-compaction bioreactors that were not available at the time of MSKCC’s evaluation. Adenovirus: Finvector, a contract manufacturer based in Finland, presented its work with the iCELLis system at industry conferences including the March 2015 ISBiotech Spring Meeting (5) and the annual conference of the American Society for Gene and Cell Therapy (6). The company scaled a transient transfection adenovirus process using HEK293 cells from an iCELLis Nano system to a 100-m2 iCELLis 500 bioreactor, achieving a reported 6 × 1015 viral particles/batch. Additional scaling to a 500 m2 iCELLis could increase the yield up to fivefold. Recombinant Adenoassociated Virus (rAAV): At the 2013 European Society for Gene and Cell Therapy (ESGCT) meeting, researchers from the University 10-Layer Cell Stacks (CS10) 104 157 209 314 524 786 HyperStack 36 37 56 74 111 185 278 College London (UCL) presented results of three plasmid rAAV transfections into HEK 293T cells, again comparing an iCELLis Nano bioreactor with a Cell Factory control (7). Conclusions reached in this study included that the . . . iCELLis Nano system appears to be suitable for the development of industrial scale production of rAAV vectors by transfection. Optimization of the culture parameters and harvesting method will likely allow for a similar productivity compared to conventional cell culture platform (CF10 harvested by freeze/thaw technique). Linear scalability is extrapolated to produce viral yields of at least 2–5 × 1015 vg (viral genomes) per run at large scale in a 133 m2 iCELLis bioreactor. In a poster at the same conference, Rentschler Biotechnologie presented results of an initial experiment with a two plasmid rAAV construct in HEK 293T cells. That team observed similar cell counts and metabolite profiles between iCELLis and their control, concluding, Overall, this first set of data is very promising, especially when considering that the underlying process was optimized for 2D culture vessels. The controlled iCELLis bioreactor system offers Table 3: Comparing iCELLis results with yield extrapolation to iCELLis 500-m2 fixed-bed Vector Retrovirus Adenovirus rAAV Group MSKCC MSKCC Finvector UCL Rentschler 46 BioProcess International Cell Line HEK 293Vec PG13 HEK 293 HEK293T HEK293T 13(8)sp Scale nano nano 100 nano nano S eptember 2015 Surface Area 2.65 m2 2.65 m2 100.00 m2 0.53 m2 0.53 m2 Vector Yield 2.53 × 1012 2.9 × 1010 6 × 1015 2.3 × 1013 2.2 × 1010 Projected Yield at 500 m2 4.78 × 1014 5.48 × 1012 3 × 1016 2.17 × 1016 2.07 × 1013 Scaling Factor 189× 189× 5× 943× 943× Sponsored Supplement many possibilities for process development. For instance the results of the growth experiments . . . show that lower seeding densities could facilitate the seed train for large-scale production. Moreover, the fact that the cells could be grown to more than 2E+06 cells/cm 2 by simple medium exchange indicates the potential for process optimization and, therefore, possible improvement of productivity. Lentivirus: Viral-vector production in iCELLis with lentivirus is currently being pursued by several commercial entities for which publicly disclosed information is not yet available. At the 2015 ASGCT meeting, however, Finvector did present initial lentivirus results, indicating successful transfection and virus yield (8). Exosomes: An Emerging Therapeutic Approach Therapeutic application of exosomes is an emerging area of research and development. Exosomes are lipid bilayered nanovesicles secreted by cells. First described in the 1970s, exosomes are attracting recent commercial interest due to the recognition that they play important roles in paracrine and autocrine signaling (9). Exosomes secreted by stem cells may provide the same functional benefit as their parent cells but with reduced immunogenicity, simplified handling (e.g., long term stability without cryopreservation) and biomanufacturing through traditional cell culture techniques (10). Capricor Therapeutics Inc. (Los Angeles, CA) and ReNeuron Group plc (Guilford, UK) have both announced exosomedevelopment programs. For production of exosomes, the iCELLis bioreactor can provide a continuous process environment for long-term cultivation of stem cells and harvest of exosomes through perfusion culture. Conclusion The varied nature of cell therapies — including gene-modified cell therapies — requires a range of manufacturing technologies to address different process and scale requirements for commercial-scale production volumes. Specifically, viral-vector production Sponsored Supplement processes require large surface areas for adherent cell culture. Fixed-bed bioreactors represent an ideal technology for supporting large-scale production of viral vectors. Pall Life Sciences’ iCELLis fixed-bed bioreactor is already being used worldwide for vector and virus production applications. The system provides surface areas up to 500 m 2 in a compact, closed, and single-use format, with possible yields reaching >1016 viral particles per batch. References 1 Nathwani, et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. New Engl. J. Med. 2011: 2357– 2365. 2 University of Pennsylvania and Novartis Form Alliance to Expand Use of Personalized T Cell Therapy for Cancer Patients. University of Pennsylvania: Philadelphia, PA, 6 August 2012; www.uphs.upenn.edu/news/News_ Releases/2012/08/novartis. 3 Rowley J, et al. Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy. BioProcess Int. 10(3) 2012: S16–S22. 4 Wang, et al. Large-Scale Clinical-Grade Retroviral Vector Production in a Fixed-Bed Bioreactor. J. Immunother. April 2015: 127–135. 5 Lesch. Process Development and Clinical Production of Viral Vectors for Phase I and Beyond. IS Biotech: Washington, DC, 2015. 6 Lesch, et al. iCELLis Fixed-Bed Technology for Adherent Cells in an Efficient, Scalable System for Viral Vector Production. American Society for Gene and Cell Therapy, New Orleans, LA, 2015. 7 Lennaertz A, et al. Adeno Associated Virus Production Using a Disposable, FixedBed Bioreactor from Bench-Scale to Industrial Scale. BMC Proceedings 7(S6) 2013: 59; www. biomedcentral.com/1753-6561/7/S6/P59. 8 Pegel A, et al. Setting Up TransfectionBased Recombinant AAV Production in the iCELLis Single Use Fixed-Bed Bioreactor. Rentschler Biotechnologie GmbH: Laupheim, Germany, 2013; http://rentschler.de/fileadmin/ Downloads/Poster/14760_REN_POS_Wissen_ Poster_841x1189mm_4c_4_Ansicht.pdf. 9 Ibrahim, et al. Exosomes As Critical Agents of Cardiac Regeneration Triggered By Cell Therapy. Stem Cell Rep. 2014: 606–619. 10 CTX-Derived Exosomes. ReNeuron Group plc: Guildford, UK, 2015; www.reneuron. com/products/ctx-derived-exosomes. • For copies of posters cited in this article, please contact the author: Matt Kremer is sales manager for new markets in cell culture technologies at Pall Life Sciences, 1-215-7565837; matthew_kremer@pall.com. Featured Products iCELLis® Fixed-Bed Reactor Applications: Efficient, scalable viral vector production for gene therapy Features: The iCELLis fixed-bed bioreactor technology has been validated from bench to process for production of human and animal vaccines, viral vectors, and recombinant proteins. These systems are available in two scalable platforms: the large-scale iCELLis 500 and the scaled-down iCELLis Nano bioreactor. They are different from any other platform for adherent cell cultures (e.g., roller bottles, cell factories, or stirred-tank reactors with microcarriers). With a compact, fixed bed of proprietary macrocarriers made of polyester microfibers, these systems provide ≤500 m² of growth surface area in just 25 L of fixed bed. They can be inoculated at very low cell density, simplifying seed-train operations with no intermediate reactor required. The iCELLis systems were designed for efficient and innovative media circulation and oxygenation. A builtin magnetic drive impeller evenly circulates media through the fixed bed to ensure low shear stress and high cell viability. These systems achieve and maintain high cell densities that equal or exceed the productivity of larger stirred-tank units. With cells trapped inside the fixed bed, no perfusion equipment is required. Contact Pall Life Sciences bioreactors@pall.com S eptember 2015 13(8)sp BioProcess International 47 S e c t i o n F o u r PERSPECTIVES Ask the Experts Core Technologies Expand Opportunities for Cell Therapy Manufacturing by S. Anne Montgomery P all Life Sciences has long been known for its expertise in processing and filtration equipment for the biopharmaceutical industry. In recent years, the company has broadened its offerings in upstream manufacturing by expanding its core capabilities in the single-use, bioreactor, and microcarrier arenas, with unique and innovative technologies for cultivation of cells to be used as therapies. To explain how its scientific and technology expertise is a logical fit with this burgeoning area of the industry, three experts from various Pall business segments were asked to comment on the company’s process development capabilities, microcarrier technologies, and the Xpansion bioreactor system, specifically. Contributing their insights to this discussion are Fabien Moncaubeig (head of bioprocess development services at Pall Life Sciences in Brussels, Belgium), Mark Szczypka, (senior director, applications and new product development, Pall Life Fabien Moncaubeig 54 BioProcess International 13(8)sp Europe Brussels, Belgium — Process Development, Application Support Portsmouth, UK — Research and Development USA Westborough, MA — Process Development, Research and Development, Application Support Ann Arbor, MI — Process Development, Research and Development Sciences, Ann Arbor, MI, USA ), and Thierry Bovy (global product manager, Xpansion multiplate bioreactor systems, Pall Life Sciences, Brussels, Belgium). Process Development Capabilities — with Fabien Moncaubeig Fabien Moncaubeig introduces Pall Life Sciences’ bioprocess development services and Pall’s expansion into cell therapy bioprocessing. He then offers thoughts about cell therapy manufacturing technologies in general and the future of microcarrier-based platforms in particular. Pall Life Sciences approaches its cell culture and downstream bioprocess development services in two ways. Our on-site support uses a team of specialists who are experienced scientists and assist our customers at their sites in their labs. The team offers extensive training on our technology but also acts in an advisory capacity to S eptember 2015 our partners throughout the entire development process by analyzing results and designing adequate experimental plans with them. A second option is to outsource our customers’ complete process development (from upstream to downstream) to our labs. We offer contract development services in Europe and in the United States in four different locations: Brussels, Belgium; Portsmouth, UK; Ann Arbor, MI; and Westborough, MA. Historically, our process development activity in the cell therapy field has been driven by collaboration to codevelop our bioreactors (2D and suspension) and our microcarriers. Thanks to our partners, we have acquired significant expertise in optimizing and scaling up stem cell processes in 2D and 3D for both autologous and allogeneic Sponsored Supplement therapies. Given the demand from the field for bioprocess experts to help industrialize processes, we continued offering this service as a contract development organization (CDO). In the past few years, we have worked with a number of cell types from various sources, including mesenchymal stem cells (MSCs), human embryonic stem cells (hESCs), induced pluripotent stem cells (iPSCs), and hepatocytes. We are also industrializing viral vector production processes using adherent or suspension cultures at scales up to a thousand liters. How different are technologies for cell therapy bioprocessing from those of more traditional bioprocessing? The main difference resides in the product of interest. In cell therapy processes, the cell is your product, whereas for vaccines, viral vectors, or monoclonal antibodies (MAbs), cells are used only to secrete the product of interest. As a consequence, the cell therapy industry has been trying to fill a technology gap to allow efficient cell amplification and recovery whether cells are grown in suspension or adherent cultures. Also, unlike the cell lines used in traditional bioprocesses that have been selected and screened for years, stem cells are far more sensitive to their environment. This is why we have adapted these cell culture technologies. How do you see Pall‘s role in advancing cell therapies to commercialization? Does Pall have a complementary or competitive relationship with contract manufacturing organizations that focus on cell therapies? Our motivation is to increase process efficiency to help our partners reduce production costs. We are in an ideal position, thanks to a wide portfolio of innovative technologies from which we can guide our customers and give neutral recommendations. Whatever type of cells they cultivate and the culture mode required, we have a suitable platform for them: adherence in 2D or 3D or suspension in rocking or stirred bioreactors. Then we can support them to minimize their learning curve during process development and shorten their timing to enter clinical trials. Sponsored Supplement Pall’s intention is not to enter the CMO business, but to facilitate implementation of our bioreactors and downstream solutions. Our contract manufacturing organization (CMO) partners take over our process development once it’s optimized and scaled up to turn it into a GMP compliant process ready for clinical phase productions. Arguably, the most promising large-scale technologies for expansion of stem cells use microcarriers. What are key considerations for choosing a microcarrier-based cell-expansion platform? Companies that choose to use microcarrier platforms should determine the bioreactor-scale requirements for each clinical trial phase to ensure that process development milestones are compatible with demand for clinical material. A key factor for success is to acknowledge the level of expertise required for industrialization of microcarrier-based processes. When choosing the best bioreactor for a cell therapy process, what are key considerations? What role do you think automation can play? One of the key considerations is the type of culture: whether it is suspension or adherent. The next is to understand what the final scale targeted for each clinical and commercial phase will be and determine how you will produce your product. Manufacturers of allogeneic therapies will look for bioreactors that can produce a high number of cells, even if that requires more complex systems such as stirred tank bioreactors and microcarrier combinations. By contrast, makers of autologous therapies will lean toward simple, automated systems requiring small footprints and minimizing the number of operations per patient. The level of automation of largescale stirred-tank bioreactors is already quite high. On the other hand, there is a lot to do for autologous therapies in terms of automation. This will be key for minimizing production costs and batch failure rates while not compromising the costs of these future therapies. Other key considerations are the time and resources a company can afford to spend on its process transfer into its new platform. Not all bioreactors require the same amount of effort to reach desired cell doubling and quality. Bioprocessing has almost completely shifted to serum-free, protein-free, animal-free, and chemically defined media. This is much less the case for cell therapy processes in general. What are your thoughts regarding serum/serum-free (and so on) for cell therapies? Serum-free processes are very likely to become a standard in the cell therapy market; first because of regulatory pressures, and also because they offer better lotto-lot reproducibility. Finally, using animal sera introduces limitations such as significant supply and pricing fluctuations. Although pricing of serum-free media for stem cells might be considered prohibitive for industrial uses today, it’s fair to anticipate that increasing demand will drive prices down, resulting in scenarios similar to vaccine or monoclonal antibody markets, where costs of serum-free and serum containing media have become comparable. As an alternative to serum, platelet lysate is gaining popularity because it could help transition from serum to serum-free processes with its economic, regulatory, and ethical advantages. Mark Szczypka Focus on Microcarriers — with Mark Szczypka Traditional stem cell culture involves two-dimensional (2D) surfaces under static conditions. Mark Szczypka S eptember 2015 13(8)sp BioProcess International 55 responds to questions about this technology’s suitability for expanding cells to larger scales. Two-dimensional flatware systems have traditionally been the initial platform of choice for most investigators. There is an intrinsic familiarity with these systems because they have been in use for many years, and a significant body of cell characterization data has been collected in 2D systems. The industry has been somewhat reluctant to transition into threedimensional culture systems because cells are exposed to conditions not encountered in flatware. Cells are exposed to shear stress in dynamic cultures, and the configuration for growth can be different from what is observed on flat surfaces. For example, aggregates can form when fibroblastlike, adult stem cells are grown on microcarriers to high cell densities. Fortunately, significant amounts of data collected over the past decade demonstrate that adult stem cells can be grown successfully on microcarriers. This is beneficial because conventional 2D systems suffer from significant limitations when expanding large numbers of cells. Microcarriers provide the most economical and practical solution for generating multibillions to trillions of cells. Even the most optimized 2D systems — such as Cell Cube, HYPERstack, and Cell Factory systems as well as hollow-fiber devices — require significant labor efforts because of the large surface-area requirement for generating sizable numbers of cells. This requirement necessitates a significant number of units to generate the requisite cell numbers. As unit numbers increase, consumables and labor costs also increase significantly. Additionally an associated increase in the number of manual interventions that are “open” handling operations increases the risk of contamination of the cultures. Microcarriers used in standard or single-use bioreactors provide an ideal solution for these challenges. A single 200-L bioreactor containing microcarriers can replace thousands of cell factories and several hundred hollow-fiber units. This advantage affords the opportunity to significantly decrease the number of production runs required and associated labor to produce large numbers of cells. Some problems reported with microcarriers include difficulties with adherence of cells to their surfaces and with dissociating cells from them later. How are these issues being addressed? Cell attachment, adherence, growth, and harvest are influenced by multiple parameters including microcarrier surface composition, media formulation, vessel configuration used for culture, methods used for dissociation of cells from microcarriers, and processes used to separate cells from microcarriers. Multiple avenues are currently being pursued to address these perceived challenges. Microcarriers with unique surface characteristics that promote rapid cell attachment and tight adherence are commercially available. New microcarriers equipped with novel surfaces are under development. Additionally, media developers have recognized the need for optimization Figure 1: Estimated time periods for microcarrier process development and optimization Cell Characterization Microcarrier Screening and Selection Weeks 2–4 56 BioProcess International 4–6 13(8)sp Small-Scale Spinners 10–20 S eptember 2015 Bioreactor Process Development Scale-Up (20 to 200 Liters) 12–24 12–36 of medium formulations to support robust cell attachment and growth of stem cells in dynamic cultures. Simple and robust methods for cell dissociation from microcarriers are also being pursued. Microcarrier surfaces that facilitate cell detachment without enzymatic harvest are being developed, and standard methodologies for cell harvest from microcarriers are being optimized. Some commercially-available separation technologies for separating detached cells from microcarriers are available, and additional processes for efficient separation of dissociated cells from microcarriers are under development by several groups — including Pall Life Sciences. It is worth noting that separation technology at the thousand-liter scale has been accomplished by multiple groups both for stem cells and standard cell lines. How comparable are cell yields using microcarrier-based systems with yields achieved with other technologies such as stacked trays? Cell yields/numbers achieved in microcarrier-based systems are comparable to yields achieved through other technologies. Under optimized conditions it is possible to eclipse yields achieved in other systems; however, when optimizing these conditions, it is imperative that stem cell identity and functionality be keenly scrutinized. The overall impact of expanding stem cells needs to be further evaluated, but a significant amount of data have been generated by many groups to indicate that cell identity and function appear to be unchanged when cells are propagated under these conditions. Can you explain the differences in porosity among microcarriers that are available on the market today? Some suppliers and users advocate macroporous options, but you prefer rigid surfaces. Why? Microcarriers can be manufactured to be nonporous, nanoporous, microporous, and macroporous. Various options are commercially available. Nonporous and nanoporous microcarriers are typically rigid substrates that can undergo surface modifications to promote cell Sponsored Supplement attachment, adherence, and growth. Such microcarriers are ideal for cell harvest because standard enzymatic methods used to harvest cells from flatware can be ineffective for cell dissociation. Cells can be separated using simple methods based on differential settling, which depends on the differences in relative density and size of cells and microcarriers. Standard screening techniques also can be used because the rigid properties prevent microcarriers from obstructing or fouling screens. Microporous microcarriers have micron-sized pores that allow for efficient cell attachment and growth. But this property also can impede or inhibit efficient, reproducible cell harvest from microcarriers. These properties are likely to be due to the ability of cells to extend filapodia into a microcarrier surface. Although this phenomenon can promote efficient cell adherence, it can also inhibit cell dissociation from the microcarrier surface. Many microporous microcarriers are also elastic and sticky. Microcarriers with these inherent properties tend to readily adhere to exposed surfaces and cause sieving screens to foul, complicating cell harvest. Macroporous microcarriers have pores large enough to allow cells to grow within the interior of the microcarrier. The potential benefit of this configuration is that cells are protected from shear stress encountered in dynamic cultures. However, because cells are propagated within “niches” for long periods of time, the maintenance of appropriate cell identity and function can be compromised. Harvesting cells from microcarrier pores can also be quite problematic. There are reportedly methods for entirely digesting the microcarriers and liberating cells, but such processes are messy. By-products generated from the digestion of microcarriers need to be removed through downstream processing so that unwanted substances do not accompany cells when they are injected into patients. This can cause significant problems with cell harvest and final product purity. Sponsored Supplement Pall’s SoloHill microcarriers are all fabricated of rigid polystyrene materials — the same base material from which traditional 2D cultureware is constructed. This facilitates adaptation and results in easier cell dissociation, and thus enables more reproducible and efficient removal of cells from microcarriers using standard enzymatic harvesting coupled with sieving methods. Using microcarriers to expand cells for cell therapy is a great mechanism for providing larger surface areas for growth of such cells. What do you see as other advantages of using this technology? What, if any, are the downsides? A significant benefit in using microcarriers to expand cells for cell therapy is that the process is amenable to closed systems and can be performed cost effectively. Labor costs and the number of manufacturing runs needed per year to meet the therapeutic need are significantly lower because lot sizes are larger. Process validation and equipment maintenance can be streamlined, and flexibility is realized by using single-use systems in GMP manufacturing suites. Once a microcarrier manufacturing platform is validated, the process can be implemented for other stem cells types after some optimization. The greatest potential risk associated with using microcarriers in large reactors is for failure of a manufacturing run. When scaling with microcarriers, the loss of a single run due to operator error, equipment failure, or facility problem can represent significant loss of revenue. Therefore, it is important to design multiple redundancies within the manufacturing setting and use thorough risk assessment processes before implementing this technology. These measures will allow manufacturers to anticipate and prevent failures and carefully plan and construct efficient facilities. How amenable are microcarriers to implementation in single-use systems? How disposable, or reusable, are they, themselves? Microcarriers are a natural fit for single-use systems because they can be used as single-use It is important to design multiple redundancies within the manufacturing setting and use thorough RISK ASSESSMENT processes before implementing microcarrier technology. components in cell manufacture. However, reasons not to reuse microcarriers include regulatory concerns, maintenance of GMP manufacturing compliance, potential impact on manufacturing reproducibility, challenges of process validation, and unknown microcarrier stability after repetitive use. Additionally, nonporous, rigid microcarriers can be gamma-irradiated without discernible loss of function. This enables direct, closed-system transfer of microcarriers into sterile reactors for immediate use and makes them ideal partners for single-use systems. One perception in the industry today (right or wrong) is that using microcarriers requires many, many months of process development to get a truly optimized process. What has led to that perception? And do you see the paradigm changing? Process development to meet large-scale manufacturing needs in any platform is by nature a fairly lengthy process. This is especially true for cell therapy products. Difficulties with cell harvest, handling of large numbers of units, and time constraints required for maintenance of cell health and identity are challenges encountered in all platforms. Processes that appear to be straightforward can manifest unforeseen challenges. Manufacturers often encounter difficulties even when they use seemly straightforward processes such as 2D cultures. Microcarrier development can be streamlined like any other process development activity. Small-scale studies help expedite identification of S eptember 2015 13(8)sp BioProcess International 57 conditions that are optimal for cell growth and harvest. Fortunately, many process parameters are scalable. Although optimization is needed as scale is increased, unit operations that are optimized at each scale can be used in subsequent process development activities. Using a carefully planned, methodical strategy for development of a microcarrierbased process is the best way to ensure success. Paying close attention to details used for cell propagation also is important. Parameters used for cell harvest before seeding dynamic cultures should be optimized to ensure reproducibility and efficiency. Although it is good practice to consider optimizing these parameters in standard systems, investigators rarely perform detailed characterization of cell harvest in 2D systems because the level of detail is usually not required for successful cell propagation in static cultures. What are the major industry limitations to implementing microcarrier technology at large scales? This is difficult to assess accurately. From my perspective there are few major limitations for adopting the technology. Each of the perceived or real logistical challenges associated with large-scale microcarrier cultures has been addressed by manufacturers of vaccines and biologics. Additionally, at least one group has scaled adult stem cell cultures into 1,000-L stirred vessels. Therefore, widespread adoption of the technology for expansion of stem cells should be eminently possible. Development of internal expertise may be perceived as one possible impediment to implementation of the technology, but cell culture scientists, engineers, and manufacturing associates can acquire the skills needed to perform microcarrier culture. As mentioned earlier, acquiring the skills is a matter of diligence and attention to detail. To facilitate this, Pall has developed SoloHill Microcarrier Training Courses, designed to help researchers get the most out of their SoloHill microcarrier processes. Each course can be customized to the specific needs of an organization and includes 58 BioProcess International 13(8)sp such areas of focus as adapting flatware and roller bottle processes to microcarriers, handling microcarriers and optimizing attachment conditions, small scale microcarrier processes in spinner flasks, glass bioreactor microcarrier processes from 2 L to 15 L, single-use microcarrier processes from 2 L to 20 L, and strategies for process optimization. From a regulatory perspective, data need to be generated that demonstrate functionality of cells grown in meaningful potency assays, and cell identity tests need to be established for cells generated in any platform. Increasingly stringent regulatory requirements for controlling particulate load injected into patients present another unique challenge for cell therapy companies. This requirement is the same for all manufacturing platforms, so it is not unique to microcarrier cultures. Regardless, a solution for controlling particulates that conforms to regulatory requirements must be found for all platforms. Thierry Bovy Cell Therapy Manufacturing: The Xpansion System — with Thierry Bovy Thierry Bovy was previously responsible for cell-therapy manufacturing at a contract manufacturer. He comments here on the importance of the role CMOs are playing in this burgeoning industry and on the role of automated platforms in driving commercial cell therapy manufacturing. S eptember 2015 CMOs partner with cell therapy companies along the development pathway with the goal of reducing time to market. They are dependable and provide GMP accredited infrastructure, qualified equipment, and skilled personnel during a period that is mutually agreed upon. They can represent a more cost-effective alternative to building or acquiring brick and mortar as well as hiring and paying personnel that would be less occupied between manufacturing campaigns. From a financial perspective, this transforms the CAPEX investments in expenses, which is easier to defend in front of investors — especially in the early phases of a clinical trial. With time, the CMOs accumulate experience that future partners can benefit from. They also provide regulatory expertise that is required to file dossiers to the regulatory agencies. What major hurdles did you experience in that work? Have those earlier experiences helped you in your work now at Pall? My department was manufacturing autologous and allogeneic fresh cell therapy products for the European market. Each process was different and had different challenges, but every process had a very short shelf life, usually only a few hours. This meant that less than a half day was allotted to filling the primary container and returning it to a patient for injection, including the release of the product and shipment. The process had to be robust and safe enough in absence of sterility testing to provide confidence to a pharmacist authorizing human injection. Also, shipping logistics was a major hurdle that had to be cleared. Manufacturing of the cells had to be aligned to the airline schedules to make sure that physicians would receive the cellular doses during regular business hours. Scale-out or scale-up: What are the major drivers when choosing which path to take in industrializing cell therapies? The batch size and demand ultimately dictates the manufacturing strategy used for a given therapy. The increase in production for autologous therapies corresponds to a scaling out, Sponsored Supplement whereas allogeneic treatments allow for scaling up, provided that the cell types used are amenable to it. In the case of autologous treatments, the full process costs are borne by the unique treatment. The quality control tests have the largest contribution in the final product cost, with limited possibilities to reduce their burden. All the efforts to industrialize an autologous process should focus on automation of the process to reduce labor costs. The allogeneic model is the closest to the classical pharmaceutical one. A “universal” donor will provide tissue from which stem cells will be isolated and multiplied in vitro to such extent that clinical doses of cellular material will be made available off the shelf for a large population. The optimization of the cost will be linked to the balance between batch size, shelf life, demand, and technology. Indeed, market demand will dictate the number of cellular doses to generate, the shelf life and size of the lots to manufacture to avoid scrapping expired material, and the technology costs achievable at the various scales. Current planar technologies could generate up to 5 × 1011 cells per lot, and three dimensional systems such as microcarriers would push this limit to 1013 cells per lot (1). The cost per million of cells produced will therefore be a combination of all these parameters. What are the major platforms for industrializing autologous and allogeneic cell therapies? What about the upside and downside of each? Scale up for adherent cells is associated with the extension of the culture surface or of the volume of the vessels for cells grown in suspension. Autologous therapies typically require fewer cells per dose and therefore involve classical culture vessels of limited surface or volume, such as T-flasks or small-scale multitray stacks. Automation represents an interesting avenue to explore to support the intensification of these therapies. Most allogeneic processes still involve the use of planar technologies, mainly multitray stacks. The surface per single unit quickly becomes a limitation, and multiple containers Sponsored Supplement to traditional 2D platforms. What benefits of the Xpansion system have made it so attractive? What are its scaling options? The Xpansion Even the BEST TECHNICIAN still represents a major source of CONTAMINANTS and variability in a GMP environment. have to be manipulated to increase the culture surface for the cells. This scale up is very labor intensive. It requires extensive class A and B working and incubation spaces because some of the process steps are considered open and performed under laminar flow hoods. Automation possibilities are limited with this type of culture vessel. Hollow-fiber devices can help cope with some perceived limitations of open systems, but they are not scalable either and require numerous automated systems to generate multibillions of cells. More and more cell therapy companies consider microcarriers to be economically sound alternatives to traditional flatware systems when trillions of cells have to be generated per lot. There are of course challenges associated with scale up and downstream processing, but this technology is fast becoming one of the readily available, favorable technologies that can overcome the obstacles associated with planar technologies. Can you offer some thoughts regarding automation’s role in cell therapy bioprocessing? Even the best technician still represents a major source of potential contaminants and variability in a GMP environment. A robot would mitigate these risks, will work 24/7, and will therefore dramatically reduce labor costs. Automation also represents the best avenue to explore to reduce costs in an autologous process. When growing adherent cells (e.g., stem cells), a surface must be provided on which they can attach and grow. Xpansion technology seems to be gaining acceptance as an alternative multiplate bioreactor is part of Pall Life Science’s single-use bioreactor family, designed for shear-sensitive adherent cell applications such as cultivation of stem cells. It was developed for the safe, large-scale production of traditional 2D cell cultures, providing the same microenvironment to the cells as in traditional multitray stacks. The Xpansion bioreactor’s multiplate structure comprises a large cell growth surface area (up to 122,400 cm 2). The 80-cm high, 200plate bioreactor provides the same growth surface as a 4-m high set of 10 traditional stacks. The compact design enables elimination of the gas phase between the plates. This gas phase is replaced by an automatically controlled aeration system that provides advanced gas diffusion. Control is automatic through disposable pH and dissolved oxygen (DO) sensors. Temperature monitoring and agitation control are also included. Xpansion bioreactors offer the capability of monitoring cell morphology and density through use of an optional digital holographic microscope. Scalability is another key feature. The Xpansion multiplate bioreactors are available in four sizes, providing an increasing surface for cell growth from 6,120 cm² to more than 122,000 cm² per single-use unit, without modification to the footprint of the bioreactor. The Xpansion 10-plate bioreactor, the smallest version, is normally used for technical evaluation whereas the 50-, 100-, and 200-plate versions are generally used for manufacturing. Once the parameters such as pH and dissolved oxygen are optimized using the smallest size, the scale-up from 10 to 200 plates is fairly linear and straightforward. c S. Anne Montgomery is cofounder and editor in chief of BioProcess International, amontgomery@bioprocessintl.com. S eptember 2015 13(8)sp BioProcess International 59 “ Working together to transform the lives of patients worldwide.” Pall Life Sciences Advances in cell therapy research are leading to exciting new treatments for many diseases, and speed-to-clinic and speed-to-market are primary commercialization drivers. When timelines are tight, experience matters. Pall Accelerator Development Services help ease the path to today’s newest bioreactors and methods. Our innovative technologies, modern lab facilities and experienced scientists combine to optimize your processes, and help accelerate your learning curve. The result? Timeline reduction, process efficiency, compliance, and future-readiness. Pall’s business philosophy is about collaboration and helping you achieve your goals. Together, we can advance the development of cutting-edge cell therapies... and dramatically improve the lives of patients worldwide. Pall Life Sciences Your vision. Our expertise. Their future. www.pall.com/celltherapy © 2015 Pall Corporation. Pall and are trademarks of Pall Corporation. ® indicates a trademark registered in the USA. GN15.6331 Pall Life Sciences Your vision. Our expertise. Their future. www.pall.com/celltherapy Corporate Headquarters Port Washington, NY, USA +1.800.717.7255 toll free (USA) +1.516.484.5400 phone biopharm@pall.com e-mail European Headquarters Fribourg, Switzerland +41 (0)26 350 53 00 phone LifeSciences.EU@pall.com e-mail Asia-Pacific Headquarters Singapore +65 6389 6500 phone sgcustomerservice@pall.com e-mail