WEEK 11 2008
Care of the child with
Special needs
Associated with
Genetic Variations
Genetics is the new frontier of biology and
medicine.
HUMAN GENOME PROJECT has changed the
world of medical care.
Now health promotion, disease prevention and
treatment can be specific for target populations.
Resources Used for this ppte
Elsevier support materials
Hockenberry & Wilson Text for 277
Hockenberry & Wilson Text CD
Mosby’s Electronic Image Collection 2001
Previous ppte versions for 277
Contributions from partners teaching
materials
Various Web sources as indicated for
genetics topics
Excerpts from "Genetic Counseling" by F.C. Fraser, The American Journal of
Human Genetics, 1974, pp. 636-659.
(W. Wertelecki, M.D.) 2003 www.ibis-birthdefects.org, All rights reserved. 23 Mar
2006 This site offers information mostly for educational purposes.
Photo Stockphotos, Free photos
http://www.dnaftb.org/dnaftb/19/concept/index.html
Centers for Disease Control and Prevention,1600 Clifton Rd, Atlanta, GA
30333, U.S.A
Tel: (404) 639-3311 / Public Inquiries: (404) 639-3534 / (800) 311-3435
Medgen.genetics.utah.edu/back.htm ( excellent link to lots of resources)
National March of Dimes
GENETIC VARIATIONS
Caring for the child with special
needs
Topics for this week
Preparatory Readings :
Abnormal Sexual Maturation
Obesity
Hyperlipidemia
Metabolic syndromes
Classroom Focus: Glossary of KEY terms for Chapter 5
Genetic patterns, expressions, metabolic errors
Osteogenesis Imperfecta and structural anomalies
Down Syndrome
Fragile X
Foetal Alcohol Syndrome
Arthritis (JRA)
Hemophilia
Sickle Cell
Hypo and hyperthyroidism
Thalasemia
A Beginning Understanding of
Genetics and Congenital
Anomalies / Birth Defects
HUMANS ARE DIPLOID BEINGS:
2n = 46
22 pairs of autosomes and 2 sex
chromosomes
There is a duplicate copy of every chromosome
within somatic cells ( non reproductive cells) of
the body, gained through exact duplication by
mitosis.
If the chromosome material does not migrate
evenly, nondisjunction occurs and one half of the
cells has 45 chromosomes and die, the other
half gain one and are 47, which is compatible for
life, and so continue to replicate in the trisomy
fashion (extra chromosome).
Somatic cells contain 44 autosomes ( 22pairs of
chromosomes that do not greatly influence
gender differentiation, and two sex
chromosomes- xx=females, xy= males).
The gametes - male and female reproductive
cells have a haploid number (n=23), so the body
seeks to combine these pairs to create 46.
Mitotic nondisjuction results in an individual with
mixed cell pairings(mosaicism) in which there
will be two distinctly different cell populations
Birth defects or congenital anomalies are present
at birth but Not all are genetic
Occur in 2-4% of live born
children around the world
across all populations.
Expressions can occur
at any time in life( Tay- Sach’s
Disease, Autism Spectrum
Disorder, Diabetes,
Huntington’s Chorea).
Normal stages of cellular and
system growth may stall and
become a defect in
subsequent stages of growth
and development (cleft lip and
palate).
.
Orphan Disease
Any congenital disorder that is so
rare that only a few individuals
have it.
puts great pressure on the family
to find resources, create interest in
the child’s condition so as to
promote research, pharmaceutical
and other interventions.
Many families spend their savings
looking for a cause and a cure but
often to little positive outcome.
No critical mass to effect change
or influence support and
legislation.
Families are usually solitary, often
without any support or resources,
unknown aspects of the future
create tension and daily care
needs and interventions can not
be predicted.
Birth defects may have multiple
causes
Many have polygenic
( complex) inheritance
patterns ( cataracts,
pyloric stenosis)
A syndrome is defined as
a pattern or malformation
resulting from a single
cause (Down Syndrome)
An Association refers to a
nonrandom pattern of
malformations of
unknown etiology
(VACTERL p.107)
Many (if not all) diseases and variations in
Morphogenic Development (human-specific) have
a genetic component
CAUSES :
*Classic single gene Mendelian Patterns of Inheritance apply to govern frequency of
expression( penetrance and expressivity)
*Incomplete replication and division of cells of zygote ( mitotic nondisjunction)
*Maternal and Paternal disease, hereditary variations, age of ova, chemical interrupters and
teratogens in the body
*Alteration in Chromosome number resulting from unequal movement of genetic materials and
incomplete strand separation of genetic materials during meitotis (gamete formation).
Variable Structure and Placement of the chromosome causes variations
*Gene location- whether on an autosome or sex chromosome directs whether the expression is
recessive or dominant
*Environmental Triggers, pollutants, Hormone interrupters, lifestyle choices, deficiencies,
unknown
GENES are the segments of DNA
that contain the species-specific
information.
Control physiological functions
Key in the characteristics
Encode proteins through structural genes
Mutations can occur through structural genes
alterations and replication of the altered synthesis of
proteins. Many mutations are life threatening.
Congenital conditions present at birth may not be
hereditary in origin but a fresh mutation / anomaly
/variation.
Environmental chemicals can alter the functions
and interactions of genes
An example of the DNA linkages for
one gene – creates a gene map
PubMed Entrez BLAST OMIM Taxonomy Structure Search Homo sapiens Build 36.2 (Current)BLAST The Human Genome Chromosome: [ 1 ] 2 3 4 5 6 7 8 9 10
11 12 13 14 15 16 17 18 19 20 21 22 X Y MT Query: CCV [clear] Master Map: Genes On CytogeneticSummary of Maps Region Displayed: 1pter1p36.23 Symbol Links Cyto Description LOC653340
1p36.33 similar to hypothetical gene supported by AK024248; AL137733 LOC728481
1p36.33 similar to
similar to RPL23AP7 protein OR4F29 hm 1p36.33 olfactory receptor, family 4, subfamily F, member 29 HES4 OMIM pr hm 1p36.33 hairy and enhancer of split 4
(Drosophila) LOC728578 pr 1p36.33 hypothetical protein LOC728578 LOC643921
1p36.33 similar to protein tyrosine phosphatase, non-receptor type 11
(Noonan syndrome 1) UBE2J2 pr hm sts 1p36.33 ubiquitin-conjugating enzyme E2, J2 (UBC6 homolog, yeast) CENTB5 pr hm sts 1p36 centaurin, beta
5 PUSL1 pr hm sts 1p36.33 pseudouridylate synthase-like 1 DVL1 OMIM pr hm sts 1p36 dishevelled, dsh homolog 1
(Drosophila) MXRA8 pr hm 1p36.33 matrix-remodelling associated 8 AURKAIP1 OMIM pr hm sts 1p36.33 aurora kinase A interacting protein
1 VWA1 pr hm sts 1p36.33 von Willebrand factor A domain containing 1 ATAD3B pr hm sts 1p36.33 ATPase family, AAA domain containing
3B SSU72 pr hm sts 1p36.33 SSU72 RNA polymerase II CTD phosphatase homolog (S. cerevisiae) GNB1 OMIM pr hm sts 1p36.33 guanine nucleotide binding
protein (G protein), beta polypeptide 1 HES5 OMIM pr hm 1p36.32 hairy and enhancer of split 5 (Drosophila) PRDM16 OMIM pr hm 1p36.23-p33 PR domain
containing 16 WDR8 OMIM pr hm sts 1p36.3 WD repeat domain 8 KIAA0495 pr sts 1p36.32 KIAA0495 LOC728750 pr 1p36.32 hypothetical protein
LOC728750 C1orf174 pr hm sts 1p36.32 chromosome 1 open reading frame 174 ICMT OMIM pr hm sts 1p36.21 isoprenylcysteine carboxyl
methyltransferase GPR153 pr hm sts 1p36.31 G protein-coupled receptor 153 ESPN OMIM pr hm sts 1p36.31p36.11 espin PLEKHG5 pr hm sts 1p36.31 pleckstrin homology domain containing, family G (with RhoGef domain) member 5 NOL9 pr hm sts 1p36.31 nucleolar
protein 9 DNAJC11 pr hm sts 1p36.31 DnaJ (Hsp40) homolog, subfamily C, member 11 CCV OMIM sts 1p36 cataract, congenital, Volkmann
type PARK7 OMIM pr hm sts 1p36.33-p36.12 Parkinson disease (autosomal recessive, early onset) 7 Summary of Maps:Map 1: Genes On SequenceTable
ViewRegion Displayed: 0-9,100K bp Download/View Sequence/EvidenceTotal Genes On Chromosome: 2782 [27 not localized]Genes Labeled: 50 Total Genes in
Region: 153Map 2: Phenotype (includes QTLs)Table ViewRegion Displayed: 0-9,100K bp Download/View Sequence/EvidenceTotal Phenotypes On Chromosome:
235Phenotypes Labeled: 10 Total Phenotypes in Region: 10Map 3: OMIM MorbidTable ViewRegion Displayed: 1pter-1p36.23Total Markers On Chromosome:
252Markers Labeled: 46 Total Markers in Region: 46Map 4: Genes On CytogeneticTable ViewRegion Displayed: 1pter-1p36.23Total Genes On Chromosome:
2980Genes Labeled: 30 Total Genes in Region: 187
Human genome overview page (Build 36.2)
Human genome overview page (Build 35.1)
Map Viewer Home Map Viewer Help
Human Maps Help
FTP
Data As Table View
Compress Map Region Shown:
You are here:
default
master
Disclaimer | Write to the Help Desk
NCBI | NLM | NIH
Look at the child you are caring for. See what their bodies can
tell youear, head shape, placement of eyes, set of ears, is face
symmetrical? Muscle tone, cry pitch, volume, finger prints, whirl
of the hair, set of teeth and palate, nailbeds, hands and feet
Genetic Variations Create
Special needs
No one is immune to possible alteration
Some major others minor
Every system and bodily function can be affected
May be a surprise or expected
Places stress on family dynamics & resources
Dreams of normality disappear
Parents may “maladapt” or “bonadapt”
Nursing responsibilities and roles
are directly related to providing
accurate information, emotional
support, and advocacy for children
and families with special needs
Cultural and ethnic groups
may have different genetic
burdens that cause
morbidity and mortality.
Tay-Sachs Disease and
Infantile Spinal Sclerosis
affects infants of Jewish
heritage. Genetic defect
can be screened- causes a
lipid storage metabolic
anomaly in which nerves
are destroyed and infant
regresses to a blind, deaf,
decerebrate, rigid, and
seizure state. Lethal
Metabolism of Dietary intake is modified by specific
enzymes, without which there may be health problems .
Some ethnic populations do have variations in enzymes and
so must consume the diet most beneficial to their physiology.
There are many multifactorial
causes -EXAMPLES
Phenyketonuria(PKU) – genetically predetermined absence of
phenyl hydoxylase present at birth- interrupts metabolism of
Phenylalanine an amino acid found food sources such as milkcauses the build up of phenyl toxins – leading to mental retardation
and death
- due to the lack of gene to produce the specific digestive enzymeinterrupts metabolic cycle, builds up incomplete products of
metabolism and thus the body becomes toxic, increased
incompatibility with life- triggered by food sources / feedings - Can
test at birth as is common enough to be of concern to the
population.
Treatment is to eliminate the offending protein sources via a special
elimination diet and special formulas, and in some cases provide the
enzyme supplement orally. Lifelong condition
PKU
PKU (Phenylketonuria)
Autosomal-recessive – absence of
enzyme needed to metabolize amino acid
Children with PKU – blond hair, blue eyes,
fair skin due to missing tyrosine
S/S: failure to thrive, vomiting, irritability
Blood sample for DX – baby should be
>24 h old
Management: restriction of dietary protein
PKU contd.
Blood sample for DX – baby should be
>24 h old
Management: restriction of dietary protein
20-30 mg of phenylalanine/kg of body
weight
Diet is supplemented with minerals,
vitamins, vegetable oil (for calories)
Glactosemia
Inborn error in metabolism of dietary
lactose. Must eliminate and avoid all
sources. Life long error in multiple systems
P.333-340 Many variations exist in the
production and lack of production of
enzymes and amino acids necessary for
growth .Often mechanism is to avoid the
offending substance, adding artificial
enzymes orally or by injection. Breaks in
the chain of chemical by product removal
and release of energy are also affected.
Mitochondrial Disorders
Outside the nucleus of each
cell are DNA strands called
mitochondrial DNA. Their main
purpose is to regulate the
cellular metabolism and
produce energy within the
cytoplasmic cellular organelle.
This is a female contribution to
cell activity. Mitochondrial
mutations are then all linked to
the mother’s genetic
contribution.
The male contributes
mechanims and material for
the nucleus metabolism.
Mitochondrial DNA Mutations
Mitochondrial DNA Mutations
are responsible for various
movement disorders,
hypotonia, respiratory
dyskinesia, seizures,
regression, dementia,
encephalopathy, heart block.
Many disorders are so
specific that they can not be
easily detected and should be
treated symptomatically for
comfort and reduction of
symptoms. No cure is possible
for most of these. Family
support is important.
Cancer genetics
There is evidence that certain types of Cancers
are more prevalent in selected families, common
types are breast, colon , ovarian and leukemias.
Premature Family deaths have been investigated
in several families and were found to be linked to
oncogenes that were associated with age and
hormonal changes and the onset- some
triggered by environmental and lifestyle
conditions such as diet. Preventive screening
and prophylactic surgery ( removal of the
targeted organ /system) have saved lives.
Cancer
The absence of – or destruction of
antioncogenes or
oncosuppressors will not facilitate
defence against unregulated cell
division.This leads to wild and
irregular tumor growth such as in
Li-Fraumeni Syndrome , an
autosomal dominant trait which is
responsible for osteosarcoma,
brain tumors, adrenalcortical
carcinomas in childhood.
Retro viruses can trigger cell
mutations.
Gene surveillance and screening
for inherited cancer is available for
retinoblastoma, medullary thyroid
carcinoma, & Wilm’s tumor( germ
cell mutations of the cancer gene
transmitted to the foetus) .
Multifactoral (Complex) Disorders
Multifactorial conditions
created through interaction of
miscued genes with various
triggers or absence of
essential proteins and
enzymes, cell cues and even
oxygen.
Cleft lip/ palate, congenital
heart defects, congenital hip
dislocation and pyloric stenosis
are all examples.
Screening and DNA analysis
of the affected child can yield
information that may be
essential to the health of the
affected child and for
consideration re: other
pregnancies.
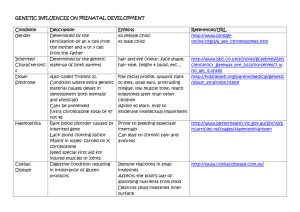
INTRAUTERINE ENVIRONMENT
The combination of genetic factors, male
and female parental contributions to the
placental placement and quality of the
foetal implantation( respectively) can have
many repercussions on the health, growth,
cellular differentiation and maturation of
the fetus in utero.
Intrauterine Growth Retardation is
associated with many genetic anomalies.
Teratogens
Need to eliminate teratogens*regulate drug ingestion
( prescribed, recreational, illicit),
*eliminate household and
occupational inhalants/ solvents,
other chemicals such as hormone
blockers or estrogen enhancers,
PCB’s, Dioxin, radiation,
*avoid viral illnesses such as rubella
and Cytomegalovirus, or
toxoplasmosis,
*prevent hyperthermia, physical
trauma,
*know and intervene in maternal
hereditary conditions, metabolic
conditions.
*Add cell mitigating nutrientsie.biotin
Normative variations & influences
Heredity helps to provide the building blocks for human growth and
development.
Genetic time keepers and hormones interact to promote the
appropriate interaction and bodily change.
Adolescence is hormone mediated, bathing every cell with sex
hormones, Onset of puberty keyed onset by hormonal regulators
that have been inherited. Now a mixture of environment & heredity.
Hormone interrupters and free estrogens( in plastics, foods, cleaning
agents) as well as hormone blocking agents( Pollutants, teratogens
dioxins and furans, PCBs, heavy metals)
Family patterns of risk-taking, talents and interests, sexuality,
physical skills and activity, maturity onset, patterns of gender
expression, growth (height/weight), breast development, hair
texture, amount, placement, gain and loss, distribution of body fat,
skin colour, extra teeth at birth, number of digits, vision and hearing,
centre of gravity and co-ordination, memory, metabolism,
Hypercholesterolemia, right from birth, the balance of “Good and Bad
Cholesterol” may be a challenge. Family history for heart disease,
stroke, BMI, activity level and dietary intake of saturated fats is
essential to healthy childhood and adulthood. P. 117, 1488-1493
These boys are the same age. What is their future?
Puberty and Heredity
gender linked expression influenced by gender.
X X or X Y , if more or less sec chromosomes,
then phenotype looks different,( Klinefelter and
Turner Syndromes )
Puberty & maturation not necessarily linked to
reproductive capability if an anomaly that is not
compatible with life. Drugs may interfere.
Species specific pattern of reproductive
capability is planned to assure future
generations and ongoing passage of gene pool.
Earlier onset of menses may relate to genes,
food amount, type, source,
Growth as an indicator of health
Stature and weight must
be plotted at each wellchild visit. A plateau may
indicate a growth
slowdown.
Some plateaus are
natural, drop of percentile
placement on chart may
identify a hereditary link.
Lack of growth hormone
may be a cause, as may
hereditary blueprint
founded in family tree.
Debate:
Obesity in children is a social health crisis that can be attributed to
overindulgent parents, affluence and the genetic predisposition of
children to store extra calories in preparation for periods of anorexia.
Obesity
Significant increase in past 10 years
Focus on health not weight
Family involvement not just individual child
Education about balance between diet &
activity. Need to limit trans fats, sodium
and calories
Some part of the Obesity Puzzle
may be genetic- but not 100% of
the explanation!
This cyclic diagram
shows the interaction
of inheritance with
lifestyle, metabolism,
hormones, behaviour,
availability, balanced
(or not) with exercise,
correct food selection,
metabolism rate,
psychosocial
wellbeing
Solutions to Obesity ?
Need family + societal responsibility
Add exercise to daily routines with
community support e.g. safe walking trails
In Quebec have started a “walking school
bus” program in some areas
Advocate for healthy school lunch
programs
Healthy alternatives in restaurants
Do not confuse Cushing Syndrome
with Obesity
The distribution of body
fat in situations
associated with the onset
of Steroid imbalance as
per Cushing Syndrome
may make it difficult to
differentiate it from
obesity. One must correct
the cause of the
imbalance and remediate
the damage to the body .
Many are made fun of at
school
Other Metabolic errors of
metabolism
Diabetes Mellitus 1705-1727 indepth management is essential as the
growth and development of the child creates highs and lows in case
management,Parents need support to promote the child’s self-care,
in aspects that the child has learned. Need to be
educated immediately and allowed to practice with
equipment.
Insulin Dependent Diabetes is increasing as is
Type 2
diabetes. a
wonderful experience in self-control and self-care.
Diabetes contd.
Parents can help
child master skills for
finger pick, glucometer
testing, insulin calculation
& drawing up, menu
selection and exercise
balance.
Skin care vital, exercise
and self-esteem need
boosting. Summer Camp
often vital to healthy
adjustment
Juvenile Rheumatoid
Arthritis( JRA)
also called Juvenile Idiopathic Arthritis (
JIA). p. 1791-1800
Most youth with JRA ask “why ME?”
mostly female affected, elevated
ESR,CRP,RF positive,
Potential painful joint swelling, limited
mobility, inflammation of joints and eyes,
fever, rash, splenomegaly & hepatomegaly,
sleep disturbance, exercise intolerance,
weight gain and moonface from prednisone,
bloating, CARE – eye protection,
antirheumatic drugs, NSAIDS,,
Methotrexate, Folic Acid daily, avoid sulfa
drugs, Steroids, protective isolation,
physio,massage,swim,splints, heat, rest
periods, relaxation, school, environmental
adaptations
HUMAN SOMATIC CELLS
( all those cells with nuclei except the ova
and spermatozoa)
Contain22,000 - 25.000
genes; Genome project
revealing 35,000
Distributed in sequences
in the form of a tight coil the DNA helix molecules
Four chemicals make up
the helix ( Adenine links
with Thymine, Guanine
links with Cytosine )
Along 46 chromosomes that very in size,
shape, and groupings. The larger the
chromosome, the more Genetic material it
holds. Addition of any chromosomes to
the allotted 46, brings added genetic
material, something that is not compatible
with life
BOX 5.1 pp 104-105
Key Genetic Terms.
The KARYOTYPE is created from the DNA sample- Blood, Lymphocytes,
Buccal Mucosa, amniotic fluid.
Technicians grow colonies in sterile medium/gel.
DNA binds with various polymers to allow for visualization of specific
sections through repeated electron microscopic photographs of the cellular
material.
Various stains and contrasting agents and enzymes bind with specific DNA,
allowing the distribution of the DNA profile to be displayed on a set grid.
The various chromosome images are photographed , then cut up and
arranged in order of chromosome groups from 1 to 22 plus the sex
chromosomes. X & Y ( see p. 126)
The images of each chromosome are further
analyzes through various electrospectrographic
imaging processes and the individual’s
chromosomal portrait is revealed, compared with
the expected norm and known profiles of
disorders and diagnoses made based on the
differences variations or similarities observed.
( Family clusters, relatives affected similarities
with others not related, but with the same
characteristics, health problems or other
population-specific variables)
NORMAL MALE KARYOTYPE
This is an example of a spectral karyotype .
The slide on the Rt is of
Klinefelter Syndrome
SINGLE GENE DISORDERS
Chromosome anomalies and variations can affect a large
number of genes . The more chromosomes that are
anomalous, and the more genes that are affected usually
translate into the increasing severity and extent of the
genetic anomalies seen. A single gene will not destroy a
chromosome’s structure or number, but can result in
severe physical and mental disorders.
Single gene disorders follow the Mendelian patterndominant or recessive expression in any system of the
body. These disorders are not detectable by regular DNA
analysis and Karyotyping. Need special diagnostics.
Depending on the power of the gene, there may be
strong or masked expression in the phenotype. Variable
Expressivity can be mild to severe ( e.g.allergy).
The larger the
chromosome defect,
the less compatible
with life - look at the
photographic layout of
chromosomes -a
KARYOTYPE - to see
differences in size,
shape, and centromere
position between the
arms.
Centromere positions can
also cause defects if the
chromosome is a
complex one and
materials are not linked
symetrically
Acrocentric( high ),
Subcentric (Low),
or metacentric(equal
length) uniting the
chromosomes to form the
shape.
Nondisjunction during meiosis
(Trisomy G21(G group)
results in Down
Syndrome
compared to Trisomy D13
( Patau Syndrome/ less
compatible with life)
CHROMOSOMES CAN BREAK
Chromosomes can be damaged by a variety of
CLASTOGENS (chromosome breaking agents) :
radiation, chemical, heat, viral.
Damage is called A STRUCTURAL ANOMALY
May be temporary or become permanent in the somatic
cells. The germ cells (reproductive) may be damaged
and pass that impact on the future generations.
DELETION- loss of a chromosomal arm of other part.
Chromosome 18 known for deletion potential.
MICRODELETIONS are common and cause typical PHENOTYPESor patterns,
Prader-Willi Syndrome,
Angelman Syndrome( Happy Puppet ) ,
Di George Syndrome
TRANSLOCATION
Fragile X
End points become sticky,
gathering rearranged
fragments from other
chromosomes through
TRANSLOCATION.
Translocation becomes a
problem in future generations if
there is more or less genetic
material to migrate in cell
division.
Fragile X syndrome is an
example of the loss of
chromosomal material from the
X chromosome that results in
physical characteristics,
behaviours, and mental
delays.
Marfan Syndrome has a distinctive
phenotype
The PHENOTYPE is the
human example seen in
real life- the variations
from the normal, the
association of different
diagnostic findings,
characteristics, functions,
missing elements, signs
and symptoms. General
clinical characteristics
and life expectancy can
be derived from how they
appear.
What others see,
may reflect your genetic makeup.
SPECIFIC ABNORMAL
KARYOTYPES YIELD
SPECIFIC PHENOTYPES
(Aneuploides)
Trisomy G(21) Down
Syndrome, Trisomy E916-18)
Edwards Syndrome, Trisomy
D( 13-15) Patau Syndrome
Sex Chromosome
Aneuploides: Klinefelter
Syndrome( XXY ) p.112-114,
Jacobs Syndrome (XYY),
Turner Syndrome (X0), Triple
X (XXX)
AUTOSOMAL DOMINANT AND
RECESSIVE INHERITANCE
The outcomes relate to the offspring of
affected parents. Gene expressions may
be found in genotypes. How they are
expressed is the phenotype.
Homozygote = normal Genotype +/+
Heterozygote= Affected Genotype +/Homozygote affected= Severe Genotype -/
Some characteristics are strongly
expressed even with a small gene
variation
Some major genetic
variations are not
compatible with life as
mutant alleles are
strongly expressive.
Some affected alleles
are linked to the male
and female gender
chromosome- either
dominant or recessive
.
Turner syndrome xo
In This condition –
lack of the second x chromosome
Creates a pseudo female but unable to reproduce.
Life is compatible but note the skeletal variations
Neck, shoulders, chest, feet hands.
Also note the adult hand to side for comfort!
Turner Syndrome in an adolescent girl
Short stature,
webbed neck,
increased abduction
of hands
broad barrel chest,
asexual development
Turner Syndrome
X O – Monosomy ; half the sex chromosome material. Physiological foetal
development copes by making tissue for all systems, but cuts down on
cellular cues for development of reproductive organs & ova.
Phenotype female- common assessment findings in all with Turner
Syndrome- web neck, barrel/shield chest, infertile, short, decreased
estrogen, ovarian cysts, urinary tract variations, delayed puberty if at all.
Treatment with growth hormone, steroids, estrogen replacement. Some
may choose cosmetic surgery. follow personal talents and interests ,
Social preparation, support in making friends- explanation to others about
condition if they ask. Need ways to be and look feminine.
Career selection to match interests and capabilities.
CONCEPTION AND VARIATION
Severely affected homozygous genotypes
will have a matching disease profile of
severity. This may influence reproduction
capacity and definitely negatively impacts
on heredity.
Not likely that severely affected individuals
will reproduce. More likely that a
heterozygous mate or a normal
homozygous person will procreate.
AUTOSOMAL DOMINANT
PATTERN
Autosomal dominant disorders
Parents are affected +/- :
unaffected normal + +
+
[Affected parent +/-]
[ Normal Parent,++] + ++ ++ ++ +
Apply Punnett Squares
Outcomes 50% normal 50%
affected
Autosomal dominant disordersPolydactyly, Achondroplasia,
Neurofibromatosis, Marfan
Syndrome, Huntington’s Chorea
Affected child has affected parent
50% chance for children to be
affected if parent is affected
Phenotypically unaffected & are
free of the disease
AUTOSOMAL RECESSIVE
PATTERN
Autosomal recessive disorders
Recessive inheritance relies on the gene
arrangements that allow for the
development of a heterozygous carrier
state when the individual has a defective
gene but it is masked by the other paired
allele, and so there is no expression.
people seem normal. Problems arise when
two carriers procreate and contribute the
defective allele creating a homozygous
affected condition.
Autosomal recessive disorders
Version 1 ( both parents carriers)
Parent (carrier) +/+ + ++ +Apply Punnett squares
- +- -Outcomes per each pregnancy
25% normal (++)
50% carriers ( +-)
25% affected ( --)Version
Autosomal recessive disorders
Version 2.
Parent1 affected ( -/-)Parent 2 normal ( +/+)
apply Punnett squares
_
_
+
+- ++
+- +Outcomes for each pregnancy = 100% carriers( +-)
Autosomal recessive disorders
Conditions may be
extreme for those
affected: Tay-Sach’s,
Sickle Cell Disease,
Cystic Fibrosis,
Thalassemia,
Congenital adrenal
Hyperplasia,
galactosemia, PKU,
MUCCOPOLYSACCH
ARISOSIS,
Sex-linked Inheritance Patterns
When affected genes are linked to either of the
sex chromosomes, this is called sex-linked
inheritance. As few genes are found on the Y
chromosome. These are associated with
masculine characteristics such as baldness,
hairy ears, bushy eyebrows and are passed
from father to son.
X-linked chromosomes can be passed from
fathers to daughters as can those from the
mother. The mother can pass x’s to sons as well
( Fragile x syndrome) myotonic dystrophy, and
Huntington disease.
Sex-linked Inheritance Patterns
contd.
Women who are homozygous for the
defective allele(xx) will express as the
phenotype, but if heterozygous ( Xx), then
they will be asymptomatic as the dominant
X will mask the recessive x.
Men with a defective x allele will have no
expression in themselves( Yx), but will
transmit the defective x to their daughter
(Xx) as they have only one x to contribute
Sex-linked Inheritance Patterns
contd
Apply Punnett squares
X X
Y YX YX all sons normal
x xX xX
all daughters carriers
___________________________________
x X
Y Yx YX Yx affected ( 50% ) YX Normal ( 50%)
X Xx XX Xx Carrier
XX Normal
X-LINKED DOMINANT PATTERN
X-LINKED RECESSIVE PATTERN
SINGLE GENE DISORDERS
Chromosome anomalies and variations can affect a large
number of genes . The more chromosomes that are
anomalous, and the more genes that are affected usually
translate into the increasing severity and extent of the
genetic anomalies seen. A single gene will not destroy a
chromosome’s structure or number, but can result in
severe physical and mental disorders.
Single gene disorders follow the Mendelian patterndominant or recessive expression in any system of the
body. These disorders are not detectable by regular DNA
analysis and Karyotyping. Need special diagnostics.
Depending on the power of the gene, there may be
strong or masked expression in the phenotype. Variable
Expressivity can be mild to severe ( e.g.allergy).
Sometimes Letters get mixed up or
collapse or move and break away
•
The structural arrangement
of DNA, which looks
something like an
immensely long ladder
twisted into a helix, or coil.
The sides of the "ladder"
are formed by a backbone
of sugar and phosphate
molecules, and the "rungs"
consist of nucleotide bases
joined weakly in the middle
by hydrogen bonds.
•
http://www.dnaftb.org/dnaft
b/19/concept/index.html
Duplication of chromosomal
materials
CHROMOSOMES may
DUPLICATE segments, join in
circles, or continuous chains,
microduplication results in
various syndromes, but not
generally obvious or
incompatible. The family on the
left , mother, daughter and
grandson, all show
compounding effect of
microduplication – condition
becomes more pronounced
with each generation.
Genetic Disorders are classified as
GENE MUTATIONS
(Sickle Cell Disease, Galactosemia,
Cystic Fibrosis)
or
CHROMOSOMAL ABNORMALITIES
(Turner Syndrome, Cri du chat Syndrome , Down Syndrome)
Genes can mutuate by a repeated pattern or gene-expanding miscue
called premutations.
Gene mutations by deletion will have different expression if male or
female, even though the same chromosome is involved( 15- Prader
Willi if deletion from a Male chromosome, but Angelman Syndrome if
deletion is from Female chromosome 15)(p.123)
One parent can contribute both mutated alleles through uniparental
disomy in which a trisomy allele is discarded leaving two defective
alleles that carry the defect, but do not affect the parent.
A few Infant cues to observe for in
congenital variations
Infant and neonatal assessments may also present cues
for consideration of genetic anomalies and variationsbirthweight, pitch of cry, low hairline at nape, tufts of hair
at coccyx, web neck, cataracts, retinal reflection( white),
sclera blue, tonicity of muscles, number and positioning
of fingers/toes, webbing, ear set, patent nares, rectum,
and esophagus, birthmarks, hip dyslocation, reflexes,
speech, cognition & developmental milestones,
behaviour, attachment, genitalia( distinct or ambiguous),
Skeletal proportion, eyes- palmar crease and foot shape
etc.
Each anomaly is unique and has typical findings for the
disorder and mutation, syndrome, & disorder.
Congenital Hypothyroidism (CH)
Transient or permanent
Deficiency in thyroid hormone or blocking of excretion
Life threatening
Preterms have CH due to immature thyroid development
as well as immature pituitary and hypothalamus.
This type resolves as premi matures and grows.
Enlarged thyroid tissue –goiter- can block airway
Maternal release - exogenous source-becomes depleted after birth
Infant soon shows signs of Hypothyroidism
Assessment findings/clinical manifestations
POOR FEEDING
LETHARGY
HOARSE CRY
CONSTIPATION
PROLONGED JAUNDICE
CYANOSIS and APNEA
BRADYCARDIA
LARGE FONTANELS
HYPOTONIA
LABOURED BREATHING
CLASSICAL FACIAL FEATURES @6wks after birth
Depressed nasal bridge,short forehead,puffy eyelids
Cool to touch,mottled,grayish skin tone
coarse dry lusterless brittle hair
Skin thick and leathery
Abdominal distention, umbilical hernia
Anemia,bradycardia,bradypnea
Hyporeflexia, hypotonia
Abnormal deep tendon reflexes- “frozen”
Wide patent cranial sutures
Immature bone age
Constipation
Lazy feeder,sluggish
SEVERE MENTAL RETARDATION
DIAGNOSTICS
Heel stick Blood spot for Thyroxine (T4)
Serum level of TSH
May follow with free T3 & T4 and Thyroid bound globulin
Nuclear medicine scan for “I” uptake
CT scans for thyroid tissue
Bone scans – growth plate
May need repeated measures
Low T4 and high TSH = CH
PROGNOSIS / OUTCOME
Early diagnosis and thyroid replacement essential
Synthroid or Levothyroid
LIFETIME OF TREATMENT NEEDED
Regular bloodwork for levels & adjustment
Can grow and learn well while on replacement
NURSING CARE
EARLY IDENTIFICATION
Look for an unusually “good”baby- sleeps all the time,never cries
Teach about condition hypo/hyper thyroid function
mandatory medication
Breast feeding fine – should watch what mother is eating as may
be eating thyroid-suppressing foods or taking own thyroxine medications
Be aware of overdose-rapid thready pulse,irritability,
insomnia,fever,dyspnea,sweating,Tremors, weight loss
Withold one dose of medication if pulse above benchmark ,call MD STAT
Teach how to take temp. pulse, resp. & Weigh child at home
keep log take to checkups
Genetic screening as an Autosomal recessive trait
URGENT NEED TO KEEP PARENTS SUPPORTED
CHILDREN DEVELOP WELL WHILE ON REPLACEMENT
CAN ACHIEVE AS LONG AS ON MEDS
A HORMONE REPLACEMENT THAT REGULATES LIFE
Later , the child may need encouragement to keep taking the tiny pill
It’s so small, how important can it be?
Watch for physiological and cognitive signs of need to modify dose
KEY Assessment S & S of
HYPOTHYROIDISM
Feeding
Energy
level
Bowels
Heart
rate
Muscle tone
Breathing patterns
contd.
Classical Features
Skin
thick & leathery
“frozen” DTR
Wide patent cranial sutures
Coarse dry lusterless brittle hair
Cool to touch, mottled, grayish skin tone
SEVERE MENTAL RETARDATION
Diagnostics
T4
& TSH
May repeat with free T3 and T4
Iodine uptake
CT scan
Bone scan
Low T4 + TSH = CH
Nursing Care
*Early
identification is the key
Teaching re: meds & expectations
Breastfeeding
Overdose signs/symptoms
Monitoring of TPR & weight
Prognosis
Hyperthyroidism
Most cases occur between 6 – 15 years of
age, peaks at 12 – 14 years
Graves disease – enlarged thyroid gland
Appears more often in girls
Exophthalmos is classic sign
Goiter – may be present at birth
If so, emergency management of airway
Hyperthyroidism
Signs & Symptoms
May develop gradually
Excessive motion – irritability
Tremors, insomnia
Growth & bone age accelerated
Therapeutic Management
Goal: reduce secretion of hormone through
drug therapy, surgery, radioiodine therapy
Comparison: Hypo vs. Hyper
Hypo
Intolerance to cold
metabolic rate
appetite with wt gain
Fatigue
Weight gain
Constipation
TX: replacement of missing
hormone
Hyper
Intolerance to heat
metabolic rate
appetite with no weight
gain
Fatigue with insomnia
Weight loss
Diarrhea
TX: surgery, meds
MUSCULAR DYSTROPHY
Muscular Dystrophy pp. 1836-1840
Duchenne is most common.
X-linked inheritance, affects males, onset begins at
start of school years- 3-6 years.
Lordosis, waddling gait, frequent falls, hard to
run/climb, toe walking, difficulty getting up from
floor( Gower sign),
fat deposits replace muscle strands due to missing
enzyme( dystrophin and enzyme CTGalNac
transferase) in large muscle groups calf muscles
hypertrophy, loss of mobility low EMG result.
MD
PATIENTS ARE EVENTUALLY IN A
wheelchair, cardiac and respiratory failure no cure yet. Complications include obesity,
pneumonia, contractures, sleep apnea,
Prednisone assisted with muscle strength retention, physio, chest physio, ROM, Family
support, IPPV breathing machine, friendship retention, camp,
Autism Spectrum Disorder
February 20th 2007, Dube family of Windsor helps HSC Geneticists to discover
missing chromosomal pieces that may explain the pervasive behavioural traits
of ASD . Early detection and intense behavioural intervention /cuing and
training is essential to teach these children Many variations ;Asperger’s
Syndrome is a higher functioning ASD. Difficulty with communication, reading
behaviours of others, learning . Difficulty talking and explaining. Handling
change is hard. Repeated behaviour, self-stimulating continues for a long time.
OSTEOGENESIS
IMPERFECTA(OI)
Genetic ( mostly Autosomal Dominant) anomaly in the
skeletal system. A defect in the coding of polypeptides
for collagen and bone matrix as well as bone
mineralization defect with susceptibility to break/crumble
4 major types depending on :
Degree of bone fragility( mild, moderate, severe)
Deformities
Dentinogenesis ( malformed grey teeth)
Deafness
Fractures
Blue sclerae
Ostegenesis imperfecta
OI –an autosomal
dominant genetic
disorder causing lack of
type 1 procollagenprecursor to bone
mineralization, causes
deformities and brittle
bones. No cure
various types
Assessed by bright blue
sclerae, transluscent
grey teeth, hearing loss,
spontanoeus fractures,
bone deformity, lack of
growth, severe disability.
Splints, protect from
falls or bumps,
physio,parent support,
live within limits
Features/ manifestations of OI
Facies- triangular face, sclera colour variation, dental
colouration and thin enamel- dental & Jaw fractures
Short stature
Barrel chest
lung deformities
Scoliosis, lordosis, kyphosis
Long bone fractures common
Epiphyseal plate malformation
Joint deformities with joint/muscle laxity
May have fewer fractures after puberty
Can be misidentified as child abuse
Types of usual fractures.
Children with OI have
different variations on
fractures. Disintegration
of calcium matrix,
greenstick fractures
Need to be aware that
may break at epiphyseal
point- This could retad
growth and create
horizontal versus vertical
bone growth.
Malforms skeleton
Treatments for OI
Supportive: splints, braces, ROM, head and
trunk control exercises/play,swimming,surgeries
as needed- rod supports,
Bone Marrow transplant
Bisphosphonate ingestion may be helpful
IV pamidronate therapy to prevent bone
resorption
Goals- to prevent contractures, fractures, bone
pain, deformities, malalignment, disuse
syndrome, osteoporosis
Nursing care FOR OI
Wholistic- safety proofing environment- anticipatory
guidance to protect from injury
Skin care re braces and splints
Slow easy movement, no strain, pulling or stretching of
joints, or bones, no roughhousing
Clarity that not child abuse, history present at all times
Schooling must be cautious, choose low impact less
physically demanding job, no problem with IQ
Regular hearing tests– May need hearing aid or learn
signing
Dental care – plastic coating to protect &soft
toothbrushes, no harsh foods( nuts)
Genetic counselling and support for parents
DOWN SYNDROME
DOWN SYNDROME
Most Common Chromosomal abnormality
1 in 600 births, slightly higher Caucasian
prevalence but distributed across all races.
Numerous theories of causation>Maternal age= old egg, slowed cell
division and chromsome migration so have
trisomy – on #21 Other variations are
translocation of #21(hereditary) and others
may be due to mosaicism
Manifestations
A syndrome with a cluster of phenotypical findings( p. 1000 Box)
Single epicanthic folds, High narrow palate, large protruding tongue,
short broad hands, straight horizontal palmar crease,
stubby fingers, altered fingerprints, and hair whorl,
Larger great toe with separation between first and others,
flat occiput and enlarged anterior fontanel- myopia, strabismus, frequent OM
Small nose, saddle bridge, small rounded ears, short pinna, hearing loss
short broad neck with extra skin folds, dry skin, mottling,
Chest shortened ribcage,possible torticollis, atlantotaxial instability( jaw, neck)
Lax flabby abdomen muscles, umbilical hernia,
Hypotonia and hyperflexibility- hip dislocation, subluxation, GI anomalies
small penis, cryptorchidism, immature labia Secondary sex characteristics late onset
most males are infertile, females may reproduce but usually offspring has defects.
May have low thyroid functioning, low metabolism rate and decreased uptake of
vitamins. Low birth weight, Low percentile for age in height, may gain weight if not
active.
Mental abilities and intelligence are variable
Immune variations- prone to infections. May have cardiac involvement as well.
Palmar Creases
Therapeutic Management
Mother has option for prenatal amniocentesis and ultra sound : decide on options- to carry to term or
terminate the pregnancy
No Cure, but can have good quality of life- Normalization and Integration into society & Life expectancy to
adulthood.
Needs to have ongoing total assessments especially for hearing, vision, cardiac, thyroid, growth
hormone.
Can have surgery for neck stabilization. For strabismus and septal defects and other concerns that arise.
Assistance with family coping skills, attachment behaviours, grief, developmental anticipatory guidance
and teaching /learning enrichment through Head Start enrichment type of programming.
Swaddling and keeping child warm at the beginning of life will encourage calorie conservation and weight
gain. Observe closely for depressed respiratory activity, inadequate mucus drainage, inability to breathe
with enlarged tongue,
Prevent infection as immune system may be compromised
Encourage breast milk but may need help of lactation consultant to assist with feeding position and
latching
Later , watch for obesity- nutrition needs to be balanced, add vitamins (C. B complex, ADEK)
Skin care should be gently. Becomes dry, course and thick – needs lubrication
May burn more easily and respond to the cold more severely- keep protected for each extreme.
Self esteem and gender identity are important
Assistance with developmental progress through enrichment programmes, normalization activities and
social integration. Sports teams can provide great friendships and affiliation needs can be met.
CASE STUDY
Down Syndrome infant with DDH
A BEVIS CARE PLAN
Introduction
The Rezpeloski family have just
immigrated from Croatia: Mom is 46, Dad
is 52, Oldest Daughter is 24, Son is 16,
and youngest daughter is 13. They have
just been surprised by the birth of their
latest son, who is about 8 months old.
They have been waiting for a refuge
hearing for eighteen months .
Joseph is the baby’s name. He was 3.6 kg. At
birth and had an APGAR of 4 as he was
difficult to get started breathing, had
hypotonic muscle tone and did not cry
independently. On examination, it was
thought that he also had DDH , and was
noted to have had straight palmar
creases,single epicanthic folds, large tongue
and stubby hands and broad wide feet.
At present, Mr &Mrs. R. state through their
interpreter that they feel overwhelmed and worry
about the rare of Joseph should they have to
leave the country. They state that they could not
get the help they need in Croatia to provide the
care . Their other children are already in the
mode that they are staying in Canada and have
made many friends, and started their studies .
Mr. & Mrs. R. are eager to begin working at their
professions – a civil engineer, and a nurse
respectively but must await permission from the
government. It is very difficult to live on their
immigrant allotment.They are trying to do things
honestly and be above board, but are finding it
tempting to cheat as they do need money and
are very proud of their past .
As they were political leaders for the
democracy movement, they were
targets of retribution by the anarchist
party. They had to flee to save their
lives. They do have papers and
validated legal accounts of their riskfilled lives. Even though Mrs. R. is a
nurse, she is still upset about her late in
life pregnancy and the outcome. She
thought she was premenopausal and
was not likely to get pregnant and they
could not afford profilactic measures.
Real Life
Their daily life is filled with daily care of a difficult
feeding baby, who is not very cuddly, but can not
turn over yet and is constipated , leading to
many upsetting days of tummy cramps and
straining . Joseph is responsive to smiles and to
his music box. He is rarely in his crib, but still
seems to have an odd shaped head and seems
to have problems holding his head up.The older
children do not want to have anything to do with
the care of Joseph. They will babysit once in
awhile.
What This Means
The parents are disappointed in their children’s
reactions to their new sibling . They are worried
that their oldest daughter wants to leave and get
married to a new friend she had become close
too- perhaps to assure she can stay in Canada?
Their older son is enjoying his schooling, but
wants to know when he can get his papers as he
wants his own money- He can’t live on $30.00 a
week.
Next Steps
Develop a list of the problems for family.
List the actual and potential health concerns for
Joseph.
Discuss the adaptation and maturation elements
related to this family.
Create a care plan for the most urgent problem
Discuss the care for DDH, compared with other
anomalies
State the common and distinct nursing
interventions related to this situation.
FRAGILE X SYNDROME
FRAGILE X SYNDROME
A common inherited cause of Mental
Retardation, second most common after Down
Syndrome
Due to an abnormal mutated gene in the lower
end of the long arm of the X chromosome that is
caused by a repeated premutation effect of 2
base pairs with reduced penetrance.
Males more severely affected and all are
affected , none are carriers: females are carriers
Fragile X Syndrome
most common cause of inherited mental
impairment.
Impairment can range from learning disabilities
to more severe cognitive or intellectual
disabilities.
FXS is the most common known cause of autism
or "autistic-like" behaviors
Unusual in that either parent can carry the gene
Treatment
Early intervention in skills development:
cognitive, motor, language, ADL’s
Often have aggression, anxiety,
hyperactivity and/or limited attention span
Pharmacological interventions may be
needed for behavioural problems
Conduct Disorder
Ref. American Psychiatric Society
TYPE A. A repetitive and persistent
pattern of behavior in which the basic
rights of others or major age-appropriate
societal norms or rules are violated, as
manifested by the presence of three (or
more) of the following criteria in the past
12 months, with at least one criterion
present in the past 6 months:
Aggression to people and animals
Destruction of property
Deceitfulness or theft
Serious violations of rules
TYPE B. The disturbance in behavior causes clinically
significant impairment in social, academic, or
occupational functioning.
TYPE C. If the individual is age 18 years or older, criteria
are not met for Antisocial Personality Disorder.
Specify type based on age at onset:
· Childhood-Onset Type: onset of at least one criterion
characteristic of Conduct Disorder prior to age 10 years
· Adolescent-Onset Type: absence of any criteria
characteristic of Conduct Disorder prior to age 10 years
Specify severity:
· Mild: few if any conduct problems in excess of
those required to make the diagnosis and
conduct problems cause only minor harm to
others
· Moderate: number of conduct problems and
effect on others intermediate between "mild" and
"severe"
· Severe: many conduct problems in excess of
those required to make the diagnosis or conduct
problems cause considerable harm to others
Treatment
Co-morbidity with ADHD and Tourette’s
suggesting neurological component
Treatment similar to ADHD plus behaviour
modification and counseling involving
child/parent/school
FETAL ALCOHOL SYNDROME
Fetal Alcohol Syndrome
Ref. American Psychiatric Society
Alcohol more dangerous than cocaine—readily
passes into fetal brain
Binge drinking more serious than occasional
drinking depends on liver’s ability to detoxify the
alcohol
No amount of alcohol is safe
Effects unpredictable depends on amount of
alcohol, degree and stage of fetal development
Manifestations
Long Face
Prominent jaw
large protruding ears
Large testes
Developmental delay
Language and reading delay
Autism-like behaviour
Anxiety ,withdrawal, depression
Behavioural difficulties
Intellectual underdevelopment
Foetal Alcohol Syndrome is entirely preventable.
NO ALCOHOL CONSUMPTION DURING PREGNANCY pp 410-412
Diagnosed based on typical facial features. Thin upper lip, short upturned
nose, hypoplastic maxilla,smooth philtrum (upper lip),short palpebral fissure,
Mental retardation, motor retardation and un co-ordination, Microcephaly,
hypotonia, hearing disorders, irritable difficult to settle, hyperactive,low
weight to long length/tall,thin, growth lag, LBW
Which of these children had FAS? How can you tell?
Poor feeders,
Reduce noxious stimulilight cold, noise, smell.
Stable schedule
Special schooling supports,
Impulse control skills, anger management
Play group to learn skills,
avoid alcohol
Parent support group
Therapeutic Management for
FAS/FAE
NO CURE
Genetic screening and counselling
Medications to control violent temper
tantrums as no behavioural control
CNS stimulants to calm down and assist
with coping and reduce hyperactivity.
Predisposed to mitral valve prolapse,
recurrent OM, seizures, and skeletal
concerns.
Goal- ZERO FAS / FAE
All Fetal Alcohol
Syndrome and fetal
alcohol effects CAN BE
ELIMINATED with ZERO
Alcohol consumption
during pregnancy. This
life-long condition causes
profound effectsbehavioural, cognitive,
metabolic, facial
anomalies.
Nursing Management for FAS/ FAE
Family support for the management of temper
Promote schooling with special behavioural expectations
built into the programme to help the child manage
behaviour and learn how to think of consequences
Genetic counselling
Networking for families
May have school board liaison as child usually is in
difficulty and also needs help to learn.
Don’t forget well child visits for age- immunizations and
health assessments from head to toe.
Thoughts and reactions related to infants who
are not perfect:
Impact on mother
Impact on father
Siblings’ thoughts
Grandparents reactions
Staff reactions and responses
Community reactions and responses
loss, meaning of child, beliefs, guilt,anger,
sadness, shock, blame, some reason, cope
SICKLE CELL ANEMIA
Sickle Cell Anemia
Normal Hgb is replaced with a “sickle
shape”
Most common in AA
Sickled cells obstruct
normal blood flow to and
from organs
What signs would be expected?
*Box 35-4 page 1548
Sickle Cell Anemia erythrocytes are displayed below . The
unaffected Red blood cells of a carrier ( Left) and the
cycled form- not in crisis ( right). When a crisis does occur,
the cells become even more distorted and clump together
.
more
Sickle Cell Disease is a serious chronic inherited hemolytic anemia that results
from defective hemoglobin strands in the erythrocytes that is susceptible to
collapse and sickle shape when experiencing physiological threats such as
hypoxia, fever, dehydration, infection, stress. Most who are affected have
African ancestors. It is a disease of colour. Sickling is painful, blocks
circulation, obstructs blood vessels(vaso-occlusive crisis) creating ischemia to
distal parts(dactylitis,priapism, stasis leg ulcers(sequestration), organ infarcts,
kidney failure) potential for sepsis . Treatment involves oxygen, IV and p.o.
hydration, electrolyte replacement, bedrest, opioids, penicillin, protective
isolation. p. 1520-1529
Sickle Cell Anemia
Genetic, hemolytic
1 in every 375 births
Mainly African descent
Splenomegally, hepatomegally, hematuria,
enuresis, bone weakness, dactylitis, CVA,
MI
Delayed growth
Transmission of Sickle-Cell
Anemia
Probability of Abnormal Hgb in
Offspring
Genotype of
Parents
Normal
Trait
Disease
1 parent with trait
50%
50%
0
2 parents with trait
25%
50%
25%
1 parent with trait,
1 parent with
disease
0
50%
50%
0
0
100%
2 parents with
disease
SICKLE CELL ANEMIA S & S
“Crises”
Vaso-occlusive crises (VOC’s)
-handfoot syndrome (dactylitis)
Sequestration crises
Aplastic crises
Megaloblastic anemia
Vaso-Occulsive Crisis
Painful, fever
Dactylitis
Arthralgia
Rx
Hydration, bedrest, antibiotics, transfusions,
O2
Splenic Sequestration
Blood pools in spleen results in shock
Rx
Transfusions
splenectomy
Aplastic Crisis
Decreased RBC production with bone
marrow failure
Tachycardia, fever ,CHF
Rx
symptomatic
Nursing Management
Minimize tissue de-oxygenation
Promote hydration
Minimize crises
Supportive therapies
Genetic counselling
Support family – suffer from guilt
Impact on Family
May create tension- gender of child and ordinal position
may impact on emotions.
Parents may be grieving- can go on for a lifetime as
milestones are missed. Focus on losses or over
compensate and push to make variation invisible.
Shame, guilt- did I/we do something wrong? We are
being punished. Pressure on siblings to help care.
Roles , family tasks, and personal development may be
sacrificed- resentment may build – blame &escape
divorce. Unable to cope so move out – may place child
in care of others- group homes .
Adjust to and build supports and resources/networks of
parents who have coped successfully and lead the way
with sound suggestions and guidance.
THALASEMIA
Thalassemia
Inherited blood disorder –Mediterranean
4 forms – minor, trait, intermedia & major
Autosomal-recessive
Defective synthesis of HgA, structurally impaired
RBCs, shortened lifespan of erythrocyte
Hypoxia & iron overload, headache, irritability,
bone pain, lethargy, anorexia, splenomegaly
Decreased growth & sexual immaturity
Hemosiderosis, hemochromatosis
Thalassemia pp.1529-1534 important in this region with
high immigrant populations
Common to the cultures of the
Mediterranean Sea and surrounding areas.
Autosomal Recessive trait from both
carrier parents.
Trait allows for resistance to malaria, Defect
is in the hemoglobin chain. Alpha type
chain and Beta types. Insidious onset,pale,
feverish, fatigued, bone pain and deformities as
bone matrix tries to compensate, growth failure,
short, forehead bossed.Hemoglobin not able
to carry O2, forms severe hypoxia, anemia,
but with iron overload( Hemosiderosis) from
repeated transfusions. Desfuroxime, Deferoxamine
Must be given to chelate out excess iron from transfusion
EFFECTS OF THALASSEMIA
What signs can you see in this
photo?
Try to describe the
findings you would
have concerning this
woman living with
Thalassemia.
Nursing Considerations for
Thalassemia
Transfusions
Chelation therapy
Bone marrow transplant
Promote compliance with treatment
Support for child & family
Observe for complications
Testing of family members
Genetic counseling
S & S for Thalassemia
Pale skin
Fatigue
Pica
Headaches, dizziness & lightheadedness
Irritability
Decreased attention span, slowed thought
processes, apathy and depression
B Thalassemia
Hereditary
Decreased RBC’s with low HGB,
Hematocrit
Fever, poor feeding, splenomegally,
hypoxia
hemochromatosis
RX
Blood transfusions
Iron chelation therapy
Prophylactic antibiotics
Diet
Emotional support
Bone marrow transplant
HEMOPHILIA
Hemophilia
Congenital deficiency of coagulation
proteins
Primary rx: replace missing clotting factor
-factor VIII concentrate
Bleeding may occur spontaneously in joints
Hemophilia
Hereditary , lack factor VIII, IX
Type A, Type B
Prolonged partial thromboplastin times
S&S
Bruising
Bleeding
Hemarthrosis
INJURIES OCCUR IN THE
JOINTS AND MUSCLES
HEMARTHROSIS
HEMATOSIS
HEMATOMA
HYPHEMA
PROPER POSITIONING FOR
ELEVATING BLEEDING LIMB
Complications
Intracranial bleed
GI hemorrhage
Hematomas
Obstruction of airway
Nursing
RICE
Prevent hemarthrosis
Physical therapy
Holistic care with multidisciplinary team
How does this affect G & D??
RX
Recombinant DNA factor VIII
DDAVP
NSAIDS
Rest, ice, elevation, splints, physio, ROM
Prevent injury, infection
Parental and patient education
Transfusion of Platelets or clotting factor
IMMUNE SYSTEM
Because there are so many aspects of
genetics integrated into our lives, let us
consider the Immune system – Allergies,
Anaphylaxis
Stevens-Johnson Syndrome
A strong immunological basis.
Must protect from infection,
fluid loss, loss of proteins,
Bedding must be sterilized and
non-clinging
Air or water mattress
Warmth must be maintained
passively- radiant warmer,
Tissue sloughing internally
as well
Hereditary Cancer
T-Cell Leukemia
Self-esteem NB
Alopecia from chemo
Wig
Weight from steroids
Moon face
Survivor aftermath
from treatments
Ethical Concerns
Questions abound
What is the quality?
Whose right to live
or die?
In pediatrics, many opportunities for moral and
ethical dilemmas. There are many participants
with various levels of autonomy, power, differing
values and beliefs, laws,mores,traditions,and
multiple cultures and family types,value systems
as well as personal values.
No easy time making complex decisions
Don’t rush– takes teamwork and
Requires taking the viewpoint of others in ethical
deliberation and discussions
Ethical practice
Nurses must do no harm, be just and fair, facilitate
multiple expressions of meaning and intent, values,
benefits, risks, probabilities , choices and alternatives.
Consider legal position and desire of guardians, child ‘s
viewpoint and thoughts ( if able to express them ),
Impact of decision on cultural acceptability and status,
what’s best
Nurses follow Standards of Practice to guide
professional action, hospital policy, laws, past outcomes
as guides, capability to effect good
Health perception
Nutritional-metabolic
Elimination
Sleep-rest
Cognitive-perceptual
Self-perception-self-concept
Role-relationship
Sexuality-reproductive
Coping-stress tolerance
Value-belief
CCKREM PVF
Communicating
Choosing
Knowing
Relating
Exchanging
Moving
Perceiving
Valuing
Feeling
Nursing Process in Pediatrics
Observations & Assessments vital
Growth and development ongoing
Data from signs more frequent
Symptoms are usually nonverbal
Data sorting by life threat ABC’s
Plan related to desired outcome
Include family in plan
SMART and KISS
Age related appropriate care
Play intervention needed
Nursing Tools integrated
Evaluation & modifications usual
Review & Recycle
Right task
Right circumstances
Right person
Right directions
Right communication
Right supervision
The RN remains accountable for
care rendered by other parties as
long as child is “in care”
COMMUNICATING WITH
PARENTS
DOCUMENTATION OF CARE
Times are vital- feeds, elimination, meds, visits,
sleep,interventions, crying, settling, play, pain,intake,lab
work
Giver of care- RN, RPN, Parent,SN
Amounts- intake output, tears,pain, IV flow rate,O2,
medications, activity,sleep, cuddles , treatments (enema) ,
medications
Visits -family, MD,teacher, classmates etc. to other
facilities, playroom
Signs of improvement, change, pain, mood , worries,
separation anxiety
Assessments and reassessments( times)
Play quality and quantity.Type, response
Fears of all parents- something will be
wrong or my child will be hurt.
Count fingers and toes. Look carefully at
configuration of face and hands, set of ears
on face, symmetry, drooping or signs of
FAS . Examine all of back,
Superstition may be involved- born wrong
day under the wrong sign etc.
Premature , look “ugly” miss dream look
Later imperfections may come as a shock if nothing
noted at birth
Late onset of genetic concerns create anxiety and
ethical dilemmas : if in adolescence- Problem
with identity
Cosmetic surgery doing marvelous things
need to look beyond the surface and find the child
Camps provide experiences with children coping
with similar problems “just like me”
Have same needs as healthy child with some
modifications
Developmental DYSPLASIA of the hip
Dislocation or subluxation of hip(s)
Asymmetry of gluteal folds, thigh folds,
Limited hip abduction in flexion
Uneven knee joints, one seems shorter than
the other ( Allis or Galeazzi’s sign
Ortolani’s click heard on affected hip during
abduction – newborn to 4 weeks.
Unequal ROM during movement and play
May be sign later in childhood of hip diseaseSubluxation, Legg Perthes, May need pinning,
Link developmental milestones to assessments
Know firsts at various ages- first smile, head lift on tummy,turning
over,thumb to mouth,toe in mouth, rolling over, sitting up,
scooting,cruising,standing alone , steps, walking, Climbing up
spoonful of food, holding spoon, cup, feed self, sleep through the
night , notices self in mirror,words, stranger anxiety ,stairs ,climb out
of crib etc.
Some parents mourn lack of movement toward milestones due to
delays
Children’s Rehabilitation Centre fosters potential and wholistic care
and education for the imperfect child.
WAYS TO HELP
Stay with parent, encourage examination and care of
infant as much as normal
reinforce explanation of specialists
Listen to parents and others, reflect their feelings
back to them – tears are OK
Take photos ( if desired )with complimentary settings
( as per Anne Geddes )
Help link with others who have coped with
experiences in a similar situation
Avoid giving false hope, take one day at a time :Point
out the positive signs and gains
Call and keep in touch through clinics
BEVIS would ask nurses to think of
Protective
Nurturative ( Nurtrative)
Generative interventions
Nurturative Interventions are those that
promote wellness, adaptation, maturation .
Feeding, cuddling, bathing, soothing,
warming,
Protective Interventions protect from injury
and illness –prevention& Health promotion
Generative interventions relate to
rehabilitation and reconstruction
There may be an emotional roller coaster for all family
members . As one member begins to cope, others will
be at a different point in adjustment.
It is important to keep each other informed of how one
is feeling and what one is thinking.
Siblings need time to ask questions and discover what
they can do and be reassured that they are still loved
and have their place – Loved & special too.
Families may want to make a memory box/
scrap book and record their feelings and
contacts as they move through stages of
adaptation and adjustment to the new
reality .
Sources of support and tension release
arts ,faith,exercise and sport,
friends,work
Parents will still need support with the
expectations of parenting. Infant with
special needs also had the basic needs that
must be met. Total dependence early on
May find that parents are both fatigued as
both are trying to maintain the emotional,
structural and functional roles of the family
unit as well as keep up with work and other
obligations.
Ask about sleep quality, amount, cycle,
ability to dream, restfulness and napping
of whole family .
Dietary intake of family may reveal deficits
– lack of metabolic necessities
Return of Intimacy may be delayed – may
need to gently ask about return to sexual
relations – fear, depression, anger, fatigue
When families feel burdened they look for
a light at the end of the tunnel and for
hope .
Some are creative , resilient ,
reach out and cope.
Others may explode in unresolved
feelings and tensions, dissolve and
disengage, separate and find little support
for a positive and healthy lifestyle.
GENETIC SCREENING AND
COUNSELLING
HUMAN SOMATIC CELLS
( all those cells with nuclei except the ova
and spermatozoa)
Contain 22-25.000 genes; Genome project revealing
35,000
Distributed in sequences in the form of a tight coil - the
DNA helix molecules
Four chemicals make up the helix ( Adenine links with
Thymine, Guanine links with Cytosine )
Along 46 chromosomes that very in size, shape, and
groupings. The larger the chromosome, the more
Genetic material it holds. Addition of any chromosomes
to the allotted 46 brings added genetic material
HIGH RISK FACTORS FOR PRENATAL
DIAGNOSIS AND GENETIC COUNSELING
Maternal Age- elderly primipara
Elevated or lower trisomy profile screening outcomes
Previous birth with chromosomal anomalies
Previous stillbirth or neonatal death
Structural anomaly of either parent
Inherited diseases- Metabolic disorders, CF, Sex-linked recessive
disorders
Balanced translocation in parents
Infection – rubella, toxoplasmosis, cytomegalovirus
Medical disease in mother- diabetes, thyroid, PKU
Teratogen exposure- drugs, job, alcohol,home,recreation
Abnormal ultrasound
Ethnic or racially linked disorders- Sickle Cell, CF, Thalassemia
Close familial relationship
Roles and
advancements
Genetic Counseling is a major role for nurses.
Nurses can take a pedigree and family history
Explain the testing processes , the risks and purposes, as well as the care
needed before,during, and after
Careful and simplified explanation of risks of having an affected child
Options and interventions for care or termination
Teach about condition after being related by geneticist or M.D.
Emotional support, advocacy, and help build a network of resources
Early Prenatal diagnosis allows the personalization of care such as
termination, foetal surgery, genetic modification through
genetic engineering and implants
slide of child with achondroplasia
GENETIC COUNSELLING
This is a specialty service
Must be undertaken in conjunction with Genetic
screening and follow up with geneticist and
family physician and obstetrician.
Must begin with a clear history and profile of the
family, capabilities, risks, past history, present
threats &worries, understanding of the situation.
A communication process at a time of great
unknowns and high tension. Moral and ethical
conflict may occur with parent(s). Educational
and supportive. Networking and resource
development
GENETIC COUNSELLINGcontd.
Focus on clarity of information, understanding and answering
questions, ways to alleviate guilt and blame, understanding of risks
and options/choices and decisions,
Leave final choice to the parents, support them in their decisionmaking
Link to services and resources to assist in care of infant and child
through life. p.137
Assist in understanding the risks of subsequent pregnancies.
Probabilities may not be understood and so put future fetal health at
risk.
Look at the parents- be observant for clues to genetic disorders that
may not have been diagnosed before- p. 135 Box 5-7
Barriers to effective communication
Personal bias prejudice, blame
Time limit
Over-exposure to technical information
Use of jargon and scientific terms
No return demonstration or explanation
Coercion and threats
Genetic Counselling
Prior to conception
After conception
After birth
When offspring or
relatives request
Adoption
Past maternity issues
stillbirths, miscarriages
congenital anomalies
Dietary malnutrition
At risk population
(W. Wertelecki, M.D.) 2003 www.ibis-birthdefects.org, All rights reserved. 23 Mar 2006 This site offers information
mostly for educational purposes. This site is not intended to alter health care protocols nor to serve as a sole source of
medical information. Always seek the advice of your local health care provider.
[All of the following material is a direct transcription of the article content with deletions for spacing and additional
comments in italics and brackets by S.McMahon for the purpose of nursing education in the course 277. ]
Genetic Counseling, - Excerpts from "Genetic
Counseling“ by F.C. Fraser, The American Journal of
Human Genetics, 1974, pp. 636-659
Definition
... Genetic Counseling is a communication process, dealing
with the human problems associated with occurrence, &
risk of occurrence of a genetic disorder in a family &
attempts to help by one or more appropriately trained
persons ( an interdisciplinary Team . S.McM ):
OUTCOMES OF GENETIC COUNSELLING /
SUPPORT will assist the
Counselee to:
(1) comprehend the medical facts, including the
diagnosis, the probable course of the disorder, and the
available management;
(2) appreciate the way heredity contributes to the
disorder, and the risk of recurrence in specified relatives;
(3) understand the options for dealing with risk of
recurrence;
(4) choose the course of action which seems appropriate
to them in view of their risk and their family goals and act
in accordance with that decision; and
(5) make the best possible adjustment to the disorder in
an affected family member and/or to the risk of
recurrence of that disorder ...
Description
Most counseling involves the occurrence of a particular
disease in a child and the concern of the parents as to
whether their future children might be similarly affected.
Parents may also want to know about the risk for the
affected child's children or the children of unaffected
sibs.
Other situations ... a person contemplating marriage who is
concerned about some aspect of the future spouse's
family history. There may be a specific disease in a near
relative, ancestors from a different racial group, or the
future spouse may be a relative ...
Some counseling problems are so complex or require
sufficiently specialized tests that the services of a
professional genetic counselor are required ...
Counselling Process
{Note: Counseling and counselling are accepted publication versions, the first is American , the latter is Canadian/
British . The Canadian version is preferred when in citations of original Canadian works.}
The process of genetic workup and counseling
may be considered in several stages:
validation of the diagnosis,
obtaining the family history,
estimation of the risk of recurrence,
helping the family to reach a decision and
take appropriate action,
and follow-up.
Training
... Some would claim that the proper qualification for a genetic counselor should include
an M.D. degree with training in one or more specialties ...
A Ph.D. degree, or at least a M.Sc. degree in human genetics plus 2 years experience
in a medical genetics unit ...
some training in psychiatry, social service, and pastoral psychology...
There is a consensus that a medical genetics unit ought to have a M.D. on its staff to
take responsibility for the medical acts that occur as part of the unit's activity ...
(Today, master’s prepared nurses with a clinical expertise in ethics, pediatrics, psychiatry,
genetics and genetic counselling and follow up family-centred care are vital members of
the genetics team. Most teams are affiliated with pediatric magnet centres such as The
Hospital for Sick Children in Toronto, L’hopital du Montreal, also at the Isaac Walter
Killam Hospital in Nova Scotia, through Queen’s University and The University of
Alberta, just to name a few. Post basic courses in genetics and counselling are
required. Some provinces and states have courses and advanced degree recognition at
various levels of extended nursing practice. Nurses are also actively involved in
research and organization of special needs programmes for the children with special
requirements to sustain the potential of a high standard quality of life. S.McM )
Quality Control
(It is vital that centres be accredited and provide accurate and honest information to
all clients who seek this service. Nurses can be the advocate for the client in the
process of counselling and screening S.McM )
(W. Wertelecki, M.D.) 2003 www.ibis-birthdefects.org, All rights reserved. 23 Mar 2006 This site
offers information mostly for educational purposes. This site is not intended to alter health care
protocols nor to serve as a sole source of medical information. Always seek the advice of your local
health care provider.
Financial Support
The source of payment for genetic counseling services is a problem of
increasing concern ...
... Most medical care programs do not recognize how time
consuming a good genetic counseling procedure can be ... Family
may take from 2 to 8 hours or more, and the usual consultation fee
charged to the medical care program does not begin to cover this.
( In Ontario, the basic costs of genetic screening are covered under
the provincial plan. Some additional services and tests may be billed
as these may not be done sufficiently frequently to have a costrecover figure integrated into the health plan, or may be too new and
still experimental and so are part of the genetic screening research
protocol rather than as a diagnostic care-therapy initiative. Recently,
there has been an great increase in the number of disorders
screened routinely at birth through cord blood- without cost.
Banking of cord blood still is a private cost assumed by the parents.
S.McM )
Counselling Simple and Complex Cases
Perhaps the optimal system would be a
multitiered one such as exists in other
branches of medicine. Simple cases might
be counseled by the primary physician at
the local level--when primary physicians
have acquired the knowledge to handle
the simple cases and to screen out those
that are not so simple.
Individuals needing sophisticated
biochemical tests and children with
esoteric syndromes or other complex
problems could be referred to the genetics
unit at the medical center ... These more
complex cases might be seen by a
professional genetics counselor, either at
the medical center if the family lives near
one or at the local community level by
means of traveling clinics.
Organization Of Genetic Counseling Services
... It has been estimated that by the mid-1980's, in North America, one
person with training in medical genetics will be needed for every
200,000 persons ...
( In 2008, today, the number has increased in Canada and in many
countries as the governmental priorities shift to cost-effectiveness
and reduction of health care burden. Ethical watchdogs are required
to facilitate the human-focus of care and research over the
corporate or government agenda. Eugenics, an abandoned
programme in many countries, was an historic movement that
demonstrated the extremes to which birth control and infanticide
could be put in order to secure strong genetic strains and lines.
Today nurses must be aware of political movements around the
world that have other goals in sight for the new science of genomics,
cloning and human benefit. The legal system and ethical study is
rich with the many scenarios and debates related to quality of life –
and for whom. Canada is a leader in genomic research, helping to
identify several genetic links to chronic diseases such as the
organism that caused SARS, C.F. and Autism. S. McM )
Since counseling is vitally dependent upon research on the
identification and delineation of new syndromes, the
improvement of data on penetrance and expressivity,
and the refinement of recurrence risk estimates, it is
desirable that the counseling service be associated with
an ongoing research program.
The personnel of the group should include a M.D. trained in
genetics… a number of others with either M.D. or Ph.D.
degrees ...
Psychodynamics of Counseling
Too little is known about the psychodynamics' of genetic
counseling (More research is needed ... S.McM )
Directive or Nondirective Counseling?
Opinions differ widely on how directive genetic counseling
should be. Some counselors (a minority) believe that
counseling should stop at the point where an estimate of
risk is given and that the parents should make up their
own minds what to do, without benefit (or otherwise) of
further advice from the counselor ... Although this has
been … termed "behavior control" by some ethicists, the
counselor is no more controlling behavior than is a
surgeon who recommends an elective operation ... Many
counselees come for advice and are disappointed if they
do not get it about their relative advantages and
disadvantages. ...( the final decision is still that of the
counselee and family. S.McM )
Group counseling may be effective when used in group
screening programs for high-risk populations
Family Follow-up
… tends to be neglected
... Family follow-up may be useful for two reasons: (1) to
counsel high-risk (and reassure low-risk) relatives, and
(2) to employ preventive measures ...
Methodology
The term "genetic counseling" was coined by Sheldon
Reed to replace the term "genetic hygiene" which had
unpleasant eugenic implications ... The literature on
genetic counseling dealt mainly with methods of arriving
at recurrence risk estimates. Gradually, however, the
psychological complexities involved in the counseling
situation began to be recognized, and now an increasing
number of articles on the impact of genetic diseases on
the family and the techniques, philosophy, psychodynamics, and ethics of counseling are appearing ...
Genetic Workup and Estimate of Risk
... The concerns vary widely from case to case ... At the other end of
the spectrum are parents who may be stunned by the birth of a
defective child, who may consider it an act of retribution or an
expression of their own imperfection, who may be torn with guilt or
feel that they are doomed to have imperfect children. Exploring
these feelings may be far more important than providing a statistical
estimate of the risk ...
... Follow-up studies suggest that many parents need counseling
because they have overestimated the magnitude of the risk and
therefore need reassuring ...
... Evaluating family history. A detailed family history is taken from
the counselees ... It is generally useful to record "cultural"
characteristics of the family, including racial background, religion,
occupation, and grandparents' birthplace. Age and state of health of
first-, second-, and third-degree relatives are recorded. Miscarriages
and stillbirths are recorded for the near relatives ...
... Estimating risk. ... Extensive research is needed to provide better
information on the clinical risks associated with many genetic
diseases
Validation of Diagnosis
... Family may be referred to the medical genetics unit for
diagnosis by biochemical or cytogenetic tests or in the
hope that the geneticist will recognize a syndrome not
generally known to the medical profession
... Genetics unit will need the services of someone skilled
in "syndromology," who may need to obtain further tests
such as radiographs ...
... Often the diagnosis already has been made by a
physician before referral to a medical genetics unit, and
the counselee wants to be informed about the
implications of the disease for the family ..
(Photographs of family members may be helpful in
establishing a pattern of findings significant of genotypes
and karyotypes . S.McM )
end of article
FAMILY HISTORY & PEDIGREE
Interview -a vital role for nurses
How families express their genetic
inheritance is often recorded through a
family history, relationships to each
other, religion, geographic roots,
migration.
photographs, stories of all the
members, placed in the context of age,
quality of life , physical and mental
status
mortality and morbidity/unexplained
death, miscarriages or spontaneous
abortions,or stillbirths + deaths across
the lifespan
cognition, talents, occupation and
schooling
Adoption
A REAL PEDIGREE
LOOKS AS IF
WOULD BE possible
to use as teaching
tool as well as
diagnostic one.
INTERVENTIONS
PREVENTION- immunization- prevent Rubella.
Genetic Screening if in high risk populations or family history
No STD’s or STI’s: HIV/AIDS Screening and treatment.
NO ALCOHOL consumption during pregnancy.
Early identification of at-risk individuals –lifestyle and repeated
pregnancies or abortions.
Education to target populations to encourage genetic screening
before any pregnancy.
INTERVENTIONS contd.
Avoidance of harmful drugs
such as no aspirin or
antimalarial medication if
G6PD confirmed.
No ethanol, barbiturates,
anticonvulsants,
sulfonamides,nor oral
contraceptives if Acute
Intermittent Porphyria is
diagnosed.
Malignant hyperthermia and
myotonic dystrophy must not
have some anesthetic agents.
Folic Acid and Biotin Additives
needed
Folic Acid and Biotin
added to diets during
pregnancy to prevent
neural tube disorders.
Dietary modificationavoid foods that are
unable to be metabolized
due to lack of enzymes.
Supplemental enzymes
p.o. for digestion.
INTERVENTIONS
Surgical management- removal-excision, reconstruction,
enucleation, mastectomy,
Amputation, cosmetic surgery, correctional surgery.
Blood immuglobulin Rhogam- Rh- incompatability
testedduring pregnance and with infant at birth
Transplantation – organs, genes
Genetic Substitution, implants, enzyme and viral
transfer of recombinant DNA
Replacement of the gene product with artificial
Environmental modification- no light,
Computer assistance to remediate for deficits and
defects- vision, hearing, mobility, learning, speaking
Support groups with special teams of professionals
AGENCIES AND NETWORKS
Support services available from moment of diagnosis
Special centres provide wholistic care teams and umbrella services for
whole family- nuclear and extended.
May be focused on one variation- Autism, Down, CF, Sickle Cell
Assist with practical supports, equipment, web links, education, economics,
access, fundraising to sponsor special projects and equipment, legislation
and policy development, advocacy, research, seamless services across the
lifespan, opportunities to shine and thrive.
Lots of contacts and information on-line. Try some of the websites listed in
your text reference listing.
Present at all levels - community, neighbourhood, local municipality, school
boards, sports groups, to regional networks, to provincial and national plus
international levels of governance and activity
Can you identify the variations.
How would you chart these?
Responsive and sensitive Care what every child and family needs
when there is a genetic variation