Epilepsy and seizure - The University of Hong Kong
advertisement

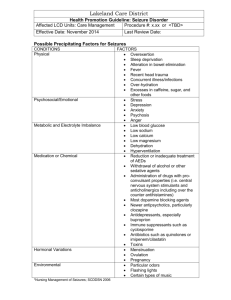
Epilepsy and seizure Specialty Clerkship Student Seminar Group B2 Chan Ying Ting, Purdy Siu Lok Man, Joanne Lee Wai Yip, Jacky OUTLINE 1. 2. 3. 4. 5. 6. 7. Prevalence and genetics Etiology Terminology Classification of seizure Epilepsy syndromes Case studies General management of seizures PREVALENCE IN CHILDHOOD 75% with onset before age of 20 years Prevalence: 4.1/1000 in children up to 11 years old(National Child Development study,1983); 4.71/1000 in children up to 19 years old(Oklahoma study,1989) Incidence: 49/100,000 population Offspring risk for epilepsy to age 20 General population:1% Mother with epilepsy: 6% Father with epilepsy: 2.4% Sibling risk for epilepsy to age 20 General population: 1% Proband with epilepsy: 3% Proband with 1 parent affected: 8% ETIOLOGY Febrile Febrile convulsion Convulsion with fever Intracranial infection Non-febrile Metabolic disturbance Trauma Poisons / toxins / recreational drugs Cerebral dysgenesis / malformation Cerebral damage / cerebral tumor Neurocutaneous syndromes TERMINOLOGY Seizure—transient involuntary alteration of consciousness, behavior, motor activity, sensation and/or autonomic function due to abnormal discharge of cortical neurons; an episodic event, may have provoking factors, e.g. anoxia, alcohol, drugs Convulsion– seizure with prominent alteration of motor activity Epilepsy—a disorder with recurrent seizures(2 or more), unprovoked by a specific event such as fever, trauma, infection, or chemical change, stereotypic Aura—a component of seizure which occurs before consciousness is lost and for which memory is retained afterwards; it localizes attack to the point of origin in the CNS Automatisms--coordinated adapted involuntary motor activity occurring during the state of clouding of consciousness; usually followed by amnesia of the event Tonic seizure: excessive motor outflow, giving rise to a tetanic state of the muscles involved. Atonic seizure: muscle tone drops to a very low values resulting in a sudden fall of the body Clonic seizure: a tonic seizure with periodic interruptions Tonic-clonic seizure: starts as a generalized tonic seizure and then interrupted during clonic phase and ending in complete relaxation. Myoclonic seizure: short involuntary contraction of one or more muscles (local or generalized) CLASSIFICATION OF SEIZURE Seizures Partial Simple Complex Generalized Partial with secondary generalization * ILAE classification of seizures 1981 Absence seizures Myoclonic seizures Clonic seizures Tonic seizures Tonic-clonic seizures Atonic seizures Partial vs Generalized Partial: if only one hemisphere is involved Simple—no impairment of consciousness, features depend on the region of the brain that is affected Complex—consciousness impaired, may have automatisms e.g. chewing, wandering off, dressing, undressing Generalized: most or both hemispheres are involved, loss of consciousness Primary VS secondary Simple Partial Seizures Preserved consciousness (“aura”) Symptoms related to involved brain regionsa Frontal lobe: movement, thought, speech Temporal lobe: memory, speech, smell, taste, abdominal sensations Parietal lobe: body sensations Occipital lobe: vision Complex Partial Seizures Altered consciousness Unresponsive or less responsive, staring Impaired memory after seizure -Automatisms: hand and mouth movements (lip smacking, grabbing) Hypermotor: wild flailing movements (frontal) Generalized Seizures Most have abnormal, unnatural movements Tonic (stiffening) Clonic (repetitive jerking) Tonic-clonic (“grand mal”) Atonic (limp) Myoclonic (irregular jerking, may retain awareness) Atonic (falling suddenly) Absence (“petit mal”): staring, may blink, arrest of activity EPILEPSY SYNDROMES Genetic causes: Familial neonatal convulsions Benign familial convulsions of infancy Benign partial seizures of infancy Febrile seizures Epilepsy syndromes: (1) Infantile spasms(West syndrome) (2) Lennox-Gastaut syndrome (3) Absence epilepsies (4) Juvenile myoclonic epilepsy (5) Benign rolandic epilepsy Seizures Age Clinical Manifestations (years) Development Primary generalized epilepsy Any Generalized tonic-clonic without fever Usually normal Childhood absence epilepsy 3-12 Blank stare with change of facial expression. De novo automatisms,e.g. rubbing face or hands Perseverative automatisms Normal Benign rolandic epilepsy 2-12 Focal seizure with motor and/or sensory manifestations in oropharyngeal region Normal Infantile spasm 3m3yrs Clusters of flexion or extension spasms, loss of visual/social interaction. Atypical spasms like head nodding, shoulder shrugging, eye rolling, facial grimace Usually abnormal Lennox-Gastaut syndrome 2-5yrs Atypical absences, atonic seizures, tonic seizures, sometimes GTC and partial seizures. May evolve from infantile spasms. Developmental delay at onset common but progressive deterioration may occur. (1)Infantile spasms (West syndrome) Onset: between 4 and 6 months of age ‘salaam spasms’ Flexor spasms last 1-2 s and are often multiple, occurring in bursts of 20-30 spasms, frequently on waking Infants will have developmental delay and later learning disability or epilepsy. Treatment: vigabatrin or corticosteroids. (2)Lennox-Gastaut Syndrome Affects children of 2-5 years old Multiple presentation of seizures Later, neuro-developmental arrest or regression and behaviour disorder Treatment: Sodium valproate Poor prognosis (3)Childhood Absence Epilepsy Onset at 3-12 years Peak at 6-7 years Second peak at 11-12 years Females more than males Family history in 15-44% Rarely associated with developmental problems. Can be induced by hyperventilation. Treatment: Sodium valproate Good prognosis with 95% remission in adolescence. Risk of generalized TC seizures is 3040%(increased risk if begin after the age of 8 years) (4)Juvenile Myoclonic Epilepsy (JME) Autosomal dominance with variable penetrance A common cause of tonic-clonic seizures in teenagers and young adults(myoclonus paricularly in morning) Myoclonic seizures precede tonic-clonic seizures by 2-3 years; tonic-clonic seizures typically occur when patient reaches 10-17 years Prognosis excellent but requires lifelong treatment (5)Benign Rolandic Epilepsy Most common partial epilepsy Onset 2-12 years M:F 1.5:1 Usually occuring in sleep-wake transition states 10-13% have a single seizure 20% have frequent seizures 65% nocturnal 15% nocturnal or diurnal 10-20% waking state only Typical presentation: On waking, fully conscious, mouth to one side, salivating and focal twitching of one side of the face Duration 1-2 mins; Child may recall a sensation of numbness, pins and needles or “electricity” in the tongue, gums or cheeks; - Remains conscious but aphasic post-ictally - Secondary generalization may be seen - Remits spontaneously in adolescence; no sequelae - No medication if infrequent seizures. CASE STUDIES History M/9 Normal development until 9 years old Encephalitis at age 9 with coma and 2 generalized seizures P/E: stupor, ?visual hallucinations, ataxia CT and MRI normal Medullobastoma discovered at age 11, died at age 16 Complex partial seizure evolving into secondary generalized seizure Seizure started as complex partial motor seizure because there is impaired consciousness Classical Jacksonian march is observed but it’s not a Jacksonian seizure since it’s not a simple partial seizure Then evolves into a generalized tonicclonic seizure History M/20 Caesarian section, anoxia, hemiconvulsions at birth, normal development Seizure onset again at 9 years old CT atrophic parieto-occipital zone; MRI large right parieto-occipital lesion probably due to birth trauma Tx: carbamazepine, clobazam, vigabatrin, clorzepate, lamotrigine Lives independently with a job Complex partial seizure with automatisms Aura (secs-mins): unresponsive, dreamy (may also have hallucinations, affect changes, déjà vu) Automatism (occur in 90%): turning head to left, lip smacking (basically any continuation of an activity that was going on when the seizure occurred) Alterations of mood, memory, perception (hence complex partial) Posticteral drowsiness: confused and disoriented for minutes afterwards History F/28 Previously healthy, no neurologically relevant diseases or family history Age of onset 7 MRI shows slight dilatation of R lateral ventricle; discrete hyperintense signals in frontal lobes SPECT: low-flow area in L temporal and frontal lobes and R temporal lobe Simple then complex seizure with secondary generalization Starts off as simple partial seizure with autonomic involving ie. epigastric rising sensation Although she is unable to speak, she raises as left hand as if to signal that she can understand This is followed by a complex partial seizure as there is automatism (hand-rubbing and lip smacking) and unresponsiveness Finally there is a tonic-clonic generalized seizure History F/8 Previously health, no neurologically relevant diseases, remote family history of epilepsy P/E normal, neruoimaging not done Frequent daily spells with LOC since 8 interictal EEG: generalized regular 3Hz spike with some polyspikes; normal background activity Absence seizure - typical Onset in childhood Child stops activity, stares, blink/roll eyes, unresponsive Usually lasts 5-10 secs but may occur hundreds of times/day Usually there is an additional feature like automatism, mild clonus, or change in tone (eg. drop attacks) May be induced by hyperventilation History M/17 Newphew of father and son of the newphew both have epilepsy Onset of seizure at age 15, treated with carbamazepine but still has daily convulsions Myoclonic – juvenile myoclonic epilepsy Sudden, brief, generalized muscle contractions Most common type is the juvenile myoclonic epilepsy (Janz syndrome) which occurs after puberty and doesn’t remit with age Also occurs in degenerative and metabolic disease Another type is themyoclonic absence type History M/6 Good past health and no family history P/E: hypotonic muscles CT showed mild diffuse cerebral atrophy Refractory to all available antiepileptic drugs Generalized clonic seizure Clonic seizure is quite rare, even in this caes there is a very short tonic start Differentiate from myoclonic seizures by the sustained rhythmical nature of the jerks History M/14 Good past health, no family history Onset at 2 years 9 months One month before onset, he had severe measles Refractory to all available antiepileptic drugs Generalized tonic seizure This particular case is unusual in that despite the tonic bending posture, the boy keeps on walking without falling History M/7 Normal pregnancy and delivery and no family history P/E: marked ataxia, severe MR, dull, protruded tongue, hypertonia, hyperreflexia, spontaneous mild jerks of limbs CT showed central and peripheral cerebral atrophy Generalized tonic-clonic seizure Many generalized tonic-clonic seizure have more than one tonic and clonic phase Tonic phase: contraction of muscles – flexion/extension of limbs, twitching of eyelids, respiratory muscles in spasm (cyanosis), LOC Clonic phase: violent jerking of face and limbs, tongue biting, incontinence In this case there is a pronounced tonic stretching of the limbs follow by a clonic phase History M/4 Premature birth with injury Febrile convulsions in paternal cousin P/E: sever MR CT showed moderate diffuse cerebral atrophy Generalized Atonic seizure Many seizures in this type of children are a mixture of atonic, tonic, and myoclonic elements Differentiation depends on analysis by combine EEG and EMG to see which element is more predominate GENERAL MANAGEMENT OF SEIZURES Aim To confirm it is a genuine seizure attack Etiology of the seizure attack Epilepsy + classification Convulsion with a febrile illness Simple febrile convulsion CNS infection Severity of the attack / any associated injury Management plan Prognosis Is it a genuine seizure attack? Symptoms before onset of seizure (prodrome) Aura (sensation / motor) Behavioural change (mood / behaviour) Presence of prodrome strongly suggest partial onset seizures Symptoms during the seizure Loss of consciousness? Temporal relationship with other symptoms LOC right from the beginning? Secondary generalization? Other symptoms Vocal symptoms Motor symptoms Respiration Autonomic symptoms Symptoms following seizure Amnesia of the event Confusion / lethargy / sleepiness Headache or muscle ache Transient focal weakness (Todd’s paresis) Nausea or vomiting The child is febrile Simple febrile convulsion? Check for the criteria CNS infection? Ask more on associated symptoms of meningitis / encephalitis Epilepsy? Any provoking factors Trauma Toxin / drug / alcohol consumptions Flashes / sleep deprivation / physical exhaustion Causes Previous CNS insult / developmental milestones Preceding neurological deficits Intracranial SOL / increased ICP Associated symptoms of neurocutaneous syndrome Family history Systemic screening Pediatric history Past health Drug history Birth history Immunization history Developmental history Social history Known history of epilepsy currently on medication Possible causes of breakthrough seizure Poor drug compliance Sleep deprivation Infection / fever Recent change of drug regimen Physical examination Febrile General condition / vital signs septic? Anterior fontonelle pressure / GCS / neck stiffness / kernig’s sign / papilloedema CNS infection? Rash / focal signs of infection source of febrile illness Epilepsy Dysmorphism / head circumference Skin features (adenoma sebaceum / shagreen patch / multiple cafe-au-lait spots / nevus flammeus) Neurological examination any focal neurological deficits Investigations Blood test Sepsis Metabolic derangement Urine toxicology screening (optional) EEG Standard investigations for first unprovoked seizure LP (optional) Only if suspect CNS infectionDrug level (if known history of epilepsy on medication) Imaging (CT / MRI) MRI is a better choice if available Those with Significant cognitive or motor impairment of unknown etiology Unexplained neurological abnormalities Partial onset seizure Suspicious EEG abnormalities Children under 1 year of age Emergency imaging in those with post-ictal focal deficit not resolving / not returning to baseline within several hours after seizure Seizure Precautions Turn child on side Do not restrain- protect child from injury Stay with child Do not put objects in mouth Loosen tight clothes Principles of drug treatment in epilepsy 1. A balance between seizure control and drug side-effects. 2. Presence of 2 or more seizures, should consider drug therapy, especially those with short fit interval(usu. <1 year). 3. Absence seizure and myoclonic seizure, once diagnosed should be treated with drugs. 4. Start with lowest dose monotherapy and titrate upwards until seizure control is attained or side effects are experienced. 5. Choice of drugs depends on type of seizures or epileptic syndromes, age of patient and potential drug adverse effects. 6. If polypharmacy has to be used, beware of drug interactions. 7. In case of well controlled epilepsy, regular clinical supervision is the only essential measure. Type of Epilepsy First line drug Generalized tonic-clonic Sodium valproate Myoclonic Sodium valproate Absence Sodium valproate Partial, complex partial or Sodium valproate partial secondarily Carbamazepine generalized Infantile spasm Vigabatrin Corticosteroid Lennox Gastaut Sodium valproate syndrome Sodium valproate(Epilim) Started at 15-20mg/kg/day in divided doses(max. daily dose: 60mg/kg/day) S/E: sedation, drowsiness, increased appetite and weight, idiosyncratic liver failure Drug interactions: serum levels decrease with carbamazepine, phenobarbital and phenytoin Carbamazepine(Tegretol) Mainly for treatment of partial seizures with and without secondary generalization. Three and sometimes four dose per day are better tolerated than twice daily dosing regimens. Maintenance dose: 10-30mg/kg/day.(Adult dose 600-2400mg/day) Therapeutic serum level: 8-12mcg/ml S/E: visual disturbance (recurrent diplopia, blurred vision), ataxia [rare: lupus-like syndrome, aplastic anemia, liver toxicity] Phenobarbital Dosage is age-dependent Maintenance dose: 2-6 mg/kg/day S/E: sedation in teenagers(but tolerance usually develops), irritability in children(up to 1/3 of cases), hyperactivity, sleep disorders and cognitive abnormalities Drug interactions: Induces P450 Phenytoin (Dilantin) Absorption incomplete and erratic in neonates and young children. Half-life in infants is usually long and variable Maintenance dose: 4-8mg/kg/day Serum therapeutic level: 10-20mcg/ml Side-effects: Cosmetic—gum hypertrophy, hirsutism Toxic level—behavioral change, nausea, emesis, nystagmus,ataxia Serious –pancytopenia, Steven-Johnson syndrome Others--lymphadenopathy Vigabatrin Licensed in UK in 1989 For treatment of refractory partial seizures and infantile spasm Paediatric dose: 40-80mg/kg/day S/E: transient sedation and dizziness, behavioral and emotional changes Drug interactions: Not significant Newer anti-epileptic drugs-S/E Gabapentin— rare: insomnia Lamotrigine—skin rash(3-15%) Topiramate—anorexia, renal calculi Main indications for therapeutic drug monitoring 1. To verify patient’s compliance 2. Presence of “breakthrough seizures” in previously well-controlled cases 3. Persistent seizures despite therapy 4. Suspicion of side effects 5. When several anticonvulsants are being used 6. Use of drugs with narrow therapeutic window 7. Very young age when blood levels fluctuates Discontinuation of drug treatment 1. Duration of treatment varies with different types of seizures and age of onset 2. In most epileptic syndromes, a normal EEG is not a prerequisite for discontinuation of treatment. 3. Epileptic syndroms of lesional origin and those synfromes known to be refractory to treatment should be treated for long periods(more than 5 years). 4. Termination of treatment can be considered after a seizure free period of 2 to 4 years. 5. Gradual withdrawal over a period of 3 to 6 months is advised. Reference(s) Video Atlas of Epileptic Seizures and CD-Rom. Commission on Classification and Terminology of ILAE: 1981. Manual of Child Neurology. The Hong Kong Society of Child Neurology & Developmental Paediatrics. (1st edition). Pages 97110;117-134. Manual for Paediatric Interns and Residents. HKU Department of Paediatrics and Adolescent Medicine. (3rd Edition September 2003).Pages 80-83. Illustrated Textbook of Paediatrics. Tom Lissauer and Graham Clayden. (2nd Edition). Mosby. Chapter 25:365-384. Clinical Guideline on Management of Febrile Convulsion. Hong Kong Journal of Paediatrics(new series) 2002;7:143-151. Thank you!