OUTLINE 535 SESSION 5 (2007) cis
advertisement
 cis")
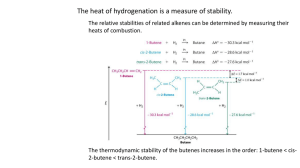
OUTLINE 535 SESSION 5 (2007) Page 42 Synthesis of Alkenes, Control of Stereochemistry 1. A word about the control of stereochemistry of disubstituted alkenes 1.1. Two choices cis or trans 1.2. Trans is easier to get. 1.2.1. Trans is most stable by approx. 2 kcal/mol. 1.2.2. This translates to usable ratios of 1.2.3. ∆G = -RTlnKeq 1.2.4. -2 kcal/mol = -2 x 10-3 kcal/(moleK)(298 K)ln Keq 1.2.5. 28.6/1 or ~97 % trans 2. Alkenes from Alkynes 2.1. Lindar catalyst-from alkene gives the cis alkene. 2.2. Dissolving metal (Na, Li) and alkyne gives the trans alkene. 3. From ketones and aldehydes 3.1. Non-stabilized Wittig generated with Li+ counter ions give trans disubstituted olefins. 3.2. Horner-Emmons reagents give trans olefin. 4. for above see: Roush, W. J. Am. Chem. Soc. 1980, 102, 1390. Stec Ac. Chem. Res. 1983, 16, 411. 5. Cis selective non-stabilized Wittig olefination. 5.1.1. These reactions have early transition states 5.1.2. Salt free conditions 5.1.2.1. Those below and 5.1.2.2. KHMDS/ 10%HMPA/ THF 42 OUTLINE 535 SESSION 5 (2007) Page 43 6. Stabilized ylides tend to be trans selective 6.1.1. This reflects the stability of the trans-oxaphosphatane 6.1.2. trans-oxaphosphatane is more stable than cis 6.1.3. Both reactions are under kinetic control 7. (KINETIC versus THERMODYANMIC CONTROL) 7.1. non-equilibrium conditions opens the way to possible kinetic control. Thermodynamic control of product distribution is always reflective of the relative free-energies of the products. Ph Ph P O Ph H H Ph Ph P O H Ph H Steric Problems Occur here between the ethyl and the phenyl substituents 8. Stabilized ylides possess ÿ-CO2R or ÿ--CO2R or ÿ-CN groups adjacent to the anion center. 8.1. or any other group that stabilizes negative charge 8.2. Stabilized ylides -> trans alkenes 43 OUTLINE 535 SESSION 5 (2007) Page 44 8.3. Unstabilized ylides in the absence of oxophilic cations (Mg and Li) -> cis alkenes 8.4. There are exceptions to the above 9. THE NEXT SECTION OF THIS LECTURE IS DESIGNED TO CONVICE YOU THAT TRISUBSTITUTED ALKENES ARE CHALLENGING TO SYNTHESIZE. 9.1. A good question at this point might be: why should they be synthetically complex? 9.2. When there are two equivalent substituents on the same side of the alkene, cuprate mediated non-polar additions to alkynes is a general solution. See: Kozlowski, J. A. "Organocuprates in the Conjugate Addition Reaction." In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4; pp 169. 10. 1970's Normant developed the stereospecific addition of organocopper reagents to alkynes. R' R CuLi RCuMgXY Et2O E+ R E R' R' -50 to -20 15 min 10.1. The above protocol looks good in theory 10.2. In practice Ph Ph Ph iPr2CuMgBr Et2O iPr iPr 68% -50 to -20 15 min 2. aqueous quenching 32% 10.3. Problems 10.3.1. In general the regiochemistry gets worse if the alkyne is di-substituted. 10.3.2. There are a few cases in which groups on the alkyne effectively direct the regiochemistry of the addition. For these see the citation above. 44 OUTLINE 535 SESSION 5 (2007) Page 45 Synthesis of Cecropia-C18 Juvenile Hormone O O H O Molecular structure of Cecropia-C18 Juvenile || Cecropia Moth, the silk moths (Hyalophora cecropia). In the next three syntheses of the Cecropia-C18 Juvenile Hormone think about general principles and specific reactions. Do not conceptualize the following information in terms of your instructor wanting you to memorize or even study three synthetic approaches to the Cecropia-C18 Juvenile Hormone. 10.4. That the synthesis appears to be convergent is the first thing you should notice. The solution to the molecular complexity in the form of trisubstituted alkenes is the second thing you notice in the synthesis below. 45 OUTLINE 535 SESSION 5 (2007) Page 46 OH OH H H Trans double bond in pericyclic transition state Cis double bond in pericyclic transition state Lower energy 10.5. The retro-ene reaction runs with the double bond in an S-cis relationship to the cyclopropane ring. The pericyclic transition state is tight, C—H—C proximity at near bond distance needs to be attained. This favors the developing cis-double bond in the 6 electron transition state. You can convince yourself with a model. 10.5.1. Read about the ene reaction Snider, B. B. "Ene Reactions with Alkenes as Enophiles." In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 5; pp 1. 10.5.2. Ripoll, J.-L.; Vallee, Y. "Synthetic applications of the retro-ene reaction." Synthesis 1993, 659-77. THPO PPh3 1. nBuLi / THF / 0 °C 2. cooled to -78 °C then A 3. -25 °C then 2 equiv. sec-BuLi 4. p-formaldehyde at 0 °C. O THPO 1. MnO2 / Hexane 2. PPh3CH2 / benzene 3. hydrazine/ EtOH / H2O2 / CuCl2(cat) OH A THPO 11. The end game of cecropia synthesis 11.1. Yield for the removal of the THP group was not reported. I suspect it was low due to non-orthogonality issues with trisubstituted alkenes. These are acid sensitive and you need acid to remove the THP group. 11.2. The rest of the synthesis was accomplished by Corey-Ganem oxidation to the carboethoxy functionality. 46 OUTLINE 535 SESSION 5 (2007) Page 47 12. The following approach to Cecropia C18 Juvenile hormone is instructive and provides a more general solution to stereocontrol in trisubstituted olefins than the synthesis above. 12.1. General is good! 12.1.1. Corey, E.J.; Katzenellenbogen, J.A.; Gilman, N.W.; Roman S.A.; Erickson,B.W. J. Am. Chem. Soc. 1968, 90, 5618-5620. 13. This synthesis of juvenile hormone centers around the stereoselective reduction of alkynes by LAH with the hydroxide as the directing group. 13.1. The rest of the synthesis was designed around this idea. 13.1.1. The intermediates have C-M functionality (carbon/metal bonds), this means it should be able to construct C-X (oxidation) or C-C bonds (substitution) with electrophiles. OH Al O CH2OH CO2Me OMe CH2OH 14. The triple bonds were operated on stereospecifically make double bonds. 14.1. Stereocontrol from lineland to flatland. 14.1.1. Alkene to alkyne is usually less complex. 14.1.2. The retrosynthesis simplifies the target (from the standpoint of molecular complexity) by the following criteria 47 OUTLINE 535 SESSION 5 (2007) Page 48 14.1.2.1. The triple bond has no stereochemical complications 14.1.2.2. sp C-H bond is easily deprotonated for anionic disconnections OCH3 NH3/Li OCH3 1 equiv O3/CH3OH DMS CO2CH3 CHO NaBH4 CO2CH3 OH THF/tAmyl alcohol OH 1. TsCl C5H5N 1. TsCl C5H5N 2. LAH ether CH2OH 2. propargylTHP ether (tetrahydropyranyl) 3. HCl/MeOH 1. PBr3 2. 3-lithio-1-trimethyl silylpropyne 3. AgNO3, KCN 4. BuLi then CH2O CH2OH 1. LAH, NaOCH3 THF 2. I2 3. Me2CuLi 4. MnO2/hexane 5. MeOH, NaCN 1. LAH, NaOCH3 THF X OH 2. I2 3. Et2CuLi X=I X=Et CO2Me 14.2. The enol ether reacts instead of the other double bond during ozonolysis in the second step. 14.2.1. Why? 14.3. Reduction the aldehyde in the presence of the ester is usually a simple matter. 14.3.1. Why? 14.4. Birch reduction: 14.4.1. Is tough to predict the products. Normally a mixture of products results. 14.4.2. Regioselective protonation of the birch intermediate. 14.4.3. Birch reduction preserves the conjugated double bond. 14.4.4. LOOK UP the Birch Reduction. Get comfortable with the mechanism and the kind of molecules you can make with this procedure. Realize that Birch reduction and the dissolving metal reduction of alkynes are related. 48 OUTLINE 535 SESSION 5 (2007) Page 49 14.4.5. Mander, L. N. (1991). Partial Reduction of Aromatic Rings by Dissolving Metals and by Other Methods. Comprehensive Organic Synthesis. B. M. Trost and I. Fleming. Oxford, Pergamon Press. 8: 489. 14.4.6. Rabideau, P. W. and Z. Marcinow (1992). “The Birch Reduction of Aromatic Compounds.” Org. React. (N.Y.) 42: 1-334. 14.4.7. Birch, A. J. and D. H. Williamson (1976). “Homogeneous Hydrogenation Catalysts in Organic Synthesis.” Org. React. (N.Y.) 24: 1. 14.5. ozone14.5.1. Made by striking an arch across an atmosphere of O2. 14.5.2. Electrophilic 1,3-dipolar cycloaddition. 14.5.3. Mechanism on next page. 14.5.4. Slow addition allows for kinetically differentiate between the two double bonds in the birch reduced product. O O R O O O O O O O R O O R' R' R' S R O O O R' R R R' S O O R O O O R' R S DMSO O + R' O 14.6. Borohydride reduction 14.6.1. Usually done in methanol or ethanol 14.6.2. Takes place from the more reactive boroester at about pH = 10 or so 14.6.2.1. Careful, pH refers strictly to aqueous solution. To facilitate discussion, this rule is broken all the time. 14.6.3. Very mild reducing agent, by moderating temperature you can selective reduce aldehydes and ketones in the presence of esters and enones. 49 OUTLINE 535 SESSION 5 (2007) Page 50 14.7. Reductive defunctionalization. 14.7.1. Tosylation is sophomore org chem. It is a method by which alcohols are activated toward substitution. If necessary, look it up. 14.8. THP protection of -OH functionality. 14.8.1. THP ethers are stable in the presence of base and almost any reducing agent. Do you know why? 14.8.2. In the presence of acid and excess alcohol THP ethers are deprotected. 14.8.3. The authors took a beating on the yield here (30%) what else can happen to the substrate in the presence of H+ and MeOH 14.9. At this point the material is >1:1000 isomerically pure cis! 14.10. LAH reduction of propargyl alcohols 14.10.1. Results in the formation of an alkenylaluminum species. 14.10.2. At this point that this alkenylaluminum is quenched with iodide, the stereochemistry of alkene is set. 14.10.2.1. Remember, In general, don’t try to control stereochemistry at anions. 2 3 14.10.2.2. Neither sp nor sp anions are good at trapping electrophiles stereospecifically. E A C E+ B A A E+ C B B C A E B C 14.10.2.3. These reactions are not stereoselective 14.10.3. The cuprate is known to conserve the stereochemistry of addition to akenylhalides 50 OUTLINE 535 SESSION 5 (2007) Page 51 14.10.3.1. Cuprate reagent reacts with alkenyliodides because it can offer the alkenyl iodide a d orbital-based mechanism. Et I Et I Et R R Cu Et Li R Li R π-complex Et Et Cu Cu Oxidative Addition LiI Et Cu Copper III Reductive Elimination Et Copper I 14.10.3.2. In the oxidative addition step the copper has gone from oxidation I to oxidation state II 14.11. 3-lithio-1-trimethylsilylpropyne 14.11.1. TMS as a surrogate proton 14.12. Silver + desilylation 14.13. Homologation with formaldehyde 14.14. Corey Ganem oxidation. O O O Mn O Mn O H O Mn 4+ O + H O NaCN H O Mn Mn 2+ OH OH CN H 51 OUTLINE 535 SESSION 5 (2007) Page 52 15. Final oxidation of the last alkene 15.1. N-bromosuccinamide/dimethoxyethane water mixture 15.1.1. Intermediate bromohydrin 15.1.2. Isopropoxide in isopropyl alcohol 15.1.3. Epoxide final product 16. Testing final product: 0.05 µg of the product injected in 50 µg of chemically pure olive oil blocked silk worm maturation. 17. The Take Home Lesson and how to study this material 17.1. You need to determine the points in the synthesis of the natural product that gave rise to stereoselective olefin synthesis and why. 17.2. You need to remember how the carbon-carbon bonds were formed and be able to apply the method to general problems you will get throughout the semester. 18. Another synthetic approach to the Cecropia Juvenile hormone: 18.1. Still, W. C.; McDonald, J. H., III; Collum, D. B.; Mitra, A. Tetrahedron Lett. 1979, 20, 593. 18.2. The main focus is the control of the stereochemistry of the trisubstituted alkene. 18.3. Retrosynthetic analysis: O OCH3 O H 2 1. HO HO O O 3 * * O HH O HH O 4 O 2,3-sigmatropic rearrangement 52 OUTLINE 535 SESSION 5 (2007) Page 53 18.3.1. In this retrosynthetic analysis the molecule is simplified from two trisubstituted alkenes to two methylene substructures. The stereochemistry of the allylic alcohols at the carbinol carbons does not matter. These are marked with * in structure 4. 18.3.2. The material at this point is a mixture of diastereomers. 6 4 Br O O + O 5 O 7 O O + Br O 8 Br 9 18.3.3. Look at 8 and 9. 18.3.3.1. Do you know why the enolate preferentially substitutes the Br on the sp3 carbon atom instead of the sp2 carbon atom? 18.3.3.2. Do you know why 5 had to be protected at the alcohol oxidation state with a ketal? 18.3.3.3. Do you know why dianion 6 preferentially reacted with aldehyde 5 at the carbanion instead of the alkoxide? 18.3.3.4. You should know what is driving the reaction from 12 to 3 forward. 18.3.3.5. You should know how to get from 3 to 1 from our previous discussions. + Br OLi O O Br Br O aqueous workup 8 9 O THF 10 Li Br 7 Br OH 1) LAH 2) K2CrO3 O Br 7 1)t-BuLi 2) 5 O OH OH O 11 53 OUTLINE 535 SESSION 5 (2007) 11 Page 54 1) BuLi 1) KH, / THF 2) Bu3SnCH2I O O O SnBu3 SnBu3 O 12 3 2) aqueous workup 18.4. The basis of the stereoselectivity of the 2,3-sigmatropic rearrangement. R R O H Me H Me H Me H H H R O H H O H H R Me Steric interaction between these two large pseudoequatorial substituents O H H trisubstituted alkene 18.4.1. This type of interaction is called 1,2-allylic strain. 18.4.2. The production of the E-alkene is more facile than the production of the Z-alkene. 18.4.3. In the transition state above R and the Me group are on opposite sides of the bond. They are anti. This anti relationship leads to a trans relationship between these two groups. 18.4.4. The transition state below is inhibited by interaction between the R and the Me groups. If this transition occurred preferentially then the syn Me and the R groups would be cis to one another in the product. 54 OUTLINE 535 SESSION 5 (2007) Page 55 R' R' H H looking down this bond generates the Newman projections below O O R R R H H H O R' H H H O H R' R' H Strain-Minimized t-State 1,3-allylic strain R O H R 1,3-allylic strain 1,3-tranannular interactio 18.4.5. The diagram above outlines another way to look at the transition that determines the stereochemistry in Still’s 2,3-sigmatropic shift. Could you see this better if you made a model? 18.4.6. Read the following review on the 2,3-sigmatropic rearrangement and its utility as a synthetic tool. 18.4.6.1. Bruckner, R. 2,3-Sigmatropic Rearrangements; Comprehensive Synthetic Organic Chemistry Trost and Flemming Eds.; Pergamon Press: Oxford, 1991; Vol. 6, pp 873. 55