Chem 781-noel
advertisement

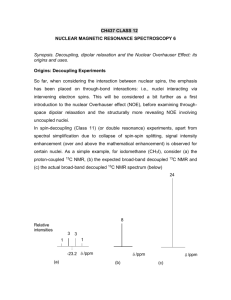
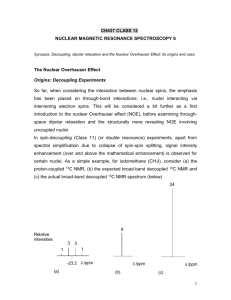
Chem 781 – Part 8 NOE and through space correlation Need for through space information • All 2D methods discussed so far utilized scalar couplings to establish through bond connectivities => determination of two dimensional structure • The magnitude of scalar coupling could be used to determine dihedral angles and establish configuration and conformational information. • For many stereo chemical problems groups more than three bonds apart have to be related. For example to assign the methyl groups of camphor coupling constants are not useful. Through space interaction (direct distance information) is often needed CH3 H3C O • => Dipolar coupling does not depend on bonds but only on proximity through space (∼ r-3) Review Dipolar coupling: DAB 1 h 3 cos2 1 A B 2 3 2 4 rAB Depends on the nature of the nuclei (γ), the distance between them (r) and the angle between the dipolar field and the internuclear vector (θ) • Can be measure directly in solids, but not always in a trivial manner, as θ will vary in powders • Typically vanishes in solution because of 3 cos2 θ -1 term averages to zero • Measurement of D is possible in solution when an aligning solvent is used (liquid crystals, stretched polymenrs) • Indirect measurement possible through effects of dipolar coupling on relaxation: Measurement of T1 (first order) or NOE (second order effect) 1D NOE experiment – C-13 NO2 A H H-1 HC HB HD Regular proton Decoupled C-13 C-13 weak pulse at position H A B1 /2 = 10 - 30 Hz decouple only one proton H-1 Selectively decoupled C-13 C-13 A B C D • decoupling (saturating) one proton at a time without affecting the other protons can be achieved by irradiating with very low power (γB1/2π ≈ 10-30 Hz) at the frequency of one specific hydrogen • This results in a spectrum where only carbons close to the irradiated proton show enhancement. Proton-proton NOE • In a similar manner, NOE’s can be measured between protons and can be used to assess H-H distances. • As the effects often are small typically the difference to a non irradiated spectrum is typically determined. NO2 H A H C HB weak pulse at position H A B A B1 /2 = 10 - 30 Hz HD p = 2- 10 s A B C D A B C D A B C D reference experiment (no saturation pulse or pulse centered outside spectral region) difference spectrum A-B NOE and relaxation • NOE arises from a change in population caused by relaxation. Consider a pair of protons close in space but not necessarily coupled: • four transitions can give rise to an NMR signal, corresponding to transitions involving flip of one spin (single quantum transitions • relaxation can occur across six transitions, the four single quantum transitions and the double- and zero quantum transitions, with the transition rates (probabilities) W1, W0 and W2. HA HB 1 A B W1A W1B A-B) W0 A+B) A W2 W1A B W1B 1 HA HB Maximum possible NOE • The maximum possible NOE is given by the ratio of cross- to total relaxation: W2 W0 cross relaxation A W1 W1B W0 W2 total spin lattice relaxation 1 T1 • Relaxation terms W1 and W0 unique to dipolar coupling • W1 terms can depend on many contributions besides dipolar coupling. • Maximum NOE observed when dipolar coupling dominant intercaction Short Theory of Relaxation Relaxation is induced by random fields caused by molecular motion: molecular motion(frequency) • orientation dependent interaction (changing field) Some important interactions are: • Anisotropy of the chemical shielding (tertiary carbons) • dipolar (through space) nuclear spin-spin coupling Important for protons and protonated heteronuclei (C-H, N-H) • dipolar nuclear spin - electron spin coupling : important when paramagnetic molecules are present (like oxygen) • quadrupolar coupling: orientation of non spherical nuclei in electric field. Only for I > ½ nuclei, but then almost always dominant Molecular motion: Brownian motion for spherical molecules: Drot 1 kT 6 C 8r 3 solv τc: Correlation time R: radius of molecule η: viscosity As the motion is random, Drot represents an average rate of motion, with a certain distribution of frequencies present in the ensemble of molecules determined by the Boltzmann distribution. Spectral Density c J ( ) 1 2 c2 Gives the fraction of molecules that are reorienting with the frequency ω The relaxation rates Wi will be most efficient when the tumbling at the transition frequency reaches a maximum. One can already see that for very fast or very slow tumbling the single quantum relaxation rate W1 at frequency ω0 will become very inefficient Spectral densities J(): number of molecules at a certain frequency (spectral density) Drot << 0 (large molecule, low T, high viscosity) Drot = 0 Drot >> 0 (small molecule, high T, low viscosity) 0 0 20 Dipolar relaxation and NOE From a lengthy and tedious quantum mechanical deviation one can obtain values for the relevant relaxation rates: W1A = 3/20 DAB2 A J(ωA); W1B = 3/20 DAB2 A J(ωB) W0 = 1/10 DAB2 A J(ωA - ωB) = 1/10 DAB2 A J(0) for A,B both 1H W2 = 6/10 DAB2 A J(ωA+ωB) = 6/10 DAB2 A J(2ω) for A,B both 1H Note that the relaxation rates depend on the square of the dipolar coupling constant, and thus are related to inter nuclear distance by rAB-6. max AB 5 4 10 23 4 2 2 0 c 2 2 o c 4 4 0 c 4 4 0 c Maximum NOE for a homonuclear spin system NOE versus τc Lets consider some extreme cases: very small molecules: Drot >> ω0 or τc ω0 << 1: ηABmax = + ½ very large molecules or viscous solutions: Drot << ω0 or τ c ω0 >> 1: ηABmax = -1 somewhere in between there will be a case with ηABmax = 0 !!! • The above leads to the apparently ridiculous result that the NOE does not depend on the distance A-B anymore. But note that we were talking of maximum NOE after achieving a steady state, considering an isolated pair of protons. • For an isolated two spin system it is not the NOE itself that depends on the distance, but the rate at which the NOE is generated, σ = W2 - W0. Therefore measurement of NOE buildup will be a more appropriate experiment in most cases. • If there where only two protons in the universe, irradiating one would always eventually result in the same NOE no matter what the distance. But if they are 1 light years apart it will take the age of the universe to build up that NOE • Usually more than two isolates spins are present and it is the competing effect of different dipolar couplings which will lead to different NOE values Example linear four spin system 0.8 % 49.2% 44.3 % A 4 B 2 45% C 1 D 49 % 0.7 % The maximum steady state NOE will be determined by the competition of relaxation pathways between different pairs of nuclei. • steady state NOE can be deceiving for isolated protons • The strength of the NOE will not be symmetrical. • In practice that means that more than one NOE should be considered before interpreting any result. Measuring Transient NOE sel a 1 H 2 b c 2 sel 2 1 H 2 x x,-x x + - Gradient a) Basic transient 1D-NOE difference experiment involving a selective 180⁰ pulse. b) Pulsed Field Gradient Spin Echo 1D NOE experiment c) NOE buildup curve for a linear four spin system. For short mixing times (<200 ms) the NOE is roughly linear with the cross relaxation rate σ Two dimensional NOESY x,-x,x,-x x x,x,-x,-x τ t1 acquisition(x,-x,-x,x) • The Sequence is identical to DQF COSY with a mixing time τ added. • A different phase cycle is employed selecting the z-magnetization present after the second 90̊ pulse (term I in the description of the COSY experiment 90x IzA ΩAt 1 90x(select z) -IzA cos(ΩA t 1) Two Dimensional NOESY (II) • During the mixing time τ cross relaxation and chemical exchange with spin B can take place with the rates σAB and kAB , respectively. • Both processes will create z-magnetization of spin B modulated with the chemical shift of spin A during t1 NOE(W2-W0) τ, exchange(kAB) τ σAB -IzA cos(ΩA t 1)(1-1/T1τm) + (W2-W0-kAB)τm IzBcos(ΩA t1) Diagonal peak cross peak z z z t1 90 y y x x z z z NOE(W2) 90x 90x y x N or OE ex (W ch 0 ) an ge (k AB y y x x z z ) 90x y x y x Result of 2D NOESY experiment: case I: σAB dominated by W0 (large molecules) OR chemical exchange: negative σAB => Diagonal and cross peak have the same sign (both normally phased positive, negative NOE gives positive cross peaks) case II: σAB dominated by W2 (small molecules): positive σAB => Diagonal and cross peak have opposite sign. Phase correction typically makes diagonal positive and cross peaks negative, positive NOE gives negative cross peaks) Properties of 2D NOESY spectrum • Cross peaks between protons which are in close spatial proximity • For short enough mixing times (small molecules 400 -800 ms, large molecules 100-200 ms) the cross peak intensity is proportional to the cross relaxation rate and thus to rab-6. • Correlation time τC t is usually not known. Therefore only relative distances can be determined. Usually known fixed distances can be used for calibration (geminal CH2, aromatic CH=CH, ...) • For small molecules NOE and chemical exchange will give cross peaks of opposite sign and can be distinguished. • As sequence is identical to DQF-COSY, coupling can cause artefacts in spectrum. These will be anti-phase in nature Limitation of NOESY experiment: • If the molecular weight is around 1000 g mol-1 Drot is of the order of the lamor frequency and W0 and W2 become very similar or equal. • NOE’s will be very weak or zero no matter how close the distance (5 + ω02τc2-4ω04τc4 = 0). NOESY experiments will fail in that case. Principle of ROESY experiment The zero NOE point is given by ω0τc = and thus depends both on ω0 and τc For a given molecule both parameters can be varied to reach a region where NOE ≠0: • One can vary τc by changing temperature or solvent (viscosity). This option is often limited due to sample considerations • change ω0: Using a different magnetic B0 field. On first sight, this option appears even more limited in practice as extremely weak magnets would give poor sensitivity and resolution. • However, relaxation taking place during a spin lock depends only on the effective field, which is the applied B1 field, which is much weaker than the static B0 field. ROESY Experiment x y t1 acquisition Spin lock pulse ωeff = ω 1SL = γHB1 typically 3-5 kHz Drot >> ω eff (or τc • ω eff << 1) will be always be fulfilled and ROE’s will always be positive. • ROE’s will be observable for molecules where NOESY gives no signal • For large molecules (or low temperature) ROE peaks and exchange peaks will have opposite sign and can be distinguished (as ROE will always be dominated by W2) ROESY vs. TOCSY The ROESY experiment utilizes the same pulse sequence as the TOCSY experiment. The only difference are the strength and length of the spin lock pulse: • TOCSY: γB1/2π 10 kHz, τPSL = 15 - 80 ms, pulse is too short for effective ROESY • ROESY: γ B1/2 π 1.5 kHz, τ PSL = 400 ms, field is too weak for effective TOCSY • sign of TOCSY peaks is always same as diagonal (positive), for ROESY peaks it is opposite to diagonal (negative) • TOCSY spectrum spectra can exhibit weak ROESY peaks, an ROESY spectra can exhibit weak TOSY peaks. Assignment of methyl groups in camphor 8 CH3 9 CH3 H H O H 4 CH3 10 5 H 7 6 H H H • CH3(8) is on the side of the C=O group • CH3(9) on the opposite side of the molecule • Assignment of diastereo topic protons 4, 6, 7 NOE and conformational analysis NOE’s can be used to supplement coupling constants in the measurement of dihedral angles. Particularly useful for identifying staggered conformations Example side-chain angle in proteins/peptides - CO NH > HMBC 2CO>3CO CO NH < 2CO~3CO, weak CO NH ~ 2CO<3CO Secondary structure in proteins H N N H i-H i+1 H N i H -N-C -H = -117 o ( ) o N i -Hi+3 H -N-C -H = -199 o ( = -139 ) o -HNi+1 Structure determination of Macromolecules Assignment J 6 NOE/ROE ~ 1/r (NOESY,ROESY) J coupling (E.COSY,DQ/ZQ,FIDS, quant. J) Distance Restraints Dihedral Restraints Structure Calculation (Simulated Annealing, Distance Geometry, Molecular Dynamics) 3D -Structure Example: Metallothionein beta-N domain: 33 residues Example Metallothionein • Using NOEs, H-H coupling constants and H-Cd couplings • Calculate many structures starting from different random starting points • Keep 30 lowest energy structures Some special considerations when working with biological macromolecules • molecular weight is 5000 - 30000 g/mol: • short T2 => broad lines • Experiments utilizing multiple steps of one-bond correlations work better than those utilizing long range couplings. In-phase transfer preferred over antiphase transfer (TOCSY vs. COSY) • heavy overlap of lines due to complexity of molecule => highest magnetic field possible should be used, two dimensional experiments reach their limits • usually computer software needed to keep track of assignment data • Often more than two dimensions are needed to resolve all signals. • Proteins can be produced using expression media, and enrichment with 13C and/or 15N which will allow experiments not possible with natural abundance samples Two dimensional NMR often insufficient CaM/C21W HN-HSQC Three – or more dimensional NMR necessary 1 H [ppm] 2.0 10 9 8 7 6 5 4 3 2 1 0 ppm w 11 w (15N ) 4.0 1( H) 5.0 4.0 3.0 1 1D 2.0 1.0 w (1H) H [ppm] 2D 3D Example HNCO 3D NMR for separating NOE peaks Slice of 3D NOESY-HSQC 2D NOESY 1 13 H [ppm] C = 20.3 ppm I74 H(CH3) 1 0.0 H [ppm] V68 H V15 H P67 H K42 H’ L29 H V93 H A85 H D53 H’ L82 H 2.0 2.0 V68 H V56 H V93 H Y23 H ’ Y76 H’ L82 H Y76 H Y23 H S90 H’ 4.0 S88 H V56 H V93 H S90 H S86 H A85 H 4.0 3.0 1 H [ppm] 2.0 N79 H V68 H V93 H’ V15 H’ V68 H’ V56 H’ 5.0 4.0 P26 H 1.0 1.2 1 0.8 H [ppm] 0.6 Example Calmodulin • 148 Amino acids • 3 D experiments necessary • Takes many months to complete assignment and structure determination

![Job Evaluation [Opens in New Window]](http://s2.studylib.net/store/data/009982944_1-4058a11a055fef377b4f45492644a05d-300x300.png)