10th_Anniversary_dm250_v2
advertisement

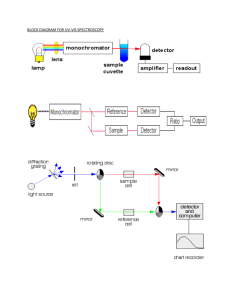
Modelling Metal Foam Formation in Helium Nanodroplets David McDonagh, The Centre for Interdisciplinary Science Project Supervisor: Professor Andrew Ellis, Department of Chemistry ABSTRACT: Conflicting experimental evidence for metal foam formation in aluminium-doped helium nanodroplets highlights the need for a deeper understanding of the favourable conditions for metastable complexes to form. Potential energy surface calculations of dimers were superimposed to simulate the environment of both magnesium and aluminium-doped nanodroplets. UV-Vis calculations were then carried out at predicted local minima and compared to experimental values. In the magnesium system, UV-Vis calculations indicate a preferred metastable state partially stabilised by favourable helium-helium interactions. The same methods applied to the aluminium system found no local minima, and UV-Vis calculations at the suspected distance of one bubble diameter produced a red-shift significantly beyond that found by experiment. These results indicate metastable complexes are unlikely to form in aluminium-doped droplets, and double-peaked UV-Vis spectra for this system is likely a result of a distortion of the bubble cavity. Introduction From a lack of Aln+ clusters in mass spectrometric analyses of aluminium-doped droplets, and a comparable UV-Vis spectrum to the magnesium system, it has been suggested that the same behaviour may be true for aluminium[5]. However, more recent experimental work appears to contradict these findings[6]. Methods To assess the differences in these two systems, a more detailed analysis of the potential energy surface of magnesium-doped nanodroplets was carried out, and UV-Vis calculations were run to assess the likelihood of each metastable state from experimental values. This was then repeated for aluminium-doped nanodroplets. Mg2, MgHe and He2 potential energy surfaces were calculated at the Coupled Cluster level of theory up to double excitations, with triple excitations treated using perturbation theory (CCSD(T)). The augmented correlation consistent polarised valence quadrupole zeta basis set (aug-cc-pVQZ) was used in each case, with the addition of bond functions developed by F. Tao & Pan (1992) for weakly bound systems found to be more consistent with literature values for MgHe. Superimposing dimer potentials was done assuming a spherical bubble cavity, shown in figure 1. UV-Vis spectra for the 3 3p transition were calculated at the Configuration Interaction level of theory up to double excitations, with results shown in table 1. To accurately model the aluminium open-shelled systems, Density Functional Theory (DFT) was used, where the three parameter hybrid functional developed by Becke (1993) with the 6-311+G* Pople basis set was found to be most consistent with literature values. In light of this, UV-Vis spectra were calculated using the same functional and basis set, using Time-Dependent Density Functional Theory (TD-DFT). All calculations were carried out in the Gaussian 03 Software Package, with orbitals rendered in Chemdraw. 0 5 Embedded Mg Dimer Potential -10 -5 Figure 1: The embedded potential energy curve predicted for two magnesium atoms solvated in a helium nanodroplet. Local minima are observed at 10.2 Å and 13.7 Å, with binding energies of approximately 9 cm-1 and 5 cm-1, respectively. -15 Energy (cm-1) Embedded Mg dimer potential Free Mg dimer potential -20 A growing body of research now involves the study of molecular complexes in helium nanodroplets, most notably due to the free rotation and translation of molecules not available in other cooling techniques, and the solvent providing a medium transparent to ultraviolet and infrared radiation[1]. The degree of solvation has been found to differ depending on the molecule used, where the level of interaction between helium atoms can result in a surface or solvated state, in the latter case resulting in clustering of the dopant species[2]. In magnesium-doped nanodroplets, however, an interesting case is observed. Resonant-twoionisation (R2PI) spectra indicate magnesium atoms enter a solvated state, but remain delocalised at a preferred distance, producing a metastable complex or ‘foam’[3]. Further evidence for this is found in potential energy surface and helium density calculations, indicating helium atoms between dopants provide several potential energy barriers, preventing cluster formation[4]. A helium density profile of an embedded magnesium dimer, Przystawik et al. 2008 6 8 10 12 14 16 Interatomic Distance (Å) Mg gas phase (nm) Embedded Mg (nm) Two embedded Mg atoms at 10.2 Å (nm) Two embedded Mg atoms at 13.7 Å (nm) Experimental 285.212 279.0 282.5 282.5 CIS(D) 285.628 278.561 281.420 279.904 Table 1: UV-Vis spectra for the 3p 3s transition. The experimental gas phase value is taken from NIST (2014), and the embedded magnesium data is taken from Przystawik et al. (2008). The embedded environment was simulated using a number explicit solvent molecules based on helium density calculations for this system[4]. Discussion The potential energy curves calculated for magnesium-doped droplets are in good agreement with literature values[3], and several local minima are observed. Through extending this to three dopant atoms, the 10.2 Å minimum was found to be the most favourable metastable state. Calculated UV-Vis spectra for this system appear to confirm this, where transitions with dopants placed at a distance of 10.2 Å show a red shift most consistent with experimental values[3]. Interestingly, in using a solvation shell density calculated from previous work[4], solvent atoms in neighbouring shells are placed at the helium-helium equilibrium distance when dopants are placed at 10.2 Å. This could therefore act in further stabilising this metastable state. The most accurate dimer potentials for aluminium-doped nanodroplets show no evidence of local minima, and calculated UV-Vis values show a redshift significantly beyond that observed experimentally. This work hence finds evidence in agreement with recent mass spectrometric analysis that aluminium atoms cluster in helium nanodroplets[6]. A more detailed analysis of the spectra of aluminium-doped nanodroplets with single dopant atoms is recommended to assess whether the doublepeaked spectrum for this system is due to delocalisation at distances greater than the bubble diameter, or a distortion of the bubble cavity. References: [1] Toennies, J.P., & Vilesov, A. F. (2004). Angewandte Chemie, 43 (20), 2622-2648 [2] Ancilotto, F., & Barone, V. (1997). Chemical Physics Letters, 274 (1-3), 242-250 [3] Przystawik, A. et al. (2008). Physical Review A, 78(2) [4] Hernando, A. et al. (2008). Physical Review B, 78(18) [5] Krasnokutski, S. A., & Huisken, F. (2011). Journal of Physical Chemistry A, 115 [6] Spence, D. et al. (2014). International Journal of Mass Spectrometry, 365