In Situ Measurements of X-Ray-Induced Silver Diffusion into a Ge Se Thin Film
advertisement

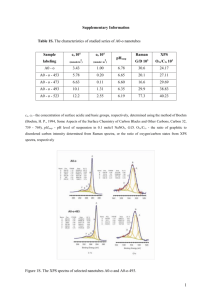
Journal J. Am. Ceram. Soc., 91 [3] 760–765 (2008) DOI: 10.1111/j.1551-2916.2007.02178.x r 2007 The American Ceramic Society In Situ Measurements of X-Ray-Induced Silver Diffusion into a Ge30Se70 Thin Film Andriy Kovalskiy, Alfred C. Miller, and Himanshu Jainw Department of Materials Science & Engineering, Lehigh University, Bethlehem, Pennsylvania 18015 Maria Mitkova Department of Electrical and Computer Engineering, Boise State University, Boise, Idaho 83725 the atomistic mechanism of photodoping, which strongly depends on the composition of the host film, is still controversial even for stoichiometric GeSe2. Ag-induced phase separation, accompanied by the formation of Ag2Se and a compositiondependent ternary phase, is the most well-known approach. Even then, the final ‘‘destination’’ of free electrons resulting from Ag photoionization remains unknown. Therefore, in this work, we characterize the evolution of X-ray photon-induced silver diffusion by measuring in situ the chemical structure and composition of the near-surface layer of a Ge30Se70 chalcogenide glass film at various stages of photodiffusion by high-resolution X-ray photoelectron spectroscopy (XPS). This method provides information about electron redistribution due to structural transformations caused by the interaction of Ag with a ChG matrix. These measurements also offer the possibility of in situ monitoring of the effects of photodoping such that X-rays simultaneously serve as a probe beam and at the same time also as a source of irradiation that causes silver diffusion into the ChG film. To our knowledge, this work is the first attempt to investigate silver diffusion in germanium selenides by in situ high-resolution XPS. Previous low-resolution XPS studies could only indicate a qualitative trend of the changes in electronic structure due to silver diffusion into thin Ge–Se films.18–20 By comparison, the present analysis of core-level spectra provides quantitative data on electron redistribution in the matrix, and hence a clearer picture of photodiffusion in these ChG films. Our entire experiment, from the very beginning (thin film deposition) to the very end of the XPS measurements, has been conducted under ultra-high vacuum. Thus, we have observed ‘‘pure’’ photodoping effects without any influence of contamination from oxygen, which can significantly alter the process of silver diffusion as suggested earlier.21 High-resolution X-ray photoelectron spectroscopy is used to identify the mechanism of X-ray-induced Ag diffusion into Ge30Se70 chalcogenide glass thin films, which are prepared in situ to avoid oxygen contamination. From the analysis of Ge 3d, Se 3d, and Ag 3d core levels, and valence band spectra, changes in the electronic structure are determined as Ag diffuses gradually with increasing irradiation. The ternary phase based on Ge2Se6 units, which contains homopolar Ge–Ge bonds, forms when diffusion approaches equilibrium where Ag content B30 at.%. The formation of a Se-rich composition is indicated in the near-surface region at the initial stage of the process, but the previously assumed Ag2Se phase is not detected. I. Introduction S ILVER photodiffusion into chalcogenide glass (ChG) thin films has a variety of applications in microlithography for fabricating micro-electro-mechanical systems,1 very large-scale integrated circuits,2 programmable metallization cell structures for nonvolatile memory,3 etc. Thin films of the Ge–Se system are strong candidates for such applications because of their wide glass-forming region, relatively high glass transition temperature Tg, and high doping ability. To optimize the processing parameters and composition of amorphous Ge–Se films, it is necessary to understand the evolution of silver diffusion and the final structure of the Ag-doped product during exposure to light. Different experimental methods including Raman spectroscopy,4 Rutherford spectroscopy,4–7 electrical resistance measurements,8 secondary ion mass spectroscopy,9 modulated temperature scanning calorimetry,10 Moessbauer spectroscopy,9 X-ray diffraction,3,11 electron diffraction,12 optical spectroscopy,11 ellipsometry,13 grazing-incidence X-ray scattering,6 differential anomalous scattering,6 extended X-ray absorption fine structure (EXAFS),6,8 transmission electron microscopy,14 microlithography technique7, atomic force microscopy, and electron energy-loss spectroscopy16 have been used to obtain structural information connected with photodoping. In spite of this remarkable list of available data, the mechanism of electronic and structural transformations accompanying the photodiffusion of Ag into the GexSe1x matrix remains unclear. The amorphous character of the host limits the usefulness of these methods and the reliability of the data. Among the existing models, the one considering photodoping as a solid-state reaction triggered by light absorption or photoionization of hot Ag electrons appears to be the most promising.17 At the same time, II. Experimental Procedure A Ge30Se70 thin film (thickness>120 Å) was thermally deposited on an HF-etched Si substrate inside an XPS chamber under a vacuum of B107 torr in darkness, using bulk glass as the starting material. On top of the Ge30Se70 film, a silver film (B100 Å) was thermally deposited in a vacuum of B108 torr. There was no exposure to any oxygen-containing environment between the depositions. High-resolution XPS spectra were obtained using a Scienta ESCA-300 spectrometer (AG Scienta AB, Uppsala, Sweden) with a monochromatic Al Ka X-ray (1486.6 eV) spot that was B3–4-mm long and B250-mm wide. Data acquisition was restricted electronically to a smaller region within the X-rayilluminated area. The angle (y) between the surface and the detector was 901; for this condition, the maximum depth of analysis was B100 Å. The spectrometer was operated in a mode that yielded a Fermi-level width of 0.4 eV for Ag metal. The residual pressure inside the analysis chamber was lower than M. Hall—contributing editor Manuscript No. 23232. Received May 18, 2007; approved September 3, 2007. Presented at the joint meeting of the ACerS Glass and Optical Materials Division and the University Conference on Glass held in Rochester, New York, May 20–23, 2007. w Author to whom correspondence should be addressed. e-mail: h.jain@lehigh.edu 760 March 2008 In Situ Measurements of X-Ray-Induced Silver Diffusion 108 torr. The energy scale was calibrated using the Fermi level of pure Ag. The XPS data consisted of survey scans over the entire binding energy (BE) range and selected scans over the valence band, Auger peaks, or core-level photoelectron peaks of interest (Ge 3d, Se 3d, and Ag 3d5/2). An energy increment of 1.0 eV was used for recording the survey spectra and 0.05 eV for the case of core-level spectra and Auger peak. The core-level and Auger peaks were recorded by sweeping the retarding field and using a constant pass energy of 150 eV. The reproducibility of the measurement was checked on different regions of a film. The surface charging from photoelectron emission was neutralized using a low-energy (o10 eV) electron flood gun, which minimized any distortion and shift of spectra. The gold 4f7/2 core-level position at 84.0 eV was used as a reference. To analyze the evolution of spectra with time of irradiation, 10 sets of measurements were made under the same vacuum conditions. The duration for one complete set of measurements was B40 min. Data analyses were conducted with casaXPS software package. For analyzing the core-level spectra, a Shirley background was subtracted and a Voigt line shape was assumed for the peaks. The concentrations of appropriate chemical elements were determined from the area of core-level peaks taking into account appropriate sensitivity coefficients. Each Ge 3d and Se 3d core-level spectrum was fitted to obtain sets of doublets representing the spin orbit splitting of d and p electron levels. The number of doublets within a given peak was determined by the goodness of fit. Only doublets that significantly improved the goodness of fit were considered. Splitting parameters (intensity ratio and peak separation) for the doublets of a particular element were chosen from the measurements of spectra of pure Ge and Se elements and their stoichiometric compounds in the same spectrometer. The full-width at halfmaximum (FWHM) was assumed to be the same for the peaks within one doublet. However, different FWHM values were allowed for independent doublets of the same core-level peak. The same mix of the Gaussian (90%) and Lorentzian (10%) parts of the Voigt function was chosen for all doublets of a given core level. Our fitting procedure gave asymmetry values close to zero for all peaks. With these constraints, the uncertainty in peak position and area of each component was 70.05 eV and 72%, respectively. The exact positions of the component peaks depend on the corresponding environment of the atom, the oxidation number of the atom within this environment, the electronegativity of the neighboring atoms, and the type of chemical bonding. The core-level spectra were recorded at the same place several times one after the other. III. Results and Discussion The study of the silver photodiffusion process in a Ge30Se70 film by a high-resolution XPS method requires comparison of the shape and structure of Ge 3d and Se 3d core-level spectra for the as-prepared (i.e. undoped) and Ag-photodoped samples, as well as an analysis of the Ag 3d5/2 core-level spectrum. Modified Auger parameter a0 5 (X1C)A (where X is the energy of the X-rays, C is BE of the appropriate core level, and A is the kinetic energy of Auger electrons) was used to determine significant structural changes (phase transformations, phase separations, and so on) around the given element.22 The results of compositional analysis of the surface region showed a gradual decrease in the concentration of silver, clearly indicating radiation-induced diffusion. Ultimately, the concentration reached a plateau when the diffusion stopped. Even though the thickness of the Ag layer was close to the analysis depth (B100 Å), we were able to examine core-level spectra for all three elements because of active diffusion of silver inside of the sample and lack of evidence for metallic silver on the top of the sample during the first set of the measurements. It is important to note that X-ray irradiation of the pure Ge30Se70 thin films inside the XPS chamber for 400 min did not produce any detectable changes in the XPS spectra of the sam- 761 Fig. 1. Fitting of X-ray photoelectron spectroscopy (XPS) Ge 3d core-level spectrum for a thermally evaporated Ge30Se70 thin film. ple. Thus, all the changes in XPS spectra discussed below are related to X-ray-induced silver diffusion only. First, we would like to discuss the initial Ge30Se70 material. The Ge 3d core-level peak is situated at B30.8 eV. It roughly coincides with the earlier data obtained by low-resolution XPS for GeSe2 (B31.1 eV).23 The difference (B0.3 eV) is explained by the lower precision of the measurement and referencing procedures in the latter case and does not relate to the variation of chemical composition. The fitting of the Ge 3d core-level peak for the nondoped sample (Fig. 1) reveals two pairs of peaks corresponding to two different environments of Ge atoms, despite the fact that we are dealing with a Se-enriched composition Ge30Se70 (in comparison with stoichiometric Ge33Se67). The major pair, positioned on the high-energy side (the parameters of the peak components are presented in Table I), as expected, is associated with Ge within GeSe4 tetrahedra (regular structural fragments, containing only Ge-–Se bonds), which are the main structural blocks of Ge30Se70 thin films24 as well as bulk stoichiometric GeSe2 glass.25 The existence of a minor pair at lower energies suggests that in thin films of Ge30Se70 composition, ‘‘wrong’’ Ge–Ge bonds (and, respectively, additional Se–Se bonds) are formed besides dominant Ge–Se bonds. Such homopolar bonds in vacuum-evaporated Ge30Se70 thin films were detected earlier by the Raman spectroscopy method.24 The concentration of the ‘‘wrong’’ homopolar bonds strongly depends on the conditions of thermal deposition. The analysis of spectra suggests that nearly 89% of all Ge atoms are within GeSe4 and B11% of them are within the units containing Ge– Ge bonds (the precision of such an estimation depends on the absolute concentration and the accuracy of the sensitivity factors, which does not exceed 3–5 at.%). Statistically, Ge covalently linked with one Ge and three Se is the most likely possibility for the formation of Ge–Ge homopolor bonds. In this case, we consider the option when Ge–Ge bonds are a part of ethane-like units Ge2(Se1/2)6 (or some modification of these units) revealed previously in GeSe2.25 The Se 3d core-level peak in Ge30Se70 is positioned at B54.3 eV, while Ueno23 observed it for GeSe2 at B54.7 eV. The possible reason for the 0.4 eV difference is already explained above. Examination of the fitted Se 3d core level spectrum for the Ge30Se70 thin film (Fig. 2) confirms the existence of a Se– Se-containing minor component (B17% of all Se atoms), most probably associated with Se in the Ge–Se–Se–Ge fragments, which is situated at a BE (see Table I) higher than the major pair (B83% of all Se atoms) corresponding to Se atoms within regular Ge–Se–Ge units. The concentration of Se–Se in the asdeposited film is two times larger than the statistically calculated value for Ge30Se70 glass, indicating an over-existence of the ‘‘wrong’’ homopolar bonds after thermal deposition. 762 — — — — — — — 100 — 29.4 50.5 20.1 2.5 40.0 44.0 16.0 2.8 Fig. 2. Fitting of X-ray photoelectron spectroscopy (XPS) Se 3d core-level spectrum for a thermally evaporated Ge30Se70 thin film. The data for core levels include peak position, area fraction, and FWHM. FWHM, full width at half maximum. — — — 29.9 47 0.99 29.9 57 0.89 — — 30.3 12 0.86 30.3 53 0.94 30.3 43 0.94 — 30.7 88 0.71 — 71.1 28.9 2.5 — Se (at. %) Ge (at. %) Se/ Ge Ag (at. %) Vol. 91, No. 3 with Ag after 10 consecutive measurements Pure Ag with Ag after 1st measurement 368.3 368.0 367.5 18 77 5 342.9 1173.8 179.1 1361.3 1134.6 720.0 0.81 0.54 0.63 368.4 367.9 367.6 12 85 3 342.9 1174.0 179.0 1361.5 1134.6 719.9 0.61 0.59 0.65 368.3 — — 726.022 51.4 6 1.2 51.6 4 1.2 — 53.0 16 0.86 52.8 2 0.49 — — — — — — — 54.7 54.2 17 83 0.78 0.82 Peak position (eV) Area fraction (%) FWHM (eV) 53.8 78 0.86 53.8 94 0.90 — a0 Ge3d Ge-Aug Ag3d5/2 III Ag3d5/2 II Ag3d5/2 I Se3d5/2 V Se3d5/2 IV Se3d5/2 III Se3d5/2 II Se3d5/2 I Ge3d5/2 III Ge3d5/2 II Ge3d5/2 I Chemical composition Table I. Fitting Parameters of High-Resolution XPS Core Level Spectra Se-Aug a0 Se3d 344.0 1173.4 179.7 1361.2 — Ag-Aug — a0 Ag3d Remarks Ge30Se70 without Ag Journal of the American Ceramic Society—Kovalskiy et al. Fitting of any single core-level peak is less precise in comparison with fitting of a complex spectrum containing overlapped components associated with spin-orbit splitting (as in the case of Ge 3d and Se 3d). However, an analysis of Ag3d5/2 core-level spectrum just after the deposition of an ultra-thin Ag layer (B100 Å) (Table I) clearly shows the existence of three different chemical states of silver (Fig. 3). To verify the origin of the components, we compare the results for the same core level after 10 consecutive measurements of the whole set of spectra (Ge 3d, Se 3d, Ag 3d5/2, Auger peaks for all three elements, and valence band spectrum) until an equilibrium state is established and no more changes in structure and hence diffusion in sample are detected. There are strong reasons to believe that already at the beginning of X-ray irradiation (i.e., the first XPS measurement), the silver layer on top of the ChG film is neither metallic (as in bulk Ag metal, usually situated at the 368.3 eV position,26 Fig. 4) nor fully ionized (such as in AgCl). First, the position of the Ag 3d5/2 peak for non bonded Ag1 ions is expected at a much higher binding energy (BE 5 369.8 eV)27 than our highest component Ag(II) at 368.3 eV (Fig. 3, Table I). Second, this component does not disappear completely even at the equilibrium state after 10 measurements, thus excluding its origin in metallic silver. The FWHM of this component (0.81 eV at the beginning of X-ray irradiation) is also too large for metal– metal interactions as in a pure Ag layer. Therefore, a probable Fig. 3. Fitting of X-ray photoelectron spectroscopy (XPS) Ag 3d5/2 core-level spectrum obtained just after deposition of a 10-nm Ag film onto a thermally evaporated Ge30Se70 thin film. March 2008 In Situ Measurements of X-Ray-Induced Silver Diffusion Fig. 4. Comparison of Ag 3d5/2 core-level spectra for pure metallic Ag and an Ag/Ge30Se70 couple. chemical state of Ag associated with 368.3 eV (B18 at. % of all Ag atoms) is metastable partly ionized Ag1d ion at the nearsurface region; compared with an isolated Ag1 ion, its charge distribution is influenced by the electronic density of the ChG matrix. The existence of such silver species in a Se-rich metastable composition based on Ag1d–Se interactions very close to the surface is supported by the significant variation of the Se/Ge ratio with the irradiation time (2.8 and 2.5 at the beginning of irradiation and in the equilibrium state, respectively). This agrees with the earlier conclusions that Ag in ChG exists as Ag1 ions incorporated into the glass matrix.20,28 The significant decrease in the FWHM of the 368.3 eV component from 0.81 to 0.61 eV after prolonged irradiation may be an evidence of the stabilization of the Ag1d–Se interactions as silver diffuses into the ChG film. Both Ag 3d5/2 components at lower BE can be linked only with some Ag-containing phases. Their stability is confirmed by the low FWHM values. More detailed structural information can be proposed only after analyzing the influence of silver on the structure of Ge 3d and Se 3d core-level spectra. The time-dependent effect of Ag diffusion into the film is clearly seen when comparing the valence band spectra of the samples just after Ag deposition and in an equilibrium state (Fig. 5). For clarity, intermediate spectra are omitted from the figure. The structure of Ag 4d peak at B5.5 eV is much more Fig. 5. Time evolution of X-ray photoelectron spectroscopy (XPS) valence band spectrum with Ag diffusion into a Ge30Se70 thin film. 763 Fig. 6. Normalized X-ray photoelectron spectroscopy (XPS) Ge 3d core-level spectra for a thermally evaporated Ge30Se70 thin film before and after diffusion of Ag. complex at the beginning of Ag diffusion, revealing a distinct additional component at the low-BE side. Narrowing of the peak in the equilibrium state is evidence supporting stable ternary phase formation, when the diffusion process is accomplished. Ag diffusion causes an B0.5 eV shift (0.4 eV was reported by other authors20) of the Ge 3d core-level peak to a lower energy (Fig. 6). This considerable change is a consequence of structural rearrangement with the complete disappearance of GeSe4 tetrahedral units, which are supposed to be the main structural fragments of a Se-rich Ge30Se70 thin film.24 Instead of GeSe4, the ternary composition, which contains Ge–Ge bonds, most probably within Ge2Se6-type structure, is proposed to be formed after Ag diffusion. The fit of the Ge 3d core-level spectrum revealed two pairs of peaks, confirming two distinct chemical states of Ge atoms in the matrix. We suggest that one of them, at a higher BE, is ‘‘pure’’ Ge2Se6 and most probably the other is a 29 Ge 3 coordination defect, typical for chalcogenide glasses, within poorly developed ethane-like units. This assignment is based on the estimated chemical shift, caused by a change in the chemical environment and/or oxidation number of Ge atoms, with respect to the position of Ge within the GeSe4 tetrahedra. A considerable increase in the concentration of Ge–Ge bonds in the matrix after silver diffusion was observed earlier by Raman spectroscopy in Ge30Se70 and even more so in Se-rich Ge20Se80 thin films after Ag diffusion.4,30 However, the relatively large FWHM (B1.0 eV) value for both Ge 3d components still requires an explanation. It could be because of the different charge states of Se atoms linked with Ge, thus introducing a distortion in electronic density around Ge atoms; we suppose that Ge2Se6 units are not face shared as in the case of Ge-rich glass.31 Most of the Se atoms probably exist in the form of SeAg1 complexes,17 but some of them could form regular covalent Ge–Se–Ag fragments. The change in the modified Auger parameter a0 can be considered to be an index of structural transformations involving participation of the selected chemical elements.22 For Ge, we observe an increase of this parameter, giving evidence of additional screening of the core-level electrons due to the presence of Ag in the structure. The longer the time of irradiation, the larger the Ag concentration in the matrix and the larger the value of a0 (see Table I). The chemical shift of Se 3d core-level spectrum of a Ge30Se70 thin film to lower BE (B0.4 eV, Fig. 7) after Ag diffusion is caused by the complete disappearance of Se–Se bonds (which should be at a higher BE because of the higher electronegativity of Se atoms) and also Ge–Se–Ge fragments. The value and direction of the shift fully agree with previous data.20 Taking into 764 Journal of the American Ceramic Society—Kovalskiy et al. Vol. 91, No. 3 still remain unclear. The most intriguing among them is the fate of the electron that becomes available as a result of ionization of an Ag atom to an Ag1 ion. Photodiffusion in the present sample occurred as a result of X-ray irradiation, and we may anticipate qualitatively similar steps from exposure to lower energy photons. Yet, future experiments are planned to verify this prediction. IV. Conclusions Fig. 7. Normalized X-ray photoelectron spectroscopy (XPS) Se 3d core-level spectra for a thermally evaporated Ge30Se70 thin film before and after diffusion of Ag. account that for the studied chemical composition the major Se 3d5/2 component at BE 5 53.8 eV should necessarily contain a Ge–Se bond, we can assume that this component is most probably associated with Se within the Ge–Se–Ag1 fragments. A possible explanation for the significant (B16 at.% of Se atoms) second component at BE 5 53.0 eV (for Se 3d5/2) is Se within a covalent Ag–Se bond. The covalent origin of Ag–Se bonding was earlier predicted by appropriate ab initio band structure calculations of Ag2Se.32 However, we cannot directly relate this component to phase-separated Ag2Se, normally observed at a higher BE B53.6 eV.33 The significant decrease of binding energy for the third component of the Se 3d core-level spectrum (51.6 eV) could be associated with lowering of oxidation number and can be assigned to onefold coordinated Se, usually expected in Se-rich ChG compositions. With increasing time of irradiation, the concentration of both minor components decreases (Fig. 8) because of structural rearrangement with the formation of an equilibrium ternary composition close to Ag30(Ge0.30Se0.70)70. The above steps provide a detailed insight into silver photodiffusion into a Ge30Se70 thin film. However, a few questions Fig. 8. Time evolution of X-ray photoelectron spectroscopy (XPS) Se 3d core-level spectra with Ag diffusion into a Ge30Se70 thin film (normalized data). High-resolution XPS is shown to be an excellent tool for identifying the various stages of X-ray-induced silver photodiffusion into a Ge30Se70 thin film. By preparing a thin-film sample within the spectrometer, oxygen contamination is avoided, and evolution of radiation-induced structural changes is observed for the first time by conducting in situ measurement as a function of time. The analysis of core-level spectra indicates the formation of a Se-enriched layer consisting of Agd1–Se at the initial stage of silver diffusion. This layer disappears with time of irradiation. Ultimately, under equilibrium, a ternary phase based on Ge2Se6 units is formed, which is characterized by Ag concentration at B30 at.%. A separated Ag2Se phase, widely described in the literature as one of the products of Ag diffusion into ChG thin films, was not detected in the present study, which was conducted completely under vacuum. The case when a ChG thin film was exposed to an O2-containing atmosphere will be discussed in a future paper. Acknowledgments The authors thank the U.S. National Science Foundation, through the International Materials Institute for New Functionality in Glass (IMI-NFG), for providing partial financial support for this work (NSF Grant No. DMR0409588). References 1 A. Kovalskiy, H. Jain, M. Vlcek, A. Fiserova, C. M. Waits, and M. Dubey, ‘‘Development of Chalcogenide Glass Photoresists for Gray-Scale Lithography,’’ J. Non-Cryst Solids, 352 [6–7] 589–94 (2006). 2 M. N. Kozicki, S. W. Hsia, A. E. Owen, and P. J. S. Ewen, ‘‘PASS–a Chalcogenide-Based Lithography Scheme for I.C. Fabrication,’’ J. Non-Cryst Solids, 137–138 [2] 1341–4 (1991). 3 M. N. Kozicki, M. Park, and M. Mitkova, ‘‘Nanoscale Memory Elements Based on Solid-State Electrolytes,’’ IEEE Trans. Nanotech., 4 [3] 331–8 (2005). 4 M. Mitkova and M. N. Kozicki, ‘‘Silver Incorporation in Ge–Se Glasses Used in Programmable Metallization Cells,’’ J. Non-Cryst. Solids, 299–302 [B] 1023–7 (2002). 5 J. Rennie, S. R. Elliott, and C. Jeynes, ‘‘Rutherford Backscattering Study of the Photodissolution of Ag in Amorphous GeSe2,’’ Appl. Phys. Lett., 48 [21] 14302 (1986). 6 A. Fischer-Colbrie, A. Bienenstock, P. H. Fuoss, and M. A. Marcus, ‘‘Structure and Bonding in Photodiffused Ag–GeSe2 Thin Films,’’ Phys. Rev. B, 38 [17] 12388–403 (1988). 7 W. Leung, N. Cheung, and A. R. Neureuther, ‘‘Photoinduced Diffusion of Ag in GexSe1x Glass,’’ Appl. Phys. Lett., 46 [6] 543–5 (1985). 8 J. M. Oldale, J. Rennie, and S. R. Elliott, ‘‘A Comparative Study of Silver Photodoping in Chalcogenide Films by Means of Extended X-Ray Absorption Fine Structure and Kinetics Measurements,’’ Thin Solid Films, 164, 467–73 (1988). 9 J. Calas, R. El Ghrandi, M. Averous, and G. Galibert, ‘‘Study of Isotropic Silver Dissolution in Vitreous GeSex Thin Films from Secondary Ion Mass Spectrometry Measurements,’’ Mater. Sci. Eng. B, 13 [4] 309–17 (1992). 10 M. Mitkova, Y. Wang, and P. Boolchand, ‘‘Dual Chemical Role of Ag as an Additive in Chalcogenide Glasses,’’ Phys. Rev. Lett., 83 [19] 3848–51 (1999). 11 J. H. S. Rennie and S. R. Elliott, ‘‘Kinetics of Photo-Dissolution of Silver in a-GeSe2,’’ J. Non-Cryst. Solids, 77–78 [2] 1161–4 (1985). 12 C. H. Chan and K. L. Tai, ‘‘Electron Diffraction Studies of Ag Photodoping in GexSe1x Glass Films,’’ Appl. Phys. Lett., 37 [7] 605–7 (1980). 13 V. Honig, V. Fedorov, G. Liebmann, and P. Suptitz, ‘‘Ellipsometric Investigation of Ag-Doping Profiles in Amorphous Chalcogenide Thin Films,’’ Phys. Status Solidi A, 96 [2] 611–9 (1986). 14 J. S. Romero, A. G. Fitzgerald, and M.J Rose, ‘‘A Transmission Electron Microscope Study of Metal/Chalcogenide Amorphous Thin Films,’’ Appl. Surf. Sci., 234 [1–4] 369–73 (2004). 15 M. Mitkova, M. N. Kozicki, and J. P. Aberouette, ‘‘Morphology of Electrochemically Grown Silver Deposits on Silver-Saturated Ge–Se Thin Films,’’ J. NonCryst. Solids, 326–327, 425–9 (2003). March 2008 16 In Situ Measurements of X-Ray-Induced Silver Diffusion A. E. Meixner and C. H. Chen, ‘‘Studies of Ag Photodoping in Glassy GexSe1x Films by Electron-Energy-Loss Spectroscopy,’’ Phys. Rev. B, 27 [12] 7489–94 (1983). 17 G. Kluge, ‘‘A New Interpretation of the Photodoping Effect in Amorphous As- and Ge-Chalcogenides,’’ Phys. Status Solidi A, 101, 105–14 (1987). 18 S. Rajagopalan, K. S. Harshavardhan, B. Singh, and K. L. Chopra, ‘‘Photo and Electrochemical Doping in Ge-Based Chalcogenides,’’ J. Phys., 42 [C-4, 2] 911–4 (1981). 19 S. Zembutsu, ‘‘X-ray Photoelectron Spectroscopy Studies of Ag Photodoping in Se–Ge Amorphous Films,’’ Appl. Phys. Lett., 39 [12] 969–71 (1981). 20 T. Ueno and A. Odajima, ‘‘X-ray Photoelectron Spectroscopy of Ag- and Cu-Doped Amorphous As2Se3 and GeSe2,’’ Jpn. J. Appl. Phys., 21 [2] 230–4 (1982). 21 G. Chen and J. H. Horton, ‘‘Photochemically Induced Oxidation and Silver Deposition on Amorphous Germanium Sulfide Films, Studied Using Atomic Force Microscopy and Ion Beam Methods,’’ Thin Solid Films, 347 [1–2] 208–13 (1999). 22 M. Gester, J. A. D. Matthew, S. M. Thompson, K. Ounadjela, and G. Beamson, ‘‘High-Resolution X-Ray Photoelectron Spectroscopy on Giant Magnetoresistive CoAg Granular Films Sandwiched between Pt Layers,’’ Surf. Interface Anal., 26 [1] 30–8 (1998). 23 T. Ueno, ‘‘X-ray Photoelectron and Auger Electron Spectroscopic Studies of Chemical Shifts in Amorphous Ge–Se System,’’ Jpn. J. Appl. Phys., 22 [9] 1349–52 (1983). 24 H. Takeuchi, O. Matsuda, and K. Murase, ‘‘Reversible Mesoscopic Structural Transformations in Vacuum Evaporated Amorphous Ge30Se70 Film Studied by Raman Scattering,’’ J. Non-Cryst. Solids, 238 [1–2] 91–7 (1998). 25 765 P. Boolchand, J. Grothaus, M. Tenhover, M. A. Hazle, and R. K. Grasselli, ‘‘Structure of GeSe2 Glass: Spectroscopic Evidence for Broken Chemical Order,’’ Phys. Rev. B., 33 [8] 5421–34 (1986). 26 M. Schnippering, M. Carrara, A. Foelske, R. Koetz, and D. J. Fermin, ‘‘Electronic Properties of Ag Nanoparticle Arrays. A Kelvin Probe and High Resolution XPS Study,’’ Phys. Chem. Chem. Phys., 9 [6] 725–30 (2007). 27 Z. Mousavi, J. Bobacka, A. Lewenstam, and A. Ivaska, ‘‘Response Mechanism of Potentiometric Ag1 Sensor Based on Poly(3,4-Ethylenedioxythiopene) Doped with Silver Hexabromcarbonate,’’ J. Electroanalyt. Chem., 593 [1–2] 219– 26 (2006). 28 T. Kawaguchi, ‘‘A Structural Study of Ag–Rich Ag–As–S Glasses,’’ Jpn. J. Appl. Phys., P.1, 37 [1] 29–33 (1998). 29 S. Blaineau, P. Jund, and D. A. Drabold, ‘‘Physical Properties of a GeS2 Glass Using Approximate Ab Initio Molecular Dynamics,’’ Phys. Rev. B, 67 [9] 94204-1– 6 (2003). 30 M. Mitkova, M. N. Kozicki, H. C. Kim, and T. L. Alford, ‘‘Local Structure Resulting from Photo and Thermal Diffusion of Ag in Ge–Se Thin Films,’’ J. NonCryst. Solids, 338–340, 552–6 (2004). 31 V. Boev, M. Mitkova, E. Lefterova, T. Wagner, S. Kasap, and M. Vlcek, ‘‘Glass Formation in the Ge–Se–AgI Ternary,’’ J. Non-Cryst. Solids, 266–269, 867–71 (2000). 32 C. M. Fang, R. A. de Groot, and G. A. Wiegers, ‘‘Ab Initio Band Structure Calculations of the Low-Temperature Phases of Ag2Se, Ag2Te and Ag3AuSe2,’’ J. Phys. Chem. Solids, 63 [3] 457–64 (2002). 33 W. Wang, Y. Geng, Y. Qian, M. Ji, and Y. Xie, ‘‘A Novel Room Temperature Method to Nanocrystalline Ag2Se,’’ Mater. Res. Bull., 34 [6] 877–82 (1999). &